

Abstract
Background
Several clinical studies have compared single with tandem (also called double) autologous stem cell transplantation (ASCT) as first‐line treatment in patients with symptomatic multiple myeloma (MM), one of the leading indications for ASCT worldwide.
Objectives
The present Cochrane Review compares tandem autologous stem cell transplantation (TASCT) with single autologous stem cell transplantation (SASCT) as first‐line treatment in patients with symptomatic MM with respect to overall survival (OS), event‐free survival (EFS), quality of life (QoL) and treatment‐ or transplantation‐related mortality.
Search methods
We systematically identified controlled trials published between January 1995 and May 2011 in two bibliographic databases (MEDLINE and CENTRAL) and in clinical trial registries.
Selection criteria
One researcher screened references for controlled trials to determine eligibility for the systematic review (SR) according to pre‐specified inclusion and exclusion criteria, reflecting characteristics of disease and the interventions. We required a minimal set of details to be reported for observational studies for the studies to be included.
Data collection and analysis
We critically evaluated eligible trials with respect to quality of design and actual performance. One researcher extracted individual trial results, which were checked by another researcher. We recapitulated the results of the individual trials in a standardised way for the SR in order to allow a systematic assessment of potential sources of bias.
Main results
Overall, we identified 14 controlled studies. One registered randomised controlled trial (RCT) is still recruiting patients at the time of this review and no clinical results have been published. Two registered RCTs have remained unpublished despite their termination. Publications on one RCT had been retracted. We excluded five observational studies since neither patients nor treatment regimens were sufficiently characterised to allow an assessment of potential confounding by indication. We conducted a SR of study designs, definition of endpoints, treatment regimens and baseline characteristics of patients in the five included RCTs (two full‐text publications, three conference presentations) enrolling1506 patients in total. Because we identified substantial clinical and methodological heterogeneity, we refrained from conducting a formal meta‐analysis.
While we included only previously untreated, symptomatic patients with MM the treatment regimens differed notably with respect to acute toxicity, between trials and also between study arms. Compared to state of the art treatment standards, the treatment regimens applied in all trials have to be considered as below standard from a contemporary perspective in at least one component.
Three trials were likely to have the potential of being highly biased while two RCTs had a moderate potential for bias. The observed treatment effects in the set of included trials may have been influenced by a steep decrease in compliance with the second ASCT and the concomitant selection of patients. In addition, OS data were confounded by the treatment subsequent to first‐line therapy.
OS was statistically significantly improved in one trial only. While EFS was prolonged in four of the five trials, the median prolongation ranged between three to 12 months, with an uncertain direction of bias in the individual trials. QoL was not reported in any study. Results concerning treatment‐ or transplantation‐related mortality could not be adequately assessed due to substantial differences in definitions between trials and low reporting quality.
Authors' conclusions
We did not consider any study to be sufficiently informative for contemporary treatment decisions concerning the question single versus tandem ASCT in view of inherent biases. In addition, none of the trials integrated the so‐called "novel agents" which are now considered standard treatment for MM. To improve the quality of future studies, sample size calculations should consider the potentially steep decrease in compliance with the second ASCT. Reporting of results of treatment‐ or transplantation‐related mortality should clearly specify the type and number of events (the numerator) in a well‐defined population (the denominator).
Keywords: Adult; Aged; Humans; Middle Aged; Antineoplastic Agents; Antineoplastic Agents/administration & dosage; Bone Marrow Transplantation; Bone Marrow Transplantation/methods; Bone Marrow Transplantation/mortality; Bone Marrow Transplantation/standards; Combined Modality Therapy; Combined Modality Therapy/methods; Combined Modality Therapy/mortality; Combined Modality Therapy/standards; Disease‐Free Survival; Hematopoietic Stem Cell Transplantation; Hematopoietic Stem Cell Transplantation/methods; Hematopoietic Stem Cell Transplantation/mortality; Hematopoietic Stem Cell Transplantation/standards; Multiple Myeloma; Multiple Myeloma/drug therapy; Multiple Myeloma/surgery; Quality of Life; Randomized Controlled Trials as Topic; Transplantation, Autologous; Transplantation, Autologous/methods; Transplantation, Autologous/mortality; Transplantation, Autologous/standards
Plain language summary
High‐dose chemotherapy plus single vs tandem autologous transplantation as initial treatment for multiple myeloma
Multiple myeloma is a cancer of antibody‐producing cells in the bone marrow. It causes bone destruction and patients are usually at a higher risk for infections and renal damage. Autologous stem cell transplantation has been established as standard initial treatment for fit patients with symptomatic multiple myeloma. During autologous stem cell transplantation, blood‐forming stem cells are removed from the patient prior to intense chemotherapy and later given back to the same patient. The chemotherapy is aimed at killing tumour cells (the higher the dose the more tumour cells are killed) but also affects normal blood‐forming cells that are needed to fight infections, transport oxygen and control bleeding. By giving the patient back his or her own blood‐forming cells, the recovery from the chemotherapy is notably faster and better. Since it is unclear whether autologous stem cell transplantation as initial treatment of multiple myeloma should be performed once or twice, we systematically searched for publications addressing the question whether the acute toxicity of autologous stem cell transplantation is counterbalanced by a long‐term benefit for the patient. Several studies in which patients undergoing one treatment with autologous stem cell transplantation were compared to patients undergoing autologous stem cell transplantation twice were identified. Only five of 14 studies identified could be analysed in the present systematic review. We were interested in long‐term benefit for patients with respect to overall survival or so called event‐free survival, that is survival without disease progression. Quality of life and treatment‐related mortality should also be analysed in clinical studies.
When the included studies were analysed with respect to treatment regimen and design characteristics, all turned out to have methodological problems which do not allow us to draw firm conclusion from the findings. Since the way to treat multiple myeloma has changed since the performance of the included trials, conclusions cannot be drawn with respect to contemporary treatment decisions. We also noted that reporting of completed trials needs to be improved.
Background
Description of the condition
Multiple myeloma (MM) is a haematological malignancy that contributes to approximately 15% of haematological malignancies and close to 27% of all haematopoietic cell cancer‐related deaths (Munshi 2008). The incidence increases steadily with age and 75% of patients are older than 60 years (Durie 2001; Alexander 2007).
Myeloma cells derive from antibody‐producing B‐cells, therefore MM is characterised by the presence of often non‐functional monoclonal immunoglobulins in the serum or urine, or both (Laubach 2011). Patients frequently present with bone pain or increased susceptibility to infections, or both. Among other factors, the release of calcium from bone into the blood stream leads to hypercalcaemia; and the generally high protein concentration in serum may result in renal failure. End‐organ damage is the decisive factor that indicates the necessity of starting aggressive treatment and it has a major impact on the quality of life (Anderson 2008).
Over decades, MM has been classified according to the Durie‐Salmon (DS) staging system which aims at correlating clinical features with an estimate of myeloma cell mass (Durie 1975). Recently, a new international staging system (ISS) based solely on two readily available laboratory tests (β2‐microglobulin and albumin) has been developed (Greipp 2005).
Despite progress in the understanding of MM, especially regarding pathophysiology, its main causes remain unknown (Durie 2001; Alexander 2007). There seems to be more than one genetic pathway to MM and in addition to translocations, karyotypic instabilities are often observed. Hyperdiploid or non‐hyperdiploid tumours carry different numbers of chromosomes and the two classes differ with respect to chromosomal content, stromal dependency and aggressiveness of the tumours (Fonseca 2009).
The clinical course of MM is very heterogeneous with overall survival (OS) ranging from months to decades depending on the stage of disease or the presence of prognostic factors for example (Gertz 2007). Of unfavourable prognosis are an especially bad performance status, older age, high plasma cell labelling index, and high β2‐microglobulin or lactate dehydrogenase levels (Fonseca 2007). More recently, cytogenetic changes such as chromosome 13 and chromosome 11 abnormalities are also proposed to have a strong correlation with a worse outcome (Fonseca 2009; Nahi 2011). The high frequency of patients who either are or become refractory to treatment, a feature also markedly impacting on treatment outcome, is thought to be related to the strong interactions between tumour cells and the surrounding stroma (Podar 2009).
Patients with stage I, II, and III disease according to the ISS have a median OS of 62, 44 and 29 months, respectively (Greipp 2005). An improvement of OS was demonstrated in the last decade, both in the relapsed setting as well as from diagnosis, with a doubling in OS for patients who were treated with one or more of the novel agents (thalidomide, lenalidomide or bortezomib) (Kumar 2008; Johnsen 2010; Turesson 2010).
Therapeutic strategies in MM not only focus on prolonging OS but also to induce tumour response, inhibit tumour progression and delay disease‐related complications (Anderson 2008, Durie 2010). The surrogacy of tumour response with survival is still under discussion, however (Durie 2008; Harousseau 2009; Durie 2010). Validation of response as a patient‐relevant outcome is mainly hampered by the dynamic changes in treatment from the high‐dose combination chemotherapy approaches of the 1990s to integration of novel agents (Durie 2010).
One treatment approach for symptomatic MM was to intensify chemotherapy, either by increasing doses or by using combinations of multiple drugs (reviewed in Hahn 2003). The first high‐dose chemotherapy schemes resulted in high toxicity and extensive myelosuppression without substantial prolongation of survival (Barlogie 1986; Myeloma Trialists 98). Outcomes after intensive treatment were improved only by autologous stem cell transplantation (ASCT).
Description of the intervention
Eligibility for transplantation remains one of the main determinants of the treatment strategy until today (for example Kortüm 2010; NCCN 2011). A number of randomised trials have been performed comparing high‐dose chemotherapy supported by ASCT with conventional chemotherapy as first‐line treatment (cited in Levy 2005; Koreth 2007). The subsequent meta‐analyses did not provide evidence for an extended OS after ASCT, but progression‐free survival (PFS) was significantly improved ( Levy 2005; Koreth 2007). Importantly, the size and treatment effects with respect to both OS and PFS of single autologous stem cell transplantation (SASCT) over combination chemotherapy were quite variable in the individual trials (indeterminate, favourable and even unfavourable in one trial). Accordingly, the meta‐analytical result indicated statistically significant heterogeneity (Levy 2005; Koreth 2007). Although transplantation‐related mortality could be reduced to less than 5% in the majority of ASCT trials, it remained consistently higher after a transplantation regimen compared to chemotherapy alone, both in RCTs as well as in daily practice (Jantunen 2006a; Gertz 2008; Jones 2008).
How the intervention might work
Assuming a favourable outcome of SASCT compared to chemotherapy, in the early 1990s some investigators concentrated on tandem autologous stem cell transplantation (TASCT) (Harousseau 1992; Barlogie 1997). TASCT corresponds to a second prospectively planned ASCT within a few months after the first ASCT. TASCT was introduced to further improve or consolidate disease control as achieved after SASCT in view of a documented dose‐response relationship of melphalan with improved outcome (Awedan 2002; Giralt 2010).
Why it is important to do this review
MM is among the leading indications for ASCT in Europe and the USA (Copelan 2006; Gratwohl 2007). Two randomised studies comparing TASCT with SASCT as initial treatment in patients with symptomatic MM have been published as full text publications (IFM94; Bologna96). Further results from RCTs were presented at conferences (DSMM‐I; GMMG‐HD2; MAG95). As with RCTs comparing SASCT with combination chemotherapy (see Levy 2005; Koreth 2007), a statistically significant improvement of either survival endpoint was not consistently replicated in the other trials addressing the same question. This is a classical starting point for a meta‐analysis aiming at determining clinical or methodical reasons for the observed heterogeneity, either with respect to statistical significance or with respect to the size of the treatment effect (Egger 2001; Higgins 2011; Pignon 2001).
Although TASCT has been used as first‐line treatment of MM for more than 16 years now, there is continued controversy about its value. There is concern that the increased short‐term risks associated with double intensified treatment might call into question the benefit of long‐term disease control, in all or specific subgroups of patients (Fermand 2007; Barlogie 2007; Tricot 2008). Thus, an 'acceptable level of toxicity' for the primary MM treatment is still unclear. Usually, higher toxicity is accepted for a curative treatment compared to a palliative treatment (EMA/CHMP/EWP/520088/2008).
On the one hand, TASCT is an integral component of a "Total Therapy" programme (TT), which currently includes novel agents in both the induction and maintenance treatment regimen (Barlogie 1997; Total therapy). Barlogie and colleagues support TT as a key to the cure of MM (Barlogie 2009). Some investigators, however, have started to regard MM as a chronic disease and principally question the value of an intensive (and thereby toxic) treatment approach (Rajkumar 2008; Attal 2009; Gertz 2009).
Objectives
We systematically reviewed the evidence on the effects of TASCT compared to SASCT as first‐line treatment in MM on overall survival (OS), event‐free survival (EFS) and treatment‐ or transplantation‐related mortality. By systematically reviewing both prospectively planned features of the included trials and actual experiences recorded during their performance, we aimed to contribute to a discussion on clinical and methodological issues critical for the interpretation of the available evidence.
Methods
Criteria for considering studies for this review
Types of studies
Both randomised and non‐randomised trials comparing first‐line TASCT with SASCT in patients with symptomatic MM were included into the systematic review according to pre‐specified inclusion and exclusion criteria (Table 1). All of the inclusion criteria had to be fulfilled and none of the exclusion criteria for including a study. Duration of follow‐up was not a criterion for selecting trials.
1. Searching for trials: Inclusion and exclusion criteria for eligibility.
| General aspects | |
| Accepted study designs | Studies with contemporary control groups (RCT or observational) |
| Accepted date of publication | 1995‐2011 |
| Accepted publication type | RCTs: full text, abstract, conference presentation Observational studies: full‐text only |
| Inclusion criteria | |
| I1 | Patients with previously untreated symptomatic MM (excluding the prognostically very different plasma cell leukaemia (International Myeloma Workshop 2003)) comprising at least 85% of the study population |
| I2 | Intervention group: tandem autologous stem cell transplantation |
| I3 | Control group: single autologous stem cell transplantation |
| I4 | At least one of patient‐relevant outcomes reported (OS, EFS, transplantation‐ or treatment‐related mortality) |
| I5 | Observational studies: comparability of study groups with detailed information for at least 5 relevant prognostic criteria per arm |
| Exclusion criteria | |
| E1 | Other research questions (e.g. basic research, other therapeutic approaches, other diseases, prognostic studies) or publication type (e.g. narrative review, editorial, letter without original data) |
| E2 | Duplicate publication without additional information1 |
| E3 | Report without quantifiable/assignable outcome measures of first‐line treatment in patients with MM |
| E4 | No acceptable study design (or, in addition, publication type for observational studies) |
| E5 | Low number of evaluable patients per arm (< 25 patients per arm for RCTs, < 50 for observational studies)2 |
| E6 | Other Language of publication than English, French or German |
1 For cumulative reports such as registry data, only the last publication was included. Abstract presentations of included RCTs published in full‐text were only included if they contained additional data (e.g. long‐term follow‐up).
2 In view of the lower relevance of non‐randomised studies for assessment of intervention effects, larger studies were required for inclusion.
The reason for including comparative observational studies in the search for the systematic review was to prepare for customary reproaches against RCTs, that is their alleged artificiality or distance from clinical practice. In order for observational studies to be included, however, they had to report on a contemporary control group and both groups needed to be described with sufficient detail to allow assessment of potential 'confounding by indication' (Kunz 1998; Klungel 2004; Klein‐Geltink 2007; Vandenbroucke 2008). At least baseline characteristics, the treatment regimen and outcome data for the two groups had to be reported separately for both groups.
In view of a potentially critical publication bias if only full‐text articles were accepted, conference presentations of RCTs were eligible (Krzyzanowska 2003; Curt 2008; Ramsey 2008; Tam 2008) while full‐text publications only were acceptable for observational studies.
Types of participants
We included patients with MM at first diagnosis. Prior treatment was accepted if patients had been treated conventionally (melphalan, prednisone) for a limited time span (up to six months) (see Differences between protocol and review). No age restriction was applied.
We excluded patients with a localised form of myeloma as it has different biological behaviour and requires different treatment. We also excluded studies in patients with preliminary stages of MM and the closely related monoclonal gammopathy of unknown significance as well as smoldering myeloma, for which treatment is not recommended.
Types of interventions
We included all studies comparing first‐line TASCT with SASCT.
We included all trials assessing TASCT regardless of the timing of transplantation as long as the second transplantation was prospectively planned as first‐line treatment, before disease progression. We accepted all types of induction, conditioning and maintenance regimens and included studies irrespective of the source of stem cells.
