

Abstract
Germline-targeting (GT) HIV vaccine strategies are predicated on deriving broadly neutralizing antibodies (bnAbs) through multiple boost immunogens. However, as the recruitment of memory B cells (MBCs) to germinal centers (GCs) is inefficient and may be derailed by serum antibody-induced epitope masking, driving further B cell receptor (BCR) modification in GC-experienced B cells after boosting poses a challenge. Using humanized Immunoglobulin (Ig) knockin mice, we found that GT protein trimer immunogen N332-GT5 could prime inferred-germline precursors to the V3-glycan-targeted bnAb BG18, and that B cells primed by N332-GT5 were effectively boosted by either of two novel protein immunogens designed to have minimum cross-reactivity with the off-target V1-binding responses. The delivery of the prime and boost immunogens as mRNA-LNPs generated long-lasting GCs, somatic hypermutation, and affinity maturation, and may be an effective tool in HIV vaccine development.
One sentence summary:
Germline-targeting mRNA-LNP prime-boost immunogens elicit V3-glycan-targeting broadly neutralizing antibody BG18 precursors.
Broadly neutralizing antibodies (bnAbs) to conserved epitopes on the HIV-1 envelope spike protein (Env) could guide the design of a vaccine capable of inducing a protective humoral immune response (1). For most bnAbs, germline precursors lack affinity to Env (2–9). One approach to overcoming this deficit is germline targeting (GT): recruiting potential bnAb germline precursors to germinal centers (GCs) and then introducing a series of increasingly native-like boost immunogens to guide precursor maturation toward affinity to native Env (3, 10–13). This approach requires the design of an array of immunogens to activate precursors and then walk those precursors through the desired evolutionary path. Recently, a trial of the eOD-GT8 60mer, which induced VRC01-class bnAb precursors in the overwhelming majority of participants, provided clinical proof of principle for the germline-targeting priming step (14); several other GT immunogens have entered or are entering clinical trials (15).
The efficacy of GT priming is highly dependent on bnAb precursor frequencies (16–18). An approach to overcoming the limited number of bnAb precursors in the human B cell repertoire using a broad, potent V3-glycan-targeting bnAb, BG18, as a test case was previously reported (19). The V3-glycan epitope, or N332 high mannose patch, which includes part of the CCR5 co-receptor binding site and an associated forest of glycans (20), is a common target for bnAbs isolated from HIV-1 infected cohorts, suggesting a reasonably widespread capacity among neutralizers to develop antibodies to this site (21). V3-glycan-targeting bnAbs isolated from human elite neutralizers, such as PGT121 and PGT128, show substantial neutralization potency and achieve a meaningful level of virus coverage (22, 23). BG18, which was identified from a donor “controller” infected with HIV-1 clade B, is the most potent known V3-glycan bnAb (24). Sequence evolution to achieve the mature BG18 bnAb HCDR3—key to its interaction with the target epitope—may be relatively straightforward despite high SHM, as the maturation process requires no indels (24–26), and even minimally mutated BG18 variants exhibit substantial neutralization breadth (19). In a prior study, to create an expanded precursor target space, features of the interaction between the mature BG18 antibody and its target epitope were characterized and precursors in the human repertoire exhibiting those features were then identified via bioinformatics, extending the potential target pool. Immunogens that target this broader bnAb precursor set could then be generated using directed evolution, ultimately producing a series of GT Env trimers, including N332-GT5 (19)—which is currently being tested in the HVTN144 phase 1 clinical trial (ClinicalTrials.gov Identifier: NCT06033209).
Improved responses through sequential immunization approaches, including neutralization, have been demonstrated in preclinical mouse models for several GT HIV immunogen series (27–30). However, boosting may simultaneously drive the maturation of off-target responses (31). Conceptually, GT immunogens should progressively resemble native Env (13), but which immunogens best accommodate binding to the evolving precursor set and drive antibodies along a path to the mature-like state while avoiding undesirable responses is an empirical question. Humanized mouse models have proven critical to the preclinical development of GT immunogens (11, 16, 30, 32–38), and we have developed a CRISPR/Cas9 method to rapidly generate mice with B cells expressing human Ig V(D)J sequences (39). These models provided support for the use of N332-GT2 as a potential priming immunogen (19); here, we use them to preclinically validate a next-generation prime, N332-GT5 (referred to below as GT5). Furthermore, we demonstrate the efficacy of two novel boost immunogens, B11 and B16, both of which have fewer germline-targeting mutations than the N332-GT5 prime, and which were designed to limit cross-reactivity with V1 loop-directed antibodies.
The transformational possibilities of mRNA vaccines have been made inescapable by the current SARS-CoV-2 pandemic (40, 41). mRNA can be produced rapidly at scale (42, 43), and thus potentially more easily adapted to variants. Here, using our established BG18gH mouse model, we found that an mRNA-LNP N332-GT5/B11 or B16 prime-boost regimen was highly effective. The relative ease of adaptation and production of mRNA immunogens may allow this technology to play an important role in the development of sequential boosters for an HIV vaccine.
RESULTS
N332-GT5 trimer protein efficiently activates BG18 precursors
We made use of our pre-existing humanized mouse model BG18gH (19, 39), in which B cells carry the inferred germline (iGL) heavy chain of BG18 (24), to investigate the immunobiology of N332-GT5-induced activation.
As BG18gH expresses the human heavy chain (HC) in association with a variety of mouse light chains (LC), we wanted to determine which proportion of B cells, if any, could specifically bind N332-GT immunogens. To assay the binding potential of the naïve repertoire in heterozygous BG18gH mice, we analyzed the frequency of GT5-binding B cells from peripheral blood by flow cytometry; in contrast with WT mice, in which GT5 binders were negligible, 17.4% of the naïve B cell repertoire in BG18gH bound GT5 (Fig. 1A–B), comparable to the frequencies established for GT2 in this model (12%) (19). 16.9% of the naïve B cells in BG18gH mice expressed the KI human BG18 HC, associating with diverse murine light chains (Fig. S1A–B). Sequenced GT5 binders near-exclusively expressed the KI human BG18 HC (HBG18gl = 92%) (Fig. S1C). While a variety of native murine LCs were found in conjunction with HBG18gl, IGKV10–96, IGKV1–135, IGKV3–2, IGKV6–15, and IGKV12–44 were most common (Fig. 1C, S1D). We additionally expressed epitope-specific Fabs isolated from naïve B cells and measured their dissociation constants from GT5 and its predecessor GT2: the geometric mean KD was far lower for GT5 (9.3 nM) than GT2 (343 nM) (Fig. 1D). Taken together, these data indicated that the humanized naïve B cells in the BG18gH mouse model were capable of binding GT5, and with higher affinity than GT2.
Figure 1. N332-GT5 trimer protein can specifically and efficiently activate BG18gH B cells.
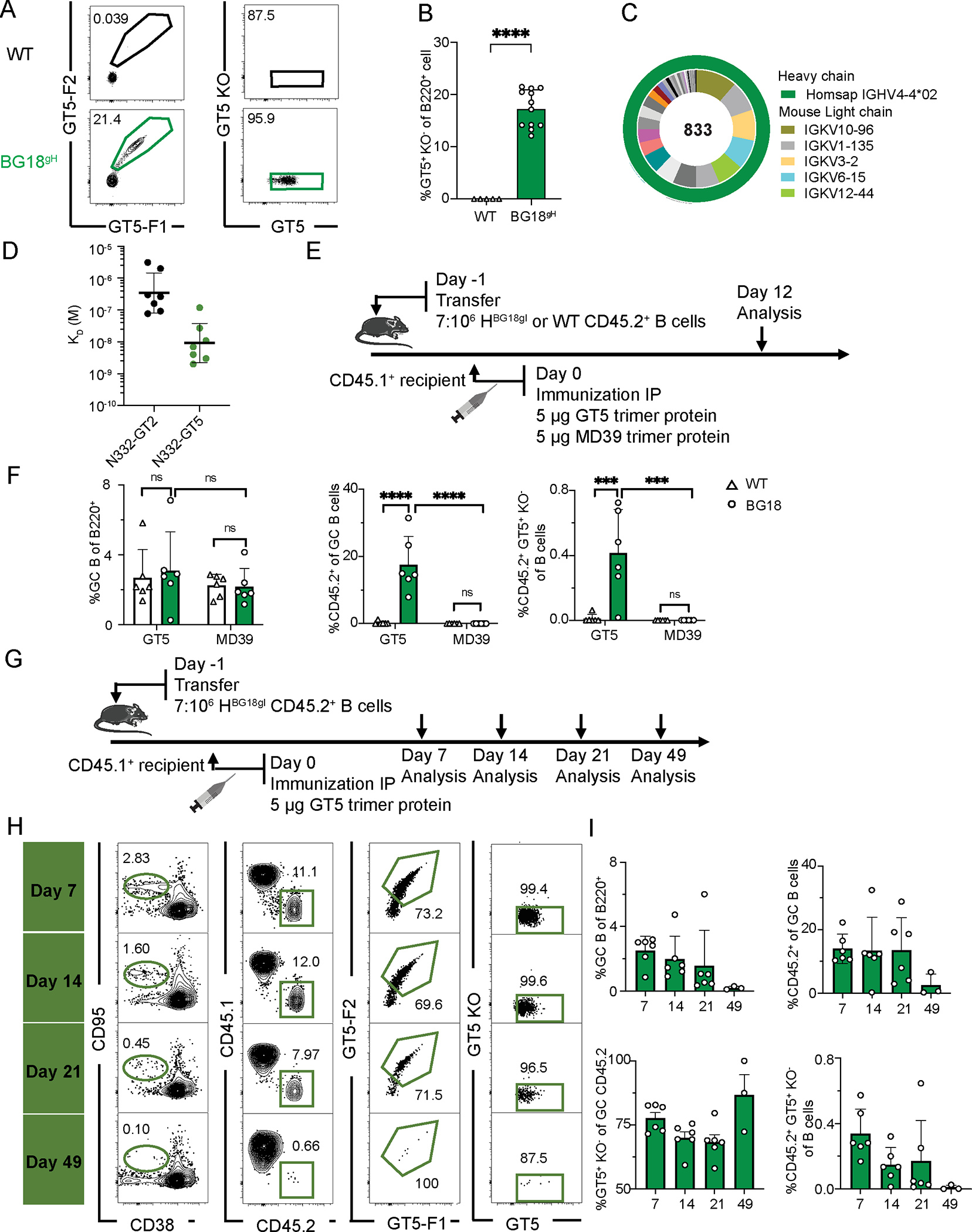
(A) Gating strategy to identify epitope-specific (N332-GT5+/N332-GT5-KO−) B cells in BG18gH and WT mice. (B) Frequency of epitope-specific B cells in non-immunized BG18gH (n=12) and WT (n=5) mice. Welch’s t-test applied; error bars are SD. ****p < 0.0001. (C) Distribution of VH and VL genes in epitope-specific (GT5+KO−) naive B cells in naive BG18gH mice. (D) SPR dissociation constants (KD) for N332-GT2 and GT5 trimer binding to epitope-specific Fabs derived from naive BG18gH cells. Each symbol corresponds to a different Fab and represents one or two measurements. Bars indicate geometric mean and geometric SEM; n=7. (E) Schematic of BG18gH and WT B cell adoptive transfer recipients immunized with GT5 and MD39 trimer protein. Data was collected from one experiment. (F) GC B cells, CD45.2+ B cells in GC and epitope-specific (GT5+KO−) CD45.2+ cells as a percentage of total B cells in each condition 12 dpi WT (GT5 trimer), BG18gH (GT5 trimer), WT (MD39 trimer), BG18gH (MD39 trimer); n=6 in each group. 2-way ANOVA with Bonferroni multiple comparison test applied; bars indicate mean + SD, nsp >0.05, ***p < 0.001, ****p < 0.0001. (G) Schematic showing immunization with GT5 trimer protein. Samples were collected at day 7, 14, 21 and 49 for analysis. At least two independent experiments were performed, and representative data from one is shown. (H) Gating strategy showing GC B cells, CD45.2+ B cells in GC, CD45.2+ GT5 binders in GC and epitope binding specificity 7, 14, 21 and 49 dpi. (I) Frequency of GC B cells, CD45.2+ GC B cells, GT5+KO− cells in GC CD45.2+ cells gated as in (H) and CD45.2+GT5+KO− cells in total B cells 7, 14, 21 and 49 days after immunization with GT5 trimer protein. Each symbol represents a different mouse. Bars indicate mean + SD. n = 3 (day 49); others n=6.
To determine the efficacy of GT5, we used adoptive transfer from the CD45.2 KI BG18gH or WT mouse lines into CD45.1 WT animals to establish a ratio of approximately seven CD45.2 (BG18gH or WT) B cells to every million CD45.1 WT B cells—the frequency previously used to test GT2 in this model (19). As a primary driver of the need for engineered GT immunogens is the lack of human bnAb precursor affinity for native Env (4–9), CD45.1 recipient mice were then immunized with either 5 μg of the HIV-1 Env native-like BG505-MD39 SOSIP (hereafter referred to as MD39) or 5 μg of the GT5 trimer protein; spleens were examined by flow cytometry 12 days post-immunization (dpi) (Fig. 1E). While both immunogens produced GCs (GC, CD38−CD95+) of statistically indistinguishable sizes, only GT5 robustly recruited CD45.2 BG18gH B cells to those GCs (17.3%) (Fig. 1F, left and center). Additionally, a significantly higher fraction of transferred cells were both antigen-positive and negative for the knockout probe (which lacks the N332 binding site) after GT5 immunization (Fig. 1F, right panel), demonstrating that GT5 specifically activates BG18 iGL precursors.
Depending on how the category “BG18 precursor” is defined, frequency in the human repertoire ranges from an estimated 1 in 54 million (Type I) to 1 in 7 million (Type II) (19); with the BG18gH Type I precursor B cells we investigated the comparative effectiveness of 5 μg of GT2 or GT5 trimers at a stringent precursor frequency of 0.7:106, or 1 in 1.4 million (Fig. S2A). While GCs were of comparable sizes, significantly more CD45.2 cells were found in GCs 9 days post-GT5 immunization than post-GT2 (Fig. S2B–C). The GT5 trimer, in line with its higher affinity for naïve BG18gH cells, can more efficiently activate BG18 precursors at lower frequencies.
GT2 multivalent ferritin nanoparticles (NP) were previously tested in BG18gH adoptive transfer experiments in addition to protein trimers (19); we therefore performed a comparative study of BG18gH activation by GT2-NP and GT5-NP (1 μg each) in adoptively transferred mice (7:106 and 70:106) (Fig. S3A). Overall, GT5-NP and GT2-NP have statistically indistinguishable profiles in terms of GC activation and CD45.2 recruitment 9 dpi (Fig. S3B–C). GT5 NP elicited notably higher epitope-specific IgG titers in BG18gH-transferred mice than in WT controls (Fig. S3D), even higher than GT5 trimer protein (Fig. S3E). GT5-NP thus effectively activated BG18 precursors even at low precursor frequencies and doses.
To examine the longer-term GC response to GT5 trimer protein immunization, we immunized BG18gH recipient mice (7:106) with 5 μg of the GT5 trimer, performing analyses 7, 14, 21, and 49 dpi (Fig. 1G). The GC reaction was detectable until 49 dpi, though it was quite attenuated at that point—as was CD45.2+ occupancy, though the majority of those CD45.2+ GCs remained GT5+ throughout (Fig. 1H–I). The GT5 trimer protein and nanoparticle could therefore specifically activate humanized BG18gH B cells at rare frequencies, and that activation was robustly maintained over an extended period post-immunization.
N332-GT5 protein trimer drives affinity maturation in BG18 precursors
The development of mature bnAbs from their germline predecessors requires an extensive process of affinity maturation via class switch and somatic hypermutation (SHM) (10). Having established that N332-GT5 can activate rare BG18gH precursors, we next sought to investigate the affinity maturation process.
Adoptively transferred CD45.1 mice (7 HBG18gl CD45.2: 106 WT CD45.1) were immunized as above (5 μg GT5 trimer protein), and serum IgG titers were analyzed 16 and 42 dpi. In BG18gH recipients, IgG titers were significantly higher than in controls at both timepoints (Fig. 2A, S2D). The serum polyclonal antibody (pAb) responses 16 and 42 dpi were characterized by negative-stain electron microscopy polyclonal epitope mapping (nsEMPEM): both the BG18 and WT mouse serum antibodies approached the V3-glycan epitope of GT5 but used distinct binding poses, similar to observations previously made for GT2 (Fig. 2B, S2E). The polyclonal Fabs from both BG18gH and WT mice displayed off-target binding to the base epitope after GT5 trimer protein immunization, which is frequently observed after BG505 SOSIP protein immunization (Fig. 2B) (44). The serum BG18 antibodies induced by GT2 NP and GT5 NP approaching the V3-glycan epitope appeared to be indistinguishable (Fig. S3F); interestingly, no off-target polyclonal Fabs to the base epitope were detected in nanoparticle-immunized mice (Fig. S3F–G). To investigate the V3-glycan responses in more detail, we performed cryo-electron microscopy EMPEM (cryoEMPEM), as previously described (45), on representative samples from either BG18gH or WT mice and obtained ~3.2 Å and 4.2 Å maps, respectively, of each V3-glycan pAb-trimer complex (Fig. 2C). Overlay of the two maps corroborates the distinct binding angles of WT- vs BG18gH-elicited responses to the V3-glycan epitope (Fig. 2C). Atomic models generated using the maps as constraints revealed that BG18 pAbs contact GT5 protein at the conserved GDIR motif at the base of the V3 loop via long HCDR3s, while the WT pAb-GT5 interaction focuses on the V1 loop and engineered glycan hole (Fig. 2D). Furthermore, HCDR3 of a previously determined atomic model of BG18gl monoclonal antibody agrees well with the side chain densities of the EM map, providing additional evidence that BG18gl B cells are activated by GT5 (Fig. 2E). The HCDR3 interaction with the base of the V3 loop is a key characteristic shared by mature BG18 and typical V3 glycan-targeted bnAbs (20, 25, 46–48).
