

Summary
Mitochondria import nearly all their ~1,000–2,000 constituent proteins from the cytosol across their double-membrane envelope1–5. Genetic and biochemical studies have shown that the conserved protein translocase, termed the TIM23 complex, mediates import of presequence-containing proteins (preproteins) into the mitochondrial matrix and inner membrane. Among about ten different subunits of the TIM23 complex, the essential multi-pass membrane protein Tim23, together with the evolutionarily related protein Tim17, has long been postulated to form a protein-conducting channel6–11. However, the mechanism by which these subunits form a translocation path in the membrane and enable the import process remained unclear due to a lack of structural information. Here, we have determined the cryo-electron microscopy (cryo-EM) structure of the core TIM23 complex (heterotrimeric Tim17–Tim23–Tim44) from Saccharomyces cerevisiae. Contrary to the prevailing model, Tim23 and Tim17 themselves do not form a water-filled channel, but instead have separate, lipid-exposed concave cavities that face in opposite directions. Our structural and biochemical analyses show that surprisingly, the cavity of Tim17, not Tim23, forms the protein translocation path whereas Tim23 is likely to play a structural role. The results further suggest that, during translocation of substrate polypeptides, the nonessential subunit Mgr2 seals the lateral opening of the Tim17 cavity to facilitate the translocation process. We propose a new model for the TIM23-mediated protein import and sorting mechanism, a central pathway in mitochondrial biogenesis.
Cryo-EM analysis of yeast TIM23
In S. cerevisiae, the TIM23 complex is formed by up to eleven proteins: Tim23, Tim17, Tim44, Tim50, Tim21, Pam16, Pam17, Pam18, Mgr2, mitochondrial Hsp70 (mtHsp70), and Mge1 (ref. 12–14) (Extended Data Fig. 1a). To overcome limited sample quantities obtained from purification of the endogenous complex, we co-overexpressed almost all TIM23 subunits, including Spot-tagged Tim17 (Extended Data Fig. 1b,c). Compared to the endogenous complex, the apparent stoichiometries of copurified Tim50, Tim44, Pam16, and Pam18 with respect to Tim17 and Tim23 were substantially higher in the overexpressed complex. This variability is probably caused by concentration-dependent dissociation of these components from the Tim17–Tim23 core due to their weak association15–19.
Our initial cryo-EM analysis of the overexpressed TIM23 complex yielded only a low (~8-Å)-resolution disc-like reconstruction with a small protrusion, which we speculated to be the C-terminal domain (CTD) of Tim44 (Extended Data Fig. 1 d–h). The low resolution seemed due to the small molecular weight (~50–60 kDa) of ordered parts. We therefore developed a monoclonal antibody against Tim44-CTD and used its antigen-binding fragment (Fab) as a fiducial marker. The Fab dramatically increased the map resolution, up to 2.7 Å with a sample containing Tim17, Tim23, Tim44, Pam16, and Pam18, allowing atomic modeling of a core composed of Tim17, Tim23, and Tim44 (Fig. 1, Extended Data Table 1, and Extended Data Fig. 2). The Tim44-CTD model could be superposed with its previous crystal structure20, indicating that Fab binding did not affect its overall structure (Extended Data Fig. 2i). We note that the structure of Tim17–Tim23–Tim44 was essentially invariant for all samples we analyzed, although their non-core subunit composition varied21. Other copurified subunits, such as Tim50, mtHsp70, Pam16, and Pam18, were invisible or appeared only as low-resolution features in our reconstructions, likely due to their low occupancy and/or conformational flexibility.
Figure 1. Cryo-EM structure of the core TIM23 complex from S. cerevisiae.
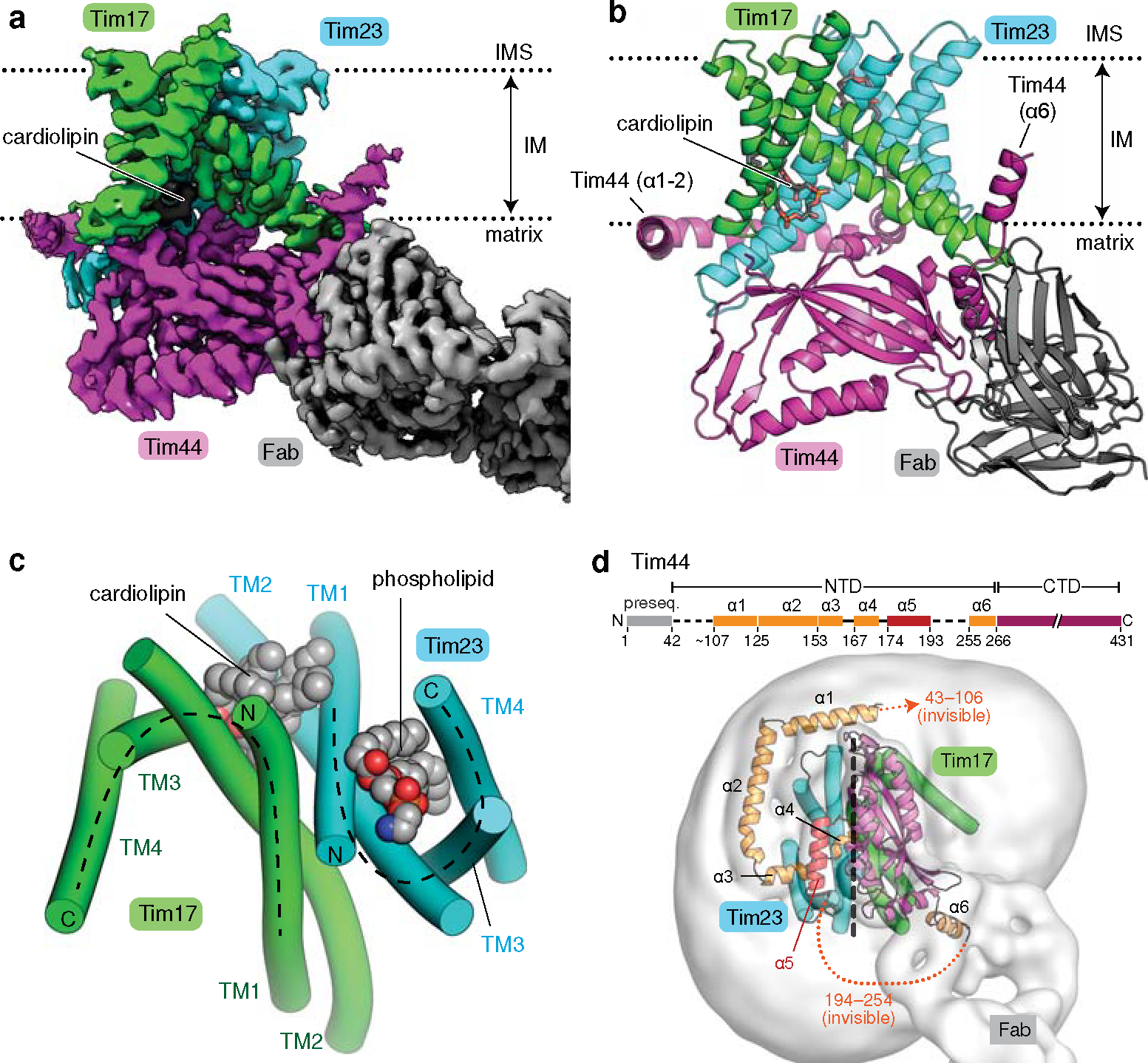
a, Cryo-EM reconstruction of Fab-bound core TIM23 complex. b, As in a, but showing the atomic model. We note that the amphipathic helix α6 of Tim44 is tilted upwards because it sits on a curved micelle edge (see d). c, Arrangement of the TMs of Tim17 and Tim23 in a view from the IMS. TMs are represented as cylinders (arranged in the N to C order), and lipids are shown as spheres. The topology of the cavities is highlighted with dashed lines. d, Organization of Tim44 domains. Upper panel, linear diagram; lower panel, view from the matrix. The detergent micelle is shown with a lowpass-filtered cryo-EM map (gray semitransparent surface). Tim44 is shown as a ribbon diagram (orange, amphipathic helices; red, α5; purple, CTD). Dashed line, plane of the Tim17-Tim23 interface.
The Fab also allowed us to analyze a core TIM23 complex formed by endogenous Tim17 and Tim23. Because Tim44 tended to dissociate during purification of this complex, we overexpressed Tim44 by ~12-fold to increase its occupancy without causing cell growth defects (Extended Data Fig. 3 a–d). A 2.9-Å resolution structure of this complex was essentially identical to the structures obtained by the overexpression approach (Extended Data Fig. 3 e–j). Thus, overexpressed core Tim subunits assemble like the endogenously expressed subunits.
Structure of Tim17–Tim23–Tim44
Our structure shows that Tim17, Tim23, and Tim44 form a 1:1:1 heterotrimer (Fig. 1; also see Supplementary Discussion). Tim17 and Tim23 are arranged with pseudo-two-fold rotational symmetry along the membrane normal. The four transmembrane segments (TMs) of both Tim23 and Tim17 are arranged in a tilted fashion to generate a cavity that is open toward the lipid phase (Fig. 1c). Tim17 and Tim23 associate in a back-to-back arrangement with their cavities facing away from each other. At the interface, the TMs 1–2 of Tim17 and Tim23 are closely packed against each other with several invariant glycine residues (Extended Data Fig. 4 a–c). This arrangement explains the decreased stability of the TIM23 complex upon mutation of these glycine residues22,23.
TMs 1–2 of Tim17 and Tim23 create a shallow depression on the matrix side, where Tim44-CTD docks (Fig. 1b). Our structure reveals that the relatively flat structure of Tim44-CTD aligns vertically along the plane of the Tim17-Tim23 interface, such that one side of Tim44-CTD is oriented towards Tim17 and the other side towards Tim23 (Fig. 1d). This arrangement is in excellent agreement with previous in-vivo photocrosslinking data22,24,25 (Extended Data Fig. 4 d and e).
Tim44 is peripherally associated with the matrix leaflet of the IM through amphipathic helices (α1–α4 and α6) in its N-terminal domain (Fig. 1d). These helices encircle the Tim17–Tim23 heterodimer around the Tim23 side, enabled in part by the α5 segment (residues 174–193) attached to the Tim23-facing side of Tim44-CTD. Mutations in α5 have been shown to cause severe functional defects and destabilization of the complex26, suggesting an important role of α5 in organizing the Tim44 domains. Although invisible in our structure, an ~60-amino-acid long N-terminal segment preceding the α1 helix appears to be directed towards the Tim17 side (Fig. 1d). This segment is important for an interaction with Pam1618,27 and may explain the observed interaction of Pam18, the partner of Pam16, with Tim1712,18.
Our structure shows how the TIM23 complex interacts with cardiolipin, a negatively-charged mitochondrial-specific lipid that is known to be critical for the stability of TIM23 28–30 (Fig. 1 a–c). A well-resolved density for cardiolipin was seen in a crevice formed between Tim17 and Tim23 (Extended Data Fig. 2h). Multiple arginine side chains from Tim17 (Arg105) and Tim44-CTD (Arg347, Arg370, and Arg428) are positioned near the two phosphate groups of the cardiolipin molecule. Destabilization of the complex observed with a Tim17-R105A mutant might be due to decreased affinity towards cardiolipin22.
Cavities of the Tim17 and Tim23 subunits
Contrary to the prevailing model, our structure suggests that neither Tim23 by itself nor together with Tim17 forms a water-filled channel. Instead, one of the two lipid-exposed cavities of the Tim17–Tim23 heterodimer likely constitutes the translocation path. Moreover, the laterally open cavity would be compatible with the IM protein integration (sorting) activity of the TIM23 complex. This idea is consistent with recent cryo-EM structures of the TIM22 complex31,32, a membrane insertase for certain multi-pass IM proteins, such as mitochondrial carrier proteins. Although the TIM22 complex is functionally and structurally distinct from the TIM23 complex, its central subunit Tim22 belongs to the same protein family as Tim17 and Tim23 and adopts the same fold (Extended Data Fig. 5 a and b).
Both Tim17 and homologous Tim22 form a cavity that is large enough to accommodate a polypeptide chain (Extended Data Fig. 5b). On the other hand, due to the inwardly tilted position of Tim23-TM4, the cavity of Tim23 seems too restricted to accommodate even an extended polypeptide (Extended Data Fig. 5a). Furthermore, the cavity of Tim23 is occupied by a well-ordered phospholipid (Extended Data Fig. 2g), and its vertical axis is largely obstructed on the matrix side by a segment of Tim44 (positions 153–171; referred to as α3–α4) (Extended Data Fig. 5c). A partial truncation (Δ150–164) or mutation (154–157 to Ala4) of the α3–α4 segment causes functional defects and impairs association of Tim44 with Tim17–Tim23 (ref. 26), suggesting the importance of α3–α4 for structural integrity of TIM23.
Mutational and computational analyses
The above observations indicated that the cavity of Tim17, rather than that of Tim23, is more likely to be used as the path for protein translocation. Supporting this idea, we found that amino acids lining the cavity of Tim17 are highly conserved whereas those in Tim23 are markedly variable (Fig. 2a). On the surface of the Tim17 cavity, multiple conserved acidic amino acids form a negatively charged patch that extends ~8 Å deep into the membrane from the IMS (Fig. 2 b and c), which is analogous to the functionally important negatively-charged surface observed in the Tim22 structure31,32 (Extended Data Fig. 5d). As a result, the apolar region of the Tim17 cavity seems to span only ~10–15 Å of the membrane (Fig. 2c). These features may cause local membrane thinning in the cavity, reducing the energy barrier for polypeptide translocation. This idea was further supported by all-atom molecular dynamics (MD) simulations, in which we consistently observed formation of a substantial depression in the IMS membrane leaflet at the TIM17 cavity with the conserved acidic amino acids largely exposed to the aqueous phase (Fig. 2 d and e, and Extended Data Fig. 5 e–i).
Figure 2. The cavity of Tim17 forms the path for protein translocation.
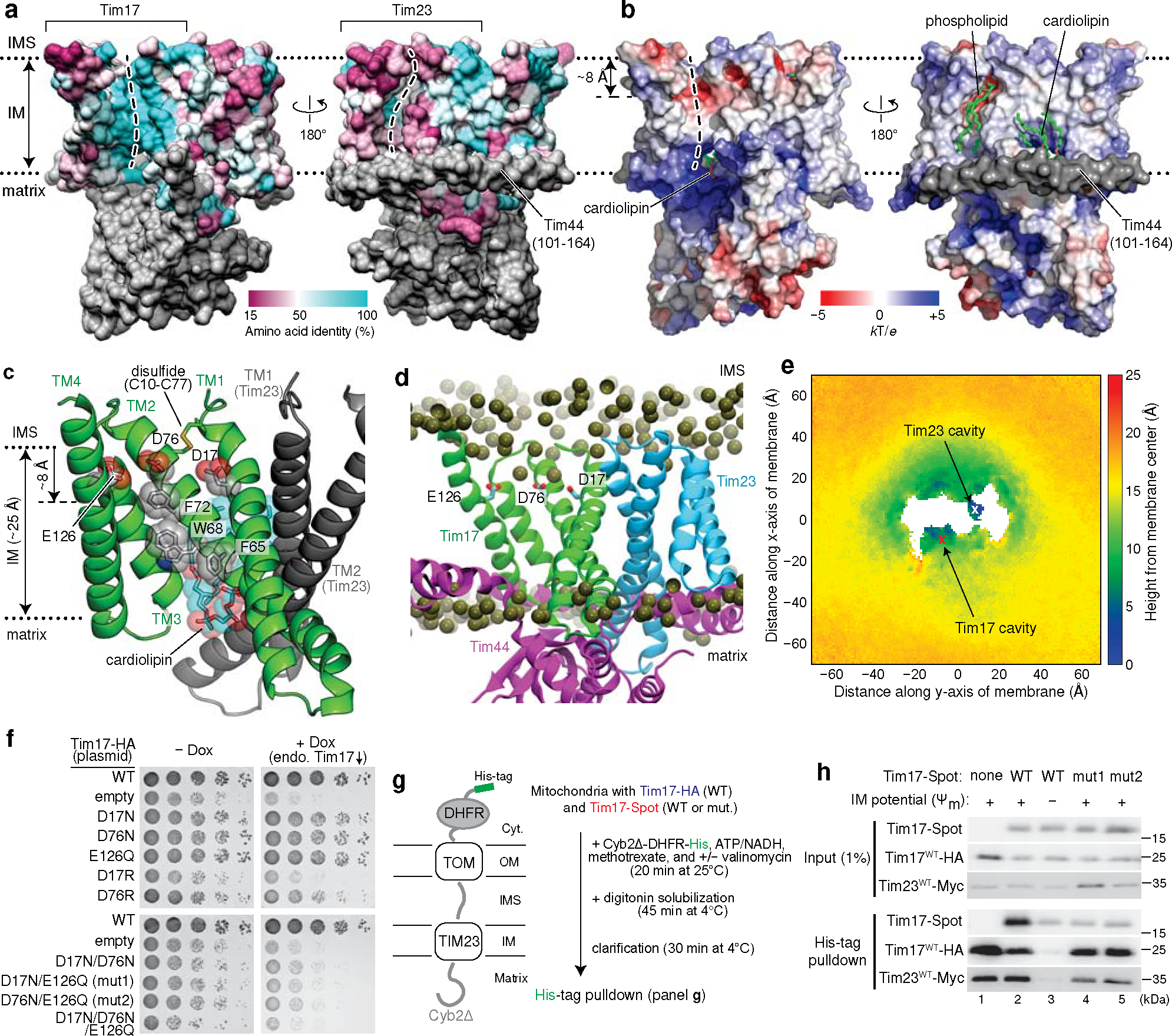
a, Mapping of amino acid sequence conservation of Tim17 and Tim23 onto the structure. Left, view into the Tim17 cavity; right, view into the Tim23 cavity. Dashed line, path along the cavity. b, Surface electrostatics of the TIM23 complex. The views are equivalent to those in a. α1–3 (residues ~107–164) of Tim44 (modelled as polyalanine) were not included in the calculation. c, Conserved acidic and aromatic amino acids lining the Tim17 cavity. Note that Tim17 contains a disulfide bond between Cys10 and Cys77, as shown previously47. d and e, All-atom MD simulation of Tim17–Tim23–Tim44 in a model mitochondrial membrane. In d, positions of lipid headgroup phosphorous atoms are shown as spheres. Panel e shows time-averaged thickness of the lipid hydrophobic layer from the membrane center to the IMS along the membrane plane measured by positions of lipid glycerol atoms (a view from the IMS; also see Extended Data Fig. 5 e–i). We note that a low value at the Tim23 cavity is due to the coordinated phosphatidylethanolamine (Fig. 1c). f, Indicated acidic amino acids lining the Tim17 cavity were mutated, and their functionality was tested by a cell growth assay. The addition of doxycycline (Dox) represses expression of chromosomal WT Tim17. Note that slower growth with the ‘empty/−Dox’ condition is likely due to hypomorphic expression of chromosomal TIM17 under a tetracycline promoter. g, Schematic for pulldown experiments of stalled Cyb2Δ-DHFR. h, Cyb2Δ-DHFR was pulled down with nickel resin after forming a stalled translocation intermediate, and samples were analyzed by immunoblotting. In lane 3, ΔΨm was dissipated by addition of valinomycin. Data in f and h are representative of three independent experiments.