Types of outcome measures
Ideally, both prolongation of the objective endpoint OS and improved quality of life should be used for demonstrating patient benefit (Anderson 2008; Durie 2010). Quality of life measures were, however, only to be included if they were assessed by validated instruments. Considerable confounding of OS was expected in all included RCTs as all were performed in the years during which the so‐called 'novel agents', with considerable efficacy in MM (thalidomide, lenalidomide or bortezomib), were developed for MM (Kumar 2008). Therefore, EFS can be considered as an indicator of efficacy as long as a deleterious effect on OS could be excluded, in an analogy to drug development (CPMP/EWP/205/95 2005). It is important to note that EFS also includes laboratory parameters which signal progression but need not immediately affect the patients' health status. Since the criteria for progression became more sophisticated over the time period during which the included trials were performed, for example by increasing sensitivity with respect to progression using immunofixation, there is some heterogeneity of definitions used in all included trials (Attal 1996,; Bladé 1998; Durie 2006). The outcome in clinical trials in oncology is often also assessed by measuring response to therapy. In view of the different definitions for response in the relevant time period of TASCT studies, and the ongoing discussion on whether a (best) response analysis allows a demonstratration of (durable) benefit for all patients, response was not assessed for the present review (Harousseau 2009a; Moreau 2011) (see Differences between protocol and review). Treatment‐ or transplant‐related mortality, sometimes indiscriminately abbreviated as TRM in the original reports, was to be analysed as a measure of safety (see Differences between protocol and review).
Search methods for identification of studies
Evidence on the efficacy and safety of TASCT compared to SASCT in the first‐line treatment of MM was identified by systematic electronic searching for literature in two bibliographic databases (Cochrane Central Register of Controlled Trials (CENTRAL); MEDLINE) according to the recommendation by The Cochrane Collaboration (Dickersin 1994; Higgins 2011). We had to exclude the database EMBASE due to budgetary restrictions and considered only publications in English, French and German. Since CENTRAL is updated with randomised controlled trials from EMBASE through retrospective searches conducted by the UK Cochrane Centre, up to 2008, EMBASE was at least partially covered through CENTRAL in our review.
Since observational studies, for example registry reports, were to be included in addition to RCTs, the search filter was adapted accordingly (see Appendix 1 and Appendix 2). Overall, the sensitivity of the presented search was lower for observational studies compared to RCTs due to the choice of bibliographic databases (CENTRAL is enriched for RCTs only).
The systematic search was restricted in the years of publication with the starting point of 1995 in order to approximate the establishment of TASCT as first‐line treatment of MM (Barlogie 1997). A pilot search was performed in 2007. The systematic search of the two bibliographic databases MEDLINE and CENTRAL was performed on 30th of April 2010. The search was updated on the 23rd of May 2011.
Electronic searches
Bibliographic databases (1995 to 2011)
See Appendix 1 for MEDLINE, and Appendix 2 for CENTRAL search strategy.
Searching other resources
Databases of ongoing trials
Further potential sources for identifying trials were asking clinical experts (personal communications by Alexander Greb, Roland Schnell) or by browsing reference lists and the following websites using keywords such as "myeloma AND transplantation".
www.myeloma.org.
www.onkodin.de.
www.clinicaltrials.gov.
www.controlled‐trials.com.
In addition, the Deutsches Krebsstudienregister (German Cancer Trials Registry) and Google were searched using the names of the first or senior authors' of the included trials.
Handsearches
No additional handsearches were performed.
Contact
Authors of the all RCTs were contacted at least once by email or telephone, or both, for further information.
Data collection and analysis
Information from included studies is either presented by study in the Characteristics of included studies or by topic: baseline characteristics, treatment regimen, definition of endpoints, in Table 2Table 3; Table 4; and Table 5, respectively. Quality of reporting and extent of uncertainty due to missing information was captured for all included studies (Table 6).
2. Baseline characteristics of patients.
| Baseline characteristics | N |
Age [y] |
β2 micro‐globulin [mg/l] |
Sex [% male] |
Durie Salmon stage n (%) |
Status at Rdx | Prognostic factors reported (n) |
Note |
|
IFM94 TASCT |
200; [203* in LTFU] |
52 ± 6 [mean] |
5±9 | 55 | I: 14 (7) II: 31 (16) III: 155 (78) |
Untreated | Age, Alb, B2M,
BM cyt., Ca, Crea, CRP, DS, Hb, LDH, M protein Sex (12) |
"No significant differences between groups" |
|
SASCT IFM94 |
199 [198* in LTFU for EFS] |
52 ± 6 [mean] |
5±6 | 56 | I: 17 II: 23 III:159 |
|||
|
MAG95 TASCT |
114 | 50
[22‐56] (overall) |
2.8 [0.9‐65] (overall) |
NR | I: NR II: ‐ (12) III: ‐ (85) (overall) |
Newly diagnosed | Age, B2M, DS, M protein (4) |
"No significant differences between groups", not reported per treatment arm |
|
MAG95 SASCT |
113 | |||||||
|
Bologna96 TASCT |
158 | 53± 6 | 4.6±7.8 | 60 | I: 31 (20) II: 29 (18) III: 98 (62) |
Previously untreated | Age; B2M; BM cyt.; CRP; Crea.; DS; Hb; Pt; M protein; Sex (10) |
"No statistically significant differences between groups" |
|
Bologna96 SASCT |
163 | 52± 6 | 4.2±5.2 | 61 | I: 32 (20) II: 23 (14) III: 108 (66) |
|||
|
GMMG‐HD2 TASCT |
180* | 56 (med.) | 2.8 [med.] |
NR | I: (excl.) II: III: (63% in interim analysis) |
At least SD after induction | Age, Alb, B2M, CRP, Hb, (5) |
Reported factors similar after visual inspection; total number of included patients unclear (N=358‐485); |
|
GMMG‐HD2 SASCT |
178* | 55 (med.) | 2.5 [med.] |
NR | I: (excl.) II: III: (70% in interim analysis) |
|||
|
DSMM‐I TASCT (immature) |
98 | 54 (30‐60) | [>3.5 g/dl] 31.6% |
63 | I: (excl.) II: 28% III: 72% |
At least SD after induction and sufficient number of stem cells collected | Age, B2M, Crea, DS, LDH, M protein, bone, sex (8) |
"No statistically significant differences between groups" |
|
DSMM‐I SASCT (immature) |
100 | 54 (34‐61) |
[>3.5 g/dl] 29% |
63 | I: (excl.) II: 38% III: 62 % |
rounded figures; Alb: serum albumin; B2M: β2 microglobulin; BM cyt: bone marrow cytosis; Ca: serum calcium; Crea: serum creatinine; CRP: C‐reactive protein; DS: Durie‐Salmon stage; Hb: haemoglobin; LDH: serum lactate dehydrogenase, LTFU: long‐term follow‐up; Pt: platelets; Rdx: randomisation
˜ median OS read of KM curves; * evaluated patients
3. Treatment regimen in included studies.
| Study | Stem cell mobilisation/ source (type; minimal number per transplant | Induction | Conditioning ASCT | Compliance ASCT | Maintenance [Compliance] | Salvage | Cross‐over (SASCT to TASCT) | Notes |
|
IFM94 TASCT |
G‐CSF for PBSC or BM harvest after induction; (PBSC or BM; 2*106 CD34+/kg) |
3‐4 x VAD |
1. Mel 140 2. Mel 140 + 8 Gy TBI |
1. 88% 2. 78% |
Interferon α; [49%] |
59%, e.g. 21% Thal., 17% SCT |
/ | 2nd rdx BM/PBSC no clinically relevant interaction; different dose intensity up to first ASCT; |
|
IFM94 SASCT |
as TASCT | as TASCT | Mel 140 + 8 Gy TBI | 85% | Interferon α; steroids; [57%] |
68% e.g. 12% Thal., 17% SCT |
NR | |
|
MAG95 TASCT |
CYP, G‐CSF; PBSC (PBSC; NR) |
HD steroids and ? | 1. Mel 140 2. Mel 140 +VP16 +12 Gy TBI |
1st: 100% 2nd: 92% |
NR or none | NR | / | Stem cell collection after high‐dose steroids; clinically relevant interaction between purging and ASCT arm; notably different dose intensity and dose density up to and including first ASCT |
|
MAG95 SASCT |
as TASCT |
HD steroids and 3 x “VAD‐like” |
BCNU +VP16,
+Mel 140 +CYP + 12 Gy TBI |
94% | NR | NR | NR | |
|
Bologna96 TASCT |
CYP G‐CSF (PBSC 2*106 CD34+/kg) |
4 x VAD | 1. Mel 200 2. Mel 120 + oral BU (4 mg/kg d‐5 to d‐3) |
1st: 90% 2nd: 65% [e.g. refused by patient: 7%] |
Interferon α; [55%] |
10% SCT 55% novel agents |
/ | Same dose intensity and density up to and including first ASCT; |
|
Bologna96 SASCT |
as TASCT | as TASCT | Mel 200 |
85% | Interferon α; [77%] |
33% SCT; 50% novel agents |
NR | |
|
GMMG‐HD2 TASCT |
HD CYP/IFO G‐CSF (PBSC; NR; purging optional) |
VID or VAD (up to 6 cycles; until CR or plateau) | 1. Mel 200 2. Mel 200 |
1st: 93%* 2nd: 52% [refused by patient: 28%) |
Interferon α; [NR] |
> 20% salvage SCT; > 40 % novel agents ) |
/ | No interaction reported for optional randomisation VID vs VAD (VAD less toxic) and ASCT; same dose intensity and density up to and including first ASCT |
|
GMMG‐HD2 SASCT |
as TASCT | as TASCT | Mel 200 |
88% | Interferon α; [NR] |
(no info per arm reported | NR | |
|
DSMM‐I TASCT |
IEV + G‐CSF
(PBSC 2*106 CD34+/kg) |
4 x ID | 1. Mel 200 2. Mel 200 |
1st: NR 2nd: NR |
Interferon α; [NR] |
NR | / | Different dose intensity up to and including first ASCT |
|
DSMM‐I SASCT |
as TASCT | as TASCT | BU CYP + 9 Gy TMI | 72% | Interferon α; [NR] |
NR | 18% (time point not reported) | |
| Current standard and reporting recommendations | HD CYP + G‐CSF; unselected graft |
novel agent plus dexamethasone | Mel 200; no TBI |
Consider to increase number of drop‐out for sample size calculation | Report compliance per arm | Report number of patients and outcome per arm | Report number of patients and time point/status at cross‐over per arm; |
BM: bone marrow; BCNU: carmustine; BU: busulfan; CD34: cluster of differentiation (stem cell marker); CR: complete response; CYP: cyclophosphamide; G‐CSF: granulocyte‐colony stimulating factor; Gy: Gray; HD: high‐dose; IFO: ifosfamide; IEV: Ifosfamide, epirubicin, etoposide; ID: idarubicin, dexamethasone; Mel: Melphalan; NR: not reported; PBSC: peripheral blood stem cells; rdx: randomisation; SCT: stem cell transplant; TBI: total body irradiation; TMI: total marrow irradiation; VAD: vincristine, adriamycin, dexamethasone; VID: vincristine, idarubicine, dexamethasone VP16 etoposide * of evaluated patients
4. Definition of survival outcomes.
| Study | d0 | OS | EFS | Response criteria | Progression | Relapse |
Follow up (med.) |
Note |
| IFM94 | Diagnosis | Death | Progression relapse death |
IFM (pre‐EBMT) methods without immunofixation, i.e. overestimation of CR | 25% increase in the para‐protein level after two cycles of the initial chemotherapy; | After CR
reappearance of M‐protein, recurrence of bone marrow infiltration, or both after response 50% increase above the plateau of M‐protein in two samples obtained 4 weeks apart |
75 mo / 11.6 y |
Including period of induction and mobilisation, lower sensitivity with respect to detecting relapse without immunofixation |
| MAG95 | After HD steroids before mobilisation | NR |
NR |
NR | NR | NR | 73 mo | Including period of induction and mobilisation |
| Bologna96 | Start of treatment | Death | Progression relapse death |
EBMT+ nCR (=CR with positive immunofixation) |
Responders: > 25% increase in M protein from nadir and/or appearance of new bone lytic lesions. non‐responders: > 25% increase in M protein from baseline and/or appearance of new bone lytic lesions. |
After CR/nCR reappearance of M protein on immunofixation or routine electro‐phoresis, respectively, and/or appearance of new bone lytic lesions |
70 mo (survivors only) | Including period of induction and mobilisation |
| GMMG‐HD2 | Unclear |
NR |
NR |
NR | NR | NR | Unclear 2003: 36 months 2005: 24 months 2007: NR |
Reference point for time‐to event and follow‐up unclear; likely excluding induction but including mobilisation in selected patients |
| DSMM‐I | Unclear |
NR |
NR |
NR | NR | NR | 48 mo total?? | Reference point for time‐to event and follow‐up unclear; so called TRM of 4% reported for TASCT arm but OS at 100% for 500 days of FU. |
CR: complete response; EBMT: European Bone Marrow Transplantation; FU: follow‐up; IFM: Intergroupe francophone du myelome; med: median; mo: months; nCR: near complete response
5. Defition of treatment‐ or transplant‐related mortality.
| Study | Definition used |
How reported in which population |
Start point |
Period | Other causes of death reported separately |
| IFM94 | Treatment‐related mortality: "toxic effects of transplantation (sepsis)" "toxic effects to VAD (sepsis)" |
number of deaths; % per arm; ITT | Unclear |
NR |
"Death due to myeloma"; "cardiovascular or thrombo‐ embolic disease" "another cancer" "unknown cause" "suicide" |
| MAG95[1] | Interim analysis: "Toxic death"1
(performed with 85% of final population) Final analysis "Early death" "death within 9 months post randomisation including toxic death and fatal progressive disease" 2 |
% per arm; population NR (for both definitions) |
Rdx | 9 months | NR |
| Bologna96 | "Transplantation‐related mortality included any death within 90 days and attributable to high‐dose therapy" | % per arm; population NR |
SCT | 90 days | N of deaths as reason for non‐compliance (no cause of death reported) |
| GMMG‐HD2 | So‐called TRM (treatment or transplantation?) reported without the clarification of population |
% per transplantation per arm; population NR3 | NR | NR | NR |
| DSMM‐I | So‐called TRM (treatment or transplantation?) | % per arm; population NR | NR | NR* |
NR |
1 toxic death lower in TASCT compared to SASCT arm (7% versus 9%)
2 notable difference in dose intensity between TASCT and SASCT resulting in 7% versus 12% early mortality.
3 only 56% compliance with second ASCT
* plateau of 100% in Kaplan‐Meier curve from d1‐d400 in TASCT arm despite so‐called TRM of 4%; reported safety population for SASCT N=80; TASCT N=118 since 20 patients crossed over from SASCT to TASCT arm
6. Quality criteria for the assessment of bias.
| Quality criteria | positive | negative | unclear | Consequence of negative classification |
| RCT | ||||
| Central randomisation | Performed with adequate methods described | Inadequate methods described | Not Reported (NR) | Confounding |
| Concealment of allocation | Performed with adequate methods described | Inadequate methods described | NR | Confounding |
| Statistical power | Powered for survival | Powered for response | NR | Underpowered for survival |
| Observational studies | ||||
| Selection of patients | Based on transparent criteria; plausible matching criteria | Indicative of strong selection bias; resulting in incomparable groups | NR | Confounding |
| Study size | Justified | Fewer patients than planned | NR | Underpowered for OS |
| RCT and observational study | ||||
| Confounding at baseline | Reporting of at least 5 key prognostic factors per arm allowing assessment of similarity of groups | Relevant difference in key prognostic factors obviously present clinically but not addressed | </= 5 factors reported or no information per arm | Confounding |
| Comparability of treatment | Treatment identical up to and including first ASCT | Notable difference with onset and duration of toxic treatment | Unclear | Lead‐time bias/ confounding |
| Subsequent treatment | Information available with type and outcome per arm | No information available | NR or no info per arm | Confounding |
| ITT analysis | Yes, including definition reported | No | Analysis or definition NR | Selection and attrition bias with loss of power1 |
| Compliance with 2nd TASCT | High (> 85%) | Low (<70%) | 70‐85% or NR |
Attrition bias1 or loss of power2 |
| Completeness of follow‐up | <15 % loss to follow‐up; censoring rules defined | Incomplete follow‐up (> 15%) with unknown consequence for analysis | Loss to follow‐up or censoring rules NR | Attrition bias1 or loss of power2 |
| Cross‐over | Reported and < 15% | Reported and > 15% | NR | Null bias |
| Definition of endpoints | Comprehensive reporting of accepted/ established endpoint definitions | Use of biased endpoint (e.g. different period of observation for study arms) | Incomplete reporting* | Lack of scientific validity |
| Standardisation of diagnostic procedures or central review | Mentioning of standardisation efforts, use of accepted staging systems | Reporting of centre effects or major changes in diagnostic criteria over the course of the study | NR | Information bias (if systematically different between arms) |
| Data maturity (Tricot 2009) | > 4 years follow‐up (FU) | Less than 4 years FU | NR | Insufficient follow‐up |
| Publication type& | Peer‐reviewed full‐text |
Conference presentations | Not Applicable (NA) | Error‐prone due to less proof‐reading (at the least) |
NR not reported
1 if informative censoring
2 if at random
* of endpoint definitions with major impact on denominator (e.g. so‐called TRM)
Selection of studies
Studies were selected in two steps by one researcher (Figure 1). An independent meta‐analysis published in 2009 and a health technology assessment (HTA) report published in 2011 were regarded as control for the comprehensiveness of the search (Kumar 2009; IQWIG N05‐03C). Evidently irrelevant studies (such as animal experiments, other treatment approaches for MM or narrative reviews) were excluded by screening the title and abstract of the reference identified. The decision was based on the full‐text publication in the case of uncertainty. Selected studies were assessed with an eligibility form as to whether they meet all inclusion criteria and none of the exclusion criteria (Table 1).