Figure 2. N332-GT5 trimer protein drives BG18gH somatic hypermutation and affinity maturation.
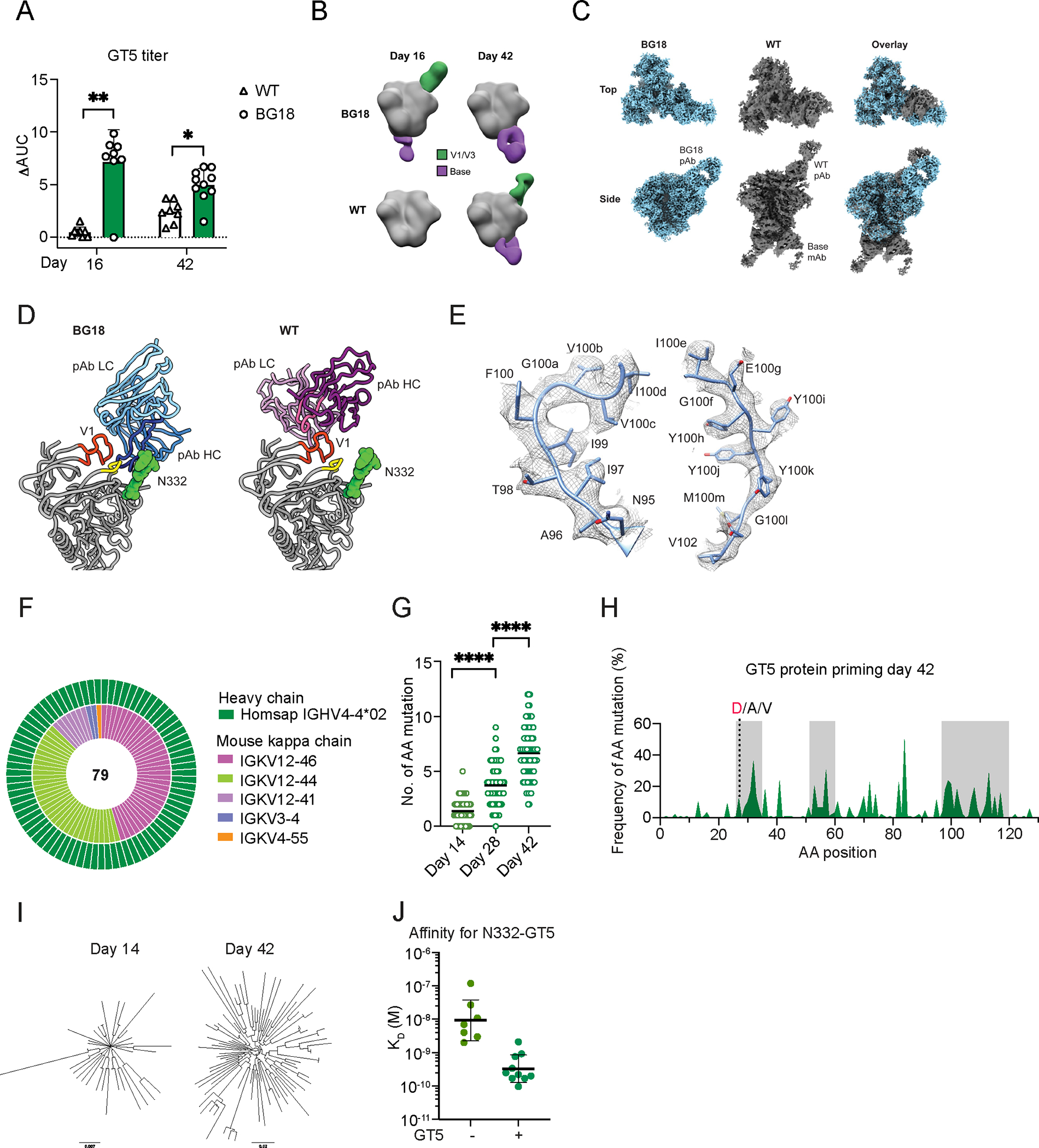
(A) ELISA quantification of N332-GT5-specific IgG 16 and 42 dpi by N332-GT5 protein trimer. ΔAUC (Delta area under the curve) compared for N332-GT5 and N332-GT5-KO. Each symbol represents a different mouse: WT (triangle), BG18gH (circle). Samples are pooled from 2–3 experiments with 8–10 mice per group. 2-way ANOVA with Bonferroni multiple comparison test used; bars indicate mean + SD. *p < 0.05, **p < 0.01. (B) Composite nsEMPEM maps of serum pAb responses in immunized animals, 16 and 42 days after GT5 immunization. Sera were pooled for each timepoint: BG18gH (GT5), n=10; WT (GT5), n=8. (C) Cryo-EM reconstructions of polyclonal complexes presented in (B). pAbs from both timepoints were combined. Estimated resolutions: BG18 ~3.2 Å, WT ~ 4.2 Å. (D) Atomic models of BG18 or WT pAb backbones with their respective epitopes highlighted. V1 (HXB2 133–155) in orange, co-receptor GDIR motif (HXB2 324–327) in yellow. CDRH3 of each pAb highlighted in dark blue (BG18) or bright pink (WT). (E) Cryo-EM map of CDRH3 of BG18 mouse pAb with inferred germline BG18 monoclonal model (PDB 6dfh) relaxed into the density. (F–J) Sequencing and affinity of BCRs from epitope-specific CD45.2+ cells. (F) Nested pie chart of paired single-cell BCR sequences 42 dpi. Outer layer shows BG18 IGHV; inner layer murine IGKV. (G) Heavy chain AA mutations across all sites 14, 28 and 42 dpi. Statistical analysis made using Kruskal-Wallis test. Bars indicate mean; ****p < 0.0001. (H) Per site heavy chain AA mutation frequency 42 dpi. Red letters represent key BG18 mature or mature-like mutation; grey letters show other mutations on the key residue sites. CDRs are boxed in grey. (I) Phylogenetic trees of HCs 14 and 42 dpi. Tree scale (0.007 at left and 0.02 at right) indicates the number of substitutions per site. (J) SPR dissociation constants (KD) for Fabs derived from GT5+KO− naive B cells in non-immunized BG18gH mice and GT5+KO− GC CD45.2+ B cells in BG18gH adoptive transfer recipients 42 dpi. Each symbol corresponds to a different Fab and represents one or two measurements. Bars indicate geometric mean and geometric SEM; n=7 or 10 Fabs. The Kd values for non-immunized mice are reproduced from Fig. 1D for comparison.
All CD45.2 GT5 binders in the GC 42 dpi expressed BG18gH HC, and murine LCs were restricted to a small number of VK genes, with mouse IGKV12–46 and 12–44 predominant at both 14 and 42 dpi (Fig. 2F, S2F). SHM was ongoing throughout the lifespan of the GCs: by 14 dpi, sequenced HCs had acquired an average of 1.4 amino acid (aa) mutations/2.6 nucleotides (nt); by 28 dpi, 3.7 aa (6.5 nt); by 42 dpi, 6.7 aa (10.8 nt) (Fig. 2G, S2G). Mutations were enriched in CDRs (Fig. 2H), suggesting intense selection in the GCs. In the BG18gH HCDR3, the interface contact residues present in the germline sequence were identical (3 of 4) or chemically similar (1 of 4) to the mature BG18 sequence. Therefore, our analysis of SHM in the HCDR3 was mainly to determine if the prime or boost immunogens maintained the bnAb-like aas at the interface. In addition to the HCDR3, the HCDR1 is highly mutated in mature BG18, and these mutations provide essential contacts to the N332 glycan (19). Mutations G27D, one of the three key HCDR1 mutations that interact with the N332 glycan, were detected post-prime (Fig. 2H). Additionally, 60% of the sequences contained polar or charged SHM in the HCDR1, which could potentially form similar interactions to the N332 glycan as those mutations present in mature BG18 (Fig. S5A). SHM drove substantial diversification of BG18gH HCs by 42 dpi (Fig. 2I); this diversification was associated with a substantial affinity gain on the part of epitope-specific Fabs for GT5 (Fig. 2J). In sum, N332-GT5 protein priming drove substantial diversification in BG18 precursors, resulting in a diverse pool of high-affinity variants.
B11 and B16 protein trimers can boost BG18 precursors after N332-GT5 priming
Activation of precursors is only the first step toward bnAb induction; activated precursors must be recruited or maintained in GCs by subsequent more native-Env-like boost immunogens (10–13, 29). The N332-GT5 immunogen has 20 germline-targeting mutations and a V1 loop glycan hole to enhance germline BG18 binding; to usher precursors through the next stage, two boost immunogens were developed, B16 and B11. Both immunogens were designed to have minimal cross-reactivity with V1 loop-directed mouse antibodies. In the design of B16, mammalian display directed evolution was used to identify V1 loop mutations that specifically knocked out the V1 loop-directed serum response in N332-GT2 and N332-GT5 immunized WT mice while maintaining high affinity for a BG18-class antibody isolated 42 days post-prime. This resulted in a BG505 SOSIP MD39-based trimer with eight V1 loop mutations and one V3 loop mutation. For B11, the purpose was to create a more native-like boost and in the process knockout binding to the V1 loop competitor antibodies by restoring the V1 loop glycosylation sites at positions N133 and N137. Multiple germline-targeting mutations were reverted to native; five were retained in the V1 loop and one in the V3 loop to maintain affinity for BG18-class antibodies. While these modifications did prevent binding of N332-GT2 primed mouse antibodies (Fig. 3A–B), glycan analysis later determined that the V1 loop glycans were not well occupied on the recombinant protein (Figure. S4).
Figure 3. Boosting with B11 or B16 protein trimers increases GC-resident BG18gH B cells.
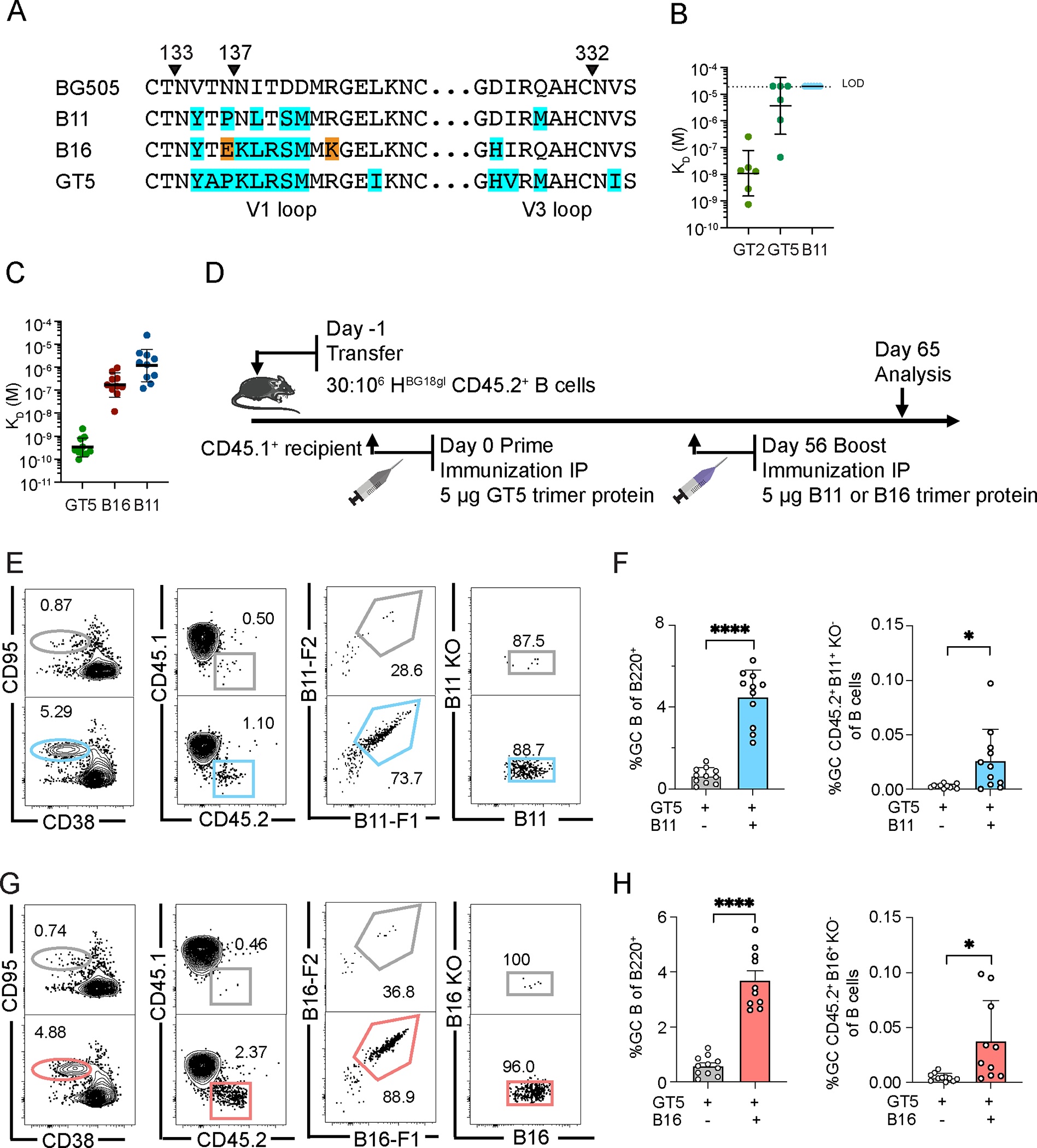
(A) Mutations in B11 and B16 relative to BG505 are highlighted with the equivalent regions on GT5 shown for comparison. Orange indicates mutations that are only present in B16 that were selected for reduced binding to WT mouse serum that was immunized with GT2 or GT5. (B) SPR dissociation constants (KD) for GT2, GT5 and B11 trimer binding to epitope specific (GT2+KO−) Fabs derived from WT GC B cells 10 dpi of GT2 trimer protein; n=6 Fabs. Dashed line indicates limit of detection (LOD). (C) SPR dissociation constants (KD) for GT5, B16 and B11 trimer binding to epitope-specific (GT5+KO−) Fabs derived from GC CD45.2+ B cells 42 days after GT5 protein immunization of BG18gH B cell adoptive transfer recipient mice. Each symbol corresponds to a different Fab and represents one or two measurements. Bars indicate geometric mean and geometric SEM; n=10 Fabs. GT5 prime-only KD values are those presented in Fig. 2J. (D) Schematic showing BG18gH B cell adoptive transfer recipients primed with GT5 trimer protein on day 0, followed by boosting with B11 and B16 trimer protein on day 56. Samples from all treatments were collected on day 65 for analysis. (E) Representative flow cytometry plots of GC B cells, CD45.2+ cells in GC, B11 binders of GC CD45.2 cells and epitope binding specificity on day 65. (F) GC and B11-specific CD45.2+ GC B cells as a percentage of total B cells on day 65. Each symbol represents a different mouse: BG18gH (GT5 prime), BG18gH (B11 boost), n=11. Bars indicate mean + SD. Analyzed using Welch’s t test. *p <0.05, ****p < 0.0001. We have pooled data from two independent experiments. (G) Representative flow cytometry plots of GC B cells, CD45.2+ cells in GC, B16 binders of GC CD45.2 cells and epitope binding specificity on day 65. (H) GC and B16-specific CD45.2+ GC B cells as a percentage of total B cells on day 65. Each symbol represents a different mouse: BG18gH (GT5 prime), n=11; BG18gH (B16 boost), n=10; analysis otherwise as in (F). *p <0.05, ****p < 0.0001. We have pooled data from two independent experiments.
After GT5 priming, the BG18-class precursor antibodies gained detectable affinity for B16 and B11 boosters, though gains were far smaller than for GT5 (Fig. 3C). Given the predictions in the literature that memory B cell reentry to GCs is infrequent (49), we slightly increased the precursor frequency (~4) times to establish a slightly higher but still stringent starting frequency (30:106). Recipient mice were immunized with 5 μg of the GT5 trimer protein; 56 days post-prime, they received a 5 μg boost immunization of either the B11 or B16 trimer protein; analysis was performed at 65 days post-prime (Fig. 3D). Boosted GCs at day 65 were several-fold larger than those found in prime-only animals (B11: 4.49%, B16: 3.70%; prime-only comparison groups were at 0.64% and 0.58%, respectively). Epitope specific CD45.2 B cells in GCs increased significantly as a percentage of total B cell population after boosting (Fig. 3E–H). Boosting by B11 or B16, therefore, was capable of substantially increasing the size of GCs while maintaining BG18 precursor participation.
BG18gH B cells undergo extensive SHM after a B11 or B16 protein boost
The goal of boost immunizations in the GT-HIV-vaccine schema is to maintain or re-recruit B cells to GCs to drive SHM and improve affinity on a route that recapitulates the development of the mature bnAb (11, 13). To determine whether the B11 and B16 protein boosters effectively accomplished this, B11 and B16 CD45.2 binders from a new set of immunizations were isolated for BCR sequencing 77 days post-prime/35 days after boosting (Fig. 4A). All paired LCs found after B11 boosting were IGKV12–46 or 12–44. While these were also found after B16 boosting, IGKV3–5, which was not prevalent after either the GT5 prime or B11 boosting, dominated the post-B16-boost sample; a fourth LC, IGKV3–10, was also observed (Fig. 4B). Priming produced only an average of 6.7 aa (10.8 nt) SHM at 42 dpi, while boosting with B11 led to a mean of 8.5 aa (13.8 nt) and B16 of 9.3 aa (14.0 nt) by day 77 (Fig. 4C–D)—the length of the post-prime CD45.2 responses necessitated the earlier prime collection date. Mutations accumulated in the CDRs after boosting (Fig. 4E). SHM drove substantial diversification in boosted cells by day 77 (Fig. 4F); this diversification led to the acquisition of increased affinity for B11 post B11-boost in approximately half of the Fabs measured, and for a similar portion of Fabs for B16 post-B16-boost (Fig. 4G). The lack of affinity gain in a subset is likely a factor of the heterogeneity of GC B cells throughout the response (50), which can include the maintenance of low-affinity cells (51).