To examine the functional importance of the negatively charged surface in Tim17, we mutated the acidic residues and tested cell viability since TIM23-mediated protein translocation is essential for yeast cell growth (Fig. 2f, and Extended Data Fig. 6 a–g). Although mutating Asp17, Asp76, and Glu126 individually to a neutral Asn/Gln residue did not affect cell growth, double or triple mutations severely impaired viability. In addition, single charge-reversal mutations caused a modest to severe growth defect.
Defects by Tim17 mutations were not due to reduced protein expression as stronger expression did not alleviate the growth impairment (Extended Data Fig. 6 d–g). Co-immunoprecipitation experiments suggested that functionally defective Tim17 mutants (D17N/E126Q and D76N/E126Q; referred to as mut1 and mut2, respectively) assemble properly into the TIM23 complex like WT Tim17 (Extended Data Fig. 6h). To probe translocation defects in these mutants, we generated a stalled translocation intermediate in vitro with mitochondria expressing both WT and mutant Tim17 using a model preprotein fused to a dihydrofolate reductase (Cyb2Δ-DHFR; ref. 33) (Fig. 2g, h and Extended Data Fig. 6 i–k). When Cyb2Δ-DHFR was pulled down after forming the intermediate under the energized condition (+Ψm), WT Tim17 could be markedly enriched (Fig. 2h, lane 2 versus 3). By contrast, mutant Tim17 did not copurify beyond the extent seen under a membrane potential-depleted condition with valinomycin (WT/−Ψm; lanes 4 and 5), whereas the co-expressed WT Tim17 copies were enriched. Thus, mutations in the Tim17 cavity seem to impair preprotein engagement.
We note that Tim23 also has a negatively charged, polar patch in its cavity, mainly formed by Asp96, Asn160, Asn163, and Asp167 (Extended Data Fig. 6l). Unlike Tim17, however, simultaneous mutations of these amino acids to Asn or Ala did not detectably compromise growth (Extended Data Fig. 6m), providing additional evidence that Tim23 is not the route for protein translocation. The cavity of Tim17 also contains conserved, surface exposed aromatic amino acids (Fig. 2c). Mutations of these amino acids to valine did not cause growth defects but the F65N mutation greatly reduced cell viability (Extended Data Fig. 6 a and c). Thus, hydrophobicity of these residues may be more important for function than their amino acid identity per se.
Preproteins pathway along Tim17 and Mgr2
During the course of this study, AlphaFold2 was published34. Provided with the information that Tim17, Tim23, and Tim44 form a 1:1:1 heterotrimer, AlphaFold2 could predict a structure similar to our cryo-EM structure (Extended Data Fig. 7a). Furthermore, a pairwise heterodimer structure prediction study of the yeast proteome found that Mgr2, a small nonessential subunit of TIM23, coevolved with Tim17 and might form a channel-like structure with Tim17 (ref. 35) (Fig. 3a and Extended Data Fig. 7b–d). In this prediction, the two TMs of Mgr2 seal the lateral opening of the Tim17 without perturbing the overall structure of the Tim17–Tim23–Tim44 heterotrimer. This arrangement of Mgr2 is compatible with our model that Tim17 forms the polypeptide translocation path and previous biochemical data showing that Mgr2 is involved in regulating lateral release of clients’ hydrophobic sort signals into the IM14,36.
Figure 3. Preproteins are transported through the Tim17 cavity that is further sealed by Mgr2.
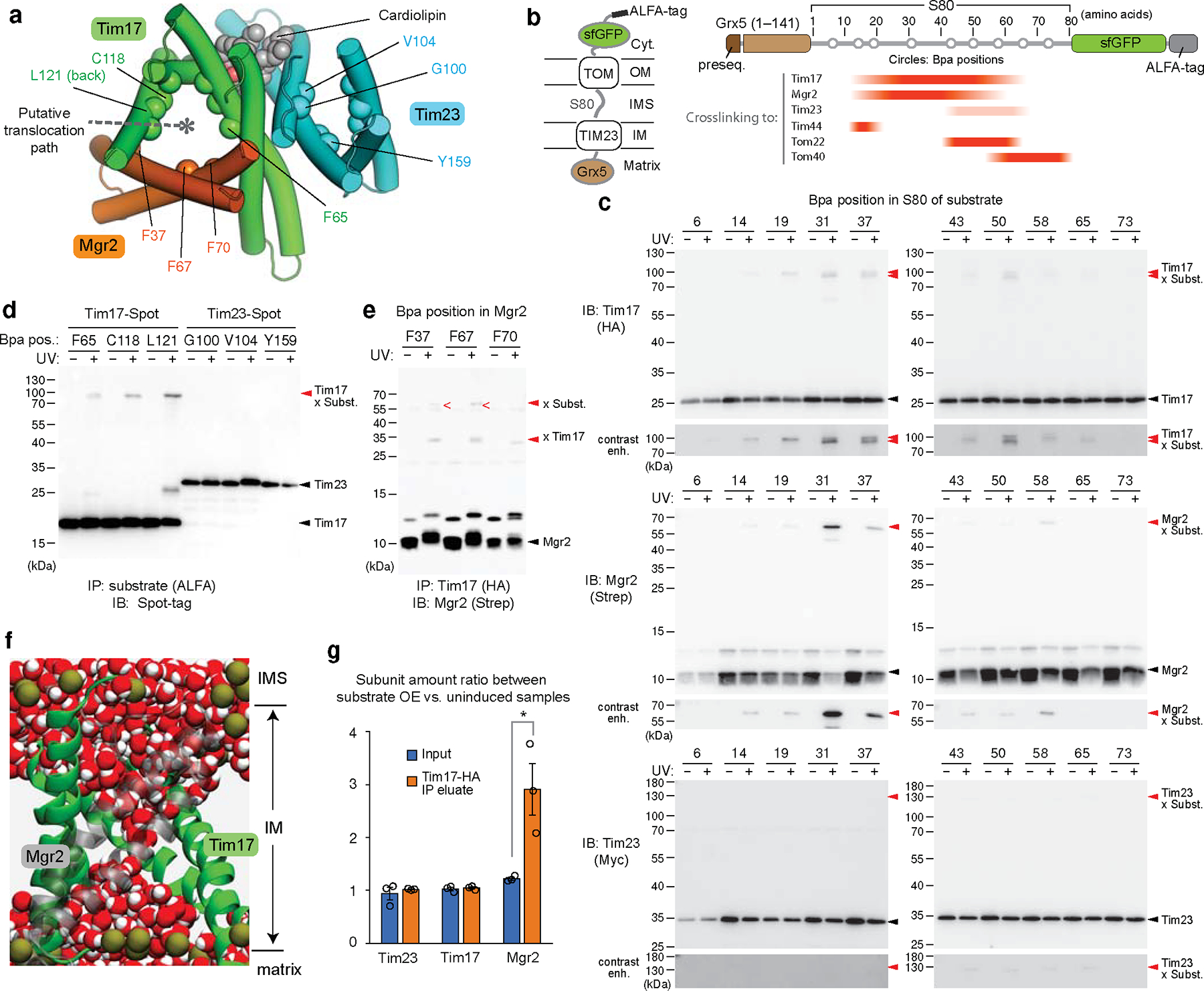
a, Hybrid model (view from IMS) containing the cryo-EM structure of the core TIM23 complex and the structure of Mgr2 predicted by AlphaFold2. For clarity, Tim44 is not shown. Spheres indicate the positions of amino acids that were replaced by Bpa for photocrosslinking (see d and e). Note that L121 is located behind the α-helix and not directly visible in this cartoon. b, Schematic for the stalling substrate Grx5-S80-sfGFP used for Bpa photocrosslinking and summary of results (see c and Extended Data Fig. 8e). c, Bpa was introduced to the indicated position in the S80 region of Grx5-S80-sfGFP. Crosslink adducts were analyzed by ALFA-tag immunoprecipitation (IP) of digitonin-solubilized lysates, followed by immunoblotting (IB) for indicated subunits of TIM23 (for crosslinking to Tim44, Tom22, and Tom40, see Extended Data Fig. 8e). Lower panels show contrast enhanced (contrast enh.) images of crosslinked bands. d, Similar to c, but Bpa was introduced to indicated positions in the Tim17 and Tim23 cavities. Photocrosslinking was performed with overexpression of Grx5-S80-sfGFP (subst.). Also see Extended Data Fig. 9a,b. e, Similar to d, but Bpa was introduced to Mgr2. Subst., Grx5-S80-sfGFP. f, MD simulations predict permeation of water (red/white spheres) across the cavity formed by Tim17 (green) and Mgr2 (semi-transparent gray) if not occupied by a polypeptide or lipids. For clarity, Tim23, Tim44 and lipids are not shown. Dark gold spheres, phosphorous atoms of lipids. g, Amounts of indicated subunits in the Tim17-HA IP experiments were quantified and ratios between samples overexpressing (OE) Grx5-S80-sfGFP and uninduced samples were plotted. Data are presented as mean values +/− SEM (n=3 independent experiments). Asterisk, p-value=0.03 (one-way ANOVA). Also see Extended Data Fig. 9j. Data in c, d, and e are representative of two (e) or three (c and d) independent experiments.
The non-essentiality of Mgr2 indicates that formation of the channel-like structure is not required for preprotein translocation. However, such a structure may contribute to the efficiency and fidelity of the process. Indeed, mgr2Δ is known to reduce preprotein translocation efficiency and abolish detection of a stable stalled translocation intermediate (i.e., the TOM–TIM23–substrate supercomplex) in blue-native (BN) PAGE14,37. The latter observation can be explained by dissociation of TIM23 from the substrate upon detergent solubilization due to the laterally open cavity of Tim17 without Mgr2. We could also recapitulate this in our immunoprecipitation experiments using a model substrate containing an N-terminal matrix protein (Grx5), an ~80-amino-acid-long hydrophilic segment (S80), and a C-terminal superfolder green fluorescent protein (sfGFP) (Extended Data Fig. 8 a–d). Upon expression, a population of Grx5-S80-sfGFP accumulated as a stalled intermediate spanning both TOM and TIM23 complexes due to folding of sfGFP. Compared to WT, mgr2Δ strongly diminished copurification of TIM23 with Grx5-S80-sfGFP.
To demonstrate that Tim17 and Mgr2 indeed form the translocation path, we performed a series of in-vivo photocrosslinking experiments. We first incorporated the photocrosslinkable p-benzoyl-L-phenylalanine (Bpa) into specific positions along the S80 segment of Grx5-S80-sfGFP (Fig. 3 b and c, and Extended Data Fig. 8 e–i). In agreement with the expected topology, positions near sfGFP crosslinked to Tom40 whereas positions near Grx5 crosslinked to Tim44. Importantly, intermediate positions, residues ~19–60, crosslinked to both Tim17 and Mgr2, suggesting that this region spans the cavity formed by Tim17 and Mgr2. By contrast, the substrate crosslinked only very weakly with Tim23 between positions 43 and 65. This crosslinking could be due to an interaction with the ~90-amino-acid-long N-terminal IMS domain of Tim23.
To further show that Tim17, not Tim23, is used for translocation, we introduced Bpa into the cavities of Tim17 and Tim23, at eight different positions each about halfway across the membrane. Consistent with our hypothesis, all eight positions in Tim17 could crosslink to the substrate albeit with different efficiencies, whereas no crosslink was observed with any Tim23 positions tested (Fig. 3d and Extended Data Fig. 9a–d). Next, we incorporated Bpa into Mgr2. With overexpression of Grx5-S80-sfGFP, we could detect two crosslink adduct bands of Mgr2, one with the stalled substrate and the other with Tim17 (Fig. 3e and Extended Data Fig. 9e–h). Thus, all crosslinking results consistently support our model that preproteins are transported through the cavity of Tim17, the lateral opening of which can be further sealed by Mgr2.
Lastly, to characterize properties of the predicted Tim17–Mgr2 pore structure, we performed additional MD simulations. We focused on the Tim44–Tim23–Tim17–Mgr2 subcomplex, since co-assemblies with Tim50, Pam16, and Pam18 could not be reliably modeled. We also note that the only functionally essential TMs are those from Tim17 and Tim2313,17,19. Because the short time scale (1 μs) of the MD simulations cannot capture the dynamics of the Tim17–Mgr2 interaction, we ran simulations in two different conditions: with and without lipids in the Tim17–Mgr2 cavity. When we hypothesized that the pore was not occupied by lipids, a water channel across the membrane was formed (Fig. 3f), which was not observed in Tim17 alone (Extended Data Fig. 5e) or when we assumed that the Tim17–Mgr2 cavity was occupied by lipids (Extended Data Fig. 9i). Considering that a water channel would likely compromise the proton permeability barrier, Mgr2 may dynamically associate with Tim17 when the cavity is engaged with a polypeptide substrate. Alternatively, the cavity may be occupied by lipids or an uncharacterized segment of a Tim subunit. To test translocation-dependent association of Mgr2 to Tim17, we measured co-purification of Mgr2 with Tim17 under basal and substrate-overexpressed conditions. Indeed, a significantly higher amount of Mgr2 copurified upon overexpression of Grx5-S80-sfGFP (Fig. 3g and Extended Data Fig. 9j), suggesting a cooperativity between Mgr2 and the substrate polypeptide in binding to Tim17.
Discussion
Our findings have major implications for the mechanism of TIM23-mediated mitochondrial protein import (also see Supplementary Discussion). First, our data show that Tim17, not Tim23, constitutes the translocation path for preprotein transport (Fig. 4). It has been generally assumed that the path would be formed mainly by Tim23, either alone (in the form of a homomultimer) or together with Tim17 (ref. 1–4). Our study now suggests that Tim23 plays primarily a structural role as a platform to associate Tim17, Tim44, and other subunits. Tim23 has also been shown to interact with Tim50, the putative presequence receptor15,38–41. It is noteworthy that certain protists only possess one homolog of Tim17/Tim22/Tim23 that is substantially more related to Tim17 (e.g., Giardia and Cryptosporidium) or Tim22 (e.g., Trypanosoma and Trimastix) than to Tim23 (ref. 42–45), consistent with the protein transport process being mediated by Tim17 and Tim22.
Figure 4. Model for the polypeptide transport mechanism of the TIM23 complex.
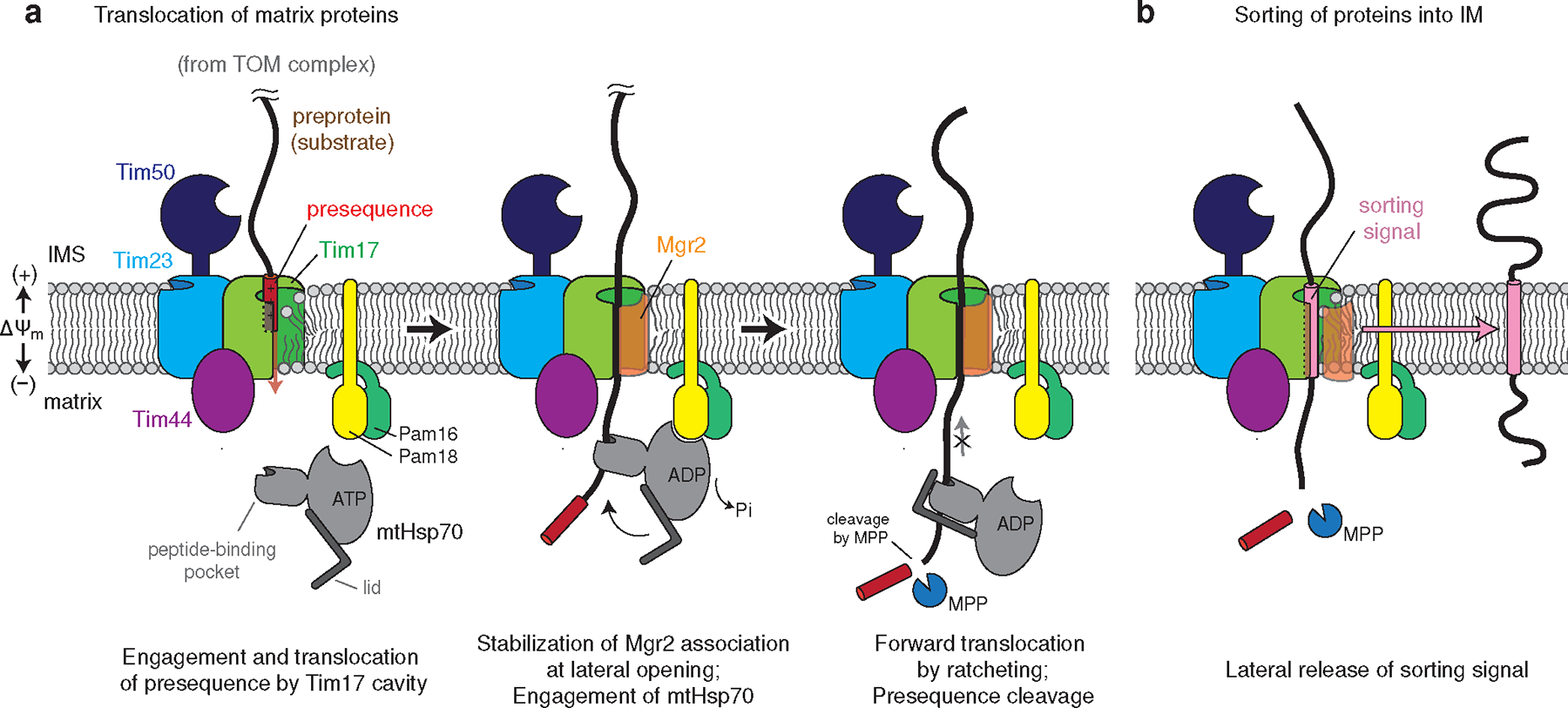
a, The N-terminal presequence first inserts into the cavity of Tim17. This step may be facilitated by Tim50, which may recruit the presequence to the TIM23 complex, and the negatively charged surface of the Tim17 cavity. Initial translocation of the presequence is likely powered by membrane potential (ΔΨm) across the inner membrane48,49. During translocation, the nonessential subunit Mgr2 stably associates with Tim17, sealing the lateral opening, and the preprotein is grasped by mtHsp70 as it emerges into the matrix. The J-domain of Pam18 activates ATP hydrolysis, leading to tight binding of mtHsp70 onto the polypeptide. The tight association of mtHsp70 will promote forward translocation of the polypeptide into the matrix while preventing backsliding. The presequence will be cleaved by the mitochondrial processing peptidase (MPP; not shown). For clarity, the tethers between the core TIM23 complex and Pam16–Pam18 and between Tim44 and mtHsp70 (ref. 26) are not shown. b, In the case of preproteins containing a TM sorting signal, dissociation of Mgr2 allows the signal to be released into the lipid phase through the open lateral gate of the Tim17 cavity.
