

Keywords: coenzyme Q, CoQ, CoQ deficiency, mitochondrial disease, ubiquinone
Abstract
Coenzyme Q (CoQ), also known as ubiquinone, comprises a benzoquinone head group and a long isoprenoid side chain. It is thus extremely hydrophobic and resides in membranes. It is best known for its complex function as an electron transporter in the mitochondrial electron transport chain (ETC) but is also required for several other crucial cellular processes. In fact, CoQ appears to be central to the entire redox balance of the cell. Remarkably, its structure and therefore its properties have not changed from bacteria to vertebrates. In metazoans, it is synthesized in all cells and is found in most, and maybe all, biological membranes. CoQ is also known as a nutritional supplement, mostly because of its involvement with antioxidant defenses. However, whether there is any health benefit from oral consumption of CoQ is not well established. Here we review the function of CoQ as a redox-active molecule in the ETC and other enzymatic systems, its role as a prooxidant in reactive oxygen species generation, and its separate involvement in antioxidant mechanisms. We also review CoQ biosynthesis, which is particularly complex because of its extreme hydrophobicity, as well as the biological consequences of primary and secondary CoQ deficiency, including in human patients. Primary CoQ deficiency is a rare inborn condition due to mutation in CoQ biosynthetic genes. Secondary CoQ deficiency is much more common, as it accompanies a variety of pathological conditions, including mitochondrial disorders as well as aging. In this context, we discuss the importance, but also the great difficulty, of alleviating CoQ deficiency by CoQ supplementation.
CLINICAL HIGHLIGHTS.
Coenzyme Q10 (CoQ10) was discovered more than half a century ago for its key role in mitochondrial respiration. It also participates in several other important cellular functions such as reactive oxygen species (ROS) generation during mitochondrial respiration, protection against oxidation of membrane lipids, and the redox balance of the cell. Mutations in the genes required for the biosynthesis of CoQ10 lead to primary CoQ10 deficiency (PCD) and present with heterogeneous clinical symptoms ranging from birth- or infantile-onset multisystem disorders to isolated symptoms involving single organs or systems. Overall, PCD frequently resembles mitochondrial disease syndromes. However, it is unknown whether the same pathophysiology underlies each symptom. For example, some studies in mice suggest that it is increased oxidative stress due to low CoQ, and not damaged mitochondrial function, that is responsible for renal symptoms. In addition to PCD, a variety of diseases and conditions have been found to be associated with secondary CoQ10 deficiency (SCD), which refers to all the conditions in which the etiology of the CoQ10 deficiency is not a molecular lesion in the CoQ10 biosynthetic pathway. These include mitochondrial disorders, multiple system atrophy, ataxia due to APTX mutations, mutations in ETFDH, and Parkinson’s disease. In addition to patients with documented CoQ10 deficiency and/or mutations of biosynthetic genes, CoQ10 is frequently recommended to mitochondrial disease patients as well as for treating a wide range of other conditions (e.g., heart failure and neurodegenerative diseases). Oral CoQ10 supplementation is the only currently available treatment option for CoQ10 deficiency. However, a recent systematic review of all PCD patients who have been treated with CoQ10 suggests that oral supplementation is virtually without effect, despite the fact that the lack of CoQ10 is the primary cause of these patients’ symptoms. Future research will be necessary to develop effective therapies to treat or prevent CoQ10 deficiency.
1. INTRODUCTION
Coenzyme Q (CoQ), also known as ubiquinone (UQ), is a lipophilic molecule that is essential for several distinct cellular processes, including energy production, and is thus essential for life. It is one of the most conserved molecules across all kingdoms of life. Frederick Crane and colleagues (1) at the Enzyme Institute of the University of Wisconsin in Madison first isolated it in 1957 from beef heart mitochondria as a yellow-orange lipophilic substance with redox properties, and it was proposed to function as a coenzyme for mitochondrial electron transfer. As such, it was given the name coenzyme Q. Its other name, ubiquinone, which was officially given to the substance in 1975 by the IUPAC-IUB Commission on Biochemical Nomenclature, refers to the fact that it has a ubiquitous presence from bacteria to humans.
The chemical structure of CoQ was determined by Karl Folkers and coworkers at Merck. Its full chemical name is often given as 2,3-dimethoxy-5-methyl-6-multiprenyl-1,4-benzoquinone. Another possible formalism is 2-methyl-3-multiprenyl-5,6-dimethoxy-1,4-benzoquinone, which we are following in this review, including in the figures. CoQ is composed of a redox-active benzoquinone ring conjugated to an isoprenoid unbranched side chain whose length is species specific, ranging from 6 to 10 isoprenoid repeats (FIGURE 1). For example, in humans, the side chain is 10 isoprene subunits long and the molecule is therefore abbreviated as CoQ10. Rodents and Caenorhabditis elegans (C. elegans) mainly produce CoQ9, and Saccharomyces cerevisiae (S. cerevisiae) and Escherichia coli (E. coli) produce CoQ6 and CoQ8, respectively. Some species make more than one form of CoQ. For example, although CoQ9 is the main form in mice, small amounts of CoQ10 also occur in most tissues, with tissue-specific ratios of the two forms. Why different organisms have CoQ with varying side chain lengths is not understood. The CoQ benzoquinone ring is the functional group of the molecule, capable of reversible oxidation-reduction states without change in structure. The benzoquinone ring of quinone can exist in nine different redox states (2, 3). However, functionally there are three redox states of CoQ, that is, fully oxidized (CoQ, UQ), partially reduced (a semiquinone anion radical with a reactive unpaired electron, CoQ•−, UQ•−), and fully reduced (CoQH2, UQH2) (4, 5) (FIGURE 1). The redox chemistry of CoQ, which is able to accept/donate one or two electrons at a time, is at the core of its best-understood biological functions (6). The isoprenoid tail is responsible for the extreme hydrophobicity of CoQ and its solubility in membrane bilayers (6).
FIGURE 1.

Structure and redox states of coenzyme Q (CoQ). CoQ exists in 3 redox states: the fully oxidized form (CoQ) accepts 2 electrons to form CoQH2 or accepts 1 electron to form the ubisemiquinone intermediate, followed by acceptance of an additional electron to form CoQH2. The number of isoprene units in the tail varies between species from 6 to 10.
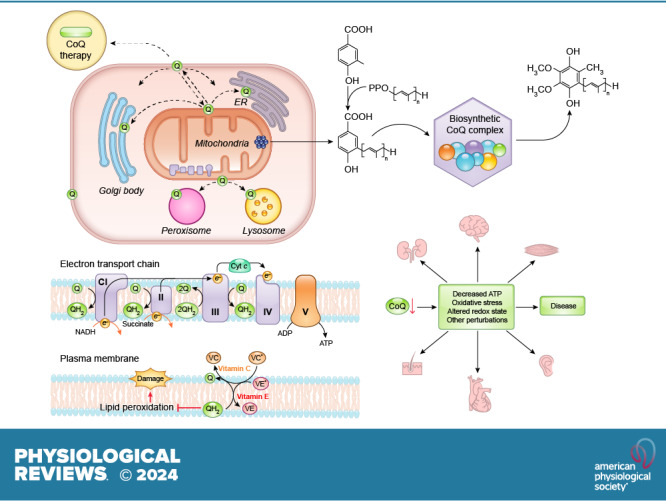
CoQ is likely found in all eukaryotic lipid membranes (7, 8). The best-known function of CoQ is to act as an electron carrier in the electron transport chain (ETC) in the inner membrane of mitochondria (IMM). In fact, mitochondria are the most enriched in CoQ among all subcellular compartments (9, 10). Other functions described for CoQ include participation in trans-plasma membrane electron transport, regulation of the mitochondrial permeability transition pore (mPTP), and activation of uncoupling proteins (UCPs), as well as an important dual role as pro- and antioxidant (7, 11–13). It has been proposed that CoQ may also play a role in the physicochemical properties of the lipid membranes in which it resides, but this is not yet well established or understood (14–23). In addition, new aspects of the function of CoQ are regularly reported. For example, a recent study suggests that the ratio of reduced to oxidized CoQ (CoQ/CoQH2) helps metabolic adaptation by acting as a sensor of the efficiency of the mitochondrial ETC (24). CoQ deficiency, no matter its cause, is currently defined as a decrease in the CoQ content in cells or organisms that can potentially impair many cellular functions, with mitochondria respiration expected to be the most vulnerable.
All cells rely on endogenous synthesis for their CoQ supply. CoQ biosynthesis is a complex and highly conserved pathway in which at least 10 proteins are involved. In eukaryotes, CoQ is synthesized from precursors in the IMM, from which it is then distributed to other subcellular compartments (25–27). To date, it is well established that several CoQ biosynthetic pathway components are recruited into a supramolecular complex that catalyzes sequential reactions that modify the aromatic ring (25–28). In animals, complete loss of CoQ biosynthesis is embryonic lethal in most species (8, 29–32). However, see the description in sect. 5.2 of the special case of clk-1 mutants of the nematode C. elegans, which can survive with a mixture of dietary CoQ and the CoQ biosynthetic intermediate demethoxyubiquinone (DMQ) (33–36). In humans, deleterious mutations in genes required for CoQ biosynthesis frequently cause severe multisystem disease due to impaired mitochondrial respiration (11, 26, 37–39). After diagnosis, the patients are generally treated with oral CoQ10 supplementation. Unfortunately, there is only very weak evidence for the efficacy of the treatment (40). Efforts are underway to develop methods for more effective CoQ10 delivery to overcome its extremely poor water solubility and limited oral bioavailability (41, 42). Moreover, in view of its essential role in mitochondrial respiration and its antioxidant capabilities, CoQ10 has been recommended to treat conditions with no evidence of CoQ10 deficiency as a causative factor, such as congestive heart failure, neurodegenerative diseases, cancer, and more (43). However, in our view, whether CoQ10 supplementation truly provides benefits to any type of patient remains in need of a clear demonstration.
2. THE FUNCTION OF CoQ AS MITOCHONDRIAL ELECTRON TRANSPORTER
2.1. Requirement of CoQ in Aerobic Respiration
After its discovery, the most crucial function that CoQ has been shown to perform is as an electron carrier in the mitochondrial ETC (44–46). This key function of CoQ became evident in the late 1960s when it was demonstrated that depletion of CoQ10 from beef heart submitochondrial particles (SMPs) by pentane extraction caused inhibition of both the NADH and succinate oxidase activities and that the activities were restored upon reconstitution of extracted SMPs with CoQ at physiological concentrations (47, 48).
Mitochondrial complex I (CI) is the nicotinamide adenine dinucleotide (NADH)-CoQ oxidoreductase. Reduction of CoQ by electrons from CI is the last step of the electron transfers between sites across CI, which, as a whole, powers proton (H+) translocation across the IMM into the intermembrane space (FIGURE 2). CoQ also accepts electrons from complex II (CII is the succinate CoQ reductase), a process that does not translocate protons across the inner membrane directly but participates in respiratory chain function by increasing the size of the pool of reduced CoQ (CoQH2). CoQH2 resulting from electron transfer from CI and CII and other metabolic enzymes enters complex III (CIII is the cytochrome bc1 complex), where it transfers electrons to cytochrome c (cyt c) and thus becomes reoxidized (FIGURE 2). In CIII, CoQ undergoes the Q cycle (see below), whose net result is the translocation of 4 protons across the IMM for the full oxidation of each CoQH2 molecule (46). This ends the role of CoQ in electron transport and in creating the mitochondrial transmembrane potential and proton gradient (49).
FIGURE 2.
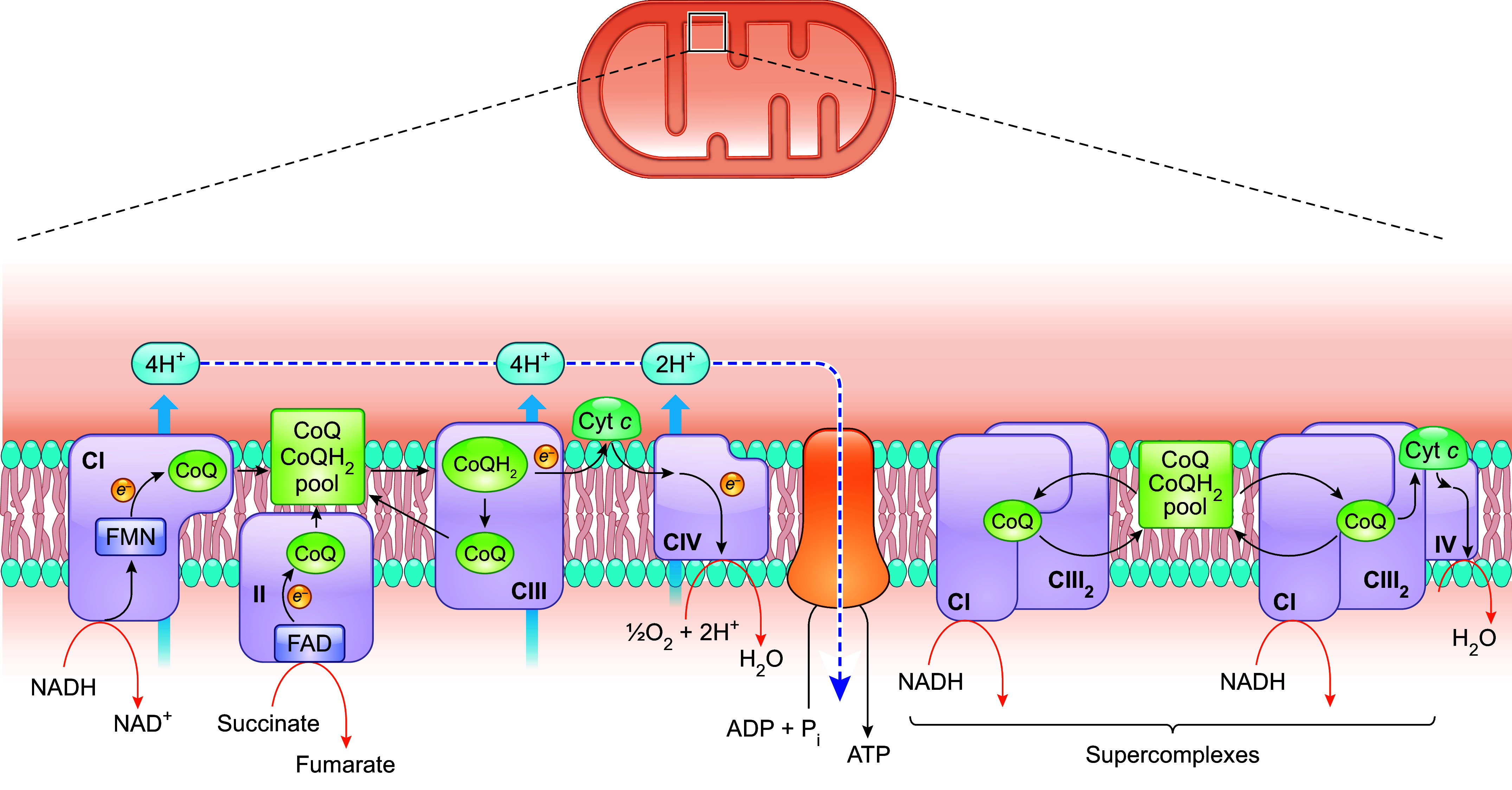
Functions of CoQ in the mitochondrial respiratory chain. CoQ is a pivotal component of the mitochondrial electron transport chain, acting as a mobile electron carrier shuttling electrons from CI and CII to CIII. During this process, CoQ cycles between reduced and oxidized states. In addition to moving randomly and colliding with CI and CII, CoQ is also present in CI- and CIII-containing respiratory supercomplexes (SCs), formed by the dynamic association of ETC complexes. In SCs, CIII is normally observed as a dimer (CIII2). All CoQ in the IMM likely behaves as a single functional pool, that is, CoQH2 can diffuse out of the CI and CIII assembled in SCs and become oxidized by CIII found outside of SCs. Conversely, CoQH2 generated independently of SCs can diffuse in, and be oxidized by, CIII attached to CI assembled in SCs. See glossary for other abbreviations.
It has been suggested that only 10–32% of total mitochondrial CoQ is bound to membrane proteins (50, 51). The classic liquid-state or random-collision model postulates that in the IMM there exists a bulk CoQ pool that is accessible to all the dehydrogenases that need to donate reducing equivalents to CoQ. In this model, CoQ diffuses freely within the lipid bilayer and electron transfer occurs after random collisions between CoQ and the enzymes that are themselves diffusing in the plane of the IMM, including the ETC complexes (52). This view, however, has been partially abandoned, after the discovery that individual ETC complexes can assemble into a variety of supramolecular structures known as supercomplexes (SCs) (53–57). The major SCs identified comprise CI/CIII2 (CI associated with a CIII dimer), CI/CIII2/CIV, and CIII2/CIV. The SC formed by CI, CIII2, and CIV is also known as the respirasome because, in principle, it has all the elements required to carry out respiration (53). There is evidence that molecules of CoQ are present in SC assemblies, more specifically in the lipid boundary between CI and CIII, and that the electron transfer between the two complexes occurs through CoQ trapped within (53, 58, 59). In fact, purified SCs (CI/CIII2 and CI/CIII2/CIV) were shown to be functional, being able to transfer electrons without the addition of any external CoQ (60).
One early hypothesis was that the purpose of SCs might be to mediate substrate channeling to enhance metabolic efficiency. This would mean that each SC sequesters its own subpopulation of the mobile electron carriers (CoQ and/or cytochrome c) and electron transfer between two sequential enzymes (CI-CIII and CIII-CIV) occurs by successive reduction and reoxidation of the mobile intermediates that are enclosed in internal channels connecting one enzyme active site to another (50, 61). Such channels would prevent the reaction intermediates from diffusing into the bulk membrane pool, thus minimizing the distance that the intermediates must travel between active sites, with an overall effect of increasing electron transport efficiency (50, 60). However, no robust evidence has yet been found that indicates the presence in SCs of confined spaces that connect active sites and retain mobile redox cofactors within (57, 62, 63). Rather, enzyme kinetic analyses and experiments where the addition of an alternative CoQH2 oxidase (AOX) to bovine heart mitochondrial membranes caused a substantial increase of the electron flux through the CI/CIII2/CIV suggest that there is no sealed-in CoQ pool in SCs (64, 65). AOX from plants directly oxidizes CoQH2, using oxygen (O2) as the terminal electron acceptor. The fact that it can compete with the CIII/CIV pathway for electrons indicates that it has access to the CoQH2 pool. In mammalian mitochondria, CI is shown to be mostly associated with other complexes in SCs (54). Therefore, if there is direct substrate channeling of CoQ in SCs, the presence of AOX outside of the SC structure should have a negligible effect on electron flux from CI to oxygen. The fact that the addition of AOX was found to increase the NADH oxidation rate suggests that CoQH2 can diffuse out of SCs to react with AOX (65). Furthermore, functional and structural characterization of mammalian SCs (from ovine heart mitochondria) demonstrated the existence of CoQ in three of four possible CoQ-binding sites in CI/CIII2 SCs: the two Qi sites and one of the two Qo sites of the two CIII. The study also showed that CoQ trapping in the SC actually reduces CI activity, which is also inconsistent with the substrate channeling hypothesis (59).
Even without direct channeling within SCs, the assembly of SCs could still decrease the traveling distance for the electron carriers and thus facilitate more efficient electron transfer (59, 66). As discussed in sect. 3.1, the ETC is the major site of ROS production in the cell. During respiration, electrons can escape from the ETC and be captured by molecular oxygen, thus generating superoxide (O2•−). CoQ in the ubisemiquinone state is known to be one of the sources of electron leakage. Overall, although the exact nature and role of SC formation are not yet clear, an often-accepted view is that it is beneficial. By facilitating electron transfer, it potentially increases respiration rate and lowers electron leakage to molecular oxygen, thus boosting OXPHOS efficiency and minimizing ROS generation (67, 68). Furthermore, a role in supporting the structural stability of the individual complexes has been proposed (56). Supporting evidence shows that respiratory activity is enhanced when more SCs are formed and an organism’s fitness is compromised when SCs formation is impaired (69–71).
As mentioned above, in mammalian mitochondria, it is believed that all or most of CI (≥90%) is associated with other complexes in SCs (54). Whether CII participates in any SC formation is still an open question (72). It has been proposed that, given the likelihood of free CoQ diffusion in and out of SCs, the overall electron flux through the ETC occurs by a mixture of electron transfer in SCs and random collision events between the two mobile electron carriers (CoQ and cyt c) and individual ETC complexes. This is consistent with data that show that all CoQ in the IMM (whether or not associated with SCs) behaves as a single functional pool (57, 59, 73). In other words, CI, CII, and other enzymes that deliver electrons to CoQ (see sect. 2.2) compete for the same CoQ pool (64, 74). However, it is worth noting that this is still a matter of controversy (50, 56, 65). Furthermore, as discussed further in sect. 2.3, CoQ deficiency is usually found to be associated with a partial loss of both CI- and CII-mediated respiration, which is not in support of the existence of two segregated CoQ pools.
2.2. Other Electron Transport Pathways That Deliver Electrons to Mitochondrial CoQ
In addition to the electrons that the two respiratory complexes CI and CII transfer to CoQ, it also receives electrons from at least seven other dehydrogenases that are associated with the IMM, either on the intermembrane space side or on the matrix side. These include 1) the mitochondrial glycerol 3-phosphate dehydrogenase (G3PDH), a part of the glycerophosphate shuttle (75), 2) the mitochondrial dihydroorotate dehydrogenase (DHODH), an enzyme involved in a key step in the production of pyrimidine nucleotides (76), 3) the electron transport flavoprotein dehydrogenase (ETFDH), a key enzyme of fatty acid β-oxidation and amino acid catabolism, 4) proline dehydrogenase (PRODH) and proline dehydrogenase 2 (PRODH2), both of which are involved in proline, glyoxylate, and arginine metabolism, 5) choline dehydrogenase (CHDH), which is primarily found in liver and kidney in humans and catalyzes the oxidation of choline to glycine betaine (77), and 6) sulfide-quinone oxidoreductase (SQOR), which is essential for detoxification of hydrogen sulfide (H2S) (78). Like CII, these dehydrogenases reduce flavin adenine dinucleotide (FAD) to FADH2, which then transfers electrons to CoQ, but these processes are not coupled to proton translocation to the mitochondrial intermembrane space (IMS) because FADH2 and CoQ have similar reduction potentials and therefore these transfers do not result in a sufficiently large gain in Gibbs free energy to power proton translocation. To date, there is not much known about how tightly the rates at which these metabolic pathways function are linked to the level of CoQ in the IMM or whether an impact on these pathways contributes to the pathophysiology of CoQ deficiency. Below we briefly describe two of the enzymes whose activities have been reported to be affected by CoQ deficiency.
Eukaryotic cells devoid of mitochondrial DNA (ρ0) need supplementation with uridine to sustain viability. This is because DHODH, which catalyzes a crucial step in intracellular de novo pyrimidine biosynthesis (conversion of dihydroorotate to orotate), needs CoQ as a cofactor (FIGURE 3). The activity of DHODH is inhibited in ρ0 cells because of the loss of the ETC and the resultant lack of oxidized CoQ to which electrons can be transferred. Uridine is a downstream product of DHODH and therefore needs to be provided to ρ0 cells to compensate for the lack of endogenous pyrimidine biosynthesis, which is essential for RNA/DNA synthesis (76, 79). Uridine was reported to improve the growth rate of human COQ2 mutant fibroblasts that have <20% residual CoQ10, suggesting the possibility of a deficit of pyrimidine biosynthesis in these cells (80, 81). In contrast, no exogenous addition of uridine to the culture medium was needed for Pdss2/Coq7 double-knockout mouse embryonic fibroblasts (MEFs), despite being completely devoid of detectable CoQ, and in fact these cells showed no sign of any growth defect under standard culture conditions in medium that contained sufficient glucose (82, 83). Normal culture medium contains a minimal amount of CoQ10. Thus, in contrast to ρ0 cells, Pdss2/Coq7 double-knockout cells sustain some ETC activity at an extremely low level despite a complete lack of CoQ biosynthesis (83). This low level of CoQ and respiratory function appears to allow for adequate pyrimidine synthesis, suggesting a very minimal requirement for mitochondrial respiratory function to maintain sufficient DHODH activity for cells to survive.
FIGURE 3.

CoQ is a cofactor for mitochondrial dihydroorotate dehydrogenase (DHODH). DHODH catalyzes the oxidation of dihydroorotate to orotate during the fourth step of the de novo biosynthesis of pyrimidine. The reaction is coupled to the reduction/oxidation of flavin mononucleotide (FMN) and CoQ. Orotate diffuses back to the cytosol to be converted by uridine monophosphate synthase (UPMS) to uridine 5-monophosphate (UMP), the precursor of all pyrimidine nucleotides. Preexisting uridine can be phosphorylated to UMP by uridine kinase (UCK), thereby bypassing the need for the DHODH-catalyzed step. See glossary for other abbreviations.
The IMM flavoprotein protein sulfide-quinone oxidoreductase (SQOR) is the first enzyme to act in the mitochondrial metabolism of hydrogen sulfide (H2S). It catalyzes two-electron oxidation of H2S and utilizes CoQ as the electron acceptor, thus coupling the reaction to CoQ in the ETC (84–86) (FIGURE 4). The oxidized sulfur is transferred to a small-molecule acceptor, which is predicted to be primarily glutathione (GSH) under physiological conditions (84). Glutathione persulfide (GSSH) produced by SQOR is converted to sulfite () which is further catabolized by thiosulfate sulfurtransferase (TST, also known as rhodanese) or sulfite oxidase (SUOX) to produce thiosulfate () or sulfate () (84). This sulfide oxidation pathway plays a key role in governing cellular H2S levels (85). H2S has toxic properties but also functions in regulating homeostasis as a cell signaling molecule (78). In human skin fibroblasts, a ≤50% reduction in CoQ10 levels was shown to cause an impairment of SQOR-driven oxygen consumption (87). Moreover, accumulation of H2S, a direct consequence of impaired sulfide oxidation, was reported for CoQ-deficient fission yeast and mouse tissues (87–89). Other abnormalities related to H2S accumulation include depletion of GSH, reduction of thiosulfate (), increased protein sulfhydration, and increased blood levels of C4-C6 acylcarnitines, consistent with inhibition of short-chain acyl-CoA dehydrogenase (SCAD), a known toxic effect of H2S (87, 89–91). Interestingly, among the mouse tissues examined, including the kidney, brain, and muscle, the kidney showed the most pronounced accumulation of H2S (87, 89). High levels of sulfide were observed in the kidney of two different CoQ9-deficient mouse models (Pdss2kd/kd and Coq9R239X) which have <15% residual CoQ9 levels, whereas in the cerebrum of Coq9R239X mice (with 10–15% residual CoQ9) and the whole brain of Pdss2kd/kd mice (with ≈30% residual CoQ9), the levels of sulfides were shown to be similar to those in wild-type control mice (87, 89). Somewhat surprisingly, CoQ deficiency decreases SQOR levels, worsening the effect on sulfide metabolism (87, 89, 90). Conversely, supraphysiological levels of CoQ10 (>2,300 fold!) were shown to upregulate SQOR expression in cultured skin fibroblasts, and a similar effect was observed in the liver of wild-type mice after supplementation with CoQ10H2 (92). Moreover, the amount of reduced SQOR in mutant HeLa cells with ≈50% residual CoQ10 was shown to be elevated after CoQ10 supplementation (90). Long-term CoQ10 treatment was shown to partially rescue decreased SQOR protein levels in the kidney of Pdss2kd/kd mutant mice despite only a small rise in CoQ10 levels, suggesting a high sensitivity of SQOR levels to CoQ levels (90). The mechanisms underlying the connection between the levels of CoQ and SQOR expression are not understood. It also remains to be elucidated how altered sulfide metabolism participates in the development and progression of kidney disease due to CoQ deficiency and what possible significance the CoQ-SQOR connection could have for CoQ10 supplementation therapy.
FIGURE 4.

CoQ levels modulate sulfide-quinone oxidoreductase (SQOR) activity. SQOR catalyzes the initial oxidation of hydrogen sulfide (H2S) and utilizes CoQ as the electron acceptor. Sulfur is primarily transferred to glutathione (GSH) under physiological conditions, forming glutathione persulfide (GSSH), which is then oxidized by sulfur dioxygenase (SDO), to produce sulfite () and regenerate GSH. SDO is also known as ethylmalonic encephalopathy protein1 (ETHE1), as its mutations are associated with ethylmalonic encephalopathy, an infantile metabolic disorder. GSSH is also a substrate for thiosulfate sulfurtransferase (TST), which converts to thiosulfate (). Alternatively, is converted to sulfate () by sulfite oxidase (SUOX) residing in the intermembrane space. A further effect of H2S accumulation is inhibition of the enzymatic activity of short-chain acyl-CoA dehydrogenase (SCAD) that catalyzes the first reaction in the β-oxidation of short-chain fatty acids. Elevated blood butyrylcarnitine (C4) is the hallmark biomarker of SCAD deficiency. In addition to acting as a cofactor of SQOR, CoQ levels regulate SQOR transcriptionally by an unknown mechanism. See glossary for other abbreviations.
2.3. CoQ Concentration and Respiratory Capacity
Most CoQ (>84%) is believed to be free in the bilayer (74). A direct measurement of the amount of CoQ associated with mitochondrial membrane proteins in five different mammalian species (namely mouse, rat, rabbit, pig, and cow) has shown values between 10% and 32% of total CoQ to be protein bound (51, 93). Kinetics studies of CoQ reduction, performed in vitro on mitochondria or submitochondrial particles (inverted vesicles of the IMM), suggest that mitochondrial CoQ concentration is limiting for NADH oxidation by CI. That is, endogenous CoQ concentration in the mitochondria appears to be lower than that allowing maximal NADH oxidation rates (94–97). On the other hand, the normal concentration of CoQ in the IMM appears to be saturating for succinate oxidation by CII (94, 96, 97). Furthermore, in agreement with the kinetic data, it was shown that the incorporation of excess CoQ10 into native beef heart SMPs (by cosonication) induces an increase in NADH oxidation rate, but no rate increase was found for succinate oxidation (97). These are important observations because if the normal endogenous CoQ concentration is limiting, then increasing it could improve respiration, possibly even in the presence of defects in mitochondrial function. However, it should be added that most of the kinetic studies required lyophilization and the use of organic solvents (e.g., pentane) to extract and reconstitute CoQ back into membranes. These are harsh treatments that may seriously perturb the native membrane environment. For example, they could cause SC disassembly. Furthermore, the kinetic studies were mostly conducted with beef heart mitochondria. Thus, how much these in vitro observations are relevant to the in vivo situation and mitochondria of different organisms and tissues needs to be further established.
Under in vitro culture conditions, adding CoQ10 to cells with normal CoQ levels has only sometimes been found to have positive effects on mitochondrial respiration. For example, one study reported that supplementation with a water-soluble CoQ10 formulation resulted in an elevation of uncoupled cellular respiration in T67 human glioma and H9C2 rat myoblast cell lines, measured with a respirometry chamber (41). Other studies showed that treatment with CoQ10 had no effect on mitochondrial respiration in human skin fibroblasts and in a rat pancreatic beta cell line (INS-1), measured with a Seahorse XF Analyzer (98, 99).
Extensive studies have been conducted on the effect of CoQ deficiency on mitochondrial respiration. Overall, as expected, CoQ deficiency impairs respiratory function, but this is only observed under conditions of severe CoQ deficiency. In E. coli, CoQ8 functions in the aerobic respiratory chain in the cytoplasmic membrane, where it serves to transfer electrons from various substrate-specific dehydrogenases to two terminal oxidases, cytochrome bo3 and cytochrome bd (100). E. coli mutants without CoQ8 biosynthesis (ΔubiA, ΔubiB, ΔubiE, ΔubiF, ΔubiH, ΔubiG) or with a very low level of CoQ8 (<15%) (ΔubiX) grow poorly on nonfermentable succinate, which is indicative of a respiratory defect (101–105). In contrast, ΔubiI and ΔubiK mutants that produce 15–20% of the normal level of CoQ8 showed no growth defect on nonfermentable carbon sources, whereas the ΔubiIΔubiK double mutant, which produces no CoQ8, cannot grow at all on succinate (106–109). As discussed in sect. 5.1.1., yeast mutants lacking CoQ6 biosynthesis are also respiration defective. Interestingly, some findings suggest that CoQ is actually required to stabilize CIII, but how much the effect on CIII stability contributes to the mutant phenotype is not clear (110).
In mammalian cells, a decrease of CoQ levels below ≈60–70% of normal levels was shown to cause an inhibition of CoQ-dependent ETC activities (CI-III and CII-III) as well as a reduction in respiratory capacity and ATP levels. These observations were mostly made in dermal fibroblasts obtained from patients or in mouse embryonic fibroblasts (MEFs) from mutants with defective CoQ biosynthesis (80, 83, 111–118). For other cell types, a pronounced depression of respiration was shown for mature murine brown adipocytes and T67 human glioma cells whose CoQ content was depleted to a similar degree (≈50–60% reduction of CoQ) by treatment with a COQ biosynthesis inhibitor (119, 120). It is worth noting that it is likely that the requirement for CoQ, especially for functions other than mitochondrial respiration, varies considerably among different cell types and under different physiological and pathological conditions. Therefore, the conclusions of any study about CoQ must be viewed in the context of cell types and experimental conditions.
Studies at the tissue level are confined to the measurements of CoQ-dependent ETC functions in whole tissues or mitochondria from genetic CoQ deficiency models in mice. The effects of reduced CoQ production on ETC function have been reported for the heart, skeletal muscle, brain, kidney, and liver, which revealed significant variation in the sensitivity to CoQ deficiency across different tissues (30, 82, 90, 121–124). In the liver, an almost complete depletion of CoQ obtained by genetic means in hepatocytes causes only mild or moderate impairment of ETC function (30, 82). However, in the kidney, brain, and heart, which are known to be more energy demanding, a greater loss of respiratory function was found to always accompany severe CoQ deficiency (121, 123). Nonetheless, full respiratory function was observed in mouse kidney mitochondria with less than half of the normal level of CoQ, which is in contrast to what was observed in the heart, where ≈35% of wild-type CoQ levels were found to only sustain about one-half of full oxidative phosphorylation capacity (state 3 respiration) (121, 123). It still remains poorly understood how CoQ deficiency affects individual tissues and cell types. The variation in the relation of CoQ level to mitochondrial respiration may reflect, at least in part, tissue differences in other CoQ functions besides its role in the ETC. The heart is one of the most energy-consuming organs in the body and rich in mitochondria. Thus, likely there is an unusually high proportion of cellular CoQ associated with the ETC in cardiomyocytes. One therefore expects a high correlation between CoQ levels and mitochondrial respiration in cardiomyocytes. In contrast, as further discussed in sect. 3.3.1.2, studies of Pdss2kd/kd mutant mice showed that oxidative stress, apparently caused by impaired H2S oxidation, is most prominent in the kidney, and kidney failure is the primary phenotypic consequence of CoQ deficiency in this strain. A small increase in tissue CoQ10 level after long-term supplementation is sufficient to alleviate oxidative stress and kidney pathology of the mutant (90, 124). We postulate that, although the kidney is also relatively enriched in mitochondria, respiration is not the main consumer of CoQ. As CoQ is made in mitochondria, in the kidney the requirement of CoQ for ETC function might be relatively easily met but not the requirements for CoQ functions that require CoQ export from the mitochondria and distribution to other membranes, which might suffer more. And this might be the case for the place where the antioxidant function of CoQ is so crucially needed. For the liver, whose respiratory function appears to require very little CoQ, the explanation could be in the fact that it is in hepatocytes that dietary CoQ accumulates and is incorporated into lipoproteins (see sect. 3.2.5).
3. DUAL PROOXIDANT AND ANTIOXIDANT ROLES OF CoQ
A free radical is an atom or molecule that contains one or more unpaired electrons. Because of the possession of odd electrons, free radicals are usually unstable, short lived, and highly reactive (125). They can be stabilized by losing or gaining electrons through interactions with other atoms or molecules (to which they provide or from which they steal an electron). This, in turn, can alter the chemical properties of the entities with which they interact. In biological systems, free radicals are mostly oxygen- or nitrogen-containing species, namely reactive oxygen species (ROS) and reactive nitrogen species (RNS), respectively. Their production is part of normal metabolism and an inevitable consequence of aerobic life (126). Under normal physiological conditions, the intracellular levels of ROS and RNS are maintained at low concentrations. Conversely, when produced in excess, their highly reactive nature makes them potentially harmful through their ability to damage macromolecules, such as lipids, proteins, and DNA, which can lead to irreparable cell damage and death (127, 128). ROS and RNS have also been recognized as signaling molecules involved in regulating various physiological processes (129, 130). Therefore, for cell health and survival, a delicate balance must be maintained between ROS and RNS production and elimination (131, 132). In general, an antioxidant is defined as any substance that is capable of neutralizing reactive free radicals into a relatively stable unreactive form. Cells are equipped with antioxidant defense systems, consisting of both ROS-scavenging enzymes (such as superoxide dismutase and catalase) and various nonenzymatic compounds, to neutralize ROS or RNS directly or through enzymatic reactions (133).
The principal ROS produced spontaneously or enzymatically in biological systems is the superoxide anion radical (O2•−), which results from the one-electron transfer to an oxygen molecule. The discovery of superoxide dismutase (SOD), a unique enzyme that converts O2•− into hydrogen peroxide (H2O2), helped launch the free radical theory of aging, which is centered on the accumulation of ROS-caused damage with time (134). ROS are generated by various sources, among which the mitochondrial ETC is one of the principal endogenous ROS generators. There are 12 sites in the mitochondria, with links to the ETC, that have been identified in mammalian cells to be capable of leaking electrons to oxygen and generating O2•− (135). CoQ is one of the major ROS-generating sources in the ETC. During CoQ-mediated electron transport a partially reduced state of CoQ, ubisemiquinone (CoQ•−) is produced as an intermediate that can donate one electron to molecular oxygen, resulting in the formation of O2•− at the CoQ binding sites of ETC complexes. Yet it remains to be established to what extent the amount of CoQ and its redox state contribute to total mitochondrial ROS in a given cell or cell type in a particular physiological state. On the other hand, CoQH2, the fully reduced form, can neutralize free radicals or regenerate other antioxidants, by giving up its own electrons, especially in the lipid membranes where it resides. In fact, CoQ is widely hailed as an antioxidant, and this property along with its key role in mitochondrial bioenergetics is the rationale given for providing CoQ10 as a health supplement. In this section, we summarize findings and analyses in support of the dual pro- and antioxidant role of CoQ.
3.1. Roles of CoQ in Mitochondrial ROS Generation
Superoxide (O2•−) is produced by one-electron reduction of molecular oxygen. SOD converts O2•− to H2O2, which is believed to play a central role in redox signaling. However, the reactivity of H2O2 itself can in turn lead to the formation of other reactive species, such as the very damaging hydroxyl radical (•OH) (136). O2•− also reacts with nitric oxide (NO•) to produce peroxynitrite (ONOO•), a toxic RNS. In fact, the reaction rate constant of O2•− with NO• (6.7 × 109 M−1 s−1) is several times faster than the rate constant of the action of SOD on O2•− (1.6 × 109 M−1 s−1) (137). Thus, changes in NO• levels can potentially affect O2•− levels and hence the cellular redox state. Conversely, excessive O2•− can have an impact on the level of NO• as a signaling molecule and on nitrosative stress as a result of increased production of ONOO• (138, 139).
In most cells, the ETC is the major O2•− production site, except in phagocytes, where ROS are deliberately produced by NADPH oxidases (NOX) to produce an oxidative burst designed to kill pathogens in the phagosome (140–142). It is commonly repeated that mitochondria generate ∼90% of cellular ROS and during mitochondrial respiration ∼0.2–2% of the molecular oxygen consumed is reduced to O2•− (143–146). However, the actual numbers are still debated. One commonly used method to measure total ROS produced by isolated intact mitochondria is to use Amplex Red dye, which, in the presence of H2O2, can be oxidized by horseradish peroxidase to give rise to a fluorescent oxidation product, resorufin (147). Although O2•− does not readily cross membranes, SOD is provided at a high concentration in the assay’s medium to ensure that all O2•− produced is actually converted to H2O2. It is because of this method that in the text below we sometimes refer to O2•−/H2O2 generation, although the species that is expected to be formed at a site of interest is O2•−.
Studies with isolated ETC complexes and mitochondria have identified a number of sites of ROS production including the CoQ binding sites of CI and CIII. CII is not normally a substantial source of ROS production by mitochondria. In conditions under which ROS production is induced from mammalian CII, it is the flavin site, not the CoQ binding site IIQ, that is the most likely source of electron leak (148–150). Interestingly, it is also worth noting that among the other IMM dehydrogenases, mitochondrial G3PDH (mGPDH) was shown to be capable of producing significant amounts of ROS, with CoQ suggested to be the source of ROS in this process (150, 151).
Reduction and oxidation of CoQ in mitochondria occur in two sequential one-electron steps (152). Inevitably, the process involves the creation of a partially reduced form of CoQ (CoQ•−) as an intermediate (FIGURE 1) (153, 154). As mentioned, CoQ•− is a source of mitochondrial O2•− because of its propensity to donate its unpaired electron to O2. Indeed, CoQ•− signals were detected at the CoQ binding sites of the ETC complexes CI, CII, and CIII by electron paramagnetic resonance (EPR) (152, 154–157). The capability of CoQ to participate in O2•− formation was first demonstrated in beef heart SMPs from which CoQ10 was extracted and then replenished (153). SMPs are inverted (inside out) vesicles of the IMM (FIGURE 5). As they maintain the structural integrity of the IMM and have the substrate binding sites exposed to the outer surface, they have been a valuable tool for mitochondrial functional studies. Later studies also used electron transport inhibitors specific to particular ETC complexes or sites (such as antimycin A and potassium cyanide) and, more recently, electron leak suppressors for specific CoQ sites (147, 158–160). These studies further established the contribution of different CoQ binding sites to ROS production by mitochondria during the oxidation of different substrates. Yet, not surprisingly given the chemo-physical complexity of the reactions involved, many uncertainties remain.
FIGURE 5.
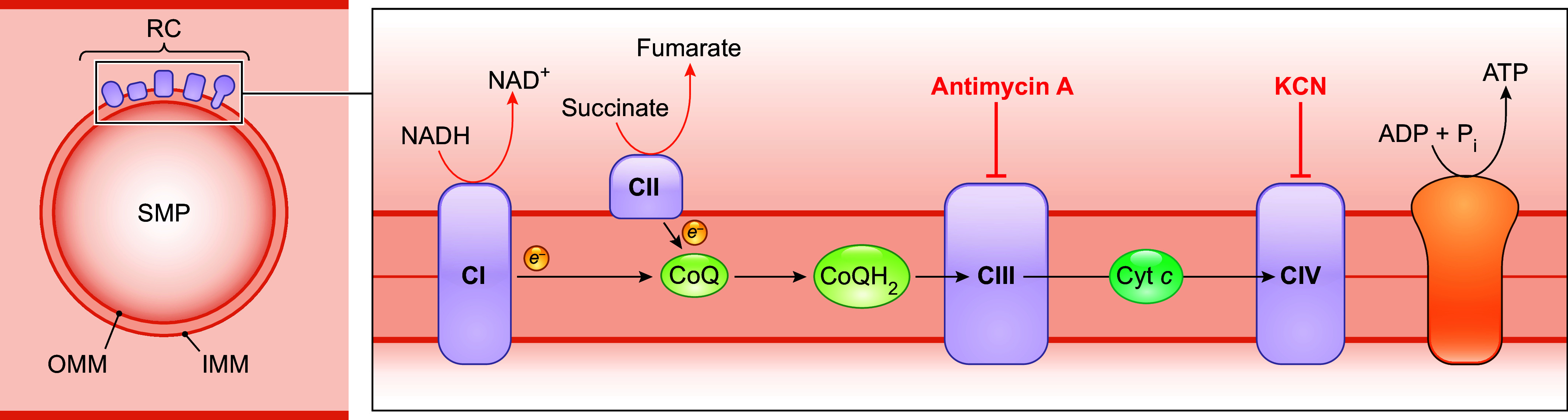
Diagram of a submitochondrial particle (SMP). A SMP is an inside-out vesicle of the inner mitochondrial membrane (IMM). It retains all the respiratory chain (RC) components, and the inversion of the IMM exposes CI and CII to the medium, allowing unrestricted access to oxidation substrates, including NADH, which could not pass through the IMM. See glossary for other abbreviations.
3.1.1. Role of CoQ in ROS production from complex I.
3.1.1.1. ros production by complex i during forward electron flow.
At CI, electrons move from NADH to the flavin mononucleotide (FMN, the IF site) to iron-sulfur clusters, and finally to CoQ (FIGURE 6). CI from the yeast Yarrowia lipolytica and E. coli were shown to contain 0.2–1 CoQ molecules per complex (161). The CoQ reduction site (the IQ site) is located at the junction of the hydrophobic membrane arm and the hydrophilic matrix arm (162). CI-linked substrates (i.e., glutamate or pyruvate in combination with malate) that feed electrons from NADH to the respiratory chain in the forward direction (starting from the IF site) are generally considered to give low rates of O2•− production, and ROS production under these conditions largely originates from the IF site (FIGURE 6). In other words, the IQ site normally does not dominate ROS production from CI, although this is debated (147, 163–165).
FIGURE 6.
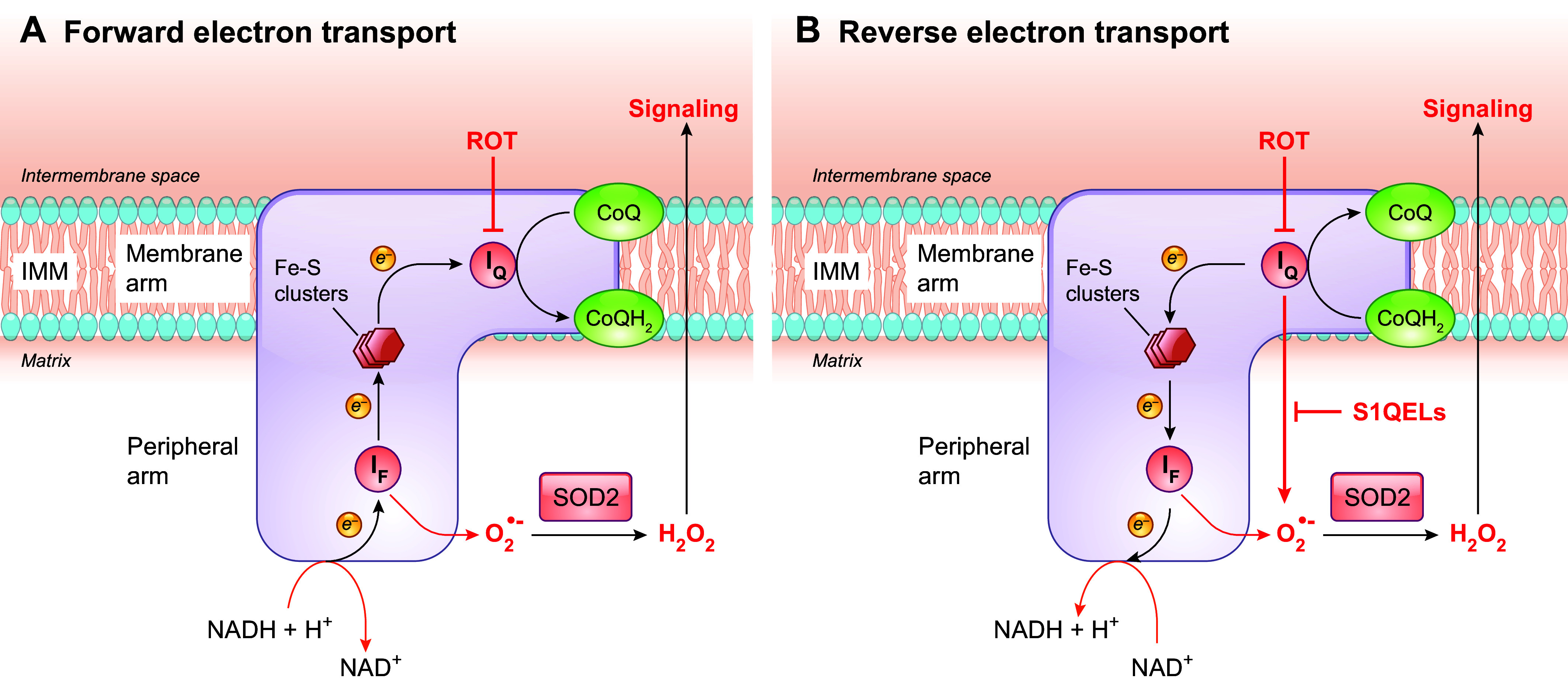
ROS production sites in CI. A: with forward electron transport, NADH is oxidized at the flavin mononucleotide (FMN, the IF site). Electrons are then passed via several iron-sulfur (Fe-S) clusters to the CoQ binding site (IQ), where CoQ is reduced before it dissociates from CI. B: reverse electron transport occurs when electrons from an overreduced CoQ pool flow back to CI and reduce NAD+. The IF site has been considered to be the main site of ROS production from CI under the oxidation of NADH-linked substrates. ROS production during reverse electron transport mainly originates from electron leakage from reduced CoQ formed at the IQ site, but the IF site has also been shown to contribute. Rotenone (ROT) blocks the flow of electrons by inhibiting the binding of CoQ to IQ, whereas S1QELs suppress electron leak from CoQ•− to oxygen at the IQ site specifically. They do this without interfering with normal electron flow, and therefore this is expected to affect ROS generation during reverse electron transport. The O2•− produced by CI is released into the matrix, where superoxide dismutase 2 (SOD2) converts it to H2O2. See glossary for other abbreviations.
3.1.1.2. ros production by complex i during reverse electron flow.
CI can also produce ROS when electrons flow through CI in the reverse direction. That is, electrons flow back from CoQH2 to CI and reduce NAD+ to NADH. Reverse electron transport (RET) has been known since the 1960s. It was first associated with ROS production in well-coupled SMPs, and it was later also shown to take place in isolated mitochondria from different tissues (166, 167). The conventional substrate to drive RET is the CII substrate succinate. In fact, in the setting of isolated mitochondria, succinate-induced RET produces the highest rate of ROS production (167, 168). Although the question of exactly where ROS are produced during RET is still controversial, it has been largely accepted that the CoQ binding site in CI, the IQ site, is one of the prime loci of electron leak during RET (FIGURE 6) (135). Superoxide production by CI during RET is sensitive to the classic IQ site inhibitors, such as rotenone and piericidin A (147, 169, 170). These inhibitors block the binding of CoQ to IQ, thus preventing the possibility of electron escape from CoQ•− to oxygen (171). However, their use also inhibits reverse electron flow into CI, potentially affecting O2•− production from other sites as well. In fact, more recently, both IF and IQ sites were shown to generate ROS in mitochondria isolated from rat skeletal muscle when respiring on succinate (also see sect. 3.1.1.3) (172). Notably, in recent studies by Martin Brand’s group, a novel approach was developed that allows estimation of the rate of rotenone-sensitive O2•− production from the site IQ while considering any change of ROS production by the two other key sites (the IF site of CI and the Qo site of CIII). With this approach it was estimated that in rat skeletal muscle mitochondria under succinate oxidation, ≈83% of O2•− originates from the IQ site, whereas that site made little or no contribution when the substrates were glutamate plus malate (163). Furthermore, in a step toward understanding ROS production in muscles in vivo, it was shown ex vivo that under conditions that mimic those in resting muscles a quarter of the total O2•− production of rat skeletal muscle mitochondria could be attributed to the IQ site, whereas the IF site of CI became the dominant contributor (≈99%) under conditions mimicking intense exercise, when total O2•− production is much lower (173).
RET is energetically uphill (i.e., against the difference of redox potentials). For IQ to generate O2•− at high rates, a highly reduced CoQ pool (to provide the electrons) and a high protonmotive force (PMF) that drives protons back into the matrix through CI are necessary (167, 174, 175). These conditions are thought unlikely to occur often under normal physiological conditions. Moreover, mitochondrial succinate levels and succinate dehydrogenase (SDH) activity in normal cells are low (176). Therefore, succinate-driven RET had initially been proposed to be minimal under normal conditions but capable of being triggered by particular stresses and thus leading to damage. One of the most cited examples is ischemia-reperfusion (I/R) injury. Succinate and other metabolic substrates accumulate during ischemia, but upon reperfusion succinate is rapidly oxidized, leading to a burst of ROS production through RET, which may contribute significantly to reperfusion injury (168, 177, 178). It is noteworthy, however, that the role of succinate-driven RET in I/R injury still remains to be fully elucidated, as it has been challenged by some studies (179). RET-dependent ROS is now thought to exist also beyond pathological conditions. It has been associated with various cellular processes, including differentiation of myoblasts into myotubes, initiation of macrophage inflammatory responses, oxygen sensing by the carotid body chemoreceptors, and uncoupling of mitochondria in brown adipose tissue (180–183). Of particular interest, ROS generation from RET has been implicated in metabolic adaptation, with the CoQ redox status acting as a sensor to adjust the respiratory chain organization for optimal efficiency. During a metabolic shift from glucose to fatty acids, which increases electron flux through FAD, it has been shown that accumulation of reduced CoQ (CoQH2) induces RET and results in the local generation of ROS that oxidizes CI proteins. These events, in turn, lead to CI degradation, which liberates CIII from CI + CIII SCs to receive FADH2 electrons from CII in order to adapt to substrate utilization (24).
3.1.1.3. findings with specific suppressors of the iq site electron leak.
Additional insight on CI ROS generation and CoQ has been provided by more recent studies with novel site-selective suppressors of electron leak. As mentioned above, the classic IQ site inhibitors, such as rotenone, block the electron transfer from, or to, CoQ through the IQ site (171). This inevitably perturbs regular electron flow and thus affects ROS production from other sites as well. In the forward direction under oxidation of NADH-linked substrates, rotenone suppresses electron flow to CIII but raises ROS production from the CI sites upstream of IQ as a result of increased FMN and iron-sulfur center reduction. Conversely, during succinate oxidation, by inhibiting electrons flowing back to CI, rotenone reduces ROS production at the IQ site as well as at the IF site (172). In contrast, the newly developed S1QELs (suppressors of site IQ electron leak) can specifically suppress electrons leaking from the IQ site without interfering with normal electron flow and respiration, making them a better tool for studying ROS generation from the IQ site (172, 184). With this tool, it was shown that S1QELs can suppress ROS generation from CI without affecting reverse electron flow during RET, providing better evidence for IQ being a source of mitochondrial ROS when CoQ becomes overly reduced by electrons from CII or other enzymes. Moreover, with this tool, it was shown that ≈12% of the total rate of H2O2 release in C2C12 mouse myoblasts comes from the IQ site. After differentiation into myotubes, total ROS release was increased, and the relative contribution of the IQ site doubled (185). A similar study with several other cell lines further showed that although the absolute cellular H2O2 production rates vary considerably, the relative contribution of the IQ site to total H2O2 release is similar (range 11–26%) among the diverse cell types under unstressed conditions (186). In isolated mitochondria from rat muscle incubated in media mimicking the cytosol of resting muscle, ≈12–18% of total ROS emission was sensitive to S1QELs, consistent with the above-mentioned measurements made using endogenous reporters of H2O2 levels (173, 185). S1QELs have also demonstrated a protective effect against stress-induced stem cell hyperplasia in the Drosophila intestine and in mice I/R injury models (184). These findings argue for the physiological significance of ROS production at the IQ site. Whether all IQ site ROS production is via RET is not yet clear (165, 187).
Emerging studies suggest that RET could be favored by other conditions besides a high concentration of succinate. This includes an elevation of the activity of the other metabolic pathways that feed electrons to the CoQ pool (see sect. 2.2) and a slowdown of CoQH2 reoxidation at CIII (24, 75, 160, 163, 188). For example, the IQ site has been shown to contribute substantially (≈33%) to the total O2•− production rate when glycerol 3-phosphate is provided as a respiratory substrate (163).
Finally, it should be mentioned that different subpopulations of CoQ•− have been reported to be associated with CI (189). Rotenone-sensitive and -nonsensitive CoQ•− were first described to be detectable in bovine heart purified CI upon reduction by NADH and in SMPs from bovine heart mitochondria under oxidation of NADH or succinate (157, 190). Later studies with bovine heart SMPs and isolated CI from different species also demonstrated the presence of at least two types of CI-associated CoQ•− species with distinct spin relaxation behaviors: namely, the fast-relaxing ubisemiquinone (SQNf) and the slowly relaxing ubisemiquinone (SQNs) (154, 156, 191–196). The SQNf signal is sensitive to uncouplers and rotenone and was more obvious in the presence of the ATP synthase inhibitor oligomycin (156, 194, 196). The presence of two different EPR-detectable CI-associated CoQ•− species has been taken to indicate the presence of two spatially separated CoQ binding sites in CI. However, this is still debated (154, 156, 161, 192, 195, 197).
3.1.2. The role of CoQ in ROS production from complex III.
3.1.2.1. the q cycle.