Figure 4. B11 and B16 protein boosters increase BG18gH B cell SHM and affinity maturation.
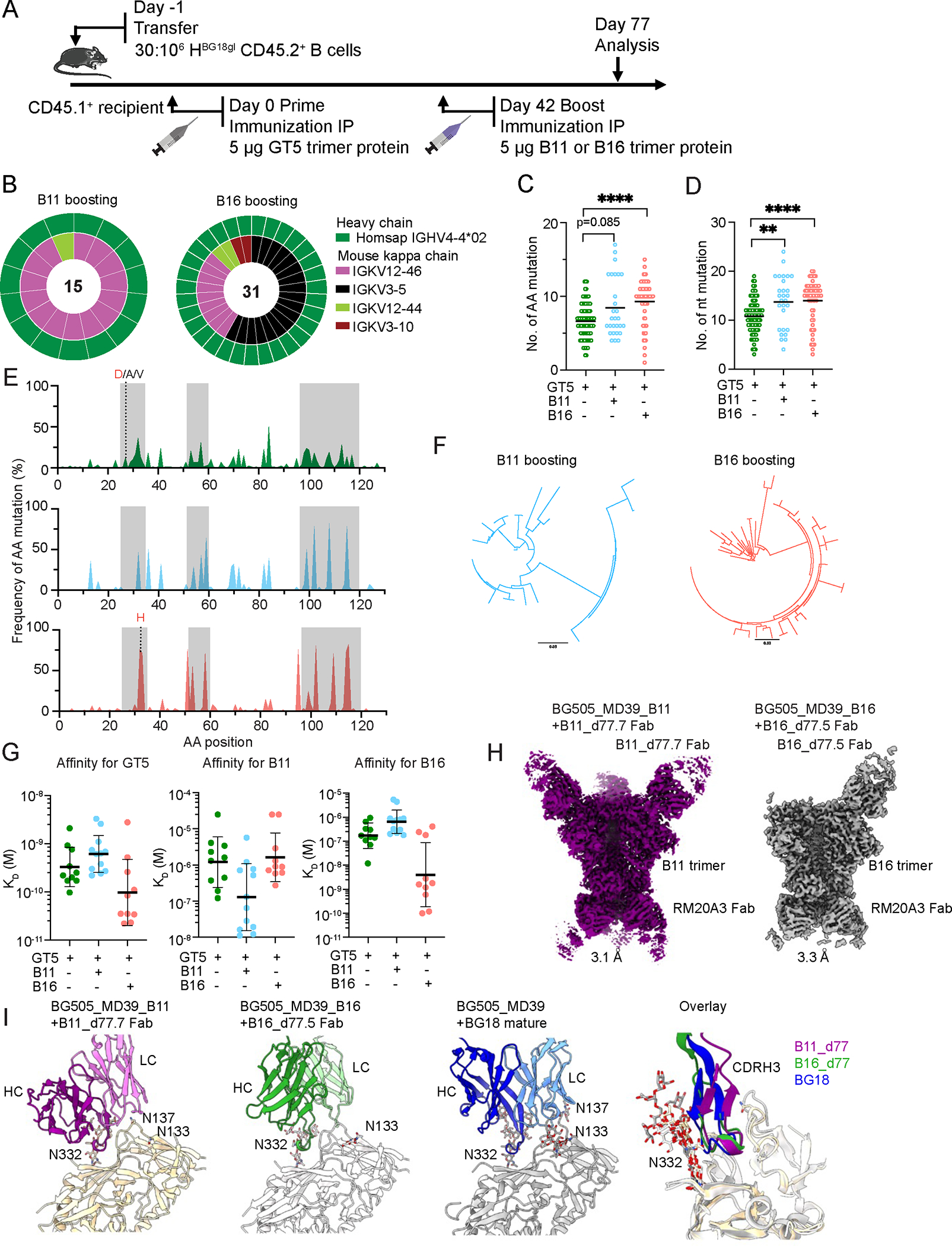
(A) Schematic showing BG18gH B cell adoptive transfer recipients primed with GT5 trimer protein on day 0 and boosted with B11 or B16 trimer protein on day 42. Prime-only samples (green) were collected for analysis on day 42; boost samples (B16 = red; B11 = blue) were collected on day 77. Day 42 prime-only sequences are those presented in Figs. 2G & H and fig. S2G.(B) Nested pie chart showing paired BCR sequences on day 77 from Ag+ (antigen positive) KO− CD45.2+ post-boost B cells. Outer layer represents BG18 IGHV; inner layer represents murine IGKV. (C–D) Heavy chain AA (C) and nt (D) mutations across all sites (source: BCRs sequenced from epitope-specific CD45.2+ B cells on day 42 for prime-only and 77 for boosted mice). Statistical analysis performed using Kruskal-Wallis test. Bars indicate mean. **p <0.01, ****p < 0.0001. (E) Per site heavy chain AA mutation frequency. Red letters represent key BG18 mature or mature-like mutation; grey letters show other mutations on the key residue sites. CDRs are boxed in grey. GT5 prime day 42 (top); B11 boost day 77 (middle); B16 boost day 77 (bottom). (F) Phylogenetic trees of post-boost HCs. Scale (values: 0.03) shows substitutions per site. (G) SPR dissociation constants (KD) for GT5, B11 and B16 trimer binding to Fabs derived from epitope-specific CD45.2+ B cells. Each symbol corresponds to a different Fab and represents one or two measurements. Bars indicate geometric mean and geometric SEM; n=10 or 12. Isolation on day 42 for prime-only and 77 for boosted mice. GT5 prime-only affinity data is as presented in Figs. 2J & 3C. (H) Cryo-EM reconstructions of Fabs derived from epitope specific CD45.2+ BG18 cells at day 77 after either B11 or B16 boost in complex with their respective immunogens. Estimated Fourier Shell Correlation resolutions are listed. (I) Comparison of cryo-EM models of B11 or B16 boost-derived Fabs with mature BG18 (PDB 6dfg). PNGS proximal to the binding epitopes and present in the trimer (B11, B16 or MD39) are highlighted.
Three features of mature BG18 that enable it to neutralize a broad panel of viruses are: 1) its interactions to the conserved patch at the base of the V3 loop primarily through the HCDR3 but also the LCDR2; 2) its ability to form an extensive array of polar interactions to the conserved N332 glycan from both the HCDR1 and HCDR3; and 3) its ability to accommodate diverse V1 loops, which typically have one or more glycosylation sites. The HCDR3-dominant binding mode is encoded in the germline sequence and is maintained after immunization (Fig. S5B), while the other features must be acquired through SHM. To gain structural insight into the progress of affinity maturation we determined cryo-EM structures of two post-boost Fabs in complex with their respective boosting trimers (Fig. 4H). One Fab isolated post-B11-boost (B11_d77.7_Fab), which used a IGKV12–46 LC, had 14 HC mutations and 7 VK mutations and was complexed with the B11 trimer. The other Fab was isolated post-B16-boost (B16_d77.5_Fab), used the B16-specific LC IGKV3–5, had 13 HC mutations and 11 VK mutations, and was complexed with the B16 trimer. The B11_d77.7_Fab complex revealed a typical BG18-like HCDR3-dominant binding mode, but with some rotation of the binding angle to accommodate the mouse kappa LC rather than the human lambda LC (Fig. 4I). Compared to previously solved structures of germline precursor Fab complexes, the B11_d77.7 Fab showed improved polar interactions to the N332 glycan, mostly via backbone atoms and some sidechains such as an S32N mutation in the HCDR1, although the interactions were not as extensive as those in mature BG18 (Fig. S6A–D). Additionally, while the B11 trimer had low V1 loop glycan occupancy and we did not attempt to model the glycans at N133 and N137, partial density for the first N-acetylglucosamine sugar could be observed, indicating that the fraction of N133/N137 that is glycosylated does not clash with the B11_d77.7 Fab (Fig. S6E). Furthermore, alignment of BG505 SOSIP to the B11 trimer indicates that the glycans on a WT V1 loop do not clash with the B11_d77.7 Fab (Fig. S6F). We did not observe interactions to the N392 glycan as can be seen in structures of mature BG18 (19, 25). The effect of SHM in the LC was more difficult to assess, as the human bnAb BG18 uses a lambda LC whereas B11_d77.7 uses a mouse kappa LC. However, the LCDR3 appeared to make important interactions to the V1 loop, such as D93 and W92, which form salt bridge and cation-pi interactions, respectively to R151 in the V1 loop (Fig. S6G). Overall, high affinity variants arose post-boost, and the structure indicated that SHM improved the interactions to conserved glycans and avoided clashes with others, though not to the extent of mature BG18. The B16_d77.5_Fab complex also showed an HCDR3 interaction that overlapped with mature BG18 (Fig. 4I). The IGKV3–5 LC had a more twisted angle of approach compared to the IGKV12–46 LC of B11_d77.7, which was likely caused by its extended LCDR1 conformation (Fig. S6G). Interestingly, the LCDR1 of B16_d77.5_Fab shows partial mimicry with the LCDR2 of mature BG18 with both LCDRs positioned between the V1 and V3 loops of gp120 increasing the 9 buried surface area on the conserved V3 loop (Fig. S6H). As noted above, B16 selects for IGKV3–5 LCs and there are only 2 unique mutations on the B16 V1 loop compared to N332-GT5 and B11, both of which select for IGKV12 LCs. While R151 makes key interactions to the LCDR3 of the IGKV12 LC in B11, it is mutated to Lys in B16 (R151K) and not available to interact with the LCDR3 as it is forming a salt bridge with its V1 loop neighbor E136 (also a unique B16 mutation) (Fig. S6G). Thus, reduced interactions to the IGKV12 LCDR3 may favor binding to the IGKV3–5 LC. Overall, both antibodies isolated post-boost show features in common with mature BG18 but neither antibody shows all of the features required of a bnAb.
N332-GT5 membrane-anchored mRNA priming produces long-lasting precursor activation
The effectiveness of the mRNA-1273 and BNT162b2 SARS-CoV-2 vaccines have brought the promise of mRNA-based vaccines to the fore (40, 41). Having established GT5 trimer protein as a robust activator of BG18gH precursors, we sought to determine whether it could be effectively delivered as a membrane-anchored mRNA. Using a membrane-bound gp151 trimer format reported previously (35, 52), we performed immunization experiments as above with 10 μg GT5 membrane-bound trimer mRNA (GT5-mRNA) and analyzed responses 14, 28, and 56 dpi (Fig. 5A). GT5-mRNA vaccination produced robust GCs, which were robust out to 56 dpi (mean 14 dpi = 2.64%, 28 dpi = 5.65%, 56 dpi = 6.44). GCs were comprised of robust fractions of CD45.2 BG18gH B cells, which drop off at day 56 (mean 14 dpi = 23.9%, 28 dpi = 26.7%, 56 dpi = 7.3%) (Fig. 5B–C). Approximately half of CD45.2s in GCs were epitope specific binders (GT5+KO−) at all dates analyzed (mean 14 dpi = 53.3%, 28 dpi = 58.8%, 56 dpi = 45.0%) (Fig. 5B–C). Therefore, GT5 delivered by mRNA as a membrane-anchored immunogen triggered a vigorous and durable humoral response.
Figure 5. Membrane-anchored N332-GT5 mRNA induces robust and durable humoral immune response in mice adoptively transferred with BG18gH B cells.
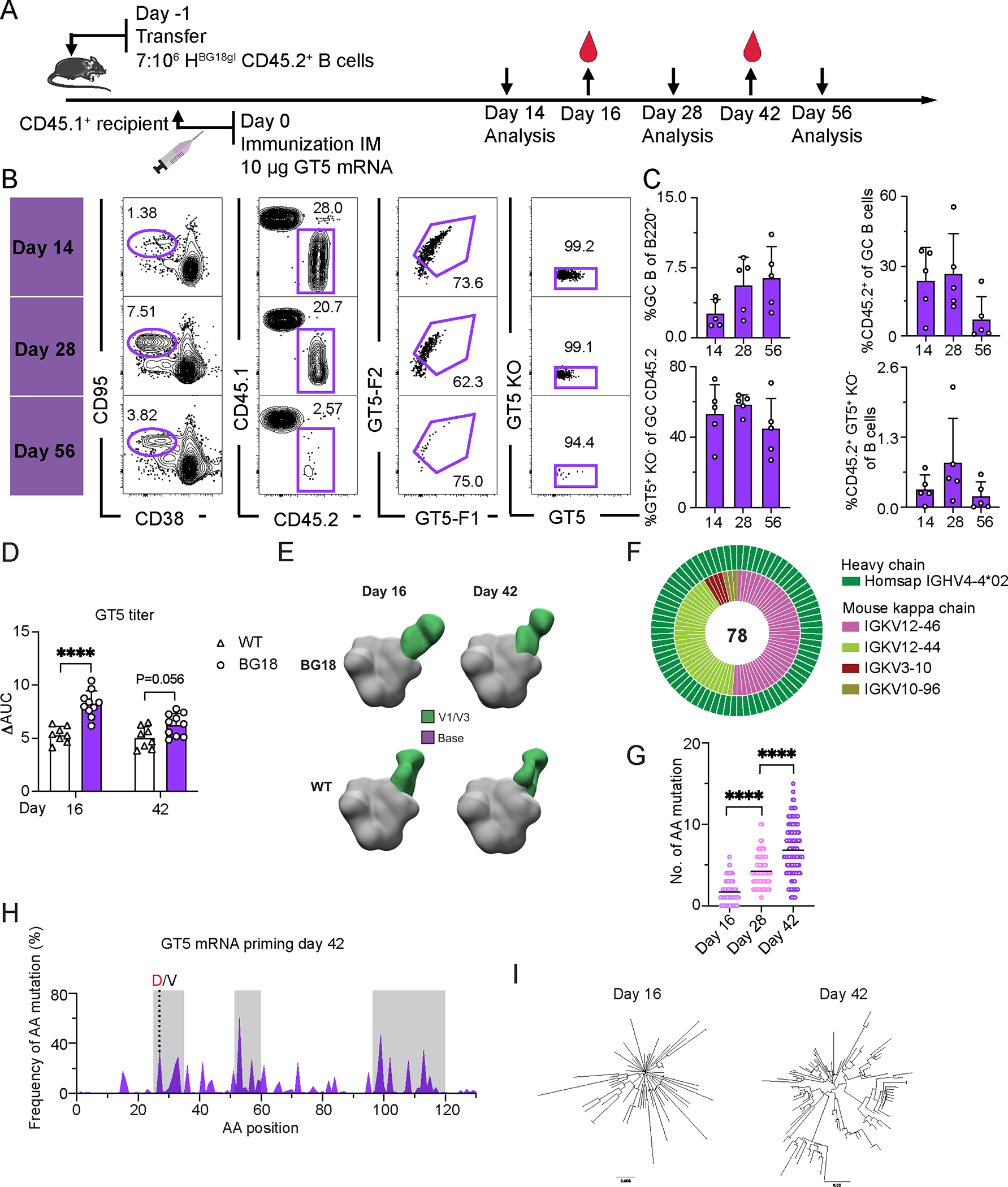
(A) Schematic showing the prime immunization regimen with membrane-anchored GT5 mRNA. FACS analyses were performed on days 14, 28, and 56 from inguinal or popliteal lymph nodes. Serum was collected on days 16 and 42. At least two independent replicates were performed on days 14, 16, 28, and 42; day 56 is from a single run. Representative data from one experiment is shown for flow cytometry below. (B) Gating strategy for GC size, CD45.2+ cells in GC, CD45.2 cells binding to GT5 probe and binding specificity. (C) Frequency of GCs, CD45.2+ B cells in GC, epitope-specific CD45.2 GC B cells and the CD45.2 epitope specific binders in total B cells. Each symbol represents a different mouse. Bars indicate mean + SD. n=5. (D) ELISA quantification of N332-GT5-epitope specific IgG in WT or BG18gH adoptively transferred mice 16 and 42 days after GT5 mRNA immunization. ΔAUC compared for N332-GT5 and N332-GT5-KO. Each symbol represents a different mouse: WT (triangle), BG18gH (circle). Samples are pooled from 2–3 experiments with 8–10 mice per group. Analysis used a 2-way ANOVA with Bonferroni multiple comparison test. Bars indicate mean + SD. ****p < 0.0001. (E) Composite figures from nsEMPEM analysis of polyclonal responses of serum collected from mice adoptively transferred with CD45.2 BG18gH or WT B cells. Antibody targeting V1/V3 (green) and base (purple) colorized. BG18gH (GT5 mRNA); WT (GT5 mRNA). Sera were pooled for each time point: BG18gH (GT5 mRNA), n=10; WT (GT5 mRNA), n=8. (F) Paired BCR V region sequence isolated from CD45.2 epitope specific binders (GT5+KO−) 42 days after GT5 mRNA immunization. Outer layer represents BG18 IGHV; inner layer represents murine IGKVs. The number of sequences is in the pie chart center. (G) Heavy chain AA mutation number across all sites. Statistical analysis was made using Kruskal-Wallis test. Bars indicate mean. ****p < 0.0001. (H) Per site heavy chain AA mutation frequency 42 dpi. Red letters represent key BG18 mature or mature-like mutation; grey letters show other mutations on the key residue sites. CDRs are boxed in grey. (I) Phylogenetic trees of heavy chains 16 and 42 dpi. Tree scale (left, 0.008 and right, 0.03) indicates substitutions per site.