Second, TIM23-mediated protein translocation is unlikely to require an aqueous channel. Aside from the TMs of Tim17 and Tim23, all remaining TMs from other TIM23 complex subunits are nonessential for protein import13,17,19. Although Tim17 and Mgr2 together seem to form a channel-like structure, Mgr2 is not essential at a normal growth temperature. This suggests that in the absence of Mgr2, substrate polypeptides can still move across the cavity of Tim17 with part of the polypeptide exposed to lipids. The conserved acidic amino acids lining this ‘half-channel’-like Tim17 cavity likely lower the energy barrier for polypeptide transport by local membrane thinning46 (Fig. 4). In addition, the negatively-charged surface of Tim17 might guide insertion of presequences, which typically form a positively charged amphipathic helix. The lipid-exposed translocation path also explains the ability of the TIM23 complex to detect and integrate TM sorting signals into the IM during translocation.
Accumulating data suggest that Mgr2, although not essential, plays an important role in facilitating the translocation process14,37. Our crosslinking results indicate that Mgr2 is positioned at the lateral opening of the Tim17 cavity during preprotein translocation. Current evidence also suggests that Mgr2 is a relatively dynamic component of the TIM23 complex that becomes stably associated upon preprotein engagement (this study and ref. 14). Our MD simulations showing formation of a continuous water wire across an empty Tim17–Mgr2 pore suggest that the pore structure might form only during the translocating state or would be occupied by lipids in the idle state. Dynamic Mgr2 recruitment is also consistent with previous observations that membrane integration of TM sorting signals is promoted by Mgr2 deletion and suppressed by Mgr2 overexpression14,36, which are difficult to explain with a stable structure. Nevertheless, the detailed nature of the Tim17–Mgr2 structure remains to be understood, including the exact functions of Mgr2, dynamics in the native membrane, and possible regulatory mechanisms during the translocation process (see ‘Limitations of our study’ section in Supplementary Discussion). This will also need an experimental high-resolution structure of an Mgr2-containing TIM23 complex in various functional states.
In summary, our study proposes a new model in which Tim17 holds a central position in TIM23-mediated protein translocation. Given the high degree of sequence conservation, the model we presented here is likely conserved in most eukaryotes. Our work provides a structural framework for interpreting previous functional data on TIM23 and lays the foundation for future investigations.
Methods
Plasmids and yeast strains
A list of yeast strains and plasmids used in this study is given in Supplementary Tables 1 and 2, respectively. Primers and additional DNA sequences are listed in Supplementary Tables 3 and 4, respectively.
For purification of the endogenous TIM23 complex (Extended Data Fig. 1b), the S. cerevisiae strain yMLT62 (a gift from J. Thorner) was modified to generate the strain ySS078, which expresses C-terminally 2xSpot-tagged Tim17 from the TIM17 locus (Saccharomyces Genome Database [SGD] ID: S000003679). The tag also contains a 16-amino-acid-long linker and an HRV 3C protease cleavage site (amino acid sequence: …PLQA [Tim17]–ASGTLEVLFQGPT-ASGPDRVRAVSHWSSGGGSGGGSTPDRVRAVSHWSS*; Spot-tags are underlined, and the asterisk indicates the C-terminus). To attach the tag to Tim17, a PCR product was first generated containing a 74-bp 5’ homology arm immediately before the TIM17 stop codon, the tag, a hygromycin resistance cassette, and a 74-bp 3’ homology arm downstream of TIM17 (Supplementary Table 4, ‘TIM17–3C-Spot’). DNA was introduced to yeast cells by a standard lithium acetate/polyethylene glycol transformation method. Colonies were selected on a YPD (1% yeast extract, 2% peptone, 2% glucose, and 2% bacto-agar) agar plate containing 400 ug/mL hygromycin B, and chromosomal integration was confirmed by PCR using primers EP_446 and EP_447.
For the overexpression of all TIM23 complex subunits from an inducible GAL1 promoter (Extended Data Fig. 1c), the yeast strain ySS025 was used. This strain expresses a chimeric transcription activator (Gal4-ER-VP16) that drives expression of genes under GAL promoters in response to β-estradiol added to the medium50. To construct this strain, we first generated two integration plasmids (pSS011 and pSS015) using the MoClo Yeast Tool Kit (YTK) (ref. 51). The coding sequences (CDS) for Tim23 (SGD:S000005300), Tim17 (SGD:S000003679), Tim50 (SGD:S000005984), Tim44 (SGD:S000001284), Tim21 (SGD:S000003265), Pam16 (SGD:S000003640), Pam17 (SGD:S000001773), Pam18 (SGD:S000003998), and Mgr2 (SGD:S000006019) were first amplified via PCR using genomic DNA (gDNA) of S. cerevisiae BY4741 as a template and cloned individually into a pYTK001 entry plasmid (internal BsaI and BsmBI sites in CDSs were removed). Each CDS part was assembled into expression cassettes in pYTK095 together with connectors, promoter (pGAL1), and terminator parts from MoClo YTK and a part for 2xSpot-tag (pYTK-e201). The Tim23, Tim17-Spot, Tim50, Tim44, and Tim21 expression cassettes were inserted into pYTK096 by BsmBI Golden Gate cloning, resulting in pSS011. Similarly, the Pam16, Pam17, Pam18, and Mgr2 expression cassettes were inserted into pYTK-e106 (Supplementary Table 2), resulting in pSS015. The integration plasmids were linearized with NotI and used to transform yMLT62. Colonies were selected on synthetic complete agar medium lacking uracil (SC[−Ura]) for pSS011 or lacking leucine (SC[−Leu]) for pSS015. Chromosomal integration was confirmed by PCR as described previously51.
For purification of the Tim17/23/44–Pam16/18 complex (Extended Data Fig. 2), the yeast strain ySS055 was used. This strain was made from yMLT62 by successively integrating two plasmids, pSS077 expressing Tim17-Spot, Tim23, and Tim44, and pSS082, expressing Pam16 and Pam18, all under GAL1 promoters. pSS077 and pSS082 were assembled by the MoClo approach, as described above. We found that a 2-amino-acid-long GlySer tail at the C-terminus of Tim44, which was introduced during the construction of the YTK part plasmids, reduces the affinity between Tim44 and Tim17–Tim23. Thus, we removed the GlySer tail from Tim44 (hereafter Tim44[−GS]) by mutagenesis during the construction of pSS077.
For generation of strain (ySS176) that moderately overexpresses Tim44 (Extended Data Fig. 3), an integration plasmid (pSS219) was generated using MoClo YTK from pYTK096, pYTK018 (pALD6), Tim44(−GS) CDS, and pYTK051 (tENO1). This integration plasmid was introduced into ySS078, generating ySS176. Strains ySS183 (tim21Δ) and ySS184 (pALD6-TIM44, tim21Δ) were made by deleting chromosomal TIM21 (tim21Δ::LEU2) by transformation with a PCR product, which was amplified from pYTK076 (LEU2 marker) using primers SS_2578 and SS_2579. Chromosomal deletion was confirmed by PCR with sequencing primers SS_1942 and SS_2580.
To generate yeast strains with TIM17 or TIM23 under a repressible Tet promoter (yYC17a and yYC23a, respectively), the tetracycline response element (TRE) and KanMX cassette were PCR amplified from the genome of yeast strain TH_5187 (Horizon Discovery; Cat. #YSC1180–202219224) with primers designed for Tim17 (SS_1700 and SS_1701) and Tim23 (SS_1696 and SS_1697). PCR fragments were then transformed into the R1158 strain (Horizon Discovery; cat. #YSC1210) and selected on YPD agar plates with 300 μg/mL G418. Chromosomal integration was confirmed by PCR with sequencing primers (for Tim17, SS_1702 and SS_1703; for Tim23, SS_1698 and SS_1699). The ySS150 strain was constructed by transformation of the W303–1A yeast strain with the URA::PCMV-tTA cassette from R1158, which was amplified by PCR using primers EP_420 and EP_423. Using the same strategy as described above, yYC17b (ySS150 PTIM17::KanMX-tetO7-PCYC1) and yYC23b (ySS150 PTIM23::KanMX-tetO7-PCYC1) were generated from ySS150 for photocrosslinking experiments.
The yeast strain ySS121 (BY4741 TIM23-Myc::HphMX TIM50-HA::NatMX) was made by successively transforming BY4741 with PCR products ‘TIM23-myc’ and ‘TIM50-HA’ (Supplementary Table 4). The Myc-tag contains a 16-amino-acid-long linker (amino acid sequence: …LLEK [Tim23]–ASGTLEVLFQGPTASG-EQKLISEEDLNGEQKLISEEDL*; Myc-epitopes are underlined). The HA-tag for Tim50 contains a 25-amino-acid-long linker (amino acid sequence: …AESK [Tim50]–AGGATTASGTGENLYFQGTASGGGS-YPYDVPDYAGSYPYDVPDYA*; HA-epitopes are underlined). Chromosomal integration was confirmed by PCR using primers EP_442 and EP_443 for the TIM23 locus, EP_450 and EP_451 for the TIM50 locus. A plasmid expressing Spot-tagged Tim17 under the endogenous promoter was made by inserting DNA encoding 426 bp upstream to 241 bp downstream of the CDS into the plasmid pYTK-e112 (a CEN/ARS plasmid with a LEU2 marker; Supplementary Table 2) between its EcoRI and PstI sites (PCR with YC_1760 and YC_1761). A Spot-tag was introduced to the C-terminus of Tim17 by PCR, generating pSS122 (amino acid sequence: …PLQA [Tim17]–TSPDRVRAVSHWSSGGGSGGGST-PDRVRAVSHWSS*; Spot-tags are underlined). Mutations in Tim17 were inserted by PCR mutagenesis. The ySS121 yeast strain was transformed with a pSS122 plasmid, encoding WT or respective mutant Tim17 (Extended Data Fig. 6h).
The Cyb2Δ-DHFR pulldown and BN-PAGE experiments (Fig. 2 g and h and Extended Data Fig. 6k) were performed in yeast strain yYC02 (BY4741 TIM17-HA::NatMX TIM23-Myc::HphMX), which was made by successively transforming BY4741 with PCR products ‘TIM23-myc’ and ‘TIM17-HA’ (Supplementary Table 4). The Myc-tag was introduced to Tim23, as described for ySS121. The HA-tag for Tim17 contains a 25-amino-acid-long linker as described for Tim50-HA above. Chromosomal integration was confirmed with EP_446 and EP_447 for the TIM17 locus. Similarly, ySS171 (W303–1A TIM17-HA::NatMX TIM23-Myc::HphMX) was constructed for Mgr2 growth complementation, co-IP assays, and photocrosslinking experiments. To additionally express Spot-tagged Tim17 (WT, D17N/E126Q, or D76N/E126Q) in yYC02, the C-terminally 2xSpot-tagged versions were cloned from respective pSS122 plasmids into the integration vector pYTK-e106 between the EcoRI and PstI sites. The integration plasmids were then introduced to the HO locus of yeast strain yYC02, resulting in yYC03 (WT), yYC04 (D17N/E126Q), and yYC05 (D76N/E126Q).
Strain ySS199 was made by deleting chromosomal MGR2 (mgr2Δ::URA3) in ySS171 by transformation with a PCR product, which was amplified from pYTK074 (URA3 marker) using primers YC_2425 and YC_2426. Chromosomal deletion was confirmed by PCR with sequencing primers YC_2427 and YC_2521.
Expression of Tim17 and Tim23 under the native promoters used plasmid pYC17a (Fig. 2f and Extended Data Fig. 6a–g) and pYC23 (Extended Data Fig. 6m), respectively. First, a BamHI cleavage site was introduced before the PstI site of pYTK-e115 (a CEN/ARS plasmid with a NatMX marker) by PCR-based mutagenesis. Then, the WT TIM17 gene, including its native promoter and terminator, was amplified by PCR (YC_1760 and YC_1761) from the gDNA of BY4741 and cloned into this plasmid between the EcoRI and BamHI, resulting in pYC17a (see Supplementary Table 4). A similar strategy was used to generate pYC23 for Tim23 (primers YC_1762 and YC_1763 for PCR) (see Supplementary Table 4). When indicated, 2xHA tags (amino acid sequence: …PLQA [Tim17]–TSYPYDVPDYAGSYPYDVPDYA*; HA tags are underlined) were added to the C-terminus by PCR. Point mutations were introduced by PCR mutagenesis.
Tim17 expression under a cyanamide-inducible DDI2 promoter 52 was performed with plasmid pYC17b. This plasmid is derived from pYTK-e122 (a 2μ plasmid with a LEU2 marker; Supplementary Table 2) with insertion of a 938-bp segment upstream of the DDI2 CDS (PCR using primers YC_1981 and YC_1982 and gDNA of BY4741), TIM17 CDS, and TIM17 terminator between the EcoRI and PstI sites of pYTK-e122.
The Mgr2 expressing integration plasmid (pSS241; Fig. 3g and Extended Data Fig. 8b–d and Fig. 9j) was generated using MoClo YTK from pYTK-e105, pYTK018 (pALD6), and pYTK001-Strep-Mgr2 (Supplementary Tables 2 and 4). The plasmid (pSS239) expressing Mgr2 under the endogenous promoter was made by inserting DNA encoding 224 bp upstream to 317 bp downstream of the CDS into the plasmid pYTK-e105 between its EcoRI and PstI sites (PCR with SS_2455 and SS_2456). pSS254 was made by introducing a Strep-tag to the N-terminus of Mgr2 in pSS239 by PCR. The integration plasmids pSS239, pSS254, and pSS241 were then introduced to the HO locus of a ySS199, resulting in ySS207, ySS204, and ySS205, respectively.
A plasmid (pYC005) expressing Grx5-S80-sfGFP-ALFA under a GAL1 promoter was constructed by standard molecular cloning techniques into pYTK-e112 (Supplementary Table 2). The Grx5 sequence was derived from yeast GRX5 (amino acids 1–141), and the S80 sequence was derived from yeast CYB2 (amino acids 32–115 but deleting 47–65). TAG amber codons were introduced by site-directed mutagenesis for photocrosslinking experiments. Grx5-S99(TM)-sfGFP-ALFA (pYC006) was constructed similarly except that the S99(TM) sequence was derived from amino acids 32–115 of yeast CYB2. For the Grx5-S80-sfGFP-ALFA used in Extended Data Fig. 8a, we note that the construct (pYC007) contains an additional 20-amino-acid-long linker between sfGFP and ALFA-tag (i.e., [sfGFP]-GSGENLYFQGGSGSGENLYFQGG-[ALFA-tag]). DNA sequences of these constructs are included in Supplementary Table 4.
Expression of Tim17 and Tim23 with Bpa incorporated at their cavities was performed using pYC003 (CEN/ARS/HIS3 plasmids) and pYC008 (2μ/ARS/HIS3 plasmids), respectively. To generate these plasmids, expression cassettes encoding Tim17-Spot (from pSS122) and Tim23-Spot (from pYC23, but replacing the HA-tag with 2xSpot-tag [Tim23-TS-PDRVRAVSHWSSGGGSGGGSTPDRVRAVSHWSS*]) were inserted between the EcoRI and PstI sites of pYTK-e113 and pYTK-e123. TAG amber codons were introduced by site-directed mutagenesis. Similarly, Strep-Mgr2 with a TAG amber codon was expressed from pYTK-e113-derived plasmid pSS277, encoding Strep-Mgr2 under the ALD6 promoter.
Expression of Tim44-CTD from E. coli was performed using the plasmid pSS070. The coding sequence of Tim44-CTD was amplified via PCR using the gDNA of BY4741 as a template and inserted into a pETDuet-1 vector between the BamHI and XhoI sites (Supplementary Tables 2 and 4). To enable affinity purification of the protein, a His-tag (GSSHHHHHHSQDP) and an HRV 3C protease cleavage site (LEVLFQGP) were attached to the N-terminus of Tim44-CTD. For E. coli expression of Cyb2Δ-DHFR, DNA encoding the first 165 amino acids from yeast CYB2 (with a.a. 47 to 65 deleted and the endogenous Cys14 and Cys105 mutated to Ser) fused with DHFR was synthesized. This fragment was introduced to pET32a (EMD Millipore) before the His-tag with NdeI and XhoI to generate pYC002.
Purification of the TIM23 complex
Yeast cells were grown in YPEG medium (1% yeast extract, 2% peptone, 2% ethanol and 3% glycerol) in shaker flasks at 30°C. Cells were harvested upon reaching an optical density of 600 nm (OD600) of ~1.5–2. For purifications shown in Extended Data Fig. 1b and 1c, yeast strains ySS078 and ySS025 were used, respectively. For purification Extended Data Figs. 2a and 3d yeast strains ySS055 and ySS184 were used, respectively. When the TIM23 complex was overexpressed, yeast cells were induced with 50 nM β-estradiol upon reaching an OD600 of ~0.7–1.0. After 13–15 h of induction, cells were harvested by centrifugation (3,000 g for 5 min).