CIII, which is also often called the cytochrome bc1 complex (cyt bc1), harbors two separate CoQ binding sites: Qo (also called Qp) and Qi (also called Qn), which face the compartments on opposite sides of the IMM. Qo is located close to the outer surface of the IMM in mitochondria and on the periplasmic side in bacteria, whereas Qi is facing the matrix (mitochondria) or cytoplasm (bacteria) (198). The Qo and Qi sites are connected electronically by two cyt b hemes, bL and bH (3). As mentioned above, CIII catalyzes a reaction of net oxidation of CoQH2 and reduction of cyt c. Oxidation of CoQH2 occurs at the Qo site and is accompanied by a CoQ reduction reaction at the other site (Qi) (199, 200). The electron transfer reactions are coupled with proton movement. That is, protons are taken up by CoQ at the Qi site, carried across the membrane by CoQH2, and released at the Qo site (200). The mechanism by which electrons are transferred from CoQH2 to cyt c and by which, at the same time, protons get translocated into the intermembrane space is known as the Q cycle (FIGURE 7). This mechanism was originally proposed by Peter Mitchell almost a half-century ago but has since been modified by several groups (46, 201–203). In essence, in the version generally adopted nowadays, the Qo site oxidizes two CoQH2 molecules in two successive steps, which provides two electrons needed to fully reduce one CoQ molecule at the Qi site. In the first step, following the binding of one CoQH2 molecule to the Qo site, the transfer of two electrons from that molecule is bifurcated. That is, one electron moves through the “Rieske” iron-sulfur protein (RISP), a component of CIII, and the cyt c1 heme before it is accepted by cyt c. The other electron enters the Q cycle, where it is routed through the cyt bL and cyt bH hemes and moves across the membrane to reach the Qi site, where it acts as an electron donor to reduce CoQ to CoQ•−. The second step is the repeat of the first, where a new CoQH2 binds to the Qo site and again one electron is sent through the cyt b chain but now it encounters a CoQ•− at site Qi. Therefore, at the end of a complete Q cycle, as a net result two CoQH2 molecules are oxidized at the Qo site and four electrons move through the Q cycle, resulting in the passaging of two electrons to cyt c and sequential reduction of one CoQ molecule to CoQH2 at the Qi site before it is released to the CoQ pool (198). Concurrently, there is a net release of four protons (H+) into the intermembrane space from the two CoQH2 molecules oxidized at the Qo site and uptake of two H+ from the mitochondrial matrix into the Qi site.
FIGURE 7.
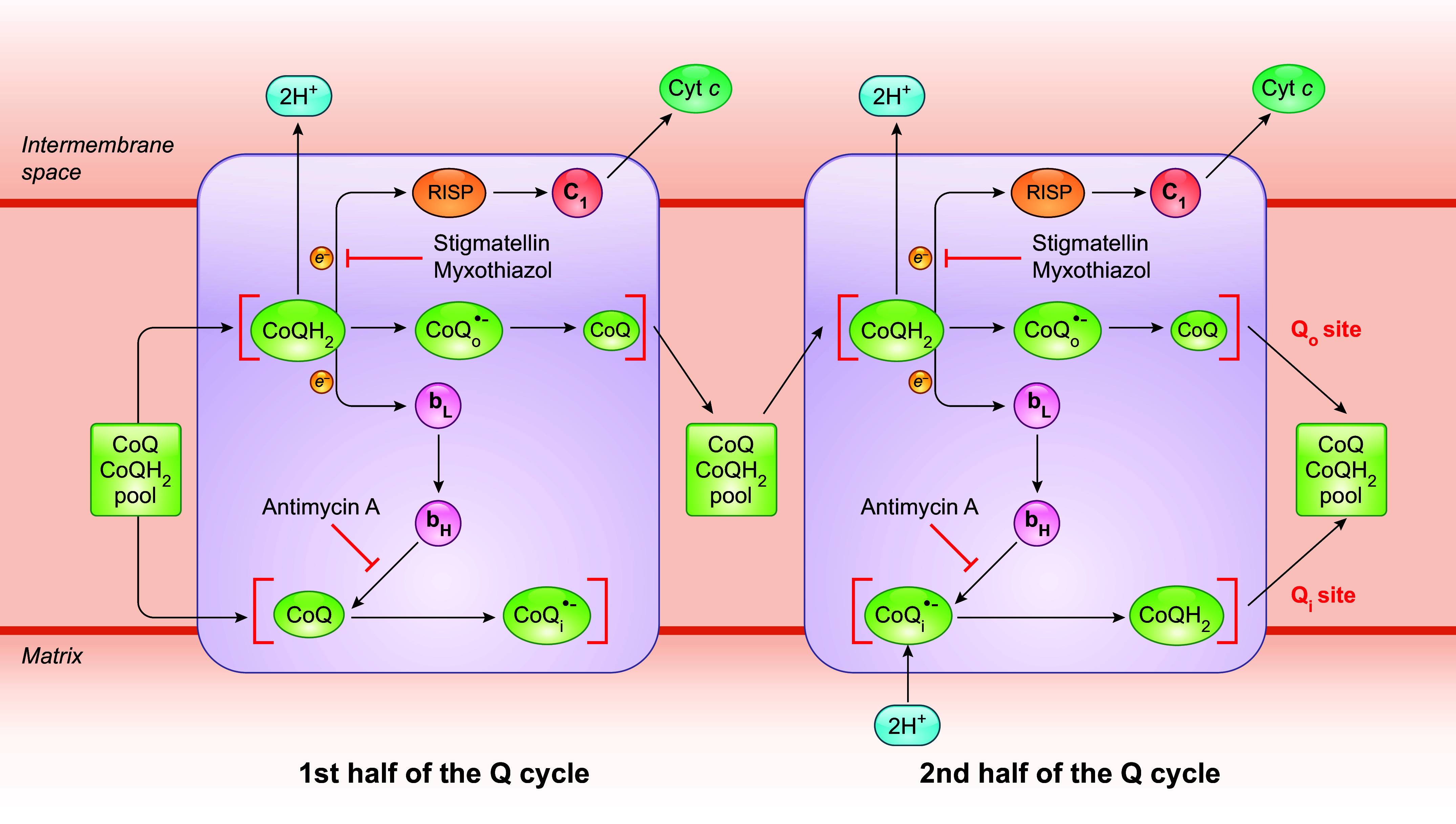
Schematics of the mechanism of the Q cycle. The Q cycle mechanism defines 2 reaction sites in CIII: CoQH2 oxidation (Qo) and CoQ reduction (Qi). The Qo site is located between the Rieske iron-sulfur protein (RISP) and heme bL, toward the intermembrane space, whereas the Qi site is close to the matrix side. It takes 2 CoQH2 oxidation cycles to complete the Q cycle. At first, a CoQH2 moves into the Qo site and undergoes oxidation, with 1 electron being transferred to RISP and then to cyt c via cyt c1. The other electron passes through 2 b-type hemes (bL and bH) across the membrane to the Qi site, where a bound CoQ is reduced to CoQ•− and finally to CoQH2. CoQ and CoQH2 are recycled back to the CoQ pool from the Qo and Qi sites, respectively, after being fully oxidized or reduced. Oxidation of each CoQH2 molecule releases 2 protons into the intermembrane space, and in the second half of the cycle 2 protons from the matrix are used to reduce CoQ•−. Given that it takes 2 electrons to fully reduce a CoQ molecule, a CoQ•− intermediate is expected to be formed at the 2 distinct CoQ binding sites. Stigmatellin and myxothiazol are Qo site inhibitors, whereas antimycin A blocks electron transfer from bH to the CoQ molecule at the Qi site. See glossary for other abbreviations.
3.1.2.2. mechanism of coqh2 reoxidation at the qo site.
A key feature of the mechanism of the Q cycle is that there are two distinct CoQ reaction sites: a CoQH2 oxidation center (the Qo site) and a CoQ reduction center (the Qi site). There has been much research aimed at understanding electron bifurcation at the Qo site, which is believed to be the only known reaction of its kind in biology. One model of Qo site catalysis postulates two sequential electron transfer steps: the first electron transfer from CoQH2 to the [2Fe-2S] cluster (ISC) of RISP, leading to the generation of a CoQ•− radical intermediate, CoQo•−, followed by the oxidation of this intermediate by heme bL in the second reaction (3, 152, 204). So far, native CoQ or CoQH2 molecules have not been resolved at the Qo position in X-ray crystallography studies (205, 206). Characterization of the bindings of different Qo inhibitors, through observations of their effects on the absorption spectrum of heme bL or the EPR spectrum and redox properties of the ISC, and later crystallographic studies of cyt bc1 complexes, suggests that separate functional domains might be present within the Qo site. For example, although myxothiazol, stigmatellin, and 5-undecyl-6-hydroxy-4,7-dioxobenzothiazol (UHDBT) all bind to the Qo site and prevent CoQH2 oxidation, myxothiazol binds to the proximal region near cyt bL, whereas the binding site of UHDBT is at a greater distance from cyt bL and interacts specifically with the ISC of RISP and stigmatellin overlaps both the distal and proximal positions (204, 207–209). Based on studies with the inhibitors, two subsites within the Qo site have been proposed: the proximal part of the site, close to the bL heme (≈7 Å from the heme bL and ≈12 Å from the ISC), and the distal part of the site, closer to RISP (≈7 Å from the ISC and ≈12 Å from heme bL) (208, 209).
Although the binding of a CoQH2 molecule to the different regions of the Qo site still remains to be formally confirmed, the possibility of two separate CoQ binding regions in the Qo pocket leads to a hypothesis that a CoQo•− intermediate generated after the first electron transfer from CoQH2 to RISP might diffuse from one subsite to another before the second electron transfer from the CoQo•− to heme bL occurs. To be more specific, it is postulated that CoQH2 is first bound in the distal part of the Qo pocket where CoQH2 transfers one electron to RISP, generating a CoQo•− intermediate. After having formed, the partially oxidized intermediate moves into the pocket at the proximal end of the site, near heme bL. The movement, together with a conformation change of the site, provides the mechanistic barrier for preventing any CoQo•− formed from further interaction with the oxidized ISC of RISP (208). Separately, an alternative double-occupancy model proposes that the Qo site can accommodate two CoQH2 molecules at the proximal and distal regions simultaneously (209–211). However, this hypothesis has been challenged by crystal structure studies suggesting there might not be enough room in the Qo site to accommodate two CoQH2 at the same time (205).
Experimental detection of CoQ•− bound at the Qo site has proven difficult. It has been variously interpreted as indicating a high instability of CoQo•−, or extreme difficulty in its detection, possibly due to magnetic coupling between CoQo•− and the reduced ISC of RISP, or the possibility that, if both electron transfers to RISP and heme bL occur simultaneously, no CoQo•− intermediate would actually be formed (3, 152, 204, 212). However, several recent studies have reported successful CoQo•− detection when its reoxidation is blocked, by the use of either a Qi site-specific inhibitor or a heme bH knockout by genetic means (155, 213, 214). These findings, though not accepted by everyone, argue against a Qo site model of simultaneous two one-electron transfers from CoQH2.
3.1.2.3. coq reduction at the qi site.
The Qi site is at the end of the cyt bL-cyt bH electron transfer chain and is situated near the matrix side of mitochondria and the cytoplasmic side of the bacterial membrane, where protons are taken up during catalysis for reduction of CoQ (215). In contrast to the situation with the Qo site, X-ray and cryo-electron microscopy (cryo-EM) structural studies of the cyt bc1 complexes have documented a CoQ occupancy within the Qi site (198, 206, 216–218). Mechanistically, heme bH reduces CoQ to CoQi•− after an electron is transferred from the first CoQH2 that moves to the Qo site and reduces CoQi•− to CoQH2 after a second oxidation event. As the CoQi•− intermediate that is formed after every first CoQH2 oxidation at Qo needs to remain at the Qi site until the second CoQH2 oxidation takes place, CoQi•− is predicted to be more tightly bound than CoQo•−. This has been regarded as a plausible explanation of why CoQi•− intermediate is easier to detect than CoQo•−. Antimycin A, the best-known Qi site inhibitor, blocks the electron transfer from cyt bH to the Qi site and thus inhibits the reduction of the CoQ pool.
3.1.2.4. ubisemiquinone is the source of mitochondrial ros generated by complex iii.
We have discussed that an unstable CoQ•− can directly reduce O2, forming O2•−, and that the CoQ•− formed at the Qo site is less stable than that at the Qi site (219, 220). Moreover, only the CoQ•− at the Qo site is thought to have a sufficiently low redox potential to be able to give an electron to oxygen (221). Thus, the Qo site, not the Qi site, is the most likely electron donor in the production of O2•− at CIII. This has been demonstrated experimentally in various types of preparations (intact mitochondria, SMPs, and isolated cyt bc1 complexes) by the use of inhibitors that bind specifically to only one of the two distinct CoQ-binding sites (3, 147). Generally, Qi site inhibitors (the most classic one being antimycin A) are found to induce high rates of ROS production, whereas Qo site inhibitors (e.g., stigmatellin and myxothiazol) suppress antimycin A-induced ROS production (209, 222, 223). These findings can be best explained by the Q cycle mechanism of CIII. In the presence of antimycin A, hemes bH and bL cannot be reoxidized by electron transfer to the Qi site. Thus, CoQ•− formed upon the first oxidation of the first CoQH2 at the Qo site is unable to donate electrons to hemes bL, resulting in longer residence of CoQo•− at the Qo site and greater probability of electron transfer to oxygen resulting in O2•− formation (224). Qo site inhibitors stigmatellin and myxothiazol on the other hand prevent the formation of CoQ•− at the Qo site and thus eliminate the stimulation of O2•− production by antimycin A. But, notably, in contrast to stigmatellin, which completely blocks O2•− production by CIII, myxothiazol only partially (by ≈70%) prevented antimycin A-induced O2•− production (225, 226). Furthermore, the effect of myxothiazol on its own also leads for O2•− formation. The rate of myxothiazol-induced O2•− production is lower than that observed with antimycin A and is highly sensitive to stigmatellin as well (209, 225, 227). Stigmatellin, as mentioned above, binds to the Qo site in the distal part of the site, near RISP. A crystal structure with bound stigmatellin shows it binding in the same position as CoQH2, to a histidine ligand of the ISC of RISP via a hydrogen bond. This would be expected to exclude CoQH2 and prevent CoQo•− formation [Crofts (198)]. Myxothiazol, however, is binding to the proximal part of the Qo site, near cyt bL (209, 216). This suggests that myxothiazol does not entirely exclude CoQH2 from binding at Qo. Overall, these findings suggest a model in which the distal part of the Qo site pocket is the main source of CoQ•− formation at Qo but both the distal and proximal parts of the CoQ binding sites transiently contain CoQ•−, with the potential to reduce oxygen and contribute to CIII ROS production.
With regard to the relative contribution of the Qo site to mitochondrial ROS, it was shown that, in rat skeletal muscle mitochondria, the Qo site made only a modest contribution (≈10%) to the total O2•− production under succinate oxidation but accounted for ≥30% of the total production rate when CI substrates (glutamate or malate) or substrates of β-oxidation (palmitoylcarnitine plus carnitine) were being oxidized. This estimate was made possible by using the state of cyt bL reduction as an endogenous reporter for the rate of O2•− formation at the Qo site (163, 228). Under ex vivo conditions that mimic rest or mild aerobic exercise, ≈15% of total O2•− was produced from the Qo site, whereas very little or no ROS was produced from the Qo site under conditions that mimic intense aerobic exercise in skeletal muscles (173). These findings are in good agreement with the reported value obtained with several S3QELs (Suppressors of site IIIQo electron leak), which selectively suppress O2•− formation from the Qo site but do not block electron flow or affect OXPHOS (185, 229). S3QELs were also used to assess the contribution of Qo-derived O2•− to cell physiology and pathology. In various cell types, treatment with S3QELs was shown to suppress the total rate of extracellular H2O2 release by a similar extent within a 13–30% range, despite the fact that the absolute cellular H2O2 production rates vary greatly among the diverse cell types (141, 185, 186). In vivo studies with Drosophila reported that feeding S3QELs protects against ROS-induced stem cell hyperplasia in the intestine, and S3QELs also decrease diet-induced intestinal barrier disruption in both flies and mice, suggesting a key role for Qo ROS in these pathologies (184, 230).
Finally, it is important to note that, in contrast to most mitochondrial ROS forming sites, which release O2•− into the matrix, the Qo site emits at least some O2•− directly into the IMS (145, 160, 186, 231, 232). As mentioned above, O2•− cannot easily diffuse through the IMM, so after being released into the matrix it is mostly confined to the matrix, where it can directly oxidize the Fe-S clusters of enzymes such as aconitase or be converted to H2O2 by SOD2 (233). H2O2 can then diffuse out of the mitochondria and function as a signaling agent. A minor fraction of cytosolic SOD1 is found in the IMS, where it can catalyze the dismutation of O2•− into H2O2 (234–236). Unlike the IMM, the OMM is porous, allowing the easy passage of small molecules including H2O2. Moreover, some of the O2•− in the IMS may also escape into the cytosol via voltage-dependent anion channels in the OMM (237). Consequently, O2•− and H2O2 produced in the IMS should have easier access to the cytosol, where they can act as signaling molecules. For example, several studies showed that ROS from the Qo site is required for the stabilization of the HIF-1α protein during hypoxia, although conflicting findings have also been published (238, 239).
3.2. The Antioxidant Function of CoQ
It is well known that by virtue of its chemical properties, CoQ can act as an antioxidant. In its reduced state, it can give up its electrons to free radicals, thus stabilizing them and neutralizing their reactivity (240, 241). CoQ is also known for its ability to help regenerate other antioxidants such as vitamin E back to their active states. CoQ appears present in all cellular membranes, where its reduced form (CoQH2) can be restored from the oxidized form (CoQ) by various enzymatic mechanisms, and its primary antioxidant role is believed to act on lipid radicals generated when lipids are peroxidized (12). In fact, significant amounts of CoQH2 can be measured in various membrane fractions (including the plasma membrane and endomembranes) (242, 243). The antioxidant activity/capacity of CoQ is doubtlessly dependent on both the total amount of CoQ and the ratio between reduced and oxidized forms (CoQH2/CoQ). In the IMM, where CoQ passes through oxidation and reduction reactions during electron transport, the balance of reduced and oxidized CoQ is maintained by the activity of the respiratory chain. Yet what affects total CoQ content and the ratio between CoQH2 and CoQ in the IMM remains largely elusive. Outside the mitochondria, the plasma membrane redox system (PMRS), also called the trans-plasma membrane electron transport (PMET) system, is the best-understood mechanism that involves the redox cycling of CoQ.
3.2.1. Redox cycling of CoQ in the plasma membrane redox system.
The PMRS operates in all living cells, from bacteria to humans, although its components may vary depending on cell type (244, 245). Mainly, it allows electrons from intracellular substrates to flow outward to extracellular electron acceptors, by a process that is centered on CoQ (7, 246, 247). Physically, the classic description of the PMRS in mammalian cells consists of cytosolic and plasma membrane-associated oxidoreductases that transfer electrons, derived from NADH or NADPH, to the membrane-embedded intermediate electron carrier CoQ and finally to extracellular electron acceptors such as oxygen (248) (FIGURE 8). The enzymes involved in CoQ-dependent PMRS activity include NADH-cytochrome b5 reductase (CYB5R), NAD(P)H:quinone oxidoreductase 1 (NQO1), formerly known as DT-diaphorase, and the recently identified ferroptosis suppressor protein 1 (FSP1) (249–253). CYB5R, present at the inner surface of the plasma membrane (also in mitochondria and the endoplasmic reticulum), catalyzes the one-electron reduction of CoQ by NADH, resulting in the formation of ubisemiquinone (CoQ•−), which can be further reduced to CoQH2, whereas NQO1 reduces CoQ with either NADH or NADPH by a two-electron reaction, directly to CoQH2 (245, 254). NQO1 is known to be primarily cytosolic but can be translocated to the inner surface of the plasma membrane under stress conditions (255). FSP1 (also known as AIFM2), like NQO1, utilizes both NADH and NADPH as electron donors to reduce CoQ. The NH2 terminus of FSP1 contains a canonical myristoylation site that is essential for its plasma membrane localization (252, 253, 256). The reduced CoQH2 can then shuttle electrons to the cell surface NADPH/NADH oxidase (NOX) (external NOX, ENOX) that is able to reduce oxygen to yield O2•– or use the oxidized form of ascorbate (Asc), the ascorbyl free radical (AFR), as the terminal electron acceptor (246, 257–259). Other PMRS activities that do not involve CoQ include electron transfer by the transmembrane NOX proteins, the duodenal cytochrome b (DCYTB), or the NADH:ferricyanide reductase [known as the voltage-dependent anion-selective channel (VDAC)] (260–265) (FIGURE 8). Of note, it was suggested that CoQ might also function as a physiological substrate of VDAC (261).
FIGURE 8.
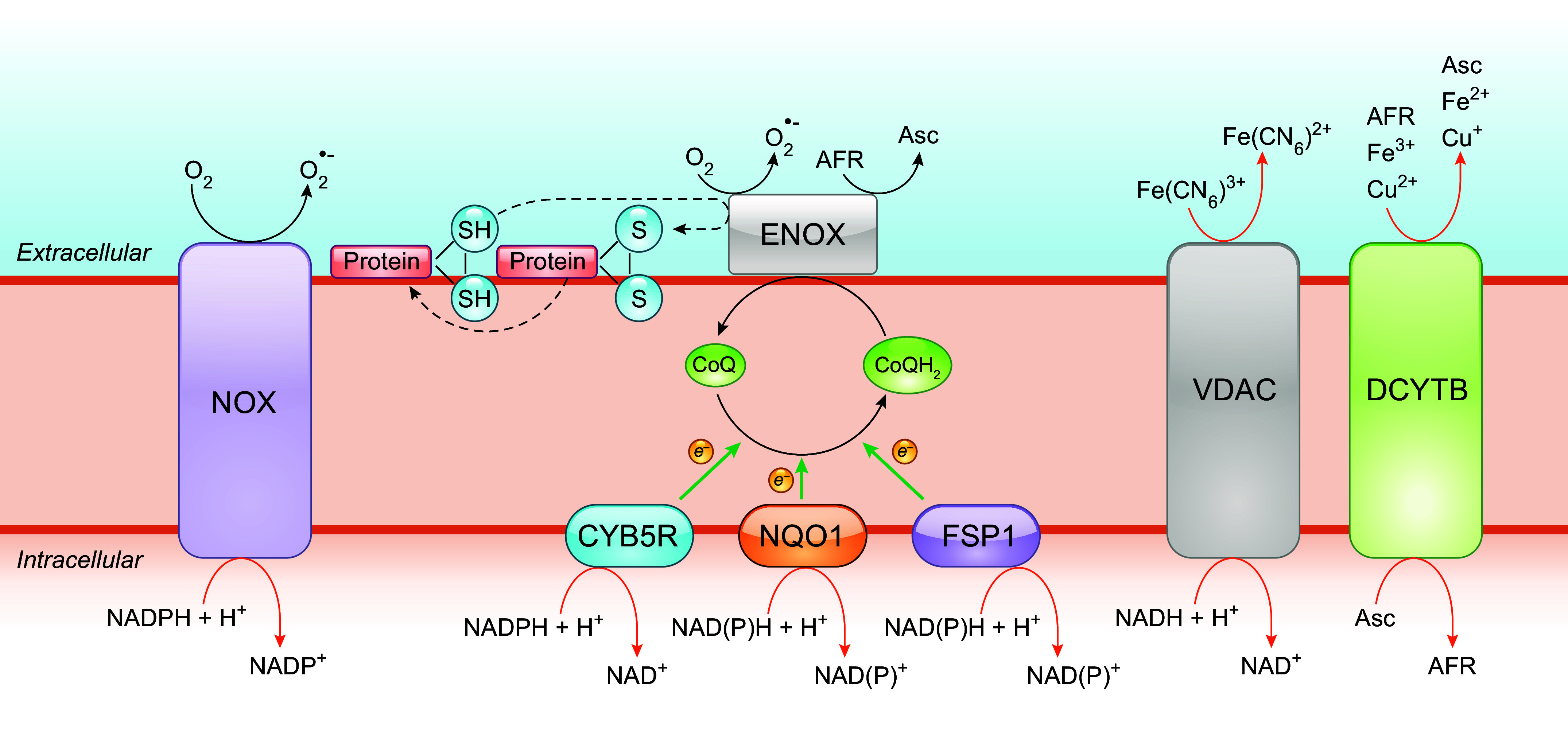
Role of CoQ in the plasma membrane redox system (PMRS). The PMRS consists of multiple component operations that result in electron transfer from cytosolic reducing equivalents to extracellular electron acceptors. NADH-cytochrome b5 reductase (CYB5R), NAD(P)H:quinone oxidoreductase 1 (NQO1), and ferroptosis suppressor protein 1 (FSP1) are CoQ reductases that oxidize NADH or NADPH to reduce CoQ. The cell surface protein ENOX is the terminal oxidase by catalyzing electron transport from CoQH2 to extracellular electron acceptors, including oxygen (O2) and ascorbyl (monodehydroascorbate) free radical (AFR). Besides oxidizing CoQH2, ENOX also possesses an alternative activity, which is catalyzing protein disulfide-thiol interchange. Other enzymes of the PMRS include the NADPH/NADH oxidase (NOX) that directly catalyzes the 1-electron transfer from cytosolic NADPH to molecular oxygen, the voltage-dependent anion-selective channel (VDAC) that reduces extracellular ferricyanide using NADH as electron donor, and the duodenal cytochrome b (DCYTB) that utilizes ascorbate (Asc) in the cytosol as an electron donor to reduce either extracellular ferric iron (Fe3+), cupric copper (Cu2+), or AFR. See glossary for other abbreviations.
One of the recognized roles of the PMRS, perhaps the most important one, is the control of the cytosolic NAD+-to-NADH ratio, thus modulating the cellular energy balance and redox homeostasis (see also sect. 4.4) (245). The PMRS is also implicated in iron uptake and immune cell function (246). Furthermore, the PMRS allows for the reduction of CoQ at the expense of intracellular reducing equivalents. This reduced pool of CoQ is believed to play an important role in antioxidant protection, mainly against membrane lipid peroxidation and also via regenerating other antioxidants (see below).
3.2.2. Antioxidant role of CoQ by regenerating vitamin C and E.
3.2.2.1. role of coq in vitamin c regeneration.
As indicated above, in addition to oxygen, oxidized vitamin C (VC) is one of the extracellular targets of the PMRS. Vitamin C, also known as ascorbic acid or ascorbate (Asc), is the most abundant water-soluble antioxidant in the extracellular fluid. Because of its low redox potential (+0.282 V at pH 7), Asc can readily donate electrons to stabilize free radicals (266). Asc reacts with all kinds of biologically generated radicals (267). It is a particularly effective scavenger of aqueous peroxyl radicals (•OH), with a rate constant of 7.9 × 109 M−1 s−1 to 1.1 × 1010 M−1 s−1 (268, 269), although, according to its rate constant toward O2•− (2.7 × 105 M−1 s−1), it is not an effective scavenger of O2•−. Nonetheless, the reaction between O2•− and Asc is likely to happen in vivo given the abundance of Asc in tissues (270–272). For its antioxidant action, Asc preferably serves as a one-electron donor, generating a relatively stable AFR (also written as Asc•−) (FIGURE 9) (273, 274). Losing the second electron from AFR leads to its transformation into dehydroascorbic acid (DHA). Asc cycles predominantly between the fully reduced form and AFR. DHA is produced mainly by disproportionation of AFR, reactions of 2 AFR to yield 1 DHA and 1 ascorbate molecule (FIGURE 9) (275). More importantly, as AFR is the major product of Asc oxidation, the ability to recycle AFR back to the reduced form is most crucial for the regeneration of antioxidant Asc. As indicated above, in addition to oxygen, AFR is a terminal electron acceptor in the PMRS as well. Thus, the CoQ-dependent PMRS serves to reduce AFR in the extracellular space and restore the Asc pool. But other mechanisms have also been identified. DHA produced extracellularly can be transported into the cell, where it can be reduced back to Asc, for example by glutathione-dependent DHA reductase (275, 276). In human erythrocytes, it was shown that DCYTB can contribute to extracellular Asc recycling by using intracellular Asc as an electron donor (FIGURE 9) (277–279). Moreover, Asc export to the extracellular space has been reported, which could help replenish the Asc pool (277, 278).
FIGURE 9.
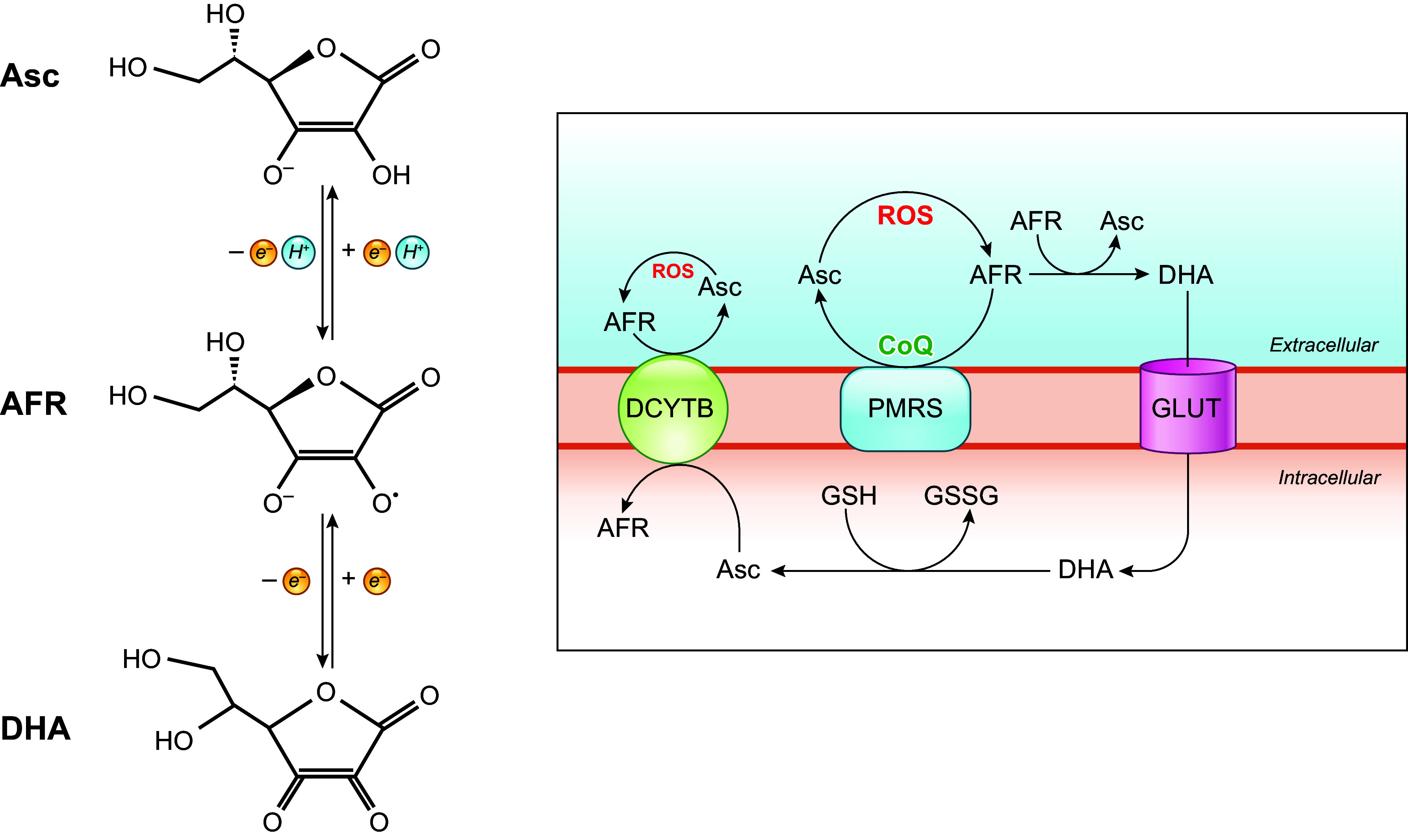
Redox metabolism of ascorbate. Ascorbate (Asc) can undergo 2 consecutive 1-electron oxidations that generate the ascorbyl free radical (AFR) as an intermediate and the complete oxidation product dehydroascorbate (DHA). Free radical-mediated oxidative stress results in the oxidation of Asc, yielding AFR. The CoQ-dependent plasma membrane redox system (PMRS) transfers reducing equivalents from intracellular electron donors to AFR outside of the cell, converting AFR back to reduced Asc. Two molecules of AFR can react with each other to form 1 DHA and 1 Asc. DHA made extracellularly can be transported through glucose transporters (GLUT) into the cell, where it can be recycled back to Asc using glutathione (GSH) as a reductant, yielding glutathione disulfide (GSSG). Extracellular AFR can also be reduced by duodenal cytochrome b (DCYTB) using intracellular Asc as an electron donor in some species and tissues. See glossary for other abbreviations.
A number of experimental findings point to an important role of the CoQ-dependent PMRS in ascorbate regeneration. CoQ extraction (with heptane) from lyophilized plasma membranes (from pig liver or K562 human leukemia cells) results in inhibition of NADH-ascorbate free radical (AFR) reductase (250). Incorporation of CoQ10 stimulates NADH-AFR reductase activity, and supplementation of K562 cells with CoQ10 is associated with a dose-dependent increase of extracellular ascorbate stabilization, which indicates a higher rate of plasma membrane AFR reduction (280–282). Furthermore, a yeast coq3 mutant defective in CoQ6 production was documented to have diminished NADH-AFR reductase activity and reduced extracellular ascorbate stabilization, and both activities were rescued after restoration of CoQ6 levels (283, 284). However, the physiological and pathological significance of the plasma membrane CoQ in contributing to extracellular antioxidant defense by allowing ascorbate regeneration needs further study and clarification.
3.2.2.2. role of coq in vitamin e regeneration.
CoQ is also believed to play a role in regenerating another primary exogenous antioxidant: vitamin E (VE), also known as α-tocopherol. Like CoQ, VE is lipophilic and located in membranes and lipoprotein fractions (285). A role for VE in membrane structural stabilization has been generally recognized, although how it is achieved is not yet well defined (286). Another important function of vitamin E is to act as an antioxidant protecting membrane lipids against peroxidation by scavenging lipid radicals.
Lipids in the form of polyunsaturated fatty acids (PUFAs) are the major components of cellular membranes. Owing to their multiple carbon–carbon (C=C) double bonds, PUFAs are especially labile to oxidation, given the relative ease with which hydrogen atoms can be removed from the bis-allylic methylene (–CH2–) between double bonds (287). Free radicals (such as OH•) can rip off hydrogen from the methylene group in PUFAs, resulting in the formation of a carbon-centered lipid radical that upon rearrangement forms a stable carbon-centered radical (L•). As L• has an unpaired electron on carbon, this makes it reactive and inclined to react with oxygen, yielding a lipid peroxyl radical (LOO•). LOO• has a long half-life (10 s) and in turn can abstract a hydrogen atom from another adjacent lipid molecule to produce a lipid hydroperoxide (LOOH), while producing a new L•. This new L• can then initiate a new oxidation reaction (288). Thus, the whole process becomes self-propagating and will keep going until the substrate is consumed or termination occurs (289). The process by which lipids are attacked by radicals leading to the formation of LOOH is called lipid peroxidation (FIGURE 10). It is one of the most important types of oxidative damage in biological systems. Because the process is self-perpetuating, the effect of the original free radical will be amplified, leading to numerous modified lipids. This oxidative degradation of membrane lipids can perturb the bilayer structure and alter membrane properties such as fluidity, permeability, conductivity, and ion transport (290). Furthermore, although LOOH is stable compared to lipid radicals, by the process of Fenton chemistry when interacting with a metal ion, it can decompose into a lipid alkoxyl radical (LO•) that may continue to propagate new peroxidation reactions. In addition, the decomposition of LOOH can form various aldehydes that are potentially toxic. For example, the LOOH breakdown products malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) are capable of damaging membrane-bound receptors and enzymes, which in turn can trigger cell death (291, 292). There are many excellent reviews on various aspects of lipid peroxidation, such as Refs. 289, 293–295, to which readers can turn for more information.
FIGURE 10.

The lipid peroxidation process. It is initiated by the radical-mediated abstraction of a hydrogen atom from a bis-allylic methylene group in a lipid (LH). This leads to the formation of a carbon-centered lipid radical (L•), which undergoes molecular rearrangement and then reacts with oxygen to form a peroxyl radical (LOO•). In turn, LOO• propagates a chain reaction through the formation of a new L• by hydrogen abstraction from other lipids, while it itself is converted to a lipid hydroperoxide (LOOH). Decomposition of LOOH yields lipid alkoxyl (LO•) and the formation of aldehydes such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) and other secondary and end products of lipid peroxidation.
The chain reaction of lipid peroxidation is terminated by lipid radicals that take part in the chain reaction, quenching each other or combining with a similar radical to form nonradical products, or by antioxidants. The intermediate product LOO• is responsible for the propagation of the lipid peroxidation process. VE, by virtue of its ability to donate electrons to LOO• species, is able to neutralize LOO• while it itself is oxidized to a vitamin E radical (VE•), also known as an α-tocopherol radical (FIGURE 11) (296–298). VE• is not very reactive and can be regenerated to VE. Thus, VE is known as a chain-breaking antioxidant, as it intercepts LOO• and thus interrupts lipid peroxidation cascades. The cell’s ability to regenerate VE from its radical form back to its reduced native state determines the antioxidant activity of VE. The one-electron redox potential for CoQH2 [CoQH2/CoQ•− = −0.2 V] is more negative than that for vitamin E [VE/VE• = 0.5 V], and therefore CoQH2 is capable of reducing oxidized VE, thus regenerating its activity (266). VE can also be regenerated by Asc, but it has been claimed that regeneration by CoQ is favored (10). Other potential fates of VE• include reacting with another VE• or LOO• forming nonradical stable products or being further oxidized to VE quinone (288, 299).
FIGURE 11.

Antioxidant action of vitamin E (VE) against lipid peroxidation. VE scavenges lipid peroxyl radical (LOO•) before it attacks other lipids (LH) to form lipid hydroperoxide (LOOH) and a new lipid radical (L•), by which it terminates lipid peroxidation chain reactions. This leaves behind the vitamin E radical (VE•). VE• can be converted back to the reduced antioxidant form by CoQ or vitamin C (VC). VE• can also react with another LOO•, forming poorly reactive nonradical adducts, decay by reaction with another VE• molecule to give inactive dimers, or be completely oxidized to vitamin E quinone (VEQ). VC•, vitamin C radical. See glossary for other abbreviations.
By electron spin resonance (ESR) and stopped-flow spectroscopy, it was shown that VE• disappears after the addition of CoQ10H2 (but not CoQ10) (300, 301). Moreover, mitochondrial CII integrated into proteoliposomes slowed the accumulation of the VE• ESR signal (induced by lipoxygenase-catalyzed linolenic acid oxidation) only when succinate and CoQ10 were both also incorporated into these liposomes (302). Similarly, in rat liver SMPs, the simultaneous addition of NADH and CoQ1 was shown to produce a maximum decrease of VE• signals (303). These observations can be interpreted if CoQH2 reduces VE• and thus mitochondrial enzyme-linked reduction of CoQ to CoQH2 prevents accumulation of VE•.
3.2.3. Direct antioxidant properties of CoQ by scavenging lipid radicals.
Shortly after the discovery of CoQ in 1961, it was suggested that CoQ itself can act as an antioxidant against lipid peroxidation. It was proposed that it could directly quench LOO• as follows (304):
| (1) |
| (2) |
where CoQH2 undergoes one electron oxidation to give LOOH and CoQ•− (reaction 1). The reaction of CoQ•− with another LOO• produces LOOH and a fully oxidized CoQ (reaction 2). Because, as VE, CoQH2 eliminates LOO• by direct scavenging, thus terminating the propagation of peroxidation, it is considered to be primarily a chain-breaking antioxidant (12). Of note, the possibility that CoQH2 may also be able to quench L•, thus inhibiting the initiation of lipid peroxidation, has also been considered but is still under study (240, 305).
The chemical reactivity of CoQH2 with peroxyl radicals was demonstrated in solvent solutions, and its reactivity toward peroxyl radicals was shown to be lower than that of VE (300, 306, 307). This means that VE scavenges peroxyl radicals faster than CoQ. In liver microsomal and mitochondrial membranes, it was shown that, in comparison to VE, treatment with externally added CoQ9 and CoQ10 showed much weaker inhibition of lipid peroxidation induced by Fe2+ plus ascorbate (or by Fe2+ plus NADPH or tert-butyl hydroperoxide) (303). Considered together with the reported observation that the concentrations of CoQ in membranes are usually close to, or lower than, those of VE, CoQ seems unlikely to be the major lipid-soluble antioxidant in cellular membranes but might instead defend against lipid peroxidation by regenerating VE (303, 308). However, it is noteworthy that in beef heart SMP preparations that contain no detectable amount of VE, inhibition of MDA formation was reported to be observed again after reincorporation of CoQ10, indicating some direct effect of CoQ (309).
3.2.4. Other experimental evidence for the antioxidant effects of CoQ.
Most available direct evidence for an antioxidant effect of CoQ came from studies using in vitro subcellular systems such as liposomes and SMPs. Liposomes are small artificial spherical structures, with a lipid bilayer primarily made by the self-assembly of phospholipids. Thus, they have been actively used as a simple model of cell membranes. Lipophilic molecules such as CoQ can be incorporated into the lipid bilayers of liposomes. Indeed, the addition of CoQH2 to liposomes was shown to inhibit the formation of lipid peroxidation induced by different free radical generators. For example, in liposomes made of phosphatidylcholine (PC, a major type of phospholipid of cell membranes), PC hydroperoxide formation induced by 2,2′-azobis(2,4-dimethylvaleronitrile) (AMVN), a lipophilic free radical initiator, was decreased when CoQ10H2 was added (at a molar ratio to phospholipids similar to that in human tissues), and no effect was seen after addition of the oxidized form CoQ10 (304). Similarly, in reconstituted egg yolk PC liposomes containing NADH-cytochrome b5 reductase or DT-diaphorase and a high amount of CoQ10 (50 µM), incubation with NADH led to an increased pool of CoQ10H2 and a simultaneous reduction of lipid peroxidation induced by exposure to free radical-generating compounds, such as 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH) or AMVN (249, 310).
A correlation between CoQ and lipid peroxidation levels was also demonstrated in SMPs. As they retain OXPHOS activity when provided with oxidizable substrates (NADH or succinate) to provide reducing equivalents, they are capable of generating CoQH2 from CoQ and the conversion of CoQ to CoQH2 can be further maximized by simultaneously blocking its oxidation at CIII (FIGURE 5). For example, CoQ can be kept in a highly reduced state (>80%) in SMPs under oxidation of NADH or succinate in the presence of CIII inhibitors (e.g., antimycin A) or CIV inhibitors (e.g., potassium cyanide, KCN) (240).
MDA is one of the most frequently used biomarkers of lipid peroxidation. In bovine heart SMPs, it was shown that MDA formation induced by the addition of NADH or NADPH in the presence of ADP and Fe3+ was inhibited by succinate, and the effect of succinate was further enhanced by antimycin A and KCN (311). Similar findings were reported in studies using ADP/Fe3+ plus Asc to induce lipid peroxidation (309). In contrast, the antiperoxidative effect of CoQ was not observed in beef heart SMPs from which most CoQ was removed, but it appeared again upon reincorporation of CoQ (240, 309, 311, 312).
Finally, given the high hydrophobicity and membrane location of CoQ, most in vitro studies of its antioxidant properties have been focused on lipid peroxidation. However, a protective effect of endogenous CoQ against protein and DNA oxidation has also been suggested. Incubation of isolated rat liver mitochondria with ADP/Fe3+/Asc resulted in substantial protein oxidation (measured by protein carbonylation) and an increase in the amount of 8-hydroxy-2-deoxyguanosine (8-OHdG), a product of oxidative damage to DNA. Both were shown to be inhibited by the addition of succinate and antimycin A (313). Similarly, increasing the pool of CoQH2 in beef heart SMPs in the presence of succinate and antimycin A was reported to inhibit the stimulation of both lipid peroxidation [measured with the thiobarbituric acid reactive substances (TBARS) assay] and protein carbonylation by ADP/Fe3+/Asc (314). However, these studies could not determine whether the oxidative damage to proteins and DNA was independent of lipid peroxidation, and thus whether the effect of CoQH2 was direct or indirect (240, 313).
3.2.5. CoQ in lipoproteins.
An additional antioxidant role of CoQ in mammals appears to be to protect lipoproteins from oxidation. Lipoproteins are lipid-protein complexes that are synthesized in the liver and secreted into the circulation, where they function as vehicles for lipids. A small amount of CoQ is found in the blood, where it is carried by lipoproteins, mostly in low-density lipoproteins (LDL) and very low-density lipoproteins (VLDL), in which it has been shown to exist predominantly in its reduced form (315–317). Several in vitro investigations into lipoprotein oxidation reported a protective effect of the presence of CoQH2. For example, exposure of human plasma or lipoproteins isolated from human subjects to peroxyl radicals [such as 2,2′-azobis(2-amidinopropane) dihydrochloride or 2,2′-azobis(2,4-dimethylvaleronitrile)] led to rapid oxidation of CoQ10H2 (315, 318). Moreover, CoQ10H2 was shown to be the first consumed after exposure to such ex vivo oxidation, and the onset of lipoprotein lipid oxidation (measured by HPLC detection of lipid hydroperoxides) corresponded closely with the complete conversion of CoQ10H2 to CoQ10. That is, lipid peroxidation of lipoproteins proceeded rapidly only after most CoQ10H2 was consumed (318–320). Furthermore, an increase of CoQ10 in plasma (≈2- to 7-fold increase) or plasma lipoproteins (≈3- to 4-fold increase) from dietary CoQ10 supplementation was demonstrated to enhance the resistance to ex vivo-induced oxidation of lipoproteins (315, 317, 321–323). Thus, it was long proposed that CoQ is crucial for lipoprotein protection against oxidation (318). But whether the action is direct or indirect via regeneration of VE is not clear. The average number of CoQ10H2 molecules per native LDL was reported to be 0.5–1.0 versus 6–12 for VE (315, 322). Correspondingly, the mean CoQ10 level in human serum was reported to be >10 times less than that of VE (316).
In vivo, LDL oxidation is thought to occur mainly in the subendothelial space in the vascular wall and is widely regarded as a key factor in the pathogenesis of atherosclerosis (324). However, the importance of CoQ for preventing LDL oxidation in vivo and thus reducing the risk of atherosclerosis is unknown. It was shown that in apolipoprotein E-deficient mice fed a high-fat diet, which is a widely used murine model for atherosclerosis, supplementation of CoQ10 resulted in a variable increase of the total aortic content of CoQ and in reduced aortic LOOH accumulation as well as in a smaller area of atherosclerotic lesions in the aorta (317, 323). This suggests a benefit of CoQ in inhibiting oxidized LDL (ox-LDL)-related pathophysiology. However, several studies also reported a lack of any effect of CoQ10 supplementation on arterial lesions or endothelial function, despite decreased aortic lipid oxidation and increased resistance of plasma lipids to ex vivo oxidation (319, 321). CoQ is packaged into LDL and VLDL in the liver, and the liver is one of the few organs that readily takes up oral CoQ10 (8, 30, 42, 82, 123, 325). Thus, although it might be possible to significantly boost CoQ10 levels in lipoproteins, more research is needed to determine whether CoQ10 supplementation can inhibit the development or progression of atherosclerosis in humans.
3.2.6. Summary.
The chemical properties of CoQ and its abundant distribution in membranes where its active antioxidant form can be continuously regenerated by enzymatic mechanisms suggest that CoQ is an important participant in redox control. Moreover, the fact that CoQ is actually in the immediate vicinity of the site of ROS production in the IMM is also thought to contribute to its function as an antioxidant. As we have seen, there is a fair number of early in vitro studies that point to CoQ’s antioxidant bona fides. Perhaps the most direct evidence is provided by its ability to inhibit lipid peroxidation in membrane vesicles when its reduced form is readily regenerated. However, it remains unclear how important the antioxidant properties of CoQ are in vivo, compared to other antioxidant mechanisms, and how exactly it acts. In the extraction and reincorporation studies with artificial membrane systems such as liposomes and SMPs, the amount of CoQ reincorporated was likely not physiological. Moreover, most of the in vitro studies examined the effect of CoQ on lipid peroxidation induced by direct exposure to oxidant compounds. The level of oxidative insults to which tissue cells are exposed under normal conditions is likely many orders of magnitude lower than that produced by such artificial conditions. In summary, although CoQ has an intrinsic free radical-scavenging potential, its ability to regenerate other antioxidants, especially vitamin E, might be of greater importance. Overall, despite the reputation of CoQ as an antioxidant, its action as a prooxidant is actually better documented.
3.3. CoQ Level and Oxidative Stress
Generally speaking, oxidative stress is defined as a state in which ROS levels are high enough to lead to a deleterious level of molecular damage. As outlined in the sections above, the capability of CoQ to function as a source of mitochondrial ROS as well as an antioxidant itself is well recognized. However, the relative importance of these two functions in vivo, in different tissues and under different conditions, is yet to be clearly understood. On this front, a better understanding is needed for elucidating the pathophysiology that accompanies CoQ deficiency in patients (see sect. 6), as well as for addressing the rational use of CoQ10 as an antioxidant health supplement.
3.3.1. Less CoQ and oxidative stress.
As we have seen, mitochondria are considered to be the major source of ROS in most cell types, and CoQ•−, the reactive intermediate of CoQ redox cycling, is one of the sources of mitochondrial ROS (146, 326). An inadequate CoQ concentration in the IMM could mean insufficient CoQ available to carry electrons to CIII. This could have consequences for O2•– generation at CI, at CIII, and during electron transfer from other CoQ-dependent hydrogenases (such as DHODH and G3PDH). All this will depend on the type of cells, the sources and usage of respiratory substrates, and the severity of the CoQ shortage (327). Less CoQ redox cycling at CIII likely decreases O2•– release at that site. However, a reduction of CoQ-dependent CI + III activity may increase the NADH-to-NAD+ ratio in the mitochondrial matrix, leading to the hyperproduction of O2•– at the FMN site of CI (see sect. 3.1.1) (166). Moreover, an overall decrease of respiratory chain activity caused by CoQ deficiency may influence mitochondrial ROS production by an effect on membrane potential or induction of some compensative or adaptive alterations of ETC function, such as altered supercomplex structure and mPTP opening. For example, under conditions in which mitochondrial respiration is strongly inhibited, CV (the F0F1-ATP synthase) can run in reverse, hydrolyzing ATP and extruding protons out of the matrix (328). A study with human SH-SY5Y neuronal cells has suggested that this can result from CoQ deficiency (at an ≈50% decrease in CoQ10 level) (329). It is still unknown whether this occurs in other cell types and under milder, or more severe, CoQ deficiency. Finally, as discussed above, CoQ is widely believed to be an endogenous antioxidant, acting directly through its radical-scavenging chemical property and indirectly by regenerating the antioxidant form of VE and VC. Hence, the consequences of CoQ deficiency on the cellular redox state and oxidative damage, determined by the balance between ROS production and clearance, are unlikely to be straightforward and universal.
3.3.1.1. sensitivity to oxidative stress of fully coq-deficient yeast mutants.
In the yeast S. cerevisiae, a common feature of fully CoQ-deficient coq deletion mutants (including coq1Δ, coq2Δ, coq3Δ, coq5Δ, coq6Δ, coq7Δ, coq8Δ, and coq9Δ) is hypersensitivity to treatment with the PUFA linolenic acid (αLnn) (330–333). Exogenously supplemented αLnn can readily incorporate into the yeast cell membrane, and autoxidation of linolenic acid gives rise to LOOH and many secondary products, some of which are toxic, causing cell death (334, 335). All the coq mutants lacking CoQ display lower survival after exposure to PUFAs, which are known to undergo an autocatalytic oxidation process. In contrast, they are not affected by monounsaturated oleic acid that does not autooxidize easily and is much less vulnerable to peroxidation (331–333). In contrast, the cor1 and atp2 mutants, with defects in CIII and CV, respectively, remain resistant to PUFA treatment (331). Consistently, quantification of lipid peroxides and late-stage aldehyde products (TBARS) as markers of lipid peroxidation showed higher levels in αLnn-treated coq3Δ yeast compared to the wild type (332). These findings indicate a protective role of CoQ against the toxicity of PUFA oxidation, which is further supported by the observation that the sensitivity of coq3Δ yeast to αLnn can be reduced by vitamin E and the antioxidant butylated hydroxytoluene (BHT) (331, 332). These studies, however, did not examine the effect of exogenous CoQ on the lipid peroxidation sensitivity of the mutants. In addition, surprisingly, vitamin C showed no effect on αLnn sensitivity (331). Likely, the lipid peroxidation stress imposed by exogenous supplementation of αLnn is much higher than what the cells normally encounter. Thus, these studies cannot establish unambiguously the importance of CoQ in membrane protection under normal physiological conditions. The yeast coq1-null mutant (coq1Δ) was shown to exhibit enhanced sensitivity to H2O2 killing and elevated O2•– levels compared to wild-type yeast (330). However, a similar hypersensitivity to H2O2 was also reported for the mutants with deletions of subunits of mitochondrial CIII (cor1Δ, cyt1Δ), suggesting that the elevated O2•– and the sensitivity to H2O2 are more likely to be attributable to a consequence of respiratory dysfunction rather than a lack of the antioxidant role of CoQ (330). Finally, the coq10Δ yeast mutant, which has a near wild-type content of CoQ6 in the stationary phase but inefficient CoQ6 biosynthesis during the log phase (≈30% relative to wild type), was also found to be sensitive to αLnn exposure during mid-log phase (though less than coq3Δ mutant), and, consistently, αLnn-treated coq10Δ mutants showed significantly increased lipid peroxidation levels compared to the wild type (331). Whether this implies insufficient CoQ6 biosynthesis in the coq10 mutant for protection against PUFA stress or whether there is a requirement for Coq10 for the antioxidant function of CoQ remains to be resolved.
3.3.1.2. coq deficiency and oxidative stress in mammalian cells and tissues.
Using cultured skin fibroblasts from patients that carry genetic lesions in the CoQ biosynthetic pathway, Quinzii et al. were the first to assess the consequences of different CoQ deficiency levels on the cells’ ROS level and oxidative stress status. They found that in COQ2 mutant fibroblasts that have 30–40% normal level of CoQ there were signs of excess ROS, as measured by MitoSOX (a mitochondrial fluorescent probe with high reactivity to O2•–) and lipid peroxidation markers (MDA and 4-HNE), but the markers of oxidative stress were absent in the patient cells with 12–18% (due to PDSS2 or COQ9 mutation) or >50% (due to ADCK3 mutations) of CoQ10 (113, 336). See sect. 5.1 on biosynthesis for more information about the function of these genes. A similar finding was reported for CoQ10 deficiency induced by inhibition of CoQ synthesis with 4-nitrobenzoic acid (4-NB, a competitive inhibitor of COQ2). Lowering the CoQ10 level to 40–50% (in controls and ADCK3 mutant fibroblasts) led to an increase in MitoSOX fluorescence (336). A later study also showed a higher ROS level (measured with both 2′-7′-dichlorodihydrofluorescein diacetate (DCFH-DA) and MitoSOX] in ADCK3 mutant fibroblasts with ≈50% CoQ10, compared to nonmutant controls (337). And the measures of increased ROS levels were associated with increased sensitivity to H2O2 as well as elevated levels of lipid peroxidation and protein tyrosine nitration, a marker of nitro-oxidative damage (337). Taken together, an inverted U-shaped relationship is postulated for levels of CoQ and oxidative stress: an increase of oxidative stress is only manifested under moderate CoQ10 deficiency (30–50% residual CoQ10), whereas a more severe or milder deficit of CoQ10 (<30%, or >50% of normal level) is not accompanied by overproduction of ROS (FIGURE 12) (113, 336). Such a relationship might be due to the dual pro- and antioxidant properties of CoQ.
FIGURE 12.
Relationship between the levels of CoQ and ROS in human skin fibroblasts. Severe (<30% of residual CoQ10) and moderate (>50% of residual CoQ10) defects are not associated with induction of oxidative stress. However, an intermediate defect (30–50% of residual CoQ10) results in an increase of cellular ROS levels. WT, wild type. See glossary for other abbreviations.