To distinguish the role of the GT5 prime from the delivery format, we also performed immunizations comparing membrane-anchored MD39 and GT5 delivered by mRNA (Fig. S7A). While both primes produced robust GCs at 12 dpi, only GT5 mRNA was capable of recruiting BG18gH precursors to GCs (Fig. S7B). Thus, GT5 mRNA, but not MD39 mRNA, engaged BG18gH precursors for activation.
Both the BNT162b2 and mRNA-1273 vaccines for SARS-CoV-2 generate a transmembrane-anchored spike protein (53, 54); to determine what effect of membrane anchoring might have on immunogenicity in our system, we repeated the membrane-anchored mRNA immunizations but included a direct comparison to soluble GT5 mRNA (Fig. S8A). GC reactions were equivalent in mice primed with either soluble or membrane-anchored GT5 (Fig. S8B–C). CD45.2 recruitment to GCs also did not vary significantly on the soluble–membrane-anchored axis (Fig. S8B–C). Both GT5 mRNAs elicited higher GT2-specific IgG titers in BG18gH B cell adoptive transfer recipients than WT mice 14 dpi (Fig. S8D). There did not, therefore, seem to be a substantial difference between the membrane-bound and soluble forms in BG18 iGL precursor priming.
We therefore returned to the membrane-bound GT5 immunogen to determine its effects on affinity maturation. Serum analysis 16 dpi found GT5 epitope-specific IgG titers were significantly higher in BG18 recipient mice than WT; by 42 dpi the difference was no longer significant (Fig. 5D, S7C). As was the case with the GT5 protein prime, nsEMPEM analysis found that serum antibodies from BG18gH and WT mice at 16 and 42 dpi approached the V3-glycan epitope with different binding poses; in contrast to the protein prime, off-target binding to the base epitope was not detected in serum after GT5-mRNA immunization (Fig. 5E). The most commonly detected murine LCs among the binders changed from 16 to 42 days, suggesting some selection (Fig. 5F, S7D), and the two most prevalent murine LCs at 42 days were identical to those 42 days after the GT5 protein prime (Fig. 5F and 2F). HC SHM progressed over time (16 dpi = 1.7 aa/2.6 nt; 28 dpi = 4.2 aa/7.0 nt; 42 dpi = 6.8 aa/10.6 nt) (Fig. 5G, S7E), accumulating mutations in CDRs, and the key CDR1 mutation G27D was also substantially enriched (Fig. 5H). At 42 dpi, HCs had diversified substantially (Fig. 5I).
Thus, mRNA-delivered membrane-anchored GT5 was immunogenically similar, but potentially preferable, to protein trimer: it generated long lasting GCs and a similar mutational fingerprint, but the nsEMPEM results suggested less off-target binding to the base.
An mRNA prime followed by an mRNA boost generates a long-lasting humoral response
Having established both the efficacy of the B11 and B16 protein trimers as post-GT5 booster immunogens, as well as the improved immunogenicity of GT5 mRNA priming relative to GT5 protein trimers, we next investigated the response to an mRNA-delivered prime-boost regimen with membrane-anchored trimers, establishing a higher-frequency recipient model as with the protein boosters (Fig. 6A). GCs were robust in boosted mice at day 58 (B11 boost = 2.31%, B16 = 2.11%, vs GT5 prime only at 0.30 % or 0.34%, respectively), and maintained until day 78 (B11 = 1.44%, B16 = 0.74%, vs. prime only at 0.14% or 0.15%) (Fig. 6B–E). We then analyzed antigen binding among CD45.2 GC B cells; two apparent subpopulations were detected after B16 boosting (Fig. 6D), which may be attributable to alternate probe binding by BCRs using different murine light chains. Relative to the prime, B11 and B16 boosts increased the number of antigen-specific GC CD45.2 B cells at both day 58 and day 78, though these increases were significant for both boosts only on day 58 (Fig. 6B–E). mRNA-LNP is thus an effective alternate delivery system for prime-boost regimens.
Figure 6. An mRNA prime followed by an mRNA boost generates a long-lasting HIV humoral immune response.
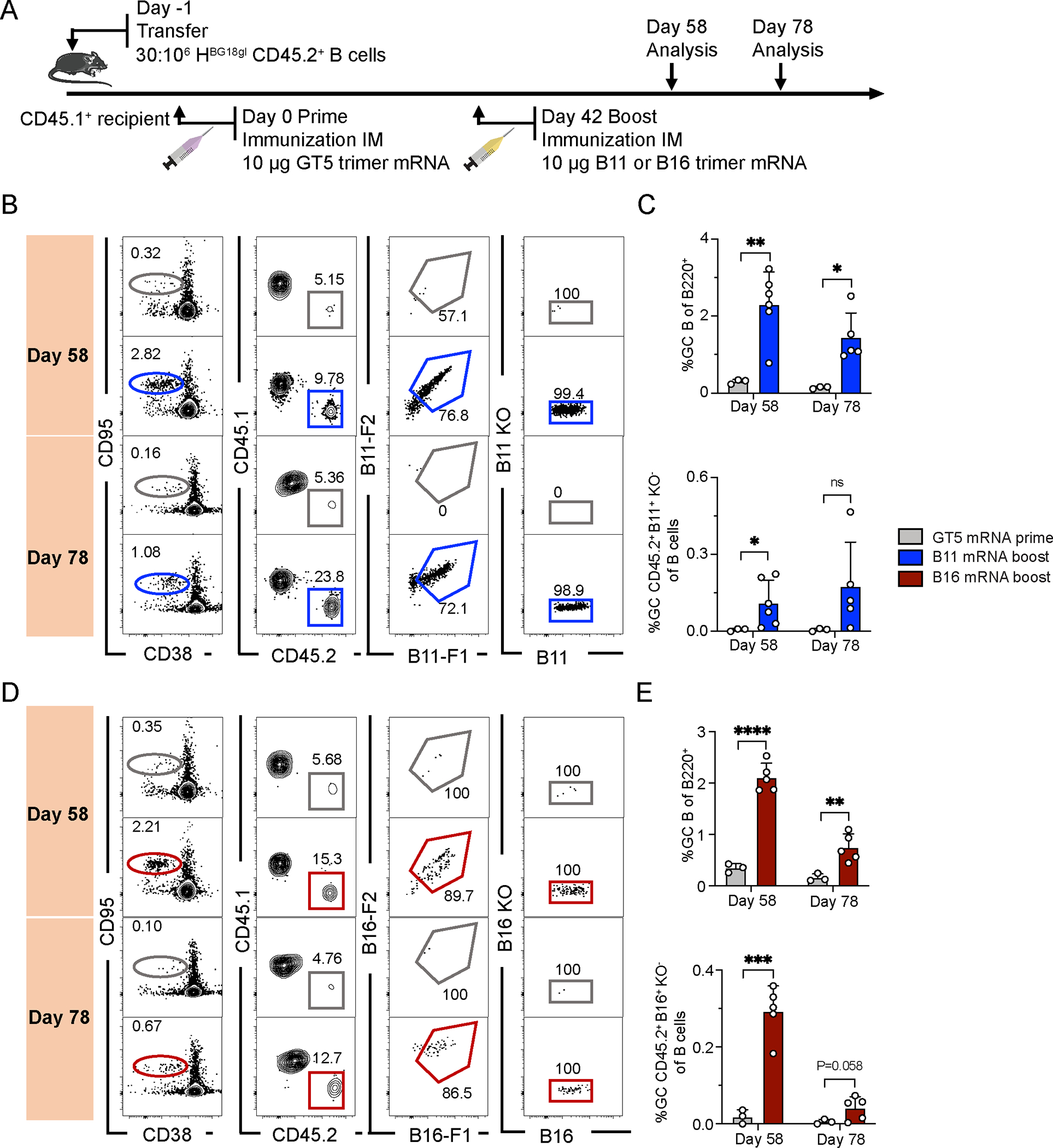
(A) Schematic of BG18gH B cell adoptive transfer recipients primed with GT5 mRNA on day 0, followed by boosting with B11 and B16 mRNA on day 42. Samples were collected on days 58 and 78 for analysis. Data was collected from one experiment. (B) Gating strategy for GC, CD45.2 cells in GC, B11-binders of CD45.2 cells in GC and epitope-binding specificity. (C) Quantification of GC size and B11-specific CD45.2+ GC B cell binders in total B cells. Each symbol represents a different mouse: BG18gH (GT5 mRNA prime), n=3; BG18gH (B11 mRNA boost), n=5 or 6. (D) Gating strategy for GC, CD45.2 cell in GC, B16 binders of CD45.2+ cells in GC and epitope-binding specificity. (E) Quantification of GC size and B16 specific CD45.2+ GC B cell binders in total B cells. Each symbol represents a different mouse: BG18gH (GT5 mRNA prime), n=3; BG18gH (B16 mRNA boost), n=5. Multiple t-tests without consistent SD assumptions used in C and E. Bars represent mean + SD, nsp >0.05, *p <0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
mRNA prime-mRNA boost regimen produces SHM accumulation and affinity maturation
To characterize the diversification driven by mRNA prime/mRNA boost sequences, we isolated CD45.2+ B cell epitope-specific binders 36 days post-boost (day 78) for single-cell BCR sequencing. LC usage was restricted to a subset of mouse kappa chains (Fig. 7A). HCs exhibited higher SHM frequencies than after GT5 mRNA priming alone (prime only, at day 42 = 6.8 aa/10.6 nt; boosting, day 78 B11 = 11.3 aa/16.5 nt; B16 = 12.3 aa/18.7 nt) (Fig. 7B–C); CDRs were, as noted after protein boosting, enriched for aa mutations (Fig. 7D). In addition to the known bnAb HCDR1 mutations, other mutations to polar and charged residues were common in the HCDR1. For example, the S32N mutation, which interacts with N332 in the B11_d77.7 Fab-B11 trimer complex, was found in all groups of mice and occurred at higher frequencies in the B11-mRNA (65% of sequences) and B16-mRNA (43%) boost groups than the GT5 mRNA prime group (20%) (Fig. S9A). The HCDR3-dominant binding mode is encoded in the germline sequence and maintained after immunization (Fig. S9B). This suggests there may be more common ways of forming interactions with the N332 glycan than the known BG18 bnAb mutations.
Figure 7. B11 and B16 mRNA boosters drive BG18gH SHM and affinity maturation.
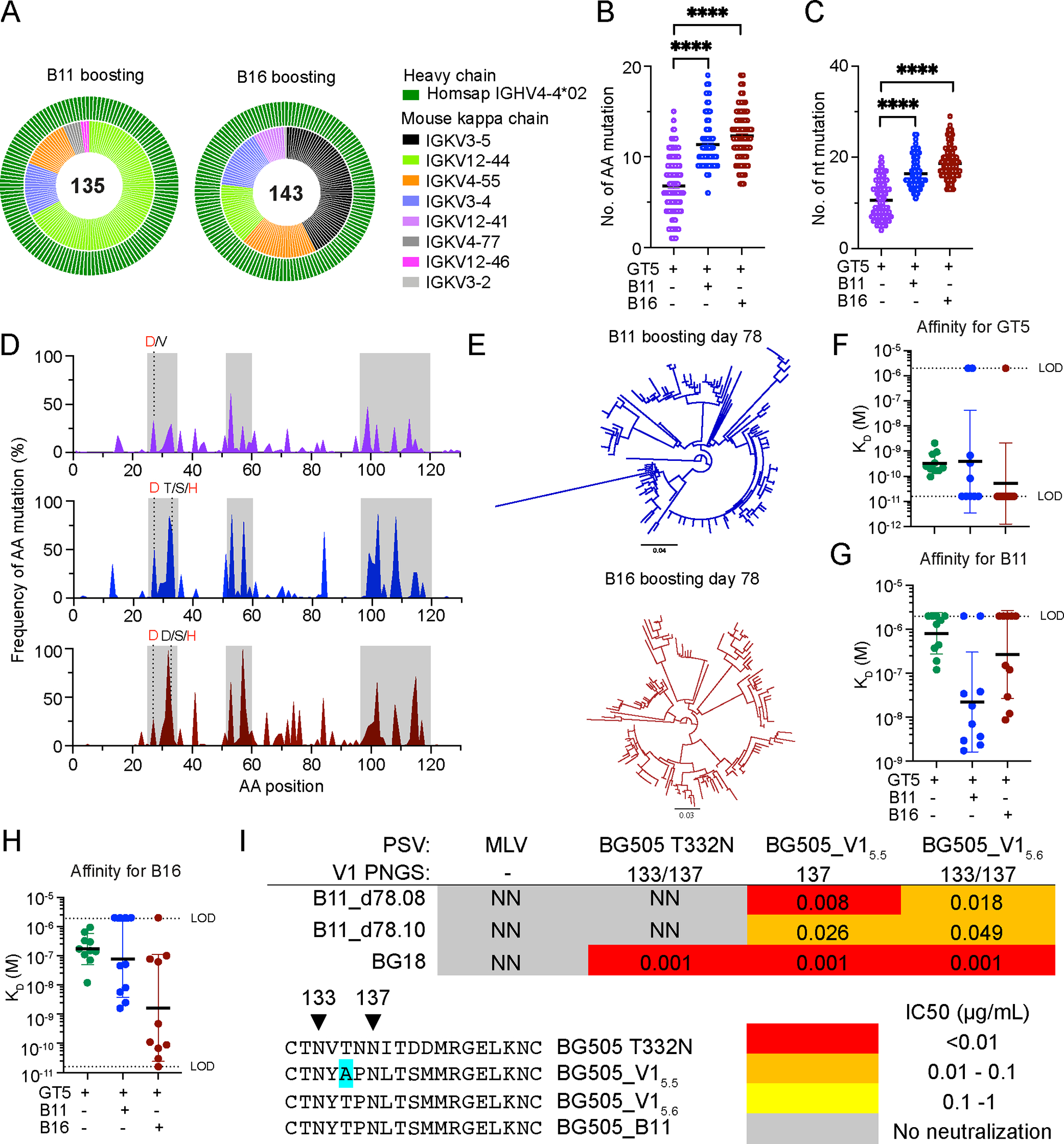
(A–G) BCRs isolated from epitope specific CD45.2+ B cells 42 days after GT5 (GT5+KO−) mRNA prime and 36 days after B11 (B11+KO−) or B16 (B16+KO−) mRNA boosting (day 78). GT5 mRNA prime day 42 shown in purple; GT5 + B11 mRNA day 78 in blue; GT5 + B16 mRNA day 78 in red. (A) Day 42 prime-only BCR sequence data is as presented in Figs. 5G & H and S7E, and shown here for comparison. Outer circle represents BG18 IGHV; inner layer murine IGKVs. (B–C) Heavy chain (B) AA and (C) nt mutations across all sites. Analyzed using Kruskal-Wallis test; bars indicate mean. ****p < 0.0001 (D) Heavy chain per site AA mutation frequency. Red letters represent key BG18 mature or mature-like mutation; grey letters show other mutations on the key residue sites. CDRs are boxed in grey. (E) Phylogenetic trees of HCs. Tree scale (0.04 at top and 0.03 at bottom ) indicates substitutions per site. (F–H) SPR dissociation constants (KD) for GT5 (F), B11 (G) and B16 (H) trimer binding to Fabs derived from epitope specific CD45.2+ B cells of BG18gH B cell adoptively transferred recipient mice 42 days after GT5 protein prime or 36 days after B11 and B16 mRNA boosting. Each symbol corresponds to a different Fab and represents one or two measurements. Bars indicate geometric mean and geometric SEM; n=10; dashed lines indicate limit of detection (LOD). Prime-only affinity data used for comparison is as presented in Figs. 2J, 3C, and 4G. (I) Neutralization potency (IC50) of antibodies elicited 36 days post B11 mRNA boost against native (BG505 T332N) and V1 loop-modified pseudoviruses. IC50 is reported as the mean IC50 of two biological replicates. The positions of V1 loop PNGS on the pseudoviruses are indicated in the table. The sequences of the modified V1 loops are shown.
As with protein boosting, SHM drove substantial diversification (Fig. 7E), and the affinity maturation that resulted from said diversification was also substantial (Fig. 7F–H). We tested two of the antibodies isolated 36 days post B11 mRNA boost (B11_d78.08 and B11_d78.10) that had high affinity for B11 (~2 nM KD) for their ability to neutralize V1 loop modified pseudoviruses based on the BG505 isolate. BG505_V15.6 contains the B11 V1 loop; BG505_V15.5 has the same V1 loop, except the N133 glycosylation site has been removed by mutagenesis while the rest of the Env is matched to the BG505_T332N isolate. Thus, the BG505_V15.6 Env is more native-like than the B11 boost immunogen as B11 has the V3 germline targeting mutation Q328M in addition to V1 loop mutations. Both antibodies neutralized the two V1 modified pseudoviruses, but they were not able to neutralize the WT BG505_T332N virus (Fig. 7I)., indicating that the mRNA-boosted antibodies are functional but have not yet obtained the quality of SHM required to neutralize a fully native pseudovirus.
Therefore, an mRNA prime/mRNA boost sequence produced directionally similar, but slightly more extensive, SHM: the median number of day 77 aa mutations following a protein boost was 7 for B11 and 10 for B16 (Fig. 4C), vs 11 and 12 mutations, respectively, on day 78 of the mRNA regimen (Fig. 7B). This resulted in higher affinity gains (Geometric mean: 22.0 nM for B11 and 1.6 nM for B16; Fig. 7G–H) relative to a protein/protein regimen (127.3 nM for B11 and 4.1 nM for B16; Fig. 4G). Nonetheless, native pseudovirus neutralization will require further BCR modification.
