

Abstract
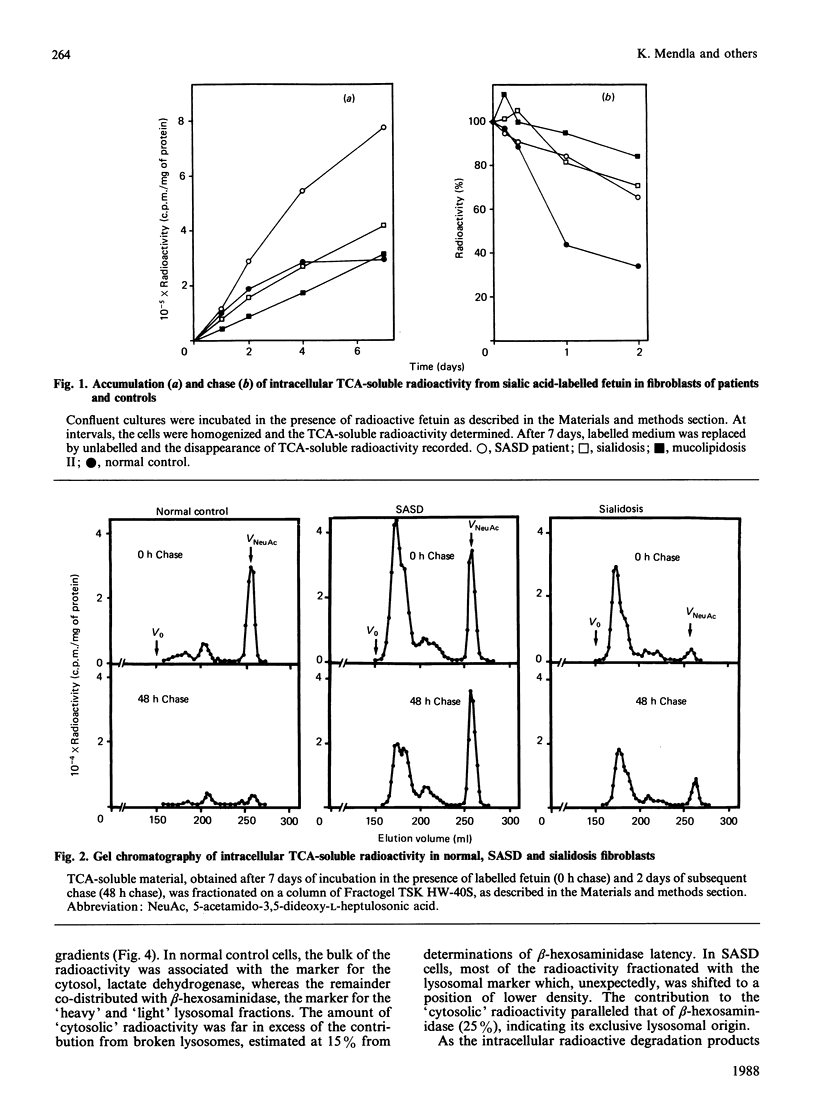
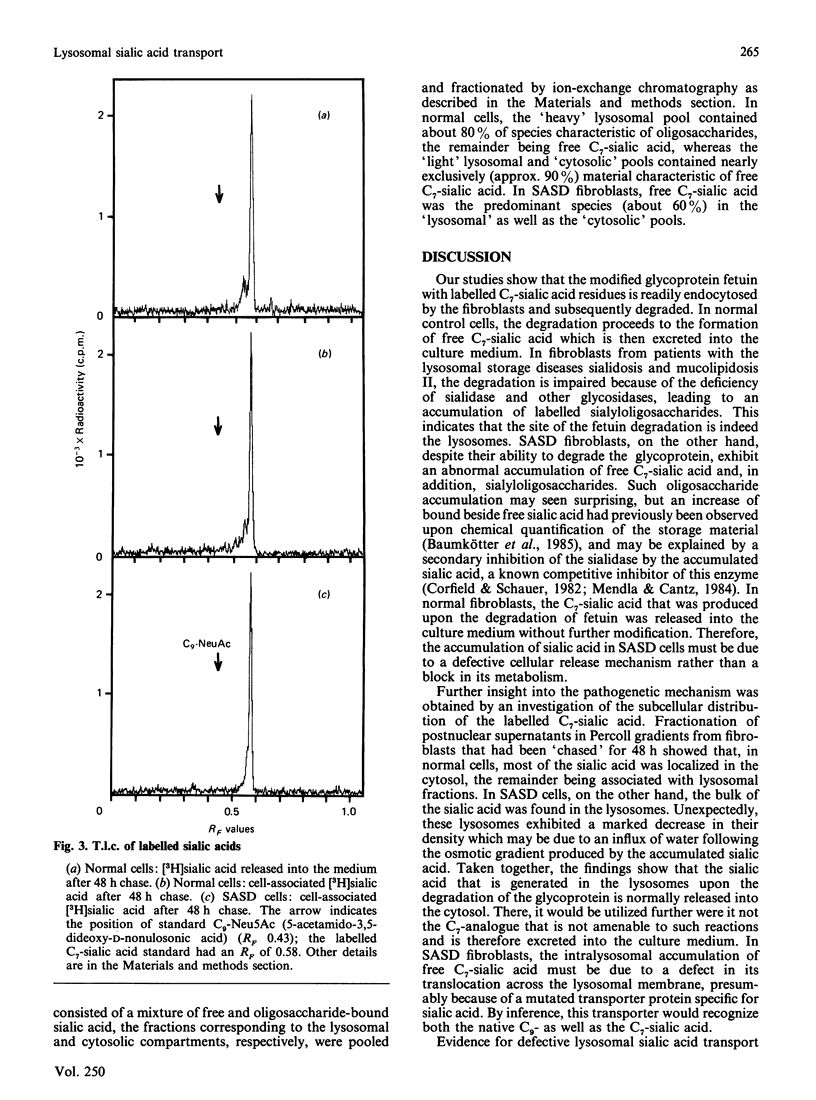
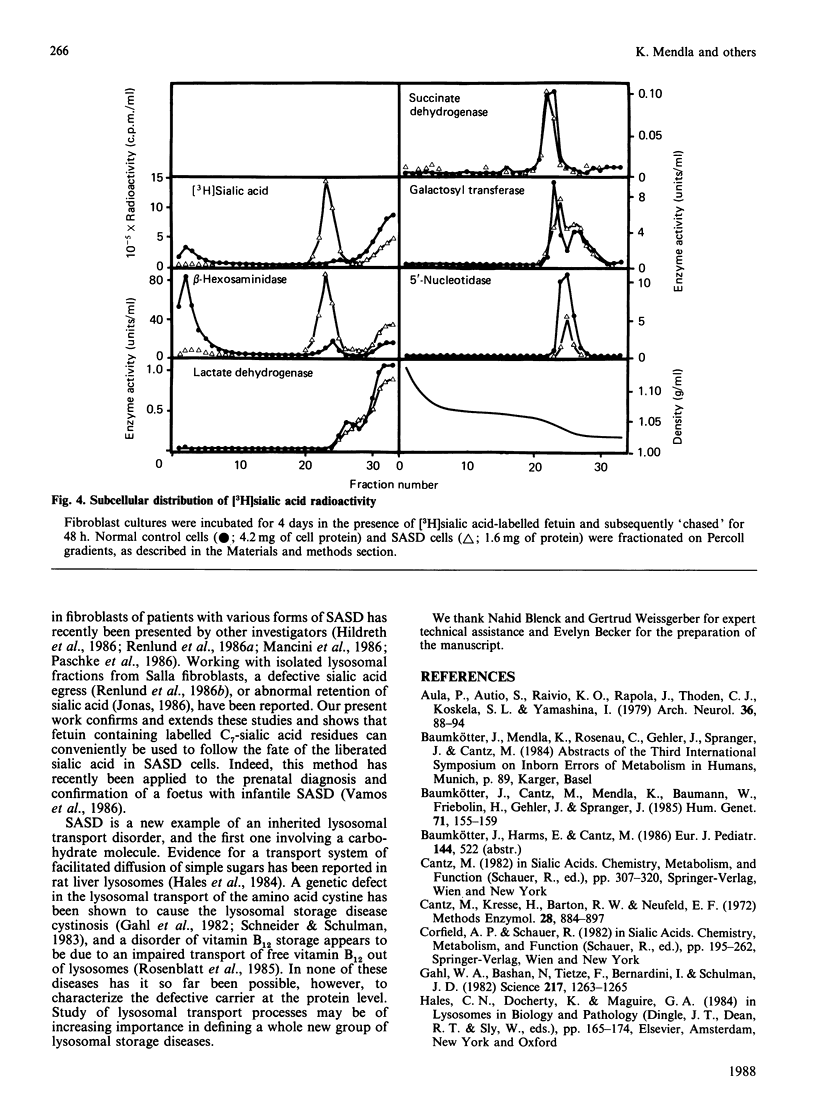
Fibroblasts from patients with sialic acid storage disease (SASD), sialidosis, mucolipidosis II, and from normal controls, were incubated in the presence of the glycoprotein fetuin that was tritium-labelled in its sialic acid residues by the periodate/[3H]borohydride reduction method, and the fate of the intracellular radioactive sialic acid (C7-sialic acid) followed in pulse-chase experiments. The model glycoprotein was readily endocytosed and degraded, more than 90% of the radioactivity being trichloroacetic acid (TCA)-soluble after 4 days of incubation. In all of the patients' fibroblasts, there was an increased accumulation of TCA-soluble radioactivity and, upon chase, a much lower rate of elimination than in normal controls. Gel chromatography of the material from the chase experiment showed that, in normal cells, most of the radioactivity at zero time behaved as free C7-sialic acid. This, as well as material of larger size (sialyloligosaccharides), was very much diminished by 48 h. In cells from two patients with SASD, there were large peaks both in the sialic acid and oligosaccharide positions; whereas the oligosaccharides were somewhat decreased by the end of the chase period, the sialic acid was essentially unchanged. In sialidosis fibroblasts, the radioactive material consisted of oligosaccharides, but very little C7-sialic acid; the elimination of the oligosaccharides was retarded. In normal cells, about 80% of the radioactivity released into the medium after 48 h chase behaved as free C7-sialic acid upon gel chromatography and t.l.c. Subcellular fractionation in Percoll gradients showed that the radioactive C7-sialic acid remaining in normal cells after 48 h of chase was mainly localized in the cytosol. In SASD cells, on the other hand, it was associated with lysosomal fractions which, unexpectedly, exhibited an abnormally low density. Our findings demonstrate that SASD fibroblasts degrade the sialoglycoprotein but, unlike normal cells, accumulate the liberated C7-sialic acid along with sialyloligosaccharides in their lysosomes. The results therefore support the concept of a defective transport system for sialic acid in the lysosomal membrane of patients with SASD.