Whether such a relationship can be expected in other cell types has not been systematically investigated. Induction of CoQ deficiency in wild-type cells relies on pharmacological inhibition of CoQ biosynthesis with compounds that interfere with the normal CoQ biosynthesis pathway. Most of the studies use reagents such as para-aminobenzoic acid (pABA) and 4-NB, which are competitive inhibitors of the COQ2 enzyme. With such an approach, it can be challenging to produce different CoQ deficiency levels or a very severe loss in CoQ without negatively affecting cell viability. In mouse adipocyte 3T3-L1 cells, it was shown that an ≈25–50% decrease of mitochondrial CoQ9, due to treatment with 4-NB or knockdown of the CoQ biosynthetic gene Coq7 or Coq9, led to increased mitochondrial peroxiredoxin (PRDX) dimerization, which was used as an indicator of H2O2 burden, as well as increased concentration of mito-2-hydroxyethidium, an O2•–-specific oxidation product of MitoSOX (338). The effect of CoQ deficiency at levels outside of this range was not investigated. In pABA-treated human myeloid leukemia HL-60 cells, ≈50% decrease in the level of cellular CoQ10 was also shown to display, in addition to a decrease of CoQ-dependent respiratory activity, a higher level of O2•– compared to a no-treatment control (339). Moreover, a similar level of reduction of CoQ10 levels in 4-NB-treated T67 human glioma cells (by ≈50%) was demonstrated to cause higher cellular and mitochondrial oxidative stress (demonstrated by increased DCFH-DA and MitoSOX signals) as well as an elevation of nitric oxide (NO•) levels (338). In both cell lines, treatment with exogenous CoQ10 reversed the levels of oxidative stress markers to control values, supporting a causal link between the CoQ decrease and the higher ROS reporter intensity (120, 339). In human neuronal SH-SY5Y cells, increased mitochondrial oxidative stress (measured by MitoSOX probe), but no sign of increased lipid peroxidation (visualized by C11-BODIPY staining), was observed at both 46% and 76% residual CoQ10 levels produced by treatment with pABA. This type of cell appears to display higher ROS production at slightly higher CoQ levels than skin fibroblast. Conceivably, endogenous CoQ levels and the contributions of CoQ to ROS production and cellular antioxidant status differ in different cell types and under different conditions. It should be noted that so far all the in vitro studies with CoQ-deficient or supplemented cells were conducted under standard cell culture conditions where the O2 levels are hyperoxic with respect to the 1–6% experienced by most mammalian cells in vivo (340). However, the level of oxygen that cells are exposed to is known to be able to impact biochemical reactions and cellular activities, especially energy metabolism and ROS production. It is possible therefore that O2 levels affect experimental outcomes and the conclusions drawn from them (340–342). Thus, the potential implications of in vitro findings for in vivo conditions remain to be evaluated. It is also not known how intracellular O2 levels could affect CoQ levels. Interestingly, a recent study showed that culturing human endothelial cells under hypoxia (1% O2) leads to a marked decrease in CoQ10 content (343).
With isolated mouse heart mitochondria, an ≈90% decrease in CoQ levels has been shown to cause a significant reduction in ROS production rate. The mitochondria used in the study were isolated from the heart of inducible Coq7 knockout (KO) mice (123). The hearts were harvested from ≈8-mo-old mice, ≈6 mo after the gene was excised. Mitochondrial ROS production rate was assessed by the commonly used Amplex Red/HRP assay. A decrease of net O2•− release was seen with both CI and CII substrates in the presence of the CI CoQ site inhibitor rotenone or the Qi inhibitor antimycin A, suggesting lower ROS production at both CI and CIII and in both forward and reverse electron transfer modes (123). Moreover, it was shown that despite a global deficit of CoQ in the mutant mice (here an average of 10–30% CoQ left), no sign of systemic oxidative stress was detected, as indicated by normal levels of plasma F2-isoprostanes (a result of lipid peroxidation) and plasma content of 8-OHdG (an oxidative DNA damage marker) (123). An increase in the expression of antioxidant enzymes often occurs in response to elevated oxidative stress. However, in the kidneys and heart of the Coq7 KO mice that contain only 10–15% of CoQ relative to that in non-KO control mice, a lower level of the cytoplasmic superoxide dismutase 1 (SOD1) was detected, as well as a markedly decreased level of catalase in the kidney (123). These are likely to be compensatory responses that may be induced by low cytoplasmic ROS and that help maintain ROS at physiological levels necessary for optimal cell function. Thus, overall, a very low level of CoQ in these mice appears to be associated with a lower oxidative stress state, most likely because of reduced mitochondrial ROS production. It is noteworthy, however, that Coq7 mutants accumulate the biosynthetic intermediate DMQ, which might have phenotypic consequences on ROS metabolism.
The effect of CoQ deficiency on oxidative stress has also been examined in the kidney of Pdss2kd/kd mice harboring a homozygous kidney disease (kd) mutation in Pdss2. The partial loss-of-function mutation of Pdss2 was identified as a spontaneous mutation in a colony of inbred mice and was designated kd because renal failure is the most prominent disease manifestation in mutant kd/kd homozygotes (30, 344–346). Pdss2kd/kd kidney displayed an increase of dihydroethidine fluorescence, indicating elevated ROS production, and this is associated with 20–30% residual CoQ9 content (124). In contrast, levels of fluorescence comparable to the wild type were not observed in other tissues of the mutant (brain and muscle), despite similar reductions in CoQ9 levels (124). Moreover, two separate markers of oxidative stress, anti-nitrotyrosine and anti-4-HNE immunostaining, were also found to be elevated only in the glomerulus of Pdss2kd/kd mice where tissue damage occurs (124). Lower CoQ-dependent ETC complex activities (CI-CIII and CII-CIII) were noted in Pdss2kd/kd kidney but also in other tissues (124). Taken together, it has been postulated that in the kidney (in particular in the glomeruli) CoQ deficiency is more liable to lead to high oxidative stress than in other tissues. This would explain why Pdss2kd/kd mutants mainly develop a renal phenotype, despite widespread CoQ9 deficiency (30, 124). A beneficial effect in preventing renal lesions in these mice of treatment with CoQ10 or with probucol, a hypolipidemic agent with antioxidant action, lends further support to this idea (347, 348). This is in stark contrast to findings in other mouse mutants of CoQ biosynthetic genes. No sign of oxidative damage in the kidney (measured as 8-OHdG levels) and normal appearance of kidney glomeruli and tubules were found in inducible Coq7 knockout mice at 6 mo of age, despite a severe depletion of total CoQ (≈10% of residual CoQ levels). And no renal dysfunction (as measured by blood urea nitrogen) was observed in Coq7 knockout mice treated with 2,4-dihydroxybenzoic acid (2,4-DHB), which showed ≈ a 30% residual CoQ level in the kidney (see sect. 5.4 for additional details about 2,4-DHB) (123). A study with a Coq9 mutant mouse (Coq9R239X) reported that despite a severe loss of total CoQ (<30% of the normal level) across tissues and fatal mitochondrial encephalomyopathy, the phenotype showed no renal involvement (349). This study also reported increased 8-OHdG and 4-HNE staining in the brain, suggesting elevated oxidative stress, but the possible role of ROS in the brain pathology observed has not yet been elucidated (349). The reasons for these phenotypic discrepancies are not understood. A more recent study showed that an increase of the lipid peroxidation product MDA was associated with ≈25% residual CoQ levels (CoQ9 + CoQ10) in a brown adipose tissue (BAT)-specific in vivo model of CoQ deficiency obtained by targeting Pdss2 (119). Overall, it appears that in certain situations, but not all, CoQ deficiency causes elevated ROS generation and increased oxidative damage. Why this is and its role in CoQ deficiency-associated pathophysiology are not yet clear.
3.4. Utility of Exogenous CoQ10 Supplementation
3.4.1. In vitro and in vivo CoQ10 supplementation.
For in vitro supplementation, CoQ that has been dissolved in an organic solvent such as ethanol is often directly added to culture media. When added in this manner it can be taken up efficiently by cells, after which it can reach mitochondria and most likely other cellular compartments as well (41, 42, 82, 110, 350–353). In fact, a dramatic increase of CoQ10 level is often observed in cells after culture in the presence of CoQ10 (TABLE 1). The respiration deficiency of CoQ-deficient cells can be rescued by exposure to CoQ, demonstrating the ability of exogenous CoQ to reach the mitochondrial inner membrane (42, 82, 115, 351, 358–360). The cellular mechanisms whereby CoQ is taken up from the culture medium and transported to various locations are not well known. In mammalian cells, the class B scavenger receptor CD36 and Niemann–Pick C1-like 1 (NPC1L1, an intestinal cholesterol transporter) have been shown to be involved in exogenous CoQ uptake (42, 356, 361). In yeast, several genes have been identified that affect CoQ uptake and intracellular trafficking. The products of most of these genes are associated with endosomal trafficking, but their exact roles are yet to be understood (352, 360, 362).
Table 1.
Reported effects of in vitro CoQ10 supplementation on cellular CoQ10 levels
| Cell Type | Total Dosage | Treatment Effects |
Refs. | |
|---|---|---|---|---|
| On CoQ10 levels | On ETC function | |||
| Cells with intact CoQ biosynthesis | ||||
| SH-SY5Y neuroblastoma cells | 5 µM 3 days | ↑↑↑ (cells) | ND | (354) |
| Human astrocytoma cells & rat embryonic cardiomyocytes | 10 µM 1 day | ↑↑↑ (cells) | NE | (41) |
| Breast cancer cell lines | 9 µM 2 day | ↑↑↑ (mito) | NE | (355) |
| Mouse brown adipocytes | 10 µM 3 h | ↑↑↑ (cells) | NE | (356) |
| Human dermal fibroblasts | 5 µM 1 day | ↑↑↑ (cells) | NE | (351) |
| Mouse embryonic fibroblasts | 2.5 µM 3 days | ↑↑ (cells) | ND | (42) |
| Cells with defective CoQ biosynthesis | ||||
| Human dermal fibroblasts | 5–10 µM 1–7 days | ND | ATP ↑, respiration↑, CI-CIII ↑, CII+CIII ↑ | (80, 115, 351, 357) |
| Mouse embryonic fibroblasts | 2.5–10 µM 3–4 days | ↑↑ (cells)↑↑ (mito) | Respiration↑ | (42, 83) |
Cell treatment with CoQ10 was carried out in the listed studies by adding CoQ10 dissolved in a solvent (such as ethanol) to the culture media. CI-CIII, complex I-III activity, CII-CIII, complex II-III activity; ETC, electron transport chain; mito, mitochondria; ND, not determined; NE, no effect. ↑, <2-fold increase; ↑↑, 2- to 10-fold increase; ↑↑↑, ≥10-fold increase.
To investigate the effects of CoQ supplementation in vivo, the most commonly used route is oral. Specifically, CoQ10 supplementation in food or water has been used in most rodent studies, as the human isoform can be easily distinguished from the rodents’ predominant endogenous isoform (CoQ9). Although direct experimental evidence is still lacking, it is generally believed that, like other dietary lipids, orally administered CoQ10 is absorbed into the intestinal enterocytes. In the enterocytes, CoQ10 is packaged into chylomicrons that, once formed, are released from the enterocytes into the lymphatic system, and eventually into the bloodstream (363, 364). CoQ10 can go back into the circulation after liver uptake of chylomicron remnants and assembly and secretion of CoQ10-containing lipoproteins (363, 364). However, because of its high hydrophobicity and large molecular weight, the gastrointestinal absorption of CoQ10 is known to be slow and poor, resulting in low bioavailability (364, 365). In fact, animal studies suggest that typically <5% of orally administered CoQ10 can be absorbed and reach the bloodstream (366). TABLE 2 lists the reported effects of CoQ10 supplementation in mice on tissue CoQ levels and ETC function. Consistent with the role of the liver in the metabolism of oral CoQ, it is in the liver that a significant response is most often observed. For other tissues, much more variable responses have been reported. Finally, it is worth noting that, to overcome the low oral bioavailability of CoQ10 and the disappointing outcomes of patient treatment with CoQ10 (40, 370–372), novel CoQ10 formulations have been proposed. Here are a few examples: CoQ10 nano-liposomes, lipid-CoQ10 conjugate nanodispersion (BPM31510), multicomposite CoQ10 terclatrate (Q-TER), CoQ10/β-cyclodextrin complexes, and nano-micellar CoQ10 formulations (Ubisol-Q10 and micellular formation with caspofungin) (42, 373–378). These formulations were demonstrated to have higher delivery efficiency in vitro compared to the addition of free CoQ10 to the culture medium, and some of them have already been shown to deliver better bioavailability of CoQ10 in vivo (41, 42, 365, 377–380). Administration via alternative routes, especially intravenous, which is possible with some formulations, is particularly appealing with respect to overcoming the poor and variable absorption of oral CoQ10 (42, 381, 382). However, the effects of these treatments on tissue CoQ levels and on in vivo efficacy in relieving deficiency and deficiency symptoms remain to be determined.
Table 2.
Reported effects of CoQ10 supplementation on tissue CoQ10 levels
| Model Type | CoQ10 Formulation | Total Dosage | Treatment Effects |
Refs. | |
|---|---|---|---|---|---|
| On CoQ10 Levels | On ETC Function | ||||
| Oral feeding | |||||
| Wild-type mice | |||||
| CoQ10 in chow | 0.5% CoQ10, 3 mo | Kidney ↑, liver ↑↑, brain ↔ | ND | (90) | |
| CoQ10 in chow | 93 or 371 mg/kg body wt/day, 3.5 or 17.5 mo | Heart ↑, liver ↑↑, kidney ↑↑, skeletal muscle ↑↑, brain ↔, mito (liver, heart, kidney, skeletal muscle) ↑ | Respiration of liver mito. ↔ | (367) | |
| Liposomal CoQ10 in drinking water | ∼300–400 mg/kg body wt/day, 3–7 mo | Liver ↑↑↑, muscle ↔, heart ↔, kidney ↔ | NE | (82, 123) | |
| CoQ10 in chow | 2.81 mg/g CoQ10 in food, 3 wk | Liver and liver mito. ↑↑↑ | NE | (8) | |
| Liposomal CoQ10 in drinking water | 0.4 mg/mL, 12–13 wk | Liver ↑↑↑; ovaries ↑↑, kidney ↔ | oocyte mito. respiration and ATP ↑ | (325) | |
| Liquid CoQ10 | 15 mg/kg/day for 3 mo | Liver ↑↑; muscle ↔, | ND | (117) | |
| CoQ deficiency mouse models | |||||
| Pdss2kd/kd | CoQ10 in chow | 0.5% CoQ10 in food, 3 mo | Kidney ↑, liver ↑↑, brain ↔ | Kidney CI-III and CI-III ↔ | (90) |
| Pdss2kd/kd | Liposomal CoQ10 in drinking water | ∼200 mg/kg body wt/day, 3.5 mo | Kidney ↔ | ND | (347) |
| Coq7 KO | Liposomal CoQ10 in drinking water | ∼300–400 mg/kg body wt/day, 3–7 mo | Liver ↑↑↑, muscle ↔, heart ↔, kidney ↔ | Respiration of liver mito. ↑ | (82, 123) |
| Coq7+/ | CoQ10 in chow | 2.81 mg/g CoQ10 in food, 3 wk | Liver and mito ↑↑↑ | Liver CI-CIII ↑ | (8) |
| Coq9R239X | Liquid CoQ10 | 240 mg/kg body wt/day, 2 mo | Liver ↑↑, muscle ↑↑, heart ↔, kidney ↔, brain ↔, kidney ↔ | Cerebrum mito. CI-CIII ↔, CII-CIII ↔ | (368) |
| Coq9R239X | Liquid CoQ10H2 | 240 mg/kg body wt/day, 2 mo | Liver ↑↑, kidney ↑, muscle ↑↑, heart ↑, kidney ↑, brain ↑, cerebrum mito. ↑ | Cerebrum mito. CI-CIII ↑ CII-CIII ↑ | (89, 368) |
| ADCK2+/− | Liquid CoQ10 | 15 mg/kg/day for 3 mo | Liver ↑↑; muscle ↑ | ND | (117) |
| Intraperitoneal injection | |||||
| Wild-type mice | Intralipid-solubilized CoQ10 | 200 µL of 1.45 mM once | Liver ↑↑↑, BAT ↑↑, skeletal muscle ↔, serum ↔ | NE | (356) |
| CoQ10 in 10% Tween 20 | 0.1 mg/g/day for 3 days | Heart mito. ↔ | NE | (369) | |
| Intravenous administration | |||||
| Wild-type mice | Micellar CoQ10 solution prepared with caspofungin | 12.0 mg/kg body wt/day, 10 days | Plasma ↑↑↑, liver↑↑↑, heart ↑↑, muscle ↑↑, kidney↑, spleen↑↑, lung ↑↑, brain ↑, heart mito ↑ | ND | (42) |
BAT, brown adipose tissue; CI-CIII, complex I-III activity; CII-CIII, complex II-III activity; ETC, electron transport chain; mito., mitochondria; ND, not determined; NE, no effect. ↑<2-fold increase of CoQ10 levels; ↑↑2- to 10-fold increase of CoQ10 levels; ↑↑↑≥10-fold increase of CoQ10 levels; ↔no change in CoQ10 levels.
3.4.2. Physiological effects of added exogenous CoQ.
As stated in sect. 2.3, boosting CoQ levels in cells without CoQ deficiency is not associated with increased respiratory rate, indicating that the normal CoQ amount is sufficient for mitochondrial respiratory function. On the other hand, increasing CoQ content may potentially augment antioxidant protection against oxidative stress-related damage. Many studies with cultured cells describe cytoprotective effects against oxidative stress of treatment with exogenous CoQ10. For example, in human skin fibroblasts, human umbilical vein endothelial cells (HUVECs), and rat pheochromocytoma (PC12) cells, treatment with CoQ10 ameliorates H2O2-induced cytotoxicity, suppresses senescent phenotypes induced by H2O2, or attenuates the increase of DCFH-DA fluorescence and MDA formation after H2O2 stress (383–385). In human neuroblastoma SH-SY5Y cells, protective effects of CoQ10 pretreatment were described for the toxicity of paraquat (1,1′-dimethyl-4,4′-bipyridinium), a redox cycler widely used to stimulate O2•− production in mitochondria (386). However, where exactly the exogenous CoQ10 intervenes in the cytotoxic actions of H2O2 or paraquat is not clear. Another example is a finding with isolated rat hepatocytes. Here it was shown that CoQ10 prevented cytotoxicity induced by CI inhibition by rotenone but not by the CIII inhibitor antimycin A and that the cytoprotection against rotenone was abolished after preincubation of the cells with dicumarol to inhibit DT-diaphorase (NQO1) (387). As expected, both rotenone and antimycin A treatments increase overall ROS levels as measured by DCFH-DA (387). This finding suggested that NQO1 may have a major role in maintaining CoQH2 levels in the cell. Note that reduction of CoQ by NQO1 oxidizes NADPH, which could alter the cellular NAD(P)+/NAD(P)H redox balance, thus affecting many processes.
Because of its extreme hydrophobicity, effective treatment with CoQ10 for whole animals has been challenging. In short, CoQ10 can only be given orally, but the oral bioavailability of CoQ10 is insufficient to ensure uptake in most organs. Hepatic uptake of oral CoQ10, however, can be substantial (8, 123, 388, 389). For these reasons, here we only take note of some studies that examined oxidative stress-related pathologies induced in the liver and the effect of CoQ10 supplementation on them. One of the earliest studies showed that injection of bacterial endotoxin (lipopolysaccharide) induces liver damage in mice and that this is associated with elevated liver lipid peroxidation. A drop in the levels of CoQ9H2 and vitamin E was also observed, presumably because of their oxidation. Administration of CoQ10 together with the endotoxin was shown to suppress lipid peroxidation, prevent the decrease in CoQ9H2 and vitamin E, and increase the survival rate of treated mice compared to endotoxin-only control mice (390). Oral CoQ10 supplementation in mice or rats was also shown to inhibit hepatic lipid peroxidation and liver toxicity induced by chemical exposure, such as to carbon tetrachloride, acetaminophen, or valproic acid (391–393). On the other hand, although the levels of lipid hydroperoxides were shown to increase significantly in the liver of aged PUFA-fed rats, this was not improved by CoQ10 supplementation (389). Thus, whether CoQ10 has a protective effect on liver lipid peroxidation remains ambiguous. Exploring the effects of exogenous CoQ on the redox status in other tissues will only become feasible when new methods to boost CoQ levels throughout the animals will become widely used (42).
4. OTHER FUNCTIONS OF CoQ
In this section, we focus on a number of CoQ functions that are less known and characterized compared to transporting electrons in the inner membrane of mitochondria and roles in ROS metabolism.
4.1. Role in Protection from Ferroptosis
The plasma membrane redox system (PMRS) was discovered in 1960 (245). CoQ at the plasma membrane is a vital part of the PMRS (see sect. 3.2.1). The ferroptosis suppressor protein 1 (FSP1, also known as AIFM2) was recently identified as a new member of the CoQ oxidoreductases that can regenerate CoQH2 in the plasma membrane. In this role, FSP1 can inhibit ferroptosis, a distinct form of regulated cell death that is characterized by excessive iron-dependent oxidation of PUFAs in cell membranes (252, 253, 256). Although many details of ferroptosis are yet to be worked out, this special form of cell death has been implicated in various pathophysiological conditions and diseases (394). Oxidation of PUFA during oxidative stress generates LOOH, which is considered a pivotal trigger of ferroptosis. The excessive accumulation of LOOH can lead to an unusual increase in the tension and cation permeability of the membrane, ultimately altering the ion content of the cell and rupturing the membrane (395).
Glutathione peroxidase 4 (GPX4), which is present in both the cytosol and mitochondria, is arguably the most important enzyme that keeps ferroptosis in check (394). The FSP1/CoQ pathway provides another distinct defense mechanism against ferroptosis at the plasma membrane (252, 256) (FIGURE 13). FSP1 reduces CoQ to CoQH2, using electrons from NAD(P)H (252, 256). CoQH2 can scavenge the lipid peroxyl radical (LOO•) generated during membrane PUFA oxidation and thus interrupt the spread of the lipid-radical chain reactions (see sect. 3.2.3). The G156A mutation in FSP1, which does not affect FSP1 expression or localization but impairs FSP1-mediated reduction of CoQ10, was shown to abolish the antiferroptotic activity of the protein (252). FSEN1, a small-molecule inhibitor of FSP1 that targets FSP1 CoQ oxidoreductase activity specifically, sensitized cancer cells to ferroptotic death induced by loss of GPX4 activity (396). Also, lowering CoQ10 levels was shown to hinder the ability of reexpression of FSP1 to rescue the GPX4 inhibitor RSL3-induced ferroptotic death of FSP1 knockout cells (252, 256). These cell culture findings were further corroborated by the in vitro finding that it is only in combination with CoQ10 that FSP1 significantly delays the autoxidation of the polyunsaturated lipids of chicken egg phosphatidylcholine liposomes (256).
FIGURE 13.
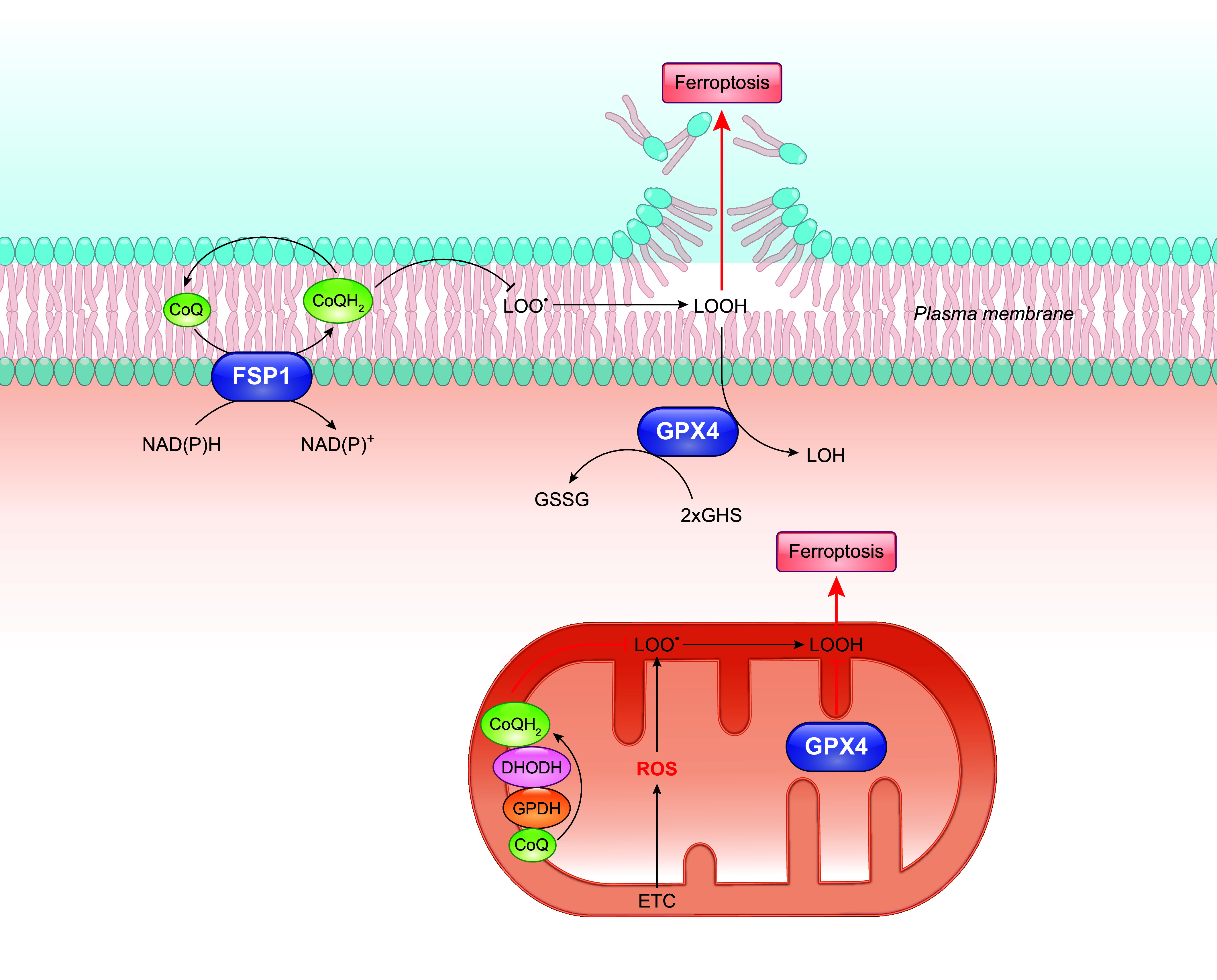
Role of CoQ in ferroptosis regulation. Ferroptosis suppressor protein 1 (FSP1) acts as an oxidoreductase mainly localized on the plasma membrane and reduces CoQ to CoQH2 using electrons from reduced nicotinamide adenine dinucleotide (NADH) or reduced nicotinamide adenine dinucleotide phosphate (NADPH). By directly scavenging lipid peroxy radicals (LOO•) generated from lipid peroxidation, the fully reduced form of CoQ, CoQH2, prevents excessive lipid peroxidation and thus inhibits ferroptosis. In the cytosol and mitochondria, glutathione peroxidase 4 (GPX4) converts glutathione (GSH) to oxidized glutathione (GSSG) while reducing lipid hydroperoxides (LOOH) to lipid alcohols (LOH), which is the main mechanism to regulate ferroptosis. Mitochondrial ROS production likely contributes to ferroptosis. On the other hand, CoQH2 generation from respiration and other dehydrogenases such as dihydroorotate dehydrogenase (DHODH) and glycerol-3-phosphate dehydrogenase (GPDH) likely enhance defense against ferroptosis by inhibiting lipid peroxidation. See glossary for other abbreviations.
Superoxide generated from the ETC in the mitochondrial matrix can be converted to H2O2 by SOD2. H2O2 subsequently can be converted to hydroxyl radicals (•OH) by the Fenton reaction. •OH is one of the most reactive species known and can rapidly abstract hydrogen atoms from PUFA, triggering lipid peroxidation. Therefore, mitochondrial ROS production likely contributes to ferroptosis (397). On the other hand, CoQH2 generation from the ETC should enhance protection against ferroptosis by inhibiting lipid peroxidation (398, 399). Reduction in the level or activity of dihydroorotate dehydrogenase (DHODH) or mitochondrial glycerol-3-phosphate dehydrogenase (mGPDH, also referred to as GPD2) decreases the CoQH2-to-CoQ ratio, as expected, and this was shown to be associated with increased sensitivity to induction of ferroptosis by inhibition of GPX4, indicating an important role of these two IMM enzymes in the regulation of ferroptosis via the production of CoQH2 (398, 399). In addition, treatment with MitoQH2 (a mitochondria-targeted analog of CoQH2) attenuated the sensitizing phenotype resulting from the knockout of DHODH or mGPDH (398, 399).
The role of CoQH2 in ferroptosis is further supported by the observed sensitization to GPX4 inhibitors in cells with impaired CoQ10 biosynthesis (due to pharmacological inhibition of COQ2) or overexpression of the alternative oxidase (AOX), which oxidizes CoQH2 (252, 398, 400). On the other hand, how boosting CoQH2 levels affects ferroptosis is unclear. Overexpression of FSP1 was shown to inhibit ferroptosis and improve the loss of cell viability induced by RSL3 in H1703 and H446 lung cancer cells that have relatively low levels of FSP1 (252). A ferroptosis-inhibiting effect was also reported by overexpressing mGPDH (401). However, a direct link between the inhibition of ferroptosis and the level of reduction of CoQ remains to be clarified. MitoQH2 appears to protect against RSL3-induced cell death in HeLa and RPMI7951 (melanoma) cells, but only very little protection was observed in HT-1080 fibrosarcoma cells (252, 399). Idebenone, a short acyl side chain soluble CoQ10 analog, was shown to inhibit RSL3-induced ferroptosis in U2OS osteosarcoma cells [Bersuker et al. (252)]. Given potential variations in the integration of different ferroptosis defense mechanisms among cell types as well as variations in redox metabolism, it seems that the range of CoQ levels required for optimum defense against ferroptosis could be quite different among cell types and with different ferroptosis inducers.
4.2. CoQ in the Regulation of the Mitochondrial Permeability Transition Pore
The mitochondrial permeability transition pore (mPTP, also referred to as PTP or MTP) is a voltage-gated, nonselective, large IMM conductance channel. The IMM has a low permeability to ions and solutes in order to maintain the proton electrochemical gradient generated during electron transfer to drive ATP synthesis. The energy stored in the form of the transmembrane proton gradient is also used for other mitochondrial processes, such as ion homeostasis, protein import, etc. mPTP opening allows for all low-molecular weight molecules to equilibrate across the IMM, including H+. Transient mPTP opening is implicated in ROS signaling and mitochondrial Ca2+ homeostasis, whereas sustained opening can cause a collapse of the mitochondrial membrane potential (ΔΨm), uncoupling of OXPHOS, Ca2+ release into the cytosol, and eventually rupture of the outer membrane and cell death (402–404).
The mitochondrial membrane potential (ΔΨm) maintained by the H+ gradient across the IMM is intrinsically related to ROS generation from the respiratory chain. Indeed, high ΔΨm promotes ROS generation, whereas ROS production tends to decrease at lower ΔΨm (327, 405). It has been proposed that when ΔΨm is high some transients of the respiratory chain electron transport, capable of reducing O2 to O2•−, such as CoQ•−, become long-lived, thereby increasing the possibility of electron leak and ROS production (405). On the other hand, increased mitochondrial ROS can activate mPTP opening, which can lead to further ROS production and release (326). Thus, potentially, CoQ deficiency could induce mPTP opening through increased production of ROS (see sect. 3.3.1). However, this has not been supported by evidence so far. mPTP opening was found not to be significantly altered in the podocytes of mouse Pdss2 mutants whose CoQ content is only ≈30% that of control mice (406–409).
On the other hand, studies with isolated mitochondria have documented that some CoQ analogs can directly act on mPTP opening. For example, the addition of CoQ2, CoQ10, or decyl-ubiquinone (a CoQ derivative with an n-alkyl side chain) to rat liver mitochondria was shown to increase the capacity to retain calcium, suggesting an inhibition of the mPTP (410, 411). In contrast, in isolated rabbit heart mitochondria, the addition of CoQ2 was found to stimulate mPTP opening (411). In newborn rat cardiomyocyte mitochondria, the addition of decyl-ubiquinone caused an increase in respiration and a concomitant decline in membrane potential, with both effects being inhibited by Cyclosporine A (CsA) (412).
4.3. CoQ in the Regulation of Uncoupling Proteins
Uncoupling proteins (UCPs) are mitochondrial inner membrane proteins that function as H+ leak channels. By allowing H+ to flow back from the mitochondrial IMS to the matrix, UCPs catalyze the dissipation of the proton electrochemical gradient built up by the respiratory chain. Thus, one key consequence of UCP activation is the uncoupling of mitochondrial respiration from ATP synthesis, leading to energy dissipation as heat (413). In fact, the first UCP protein discovered, UCP1, is expressed mainly in brown adipose tissue (BAT), where it plays a well-defined role in nonshivering thermogenesis (414). The roles of other UCPs that are found throughout mammalian tissues (UCP2 in a wide variety of tissues, UCP3 predominantly in skeletal muscle, UCP4 and UCP5 in the brain) are still largely tentative, such as being possibly important in the immune response, in insulin secretion, and in the control of mitochondrial ROS generation (413, 415–418).
UCP-mediated uncoupling can be activated by free fatty acids (FFA) by a mechanism that is not fully elucidated (419). FFA-induced UCP activity is sensitive to purine nucleotides (PNs), which were first recognized as inhibitors of UCPs (420). In fat tissue, PN-dependent inhibition of respiratory rate accompanied by the restoration of ΔΨm has been considered diagnostic of UCP function (421). However, the PN sensitivity has been mostly demonstrated in artificial membrane models, whereas conflicting results were obtained with mitochondrial preparations where poor or even no sensitivity to PNs has been observed for the FFA-induced proton leak (413, 422–424). Moreover, the question arises as to how UCPs could ever effectively conduct proton in vivo, given the apparent affinity of UCPs for PNs and the high concentrations of nucleotides in vivo (in the millimolar range in cells) (425, 426).
A link between CoQ and UCP was first proposed more than two decades ago. Echtay and colleagues showed that UCPs (UCP1, 2, and 3) expressed in E. coli can be incorporated into liposomes to yield H+ transport activity. But no H+ influx into the vesicles was observed in the presence of an FFA (e.g., lauric acid) unless oxidized CoQ (CoQ6 or CoQ10) was provided, and the activated H+ uptake was sensitive to nucleotides (ADP or ATP) (427, 428). The findings were interpreted to indicate an obligatory role of CoQ in FFA-induced H+ transport by UCPs. This, however, was refuted in a subsequent study that used the same model system but with optimized conditions and showed that CoQ had no effect at all on lauric acid-dependent H+ transport by UCPs (429). Moreover, a yeast study reported that the activity of mouse UCP1 is similar whether expressed in CoQ-replete or CoQ-deficient mitochondria (714). Thus, it remains unclear whether there is any effect of CoQ on UCPs. Interestingly, a different mechanism by which CoQ could be involved in the regulation of UCP activity has been proposed. In experiments with isolated mitochondria, it was observed that the CoQ redox state modulates the sensitivity of the FFA-induced UCP activity to PNs (413, 423). A pioneering study with rat muscle mitochondria showed that GTP (a PN) was not able to inhibit lauric acid-induced UCP2/3-mediated uncoupling under respiratory state 3 (phosphorylating). However, an inhibitory effect was observed after the addition of the CII inhibitor malonate but not after the addition of the CIII Qi site inhibitor antimycin A. The different sensitivities to PN were interpreted in the context of the differences in the mitochondrial CoQH2-to-CoQ ratios brought about by the addition of malonate and antimycin A. That is, CII inhibition decreases the level of reduced CoQ, whereas antimycin A prevents oxidation of CoQH2, even though it also slows respiration (423). Subsequently, it was further reported that malonate can prevent the suppression of UCP inhibition by PN when antimycin A is present, consistent with its effect on decreasing the ratio of reduced to oxidized CoQ (413).
Similar results were obtained in later studies with BAT UCP1, various mitochondria under phosphorylating or nonphosphorylating conditions (from microorganisms, plants, or mammals), and different PNs (422, 430–432). The level of CoQ reduction under which the inhibition by PN becomes effective varies depending on conditions. For example, under phosphorylating, that is, coupled, conditions, relief of GTP inhibition was observed with a CoQ level of reduction above 85–88% in rat BAT mitochondria and above 57–64% in rat skeletal muscle mitochondria (423, 430). Of note, the endogenous CoQ redox state was shown to have no effect on the FFA-induced UCP activity in the absence of PNs (430–432). Considering these and taking into account the apparent affinity of UCPs for PNs and PN concentrations in vivo (in the millimolar range) (425, 426), it has been suggested that CoQ is involved in regulating the activity of UCP by modulating UCP sensitivity to PNs. The idea is that the high availability of CoQH2 in mitochondrial membranes promotes UCP activation by relieving the inhibition from PNs. Yet, under conditions leading to low CoQH2, such as low availability of respiratory substrates, PN sensitivity is turned on, leading to inactive UCP and more efficient ATP production (413, 421). This further implicates that different levels of mitochondrial CoQ reduction in different tissues or organisms could be a determinant of intrinsic UCP activity and metabolic efficiency. The exact mechanism for how CoQH2 prevents PN to inhibit FFA-activated UCPs remains to be elucidated. One postulate is that CoQH2 interferes directly with PN binding to UCPs (FIGURE 14) (421).
FIGURE 14.

A tentative model of involvement of CoQ in UCP regulation. UCP-mediated uncoupling can be activated by free fatty acids (FFA), an effect that is sensitive to purine nucleotides (PNs). The CoQ redox state has no effect on basal and FFA-induced UCP-catalyzed H+ conductance in the absence of PNs, but it modulates the sensitivity of UCP to inhibition by PNs. At a given fatty acid concentration, increased CoQ reduction by the respiratory chain decreases the binding affinity of PN to UCP, possibly by directly interfering with PN binding to UCPs due to structural similarities, thereby promoting UCP activation. Conversely, at lower CoQH2 levels, no negative regulation occurs and UCP activity is inhibited by PNs. Additionally, it has been proposed that altered levels of CoQ or its redox state potentially could affect ROS-mediated UCP activation through its antioxidant activity against lipid peroxidation. See glossary for other abbreviations.
ROS-mediated UCP activation implicates a different pathway by which CoQ could play a role in the regulation of UCP activation. Incubating rat skeletal muscle and BAT mitochondria with xanthine plus xanthine oxidase to generate O2•− was shown to increase mitochondrial proton conductance through the effects on UCPs (433). Later it was shown that the effect of O2•− on UCP-mediated proton conductance is mediated through lipid peroxidation products, such as 4-hydroxynonenal (4-HNE) (434). CoQ could be involved as a site of ROS production during electron transport or CoQH2 could help to prevent lipid peroxidation (see sects. 3.1 and 3.2). However, as far as we know, altered levels of CoQ or its redox state have not been documented to affect UCP activation by O2•−. It also should be noted that activation of UCPs by O2•− or HNE is still in dispute, as conflicting results have been reported (435, 436). The observed effects of O2•− on stimulating uncoupling appear to be tissue specific by mechanisms unknown (437). Finally, and noteworthily, a more recent study reported that in UCP1-positive murine brown and beige adipocytes and in mouse brown adipose tissue, a 30–75% reduction of CoQ levels triggered a robust suppression of UCP1 expression that can be rescued by exogenous CoQ supplementation (119).
We have seen how CoQ has been implicated in modulating the activity of mPTP and UCPs, both of which are important players in the maintenance of the membrane potential (ΔΨm) across the IMM. It might be worth noting that there are several reports that measure ΔΨm in CoQ-deficient cells by using the fluorescence dye tetramethylrhodamine ethyl ester (TMRE), whose accumulation in mitochondria is strongly potential dependent. In human COQ9 mutant cells with ≈20% residual CoQ10, a decrease of TMRE fluorescence was reported but no increase of mitochondrial ROS was found (as assessed by MitoSox staining intensity) (113). In contrast, COQ2 mutant cells, which have ≈36% residual CoQ10, display an increase in MitoSox fluorescence but no change in TMRE fluorescence (113). Inhibition of COQ2 by 4-NB in wild-type or ADCK3 mutant cells that lowers CoQ10 to below 50% increases the MitoSOX signal but does not always affect the TMRE signal in the mitochondria (336). With a similar approach, a ≈50% reduction in CoQ10 level in the T67 human glioma cells was found to be associated with a slight elevation of ΔΨm (338). Together these findings indicate a complex and bidirectional relationship between CoQ deficiency and the effect on ΔΨm.
4.4. Involvement of Plasma Membrane CoQ in the Regulation of the Cytosolic Redox State
As mentioned in a previous section, CoQ is a vital part of the constitutive PMRS (FIGURE 8). In addition to its antioxidant role in maintaining antioxidant levels in and around membranes, PMRS plays a role in the modulation of cytosolic NAD(P)+/NAD(P)H levels and the generation of NAD+ that is required to support glycolytic ATP production. This role is shown to be particularly essential for cell survival under conditions of mitochondrial energetic failure (438–440). Cells with damaged mitochondria, such as rho zero (ρ0) cells (devoid of mitochondrial DNA), upregulate PMRS activity, by inducing the expression of PMRS enzymes (438, 439, 441). The resulting elevated electron transfer rate across the plasma membrane, in addition to potentially strengthening antioxidant regeneration (CoQ, vitamin E, and vitamin C), generates more NAD+ from NADH (439, 441). Thus, by means of the PMRS, CoQ is involved in balancing the cytosolic NAD(P)+-to-NAD(P)H ratio, which is a key component of the redox metabolic state. But whether or when the availability of CoQ becomes limiting for the overall rate of electron flux across the plasma membrane is unknown. Interestingly, by inhibiting CoQ10 synthesis with 4-NB in T67 human glioma cells to ≈50% of normal levels, an increase in the overall activity of plasma membrane NADH oxidoreductase (measured as diphenyleneiodonium-sensitive oxygen consumption) was observed. This was likely mostly caused by the upregulation of the dehydrogenases of PMRS as a response to mitochondrial impairment (120). CoQ10 supplementation appeared to reverse the effect on NADH oxidoreductase but further increased the overall activity of plasma membrane NADH oxidoreductase, indicating that a ≈50% level of CoQ10 is not sufficient to support maximum stimulation of the PMRS (120).
4.5. Localization and Effects of CoQ in Lipid Membranes
Biological membranes are composed primarily of two layers of phospholipids, which are amphipathic molecules with a hydrophilic head group and a hydrophobic tail. Various biophysical methods, such as NMR spectroscopy, fluorescence, linear dichroism, differential scanning calorimetry, and neutron diffraction have been used to determine CoQ localization within lipid bilayers. However, no unequivocal conclusion about this can yet be drawn. Typically, two possible orientations are considered: one where CoQ molecules with tails of at least three isoprenoid units are fully embedded in the center of the bilayers lying flat parallel to the mid-membrane plane and one where the quinone ring resides near the region of the phospholipid head groups, with the isoprenoid chain encapsulated inside the bilayer toward the center (14–17, 20, 442–449). Other proposals include the formation of head-to-head aggregates, which are hypothesized to facilitate the location of the polar quinone ring in the hydrophobic middle of the bilayer (15, 17, 450). Empirical evidence is presented for both main models, leaving the question unresolved. Finally, an interesting variation is the suggestion that the CoQ redox state might affect where exactly the molecule is located, with one study suggesting that CoQ10H2 appears to be closer to the phospholipid head groups and the polar surface of the membrane than the oxidized form (17).
Does CoQ localization in lipid membranes affect membrane properties? A study with CoQ-deficient E. coli reported that ΔubiG mutants are more sensitive to high-salt stress and that supplementation with CoQ10 restores the mutants’ tolerance (19). More surprisingly, in response to hyperosmotic salt stress, the study reported to observe ≈100-fold increase in endogenous CoQ8 levels. Furthermore, in vitro liposome experiments showed greater hyperosmotic stress after the addition of CoQ10. However, a more recent study seems incompatible with the idea of a membrane-stabilizing and osmoprotective role of CoQ. Indeed, the function of the membrane protein ProP, a known protonmotive force-dependent osmosensory and osmoregulatory transporter, was found to be impaired in ΔubiG mutants (23). This suggests that the effect of CoQ8 deficiency on ProP function was the result of impaired respiration, not of altered physical properties of the membrane. Furthermore, this study showed that in wild-type E. coli respiration was significantly inhibited after osmotic upshift but was restored after prolonged culture in a high-osmotic medium. Taken together, the latter study proposed a key role of impaired respiration in the osmosensitivity induced by CoQ deficiency (23).
Nevertheless, multiple studies conducted in artificial liposomes reported other specific effects of CoQ10 on bilayer properties. In liposomes made of the phospholipid palmitoyl-2-oleoyl-sn-glycero-phosphocholine (POPC), the inclusion of CoQ10 leads to a membrane condensing effect, increased resistance toward rupture, and decreased membrane permeability (451). Also, in several model membranes, the addition of CoQ10 was shown to increase lipid packing, which could affect membrane rigidity and other physical properties such as mechanical stability and permeability (21–23). It might also be worth mentioning that solanesol, a nonquinone isoprenoid that has 9 isoprene subunits like CoQ9 but lacks the quinone head group, was used to determine whether the quinone ring is essential for the effects induced by CoQ10. Contradictory findings about this were reported. Solanesol was shown to be as effective as CoQ10 in protecting against collapse due to high-salt stress in liposomes prepared from a lipid mixture but had no, or very little, effect on lipid packing, membrane density, or permeability in POPC liposomes and in an IMM membrane model (19, 21, 22, 451).
Although the liposomes used in these investigations were built from biologically relevant lipids (such as POPC), the lipid composition did not exactly reflect that of native membranes. Between different species and tissues but also among different subcellular compartments, lipid content and compositions vary considerably, and thus so might the interactions with CoQ. For example, it has been suggested, but remains to be investigated, that CoQ in the IMM, where the content of the well-known membrane stabilizer cholesterol is low and protein load is high, might create a mechanical barrier for transmembrane proton leaks (16).
5. CoQ BIOSYNTHESIS
Endogenous synthesis is the main source of cellular CoQ. The biosynthesis of CoQ is highly conserved, especially among eukaryotes, and has been investigated in different model organisms. To date, it has been best characterized in E. coli and the budding yeast S. cerevisiae, but the knowledge obtained from the model systems has been shown to apply closely to higher organisms including humans. The process of CoQ biosynthesis begins with the formation of the lipophilic isoprenoid side chain and the synthesis of 4-hydroxybenzoic acid (4-HB), which is the universal precursor of the aromatic head group of CoQ. 4-HB is derived from different precursors depending on the species. The isoprenoid side chain is made in similar ways in all eukaryotes but by a different pathway in E. coli (452, 453). Briefly, after the attachment of the isoprenoid tail to the quinone ring, the ring group undergoes a series of modifications to yield the final product. These steps are catalyzed by several proteins in sequential steps. It has now been widely accepted that most of the pathway components need to assemble into a multisubunit protein complex for the biosynthetic reactions to occur efficiently and produce CoQ (25–28, 454). However, many molecular, structural, and mechanistic details remain to be elucidated. A related open question is how the whole process and thus the final yield of CoQ is regulated. Below we summarize the main understanding of CoQ biosynthesis in budding yeast S. cerevisiae and Mus musculus (mouse), which are the most detailed and most relevant models for humans, respectively. For additional information, as well as for CoQ biosynthesis in E. coli and other organisms, the reader can refer to many comprehensive review articles (7, 10, 11, 25–27, 37, 109, 455–465).
5.1. CoQ Biosynthetic Pathways in Eukaryotes
CoQ synthesis in eukaryotes takes place in the mitochondria, more specifically on the matrix side of the IMM. Most eukaryotic genes and proteins required for CoQ biosynthesis are given COQ names plus a number based on the order in which they were identified. They are nucleus encoded in all well-studied species and contain mitochondrial targeting sequences. The main precursor of the aromatic quinone ring used by both yeast and mammals is 4-HB, which can be derived from tyrosine, but some steps of its synthesis pathway remain unidentified (464, 466). Human cells can also use phenylalanine as a precursor for 4-HB because phenylalanine hydroxylase is present in human cells. However, this enzyme is absent in yeast (456). Furthermore, mammalian cells may also be able to use polyphenols in CoQ biosynthesis, such as kaempferol (467). The pathways by which polyphenols enter into the biosynthesis of CoQ remain to be fully delineated. See Ref. 458 for more details.
In eukaryotes, isoprenoid side chains are synthesized via the mevalonate pathway, which begins with acetyl-CoA and produces isopentenyl pyrophosphate (IPP). Isomerization of IPP generates dimethylallyl pyrophosphate (DMAPP or DPP). Catalyzed by farnesyl diphosphate synthase (FDPS), these two metabolites generate farnesyl pyrophosphate (FPP), which is predominantly localized to peroxisomes and is the precursor of several biomolecules with distinct functions including CoQ, dolichols, prenylated proteins, and sterols (455). In the mitochondria, condensation of FPP with several IPP molecules catalyzed by a trans-prenyl-transferase ultimately delivers an isoprenoid side chain of a specific length for CoQ biosynthesis. To date, it is not fully understood how the building block of the isoprenoid tail and the ring precursors are transported into mitochondria for CoQ biosynthesis. A recent study identified Hem25p, a mitochondrial glycine transporter, required for mitochondrial IPP transport for CoQ biosynthesis in the yeast S. cerevisiae. However, its mammalian homolog SLC25A38 does not function as an IPP transporter (468, 469).
How CoQ moves out of the mitochondria to other various membranous organelles is not yet understood. However, several possible routes have been considered to have a role in the intracellular distribution of CoQ. One or a combination of them could be responsible for CoQ transport in different eukaryotic cells. Here are a few proposals. An in vitro study that followed the appearance of newly synthesized, labeled CoQ10 in human leukemia cells (HL60) suggested that the endomembrane system might be involved (353). STARD7, a lipid-binding protein that is known to be required for phosphatidylcholine delivery to the mitochondria, has been shown to play a role in CoQ transport to the plasma membrane and protection of ferroptosis (400, 469). Saposin B, a small protein with lipid binding properties involved in glycosphingolipid metabolism, binds to CoQ, and its precursor prosaposin level was shown to affect the levels of CoQ in human HepG2 and CaCo-2 cells (470–473). Yeast studies showed that the bridgelike structure that forms at the membrane contact site between the endoplasmic reticulum (ER) and the outer mitochondrial membrane, named ERMES, is necessary for CoQ export from the mitochondria (474). Finally, it is worth noting that, in a mouse model, deficiency of COQ7 was shown to affect CoQ distribution within mitochondrial membranes, more specifically between the IMM and OMM (8). Readers interested in this topic can refer to Ref. 360 and the references therein for further information.
5.1.1. The budding yeast S. cerevisiae.
As mentioned, in S. cerevisiae the benzoquinone ring precursor for CoQ 4-HB can be derived from the metabolism of tyrosine. Five aminotransferases (Aro8, Aro9, Bat2, Bna3, and Aat2) have recently been shown to each be able to catalyze the deamination of tyrosine to 4-hydroxyphenylpyruvate (4-HPP), the first step of 4-HB production from tyrosine (456, 475). The last step is the oxidation of 4-hydroxbenzaldehyde (4-HBz) to 4-HB, which is catalyzed by the aldehyde dehydrogenase Hfd1, mainly located in the OMM (456, 476, 477). Other details of the tyrosine-to-4-HB pathway are not yet clear. Of note, expression of ALDH3A1, a human homolog of yeast Hfd1, restored CoQ6 biosynthesis in the hfd1 yeast mutant (456). However, it remains to be determined whether CoQ biosynthesis in mammals requires ALDH3A1 (478). In addition to tyrosine, 4-HPP can also be derived from the shikimate pathway, also known as the chorismate biosynthesis pathway, which is not present in animals (479, 480). Chorismate is utilized as a substrate in a number of biosynthetic pathways of aromatic compounds. One branch generates para-aminobenzoic acid (pABA) via a two-step reaction catalyzed by Abz1 and Abz2, respectively (481, 482). pABA, a well-known precursor of folic acid (an essential B vitamin), can serve as an alternative ring precursor of CoQ biosynthesis in yeast but not in E. coli or mammalian cells (482, 483). The CoQ intermediates derived from pABA contain an amino group (–NH2) at the C4 position instead of a hydroxyl group (–OH), which needs to be replaced to yield CoQ. The conversion of the NH2 into an OH is proposed to occur on the early intermediate 3-hexaprenyl-4-amino-5-hydroxybenzoic acid (HHAB) and relies on the Coq6 enzyme (484–486). Finally, like E. coli and mammals, S. cerevisiae is able to use para-coumarate and resveratrol as CoQ ring precursors (487). The discovery of the ability of yeast to use different sources as the ring precursor of CoQ biosynthesis raises the question of how the cells interpret particular media conditions and adapt to different nutrient mixtures. Competition experiments using 13C-aromatic ring precursors suggest that pABA and 4-HB provided exogenously are equally efficient at generating CoQ6 (482). However, their relative contribution to CoQ6 production under physiological conditions is not known.
At least 12 gene products, namely Coq1–Coq9, Coq11, Arh1, and Yah1, have been identified in S. cerevisiae to be necessary for making CoQ6 from the ring and side chain precursors (FIGURE 15) (25–27, 483, 489–495). Coq2 is an integral membrane protein, whereas other Coq polypeptides (Coq1, Coq3–Coq11) are localized to the matrix side of the IMM. Coq1 (trans-prenyl-transferase) synthesizes the hexaprenyl diphosphate tail. Coq2 (polyprenyl diphosphate transferase) then attaches the tail to aromatic ring precursors such as 4-HB. At this time, four Coq proteins have been identified to participate in the subsequent modifications of the CoQ benzoquinone ring. They include Coq3 (O-methyltransferase), Coq5 (C-methyltransferase), and Coq6 and Coq7 (hydroxylases). The function of Coq6 requires ferredoxin (Yah1) and ferredoxin reductase (Arh1), both of which are mitochondrial iron-sulfur [2Fe-2S] redox proteins with a known role in heme A biosynthesis and in Fe-S cluster assembly (483, 489). Their function in CoQ6 biosynthesis is predicted to be as electron donors for Coq6’s activity. coq1–coq9 deletion mutants lack CoQ6 and are therefore respiration incompetent, as they are unable to grow on nonfermentable carbon sources such as glycerol or ethanol (25, 496, 497).
FIGURE 15.

The CoQ biosynthesis pathway in the yeast S. cerevisiae and humans. The proteins are in blue (S. cerevisiae) or green (humans). Dotted arrows designate multiple-step reactions, and the steps that are specific to yeast are shown in blue. R indicates the poly-isoprenoid tail. The synthesis of the isoprenoid depends on the mevalonate pathway, which produces the biosynthetic precursors of isoprenoids. Coq1 in yeast and a heterotetrameric protein formed by PDSS1 and PDSS2 in humans determine the number of isoprene units in the polyisoprene tail. The main ring precursor used by both yeast and humans is 4-hydroxybenzoic acid (4-HB), synthesized from tyrosine in the cytosol. The first and last steps of this pathway have been defined in the yeast, where 5 aminotransferases, Aro8, Aro9, Bat2, Bna3, and Aat2, redundantly convert tyrosine to 4-hydroxyphenylpyruvate (4-HPP) and the last step is catalyzed by Hfd1. After its transport into mitochondria, Coq2/COQ2 attaches the isoprenoid tail to 4-HB. Subsequent to this step, the CoQ ring undergoes several sequential modifications before yielding CoQ. The intermediates detected in yeast are shown. A recent study using a human embryonic kidney cell line (HEK293) showed that the decarboxylation and hydroxylation of position C1 occur in a single oxidative decarboxylation step and it is catalyzed by COQ4. The reaction catalyzed by COQ4 preferentially occurs before the C5 hydroxylation by COQ6; however, the alternative sequence of reactions, that is COQ6 and COQ3 may act before COQ4, is also possible (488). Yeast can also utilize para-aminobenzoic acid (pABA) for CoQ synthesis, which is made from chorismate in 2 steps catalyzed by Abz1 and Abz2. The nitrogen-containing intermediates generated from its utilization are also depicted. Coq6 and Coq9 are able to deaminate the ring C4 position on the intermediate derived from pABA. Whether and where Coq4 is involved before the deamination step remains to be demonstrated. pABA is also an intermediate in the synthesis of folate in the yeast. Two additional compounds that can also serve as ring precursors both in S. cerevisiae and mammals are shown at top: para-coumaric acid and resveratrol. The absence of Coq6 activity leads to the accumulation of 3-hexaprenyl-4-aminophenol (4-AP) and 3-hexaprenyl-4-hydroxyphenol (4-HP) when yeast cells are grown in pABA and 4-HB, respectively. The Coq/COQ proteins with unclear molecular functions are listed in a box. CoQ and intermediates are shown in their reduced forms. DDMQH2, 3-hexaprenyl-5-methoxy-1,4-benzenediol; DMeQH2, 2-methyl-3-hexaprenyl-5-methoxy-1,4,6-benzenetriol; DMQH2, 2-methyl-3-hexaprenyl-5-methoxy-1,4-benzenediol; FPP, farnesyl diphosphate; GPP, geranyl pyrophosphate; HAB, 3-hexaprenyl-4-aminobenzoic acid; 4-HBz, 4-hydroxbenzaldehyde; HHAB, 4-amino-3-hexaprenyl-5-hydroxybenzoic acid; HHB, 3-hexaprenyl-4-HB; IPP, isopentenyl pyrophosphate. See glossary for other abbreviations.