1.
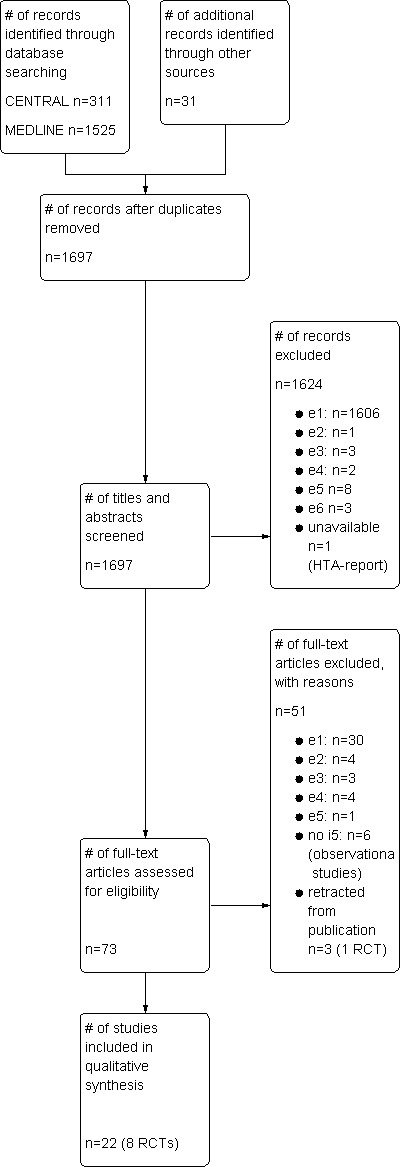
Study flow diagram.
Data extraction and management
Data extraction tables were developed in order to provide a clear framework for the verifiable collection of data items. Data items included both elements from prospective planning of the study (for example the hypothesis for sample size calculation) as well as the actual experience of the trial (for example compliance with intended treatment). The following information was collected by one researcher (FN) and the correctness of the extracted data was checked for accuracy by another researcher (RS).
Information on study basics such as the setting of the trial (time period, location), design, basis for sample size calculation, an intention‐to‐treat analysis (ITT) and number of patients dropping out, notable inclusion or exclusion criteria, type of reported outcomes relevant for the present review, and type of publication are described in the Characteristics of included studies. The most important aspects for interpretation of the individual study (as discussed also in the bias section) are also repeated as 'Notes'.
Information from the included trials regarding the following characteristics were grouped per topic (rather than by individual trial) in order to facilitate the comparison of one feature across all included trials.
Baseline characteristics of patients (n, age, β2‐microglobulin, sex, stage, status at randomisation, type and number of prognostic factors reported) were extracted for all included studies (Table 2).
Intervention details (type of stem cell mobilisation and source of stem cells, induction, conditioning, maintenance and salvage treatment) were noted with the aim of highlighting clinically relevant differences in the treatment regimen between study arms and between studies, and their deviance from current treatment standards. The compliance with the intended treatment schedule and the extent of cross‐over (if reported) were also extracted (Table 3).
Definitions of outcome measures (OS; EFS: starting point of the analysis for time‐to‐event data, definition of relevant events including the extent of follow up (Table 4); so‐called TRM: definition of numerator (cause of death) and denominator (type of population), period of observation and additional causes of death reported in the study) were summarised for all included studies (Table 5). See also Differences between protocol and review.
Results concerning both benefit (OS, EFS) and harm (so‐called TRM or early mortality) of the individual trials were extracted.
Assessment of risk of bias in included studies
The quality of included trials was critically evaluated according to methodological criteria listed in Table 6. Several of these criteria were shown empirically to have an impact on the validity of trial results and are listed, for example in the CONSORT statement, the Cochrane Handbook for Systematic Reviews of Interventions using RCTs or in the STROBE statement for observational studies (Juni 2001; Moher 2001; von Elm 2008; Higgins 2011). In addition, characteristics regarded as essential for the interpretation of trials studying TASCT were discussed, for example acute toxicity of the treatment regimen or compliance with the treatment plan. A colour code was applied for the risk of bias table (Figure 2) in order to indicate compliance with (green) or missing (red) high quality standards (Table 6), while lack of information was coded with yellow. The quality of all studies which were deemed eligible for the review were systematically assessed by one researcher (FN) and discussed with the senior author (RS).
2.
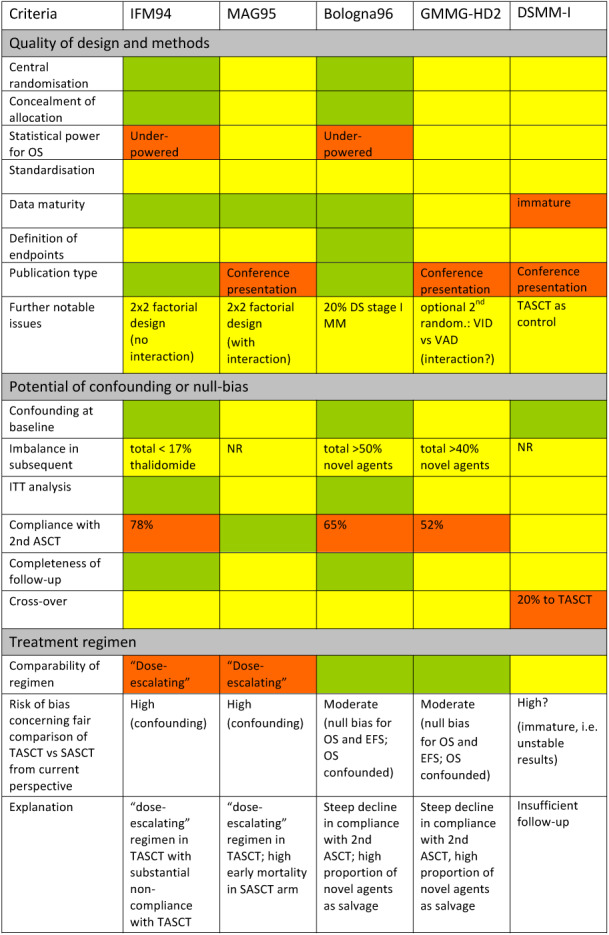
Overview of Risk of bias in the set of included trials: Green colour indicates sufficient information available to verify adequate methods or similar features in both study arms. Red colour is used to highlight characteristics which were systematically different between study arms and therefore may result in a biased comparison (both conservative or anti‐conservative). Yellow colour indicates lack of (sufficiently detailed) information.
Measures of treatment effect
No meta‐analysis has been performed for the comparison of TASCT with SASCT (see Differences between protocol and review). Results from individual studies are presented in tables aimed at standardising highly variable reporting (Table 7). In addition, results are summarised as a narrative (Effects of interventions).
7. Effect of TASCT compared to SASCT on OS, EFS and "TRM".
| Follow‐up months (mo) |
Kaplan‐Meier curve | Median OS | Kaplan‐Meier curve | Median EFS | Treatment‐ or Transplant‐related mortality | Notes | ||||
| Study | OS | TASCT | SASCT | EFS | TASCT | SASCT | TASCT | SASCT | ||
| IFM94 | 75 mo (median) |
TASCT superior; separation of curves after ca. 26 mo; |
58 mo* | 48 mo* P=0.01 |
TASCT superior; separation of curves after ca. 18 mo; |
30 mo* | 25 mo* P=0.03 |
Treatment‐related deaths (including induction) N= 12 (6%) Transplantation‐related deaths (sepsis) N=5 |
Treatment‐related deaths (including induction) N= 8 (4%) Transplantation‐related deaths (sepsis) N=3 |
Five additional causes of death listed; Long term follow‐up (11.6 years) OS P=0.08 EFS P=0.06 |
| MAG95 | 73 mo (median) |
SASCT inferior; (overall transient) separation of curves after cluster of events in control arm after 2‐4 and 35 months; |
75 mo | 57 mo | No KM‐curve | 34 mo | 31 mo | Toxic death (interim data§): 7% "Early mortality" N=8 (7%) |
Toxic death (interim data§): 9% "Early mortality" N= 13 (12%); |
Final OS survival proportions similar |
| Bologna96 | 70 mo (median for survivors) | no KM‐curve (inconsistent median OS and 7 year OS (TASCT 43 vs SASCT 46%) indicate crossing curves) |
71 mo | 65 mo | TASCT superior; separation of curves after 18 mo; 5 y EFS: 29% vs 17% |
35 mo* | 23 mo* P=0.001 |
Transplantation‐related deaths Table 5 4% |
Transplantation‐related deaths Table 5 3% |
Compliance with second ASCT: 65% |
| GMMG‐HD2 | Unclear 2003: 36 mo 2005: 24 mo 2007 (final): NR |
ITT: superimposable curves (multiple crossings); PP: SASCT slightly superior |
ca. 77 mo | ca. 72 mo | ITT: TASCT transiently superior; crossing of curves at 38 mo; PP: similar to ITT |
ca. 29 mo | ca. 25 mo | So‐called TRM Table 5 1st 2% 2nd 3% (population unclear) |
So‐called TRM Table 5 2% (population unclear) |
Compliance with second ASCT: 52% of evaluable patients |
| DSMM‐I | ca. 48 months (reported as median follow‐up; according to KM: total follow‐up) | TASCT initially superior; crossing of KM curves at 36 mo; 4 yr OS 72% in both arms |
Not reached | Not reached | TASCT initially superior; crossing of KM curves at 22 mo |
36,4 mo | ca. 43.4 mo | So‐called TRM (excluding induction)
Table 5 4% (population?); (OS at 100% for 400 d according to KM‐curve) |
So‐called TRM (excluding induction)
Table 5 3% (population?) |
high proportion of cross‐over; |
* statistically significant difference; ca. = read from Kaplan‐Meier plots
§ interim data with 85% of enrolled patients
Results
Description of studies
Results of the search
Overall, 1697 potentially relevant articles, abstracts or conference presentations were identified by systematic searching for both randomised and non‐randomised controlled studies in CENTRAL and MEDLINE. The comprehensiveness of our search was confirmed since all studies that were included either in the recently published meta‐analysis or the HTA report on TASCT (Kumar 2009; IQWIG N05‐03C) were also identified by our search. A flow chart of the systematic search for eligible studies is presented in Figure 1 according to the recommendations of the PRISMA statement (Moher 2009).
Twenty‐two references dealt with our research question and were eligible for the systematic review according to pre‐specified inclusion and exclusion criteria (Table 1), representing eight RCTs in total. One additional RCT had been retracted from publication and was therefore not included (Abdelkefi 2008; Abdelkefi 2009). Five RCTs were eligible for inclusion in a meta‐analysis while three were ineligible, for various reasons. One RCT was identified as ongoing (currently recruiting patients) so that no data were yet available NCT01109004. Two trials (DM00‐196 and N0265041749) were reported as completed or terminated in clinical trial registries but no publications with clinical outcome data were available. One abstract on DM00‐196 focused on the reasons for withdrawal from the treatment protocol (Wilson 2003). N0265041749 had been registered as completed in the UK national registry but remained unpublished. The principal investigator of N0265041749, when contacted for further information, explained that the study had been closed due to insufficient recruitment. No further information was obtained after having contacted the authors of the other RCTs.
All of the five otherwise eligible comparative observational studies (Lahuerta 2003; EBMTR; Kim 2009; Koren 2010; NMSG) were excluded from the systematic review since they did not sufficiently describe the included patient population and applied treatment regimen (see Excluded studies).
Included studies
Description of individual trials
In the following sections, the characteristics of the included studies with outcomes relevant to the systematic review are briefly summarised individually (Characteristics of included studies). Further information on the trial design and performance is summarised below (comparison of characteristics of studies) and listed in detail (Table 2, Table 3, Table 4, Table 5). The results of individual studies are described in the section Effects of interventions and Table 7, both sections aiming at standardising highly variable reporting.
IFM94: Attal and colleagues selected 399 patients younger than 60 years with symptomatic disease predominantly of Durie‐Salmon (DS) stage II and III. Patients were randomised before induction treatment by a 2 x 2 factorial design to: a) TASCT or SASCT and b) bone marrow or peripheral blood stem cells. Treatment groups were reported to have similar baseline characteristics (Table 2). Transplantation regimens differed between the first and second ASCT in the TASCT arm (Table 3).
MAG95: patients with MM (stage II and III) younger than 56 years were selected for MAG95 (Table 2); 227 patients were randomised before induction treatment by a 2 x 2 factorial design between: a) TASCT or SASCT and b) selection of CD34+ positive cells from peripheral blood stem cell grafts, or no selection (Table 3). Treatment groups were reported to have similar baseline characteristics (Table 2). Transplantation regimens differed between arms and between the first and second transplantation in the TASCT group (Table 3).
The Bologna96 trial also assessed the concept of TASCT compared to SASCT in 321 patients younger than 60 years with symptomatic MM stage I, II and III. Patients were randomised before induction treatment to either one or two ASCT with similarly dosed preparative regimens (Table 3). Treatment groups were reported to have similar baseline characteristics (Table 2).
The GMMG‐HD2 included between 358 and 485 patients with DS stage II and III MM younger than 66 years (numbers inconsistent between different conference presentations). While only patients with at least stable disease were randomised between one or two ASCT with identical preparative regimens (Table 3), an optional randomisation between two induction regimens was reported for this trial. It remained unclear whether the randomisations were performed and analysed under the same protocol. Treatment groups were reported to have similar baseline characteristics (Table 2).
DSMM‐I evaluated a novel conditioning regimen including bone marrow irradiation preceding SASCT and compared it to the 'standard' TASCT regimen as control (Table 3). Overall, 198 patients younger than 60 years with SD stage II and III disease were randomised if they had at least stable disease after induction and had successfully mobilised a sufficient number of stem cells. Treatment groups were reported to have similar baseline characteristics (Table 2).
Comparison of characteristics of studies
In the following section, general observations on the set of included trials are discussed with reference to more detailed tables on baseline characteristics (Table 2), treatment regimen (Table 3), and definition of endpoints (Table 4; Table 5).
Baseline characteristics of included patients
Information on allocation was limited for the majority of studies. All authors documented the similarity of study groups however, either by referring to P values or by presenting similar values for selected prognostic factors. The only prognostic factors described for all studies were age, β2‐microglobulin and stage (Table 2). For MAG95, baseline characteristics of treatment arms were not reported separately. While MAG95 included the youngest patient population of the set of included trials, the Bologna96 trial included the highest proportion of patients with symptomatic stage I disease. In DSMM‐I and GMMG‐HD2, only patients responding to induction treatment were included. No obvious hierarchy concerning baseline risk between studies could be determined in view of the heterogeneity of eligible age range, stage or prognostic factors.
Treatment plan and compliance
None of the included trials assessed exactly the same treatment strategy. All trials deviated from what would be considered standard today in at least one component of the ASCT regimen (SASCT or TASCT). Different combinations or sequences of induction, mobilisation, conditioning or maintenance regimen were used. Rather than repeating one ASCT regimen, the TASCT regimens used in IFM94 and MAG95 could be described as dose‐escalating since the intensity of the preparative regimen used for the initial ASCT was lower than for the second ASCT. Compliance was usually high with the first or only transplantation in all trials but was highly variable for the second transplantation (Table 3). Causes of non‐compliance were either myeloma‐, toxicity‐ or procedural‐related, or were based on patients' preferences: patients developed contra‐indications for second transplantation while on the trial, or an insufficient number of stem cells precluded further ASCT. A notable number of patients refused further treatment or were denied coverage. Among the reported reasons for non‐compliance, refusal by patients was the most common reason for not performing the second transplantation. The lowest compliance with second transplantation was observed in Bologna96 (65%) and GMMG‐HD2 (52% of evaluable patients).