DISUCUSSION
The incredible diversity of HIV (55), and the series of unlikely coevolutionary events underpinning the development of the known HIV bnAbs (56, 57), are major impediments to vaccine development. HIV bnAb germline precursors generally lack affinity for the HIV Env, and they have accumulated an unusually high degree of somatic hypermutations (3, 58). In this study, we evaluated the efficacy of germline-targeting immunogen N332-GT5—which was effective in macaques (59) and is now being tested in a clinical trial (HVTN144; NCT06033209)—and its first boost candidates, B11 and B16. We found that N332-GT5 could initiate the activation and expansion of the BG18 germline precursors, and that B11 and B16 could begin the shepherding of those activated precursors along the path to target the native Env spike, including when the immunogens were delivered as mRNA.
The underlying biology of GT prime-boost, and thus the route necessary to achieve it, remains opaque. Fundamentally, it is unclear as to whether the high rates of SHM present in mature bnAbs are achieved through 1) re-recruitment of GC-experienced memory B cells (MBCs) to GCs or 2) the “refueling” of ongoing GCs by increasing the availability of T cell help, thus enhancing retention, expansion, and further BCR modification for those B cells recruited during the prime (60). Secondary GCs that form after re-challenge seem to primarily recruit naïve B cells (49); among MBCs, those that have accumulated more mutations or undergone class-switch recombination may be more strongly disfavored for entry to secondary GCs (61–63), though this is not universally observed (64, 65). Furthermore, the affinity, concentration, and epitope-specificity of circulating antibodies are all known to affect whether and which B cells enter GCs (62, 66–69), and how and when they exit (70, 71). In our system, continuing SHM was observed post-boost despite the fact that high-affinity antibodies to the same epitope can block at least naïve BG18gH B cells from GC participation (69). We cannot confidently distinguish between the contributions of re-recruitment and refueling at this time, but in a scenario where boosting occurred through re-recruitment of MBCs to GCs, either the BG18 antibody blockade only affects naïve B cells, or the epitopes presented by the prime and boost immunogens, altered by glycosylation and GT mutation number, were sufficiently distinct to allow passage and avoid the stasis of “original antigenic sin”; in a refueling scenario, circulating antibody feedback must have been insufficient to terminate ongoing GCs, though our data does not rule out potential effects on differentiation by circulating antibody. Of note, our findings are in line with recent clinical trial results for the eOD-GT8 60mer GT immunogen, where SHM levels clearly suggested further maturation of GC-experienced B cells after boosting (14).
The generally immune-evasive glycosylation of HIV-1 has been a source of frustration for vaccinology (72)—thus the introduction of the V1 loop glycan hole in N332-GT5 to ease binding to precursors; ultimately, however, a BG18-like bnAb will need to accommodate V1 loop glycosylation. The starting HCDR3 length in our knockin model system is likely an important determinant of the efficacy of boosting with B11 and B16 immunogens, in which some glycosylation sites are restored in V1. Our nsEMPEM assay demonstrated that WT and BG18 lineage-derived serum antibodies approached the V3-glycan epitope at distinct angles after N332-GT5 priming. Furthermore, the high resolution structure obtained after protein boosting revealed that the BG18-lineage antibody contacted the V3 loop via its long HCDR3—specifically approaching the Gly-Asp-Ile-Arg (GDIR) motif, a key feature shared by most bnAbs targeting the V3-glycan epitope (73)—whereas the WT antibody instead bound the V1 loop—likely due to the fact that WT mice have a shorter mean HCDR3 length, distributed in a narrower range, than humans (74)—or mouse models bearing humanized BCRs. Importantly, the structural analysis demonstrated that modifying the V1 loop and removing key glycans makes the region more immunogenic—including in WT mice—but the engineered affinity for BG18gH is high enough to enable BG18-class responses to out-compete the V1-loop specific, non-BG18-class responses. There was, however, non-competing off-target binding to base observed after the protein trimer prime in both the BG18-lineage and WT samples.
Notably, the nsEMPEM data after mRNA prime indicated no detectable off-target binding to base. This decreased off-target response may be due to the limited exposure of the base when the Env spikes are presented on a cell membrane; membrane-anchored SOSIP mRNA design could, therefore, potentially focus the antibody response more on the V3-loop epitope (35, 52). As we and others have previously observed, circulating antibodies to an antigen may in some instances improve responses by forming immunocomplexes and enhancing antigen trafficking (75), but may also mask the epitope or cause antigen consumption (69). Furthermore, membrane-bound mRNA has additional points in its favor, such as the strong immunogenicity of antigens presented and anchored in the cell membrane and their contributions to immunological synapse formation (76–78). Furthermore, mRNA may lead to antigen production in mice for days after administration (79), and this and other forms of extended antigen availability, such as slow delivery, are associated with robust and diverse responses (80, 81). However, the availability of antigen after protein and mRNA immunization is difficult to directly compare. Furthermore, the doses of mRNA used here in mice are, by weight, much higher than those used in humans in the closest point of comparison, the two SARS-CoV-2 mRNA vaccines (40, 41). Though higher antigen concentrations can weaken selection and allow non-preferred competitors to persist in GCs (82), in the case of GT immunization, this relaxation of competitive exclusion and robust T cell help may allow otherwise less competitive bnAb precursors more chances to enter the GCs, and increase the likelihood that they stay in.
This points to a broader issue in the comparison of mRNA-LNPs and protein trimers—we have a far more comprehensive understanding of the effects, in terms of antigen availability in the LN, of varying dosages and differing formulations for protein immunogens than we do for mRNA-LNPs. Nonetheless, several other recent studies in the field have supported the use of mRNA. During the preparation of our manuscript, another murine study reported using repetitive mRNA delivery of trimer-nanoparticles to induce antibodies capable of neutralizing some heterologous HIV viruses (83); additionally, a multiclade HIV-1 env-gag mRNA vaccine has shown promising results in rhesus macaques (84). Finally, regarding the boost stage, whether the state of glycosylation is comparable after membrane-bound expression to the under-glycosylation observed in the B11 protein trimers is both unknown and critical to understanding whether the boost phase response stringently selects for the desired binding posture.
In sum, we delivered the germline-targeting N332-GT5 trimer and two novel boost immunogens, B11 and B16, in both protein and mRNA-LNP formats and, using a stringent knockin model, found that prime-boost regimens deploying either protein trimers or mRNA-LNP could drive affinity maturation. The immunogenicity of mRNA-LNP and the potential decrease in base off-target binding suggest that mRNA prime-boost strategies may resolve some of the major challenges of HIV vaccine development.
Materials and Methods:
Table of reagents
| Antibodies | Supplier | Cat number |
|---|---|---|
|
| ||
| BV786-B220 | BD Bioscience | 563894 |
| Alexa Fluor 594-B220 | BioLegend | 103254 |
| Alexa Fluor 700-Gr1 | BioLegend | 108422 |
| Alexa Fluor 700-CD3 | BioLegend | 100216 |
| Alexa Fluor 700-F4/80 | BioLegend | 123130 |
| BV510-CD38 | BD Bioscience | 740129 |
| PE-Cy7-CD95 | BD Bioscience | 557653 |
| PerCP Cy5.5-CD45.1 | BioLegend | 110728 |
| PE-CD45.2 | BioLegend | 109808 |
| BV786-CD45.2 | BD Bioscience | 563686 |
| BUV395-IgG1 | BD Bioscience | 742481 |
| APC-Cy7-IgM | BioLegend | 406516 |
| BV650-IgD | BioLegend | 405721 |
|
| ||
| Commercial chemicals | ||
|
| ||
| LIVE/DEAD™ Fixable Blue | Invitrogen | L23105 |
| Dead Cell Stain Kit | ||
| Alexa Fluor 647-Streptavidin | BioLegend | 405237 |
| BV421- Streptavidin | BD Bioscience | 563259 |
| Alexa Fluor 488-Streptavidin | BioLegend | 405235 |
| Alexa Fluor 594-Streptavidin | BioLegend | 405240 |
| Sigma Adjuvant | Sigma | S6322 |
| ACK lysing buffer | Lonza | BP10–548E |
Experimental model and subject details
Healthy adult male or female C57BL/6J (CD45.2+/+) mice heterozygous for the BG18gH KI, generated as previously reported (39), and 8–12 week-old male B6.SJL-Ptprca Pepcb/BoyJ mice (CD45.1+/+) purchased from Jackson labs were used in this study. Mice were housed at the animal facility, with free access to food and water, controlled temperature and a 12:12 h light-dark cycle. Mice were not involved in previous procedures and were drug and test naive. The mouse maintenance and experiments were performed following the approved protocols by the Institutional Animal Care and Use Committee (IACUC) of MGH, an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International-accredited facility, under Animal Study Protocols 2016N000286 and 2016N000022.
B cell adoptive transfer
Lymphocytes from BG18gH or WT mice were isolated from spleen. After collection, spleens were crushed in FACS buffer and filtered with a 70 μm cell strainer, after which the cell suspension was subjected to pan B isolation kit (Miltenyi Biotec) according to the manufacturer’s protocol. Finally, the B cell pellets were suspended in DPBS. Cell viability and concentration were analyzed by the cell counter, then naive B cells were transferred to CD45.1 recipient with a specified frequency via intravenous (IV) injection.
Development of B11 and B16
For the design of B11 we started from the BG505 SOSIP MD39 trimer and added five V1 loop mutations from N332-GT5 but left the two V1 loop glycosylation sites (N133 and N137) intact (CTNYTPNLTSMMRGELKNC). Additionally, the Q328M mutation was incorporated at the base of V3 to enhance binding to the BG18 HCDR3. For the development of B16, four mammalian display combinatorial libraries were assembled. The starting template sequence was based on the BG505 SOSIP MD39 trimer but with a V1 loop derived from N332-GT1/2 and the N133 glycosylation site was restored (CTNYTPKLRSNMRGELKNC). Then for library 1, the degenerate codons NNK, NNK, AWS, SAC were introduced at positions 139, 140, 141, 325, respectively. For library 2, the degenerate codons NNK, AWS, NNK, SAC were introduced at positions 136, 141, 151, 325, respectively. For library 3, the degenerate codons NNK, NNK, AWS, SAC were introduced at positions 138, 139, 141, 325, respectively. For library 4, the degenerate codons NNK, AWS, NNK, SAC were introduced at positions 139, 141, 151, 325, respectively. Each library was integrated into an rtTA3G-knock-in 293T cell line using lentivirus and the four library cell pools were combined to form one larger library. As a positive selection reagent, cells were stained with 200 nM d42.11, an antibody isolated 42 days after immunization with N332-GT2 in mice adoptively transferred with BG18gH knock-in B cells (19) As a negative selection reagent, cells were stained with 1 uL of mouse serum isolated from wt mice 42 days after immunization with either N332-GT2 or N332-GT5. After three rounds of fluorescence-activated cell sorting the library DNA was isolated and sequenced; it was found to be highly enriched in one sequence derived from library 2 that contained the V1 loop sequence (CTNYTEKLRSMMKGELKNC) and H325 in the V3 loop. All immunogen amino acid sequences are displayed in the supplemental figures (Fig. S10, S11).
Protein expression and purification
C-terminal His-tagged BG505-based trimer variants cloned into the pHLsec vector were expressed and purified essentially as described previously (52). DNA grown in DH5alpha cells was maxi-prepped with a BenchPro 2100 (Thermo Fisher Scientific). The purified DNA constructs were transfected by incubating 300 uL PEI max with 70 ug DNA and 25 ug protease encoded DNA into 15 mL OptiMEM. After 30 min, the mixture was added to 293F cells grown in 293 Freestyle media at a density of 1 million cells/mL. After 6 days, the supernatant was clarified by centrifugation and the trimer was purified from the supernatant using a HIS-TRAP column, starting with a wash buffer (20 mM Imidazole, 500 mM NaCl, 20 mM Na2HPO4) and mixing with elution buffer (500 mM Imidazole, 500 mM NaCl, 20 mM Na2HPO4) using a linear gradient. The trimer fraction was collected and further purified on an S200Increase 10–300 column (GE) in HEPES buffered saline (HBS) (10 mM HEPES, 150 mM NaCl). The oligomeric state of the trimers were then confirmed by size exclusion chromatography-multi-angle light scattering using the DAWN HELEOS II multi-angle light scattering system with Optilab T-rEX refractometer (Wyatt Labs). The trimers were frozen in thin-walled PCR tubes at 1 mg/mL using liquid nitrogen and stored at −80°C. The GT2-NP was expressed and purified as described previously (19) and the GT5-NP was expressed and purified using similar methods but with co-transfecting GT5-NP and furin DNA in a 2:1 ratio. For biotinylated probes, proteins were expressed with both a his-tag and avi-tag, purified by Ni++ affinity chromatography followed by SEC, and biotinylated using a BirA biotin-protein ligase reaction kit (Avidity, Cat# BirA500) according to the manufacturer instructions. Paired HC and LC Fab variable region sequences were gene synthesized and inserted into human Fab HC constant region expressing vector pFabCW and human kappa expressing vector pCW-CLig-hk. Fabs were expressed in 500 mL FreeStyle™ 293F cell cultures. For transfection, 300 μg of HC and 150 μg of LC plasmids were mixed with 225 μg polyethylenimine (PEI; 1:3 DNA:PEI ratio) in 5 mL of Opti-MEM™ reduced serum medium (Thermo Fisher Scientific, Cat# 31985070) for 30 min, then added to 293F cells. Supernatant was collected after 5–6 days. Harvested supernatants were filtered through 0.45 or 0.25 μm membrane filters and batch bound to CaptureSelect CH1-XL Affinity resin (Thermo Fisher Scientific, Cat# 1943462005). Resin was washed with PBS, and captured Fabs were eluted with 50 mM NaOAc pH 4.0, buffer exchanged into 1× PBS, and concentrated using a 30k MWCO concentrator. For expression of IgG the HC variable region was cloned into the pCW-CHIg-hG1 vector. Transfection was carried out as described above but batch binding occurred overnight at 4°C to Protein A resin (Thermo Fisher Scientific, Cat# 20334) while on a rocker. Unbound supernatant was allowed to flowthrough, and the resin was washed with PBS until protein A280 reading of the flowthrough measured by a nanodrop reached background levels. Protein A bound IgG was eluted with 0.1 M Glycine pH 2.7. Eluted mAbs were buffer exchanged into 1× PBS and concentrated using a 50k MWCO concentrator (Millipore).
Immunization
5 μg trimer protein or 1 μg nanoparticle protein immunogens diluted in PBS at the volume of 100 μl/per mouse, were mixed with Sigma adjuvant at the ratio of 1:1, then shaken on the shaking table for 30–60 mins. Each mouse was given 200 μl immunogen/adjuvant complex via intraperitoneal (IP) injection. 10 μg mRNA lipid nanoparticle (LNP) immunogen was diluted in PBS at the volume of 100 μl/per mouse. Each mouse was given 100 μl mRNA immunogen via intramuscular (IM) injection in the thigh muscles of the two side hind limbs.
Protein probe preparation
For flow cytometric probe binding, monomer protein probes (GT5, B11, and B16) were biotinylated by BirA enzymatic reaction (Avidity, Inc) with Avi site according to the manufacturer’s protocol. Knockout probes which lack the N332 binding site were designed and generated analogous to the respective WT probes. Biotinylated monomeric probes were pre-reacted with fluorescently labeled streptavidin (SA-488, SA-647 or SA-BV421) in a 4:1 molar ratio in independent tubes for at least 30 mins and then combined with fluorescently labeled antibodies for flow cytometry staining.
Flow cytometry
Single-cell suspensions were generated by mechanical dissociation of whole spleens or lymph nodes. ACK buffer was used to remove red blood cells and then cell pallets were suspended in FACS buffer (2% FBS/DPBS). Cell viability was stained with Live/Dead Blue in PBS at 4°C for 15 mins and then cells were Fc blocked (clone 2.4G2, BD Biosciences) in FACS buffer at 4°C for another 10 mins. Cells were preincubated with freshly fluorescently labeled protein probes for 20 mins before the cocktail of surface antibodies merged and stained for another 20 mins at 4 °C. Samples were acquired and events were recorded on a BD LSR Fortessa for flow cytometry analysis. Cell sorting was done on a BD FACS Aria Fusion or Aria II at a flow rate of < 2000 events/second for single cell plate sorting or ~4000 events/second for tube sorting using an 85 um nozzle. Data was analyzed by FlowJo software (Tree Star, Inc.). B cells were single-cell sorted into 96-well plates, rapidly frozen on dry ice, and stored at −80 °C before processing.
BCR Sequencing
After individual CD45.2+ IgD− IgM− epitope specific B cells were sorted, the transcript was amplified through RT-PCR. Briefly, first strand cDNA synthesis was acquired using SuperScript III Reverse Transcriptase (Invitrogen) according to manufacturer’s protocol. Two rounds of nested PCR were performed as 25 μl reactions with HotStarTaq Master Mix (QIAGEN), using IgG- and IgK-specific primer pools and thermocycling conditions described previously (85). For BG18gH cells, the forward primers specific to the leader sequence of the BG18 HC genes was used (5’ GCTGGATTTTCCTTGCTGCTAT 3’). PCR products were run on a E-Gel Precast 2% Agarose Gels with SYBR Safe (Thermo Fisher Scientific) and wells with bands of the expected size were submitted to GENEWIZ company for Sanger sequencing. HC products were sequenced using the HC reverse primer (5’ GCTCAGGGAARTAGCCCTTGAC 3’) from the second PCR reaction. The LC was sequenced using the LC reverse primer (5’ TGGGAAGATGGATACAGTT 3’) from the second PCR reaction.