It has been well established that a subset of CoQ6 pathway components in S. cerevisiae form a supramolecular protein complex that carries out all the reactions subsequent to the Coq2-catalyzed prenylation. This large multi-Coq protein complex in the mitochondria containing Coq3–9 and Coq11 has been termed the CoQ synthome or complex Q. To avoid confusion, we spell it out as “the biosynthetic CoQ complex” for the rest of this review. The complex is necessary for the stability and function of its individual constituents, which is supported by the findings that 1) decreased levels of other Coq polypeptides are generally found in strains harboring coq gene deletions but no or a smaller effect was observed on the expression of other Coq proteins in mutants harboring point mutations instead of deletions and 2) only the early intermediates of CoQ6 biosynthesis, 3-hexaprenyl-4-hydroxybenzoic acid (HHB) and 3-hexaprenyl-4-aminobenzoic acid (HAB), produced by the prenylation of 4-HB and pABA, respectively, are readily detected in each of the coq3–coq9 null yeast mutants. The intermediates that should accumulate because they are the substrates of the missing enzymatic reactions are not accumulating in detectable amounts (TABLE 3) (25, 465, 485, 492, 498–506). A high-molecular weight protein complex comprising Coq proteins was detected by size exclusion chromatography, and several Coq polypeptides were shown to comigrate in blue-native polyacrylamide gel electrophoresis (BN-PAGE) as a high-molecular weight band (498, 499, 503, 506). Nevertheless, further characterization of the CoQ biosynthetic complex remains necessary to determine its exact composition, subunit stoichiometry, and three-dimensional (3-D) structure and the likely dynamic nature of its assembly.
Table 3.
Yeast S. cerevisiae CoQ biosynthetic mutants
| Gene | CoQ6 Synthesis | Respiratory Growth | CoQ Intermediates |
|
|---|---|---|---|---|
| − | + Coq8 OE | |||
| coq1 | Absent | Absent | Not found | Not found |
| coq2 | Absent | Absent | Not found | Not found |
| coq3 | Absent | Absent | HHB↑, HAB↑* | HHB↑, HAB↑* |
| coq4 | Absent | Absent | HHB↑, HAB↑* | HHB↑, HHAB↑* |
| coq5 | Absent | Absent | HHB↑, HAB↑* | DDMQ6↑, DDMQ6↑* |
| coq6 | Absent | Absent | HHB↑, HAB↑* | 4-HP↑, 4-AP↑* |
| coq7 | Absent | Absent | HHB↑, HAB↑* | DMQ6↑, DMQ6↑* |
| coq8 | Absent | Absent | HHB↑, HAB↑* | CoQ6 |
| coq9 | Absent | Absent | HHB↑, HAB↑* | DMQ6↑, 4-HP↑, 4-AP↑* |
| coq10 | Impaired | Impaired | HHB↑, DMQ6↓ HAB↑ * | HHB↑, HAB↑* |
| coq11 | Impaired | Not affected | Not found | Not found |
*In the presence of pABA as the labeled precursor instead of labeled 4-HB. For references, see sect. 5.1.1. 4-AP, 4-aminophenol; 4-HP, 3-hexaprenyl-4-hydroxyphenol; DDMQ6, demethyldemethoxy-CoQ6; DMQ6, demethoxy-CoQ6; OE, overexpression. See glossary for other abbreviations.
Among the Coq proteins whose functions are not fully understood, namely Coq4, Coq8, Coq9, Coq10, and Coq11, Coq4 and Coq8 are proposed to be involved in the organization or maintenance of the biosynthetic CoQ complex, though this has not yet been directly demonstrated (484, 498, 499, 501, 506). A recent study, however, shows that, in addition to the proposed structural role in the organization of the complex, human COQ4 acts as an oxidative decarboxylase, substituting in a single step the carboxylic acid group with a hydroxyl group on carbon C1 of CoQ precursors (488). For Coq8, its overexpression (OE) in coq3Δ, coq5Δ, coq6Δ, coq7Δ, or coq9Δ cells has been shown to elevate the steady-state levels of other Coq proteins and to stabilize the biosynthetic CoQ complex and, as a result, to allow the accumulation of the corresponding CoQ biosynthetic intermediates (484, 501). Furthermore, Coq8 might also enable CoQ intermediate extraction from the IMM to facilitate their availability to CoQ aromatic ring-modifying enzymes (507, 508). Finally, it has been proposed that Coq8 and its ATPase activity may have a role in the organization of the biosynthetic CoQ complex into discrete domains within mitochondria, observed as puncta located near endoplasmic reticulum (ER)-mitochondria contact sites (502). Such localization might be required for orchestrating CoQ synthesis in the IMM and its export from mitochondria (474, 502).
Coq9 is a member of the biosynthetic CoQ complex and is required for optimal function of Coq6 and Coq7 (484, 485, 493, 498). In fact, a physical association between human COQ9 and COQ7 was demonstrated in vitro in a cell-free protein expression system, and conserved residues around the lipid-binding site of COQ9 are shown to be important for maintaining the interaction with COQ7 (509). Of all Coq proteins, Coq11 is the most different and, so far, the least understood. Unlike the other coq mutants lacking one of the components of the biosynthetic CoQ complex, coq11Δ cells have no decrease but even a slight elevation in the levels of other Coq proteins and also lack evidence for destabilization of the biosynthetic CoQ complex (502, 510). Moreover, coq11Δ cells retain the ability to make CoQ6, though their de novo CoQ6 biosynthesis is significantly impaired. These findings, taken together, have prompted speculation that the absence of Coq11 does not impair but enhances the CoQ biosynthetic machinery and that the net loss of CoQ biosynthesis in coq11Δ mutants suggests a function for Coq11 that is related to other aspects of CoQ metabolism rather than to biosynthesis per se. On this note, Coq11 was identified as part of the mitochondrial organization of gene expression (MIOREX) complexes, which are large assemblies of ribosomes comprising factors that are involved in mitochondrial gene expression (511). No functional homolog of Coq11 has so far been found in animal genomes. In silico analysis indicates that it belongs to the atypical short-chain dehydrogenase/reductase (SDR) superfamily and the closest higher eukaryotic Coq11-like protein is NDUFA9, a subunit of CI (492).
Along with Coq1 and Coq2, Coq10 appears to not be part of the biosynthetic CoQ complex (498). Despite reduced de novo CoQ6 synthesis efficiency, nearly normal steady-state levels of CoQ6 were detected in coq10Δ cells. Nevertheless, the defect in respiratory function as well as a higher sensitivity to PUFA indicate compromised CoQ function (331, 512). Structural determination of a Coq10 homolog, CC1736 from the bacterium Caulobacter crescentus, identified a lipophilic START (steroidogenic acute regulatory-related lipid transfer) domain, which is known to bind lipid in a hydrophobic binding pocket and to be involved in the nonvesicular intracellular transport of lipids and sterols (512). Thus, it has been hypothesized that Coq10 functions as a chaperone to facilitate CoQ delivery to its sites of action, especially in the IMM (331, 512, 513). How loss of Coq10 affects de novo synthesis of CoQ6 is not understood, though destabilization of the biosynthetic CoQ complex in the absence of Coq10 was suggested by some observations (331, 498, 501, 502, 510, 512). Finally, it is worth noting that a more recent study reported a functional interaction between Coq10 and Coq11. Specifically, deletion of COQ11 alleviates the CoQ deficiency phenotype of coq10Δ mutants and this was associated with a significant elevation of mitochondrial CoQ6 (510). Furthermore, the presence of Coq11 appears to be needed for Coq8 overexpression to be able to confer a beneficial effect in the coq10Δ mutant (510).
5.1.2. Mouse CoQ biosynthesis mutants.
Except for coq11, homologs of the yeast coq genes are all present in mammals (408, 512, 514–520). In rodents and humans, the enzymes responsible for the first committed step of CoQ, the assembly and elongation of the isoprenoid side chain, are heterotetramers of two protein subunits rather than monomeric enzymes as in budding yeast (Coq1) and E. coli (IspB) (521). The genes encoding the two subunits are designated Pdss1 and Pdss2 in mice and PDSS1 and PDSS2 in humans. In mice, complete elimination of CoQ biosynthesis causes embryonic lethality, as has already been demonstrated with mutations in four Coq genes: Pdss2, Coq3, Coq7, and Coq8b (8, 29–31, 522). This is not surprising since a lethal phenotype is typical for mice with mutations in genes encoding essential ETC components, e.g., the “Rieske” iron-sulfur protein (Risp) of CIII, cyt c, and CIV assembly factor Cox10 (523–525). TABLE 4 lists all the CoQ deficiency mouse models reported in the literature so far.
Table 4.
Mouse CoQ biosynthesis mutants
| Mouse Mutant | CoQ and CoQ Intermediates | Gross Phenotype |
|---|---|---|
| Spontaneous mutation | ||
| Pdss2kd/kd | Widespread moderate to severe CoQ deficiency | Nephrotic syndrome, kidney failure, impaired motor behavior, plasma lipid abnormalities, death by 9.5 mo of age |
| Germline mutation | ||
| Coq3+/− | Normal CoQ9 levels | Wild-type appearance, normal life span |
| Coq7+/− | Normal level of total CoQ9, mild CoQ9 loss in the IMM | Wild-type appearance, 30% longer life span |
| Coq8a/ADCK3−/− | Moderate CoQ9 loss in skeletal muscles, kidney, and liver, not in cerebellum and serum | Progressive cerebellar ataxia and mild exercise intolerance, normal life span |
| Coq9R239X | Global and severe CoQ9 loss | Poor overall growth, impaired motor function, progressive paralysis, death between 3 and 6 mo of age |
| Coq9Q95X | Moderate CoQ9 loss in the kidney and cerebrum, severe CoQ9 loss in skeletal muscle | Adult-onset mild myopathy |
| Conditional knockout | ||
| Pdss2 podocyte KO Nphs1-cre;Pdss2loxP/loxP | ND | Nephrotic syndrome |
| Pdss2 renal tubular epithelium KO PEPCK-cre;Pdss2loxP/loxP | ND | Absence of overt phenotype |
| Pdss2 Purkinje cell KO Pcp2-cre;Pdss2loxP/− | ND | Ataxia-like symptoms at 9.5 mo |
| Pdss2 Purkinje cell KO Pax2-cre;Pdss2 loxP/− | ND | Death within the first 36 h of life |
| Pdss2 liver KO Alb-cre;Pdss2 loxP/loxP | Severe CoQ9 loss in the liver | Absence of overt phenotype, altered amino acid and DNA metabolism |
| Pdss2 dopaminergic neuron KO DAT-cre;Pdss2loxP/loxP | ND | Loss of TH-positive neurons, motor deficit |
| Coq6 podocyte KO Nphs2-cre;Coq6loxP/loxP | Renal dysfunction, death at ≈10 mo | |
| Coq7 liver KO Alb-cre;Coq7 loxP/loxP | Severe CoQ9 loss and DMQ9 accumulation in the liver | Absence of overt phenotype, normal life span |
| Coq7 whole body inducible KO CAG-creERT2;Coq7 loxP/loxP | Global and severe CoQ9 loss and DMQ9 accumulation | Weight loss, loss of coat hair, kidney dysfunction, elevated blood lactate, short life span |
| Coq8a/ADCK3 Purkinje cell KO (Pcp2-Cre; Coq8aloxP/loxP) | ND | Ataxia |
| Coq8b/ADCK4 podocyte KO (Nphs2-Cre; Coq8bloxP/loxP) | ND | Renal disease, death at ≈12 mo |
IMM, inner mitochondrial membrane; KO, knockout; ND, not determined; TH, tyrosine hydroxylase. For references, see sect. 5.1.2.
5.1.2.1. pdss2 mouse mutants.
The first reported mutation in Pdss2 appeared spontaneously in an inbred strain of mice and was designated the “kidney disease” (kd) allele because the most prominent phenotype of homozygous mutant mice is kidney dysfunction leading to renal failure (344). The kd allele is a missense mutation resulting in the amino acid change V117M (30). Pdss2kd/kd mice develop nephrotic syndrome recognizable at ≈10 wk of age, with proteinuria, excessive drinking, and visceral epithelial abnormalities, accompanied by collapsing glomerulopathy. The mice die by 9.5 mo from renal failure (526, 527). Plasma lipid abnormalities such as high levels of serum triglycerides and cholesterol have also been previously observed to develop with age in Pdss2kd/kd mice (526). No overt manifestations were observed in all other tissues examined, including brain, retina, liver, and skeletal muscles (30, 87). Widespread CoQ deficiency was observed in Pdss2kd/kd mice: in the liver (≈60–70% of the mean of control mice), brain (20–28%), kidney (14–28%), and muscle (20–35%) (30, 124). The currently prevailing idea is that kidney disease in Pdss2kd/kd mice, and perhaps in some other CoQ deficiency conditions as well, results from podocyte dysfunction and is caused by oxidative stress mediated by impairment of the sulfides oxidation pathway (87, 90, 124).
Tissue-specific conditional Pdss2 KO models targeting renal glomeruli or tubules demonstrated that renal glomerular podocytes indeed have a particularly high sensitivity to Pdss2 deletion. Specifically, Podocin-cre;Pdss2loxP/loxP mice with podocyte-specific deletion of Pdss2 were shown to exhibit a phenotype similar to Pdss2kd/kd mice, whereas no renal abnormality was observed in PEPCK-cre;Pdss2loxP/loxP mice that express Cre predominantly in the renal tubular epithelium (30). More recently, a single-nucleus RNA-Seq study on kidneys of Pdss2kd/kd mice revealed a podocyte-specific perturbation of the Braf/Mapk pathway, which is linked to altered PUFA metabolism and elevation of Gpx4, which, as mentioned in sect. 4.1, encodes a glutathione peroxidase playing a key role in protecting against lipid peroxidation and ferroptotic cell death. Moreover, it was shown that GDC-0879, a selective B-Raf inhibitor, can ameliorate kidney injury in Pdss2kd/kd mice, pointing to a new pathway that may contribute to the renal pathophysiology induced by CoQ deficiency (406). Models with Pdss2 specifically ablated in the cerebellum, dopaminergic neurons, liver, or monocytes were also reported. Pcp2-cre;Pdss2 loxP/− mice with postnatal Pdss2 deletion in cerebellar Purkinje cells and retinal bipolar neurons exhibited a significant loss in Purkinje cells by 6 mo of age and developed ataxia-like symptoms at 9.5 mo (528). On the other hand, Pax2-cre;Pdss2loxP/− mice with a targeted deletion of Pdss2 in the midbrain-hindbrain at embryonic day 9.5 died shortly after birth (528). Conditional Pdss2 KO in dopaminergic neurons was shown to cause loss of tyrosine hydroxylase (TH)-positive neurons from the substantia nigra and consequently have decreased motor coordination and locomotive activities (529). Of note, although Pdss2kd/kd mice suffer mostly from kidney dysfunction, they were also reported to display motor phenotypes (529). On the other hand, mice with liver-specific KO of Pdss2 (Alb-Cre;Pdss2 loxP/loxP) showed no overt manifestations of liver dysfunction, despite very low CoQ levels (30). Significantly elevated plasma cholesterol was seen in both the podocyte- and liver-specific Pdss2 KOs, indicating that Pdss2 gene defects in hepatocyte and podocytes both contribute to the plasma lipid abnormalities observed in Pdss2kd/kd mice (30).
5.1.2.2. coq6 mouse mutants.
A podocyte-specific Coq6 KO mouse model (Nphs2-cre;Coq6loxP/loxP) was generated and described to recapitulate aspects of the pathology of focal segmental glomerulosclerosis (FSGS) observed in patients with COQ6 mutations (530). Nphs2-cre;Coq6loxP/loxP mice showed an onset of proteinuria at 5 mo, and progressive FSGS was observed in 10-mo-old mice, which then became moribund. 2,4-DHB significantly inhibited disease progression and improved life expectancy. This is a surprising result. Indeed, 3,4-3,4-dihydroxybenzoate (3,4-DHB), but not 2,4-DHB, was shown to rescue yeast COQ6 deletion mutants, which is also the logical outcome (see sect. 5.1.2.3 and sect. 5.4 for additional details about 2,4-DHB). No mechanism for the reported benefit of 2,4-DHB treatment in this conditional Coq6 KO model has been proposed, and the effect of 2,4-DHB treatment on the tissue levels of CoQ was not determined in the study.
5.1.2.3. coq7 mouse mutants.
Both heterozygous Coq3+/− and Coq7+/− mice look superficially wild type and show normal tissue levels of CoQ9. However, in contrast to Coq3+/− mice, a variety of phenotypes were found in Coq7 hemizygotes (Coq7+/−), including respiratory chain deficiency, decreased ATP production, higher mitochondrial oxidative stress, increased expression of HIF-1α, and more, all of which were not observed with Coq3+/− mice (8, 122, 531). Coq7+/− mice also display an increased life span of up to 30% (532). The total amount of CoQ was the same in the mitochondria of Coq7+/− mice as in wild-type control mice, but CoQ quantification in mitochondrial membrane fractions revealed a decrease in the IMM with a concomitant elevation in the OMM. This local CoQ deficiency in the IMM is the most likely underlying cause of the phenotypic abnormalities seen in Coq7+/− mutant mice (8). These findings suggest that, at least under certain conditions, a mild reduction in CoQ level in the IMM is sufficient to induce mitochondrial dysfunction leading to phenotypic consequences.
A global conditional KO mouse model for Coq7 was generated using a transgene expressing a tamoxifen-dependent CRE recombinase (CreERT2) (123). Induction of Coq7 KO by tamoxifen injection at ≈2 mo of age led to a global, gradual loss of CoQ, impairment of mitochondrial function, gradual development of disease phenotypes, and shortened life span. The mice’s phenotypes, examined at 6 mo after KO induction, include severe growth retardation, loss of coat hair, kidney dysfunction, the elevation of blood lactate, and decreased levels of fasting blood glucose and nonfasting plasma triglycerides. Although the ultimate cause of death is unknown, it is unlikely that the mice died from heart failure, as at ≈8 mo after KO induction, shortly before the mice started to die, echocardiographic examination of the mutant and control mice found similar baseline parameters of systolic and diastolic function, despite the fact that there was hardly any CoQ left in the heart (123). At ≈2 mo after Coq7 KO induction there was already an ≈80% reduction in CoQ9 levels in all the tissue examined, including the heart, kidney, skeletal muscles, and intestine. At 6 mo, the level of CoQ in the strongly affected tissues averaged only 10–15% of controls. However, the mutant mice survived for almost an additional 6 mo, with a median survival of ≈276 days.
2,4-DHB is a structural analog of 4-HB. It can be used as an alternative ring precursor of CoQ when provided and allows for the bypass of a COQ7 defect (see sect. 5.4 for additional details about 2,4-DHB). 2,4-DHB treatment was shown to lead to a dose-dependent phenotypic rescue of Coq7 KO mice (115). More strikingly, at the dose of 1 g/kg body wt/day, provision of 2,4-DHB starting 2 wk after completion of tamoxifen injection led to only partial restoration of tissue CoQ levels but that nonetheless was sufficient for an almost complete rescue of the mutant phenotypes. In the heart, kidney, and skeletal muscles, the CoQ9 levels of treated KO mice were increased to ∼30–40% of the wild-type levels. However, except for their lower body weight, treated KO mutants were visually indistinguishable from the wild type and could live a full life span (123). These findings indicate that CoQ content in most tissues is probably maintained at largely excess levels for viability, at least under laboratory conditions. Furthermore, a late-onset 2,4-DHB treatment starting as late as 6 mo after KO induction, a time point when untreated KO mice already displayed severe, sublethal, phenotypes, was also able to successfully rescue the KO mice, demonstrating that most or all CoQ deficiency phenotypes are reversible (123).
5.1.2.4. coq8 mouse mutants.
Two coorthologs of yeast COQ8 have been identified in mice: Coq8a (also known as Adck3) and Coq8b (also known as Adck4) (507). A study reported that Coq8a KO (Coq8a−/−) mice are viable and show normal growth and life span. However, they nonetheless develop a slowly progressive cerebellar ataxia and mild exercise intolerance. A specific defect in the cerebellar Purkinje cell layer was found in the central nervous system of Coq8a−/− mice. Histological analyses revealed abnormal mitochondrial morphology in the skeletal muscle, but mitochondrial respiration and metabolites of central carbon metabolism (e.g., lactate) were not altered. A variable reduction in CoQ levels was observed in the skeletal tissue, kidney, and liver of Coq8a−/− mice, ranging from 20% to 50% decrease at 7 mo of age. However, cerebellar CoQ levels were normal, suggesting that CoQ loss is most likely restricted to Purkinje cells, which only make up a small fraction of the cerebellum (<0.1%) (507). The study also reported loss of the biosynthetic CoQ complex proteins (COQ3–COQ9), in several tissues, to varying degrees. The mild and tissue-specific phenotype of Coq8a−/− mice is consistent with a regulatory function of COQ8 in CoQ biosynthesis, with its role in assisting in biosynthetic CoQ complex formation and stability, and with the existence of two close homologs (COQ8A and COQ8B), which might slightly differ in biochemical activity and in tissue-specific expression but nonetheless complement each other’s loss. It has been speculated that more distantly related COQ8A/B homologs (ADCK1, ADCK2, and ADCK5) could also contribute (507).
The high sensitivity of Purkinje neurons to Coq8a mutation is further corroborated by the finding that deletion of COQ8A in Purkinje cells was sufficient to cause ataxia in a conditional mouse model (533). Of particular importance is to note that cerebellar ataxia is a typical symptom of human patients harboring mutations in COQ8A, which is well recapitulated in these mouse models (507, 533). In contrast, whole body KO of Coq8b in mice is lethal. As COQ8B mutations have been implicated in steroid-resistant nephrotic syndrome (SRNS) in patients, a podocyte-specific Coq8b KO model (Nphs2-Cre;Coq8bloxP/loxP) was generated and the mice were shown to present with glomerulopathy and renal dysfunction that started at ∼4 mo (522, 534). The kidney function of the mutant mice continued to decline with time and progressed to renal failure and death by 12 mo (522). It was also observed that the protein levels of COQ3, COQ5, and COQ9 were decreased in Coq8b KO podocytes. Surprisingly, treatment with 2,4-DHB was shown to rescue the disease phenotypes and the shorter survival of Nphs2-Cre;Coq8bloxP/loxP mice (522). As mentioned above, 2,4-DHB is an alternative ring precursor that allows for the bypass of a COQ7 defect (see sect. 5.4 for additional details). Thus, the beneficial effects of 2,4-DHB treatment may indicate a deficiency of COQ7 activity in Coq8b KO cells. CoQ level was not determined in the kidney of Nphs2-Cre;Coq8bloxP/loxP mice; thus we do not know about the severity of CoQ deficiency in the affected podocytes and whether they accumulate DMQ (the substrate of COQ7).
5.1.2.5. coq9 mouse mutants.
Two Coq9 knockin models of different severity were described. Coq9R239X mice carry a homozygous truncation mutation that is a homolog to the human mutation R244X identified in the first COQ9 patient reported (349, 518). Severe CoQ deficiency was found in all tissues tested, including cerebrum, cerebellum, heart, kidney, liver, and skeletal muscles, i.e., there was ≥80% reduction of CoQ levels in Coq9R239X tissues compared to wild type (349). Coq9R239X mice exhibit predominant encephalomyopathy as a phenotype, and in the brain significant impairment of mitochondrial respiratory function, neuronal death, and profound demyelination were found in the hindbrain area, resulting in early death between 3 and 6 mo of age. Similar to what was found in the skin fibroblasts from the COQ9 (R244X) patient, Coq9R239X tissues showed a severe reduction in COQ7 protein levels and accumulation of DMQ, the substrate of the COQ7 enzyme, indicating the dependence of full COQ7 expression level and activity on the presence of COQ9 (349, 518). In comparison, Coq9Q95X mice have higher residual CoQ levels than Coq9R239X mice. In the cerebrum and kidney, 40–50% residual CoQ9 levels were detected in Coq9Q95X mice, whereas the CoQ9 levels declined by 85–90% in the same tissues of Coq9R239X mice (349, 535). In muscle, however, CoQ levels are similar in the two models: both have 10–20% CoQ compared to their respective wild-type controls. Consistent with less severe loss of CoQ, Coq9Q95X mice have a much milder phenotype. The female mice, but not the males, showed signs of mild mitochondrial myopathy at 6 mo of age (535). Perturbation of the mitochondrial sulfide oxidation pathway was observed in both Coq9Q95X and Coq9R239X tissues, and the decrease in SQOR protein levels and activity appears to correlate with the severity of the CoQ loss (89). Finally, only Coq9R239X but not Coq9Q95X mice were found to respond to 2,4-DHB treatment, which is consistent with a more pronounced reduction of COQ7 protein levels and more severe loss of CoQ in Coq9R239X mutants (121, 535).
5.2. CoQ Deficiency in the Invertebrate Model C. elegans
Here we propose to review some studies in the C. elegans invertebrate model to give a flavor of the research on CoQ in the wider field of biology and because of the links of this research to the aging process and the ROS theory of aging. An exhaustive description of CoQ research in other invertebrates and plants would go beyond the scope of the present review.
The nematode C. elegans synthesizes CoQ9, but it also acquires CoQ8 from its bacterial food source (33, 35). Knockout mutations in C. elegans CoQ biosynthesis genes such as coq-1, coq-2, coq-3, and coq-8 result in animals that can only survive for one generation on the normal bacterial food supply (E. coli strain OP50 that produce CoQ8) but whose progeny cannot develop normally and become fertile (36, 536–538). The mechanism by which the first homozygous generation can survive appears to be via sufficient gene product and CoQ being deposited in the egg by the heterozygous mother to last one generation, underscoring how very little CoQ is actually necessary for survival (539). The situation is different for mutants of the clk-1 gene (the ortholog of yeast COQ7) (540). clk-1, which stands for clock, was identified in a forward genetic screen for mutants with slow and irregular development and behavior but a long life span (541). In fact, clk-1 was one of the very first longevity genes ever identified and studied at the molecular level (540). Unlike other C. elegans coq deletion mutants, clk-1 mutant strains can grow indefinitely on the normal bacterial food supply (the E. coli strain OP50) but cannot grow on bacteria that do not produce CoQ8 (35). In clk-1 mutants, the biosynthetic intermediate demethylubiquinone-9 (DMQ9; FIGURE 15) accumulates instead of CoQ9 (33, 35). This strongly suggests that DMQ9 has functional properties that partially compensate for the loss of CoQ9, which allows these mutants to grow as long as they can obtain dietary CoQ8 (see also Ref. 83 for a study of the same situation in mammalian cells). However, how and where in the cell DMQ functions to allow survival is not yet known (see sect. 5.3). The properties of DMQ9 clearly cannot fully compensate for the loss of CoQ9, as clk-1 mutants cannot grow without CoQ8 (32, 35, 542). On the other hand, although the presence of endogenously synthesized DMQ9 supplemented with dietary bacterial CoQ8 allows for viability, this cannot fully replace CoQ9 functionally, as the loss of clk-1 produces a long list of complex phenotypes including slow embryonic and postembryonic growth rates, slow rhythmic behaviors, lower brood size, and increased life span (540, 543, 544). A particularly interesting type of phenotype shown by clk-1 mutants is loss of normal acclimation to changes in temperature (543, 545). Interestingly, the life span of clk-1 mutants has been reported to be unresponsive to another environmental parameter, failing to display life span changes in response to food availability (546).
All these phenotypic effects of clk-1 mutations must result from the profound alteration in CoQ content of the mutants (no CoQ9, some CoQ8, and plenty of DMQ9). The clk-1 phenotype, although resulting in lower oxygen mitochondrial respiratory chain function and a long life span, is quite different from the phenotype of other longevous mutants that affect mitochondrial function (544, 547, 548). Thus, it is likely that it is not low mitochondrial function per se that results in these complex phenotypes. Of course, as reviewed in sects. 3 and 4, CoQ has many other functions besides participating in energy generation. Any one of them could be involved in the complex clk-1 phenotype. CoQ is also expected to be deeply involved in ROS biology, as reviewed in sect. 3, and ROS are signaling molecules that modulate numerous signal transduction pathways. The CLK-1 protein could also have additional functions besides CoQ biosynthesis, as has been suggested (549). However, this does not seem to be the case, as providing clk-1 mutants with 2,4-DHB, which is an alternative, unnatural, biosynthetic precursor of CoQ synthesis (see sect. 5.4), fully rescues the clk-1 mutant phenotypes in the total absence of the CLK-1 protein (550).
Finally, it is notable that a genetic screen for suppression of the slow growth phenotype of the clk-1(e2519) mutant, which is a partial loss of function allele with a glutamic acid-to-lysine substitution at position 148 of the protein, has identified several suppressor mutations. But despite the high number of genomes screened, all the identified suppressor mutations were mapped to tRNAGlu genes whose anticodons were altered to read the substituted Lys codon of clk-1(e2519) (32). Furthermore, when the null mutation qm30 was used in a similar screen, no suppressor was found. These findings strongly suggest that no genetic change in C. elegans can bypass the need for endogenously synthesized CoQ9. The irreplaceability of endogenously synthesized CoQ9 suggests that exogenous CoQ8 (from the diet) cannot reach all cells or subcellular locations where CoQ is needed, or the length of the side chain is more crucial than anticipated. But it also means that where CoQ9 is missing, no process can be genetically tweaked to compensate for the absence of CoQ9. Another insight that the study of clk-1(e2519) phenotype suppressors provided is that the effects of lacking CLK-1 and CoQ are cell autonomous. The suppressor mutations mapped to five distinct tRNAGLU genes, and although the mutation in these genes were always the same (a C-to-T transition in the anticodon at position 36 of the gene), they suppressed different subsets of clk-1 phenotypes to different degrees. This means that the different tRNAGLU genes are expressed to different levels in different cell types and thus participate there more or less in the translation of proteins (here CLK-1). And the differences in the phenotypic rescue by the different suppressors mean very little, or no CoQ synthesized in any cell has the opportunity to move to another cell.
5.3. The CoQ Biosynthetic Intermediate DMQ
DMQ is the only late intermediate capable of accumulating in mutants with COQ gene defects. Its accumulation results from reduced activity of COQ7 (31, 33, 542, 551, 552). DMQ differs from CoQ only by missing one of the two methoxy groups (FIGURE 15). There have been considerations of its possible role in the phenotypes of COQ7 mutants, by fulfilling (or interfering with) some of the functions of CoQ. In the nematode C. elegans, the clk-1 (coq7 ortholog) mutant is the only coq mutant that can be maintained as a homozygous line (see sect. 5.2 for more information about clk-1 worm mutants). All other C. elegans mutants completely devoid of endogenous CoQ or DMQ are not viable, even when feeding E. coli able to produce CoQ8, without which even clk-1 mutants are not viable (35, 36). The large amount of DMQ9 present in clk-1 mutants most likely determines the viability of the clk-1 mutants with access to dietary CoQ8, but it is not yet clear how (33, 34). An in vitro study examining the effects of the addition of DMQ9 on CoQ-dependent electron transport activity by replenishing CoQ9-depleted mitochondria membranes with DMQ9 (prepared from clk-1 worms) suggested that DMQ9 competes with CoQ in the respiratory chain of worms, specifically in the electron transport from CI to CIII (35). However, this competition is likely not seriously deleterious, as it was shown that the presence of DMQ does not contribute much, or anything, to the Clk-1 phenotype (32, 553). This is best demonstrated by missense tRNA suppressors of the clk-1(e2519) point mutation that restores endogenous CoQ9 biosynthesis only to a very small extent (but still allows for the accumulation of large amounts of DMQ9) yet leads to full rescue of the Clk-1 phenotype (32). Along these lines, as mentioned above, in liver-specific CoQ deficiency mouse models it was found that a large depletion of CoQ in hepatocytes causes only mild impairment of respiratory chain function for both Pdss2 KO livers that make no DMQ and DMQ-accumulating Coq7 KO livers (30, 82).
As mentioned above, total KO of Coq7 in mice is embryonic lethal, similar to mutations in other CoQ biosynthetic enzymes (8, 29, 31, 522, 528). However, mouse embryonic fibroblasts in which COQ7 activity is absent are viable in vitro. They lack CoQ9 and show an accumulation of DMQ9 (31, 82). The viability of Coq7 KO cells under in vitro culture conditions is in fact due to the minimal amount of CoQ10 present in the normal culture media (83), similar in this way to C. elegans clk-1 mutants that require bacterial CoQ8 to survive. However, although mitochondrial oxidative phosphorylation was compromised in Coq7 KO MEFs, it was to a much lesser extent compared to Coq7/Pdss2 double-KO cells that have neither CoQ nor DMQ (83). This demonstrates that DMQ is actually capable of some respiratory electron transport. However, the fact that Coq7 KO cells, like other ETC mutants, cannot grow in the respiration-dependent (galactose) medium and their lethality in galactose can be rescued by provision of exogenous CoQ indicates that DMQ is a much less efficient electron transporter than CoQ and that the C6-methoxy group in the CoQ ring is crucial for its electron transport function in the respiratory chain (82, 83).
Another possible function of DMQ that has been considered is to act as an antioxidant. This could explain why oxidative stress is not elevated in Coq7 mouse KO and the expression of antioxidant enzymes is low (123). However, so far this has not been thoroughly tested. In yeast cells, DMQ6 was shown not to be effective in protecting against oxidative stress generated by H2O2 or PUFA linolenic acid (554). In nematode, it was shown that DMQ9 present in the plasma membrane is unlikely to be active in redox reactions (555).
5.4. Chemical Analogs of 4-HB That Can Bypass Deficiencies in Some Steps of CoQ Biosynthesis
Some structural analogs of 4-HB, specifically vanillic acid (VA), 3,4-dihydroxybenzoate (3,4-DHB), and 2,4-dihydroxybenzoate (2,4-DHB), were first shown in yeast to be usable as alternative CoQ biosynthetic precursors (484, 489). Because their aromatic part already carries the chemical groups that are normally added by specific enzymes, such as COQ6 and COQ7, their use allows for bypass of the specific synthesis blocks due to mutations in these enzymes and thereby enables endogenous CoQ biosynthesis despite the absence of the enzymes (FIGURE 16). In yeast, this requires that the biosynthetic CoQ complex can still form, which can be achieved by overexpressing COQ8 or by expressing a stable yet inactive mutant enzyme to prevent a complete collapse of the complex. In the case of COQ6, it was shown in yeast Δcoq6 cells expressing enzymatic inactive but structurally stable yeast or human COQ6 protein that treatment with VA or 3,4-DHB leads to a partial restoration of endogenous CoQ6 biosynthesis (489, 556). Another hydroxylated form of 4-HB, 2,4-DHB, differs from 4-HB by already having a hydroxyl group at the position of the aromatic ring that COQ7 normally hydroxylates, thereby obviating the need for Coq7 for CoQ6 biosynthesis (484).
Figure 16.

The chemical structure of 2,4-dihydroxybenzoic acid (2,4-DHB), 3,4-dihydroxybenzoic acid (3,4-DHB), and vanillic acid (VA) and their use as alternative benzoquinone ring precursors for CoQ biosynthesis. Of note, as mentioned in FIGURE 15. the order of Coq3-, Coq4-, and Coq6-catalyzed reactions has not been clearly established. A representative scheme of their use in the CoQ biosynthetic pathway is shown. However, it is possible that different sequences of reactions coexist or that the sequence of reactions changes depending on the availability of different precursors. See glossary for other abbreviations.
These findings have been verified in metazoans (123, 479, 550, 557). For example, VA is shown to increase CoQ10 production in a CRISPR COQ6 KO in human HEK293 cells (558), and 2,4-DHB has been shown to successfully restore CoQ biosynthesis in mouse and human cells with partial or total loss of COQ7 (112, 115, 123). Importantly, 2,4-DHB administration has been demonstrated to reverse disease phenotypes in C. elegans clk-1 mutant, conditional Coq7 knockout (KO) mice, and a Coq9 mouse model with particularly low COQ7 activity (Coq9R239X) (121, 123, 559). But 2,4-DHB does not show a CoQ-increasing effect in all conditions in which there is a partial loss of COQ7 (99, 115). We postulate that in mammalian cells in which a loss of COQ7 does not result in a complete loss of the biosynthetic CoQ complex, 2,4-DHB likely would benefit only very severe CoQ deficiency in which there is very little remaining COQ7 activity. This is because if there is a residual native CoQ biosynthetic pathway using 4-HB as the aromatic ring precursor, this residual pathway would compete for the same CoQ pathway enzymes (the other enzymes in the pathway besides COQ7). So, treatment of COQ7 mutants with 2,4-DHB does not necessarily result in increased CoQ production because lower CoQ production from 4-HB might offset the gain from CoQ produced via 2,4-DHB. The observed lesser accumulation of DMQ in the mutant cells and a decrease of CoQ in normal cells after 2,4-DHB treatment are consistent with this model (112, 115, 123). Future studies are needed to understand the mechanisms at play to be able to explore the possibility of using 2,4-DHB as a treatment for certain types of CoQ deficiency.
6. HUMAN PRIMARY CoQ10 DEFICIENCY
6.1. Human Patients with Primary CoQ10 Deficiency
In animals, CoQ is synthesized in all tissues, and dietary intake is not known to have any impact. The presence of CoQ in the blood reflects mostly the amount carried in lipoproteins, not intracellular levels in the cells of solid tissues. Primary CoQ10 deficiency is a clinically heterogeneous and rare disorder that is caused by mutations in genes implicated in the CoQ10 biosynthesis pathway (11, 26, 27, 39, 460). The first case of CoQ deficiency was described in 1989 (560). In the last few decades, with the increasing availability and affordability of genomic sequencing technology, whole genome or exome sequencing is increasingly becoming the first-line diagnostic test for patients suspected of having genetic disorders, including primary CoQ deficiency. This has accelerated the discovery of novel primary CoQ deficiency disease variants. The prevalence of primary CoQ10 deficiency was conservatively estimated to be a total of 123,789 individuals worldwide and 1,462 in the United States (561). Disease-causing mutations have now been reported for PDSS1, PDSS2, COQ2, COQ4, COQ5, COQ6, COQ7, COQ8A, COQ8B, and COQ9 genes, with a total of 383 patients from 276 families reported so far in the literature (TABLE 5) (38–40, 562).
Table 5.
Primary CoQ10 deficiency patients reported in the literature
| Gene | No. of Patients (no. of Families) | No. of Pathogenic Variants | Age of Onset (range) | Common Clinical Manifestations |
|---|---|---|---|---|
| PDSS1 | 3 (2) | 3 | Infancy to 2 yr | Encephalopathy, development delay, SRNS, SND |
| PDSS2 | 7 (5) | 5 | Infancy to 2 yr | Kidney disease mainly SRNS, ataxia, SND |
| COQ2 | 31 (23) | 23 | Infancy to 68 yr | Kidney disease mainly SRNS, encephalopathy |
| COQ4 | 35 (26) | 22 | Infancy to 9 yr | Encephalopathy, development delay, seizure, hypotonia, cardiomyopathy |
| COQ5 | 5 (3) | 3 | Infancy to childhood | Ataxia, dysarthria, encephalopathy, development delay |
| COQ6 | 33 (24) | 16 | 2 mo to 16 yr | SRNS, SND |
| COQ7 | 32 (25) | 9 | Infancy to 15 yr | Spasticity, limb weakness, hypotonia, neuropathy, difficulty walking, hearing loss |
| COQ8A | 133 (104) | 89 | Infancy to 75 yr | Cerebellar ataxia, development delay, muscular symptoms |
| COQ8B | 97 (60) | 38 | 10 days to 32 yr | Kidney disease, mainly SRNS |
| COQ9 | 7 (4) | 5 | Infancy to 9 mo | Encephalopathy, seizure, renal tubulopathy, development delay |
NS, nephrotic syndrome; SND, sensorineural deafness; SRNS, steroid-resistant nephrotic syndrome. Infancy: 0–1 yr. For references, see sect. 6.1.
Generally speaking, typical primary CoQ10 deficiency patients present symptoms that resemble those of inborn mitochondrial respiratory chain disorders, including early onset, multiorgan involvement, and prevalent neurological and muscular manifestations. However, there is great heterogeneity in the clinical manifestations of CoQ deficiency, which is not fully understood. Nonetheless, mutations disrupting an enzymatic activity in the CoQ biosynthetic pathway or a key organizer of the CoQ biosynthetic complex lead more frequently than not to severe disease outcomes. Fatal infantile multisystem disease has been reported for PDSS2, COQ2, COQ4, COQ7, and COQ9 (111, 115, 518, 563–565). However, in the majority of patients, primary CoQ10 deficiency affects only a few organs or tissues. Mutations in PDSS2, COQ2, COQ6, and COQ8B/ADCK4 are frequently associated with glomerular disease, mainly steroid-resistant nephrotic syndrome (SRNS) (534, 566, 567). Most SRNS cases are accompanied by focal segmental glomerulosclerosis (FSGS) and are characterized by childhood onset of proteinuria and a high risk of progression to kidney failure. To date, PDSS2 mutations have been identified in seven patients, and all of them have nephrotic syndrome (NS) presenting at a young age. For most of them (6/7) other organs are also affected (407, 563, 568–570). Twenty-two out of 31 identified COQ2 patients present with renal dysfunction (mainly SRNS). In most of these cases (17/22) renal symptoms had already occurred within the first 2.5 yr of life, and 14 showed no sign of extrarenal involvement (81, 516, 564, 565, 571–581). Out of a total of 34 identified COQ6 patients, 29 (≈85%) have renal manifestations, mainly isolated SRNS, and about one-third (≈35%) have sensorineural deafness with or without a renal phenotype (408, 569, 577, 582–588). Of the 100 COQ8B/ADCK4 patients who have been described, almost all present with renal dysfunction, with SRNS being the most frequent disease manifestation (36/97). Thus, mutations in the COQ8B/ADCK4 gene account for the highest number of primary CoQ10 deficiency patients with kidney disease. Extrarenal symptoms were scarce in COQ8B/ADCK4 patients, and their symptoms are relatively milder, mostly likely owing to less disruption of the CoQ10 biosynthetic machinery and a selective glomerular phenotype (534, 566, 567). The kidney focus of the phenotype suggests that COQ8B/ADCK4 might be particularly limiting for CoQ10 production in the kidney. But it is unknown whether the pathogenic variants of COQ8B actually cause a more severe CoQ deficiency in the kidney, in particular in the glomeruli.
Cerebellar ataxia is one of the most common presentations of primary CoQ10 deficiency, which has been considered to reflect a higher susceptibility of cerebellar Purkinje cells to CoQ deficiency and mitochondrial dysfunction. Ataxia is not observed in patients with COQ8B mutations, but the dominant clinical feature associated with COQ8A/ADCK3 mutations is progressive cerebellar ataxia, variably combined with muscular and other neurological symptoms. COQ8A-associated ataxia is also known as autosomal recessive cerebellar ataxia 2 (ARCA2) or autosomal recessive spinocerebellar ataxia (SCAR9). Overall, it is highly heterogeneous in terms of age at onset and degree of severity, ranging from severe childhood-onset to milder adult-onset forms. In the available literature, we found 133 patients reported to harbor COQ8A/ADCK3 mutations, and all of them suffer from cerebellar ataxia of varying degrees of severity (589–620). It should be noted, however, that, because of the known high prevalence of ataxia in COQ8A/ADCK3 patients, individuals with unexplained ataxia are more likely to be tested for mutations in COQ8A than in other COQ genes. Besides COQ8A, all four COQ5 patients for whom clinical data are available are also found to show a cerebellar ataxic phenotype (621, 622).
COQ4 patients often present with a broad spectrum of symptoms, and most (≈83%, 29/35) have an age of onset of <8 mo of age. About half of the patient cases (14/35) were reported to have poor outcomes, with death within 2 yr of life (111, 114, 623–628). The high clinical severity of mutations in the COQ4 gene is consistent with its predicted central role in the physical organization of the CoQ biosynthetic complex. Mutations in PDSS1 and COQ9 have been reported in a few numbers of patients, with variable phenotypes ranging from infancy-onset multisystemic disorder to a milder disease (516, 518, 629–632). Interestingly, COQ7 patients appear to frequently suffer from neuropathy and muscular disorders such as spasticity, limb weakness, pure motor neuropathy, and difficulty in walking (99, 112, 115, 357, 633–639). The mechanism that underlies the high frequency of motor axonal damage in COQ7 patients is unknown.
Variable tissue- or cell type-specific expression levels of COQ genes, varying degrees of pathogenicity of different mutations, and differences in tissue requirement for CoQ are the likely causes of the high variability of disease presentation and severity of primary CoQ10 deficiency. For example, unlike other tissues, glomerular podocytes are shown to express COQ8B at high levels but their expression of COQ8A is very low (566). This could influence the largely kidney-limited phenotypes of COQ8B patients. It also remains to be clarified whether DMQ accumulation in COQ7 mutants has any role in the phenotypes that distinguish COQ7 patients (83). It is worth noting that, in contrast to yeast coq7 null mutants, mammalian cells and tissues accumulate DMQ in the complete absence of COQ7 and show no significant loss of other COQ proteins, indicating possible differences in the constituent proteins or assembly control of the CoQ biosynthetic complex between phyla (82, 115, 123).
6.2. Treatment of Primary CoQ10 Deficiency
After diagnosis, primary CoQ10 deficiency patients are immediately given oral CoQ10 as replacement therapy. Exogenous CoQ10 supplementation is the only treatment option currently available for CoQ deficiency and, to the best of our knowledge, is only available in an oral formulation (640). Its effectiveness, however, is highly controversial (38–40). Overall, in the cases where a positive response to CoQ10 supplementation treatment was described, the reported responses were very partial improvement of only some symptoms, and the reports, which necessarily lack controls, are often plagued by a lack of details and follow-up (40). On the other hand, animal studies suggest that most phenotypes due to severe CoQ deficiency can be completely rescued by a partial restoration of endogenous CoQ biosynthesis (121, 123). Hence, there is a need to develop alternative strategies for providing CoQ10 or different treatment strategies that act by boosting the residual activity of the CoQ biosynthetic pathway. For some specific primary forms of CoQ deficiency, that are due to mutations in COQ6, COQ7, or COQ8B, modified precursors of the quinone ring of CoQ10, for example, 2,4-dihydroxybenzoic acid (2,4-DHB), have been considered as a potential alternative treatment option (115, 123, 489, 522, 530, 535, 556). As mentioned in sect. 5.4, these molecules, by providing the lacking chemical group on the quinone ring due to defects in specific COQ enzymatic activities, allow the restoration of CoQ biosynthesis in the cells lacking a specific COQ gene product (484). However, further work is needed to explore and clarify the possibilities of their use in treating primary CoQ deficiency, especially given that the patient cells still always retain some degree of residual activity of the affected proteins, and that this residual activity can be inhibited by the alternative pathway triggered by the presence of alternative precursors (99, 115).
7. SECONDARY CoQ DEFICIENCY
Secondary CoQ deficiencies caused by defects that are not directly related to the CoQ synthesis machinery have been reported to be associated with various diseases (641, 642). Moreover, we can expect that more cases will be reported with the increasing awareness of the existence of such secondary deficiencies and their potential role as aggravating factors in disease pathophysiology. Below, we list and summarize secondary CoQ deficiencies described in the available literature (FIGURE 17). We have grouped them based on the link between the primary cause of disease and the cellular CoQ biosynthetic machinery.
Figure 17.
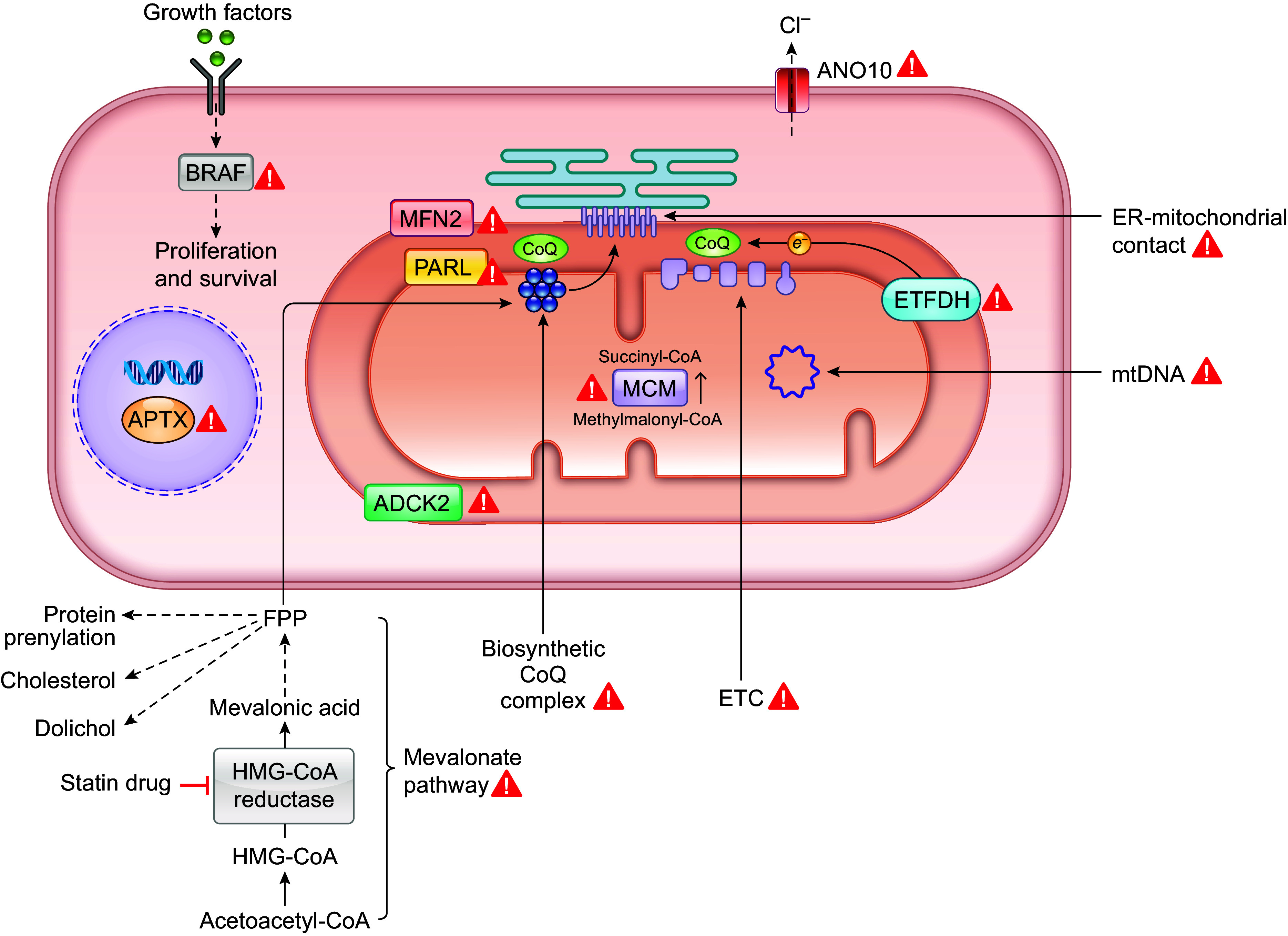
Gene products and pathways whose defects are shown to cause secondary CoQ deficiency. See text for details. See glossary for abbreviations.
7.1. CoQ Deficiency Induced by Inhibition of the Mevalonate Pathway
The most well-known condition associated with secondary CoQ deficiency is the use of the cholesterol-lowering drug statin. The function of statin drugs is to inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, a rate-limiting enzyme of the mevalonate pathway that synthesizes farnesyl pyrophosphate (FPP) as an intermediate. FPP is a direct precursor of squalene, an isoprenoid containing six isoprene subunits that is the precursor of cholesterol biosynthesis. But FPP is also the source of the isoprenoid side chain of CoQ and a substrate for a number of other biosynthetic processes including the biosynthesis of dolichol and isoprenylated hemes and proteins (643). Therefore, inhibition of the mevalonate pathway that leads to reduced FPP production likely inhibits CoQ biosynthesis as well, in addition to lowering cholesterol levels. However, it is not completely understood how the metabolic flux of FPP toward the biosynthesis of different isoprenoids is regulated. According to the flow diversion hypothesis, the regulation is mediated by the different affinities of the branch-point enzymes for FPP. This predicts that the first committed enzyme of cholesterol synthesis (squalene synthase) should have a lower affinity for FPP compared to the branch-point enzymes involved in CoQ and dolichol synthesis. This, in turn, would mean that an alteration of the FPP level would mainly affect the synthesis of cholesterol since the other branch-point enzymes are saturated even at low substrate concentrations (643).
Nevertheless, since its introduction to the market decades ago, statin therapy has been reported to lower blood CoQ10 levels (644–649). However, notably, one randomized crossover study failed to find any decrease in blood CoQ10 levels in healthy volunteers (650). Animal studies reported similar findings of reduced blood levels of CoQ after statin treatment (651, 652). However, the blood CoQ levels are thought to be mainly determined by circulating lipid levels, especially lipoproteins. Thus, a change in blood CoQ level may not reflect the CoQ biosynthetic status in tissues but only inform on the liver, where CoQ is incorporated into lipoprotein particles. In fact, in some studies a parallel decrease in CoQ and blood lipids was observed (648, 653).
One of the most well-known adverse effects of statin drugs is myopathy. The reported incidence of statin-induced myopathy ranges from 0.1–0.2% to as high as 25% (654, 655). The mechanism of statin-induced myotoxicity is not fully understood. CoQ deficiency, possibly resulting from reduced availability of FPP for the synthesis of the isoprenoid side chain of CoQ, has been hypothesized to be a possible cause. However, it is unclear whether statin therapy actually reduces CoQ in muscles. Among the few studies that have examined the muscle levels of CoQ10 in patients receiving statin treatment, one reported an ≈34% decrease of muscle CoQ10 levels in patients treated with simvastatin, but no significant effect, or even an increase, in skeletal muscle CoQ10 concentrations, was reported in three other studies, including one where the CoQ10 level was examined in patients with statin-associated myopathy (656–659). Another recent randomized trial with 37 patients taking simvastatin found no change in muscle CoQ10 levels after 8 wk of supplementation with CoQ10, despite a 4.8-fold increase in plasma CoQ10 concentration (660). The data from animal studies are also inconsistent (661–663). Impaired mitochondrial function, the primary consequence expected from CoQ depletion, has also not been unambiguously shown to occur (657, 658, 664–666). Furthermore, there is currently a lack of concrete evidence to support the beneficial effects of oral CoQ10 supplementation on the myopathic symptoms of statin users (see sect. 7.5). In conclusion, the effect of statins on CoQ levels and their potential role in statin myopathy remain to be convincingly demonstrated.
7.2. Secondary CoQ Deficiency in Mitochondrial Disorders
Mitochondrial disorders have been most frequently reported to be associated with CoQ deficiency. Low levels of CoQ have been described in muscle biopsies or cultured fibroblasts from patients with various types of mitochondrial defects, especially those involving mitochondrial DNA (mtDNA) mutations or depletion. A study involving 39 patients with a clinical phenotype suggestive of mitochondrial DNA depletion syndromes (MDS) showed that 75% of patients with mtDNA depletion presented with a decreased level of muscle CoQ but only 21% of patients with other mitochondrial disorders showed decreased muscle CoQ levels (667). A study including 72 cases showed that 44% of OXPHOS disorders (n = 44) displayed different degrees of CoQ deficiency in either their muscle or their fibroblasts. However, similar rates of low CoQ values were detected in the non-OXPHOS disorder group that includes 11 mitochondrial disease patients and 17 patients with diagnoses of other nonmitochondrial diseases (668). In the same vein, a high frequency of CoQ10 deficiency (28 over 76 patients) was reported to be present in patients presenting mitochondrial myopathy. However, no clear correlation was found between clinical muscle phenotypes (such as weakness and exercise intolerance) and the severity of CoQ deficiency in the muscle (669). Furthermore, fibroblast cultures obtained from two patients with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) were reported to have an ≈40% reduction in CoQ10 levels. MELAS is a mitochondrial disorder caused by pathogenic variants in mtDNA (670). These two patients carry an A-to-G mutation at nucleotide 3243 in the tRNALeu(UUR) gene, which inhibits mitochondrial synthesis and predominantly results in CI defect. A more recent study that analyzed CoQ10 levels in muscle mitochondria from 118 patients with mitochondrial disease of a variety of genetic etiologies found low levels (2 standard derivations below the normal mean) in about one-third (31%) of the samples (671).
According to the current model, CoQ is produced in the IMM by a supramolecular protein complex on the matrix side, the biosynthetic CoQ complex (see sect. 5.1.1). Moreover, yeast studies suggest that the biosynthetic CoQ complexes cluster into discrete foci (called CoQ domains) and are positioned adjacent to the ER-mitochondria contact sites. In yeast, loss of the ER-mitochondria encounter structure (ERMES) complex that tethers the ER to the mitochondria causes a disruption of the biosynthetic CoQ complex and impairs CoQ production (474, 502, 672). The IMM has an intricate ultrastructure. It is highly folded (into cristae) to create more surface area for chemical reactions to occur. Mitochondria and IMM cristae are also believed to constantly change in shape and volume in response to the metabolic state of the cell. Thus, it is reasonable to speculate that reduced respiratory chain activity and alterations in the IMM’s size, ultrastructure, or other properties, which are common consequences of mitochondrial disorders, could perturb the formation of the biosynthetic CoQ complex and its proper positioning in the IMM and thus CoQ production. In fact, abnormal mitochondria and crista morphology are common features of mitochondrial disorders (673). Mitochondrial dysfunction is also often associated with the accumulation of abnormal mitochondria or decreased mitochondrial mass. CoQ occurs in all cellular membranes, but its concentration is highest in the mitochondria. Therefore, lower cellular steady-state levels of CoQ also reflect changes in mitochondria volume when CoQ is measured in total cell or tissue lysate and normalized to total proteins, as is often the case in most CoQ assays. Nevertheless, implied consequences on cellular health, especially on energy production, are probably not less significant even if a secondary CoQ deficiency state mainly stems from mitochondrial loss. However, it is important to distinguish the two conditions, because different treatment strategies might be necessary.