Information on maintenance or salvage treatment (treatment after relapse) was limited for the majority of trials. First, it was often not reported separately for the two arms (DSMM‐I; GMMG‐HD2; MAG95) and second, only proportions of types of salvage treatment were reported but not the outcome. Patients crossing‐over to the other treatment arm were only reported for DSMM‐I, where 18% of patients from the SASCT crossed over to the TASCT arm without information on their status at the time of cross‐over.
Assessment of treatment effect
Although heterogeneity of disease and potential differences in response criteria existed (locally or over time), no trial reported on efforts of standardisation across the usually high number of study centres (see Characteristics of included studies).
Excluded studies
Randomised trials
Sonneveld 2007 was excluded from the systematic review due to the non‐myeloablative approach of intermediate dosing of melphalan in the control arm (two times 70 mg/m² melphalan instead of one high dose of melphalan 140 mg/m²).
Observational studies
Comparative observational studies were to be included in the trials if both the population and treatment regimen were described in sufficient detail. None of the observational studies identified allowed us to adequately assess or rule out 'confounding by indication', the most critical issue for interpretation of treatment effects from observational studies (Kunz 1998; Vandenbroucke 2008). Therefore those data, mostly registry data (EBMTR; Kim 2009; NMSG) or single centre experience (Lahuerta 2003; Koren 2010), were not formally included in this systematic review.
Total therapy (no RCT)
TASCT is an integral part of 'Total therapy' (TT), developed by Barlogie and colleagues (Total therapy). TT publications were not included in the systematic review for the following reason: TT was supported by an historical comparison of a single‐armed study in a single specialised centre, both initially (TT1; Barlogie 1997) and upon development of further TT generations (Barlogie 2009). With this design, selection bias at enrolment and potentially informative censoring during study conduct can neither be assessed nor excluded. Of note, all generations of the total therapy program, except TT1, integrate the so‐called novel‐agents either before or after TASCT, or both, which is regarded as a critically different treatment approach compared to that in the set of included RCTs (Barlogie 2006).
Risk of bias in included studies
Assessment of the potential of bias was based on several methodical aspects and actual experience during (for example compliance) or after the performance of the trial (for example publication type) (Table 6; Figure 2).
Most information was available for classification of risk of bias for IFM94 and Bologna96, which were published as full‐text publications, compared to the trials available as conference presentation only (DSMM‐I; GMMG‐HD2; MAG95). The quality of the trials was rated both according to prospective elements such as the availability of an ITT analysis, the reported basis for sample size calculation or whether a detailed description of the baseline would allow the reader to rule out clinically relevant if not statistically relevant differences in baseline characteristicss (see Figure 2).
Bias was also assessed based on the time point of analysis, that is the extent of follow‐up or data maturity. There is consensus that minimal median follow‐up in MM should be more than four years (Harousseau 2009a; Tricot 2009). RCTs with follow‐up of more than four years were marked in green, while trials with less or insufficient information were coded in red or yellow (see Figure 2). Since neither conference presentations nor abstracts contained sufficient data to ascertain the validity of the results, the lack of a full‐text publication was also rated as a negative feature (Tam 2008).
The overall potential for bias (direction of bias unclear) was rated per study (Figure 2) based on the most important factors involved (Table 6). With respect to individual endpoints, OS was considered to be more biased compared to EFS and so‐called TRM since, in addition to transplantation regimen and compliance (see Other potential sources of bias), follow‐on treatment could have impacted on the effect on OS.
IFM94, MAG95 and DSMM‐I were considered as having the potential to be highly biased, with an unclear direction of bias (conservative or anti‐conservative). Most relevant issues for the classification of IFM94 were the dose‐escalating TASCT regimen (Other potential sources of bias), low compliance with the second ASCT and unclear type and effect of follow‐on treatment (Table 3). The critically limited amount of information due to its publication type (Selective reporting), together with TASCT and SASCT regimens which would be regarded as non‐standard today (Other potential sources of bias), were decisive for classifying MAG95 as potentially highly biased. In view of its relatively short follow‐up, apparently unstable results with multiple crossings at the right end of the Kaplan‐Meier curves and high cross‐over rate, DSMM‐I was also rated as having the potential to be highly biased, with an unclear direction of bias.
Since Bologna96 and GMMG‐HD2 (at least) evaluated a standard‐dose TASCT conditioning regimen from a contemporary perspective, they were (rather subjectively) rated as moderately biased despite the steep decrease in compliance with the second ASCT (see also Discussion).
Allocation
Information on allocation was limited for the majority of studies (see Characteristics of included studies). While all reports were reported as randomised, only the full‐text articles of IFM94 and Bologna96 allowed the assessment of procedural aspects of randomisation which are necessary to judge whether, for example, concealment of allocation would be achieved, or not.
Blinding
In view of the obvious differences in treatment plan, none of the studies were blinded. This is acceptable for ethical reasons but heightens the importance of objective outcome assessment.
Incomplete outcome data
The steep decrease in compliance with the second ASCT most likely results in a null bias, that is a conservative shift of the treatment effect in an ITT analysis. The overall shift of the treatment effect was, however, unclear in view of the dilution of effects by non‐compliance, unpredictable selection bias of compliant patients and further confounding by salvage treatments (Schulz 2002; Wheatley 2006).
ASCT as first‐line treatment of MM was followed by further lines of therapy as salvage treatment in case of relapse or progression (see Table 3). Since all included RCTs coincided with the clinical development of several so‐called 'novel agents' with considerable activity in myeloma, the type and outcome of follow‐on treatment is critically important for the interpretation of OS. Information on the type of salvage treatment per arm, if at all available, was insufficient for all trials. No or limited information was available concerning completeness of follow‐up or cross‐over (before an event) for most RCTs.
Selective reporting
Both the reported items and printing quality differed widely between full‐text publications, abstracts and conference presentations. Endpoints were only superficially defined in most trials, with only Bologna96 referring to censoring rules in time‐to‐event analyses(Table 4). Kaplan‐Meier (KM) curves were not available for OS in the full‐text publication of Bologna96 and not for EFS in the conference presentation of MAG95.
Both definitions of the relevant period of observation and causes of death to be listed as so‐called TRM remained unclear in the majority of trials (indicated by depicting the latter as 'so‐called TRM') (Table 5). Only interim data on 'toxic death' were available for MAG95 (see Discussion). In addition, mainly proportions (and not number of deaths in a specified population) were reported in the majority of trials, leaving room for interpretation concerning the denominator used for the calculation of the proportions, which is especially important in view of usually lower compliance with the second transplantation (Table 3).
Only those studies in which statistically significant improvements in survival outcomes were observed were published as full‐text publications (IFM94; Bologna96) while the non‐significant results of three further RCTs were available on the Internet as conference presentations only (MAG95, GMMG‐HD2, DSMM‐I). Two RCTs remain unpublished in spite of being registered as terminated (DM00‐196; N0265041749). According to the principal investigator, DM00‐196 was closed due to insufficient recruitment. The dependency of publication on the size of the treatment effect (and the associated statistical significance) is an obvious sign of publication bias which could only be detected by a meticulous literature research or interaction with experts who had been aware of the trials during their performance.
Selective reporting of outcomes, apart from uncertainty due to unclear definitions (see Incomplete outcome data; Table 4; Table 5), was not observed for the endpoints considered for this systematic review.
Other potential sources of bias
Impact of treatment plan and compliance
While the cumulative dose was higher in the TASCT arm for all studies, dose density differed notably between TASCT and SASCT in IFM94, MAG95 and DSMM‐I. When assessing the comparability of treatment toxicity between the SASCT and TASCT regimens within studies, some SASCT regimens were considerably more toxic, either overall (DSMM‐I) or at an earlier time point (with higher dose density), compared to a sort of dose‐escalating scheme in the TASCT arm with 'softer' conditioning for the first ASCT (Table 3 ). Total body irradiation (TBI) was omitted for conditioning before the first ASCT in the TASCT arm of IFM94, while the second ASCT was identical to the only ASCT in the SASCT arm. The first ASCT for the TASCT arm in MAG95 involved an overall lower dose of chemotherapy without TBI and CD34‐selection in contrast to the only transplantation in the SASCT arm. Twice as many patients died early in the group receiving CD34‐selected grafts in MAG95 (N = 14 versus n = 7). In view of the treatment plan for MAG95, with differences in the timing and intensity of treatment regimens (Table 3), a clinically relevant interaction between CD34‐selection and randomisation to the TASCT or SASCT arm was likely.
On the one hand, delayed onset of intense chemo(radio)therapy may initially favour the TASCT arm in the trials with respect to sparing patients' acute toxicity (potential lead time bias). On the other hand, insufficient dose intensity up to and including the first ASCT may leave patients who discontinue prior to second transplantation undertreated. This is especially important for IFM94, in which the compliance with the second transplantation was only 78%. The possibility of these biases was coded with a red colour (Figure 2; see also Incomplete outcome data (attrition bias).
Cross‐over from the SASCT to the TASCT arm was only reported for the DSMM‐I trial (18% cross‐over) while no information was available about the time point of crossing‐over (before or after progression). Cross‐over also confounds the reported results by potential null and selection bias.
Effects of interventions
Initially, we planned to perform a meta‐analysis of the five included studies: all trials were comparing TASCT with SASCT as first‐line treatment of symptomatic MM in patients who were either untreated or had received conventional treatment, that is non‐high‐dose chemotherapy, for a maximum of six months. Features of the treatment regimen and obvious biases introduced during study performance (compliance, interaction, course of events in control group) stopped us from conducting a formal meta‐analysis however (Differences between protocol and review). In order to summarise the results of the individual studies, the number of events and survival proportions are both listed in Table 7 and recapitulated in a standardised way in the section below. The time course observed in the Kaplan‐Meier (KM) curves (not available for OS in Bologna96, EFS in MAG95) was added in order to transmit the maximum amount of information from the individual trials into the systematic review.
Quality of life was not reported for any included study, which is deplorable in view of the known adverse effect of ASCT on quality of life (Campagnaro 2008; Jones 2008). Furthermore, late toxic effects of ASCT, such as secondary malignancies or infections, were not specifically reported in any trial (Jantunen 2006; Majhail 2008).
In IFM94 a statistically significant benefit for the tandem arm on both OS and EFS was observed after a median 75 months of follow‐up: Median EFS and OS were prolonged by TASCT from 25 to 30 months and from 48 to 58 months (P = 0.03 and P = 0.01, respectively). Seven‐year OS (21% versus 42%) and EFS (10% versus 20%) proportions also significantly favoured the tandem‐transplantation group (Table 7). A trend for improved survival was observed in the long‐term follow‐up analysis, with a median 11.6 years of follow‐up (OS P = 0.08; EFS P = 0.06). No difference between the second randomisation option (bone marrow versus peripheral blood stem cells) was observed. Exploratory subgroup analysis suggested that patients who benefited most from the TASCT were those who did not achieve at least a very good partial response after their first transplantation. It is important to note that the first ASCT in the TASCT arm was performed with a lower dose (140 mg/m² melphalan) compared to the only ASCT in the SASCT arm (140 mg melphalan and TBI) (Table 3) and may therefore be classified as underdosed from a contemporary perspective. This is of relevance in view of only 78% of patients in the TASCT arm actually undergoing the second transplantation. Treatment‐related mortality was observed in 6% of the TASCT and 4% of the SASCT arm (N = 12 versus N = 8), of which N = 5 (TASCT) and N = 3 were related to the transplantation‐related complication sepsis. Overall, seven different causes of death were specified (Table 5). Similar two‐year survival proportions were observed for patients after relapse in SASCT and TASCT, although imbalances in salvage treatment limit the interpretability of the results (see Table 3; Assessment of risk of bias in included studies).
In MAG95, after a median follow‐up of 73 months OS appeared superior in both the TASCT group compared to the SASCT group (randomisation option A) and in the group receiving unselected grafts compared to those receiving selected grafts (randomisation option B) (P = 0.09 and P = 0.33, respectively) (Table 7). The prolongation of the median OS from 57 to 75 months in the TASCT arm might be misleading due to the position of the median at the right end of the Kaplan‐Meier graph with very few patients at risk. Only a slight prolongation of median EFS was reported for the TASCT arm (34 versus 31 mo). In the subgroup of patients who had received unselected grafts, OS was statistically significantly improved in the TASCT arm compared to patients in the SASCT arm (P = 0.04), highlighting the potential interaction of CD34‐selection and mortality (Assessment of risk of bias in included studies; Awedan 2002). Only interim data were available for MAG95 concerning toxic deaths, based on 85% of the final study population. For the final analysis, early deaths which included myeloma‐related deaths (TASCT 7% versus SASCT 12%) were reported.
The time‐course of mortality as seen in the Kaplan‐Meier graph for MAG95 revealed two clusters of events in the control group which appeared to drive the overall difference between the SASCT and TASCT arms. The first of the two clusters of events in the control arm (two to four months post‐randomisation) coincided with the treatment phase of the trial with an almost doubled early mortality in the SASCT arm (Table 7). In the MAG95 trial, patients in the TASCT group received the selected graft only at the second ASCT, while the graft used for the first ASCT was unselected (Table 3; Assessment of risk of bias in included studies).
Bologna96 did not confirm the significant OS benefit as observed in the IFM94 trial for TASCT in an ITT analysis (P = 0.9). While the median OS was extended from 65 (SASCT) to 71 (TASCT) months, seven‐year survival proportions were similar (43% after TASCT versus 46% after SASCT). No Kaplan‐Meier graph was available for OS. Five‐year EFS was significantly better in the TASCT arm (29% versus 17%) (Table 7). The improvement in median EFS from 23 to 35 months in the TASCT group was statistically significant (P = 0.001) with a rather steep decrease in EFS in the control arm approximately 18 months after randomisation. The duration of relapse‐free survival was prolonged from 24 months in the control group to 42 months in the tandem‐transplantation arm. Post‐relapse survival (PRS), however, favoured the SASCT arm with a median PRS of 32 months compared to 25 months in the TASCT arm. In view of the low compliance with the second transplantation in the TASCT group (65%) (Table 3), the interpretation of results was hampered due to potential null and selection biases (Assessment of risk of bias in included studies). Subgroup analysis of patients not reaching complete remission after induction confirmed the observation of the IFM94 trial suggesting that patients who do not respond well to initial therapy benefited most from the double intensified protocol. Transplantation‐related mortality (definition see Table 5) was reported as 4% in the TASCT and 3% in the SASCT arm.
In GMMG‐HD2, the final analysis did not demonstrate a significant prolongation of the median EFS (29 versus 25 mo; no P value provided) (Table 7). The Kaplan‐Meier curves for EFS in GMMG‐HD2 in the ITT population separated only transiently between 18 months and 38 months (TASCT superior) but the final proportions of patients surviving event‐free were similar (if more than 10 patients still at risk). Kaplan‐Meier curves for OS, for the ITT population were superimposable. In view of a very low compliance with the second ASCT (52 % of evaluable patients) (Table 3), per protocol (PP) analyses were performed in addition to the ITT analyses (see also Assessment of risk of bias in included studies). While the per protocol (PP) analysis of EFS was similar to the ITT analysis, the PP analysis of OS indicated numerically inferior survival for the TASCT arm over the majority of the follow‐up period (Discussion). So‐called TRM (definition see Table 5) was reported as 2% during the first and 3% during the second transplantation compared to 2% in the SASCT group (Table 7).
In DSMM‐I, with a follow‐up of approximately 48 months (corresponding to the entire time axis in the Kaplan‐Meier graph) the Kaplan‐Meier curves appeared unstable with multiple crossings for OS and EFS and a flattening of the curves at the right end (Assessment of risk of bias in included studies). While OS, EFS and PFS all appeared superior for the TASCT group, final survival proportions were favourable for the SASCT arm (Table 7). Kaplan‐Meier graphs presented later (Einsele 2007), indicated that PFS was consistently higher for the SASCT arm. No information was reported with respect to the type of analysis (ITT, PP, subgroup analysis?). So‐called TRM (definition see Table 5) was reported as 4% for the TASCT and 3% for the SASCT arm. The definition of so‐called TRM remained unclear. Notably, OS in the TASCT arm remained at 100% for 400 days (including treatment phase?) in the TASCT arm (Table 7).
Discussion
Empirical research should always take into account previous experience, either to build on it, refute it, or at least to learn from it (Clarke 2004; Chalmers 2009). Systematic reviews or meta‐analysis are formalised tools to review available evidence on a clearly defined question. They are also an occasion to systematically and rigorously assess the quality of the available evidence (Egger 2001).
The findings of a comparison of two treatments, that is the treatment effect, may result from a) improvement of an outcome by the test treatment, b) an inferior outcome of the control treatment, c) a chance finding, or even in the setting of a RCT d) be driven by confounding factors within a small and heterogeneous patient population (Blair 2004). All four options should be addressed in the course of an exploratory systematic review. Both patient characteristics and study design (including the treatment plan) should be scrutinised for their propensity to favour one or more of the four possibilities. While a systematic review as a retrospective observational study cannot settle a controversy, the examination may help to contribute to the design of future studies addressing (an emerging) one.