Reads were quality-checked, trimmed, aligned, and analyzed using the Geneious software (Biomatters Ltd, New Zealand). Human/mouse Ig gene assignments were carried out by IMGT/V-QUEST (http://www.imgt.org).
10x Genomics single-cell BCR analysis
Spleens collected from naive BG18gH mice were crushed and filtered, and then cell suspensions were subjected to pan B isolation kit (Miltenyi Biotec) according to manufacturer’s protocol. Enriched B cells were stained with fluorescently labeled probes for 20 mins and then incubated with CD45 hashtags and antibodies against surface markers for 30 mins at 4°C. Afterwards, cells were thoroughly washed and suspended in FACS buffer with DAPI. 1500–2000 live cells were sorted from each sample and about 10,000 in total in a reaction were loaded into the 10x Genomics Chromium Controller and encapsulated in gel beads in emulsion. Single-cell gene expression, V(D)J and hashtag oligo libraries were prepared using the Chromium Next GEM Single-cell 5′ Reagent Kits v2 following the manufacturer’s protocol (10x Genomics, Pleasanton, CA). The integrity of the library was determined using the Tapestation 4200 (Agilent, Santa Clara, CA) and quantified using the Qubit fluorometry assay (AAT Bioquest, Sunnyvale, CA). BCR libraries were sequenced on the Nextseq2000 sequencer (Illumina, San Diego, CA). Sequencing data produced were analyzed using Cell Ranger followed by a customized analysis pipeline that includes the BG18 inferred germline heavy chain sequence.
ELISA
Serum antigen specific antibody titers were detected by ELISA, using anti-His Ab (2 μg/ml) to capture N332-GT2, N332-GT5 or respective KO antigen (2 μg/ml) on the plate. Mouse sera were incubated for two hours, and alkaline phosphatase conjugated anti-mouse IgG (Jackson ImmunoResearch, #115–055-071) was incubated for another hour. p-Nitrophenyl phosphate (Sigma, #N2770) dissolved in ddH2O (50 μl/well, RT, 30 min) was used for detection. Absorbance at 405 nm was determined with a plate reader (BioTek). Titers were determined from the dilution curve in the linear range of absorbance. ELISA curves were calculated and analyzed using GraphPad Prism (GraphPad).
Phylogenetic analysis
BG18 heavy chain amino acid sequences from heavy chain PCR and sanger sequencing were aligned using MUSCLE. Clonal phylogeny trees were generated using Geneious software (Biomatters Ltd, New Zealand).
Surface plasmon resonance (SPR)
Kinetics and affinities of antibody-antigen interactions were measured on a ProteOn XPR36 (Bio-Rad) using HC30M XanTex chips and 1×HBS-EP+ pH 7.4 running buffer (20× stock from Teknova, Cat. No. H8022) supplemented with BSA at 1 mg/ml. His-tag Antibody, (pAb, Rabbit, Cat. No. A00174, GenScript) was used for capturing trimer (ligand) and flowing Fab as analyte. About 7,000 response units of capture antibody were covalently immobilized on the sensor surface via EDC/NHS. About 300 to 400 RUs of antigen at 1 ug/mL were captured onto each flow cell. Analytes were passed over the flow cell at 50 μl/min for 3 min followed by a 5-min dissociation time. Regeneration was accomplished using phosphoric acid 50X or 1.7% with a 180-s contact time and injected four times per cycle. ProteOn Manager software (Bio-Rad) was used to analyze raw sensograms, including interspot and column double referencing, and to perform either Equilibrium fits or Kinetic fits with Langmuir model, or both, when applicable.
TZM-bl pseudovirus neutralization assay
Pseudoviruses were produced in HEK293T cells co-transfected using FuGENE 6 (Promega, cat# E2691) with pseudovirus Env-expressing plasmid and Env-deficient backbone plasmid (PSG3DEnv). Pseudoviruses were harvested 72 hours post-transfection, sterile filtered (0.45 μm), and concentrated (EMD Millipore, cat# UFC905024). Equal volumes of serially diluted monoclonal antibodies at appropriate concentrations were incubated with HIV pseudovirus in half-area 96-well plates (Greiner, cat# 675083) at 37°C for 1 hour. Next, 50 μL of TZM-bl cells at 200,000 cells/mL with or without diethylaminoethyl-dextran (5 μg/mL final concentration) were added to each well containing the antibody-virus mixture and incubated at 37°C for 72 hours in a humidified atmosphere of 5% CO2. After incubation, culture media was removed, and cells were lysed with 45 μL/well 1x Luciferase Culture Lysis buffer (Promega, cat# E1531) for 20 min at RT. Neutralization was measured by adding 30 μL luciferase reagent/well (Promega, cat# E1500) and measuring luminescence. IC50 was calculated using a nonlinear regression curve fit, sigmoidal, 4PL equation constrained from 0–100% in GraphPad Prism 9.3.1. IC50 is reported as the mean IC50 of two biological replicates.
EMPEM
Polyclonal Fab preparation from serum and sample preparation for electron microscopy were previously described (69). Briefly, sera (volume range 0.5–1 mL for soluble trimer and mRNA groups, 0.05 mL for nanoparticle groups) from all mice (n=8 or 10 for soluble trimer and mRNA groups, n=5 or 8 for nanoparticle groups) of an experimental group were combined. IgG was isolated using Protein G (Cytiva) and Fab digestion was performed using papain (Sigma Aldrich). = 15 μg of GT5 were incubated with 1 mg of Fab mixture (which also contains Fc and residual papain) overnight and purified the next day using a Superose 6 Increase (Cytiva) gel filtration column. Purified complexes were diluted to 0.03 mg/mL and deposited on glow-discharged carbon-coated copper mesh grids, followed by staining with 2% (w/v) uranyl formate. Imaging was performed on an FEI Tecnai Spirit T12 equipped with an FEI Eagle 4k × 4k CCD camera (120 keV, 2.06 Å/pixel), a Thermo Fisher Scientific (TFS) Talos F200C equipped with a TFS Ceta camera (200 keV, 1.98 Å/pixel) or an FEI TF20 equipped with a TVIPS TemCam F416 CMOS 4k × 4k camera (200 keV, 1.77 Å/pixel). All data were processed using Relion 3.0 using standard 2D and 3D classification procedures (86). Composite maps were generated using UCSF Chimera (87). Representative maps have been deposited to the Electron Microscopy Data Bank.
Cryo-EM
Recombinant Fabs derived from epitope specific CD45.2+ BG18 cells at day 77 either B11 or B16 boost were incubated at a 6x molar excess (Fab:trimer) with the respective immunogen (BG505_MD39,_B11 SOSIP or BG505_MD39_B16 SOSIP) overnight at room temperature. The following morning each complex was purified over a HiLoad 16/600 Superdex 200 pg gel filtration column (Cytiva). Fractions corresponding to the complex peak were combined and concentrated to 5.9 mg/mL (B11 complex) or 6.9 mg/mL (B16 complex). Samples were briefly mixed with either n-dodecyl-β-D-maltoside (DDM; final concentration 0.06 mM; Anatrace) or lauryl maltose neopentyl glycol (LMNG; final concentration 0.005 mM; Anatrace) to aid with particle dispersion and a 3 μl drop was applied to carbon or gold foil holey grids. Sample vitrification was performed using the Vitrobot Mark IV (Thermo Scientific). A similar procedure was followed for polyclonal Fab complexes from WT or BG18gH mice except that the trimer used was GT5 and the molar excess of total polyclonal Fab (SEC purified) to trimer was between 20–40X (0.2 mg GT5 and 1–2 mg polyFab). Final concentrations of purified complexes were 7.0 mg/mL (WT pAb + GT5) and 2.6 mg/mL (BG18gH pAb + GT5).
B11 + d77 Fab data collection was performed on a Thermo Fisher Scientific Arctica equipped with a Gatan K2 Summit direct electron detector (200 keV, 1.15 Å/pixel counting mode). Leginon (88) was used for automated data collection, frame alignment was performed using MotionCor2 (89) and subsequent data processing was performed using cryoSPARC v3 (90). B16 + d77 Fab, and WT pAb + GT5 images were collected on a Thermo Fisher Scientific Glacios using a Thermo Fisher Scientific Falcon 4 direct electron detector (200 keV, 0.725 Å/pixel counting mode). EPU (Thermo Fisher) was used for automated data collection, cryoSPARC Live (90) for frame alignment and CTF correction, and cryoSPARC v3 and Relion 3.1 (91) for subsequent data processing. BG18gH pAb + GT5 data collection occurred at the Pacific Northwest Center for Cryo-EM (PNCC) using a Thermo Fisher Scientific Krios and a Gatan K3 direct electron detector (300 keV, 0.40075 Å/pixel super-resolution mode). EPU (Thermo Fisher) was used for automated data collection, and Relion 3.1 for motion correction, CTF correction, and subsequent processing. Final refinements were performed in cryoSPARC 3. For both polyclonal datasets, focused classifications were performed in Relion 3.1 using a 40 Å spherical mask around the V3 patch Fab/epitope and skip-align 3D classification. Final data collection and processing stats are summarized in Supplementary Table 1.
Model building was performing by docking homology models of trimer and Fab Fv (in the case of polyclonal complexes, only a poly-alanine for antibody heavy and light chains) in UCSF Chimera, manually building and refinement in Coot 0.9.8 (92) and real space refinement using Rosetta (93) and Phenix. Final (94) models were validated using MolProbity and EMRinger in the Phenix suite, and statistics are summarized in Supplementary Table 1. All maps and models have been deposited to the Electron Microscopy Data Bank and Protein Data Bank, respectively with accession codes summarized in Supplementary Table 1.
Site-specific glycan analysis
DeGlyPHER (95) is used to ascertain site-specific glycan occupancy and processivity on the examined glycoproteins.
Proteinase K treatment and deglycosylation:
HIV Env glycoprotein was exchanged to water using Microcon Ultracel PL-10 centrifugal filter. Glycoprotein was reduced with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl) and alkylated with 10 mM 2-Chloroacetamide in 100 mM ammonium acetate for 20 min at room temperature (RT, 24°C). Initial protein-level deglycosylation was performed using 250 U of Endo H for 5 μg trimer, for 1 h at 37°C. Glycoprotein was digested with 1:25 Proteinase K (PK) for 30 min at 37°C. PK was denatured by incubating at 90°C for 15 min, then cooled to RT. Peptides were deglycosylated again with 250 U Endo H for 1 h at 37°C, then frozen at −80°C and lyophilized. 100 U PNGase F was lyophilized, resuspended in 20 μl 100 mM ammonium bicarbonate prepared in H218O, and added to the lyophilized peptides. Reactions were then incubated for 1 h at 37°C, subsequently analyzed by LC-MS/MS.
LC-MS/MS:
Samples were analyzed on an Q Exactive HF-X mass spectrometer. Samples were injected directly onto a 25 cm, 100 μm ID column packed with BEH 1.7 μm C18 resin. Samples were separated at a flow rate of 300 nL/min on an EASY-nLC 1200 UHPLC. Buffers A and B were 0.1% formic acid in 5% and 80% acetonitrile, respectively. The following gradient was used: 1–25% B over 160 min, an increase to 40% B over 40 min, an increase to 90% B over another 10 min and 30 min at 90% B for a total run time of 240 min. Column was re-equilibrated with solution A prior to the injection of sample. Peptides were eluted from the tip of the column and nanosprayed directly into the mass spectrometer by application of 2.8 kV at the back of the column. The mass spectrometer was operated in a data dependent mode. Full MS1 scans were collected in the Orbitrap at 120,000 resolution. The ten most abundant ions per scan were selected for HCD MS/MS at 25 NCE. Dynamic exclusion was enabled with exclusion duration of 10 s and singly charged ions were excluded.
Data Processing:
Protein and peptide identification were done with Integrated Proteomics Pipeline (IP2). Tandem mass spectra were extracted from raw files using RawConverter (96) and searched with ProLuCID (97) against a database comprising UniProt reviewed (Swiss-Prot) proteome for Homo sapiens (UP000005640), UniProt amino acid sequences for Endo H (P04067), PNGase F (Q9XBM8), and Proteinase K (P06873), amino acid sequences for the examined proteins, and a list of general protein contaminants. The search space included no cleavage-specificity. Carbamidomethylation (+57.02146 C) was considered a static modification. Deamidation in presence of H218O (+2.988261 N), GlcNAc (+203.079373 N), oxidation (+15.994915 M) and N-terminal pyroglutamate formation (−17.026549 Q) were considered differential modifications. Data was searched with 50 ppm precursor ion tolerance and 50 ppm fragment ion tolerance. Identified proteins were filtered using DTASelect2 (98) and utilizing a target-decoy database search strategy to limit the false discovery rate to 1%, at the spectrum level (99). A minimum of 1 peptide per protein and no tryptic end per peptide were required and precursor delta mass cut-off was fixed at 15 ppm. Statistical models for peptide mass modification (modstat) were applied. Census2 (100) label-free analysis was performed using the measured area under curve for each identified peptide/precursor in the chromatogram, with a 15 ppm precursor mass tolerance and 0.1 min retention time tolerance. “Match between runs” was used to find missing peptides between runs. Data analysis using GlycoMSQuant (95) was implemented to automate the analysis. GlycoMSQuant summed precursor peak areas across replicates, discarded peptides without NGS, discarded misidentified peptides when N-glycan remnant-mass modifications were localized to non-NGS asparagines and corrected/fixed N-glycan mislocalization where appropriate.
Supplementary Material
Acknowledgements:
We thank all Batista lab members for assistance with the experiments. We thank H.Turner, B. Anderson, C. Bowman, J.C. Ducom and A. Antanasijevic of Scripps Research and Sean Mulligan of PNCC for providing electron microscopy support.
Funding:
A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. This research was supported through the Collaboration for AIDS Vaccine Discovery (CAVD) grants OPP1084519, OPP1147787, INV-034657 (to W.R.S.), OPP1196345/INV-008813 (to A.B.W. and W.R.S.), OPP1115782/INV-002916 (to A.B.W.) and INV009585 and INV046626 (to F.D.B.) funded by the Bill and Melinda Gates Foundation. Funding was also provided by the National Institute of Allergy and Infectious Diseases (NIAID) UM1 AI144462 (Scripps Consortium for HIV/AIDS Vaccine Development) (to W.R.S., J.C.P., A.B.W. and F.D.B.) and R01 AI113867 (to J.C.P and W.R.S.); and by the Ragon Institute of MGH, MIT and Harvard (to W.R.S and F.D.B.).
Footnotes
Competing Interests: J.S. and W.R.S are inventors on patent applications filed by TSRI and IAVI regarding the immunogens described here. S. H., A.C., S.F., and W.R.S. are current or past employees of Moderna, Inc. F.D.B. has consultancy relationships with Adimab, Third Rock Ventures, and The EMBO Journal.
Data and materials availability:
Cryo-EM maps were deposited to the Electron Microscopy Data Bank (EMDB) under accession codes EMD-28941, EMD-28942 and EMD-28945 and EMD-43190, and corresponding atomic coordinates were deposited to the Protein Data Bank (PDB) under accession codes 8f92, 8f9g, 8f9m and 8vfv. Negative-stain EMPEM maps were deposited to the EMDB under accession codes EMD-28937, EMD-28938, EMD-28939, EMD-28940, EMD-29330, EMD-29333, EMD-29334, EMD-29335, EMD-43191 and EMD-43192. BCR sequence data deposition at GenBank: PP324161–PP325746; PP321345–PP322350; PP322351–PP324160. B11 glycosylation data was deposited in MassIVE repository (ID# MSV000094176 or PXD050174). Model animals available from corresponding author (F.D.B.) on request, subject to standard material transfer agreements.