Full text
PDF



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Aula P., Autio S., Raivio K. O., Rapola J., Thodén C. J., Koskela S. L., Yamashina I. "Salla disease": a new lysosomal storage disorder. Arch Neurol. 1979 Feb;36(2):88–94. doi: 10.1001/archneur.1979.00500380058006. [DOI] [PubMed] [Google Scholar]
- Baumkötter J., Cantz M., Mendla K., Baumann W., Friebolin H., Gehler J., Spranger J. N-Acetylneuraminic acid storage disease. Hum Genet. 1985;71(2):155–159. doi: 10.1007/BF00283373. [DOI] [PubMed] [Google Scholar]
- Gahl W. A., Bashan N., Tietze F., Bernardini I., Schulman J. D. Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science. 1982 Sep 24;217(4566):1263–1265. doi: 10.1126/science.7112129. [DOI] [PubMed] [Google Scholar]
- Hancock L. W., Horwitz A. L., Dawson G. N-acetylneuraminic acid and sialoglycoconjugate metabolism in fibroblasts from a patient with generalized N-acetylneuraminic acid storage disease. Biochim Biophys Acta. 1983 Oct 4;760(1):42–52. doi: 10.1016/0304-4165(83)90122-8. [DOI] [PubMed] [Google Scholar]
- Hancock L. W., Thaler M. M., Horwitz A. L., Dawson G. Generalized N-acetylneuraminic acid storage disease: quantitation and identification of the monosaccharide accumulating in brain and other tissues. J Neurochem. 1982 Mar;38(3):803–809. doi: 10.1111/j.1471-4159.1982.tb08701.x. [DOI] [PubMed] [Google Scholar]
- Hildreth J., 4th, Sacks L., Hancock L. W. N-acetylneuraminic acid accumulation in a buoyant lysosomal fraction of cultured fibroblasts from patients with infantile generalized N-acetylneuraminic acid storage disease. Biochem Biophys Res Commun. 1986 Sep 14;139(2):838–844. doi: 10.1016/s0006-291x(86)80066-3. [DOI] [PubMed] [Google Scholar]