Crude yeast mitochondria were isolated as described previously53. Cells were washed with deionized water, resuspended in prewarmed DTT buffer (100 mM Tris-H2SO4, pH 9.4, 10 mM DTT) (2 mL/g wet weight cells), and incubated at 30°C for 30 min to 1 h. Cells were then harvested by centrifugation (3000 g for 5 min), washed with lyticase buffer (1.2 M sorbitol, 20 mM potassium phosphate, pH 7.4) (7 mL/g wet weight cells), and resuspended in lyticase buffer containing a crude preparation of homemade lyticase. After incubation at 30°C for 1–2 h with lyticase, cells were harvested by centrifugation (3000 g for 5 min), washed with lyticase buffer, and resuspended in ice-cold homogenization buffer (0.6 M sorbitol, 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 2 mM PMSF) (6 mL/g wet weight cells). All subsequent steps were performed at 4°C. The yeast spheroplasts were then homogenized with 40 strokes of a Dounce homogenizer and diluted two-fold with homogenization buffer. Crude mitochondria were subsequently isolated through steps of differential centrifugation. The suspension was first centrifuged at 1,500 g for 5 min to remove cell debris and nuclei and centrifuged again at 4000 g for 5 min. The supernatant from this step was then subjected to further centrifugation at 20,000 g for 20 min (Beckman JA-17 rotor) to isolate the mitochondrial fraction. The mitochondrial pellet was washed in SEM buffer (250 mM sucrose, 1 mM EDTA, 10 mM MOPS-KOH, pH 7.2) and harvested at 17,000 g for 20 min. The supernatant was removed, and aliquots of mitochondrial pellets (~500 mg) were frozen in liquid nitrogen and stored at −80°C until use.
To purify the TIM23 complex, mitochondrial pellets were first thawed and resuspended in ice-cold lysis buffer (Buffer L; using 3 times volume of the mitochondrial pellet volume) containing 50 mM Tris pH 7.5, 200 mM NaCl, 1 mM EDTA, 10% glycerol, 5 μg/mL aprotinin, 5 μg/mL leupeptin, 1 μg/mL pepstatin A, and 2 mM PMSF. All subsequent steps were carried out at 4°C. The suspension was briefly homogenized with 10 strokes of a Dounce homogenizer. Mitochondria were then solubilized with a detergent mixture of 0.4% glycol-diosgenin (GDN; Anatrace) and 0.6% lauryl maltose neopentyl glycol (LMNG; Anatrace). After a 1.5 h incubation, the lysate was clarified by centrifugation at 20,000 g for 30–45 min (Beckman JA-17 rotor), and the supernatant was incubated with Sepharose beads conjugated with anti-BC2 nanobody for 2 h. The beads were then washed with approximately 30 column volumes of a wash buffer (Buffer W; 25 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA) containing 0.02% GDN. The TIM23 complex was eluted by incubating the beads with ~50 μg/mL HRV 3C protease for 1 h. Cleaved protein was concentrated using an Amicon Ultra (cut-off 100k; GE Life Sciences). The sample was then injected into a Superose 6 Increase 10/300 GL column (GE Life Sciences), equilibrated with Buffer W containing 0.02% GDN. Where indicated, anti-Tim44 Fab was added to the TIM23 complex at a molar ratio ~1:2 (incubated at 4°C for 15 min) prior to size-exclusion chromatography. Peak fractions were pooled and concentrated to ~5–7 mg/mL using an Amicon Ultra. 3 mM fluorinated Fos-Choline-8 (FFC8; Anatrace) was added to the purified TIM23 sample prior to cryo-EM grid preparation.
Antibody generation and purification
Monoclonal antibodies were generated against Tim44 by immunizing mice with purified Tim44-CTD (residues 210–431). BL21(DE3) E. coli was transformed with pSS070, and cells were grown at 37°C to an OD600 of 1.5 in Luria Broth supplemented with 100 ug/mL ampicillin. 0.5 mM IPTG was then added to cultures, and cells were grown for additional 3 h at 37°C. After harvesting by centrifugation, cell pellets were washed with water and stored at −80°C until use. The thawed pellets were then resuspended in 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 10% glycerol, 5 μg/mL aprotinin, 5 μg/mL leupeptin, 1 μg/mL pepstatin A, and 2 mM PMSF and lysed by sonication for 10 min on ice. All subsequent steps were carried out at 4°C. The lysate was clarified by ultracentrifugation (Beckman Type 45 Ti rotor) at 100,000 g for 1 h, and the supernatant was collected and incubated with cobalt resin (Thermo Fisher, Cat. #89966) for 2 h. The beads were then washed with approximately 30 column volumes of 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 10% glycerol, and 10 mM imidazole. Tim44-CTD was eluted by incubating beads with 4 column volumes of 50 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 10% glycerol, and 200 mM imidazole. The His-tag was subsequently removed from Tim44-CTD by incubating the beads with ~5 μg/mL HRV 3C protease for 1 h. Cleaved protein was concentrated using an Amicon Ultra (cut-off 10k; GE Life Sciences) and injected into a Superdex 200 Increase 10/300 GL column (GE Life Sciences), equilibrated with 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 10% glycerol. Tim44-CTD was concentrated to ~2 mg/mL, frozen in 50 μL aliquots with liquid nitrogen, and stored at −80°C until use.
Immunization of mice with purified Tim44-CTD and initial screening of positive hybridoma clones by ELISA was performed by Antibodies, Inc (Davis, CA). Briefly, five 7–12-week-old female Balb/c mice were immunized four times each with protein complex combined with Sigma adjuvant System (Sigma Aldrich). 40 μg of Tim44-CTD in adjuvant was used for the first immunization while 20 μg of Tim44-CTD in adjuvant was used for the subsequent three immunizations. Test bleeds from each of the mice were assayed by ELISA on a Tim44-CTD coated plate and the mouse with the highest antibody titer was intravenously boosted and then sacrificed 4 days later to obtain the spleen which was used in a fusion reaction with the SP2/0 myeloma cell line to create mouse hybridomas. All procedures were approved by Antibodies Incorporated’s Institutional Animal Care and Use Committee and performed under their current Office of Laboratory Animal Welfare assurance.
For ELISA screens of hybridoma, the biotinylated core TIM23 complex (Tim17–Tim23–Tim44) was used. After the core complex was purified (in HEPES instead of Tris buffer) and concentrated, a 2.5x molar excess of EZLink NHS-PEG4-Biotin (Thermo Fisher, Cat. #21362) was added to the sample and incubated at 4°C for 16 h. The sample was then injected into a Superose 6 Increase 10/300 GL column (GE Life Sciences), equilibrated with Buffer W. Peak fractions containing the core TIM23 complex were pooled and concentrated to ~0.5 mg/mL. Biotinylation was confirmed by western blotting using StrepTactin-HRP (Bio-Rad, Cat. #1610381). The biotinylated core TIM23 complex was subsequently immobilized to NeutrAvidin coated ELISA plates.
ELISA-positive hybridoma supernatants were further tested by co-immunoprecipitation and immunoblotting experiments to identify clones producing antibodies that could bind to the core TIM23 complex. We could identify three such clones from thirty ELISA-positive clones. One of these (clone #6) was made monoclonal by a limiting dilution method and used for cryo-EM analysis.
To purify IgG, monoclonal hybridoma (clone #6) cells were grown in high glucose DMEM supplemented with 20% v/v fetal bovine serum (Gibco, Cat. #10437028), 8% v/v macrophage-conditioned medium (Antibodies, Inc), 10% v/v NCTC-109 (Gibco, Cat. #21340039), 1x Penicillin-Streptomycin-Glutamine (Gibco, Cat. #10378016), 1x MEM NEAA (Gibco, Cat. #11140050) in a CELLine Flask (Wheaton, Cat. #WCL1000). Hybridoma cells were not tested for mycoplasma contamination. The antibody supernatant was harvested and centrifuged at 3000 g for 30 min at 4°C to remove any insoluble material. Saturated ammonium sulfate ([NH4]2SO4) was slowly added to the supernatant to a final concentration of 25% (w/v) and incubated for 5 h at 4°C. The mixture was centrifuged at 3000 g for 30 min at 4°C, and saturated (NH4)2SO4 was slowly added to the supernatant to a final concentration of ~50% (w/v) and incubated for 16 h at 4°C. The precipitate containing antibody was collected by centrifugation at 17,000 g (Sorvall SS-34 rotor) for 10 min at 4°C. The pellet was resuspended in TBS buffer containing 25 mM Tris-HCl pH 7.5, 80 mM NaCl and dialyzed against 2 changes of 2 L of TBS buffer for 48 h and 1 change of 1 L of 20 mM Tris-HCl pH 8.0 for 16 h in 10-kDa-MWCO dialysis tubing. Dialyzed samples were loaded directly onto a 5-mL HiTrap Q HP anion exchange column (GE Life Sciences) equilibrated in 20 mM Tris-HCl pH 8.0. After washing with 5 column volumes of 20 mM Tris pH 8.0, antibodies were collected during gradient elution to 20 mM Tris pH 8.0, 1 M NaCl. Fractions containing the eluted IgG were pooled (4 mg/mL).
Fab fragments were generated by digestion of IgG with papain (1:25 w/w; Sigma-Aldrich, Cat. #P3125) in buffer containing 20 mM HEPES-NaOH pH 7.0, 150 mM NaCl, 10 mM L-cysteine HCl, 10 mM β-mercaptoethanol, 10 mM EDTA, and additional 9 mM NaOH at 37°C for 3 h. Cleaved antibodies were diluted in 4 L of 20 mM Tris-HCl pH 8.0 for 16 h in 10-kDa-MWCO dialysis tubing. To collect the Fab fragments, the cleaved IgG sample was applied to a 5-mL HiTrap Q HP anion exchange column (GE Life Sciences) equilibrated in 20 mM Tris-HCl pH 8.0. After washing with 5 column volumes of 20 mM Tris-HCl pH 8.0, Fab fragments were collected during gradient elution to 20 mM Tris-HCl pH 8.0, 1 M NaCl. Fractions containing Fab fragments were pooled, diluted with buffer containing 25 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM EDTA, and 20% glycerol, and concentrated using an Amicon Ultra (cut-off 30k; GE Life Sciences) to ~4.5 mg/mL. Samples were either stored at 4°C or frozen in liquid nitrogen and stored at −80°C until use. The sequences of the Fab variable domains were determined by sequencing the cDNAs generated from RNAs that were extracted from the hybridoma cell lines as described previously54.
Cryo-EM data collection
To prepare cryo-EM grids, 3 μL of the sample were applied to a glow-discharged (PELCO easiGlow; 0.39 mBar, 25–30 mA, 40–45 s) gold holey carbon grid (Quantifoil R 1.2/1.3, 400 mesh). The grid was blotted for 3–4 s and plunge-frozen in liquid-nitrogen-cooled liquid ethane using Vitrobot Mark IV (FEI) operated at 4°C and 100% humidity. Whatman No. 1 filter paper was used to blot the samples.
The dataset in which all known subunits of TIM23 complex (TIM23full) were overexpressed (Extended Data Fig. 1c) was collected on a Talos Arctica electron microscope (FEI), operated at an acceleration voltage of 200 kV. The Tim17/23/44+Pam16/18 (Extended Data Fig. 2) and endogenous core-TIM23 complex (Extended Data Fig. 3) datasets were collected on a Titan Krios G3i electron microscope (FEI), operated at an acceleration voltage of 300 kV and equipped with a Gatan Quantum Image Filter (slit width of 20 eV). Dose-fractionated images (~50 electrons per Å2 applied over 42 or 50 frames) were recorded on a K3 direct electron detector (Gatan) using the super-resolution mode. All datasets were collected using SerialEM (ref. 55). The datasets were collected with image-beam-shift multiple recording (4 to 9 recordings per stage movement), except for the TIM23full dataset, which was collected without image shift. Coma induced by image-beam shift was corrected by beam tilt compensation in SerialEM. The physical pixel size was 1.14 Å for the Arctica dataset and 1.05 Å for the Krios G3i datasets. Target defocus values typically ranged from −0.7 to −2.6 μm.
Cryo-EM structure determination
Movies were initially preprocessed using Warp (ref. 56), by motion-correcting and estimating contrast transfer function (CTF) and defocus parameters with 7-by-5 or 5-by-5 tiling. Micrographs were manually inspected to remove micrographs that were not suitable for image analysis, largely those containing crystalline ice. Particles were automatically picked by Warp and extracted with a box size of 256 pixels. All subsequent image processing was performed in cryoSPARC v3 (for final refinement) or v2 (for other steps) 57, as described in detail below. While the final maps for the TIM23full and the endogenous core-TIM23 complex datasets were reconstructed from particles picked in Warp, the movies for the Tim17/23/44+Pam16/18 dataset were reprocessed and particles were repicked in cryoSPARC (Extended Data Fig. 2b). The particles selected from Warp were used to generate initial maps in cryoSPARC, which were subsequently used as a template for the second round of particle picking and heterogenous refinement in cryoSPARC.
(1). TIM23full complex
A summary of the single particle-analysis is outlined in Extended Data Fig. 1d. 854,545 particles were automatically picked in Warp from 2,359 movies. Particles were imported into cryoSPARC for reference-free 2D classification, and classes without clear protein features (mostly empty micelles) were removed. 504,914 particles selected from the 2D classification were subjected to ab initio reconstruction, yielding three initial models (Classes 1 to 3). Class 1 showed a disc-shaped micelle and may represent the Tim17-Tim23 complex (Extended Data Fig. 1f). Class 2 contained two distinguishing bulges (Extended Data Fig. 1 g and h). The particles in Class 2 (193,357 particles) were used for 3D reconstruction by non-uniform (NU) refinement to yield a map at 8.4-Å resolution.
(2). Fab-bound core TIM structure (Tim17/23/44 + Pam16/18)
A summary of the single particle-analysis is outlined in Extended Data Fig. 2b. The initial set of 466,320 particles automatically picked in Warp from 1,506 movies was used for 2D classification. The 268,256 particles selected from the 2D classification (excluding mainly empty micelles) were subjected to ab initio reconstruction, yielding three initial models. Clear features of the core TIM complex and the Fab fragment were visible in one of these classes. These particles (268,256 particles) selected from the 2D classification were classified by a round of heterogeneous refinement using the ab initio reconstructions. The resulting particles (174,222 particles) were used for 3D reconstruction by NU refinement, yielding a map at 2.8-Å resolution for the core+Pam16/18 dataset.
Raw movies (1,506 movies) were then imported into cryoSPARC for tile-based motion correction and CTF estimation. 1,485 micrographs were selected, and a total of 689,333 particles were picked with lowpass-filtered templates generated from the 2.8-Å-resolution 3D reconstruction of the TIM23 complex. Particles were extracted with a box size of 320 pixels, Fourier-cropped to 160 pixels, and subjected to a round of 2D classification. Selected particles from 2D classification (331,513 particles) were subjected to ab initio reconstruction, generating four initial models, and heterogeneous refinement. 188,938 particles were classified into Class 1, which showed clear features of the TIM23–Fab complex. This final set of particles (159,958 particles) was subjected to NU refinement and local CTF refinements to yield a map at 2.7-Å resolution.
(3). Fab-bound endogenous core-TIM23 complex
A summary of the single particle-analysis is outlined in Extended Data Fig. 3e. 1,251,068 particles were automatically picked in Warp from 4,188 movies. Particles were imported into cryoSPARC for reference-free 2D classification, and classes without clear protein features (mostly empty micelles) were removed. 611,378 particles selected from the 2D classification were subjected to ab initio reconstruction and heterogeneous refinement, yielding four models (Classes 1 to 4). Clear features of the core TIM23 complex and the Fab fragment were visible in Class 1. 275,146 particles from Class 1 were subjected to another round of ab initio reconstruction. This final set of particles (214,281 particles) was subjected to local CTF refinement and NU refinement to yield a map at 2.9-Å resolution.
Atomic Model Building
The initial atomic model was built de novo into the sharpened map of the core TIM23 complex dataset using Coot (ref. 58). The model for the endogenous core-TIM23 complex dataset was built after rigid-body fitting into the corresponding map using the initial atomic model and rounds of local refinement in Coot. The following sequences were not modeled in the Tim17/23/44+Pam16/18 structure because they were poorly resolved in the density maps: N to 5 (Tim17), 139 to C (Tim17), N to 85 (Tim23), 221 to C (Tim23), N to 106 (Tim44), and 194 to 254 (Tim44). Residue side chains with poor density were truncated at the β-carbon.
Model refinement was performed using Phenix (phenix.real_space_refine) (ref. 59) with the refinement resolution limit set to the overall resolution of the map. Cryo-EM maps were sharpened using the Phenix Auto-sharpen tool (auto_sharpen) (ref. 60) (Extended Data Table 1) and used for refinement. Structural validation was performed using MolProbity 61 in the Phenix package. Protein electrostatics were calculated using the Adaptive Poisson-Boltzmann Solver (ref. 62) with default parameters (with monovalent ion concentrations of 0.15 M each) built in PyMOL (Schrödinger). UCSF Chimera (ref. 63), Chimera X (ref. 64), and PyMOL were used to prepare structural figures in the paper.
Yeast growth assay
For the experiment in Extended Data Fig. 3b, yeast strains ySS078 (WT), ySS176 (pALD6-Tim44), ySS183 (tim21Δ), and ySS184 (tim21Δ, pALD6-Tim44) were used. Cells were grown in YPD and spotted on YP agar plates containing 2% glucose (YPD) or 2% glycerol (YPG). For experiments in Fig. 2f and Extended Data Fig. 6a and 6m, yeast strains yYC17a and yYC23a were transformed with plasmid pYC17a and pYC23, respectively. These plasmids encode Tim17 and Tim23 under its endogenous promoter, respectively. Cells were grown in YPD with 100 μg/mL nourseothricin and spotted on YPD/nourseothricin agar plates with or without 10 μg/mL doxycycline. For the experiment in Extended Data Fig. 6d, yYC17a was transformed with pYC17b encoding Tim17 mutants under the DDI2 promoter. Cells were grown in a synthetic complete medium containing 2% glucose and lacking leucine (SC[−Leu]) and spotted on SC(−Leu) agar plates with or without 10 μg/mL doxycycline. When indicated, cyanamide (Alfa Aesar, Cat# L2044822) was included in the medium to induce protein expression. For the experiment in Extended Data Fig. 8b, yeast strains ySS171 (WT), ySS199 (mgr2Δ), ySS207 (mgr2Δ, Mgr2), ySS204 (mgr2Δ, Strep-Mgr2), and ySS205 (mgr2Δ, pALD6 Strep-Mgr2) were used. Cells were grown in an SC medium containing 2% glucose and spotted on SC agar plates containing 2% glycerol.
For all spot assays, cells were grown at 30°C to an OD600 of 0.7–1.5 and diluted to an OD600 of 0.1 with fresh media. After 5-fold serial dilution, 10 uL were spotted. The plates were incubated at the indicated temperature (30°C unless indicated otherwise) for 2–3 days before imaging.