Summary of main results
In the set of publicly available RCTs performed between 1994 and 2002 (five of seven completed RCTs), TASCT was accompanied by a statistically significant improvement in OS compared to SASCT for the first‐line treatment of patients with MM in one study only (IFM94; statistical significance not confirmed in long‐term follow‐up). In the other four trials it appeared to have no effect on survival (Table 7). None of the studies were adequately powered for the analysis of OS and considerable confounding due to varying access to salvage treatment is likely. Of note, OS appeared numerically inferior for the per protocol population of the GMMG‐HD2 trial, which may be due to the selection of patients who proceeded to second transplantation. An observational study with matched pair design also reported on numerically inferior OS (NMSG), as well as a reported case series from a single centre experience (Koren 2010), highlighting the necessity to further study the impact of the second transplantation on long‐term outcomes.
TASCT improved EFS compared to SASCT in four of the five trials (not in DSMM‐I) (Table 7), but the effect was statistically significant only in the two trials published as full‐text articles (IFM94; Bologna96). The beneficial effect on EFS appeared to be transient in GMMG‐HD2. The results of DSMM‐I appeared to be too immature to allow firm conclusions.
Treatment‐ or transplant‐related mortality, sometimes indiscriminately summarised as so‐called TRM, was higher for the TASCT arm in four of five studies without reference to statistical significance (all except MAG95). For MAG95, both the time course of events in the Kaplan‐Meier graph and interim data indicate that here the SASCT regimen claimed more early and toxic deaths compared to the TASCT regimen.
Quality of life was not reported in any of the included studies. Response to treatment had not been assessed for the present review.
In view of the observed heterogeneity of treatment concepts and the extent of potential bias in the individual studies, we refrained from performing a formal meta‐analysis (Feinstein 1995). Critical features in the set of included trials concern major differences in the treatment plans, not only between but also within individual RCTs. Variable compliance with the planned treatment during performance of the trials and the potential confounding of OS data by subsequent treatment may also impact on the observed treatment effect. Overall, the direction of bias was considered as unpredictable (Risk of bias in included studies; Quality of the evidence; Figure 2).
Overall completeness and applicability of evidence
Publication bias
Two completed or terminated RCTs were identified in clinical trials registries, however they remained unpublished. For the only American trial (DM00‐196), an interim analysis in abstract form reported on a critical lack of compliance with the second ASCT, without follow‐up data (Wilson 2003). The UK‐based study (N0265041749) reported on similar problems with accrual, which resulted in early closure of the trial (Mahendra personal communication). Three of the five included RCTs (DSMM‐I; GMMG‐HD2; MAG95) were available as conference presentations only, emphasising both the necessity to include non‐full text information when systematically evaluating a treatment concept in oncology and the difficulty of accepting conference presentations in view of low reporting quality (Ramsey 2008; Tam 2008). Of note, only the two journal articles report statistically significant improvements in survival endpoints (IFM94; Bologna96), while the conference abstracts refer to non‐significant results or to difficulty in actually performing the second transplantation (Hopewell 2009). Overall, it appears as if only positive results of TASCT are published in widely accessible medical journals while the scientific community is not equally informed on disappointing results, concerning either practicability or clinical outcome.
On the one hand, lack of reporting of clinical outcome data is an obvious sign of publication bias, a well‐known phenomenon in clinical research (Curt 2008; Ramsey 2008). On the other hand, the problems with either accrual of patients or low compliance with the protocol illustrates the strenuous nature of a TASCT regimen.
Baseline characteristics
Even in RCTs, relevant imbalances at baseline may help to explain the relative outcome of the study arms (Blair 2004). MM is a very heterogeneous disease and the evaluable set of of participants of some of the trials was only around 100 (compliant) patients per arm (DSMM‐I; GMMG‐HD2; Bologna96), which may be insufficient to balance out chance findings (Moore 1998). In addition, no information on cytogenetic characteristics was collected in the included trials. The cytogenetic information is regarded as critical for extrapolation of results, since cytogenetic analysis belongs to the basic workup for contemporary patients with MM (Munshi 2011).
Treatment regimen
When reviewing literature, it is important to take into consideration recent improvements in individual steps of the rather complex intervention of autologous stem cell transplantation (Awedan 2002; Attal 2007; Harousseau 2009a). Preparation of patients for ASCT has changed substantially during the last two decades, including induction treatment, mobilisation or handling of stem cells and a conditioning regimen for ASCT. While induction treatment used to be a combination chemotherapy with vincristine, doxorubicin and dexamethasone (VAD), or high‐dose steroids, nowadays the so‐called 'novel agents' have replaced the classical induction regimen (Lane 2005; NCCN 2011). Furthermore, for contemporary ASCT, only peripheral blood stem cells without graft manipulation (for example purging of tumour cells) are used for ASCT in view of better engraftment characteristics when compared to bone marrow (Gratwohl 2007). TBI has been largely abandoned as a conditioning regimen in view of increased toxicity (Moreau 2002; Tricot 2009). In addition, the conditioning regimen for the second ASCT in the TASCT arm included busulfan, which has recently been associated with a high treatment‐related morbidity, possibly impacting on further access to salvage treatment (Lahuerta 2010). Today, 200 mg melphalan is considered to be the standard conditioning regimen (Giralt 2010). It is important to note that the regimen used for both TASCT and SASCT followed the learning curve of the development of the standard regimens in use today, that is all regimens would be considered substandard from a contemporary perspective in at least one or even several components (Table 3).
Overall, treatment regimens were diverse in the included trials, with notable differences in toxicity between trials but also between study arms, thereby potentially biasing the overall results (lead‐time bias). The low compliance with the second transplantation in three trials (IFM94, Bologna96 and GMMG‐HD2) (see Table 3) makes the interpretation of treatment effect difficult in view of unassessable attrition bias, even more so as the TASCT regimen in IFM94 and MAG95 followed a sort of dose‐escalating scheme. Both MAG95 and DSMM‐I applied non‐standard SASCT regimens to the control group, which also needs to be considered for the interpretation of data since treatment effects may stem from a superior test regimen or from an inferior control arm. Overall, the possibility of extrapolating the available evidence to treatment decisions for contemporary patients is questioned.
Quality of the evidence
For interpretation of the effect on OS, the following issues were regarded as critical in the course of the present systematic review: confounding by subsequent treatment (all studies), compliance with the second transplantation (IFM94; Bologna96; GMMG‐HD2), acute toxicity of control treatment (DSMM‐I; MAG95), extent of cross‐over (DSMM‐I), and time course of mortality (MAG95). The observed cluster of events in the control arm during the treatment phase of the only transplantation in the SASCT arm of MAG95 is indicative of an increased toxicity in the SASCT arm, that is an inferior outcome for the control arm.
For interpretation of the effects on EFS, the following issues were regarded as critical: extent of cross‐over (DSMM‐I), low compliance with the second transplantation (IFM94; Bologna96; GMMG‐HD2). Furthermore, it remained unclear whether any of the studies made provisions to address relevant biases introduced by irregular or asymmetric assessments for events in the course of the EFS assessments (CHMP/EWP/27994/08 2008). In addition, events for EFS were defined differently between trials (Table 4)
In the included trials both definitions of the relevant period of observation and causes of death to be listed as so‐called TRM remained unclear, and were not comparable between trials (Table 5). Another uncertainty concerning the comparability of study‐specific results stemmed from the different level of sophistication of ascribed causes of death (for example IFM94). In addition, the definition of the population in which so‐called TRM was reported as a percentage was missing for most trials. This is of relevance in view of the low compliance with second transplantation, since there may be large deviance between different populations in the analysis (for example ITT or 'as treated').
The short‐term risk would need to be counterbalanced by a convincing prolongation of long‐term survival accompanied by an improved quality of life. The per protocol population of GMMG‐HD2, a matched‐pair analysis and a single‐centre experience (GMMG‐HD2; NMSG; Koren 2010), although all methodically contestable pinpoint to a potentially inferior survival outcome for patients actually undergoing second transplantation compared to a SASCT regimen. More information is required on the long‐term benefit for patients in view of the overall strenuous treatment approach of TASCT.
Agreements and disagreements with other studies or reviews
Only one other meta‐analysis on the same topic has been published (Kumar 2009). In their conclusion, Kumar et al advised against the routine use of TASCT in view of statistically significantly increased treatment‐related mortality without significant prolongation of OS (Kumar 2009).
The discouragement of first‐line TASCT triggered substantial criticism among US experts in MM, accompanied by a controversial discussion on the methods and interpretation of data in the meta‐analysis (Giralt 2009; Mehta 2009; Tricot 2009). On the one hand, the clinicians commenting on the meta‐analysis were disappointed that important clinical aspects had not been discussed in the course of the meta‐analysis, for example the impact of salvage treatment, the use of TBI in the older trials or the different definitions of TRM in the set of included trials. On the other hand, the authors of the meta‐analysis objected to methodically problematic proposals for analyses or interpretations of available evidence by the clinicians (Kumar 2009a; Kumar 2009b).
The present systematic review differs from Kumar 2009 in several aspects. First, with respect to the set of included trials, Kumar included Sonneveld 2007 which in our view is not assessing TASCT but rather a non‐myeloablative double‐intensified protocol (Excluded studies). DSMM‐I, included in our analysis, was not mentioned by Kumar et al. While DSMM‐I yielded indefinite results for all outcomes, Sonneveld reported statistically significant results for EFS (TASCT better) and TRM (TASCT worse).
Second, the focus of our qualitative assessment of included studies exceeded the prospectively planned elements (such as reporting of sample size calculations or definitions of endpoints) of trial design considered by Kumar et al. Here, we integrated both additional design elements of the trial into the assessment (for example acute toxicity of treatment regimen) as well as actually observed effects during trial performance (such as time course of mortality, compliance with treatment) (Other potential sources of bias).
While Kumar presented meta‐analytical results, we considered the observed clinical and methodological heterogeneity as being too pronounced to gain further insights from a combined analysis.
We agree with Kumar's conclusion that a short‐term risk needs to be counterbalanced by a long‐term benefit in order to justify routine use of a medical intervention. We also consider it likely that there is only a marginal, if any, beneficial effect of TASCT on OS. Thus, we agree in questioning the value of tandem autologous stem cell transplantation as a first‐line treatment in MM. We hestitate to follow Kumar et al, however, with respect to the informative value of the increased TRM observed after TASCT. We raise the question that if an inevitably life‐threatening procedure (ASCT) was performed twice instead of once, would we not even expect an at least doubled risk of transplantation‐related mortality (Jones 2008)? In addition to the diversity of the defintions of TRM used (Table 5), the low compliance with the second transplantation (Table 3) may further complicate a definite analysis of treatment‐ or transplantation‐related mortality.
We also deplore the low precision of reporting on this important outcome of patients' safety (Table 5). It remained unclear whether treatment‐ or transplant‐related mortality was reported when the ambiguous abbreviation was used (GMMG‐HD2; DSMM‐I). Finally, we do not agree with Kumar that TRM in MAG95 might have been so high that the authors refrained from reporting it. As described above, we even consider an exceptionally high mortality in the SASCT arm of MAG95 as plausible in view of the Kaplan‐Meier graph of the final analysis, interim data on toxic death and the expected toxicity of the treatment regimen based on dose density (Other potential sources of bias; Effects of interventions).
Overall, we aimed at contributing to improvements of future trial designs on tandem transplantation rather than settling the conflicting evidence on TASCT, also considering the evolution of treatment approaches of MM in the last decade. We therefore elaborate in some detail on the difference of the treatment approaches to TASCT, then and now, and discuss further methodological issues that prevent clear‐cut interpretation of data, especially for contemporary patients (Other potential sources of bias; Implications for practice; Implications for research). We doubt that these potentially confounding issues could be adequately addressed and recompensed in an individual patient data (IPD) analysis.
We regret to conclude that the set of available RCTs do not yield sufficient evidence for contemporary treatment decisions.
Authors' conclusions
Implications for practice.
The present systematic review critically questions the validity of the included set of trials to provide a basis for contemporary treatment recommendations, especially for patients who have already achieved a good response after the first transplantation. Treatment of MM has changed dramatically in the past 15 years and there are considerable uncertainties and potential sources of bias in this rather old set of trials. In addition, the quality of reporting of trial methods and results, especially in those trials available as conference presentation only, do not allow us to draw firm conclusions.
The uncertainty in the medical community as to whether the acute toxicity of a second ASCT is counterbalanced by long‐term benefit is also reflected in the design of a number of recent and ongoing trials, and current treatment guidelines. In view of the increase in the proportion of complete responses after a second transplantation in the set of included trials, the option of a second transplantation is sometimes limited to patients who have not responded optimally to the first transplantation (for example Kortüm 2010). The dependency of a second transplantation on a suboptimal response to first ASCT was already integrated into the unpublished DM00‐196 trial, and into the current generation of studies performed by the IFM and GMMG study groups (for example GMMG‐HD5 or Moreau 2011 studies). Also, Cavo highlights the necessity of evaluating the additional benefit of a second transplantation in view of the high complete response rates already obtained with the novel induction regimen (Cavo 2010).
Implications for research.
The observation that, for various unavoidable reasons, compliance with a second transplantation decreased sharply needs to be taken into account for future trial designs. This finding is also supported with data described for 2655 patients in a TASCT program in the European Bone Marrow Transplantation Registry (EBMTR), for whom a compliance of only 55.3% was observed with the second ASCT (Morris 2004). Therefore, sample size calculations for studies assessing TASCT upfront (that is independent from response to treatment, such as the ongoing study (NCT01109004)) should take into account that patients may progress prior to or develop contraindications against a second ASCT, or simply refuse to undergo a second transplantation. This decrease in compliance cannot be considered at random thus strategies need to be developed which aim at addressing the expected selection of patients both fit enough and willing to continue to second transplantation (see also Wheatley 2006). Alternatively, patients who successfully underwent first transplantation (and for whom sufficient stem cells were collected before) could be randomised between different consolidation treatment options. While the latter option bears the risk of introducing selection bias in the absence of objective criteria to describe the relevant population, it may ultimately avoid the other biases described at length above.
In view of the difficulty in ascribing specific causes of death in a multimorbid patient population, we propose reconsideration of the definition of the important endpoint of treatment‐ or transplantation‐related mortality in order to optimise its clinical utility. Rather than defining the numerator, that is the type of events which qualify as treatment‐related (a challenging and potentially subjective process (Keirns 2008)), the reporting of all‐cause mortality in a relevant period of time (such as 'from randomisation until three months after completion of intensive treatment') is proposed. Specific causes of deaths, such as fatal infection during aplasia, could additionally be reported for didactic purposes. Early mortality reflects either too toxic or ineffective treatment, both of which are undesirable for symptomatic patients in the need of treatment.
The publication bias, as determined for the RCTs comparing TASCT with SASCT, emphasises both the necessity and the difficulties in including non‐full text information when systematically evaluating a treatment concept in oncology (Curt 2008; Ramsey 2008; Tam 2008). Reporting of trials still needs to be improved in order to allow at least an informed discussion on the potential direction of bias (Hopewell 2010).
History
Protocol first published: Issue 1, 2004 Review first published: Issue 10, 2012
| Date | Event | Description |
|---|---|---|
| 26 May 2008 | Amended | Converted to new review format. |
Notes
None
Acknowledgements
We would like to thank the Cochrane Haematological Malignancies Review Group Editorial Team for their support.