- Jonas A. J. Studies of lysosomal sialic acid metabolism: retention of sialic acid by Salla disease lysosomes. Biochem Biophys Res Commun. 1986 May 29;137(1):175–181. doi: 10.1016/0006-291x(86)91192-7. [DOI] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Lowden J. A., O'Brien J. S. Sialidosis: a review of human neuraminidase deficiency. Am J Hum Genet. 1979 Jan;31(1):1–18. [PMC free article] [PubMed] [Google Scholar]
- Mancini G. M., Verheijen F. W., Galjaard H. Free N-acetylneuraminic acid (NANA) storage disorders: evidence for defective NANA transport across the lysosomal membrane. Hum Genet. 1986 Jul;73(3):214–217. doi: 10.1007/BF00401229. [DOI] [PubMed] [Google Scholar]
- Mendla K., Cantz M. Specificity studies on the oligosaccharide neuraminidase of human fibroblasts. Biochem J. 1984 Mar 1;218(2):625–628. doi: 10.1042/bj2180625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschke E., Höfler G., Roscher A. Infantile sialic acid storage disease: the fate of biosynthetically labeled N-acetyl-(3H)-neuraminic acid in cultured human fibroblasts. Pediatr Res. 1986 Aug;20(8):773–777. doi: 10.1203/00006450-198608000-00015. [DOI] [PubMed] [Google Scholar]
- Renlund M., Chester M. A., Lundblad A., Parkkinen J., Krusius T. Free N-acetylneuraminic acid in tissues in Salla disease and the enzymes involved in its metabolism. Eur J Biochem. 1983 Jan 17;130(1):39–45. doi: 10.1111/j.1432-1033.1983.tb07114.x. [DOI] [PubMed] [Google Scholar]
- Renlund M., Kovanen P. T., Raivio K. O., Aula P., Gahmberg C. G., Ehnholm C. Studies on the defect underlying the lysosomal storage of sialic acid in Salla disease. Lysosomal accumulation of sialic acid formed from N-acetyl-mannosamine or derived from low density lipoprotein in cultured mutant fibroblasts. J Clin Invest. 1986 Feb;77(2):568–574. doi: 10.1172/JCI112338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renlund M., Tietze F., Gahl W. A. Defective sialic acid egress from isolated fibroblast lysosomes of patients with Salla disease. Science. 1986 May 9;232(4751):759–762. doi: 10.1126/science.3961501. [DOI] [PubMed] [Google Scholar]