Co-Immunoprecipitation
For the experiment in Extended Data Fig. 3c, the yeast strains ySS078 (WT), ySS176 (pALD6-Tim44), ySS183 (tim21Δ), and ySS184 (tim21Δ, pALD6-Tim44) were grown in YPEG medium at 30°C to an OD600 of ~0.8–1.0. Crude mitochondria were prepared as described above, and mitochondria were resuspended in ice-cold Buffer L and solubilized with a detergent mixture of 0.4% GDN and 0.6% LMNG (the lysate volume was approximately 10 times the mitochondrial pellet volume). All subsequent steps were carried out at 4°C. After a 1 h incubation, the lysate was clarified by centrifugation at 17,000 g for 30 min, and the supernatant was incubated with Sepharose beads conjugated with anti-BC2 nanobody for 1 h. The beads were washed four times with 1 mL of Buffer W containing 0.02% GDN. Proteins were eluted by addition of SDS sample buffer and heating samples for 30 min at 60°C before analysis by SDS-PAGE and immunoblotting. All co-immunoprecipitation experiments were performed as described in this base procedure, with the following modifications.
For the experiment in Extended Data Fig. 6h, crude mitochondria were isolated from ySS121 strains harboring plasmid pSS122 (a CEN/ARS plasmid that constitutively expresses WT or mutant Tim17 under the endogenous TIM17 promoter) and solubilized with Buffer L containing 1% digitonin. The supernatant was incubated with Sepharose beads conjugated with anti-BC2 nanobody. The beads were washed with Buffer W containing 0.1% digitonin.
For the experiment in Extended Data Fig. 8d, the yeast strains ySS171 (WT), ySS199 (mgr2Δ), and ySS205 (mgr2Δ, pALD6 Strep-Mgr2) were transformed with plasmid pYC005 (for Grx5-S80-sfGFP) or pYC006 (for Grx5-S99[TM]-sfGFP). Cells were grown in YPL medium (1% yeast extract, 2% peptone, 3% lactate, 0.1% glucose) at 30°C to an OD600 of ~0.8–1.0. Substrate expression was induced with 0.5% galactose for 4 h at 30°C. Crude mitochondria were isolated and solubilized in Buffer L containing 1% digitonin. The supernatant was incubated with Sepharose beads conjugated with anti-ALFA nanobody. The beads were washed with Buffer W containing 0.05% digitonin.
For the experiment in Fig. 3g and Extended Data Fig. 9j, the yeast strain ySS205 was transformed with plasmid pYC005 or an empty plasmid (pYTK-e112). Cells were grown in YPL medium, and substrate expression was induced with 0.5% galactose for 4 h at 30°C. Crude mitochondria were isolated and solubilized in Buffer L containing 1% digitonin. The supernatant was incubated with Protein A beads bound to rabbit anti-HA antibodies (Cell Signaling, Cat# 3724). The beads were washed with Buffer W containing 0.05% digitonin.
In-vitro mitochondrial import assay
C-terminally His-tagged Cyb2Δ-DHFR was purified with a procedure adapted from a previous study33. Briefly, E. coli BL21 (DE3) was transformed with pYC002 encoding Cyb2Δ-DHFR. Cells were grown Luria broth (LB) medium to OD600 of ~0.6, and the expression was induced with 0.5 mM isopropylthio-β-galactoside (IPTG) overnight at 10°C. Cells were lysed by sonication in 40 mM Tris-HCl pH 7.5, 300 mM NaCl, 1 mM PMSF, 10 mM DTT, 0.1% Triton X-100. After clarification (Sorvall SS-34 rotor; 1 h at 23,000 g), Cyb2Δ-DHFR was purified with cobalt resin (Thermo Scientific, Cat. #89966). The eluate was dialyzed against 0.1 M phosphate buffer pH 6.5 and further purified by a HiTrap SP HP column (GE Life Science) and a NaCl gradient elution. Purified Cyb2Δ-DHFR was concentrated to 0.25 mg/mL in storage buffer (10 mM MOPS-KOH pH 7.2, 150 mM NaCl, 1 mM EDTA, 10% glycerol), frozen, and stored at −80°C.
Crude mitochondria were prepared as described above from yeast strains yYC03, yYC04, and yYC05. As a control, mitochondria from yYC02 (strain without Tim17-Spot) were prepared in the same manner. To generate stalled translocation intermediates (Fig. 2 g and h), 200 μg of mitochondria (mitochondrial protein concentration determined by Braford assay [Pierce, Cat #23200]) were mixed with 4 μg Cyb2Δ-DHFR in 400 μL import buffer (3% w/v bovine serum albumin [BSA], 250 mM sucrose, 80 mM KCl, 10 mM MOPS-KOH pH 7.2, 5 mM MgCl2) and supplemented with 2 mM ATP, 2 mM NADH and 2 μM methotrexate (MTX; TCI America, Cat. #M16641). Where indicated, mitochondria were pre-incubated with 2 μM valinomycin (Sigma Aldrich, Cat. #V0627) for 5 min on ice to dissipate the membrane potential, before setting up the import reactions. After 20 min incubation at 25°C, mitochondria were pelleted and washed twice with sucrose-MOPS buffer (250 mM sucrose, 10 mM MOPS-KOH pH 7.2). For Fig. 2 g and h, mitochondria were resuspended in 220 uL of lysis buffer containing 10 mM MOPS-KOH pH 7.2, 10% glycerol, 200 mM NaCl, 10 mM imidazole, 5 μg/mL aprotinin, 5 μg/mL leupeptin, 1 μg/mL pepstatin A, 2 mM PMSF and solubilized with 1% digitonin for 45 min at 4°C. The lysates were cleared by centrifugation, and the supernatants were incubated with 20 μL Ni-charged resin (GenScript, Cat. #L00223) for 1 h at 4°C. The resin was washed three times with 1 mL wash buffer containing 10 mM MOPS-KOH pH 7.2, 10% glycerol, 200 mM NaCl, 20 mM imidazole, 0.02% digitonin. Bound proteins were eluted with 40 μL of 10 mM MOPS-KOH pH 7.2, 10% glycerol, 200 mM NaCl, 250 mM imidazole, and 0.02% digitonin. Samples were analyzed by SDS-PAGE and western blotting.
For in vitro import assays in Extended Data Fig. 6 i and j, reactions were set up as described above by mixing 1 μg Cyb2Δ-DHFR and 50 μg of mitochondria from wildtype BY4741 in 100 μL import buffer. Where indicated, mitochondria were pre-incubated with 2 μM valinomycin for 5 min on ice. After 20 min incubation at 25°C, mitochondria were spun down and resuspended with 0.5 mL MOPS-EDTA buffer (10 mM MOPS-KOH pH 7.2, 1 mM EDTA). Where indicated, a final concentration of 50 ug/mL proteinase K (Alfa Aesar, Cat# J62051) was added and incubated on ice for 15 min. 1 mM PMSF (final concentration) was added, and incubated on ice for another 5 min to quench PK. Mitochondria were pelleted and washed twice with sucrose-MOPS-EDTA buffer (250 mM sucrose, 10 mM MOPS-KOH pH 7.2, 1 mM EDTA). Samples were analyzed by SDS-PAGE and western blotting.
Blue-native (BN) PAGE
For the experiment in Extended Data Fig. 8a, the yeast strain ySS171 harboring pYC007 was grown in an SC(–Leu) medium containing 2% glucose. The culture was diluted to an OD600 of ~0.01 in YPL medium and grown at 30°C to an OD600 of ~0.8–1.0. Where indicated, substrate expression was induced with 0.5% galactose for 4 h at 30°C. Crude mitochondria were isolated and solubilized with a Buffer L containing 1% digitonin for 1 h at 4°C. After clarification, the lysate was incubated with anti-ALFA nanobody beads for 1 h. The beads were washed four times with Buffer W containing 0.05% digitonin. Proteins were eluted by TEV digestion for 1 h at 4°C. The samples were analyzed with a BN-PAGE gel (Invitrogen; cat# BN1004) and immunoblotting. Where indicated, 0.1% or 0.2% DDM was added to the eluate and incubated for 30 min at 4°C before loading. For Extended Data Fig. 6k, in vitro import assay was performed as described in previous section, and washed mitochondria pellets were solubilized with ice-cold Buffer L containing 1% digitonin for 1 h at 4°C. The lysates were clarified by centrifugation, and the supernatants were loaded onto a BN-PAGE gel.
Photocrosslinking experiments
For photocrosslinking assays with Bpa sites introduced into the stalled substrate (Fig. 3c and Extended Data Fig. 8e–i), the yeast strain ySS205 was transformed with SNRtRNA-pBpaRS(TRP) (ref. 65) and pYC005 expressing substrate Grx5-S80-sfGFP with a TAG mutation. For photocrosslinking assays with Bpa sites introduced into Tim17, Tim23, or Mgr2 (Fig 3 d and e and Extended Data Fig. 9a–h), in addition to SNRtRNA-pBpaRS(TRP) and pYC005, yeast strains yYC17b (PTIM17::KanMX-tetO7-PCYC1), yYC23b (PTIM23::KanMX-tetO7-PCYC1) and ySS199 (mgr2Δ) were transformed with pYC003, pYC008, or pSS277 expressing TAG mutants on Tim17-Spot, Tim23-Spot, and Strep-Mgr2 respectively. Cells were initially grown in a synthetic medium (SC[−Leu/−Trp]) for Bpa incorporation to the substrate and SC[−Leu/−Trp/−His] for others) containing 2% glucose at 30°C. The yeast cultures were then switched to appropriate SC medium containing 3% lactate, 0.1% glucose, 2 mM Bpa (Amatek, Cat# A-0067) and were grown in the dark. Substrate expression was induced by addition of 0.5% galactose at OD600 ~0.6–0.8 for 4 h at 30°C. Cells were harvested, washed with deionized water, and split into two samples. One sample was maintained on ice and kept in the dark (for ‘−UV’ controls). The other sample was transferred to a 24-well plate and UV (365nm)-irradiated for 1 h at 4°C. Cells were then resuspended in 0.5 mL of ice-cold Buffer L and lysed by beating with 0.5 mm glass beads. Cell lysate was supplemented with 1% digitonin. After solubilization for 1 h at 4°C, the lysate was clarified by centrifugation. The supernatant was incubated with appropriate beads (Sepharose beads conjugated with anti-ALFA nanobody for substrate pull-down; Protein A beads bound to rabbit anti-HA antibodies (Cell Signaling, Cat# 3724) or Sepharose beads conjugated with anti-BC2 nanobody for Tim17 pull-down) for 1 h. The beads were washed three times with 1 mL of Buffer W containing 0.05% digitonin (Buffer WN). Proteins were eluted by addition of SDS sample buffer and heating for 20 min at 60°C and 10 min at 90°C before analysis by SDS-PAGE and immunoblotting.
For urea washing of photocrosslinking adducts, after washing beads once with Buffer WN as described above, two additional washes were performed with Buffer W containing 6 M urea and 0.5% Triton X-100 (Buffer WD). This was followed by another wash with Buffer WN and elution with SDS sample buffer.
For co-IP experiments with SDS-denatured samples (Extended Data Fig. 8 h and i and Extended Data Fig. 9 d, f and h), cells were harvested after UV treatment, crude mitochondria were prepared and resuspended in a solubilization buffer containing 50 mM Tris-HCl pH 7.5 and 1% SDS. Samples were heated for 10 min at 90°C and then diluted 10-fold with Buffer D, containing 50 mM Tris-HCl pH 7.5, 200 mM NaCl, 1 mM EDTA, 10% glycerol, and 0.5% Triton X-100. The lysate was clarified by centrifugation, and pulldown was performed similarly with appropriate beads (Strep-Tactin XP 4Flow [IBA, Cat# 2–5010-010] for Strep-Mgr2; BC2T-nanobody-conjugated Sepharose for Tim17-Spot; Protein A beads incubated with mouse Tim44 hybridoma supernatant for Tim44) and washing with Buffer D2, containing 25 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 10% glycerol, and 0.5% Triton X-100. For the experiment shown in Extended Data Fig. 8g, after ALFA-tag IP was performed with digitonin buffers as described above, bound proteins were eluted with 25 mM Tris-HCl pH 7.5 and 0.4% SDS and heated for 10 min at 90°C. Samples were then diluted 10-fold with Buffer D and subjected to the second IP with anti-HA resin (Abcam, Cat# ab270603), as described above for the denaturing IP.
Immunoblotting and antibodies
Expression levels of proteins were determined by diluting overnight yeast cultures in appropriate medium and growing them to a mid-log phase at 30°C. Equal numbers of cells (1.0–2.5 ODs) were harvested and lysed with NaOH buffer (0.1 M NaOH, 50 mM EDTA, 2% SDS, 2% β-mercaptoethanol). The samples were neutralized with acetic acid and analyzed by SDS-PAGE. For the experiments in Extended Data Figs. 3a and 8c, cultures were grown in YPEG and YPD media, respectively. For the experiments in Extended Data Fig. 6 b, c, and e–g, cells were grown in YPD with 100 μg/mL nourseothricin for endogenous promoter constructs and SC(−Leu) with cyanamide at indicated concentration for DDI2 promoter constructs.
Immunoblotting experiments were performed with antibodies against Pam16 (a gift from E. Craig; 1:2,500 dilution), Pam18 (a gift from E. Craig; 1:1,000 dilution), Pgk1 (a gift from J. Thorner; 1:1,000 dilution), Myc-tag (Invitrogen, Cat# 13–2500; 1:1,000 dilution), HA-tag (for mouse IgG, Invitrogen, Cat# 26183; 1:5,000 dilution), His-tag (Invitrogen, Cat# MA1–21315; 1:1,000 dilution), Strep-tag (Genscript, Cat# A01732; 1:2,000 dilution), GFP-tag (for rabbit IgG, Genscript, Cat# A01388, 1:1,000 dilution; for mouse IgG, Roche, Cat# 11814460001, 1:1,000 dilution), Tom22 (a gift from N. Pfanner; 1:1,000 dilution) and Tom40 (a gift from N. Pfanner; 1:500). For anti-Tim44 blots, we used the non-monoclonal hybridoma supernatant of Clone #23 (1:100 dilution), which could detect Tim44 on immunoblots. For Spot-tag and ALFA-tag blots and pull-downs, we used home-made BC2-Nb and ALFA-Nb fused with a rabbit Fc domain, which was produced in HEK293S GnTI− cells (ATCC CRL-3022) by transient transfection (HEK293S cells were not tested for mycoplasma contamination or authenticated). Secondary antibodies conjugated to HRP used in this study were goat anti-rabbit (Thermo, Cat# 31460; 1:10,000 dilution), goat anti-mouse (Thermo, Cat# 31430; 1:10,000 dilution), and mouse antirabbit (Genscript, Cat# A01827; 1:10,000 dilution).
Molecular dynamics (MD)
An atomic model of the Tim17–Tim23–Tim44 complex used for the MD runs was derived from the cryo-EM atomic structure. For Tim17 and Tim23 the N- and C-termini were capped with neutral acetyl and amide groups, respectively. For Tim44, a peripheral membrane protein, the cryo-EM structure is missing the N-terminal residues 1–106 as well as residues 194–254. Of note, residues 255–265 form an amphipathic α-helix (α6) that appeared to protrude into the membrane, likely as result of the curvature of the micelle used to obtain the structure. Given the amphipathic nature of this stretch, it was realigned with the membrane interface such that it lies parallel to the plane of the membrane with hydrophobic residues pointing towards the bilayer center. Moreover, during the course of this work, structures derived using AlphaFold2 (ref. 34,66; AlphaFold2 Multimer version 2.2.0 was used), became available, including a S. cerevisiae Tim44 model (UniProt Q01852) for Tim44. There are regions of high confidence that were missing in the cryo-EM structure, in particular residues 228–254 of Tim44, which were added and placed in the plane of the membrane. A hybrid atomic model of Tim17–Tim23–Tim44–Mgr2 was generated by combining the cryo-EM structure of Tim17–Tim23–Tim44 and AlphaFold2 generated Mgr2. We first generated an atomic model of Tim17–Tim23–Tim44–Mgr2 using AlphaFold2, and this model was aligned to the cryo-EM structure. We then combined the cryo-EM structure with the Mgr2 portion of the AlphaFold2 model.
TIM23 atomic models were placed in a model yeast inner mitochondrial membrane with an overall composition of 33% YOPC, 21% YOPE, 14% POPI, 4% POPS, 2% POPA, 12% ergosterol (ERG), and 14% cardiolipin (TOCL2) (ref. 67–70) using CHARMM-GUI (ref. 71). The membrane-protein systems were placed in a TIP3P (ref. 72) water box and neutralized with 0.15 M KCl. The bound cardiolipin and phospholipid observed in the cryo-EM structure were included and modeled as TOCL2 and YOPE, respectively. An asymmetric distribution of cardiolipin (CL) has been reported as 20% on the intermembrane-space (IMS) side and 80% on the matrix side for mammalian mitochondria (ref. 73), and thus we modeled this 20:80 ratio and scaled the other components accordingly. The IMS-side composition was taken as 37% YOPC, 23% YOPE, 15% POPI, 4% POPS, 2% POPA, 13% ERG, and 6% TOCL2, while the matrix side was taken as 30% YOPC, 19% YOPE, 13% POPI, 3% POPS, 2% POPA, 11% ERG and 22% TOCL2. Given the uncertainty of the asymmetry, simulations of the Tim17-Tim23-Tim44 complex using a symmetric lipid distribution were also performed for comparison. For simulations of the hybrid model with lipids in the Tim17–Mgr2 cavity, phospholipids were placed as follows: first Mgr2 was removed and a simulation cell was built using the asymmetric lipid composition given above, then the lipids were allowed to relax for 100 ns with the TIM23 complex restrained, lastly the Mgr2 was re-introduced and overlapping lipids were removed.
All simulations used the CHARMM36m force field for proteins74 and CHARMM36 force field for lipids75. Each system was equilibrated in multiple stages using NAMD 2.14 (ref. 76). In the first stage, the proteins and lipid headgroups were restrained for 1 ns. In the second stage, the protein and bound lipids were restrained to their starting positions for 10 ns; lastly, only the protein backbone was restrained for 100 ns to allow for lipid equilibration about the complex. Finally, all restraints were released for the production runs. Simulations were run at constant temperature (310 K) and constant semi-anisotropic pressure (1 atm) enforced by a Langevin thermostat and Langevin piston barostat, respectively. A 2-fs time step was used, with long-range electrostatics calculated every other time step using the particle-mesh Ewald method77 for the first three equilibration steps; otherwise a uniform 4-fs time step was employed using hydrogen mass repartitioning78. Short range non-bonded interactions were cut off at 12 Å, with a force-based switching function starting at 10 Å. After equilibration, each system was run in duplicate using GPU-accelerated NAMD3 (ref. 76). Unless otherwise stated, the simulations were performed with a –150 mV membrane potential79 and run to 1.0 μs per replica. Analysis was carried out and images were rendered with VMD80. The membrane maps were generated with LOOS81,82, using the glycerol atoms of the phospholipids with a 1-Å bin width.