Appendices
Appendix 1. MEDLINE search strategy
#7 Search myelom*[Text Word]#8 Search kahler*[Text Word] #9 Search plasmacytom*[Text Word] #10 Search plasmocytom*[Text Word] #12 Search "stem cell transplantation"[MeSH Major Topic] #13 Search "transplantation, autologous"[MeSH Major Topic] #14 Search "transplantation, homologous"[MeSH Major Topic] #16 Search transplant*[Text Word] #17 Search graft*[Text Word] #19 Search autolog*[Text Word] #23 Search autograft*[Text Word] #24 Search auto‐graft*[Text Word] #25 Search auto‐transplant*[Text Word] #26 Search autotransplant*[Text Word] #27 Search asct*[Text Word] #28 Search abmt*[Text Word] #39 Search "multiple myeloma"[MeSH Major Topic] #40 Search #7 or #8 or #9 or #10 or #39 #50 Search (#12 or #16 OR #17) AND #19 #52 Search #13 OR #14 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #50 #53 Search #40 and #52 #55 Search (Therapy/Broad[filter] OR EBMT[All Fields] OR IBMTR[All Fields] OR (registry[tw] OR register[tw]) OR "center experience"[tw] OR "centre experience" ) AND ("humans"[MeSH Terms] AND (English[lang] OR French[lang] OR German[lang]) AND ("1995"[PDAT] : "2010"[PDAT])) #56 Search #53 and #55
Appendix 2. CENTRAL search strategy
1 MeSH descriptor MULTIPLE MYELOMA explode all trees 2 (myelom* or myeloom* or mielom*).tw,kf,ot. 3 myelomatos*.tw,kf,ot. 4 (kahler or kahler’s).tw,kf,ot. 5 (plasm*cytom* or plasmozytom*).tw,kf,ot. 6 #1 or #2 or #3 or #4 or #5 7 MeSH descriptor STEM CELL TRANSPLANTATION explode all trees 8 MeSH descriptor TRANSPLANTATION, AUTOLOGOUS explode all trees 9 MeSH descriptor TRANSPLANTATION, HOMOLOGOUS explode all trees 10 #7 or #8 or #9 11 (transplant* or graft*).tw,kf,ot. 12 autolog*.tw,kf,ot. 13 #11 and #12 14 (autograft* or auto‐graft*).tw,sh,ot. 15 (homograft* or homo‐graft*).tw,sh,ot. 16 (autotransplant* or auto‐transplant*).tw,sh,ot. 17 ((tandem* or double*) near/3 transplant*).tw,kf,ot. 18 (asct or abmt).tw,kf,ot. 19 #14 or #15 or #16 or #17 or #18 20 #10 or #13 or #19 21 #6 and #20
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bologna96.
| Methods | Central randomisation before induction treatment Multicentre (N=32) RCT addressing superiority of TASCT |
|
| Participants | TASCT N=158, SASCT N=163 age < 60 years Previously untreated, symptomatic or progressive MM Salmon‐Durie stage I‐III (Baseline characteristics Table 2) |
|
| Interventions | 1x 200 mg/m² melphalan + (140 mg/m² melphalan + 12 mg/m² oral busulfan) vs 1 x 200 mg/m² melphalan SASCT (Treatment regimen Table 3) |
|
| Outcomes | OS, EFS, transplantation‐related mortality in ITT population (Definition of endpoints Table 4; Table 5) |
|
| Period and Location | Jan 96 – Dec 01 in Italy | |
| Sample size calculation | Improvement of CR/nCR | |
| ITT analysis (n of drop‐outs) | Yes (N=3/321 in SASCT) | |
| Notes | Journal publication (peer‐review) Null‐bias (only 65% compliance with 2nd ASCT) Potential confounding by subsequent treatment (superior post‐relapse survival for SASCT arm) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central |
| Allocation concealment (selection bias) | Unclear risk | Central |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unavoidable in view of obvious difference in treatment plan |
| Other bias | High risk | See Table 6 and Figure 2 |
DM00‐196.
| Methods | Randomised phase II trial, no information on method of randomisation Single centre study comparing without reporting of basic statistical assumption (superiority/non‐inferiority) |
|
| Participants | Patients of MM not achieving CR by day 180 after initial ASCT (planned for initial ASCT N=120; interim analysis on N=31 at d180 post transplantation) No further information on age or stage |
|
| Interventions | TASCT (1. ASCT: "high dose melphalan"; 2. ASCT: melphalan/topotecan/cyclophosphamide) vs SASCT versus maintenance with thalidomide/dexamethasone | |
| Outcomes | PR and CR, relative toxicities | |
| Period and Location | USA Dec 2000 ‐ not reported | |
| Sample size calculation | NR | |
| ITT analysis (n of drop‐outs) | NR | |
| Notes | Register information; no clinical outcome data for systematic review reported Steep decrease of compliance with second ASCT (high refusal rate by patients to proceed to second transplantation, developing contra‐indications, denied coverage by insurance) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NR |
| Allocation concealment (selection bias) | Unclear risk | NR |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | NR |
| Other bias | Unclear risk | NR |
DSMM‐I.
| Methods | Randomisation of patients with at least stable disease after induction and successful stem‐cell mobilisation Multicentre (N= 46‐49) RCT addressing superiority of experimental SASCT regimen compared to trial; TASCT |
|
| Participants | TASCT N=98; SASCT N=100 ≤60 years; de novo or max. < 6 cycles of "conventional Ctx" Salmon‐Durie stage II and III (Baseline characteristics Table 2) |
|
| Interventions | 2 x 200 mg/m² melphalan TASCT compared to experimental SASCT (chemo‐radio conditioning regimen) (Treatment plan Table 3) |
|
| Outcomes | OS, EFS (primary endpoint), so‐called TRM (Definitions of endpoints Table 4; Table 5) |
|
| Period and Location | Aug. 98 – Jan. 02 in Germany and Italy (USA?) | |
| Sample size calculation | Not reported | |
| ITT analysis (n of drop‐outs) | Yes (unclear) | |
| Notes | Conference presentations with some inconsistencies Safety population differs notably from ITT population (TASCT N=118; SASCT N=80) Data likely immature Total follow‐up of only 48‐61 months according to Kaplan‐Meier graph (reported as median follow‐up) Potential confounding by cross‐over from SASCT to TASCT in 18% of patients and by subsequent treatment |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NR |
| Allocation concealment (selection bias) | Unclear risk | NR |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unavoidable in view of obvious difference in treatment plan |
| Other bias | High risk | See Table 6 and Figure 2 |
GMMG‐HD2.
| Methods | Randomisation of patients with at least stable disease after induction Multicentre (N=66) RCT comparing TASCT with SASCT without reporting of aim (superiority/non‐inferiority) Optional additional randomisation of induction regimen (VID vs VAD) |
|
| Participants | TASCT N= 180; SASCT N=178 (evaluated patients; actual number of patients depending unclear and different in different conference presentations (N=358‐485) age 18 ‐ 66 years previously untreated (or max 6 cycles of conventional chemotherapy) patients with MM Salmon‐Durie stage II and III (Baseline characteristics Table 2) |
|
| Interventions | 2 x 200 mg/m² melphalan TASCT vs 1 x 200 mg/m² melphalan SASCT (Treatment plan Table 3) | |
| Outcomes | OS, EFS (primary endpoint), so‐called TRM (ITT and PP analyses) (Definitions of endpoints Table 4; Table 5) |
|
| Period and Location | Nov (96?) 98 – Feb 02 in Germany | |
| Sample size calculation | NR | |
| ITT analysis (n of drop‐outs) | Yes (unclear); per protocol analyses available | |
| Notes | Conference presentation with some inconsistencies Unclear, how the two randomisations were analysed (separately?, interaction test?) Very low compliance (52% of evaluable population) with 2nd ASCT (null bias for OS, EFS); Potential confounding by salvage treatment (OS) Numerically inferior OS for TASCT group in PP analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NR |
| Allocation concealment (selection bias) | Unclear risk | NR |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unavoidable in view of obvious difference in treatment plan |
| Other bias | High risk | See Table 6 and Figure 2 |
IFM94.
| Methods | Central randomisation at diagnosis Multicentre (N=45) RCT assessing superiority of TASCT 2x2 factorial design: additional randomisation between bone marrow and peripheral blood stem cells |
|
| Participants | TASCT N=200, SASCT N=199; (long‐term follow‐up: TASCT N=203; SASCT N=199) age < 60 years Previously untreated patients with MM Salmon‐Durie stage I (with bone lesion), II‐III (Baseline characteristics Table 2) |
|
| Interventions | "Dose‐escalating" TASCT regimen compared to SASCT (conditioning melphalan + TBI 8 Gy) ‐ both either with bone marrow or peripheral blood stem cells (Treatment regimen Table 3) |
|
| Outcomes | OS, EFS, treatment‐ and transplantation‐related mortality in ITT population (Definition of endpoints Table 4; Table 5) |
|
| Period and Location | Oct 94 – Mar 97 in France and Belgium | |
| Sample size calculation | Improvement of CR% | |
| ITT analysis (n of drop‐outs) | Yes (NR) | |
| Notes | Journal publication (peer review) Long‐term follow‐up data for OS and EFS available No clinically relevant interaction of two randomisations Dose‐escalating" TASCT regimen not comparable to current treatment approaches (dose of first ASCT lower than only ASCT in SASCT arm) Low compliance (75%) with second ASCT in TASCT arm Potential confounding subsequent salvage treatment (inferior post‐relapse survival in TASCT group) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central |
| Allocation concealment (selection bias) | Low risk | Central |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unavoidable in view of obvious difference in treatment plan |
| Other bias | High risk | See Table 6 and Figure 2 |
MAG95.
| Methods | Randomisation before induction treatment Multicentre (number not reported) RCT without reporting of basic statistic assumption (superiority/non‐inferiority) 2x2 factorial randomisation (unselected vs CD34 selected PBSC) |
|
| Participants | TASCT N=114, SASCT N=113 age <56 years Previously untreated patients with MM Salmon‐Durie stage II and III |
|
| Interventions | "Dose‐escalating" TASCT vs SASCT (combination chemotherapy + 12 Gy TBI); both with‐ and without selection of CD34‐positive cells | |
| Outcomes | OS, median EFS; early death (toxic deaths for interim analysis, only) | |
| Period and Location | "1996" in France (no further information provided) | |
| Sample size calculation | NR | |
| ITT analysis (n of drop‐outs) | NR (NR) | |
| Notes | Conference presentations Clinically relevant interaction of two randomisations (inferior outcome after selection of CD34+) Non‐standard conditioning regimen with high dose TBI (12 Gy) with asymmetric course of toxicity between two arms (later onset of high toxicity in TASCT arm) Potential confounding by subsequent salvage treatment (OS) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NR |
| Allocation concealment (selection bias) | Unclear risk | NR |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unavoidable in view of obvious difference in treatment plan |
| Other bias | High risk | See Table 6 and Figure 2 |
N0265041749.
| Methods | Randomised trial (no further information) following stem cell collection Single centre RCT assessing superiority of TASCT |
|
| Participants | No information on planned or recruited number of patients age < 65 years Newly diagnosed (up to 3 months after diagnosis) patients with chemosensitive MM Salmon‐Durie stage II‐III |
|
| Interventions | Double vs single high‐dose melphalan (Treatment regimen Table 3) |
|
| Outcomes | OS, quality of life | |
| Period and Location | Birmingham, Jan 1999 ‐ Jan 2001 | |
| Sample size calculation | NR | |
| ITT analysis (n of drop‐outs) | NR | |
| Notes | Register information (no clinical outcomes reported) According to principal investigator study failed to recruit a "sufficient number of patients" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NR |
| Allocation concealment (selection bias) | Unclear risk | NR |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | NR |
| Other bias | Unclear risk | NR |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| EBMTR | Insufficient information on baseline characteristics (less than 5 factors without substantial missing data). Insufficient information on treatment plan, already apparent imbalances between use of TBI in the "planned" or "unplanned" group. |
| Kim 2009 | No separate information on patients receiving either one or two transplants. |
| Koren 2010 | No separate information on patients receiving either one or two transplants. |
| Lahuerta 2003 | No separate information on patients receiving either one or two transplants. |
| NMSG | Insufficient information on baseline characteristics (less than 5 factors). Insufficient information on treatment plan. |
| Sonneveld 2007 | Non‐myeloablative dosing regimen of melphalan (2 x 70 mg/m²). |
| Total therapy | No RCT. No contemporary control group. Of note: integration of novel agents in TT generation >1 is regarded as a critically different treatment approach compared to the set of included RCTs. |
Characteristics of ongoing studies [ordered by study ID]
NCT01109004.
| Trial name or title | A trial of single autologous transplantation with or without consolidation therapy versus tandem autologous transplantation with lenalidomide maintenance for patients with MM (BMT CTN 0702) |
| Methods | Randomised, multicenter (N=52), open‐label, three‐arm trial |
| Participants | Up to 750 patients with symptomatic MM who have received at least two cycles of any regimen as initial systemic therapy and are within 2 ‐ 12 months of the first dose of initial therapy and with an adequate autologous graft < 70 years |
| Interventions | TASCT plus maintenance therapy versus the strategy of SASCT plus consolidation therapy with lenalidomide, bortezomib and dexamethasone (RVD) followed by maintenance therapy or SASCT plus maintenance therapy as part of upfront treatment of MM. Lenalidomide will be used as maintenance therapy for three years in all arms. |
| Outcomes | Primary 3 year PFS, 3 year OS, Quality of life, treatment‐related mortality |
| Starting date | May 2010 ‐ May 2016 (estimated final data collection date for primary outcome measure) |
| Contact information | Mary Horowitz, MD, MS http://clinicaltrials.gov/ct2/show/NCT01109004?term=NCT01109004&lup_s=01%2F30%2F2011&lup_d=30 |
| Notes |
Differences between protocol and review
While performing the review, we had to restrict our search to publications in English, French and German and had to exclude the database EMBASE due to budgetary restrictions.
Initially, it was planned to perform a statistical meta‐analysis for the included studies. In view of notable clinical heterogeneity and several methodological issues, the presented results are limited to a narrative systematic review. Furthermore, response and time‐to‐progression were not analysed. In addition to newly diagnosed patients, studies with previously treated patients were acceptable as long as the treatment was conventional and of limited duration (up to six months). Instead of limiting our systematic search to RCTs as planned in the protocol, we also searched for observational studies with contemporary control groups.
The outcome transplantation‐related mortality was only vaguely defined in the majority of trials. Instead of including only trials reporting transplantation mortality (as initially planned), we extracted results where all kind of definitions for early mortality were used.
Fewer authors contributed to the finalisation of the review compared to the protocol.
Contributions of authors
NF: updating protocol (based on earlier version), searching, eligibility screening, data extraction and quality assessment, content input, interpretation of findings
AG: drafting of first version of the protocol, searching, eligibility screening
PB: interpretation of findings and providing a clinical perspective
JB: methodological expert
AE: providing a clinical perspective
RS: searching, eligibility screening, data checking and quality assessment, interpretation of findings, providing a clinical perspective
Sources of support
Internal sources
No sources of support supplied
External sources
New Source of support, Not specified.
Declarations of interest
No interests to declare.
The views and opinions expressed in the systematic review are those of the individual authors and should not be attributed to the institutions where they are employed.
New
References
References to studies included in this review
Bologna96 {published and unpublished data}
- Cavo M, Tosi P, Zamagni E, Cellini C, Tacchetti P, Patriarca F, et al. Prospective, randomized study of single compared with double autologous stem‐cell transplantation for multiple myeloma: Bologna 96 clinical study. Journal of Clinical Oncology 2007;25(17):2434‐41. [DOI] [PubMed] [Google Scholar]
DM00‐196 {unpublished data only}
- Giralt S, MD Anderson Study Center. A Randomized Phase II Trial of tandem transplantation vs thalidomide/dexamethasone maintenance therapy for patients with multiple myeloma failing to achieve complete remission after intensive induction therapy. http://utm‐ext01a.mdacc.tmc.edu/dept/prot/clinicaltrialswp.nsf/Index/DM00‐196 2000 [accessed 22.2.2011].
- Wilson RL, Gray ML, Giralt S. Barriers to second transplant for multiple myeloma in a randomised autologous tandem trial [IBMTR/ABMTR Participants' meeting, Keystone Colorado, January 2003]. Biology of Blood and Marrow Transplantation. 9 2003; Vol. 9, issue 2.
DSMM‐I {published and unpublished data}
- Einsele H. Stem Cell Transplantation for MM: Analysis of Prognostic Factors[S9.3] [XIth International Myeloma Foundation Workshop, Kos]. Haematologica. 2007; Vol. 90, issue s2:49 http://myeloma.org/pdfs/Kos2007_Einsele.pdf [accessed 22.2.2011].
- Einsele H, Liebisch P, Bargou R, Meisner C, Metzner B, Wandt H, et al. Single high‐dose chemoradiotherapy versus tandem high‐dose melphalan followed by autologous stem cell transplantation: preliminary analysis [Xth International Myeloma Foundation Workshop, Sydney]. Haematologica. 90 2005; Vol. 90 Suppl 1:131.
- Knop S. New approaches in first‐line therapy ‐ international perspective: 3. Germany ‐ Younger patients [1st Wurzburg Myeloma Workshop 2008]. http://mds‐forum.onkodin.de/myelom2008/content/e54/e4466/e2568/index_ger.html [accessed 22.2.2011].
- Knop S, Bauer K, Hebart H, Wandt H, Trümper L, Liebisch P, et al. A Randomized Comparison of Total‐Marrow Irradiation, Busulfan and Cyclophosphamide with Tandem High‐Dose Melphalan in Patients with Multiple Myeloma [#728] [American Society of Hematology Annual meeting 2007 Atlanta, USA]. Blood. 110 2007; Vol. 110, issue 11:http://myeloma.org/ArticlePage.action?articleId=2252 [accessed 22.2.2011].