References and Notes
- 1.Burton DR, Poignard P, Stanfield RL, Wilson IA, Broadly neutralizing antibodies suggest new prospects to counter highly antigenically diverse viruses. Science 337, 183–186 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoot S, McGuire AT, Cohen KW, Strong RK, Hangartner L, Klein F, Diskin R, Scheid JF, Sather DN, Burton DR, Stamatatos L, Recombinant HIV Envelope Proteins Fail to Engage Germline Versions of Anti-CD4bs bNAbs. PLOS Pathogens 9, e1003106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jardine JG, Julien J-P, Menis S, Ota T, Kalyuzhniy O, McGuire A, Sok D, Huang P-S, MacPherson S, Jones M, Nieusma T, Mathison J, Baker D, Ward AB, Burton DR, Stamatatos L, Nemazee D, Wilson IA, Schief WR, Rational HIV Immunogen Design to Target Specific Germline B Cell Receptors. Science 340, 711–716 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGuire AT, Hoot S, Dreyer AM, Lippy A, Stuart A, Cohen KW, Jardine J, Menis S, Scheid JF, West AP, Schief WR, Stamatatos L, Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4 binding site antibodies. J Exp Med 210, 655–663 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pancera M, McLellan JS, Wu X, Zhu J, Changela A, Schmidt SD, Yang Y, Zhou T, Phogat S, Mascola JR, Kwong PD, Crystal Structure of PG16 and Chimeric Dissection with Somatically Related PG9: Structure-Function Analysis of Two Quaternary-Specific Antibodies That Effectively Neutralize HIV-1. J Virol 84, 8098–8110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TYK, Pietzsch J, Fenyo D, Abadir A, Velinzon K, Hurley A, Myung S, Boulad F, Poignard P, Burton DR, Pereyra F, Ho DD, Walker BD, Seaman MS, Bjorkman PJ, Chait BT, Nussenzweig MC, Sequence and Structural Convergence of Broad and Potent HIV Antibodies That Mimic CD4 Binding. Science 333, 1633–1637 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soto C, Ofek G, Joyce MG, Zhang B, McKee K, Longo NS, Yang Y, Huang J, Parks R, Eudailey J, Lloyd KE, Alam SM, Haynes BF, Program NCS, Mullikin JC, Connors M, Mascola JR, Shapiro L, Kwong PD, Developmental Pathway of the MPER-Directed HIV-1-Neutralizing Antibody 10E8. PLOS ONE 11, e0157409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao X, Chen W, Feng Y, Zhu Z, Prabakaran P, Wang Y, Zhang M-Y, Longo NS, Dimitrov DS, Germline-like predecessors of broadly neutralizing antibodies lack measurable binding to HIV-1 envelope glycoproteins: Implications for evasion of immune responses and design of vaccine immunogens. Biochem Biophys Res Commun 390, 404–409 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou T, Georgiev I, Wu X, Yang Z-Y, Dai K, Finzi A, Kwon YD, Scheid J, Shi W, Xu L, Yang Y, Zhu J, Nussenzweig MC, Sodroski J, Shapiro L, Nabel GJ, Mascola JR, Kwong PD, Structural Basis for Broad and Potent Neutralization of HIV-1 by Antibody VRC01. Science 329, 811–817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burton DR, Hangartner L, Broadly Neutralizing Antibodies to HIV and Their Role in Vaccine Design. Annual Review of Immunology 34, 635–659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dosenovic P, von Boehmer L, Escolano A, Jardine J, Freund NT, Gitlin AD, McGuire AT, Kulp DW, Oliveira T, Scharf L, Pietzsch J, Gray MD, Cupo A, van Gils MJ, Yao K-H, Liu C, Gazumyan A, Seaman MS, Björkman PJ, Sanders RW, Moore JP, Stamatatos L, Schief WR, Nussenzweig MC, Immunization for HIV-1 Broadly Neutralizing Antibodies in Human Ig Knockin Mice. Cell 161, 1505–1515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jardine JG, Sok D, Julien J-P, Briney B, Sarkar A, Liang C-H, Scherer EA, Henry Dunand CJ, Adachi Y, Diwanji D, Hsueh J, Jones M, Kalyuzhniy O, Kubitz M, Spencer S, Pauthner M, Saye-Francisco KL, Sesterhenn F, Wilson PC, Galloway DM, Stanfield RL, Wilson IA, Burton DR, Schief WR, Minimally Mutated HIV-1 Broadly Neutralizing Antibodies to Guide Reductionist Vaccine Design. PLoS Pathog 12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steichen JM, Kulp DW, Tokatlian T, Escolano A, Dosenovic P, Stanfield RL, McCoy LE, Ozorowski G, Hu X, Kalyuzhniy O, Briney B, Schiffner T, Garces F, Freund NT, Gitlin AD, Menis S, Georgeson E, Kubitz M, Adachi Y, Jones M, Mutafyan AA, Yun DS, Mayer CT, Ward AB, Burton DR, Wilson IA, Irvine DJ, Nussenzweig MC, Schief WR, HIV Vaccine Design to Target Germline Precursors of Glycan-Dependent Broadly Neutralizing Antibodies. Immunity 45, 483–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leggat DJ, Cohen KW, Willis JR, Fulp WJ, deCamp AC, Kalyuzhniy O, Cottrell CA, Menis S, Finak G, Ballweber-Fleming L, Srikanth A, Plyler JR, Schiffner T, Liguori A, Rahaman F, Lombardo A, Philiponis V, Whaley RE, Seese A, Brand J, Ruppel AM, Hoyland W, Yates NL, Williams LD, Greene K, Gao H, Mahoney CR, Corcoran MM, Cagigi A, Taylor A, Brown DM, Ambrozak DR, Sincomb T, Hu X, Tingle R, Georgeson E, Eskandarzadeh S, Alavi N, Lu D, Mullen T-M, Kubitz M, Groschel B, Maenza J, Kolokythas O, Khati N, Bethony J, Crotty S, Roederer M, Karlsson Hedestam GB, Tomaras GD, Montefiori D, Diemert D, Koup RA, Laufer DS, McElrath MJ, McDermott AB, Schief WR, Vaccination induces HIV broadly neutralizing antibody precursors in humans. Science 378, eadd6502 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JH, Crotty S, HIV Vaccinology: 2021 Update. Semin Immunol 51, 101470 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbott RK, Lee JH, Menis S, Skog P, Rossi M, Ota T, Kulp DW, Bhullar D, Kalyuzhniy O, Havenar-Daughton C, Schief WR, Nemazee D, Crotty S, Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested with Germline-Targeting HIV Vaccine Immunogens. Immunity 48, 133–146.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dosenovic P, Kara EE, Pettersson A-K, McGuire AT, Gray M, Hartweger H, Thientosapol ES, Stamatatos L, Nussenzweig MC, Anti-HIV-1 B cell responses are dependent on B cell precursor frequency and antigen-binding affinity. Proc Natl Acad Sci U S A 115, 4743–4748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Havenar-Daughton C, Abbott RK, Schief WR, Crotty S, When designing vaccines, consider the starting material: the human B cell repertoire. Curr Opin Immunol 53, 209–216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steichen JM, Lin YC, Havenar-Daughton C, Pecetta S, Ozorowski G, Willis JR, Toy L, Sok D, Liguori A, Kratochvil S, Torres JL, Kalyuzhniy O, Melzi E, Kulp DW, Raemisch S, Hu X, Bernard SM, Georgeson E, Phelps N, Adachi Y, Kubitz M, Landais E, Umotoy J, Robinson A, Briney B, Wilson IA, Burton DR, Ward AB, Crotty S, Batista FD, Schief WR, A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science 366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sok D, Pauthner M, Briney B, Lee JH, Saye-Francisco KL, Hsueh J, Ramos A, Le KM, Jones M, Jardine JG, Bastidas R, Sarkar A, Liang C-H, Shivatare SS, Wu C-Y, Schief WR, Wong C-H, Wilson IA, Ward AB, Zhu J, Poignard P, Burton DR, A prominent site of antibody vulnerability on HIV envelope incorporates a motif associated with CCR5 binding and its camouflaging glycans. Immunity 45, 31–45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landais E, Huang X, Havenar-Daughton C, Murrell B, Price MA, Wickramasinghe L, Ramos A, Bian CB, Simek M, Allen S, Karita E, Kilembe W, Lakhi S, Inambao M, Kamali A, Sanders EJ, Anzala O, Edward V, Bekker L-G, Tang J, Gilmour J, Kosakovsky-Pond SL, Phung P, Wrin T, Crotty S, Godzik A, Poignard P, Broadly Neutralizing Antibody Responses in a Large Longitudinal Sub-Saharan HIV Primary Infection Cohort. PLOS Pathogens 12, e1005369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mouquet H, Scharf L, Euler Z, Liu Y, Eden C, Scheid JF, Halper-Stromberg A, Gnanapragasam PNP, Spencer DIR, Seaman MS, Schuitemaker H, Feizi T, Nussenzweig MC, Bjorkman PJ, Complex-type N -glycan recognition by potent broadly neutralizing HIV antibodies. Proc. Natl. Acad. Sci. U.S.A. 109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien J-P, Wang S-K, Ramos A, Chan-Hui P-Y, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong C-H, Phogat S, Wrin T, Simek MD, Principal Investigators PG, Koff WC, Wilson IA, Burton DR, Poignard P, Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freund NT, Wang H, Scharf L, Nogueira L, Horwitz JA, Bar-On Y, Golijanin J, Sievers SA, Sok D, Cai H, Cesar Lorenzi JC, Halper-Stromberg A, Toth I, Piechocka-Trocha A, Gristick HB, van Gils MJ, Sanders RW, Wang L-X, Seaman MS, Burton DR, Gazumyan A, Walker BD, West AP, Bjorkman PJ, Nussenzweig MC, Coexistence of potent HIV-1 broadly neutralizing antibodies and antibody-sensitive viruses in a viremic controller. Sci Transl Med 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barnes CO, Gristick HB, Freund NT, Escolano A, Lyubimov AY, Hartweger H, West AP, Cohen AE, Nussenzweig MC, Bjorkman PJ, Structural characterization of a highly-potent V3-glycan broadly neutralizing antibody bound to natively-glycosylated HIV-1 envelope. Nat Commun 9, 1251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sok D, Burton DR, Recent progress in broadly neutralizing antibodies to HIV. Nat Immunol 19, 1179–1188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Briney B, Sok D, Jardine JG, Kulp DW, Skog P, Menis S, Jacak R, Kalyuzhniy O, de Val N, Sesterhenn F, Le KM, Ramos A, Jones M, Saye-Francisco KL, Blane TR, Spencer S, Georgeson E, Hu X, Ozorowski G, Adachi Y, Kubitz M, Sarkar A, Wilson IA, Ward AB, Nemazee D, Burton DR, Schief WR, Tailored Immunogens Direct Affinity Maturation toward HIV Neutralizing Antibodies. Cell 166, 1459–1470.e11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Zhou T, Schmidt SD, Duan H, Cheng C, Chuang G-Y, Gu Y, Louder MK, Lin BC, Shen C-H, Vaccination induces maturation in a mouse model of diverse unmutated VRC01-class precursors to HIV-neutralizing antibodies with> 50% breadth. Immunity 54, 324–339. e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Escolano A, Steichen JM, Dosenovic P, Kulp DW, Golijanin J, Sok D, Freund NT, Gitlin AD, Oliveira T, Araki T, Lowe S, Chen ST, Heinemann J, Yao K-H, Georgeson E, Saye-Francisco KL, Gazumyan A, Adachi Y, Kubitz M, Burton DR, Schief WR, Nussenzweig MC, Sequential Immunization Elicits Broadly Neutralizing Anti-HIV-1 Antibodies in Ig Knockin Mice. Cell 166, 1445–1458.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian M, Cheng C, Chen X, Duan H, Cheng H-L, Dao M, Sheng Z, Kimble M, Wang L, Lin S, Schmidt SD, Du Z, Joyce MG, Chen Y, DeKosky BJ, Chen Y, Normandin E, Cantor E, Chen RE, Doria-Rose NA, Zhang Y, Shi W, Kong W-P, Choe M, Henry AR, Laboune F, Georgiev IS, Huang P-Y, Jain S, McGuire AT, Georgeson E, Menis S, Douek DC, Schief WR, Stamatatos L, Kwong PD, Shapiro L, Haynes BF, Mascola JR, Alt FW, Induction of HIV Neutralizing Antibody Lineages in Mice with Diverse Precursor Repertoires. Cell 166, 1471–1484.e18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Escolano A, Gristick HB, Gautam R, DeLaitsch AT, Abernathy ME, Yang Z, Wang H, Hoffmann MAG, Nishimura Y, Wang Z, Koranda N, Kakutani LM, Gao H, Gnanapragasam PNP, Raina H, Gazumyan A, Cipolla M, Oliveira TY, Ramos V, Irvine DJ, Silva M, West AP, Keeffe JR, Barnes CO, Seaman MS, Nussenzweig MC, Martin MA, Bjorkman PJ, Sequential immunization of macaques elicits heterologous, neutralizing antibodies targeting the V3-glycan patch of HIV-1 Env. Sci Transl Med 13, eabk1533 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang D, Abbott RK, Havenar-Daughton C, Skog PD, Al-Kolla R, Groschel B, Blane TR, Menis S, Tran JT, Thinnes TC, Volpi SA, Liguori A, Schiffner T, Villegas SM, Kalyuzhniy O, Pintea M, Voss JE, Phelps N, Tingle R, Rodriguez AR, Martin G, Kupryianov S, deCamp A, Schief WR, Nemazee D, Crotty S, B cells expressing authentic naive human VRC01-class BCRs can be recruited to germinal centers and affinity mature in multiple independent mouse models. PNAS 117, 22920–22931 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jardine JG, Ota T, Sok D, Pauthner M, Kulp DW, Kalyuzhniy O, Skog PD, Thinnes TC, Bhullar D, Briney B, Menis S, Jones M, Kubitz M, Spencer S, Adachi Y, Burton DR, Schief WR, Nemazee D, Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 349, 156–161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medina-Ramírez M, Garces F, Escolano A, Skog P, de Taeye SW, Del Moral-Sanchez I, McGuire AT, Yasmeen A, Behrens A-J, Ozorowski G, van den Kerkhof TLGM, Freund NT, Dosenovic P, Hua Y, Gitlin AD, Cupo A, van der Woude P, Golabek M, Sliepen K, Blane T, Kootstra N, van Breemen MJ, Pritchard LK, Stanfield RL, Crispin M, Ward AB, Stamatatos L, Klasse PJ, Moore JP, Nemazee D, Nussenzweig MC, Wilson IA, Sanders RW, Design and crystal structure of a native-like HIV-1 envelope trimer that engages multiple broadly neutralizing antibody precursors in vivo. Journal of Experimental Medicine 214, 2573–2590 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melzi E, Willis JR, Ma KM, Lin Y-C, Kratochvil S, Berndsen ZT, Landais EA, Kalyuzhniy O, Nair U, Warner J, Steichen JM, Kalyuzhniy A, Le A, Pecetta S, Perez M, Kirsch K, Weldon SR, Falcone S, Himansu S, Carfi A, Sok D, Ward AB, Schief WR, Batista FD, Membrane-bound mRNA immunogens lower the threshold to activate HIV Env V2 apex-directed broadly neutralizing B cell precursors in humanized mice. Immunity 55, 2168–2186.e6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parks KR, MacCamy AJ, Trichka J, Gray M, Weidle C, Borst AJ, Khechaduri A, Takushi B, Agrawal P, Guenaga J, Wyatt RT, Coler R, Seaman M, LaBranche C, Montefiori DC, Veesler D, Pancera M, McGuire A, Stamatatos L, Overcoming Steric Restrictions of VRC01 HIV-1 Neutralizing Antibodies through Immunization. Cell Rep 29, 3060–3072.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sok D, Briney B, Jardine JG, Kulp DW, Menis S, Pauthner M, Wood A, Lee E-C, Le KM, Jones M, Ramos A, Kalyuzhniy O, Adachi Y, Kubitz M, MacPherson S, Bradley A, Friedrich GA, Schief WR, Burton DR, Priming HIV-1 broadly neutralizing antibody precursors in human Ig loci transgenic mice. Science 353, 1557–1560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Ray R, Kratochvil S, Melzi E, Lin Y-C, Giguere S, Xu L, Warner J, Cheon D, Liguori A, Groschel B, Phelps N, Adachi Y, Tingle R, Wu L, Crotty S, Kirsch KH, Nair U, Schief WR, Batista FD, Multiplexed CRISPR/CAS9-mediated engineering of pre-clinical mouse models bearing native human B cell receptors. The EMBO Journal 40, e105926 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Y-C, Pecetta S, Steichen JM, Kratochvil S, Melzi E, Arnold J, Dougan SK, Wu L, Kirsch KH, Nair U, Schief WR, Batista FD, One-step CRISPR/Cas9 method for the rapid generation of human antibody heavy chain knock-in mice. The EMBO Journal 37, e99243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.El Sahly HM, Baden LR, Essink B, Doblecki-Lewis S, Martin JM, Anderson EJ, Campbell TB, Clark J, Jackson LA, Fichtenbaum CJ, Zervos M, Rankin B, Eder F, Feldman G, Kennelly C, Han-Conrad L, Levin M, Neuzil KM, Corey L, Gilbert P, Janes H, Follmann D, Marovich M, Polakowski L, Mascola JR, Ledgerwood JE, Graham BS, August A, Clouting H, Deng W, Han S, Leav B, Manzo D, Pajon R, Schödel F, Tomassini JE, Zhou H, Miller J, Efficacy of the mRNA-1273 SARS-CoV-2 Vaccine at Completion of Blinded Phase. New England Journal of Medicine 385, 1774–1785 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas SJ, Moreira ED, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Polack FP, Zerbini C, Bailey R, Swanson KA, Xu X, Roychoudhury S, Koury K, Bouguermouh S, Kalina WV, Cooper D, Frenck RW, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Yang Q, Liberator P, Tresnan DB, Mather S, Dormitzer PR, Şahin U, Gruber WC, Jansen KU, Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months. N Engl J Med 385, 1761–1773 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang Q, Zeng J, Yan J, COVID-19 mRNA vaccines. Journal of Genetics and Genomics 48, 107–114 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kowalzik F, Schreiner D, Jensen C, Teschner D, Gehring S, Zepp F, mRNA-Based Vaccines. Vaccines 9, 390 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turner HL, Andrabi R, Cottrell CA, Richey ST, Song G, Callaghan S, Anzanello F, Moyer TJ, Abraham W, Melo M, Silva M, Scaringi N, Rakasz EG, Sattentau QJ, Irvine DJ, Burton DR, Ward AB, Disassembly of HIV envelope glycoprotein trimer immunogens is driven by antibodies elicited via immunization. Sci Adv 7, eabh2791 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antanasijevic A, Sewall LM, Cottrell CA, Carnathan DG, Jimenez LE, Ngo JT, Silverman JB, Groschel B, Georgeson E, Bhiman J, Bastidas R, LaBranche C, Allen JD, Copps J, Perrett HR, Rantalainen K, Cannac F, Yang YR, de la Peña AT, Rocha RF, Berndsen ZT, Baker D, King NP, Sanders RW, Moore JP, Crotty S, Crispin M, Montefiori DC, Burton DR, Schief WR, Silvestri G, Ward AB, Polyclonal antibody responses to HIV Env immunogens resolved using cryoEM. Nature Communications 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garces F, Sok D, Kong L, McBride R, Kim HJ, Saye-Francisco KF, Julien J-P, Hua Y, Cupo A, Moore JP, Paulson JC, Ward AB, Burton DR, Wilson IA, Structural evolution of glycan recognition by a family of potent HIV antibodies. Cell 159, 69–79 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gristick HB, von Boehmer L, West AP, Schamber M, Gazumyan A, Golijanin J, Seaman MS, Fätkenheuer G, Klein F, Nussenzweig MC, Bjorkman PJ, Natively glycosylated HIV-1 Env structure reveals new mode for antibody recognition of the CD4-binding site. Nat Struct Mol Biol 23, 906–915 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pejchal R, Doores KJ, Walker LM, Khayat R, Huang P-S, Wang S-K, Stanfield RL, Julien J-P, Ramos A, Crispin M, Depetris R, Katpally U, Marozsan A, Cupo A, Maloveste S, Liu Y, McBride R, Ito Y, Sanders RW, Ogohara C, Paulson JC, Feizi T, Scanlan CN, Wong C-H, Moore JP, Olson WC, Ward AB, Poignard P, Schief WR, Burton DR, Wilson IA, A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 334, 1097–1103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mesin L, Schiepers A, Ersching J, Barbulescu A, Cavazzoni CB, Angelini A, Okada T, Kurosaki T, Victora GD, Restricted Clonality and Limited Germinal Center Reentry Characterize Memory B Cell Reactivation by Boosting. Cell 180, 92–106.e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tas JMJ, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, Chen CS, Weill J-C, Reynaud C-A, Browne EP, Meyer-Hermann M, Victora GD, Visualizing Antibody Affinity Maturation in Germinal Centers. Science 351, 1048–1054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sprumont A, Rodrigues A, McGowan SJ, Bannard C, Bannard O, Germinal centers output clonally diverse plasma cell populations expressing high- and low-affinity antibodies. Cell 186, 5486–5499.e13 (2023). [DOI] [PubMed] [Google Scholar]