Model animal studies provide direct evidence in support of the connection between mitochondrial defects and secondary CoQ deficiency. In a series of heart conditional KO models targeting essential factors required for mtDNA gene expression (Twnk, Tfam, Polrmt, Lrpprc, Mterf4), leading to OXPHOS dysfunction, a profound decrease of CoQ levels was observed as a result of a significant decrease in levels of CoQ biosynthetic pathway components (COQ3, COQ5, COQ6, COQ7, COQ8A, COQ9, and COQ10A) suggestive of disruption of the biosynthetic CoQ complex (674). Interestingly, the regulator components COQ8A and COQ8B showed strong opposite responses at both transcript and protein levels, indicating that they may be reciprocally regulated, but how, by what mechanism, and for what purpose is not known (674). Of note, this molecular response was not noted in other mouse models with mitochondrial dysfunction (675). Ablation of mitofusin2 (MFN2), an OMM GTPase that mediates outer membrane fusion as well as ER-mitochondria tethering, also causes a significant decrease of the levels of CoQ (676, 677). This was shown in the mouse heart and MEFs. No change in the levels of CoQ biosynthetic enzymes was found, but instead proteomic and metabolomics analyses showed a downregulation of the cytosolic mevalonate pathway (676). Cardiomyocytes isolated from Mfn2 knockout hearts showed mitochondrial morphological heterogeneity with the appearance of enlarged mitochondria. It remains an interesting possibility that structural alterations of ER-mitochondria contact sites that lead to aberrant assembly or organization of CoQ biosynthetic domains are causative, at least partially, of the effect of Mfn2 deletion on CoQ levels.
Presenilin-associated rhomboid-like (PARL) is a protease located in the IMM and plays an essential role in mitochondrial homeostasis (678). A significant reduction of CoQ levels was reported in the brain and testis of Parl−/− mice without changes in mitochondrial mass (675, 679). The mitochondria in Parl−/− brain or spermatocytes also showed severe and progressive ultrastructural abnormalities (swollen mitochondria with crista malformations and loss of matrix density) and assembly alterations in multiple respiratory chain complexes and, in consequence, impaired mitochondrial respiration (675, 679). Increased CoQH2-to-CoQ ratio was also observed, which is believed to be due to CIII dysfunction caused by TTC19 depletion (675, 679). TTC19 is a mitochondrial protein, embedded in the IMM, and is important for correct CIII assembly (680). In addition to TTC19, a downregulation of several CoQ biosynthetic proteins (COQ3, COQ5, COQ6, COQ7, COQ9) and SQOR was observed by a mitochondrial proteomic analysis of Parl−/− brain (675). Among these, the disruption in the COQ4 levels was the most prominent (675, 679). Given the severe mitochondrial morphological changes in the Parl−/− neurons, it is attempting to speculate that the mitochondrial structural defect results in the observed secondary CoQ biosynthesis defect. Notably, Parl ablation in skeletal muscle decreased COQ4, but it was not associated with a reduced level of CoQ (675). The apparent tissue-specific effect of PARL loss is not understood.
Mammalian mitochondria contain five atypical aarF domain containing kinase (ADCK) kinases (681). ADCK3 (COQ8A) and ADCK4 (COQ8B) have been identified as the orthologs of the yeast protein Coq8 that are required for CoQ biosynthesis (see sect. 5.1). A heterozygous nonsense mutation in ADCK2 that led to severe myopathy and liver dysfunction has been identified in a human patient with histological signs of mitochondrial myopathy associated with lipid storage in skeletal muscle (117). An Adck2 knockout mouse model was generated to understand the pathogenesis resulting from the loss of ADCK2. Adck2−/− mice are embryonically lethal, whereas heterozygous inactivation was shown to lead to mitochondrial myopathy in skeletal muscle and impaired fatty acid β-oxidation, recapitulating the phenotype of a human patient (117). A mild to moderate decrease of CoQ levels as well as CoQ biosynthesis rate, accompanied by impaired CoQ-dependent respiratory activities, were found in the MEFs and skeletal muscle (not in the brain, liver, heart, and kidney) of Adck2+/− mice. CoQ deficiency, though not very severe, is proposed to be causative of the Adck2+/− mouse myopathy and liver dysfunction, based on the observation that both the human patient and the Adck2+/− mouse mutant appear to respond to CoQ10 treatment (117). The cause of CoQ deficiency in Adck2-deficient cells is not understood. It is thought, however, to be secondary to defective lipid transport into mitochondria (117).
7.3. Secondary CoQ Deficiency in Cerebellar Ataxia
ANO10 encodes anoctamin-10, a member of a family of putative calcium-activated chloride channels. Lower CoQ10 levels in muscle were reported in two patients with adult-onset cerebellar ataxia who carry ANO10 mutations (682). Of note, skin and muscle biopsies are the only readily accessible tissue samples for CoQ measurement in human patients. For genetic diseases, low levels of muscle CoQ are used to suggest whether other organs, such as the cerebellum, might have similar CoQ deficits. It has been postulated that cerebellar ataxia in patients with ANO10 deficiency may be due to deranged calcium signaling in Purkinje cells (683). Secondary CoQ deficiency was also described in association with mutations in the ATPX gene, which encodes the DNA strand-break repair protein aprataxin (APTX) (684–687). APTX mutations cause ataxia with oculomotor apraxia type 1, a neurodegenerative disorder with early-onset cerebellar ataxia, oculomotor apraxia, and severe axonal polyneuropathy. A lower-than-normal CoQ10 level was demonstrated in the muscle and fibroblasts of APTX patients. APTX functions in both nuclear and mitochondrial DNA maintenance (688). In vitro studies described mitochondrial functional and morphological changes in ATPX-depleted human U2OS cells, such as fragmentation and reduced crista density (689). However, morphology appears to be normal in HeLa cells after shRNA knockdown of APTX, which resulted in a 76% reduction in APTX protein levels and a 29% reduction in CoQ10 level (687). Two separate studies reported a decrease of CoQ10 levels in the cerebellum of multiple system atrophy (MSA) patients in comparison with normal control subjects and other neurodegenerative diseases (715, 716). Studies also reported reduced protein expression of PDSS1 and COQ5 and decreased COQ2 and COQ7 expression in disease-affected regions of the MSA patients (717). It should be mentioned that the samples used for analyses were collected postmortem. MSA is an adult-onset, fatal neurodegenerative disease characterized by a combination of motor abnormalities (parkinsonism and ataxia are the 2 main types) and symptoms that affect the involuntary (autonomic) nervous system. The neuronal loss in the affected area (autonomic centers, basal ganglia, and cerebellar circuits) is often accompanied by oligodendrocytic accumulation of α-synuclein. However, the etiology of MSA is largely unknown. A study with neural progenitors derived from induced pluripotent stem cells (iPSCs) of MSA patients showed mitochondrial morphology changes toward a more tubulated phenotype in MSA cells, but CoQ levels were not explored (690). Ataxia is one of the most common clinical phenotypes associated with CoQ10 deficiency. Reduced CoQ10 levels may act as a separate pathogenic effector causing ataxia in these genetic or acquired conditions associated with ataxia.
7.4. Secondary CoQ Deficiency in Other Conditions
The genetic conditions that have been linked to CoQ deficiency include mutations in ETFDH and BRAF. As mentioned in sect. 2.2, ETFDH, located in the IMM, mediates electron transport from flavoprotein dehydrogenases to the CoQ pool. ETFDH defects are often associated with impaired fatty acid oxidation. One study reported CoQ deficiency in the muscles of seven patients with ETDFH mutations presenting isolated myopathy (691), but a few later studies did not always find CoQ deficiency in other patients carrying ETFDH mutations (692, 693). A single patient with cardiofaciocutaneous syndrome due to a BRAF gene mutation was shown to have a muscular CoQ10 deficiency (694). To the best of our knowledge, no other case has been reported.
Fibroblasts from patients with methylmalonic acidemia due to a deficit of methylmalonyl-CoA mutase (MCM or MUT) activity were also reported to have CoQ10 contents below normal values. However, the CoQ content measured in a muscle sample from one of the patients was not affected. Methylmalonic acidemia is an organic acidemia caused by a deficiency of the mitochondrial enzyme MCM or its cofactor cyanocobalamin, or in rare cases by a deficient activity of methylmalonyl-CoA epimerase (MCE). MCM is involved in the catabolism of propionyl-CoA, a degradation product of cholesterol, branched-chain amino acids, and the β-oxidation of odd-chain fatty acids, converting L-methylmalonyl-CoA to succinyl-CoA, which then enters the TCA cycle (695). Methylmalonic acidemia is characterized by methylmalonic acid (the metabolite of methylmalonyl-CoA) accumulation in tissues and body fluids, and severe metabolic acidosis, neurological symptoms (such as mental impairment), and kidney failure are among the severe consequences of this disease. A further study reported a decrease in renal CoQ10 content in an MCM knockout mouse model (696). However, the level of the dominant CoQ species, CoQ9, was comparable to the control level, pointing to a strange CoQ isoform-specific effect.
Finally, CoQ levels were reported to decline with age, and some age-dependent diseases were reported to be associated with secondary CoQ deficiency, which could be a risk factor in disease development or progression (697). For example, decreased levels of CoQ were reported in the brains of Parkinson disease patients (698). The potential factors responsible for eliciting the decrease in cellular CoQ levels with age could include age-related decline of mitochondrial content and functional integrity, increased CoQ demand (e.g., for higher antioxidant protection capacity), and poor nutritional status.
7.5. Treatment of Secondary CoQ Deficiency
There are two crucial questions that need to be answered about secondary CoQ deficiencies: what are the underlying mechanisms leading to the deficiencies and do these secondary deficiencies add to the pathophysiology of the diseases in which they are observed? Answering these questions is especially challenging because the relevant diseases are highly heterogeneous. A lack of reliable methods for CoQ supplementation also presents an impediment to addressing the latter question (see sect. 3.4). Moreover, it is worth remarking that, given the difficulty of obtaining appropriate controls, it is difficult to establish whether the levels of CoQ in a patient are actually abnormally low. Nevertheless, given the high prevalence of some of these diseases, it is important to understand what role CoQ plays. For example, more than 200 million people around the world are estimated to take cholesterol-lowering statin drugs, and the prevalence of mitochondrial disease has been estimated at ∼1/5,000 live births worldwide (699, 700).
Whatever the underlying mechanisms for secondary deficiency may be, the implication for clinical practice is that diseases that present it could benefit from CoQ supplementation. Indeed, a number of studies, including clinical trials, have reported the effects of CoQ10 supplementation in a number of diseases. However, overall, the results are quite variable. For example, one meta-analysis of randomized controlled trials concluded that CoQ10 supplementation can ameliorate statin-associated muscle symptoms (SAMS), whereas two other similar studies did not succeed in demonstrating that CoQ10 supplementation was beneficial for patients with SAMS (701–703). One of the early randomized clinical trials in patients with mitochondrial diseases reported a 5.5-fold increase of plasma CoQ10 levels after 60 days of CoQ10 treatment (1,200 mg/day), and this was associated with a slight increase in exercise aerobic capacity (Vo2/kg lean mass after 5 min of cycling) and an attenuation of postexercise rise in lactate, whereas no effects of supplementation were observed on other clinically relevant variables such as forearm grip strength (370). Furthermore, a phase III trial of CoQ10 in children with a deficiency of ETC complexes or a molecular diagnosis of mitochondrial disorder reported that receiving CoQ10 at 10–400 mg/kg daily for 6 mo made no significant difference in the two primary outcome measures: McMaster gross motor function and pediatric quality of life scales (https://clinicaltrials.gov/ct2/show/NCT00432744). For further details on this topic, interested readers can consult references such as Refs. 43, 649, 651, 704–709.
Supplementation with CoQ10 is arguably effective in some cases, but the reported effects are often of a very small magnitude. Assessment of the actual effect on tissue CoQ levels, except for the plasma and muscle, cannot routinely be performed on human patients. Therefore, it is not known whether any beneficial effect stems from an elevation of CoQ level at its key sites of action, including mitochondria, as is generally presumed, or even whether the observed effect correlates with restoration of CoQ levels in the affected tissues. Another obvious difficulty in assessing CoQ10 treatment outcome is that different CoQ10 formulations are employed that could differ in the extent or rate of absorption and bioavailability, but such parameters are not always monitored, not even the maximum plasma concentration of CoQ10 achieved by the treatments. Finally, it has been suggested that some individuals may have an inherently low capacity to absorb dietary CoQ10 for reasons unknown (382).
Despite very limited evidence for treatment effectiveness, taking CoQ10 supplement is still being highly recommended for statin drug users, and CoQ10 is widely prescribed to mitochondrial disease patients, often as part of a “multivitamin cocktail” (641). Therefore, it is of great clinical significance to elucidate the exact pathophysiological role of secondary CoQ deficiency in these conditions. CoQ10 supplementation has also been recommended as adjunctive therapy for various other conditions, such as heart failure, diabetes, and Parkinson’s disease (43). Currently, only mixed and contradictory findings can be found concerning the benefits of CoQ10 for these disorders (372, 710–713). Thus, it remains a priority to develop truly effective CoQ10 treatments to clarify the potential contribution of partial CoQ deficiency in any disease pathophysiology.
8. SUMMARY REMARKS
CoQ is an essential membrane component that every cell synthesizes. Except for the length of the side chain, its exact structure is conserved from bacteria to humans. In other words, the molecule is billions of years old and still plays several vital roles in the life of all, or almost all, cells of the planet. It is indispensable for energy production, and loss of its function in mitochondria is the main factor for why CoQ deficiency can result in severe multisystem human disease. It remains less clear whether, or in which circumstances, the loss of other CoQ functions, such as its antioxidant function, also participates in the pathology associated with CoQ deficiency. In the past two decades, the complex multistep process of CoQ biosynthesis has in large part been worked out in model organisms from bacteria to yeast and to animals. Along with this, patients with primary CoQ deficiency presenting with a wide spectrum of pathologies continue to be identified worldwide. Despite all this, much of the biology of CoQ remains to be understood. We know little or nothing about cellular CoQ metabolism and trafficking, such as how CoQ is delivered from mitochondria to other membranes, how the level of CoQ at the different locations is regulated to match the local needs, what determines its turnover, or how it is broken down or otherwise eliminated. Nor do we understand the paths taken by supplemented exogenous CoQ to reach mitochondria. At the organism level, beyond the link to mitochondrial function, we know little about how much CoQ is required in different cell types and tissues and whether this contributes to the variability and complexity of the clinical presentation of CoQ deficiency. Finally, we know nothing firm about the causes of secondary CoQ deficiencies and the pathophysiological role of such deficiencies in the disorders in which they are observed, including some common age-related diseases.
From a clinical point of view, CoQ deficiency is potentially curable by replacement therapy with exogenous CoQ, of which we know that it can reach tissues and mitochondria. Furthermore, given its two best-known functions, as an indispensable electron transporter in the ETC and as an endogenous membrane antioxidant, CoQ is also a molecule that could have the widest therapeutic applications beyond alleviating deficiencies. However, as one of the most hydrophobic molecules occurring naturally, delivery of CoQ via the oral route has been challenging. Despite the great interest in CoQ’s therapeutic potential, it remains to be seen whether the problem of poor oral bioavailability can ever be sufficiently overcome by better formulations for delivery. On the other hand, providing CoQ supplementation via alternative routes may offer new solutions to the problem. Furthermore, a more profound understanding of all aspects of its synthesis, metabolism, and trafficking could lead to methods to boost intracellular levels of endogenous CoQ. We predict that the future will see many advances on all these fronts. Hopefully, the present review could contribute to raising the awareness and interest of researchers in these questions. It is exciting to look forward to unraveling the biology of this essential component of cellular life and using this knowledge to treat disease or enhance health.
GLOSSARY
- 2,4-DHB
2,4-Dihydroxybenzoic acid
- 3,4-DHB
3,4-Dihydroxybenzoic acid
- 4-AP
3-Hexaprenyl-4-aminophenol
- 4-HB
4-Hydroxybenzoic acid
- 4-HBz
4-Hydroxbenzaldehyde
- 4-HNE
4-Hydroxynonenal
- 4-HP
3-Hexaprenyl-4-hydroxyphenol
- 4-HPP
4-Hydroxyphenylpyruvate
- 4-NB
4-Nitrobenzoate
- 8-OHdG
8-Hydroxy-2-deoxyguanosine
- ADCK
aarF domain containing kinase
- AFR
Ascorbyl free radical
- AIFM2
Apoptosis-inducing factor mitochondria-associated 2
- AOX
Alternative quinol oxidase
- APG1
Albino or pale green 1
- APTX
Aprataxin
- ARCR2
Autosomal recessive cerebellar ataxia 2
- Asc
Ascorbate
- ATP
Adenosine triphosphate
- BAT
Brown adipose tissue
- BHT
Butylated hydroxytoluene
- CHDH
Choline dehydrogenase
- CI
Complex I
- CII
Complex II
- CIII
Complex III
- CIV
Complex IV
- CV
Complex V
- CoQ
Coenzyme Q
- CoQ•−
Ubisemiquinone
- CoQH2
Fully reduced form of CoQ
- CoQi•−
Ubisemiquinone at the Qi site of complex III
- CoQo•−
Ubisemiquinone at the Qo site of complex III
- CsA
Cyclosporine A
- Cu2+
Cupric copper
- CYB5R
NADH-cytochrome b5 reductase
- Cyt
Cytochrome
- DAP
Dihydroxyacetone phosphate
- DCYTB
Duodenal cytochrome b
- DDMQ8H2
3-Octaprenyl-5-methoxy-1,4-benzoquinone
- DHA
Dehydroascorbic acid
- DHAR
Dehydroascorbate reductases
- DHHB
3-Hexaprenyl-4,5-dihydroxybenzoic acid
- DHODH
Dihydroorotate dehydrogenase
- DMAPP
Dimethylallyl pyrophosphate
- DMeQ8H2
6-Demethyl-CoQ8H2
- DMQ8H2
2-Methyl-3-octaprenyl-5-methoxy-1,4-benzoquinone
- E. coli
Escherichia coli
- ENOX
Cell-surface NADH oxidase
- ER
Endoplasmic reticulum
- ERMES
Endoplasmic reticulum-mitochondria encounter structure
- ERP
Electron paramagnetic resonance
- ESR
Electron spin resonance
- ETC
Electron transport chain
- ETFDH
Electron transport flavoprotein dehydrogenase
- ETHE1
Encephalopathy protein 1
- FAD
Flavin adenine dinucleotide
- Fe3+
Ferric iron
- FET
Forward electron transport
- FFA
Free fatty acid
- FMN
Flavin mononucleotide
- FPP
Farnesyl diphosphate
- FPS
Farnesyl pyrophosphate FPP-synthase
- FSGS
Focal segmental glomerulosclerosis
- FSP1
Ferroptosis suppressor protein 1
- G3P
Glycerol 3-phosphate
- G3PDH
Glycerol 3-phosphate dehydrogenase
- GLUT
Glucose transporter
- GPP
Geranyl pyrophosphate
- GSH
Glutathione
- GSSG
Glutathione disulfide
- H2O2
Hydrogen peroxide
- H2S
Hydrogen sulfide
- HAB
3-Hexaprenyl-4-aminobenzoic acid
- HGA
Homogentisic acid
- HHAB
3-Hexaprenyl-4-amino-5-hydroxybenzoic acid
- HHB
3-Hexaprenyl-4-hydroxybenzoic acid
- HMG-CoA
3-Hydroxy-3-methylglutaryl coenzyme A
- HMHB
3-Hexaprenyl-4-hydroxy-5-methoxybenzoic acid
- HRP
Horseradish peroxidase
- IIQ
CoQ-binding site of complex II
- IMM
Inner mitochondrial membrane
- IMS
Intermembrane space
- IPP
Isopentenyl diphosphate
- I/R
Ischemia-reperfusion
- ISC
Iron-sulfur cluster
- IspA
Farnesyl diphosphate synthase
- IspB
Octaprenyl diphosphate synthase
- KCN
Potassium cyanide
- L•
Lipid carbon central radical
- LDL
Low-density lipoproteins
- LO•
Lipid alkoxyl radical
- LOO•
Lipid peroxyl radical
- LOOH
Lipid hydroperoxide
- MCE
Methylmalonyl-CoA epimerase
- MCM
Methylmalonyl-CoA mutase
- MDA
Malondialdehyde
- MDS
Mitochondrial DNA depletion syndromes
- MEF
Mouse embryonic fibroblast
- MELAS
Mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes
- MEP
2C-methyl-D-erythritol 4-phosphate
- MFN2
Mitofusin2
- mGPDH
Mitochondrial glycerol 3-phosphate dehydrogenase
- MIOREX
Mitochondrial organization of gene expression
- mPTP
Mitochondrial permeability transition pore
- MSA
Multiple system atrophy
- mtDNA
Mitochondrial DNA
- MUT
Methylmalonyl-CoA mutase
- NADH
Reduced nicotinamide adenine dinucleotide
- NADPH
Reduced nicotinamide adenine dinucleotide phosphate
- NH2
Amino
- NO•
Nitric oxide
- NOX
NAD(P)H oxidases
- NQO1
NAD(P)H:quinone oxidoreductase 1
- NS
Nephrotic syndrome
- O2
Oxygen
- O2•−
Superoxide
- ODP
Octaprenyl diphosphate
- OE
Overexpression
- OH
Hydroxyl
- •OH
Hydroxyl radical
- OHB
3-Octaprenyl-4-hydroxybenzoate
- OMM
Outer mitochondrial membrane
- ONOO•
Peroxynitrite
- OPHP
3-Octaprenyl-5-hydroxyphenol/2-octaprenyl-6-hydroxyphenol
- OPMP
3-Octaprenyl-5-methoxyphenol/2-octaprenyl-6-methoxyphenol
- OPP
Octaprenylphenol
- OXPHOS
Oxidative phosphorylation
- pABA
Para-aminobenzoic acid
- PARL
Presenilin-associated rhomboid-like
- PC
Phosphatidylcholine
- PMET
Trans-plasma membrane electron transport
- PMF
Protonmotive force
- PMRS
Plasma membrane redox system
- PN
Purine nucleotide
- POPC
Phospholipid palmitoyl-2-oleoyl-sn-glycero-phosphocholine
- PQ
Plastoquinone
- PRDX
Peroxiredoxin
- PRODH
Proline dehydrogenase
- PUFA
Polyunsaturated fatty acid
- RET
Reverse electron transport
- RISP
Rieske iron-sulfur protein
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- Rot
Rotenone
- RQ
Rhodoquinone
- S. cerevisiae
Saccharomyces cerevisiae
- S. pombe
Schizosaccharomyces pombe
- S1QELs
Suppressors of site IQ electron leak
Thiosulfate
- S3QELs
Suppressors of site IIIQo electron leak
- SAM
S-adenosylmethionine
- SC
Supercomplex
- SCAD
Short-chain acyl-CoA dehydrogenase
- SCAR9
Autosomal recessive spinocerebellar ataxia
- SDO
Sulfur dioxygenase
Sulfite
Sulfate
- SOD
Superoxide dismutase
- SRD
Short-chain dehydrogenase/reductase
- SRNS
Steroid-resistant nephrotic syndrome
- SQNf
Fast-relaxing ubisemiquinone
- SQNs
Slowly relaxing ubisemiquinone
- SQOR
Sulfide-quinone oxidoreductase
- START
Steroidogenic acute regulatory-related lipid transfer
- SUOX
Sulfite oxidase
- TST
Thiosulfate sulfurtransferase
- VA
Vanillic acid
- UCK
Uridine kinase
- UCP
Uncoupling protein
- UHDBT
5-Undecyl-6-hydroxy-4,7-dioxobenzothiazol
- UMP
Uridine 5-monophosphate
- UMPS
Uridine monophosphate synthase
- VDAC
Voltage-dependent anion-selective channel
- VE
Vitamin E
- VE•
Vitamin E radical
- VEQ
Vitamin E quinone
- VLDL
Very low-density lipoprotein
- αLnn
Linolenic acid
- Δp
Protonmotive force
- ΔΨm
Mitochondrial membrane potential
GRANTS
This work was supported by the Canadian Institutes of Health Research, FDN-159916 (to S. Hekimi).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.H. conceived and designed research; Y.W. prepared figures; Y.W. and N.L. drafted manuscript; Y.W., N.L., and S.H. edited and revised manuscript; S.H. approved final version of manuscript.
REFERENCES
- 1. Crane FL, Hatefi Y, Lester RL, Widmer C. Isolation of a quinone from beef heart mitochondria. Biochim Biophys Acta 25: 220–221, 1957. doi: 10.1016/0006-3002(57)90457-2. [DOI] [PubMed] [Google Scholar]
- 2. Rich PR, Bendall DS. A mechanism for the reduction of cytochromes by quinols in solution and its relevance to biological electron transfer reactions. FEBS Lett 105: 189–194, 1979. doi: 10.1016/0014-5793(79)80608-0. [DOI] [PubMed] [Google Scholar]
- 3. Sarewicz M, Osyczka A. Electronic connection between the quinone and cytochrome c redox pools and its role in regulation of mitochondrial electron transport and redox signaling. Physiol Rev 95: 219–243, 2015. doi: 10.1152/physrev.00006.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bäckström D, Norling B, Ehrenberg A, Ernster L. Electron spin resonance measurement on ubiquinone-depleted and ubiquinone-replenished submitochondrial particles. Biochim Biophys Acta 197: 108–111, 1970. doi: 10.1016/0005-2728(70)90018-6. [DOI] [PubMed] [Google Scholar]
- 5. Wikström MK, Berden JA. Oxidoreduction of cytochrome b in the presence of antimycin. Biochim Biophys Acta 283: 403–420, 1972. doi: 10.1016/0005-2728(72)90258-7. [DOI] [PubMed] [Google Scholar]
- 6. Crane FL. Biochemical functions of coenzyme Q10. J Am Coll Nutr 20: 591–598, 2001. doi: 10.1080/07315724.2001.10719063. [DOI] [PubMed] [Google Scholar]
- 7. Morré DJ, Morré DM. Non-mitochondrial coenzyme Q. Biofactors 37: 355–360, 2011. doi: 10.1002/biof.156. [DOI] [PubMed] [Google Scholar]
- 8. Lapointe J, Wang Y, Bigras E, Hekimi S. The submitochondrial distribution of ubiquinone affects respiration in long-lived Mclk1+/- mice. J Cell Biol 199: 215–224, 2012. doi: 10.1083/jcb.201203090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crane FL, Lester RL, Widmer C, Hatefi Y. Studies on the electron transport system. XVIII. Isolation of coenzyme Q (Q275) from beef heart and beef heart mitochondria. Biochim Biophys Acta 32: 73–79, 1959. doi: 10.1016/0006-3002(59)90554-2. [DOI] [PubMed] [Google Scholar]
- 10. Turunen M, Olsson J, Dallner G. Metabolism and function of coenzyme Q. Biochim Biophys Acta 1660: 171–199, 2004. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 11. Wang Y, Hekimi S. Understanding ubiquinone. Trends Cell Biol 26: 367–378, 2016. doi: 10.1016/j.tcb.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 12. Bentinger M, Brismar K, Dallner G. The antioxidant role of coenzyme Q. Mitochondrion 7, Suppl: S41–S50, 2007. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 13. Navas P, Villalba JM, de Cabo R. The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion 7, Suppl: S34–S40, 2007. doi: 10.1016/j.mito.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 14. Stidham MA, McIntosh TJ, Siedow JN. On the localization of ubiquinone in phosphatidylcholine bilayers. Biochim Biophys Acta 767: 423–431, 1984. doi: 10.1016/0005-2728(84)90040-9. [DOI] [PubMed] [Google Scholar]
- 15. Cornell BA, Keniry MA, Post A, Robertson RN, Weir LE, Westerman PW. Location and activity of ubiquinone 10 and ubiquinone analogues in model and biological membranes. Biochemistry 26: 7702–7707, 1987. doi: 10.1021/bi00398a025. [DOI] [PubMed] [Google Scholar]
- 16. Hauss T, Dante S, Haines TH, Dencher NA. Localization of coenzyme Q10 in the center of a deuterated lipid membrane by neutron diffraction. Biochim Biophys Acta 1710: 57–62, 2005. doi: 10.1016/j.bbabio.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 17. Ausili A, Torrecillas A, Aranda F, de Godos A, Sánchez-Bautista S, Corbalán-García S, Gomez-Fernandez JC. Redox state of coenzyme Q10 determines its membrane localization. J Phys Chem B 112: 12696–12702, 2008. doi: 10.1021/jp802215s. [DOI] [PubMed] [Google Scholar]
- 18. Clarke CF, Rowat AC, Gober JW. Osmotic stress: is CoQ a membrane stabilizer? Nat Chem Biol 10: 242–243, 2014. doi: 10.1038/nchembio.1478. [DOI] [PubMed] [Google Scholar]
- 19. Sévin DC, Sauer U. Ubiquinone accumulation improves osmotic-stress tolerance in Escherichia coli. Nat Chem Biol 10: 266–272, 2014. doi: 10.1038/nchembio.1437. [DOI] [PubMed] [Google Scholar]
- 20. Galassi VV, Arantes GM. Partition, orientation and mobility of ubiquinones in a lipid bilayer. Biochim Biophys Acta 1847: 1560–1573, 2015. doi: 10.1016/j.bbabio.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 21. Eriksson EK, Agmo Hernández V, Edwards K. Effect of ubiquinone-10 on the stability of biomimetic membranes of relevance for the inner mitochondrial membrane. Biochim Biophys Acta Biomembr 1860: 1205–1215, 2018. doi: 10.1016/j.bbamem.2018.02.015. [DOI] [PubMed] [Google Scholar]
- 22. Eriksson EK, Edwards K, Grad P, Gedda L, Agmo HV. Osmoprotective effect of ubiquinone in lipid vesicles modelling the E. coli plasma membrane. Biochim Biophys Acta Biomembr 1861: 1388–1396, 2019. doi: 10.1016/j.bbamem.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 23. Tempelhagen L, Ayer A, Culham DE, Stocker R, Wood JM. Cultivation at high osmotic pressure confers ubiquinone 8-independent protection of respiration on Escherichia coli. J Biol Chem 295: 981–993, 2020. doi: 10.1074/jbc.RA119.011549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guarás A, Perales-Clemente E, Calvo E, Acín-Pérez R, Loureiro-Lopez M, Pujol C, Martinez-Carrascoso I, Nunez E, Garcia-Marques F, Rodriguez-Hernandez MA, Cortes A, Diaz F, Perez-Martos A, Moraes CT, Fernandez-Silva P, Trifunovic A, Navas P, Vazquez J, Enriquez JA. The CoQH2/CoQ ratio serves as a sensor of respiratory chain efficiency. Cell Rep 15: 197–209, 2016. doi: 10.1016/j.celrep.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 25. Tran UC, Clarke CF. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion 7, Suppl: S62–S71, 2007. doi: 10.1016/j.mito.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stefely JA, Pagliarini DJ. Biochemistry of mitochondrial coenzyme Q biosynthesis. Trends Biochem Sci 42: 824–843, 2017. doi: 10.1016/j.tibs.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Y, Hekimi S. The complexity of making ubiquinone. Trends Endocrinol Metab 30: 929–943, 2019. doi: 10.1016/j.tem.2019.08.009. [DOI] [PubMed] [Google Scholar]
- 28. Hajj Chehade M, Pelosi L, Fyfe CD, Loiseau L, Rascalou B, Brugière S, Kazemzadeh K, Vo CD, Ciccone L, Aussel L, Couté Y, Fontecave M, Barras F, Lombard M, Pierrel F. A soluble metabolon synthesizes the isoprenoid lipid ubiquinone. Cell Chem Biol 26: 482–492.e7, 2019. doi: 10.1016/j.chembiol.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 29. Nakai D, Yuasa S, Takahashi M, Shimizu T, Asaumi S, Isono K, Takao T, Suzuki Y, Kuroyanagi H, Hirokawa K, Koseki H, Shirsawa T. Mouse homologue of coq7/clk-1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem Biophys Res Commun 289: 463–471, 2001. doi: 10.1006/bbrc.2001.5977. [DOI] [PubMed] [Google Scholar]
- 30. Peng M, Falk MJ, Haase VH, King R, Polyak E, Selak M, Yudkoff M, Hancock WW, Meade R, Saiki R, Lunceford AL, Clarke CF, Gasser DL. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet 4: e1000061, 2008. doi: 10.1371/journal.pgen.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Levavasseur F, Miyadera H, Sirois J, Tremblay ML, Kita K, Shoubridge E, Hekimi S. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J Biol Chem 276: 46160–46164, 2001. doi: 10.1074/jbc.M108980200. [DOI] [PubMed] [Google Scholar]
- 32. Branicky R, Nguyen PA, Hekimi S. Uncoupling the pleiotropic phenotypes of clk-1 with tRNA missense suppressors in Caenorhabditis elegans. Mol Cell Biol 26: 3976–3985, 2006. doi: 10.1128/MCB.26.10.3976-3985.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miyadera H, Amino H, Hiraishi A, Taka H, Murayama K, Miyoshi H, Sakamoto K, Ishii N, Hekimi S, Kita K. Altered quinone biosynthesis in the long-lived clk-1 mutants of Caenorhabditis elegans. J Biol Chem 276: 7713–7716, 2001. doi: 10.1074/jbc.C000889200. [DOI] [PubMed] [Google Scholar]
- 34. Miyadera H, Kano K, Miyoshi H, Ishii N, Hekimi S, Kita K. Quinones in long-lived clk-1 mutants of Caenorhabditis elegans. FEBS Lett 512: 33–37, 2002. doi: 10.1016/S0014-5793(02)02282-2. [DOI] [PubMed] [Google Scholar]
- 35. Jonassen T, Larsen PL, Clarke CF. A dietary source of coenzyme Q is essential for growth of long-lived Caenorhabditis elegans clk-1 mutants. Proc Natl Acad Sci USA 98: 421–426, 2001. doi: 10.1073/pnas.98.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hihi AK, Gao Y, Hekimi S. Ubiquinone is necessary for Caenorhabditis elegans development at mitochondrial and non-mitochondrial sites. J Biol Chem 277: 2202–2206, 2002. doi: 10.1074/jbc.M109034200. [DOI] [PubMed] [Google Scholar]
- 37. Wang Y, Hekimi S. Molecular genetics of ubiquinone biosynthesis in animals. Crit Rev Biochem Mol Biol 48: 69–88, 2013. doi: 10.3109/10409238.2012.741564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emmanuele V, López LC, Berardo A, Naini A, Tadesse S, Wen B, D’Agostino E, Solomon M, DiMauro S, Quinzii C, Hirano M. Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch Neurol 69: 978–983, 2012. doi: 10.1001/archneurol.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alcázar-Fabra M, Rodríguez-Sanchez F, Trevisson E, Brea-Calvo G. Primary Coenzyme Q deficiencies: a literature review and online platform of clinical features to uncover genotype-phenotype correlations. Free Radic Biol Med 167: 141–180, 2021. doi: 10.1016/j.freeradbiomed.2021.02.046. [DOI] [PubMed] [Google Scholar]
- 40. Wang Y, Hekimi S. The efficacy of coenzyme Q10 treatment in alleviating the symptoms of primary coenzyme Q10 deficiency: a systematic review. J Cell Mol Med 26: 4635–4644, 2022. doi: 10.1111/jcmm.17488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bergamini C, Moruzzi N, Sblendido A, Lenaz G, Fato R. A water soluble CoQ10 formulation improves intracellular distribution and promotes mitochondrial respiration in cultured cells. PLoS One 7: e33712, 2012. doi: 10.1371/journal.pone.0033712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Y, Hekimi S. Micellization of coenzyme Q by the fungicide caspofungin allows for safe intravenous administration to reach extreme supraphysiological concentrations. Redox Biol 36: 101680, 2020. doi: 10.1016/j.redox.2020.101680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hernández-Camacho JD, Bernier M, López-Lluch G, Navas P. Coenzyme Q10 supplementation in aging and disease. Front Physiol 9: 44, 2018. doi: 10.3389/fphys.2018.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Crane FL. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion 7, Suppl: S2–S7, 2007. doi: 10.1016/j.mito.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 45. Crane FL. Electron transport and cytochromes of sub-cellular particles from cauliflower buds. Plant Physiol 32: 619–625, 1957. doi: 10.1104/pp.32.6.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mitchell P. The protonmotive Q cycle: a general formulation. FEBS Lett 59: 137–139, 1975. doi: 10.1016/0014-5793(75)80359-0. [DOI] [PubMed] [Google Scholar]
- 47. Ernster L, Lee IY, Norling B, Persson B. Studies with ubiquinone-depleted submitochondrial particles. Essentiality of ubiquinone for the interaction of succinate dehydrogenase, NADH dehydrogenase, and cytochrome b. Eur J Biochem 9: 299–310, 1969. doi: 10.1111/j.1432-1033.1969.tb00609.x. [DOI] [PubMed] [Google Scholar]
- 48. Norling B, Glazek E, Nelson BD, Ernster L. Studies with ubiquinone-depleted submitochondrial particles. Quantitative incorporation of small amounts of ubiquinone and its effects on the NADH and succinate oxidase activities. Eur J Biochem 47: 475–482, 1974. doi: 10.1111/j.1432-1033.1974.tb03715.x. [DOI] [PubMed] [Google Scholar]
- 49. Garcia-Vallve S. Contribution of each complex of the mitochondrial respiratory chain in the generation of the proton-motive force. Biochem Mol Biol Educ 32: 17–19, 2004. doi: 10.1002/bmb.2004.494032010308. [DOI] [PubMed] [Google Scholar]
- 50. Enriquez JA, Lenaz G. Coenzyme q and the respiratory chain: coenzyme q pool and mitochondrial supercomplexes. Mol Syndromol 5: 119–140, 2014. doi: 10.1159/000363364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lass A, Sohal RS. Comparisons of coenzyme Q bound to mitochondrial membrane proteins among different mammalian species. Free Radic Biol Med 27: 220–226, 1999. doi: 10.1016/s0891-5849(99)00085-4. [DOI] [PubMed] [Google Scholar]
- 52. Hackenbrock CR, Chazotte B, Gupte SS. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J Bioenerg Biomembr 18: 331–368, 1986. doi: 10.1007/BF00743010. [DOI] [PubMed] [Google Scholar]
- 53. Acín-Pérez R, Fernández-Silva P, Peleato ML, Pérez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell 32: 529–539, 2008. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 54. Schägger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J 19: 1777–1783, 2000. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Acín-Pérez R, Bayona-Bafaluy MP, Fernández-Silva P, Moreno-Loshuertos R, Perez-Martos A, Bruno C, Moraes CT, Enriquez JA. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell 13: 805–815, 2004. doi: 10.1016/s1097-2765(04)00124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Milenkovic D, Blaza JN, Larsson NG, Hirst J. The enigma of the respiratory chain supercomplex. Cell Metab 25: 765–776, 2017. doi: 10.1016/j.cmet.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 57. Letts JA, Fiedorczuk K, Sazanov LA. The architecture of respiratory supercomplexes. Nature 537: 644–648, 2016. doi: 10.1038/nature19774. [DOI] [PubMed] [Google Scholar]
- 58. Althoff T, Mills DJ, Popot JL, Kühlbrandt W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J 30: 4652–4664, 2011. doi: 10.1038/emboj.2011.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Letts JA, Fiedorczuk K, Degliesposti G, Skehel M, Sazanov LA. Structures of respiratory supercomplex I+III(2) reveal functional and conformational crosstalk. Mol Cell 75: 1131–1146.e6, 2019. doi: 10.1016/j.molcel.2019.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Acin-Perez R, Enriquez JA. The function of the respiratory supercomplexes: the plasticity model. Biochim Biophys Acta 1837: 444–450, 2014. doi: 10.1016/j.bbabio.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 61. Genova ML, Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta 1837: 427–443, 2014. doi: 10.1016/j.bbabio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 62. Zhang Y, Fernie AR. Metabolons, enzyme-enzyme assemblies that mediate substrate channeling, and their roles in plant metabolism. Plant Commun 2: 100081, 2021. doi: 10.1016/j.xplc.2020.100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rathore S, Berndtsson J, Marin-Buera L, Conrad J, Carroni M, Brzezinski P, Ott M. Cryo-EM structure of the yeast respiratory supercomplex. Nat Struct Mol Biol 26: 50–57, 2019. doi: 10.1038/s41594-018-0169-7. [DOI] [PubMed] [Google Scholar]
- 64. Blaza JN, Serreli R, Jones AJ, Mohammed K, Hirst J. Kinetic evidence against partitioning of the ubiquinone pool and the catalytic relevance of respiratory-chain supercomplexes. Proc Natl Acad Sci USA 111: 15735–15740, 2014. doi: 10.1073/pnas.1413855111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fedor JG, Hirst J. Mitochondrial supercomplexes do not enhance catalysis by quinone channeling. Cell Metab 28: 525–531.e4, 2018. doi: 10.1016/j.cmet.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Enríquez JA. Supramolecular organization of respiratory complexes. Annu Rev Physiol 78: 533–561, 2016. doi: 10.1146/annurev-physiol-021115-105031. [DOI] [PubMed] [Google Scholar]
- 67. Hernansanz-Agustín P, Enríquez JA. Generation of reactive oxygen species by mitochondria. Antioxidants (Basel) 10: 415, 2021. doi: 10.3390/antiox10030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Calvo E, Cogliati S, Hernansanz-Agustín P, Loureiro-López M, Guarás A, Casuso RA, Garcia-Marques F, Acin-Perez R, Marti-Mateos Y, Silla-Castro JC, Carro-Alvarellos M, Huertas JR, Vazquez J, Enriquez JA. Functional role of respiratory supercomplexes in mice: SCAF1 relevance and segmentation of the Q(pool). Sci Adv 6: eaba7509, 2020. doi: 10.1126/sciadv.aba7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. D’Aurelio M, Gajewski CD, Lenaz G, Manfredi G. Respiratory chain supercomplexes set the threshold for respiration defects in human mtDNA mutant cybrids. Hum Mol Genet 15: 2157–2169, 2006. doi: 10.1093/hmg/ddl141. [DOI] [PubMed] [Google Scholar]
- 70. den Brave F, Becker T. Supercomplex formation boosts respiration. EMBO Rep 21: e51830, 2020. doi: 10.15252/embr.202051830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hou T, Zhang R, Jian C, Ding W, Wang Y, Ling S, Ma Q, Hu X, Cheng H, Wang X. NDUFAB1 confers cardio-protection by enhancing mitochondrial bioenergetics through coordination of respiratory complex and supercomplex assembly. Cell Res 29: 754–766, 2019. doi: 10.1038/s41422-019-0208-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jang S, Javadov S. Current challenges in elucidating respiratory supercomplexes in mitochondria: methodological obstacles. Front Physiol 9: 238, 2018. doi: 10.3389/fphys.2018.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cogliati S, Cabrera-Alarcón JL, Enriquez JA. Regulation and functional role of the electron transport chain supercomplexes. Biochem Soc Trans 49: 2655–2668, 2021. doi: 10.1042/BST20210460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lenaz G. A critical appraisal of the mitochondrial coenzyme Q pool. FEBS Lett 509: 151–155, 2001. doi: 10.1016/s0014-5793(01)03172-6. [DOI] [PubMed] [Google Scholar]
- 75. Mráček T, Drahota Z, Houštěk J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim Biophys Acta 1827: 401–410, 2013. doi: 10.1016/j.bbabio.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 76. Boukalova S, Hubackova S, Milosevic M, Ezrova Z, Neuzil J, Rohlena J. Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim Biophys Acta Mol Basis Dis 1866: 165759, 2020. doi: 10.1016/j.bbadis.2020.165759. [DOI] [PubMed] [Google Scholar]
- 77. Salvi F, Gadda G. Human choline dehydrogenase: medical promises and biochemical challenges. Arch Biochem Biophys 537: 243–252, 2013. doi: 10.1016/j.abb.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Murphy B, Bhattacharya R, Mukherjee P. Hydrogen sulfide signaling in mitochondria and disease. FASEB J 33: 13098–13125, 2019. doi: 10.1096/fj.201901304R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503, 1989. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 80. López-Martín JM, Salviati L, Trevisson E, Montini G, DiMauro S, Quinzii C, Hirano M, Rodriguez-Hernandez A, Cordero MD, Sánchez-Alcazar JA, Santos-Ocana C, Navas P. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum Mol Genet 16: 1091–1097, 2007. doi: 10.1093/hmg/ddm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S, Hirano M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet 78: 345–349, 2006. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang Y, Hekimi S. Mitochondrial respiration without ubiquinone biosynthesis. Hum Mol Genet 22: 4768–4783, 2013. doi: 10.1093/hmg/ddt330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang Y, Hekimi S. Minimal mitochondrial respiration is required to prevent cell death by inhibition of mTOR signaling in CoQ-deficient cells. Cell Death Discov 7: 201, 2021. doi: 10.1038/s41420-021-00591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Landry AP, Ballou DP, Banerjee R. Hydrogen sulfide oxidation by sulfide quinone oxidoreductase. Chembiochem 22: 949–960, 2021. doi: 10.1002/cbic.202000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Libiad M, Yadav PK, Vitvitsky V, Martinov M, Banerjee R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J Biol Chem 289: 30901–30910, 2014. doi: 10.1074/jbc.M114.602664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Paul BD, Snyder SH, Kashfi K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol 38: 101772, 2021. doi: 10.1016/j.redox.2020.101772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ziosi M, Di Meo I, Kleiner G, Gao XH, Barca E, Sanchez-Quintero MJ, Tadesse S, Jiang H, Qiao C, Rodenburg RJ, Scalais E, Schuelke M, Willard B, Hatzoglou M, Tiranti V, Quinzii CM. Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol Med 9: 96–111, 2017. doi: 10.15252/emmm.201606356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhang M, Wakitani S, Hayashi K, Miki R, Kawamukai M. High production of sulfide in coenzyme Q deficient fission yeast. Biofactors 32: 91–98, 2008. doi: 10.1002/biof.5520320111. [DOI] [PubMed] [Google Scholar]
- 89. Luna-Sánchez M, Hidalgo-Gutiérrez A, Hildebrandt TM, Chaves-Serrano J, Barriocanal-Casado E, Santos-Fandila A, Romero M, Sayed RK, Duarte J, Prokisch H, Schuelke M, Distelmaier F, Escames G, Acuna-Castroviejo D, Lopez LC. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol Med 9: 78–95, 2017. doi: 10.15252/emmm.201606345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kleiner G, Barca E, Ziosi M, Emmanuele V, Xu Y, Hidalgo-Gutierrez A, Qiao C, Tadesse S, Area-Gomez E, Lopez LC, Quinzii CM. CoQ10 supplementation rescues nephrotic syndrome through normalization of H2S oxidation pathway. Biochim Biophys Acta Mol Basis Dis 1864: 3708–3722, 2018. doi: 10.1016/j.bbadis.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pedersen CB, Bross P, Winter VS, Corydon TJ, Bolund L, Bartlett K, Vockley J, Gregersen N. Misfolding, degradation, and aggregation of variant proteins. The molecular pathogenesis of short chain acyl-CoA dehydrogenase (SCAD) deficiency. J Biol Chem 278: 47449–47458, 2003. doi: 10.1074/jbc.M309514200. [DOI] [PubMed] [Google Scholar]
- 92. González-García P, Hidalgo-Gutiérrez A, Mascaraque C, Barriocanal-Casado E, Bakkali M, Ziosi M, Abdihankyzy UB, Sanchez-Hernandez S, Escames G, Prokisch H, Martin F, Quinzii CM, Lopez LC. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum Mol Genet 29: 3296–3311, 2020. doi: 10.1093/hmg/ddaa214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lenaz G, Genova ML. Mobility and function of coenzyme Q (ubiquinone) in the mitochondrial respiratory chain. Biochim Biophys Acta 1787: 563–573, 2009. doi: 10.1016/j.bbabio.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 94. Lenaz G, Parenti Castelli G, Fato, D’Aurelio M, Bovina C, Formiggini G, Marchetti M, Estornell E, Rauchova H. Coenzyme Q deficiency in mitochondria: kinetic saturation versus physical saturation. Mol Aspects Med 18, Suppl: S25–S31, 1997. doi: 10.1016/s0098-2997(97)00029-0. [DOI] [PubMed] [Google Scholar]
- 95. Lenaz G, Genova ML. Kinetics of integrated electron transfer in the mitochondrial respiratory chain: random collisions vs. solid state electron channeling. Am J Physiol Cell Physiol 292: C1221–C1239, 2007. doi: 10.1152/ajpcell.00263.2006. [DOI] [PubMed] [Google Scholar]
- 96. Estornell E, Fato R, Castelluccio C, Cavazzoni M, Parenti Castelli G, Lenaz G. Saturation kinetics of coenzyme Q in NADH and succinate oxidation in beef heart mitochondria. FEBS Lett 311: 107–109, 1992. doi: 10.1016/0014-5793(92)81378-y. [DOI] [PubMed] [Google Scholar]
- 97. Fato R, Bernardo SD, Estornell E, Parentic Castelli G, Lenaz G. Saturation kinetics of coenzyme Q in NADH oxidation: rate enhancement by incorporation of excess quinone. Mol Aspects Med 18, Suppl: S269–S273, 1997. doi: 10.1016/s0098-2997(97)00027-7. [DOI] [PubMed] [Google Scholar]
- 98. Luo K, Yu JH, Quan Y, Shin YJ, Lee KE, Kim HL, Ko EJ, Chung BH, Lim SW, Yang CW. Therapeutic potential of coenzyme Q10 in mitochondrial dysfunction during tacrolimus-induced beta cell injury. Sci Rep 9: 7995, 2019. doi: 10.1038/s41598-019-44475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang Y, Gumus E, Hekimi S. A novel COQ7 mutation causing primarily neuromuscular pathology and its treatment options. Mol Genet Metab Rep 31: 100877, 2022. doi: 10.1016/j.ymgmr.2022.100877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kaila VR, Wikström M. Architecture of bacterial respiratory chains. Nat Rev Microbiol 19: 319–330, 2021. doi: 10.1038/s41579-020-00486-4. [DOI] [PubMed] [Google Scholar]
- 101. Wu G, Williams HD, Gibson F, Poole RK. Mutants of Escherichia coli affected in respiration: the cloning and nucleotide sequence of ubiA, encoding the membrane-bound p-hydroxybenzoate:octaprenyltransferase. J Gen Microbiol 139: 1795–1805, 1993. doi: 10.1099/00221287-139-8-1795. [DOI] [PubMed] [Google Scholar]
- 102. Kwon O, Kotsakis A, Meganathan R. Ubiquinone (coenzyme Q) biosynthesis in Escherichia coli: identification of the ubiF gene. FEMS Microbiol Lett 186: 157–161, 2000. doi: 10.1111/j.1574-6968.2000.tb09097.x. [DOI] [PubMed] [Google Scholar]
- 103. Lee PT, Hsu AY, Ha HT, Clarke CF. A C-methyltransferase involved in both ubiquinone and menaquinone biosynthesis: isolation and identification of the Escherichia coli ubiE gene. J Bacteriol 179: 1748–1754, 1997. doi: 10.1128/jb.179.5.1748-1754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Aussel L, Loiseau L, Hajj Chehade M, Pocachard B, Fontecave M, Pierrel F, Barras F. ubiJ, a new gene required for aerobic growth and proliferation in macrophage, is involved in coenzyme Q biosynthesis in Escherichia coli and Salmonella enterica serovar Typhimurium. J Bacteriol 196: 70–79, 2014. doi: 10.1128/JB.01065-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Balle BS, Poole RK. Requirement for ubiquinone downstream of cytochrome(s) b in the oxygen-terminated respiratory chains of Escherichia coli K-12 revealed using a null mutant allele of ubiCA. Microbiology (Reading) 144: 361–373, 1998. doi: 10.1099/00221287-144-2-361. [DOI] [PubMed] [Google Scholar]
- 106. Agrawal S, Jaswal K, Shiver AL, Balecha H, Patra T, Chaba R. A genome-wide screen in Escherichia coli reveals that ubiquinone is a key antioxidant for metabolism of long-chain fatty acids. J Biol Chem 292: 20086–20099, 2017. doi: 10.1074/jbc.M117.806240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gulmezian M, Hyman KR, Marbois BN, Clarke CF, Javor GT. The role of UbiX in Escherichia coli coenzyme Q biosynthesis. Arch Biochem Biophys 467: 144–153, 2007. doi: 10.1016/j.abb.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hajj Chehade M, Loiseau L, Lombard M, Pecqueur L, Ismail A, Smadja M, Golinelli-Pimpaneau B, Mellot-Draznieks C, Hamelin O, Aussel L, Kieffer-Jaquinod S, Labessan N, Barras F, Fontecave M, Pierrel F. ubiI, a new gene in Escherichia coli coenzyme Q biosynthesis, is involved in aerobic C5-hydroxylation. J Biol Chem 288: 20085–20092, 2013. doi: 10.1074/jbc.M113.480368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Pelosi L, Ducluzeau AL, Loiseau L, Barras F, Schneider D, Junier I, Pierrel F. Evolution of ubiquinone biosynthesis: multiple proteobacterial enzymes with various regioselectivities to catalyze three contiguous aromatic hydroxylation reactions. mSystems 1: 10.1128/mSystems.00091-16, 2016. doi: 10.1128/mSystems.00091-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Santos-Ocaña C, Do TQ, Padilla S, Navas P, Clarke CF. Uptake of exogenous coenzyme Q and transport to mitochondria is required for bc1 complex stability in yeast coq mutants. J Biol Chem 277: 10973–10981, 2002. doi: 10.1074/jbc.M112222200. [DOI] [PubMed] [Google Scholar]
- 111. Brea-Calvo G, Haack TB, Karall D, Ohtake A, Invernizzi F, Carrozzo R, Kremer L, Dusi S, Fauth C, Scholl-Bürgi S, Graf E, Ahting U, Resta N, Laforgia N, Verrigni D, Okazaki Y, Kohda M, Martinelli D, Freisinger P, Strom TM, Meitinger T, Lamperti C, Lacson A, Navas P, Mayr JA, Bertini E, Murayama K, Zeviani M, Prokisch H, Ghezzi D. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am J Hum Genet 96: 309–317, 2015. doi: 10.1016/j.ajhg.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Freyer C, Stranneheim H, Naess K, Mourier A, Felser A, Maffezzini C, Lesko N, Bruhn H, Engvall M, Wibom R, Barbaro M, Hinze Y, Magnusson M, Andeer R, Zetterström RH, von Döbeln U, Wredenberg A, Wedell A. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J Med Genet 52: 779–783, 2015. doi: 10.1136/jmedgenet-2015-102986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Quinzii CM, López LC, Gilkerson RW, Dorado B, Coku J, Naini AB, Lagier-Tourenne C, Schuelke M, Salviati L, Carrozzo R, Santorelli F, Rahman S, Tazir M, Koenig M, DiMauro S, Hirano M. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J 24: 3733–3743, 2010. doi: 10.1096/fj.09-152728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Salviati L, Trevisson E, Rodriguez Hernandez MA, Casarin A, Pertegato V, Doimo M, Cassina M, Agosto C, Desbats MA, Sartori G, Sacconi S, Memo L, Zuffardi O, Artuch R, Quinzii C, Dimauro S, Hirano M, Santos-Ocaña C, Navas P. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J Med Genet 49: 187–191, 2012. doi: 10.1136/jmedgenet-2011-100394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wang Y, Smith C, Parboosingh JS, Khan A, Innes M, Hekimi S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J Cell Mol Med 21: 2329–2343, 2017. doi: 10.1111/jcmm.13154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Barriocanal-Casado E, Cueto-Ureña C, Benabdellah K, Gutiérrez-Guerrero A, Cobo M, Hidalgo-Gutiérrez A, Rodriguez-Sevilla JJ, Martin F, Lopez LC. Gene therapy corrects mitochondrial dysfunction in hematopoietic progenitor cells and fibroblasts from Coq9R239X mice. PLoS One 11: e0158344, 2016. doi: 10.1371/journal.pone.0158344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Vázquez-Fonseca L, Schaefer J, Navas-Enamorado I, Santos-Ocaña C, Hernández-Camacho JD, Guerra I, Cascajo MV, Sánchez-Cuesta A, Horvath Z, Siendones E, Jou C, Casado M, Gutiérrez P, Brea-Calvo G, López-Lluch G, Fernández-Ayala DJ, Cortés-Rodríguez AB, Rodríguez-Aguilera JC, Matté C, Ribes A, Prieto-Soler SY, Dominguez-Del-Toro E, Francesco A, Aon MA, Bernier M, Salviati L, Artuch R, Cabo R, Jackson S, Navas P. ADCK2 Haploinsufficiency reduces mitochondrial lipid oxidation and causes myopathy associated with CoQ deficiency. J Clin Med 8: 1374, 2019. doi: 10.3390/jcm8091374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Rustin P, Munnich A, Rotig A. Mitochondrial respiratory chain dysfunction caused by coenzyme Q deficiency. Methods Enzymol 382: 81–88, 2004. doi: 10.1016/S0076-6879(04)82005-6. [DOI] [PubMed] [Google Scholar]