Multiple sequence alignment
Amino acid sequences of Tim17 and Tim23 from various organisms were obtained from the UniProt Reference Clusters (UniRef100, 2021_03). For Tim17, any sequence smaller than 130 amino acids or larger than 256 amino acids were excluded. For Tim23, any sequence smaller than 166 amino acids or larger than 257 amino acids were excluded. A total of 94 sequences of Tim17 and 102 sequences of Tim23 were separately aligned using MAFFT (ref. 83). Amino acid identity was mapped onto the structure using UCSF Chimera.
Extended Data
Extended Data Fig. 1 |. Purification of the TIM23 complex from S. cerevisiae and initial cryo-EM analysis.
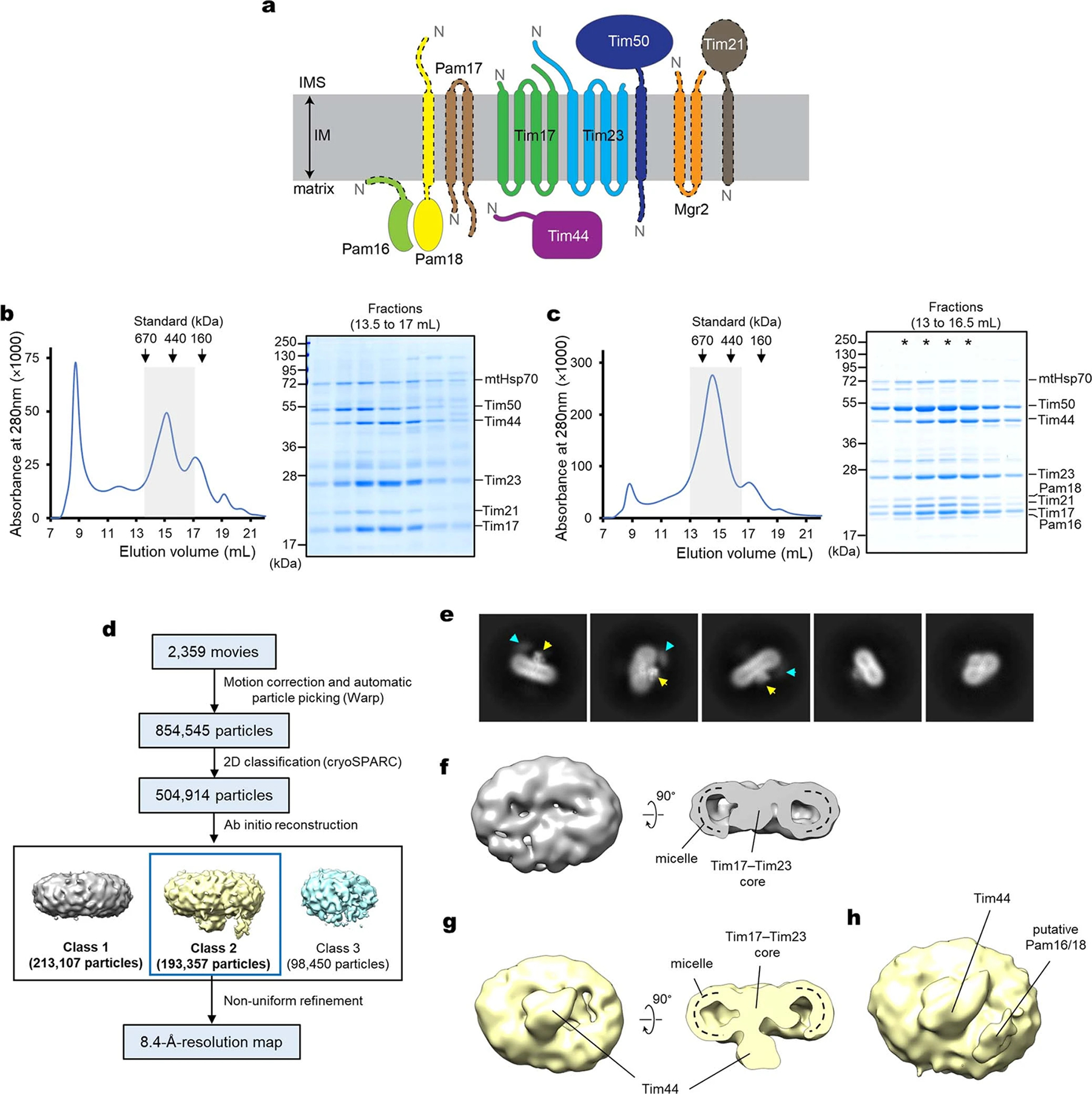
a, Schematic diagram of the membrane-associated components of the TIM23 complex. The two soluble matrix components mtHsp70 and Mge1 were not shown. The N-terminus of each subunit is indicated by an “N”. Note that Tim21, Pam17, and Mgr2 are not essential, and individual deletions of the portions of essential Pam16, Pam18, and Tim50 proteins outlined by a dashed line are viable13,17,19. b, Purification of the endogenous TIM23 complex. After affinity purification via Spot-tagged Tim17, the sample was subjected to Superose-6 size-exclusion chromatography (SEC). The gray box indicates fractions further analyzed by non-reducing SDS-PAGE and Coomassie staining (right panel). We note that copurified Tim50 often did not fully co-migrate with the other subunits in SEC, indicating its dissociation over time. c, As in b, but purification of the TIM23 complex from mitochondria co-overexpressing the nine subunits shown in a, omitting mtHsp70 and Mge1. Fractions marked with asterisks are pooled and used for cryo-EM analysis shown in d–h. d, Summary of single particle analysis of the sample shown in c. e, Example two-dimensional (2D) classes of the TIM23 complex. Putative matrix domains of Tim44-CTD and Pam16/18 are indicated by yellow and cyan arrowheads, respectively. f, Class 1 from ab-initio refinement of particles shown in d. g, As in f, but showing Class 2. h, As in the left panel of g, but shown at a lower isosurface threshold value. Data shown in b and c are representative of two experiments.
Extended Data Fig. 2 |. 2.7-Å-resolution cryo-EM structure of the TIM23 complex (Tim17/23/44 + Pam16/18).
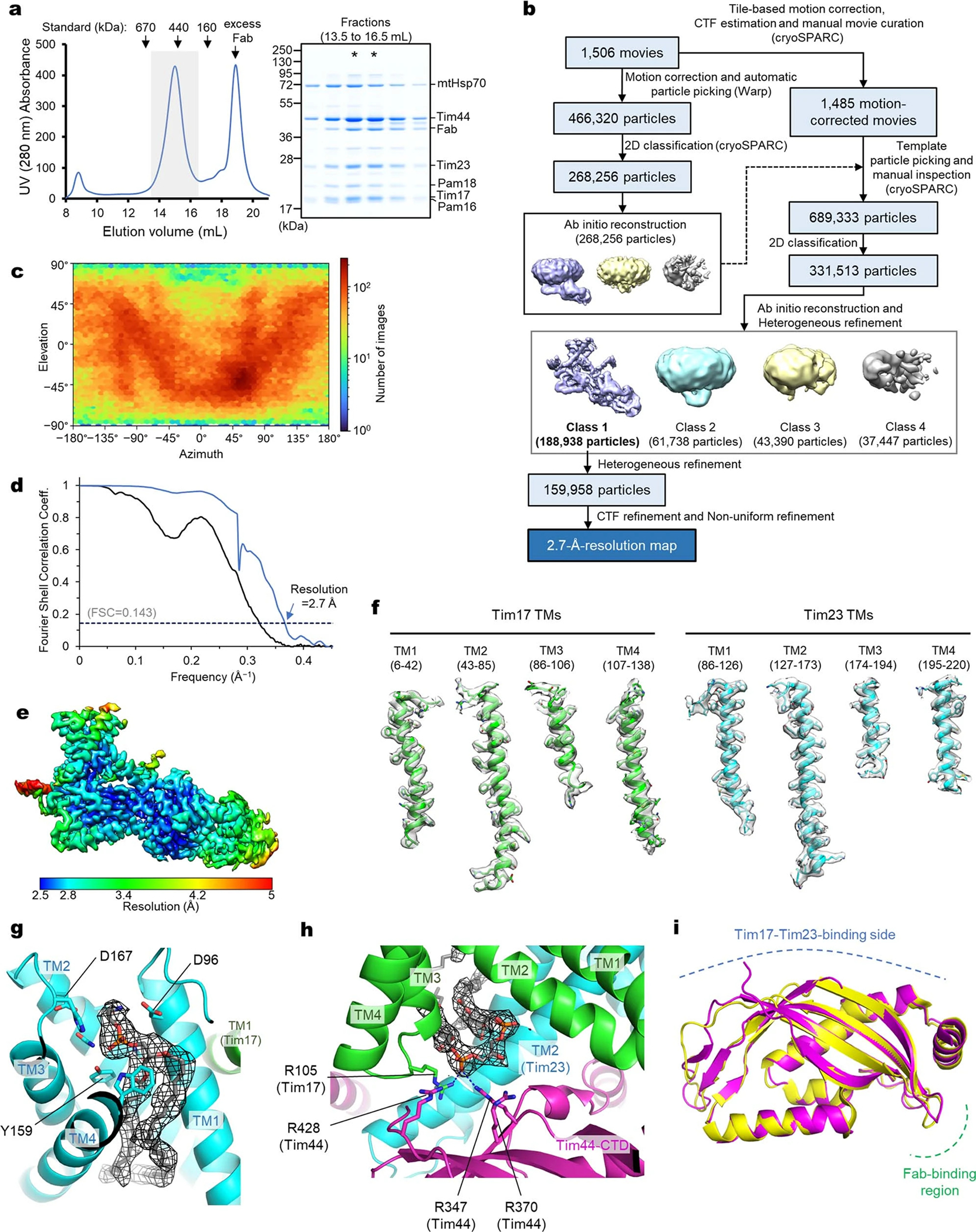
a, Purification of the Fab-bound core TIM23 complex (Tim17/23/44 + Pam16/18). Left, Superose-6 SEC elution profile; right, Coomassie-stained non-reducing SDS gel of fractions indicated in the left panel by a gray box. Fractions marked with asterisks were pooled and used for cryo-EM analysis. b, Summary of single particle analysis of the Fab-bound TIM23 complex. c–e, Particle view distribution (c), Fourier shell correlation (d), and local resolution distribution (e) for the Fab-bound TIM23 complex. f, Examples of segmented EM densities and atomic models. The amino acid ranges are indicated. The threshold values for the map range from 7.8 to 8.9σ. g, Phospholipid bound to the cavity of Tim23. The EM density of the phospholipid is shown as black mesh (at a map threshold value of 5.9σ). Side chains contacting the lipid head group are shown as sticks. We note that, although the identity of the phospholipid could not be unambiguously determined, the EM density agrees very well with phosphatidylethanolamine. h, As in Fig. 1b, but showing a zoomed-in view for the cardiolipin molecule. The EM density is shown in black mesh (at a map threshold value of 10.6σ). Polar interactions are indicated by dashed line. i, Superposition between Tim44 structures from the present cryo-EM study and a previous X-ray crystallography study (PDB: 2FXT). The root-mean-square deviation (RMSD) is 0.75 Å for Cα atoms. Data shown in a are representative of two experiments.
Extended Data Fig. 3 |. High-resolution cryo-EM structure of the endogenous core-TIM23 complex.
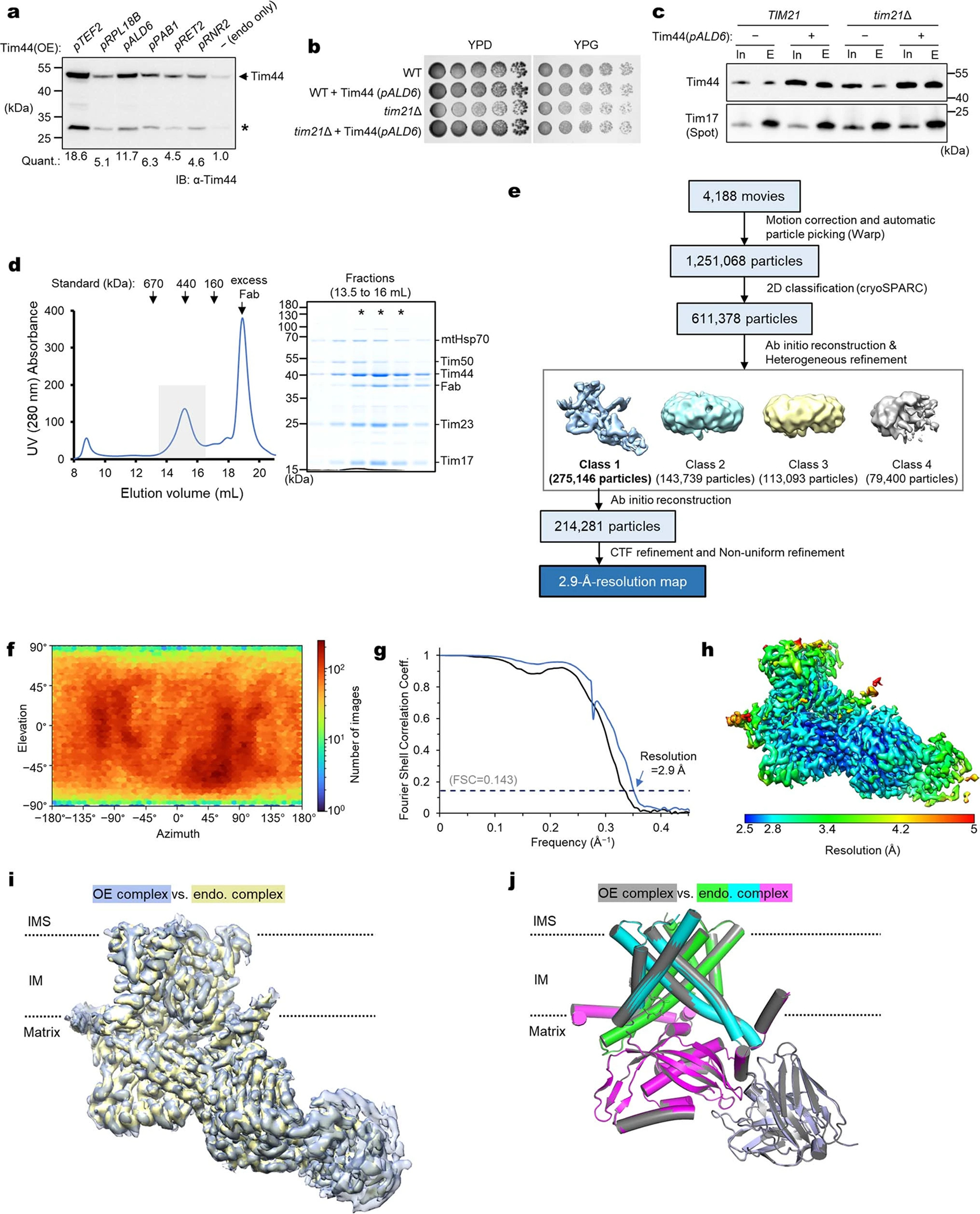
a, To increase Tim44 occupancy in the core TIM23 complex formed by endogenous Tim17 and Tim23, Tim44 was overexpressed under indicated constitutive promoters, and protein levels were analyzed in yeast whole cell lysates (Quants., relative band intensities of full-length Tim44). For the subsequent analyses, we chose the ALD6 promoter (pALD6). Asterisk indicates partial degradation product. b, ALD6-promoter-driven overexpression of Tim44 does not exhibit any cell growth defect. Because the non-essential subunit Tim21 may reduce the amount of the Tim44-containing TIM23 complex by forming a “TIM23-sort” complex12,84, we also tested a chromosomal deletion of tim21. c, Tim17 was immunoprecipitated from the mitochondrial lysate of the strains used in b and co-precipitated Tim44 was detected by immunoblotting (In, input; E, eluate). Note that a substantially increased amount of Tim44 was co-purified with Tim44 overexpression. d, Purification of the Fab-bound, endogenous core-TIM23 complex from the tim21Δ/pALD6-Tim44 strain. Left, Superose-6 SEC elution profile; right, Coomassie-stained SDS gel of fractions indicated in the left panel by a gray box. Fractions marked with asterisks were pooled and used for cryo-EM analysis. Note the substantially increased Tim44:Tim23 ratio compared to the purification shown in Extended Data Fig. 1b. e, Summary of single particle analysis of the endogenous core-TIM23 complex. f–h, Particle view distribution (f), Fourier shell correlation (g), and local resolution distribution (h). i, Superposition of the endogenous core-TIM23 cryo-EM map (blue) onto the TIM23 map (yellow) from overexpression (OE) (Extended Data Fig. 2). The map correlation coefficient was 0.985 in UCSF Chimera. j, As in i, but showing superposition of the atomic models. The RMSD value is 0.26 Å for 739 out of 760 aligned Cα atoms. Data shown in a–d are representative of two experiments.
Extended Data Fig. 4 |. Interactions among Tim17, Tim23, and Tim44.