- Rome L. H., Garvin A. J., Allietta M. M., Neufeld E. F. Two species of lysosomal organelles in cultured human fibroblasts. Cell. 1979 May;17(1):143–153. doi: 10.1016/0092-8674(79)90302-7. [DOI] [PubMed] [Google Scholar]
- Rosenblatt D. S., Hosack A., Matiaszuk N. V., Cooper B. A., Laframboise R. Defect in vitamin B12 release from lysosomes: newly described inborn error of vitamin B12 metabolism. Science. 1985 Jun 14;228(4705):1319–1321. doi: 10.1126/science.4001945. [DOI] [PubMed] [Google Scholar]
- Skoza L., Mohos S. Stable thiobarbituric acid chromophore with dimethyl sulphoxide. Application to sialic acid assay in analytical de-O-acetylation. Biochem J. 1976 Dec 1;159(3):457–462. doi: 10.1042/bj1590457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson R. E., Lubinsky M., Taylor H. A., Wenger D. A., Schroer R. J., Olmstead P. M. Sialic acid storage disease with sialuria: clinical and biochemical features in the severe infantile type. Pediatrics. 1983 Oct;72(4):441–449. [PubMed] [Google Scholar]
- Tondeur M., Libert J., Vamos E., Van Hoof F., Thomas G. H., Strecker G. Infantile form of sialic acid storage disorder: clinical, ultrastructural, and biochemical studies in two siblings. Eur J Pediatr. 1982 Oct;139(2):142–147. doi: 10.1007/BF00441499. [DOI] [PubMed] [Google Scholar]
- Vamos E., Libert J., Elkhazen N., Jauniaux E., Hustin J., Wilkin P., Baumkötter J., Mendla K., Cantz M., Strecker G. Prenatal diagnosis and confirmation of infantile sialic acid storage disease. Prenat Diagn. 1986 Nov-Dec;6(6):437–446. doi: 10.1002/pd.1970060607. [DOI] [PubMed] [Google Scholar]
- Van Lenten L., Ashwell G. Studies on the chemical and enzymatic modification of glycoproteins. A general method for the tritiation of sialic acid-containing glycoproteins. J Biol Chem. 1971 Mar 25;246(6):1889–1894. [PubMed] [Google Scholar]
- Wolburg-Buchholz K., Schlote W., Baumkötter J., Cantz M., Holder H., Harzer K. Familial lysosomal storage disease with generalized vacuolization and sialic aciduria. Sporadic Salla disease. Neuropediatrics. 1985 May;16(2):67–75. doi: 10.1055/s-2008-1052546. [DOI] [PubMed] [Google Scholar]