- 119. Chang CF, Gunawan AL, Liparulo I, Zushin PH, Bertholet AM, Kirichok Y, Stahl A. CoQ regulates brown adipose tissue respiration and uncoupling protein 1 expression. Antioxidants (Basel) 12: 14, 2022. doi: 10.3390/antiox12010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Liparulo I, Bergamini C, Bortolus M, Calonghi N, Gasparre G, Kurelac I, Masin L, Rizzardi N, Rugolo M, Wang W, Aleo SJ, Kiwan A, Torri C, Zanna C, Fato R. Coenzyme Q biosynthesis inhibition induces HIF-1alpha stabilization and metabolic switch toward glycolysis. FEBS J 288: 1956–1974, 2021. doi: 10.1111/febs.15561. [DOI] [PubMed] [Google Scholar]
- 121. Hidalgo-Gutiérrez A, Barriocanal-Casado E, Bakkali M, Díaz-Casado ME, Sánchez-Maldonado L, Romero M, Sayed RK, Prehn C, Escames G, Duarte J, Acuña-Castroviejo D, López LC. beta-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9R239X mice. EMBO Mol Med 11: e9466, 2019. doi: 10.15252/emmm.201809466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lapointe J, Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/- mice. J Biol Chem 283: 26217–26227, 2008. doi: 10.1074/jbc.M803287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wang Y, Oxer D, Hekimi S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat Commun 6: 6393, 2015. doi: 10.1038/ncomms7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Quinzii CM, Garone C, Emmanuele V, Tadesse S, Krishna S, Dorado B, Hirano M. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J 27: 612–621, 2013. doi: 10.1096/fj.12-209361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Efremov RG, Baradaran R, Sazanov LA. The architecture of respiratory complex I. Nature 465: 441–445, 2010. doi: 10.1038/nature09066. [DOI] [PubMed] [Google Scholar]
- 126. Halliwell B. The wanderings of a free radical. Free Radic Biol Med 46: 531–542, 2009. doi: 10.1016/j.freeradbiomed.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 127. Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J 219: 1–14, 1984. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Fridovich I. Oxygen: aspects of its toxicity and elements of defense. Curr Eye Res 3: 1–2, 1984. doi: 10.3109/02713688408997181. [DOI] [PubMed] [Google Scholar]
- 129. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol 194: 7–15, 2011. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 21: 363–383, 2020. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 131. Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol 4: 180–183, 2015. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sies H, Belousov VV, Chandel NS, Davies MJ, Jones DP, Mann GE, Murphy MP, Yamamoto M, Winterbourn C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat Rev Mol Cell Biol 23: 499–515, 2022. doi: 10.1038/s41580-022-00456-z. [DOI] [PubMed] [Google Scholar]
- 133. Lü JM, Lin PH, Yao Q, Chen C. Chemical and molecular mechanisms of antioxidants: experimental approaches and model systems. J Cell Mol Med 14: 840–860, 2010. doi: 10.1111/j.1582-4934.2009.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300, 1956. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 135. Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem 292: 16804–16809, 2017. doi: 10.1074/jbc.R117.789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wang Y, Branicky R, Noë A, Hekimi S. Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol 217: 1915–1928, 2018. doi: 10.1083/jcb.201708007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol Lung Cell Mol Physiol 268: L699–L722, 1995. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 138. Halliwell B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol 141: 312–322, 2006. doi: 10.1104/pp.106.077073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol Cell Physiol 271: C1424–C1437, 1996. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 140. Mailloux RJ. An update on mitochondrial reactive oxygen species production. Antioxidants (Basel) 9: 472, 2020. doi: 10.3390/antiox9060472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Wong HS, Benoit B, Brand MD. Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radic Biol Med 130: 140–150, 2019. doi: 10.1016/j.freeradbiomed.2018.10.448. [DOI] [PubMed] [Google Scholar]
- 142. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol 6: 472–485, 2015. doi: 10.1016/j.redox.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 120: 483–495, 2005. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 144. Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 59: 527–605, 1979. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 145. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277: 44784–44790, 2002. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 146. Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol 554: 165–181, 2009. doi: 10.1007/978-1-59745-521-3_11. [DOI] [PubMed] [Google Scholar]
- 147. Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol 45: 466–472, 2010. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem 287: 27255–27264, 2012. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Hadrava Vanova K, Kraus M, Neuzil J, Rohlena J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep 25: 26–32, 2020. doi: 10.1080/13510002.2020.1752002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Mráček T, Holzerová E, Drahota Z, Kovářová N, Vrbacký M, Ješina P, Houštěk J. ROS generation and multiple forms of mammalian mitochondrial glycerol-3-phosphate dehydrogenase. Biochim Biophys Acta 1837: 98–111, 2014. doi: 10.1016/j.bbabio.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 151. Orr AL, Quinlan CL, Perevoshchikova IV, Brand MD. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J Biol Chem 287: 42921–42935, 2012. doi: 10.1074/jbc.M112.397828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Pietras R, Sarewicz M, Osyczka A. Distinct properties of semiquinone species detected at the ubiquinol oxidation Qo site of cytochrome bc1 and their mechanistic implications. J R Soc Interface 13, 2016. doi: 10.1098/rsif.2016.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Beyer RE. An analysis of the role of coenzyme Q in free radical generation and as an antioxidant. Biochem Cell Biol 70: 390–403, 1992. doi: 10.1139/o92-061. [DOI] [PubMed] [Google Scholar]
- 154. Nakamaru-Ogiso E, Narayanan M, Sakyiama JA. Roles of semiquinone species in proton pumping mechanism by complex I. J Bioenerg Biomembr 46: 269–277, 2014. doi: 10.1007/s10863-014-9557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Cape JL, Bowman MK, Kramer DM. A semiquinone intermediate generated at the Qo site of the cytochrome bc1 complex: importance for the Q-cycle and superoxide production. Proc Natl Acad Sci USA 104: 7887–7892, 2007. doi: 10.1073/pnas.0702621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Magnitsky S, Toulokhonova L, Yano T, Sled VD, Hägerhäll C, Grivennikova VG, Burbaev DS, Vinogradov AD, Ohnishi T. EPR characterization of ubisemiquinones and iron-sulfur cluster N2, central components of the energy coupling in the NADH-ubiquinone oxidoreductase (complex I) in situ. J Bioenerg Biomembr 34: 193–208, 2002. doi: 10.1023/A:1016083419979. [DOI] [PubMed] [Google Scholar]
- 157. Suzuki H, King TE. Evidence of an ubisemiquinone radical(s) from the NADH-ubiquinone reductase of the mitochondrial respiratory chain. J Biol Chem 258: 352–358, 1983. doi: 10.1016/S0021-9258(18)33264-2. [DOI] [PubMed] [Google Scholar]
- 158. Hirst J. Mitochondrial complex I. Annu Rev Biochem 82: 551–575, 2013. doi: 10.1146/annurev-biochem-070511-103700. [DOI] [PubMed] [Google Scholar]
- 159. Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life 52: 159–164, 2001. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- 160. Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 100: 14–31, 2016. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 161. Tocilescu MA, Zickermann V, Zwicker K, Brandt U. Quinone binding and reduction by respiratory complex I. Biochim Biophys Acta 1797: 1883–1890, 2010. doi: 10.1016/j.bbabio.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 162. Galemou Yoga E, Schiller J, Zickermann V. Ubiquinone binding and reduction by complex I—open questions and mechanistic implications. Front Chem 9: 672851, 2021. doi: 10.3389/fchem.2021.672851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol 1: 304–312, 2013. doi: 10.1016/j.redox.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci USA 103: 7607–7612, 2006. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Gibbs ET, Lerner CA, Watson MA, Wong HS, Gerencser AA, Brand MD. Site IQ in mitochondrial complex I generates S1QEL-sensitive superoxide/hydrogen peroxide in both the reverse and forward reactions. Biochem J 480: 363–384, 2023. doi: 10.1042/BCJ20220611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, James AM, Murphy MP. Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem 293: 9869–9879, 2018. doi: 10.1074/jbc.RA118.003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Dambrova M, Zuurbier CJ, Borutaite V, Liepinsh E, Makrecka-Kuka M. Energy substrate metabolism and mitochondrial oxidative stress in cardiac ischemia/reperfusion injury. Free Radic Biol Med 165: 24–37, 2021. doi: 10.1016/j.freeradbiomed.2021.01.036. [DOI] [PubMed] [Google Scholar]
- 169. Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol 37: 101674, 2020. doi: 10.1016/j.redox.2020.101674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Treberg JR, Quinlan CL, Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J Biol Chem 286: 27103–27110, 2011. doi: 10.1074/jbc.M111.252502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Chance B, Williams GR, Hollunger G. Inhibition of electron and energy transfer in mitochondria. I. Effects of amytal, thiopental, rotenone, progesterone, and methylene glycol. J Biol Chem 238: 418–431, 1963. doi: 10.1016/S0021-9258(19)84014-0. [DOI] [PubMed] [Google Scholar]
- 172. Wong HS, Monternier PA, Brand MD. S1QELs suppress mitochondrial superoxide/hydrogen peroxide production from site I(Q) without inhibiting reverse electron flow through Complex I. Free Radic Biol Med 143: 545–559, 2019. doi: 10.1016/j.freeradbiomed.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 173. Goncalves RL, Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Brand MD. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J Biol Chem 290: 209–227, 2015. doi: 10.1074/jbc.M114.619072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J 382: 511–517, 2004. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Komlódi T, Geibl FF, Sassani M, Ambrus A, Tretter L. Membrane potential and delta pH dependency of reverse electron transport-associated hydrogen peroxide production in brain and heart mitochondria. J Bioenerg Biomembr 50: 355–365, 2018. doi: 10.1007/s10863-018-9766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Panov A, Orynbayeva Z. Determination of mitochondrial metabolic phenotype through investigation of the intrinsic inhibition of succinate dehydrogenase. Anal Biochem 552: 30–37, 2018. doi: 10.1016/j.ab.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 177. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515: 431–435, 2014. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Garcia-Dorado D, Rodriguez-Sinovas A, Barba I, Ruiz-Meana M. Letter in response to “The role of succinate and ROS in reperfusion injury—a critical appraisal” by Andrienko et al. J Mol Cell Cardiol 112: 131, 2017. doi: 10.1016/j.yjmcc.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 179. Andrienko TN, Pasdois P, Pereira GC, Ovens MJ, Halestrap AP. The role of succinate and ROS in reperfusion injury—a critical appraisal. J Mol Cell Cardiol 110: 1–14, 2017. doi: 10.1016/j.yjmcc.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Lee S, Tak E, Lee J, Rashid MA, Murphy MP, Ha J, Kim SS. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res 21: 817–834, 2011. doi: 10.1038/cr.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JH, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167: 457–470.e13, 2016. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Fernández-Agüera MC, Gao L, González-Rodriguez P, Pintado CO, Arias-Mayenco I, Garcia-Flores P, Garcia-Perganeda A, Pascual A, Ortega-Saenz P, Lopez-Barneo J. Oxygen sensing by arterial chemoreceptors depends on mitochondrial Complex I signaling. Cell Metab 22: 825–837, 2015. doi: 10.1016/j.cmet.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 183. Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, Yoneshiro T, Spinelli JB, Lu GZ, Kazak L, Banks AS, Haigis MC, Kajimura S, Murphy MP, Gygi SP, Clish CB, Chouchani ET. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 560: 102–106, 2018. doi: 10.1038/s41586-018-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Brand MD, Goncalves RL, Orr AL, Vargas L, Gerencser AA, Borch Jensen M, Wang YT, Melov S, Turk CN, Matzen JT, Dardov VJ, Petrassi HM, Meeusen SL, Perevoshchikova IV, Jasper H, Brookes PS, Ainscow EK. Suppressors of superoxide-H2O2 production at site IQ of mitochondrial complex I Protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab 24: 582–592, 2016. doi: 10.1016/j.cmet.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Goncalves RL, Watson MA, Wong HS, Orr AL, Brand MD. The use of site-specific suppressors to measure the relative contributions of different mitochondrial sites to skeletal muscle superoxide and hydrogen peroxide production. Redox Biol 28: 101341, 2020. doi: 10.1016/j.redox.2019.101341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Fang J, Wong HS, Brand MD. Production of superoxide and hydrogen peroxide in the mitochondrial matrix is dominated by site IQ of complex I in diverse cell lines. Redox Biol 37: 101722, 2020. doi: 10.1016/j.redox.2020.101722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Banba A, Tsuji A, Kimura H, Murai M, Miyoshi H. Defining the mechanism of action of S1QELs, specific suppressors of superoxide production in the quinone-reaction site in mitochondrial complex I. J Biol Chem 294: 6550–6561, 2019. doi: 10.1074/jbc.RA119.007687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Lambert AJ, Buckingham JA, Brand MD. Dissociation of superoxide production by mitochondrial complex I from NAD(P)H redox state. FEBS Lett 582: 1711–1714, 2008. doi: 10.1016/j.febslet.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 189. Hirst J, Roessler MM. Energy conversion, redox catalysis and generation of reactive oxygen species by respiratory complex I. Biochim Biophys Acta 1857: 872–883, 2016. doi: 10.1016/j.bbabio.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Kotlyar AB, Sled VD, Burbaev DS, Moroz IA, Vinogradov AD. Coupling site I and the rotenone-sensitive ubisemiquinone in tightly coupled submitochondrial particles. FEBS Lett 264: 17–20, 1990. doi: 10.1016/0014-5793(90)80753-6. [DOI] [PubMed] [Google Scholar]
- 191. Ohnishi T, Johnson JE Jr, Yano T, Lobrutto R, Widger WR. Thermodynamic and EPR studies of slowly relaxing ubisemiquinone species in the isolated bovine heart complex I. FEBS Lett 579: 500–506, 2005. doi: 10.1016/j.febslet.2004.11.107. [DOI] [PubMed] [Google Scholar]
- 192. Yano T, Dunham WR, Ohnishi T. Characterization of the delta muH+-sensitive ubisemiquinone species (SQNf) and the interaction with cluster N2: new insight into the energy-coupled electron transfer in complex I. Biochemistry 44: 1744–1754, 2005. doi: 10.1021/bi048132i. [DOI] [PubMed] [Google Scholar]
- 193. Vinogradov AD, Sled VD, Burbaev DS, Grivennikova VG, Moroz IA, Ohnishi T. Energy-dependent Complex I-associated ubisemiquinones in submitochondrial particles. FEBS Lett 370: 83–87, 1995. doi: 10.1016/0014-5793(95)00803-h. [DOI] [PubMed] [Google Scholar]
- 194. Narayanan M, Leung SA, Inaba Y, Elguindy MM, Nakamaru-Ogiso E. Semiquinone intermediates are involved in the energy coupling mechanism of E. coli complex I. Biochim Biophys Acta 1847: 681–689, 2015. doi: 10.1016/j.bbabio.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Ohnishi T, Ohnishi ST, Shinzawa-Itoh K, Yoshikawa S, Weber RT. EPR detection of two protein-associated ubiquinone components (SQNf and SQNs) in the membrane in situ and in proteoliposomes of isolated bovine heart complex I. Biochim Biophys Acta 1817: 1803–1809, 2012. doi: 10.1016/j.bbabio.2012.03.032. [DOI] [PubMed] [Google Scholar]
- 196. van Belzen R, Kotlyar AB, Moon N, Dunham WR, Albracht SP. The iron-sulfur clusters 2 and ubisemiquinone radicals of NADH:ubiquinone oxidoreductase are involved in energy coupling in submitochondrial particles. Biochemistry 36: 886–893, 1997. doi: 10.1021/bi9612982. [DOI] [PubMed] [Google Scholar]
- 197. Ohnishi T, Salerno JC. Conformation-driven and semiquinone-gated proton-pump mechanism in the NADH-ubiquinone oxidoreductase (complex I). FEBS Lett 579: 4555–4561, 2005. doi: 10.1016/j.febslet.2005.06.086. [DOI] [PubMed] [Google Scholar]
- 198. Crofts AR. The cytochrome bc1 complex: function in the context of structure. Annu Rev Physiol 66: 689–733, 2004. doi: 10.1146/annurev.physiol.66.032102.150251. [DOI] [PubMed] [Google Scholar]
- 199. Berry EA, Guergova-Kuras M, Huang LS, Crofts AR. Structure and function of cytochrome bc complexes. Annu Rev Biochem 69: 1005–1075, 2000. doi: 10.1146/annurev.biochem.69.1.1005. [DOI] [PubMed] [Google Scholar]
- 200. Sarewicz M, Pintscher S, Pietras R, Borek A, Bujnowicz L, Hanke G, Cramer WA, Finazzi G, Osyczka A. Catalytic reactions and energy conservation in the cytochrome bc1 and b6f complexes of energy-transducing membranes. Chem Rev 121: 2020–2108, 2021. doi: 10.1021/acs.chemrev.0c00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Zhang H, Chobot SE, Osyczka A, Wraight CA, Dutton PL, Moser CC. Quinone and non-quinone redox couples in Complex III. J Bioenerg Biomembr 40: 493–499, 2008. doi: 10.1007/s10863-008-9174-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Trumpower BL. The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J Biol Chem 265: 11409–11412, 1990. doi: 10.1016/S0021-9258(19)38410-8. [DOI] [PubMed] [Google Scholar]
- 203. Ding H, Moser CC, Robertson DE, Tokito MK, Daldal F, Dutton PL. Ubiquinone pair in the Qo site central to the primary energy conversion reactions of cytochrome bc1 complex. Biochemistry 34: 15979–15996, 1995. doi: 10.1021/bi00049a012. [DOI] [PubMed] [Google Scholar]
- 204. Kim H, Xia D, Yu CA, Xia JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Inhibitor binding changes domain mobility in the iron-sulfur protein of the mitochondrial bc1 complex from bovine heart. Proc Natl Acad Sci USA 95: 8026–8033, 1998. doi: 10.1073/pnas.95.14.8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Kao WC, Hunte C. Quinone binding sites of cyt bc complexes analysed by X-ray crystallography and cryogenic electron microscopy. Biochem Soc Trans 50: 877–893, 2022. doi: 10.1042/BST20190963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Xia D, Esser L, Tang WK, Zhou F, Zhou Y, Yu L, Yu CA. Structural analysis of cytochrome bc1 complexes: implications to the mechanism of function. Biochim Biophys Acta 1827: 1278–1294, 2013. doi: 10.1016/j.bbabio.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Covian R, Kleinschroth T, Ludwig B, Trumpower BL. Asymmetric binding of stigmatellin to the dimeric Paracoccus denitrificans bc1 complex: evidence for anti-cooperative ubiquinol oxidation and communication between center P ubiquinol oxidation sites. J Biol Chem 282: 22289–22297, 2007. doi: 10.1074/jbc.M702132200. [DOI] [PubMed] [Google Scholar]
- 208. Crofts AR, Barquera B, Gennis RB, Kuras R, Guergova-Kuras M, Berry EA. Mechanism of ubiquinol oxidation by the bc1 complex: different domains of the quinol binding pocket and their role in the mechanism and binding of inhibitors. Biochemistry 38: 15807–15826, 1999. doi: 10.1021/bi990962m. [DOI] [PubMed] [Google Scholar]
- 209. Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry 42: 6493–6499, 2003. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- 210. Sharp RE, Gibney BR, Palmitessa A, White JL, Dixon JA, Moser CC, Daldal F, Dutton PL. Effect of inhibitors on the ubiquinone binding capacity of the primary energy conversion site in the Rhodobacter capsulatus cytochrome bc1 complex. Biochemistry 38: 14973–14980, 1999. doi: 10.1021/bi9914863. [DOI] [PubMed] [Google Scholar]
- 211. Ding H, Robertson DE, Daldal F, Dutton PL. Cytochrome bc1 complex [2Fe-2S] cluster and its interaction with ubiquinone and ubihydroquinone at the Qo site: a double-occupancy Qo site model. Biochemistry 31: 3144–3158, 1992. doi: 10.1021/bi00127a015. [DOI] [PubMed] [Google Scholar]
- 212. Link TA. The role of the ‘Rieske’ iron sulfur protein in the hydroquinone oxidation (QP) site of the cytochrome bc1 complex. The ‘proton-gated affinity change’ mechanism. FEBS Lett 412: 257–264, 1997. doi: 10.1016/s0014-5793(97)00772-2. [DOI] [PubMed] [Google Scholar]
- 213. Zhang H, Osyczka A, Dutton PL, Moser CC. Exposing the complex III Qo semiquinone radical. Biochim Biophys Acta 1767: 883–887, 2007. doi: 10.1016/j.bbabio.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Vennam PR, Fisher N, Krzyaniak MD, Kramer DM, Bowman MK. A caged, destabilized, free radical intermediate in the q-cycle. Chembiochem 14: 1745–1753, 2013. doi: 10.1002/cbic.201300265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Xia D, Yu CA, Kim H, Xia JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science 277: 60–66, 1997. doi: 10.1126/science.277.5322.60. [DOI] [PubMed] [Google Scholar]
- 216. Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, Crofts AR, Berry EA, Kim SH. Electron transfer by domain movement in cytochrome bc1. Nature 392: 677–684, 1998. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- 217. Gao X, Wen X, Esser L, Quinn B, Yu L, Yu CA, Xia D. Structural basis for the quinone reduction in the bc1 complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site. Biochemistry 42: 9067–9080, 2003. doi: 10.1021/bi0341814. [DOI] [PubMed] [Google Scholar]
- 218. Kolling DR, Samoilova RI, Holland JT, Berry EA, Dikanov SA, Crofts AR. Exploration of ligands to the Qi site semiquinone in the bc1 complex using high-resolution EPR. J Biol Chem 278: 39747–39754, 2003. doi: 10.1074/jbc.M305913200. [DOI] [PubMed] [Google Scholar]
- 219. Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237: 408–414, 1985. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 220. Boveris A, Cadenas E, Stoppani AO. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J 156: 435–444, 1976. doi: 10.1042/bj1560435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Bergdoll L, Ten Brink F, Nitschke W, Picot D, Baymann F. From low- to high-potential bioenergetic chains: thermodynamic constraints of Q-cycle function. Biochim Biophys Acta 1857: 1569–1579, 2016. doi: 10.1016/j.bbabio.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 222. Bleier L, Dröse S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta 1827: 1320–1331, 2013. doi: 10.1016/j.bbabio.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 223. Quinlan CL, Gerencser AA, Treberg JR, Brand MD. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J Biol Chem 286: 31361–31372, 2011. doi: 10.1074/jbc.M111.267898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Muller F. The nature and mechanism of superoxide production by the electron transport chain: its relevance to aging. J Am Aging Assoc 23: 227–253, 2000. doi: 10.1007/s11357-000-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Starkov AA, Fiskum G. Myxothiazol induces H2O2 production from mitochondrial respiratory chain. Biochem Biophys Res Commun 281: 645–650, 2001. doi: 10.1006/bbrc.2001.4409. [DOI] [PubMed] [Google Scholar]
- 226. Muller F, Crofts AR, Kramer DM. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc1 complex. Biochemistry 41: 7866–7874, 2002. doi: 10.1021/bi025581e. [DOI] [PubMed] [Google Scholar]
- 227. Sun J, Trumpower BL. Superoxide anion generation by the cytochrome bc1 complex. Arch Biochem Biophys 419: 198–206, 2003. doi: 10.1016/j.abb.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 228. Quinlan CL, Treberg JR, Perevoshchikova IV, Orr AL, Brand MD. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic Biol Med 53: 1807–1817, 2012. doi: 10.1016/j.freeradbiomed.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Orr AL, Vargas L, Turk CN, Baaten JE, Matzen JT, Dardov VJ, Attle SJ, Li J, Quackenbush DC, Goncalves RL, Perevoshchikova IV, Petrassi HM, Meeusen SL, Ainscow EK, Brand MD. Suppressors of superoxide production from mitochondrial complex III. Nat Chem Biol 11: 834–836, 2015. doi: 10.1038/nchembio.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Watson MA, Pattavina B, Hilsabeck TA, Lopez-Dominguez J, Kapahi P, Brand MD. S3QELs protect against diet-induced intestinal barrier dysfunction. Aging Cell 20: e13476, 2021. doi: 10.1111/acel.13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279: 49064–49073, 2004. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 232. Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J 353: 411–416, 2001. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Gus'kova RA, Ivanov II, Kol’tover VK, Akhobadze VV, Rubin AB. Permeability of bilayer lipid membranes for superoxide (O2-.) radicals. Biochim Biophys Acta 778: 579–585, 1984. doi: 10.1016/0005-2736(84)90409-7. [DOI] [PubMed] [Google Scholar]
- 234. Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu, Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem 276: 38084–38089, 2001. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 235. Iñarrea P. Purification and determination of activity of mitochondrial cyanide-sensitive superoxide dismutase in rat tissue extract. Methods Enzymol 349: 106–114, 2002. doi: 10.1016/s0076-6879(02)49326-3. [DOI] [PubMed] [Google Scholar]
- 236. Kawamata H, Manfredi G. Different regulation of wild-type and mutant Cu, Zn superoxide dismutase localization in mammalian mitochondria. Hum Mol Genet 17: 3303–3317, 2008. doi: 10.1093/hmg/ddn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 278: 5557–5563, 2003. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 238. Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177: 1029–1036, 2007. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Chua YL, Dufour E, Dassa EP, Rustin P, Jacobs HT, Taylor CT, Hagen T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J Biol Chem 285: 31277–31284, 2010. doi: 10.1074/jbc.M110.158485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Ernster L, Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta 1271: 195–204, 1995. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 241. Beyer RE. The relative essentiality of the antioxidative function of coenzyme Q–the interactive role of DT-diaphorase. Mol Aspects Med 15, Suppl: s117–s129, 1994. doi: 10.1016/0098-2997(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 242. Takahashi T, Okamoto T, Mori K, Sayo H, Kishi T. Distribution of ubiquinone and ubiquinol homologues in rat tissues and subcellular fractions. Lipids 28: 803–809, 1993. doi: 10.1007/BF02536234. [DOI] [PubMed] [Google Scholar]
- 243. Aberg F, Appelkvist EL, Dallner G, Ernster L. Distribution and redox state of ubiquinones in rat and human tissues. Arch Biochem Biophys 295: 230–234, 1992. doi: 10.1016/0003-9861(92)90511-t. [DOI] [PubMed] [Google Scholar]
- 244. Crane FL, Sun IL, Clark MG, Grebing C, Löw H. Transplasma-membrane redox systems in growth and development. Biochim Biophys Acta 811: 233–264, 1985. doi: 10.1016/0304-4173(85)90013-8. [DOI] [PubMed] [Google Scholar]
- 245. Del Principe D, Avigliano L, Savini I, Catani MV. Trans-plasma membrane electron transport in mammals: functional significance in health and disease. Antioxid Redox Signal 14: 2289–2318, 2011. doi: 10.1089/ars.2010.3247. [DOI] [PubMed] [Google Scholar]
- 246. Ly JD, Lawen A. Transplasma membrane electron transport: enzymes involved and biological function. Redox Rep 8: 3–21, 2003. doi: 10.1179/135100003125001198. [DOI] [PubMed] [Google Scholar]
- 247. Navas P, Villalba JM, Lenaz G. Coenzyme Q-dependent functions of plasma membrane in the aging process. Age (Dordr) 27: 139–146, 2005. doi: 10.1007/s11357-005-1632-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Berridge MV, Tan AS. High-capacity redox control at the plasma membrane of mammalian cells: trans-membrane, cell surface, and serum NADH-oxidases. Antioxid Redox Signal 2: 231–242, 2000. doi: 10.1089/ars.2000.2.2-231. [DOI] [PubMed] [Google Scholar]
- 249. Beyer RE, Segura-Aguilar J, Di Bernardo S, Cavazzoni M, Fato R, Fiorentini D, Galli MC, Setti M, Landi L, Lenaz G. The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc Natl Acad Sci USA 93: 2528–2532, 1996. doi: 10.1073/pnas.93.6.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Villalba JM, Navarro F, Córdoba F, Serrano A, Arroyo A, Crane FL, Navas P. Coenzyme Q reductase from liver plasma membrane: purification and role in trans-plasma-membrane electron transport. Proc Natl Acad Sci USA 92: 4887–4891, 1995. doi: 10.1073/pnas.92.11.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Sun IL, Sun EE, Crane FL, Morré DJ, Lindgren A, Löw H. Requirement for coenzyme Q in plasma membrane electron transport. Proc Natl Acad Sci USA 89: 11126–11130, 1992. doi: 10.1073/pnas.89.23.11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, Bassik MC, Nomura DK, Dixon SJ, Olzmann JA. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575: 688–692, 2019. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Santoro MM. The antioxidant role of non-mitochondrial CoQ10: mystery solved!. Cell Metab 31: 13–15, 2020. doi: 10.1016/j.cmet.2019.12.007. [DOI] [PubMed] [Google Scholar]
- 254. Ross D, Siegel D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol 41: 101950, 2021. doi: 10.1016/j.redox.2021.101950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Navarro F, Navas P, Burgess JR, Bello RI, De Cabo R, Arroyo A, Villalba JM. Vitamin E and selenium deficiency induces expression of the ubiquinone-dependent antioxidant system at the plasma membrane. FASEB J 12: 1665–1673, 1998. doi: 10.1096/fasebj.12.15.1665. [DOI] [PubMed] [Google Scholar]
- 256. Doll S, Freitas FP, Shah R, Aldrovandi M, Silva MC, Ingold I, , et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575: 693–698, 2019. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 257. Hyun DH, Hernandez JO, Mattson MP, de Cabo R. The plasma membrane redox system in aging. Ageing Res Rev 5: 209–220, 2006. doi: 10.1016/j.arr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 258. Kishi T, Morré DM, Morré DJ. The plasma membrane NADH oxidase of HeLa cells has hydroquinone oxidase activity. Biochim Biophys Acta 1412: 66–77, 1999. doi: 10.1016/s0005-2728(99)00049-3. [DOI] [PubMed] [Google Scholar]
- 259. Morré DJ, Morré DM. Cell surface NADH oxidases (ECTO-NOX proteins) with roles in cancer, cellular time-keeping, growth, aging and neurodegenerative diseases. Free Radic Res 37: 795–808, 2003. doi: 10.1080/1071576031000083107. [DOI] [PubMed] [Google Scholar]
- 260. Avigliano L, Savini I, Catani MV, Del Principe D. Trans-plasma membrane electron transport in human blood platelets. Mini Rev Med Chem 8: 555–563, 2008. doi: 10.2174/138955708784534463. [DOI] [PubMed] [Google Scholar]
- 261. Baker MA, Lane DJ, Ly JD, De Pinto V, Lawen A. VDAC1 is a transplasma membrane NADH-ferricyanide reductase. J Biol Chem 279: 4811–4819, 2004. doi: 10.1074/jbc.M311020200. [DOI] [PubMed] [Google Scholar]
- 262. Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J 275: 3249–3277, 2008. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 263. Oakhill JS, Marritt SJ, Gareta EG, Cammack R, McKie AT. Functional characterization of human duodenal cytochrome b (Cybrd1): redox properties in relation to iron and ascorbate metabolism. Biochim Biophys Acta 1777: 260–268, 2008. doi: 10.1016/j.bbabio.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 264. Sabirov RZ, Merzlyak PG. Plasmalemmal VDAC controversies and maxi-anion channel puzzle. Biochim Biophys Acta 1818: 1570–1580, 2012. doi: 10.1016/j.bbamem.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 265. Lane DJ, Bae DH, Merlot AM, Sahni S, Richardson DR. Duodenal cytochrome b (DCYTB) in iron metabolism: an update on function and regulation. Nutrients 7: 2274–2296, 2015. doi: 10.3390/nu7042274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch Biochem Biophys 300: 535–543, 1993. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 267. Noctor G, Foyer CH. Ascorbate and glutathione: keeping active oxygen under control. Annu Rev Plant Physiol Plant Mol Biol 49: 249–279, 1998. doi: 10.1146/annurev.arplant.49.1.249. [DOI] [PubMed] [Google Scholar]
- 268. Redpath JL, Willson RL. Reducing compounds in radioprotection and radiosensitization: model experiments using ascorbic acid. Int J Radiat Biol Relat Stud Phys Chem Med 23: 51–65, 1973. doi: 10.1080/09553007314550051. [DOI] [PubMed] [Google Scholar]
- 269. Shen J, Griffiths PT, Campbell SJ, Utinger B, Kalberer M, Paulson SE. Ascorbate oxidation by iron, copper and reactive oxygen species: review, model development, and derivation of key rate constants. Sci Rep 11: 7417, 2021. doi: 10.1038/s41598-021-86477-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Nishikimi M. Oxidation of ascorbic acid with superoxide anion generated by the xanthine-xanthine oxidase system. Biochem Biophys Res Commun 63: 463–468, 1975. doi: 10.1016/0006-291x(75)90710-x. [DOI] [PubMed] [Google Scholar]
- 271. Smirnoff N. Ascorbic acid metabolism and functions: a comparison of plants and mammals. Free Radic Biol Med 122: 116–129, 2018. doi: 10.1016/j.freeradbiomed.2018.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Homma T, Takeda Y, Nakano T, Akatsuka S, Kinoshita D, Kurahashi T, Saitoh S, Yamada KI, Miyata S, Asao H, Goto K, Watanabe T, Watanabe M, Toyokuni S, Fujii J. Defective biosynthesis of ascorbic acid in Sod1-deficient mice results in lethal damage to lung tissue. Free Radic Biol Med 162: 255–265, 2021. doi: 10.1016/j.freeradbiomed.2020.10.023. [DOI] [PubMed] [Google Scholar]
- 273. Mehlhorn RJ. Ascorbate- and dehydroascorbic acid-mediated reduction of free radicals in the human erythrocyte. J Biol Chem 266: 2724–2731, 1991. doi: 10.1016/S0021-9258(18)49905-X. [DOI] [PubMed] [Google Scholar]
- 274. Buettner GR, Jurkiewicz BA. Ascorbate free radical as a marker of oxidative stress: an EPR study. Free Radic Biol Med 14: 49–55, 1993. doi: 10.1016/0891-5849(93)90508-r. [DOI] [PubMed] [Google Scholar]
- 275. Njus D, Kelley PM, Tu YJ, Schlegel HB. Ascorbic acid: the chemistry underlying its antioxidant properties. Free Radic Biol Med 159: 37–43, 2020. doi: 10.1016/j.freeradbiomed.2020.07.013. [DOI] [PubMed] [Google Scholar]
- 276. Linster CL, Van Schaftingen E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J 274: 1–22, 2007. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 277. VanDuijn MM, Tijssen K, VanSteveninck J, Van Den Broek PJ, Van Der Zee J. Erythrocytes reduce extracellular ascorbate free radicals using intracellular ascorbate as an electron donor. J Biol Chem 275: 27720–27725, 2000. doi: 10.1074/jbc.M910281199. [DOI] [PubMed] [Google Scholar]
- 278. Lane DJ, Lawen A. Ascorbate and plasma membrane electron transport—enzymes vs efflux. Free Radic Biol Med 47: 485–495, 2009. doi: 10.1016/j.freeradbiomed.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 279. Su D, May JM, Koury MJ, Asard H. Human erythrocyte membranes contain a cytochrome b561 that may be involved in extracellular ascorbate recycling. J Biol Chem 281: 39852–39859, 2006. doi: 10.1074/jbc.M606543200. [DOI] [PubMed] [Google Scholar]
- 280. Gómez-Díaz C, Rodríguez-Aguilera JC, Barroso MP, Villalba JM, Navarro F, Crane FL, Navas P. Antioxidant ascorbate is stabilized by NADH-coenzyme Q10 reductase in the plasma membrane. J Bioenerg Biomembr 29: 251–257, 1997. doi: 10.1023/a:1022410127104. [DOI] [PubMed] [Google Scholar]
- 281. Rodríguez-Aguilera JC, Navas P. Extracellular ascorbate stabilization: enzymatic or chemical process? J Bioenerg Biomembr 26: 379–384, 1994. doi: 10.1007/BF00762778. [DOI] [PubMed] [Google Scholar]
- 282. Santos-Ocaña C, Navas P, Crane FL, Córdoba F. Extracellular ascorbate stabilization as a result of transplasma electron transfer in Saccharomyces cerevisiae. J Bioenerg Biomembr 27: 597–603, 1995. doi: 10.1007/BF02111657. [DOI] [PubMed] [Google Scholar]
- 283. Santos-Ocaña C, Córdoba F, Crane FL, Clarke CF, Navas P. Coenzyme Q6 and iron reduction are responsible for the extracellular ascorbate stabilization at the plasma membrane of Saccharomyces cerevisiae. J Biol Chem 273: 8099–8105, 1998. doi: 10.1074/jbc.273.14.8099. [DOI] [PubMed] [Google Scholar]
- 284. Santos-Ocaña C, Villalba JM, Córdoba F, Padilla S, Crane FL, Clarke CF, Navas P. Genetic evidence for coenzyme Q requirement in plasma membrane electron transport. J Bioenerg Biomembr 30: 465–475, 1998. doi: 10.1023/a:1020542230308. [DOI] [PubMed] [Google Scholar]
- 285. Perly B, Smith IC, Hughes L, Burton GW, Ingold KU. Estimation of the location of natural alpha-tocopherol in lipid bilayers by 13C-NMR spectroscopy. Biochim Biophys Acta 819: 131–135, 1985. doi: 10.1016/0005-2736(85)90203-2. [DOI] [PubMed] [Google Scholar]
- 286. Wang X, Quinn PJ. The location and function of vitamin E in membranes. Mol Membr Biol 17: 143–156, 2000. doi: 10.1080/09687680010000311. [DOI] [PubMed] [Google Scholar]
- 287. Vieira SA, McClements DJ, Decker EA. Challenges of utilizing healthy fats in foods. Adv Nutr 6: 309S–317S, 2015. doi: 10.3945/an.114.006965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Miyazawa T, Burdeos GC, Itaya M, Nakagawa K, Miyazawa T. Vitamin E: regulatory redox interactions. IUBMB Life 71: 430–441, 2019. doi: 10.1002/iub.2008. [DOI] [PubMed] [Google Scholar]
- 289. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev 111: 5944–5972, 2011. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- 290. Catalá A, Díaz M. Editorial: Impact of lipid peroxidation on the physiology and pathophysiology of cell membranes. Front Physiol 7: 423, 2016. doi: 10.3389/fphys.2016.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Ayala A, Muñoz MF, Argúelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014: 360438, 2014. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, Jiang F, Peng ZY. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev 2019: 5080843, 2019. doi: 10.1155/2019/5080843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Farmer EE, Mueller MJ. ROS-mediated lipid peroxidation and RES-activated signaling. Annu Rev Plant Biol 64: 429–450, 2013. doi: 10.1146/annurev-arplant-050312-120132. [DOI] [PubMed] [Google Scholar]
- 294. May-Zhang LS, Kirabo A, Huang J, Linton MF, Davies SS, Murray KT. Scavenging reactive lipids to prevent oxidative injury. Annu Rev Pharmacol Toxicol 61: 291–308, 2021. doi: 10.1146/annurev-pharmtox-031620-035348. [DOI] [PubMed] [Google Scholar]
- 295. Niki E. Lipid oxidation that is, and is not, inhibited by vitamin E: consideration about physiological functions of vitamin E. Free Radic Biol Med 176: 1–15, 2021. doi: 10.1016/j.freeradbiomed.2021.09.001. [DOI] [PubMed] [Google Scholar]
- 296. Traber MG, Atkinson J. Vitamin E, antioxidant and nothing more. Free Radic Biol Med 43: 4–15, 2007. doi: 10.1016/j.freeradbiomed.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. McCay PB. Vitamin E: interactions with free radicals and ascorbate. Annu Rev Nutr 5: 323–340, 1985. doi: 10.1146/annurev.nu.05.070185.001543. [DOI] [PubMed] [Google Scholar]
- 298. Niki E, Saito T, Kawakami A, Kamiya Y. Inhibition of oxidation of methyl linoleate in solution by vitamin E and vitamin C. J Biol Chem 259: 4177–4182, 1984. doi: 10.1016/S0021-9258(17)43026-2. [DOI] [PubMed] [Google Scholar]
- 299. Niki E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: in vitro and in vivo evidence. Free Radic Biol Med 66: 3–12, 2014. doi: 10.1016/j.freeradbiomed.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 300. Yamamoto Y, Komuro E, Niki E. Antioxidant activity of ubiquinol in solution and phosphatidylcholine liposome. J Nutr Sci Vitaminol (Tokyo) 36: 505–511, 1990. doi: 10.3177/jnsv.36.505. [DOI] [PubMed] [Google Scholar]
- 301. Mukai K, Kikuchi S, Urano S. Stopped-flow kinetic study of the regeneration reaction of tocopheroxyl radical by reduced ubiquinone-10 in solution. Biochim Biophys Acta 1035: 77–82, 1990. doi: 10.1016/0304-4165(90)90176-w. [DOI] [PubMed] [Google Scholar]
- 302. Maguire JJ, Kagan V, Ackrell BA, Serbinova E, Packer L. Succinate-ubiquinone reductase linked recycling of alpha-tocopherol in reconstituted systems and mitochondria: requirement for reduced ubiquinone. Arch Biochem Biophys 292: 47–53, 1992. doi: 10.1016/0003-9861(92)90049-3. [DOI] [PubMed] [Google Scholar]
- 303. Kagan V, Serbinova E, Packer L. Antioxidant effects of ubiquinones in microsomes and mitochondria are mediated by tocopherol recycling. Biochem Biophys Res Commun 169: 851–857, 1990. doi: 10.1016/0006-291x(90)91971-t. [DOI] [PubMed] [Google Scholar]
- 304. Frei B, Kim MC, Ames BN. Ubiquinol-10 is an effective lipid-soluble antioxidant at physiological concentrations. Proc Natl Acad Sci USA 87: 4879–4883, 1990. doi: 10.1073/pnas.87.12.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Baschiera E, Sorrentino U, Calderan C, Desbats MA, Salviati L. The multiple roles of coenzyme Q in cellular homeostasis and their relevance for the pathogenesis of coenzyme Q deficiency. Free Radic Biol Med 166: 277–286, 2021. doi: 10.1016/j.freeradbiomed.2021.02.039. [DOI] [PubMed] [Google Scholar]
- 306. Mukai K, Morimoto H, Kikuchi S, Nagaoka S. Kinetic study of free-radical-scavenging action of biological hydroquinones (reduced forms of ubiquinone, vitamin K and tocopherol quinone) in solution. Biochim Biophys Acta 1157: 313–317, 1993. doi: 10.1016/0304-4165(93)90115-o. [DOI] [PubMed] [Google Scholar]
- 307. Shi H, Noguchi N, Niki E. Comparative study on dynamics of antioxidative action of alpha-tocopheryl hydroquinone, ubiquinol, and alpha-tocopherol against lipid peroxidation. Free Radic Biol Med 27: 334–346, 1999. doi: 10.1016/s0891-5849(99)00053-2. [DOI] [PubMed] [Google Scholar]
- 308. Sohal RS, Forster MJ. Coenzyme Q, oxidative stress and aging. Mitochondrion 7, Suppl: S103–111, 2007. doi: 10.1016/j.mito.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Forsmark P, Aberg F, Norling B, Nordenbrand K, Dallner G, Ernster L. Inhibition of lipid peroxidation by ubiquinol in submitochondrial particles in the absence of vitamin E. FEBS Lett 285: 39–43, 1991. doi: 10.1016/0014-5793(91)80720-n. [DOI] [PubMed] [Google Scholar]
- 310. Kagan VE, Quinn PJ. Coenzyme Q: Molecular Mechanisms in Health and Disease. Boca Raton, FL: CRC Press, 2001. [Google Scholar]
- 311. Takayanagi R, Takeshige K, Minakami S. NADH- and NADPH-dependent lipid peroxidation in bovine heart submitochondrial particles. Dependence on the rate of electron flow in the respiratory chain and an antioxidant role of ubiquinol. Biochem J 192: 853–860, 1980. doi: 10.1042/bj1920853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Ernster L, Forsmark P, Nordenbrand K. The mode of action of lipid-soluble antioxidants in biological membranes. Relationship between the effects of ubiquinol and vitamin E as inhibitors of lipid peroxidation in submitochondrial particles. J Nutr Sci Vitaminol (Tokyo) Spec No: 548–551, 1992. doi: 10.3177/jnsv.38.special_548. [DOI] [PubMed] [Google Scholar]
- 313. Forsmark-Andrée P, Ernster L. Evidence for a protective effect of endogenous ubiquinol against oxidative damage to mitochondrial protein and DNA during lipid peroxidation. Mol Aspects Med 15, Suppl: s73–s81, 1994. doi: 10.1016/0098-2997(94)90015-9. [DOI] [PubMed] [Google Scholar]
- 314. Forsmark-Andrée P, Dallner G, Ernster L. Endogenous ubiquinol prevents protein modification accompanying lipid peroxidation in beef heart submitochondrial particles. Free Radic Biol Med 19: 749–757, 1995. doi: 10.1016/0891-5849(95)00076-a. [DOI] [PubMed] [Google Scholar]
- 315. Mohr D, Bowry VW, Stocker R. Dietary supplementation with coenzyme Q10 results in increased levels of ubiquinol-10 within circulating lipoproteins and increased resistance of human low-density lipoprotein to the initiation of lipid peroxidation. Biochim Biophys Acta 1126: 247–254, 1992. doi: 10.1016/0005-2760(92)90237-p. [DOI] [PubMed] [Google Scholar]
- 316. Edlund PO. Determination of coenzyme Q10, alpha-tocopherol and cholesterol in biological samples by coupled-column liquid chromatography with coulometric and ultraviolet detection. J Chromatogr 425: 87–97, 1988. doi: 10.1016/0378-4347(88)80009-4. [DOI] [PubMed] [Google Scholar]
- 317. Witting PK, Pettersson K, Letters J, Stocker R. Anti-atherogenic effect of coenzyme Q10 in apolipoprotein E gene knockout mice. Free Radic Biol Med 29: 295–305, 2000. doi: 10.1016/s0891-5849(00)00311-7. [DOI] [PubMed] [Google Scholar]
- 318. Stocker R, Bowry VW, Frei B. Ubiquinol-10 protects human low density lipoprotein more efficiently against lipid peroxidation than does alpha-tocopherol. Proc Natl Acad Sci USA 88: 1646–1650, 1991. doi: 10.1073/pnas.88.5.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Choy KJ, Deng YM, Hou JY, Wu B, Lau A, Witting PK, Stocker R. Coenzyme Q10 supplementation inhibits aortic lipid oxidation but fails to attenuate intimal thickening in balloon-injured New Zealand white rabbits. Free Radic Biol Med 35: 300–309, 2003. doi: 10.1016/s0891-5849(03)00304-6. [DOI] [PubMed] [Google Scholar]
- 320. Thomas SR, Witting PK, Stocker R. A role for reduced coenzyme Q in atherosclerosis? Biofactors 9: 207–224, 1999. doi: 10.1002/biof.5520090216. [DOI] [PubMed] [Google Scholar]
- 321. Raitakari OT, McCredie RJ, Witting P, Griffiths KA, Letters J, Sullivan D, Stocker R, Celermajer DS. Coenzyme Q improves LDL resistance to ex vivo oxidation but does not enhance endothelial function in hypercholesterolemic young adults. Free Radic Biol Med 28: 1100–1105, 2000. doi: 10.1016/s0891-5849(00)00201-x. [DOI] [PubMed] [Google Scholar]
- 322. Thomas SR, Neuzil J, Stocker R. Cosupplementation with coenzyme Q prevents the prooxidant effect of alpha-tocopherol and increases the resistance of LDL to transition metal-dependent oxidation initiation. Arterioscler Thromb Vasc Biol 16: 687–696, 1996. doi: 10.1161/01.atv.16.5.687. [DOI] [PubMed] [Google Scholar]
- 323. Thomas SR, Leichtweis SB, Pettersson K, Croft KD, Mori TA, Brown AJ, Stocker R. Dietary cosupplementation with vitamin E and coenzyme Q10 inhibits atherosclerosis in apolipoprotein E gene knockout mice. Arterioscler Thromb Vasc Biol 21: 585–593, 2001. doi: 10.1161/01.atv.21.4.585. [DOI] [PubMed] [Google Scholar]
- 324. Chisolm GM, Steinberg D. The oxidative modification hypothesis of atherogenesis: an overview. Free Radic Biol Med 28: 1815–1826, 2000. doi: 10.1016/s0891-5849(00)00344-0. [DOI] [PubMed] [Google Scholar]
- 325. Ben-Meir A, Burstein E, Borrego-Alvarez A, Chong J, Wong E, Yavorska T, Naranian T, Chi M, Wang Y, Bentov Y, Alexis J, Meriano J, Sung HK, Gasser DL, Moley KH, Hekimi S, Casper RF, Jurisicova A. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 14: 887–895, 2015. doi: 10.1111/acel.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94: 909–950, 2014. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann NY Acad Sci 1147: 37–52, 2008. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Valdebenito GE, Chacko AR, Duchen MR. The mitochondrial ATP synthase as an ATP consumer-a surprising therapeutic target. EMBO J 42: e114141, 2023. doi: 10.15252/embj.2023114141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Duberley KE, Abramov AY, Chalasani A, Heales SJ, Rahman S, Hargreaves IP. Human neuronal coenzyme Q10 deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: implications for pathogenesis and treatment. J Inherit Metab Dis 36: 63–73, 2013. doi: 10.1007/s10545-012-9511-0. [DOI] [PubMed] [Google Scholar]
- 330. Thorpe GW, Reodica M, Davies MJ, Heeren G, Jarolim S, Pillay B, Breitenbach M, Higgins VJ, Dawes IW. Superoxide radicals have a protective role during H2O2 stress. Mol Biol Cell 24: 2876–2884, 2013. doi: 10.1091/mbc.E13-01-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Allan CM, Hill S, Morvaridi S, Saiki R, Johnson JS, Liau WS, Hirano K, Kawashima T, Ji Z, Loo JA, Shepherd JN, Clarke CF. A conserved START domain coenzyme Q-binding polypeptide is required for efficient Q biosynthesis, respiratory electron transport, and antioxidant function in Saccharomyces cerevisiae. Biochim Biophys Acta 1831: 776–791, 2013. doi: 10.1016/j.bbalip.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Do TQ, Schultz JR, Clarke CF. Enhanced sensitivity of ubiquinone-deficient mutants of Saccharomyces cerevisiae to products of autoxidized polyunsaturated fatty acids. Proc Natl Acad Sci USA 93: 7534–7539, 1996. doi: 10.1073/pnas.93.15.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Poon WW, Do TQ, Marbois BN, Clarke CF. Sensitivity to treatment with polyunsaturated fatty acids is a general characteristic of the ubiquinone-deficient yeast coq mutants. Mol Aspects Med 18, Suppl: S121–S127, 1997. doi: 10.1016/s0098-2997(97)00004-6. [DOI] [PubMed] [Google Scholar]
- 334. Kohlwein SD, Paltauf F. Uptake of fatty acids by the yeasts, Saccharomyces uvarum and Saccharomycopsis lipolytica. Biochim Biophys Acta 792: 310–317, 1984. doi: 10.1016/0005-2760(84)90198-x. [DOI] [PubMed] [Google Scholar]
- 335. Schneider C. An update on products and mechanisms of lipid peroxidation. Mol Nutr Food Res 53: 315–321, 2009. doi: 10.1002/mnfr.200800131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Quinzii CM, Tadesse S, Naini A, Hirano M. Effects of inhibiting CoQ10 biosynthesis with 4-nitrobenzoate in human fibroblasts. PLoS One 7: e30606, 2012. doi: 10.1371/journal.pone.0030606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Cullen JK, Abdul Murad N, Yeo A, McKenzie M, Ward M, Chong KL, Schieber NL, Parton RG, Lim YC, Wolvetang E, Maghzal GJ, Stocker R, Lavin MF. aarF Domain Containing Kinase 3 (ADCK3) mutant cells display signs of oxidative stress, defects in mitochondrial homeostasis and lysosomal accumulation. PLoS One 11: e0148213, 2016. doi: 10.1371/journal.pone.0148213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Fazakerley DJ, Chaudhuri R, Yang P, Maghzal GJ, Thomas KC, Krycer JR, Humphrey SJ, Parker BL, Fisher-Wellman KH, Meoli CC, Hoffman NJ, Diskin C, Burchfield JG, Cowley MJ, Kaplan W, Modrusan Z, Kolumam G, Yang JY, Chen DL, Samocha-Bonet D, Greenfield JR, Hoehn KL, Stocker R, James DE. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife 7: e32111, 2018. doi: 10.7554/eLife.32111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. González-Aragón D, Burón MI, Lopez-Lluch G, Herman MD, Gomez-Diaz C, Navas P, Villalba JM. Coenzyme Q and the regulation of intracellular steady-state levels of superoxide in HL-60 cells. Biofactors 25: 31–41, 2005. doi: 10.1002/biof.5520250105. [DOI] [PubMed] [Google Scholar]
- 340. Stuart JA, Fonseca J, Moradi F, Cunningham C, Seliman B, Worsfold CR, Dolan S, Abando J, Maddalena LA. How supraphysiological oxygen levels in standard cell culture affect oxygen-consuming reactions. Oxid Med Cell Longev 2018: 8238459, 2018. doi: 10.1155/2018/8238459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Stepanova A, Konrad C, Manfredi G, Springett R, Ten V, Galkin A. The dependence of brain mitochondria reactive oxygen species production on oxygen level is linear, except when inhibited by antimycin A. J Neurochem 148: 731–745, 2019. doi: 10.1111/jnc.14654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Long LH, Halliwell B. Artefacts in cell culture: pyruvate as a scavenger of hydrogen peroxide generated by ascorbate or epigallocatechin gallate in cell culture media. Biochem Biophys Res Commun 388: 700–704, 2009. doi: 10.1016/j.bbrc.2009.08.069. [DOI] [PubMed] [Google Scholar]