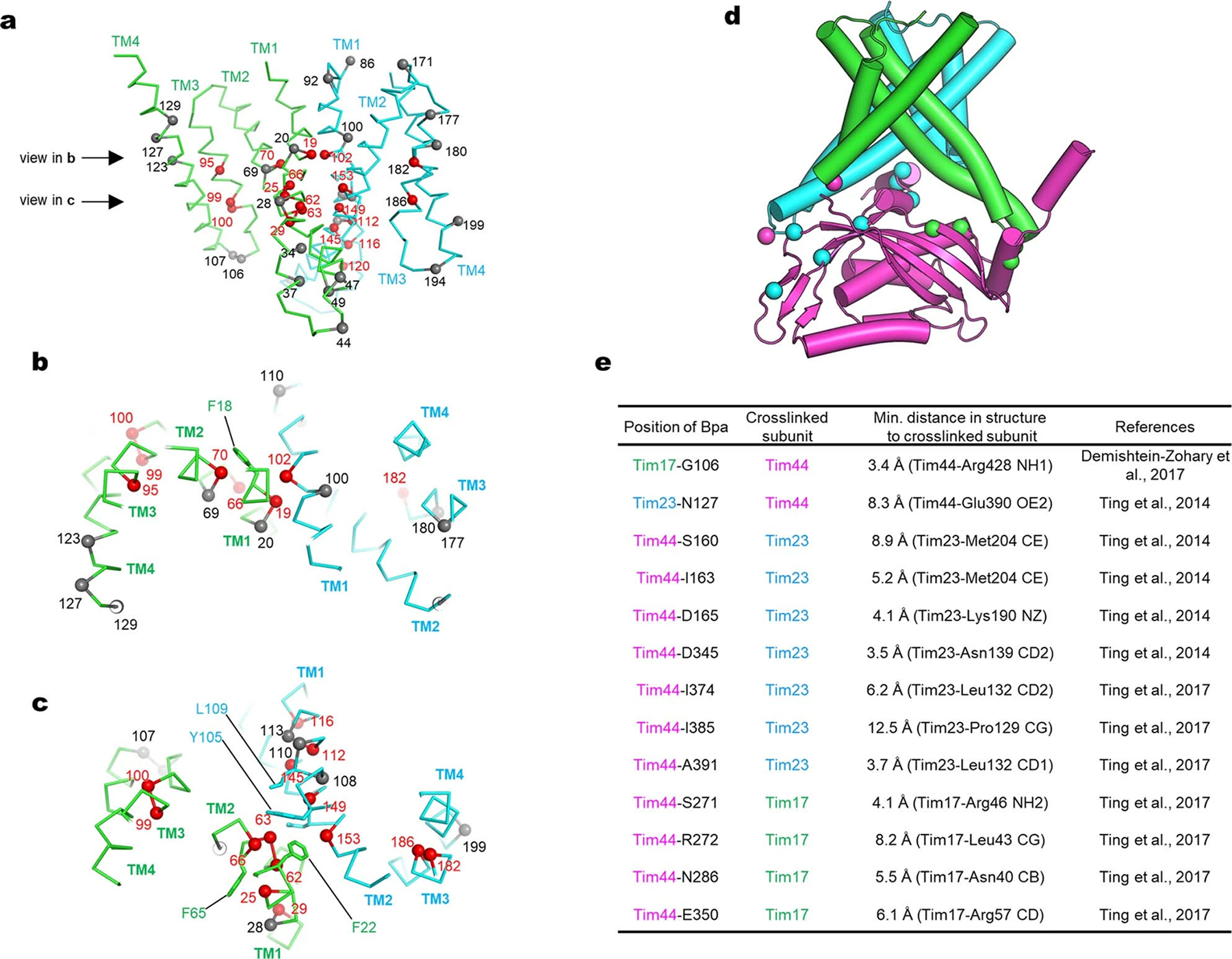
a, Side view showing the positions of glycine in Tim17 (green Cα-trace) and Tim23 (cyan Cα-trace). Positions of all glycine residues are shown as spheres (Cα atoms) with residue numbers. Red spheres indicate positions in which mutation to a non-glycine amino acid would cause steric clashes. Gray spheres indicate positions in which mutation may not cause steric clashes. b and c, As in a, but viewing down the planes indicated by arrows in panel a from the IMS. d, Mapping of amino acid positions that have been shown to produce an inter-subunit photocrosslinking adduct with p-benzoyl-L-phenylalanine (Bpa). Tim17 (green), Tim23 (cyan), and Tim44 (magenta) are shown in a cylindrical cartoon representation. Green and cyan spheres are positions in Tim44 that crosslink to Tim17 and Tim23, respectively. Magenta spheres are positions in Tim17 and Tim23 that crosslink to Tim44. See panel e for details. e, Summary of previously reported inter-subunit photocrosslinking results. The minimal distance is between the position (Cα for Tim17-G106 and Cβ atom for all other positions) into which Bpa was incorporated and the crosslinked subunit (atom is specified).
Extended Data Fig. 5 |. Structural comparisons between Tim17, Tim23, and Tim22, and membrane thinning by the cavity of Tim17.
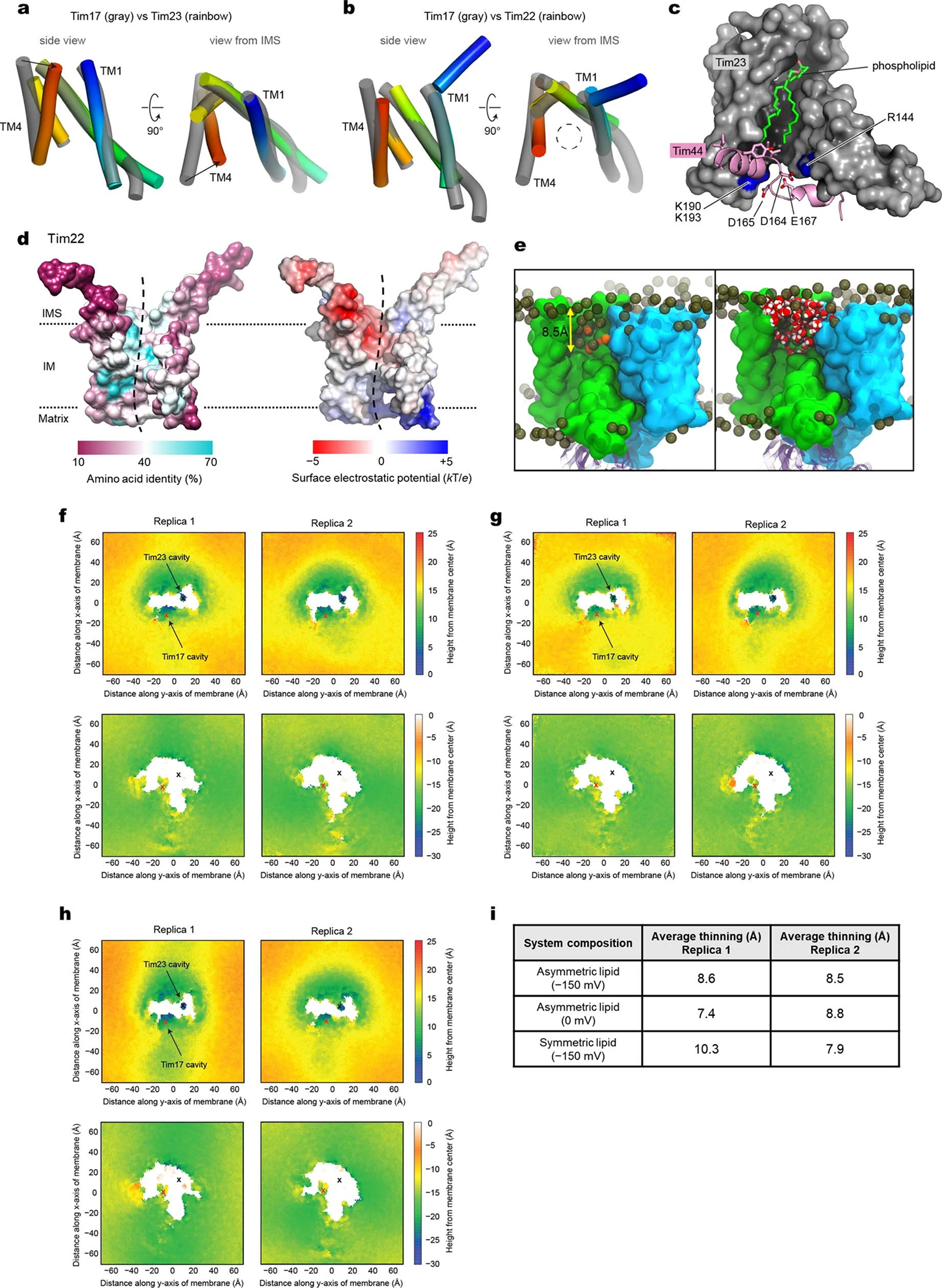
a, Comparison between the transmembrane domains of Tim17 and Tim23. TM1 to TM4 of Tim23 are shown in rainbow color (blue to red). Arrows highlight a difference between the TM4 positions. b, As in a, but comparing between Tim17 and Tim22 (PDB ID: 6LO8). Note that Tim22 has an extra IMS helix to its N-terminus. c, Side view showing the cavity of Tim23 (gray surface). Amino acid residues 153–174 of Tim44 are shown in pink. Note that residues D164, D165, and E167 interact with basic amino acids (R144, K190, and K193; blue) of Tim23. d, As in Fig. 2 a and b, but showing amino acid conservation and surface electrostatics of Tim22 (PDB 6LO8) with a side view into the concave cavity (indicated by a dashed line). e, MD simulated model showing distributions of lipids, ions, and water in the Tim17 cavity. Surfaces are rendered with Tim17 (green) and Tim23 (cyan), while Tim44 is represented with magenta transparent ribbons. Phosphorous atoms of lipids are rendered as dark gold spheres and potassium ions are rendered as orange spheres. In the right panel, extensive hydration (water is rendered in red/white spheres) was observed in the Tim17 cavity. The membrane thinning in the Tim17 cavity is approximately 8.5 Å (also see f–i). f, As in Fig. 2e, but showing two replicas of simulation with an asymmetric lipid composition and a membrane potential of −150 mV. Upper and lower panels show membrane heights from the membrane center toward the IMS and the matrix, respectively. Replica 2 is the result shown in Fig. 2e. g, As in f, but without membrane potential. h, As in f, but with a symmetric membrane composition. i, Summary of membrane thinning, calculated as the averaged difference of the height of the IMS and matrix leaflets in a 10-Å by 10-Å patch in the vicinity of the Tim17 cavity relative to the average thickness of the lipid bilayer (four patches taken remote from the protein complex in the four corners of the maps). Note that membrane thinning occurs independent of membrane potential or asymmetry of the lipid composition.
Extended Data Fig. 6 |. Mutational analysis on the cavities of Tim17 and Tim23.
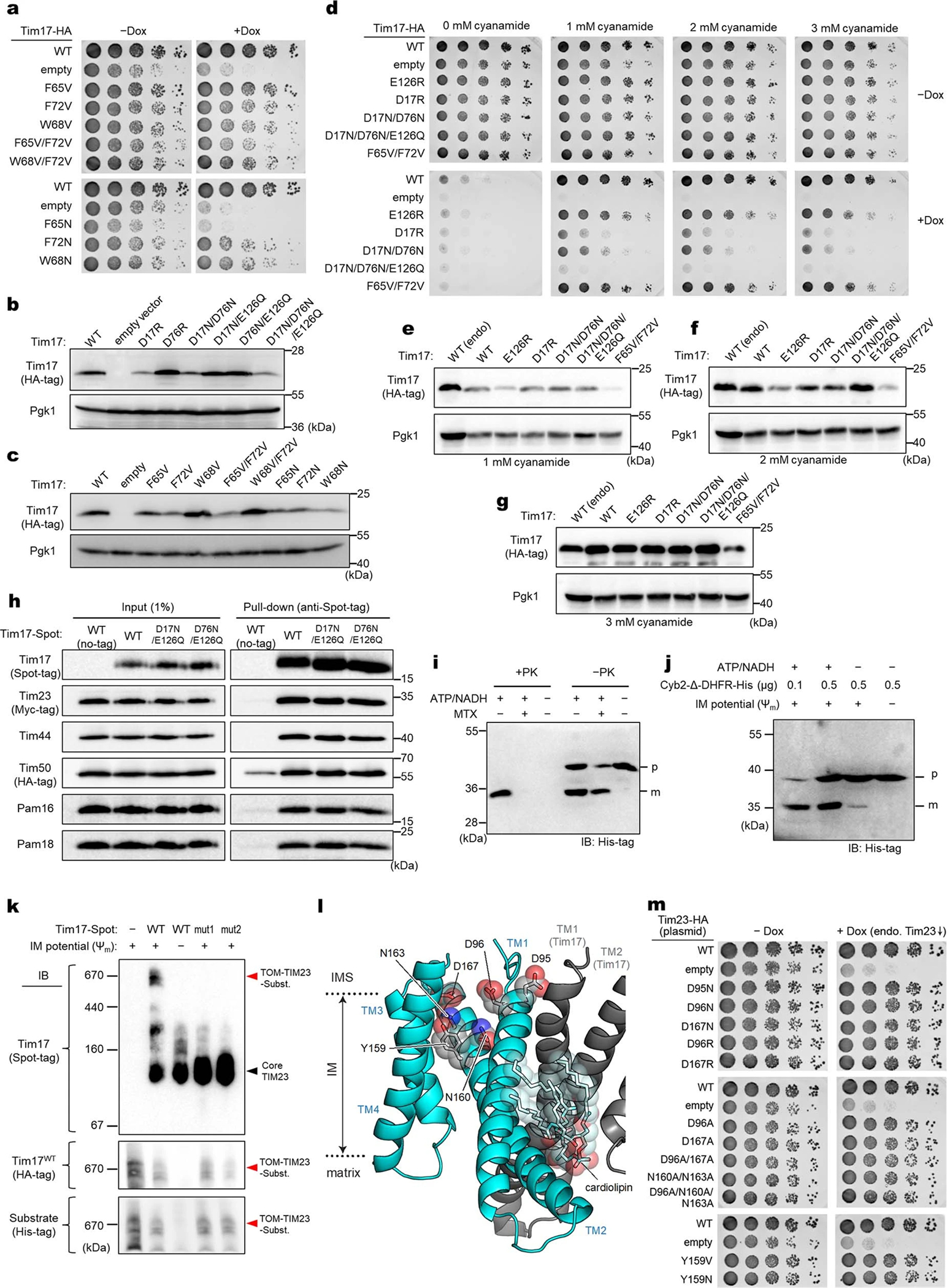
a, As in Fig. 2f, but testing mutations on aromatic residues lining the Tim17 cavity (Phe65, Phe72, and Trp68) of Tim17. b and c, Expression levels of WT and indicated mutants were measured by immunoblotting. All Tim17 variants were expressed under the endogenous promoter from a CEN/ARS plasmid. 3-phosphoglycerate kinase (Pgk1) was used as a loading control. d, As in Fig. 2f, but the experiments were performed with a 2μ plasmid expressing indicated mutants of HA-tagged Tim17 under a cyanamide-inducible DDI2 promoter. Cells were spotted on synthetic complete without leucine (SC[–Leu]) plates supplemented with varying concentrations of cyanamide. The plates contain doxycycline (Dox), where indicated. e–g, As in b and c, but measuring expression levels of Tim17 under the DDI2 promoter in the presence of varying concentrations of cyanamide. As a control, expression of WT Tim17 under the endogenous promoter (endo) from a CEN/ARS plasmid was included. h, Co-immunoprecipitation of the essential subunits of the TIM23 complex with Tim17 mutants. Mitochondria expressing indicated Tim17 variants (WT, D17N/E126Q [mut1] and D76N/E126Q [mut2]) were solubilized with digitonin, and Tim17 was pulled down with anti-Spot-tag nanobody beads. The samples were analyzed by immunoblotting with indicated antibodies. i, WT mitochondria (50 μg protein) were incubated with 1 μg of purified Cyb2Δ-DHFR-His in a 100-μL reaction volume. Where indicated, ATP/NADH (2 mM each) and/or methotrexate (MTX; 2 μM) were included. After 20-min incubation at 25°C, mitochondria were washed and resuspended in a hypo-osmotic buffer solution. After splitting the reactions, mitoplasts were treated with proteinase K (+PK) where indicated. The samples were quenched with phenylmethylsulfonyl fluoride and analyzed by SDS-PAGE and immunoblotting (IB) with anti-His-tag antibody. p, precursor form of Cyb2Δ-DHFR-His; m, mature (presequence-cleaved) form of Cyb2Δ-DHFR-His. j, Additional controls including a reaction in the presence of 2 μM valinomycin (−Ψm). Note that indicated amounts of Cyb2Δ-DHFR-His were added to 100-uL reactions and that all reactions contained MTX. k, As in Fig. 2h, but import reactions were subjected to blue native-PAGE (BN-PAGE). l, As in Fig. 2c, but view into the Tim23 cavity. The view is similar to the right panels of Fig. 2 a and b. m, As in Fig. 2f, but testing mutations of acidic, polar, and aromatic amino acids lining the Tim23 cavity. Data in a–j and m are representative of three independent experiments. Data in k is representative of two independent experiments.
Extended Data Fig. 7 |. Comparisons between the cryo-EM structure and AlphaFold2 predictions of the yeast TIM23 complex.