GMMG‐HD2 {published and unpublished data}
- Goldschmidt H. Single vs double HDT in multiple myeloma [XIth International Myeloma Workshop, Kos 2007]. http://myeloma.org/pdfs/Kos2007_Goldschmidt.pdf [accessed 22.2.2011].
- Goldschmidt H. Single vs. double high‐dose therapy in multiple myeloma: second analysis of the GMMG‐HD2 trial[PL8.02] [International Myeloma Workshop, Syndey 2005]. Haematologica. 90 2005; Vol. 90, issue s1:38 http://myeloma.org/pdfs/Sydney2005_Goldschmidt_P8.pdf [accessed 22.2.2011].
- Goldschmidt H. Neues aus den Studiengruppen ‐ GMMG (German‐Speaking Myeloma Multicenter Group) [2nd Heidelberg Myeloma Workshop 2009]. http://mds‐forum.onkodin.de/myelom2009/content/e54/e8241/e8264/index_ger.html [accessed 22.2.2011].
- Goldschmidt H. Single vs tandem autologous transplantation in multiple myeloma: the GMMG experience [IXth International Myeloma Foundation Workshop, Salamanca 2003]. Haematologica. 88 2003; Vol. 88, issue 4:S61.
IFM94 {published and unpublished data}
- Attal M, Harousseau JL, Facon T, Guilhot F, Doyen C, Fuzibet JG, et al. Single versus double autologous stem‐cell transplantation for multiple myeloma. New England Journal of Medicine 2003;349(26):2495‐502. [DOI] [PubMed] [Google Scholar]
- Barlogie B, Attal M, Crowley J, van RF, Szymonifka J, Moreau P, et al. Long‐term follow‐up of autotransplantation trials for multiple myeloma: update of protocols conducted by the intergroupe francophone du myelome, southwest oncology group, and university of arkansas for medical sciences. Journal of Clinical Oncology 2010;28(7):1209‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
MAG95 {published and unpublished data}
- Fermand J, Alberti C, Marolleau J. High does therapy supported with autologous blood stem cell transplantation in multiple myeloma: long term follow‐up of the prospective studies of the MAG group [PL 8.05] [Xth International Myeloma Foundation Workshop, Sydney 2005]. Haematologica http://myeloma.org/pdfs/Sydney2005_Fermand_P8.pdf [accessed 22.2.2011].
- Fermand JP, Marolleau JP, Alberti C, Divine M, Leblond V, Macro M, et al. American Society of Hematology, Orlando 2001 [In Single Versus Tandem High Dose Therapy (HDT) Supported with Autologous Blood Stem Cell (ABSC) Transplantation Using Unselected or CD34 Enriched ABSC: Preliminary results of a two by two designed randomized trial in 230 young patients with multiple myeloma (MM)]. Blood 2001;98 Suppl(11):815a. [abstract: #3387] [Google Scholar]
N0265041749 {unpublished data only}
- Mahendra P. A randomised study of double versus single high dose therapy with autologous stem cell transplantation in newly diagnosed patients with Stage II/III multiple myeloma (N0265041749]. http://www.nihr.ac.uk/Profiles/NRR.aspx?Publication_ID=N0265041749 2001 [accessed 22.2.2011].
References to studies excluded from this review
EBMTR {published data only}
- Morris C, Iacobelli S, Brand R, Bjorkstrand B, Drake M, Niederwieser D, et al. Benefit and timing of second transplantations in multiple myeloma: clinical findings and methodological limitations in a European Group for Blood and Marrow Transplantation registry study. Journal of Clinical Oncology 2004;22(9):1674‐81. [DOI] [PubMed] [Google Scholar]
Kim 2009 {published data only}
- Kim SJ, Kim K, Kim BS, Jo DY, Kang HJ, Kim JS, et al. Clinical features and survival outcomes in patients with multiple myeloma: analysis of web‐based data from the Korean Myeloma Registry. Acta Haematologica 2009;122(4):200‐10. [DOI] [PubMed] [Google Scholar]
Koren 2010 {published data only}
- Koren J, Spicka I, Straub J, Vackova B, Trnkova M, Pohlreich D, et al. Retrospective analysis of the results of high‐dose chemotherapy with the support of autologous blood stem cells in patients with multiple myeloma. The experience of a single centre. Prague Medical Report 2010;111(3):207‐18. [PubMed] [Google Scholar]
Lahuerta 2003 {published data only}
- Lahuerta JJ, Grande C, Martinez‐Lopez J, SJ, Toscano R, Ortiz MC, et al. Tandem transplants with different high‐dose regimens improve the complete remission rates in multiple myeloma. Results of a Grupo Espanol de Sindromes Linfoproliferativos/Trasplante Autologo de Medula Osea phase II trial. British Journal of Haematology 2003;120(2):296‐303. [DOI] [PubMed] [Google Scholar]
NMSG {published data only}
- Bjorkstrand B, Klausen TW, Remes K, Gruber A, Knudsen LM, Bergmann OJ, et al. Double versus single high‐dose melphalan 200 mg/m² and autologous stem cell transplantation for multiple myeloma: a region‐based study in 484 patients from the Nordic area. Hematology Reviews. 1 2009; Vol. 1:e2.
Sonneveld 2007 {published data only}
- Sonneveld P, HB, Segeren CM, Vellenga E, Croockewit AJ, Verhoe GE, et al. Intermediate‐dose melphalan compared with myeloablative treatment in multiple myeloma: long‐term follow‐up of the Dutch Cooperative Group HOVON 24 trial. Haematologica 2007;92(7):928‐35. [DOI] [PubMed] [Google Scholar]
Total therapy {published data only}
- Barlogie B, Anaissie E, van RF, Shaughnessy JD Jr, Szymonifka J, et al. Reiterative survival analyses of Total Therapy 2 for multiple myeloma elucidate follow‐up time dependency of prognostic variables and treatment arms. Journal of Clinical Oncology 2010;28(18):3023‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlogie B, Jagannath S, Desikan KR, Mattox S, Vesole D, Siegel D, et al. Total therapy with tandem transplants for newly diagnosed multiple myeloma. Blood 1999;93(1):55‐65. [PubMed] [Google Scholar]
- Barlogie B, Jagannath S, Vesole DH, Naucke S, Cheson B, Mattox S, et al. Superiority of tandem autologous transplantation over standard therapy for previously untreated multiple myeloma. Blood 1997;89(3):789‐93. [PubMed] [Google Scholar]
- Barlogie B, Shaughnessy J, Anaissie E, Rhee F, Alsayed Y, Waheed S, et al. Modeling for Cure:Total Therapy Trials for Newly Diagnosed Multiple Myeloma: Let the Math Speak! [American Society of Hematology ‐ Annual meeting held in New Orleans December 5‐8, 2009]. http://myeloma.uams.edu/Modeling‐for‐Cure‐ASH‐2009.pdf [accessed 01.03.2011].
- Barlogie B, Tricot G, Rasmussen E, Anaissie E, Rhee F, Zangari M, et al. Total therapy 2 without thalidomide in comparison with total therapy 1: role of intensified induction and posttransplantation consolidation therapies. Blood 2006;107(7):2633‐8. [DOI] [PubMed] [Google Scholar]
- Barlogie B, Tricot GJ, Rhee F, Angtuaco E, Walker R, Epstein J, et al. Long‐term outcome results of the first tandem autotransplant trial for multiple myeloma. British Journal of Haematology 2006;135(2):158‐64. [DOI] [PubMed] [Google Scholar]
- Barlogie B, Zangari M, Bolejack V, Hollmig K, Anaissie E, Rhee F, et al. Superior 12‐year survival after at least 4‐year continuous remission with tandem transplantations for multiple myeloma. British Journal of Haematology 2006;6(6):469‐74. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
NCT01109004 {published data only}
- Horowitz. Stem Cell Transplant With Lenalidomide Maintenance in Patients With Multiple Myeloma (BMT CTN 0702; NCT01109004). http://clinicaltrials.gov/ct2/show/NCT01109004?term=NCT01109004&rank=1 2010 [accessed 22.2.2011].
Additional references
Abdelkefi 2008
- Abdelkefi A, Ladeb S, Torjman L, Othman TB, Lakhal A, Romdhane NB, et al. Single autologous stem‐cell transplantation followed by maintenance therapy with thalidomide is superior to double autologous transplantation in multiple myeloma: results of a multicenter randomized clinical trial. Blood 2008;111(4):1805‐10. [DOI] [PubMed] [Google Scholar]
Abdelkefi 2009
- Abdelkefi A, Ladeb S, Torjman L, Ben Othman T, Lakhal A, Ben Romdhane N, et al. on behalf of the Tunisian Multiple Myeloma Study Group. Retraction of Abdelkefi A, Ladeb S, Torjman L, Othman TB, Lakhal A, Romdhane NB, et al. Blood 2008 Feb 15;111(4):1805‐10; PM. Blood 2009;113(24):6265. [DOI] [PubMed] [Google Scholar]
Alexander 2007
- Alexander DD, Mink PJ, Adami HO, Cole P, Mandel JS, Oken MM, et al. Multiple myeloma: a review of the epidemiologic literature. International Journal of Cancer 2007;120 Suppl 12:40‐61. [DOI] [PubMed] [Google Scholar]
Anderson 2008
- Anderson KC, Kyle RA, Rajkumar SV, Stewart AK, Weber D, Richardson P. Clinically relevant end points and new drug approvals for myeloma. Leukemia 2008;22(2):231‐9. [DOI] [PubMed] [Google Scholar]
Attal 1996
- Attal M, Harousseau JL, Stoppa AM, Sotto JJ, Fuzibet JG, Rossi JF, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. New England Journal of Medicine 1996;335(2):91‐7. [DOI] [PubMed] [Google Scholar]
Attal 2007
- Attal M, Moreau P, Avet‐Loiseau H, Harousseau JL. Stem cell transplantation in multiple myeloma. Hematology American Society of Hematology Education Program 2007:311‐6. [DOI] [PubMed] [Google Scholar]
Attal 2009
- Attal M, Harousseau JL. The role of high‐dose therapy with autologous stem cell support in the era of novel agents. Seminars in Hematology 2009;46(2):127‐32. [DOI] [PubMed] [Google Scholar]
Awedan 2002
- Awedan AA. High intensity regimens with autologous hematopoietic stem cell transplantation as treatment of multiple myeloma. Annals of Transplantation 2002;7(2):38‐43. [PubMed] [Google Scholar]
Barlogie 1986
- Barlogie B, Hall R, Zander A, Dicke K, Alexanian R. High‐dose melphalan with autologous bone marrow transplantation for multiple myeloma. Blood 1986;67(5):1298‐301. [PubMed] [Google Scholar]
Barlogie 1997
- Barlogie B, Jagannath S, Vesole DH, Naucke S, Cheson B, Mattox S, et al. Superiority of tandem autologous transplantation over standard therapy for previously untreated multiple myeloma. Blood 1997;89(3):789‐93. [PubMed] [Google Scholar]
Barlogie 2006
- Barlogie B, Tricot G, Rasmussen E, Anaissie E, Rhee F, Zangari M, et al. Total therapy 2 without thalidomide in comparison with total therapy 1: role of intensified induction and posttransplantation consolidation therapies. Blood 2006;107(7):2633‐8. [DOI] [PubMed] [Google Scholar]
Barlogie 2007
- Barlogie B, Rhee F, Anaissie E, Epstein J, Crowley J, Shaughnessy J. Should autologous transplant be part of the primary treatment in MM?. Haematologica. 92 2007; Vol. 92 Suppl 2:67.
Barlogie 2009
- Barlogie B, Shaughnessy J, Anaissie E, Rhee F, Alsayed Y, Waheed S, et al. Modeling for Cure:Total Therapy Trials for Newly Diagnosed Multiple Myeloma: Let the Math Speak!. American Society of Hematology ‐ Annual meeting held in New Orleans December 5‐8, 2009.
Bladé 1998
- Blade J, Samson D, Reece D, Apperley J, Bjorkstrand B, Gahrton G, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high‐dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. British Journal of Haematology 1998;102(5):1115‐23. [DOI] [PubMed] [Google Scholar]
Blair 2004
- Blair E. Gold is not always good enough: the shortcomings of randomization when evaluating interventions in small heterogeneous samples. Journal of Clinical Epidemiology 2004;57(12):1219‐22. [DOI] [PubMed] [Google Scholar]
Campagnaro 2008
- Campagnaro E, Saliba R, Giralt S, Roden L, Mendoza F, Aleman A, et al. Symptom burden after autologous stem cell transplantation for multiple myeloma. Cancer 2008;112(7):1617‐24. [DOI] [PubMed] [Google Scholar]
Cavo 2010
- Cavo M, Tacchetti P, Patriarca F, Petrucci MT, Pantani L, Galli M, et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem‐cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet 2010;376(9758):2075‐85. [DOI] [PubMed] [Google Scholar]
Chalmers 2009
- Chalmers I, Glasziou P. Avoidable waste in the production and reporting of research evidence. Lancet 2009;374(9683):86‐9. [DOI] [PubMed] [Google Scholar]
CHMP/EWP/27994/08 2008
- CHMP/EWP/27994/08. Appendix 1 to the Guideline on the Evaluation of Anticancer Medicinal Products in Man ‐ Methodological Considerations for Using Progression‐Free Survival (PFS) as Primary endpoints in confirmatory trials for registration. http://www.ema.europa.eu/pdfs/human/ewp/2799408en.pdf 2008 [accessed 22.2.2011].
Clarke 2004
- Clarke M. Doing new research? Don't forget the old. PLoS Medicine 2004;1(2):e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Copelan 2006
- Copelan EA. Hematopoietic stem‐cell transplantation. New England Journal of Medicine 2006;354(17):1813‐26. [DOI] [PubMed] [Google Scholar]
CPMP/EWP/205/95 2005
- CPMP/EWP/205/95 Rev.3. Evaluation of Anticancer Medicinal Products in Man. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/12/WC500017748.pdf 2005 [accessed 22.2.2011].
Curt 2008
- Curt GA, Chabner BA. One in five cancer clinical trials is published: a terrible symptom‐‐what's the diagnosis?. Oncologist 2008;13(9):923‐4. [DOI] [PubMed] [Google Scholar]
Dickersin 1994
- Dickersin K, Scherer R, Lefebvre C. Identifying relevant studies for systematic reviews. [Identifying relevant studies for systematic reviews.]. British Medical Journal 1994;309(6964):1286‐91. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Durie 1975
- Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer 1975;36(3):842‐54. [DOI] [PubMed] [Google Scholar]
Durie 2001
- Durie BG. The epidemiology of multiple myeloma. Seminars in Hematology 2001;38 Suppl 3(2):1‐5. [DOI] [PubMed] [Google Scholar]
Durie 2006
- Durie BG, Harousseau JL, Miguel JS, Blade J, Barlogie B, Anderson K, et al. International uniform response criteria for multiple myeloma. Leukemia 2006;20(9):1467‐73. [DOI] [PubMed] [Google Scholar]
Durie 2008
- Durie BG. Treatment of myeloma‐‐are we making progress?. New England Journal of Medicine 2008;359(9):964‐6. [DOI] [PubMed] [Google Scholar]
Durie 2010
- Durie BG. Role of new treatment approaches in defining treatment goals in multiple myeloma‐‐the ultimate goal is extended survival. Cancer Treatment Reviews 2010;36 Suppl 2:18‐23. [DOI] [PubMed] [Google Scholar]
Egger 2001
- Egger M, Smith GD, Sterne JA. Uses and abuses of meta‐analysis. Clinical Medicine 2001;1(6):478‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Einsele 2007
- Einsele H. Stem Cell Transplantation for MM: Analysis of Prognostic Factors[S9.3]. Haematologica XIth International Myeloma Foundation Workshop, Kos. 90 2007 http://myeloma.org/pdfs/Kos2007_Einsele.pdf [accessed 22.2.2011]; Vol. 90, issue s2:49.
EMA/CHMP/EWP/520088/2008
- Effficacy working party of the European Medical Agencies. Appendix 2 to the Guideline on the evaluation of Anticancer Medicinal Products in Man (CPMP/EWP/205/95 Rev. 3) on Confirmatory studies in Haematological. http://www.ema.europa.eu/pdfs/human/ewp/52008808enfin.pdf 2010 [accessed 22.2.2011].
Feinstein 1995
- Feinstein AR. Meta‐analysis: statistical alchemy for the 21st century. Journal of Clinical Epidemiology 1995;48(1):71‐9. [DOI] [PubMed] [Google Scholar]
Fermand 2007
- Fermand JP. Should autologous transplant be part of the primary treatment in MM? [XIth International Myeloma Workshop, Kos 2007]. Haematologica. 92 2007; Vol. 92, issue s2:68‐9 http://myeloma.org/pdfs/Kos2007_Fermand.pdf [accessed 22.2.2011].