- 343. Dominiak K, Galganski L, Budzinska A, Jarmuszkiewicz W. Coenzyme Q deficiency in endothelial mitochondria caused by hypoxia; remodeling of the respiratory chain and sensitivity to anoxia/reoxygenation. Free Radic Biol Med 214: 158–170, 2024. doi: 10.1016/j.freeradbiomed.2024.02.005. [DOI] [PubMed] [Google Scholar]
- 344. Lyon MF, Hulse EV. An inherited kidney disease of mice resembling human nephronophthisis. J Med Genet 8: 41–48, 1971. doi: 10.1136/jmg.8.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Neilson EG, McCafferty E, Feldman A, Clayman MD, Zakheim B, Korngold R. Spontaneous interstitial nephritis in kdkd mice. I. An experimental model of autoimmune renal disease. J Immunol 133: 2560–2565, 1984. [PubMed] [Google Scholar]
- 346. Kelly CJ, Korngold R, Mann R, Clayman M, Haverty T, Neilson EG. Spontaneous interstitial nephritis in kdkd mice. II. Characterization of a tubular antigen-specific, H-2K-restricted Lyt-2+ effector T cell that mediates destructive tubulointerstitial injury. J Immunol 136: 526–531, 1986. [PubMed] [Google Scholar]
- 347. Saiki R, Lunceford AL, Shi Y, Marbois B, King R, Pachuski J, Kawamukai M, Gasser DL, Clarke CF. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am J Physiol Renal Physiol 295: F1535–F1544, 2008. doi: 10.1152/ajprenal.90445.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Falk MJ, Polyak E, Zhang Z, Peng M, King R, Maltzman JS, Okwuego E, Horyn O, Nakamaru-Ogiso E, Ostrovsky J, Xie LX, Chen JY, Marbois B, Nissim I, Clarke CF, Gasser DL. Probucol ameliorates renal and metabolic sequelae of primary CoQ deficiency in Pdss2 mutant mice. EMBO Mol Med 3: 410–427, 2011. doi: 10.1002/emmm.201100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. García-Corzo L, Luna-Sánchez M, Doerrier C, García JA, Guaras A, Acin-Perez R, Bullejos-Peregrin J, Lopez A, Escames G, Enriquez JA, Acuna-Castroviejo D, Lopez LC. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum Mol Genet 22: 1233–1248, 2013. doi: 10.1093/hmg/dds530. [DOI] [PubMed] [Google Scholar]
- 350. Cerqua C, Casarin A, Pierrel F, Vazquez Fonseca L, Viola G, Salviati L, Trevisson E. Vitamin K2 cannot substitute Coenzyme Q10 as electron carrier in the mitochondrial respiratory chain of mammalian cells. Sci Rep 9: 6553, 2019. doi: 10.1038/s41598-019-43014-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. López LC, Quinzii CM, Area E, Naini A, Rahman S, Schuelke M, Salviati L, Dimauro S, Hirano M. Treatment of CoQ10 deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: time- and compound-dependent effects. PLoS One 5: e11897, 2010. doi: 10.1371/journal.pone.0011897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Fernández-Del-Río L, Kelly ME, Contreras J, Bradley MC, James AM, Murphy MP, Payne GS, Clarke CF. Genes and lipids that impact uptake and assimilation of exogenous coenzyme Q in Saccharomyces cerevisiae. Free Radic Biol Med 154: 105–118, 2020. doi: 10.1016/j.freeradbiomed.2020.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Fernández-Ayala DJ, Brea-Calvo G, López-Lluch G, Navas P. Coenzyme Q distribution in HL-60 human cells depends on the endomembrane system. Biochim Biophys Acta 1713: 129–137, 2005. doi: 10.1016/j.bbamem.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 354. Heaton RA, Heales S, Rahman K, Sexton DW, Hargreaves I. The effect of cellular Coenzyme Q10 deficiency on lysosomal acidification. J Clin Med 9: 1923, 2020. doi: 10.3390/jcm9061923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Greenlee H, Shaw J, Lau YI, Naini A, Maurer M. Lack of effect of coenzyme q10 on doxorubicin cytotoxicity in breast cancer cell cultures. Integr Cancer Ther 11: 243–250, 2012. doi: 10.1177/1534735412439749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Anderson CM, Kazantzis M, Wang J, Venkatraman S, Goncalves RL, Quinlan CL, Ng R, Jastroch M, Benjamin DI, Nie B, Herber C, Van AA, Park MJ, Yun D, Chan K, Yu A, Vuong P, Febbraio M, Nomura DK, Napoli JL, Brand MD, Stahl A. Dependence of brown adipose tissue function on CD36-mediated coenzyme Q uptake. Cell Rep 10: 505–515, 2015. doi: 10.1016/j.celrep.2014.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Jacquier A, Theuriet J, Fontaine F, Mosbach V, Lacoste N, Ribault S, Risson V, Carras J, Coudert L, Simonet T, Latour P, Stojkovic T, Piard J, Cosson A, Lesca G, Bouhour F, Allouche S, Puccio H, Pegat A, Schaeffer L. Homozygous COQ7 mutation: a new cause of potentially treatable distal hereditary motor neuropathy. Brain 146: 3470–3483, 2023. doi: 10.1093/brain/awac453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Jonassen T, Proft M, Randez-Gil F, Schultz JR, Marbois BN, Entian KD, Clarke CF. Yeast Clk-1 homologue (Coq7/Cat5) is a mitochondrial protein in coenzyme Q synthesis. J Biol Chem 273: 3351–3357, 1998. doi: 10.1074/jbc.273.6.3351. [DOI] [PubMed] [Google Scholar]
- 359. Gin P, Hsu AY, Rothman SC, Jonassen T, Lee PT, Tzagoloff A, Clarke CF. The Saccharomyces cerevisiae COQ6 gene encodes a mitochondrial flavin-dependent monooxygenase required for coenzyme Q biosynthesis. J Biol Chem 278: 25308–25316, 2003. doi: 10.1074/jbc.M303234200. [DOI] [PubMed] [Google Scholar]
- 360. Guile MD, Jain A, Anderson KA, Clarke CF. New insights on the uptake and trafficking of coenzyme Q. Antioxidants (Basel) 12: 1391, 2023. doi: 10.3390/antiox12071391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Nashimoto S, Takekawa Y, Takekuma Y, Sugawara M, Sato Y. Transport via Niemann-Pick C1 Like 1 contributes to the intestinal absorption of ubiquinone. Drug Metab Pharmacokinet 35: 527–533, 2020. doi: 10.1016/j.dmpk.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 362. James AM, Cochemé HM, Murai M, Miyoshi H, Murphy MP. Complementation of coenzyme Q-deficient yeast by coenzyme Q analogues requires the isoprenoid side chain. FEBS J 277: 2067–2082, 2010. doi: 10.1111/j.1742-4658.2010.07622.x. [DOI] [PubMed] [Google Scholar]
- 363. Mantle D, Dybring A. Bioavailability of Coenzyme Q. An overview of the absorption process and subsequent metabolism. Antioxidants (Basel) 9: 386, 2020. doi: 10.3390/antiox9050386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Bhagavan HN, Chopra RK. Coenzyme Q10: absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic Res 40: 445–453, 2006. doi: 10.1080/10715760600617843. [DOI] [PubMed] [Google Scholar]
- 365. Mantle D, Lopez-Lluch G, Hargreaves IP. Coenzyme Q10 metabolism: a review of unresolved issues. Int J Mol Sci 24: 2585, 2025. doi: 10.3390/ijms24032585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Zhang Y, Aberg F, Appelkvist EL, Dallner G, Ernster L. Uptake of dietary coenzyme Q supplement is limited in rats. J Nutr 125: 446–453, 1995. doi: 10.1093/jn/125.3.446. [DOI] [PubMed] [Google Scholar]
- 367. Sohal RS, Kamzalov S, Sumien N, Ferguson M, Rebrin I, Heinrich KR, Forster MJ. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic Biol Med 40: 480–487, 2006. doi: 10.1016/j.freeradbiomed.2005.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. García-Corzo L, Luna-Sánchez M, Doerrier C, Ortiz F, Escames G, Acuna-Castroviejo D, Lopez LC. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim Biophys Acta 1842: 893–901, 2014. doi: 10.1016/j.bbadis.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 369. Dadabayev AR, Yin G, Latchoumycandane C, McIntyre TM, Lesnefsky EJ, Penn MS. Apolipoprotein A1 regulates coenzyme Q10 absorption, mitochondrial function, and infarct size in a mouse model of myocardial infarction. J Nutr 144: 1030–1036, 2014. doi: 10.3945/jn.113.184291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Glover EI, Martin J, Maher A, Thornhill RE, Moran GR, Tarnopolsky MA. A randomized trial of coenzyme Q10 in mitochondrial disorders. Muscle Nerve 42: 739–748, 2010. doi: 10.1002/mus.21758. [DOI] [PubMed] [Google Scholar]
- 371. Zhu ZG, Sun MX, Zhang WL, Wang WW, Jin YM, Xie CL. The efficacy and safety of coenzyme Q10 in Parkinson’s disease: a meta-analysis of randomized controlled trials. Neurol Sci 38: 215–224, 2017. doi: 10.1007/s10072-016-2757-9. [DOI] [PubMed] [Google Scholar]
- 372. Jafari M, Mousavi SM, Asgharzadeh A, Yazdani N. Coenzyme Q10 in the treatment of heart failure: a systematic review of systematic reviews. Indian Heart J 70, Suppl 1: S111–S117, 2018. doi: 10.1016/j.ihj.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Zhou H, Liu G, Zhang J, Sun N, Duan M, Yan Z, Xia Q. Novel lipid-free nanoformulation for improving oral bioavailability of coenzyme Q10. Biomed Res Int 2014: 793879, 2014. doi: 10.1155/2014/793879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Wear D, Vegh C, Sandhu JK, Sikorska M, Cohen J, Pandey S. Ubisol-Q10, a nanomicellar and water-dispersible formulation of coenzyme-Q10 as a potential treatment for Alzheimer’s and Parkinson’s disease. Antioxidants (Basel) 10: 764, 2021. doi: 10.3390/antiox10050764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Fetoni AR, Piacentini R, Fiorita A, Paludetti G, Troiani D. Water-soluble Coenzyme Q10 formulation (Q-ter) promotes outer hair cell survival in a guinea pig model of noise induced hearing loss (NIHL). Brain Res 1257: 108–116, 2009. doi: 10.1016/j.brainres.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 376. Fetoni AR, Eramo SL, Rolesi R, Troiani D, Paludetti G. Antioxidant treatment with coenzyme Q-ter in prevention of gentamycin ototoxicity in an animal model. Acta Otorhinolaryngol Ital 32: 103–110, 2012. [PMC free article] [PubMed] [Google Scholar]
- 377. Pastor-Maldonado CJ, Suarez-Rivero JM, Povea-Cabello S, Alvarez-Cordoba M, Villalon-Garcia I, Munuera-Cabeza M, Suarez-Carrillo A, Talaveron-Rey M, Sanchez-Alcazar JA. Coenzyme Q10: novel formulations and medical trends. Int J Mol Sci 21: 8432, 2020. doi: 10.3390/ijms21228432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Dadali T, Diers AR, Kazerounian S, Muthuswamy SK, Awate P, Ng R, Mogre S, Spencer C, Krumova K, Rockwell HE, McDaniel J, Chen EY, Gao F, Diedrich KT, Vemulapalli V, Rodrigues LO, Akmaev VR, Thapa K, Hidalgo M, Bose A, Vishnudas VK, Moser AJ, Granger E, Kiebish MA, Gesta S, Narain NR, Sarangarajan R. Elevated levels of mitochondrial CoQ10 induce ROS-mediated apoptosis in pancreatic cancer. Sci Rep 11: 5749, 2021. doi: 10.1038/s41598-021-84852-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Sikorska M, Lanthier P, Miller H, Beyers M, Sodja C, Zurakowski B, Gangaraju S, Pandey S, Sandhu JK. Nanomicellar formulation of coenzyme Q10 (Ubisol-Q10) effectively blocks ongoing neurodegeneration in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model: potential use as an adjuvant treatment in Parkinson’s disease. Neurobiol Aging 35: 2329–2346, 2014. doi: 10.1016/j.neurobiolaging.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Zmitek J, Smidovnik A, Fir M, Prosek M, Zmitek K, Walczak J, Pravst I. Relative bioavailability of two forms of a novel water-soluble coenzyme Q10. Ann Nutr Metab 52: 281–287, 2008. doi: 10.1159/000129661. [DOI] [PubMed] [Google Scholar]
- 381. Niibori K, Yokoyama H, Crestanello JA, Whitman GJ. Acute administration of liposomal coenzyme Q10 increases myocardial tissue levels and improves tolerance to ischemia reperfusion injury. J Surg Res 79: 141–145, 1998. doi: 10.1006/jsre.1998.5411. [DOI] [PubMed] [Google Scholar]
- 382. López-Lluch G, Del Pozo-Cruz J, Sánchez-Cuesta A, Cortés-Rodriguez AB, Navas P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 57: 133–140, 2019. doi: 10.1016/j.nut.2018.05.020. [DOI] [PubMed] [Google Scholar]
- 383. Mine Y, Takahashi T, Okamoto T. Protective effects of coenzyme Q10 on cell damage induced by hydrogen peroxides in cultured skin fibroblasts. J Clin Biochem Nutr 69: 247–255, 2021. doi: 10.3164/jcbn.20-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Li L, Du J, Lian Y, Zhang Y, Li X, Liu Y, Zou L, Wu T. Protective effects of coenzyme Q10 against hydrogen peroxide-induced oxidative stress in PC12 Cell: the role of Nrf2 and antioxidant enzymes. Cell Mol Neurobiol 36: 103–111, 2016. doi: 10.1007/s10571-015-0224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Huo J, Xu Z, Hosoe K, Kubo H, Miyahara H, Dai J, Mori M, Sawashita J, Higuchi K. Coenzyme Q10 prevents senescence and dysfunction caused by oxidative stress in vascular endothelial cells. Oxid Med Cell Longev 2018: 3181759, 2018. doi: 10.1155/2018/3181759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. McCarthy S, Somayajulu M, Sikorska M, Borowy-Borowski H, Pandey S. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble Coenzyme Q10. Toxicol Appl Pharmacol 201: 21–31, 2004. doi: 10.1016/j.taap.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 387. Chan TS, Teng S, Wilson JX, Galati G, Khan S, O’Brien PJ. Coenzyme Q cytoprotective mechanisms for mitochondrial complex I cytopathies involves NAD(P)H: quinone oxidoreductase 1 (NQO1). Free Radic Res 36: 421–427, 2002. doi: 10.1080/10715760290021270. [DOI] [PubMed] [Google Scholar]
- 388. Kwong LK, Kamzalov S, Rebrin I, Bayne AC, Jana CK, Morris P, Forster MJ, Sohal RS. Effects of coenzyme Q10 administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic Biol Med 33: 627–638, 2002. doi: 10.1016/s0891-5849(02)00916-4. [DOI] [PubMed] [Google Scholar]
- 389. Bello RI, Gómez-Díaz C, Burón MI, Alcain FJ, Navas P, Villalba JM. Enhanced anti-oxidant protection of liver membranes in long-lived rats fed on a coenzyme Q10-supplemented diet. Exp Gerontol 40: 694–706, 2005. doi: 10.1016/j.exger.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 390. Sugino K, Dohi K, Yamada K, Kawasaki T. Changes in the levels of endogenous antioxidants in the liver of mice with experimental endotoxemia and the protective effects of the antioxidants. Surgery 105: 200–206, 1989. [PubMed] [Google Scholar]
- 391. Beyer RE. Inhibition by coenzyme Q of ethanol- and carbon tetrachloride-stimulated lipid peroxidation in vivo and catalyzed by microsomal and mitochondrial systems. Free Radic Biol Med 5: 297–303, 1988. doi: 10.1016/0891-5849(88)90100-1. [DOI] [PubMed] [Google Scholar]
- 392. Amimoto T, Matsura T, Koyama SY, Nakanishi T, Yamada K, Kajiyama G. Acetaminophen-induced hepatic injury in mice: the role of lipid peroxidation and effects of pretreatment with coenzyme Q10 and alpha-tocopherol. Free Radic Biol Med 19: 169–176, 1995. doi: 10.1016/0891-5849(94)00233-a. [DOI] [PubMed] [Google Scholar]
- 393. Alqarni F, Eweis HS, Ali A, Alrafiah A, Alsieni M, Karim S, Alkathyri MA. The effect of coenzyme Q10 on liver injury induced by valproic acid and its antiepileptic activity in rats. Biomedicines 10: 168, 2022. doi: 10.3390/biomedicines10010168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22: 266–282, 2021. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Hirata Y, Cai R, Volchuk A, Steinberg BE, Saito Y, Matsuzawa A, Grinstein S, Freeman SA. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis. Curr Biol 33: 1282–1294.e5, 2023. doi: 10.1016/j.cub.2023.02.060. [DOI] [PubMed] [Google Scholar]
- 396. Hendricks JM, Doubravsky CE, Wehri E, Li Z, Roberts MA, Deol KK, Lange M, Lasheras-Otero I, Momper JD, Dixon SJ, Bersuker K, Schaletzky J, Olzmann JA. Identification of structurally diverse FSP1 inhibitors that sensitize cancer cells to ferroptosis. Cell Chem Biol 30: 1090–1103.e7, 2023. doi: 10.1016/j.chembiol.2023.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol 220: e202105043, 2021. doi: 10.1083/jcb.202105043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, Poyurovsky MV, Olszewski K, Gan B. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593: 586–590, 2021. doi: 10.1038/s41586-021-03539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Wu S, Mao C, Kondiparthi L, Poyurovsky MV, Olszewski K, Gan B. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc Natl Acad Sci USA 119: e2121987119, 2022. doi: 10.1073/pnas.2121987119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Deshwal S, Onishi M, Tatsuta T, Bartsch T, Cors E, Ried K, Lemke K, Nolte H, Giavalisco P, Langer T. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat Cell Biol 25: 246–257, 2023. doi: 10.1038/s41556-022-01071-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Li D, Lu X, Xu G, Liu S, Gong Z, Lu F, Xia X, Jiang J, Wang H, Zou F, Ma X. Dihydroorotate dehydrogenase regulates ferroptosis in neurons after spinal cord injury via the P53-ALOX15 signaling pathway. CNS Neurosci Ther 29: 1923–1939, 2023. doi: 10.1111/cns.14150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Bonora M, Giorgi C, Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol 23: 266–285, 2022. doi: 10.1038/s41580-021-00433-y. [DOI] [PubMed] [Google Scholar]
- 403. Bauer TM, Murphy E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ Res 126: 280–293, 2020. doi: 10.1161/CIRCRESAHA.119.316306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Pérez MJ, Quintanilla RA. Development or disease: duality of the mitochondrial permeability transition pore. Dev Biol 426: 1–7, 2017. doi: 10.1016/j.ydbio.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 405. Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 416: 15–18, 1997. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 406. Sidhom EH, Kim C, Kost-Alimova M, Ting MT, Keller K, Avila-Pacheco J, Watts AJ, Vernon KA, Marshall JL, Reyes-Bricio E, Racette M, Wieder N, Kleiner G, Grinkevich EJ, Chen F, Weins A, Clish CB, Shaw JL, Quinzii CM, Greka A. Targeting a Braf/Mapk pathway rescues podocyte lipid peroxidation in CoQ-deficiency kidney disease. J Clin Invest 131: e141380, 2021. doi: 10.1172/JCI141380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Schijvens AM, van de Kar NC, Bootsma-Robroeks CM, Cornelissen EA, van den Heuvel LP, Schreuder MF. Mitochondrial disease and the kidney with a special focus on CoQ10 deficiency. Kidney Int Rep 5: 2146–2159, 2020. doi: 10.1016/j.ekir.2020.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z, , et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest 121: 2013–2024, 2011. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658, 2005. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 410. Walter L, Nogueira V, Leverve X, Heitz MP, Bernardi P, Fontaine E. Three classes of ubiquinone analogs regulate the mitochondrial permeability transition pore through a common site. J Biol Chem 275: 29521–29527, 2000. doi: 10.1074/jbc.M004128200. [DOI] [PubMed] [Google Scholar]
- 411. Gharib A, De Paulis D, Li B, Augeul L, Couture-Lepetit E, Gomez L, Angoulvant D, Ovize M. Opposite and tissue-specific effects of coenzyme Q2 on mPTP opening and ROS production between heart and liver mitochondria: role of complex I. J Mol Cell Cardiol 52: 1091–1095, 2012. doi: 10.1016/j.yjmcc.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 412. Barajas M, Wang A, Griffiths KK, Matsumoto K, Liu R, Homma S, Levy RJ. The newborn Fmr1 knockout mouse: a novel model of excess ubiquinone and closed mitochondrial permeability transition pore in the developing heart. Pediatr Res 89: 456–463, 2021. doi: 10.1038/s41390-020-1064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Sluse FE, Jarmuszkiewicz W, Navet R, Douette P, Mathy G, Sluse-Goffart CM. Mitochondrial UCPs: new insights into regulation and impact. Biochim Biophys Acta 1757: 480–485, 2006. doi: 10.1016/j.bbabio.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 414. Chouchani ET, Kazak L, Spiegelman BM. New advances in adaptive thermogenesis: UCP1 and beyond. Cell Metab 29: 27–37, 2019. doi: 10.1016/j.cmet.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 415. Mailloux RJ, Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med 51: 1106–1115, 2011. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 416. Cadenas S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg 1859: 940–950, 2018. doi: 10.1016/j.bbabio.2018.05.019. [DOI] [PubMed] [Google Scholar]
- 417. Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105: 745–755, 2001. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 418. Rupprecht A, Bräuer AU, Smorodchenko A, Goyn J, Hilse KE, Shabalina IG, Infante-Duarte C, Pohl EE. Quantification of uncoupling protein 2 reveals its main expression in immune cells and selective up-regulation during T-cell proliferation. PLoS One 7: e41406, 2012. doi: 10.1371/journal.pone.0041406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151: 400–413, 2012. doi: 10.1016/j.cell.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Klingenberg M, Huang SG. Structure and function of the uncoupling protein from brown adipose tissue. Biochim Biophys Acta 1415: 271–296, 1999. doi: 10.1016/s0005-2736(98)00232-6. [DOI] [PubMed] [Google Scholar]
- 421. Woyda-Ploszczyca AM, Jarmuszkiewicz W. The conserved regulation of mitochondrial uncoupling proteins: from unicellular eukaryotes to mammals. Biochim Biophys Acta Bioenerg 1858: 21–33, 2017. doi: 10.1016/j.bbabio.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 422. Jarmuszkiewicz W, Swida A, Czarna M, Antos N, Sluse-Goffart CM, Sluse FE. In phosphorylating Acanthamoeba castellanii mitochondria the sensitivity of uncoupling protein activity to GTP depends on the redox state of quinone. J Bioenerg Biomembr 37: 97–107, 2005. doi: 10.1007/s10863-005-4133-y. [DOI] [PubMed] [Google Scholar]
- 423. Jarmuszkiewicz W, Navet R, Alberici LC, Douette P, Sluse-Goffart CM, Sluse FE, Vercesi AE. Redox state of endogenous coenzyme q modulates the inhibition of linoleic acid-induced uncoupling by guanosine triphosphate in isolated skeletal muscle mitochondria. J Bioenerg Biomembr 36: 493–502, 2004. doi: 10.1023/B:JOBB.0000047331.25248.7a. [DOI] [PubMed] [Google Scholar]
- 424. Navet R, Douette P, Puttine-Marique F, Sluse-Goffart CM, Jarmuszkiewicz W, Sluse FE. Regulation of uncoupling protein activity in phosphorylating potato tuber mitochondria. FEBS Lett 579: 4437–4442, 2005. doi: 10.1016/j.febslet.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 425. Zackova M, Skobisová E, Urbánková E, Jezek P. Activating omega-6 polyunsaturated fatty acids and inhibitory purine nucleotides are high affinity ligands for novel mitochondrial uncoupling proteins UCP2 and UCP3. J Biol Chem 278: 20761–20769, 2003. doi: 10.1074/jbc.M212850200. [DOI] [PubMed] [Google Scholar]
- 426. Zhu R, Rupprecht A, Ebner A, Haselgrübler T, Gruber HJ, Hinterdorfer P, Pohl EE. Mapping the nucleotide binding site of uncoupling protein 1 using atomic force microscopy. J Am Chem Soc 135: 3640–3646, 2013. doi: 10.1021/ja312550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Echtay KS, Winkler E, Klingenberg M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 408: 609–613, 2000. doi: 10.1038/35046114. [DOI] [PubMed] [Google Scholar]
- 428. Echtay KS, Winkler E, Frischmuth K, Klingenberg M. Uncoupling proteins 2 and 3 are highly active H+ transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc Natl Acad Sci USA 98: 1416–1421, 2001. doi: 10.1073/pnas.98.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Jaburek M, Garlid KD. Reconstitution of recombinant uncoupling proteins: UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J Biol Chem 278: 25825–25831, 2003. doi: 10.1074/jbc.M302126200. [DOI] [PubMed] [Google Scholar]
- 430. Swida-Barteczka A, Woyda-Ploszczyca A, Sluse FE, Jarmuszkiewicz W. Uncoupling protein 1 inhibition by purine nucleotides is under the control of the endogenous ubiquinone redox state. Biochem J 424: 297–306, 2009. doi: 10.1042/BJ20091158. [DOI] [PubMed] [Google Scholar]
- 431. Swida A, Woyda-Ploszczyca A, Jarmuszkiewicz W. Redox state of quinone affects sensitivity of Acanthamoeba castellanii mitochondrial uncoupling protein to purine nucleotides. Biochem J 413: 359–367, 2008. doi: 10.1042/BJ20080333. [DOI] [PubMed] [Google Scholar]
- 432. Woyda-Ploszczyca A, Jarmuszkiewicz W. Ubiquinol (QH2) functions as a negative regulator of purine nucleotide inhibition of Acanthamoeba castellanii mitochondrial uncoupling protein. Biochim Biophys Acta 1807: 42–52, 2011. doi: 10.1016/j.bbabio.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 433. Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature 415: 96–99, 2002. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 434. Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otín M, Pamplona R, Vidal-Puig AJ, Wang S, Roebuck SJ, Brand MD. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J 22: 4103–4110, 2003. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Couplan E, del Mar Gonzalez-Barroso M, Alves-Guerra MC, Ricquier D, Goubern M, Bouillaud F. No evidence for a basal, retinoic, or superoxide-induced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J Biol Chem 277: 26268–26275, 2002. doi: 10.1074/jbc.M202535200. [DOI] [PubMed] [Google Scholar]
- 436. Shabalina IG, Petrovic N, Kramarova TV, Hoeks J, Cannon B, Nedergaard J. UCP1 and defense against oxidative stress. 4-Hydroxy-2-nonenal effects on brown fat mitochondria are uncoupling protein 1-independent. J Biol Chem 281: 13882–13893, 2006. doi: 10.1074/jbc.M601387200. [DOI] [PubMed] [Google Scholar]
- 437. Aguirre E, Cadenas S. GDP and carboxyatractylate inhibit 4-hydroxynonenal-activated proton conductance to differing degrees in mitochondria from skeletal muscle and heart. Biochim Biophys Acta 1797: 1716–1726, 2010. doi: 10.1016/j.bbabio.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 438. Larm JA, Vaillant F, Linnane AW, Lawen A. Up-regulation of the plasma membrane oxidoreductase as a prerequisite for the viability of human Namalwa rho 0 cells. J Biol Chem 269: 30097–30100, 1994. doi: 10.1016/S0021-9258(18)43779-9. [DOI] [PubMed] [Google Scholar]
- 439. Scarlett DJ, Herst P, Tan A, Prata C, Berridge M. Mitochondrial gene-knockout (rho0) cells: a versatile model for exploring the secrets of trans-plasma membrane electron transport. Biofactors 20: 199–206, 2004. doi: 10.1002/biof.5520200404. [DOI] [PubMed] [Google Scholar]
- 440. Martinus RD, Linnane AW, Nagley P. Growth of rho 0 human Namalwa cells lacking oxidative phosphorylation can be sustained by redox compounds potassium ferricyanide or coenzyme Q10 putatively acting through the plasma membrane oxidase. Biochem Mol Biol Int 31: 997–1005, 1993. [PubMed] [Google Scholar]
- 441. Hyun DH, Hunt ND, Emerson SS, Hernandez JO, Mattson MP, de Cabo R. Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J Neurochem 100: 1364–1374, 2007. doi: 10.1111/j.1471-4159.2006.04411.x. [DOI] [PubMed] [Google Scholar]
- 442. Lenaz G, Samorì B, Fato R, Battino M, Parenti Castelli G, Domini I. Localization and preferred orientations of ubiquinone homologs in model bilayers. Biochem Cell Biol 70: 504–514, 1992. doi: 10.1139/o92-078. [DOI] [PubMed] [Google Scholar]
- 443. Katsikas H, Quinn PJ. The polyisoprenoid chain length influences the interaction of ubiquinones with phospholipid bilayers. Biochim Biophys Acta 689: 363–369, 1982. doi: 10.1016/0005-2736(82)90270-x. [DOI] [PubMed] [Google Scholar]
- 444. Katsikas H, Quinn PJ. The distribution of ubiquinone-10 in phospholipid bilayers. A study using differential scanning calorimetry. Eur J Biochem 124: 165–169, 1982. doi: 10.1111/j.1432-1033.1982.tb05920.x. [DOI] [PubMed] [Google Scholar]
- 445. Katsikas H, Quinn PJ. Fluorescence probe studies of the distribution of ubiquinone homologues in bilayers of dipalmitoylglycerophosphocholine. Eur J Biochem 131: 607–612, 1983. doi: 10.1111/j.1432-1033.1983.tb07306.x. [DOI] [PubMed] [Google Scholar]
- 446. Ondarroa M, Quinn PJ. A difference infrared-spectroscopic study of the interaction of ubiquinone-10 with phospholipid bilayers. Biochem J 240: 325–331, 1986. doi: 10.1042/bj2400325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Samorì B, Lenaz G, Battino M, Marconi G, Domini I. On coenzyme Q orientation in membranes: a linear dichroism study of ubiquinones in a model bilayer. J Membr Biol 128: 193–203, 199 2. 1501247 [DOI] [PubMed] [Google Scholar]
- 448. Afri M, Ehrenberg B, Talmon Y, Schmidt J, Cohen Y, Frimer AA. Active oxygen chemistry within the liposomal bilayer. Part III: Locating vitamin E, ubiquinol and ubiquinone and their derivatives in the lipid bilayer. Chem Phys Lipids 131: 107–121, 2004. doi: 10.1016/j.chemphyslip.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 449. Gómez-Fernández JC, Llamas MA, Aranda FJ. The interaction of coenzyme Q with phosphatidylethanolamine membranes. Eur J Biochem 259: 739–746, 1999. doi: 10.1046/j.1432-1327.1999.00109.x. [DOI] [PubMed] [Google Scholar]
- 450. Castresana J, Alonso A, Arrondo JL, Goñi FM, Casal H. The physical state of ubiquinone-10, in pure form and incorporated into phospholipid bilayers. A Fourier-transform infrared spectroscopic study. Eur J Biochem 204: 1125–1130, 1992. doi: 10.1111/j.1432-1033.1992.tb16737.x. [DOI] [PubMed] [Google Scholar]
- 451. Agmo Hernández V, Eriksson EK, Edwards K. Ubiquinone-10 alters mechanical properties and increases stability of phospholipid membranes. Biochim Biophys Acta 1848: 2233–2243, 2015. doi: 10.1016/j.bbamem.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 452. Takahashi H, Aihara Y, Ogawa Y, Murata Y, Nakajima KI, Iida M, Shirai M, Fujisaki S. Suppression of phenotype of Escherichia coli mutant defective in farnesyl diphosphate synthase by overexpression of gene for octaprenyl diphosphate synthase. Biosci Biotechnol Biochem 82: 1003–1010, 2018. doi: 10.1080/09168451.2017.1398066. [DOI] [PubMed] [Google Scholar]
- 453. Asai K, Fujisaki S, Nishimura Y, Nishino T, Okada K, Nakagawa T, Kawamukai M, Matsuda H. The identification of Escherichia coli ispB (cel) gene encoding the octaprenyl diphosphate synthase. Biochem Biophys Res Commun 202: 340–345, 1994. doi: 10.1006/bbrc.1994.1933. [DOI] [PubMed] [Google Scholar]
- 454. Nicoll CR, Alvigini L, Gottinger A, Cecchini D, Mannucci B, Corana F, Mascotti ML, Mattevi A. In vitro construction of the COQ metabolon unveils the molecular determinants of coenzyme Q biosynthesis. Nat Catal 7: 148–160, 2024. doi: 10.1038/s41929-023-01087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455. Acosta MJ, Vazquez Fonseca L, Desbats MA, Cerqua C, Zordan R, Trevisson E, Salviati L. Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta 1857: 1079–1085, 2016. doi: 10.1016/j.bbabio.2016.03.036. [DOI] [PubMed] [Google Scholar]
- 456. Payet LA, Leroux M, Willison JC, Kihara A, Pelosi L, Pierrel F. Mechanistic details of early steps in coenzyme Q biosynthesis pathway in yeast. Cell Chem Biol 23: 1241–1250, 2016. doi: 10.1016/j.chembiol.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 457. Villalba JM, Navas P. Regulation of coenzyme Q biosynthesis pathway in eukaryotes. Free Radic Biol Med 165: 312–323, 2021. doi: 10.1016/j.freeradbiomed.2021.01.055. [DOI] [PubMed] [Google Scholar]
- 458. Fernández-Del-Río L, Clarke CF. Coenzyme Q biosynthesis: an update on the origins of the benzenoid ring and discovery of new ring precursors. Metabolites 11: 385, 2021. doi: 10.3390/metabo11060385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Kawamukai M. Biosynthesis and bioproduction of coenzyme Q10 by yeasts and other organisms. Biotechnol Appl Biochem 53: 217–226, 2009. doi: 10.1042/BA20090035. [DOI] [PubMed] [Google Scholar]
- 460. Awad AM, Bradley MC, Fernández-Del-Río L, Nag A, Tsui HS, Clarke CF. Coenzyme Q10 deficiencies: pathways in yeast and humans. Essays Biochem 62: 361–376, 2018. doi: 10.1042/EBC20170106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461. Meganathan R. Ubiquinone biosynthesis in microorganisms. FEMS Microbiol Lett 203: 131–139, 2001. doi: 10.1111/j.1574-6968.2001.tb10831.x. [DOI] [PubMed] [Google Scholar]
- 462. Abby SS, Kazemzadeh K, Vragniau C, Pelosi L, Pierrel F. Advances in bacterial pathways for the biosynthesis of ubiquinone. Biochim Biophys Acta Bioenerg 1861: 148259, 2020. doi: 10.1016/j.bbabio.2020.148259. [DOI] [PubMed] [Google Scholar]
- 463. Aussel L, Pierrel F, Loiseau L, Lombard M, Fontecave M, Barras F. Biosynthesis and physiology of coenzyme Q in bacteria. Biochim Biophys Acta 1837: 1004–1011, 2014. doi: 10.1016/j.bbabio.2014.01.015. [DOI] [PubMed] [Google Scholar]
- 464. Block A, Widhalm JR, Fatihi A, Cahoon RE, Wamboldt Y, Elowsky C, Mackenzie SA, Cahoon EB, Chapple C, Dudareva N, Basset GJ. The origin and biosynthesis of the benzenoid moiety of ubiquinone (coenzyme Q) in Arabidopsis. Plant Cell 26: 1938–1948, 2014. doi: 10.1105/tpc.114.125807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. González-Mariscal I, García-Testón E, Padilla S, Martin-Montalvo A, Pomares-Viciana T, Vazquez-Fonseca L, Gandolfo-Dominguez P, Santos-Ocana C. Regulation of coenzyme Q biosynthesis in yeast: a new complex in the block. IUBMB Life 66: 63–70, 2014. doi: 10.1002/iub.1243. [DOI] [PubMed] [Google Scholar]
- 466. Olson RE, Bentley R, Aiyar AS, Dialameh GH, Gold PH, Ramsey VG, Springer CM. Benzoate derivatives as intermediates in the biosynthesis of coenzyme Q in the rat. J Biol Chem 238: 3146–3148, 1963. [PubMed] [Google Scholar]
- 467. Fernández-Del-Río L, Nag A, Gutiérrez Casado E, Ariza J, Awad AM, Joseph AI, Kwon O, Verdin E, de Cabo R, Schneider C, Torres JZ, Buron MI, Clarke CF, Villalba JM. Kaempferol increases levels of coenzyme Q in kidney cells and serves as a biosynthetic ring precursor. Free Radic Biol Med 110: 176–187, 2017. doi: 10.1016/j.freeradbiomed.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Tai J, Guerra RM, Rogers SW, Fang Z, Muehlbauer LK, Shishkova E, Overmyer KA, Coon JJ, Pagliarini DJ. Hem25p is required for mitochondrial IPP transport in fungi. Nat Cell Biol 25: 1616–1624, 2023. doi: 10.1038/s41556-023-01250-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469. Liang D, Jiang X. START smuggling CoQ to fight ferroptosis. Nat Cell Biol 25: 207–208, 2023. doi: 10.1038/s41556-022-01044-1. [DOI] [PubMed] [Google Scholar]
- 470. Jin G, Kubo H, Kashiba M, Horinouchi R, Hasegawa M, Suzuki M, Sagawa T, Oizumi M, Fujisawa A, Tsukamoto H, Yoshimura S, Yamamoto Y. Saposin B is a human coenzyme q10-binding/transfer protein. J Clin Biochem Nutr 42: 167–174, 2008. doi: 10.3164/jcbn.2008024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Kashiba M, Oizumi M, Suzuki M, Sawamura Y, Nagashima K, Yoshimura S, Yamamoto Y. Prosaposin regulates coenzyme Q10 levels in HepG2 cells, especially those in mitochondria. J Clin Biochem Nutr 55: 85–89, 2014. doi: 10.3164/jcbn.13-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Kashiba M, Terashima M, Sagawa T, Yoshimura S, Yamamoto Y. Prosaposin knockdown in Caco-2 cells decreases cellular levels of coenzyme Q10 and ATP, and results in the loss of tight junction barriers. J Clin Biochem Nutr 60: 81–85, 2017. doi: 10.3164/jcbn.16-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. Takeuchi H, Sugawara K, Okamoto M, Nakamura A, Tanaka T, Fujita Y, Ishiguro K, Yamazaki H, Okada M, Mikami A, Fujisawa A, Yamamoto Y, Kashiba M. Reduced prosaposin levels in HepG2 cells with long-term coenzyme Q10 deficiency. J Clin Biochem Nutr 71: 97–102, 2022. doi: 10.3164/jcbn.21-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Eisenberg-Bord M, Tsui HS, Antunes D, Fernández-Del-Río L, Bradley MC, Dunn CD, Nguyen TPT, Rapaport D, Clarke CF, Schuldiner M. The endoplasmic reticulum-mitochondria encounter structure complex coordinates coenzyme Q biosynthesis. Contact (Thousand Oaks) 2: 2515256418825409, 2019. doi: 10.1177/2515256418825409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475. Robinson KP, Jochem A, Johnson SE, Reddy TR, Russell JD, Coon JJ, Pagliarini DJ. Defining intermediates and redundancies in coenzyme Q precursor biosynthesis. J Biol Chem 296: 100643, 2021. doi: 10.1016/j.jbc.2021.100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476. Stefely JA, Kwiecien NW, Freiberger EC, Richards AL, Jochem A, Rush MJ, Ulbrich A, Robinson KP, Hutchins PD, Veling MT, Guo X, Kemmerer ZA, Connors KJ, Trujillo EA, Sokol J, Marx H, Westphall MS, Hebert AS, Pagliarini DJ, Coon JJ. Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat Biotechnol 34: 1191–1197, 2016. doi: 10.1038/nbt.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Zahedi RP, Sickmann A, Boehm AM, Winkler C, Zufall N, Schönfisch B, Guiard B, Pfanner N, Meisinger C. Proteomic analysis of the yeast mitochondrial outer membrane reveals accumulation of a subclass of preproteins. Mol Biol Cell 17: 1436–1450, 2006. doi: 10.1091/mbc.e05-08-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478. Nees DW, Wawrousek EF, Robison WG Jr, Piatigorsky J. Structurally normal corneas in aldehyde dehydrogenase 3a1-deficient mice. Mol Cell Biol 22: 849–855, 2002. doi: 10.1128/MCB.22.3.849-855.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Pierrel F. Impact of chemical analogs of 4-hydroxybenzoic acid on coenzyme Q biosynthesis: from inhibition to bypass of coenzyme Q deficiency. Front Physiol 8: 436, 2017. doi: 10.3389/fphys.2017.00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480. Winter G, Averesch NJ, Nunez-Bernal D, Krömer JO. In vivo instability of chorismate causes substrate loss during fermentative production of aromatics. Yeast 31: 333–341, 2014. doi: 10.1002/yea.3025. [DOI] [PubMed] [Google Scholar]
- 481. Averesch NJ, Winter G, Krömer JO. Production of para-aminobenzoic acid from different carbon-sources in engineered Saccharomyces cerevisiae. Microb Cell Fact 15: 89, 2016. doi: 10.1186/s12934-016-0485-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Marbois B, Xie LX, Choi S, Hirano K, Hyman K, Clarke CF. para-Aminobenzoic acid is a precursor in coenzyme Q6 biosynthesis in Saccharomyces cerevisiae. J Biol Chem 285: 27827–27838, 2010. doi: 10.1074/jbc.M110.151894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483. Pierrel F, Hamelin O, Douki T, Kieffer-Jaquinod S, Mühlenhoff U, Ozeir M, Lill R, Fontecave M. Involvement of mitochondrial ferredoxin and para-aminobenzoic acid in yeast coenzyme Q biosynthesis. Chem Biol 17: 449–459, 2010. doi: 10.1016/j.chembiol.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 484. Xie LX, Ozeir M, Tang JY, Chen JY, Jaquinod SK, Fontecave M, Clarke CF, Pierrel F. Overexpression of the Coq8 kinase in Saccharomyces cerevisiae coq null mutants allows for accumulation of diagnostic intermediates of the coenzyme Q6 biosynthetic pathway. J Biol Chem 287: 23571–23581, 2012. doi: 10.1074/jbc.M112.360354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485. He CH, Black DS, Nguyen TP, Wang C, Srinivasan C, Clarke CF. Yeast Coq9 controls deamination of coenzyme Q intermediates that derive from para-aminobenzoic acid. Biochim Biophys Acta 1851: 1227–1239, 2015. doi: 10.1016/j.bbalip.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486. Ozeir M, Pelosi L, Ismail A, Mellot-Draznieks C, Fontecave M, Pierrel F. Coq6 is responsible for the C4-deamination reaction in coenzyme Q biosynthesis in Saccharomyces cerevisiae. J Biol Chem 290: 24140–24151, 2015. doi: 10.1074/jbc.M115.675744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Xie LX, Williams KJ, He CH, Weng E, Khong S, Rose TE, Kwon O, Bensinger SJ, Marbois BN, Clarke CF. Resveratrol and para-coumarate serve as ring precursors for coenzyme Q biosynthesis. J Lipid Res 56: 909–919, 2015. doi: 10.1194/jlr.M057919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Pelosi L, Morbiato L, Burgardt A, Tonello F, Bartlett AK, Guerra RM, Ferizhendi KK, Desbats MA, Rascalou B, Marchi M, Vázquez-Fonseca L, Agosto C, Zanotti G, Roger-Margueritat M, Alcázar-Fabra M, García-Corzo L, Sanchez-Cuesta A, Navas P, Brea-Calvo G, Trevisson E, Wendisch VF, Pagliarini DJ, Salviati L, Pierrel F. COQ4 is required for the oxidative decarboxylation of the C1 carbon of coenzyme Q in eukaryotic cells. Mol Cell 84: 981–989.e7, 2024. doi: 10.1016/j.molcel.2024.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Ozeir M, Mühlenhoff U, Webert H, Lill R, Fontecave M, Pierrel F. Coenzyme Q biosynthesis: Coq6 is required for the C5-hydroxylation reaction and substrate analogs rescue Coq6 deficiency. Chem Biol 18: 1134–1142, 2011. doi: 10.1016/j.chembiol.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 490. Tran UC, Marbois B, Gin P, Gulmezian M, Jonassen T, Clarke CF. Complementation of Saccharomyces cerevisiae coq7 mutants by mitochondrial targeting of the Escherichia coli UbiF polypeptide: two functions of yeast Coq7 polypeptide in coenzyme Q biosynthesis. J Biol Chem 281: 16401–16409, 2006. doi: 10.1074/jbc.M513267200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Poon WW, Barkovich RJ, Hsu AY, Frankel A, Lee PT, Shepherd JN, Myles DC, Clarke CF. Yeast and rat Coq3 and Escherichia coli UbiG polypeptides catalyze both O-methyltransferase steps in coenzyme Q biosynthesis. J Biol Chem 274: 21665–21672, 1999. doi: 10.1074/jbc.274.31.21665. [DOI] [PubMed] [Google Scholar]
- 492. Allan CM, Awad AM, Johnson JS, Shirasaki DI, Wang C, Blaby-Haas CE, Merchant SS, Loo JA, Clarke CF. Identification of Coq11, a new coenzyme Q biosynthetic protein in the CoQ-synthome in Saccharomyces cerevisiae. J Biol Chem 290: 7517–7534, 2015. doi: 10.1074/jbc.M114.633131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Johnson A, Gin P, Marbois BN, Hsieh EJ, Wu M, Barros MH, Clarke CF, Tzagoloff A. COQ9, a new gene required for the biosynthesis of coenzyme Q in Saccharomyces cerevisiae. J Biol Chem 280: 31397–31404, 2005. doi: 10.1074/jbc.M503277200. [DOI] [PubMed] [Google Scholar]
- 494. Do TQ, Hsu AY, Jonassen T, Lee PT, Clarke CF. A defect in coenzyme Q biosynthesis is responsible for the respiratory deficiency in Saccharomyces cerevisiae abc1 mutants. J Biol Chem 276: 18161–18168, 2001. doi: 10.1074/jbc.M100952200. [DOI] [PubMed] [Google Scholar]
- 495. Dai YN, Zhou K, Cao DD, Jiang YL, Meng F, Chi CB, Ren YM, Chen Y, Zhou CZ. Crystal structures and catalytic mechanism of the C-methyltransferase Coq5 provide insights into a key step of the yeast coenzyme Q synthesis pathway. Acta Crystallogr D Biol Crystallogr 70: 2085–2092, 2014. doi: 10.1107/S1399004714011559. [DOI] [PubMed] [Google Scholar]
- 496. Tzagoloff A, Akai A, Needleman RB. Assembly of the mitochondrial membrane system. Characterization of nuclear mutants of Saccharomyces cerevisiae with defects in mitochondrial ATPase and respiratory enzymes. J Biol Chem 250: 8228–8235, 1975. doi: 10.1016/S0021-9258(19)40840-5. [DOI] [PubMed] [Google Scholar]
- 497. Tzagoloff A, Dieckmann CL. PET genes of Saccharomyces cerevisiae. Microbiol Rev 54: 211–225, 1990. doi: 10.1128/mr.54.3.211-225.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Hsieh EJ, Gin P, Gulmezian M, Tran UC, Saiki R, Marbois BN, Clarke CF. Saccharomyces cerevisiae Coq9 polypeptide is a subunit of the mitochondrial coenzyme Q biosynthetic complex. Arch Biochem Biophys 463: 19–26, 2007. doi: 10.1016/j.abb.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499. Marbois B, Gin P, Faull KF, Poon WW, Lee PT, Strahan J, Shepherd JN, Clarke CF. Coq3 and Coq4 define a polypeptide complex in yeast mitochondria for the biosynthesis of coenzyme Q. J Biol Chem 280: 20231–20238, 2005. doi: 10.1074/jbc.M501315200. [DOI] [PubMed] [Google Scholar]
- 500. Gin P, Clarke CF. Genetic evidence for a multi-subunit complex in coenzyme Q biosynthesis in yeast and the role of the Coq1 hexaprenyl diphosphate synthase. J Biol Chem 280: 2676–2681, 2005. doi: 10.1074/jbc.M411527200. [DOI] [PubMed] [Google Scholar]
- 501. He CH, Xie LX, Allan CM, Tran UC, Clarke CF. Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochim Biophys Acta 1841: 630–644, 2014. doi: 10.1016/j.bbalip.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502. Subramanian K, Jochem A, Le Vasseur M, Lewis S, Paulson BR, Reddy TR, Russell JD, Coon JJ, Pagliarini DJ, Nunnari J. Coenzyme Q biosynthetic proteins assemble in a substrate-dependent manner into domains at ER-mitochondria contacts. J Cell Biol 218: 1353–1369, 2019. doi: 10.1083/jcb.201808044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Tauche A, Krause-Buchholz U, Rödel G. Ubiquinone biosynthesis in Saccharomyces cerevisiae: the molecular organization of O-methylase Coq3p depends on Abc1p/Coq8p. FEMS Yeast Res 8: 1263–1275, 2008. doi: 10.1111/j.1567-1364.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- 504. Baba SW, Belogrudov GI, Lee JC, Lee PT, Strahan J, Shepherd JN, Clarke CF. Yeast Coq5 C-methyltransferase is required for stability of other polypeptides involved in coenzyme Q biosynthesis. J Biol Chem 279: 10052–10059, 2004. doi: 10.1074/jbc.M313712200. [DOI] [PubMed] [Google Scholar]
- 505. Padilla S, Tran UC, Jiménez-Hidalgo M, López-Martín JM, Martín-Montalvo A, Clarke CF, Navas P, Santos-Ocana C. Hydroxylation of demethoxy-Q6 constitutes a control point in yeast coenzyme Q6 biosynthesis. Cell Mol Life Sci 66: 173–186, 2009. doi: 10.1007/s00018-008-8547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Marbois B, Gin P, Gulmezian M, Clarke CF. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim Biophys Acta 1791: 69–75, 2009. doi: 10.1016/j.bbalip.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Stefely JA, Licitra F, Laredj L, Reidenbach AG, Kemmerer ZA, Grangeray A, , et al. Cerebellar ataxia and coenzyme q deficiency through loss of unorthodox kinase activity. Mol Cell 63: 608–620, 2016. doi: 10.1016/j.molcel.2016.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Reidenbach AG, Kemmerer ZA, Aydin D, Jochem A, McDevitt MT, Hutchins PD, Stark JL, Stefely JA, Reddy T, Hebert AS, Wilkerson EM, Johnson IE, Bingman CA, Markley JL, Coon JJ, Dal Peraro M, Pagliarini DJ. Conserved lipid and small-molecule modulation of COQ8 reveals regulation of the Ancient Kinase-like UbiB family. Cell Chem Biol 25: 154–165.e11, 2018. doi: 10.1016/j.chembiol.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509. Lohman DC, Forouhar F, Beebe ET, Stefely MS, Minogue CE, Ulbrich A, Stefely JA, Sukumar S, Luna-Sánchez M, Jochem A, Lew S, Seetharaman J, Xiao R, Wang H, Westphall MS, Wrobel RL, Everett JK, Mitchell JC, López LC, Coon JJ, Tong L, Pagliarini DJ. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc Natl Acad Sci USA 111: E4697–E4705, 2014. doi: 10.1073/pnas.1413128111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510. Bradley MC, Yang K, Fernández-Del-Rio L, Ngo J, Ayer A, Tsui HS, Novales NA, Stocker R, Shirihai OS, Barros MH, Clarke CF. COQ11 deletion mitigates respiratory deficiency caused by mutations in the gene encoding the coenzyme Q chaperone protein Coq10. J Biol Chem 295: 6023–6042, 2020. doi: 10.1074/jbc.RA119.012420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. Kehrein K, Schilling R, Möller-Hergt BV, Wurm CA, Jakobs S, Lamkemeyer T, Langer T, Ott M. Organization of mitochondrial gene expression in two distinct ribosome-containing assemblies. Cell Rep 10: 843–853, 2015. doi: 10.1016/j.celrep.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 512. Barros MH, Johnson A, Gin P, Marbois BN, Clarke CF, Tzagoloff A. The Saccharomyces cerevisiae COQ10 gene encodes a START domain protein required for function of coenzyme Q in respiration. J Biol Chem 280: 42627–42635, 2005. doi: 10.1074/jbc.M510768200. [DOI] [PubMed] [Google Scholar]
- 513. Busso C, Bleicher L, Ferreira-Júnior JR, Barros MH. Site-directed mutagenesis and structural modeling of Coq10p indicate the presence of a tunnel for coenzyme Q6 binding. FEBS Lett 584: 1609–1614, 2010. doi: 10.1016/j.febslet.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 514. Vajo Z, King LM, Jonassen T, Wilkin DJ, Ho N, Munnich A, Clarke CF, Francomano CA. Conservation of the Caenorhabditis elegans timing gene clk-1 from yeast to human: a gene required for ubiquinone biosynthesis with potential implications for aging. Mamm Genome 10: 1000–1004, 1999. doi: 10.1007/s003359901147. [DOI] [PubMed] [Google Scholar]
- 515. Jonassen T, Clarke CF. Isolation and functional expression of human COQ3, a gene encoding a methyltransferase required for ubiquinone biosynthesis. J Biol Chem 275: 12381–12387, 2000. doi: 10.1074/jbc.275.17.12381. [DOI] [PubMed] [Google Scholar]
- 516. Mollet J, Giurgea I, Schlemmer D, Dallner G, Chretien D, Delahodde A, Bacq D, de Lonlay P, Munnich A, Rötig A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest 117: 765–772, 2007. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517. Casarin A, Jimenez-Ortega JC, Trevisson E, Pertegato V, Doimo M, Ferrero-Gomez ML, Abbadi S, Artuch R, Quinzii C, Hirano M, Basso G, Ocaña CS, Navas P, Salviati L. Functional characterization of human COQ4, a gene required for Coenzyme Q10 biosynthesis. Biochem Biophys Res Commun 372: 35–39, 2008. doi: 10.1016/j.bbrc.2008.04.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518. Duncan AJ, Bitner-Glindzicz M, Meunier B, Costello H, Hargreaves IP, López LC, Hirano M, Quinzii CM, Sadowski MI, Hardy J, Singleton A, Clayton PT, Rahman S. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet 84: 558–566, 2009. doi: 10.1016/j.ajhg.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519. Xie LX, Hsieh EJ, Watanabe S, Allan CM, Chen JY, Tran UC, Clarke CF. Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants. Biochim Biophys Acta 1811: 348–360, 2011. doi: 10.1016/j.bbalip.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520. Nguyen TP, Casarin A, Desbats MA, Doimo M, Trevisson E, Santos-Ocaña C, Navas P, Clarke CF, Salviati L. Molecular characterization of the human COQ5 C-methyltransferase in coenzyme Q10 biosynthesis. Biochim Biophys Acta 1841: 1628–1638, 2014. doi: 10.1016/j.bbalip.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521. Saiki R, Nagata A, Kainou T, Matsuda H, Kawamukai M. Characterization of solanesyl and decaprenyl diphosphate synthases in mice and humans. FEBS J 272: 5606–5622, 2005. doi: 10.1111/j.1742-4658.2005.04956.x. [DOI] [PubMed] [Google Scholar]
- 522. Widmeier E, Yu S, Nag A, Chung YW, Nakayama M, Fernández-Del-Río L, Hugo H, Schapiro D, Buerger F, Choi WI, Helmstadter M, Kim JW, Ryu JH, Lee MG, Clarke CF, Hildebrandt F, Gee HY. ADCK4 deficiency destabilizes the coenzyme q complex, which is rescued by 2,4-dihydroxybenzoic acid treatment. J Am Soc Nephrol 31: 1191–1211, 2020. doi: 10.1681/ASN.2019070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523. Hughes BG, Hekimi S. A mild impairment of mitochondrial electron transport has sex-specific effects on lifespan and aging in mice. PLoS One 6: e26116, 2011. doi: 10.1371/journal.pone.0026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X, Williams RS. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell 101: 389–399, 2000. doi: 10.1016/s0092-8674(00)80849-1. [DOI] [PubMed] [Google Scholar]
- 525. Torraco A, Diaz F, Vempati UD, Moraes CT. Mouse models of oxidative phosphorylation defects: powerful tools to study the pathobiology of mitochondrial diseases. Biochim Biophys Acta 1793: 171–180, 2009. doi: 10.1016/j.bbamcr.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526. Madaio MP, Ahima RS, Meade R, Rader DJ, Mendoza A, Peng M, Tomaszewski JE, Hancock WW, Gasser DL. Glomerular and tubular epithelial defects in kd/kd mice lead to progressive renal failure. Am J Nephrol 25: 604–610, 2005. doi: 10.1159/000089709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527. González-García P, Barriocanal-Casado E, Díaz-Casado ME, López-Herrador S, Hidalgo-Gutiérrez A, Lopez LC. Animal models of coenzyme Q deficiency: mechanistic and translational learnings. Antioxidants (Basel) 10: 1687, 2021. doi: 10.3390/antiox10111687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528. Lu S, Lu LY, Liu MF, Yuan QJ, Sham MH, Guan XY, Huang JD. Cerebellar defects in Pdss2 conditional knockout mice during embryonic development and in adulthood. Neurobiol Dis 45: 219–233, 2012. doi: 10.1016/j.nbd.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 529. Ziegler CG, Peng M, Falk MJ, Polyak E, Tsika E, Ischiropoulos H, Bakalar D, Blendy JA, Gasser DL. Parkinson’s disease-like neuromuscular defects occur in prenyl diphosphate synthase subunit 2 (Pdss2) mutant mice. Mitochondrion 12: 248–257, 2012. doi: 10.1016/j.mito.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530. Widmeier E, Airik M, Hugo H, Schapiro D, Wedel J, Ghosh CC, Nakayama M, Schneider R, Awad AM, Nag A, Cho J, Schueler M, Clarke CF, Airik R, Hildebrandt F. Treatment with 2,4-dihydroxybenzoic acid prevents FSGS progression and renal fibrosis in podocyte-specific Coq6 knockout mice. J Am Soc Nephrol 30: 393–405, 2019. doi: 10.1681/ASN.2018060625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Wang D, Malo D, Hekimi S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/- mouse mutants. J Immunol 184: 582–590, 2010. doi: 10.4049/jimmunol.0902352. [DOI] [PubMed] [Google Scholar]
- 532. Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev 19: 2424–2434, 2005. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533. Manolaras I, Del Bondio A, Griso O, Reutenauer L, Eisenmann A, Habermann BH, Puccio H. Mitochondrial dysfunction and calcium dysregulation in COQ8A-ataxia Purkinje neurons are rescued by CoQ10 treatment. Brain 146: 3836–3850, 2023. doi: 10.1093/brain/awad099. [DOI] [PubMed] [Google Scholar]