- 52.Willis JR, Berndsen ZT, Ma KM, Steichen JM, Schiffner T, Landais E, Liguori A, Kalyuzhniy O, Allen JD, Baboo S, Omorodion O, Diedrich JK, Hu X, Georgeson E, Phelps N, Eskandarzadeh S, Groschel B, Kubitz M, Adachi Y, Mullin T-M, Alavi NB, Falcone S, Himansu S, Carfi A, Wilson IA, Yates JR, Paulson JC, Crispin M, Ward AB, Schief WR, Human immunoglobulin repertoire analysis guides design of vaccine priming immunogens targeting HIV V2-apex broadly neutralizing antibody precursors. Immunity 55, 2149–2167.e9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corbett KS, Edwards DK, Leist SR, Abiona OM, Boyoglu-Barnum S, Gillespie RA, Himansu S, Schäfer A, Ziwawo CT, DiPiazza AT, Dinnon KH, Elbashir SM, Shaw CA, Woods A, Fritch EJ, Martinez DR, Bock KW, Minai M, Nagata BM, Hutchinson GB, Wu K, Henry C, Bahi K, Garcia-Dominguez D, Ma L, Renzi I, Kong W-P, Schmidt SD, Wang L, Zhang Y, Phung E, Chang LA, Loomis RJ, Altaras NE, Narayanan E, Metkar M, Presnyak V, Liu C, Louder MK, Shi W, Leung K, Yang ES, West A, Gully KL, Stevens LJ, Wang N, Wrapp D, Doria-Rose NA, Stewart-Jones G, Bennett H, Alvarado GS, Nason MC, Ruckwardt TJ, McLellan JS, Denison MR, Chappell JD, Moore IN, Morabito KM, Mascola JR, Baric RS, Carfi A, Graham BS, SARS-CoV-2 mRNA Vaccine Design Enabled by Prototype Pathogen Preparedness. Nature 586, 567–571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walsh EE, Frenck RW, Falsey AR, Kitchin N, Absalon J, Gurtman A, Lockhart S, Neuzil K, Mulligan MJ, Bailey R, Swanson KA, Li P, Koury K, Kalina W, Cooper D, Fontes-Garfias C, Shi P-Y, Türeci Ö, Tompkins KR, Lyke KE, Raabe V, Dormitzer PR, Jansen KU, Şahin U, Gruber WC, Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N Engl J Med 383, 2439–2450 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ndung’u T, Weiss RA, On HIV diversity. AIDS 26, 1255–1260 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Doria-Rose NA, NISC Comparative Sequencing, Schramm CA, Gorman J, Moore PL, Bhiman JN, DeKosky BJ, Ernandes MJ, Georgiev IS, Kim HJ, Pancera M, Staupe RP, Altae-Tran HR, Bailer RT, Crooks ET, Cupo A, Druz A, Garrett NJ, Hoi KH, Kong R, Louder MK, Longo NS, McKee K, Nonyane M, O’Dell S, Roark RS, Rudicell RS, Schmidt SD, Sheward DJ, Soto C, Wibmer CK, Yang Y, Zhang Z, Mullikin JC, Binley JM, Sanders RW, Wilson IA, Moore JP, Ward AB, Georgiou G, Williamson C, Abdool Karim SS, Morris L, Kwong PD, Shapiro L, Mascola JR, Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 509, 55–62 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gray ES, Madiga MC, Hermanus T, Moore PL, Wibmer CK, Tumba NL, Werner L, Mlisana K, Sibeko S, Williamson C, Abdool Karim SS, Morris L, and the CAPRISA002 Study Team, The Neutralization Breadth of HIV-1 Develops Incrementally over Four Years and Is Associated with CD4 + T Cell Decline and High Viral Load during Acute Infection. J Virol 85, 4828–4840 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liao H-X, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, Fire AZ, Roskin KM, Schramm CA, Zhang Z, Zhu J, Shapiro L, Mullikin JC, Gnanakaran S, Hraber P, Wiehe K, Kelsoe G, Yang G, Xia S-M, Montefiori DC, Parks R, Lloyd KE, Scearce RM, Soderberg KA, Cohen M, Kamanga G, Louder MK, Tran LM, Chen Y, Cai F, Chen S, Moquin S, Du X, Joyce MG, Srivatsan S, Zhang B, Zheng A, Shaw GM, Hahn BH, Kepler TB, Korber BTM, Kwong PD, Mascola JR, Haynes BF, Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496, 469–476 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steichen JM, Phung I, Salcedo E, Ozorowski G, Willis JR, Baboo S, Liguori A, Cottrell CA, Torres JL, Madden PJ, Ma KM, Sutton HJ, Lee JH, Kalyuzhniy O, Allen JD, Rodriguez OL, Adachi Y, Mullen T-M, Georgeson E, Kubitz M, Burns A, Barman S, Mopuri R, Metz A, Altheide TK, Saha S, Shields K, Schulze S, Smith ML, Schiffner T, Burton DR, Watson CT, Bosinger SE, Crispin M, Yates JR, Paulson JC, Ward AB, Sok D, Crotty S, Schief WR, Vaccine priming of rare HIV broadly neutralizing antibody precursors in rhesus macaques. Science, Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, Victora GD, T Follicular Helper Cell Dynamics in Germinal Centers. Science 341, 673–677 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, Reynaud C-A, Weill J-C, Multiple layers of B cell memory with different effector functions. Nat Immunol 10, 1292–1299 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK, Different B cell populations mediate early and late memory during an endogenous immune response. Science 331, 1203–1207 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, Shlomchik MJ, CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol 15, 631–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaji T, Furukawa K, Ishige A, Toyokura I, Nomura M, Okada M, Takahashi Y, Shimoda M, Takemori T, Both mutated and unmutated memory B cells accumulate mutations in the course of the secondary response and develop a new antibody repertoire optimally adapted to the secondary stimulus. Int Immunol 25, 683–695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McHeyzer-Williams LJ, Milpied PJ, Okitsu SL, McHeyzer-Williams MG, Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat Immunol 16, 296–305 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bergström JJE, Xu H, Heyman B, Epitope-Specific Suppression of IgG Responses by Passively Administered Specific IgG: Evidence of Epitope Masking. Frontiers in Immunology 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Forsell MNE, Kvastad L, Sedimbi SK, Andersson J, Karlsson MCI, Regulation of Subunit-Specific Germinal Center B Cell Responses to the HIV-1 Envelope Glycoproteins by Antibody-Mediated Feedback. Front Immunol 8, 738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McNamara HA, Idris AH, Sutton HJ, Vistein R, Flynn BJ, Cai Y, Wiehe K, Lyke KE, Chatterjee D, Kc N, Chakravarty S, Lee Sim BK, Hoffman SL, Bonsignori M, Seder RA, Cockburn IA, Antibody Feedback Limits the Expansion of B Cell Responses to Malaria Vaccination but Drives Diversification of the Humoral Response. Cell Host & Microbe 28, 572–585.e7 (2020). [DOI] [PubMed] [Google Scholar]
- 69.Tas JMJ, Koo J-H, Lin Y-C, Xie Z, Steichen JM, Jackson AM, Hauser BM, Wang X, Cottrell CA, Torres JL, Warner JE, Kirsch KH, Weldon SR, Groschel B, Nogal B, Ozorowski G, Bangaru S, Phelps N, Adachi Y, Eskandarzadeh S, Kubitz M, Burton DR, Lingwood D, Schmidt AG, Nair U, Ward AB, Schief WR, Batista FD, Antibodies from primary humoral responses modulate the recruitment of naive B cells during secondary responses. Immunity, S1074–7613(22)00355–7 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meyer-Hermann M, Injection of Antibodies against Immunodominant Epitopes Tunes Germinal Centers to Generate Broadly Neutralizing Antibodies. Cell Reports 29, 1066–1073.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Meyer-Hermann M, George LA, Figge MT, Khan M, Goodall M, Young SP, Reynolds A, Falciani F, Waisman A, Notley CA, Ehrenstein MR, Kosco-Vilbois M, Toellner K-M, Germinal center B cells govern their own fate via antibody feedback. J Exp Med 210, 457–464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seabright GE, Doores KJ, Burton DR, Crispin M, Protein and Glycan Mimicry in HIV Vaccine Design. J Mol Biol 431, 2223–2247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kong L, Lee JH, Doores KJ, Murin CD, Julien J-P, McBride R, Liu Y, Marozsan A, Cupo A, Klasse P-J, Hoffenberg S, Caulfield M, King CR, Hua Y, Le KM, Khayat R, Deller MC, Clayton T, Tien H, Feizi T, Sanders RW, Paulson JC, Moore JP, Stanfield RL, Burton DR, Ward AB, Wilson IA, Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat Struct Mol Biol 20, 796–803 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi B, Ma L, He X, Wang X, Wang P, Zhou L, Yao X, Comparative analysis of human and mouse immunoglobulin variable heavy regions from IMGT/LIGM-DB with IMGT/HighV-QUEST. Theoretical Biology and Medical Modelling 11, 30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cinamon G, Zachariah MA, Lam OM, Foss FW, Cyster JG, Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol 9, 54–62 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bok K, Sitar S, Graham BS, Mascola JR, Accelerated COVID-19 vaccine development: milestones, lessons, and prospects. Immunity 54, 1636–1651 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carrasco YR, Fleire SJ, Cameron T, Dustin ML in LFA-1/ICAM-1 Interaction Lowers the Threshold of B Cell Activation by Facilitating B Cell Adhesion and Synapse Formation - ScienceDirect. Immunity (2004). [DOI] [PubMed]
- 78.Fleire SJ, Goldman JP, Carrasco YR, Weber M, Bray D, Batista FD, B Cell Ligand Discrimination Through a Spreading and Contraction Response. Science 312, 738–741 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D, Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. Journal of Controlled Release 217, 345–351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cirelli KM, Carnathan DG, Nogal B, Martin JT, Rodriguez OL, Upadhyay AA, Enemuo CA, Gebru EH, Choe Y, Viviano F, Nakao C, Pauthner MG, Reiss S, Cottrell CA, Smith ML, Bastidas R, Gibson W, Wolabaugh AN, Melo MB, Cosette B, Kumar V, Patel NB, Tokatlian T, Menis S, Kulp DW, Burton DR, Murrell B, Schief WR, Bosinger SE, Ward AB, Watson CT, Silvestri G, Irvine DJ, Crotty S, Slow delivery immunization enhances HIV neutralizing antibody and germinal center responses via modulation of immunodominance. Cell 177, 1153–1171.e28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Röltgen K, Nielsen SCA, Silva O, Younes SF, Zaslavsky M, Costales C, Yang F, Wirz OF, Solis D, Hoh RA, Wang A, Arunachalam PS, Colburg D, Zhao S, Haraguchi E, Lee AS, Shah MM, Manohar M, Chang I, Gao F, Mallajosyula V, Li C, Liu J, Shoura MJ, Sindher SB, Parsons E, Dashdorj NJ, Dashdorj ND, Monroe R, Serrano GE, Beach TG, Chinthrajah RS, Charville GW, Wilbur JL, Wohlstadter JN, Davis MM, Pulendran B, Troxell ML, Sigal GB, Natkunam Y, Pinsky BA, Nadeau KC, Boyd SD, Immune imprinting, breadth of variant recognition, and germinal center response in human SARS-CoV-2 infection and vaccination. Cell 185, 1025–1040.e14 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Remmel JL, Ackerman ME, Rationalizing random walks: replicating protective antibody trajectories. Trends Immunol 42, 186–197 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mu Z, Wiehe K, Saunders KO, Henderson R, Cain DW, Parks R, Martik D, Mansouri K, Edwards RJ, Newman A, Lu X, Xia S-M, Eaton A, Bonsignori M, Montefiori D, Han Q, Venkatayogi S, Evangelous T, Wang Y, Rountree W, Korber B, Wagh K, Tam Y, Barbosa C, Alam SM, Williams WB, Tian M, Alt FW, Pardi N, Weissman D, Haynes BF, mRNA-encoded HIV-1 Env trimer ferritin nanoparticles induce monoclonal antibodies that neutralize heterologous HIV-1 isolates in mice. Cell Reports 38, 110514 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang P, Narayanan E, Liu Q, Tsybovsky Y, Boswell K, Ding S, Hu Z, Follmann D, Lin Y, Miao H, Schmeisser H, Rogers D, Falcone S, Elbashir SM, Presnyak V, Bahl K, Prabhakaran M, Chen X, Sarfo EK, Ambrozak DR, Gautam R, Martin MA, Swerczek J, Herbert R, Weiss D, Misamore J, Ciaramella G, Himansu S, Stewart-Jones G, McDermott A, Koup RA, Mascola JR, Finzi A, Carfi A, Fauci AS, Lusso P, A multiclade env–gag VLP mRNA vaccine elicits tier-2 HIV-1-neutralizing antibodies and reduces the risk of heterologous SHIV infection in macaques. Nat Med 27, 2234–2245 (2021). [DOI] [PubMed] [Google Scholar]
- 85.von Boehmer L, Liu C, Ackerman S, Gitlin AD, Wang Q, Gazumyan A, Nussenzweig MC, Sequencing and cloning of antigen-specific antibodies from mouse memory B cells. Nat Protoc 11, 1908–1923 (2016). [DOI] [PubMed] [Google Scholar]
- 86.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH, New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 88.Cheng A, Negro C, Bruhn JF, Rice WJ, Dallakyan S, Eng ET, Waterman DG, Potter CS, Carragher B, Leginon: New features and applications. Protein Sci 30, 136–150 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, Agard DA, MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 91.Zivanov J, Nakane T, Scheres SHW, Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Casañal A, Lohkamp B, Emsley P, Current developments in Coot for macromolecular model building of Electron Cryo-microscopy and Crystallographic Data. Protein Sci 29, 1069–1078 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D, Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci 23, 47–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, Adams PD, Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baboo S, Diedrich JK, Martínez-Bartolomé S, Wang X, Schiffner T, Groschel B, Schief WR, Paulson JC, Yates JR, DeGlyPHER: an ultrasensitive method for analysis of viral spike N-glycoforms. Anal Chem 93, 13651–13657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.He L, Diedrich J, Chu Y-Y, Yates JR, Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Anal Chem 87, 11361–11367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, CCL. Wong, B. Fonslow, C. Delahunty, Y. Gao, H. Shah, J. R. Yates, ProLuCID: an improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tabb DL, McDonald WH, Yates JR, DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 1, 21–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP, Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res 2, 43–50 (2003). [DOI] [PubMed] [Google Scholar]
- 100.Park SK, Venable JD, Xu T, Yates JR, A quantitative analysis software tool for mass spectrometry–based proteomics. Nat Methods 5, 319–322 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kulp DW, Steichen JM, Pauthner M, Hu X, Schiffner T, Liguori A, Cottrell CA, Havenar-Daughton C, Ozorowski G, Georgeson E, Kalyuzhniy O, Willis JR, Kubitz M, Adachi Y, Reiss SM, Shin M, de Val N, Ward AB, Crotty S, Burton DR, Schief WR, Structure-based design of native-like HIV-1 envelope trimers to silence non-neutralizing epitopes and eliminate CD4 binding. Nat Commun 8, 1655 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM maps were deposited to the Electron Microscopy Data Bank (EMDB) under accession codes EMD-28941, EMD-28942 and EMD-28945 and EMD-43190, and corresponding atomic coordinates were deposited to the Protein Data Bank (PDB) under accession codes 8f92, 8f9g, 8f9m and 8vfv. Negative-stain EMPEM maps were deposited to the EMDB under accession codes EMD-28937, EMD-28938, EMD-28939, EMD-28940, EMD-29330, EMD-29333, EMD-29334, EMD-29335, EMD-43191 and EMD-43192. BCR sequence data deposition at GenBank: PP324161–PP325746; PP321345–PP322350; PP322351–PP324160. B11 glycosylation data was deposited in MassIVE repository (ID# MSV000094176 or PXD050174). Model animals available from corresponding author (F.D.B.) on request, subject to standard material transfer agreements.