Fonseca 2007
- Fonseca R, Stewart AK. Targeted therapeutics for multiple myeloma: the arrival of a risk‐stratified approach. Molecular Cancer Therapy 2007;6(3):802‐10. [DOI] [PubMed] [Google Scholar]
Fonseca 2009
- Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia 2009;23(12):2210‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gertz 2007
- Gertz MA. Relevant prognostic features of multiple myeloma and the new International Staging System. Leukemia & Lymphoma 2007;48(3):458‐68. [DOI] [PubMed] [Google Scholar]
Gertz 2008
- Gertz MA, Ansell SM, Dingli D, Dispenzieri A, Buadi FK, Elliott MA, et al. Autologous stem cell transplant in 716 patients with multiple myeloma: low treatment‐related mortality, feasibility of outpatient transplant, and effect of a multidisciplinary quality initiative. Mayo Clinical Proceedings 2008;83(10):1131‐8. [DOI] [PubMed] [Google Scholar]
Gertz 2009
- Gertz M. Is autologous stem cell transplantation the therapy of choice for the treatment of Multiple Myeloma patients? CONTRA. 2nd Heidelberg Myeloma Workshop 2009 http://mds‐forum.onkodin.de/myelom2009/content/e54/e8239/e8258/index_ger.html [accessed 22.2.2011].
Giralt 2009
- Giralt S, Vesole DH, Somlo G, Krishnan A, Stadtmauer E, Mccarthy P, et al. Re: Tandem vs single autologous hematopoietic cell transplantation for the treatment of multiple myeloma: a systematic review and meta‐analysis. Journal of the National Cancer Institute 2009;101(13):964‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Giralt 2010
- Giralt S. 200 mg/m(2) melphalan‐‐the gold standard for multiple myeloma. Nature Reviews Clinical Oncology 2010;7(9):490‐1. [DOI] [PubMed] [Google Scholar]
GMMG‐HD5
- Goldschmidt 2009. The GMMG‐HD5 trial: bortezomib‐based induction prior to high dose therapy and autologous stem cell transplantation followed by lenalidomide‐based consolidation and maintenance therapy in patients with multiple myeloma. http://www.controlled‐trials.com/ISRCTN05745813 [accessed 26 May 2011] 2009.
Gratwohl 2007
- Gratwohl A. Activity survey and historical perspective of autologous stem cell transplantation in Europe. Seminars in Hematology 2007;44(4):220‐6. [DOI] [PubMed] [Google Scholar]
Greipp 2005
- Greipp PR, San Miguel J, Durie BG, Crowley JJ, Barlogie B, Blade J, et al. International staging system for multiple myeloma. Journal Clinical Oncology 2005;23(15):3412‐20. [DOI] [PubMed] [Google Scholar]
Hahn 2003
- Hahn T, Wingard JR, Anderson KC, Bensinger WI, Berenson JR, Brozeit G, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of multiple myeloma: an evidence‐based review. Biology of Blood and Marrow Transplantation 2003;9(1):4‐37. [DOI] [PubMed] [Google Scholar]
Harousseau 1992
- Harousseau JL, Milpied N, Laporte JP, Collombat P, Facon T, Tigaud JD, et al. Double‐intensive therapy in high‐risk multiple myeloma. Blood 1992;79(11):2827‐33. [PubMed] [Google Scholar]
Harousseau 2009
- Harousseau JL, Attal M, vet‐Loiseau H. The role of complete response in multiple myeloma. Blood 2009;114(15):3139‐46. [DOI] [PubMed] [Google Scholar]
Harousseau 2009a
- Harousseau JL, Moreau P. Autologous hematopoietic stem‐cell transplantation for multiple myeloma. New England Journal of Medicine 2009;360(25):2645‐54. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Cochrane Handbook. Cochrane Handbook for Systematic Reviews of Interventions [version 5.1.0.]. The Cochrane Collaboration, 2011.
Hopewell 2009
- Hopewell S, Loudon K, Clarke MJ, Oxman AD, Dickersin K. Publication bias in clinical trials due to statistical significance or direction of trial results. Cochrane Database of Systematic Reviews 2009, (1):MR000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hopewell 2010
- Hopewell S, Dutton S, Yu L‐M, Chan A‐W, Altman DG. The quality of reports of randomised trials in 2000 and 2006: comparative study of articles indexed in PubMed. BMJ. 340 2010; Vol. 340. [DOI] [PMC free article] [PubMed]
International Myeloma Workshop 2003
- International Myeloma Working group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. British Journal of Haematology 2003;121(5):749‐57. [PubMed] [Google Scholar]
IQWIG N05‐03C
- Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (editor). Stem cell transplantation in multiple myeloma [Stammzelltransplantation beim multiplen Myelom]. https://www.iqwig.de/download/N05‐03C_Vorbericht_Stammzelltransplantation_bei_Multiplem_Myelom.pdf 2011.
Jantunen 2006
- Jantunen E, Itala M, Siitonen T, Koivunen E, Leppa S, Juvonen E, et al. Late non‐relapse mortality among adult autologous stem cell transplant recipients: a nation‐wide analysis of 1,482 patients transplanted in 1990‐2003. European Journal of Haematology 2006;77(2):114‐9. [DOI] [PubMed] [Google Scholar]
Jantunen 2006a
- Jantunen E, Itala M, Lehtinen T, Kuittinen O, Koivunen E, Leppa S, et al. Early treatment‐related mortality in adult autologous stem cell transplant recipients: a nation‐wide survey of 1482 transplanted patients. European Journal of Haematology 2006;76(3):245‐50. [DOI] [PubMed] [Google Scholar]
Johnsen 2010
- Johnsen HE, Klausen TW, Boegsted M, Lenhoff S, Gimsing P, Christiansen I, et al. Improved survival for multiple myeloma in denmark based on autologous stem cell transplantation and novel drug therapy in collaborative trials: analysis of accrual, prognostic variables, selection bias, and clinical behavior on survival in more than 1200 patients in trials of the nordic myeloma study group. Clinical Lymphoma Myeloma and Leukemia 2010;10(4):290‐6. [DOI] [PubMed] [Google Scholar]
Jones 2008
- Jones JA, Qazilbash MH, Shih YC, Cantor SB, Cooksley CD, Elting LS. In‐hospital complications of autologous hematopoietic stem cell transplantation for lymphoid malignancies: clinical and economic outcomes from the Nationwide Inpatient Sample. Cancer 2008;112(5):1096‐105. [DOI] [PubMed] [Google Scholar]
Juni 2001
- Juni P, Altman DG, Egger M. Systematic reviews in health care: Assessing the quality of controlled clinical trials. BMJ 2001;323(7303):42‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Keirns 2008
- Keirns CC, Carr BG. From the emergency department to vital statistics: cause of death uncertain. Academic Emergency Medicine 2008;15(8):768‐75. [DOI] [PubMed] [Google Scholar]
Klein‐Geltink 2007
- Klein‐Geltink JE, Rochon PA, Dyer S, Laxer M, Anderson GM. Readers should systematically assess methods used to identify, measure and analyze confounding in observational cohort studies. Journal of Clinical Epidemiology 2007;60(8):766‐72. [DOI] [PubMed] [Google Scholar]
Klungel 2004
- Klungel OH, Martens EP, Psaty BM, Grobbee DE, Sullivan SD, Stricker BH, et al. Methods to assess intended effects of drug treatment in observational studies are reviewed. Journal of Clinical Epidemiology 2004;57(12):1223‐31. [DOI] [PubMed] [Google Scholar]
Koreth 2007
- Koreth J, Cutler CS, Djulbegovic B, Behl R, Schlossman RL, Munshi NC, et al. High‐dose therapy with single autologous transplantation versus chemotherapy for newly diagnosed multiple myeloma: A systematic review and meta‐analysis of randomized controlled trials. Biololgy of Blood and Marrow Transplantation 2007;13(2):183‐96. [DOI] [PubMed] [Google Scholar]
Kortüm 2010
- Kortüm M, Einsele H, Naumann R, Peest D, Liebisch P, Goldschmidt H. Leitlinie Multiples Myelom. http://www.dgho.de/onkopedia/Multiples%20Myelom [accessed 26.05.11] 2010.
Krzyzanowska 2003
- Krzyzanowska MK, Pintilie M, Tannock IF. Factors associated with failure to publish large randomized trials presented at an oncology meeting. JAMA 2003;290(4):495‐501. [DOI] [PubMed] [Google Scholar]
Kumar 2008
- Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008;111(5):2516‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kumar 2009
- Kumar A, Kharfan‐Dabaja MA, Glasmacher A, Djulbegovic B. Tandem versus single autologous hematopoietic cell transplantation for the treatment of multiple myeloma: a systematic review and meta‐analysis. Journal of the National Cancer Institute 2009;101(2):100‐6. [DOI] [PubMed] [Google Scholar]
Kumar 2009a
- Kumar A, Djulbegovic B. Author reply to Giralt et al and Tricot et al: Tandem versus single autologous hematopoietic cell transplantation for the treatment of multiple myeloma: a systematic review and meta‐analysis. Journal of the National Cancer Institute. 101 2009; Vol. 101, issue 13:966‐7.
Kumar 2009b
Kunz 1998
- Kunz R, Oxman AD. The unpredictability paradox: review of empirical comparisons of randomised and non‐randomised clinical trials. BMJ 1998;317(7167):1185‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lahuerta 2010
- Lahuerta JJ, Mateos MV, Martínez‐López J, Grande C, Rubia J, Rosiñol L, et al. Grupo Español de MM and Programa para el Estudio de la Terapéutica en Hemopatía Maligna Cooperative Study Groups. Busulfan 12 mg/kg plus melphalan 140 mg/m2 versus melphalan 200 mg/m2 as conditioning regimens for autologous transplantation in newly diagnosed multiple myeloma patients included in the PETHEMA/GEM2000 study. Haematologica 2010;95(11):1913‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lane 2005
- Lane SW, Gill D, Mollee PN, Rajkumar SV. Role of VAD in the initial treatment of multiple myeloma. Blood 2005;106(10):3674‐5. [DOI] [PubMed] [Google Scholar]
Laubach 2011
- Laubach J, Richardson P, Anderson K. Multiple myeloma. Annual Review of Medicine 2011;62:249‐64. [DOI] [PubMed] [Google Scholar]
Levy 2005
- Levy V, Katsahian S, Fermand JP, Mary JY, Chevret S. A meta‐analysis on data from 575 patients with multiple myeloma randomly assigned to either high‐dose therapy or conventional therapy. Medicine (Baltimore) 2005;84(4):250‐60. [DOI] [PubMed] [Google Scholar]
Majhail 2008
- Majhail NS. Old and new cancers after hematopoietic‐cell transplantation. Hematology American Society of Hematology Education Program 2008:142‐9. [DOI] [PubMed] [Google Scholar]
Mehta 2009
- Mehta J. Re: Tandem vs single autologous hematopoietic cell transplantation for the treatment of multiple myeloma: a systematic review and meta‐analysis. Journal of the National Cancer Institute 2009;101(20):1430‐1. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials.. JAMA 2001;285(15):1987‐91. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. Journal of Clinical Epidemiology 2009;62(10):1006‐12. [DOI] [PubMed] [Google Scholar]
Moore 1998
- Moore RA, Gavaghan D, Tramer MR, Collins SL, McQuay HJ. Size is everything‐‐large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998;78(3):209‐16. [DOI] [PubMed] [Google Scholar]
Moreau 2002
- Moreau P, Facon T, Attal M, Hulin C, Michallet M, Maloisel F, et al. Comparison of 200 mg/m(2) melphalan and 8 Gy total body irradiation plus 140 mg/m(2) melphalan as conditioning regimens for peripheral blood stem cell transplantation in patients with newly diagnosed multiple myeloma: final analysis of the Intergroupe Francophone du Myelome 9502 randomized trial. Blood 2002;99(3):731‐5. [DOI] [PubMed] [Google Scholar]
Moreau 2011
- Moreau P, Attal M, Pegourie B, Planche L, Hulin C, Facon T, et al. Achievement of VGPR to induction therapy is an important prognostic factor for longer PFS in the IFM 2005‐01 trial. Blood 2011;117(11):3041‐4. [DOI] [PubMed] [Google Scholar]
Morris 2004
- Morris C, Iacobelli S, Brand R, Bjorkstrand B, Drake M, Niederwieser D, et al. Benefit and timing of second transplantations in multiple myeloma: clinical findings and methodological limitations in a European Group for Blood and Marrow Transplantation registry study. Journal of Clinical Oncology 2004;22(9):1674‐81. [DOI] [PubMed] [Google Scholar]
Munshi 2008
- Munshi NC. Plasma cell disorders: an historical perspective. Hematology American Society of Hematology Education Program 2008:297. [DOI] [PubMed] [Google Scholar]
Munshi 2011
- Munshi NC, Anderson KC, Bergsagel PL, Shaughnessy J, Palumbo A, Durie B, et al. Consensus recommendations for risk stratification in multiple myeloma: report of the International Myeloma Workshop Consensus Panel 2. Blood 2011;117(18):4696‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Myeloma Trialists 98
- Myeloma Trialists' Collaborative Group. Combination chemotherapy versus melphalan plus prednisone as treatment for multiple myeloma: an overview of 6,633 patients from 27 randomized trials. Myeloma Trialists' Collaborative Group. Journal of Clinical Oncology 1998;16(12):3832‐42. [DOI] [PubMed] [Google Scholar]
Nahi 2011
- Nahi H, Sutlu T, Jansson M, Alici E, Gahrton G. Clinical impact of chromosomal aberrations in multiple myeloma. Journal of Internal Medicine 2011;269(2):137‐47. [DOI] [PubMed] [Google Scholar]
NCCN 2011
- NCCN Multiple Myeloma Panel. Multiple Myeloma, Version 3.2010. NCCN Clinical pratice guidelines in Oncology 2011; Vol. http://guidelines.nccn.org/published‐guideline/AB9CB2A2‐ABDF‐9C66‐6D92‐8F8FD1F26358/guideline.pdf [accessed 01.03.2011].
Pignon 2001
- Pignon JP, Hill C. Meta‐analyses of randomised clinical trials in oncology. Lancet Oncology 2001;2(8):475‐82. [DOI] [PubMed] [Google Scholar]
Podar 2009
- Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 2009;23(1):10‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rajkumar 2008
- Rajkumar SV. Treatment of myeloma: cure vs control. Mayo Clinical Proceedings 2008;83(10):1142‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ramsey 2008
- Ramsey S, Scoggins J. Commentary: practicing on the tip of an information iceberg? Evidence of underpublication of registered clinical trials in oncology. Oncologist 2008;13(9):925‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schulz 2002
- Schulz KF, Grimes DA. Sample size slippages in randomised trials: exclusions and the lost and wayward. Lancet 2002;359(9308):781‐5. [DOI] [PubMed] [Google Scholar]
Tam 2008
- Tam VC, Hotte SJ. Consistency of phase III clinical trial abstracts presented at an annual meeting of the American Society of Clinical Oncology compared with their subsequent full‐text publications. Journal of Clinical Oncology 2008;26(13):2205‐11. [DOI] [PubMed] [Google Scholar]
Tricot 2008
- Tricot G. Is more better in myeloma?. Blood 2008;112(9):3532. [DOI] [PubMed] [Google Scholar]
Tricot 2009
- Tricot G, Kern SE, Barlogie B. Re: Tandem vs single autologous hematopoietic cell transplantation for the treatment of multiple myeloma: a systematic review and meta‐analysis. Journal of the National Cancer Institute 2009;101(13):964‐6. [DOI] [PubMed] [Google Scholar]
Turesson 2010
- Turesson I, Velez R, Kristinsson SY, Landgren O. Patterns of improved survival in patients with multiple myeloma in the twenty‐first century: a population‐based study. Journal of Clinical Oncology 2010;28(5):830‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vandenbroucke 2008
- Vandenbroucke JP. Observational research, randomised trials, and two views of medical science. PLoS Medicine 2008;5(3):e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
von Elm 2008
- Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Journal of Clinical Epidemiology 2008;61(4):344‐9. [DOI] [PubMed] [Google Scholar]
Wheatley 2006
- Wheatley K, Hills RK, Burnett AK. Problems with up‐front randomization in clinical trials. Journal of Clinical Oncology 2006;24(34):5471‐2. [DOI] [PubMed] [Google Scholar]
Wilson 2003
- Wilson RL, Gray ML, Giralt S. Barriers to second transplant for multiple myeloma in a randomised autologous tandem trial. Biology of Blood and Marrow Transplantation IBMTR/ABMTR Participants' meeting, Keystone Colorado, January 2003.. 9 2003; Vol. 9, issue 2.