- 534. Korkmaz E, Lipska-Zietkiewicz BS, Boyer O, Gribouval O, Fourrage C, Tabatabaei M, Schnaidt S, Gucer S, Kaymaz F, Arici M, Dinckan A, Mir S, Bayazit AK, Emre S, Balat A, Rees L, Shroff R, Bergmann C, Mourani C, Antignac C, Ozaltin F, Schaefer F; PodoNet Consortium. ADCK4-associated glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol 27: 63–68, 2016. doi: 10.1681/ASN.2014121240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535. Luna-Sánchez M, Díaz-Casado E, Barca E, Tejada MA, Montilla-Garcia A, Cobos EJ, Escames G, Acuna-Castroviejo D, Quinzii CM, Lopez LC. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol Med 7: 670–687, 2015. doi: 10.15252/emmm.201404632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Asencio C, Navas P, Cabello J, Schnabel R, Cypser JR, Johnson TE, Rodríguez-Aguilera JC. Coenzyme Q supports distinct developmental processes in Caenorhabditis elegans. Mech Ageing Dev 130: 145–153, 2009. doi: 10.1016/j.mad.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 537. Gavilán A, Asencio C, Cabello J, Rodríguez-Aguilera JC, Schnabel R, Navas P. C-elegans knockouts in ubiquinone biosynthesis genes result in different phenotypes during larval development. Biofactors 25: 21–29, 2005. doi: 10.1002/biof.5520250104. [DOI] [PubMed] [Google Scholar]
- 538. Gomez F, Saiki R, Chin R, Srinivasan C, Clarke CF. Restoring de novo coenzyme Q biosynthesis in Caenorhabditis elegans coq-3 mutants yields profound rescue compared to exogenous coenzyme Q supplementation. Gene 506: 106–116, 2012. doi: 10.1016/j.gene.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539. Burgess J, Hihi AK, Benard CY, Branicky R, Hekimi S. Molecular mechanism of maternal rescue in the clk-1 mutants of Caenorhabditis elegans. J Biol Chem 278: 49555–49562, 2003. doi: 10.1074/jbc.M308507200. [DOI] [PubMed] [Google Scholar]
- 540. Ewbank JJ, Barnes TM, Lakowski B, Lussier M, Bussey H, Hekimi S. Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science 275: 980–983, 1997. doi: 10.1126/science.275.5302.980. [DOI] [PubMed] [Google Scholar]
- 541. Hekimi S, Boutis P, Lakowski B. Viable maternal-effect mutations that affect the development of the nematode Caenorhabditis elegans. Genetics 141: 1351–1364, 1995. doi: 10.1093/genetics/141.4.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542. Wang Y, Branicky R, Stepanyan Z, Carroll M, Guimond MP, Hihi A, Hayes S, McBride K, Hekimi S. The anti-neurodegeneration drug clioquinol inhibits the aging-associated protein CLK-1. J Biol Chem 284: 314–323, 2009. doi: 10.1074/jbc.M807579200. [DOI] [PubMed] [Google Scholar]
- 543. Wong A, Boutis P, Hekimi S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 139: 1247–1259, 1995. doi: 10.1093/genetics/139.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544. Felkai S, Ewbank JJ, Lemieux J, Labbé JC, Brown GG, Hekimi S. CLK-1 controls respiration, behavior and aging in the nematode Caenorhabditis elegans. EMBO J 18: 1783–1792, 1999. doi: 10.1093/emboj/18.7.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Branicky R, Shibata Y, Feng J, Hekimi S. Phenotypic and suppressor analysis of defecation in clk-1 mutants reveals that reaction to changes in temperature is an active process in Caenorhabditis elegans. Genetics 159: 997–1006, 2001. doi: 10.1093/genetics/159.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546. Greer EL, Brunet A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C-elegans. Aging Cell 8: 113–127, 2009. doi: 10.1111/j.1474-9726.2009.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Feng J, Bussière F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell 1: 633–644, 2001. doi: 10.1016/s1534-5807(01)00071-5. [DOI] [PubMed] [Google Scholar]
- 548. Yang W, Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 9: 433–447, 2010. doi: 10.1111/j.1474-9726.2010.00571.x. [DOI] [PubMed] [Google Scholar]
- 549. Monaghan RM, Barnes RG, Fisher K, Andreou T, Rooney N, Poulin GB, Whitmarsh AJ. A nuclear role for the respiratory enzyme CLK-1 in regulating mitochondrial stress responses and longevity. Nat Cell Biol 17: 782–792, 2015. doi: 10.1038/ncb3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550. Liu JL, Yee C, Wang Y, Hekimi S. A single biochemical activity underlies the pleiotropy of the aging-related protein CLK-1. Sci Rep 7: 859, 2017. doi: 10.1038/s41598-017-00754-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551. Diessl J, Berndtsson J, Broeskamp F, Habernig L, Kohler V, Vazquez-Calvo C, Nandy A, Peselj C, Drobysheva S, Pelosi L, Vögtle FN, Pierrel F, Ott M, Büttner S. Manganese-driven CoQ deficiency. Nat Commun 13: 6061, 2022. doi: 10.1038/s41467-022-33641-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552. Wang Y, Hekimi S. The CoQ biosynthetic di-iron carboxylate hydroxylase COQ7 is inhibited by in vivo metalation with manganese but remains functional by metalation with cobalt. MicroPubl Biol 2022: 10.17912/micropub.biology.000635, 2022. doi: 10.17912/micropub.biology.000635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553. Van Raamsdonk JM, Meng Y, Camp D, Yang W, Jia X, Bénard C, Hekimi S. Decreased energy metabolism extends life span in Caenorhabditis elegans without reducing oxidative damage. Genetics 185: 559–571, 2010. doi: 10.1534/genetics.110.115378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554. Padilla S, Jonassen T, Jiménez-Hidalgo MA, Fernández-Ayala DJ, López-Lluch G, Marbois B, Navas P, Clarke CF, Santos-Ocana C. Demethoxy-Q, an intermediate of coenzyme Q biosynthesis, fails to support respiration in Saccharomyces cerevisiae and lacks antioxidant activity. J Biol Chem 279: 25995–26004, 2004. doi: 10.1074/jbc.M400001200. [DOI] [PubMed] [Google Scholar]
- 555. Arroyo A, Santos-Ocaña C, Ruiz-Ferrer M, Padilla S, Gavilán A, Rodríguez-Aguilera JC, Navas P. Coenzyme Q is irreplaceable by demethoxy-coenzyme Q in plasma membrane of Caenorhabditis elegans. FEBS Lett 580: 1740–1746, 2006. doi: 10.1016/j.febslet.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 556. Doimo M, Trevisson E, Airik R, Bergdoll M, Santos-Ocaña C, Hildebrandt F, Navas P, Pierrel F, Salviati L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim Biophys Acta 1842: 1–6, 2014. doi: 10.1016/j.bbadis.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557. Herebian D, López LC, Distelmaier F. Bypassing human CoQ10 deficiency. Mol Genet Metab 123: 289–291, 2018. doi: 10.1016/j.ymgme.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 558. Acosta Lopez MJ, Trevisson E, Canton M, Vazquez-Fonseca L, Morbidoni V, Baschiera E, Frasson C, Pelosi L, Rascalou B, Desbats MA, Alcázar-Fabra M, Ríos JJ, Sánchez-García A, Basso G, Navas P, Pierrel F, Brea-Calvo G, Salviati L. Vanillic acid restores coenzyme Q biosynthesis and ATP production in human cells lacking COQ6. Oxid Med Cell Longev 2019: 3904905, 2019. doi: 10.1155/2019/3904905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559. Hidalgo-Gutiérrez A, Barriocanal-Casado E, Díaz-Casado ME, González-García P, Zenezini Chiozzi R, Acuña-Castroviejo D, López LC. beta-RA targets mitochondrial metabolism and adipogenesis, leading to therapeutic benefits against CoQ deficiency and age-related overweight. Biomedicines 9: 1457, 2021. doi: 10.3390/biomedicines9101457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci USA 86: 2379–2382, 1989. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Hughes BG, Harrison PM, Hekimi S. Estimating the occurrence of primary ubiquinone deficiency by analysis of large-scale sequencing data. Sci Rep 7: 17744, 2017. doi: 10.1038/s41598-017-17564-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562. Berardo A, Quinzii CM. Redefining infantile-onset multisystem phenotypes of coenzyme Q10-deficiency in the next-generation sequencing era. J Transl Genet Genom 4: 22–35, 2020. doi: 10.20517/jtgg.2020.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563. López LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet 79: 1125–1129, 2006. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Jakobs BS, van den Heuvel LP, Smeets RJ, de Vries MC, Hien S, Schaible T, Smeitink JA, Wevers RA, Wortmann SB, Rodenburg RJ. A novel mutation in COQ2 leading to fatal infantile multisystem disease. J Neurol Sci 326: 24–28, 2013. doi: 10.1016/j.jns.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 565. Desbats MA, Vetro A, Limongelli I, Lunardi G, Casarin A, Doimo M, Spinazzi M, Angelini C, Cenacchi G, Burlina A, Rodriguez Hernandez MA, Chiandetti L, Clementi M, Trevisson E, Navas P, Zuffardi O, Salviati L. Primary coenzyme Q10 deficiency presenting as fatal neonatal multiorgan failure. Eur J Hum Genet 23: 1254–1258, 2015. doi: 10.1038/ejhg.2014.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566. Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, , et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest 123: 5179–5189, 2013. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Yang J, Yang Y, Hu Z. A novel ADCK4 mutation in a Chinese family with ADCK4-associated glomerulopathy. Biochem Biophys Res Commun 506: 444–449, 2018. doi: 10.1016/j.bbrc.2018.10.102. [DOI] [PubMed] [Google Scholar]
- 568. Rötig A, Appelkvist EL, Geromel V, Chretien D, Kadhom N, Edery P, Lebideau M, Dallner G, Munnich A, Ernster L, Rustin P. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 356: 391–395, 2000. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- 569. Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA, Hildebrandt F; SRNS Study Group. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 26: 1279–1289, 2015. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570. Iványi B, Rácz GZ, Gal P, Brinyiczki K, Bodi I, Kalmar T, Maroti Z, Bereczki C. Diffuse mesangial sclerosis in a PDSS2 mutation-induced coenzyme Q10 deficiency. Pediatr Nephrol 33: 439–446, 2018. doi: 10.1007/s00467-017-3814-1. [DOI] [PubMed] [Google Scholar]
- 571. Salviati L, Sacconi S, Murer L, Zacchello G, Franceschini L, Laverda AM, Basso G, Quinzii C, Angelini C, Hirano M, Naini AB, Navas P, DiMauro S, Montini G. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology 65: 606–608, 2005. doi: 10.1212/01.wnl.0000172859.55579.a7. [DOI] [PubMed] [Google Scholar]
- 572. Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, Ghiggeri GM, Murer L, Barisoni L, Pastore A, Muda AO, Valente ML, Bertini E, Emma F. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol 18: 2773–2780, 2007. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 573. Dinwiddie DL, Smith LD, Miller NA, Atherton AM, Farrow EG, Strenk ME, Soden SE, Saunders CJ, Kingsmore SF. Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics 102: 148–156, 2013. doi: 10.1016/j.ygeno.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574. McCarthy HJ, Bierzynska A, Wherlock M, Ognjanovic M, Kerecuk L, Hegde S, Feather S, Gilbert RD, Krischock L, Jones C, Sinha MD, Webb NJ, Christian M, Williams MM, Marks S, Koziell A, Welsh GI, Saleem MA; RADAR the UK SRNS Study Group. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 8: 637–648, 2013. doi: 10.2215/CJN.07200712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med 369: 233–244, 2013. doi: 10.1056/NEJMoa1212115. [DOI] [PubMed] [Google Scholar]
- 576. Scalais E, Chafai R, Van Coster R, Bindl L, Nuttin C, Panagiotaraki C, Seneca S, Lissens W, Ribes A, Geers C, Smet J, De Meirleir L. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur J Paediatr Neurol 17: 625–630, 2013. doi: 10.1016/j.ejpn.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 577. Gigante M, Diella S, Santangelo L, Trevisson E, Acosta MJ, Amatruda M, Finzi G, Caridi G, Murer L, Accetturo M, Ranieri E, Ghiggeri GM, Giordano M, Grandaliano G, Salviati L, Gesualdo L. Further phenotypic heterogeneity of CoQ10 deficiency associated with steroid resistant nephrotic syndrome and novel COQ2 and COQ6 variants. Clin Genet 92: 224–226, 2017. doi: 10.1111/cge.12960. [DOI] [PubMed] [Google Scholar]
- 578. Eroglu FK, Ozaltin F, Gönç N, Nalçacıoğlu H, Özçakar ZB, Yalnızoğlu D, Güçer Ş, Orhan D, Eminoğlu FT, Göçmen R, Alikaşifoğlu A, Topaloğlu R, Düzova A. Response to early coenzyme Q10 supplementation is not sustained in CoQ10 deficiency caused by CoQ2 mutation. Pediatr Neurol 88: 71–74, 2018. doi: 10.1016/j.pediatrneurol.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 579. Starr MC, Chang IJ, Finn LS, Sun A, Larson AA, Goebel J, Hanevold C, Thies J, Van Hove JL, Hingorani SR, Lam C. COQ2 nephropathy: a treatable cause of nephrotic syndrome in children. Pediatr Nephrol 33: 1257–1261, 2018. doi: 10.1007/s00467-018-3937-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580. Xu K, Mao XY, Yao Y, Cheng H, Zhang XJ. [Clinical analysis of one infantile nephrotic syndrome caused by COQ2 gene mutation and literature review]. Zhonghua Er Ke Za Zhi 56: 662–666, 2018. doi: 10.3760/cma.j.issn.0578-1310.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 581. Wu X, Wang W, Liu Y, Chen W, Zhao L. A steroid-resistant nephrotic syndrome in an infant resulting from a consanguineous marriage with COQ2 and ARSB gene mutations: a case report. BMC Med Genet 20: 165, 2019. doi: 10.1186/s12881-019-0898-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582. Park E, Ahn YH, Kang HG, Yoo KH, Won NH, Lee KB, Moon KC, Seong MW, Gwon TR, Park SS, Cheong HI. COQ6 mutations in children with steroid-resistant focal segmental glomerulosclerosis and sensorineural hearing loss. Am J Kidney Dis 70: 139–144, 2017. doi: 10.1053/j.ajkd.2016.10.040. [DOI] [PubMed] [Google Scholar]
- 583. Cao Q, Li GM, Xu H, Shen Q, Sun L, Fang XY, Liu HM, Guo W, Zhai YH, Wu BB. [Coenzyme Q10 treatment for one child with COQ6 gene mutation induced nephrotic syndrome and literature review]. Zhonghua Er Ke Za Zhi 55: 135–138, 2017. doi: 10.3760/cma.j.issn.0578-1310.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 584. Song CC, Hong Q, Geng XD, Wang X, Wang SQ, Cui SY, Guo MD, Li O, Cai GY, Chen XM, Wu D. New mutation of coenzyme Q10 monooxygenase 6 causing podocyte injury in a focal segmental glomerulosclerosis patient. Chin Med J (Engl) 131: 2666–2675, 2018. doi: 10.4103/0366-6999.245158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585. Stańczyk M, Bałasz-Chmielewska I, Lipska-Ziętkiewicz B, Tkaczyk M. CoQ10-related sustained remission of proteinuria in a child with COQ6 glomerulopathy-a case report. Pediatr Nephrol 33: 2383–2387, 2018. doi: 10.1007/s00467-018-4083-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586. Justine Perrin R, Rousset-Rouvière C, Garaix F, Cano A, Conrath J, Boyer O, Tsimaratos M. COQ6 mutation in patients with nephrotic syndrome, sensorineural deafness, and optic atrophy. JIMD Rep 54: 37–44, 2020. doi: 10.1002/jmd2.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587. Yuruk Yildirim Z, Toksoy G, Uyguner O, Nayir A, Yavuz S, Altunoglu U, Turkkan ON, Sevinc B, Gokcay G, Kurkcu Gunes D, Kiyak A, Yilmaz A. Primary coenzyme Q10 Deficiency-6 (COQ10D6): Two siblings with variable expressivity of the renal phenotype. Eur J Med Genet 63: 103621, 2020. doi: 10.1016/j.ejmg.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 588. Wang N, Zheng Y, Zhang L, Tian X, Fang Y, Qi M, Du J, Chen S, Chen S, Li J, Shen B, Wang L. A family segregating lethal primary coenzyme Q10 deficiency due to two novel COQ6 variants. Front Genet 12: 811833, 2021. doi: 10.3389/fgene.2021.811833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589. Artuch R, Brea-Calvo G, Briones P, Aracil A, Galván M, Espinós C, Corral J, Volpini V, Ribes A, Andreu AL, Palau F, Sánchez-Alcazar JA, Navas P, Pineda M. Cerebellar ataxia with coenzyme Q10 deficiency: diagnosis and follow-up after coenzyme Q10 supplementation. J Neurol Sci 246: 153–158, 2006. doi: 10.1016/j.jns.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 590. Lagier-Tourenne C, Tazir M, López LC, Quinzii CM, Assoum M, Drouot N, Busso C, Makri S, Ali-Pacha L, Benhassine T, Anheim M, Lynch DR, Thibault C, Plewniak F, Bianchetti L, Tranchant C, Poch O, DiMauro S, Mandel JL, Barros MH, Hirano M, Koenig M. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet 82: 661–672, 2008. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591. Mollet J, Delahodde A, Serre V, Chretien D, Schlemmer D, Lombes A, Boddaert N, Desguerre I, de Lonlay P, de Baulny HO, Munnich A, Rötig A. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet 82: 623–630, 2008. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592. Gerards M, van den Bosch B, Calis C, Schoonderwoerd K, van Engelen K, Tijssen M, de Coo R, van der Kooi A, Smeets H. Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion 10: 510–515, 2010. doi: 10.1016/j.mito.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 593. Horvath R, Czermin B, Gulati S, Demuth S, Houge G, Pyle A, Dineiger C, Blakely EL, Hassani A, Foley C, Brodhun M, Storm K, Kirschner J, Gorman GS, Lochmüller H, Holinski-Feder E, Taylor RW, Chinnery PF. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J Neurol Neurosurg Psychiatry 83: 174–178, 2012. doi: 10.1136/jnnp-2011-301258. [DOI] [PubMed] [Google Scholar]
- 594. Mignot C, Apartis E, Durr A, Marques Lourenço C, Charles P, Devos D, Moreau C, de Lonlay P, Drouot N, Burglen L, Kempf N, Nourisson E, Chantot-Bastaraud S, Lebre AS, Rio M, Chaix Y, Bieth E, Roze E, Bonnet I, Canaple S, Rastel C, Brice A, Rötig A, Desguerre I, Tranchant C, Koenig M, Anheim M. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J Rare Dis 8: 173, 2013. doi: 10.1186/1750-1172-8-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595. Blumkin L, Leshinsky-Silver E, Zerem A, Yosovich K, Lerman-Sagie T, Lev D. Heterozygous mutations in the ADCK3 gene in siblings with cerebellar atrophy and extreme phenotypic variability. JIMD Rep 12: 103–107, 2014. doi: 10.1007/8904_2013_251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596. Liu YT, Hersheson J, Plagnol V, Fawcett K, Duberley KE, Preza E, Hargreaves IP, Chalasani A, Laurá M, Wood NW, Reilly MM, Houlden H. Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: clinical, genetic and biochemical characterisation. J Neurol Neurosurg Psychiatry 85: 493–498, 2014. doi: 10.1136/jnnp-2013-306483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597. Barca E, Musumeci O, Montagnese F, Marino S, Granata F, Nunnari D, Peverelli L, DiMauro S, Quinzii CM, Toscano A. Cerebellar ataxia and severe muscle CoQ10 deficiency in a patient with a novel mutation in ADCK3. Clin Genet 90: 156–160, 2016. doi: 10.1111/cge.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598. Hikmat O, Tzoulis C, Knappskog PM, Johansson S, Boman H, Sztromwasser P, Lien E, Brodtkorb E, Ghezzi D, Bindoff LA. ADCK3 mutations with epilepsy, stroke-like episodes and ataxia: a POLG mimic? Eur J Neurol 23: 1188–1194, 2016. doi: 10.1111/ene.13003. [DOI] [PubMed] [Google Scholar]
- 599. Malgireddy K, Thompson R, Torres-Russotto D. A novel CABC1/ADCK3 mutation in adult-onset cerebellar ataxia. Parkinsonism Relat Disord 33: 151–152, 2016. doi: 10.1016/j.parkreldis.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 600. Chang A, Ruiz-Lopez M, Slow E, Tarnopolsky M, Lang AE, Munhoz RP. ADCK3-related coenzyme Q10 deficiency: a potentially treatable genetic disease. Mov Disord Clin Pract 5: 635–639, 2018. doi: 10.1002/mdc3.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601. Coutelier M, Hammer MB, Stevanin G, Monin ML, Davoine CS, Mochel F, , et al. Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol 75: 591–599, 2018. doi: 10.1001/jamaneurol.2017.5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602. Jacobsen JC, Whitford W, Swan B, Taylor J, Love DR, Hill R, Molyneux S, George PM, Mackay R, Robertson SP, Snell RG, Lehnert K. Compound heterozygous inheritance of mutations in coenzyme Q8A results in autosomal recessive cerebellar ataxia and coenzyme Q10 deficiency in a female sib-pair. JIMD Rep 42: 31–36, 2018. doi: 10.1007/8904_2017_73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603. Galosi S, Barca E, Carrozzo R, Schirinzi T, Quinzii CM, Lieto M, Vasco G, Zanni G, Di Nottia M, Galatolo D, Filla A, Bertini E, Santorelli FM, Leuzzi V, Haas R, Hirano M, Friedman J. Dystonia-ataxia with early handwriting deterioration in COQ8A mutation carriers: a case series and literature review. Parkinsonism Relat Disord 68: 8–16, 2019. doi: 10.1016/j.parkreldis.2019.09.015. [DOI] [PubMed] [Google Scholar]
- 604. Hajjari M, Tahmasebi-Birgani M, Mohammadi-Asl J, Nasiri H, Kollaee A, Mahmoodi M, Ansari H. Exome sequencing found a novel homozygous deletion in ADCK3 gene involved in autosomal recessive spinocerebellar ataxia. Gene 708: 10–13, 2019. doi: 10.1016/j.gene.2019.05.016. [DOI] [PubMed] [Google Scholar]
- 605. Nair P, Lama M, El-Hayek S, Abou Sleymane G, Stora S, Obeid M, Al-Ali MT, Delague V, Mégarbané A. COQ8A and MED25 mutations in a child with intellectual disability, microcephaly, seizures, and spastic ataxia: synergistic effect of digenic variants? Mol Syndromol 9: 319–323, 2019. doi: 10.1159/000494465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606. Schirinzi T, Favetta M, Romano A, Sancesario A, Summa S, Minosse S, Zanni G, Castelli E, Bertini E, Petrarca M, Vasco G. One-year outcome of coenzyme Q10 supplementation in ADCK3 ataxia (ARCA2). Cerebellum Ataxias 6: 15, 2019. doi: 10.1186/s40673-019-0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Shalata A, Edery M, Habib C, Genizi J, Mahroum M, Khalaily L, Assaf N, Segal I, Abed El Rahim H, Shapira H, Urian D, Tzur S, Douiev L, Saada A. Primary coenzyme Q deficiency due to novel ADCK3 variants, studies in fibroblasts and review of literature. Neurochem Res 44: 2372–2384, 2019. doi: 10.1007/s11064-019-02786-5. [DOI] [PubMed] [Google Scholar]
- 608. Sun M, Johnson AK, Nelakuditi V, Guidugli L, Fischer D, Arndt K, Ma L, Sandford E, Shakkottai V, Boycott K, Warman-Chardon J, Li Z, Del Gaudio D, Burmeister M, Gomez CM, Waggoner DJ, Das S. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet Med 21: 195–206, 2019. doi: 10.1038/s41436-018-0007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609. Jiao B, Zhou Z, Hu Z, Du J, Liao X, Luo Y, Wang J, Yan X, Jiang H, Tang B, Shen L. Homozygosity mapping and next generation sequencing for the genetic diagnosis of hereditary ataxia and spastic paraplegia in consanguineous families. Parkinsonism Relat Disord 80: 65–72, 2020. doi: 10.1016/j.parkreldis.2020.09.013. [DOI] [PubMed] [Google Scholar]
- 610. Liu G, Ma D, Li J, Luo C, Sun Y, Zhang J, Hu P, Tang W, Xu Z. A novel COQ8A missense variant associated with a mild form of primary coenzyme Q10 deficiency type 4. Clin Biochem 84: 93–98, 2020. doi: 10.1016/j.clinbiochem.2020.06.010. [DOI] [PubMed] [Google Scholar]
- 611. Mutlu-Albayrak H, Kırat E, Gürbüz G. Childhood-onset autosomal recessive ataxias: a cross-sectional study from Turkey. Neurogenetics 21: 59–66, 2020. doi: 10.1007/s10048-019-00597-y. [DOI] [PubMed] [Google Scholar]
- 612. Traschütz A, Schirinzi T, Laugwitz L, Murray NH, Bingman CA, Reich S, , et al. Clinico-genetic, imaging and molecular delineation of COQ8A-ataxia: a multicenter study of 59 patients. Ann Neurol 88: 251–263, 2020. doi: 10.1002/ana.25751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613. Zhang L, Ashizawa T, Peng D. Primary coenzyme Q10 deficiency due to COQ8A gene mutations. Mol Genet Genomic Med 8: e1420, 2020. doi: 10.1002/mgg3.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614. Amprosi M, Zech M, Steiger R, Nachbauer W, Eigentler A, Gizewski ER, Guger M, Indelicato E, Boesch S. Familial writer’s cramp: a clinical clue for inherited coenzyme Q10 deficiency. Neurogenetics 22: 81–86, 2021. doi: 10.1007/s10048-020-00624-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615. Cheng HL, Shao YR, Dong Y, Dong HL, Yang L, Ma Y, Shen Y, Wu ZY. Genetic spectrum and clinical features in a cohort of Chinese patients with autosomal recessive cerebellar ataxias. Transl Neurodegener 10: 40, 2021. doi: 10.1186/s40035-021-00264-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616. Uccella S, Pisciotta L, Severino M, Bertini E, Giacomini T, Zanni G, Prato G, De Grandis E, Nobili L, Mancardi MM. Photoparoxysmal response in ADCK3 autosomal recessive ataxia: a case report and literature review. Epileptic Disord 23: 153–160, 2021. doi: 10.1684/epd.2021.1243. [DOI] [PubMed] [Google Scholar]
- 617. Ashrafi MR, Haghighi R, Badv RS, Ghabeli H, Tavasoli AR, Pourbakhtyaran E, Rezaei Z, Mahdieh N, Mohammadi P, Heidari M. Epilepsia partialis continua a clinical feature of a missense variant in the ADCK3 gene and poor response to therapy. J Mol Neurosci 72: 1125–1132, 2022. doi: 10.1007/s12031-022-01993-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618. Değerliyurt A, Gülleroğlu NB, Kibar Gül AE. Primary CoQ10 deficiency with a severe phenotype due to the c.901 C > T (p.R301W) mutation in the COQ8A gene. Int J Neurosci 134: 148–152, 2022. doi: 10.1080/00207454.2022.2095269. [DOI] [PubMed] [Google Scholar]
- 619. Paprocka J, Nowak M, Chuchra P, Śmigiel R. COQ8A-ataxia as a manifestation of primary coenzyme Q deficiency. Metabolites 12: 955, 2022. doi: 10.3390/metabo12100955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620. Hojabri M, Gilani A, Irilouzadian R, Nejad Biglari H, Sarmadian R. Adolescence onset primary coenzyme Q10 deficiency with rare CoQ8A gene mutation: a case report and review of literature. Clin Med Insights Case Rep 16: 11795476231188061, 2023. doi: 10.1177/11795476231188061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621. Malicdan MC, Vilboux T, Ben-Zeev B, Guo J, Eliyahu A, Pode-Shakked B, Dori A, Kakani S, Chandrasekharappa SC, Ferreira CR, Shelestovich N, Marek-Yagel D, Pri-Chen H, Blatt I, Niederhuber JE, He L, Toro C, Taylor RW, Deeken J, Yardeni T, Wallace DC, Gahl WA, Anikster Y. A novel inborn error of the coenzyme Q10 biosynthesis pathway: cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum Mutat 39: 69–79, 2018. doi: 10.1002/humu.23345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622. Dawidziuk M, Podwysocka A, Jurek M, Obersztyn E, Bekiesinska-Figatowska M, Goszczanska-Ciuchta A, Bukowska-Olech E, Rygiel AM, Guilbride DL, Wiszniewski W, Gawlinski P. Congenital coenzyme Q5-linked pathology: causal genetic association, core phenotype, and molecular mechanism. J Appl Genet 64: 507–514, 2023. doi: 10.1007/s13353-023-00773-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623. Chung WK, Martin K, Jalas C, Braddock SR, Juusola J, Monaghan KG, Warner B, Franks S, Yudkoff M, Lulis L, Rhodes RH, Prasad V, Torti E, Cho MT, Shinawi M. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J Med Genet 52: 627–635, 2015. doi: 10.1136/jmedgenet-2015-103140. [DOI] [PubMed] [Google Scholar]
- 624. Sondheimer N, Hewson S, Cameron JM, Somers GR, Broadbent JD, Ziosi M, Quinzii CM, Naini AB. Novel recessive mutations in COQ4 cause severe infantile cardiomyopathy and encephalopathy associated with CoQ10 deficiency. Mol Genet Metab Rep 12: 23–27, 2017. doi: 10.1016/j.ymgmr.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625. Ling TK, Law CY, Yan KW, Fong NC, Wong KC, Lee KL, Chu WC, Brea-Calvo G, Lam CW. Clinical whole-exome sequencing reveals a common pathogenic variant in patients with CoQ10 deficiency: an underdiagnosed cause of mitochondriopathy. Clin Chim Acta 497: 88–94, 2019. doi: 10.1016/j.cca.2019.07.016. [DOI] [PubMed] [Google Scholar]
- 626. Lu M, Zhou Y, Wang Z, Xia Z, Ren J, Guo Q. Clinical phenotype, in silico and biomedical analyses, and intervention for an East Asian population-specific c.370G>A (p.G124S) COQ4 mutation in a Chinese family with CoQ10 deficiency-associated Leigh syndrome. J Hum Genet 64: 297–304, 2019. doi: 10.1038/s10038-019-0563-y. [DOI] [PubMed] [Google Scholar]
- 627. Yu MH, Tsang MH, Lai S, Ho MS, Tse DM, Willis B, Kwong AK, Chou YY, Lin SP, Quinzii CM, Hwu WL, Chien YH, Kuo PL, Chan VC, Tsoi C, Chong SC, Rodenburg RJ, Smeitink J, Mak CC, Yeung KS, Fung JL, Lam W, Hui J, Lee NC, Fung CW, Chung BH. Primary coenzyme Q10 deficiency-7: expanded phenotypic spectrum and a founder mutation in southern Chinese. NPJ Genom Med 4: 18, 2019. doi: 10.1038/s41525-019-0091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628. Mero S, Salviati L, Leuzzi V, Rubegni A, Calderan C, Nardecchia F, Galatolo D, Desbats MA, Naef V, Gemignani F, Novelli M, Tessa A, Battini R, Santorelli FM, Marchese M. New pathogenic variants in COQ4 cause ataxia and neurodevelopmental disorder without detectable CoQ10 deficiency in muscle or skin fibroblasts. J Neurol 268: 3381–3389, 2021. doi: 10.1007/s00415-021-10509-6. [DOI] [PubMed] [Google Scholar]
- 629. Olgac A, Öztoprak U, Kasapkara CS, Kılıç M, Yüksel D, Derinkuyu EB, Tasci Yildiz Y, Ceylaner S, Ezgu FS. A rare case of primary coenzyme Q10 deficiency due to COQ9 mutation. J Pediatr Endocrinol Metab 33: 165–170, 2020. doi: 10.1515/jpem-2019-0245. [DOI] [PubMed] [Google Scholar]
- 630. Vasta V, Merritt JL 2nd, Saneto RP, Hahn SH. Next-generation sequencing for mitochondrial diseases: a wide diagnostic spectrum. Pediatr Int 54: 585–601, 2012. doi: 10.1111/j.1442-200X.2012.03644.x. [DOI] [PubMed] [Google Scholar]
- 631. Danhauser K, Herebian D, Haack TB, Rodenburg RJ, Strom TM, Meitinger T, Klee D, Mayatepek E, Prokisch H, Distelmaier F. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur J Hum Genet 24: 450–454, 2016. doi: 10.1038/ejhg.2015.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632. Smith AC, Ito Y, Ahmed A, Schwartzentruber JA, Beaulieu CL, Aberg E, Majewski J, Bulman DE, Horsting-Wethly K, Koning DV, Rodenburg RJ, Boycott KM, Penney LS; Care4Rare Canada Consortium. A family segregating lethal neonatal coenzyme Q10 deficiency caused by mutations in COQ9. J Inherit Metab Dis 41: 719–729, 2018. doi: 10.1007/s10545-017-0122-7. [DOI] [PubMed] [Google Scholar]
- 633. Kwong AK, Chiu AT, Tsang MH, Lun KS, Rodenburg RJ, Smeitink J, Chung BH, Fung CW. A fatal case of COQ7-associated primary coenzyme Q10 deficiency. JIMD Rep 47: 23–29, 2019. doi: 10.1002/jmd2.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634. Hashemi SS, Zare-Abdollahi D, Bakhshandeh MK, Vafaee A, Abolhasani S, Inanloo Rahatloo K, DanaeeFard F, Farboodi N, Rohani M, Alavi A. Clinical spectrum in multiple families with primary COQ10 deficiency. Am J Med Genet A 185: 440–452, 2021. doi: 10.1002/ajmg.a.61983. [DOI] [PubMed] [Google Scholar]
- 635. Liu XX, Wang N, Chen YK, Lv WQ, Hong JM, Xu GR, Zhou LY, Chen WJ, Fan DS, He J. Biallelic variants in the COQ7 gene cause distal hereditary motor neuropathy in two Chinese families. Brain 146: e27–e30, 2023. doi: 10.1093/brain/awad040. [DOI] [PubMed] [Google Scholar]
- 636. Rebelo AP, Tomaselli PJ, Medina J, Wang Y, Dohrn M, Nyvltova E, Danzi M, Garrett M, Smith S, Pestronk A, Li C, Ruiz A, Jacobs E, Feely SM, França MC, Gomes MV, Santos D, Kumar S, Lombard DB, Saporta M, Hekimi S, Barrientos A, Weihl C, Shy M, Marques W, Zuchner S. Biallelic variants in COQ7 cause distal hereditary motor neuropathy with upper motor neuron signs. Brain 146: 4191–4199, 2023. doi: 10.1093/brain/awad158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637. Sadr Z, Zare-Abdollahi D, Rohani M, Alavi A. A founder mutation in COQ7, p.(Leu111Pro), causes pure hereditary spastic paraplegia (HSP) in the Iranian population. Neurol Sci 44: 2599–2602, 2023. doi: 10.1007/s10072-023-06707-x. [DOI] [PubMed] [Google Scholar]
- 638. Smith IC, Pileggi CA, Wang Y, Kernohan K, Hartley T, McMillan HJ, Sampaio ML, Melkus G, Woulfe J, Parmar G, Bourque PR, Breiner A, Zwicker J, Pringle CE, Jarinova O, Lochmüller H, Dyment DA, Brais B, Boycott KM, Hekimi S, Harper ME, Warman-Chardon J; Care4Rare Canada Consortium. Novel homozygous variant in COQ7 in siblings with hereditary motor neuropathy. Neurol Genet 9: e200048, 2023. doi: 10.1212/NXG.0000000000200048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639. Wongkittichote P, Duque Lasio ML, Magistrati M, Pathak S, Sample B, Carvalho DR, Ortega AB, Castro MA, de Gusmao CM, Toler TL, Bellacchio E, Dallabona C, Shinawi M. Phenotypic, molecular, and functional characterization of COQ7-related primary CoQ10 deficiency: Hypomorphic variants and two distinct disease entities. Mol Genet Metab 139: 107630, 2023. doi: 10.1016/j.ymgme.2023.107630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640. Zaki NM. Strategies for oral delivery and mitochondrial targeting of CoQ10. Drug Deliv 23: 1868–1881, 2016. doi: 10.3109/10717544.2014.993747. [DOI] [PubMed] [Google Scholar]
- 641. Gueguen N, Baris O, Lenaers G, Reynier P, Spinazzi M. Secondary coenzyme Q deficiency in neurological disorders. Free Radic Biol Med 165: 203–218, 2021. doi: 10.1016/j.freeradbiomed.2021.01.017. [DOI] [PubMed] [Google Scholar]
- 642. Hernández-Camacho JD, García-Corzo L, Fernandez-Ayala DJ, Navas P, Lopez-Lluch G. Coenzyme Q at the hinge of health and metabolic diseases. Antioxidants (Basel) 10: 1785, 2021. doi: 10.3390/antiox10111785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643. Grünler J, Ericsson J, Dallner G. Branch-point reactions in the biosynthesis of cholesterol, dolichol, ubiquinone and prenylated proteins. Biochim Biophys Acta 1212: 259–277, 1994. doi: 10.1016/0005-2760(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 644. Ghirlanda G, Oradei A, Manto A, Lippa S, Uccioli L, Caputo S, Greco AV, Littarru GP. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 33: 226–229, 1993. doi: 10.1002/j.1552-4604.1993.tb03948.x. [DOI] [PubMed] [Google Scholar]
- 645. Rundek T, Naini A, Sacco R, Coates K, DiMauro S. Atorvastatin decreases the coenzyme Q10 level in the blood of patients at risk for cardiovascular disease and stroke. Arch Neurol 61: 889–892, 2004. doi: 10.1001/archneur.61.6.889. [DOI] [PubMed] [Google Scholar]
- 646. Folkers K, Langsjoen P, Willis R, Richardson P, Xia LJ, Ye CQ, Tamagawa H. Lovastatin decreases coenzyme Q levels in humans. Proc Natl Acad Sci USA 87: 8931–8934, 1990. doi: 10.1073/pnas.87.22.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647. Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 46: 1055–1057, 1993. doi: 10.1136/jcp.46.11.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648. Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 18, Suppl: S137–S144, 1997. doi: 10.1016/s0098-2997(97)00014-9. [DOI] [PubMed] [Google Scholar]
- 649. Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 49: 2231–2237, 2007. doi: 10.1016/j.jacc.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 650. Bleske BE, Willis RA, Anthony M, Casselberry N, Datwani M, Uhley VE, Secontine SG, Shea MJ. The effect of pravastatin and atorvastatin on coenzyme Q10. Am Heart J 142: E2, 2001. doi: 10.1067/mhj.2001.116762. [DOI] [PubMed] [Google Scholar]
- 651. Deichmann R, Lavie C, Andrews S. Coenzyme q10 and statin-induced mitochondrial dysfunction. Ochsner J 10: 16–21, 2010. [PMC free article] [PubMed] [Google Scholar]
- 652. Folkers K, Langsjoen P, Willis R, Richardson P, Xia LJ, Ye CQ, Tamagawa H. Lovastatin decreases coenzyme Q levels in rats. Proc Natl Acad Sci USA 87: 8931–8934, 1990. doi: 10.1073/pnas.87.22.8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653. Laaksonen R, Ojala JP, Tikkanen MJ, Himberg JJ. Serum ubiquinone concentrations after short- and long-term treatment with HMG-CoA reductase inhibitors. Eur J Clin Pharmacol 46: 313–317, 1994. doi: 10.1007/BF00194398. [DOI] [PubMed] [Google Scholar]
- 654. Bellosta S, Paoletti R, Corsini A. Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation 109: III50–III57, 2004. doi: 10.1161/01.CIR.0000131519.15067.1f. [DOI] [PubMed] [Google Scholar]
- 655. Vinci P, Panizon E, Tosoni LM, Cerrato C, Pellicori F, Mearelli F, Biasinutto C, Fiotti N, Di Girolamo FG, Biolo G. Statin-associated myopathy: emphasis on mechanisms and targeted therapy. Int J Mol Sci 22: 11687, 2021. doi: 10.3390/ijms222111687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656. Laaksonen R, Jokelainen K, Sahi T, Tikkanen MJ, Himberg JJ. Decreases in serum ubiquinone concentrations do not result in reduced levels in muscle tissue during short-term simvastatin treatment in humans. Clin Pharmacol Ther 57: 62–66, 1995. doi: 10.1016/0009-9236(95)90266-X. [DOI] [PubMed] [Google Scholar]
- 657. Laaksonen R, Jokelainen K, Laakso J, Sahi T, Harkonen M, Tikkanen MJ, Himberg JJ. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 77: 851–854, 1996. doi: 10.1016/S0002-9149(97)89180-1. [DOI] [PubMed] [Google Scholar]
- 658. Lamperti C, Naini AB, Lucchini V, Prelle A, Bresolin N, Moggio M, Sciacco M, Kaufmann P, DiMauro S. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 62: 1709–1712, 2005. doi: 10.1001/archneur.62.11.1709. [DOI] [PubMed] [Google Scholar]
- 659. Päivä H, Thelen KM, Van Coster R, Smet J, De Paepe B, Mattila KM, Laakso J, Lehtimäki T, von Bergmann K, Lütjohann D, Laaksonen R. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 78: 60–68, 2005. doi: 10.1016/j.clpt.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 660. Dohlmann TL, Kuhlman AB, Morville T, Dahl M, Asping M, Orlando P, Silvestri S, Tiano L, Helge JW, Dela F, Larsen S. Coenzyme Q10 supplementation in statin treated patients: a double-blinded randomized placebo-controlled trial. Antioxidants (Basel) 11: 1698, 2022. doi: 10.3390/antiox11091698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661. Fukami M, Maeda N, Fukushige J, Kogure Y, Shimada Y, Ogawa T, Tsujita Y. Effects of HMG-CoA reductase inhibitors on skeletal muscles of rabbits. Res Exp Med (Berl) 193: 263–273, 1993. doi: 10.1007/BF02576234. [DOI] [PubMed] [Google Scholar]
- 662. Nakahara K, Kuriyama M, Sonoda Y, Yoshidome H, Nakagawa H, Fujiyama J, Higuchi I, Osame M. Myopathy induced by HMG-CoA reductase inhibitors in rabbits: a pathological, electrophysiological, and biochemical study. Toxicol Appl Pharmacol 152: 99–106, 1998. doi: 10.1006/taap.1998.8491. [DOI] [PubMed] [Google Scholar]
- 663. Schaefer WH, Lawrence JW, Loughlin AF, Stoffregen DA, Mixson LA, Dean DC, Raab CE, Yu NX, Lankas GR, Frederick CB. Evaluation of ubiquinone concentration and mitochondrial function relative to cerivastatin-induced skeletal myopathy in rats. Toxicol Appl Pharmacol 194: 10–23, 2004. doi: 10.1016/j.taap.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 664. De Pinieux G, Chariot P, Ammi-Saïd M, Louarn F, Lejonc JL, Astier A, Jacotot B, Gherardi R. Lipid-lowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 42: 333–337, 1996. doi: 10.1046/j.1365-2125.1996.04178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665. Goli AK, Goli SA, Byrd RP Jr, Roy TM. Simvastatin-induced lactic acidosis: a rare adverse reaction? Clin Pharmacol Ther 72: 461–464, 2002. doi: 10.1067/mcp.2002.127943. [DOI] [PubMed] [Google Scholar]
- 666. Phillips PS, Haas RH, Bannykh S, Hathaway S, Gray NL, Kimura BJ, Vladutiu GD, England JD; Scripps Mercy Clinical Research Center. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med 137: 581–585, 2002. doi: 10.7326/0003-4819-137-7-200210010-00009. [DOI] [PubMed] [Google Scholar]
- 667. Montero R, Grazina M, Lopez-Gallardo E, Montoya J, Briones P, Navarro-Sastre A, Land JM, Hargreaves IP, Artuch R; Coenzyme Q Deficiency Study Group. Coenzyme Q10 deficiency in mitochondrial DNA depletion syndromes. Mitochondrion 13: 337–341, 2013. doi: 10.1016/j.mito.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 668. Yubero D, Montero R, Martín MA, Montoya J, Ribes A, Grazina M, , et al. Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 30: 51–58, 2016. doi: 10.1016/j.mito.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 669. Sacconi S, Trevisson E, Salviati L, Aymé S, Rigal O, Redondo AG, Mancuso M, Siciliano G, Tonin P, Angelini C, Auré K, Lombès A, Desnuelle C. Coenzyme Q10 is frequently reduced in muscle of patients with mitochondrial myopathy. Neuromuscul Disord 20: 44–48, 2010. doi: 10.1016/j.nmd.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 670. Cotán D, Cordero MD, Garrido-Maraver J, Oropesa-Ávila M, Rodríguez-Hernandez A, Gomez Izquierdo L, De la Mata M, De Miguel M, Lorite JB, Infante ER, Jackson S, Navas P, Sanchez-Alcazar JA. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J 25: 2669–2687, 2011. doi: 10.1096/fj.10-165340. [DOI] [PubMed] [Google Scholar]
- 671. Naess K, Bruhn H, Stranneheim H, Freyer C, Wibom R, Mourier A, Engvall M, Nennesmo I, Lesko N, Wredenberg A, Wedell A, von Dobeln U. Clinical presentation, genetic etiology, and coenzyme Q10 Levels in 55 children with combined enzyme deficiencies of the mitochondrial respiratory chain. J Pediatr 228: 240–251.e2, 2021. doi: 10.1016/j.jpeds.2020.08.025. [DOI] [PubMed] [Google Scholar]
- 672. Zung N, Schuldiner M. New horizons in mitochondrial contact site research. Biol Chem 401: 793–809, 2020. doi: 10.1515/hsz-2020-0133. [DOI] [PubMed] [Google Scholar]
- 673. Vincent AE, Ng YS, White K, Davey T, Mannella C, Falkous G, Feeney C, Schaefer AM, McFarland R, Gorman GS, Taylor RW, Turnbull DM, Picard M. The spectrum of mitochondrial ultrastructural defects in mitochondrial myopathy. Sci Rep 6: 30610, 2016. doi: 10.1038/srep30610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674. Kühl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A, Filipovska A, Larsson NG. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 6: e30952, 2017. doi: 10.7554/eLife.30952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675. Spinazzi M, Radaelli E, Horré K, Arranz AM, Gounko NV, Agostinis P, Maia TM, Impens F, Morais VA, Lopez-Lluch G, Serneels L, Navas P, De Strooper B. PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome. Proc Natl Acad Sci USA 116: 277–286, 2019. doi: 10.1073/pnas.1811938116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676. Mourier A, Motori E, Brandt T, Lagouge M, Atanassov I, Galinier A, Rappl G, Brodesser S, Hultenby K, Dieterich C, Larsson NG. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J Cell Biol 208: 429–442, 2015. doi: 10.1083/jcb.201411100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677. Filadi R, Pendin D, Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis 9: 330, 2018. doi: 10.1038/s41419-017-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678. Chan EY, McQuibban GA. The mitochondrial rhomboid protease: its rise from obscurity to the pinnacle of disease-relevant genes. Biochim Biophys Acta 1828: 2916–2925, 2013. doi: 10.1016/j.bbamem.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 679. Radaelli E, Assenmacher CA, Verrelle J, Banerjee E, Manero F, Khiati S, Girona A, Lopez-Lluch G, Navas P, Spinazzi M. Mitochondrial defects caused by PARL deficiency lead to arrested spermatogenesis and ferroptosis. Elife 12: e84710, 2023. doi: 10.7554/eLife.84710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680. Ghezzi D, Arzuffi P, Zordan M, Da Re C, Lamperti C, Benna C, D'Adamo P, Diodato D, Costa R, Mariotti C, Uziel G, Smiderle C, Zeviani M. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet 43: 259–263, 2011. doi: 10.1038/ng.761. [DOI] [PubMed] [Google Scholar]
- 681. Leonard CJ, Aravind L, Koonin EV. Novel families of putative protein kinases in bacteria and archaea: evolution of the “eukaryotic” protein kinase superfamily. Genome Res 8: 1038–1047, 1998. doi: 10.1101/gr.8.10.1038. [DOI] [PubMed] [Google Scholar]
- 682. Balreira A, Boczonadi V, Barca E, Pyle A, Bansagi B, Appleton M, Graham C, Hargreaves IP, Rasic VM, Lochmüller H, Griffin H, Taylor RW, Naini A, Chinnery PF, Hirano M, Quinzii CM, Horvath R. ANO10 mutations cause ataxia and coenzyme Q10 deficiency. J Neurol 261: 2192–2198, 2014. doi: 10.1007/s00415-014-7476-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 683. Wanitchakool P, Ousingsawat J, Sirianant L, Cabrita I, Faria D, Schreiber R, Kunzelmann K. Cellular defects by deletion of ANO10 are due to deregulated local calcium signaling. Cell Signal 30: 41–49, 2017. doi: 10.1016/j.cellsig.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 684. Quinzii CM, Kattah AG, Naini A, Akman HO, Mootha VK, DiMauro S, Hirano M. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 64: 539–541, 2005. doi: 10.1212/01.WNL.0000150588.75281.58. [DOI] [PubMed] [Google Scholar]
- 685. Le Ber I, Dubourg O, Benoist JF, Jardel C, Mochel F, Koenig M, Brice A, Lombès A, Dürr A. Muscle coenzyme Q10 deficiencies in ataxia with oculomotor apraxia 1. Neurology 68: 295–297, 2007. doi: 10.1212/01.wnl.0000252366.10731.43. [DOI] [PubMed] [Google Scholar]
- 686. Castellotti B, Mariotti C, Rimoldi M, Fancellu R, Plumari M, Caimi S, Uziel G, Nardocci N, Moroni I, Zorzi G, Pareyson D, Di Bella D, Di Donato S, Taroni F, Gellera C. Ataxia with oculomotor apraxia type1 (AOA1): novel and recurrent aprataxin mutations, coenzyme Q10 analyses, and clinical findings in Italian patients. Neurogenetics 12: 193–201, 2011. doi: 10.1007/s10048-011-0281-x. [DOI] [PubMed] [Google Scholar]
- 687. Garcia-Diaz B, Barca E, Balreira A, Lopez LC, Tadesse S, Krishna S, Naini A, Mariotti C, Castellotti B, Quinzii CM. Lack of aprataxin impairs mitochondrial functions via downregulation of the APE1/NRF1/NRF2 pathway. Hum Mol Genet 24: 4516–4529, 2015. doi: 10.1093/hmg/ddv183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688. Sykora P, Croteau DL, Bohr VA, Wilson DM 3rd.. Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc Natl Acad Sci USA 108: 7437–7442, 2011. doi: 10.1073/pnas.1100084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689. Zheng J, Croteau DL, Bohr VA, Akbari M. Diminished OPA1 expression and impaired mitochondrial morphology and homeostasis in Aprataxin-deficient cells. Nucleic Acids Res 47: 4086–4110, 2019. doi: 10.1093/nar/gkz083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690. Herrera-Vaquero M, Heras-Garvin A, Krismer F, Deleanu R, Boesch S, Wenning GK, Stefanova N. Signs of early cellular dysfunction in multiple system atrophy. Neuropathol Appl Neurobiol 47: 268–282, 2021. doi: 10.1111/nan.12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 691. Gempel K, Topaloglu H, Talim B, Schneiderat P, Schoser BG, Hans VH, Pálmafy B, Kale G, Tokatli A, Quinzii C, Hirano M, Naini A, DiMauro S, Prokisch H, Lochmüller H, Horvath R. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 130: 2037–2044, 2007. doi: 10.1093/brain/awm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692. Wen B, Li D, Shan J, Liu S, Li W, Zhao Y, Lin P, Zheng J, Li D, Gong Y, Yan C. Increased muscle coenzyme Q10 in riboflavin responsive MADD with ETFDH gene mutations due to secondary mitochondrial proliferation. Mol Genet Metab 109: 154–160, 2013. doi: 10.1016/j.ymgme.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 693. Béhin A, Acquaviva-Bourdain C, Souvannanorath S, Streichenberger N, Attarian S, Bassez G, Brivet M, Fouilhoux A, Labarre-Villa A, Laquerrière A, Pérard L, Kaminsky P, Pouget J, Rigal O, Vanhulle C, Eymard B, Vianey-Saban C, Laforet P. Multiple acyl-CoA dehydrogenase deficiency (MADD) as a cause of late-onset treatable metabolic disease. Rev Neurol (Paris) 172: 231–241, 2016. doi: 10.1016/j.neurol.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 694. Aeby A, Sznajer Y, Cavé H, Rebuffat E, Van Coster R, Rigal O, Van Bogaert P. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J Inherit Metab Dis 30: 827, 2007. doi: 10.1007/s10545-007-0612-0. [DOI] [PubMed] [Google Scholar]
- 695. Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, Huemer M, Hochuli M, Assoun M, Ballhausen D, Burlina A, Fowler B, Grünert SC, Grünewald S, Honzik T, Merinero B, Perez-Cerda C, Scholl-Burgi S, Skovby F, Wijburg F, MacDonald A, Martinelli D, Sass JO, Valayannopoulos V, Chakrapani A. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 9: 130, 2014. doi: 10.1186/s13023-014-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696. Manoli I, Sysol JR, Li L, Houillier P, Garone C, Wang C, Zerfas PM, Cusmano-Ozog K, Young S, Trivedi NS, Cheng J, Sloan JL, Chandler RJ, Abu-Asab M, Tsokos M, Elkahloun AG, Rosen S, Enns GM, Berry GT, Hoffmann V, DiMauro S, Schnermann J, Venditti CP. Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic acidemia. Proc Natl Acad Sci USA 110: 13552–13557, 2013. doi: 10.1073/pnas.1302764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 697. Kalén A, Appelkvist EL, Dallner G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 24: 579–584, 1989. doi: 10.1007/BF02535072. [DOI] [PubMed] [Google Scholar]
- 698. Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Agúndez JA. Coenzyme Q10 and parkinsonian syndromes: a systematic review. J Pers Med 12: 975, 2022. doi: 10.3390/jpm12060975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699. Buajitti E, Rosella LC, Zabzuni E, Young LT, Andreazza AC. Prevalence and health care costs of mitochondrial disease in Ontario, Canada: a population-based cohort study. PLoS One 17: e0265744, 2022. doi: 10.1371/journal.pone.0265744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700. Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 126: 1905–1912, 2003. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 701. Qu H, Guo M, Chai H, Wang WT, Gao ZY, Shi DZ. Effects of coenzyme Q10 on statin-induced myopathy: an updated meta-analysis of randomized controlled trials. J Am Heart Assoc 7: e009835, 2018. doi: 10.1161/JAHA.118.009835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 702. Wei H, Xin X, Zhang J, Xie Q, Naveed M, Kaiyan C, Xiao P. Effects of coenzyme Q10 supplementation on statin-induced myopathy: a meta-analysis of randomized controlled trials. Ir J Med Sci 191: 719–725, 2022. doi: 10.1007/s11845-021-02651-x. [DOI] [PubMed] [Google Scholar]
- 703. Kennedy C, Köller Y, Surkova E. Effect of Coenzyme Q10 on statin-associated myalgia and adherence to statin therapy: a systematic review and meta-analysis. Atherosclerosis 299: 1–8, 2020. doi: 10.1016/j.atherosclerosis.2020.03.006. [DOI] [PubMed] [Google Scholar]
- 704. Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; The Mitochondrial Medicine Society. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 11: 414–430, 2009. doi: 10.1007/s11940-009-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705. Haas RH. The evidence basis for coenzyme Q therapy in oxidative phosphorylation disease. Mitochondrion 7, Suppl: S136–S145, 2007. doi: 10.1016/j.mito.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 706. Matthews PM, Ford B, Dandurand RJ, Eidelman DH, O’Connor D, Sherwin A, Karpati G, Andermann F, Arnold DL. Coenzyme Q10 with multiple vitamins is generally ineffective in treatment of mitochondrial disease. Neurology 43: 884–890, 1993. doi: 10.1212/wnl.43.5.884. [DOI] [PubMed] [Google Scholar]
- 707. Chen RS, Huang CC, Chu NS. Coenzyme Q10 treatment in mitochondrial encephalomyopathies. Short-term double-blind, crossover study. Eur Neurol 37: 212–218, 1997. doi: 10.1159/000117445. [DOI] [PubMed] [Google Scholar]
- 708. Chan A, Reichmann H, Kögel A, Beck A, Gold R. Metabolic changes in patients with mitochondrial myopathies and effects of coenzyme Q10 therapy. J Neurol 245: 681–685, 1998. doi: 10.1007/s004150050267. [DOI] [PubMed] [Google Scholar]
- 709. Bogsrud MP, Langslet G, Ose L, Arnesen KE, Sm Stuen MC, Malt UF, Woldseth B, Retterstøl K. No effect of combined coenzyme Q10 and selenium supplementation on atorvastatin-induced myopathy. Scand Cardiovasc J 47: 80–87, 2013. doi: 10.3109/14017431.2012.756119. [DOI] [PubMed] [Google Scholar]
- 710.Parkinson Study Group QE3 Investigators, Beal MF, Oakes D, Shoulson I, Henchcliffe C, Galpern WR, , et al. randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol 71: 543–552, 2014. doi: 10.1001/jamaneurol.2014.131. [DOI] [PubMed] [Google Scholar]
- 711. Suksomboon N, Poolsup N, Juanak N. Effects of coenzyme Q10 supplementation on metabolic profile in diabetes: a systematic review and meta-analysis. J Clin Pharm Ther 40: 413–418, 2015. doi: 10.1111/jcpt.12280. [DOI] [PubMed] [Google Scholar]
- 712. Raizner AE, Quiñones MA. Coenzyme Q10 for patients with cardiovascular disease: JACC Focus Seminar. J Am Coll Cardiol 77: 609–619, 2021. doi: 10.1016/j.jacc.2020.12.009. [DOI] [PubMed] [Google Scholar]
- 713. Alehagen U, Johansson P, Björnstedt M, Rosén A, Dahlström U. Cardiovascular mortality and N-terminal-proBNP reduced after combined selenium and coenzyme Q10 supplementation: a 5-year prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. Int J Cardiol 167: 1860–1866, 2013. doi: 10.1016/j.ijcard.2012.04.156. [DOI] [PubMed] [Google Scholar]
- 714. Esteves TC, Echtay KS, Jonassen T, Clarke CF, Brand MD. Ubiquinone is not required for proton conductance by uncoupling protein 1 in yeast mitochondria. Biochem J 379: 309–315, 2004. doi: 10.1042/BJ20031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 715. Barca E, Kleiner G, Tang G, Ziosi M, Tadesse S, Masliah E, Louis ED, Faust P, Kang UJ, Torres J, Cortes EP, Vonsattel JP, Kuo SH, Quinzii CM. Decreased coenzyme Q10 levels in multiple system atrophy cerebellum. J Neuropathol Exp Neurol 75: 663–672, 2016. doi: 10.1093/jnen/nlw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716. Schottlaender LV, Bettencourt C, Kiely AP, Chalasani A, Neergheen V, Holton JL, Hargreaves I, Houlden H. Coenzyme Q10 levels are decreased in the cerebellum of multiple-system atrophy patients. PLoS One 11: e0149557, 2016. doi: 10.1371/journal.pone.0149557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717. Hsiao JT, Purushothuman S, Jensen PH, Halliday GM, Kim WS. Reductions in COQ2 expression relate to reduced ATP levels in multiple system atrophy brain. Front Neurosci 13: 1187, 2019. doi: 10.3389/fnins.2019.01187. [DOI] [PMC free article] [PubMed] [Google Scholar]