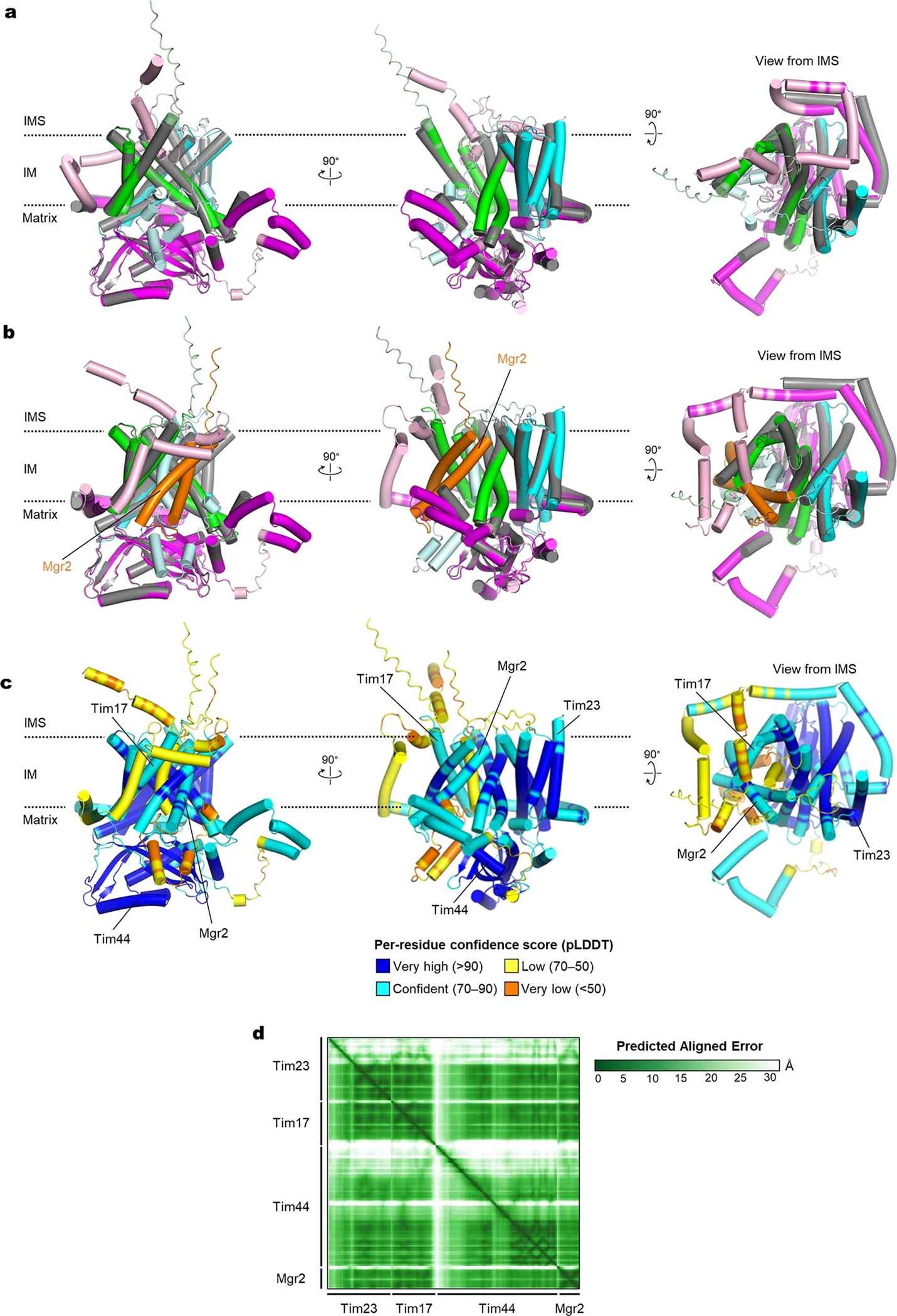
a, Superposition between the cryo-EM structure (gray) and AlphaFold2 model of Tim17–Tim23–Tim44 (in color). For the predicted structure, Tim17, Tim23, and Tim44 are in green, cyan, and magenta, respectively, and low confidence regions (pLDDT<70) are colored in lighter tones. The RMSD is 1.0 Å for 338 Cα atoms out of 480 aligned residues. Predictions were made using AlphaFold2 Multimer version 2.2.0. b, As in a, but the AlphaFold2 model additionally contains Mgr2 (orange). c, As in b, but showing per-residue confidence score of the AlphaFold2-generated Tim17–Mgr2–Tim23–Tim44 model. d, Predicted aligned error (PAE) matrix between all pairs of residues of the Tim17–Mgr2–Tim23–Tim44 AlphaFold2 model.
Extended Data Fig. 8 |. Generation of stalled translocation intermediate complexes of TIM23–TOM–substrate and probing the preprotein translocation path by UV photocrosslinking.
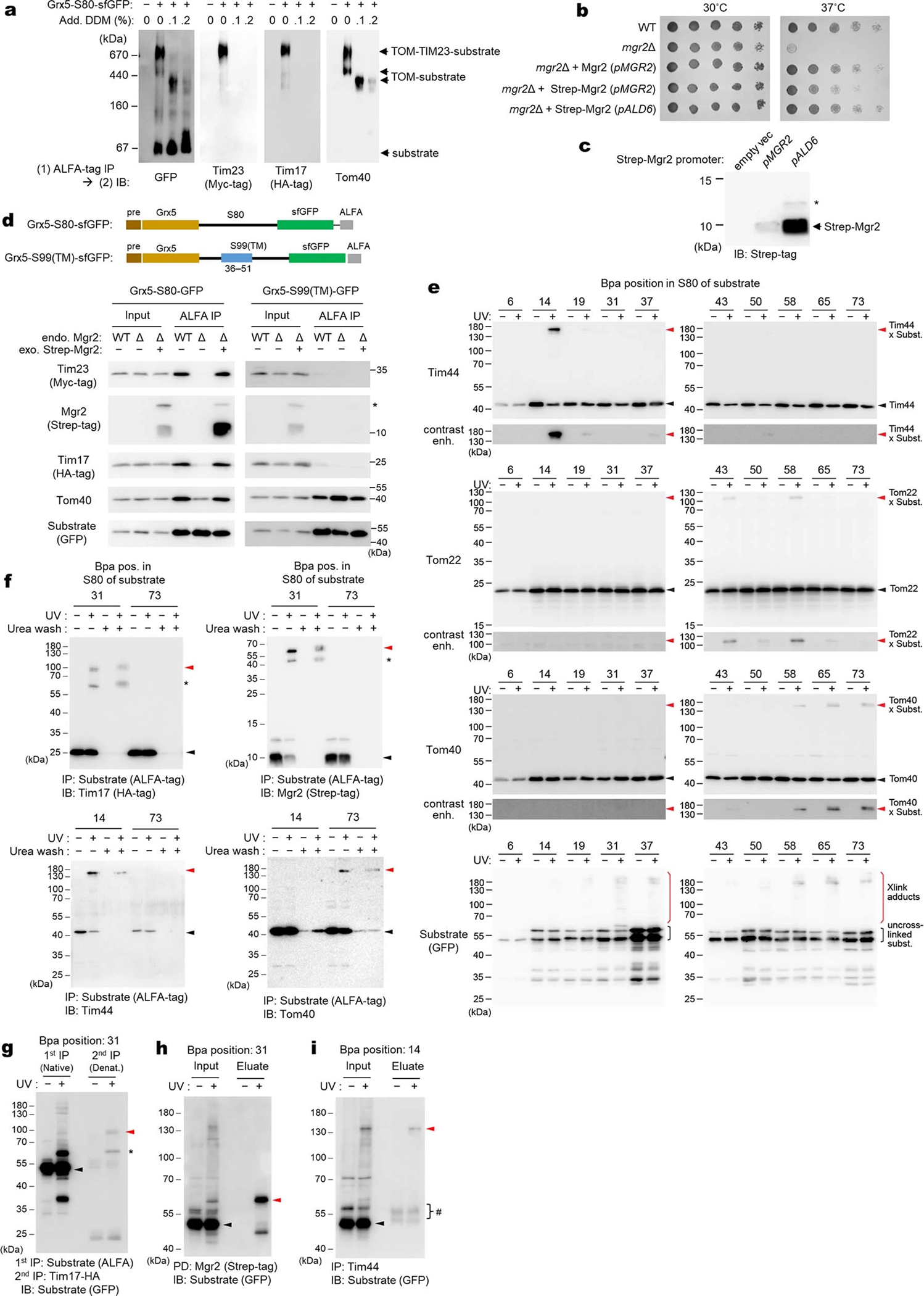
a, Formation of a supercomplex containing TIM23, TOM, and the Grx5-S80-sfGFP substrate (S80 is a hydrophilic segment derived from yeast Cyb2) was confirmed by BN-PAGE. Where indicated, expression of Grx5-S80-sfGFP (C-terminally ALFA-tagged) was induced. After solubilizing mitochondria with digitonin, ALFA-tag immunoprecipitation (IP) was performed, and eluates were subjected to BN-PAGE and analyzed by immunoblotting (IB). Where indicated, eluate was treated with additional dodecylmaltoside (DDM) to dissociate the TIM23 complex. We note that while a substantial population of Grx5-S80-sfGFP stalls in TOM and TIM23, a major population is fully imported into the matrix without stalling. Faster migration of the TOM-substrate band in the presence of DDM is likely due to changes in micelle properties85 or partial dissociation of Tom subunits. b, Testing tagging of Mgr2. To enable detection of Mgr2 in immunoblots, an N-terminal Strep-tag was added to Mgr2. Temperature sensitivity14,37 of an Mgr2-deleted strain (mgr2Δ) and strains additionally expressing Strep-Mgr2 was tested (all in a W303–1A background). Where indicated, cells were transformed with a plasmid encoding non-tagged Mgr2 under a native MGR2 promoter, or N-terminally Strep-tagged Mgr2 under the MGR2 promoter or an ALD6 promoter. Note that lesser growth rescue by MGR2 promoter-driven Strep-Mgr2 might be due to a reduced expression level by insertion of the Strep-tag at the N-terminus or partial impairment of the function of Mgr2. The partial rescue could be overcome by stronger expression of Strep-Mgr2 under the ALD6 promoter. All subsequent experiments were performed with the Strep-Mgr2 (pALD6) strain. c, As in b, but yeast whole cell lysates (from equal OD600) were analyzed by immunoblotting to test expression of Strep-Mgr2. Asterisk, a putative precursor form of Mgr2 (ref. 86). d, Co-immunoprecipitation of TIM23-TOM-substrate supercomplexes with and without Mgr2. The upper panel depicts two different stalling substrates used. Grx5-S99(TM)-sfGFP contains a 99-amino-acid-long segment with a TM helix between 36 and 51 amino acid residues (derived from Cyb2). Upon stalling, this TM is expected to be released to the IM. In lower panels, cells were induced for expression of the indicated substrate, and the mitochondrial lysates were subjected to ALFA-tag IP. Where indicated, the strain lacked endogenous Mgr2 (Δ) and/or expressed exogeneous Strep-tagged Mgr2. Asterisk, putative precursor of Mgr2. We note that Grx5-S80-sfGFP reproducibly showed reduced copurification of Tom40 in the absence of Mgr2. Although the cause of this decrease is unclear, it is unlikely due to impaired presequence engagement of Mgr2-lacking TIM23 since we did not observe a similar decrease with Grx5-S99(TM)-sfGFP. e. Photocrosslinking with Bpa-incorporated Grx5-S80-sfGFP. As in Fig. 3c, but showing the immunoblots for Tim44, Tom22, Tom40, and the substrate. f, Verification of a covalent linkage formed between Grx5-S80-sfGFP and the indicated TIM23 subunit upon photocrosslinking. Beads were split to two halves after a regular wash step of IP, and one of them was additionally washed twice with 6 M urea to remove proteins that are noncovalently associated to the substrate (note that ALFA-tagged protein retains to ALFA-nanobody beads in this denaturing condition87). Asterisk, putative partial degradation products. g, The crosslinked band (red arrowhead) between Grx5-S80-sfGFP (Bpa in position 31 of S80) and Tim17 was verified with successive IPs using the ALFA-tag and HA-tag. After the first ALFA-tag IP, proteins were denatured by SDS. Black arrowhead, the uncrosslinked substrate; asterisk, a putative partial degradation product of the crosslinked species. h and i, The crosslinked band (red arrowheads) between Grx5-S80-sfGFP and Mgr2 (h) or Tim44 (i) was verified by SDS-denatured pulldown (for Strep-Mgr2) or IP (for Tim44). Bpa sites are located in indicated positions of S80. Black arrowhead, the uncrosslinked substrate; hash, putative cross-reacted IgG. Data in a–d and f–i are representative of two independent experiments. Data in e is representative of three independent experiments.
Extended Data Fig. 9 |. Photocrosslinking experiments with Bpa-incorporated Tim17, Tim23, or Mgr2 and characterization of Mgr2 association to translocating Tim17.
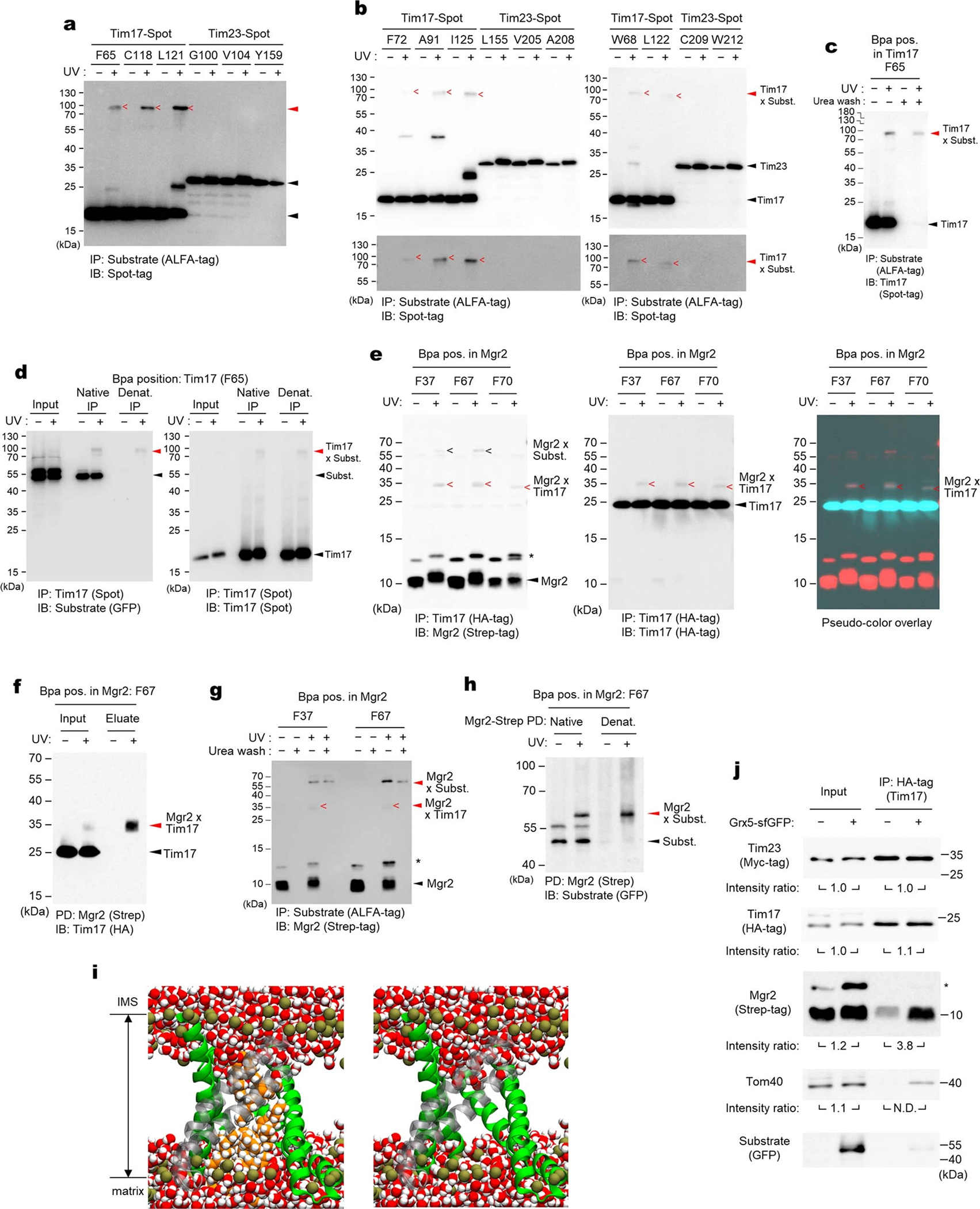
a, The blot image of Fig. 3d is adjusted for higher contrast. IP, immunoprecipitation; IB, immunoblotting. b, As in Fig. 3d, but additional positions were tested for Bpa crosslinking. “<” denotes observed crosslinking adducts with the Grx5-S80-sfGFP substrate. c, As in Extended Data Fig. 8f, but verifying the crosslinked band (red arrowheads) between Bpa-incorporated Tim17 and Grx5-S80-sfGFP. d, As in c, but the crosslinked band (red arrowheads) were validated with native and SDS-denaturing IP with Spot-tag nanobody beads. e, Validation for crosslinks between Mgr2 and Tim17. Left, the same image shown in Fig. 3e (IB for Strep-Mgr2); middle, IB for Tim17; right, Overlaid images with pseudo-coloring to show superposition of the crosslinked Mgr2-Tim17 bands between the two blots. f–h, Additional validations for crosslinking between Mgr2 and Tim17 (f) and between Mgr2 and the Grx5-S80-sfGFP substrate (g and h). Panels f and h show results of SDS-denaturing pulldown (PD) with Strep-Tactin resin. Panel g shows a result of ALFA-tag IP in combination with urea wash as in Extended Data Fig. 8f. i, Water does not cross the membrane when lipids are present in the Tim17–Mgr2 cavity (compare with Fig. 3f). MD simulations with lipids initially placed in the Tim17–Mgr2 cavity block water entry. Image taken after 0.8 μs with Tim17 rendered in green ribbons and Mgr2 in semi-transparent gray ribbons, with water (red/white spheres), phosphorous atoms (dark gold spheres), lipid molecules in the Tim17–Mgr2 cavity (orange/white spheres) rendered as van der Waals spheres. The right panel is the same as the left panel but without displaying the lipids. j, Cells induced for Grx5-S80-sfGFP expression and uninduced cells were lysed, and digitonin-solubilized mitochondrial lysates were subjected to HA-tag (Tim17) IP. Eluates were analyzed by immunoblotting. Band intensity ratios between samples with and without Grx5-S80-sfGFP expression were measured by densitometry. N.D., not determined. Data in a–b and j are representative of three independent experiments. Data in c–h are representative of two independent experiments.
Extended Data Table 1 |.
Cryo-EM data collection, refinement and validation statistics.
| Core TIM23 complex (Tim17/23/44 + Pam16/18) (EMDB-40346; PDB 8SCX) | Endogenous core TIM23 complex (Tim 17/23/44) (EMDB-27825; PDB 8E1M) | |
|---|---|---|
|
| ||
| Data collection and processing | ||
| Magnification | 64,000x | 64,000x |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e−/Å2) | 50 | 50 |
| Defocus range (μm) | −0.8 to −2.1 | −0.8 to −1.6 |
| Pixel size (Å) | 1.05 | 1.05 |
| Symmetry imposed | C1 | C1 |
| Initial particle images (no.) | 689,333 | 1,251,068 |
| Final particle images (no.) | 159,958 | 214,281 |
| Map resolution (Å) | 2.7 | 2.9 |
| FSC threshold | 0.143 | 0.143 |
| Map resolution range (Å) | 2.4–6 | 2.4–6 |
| Refinement | ||
| Initial model used | de novo + PDB-2FXT | PDB-8SCX |
| Model resolution (Å) | 2.9 | 3.0 |
| FSC threshold | 0.5 | 0.5 |
| Map sharpening B factor (Å2) | 92 | 54 |
| Model composition | ||
| Non-hydrogen atoms | 5,753 | 5,691 |
| Protein residues | 760 | 760 |
| Ligands | 2 | 2 |
| B factors (Å2) | ||
| Protein | 69 | 44 |
| Ligand | 67 | 36 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.004 | 0.005 |
| Bond angles (°) | 0.523 | 0.611 |
| Validation | ||
| MolProbity score | 1.28 | 1.53 |
| Clashscore | 5.30 | 7.25 |
| Poor rotamers (%) | 0 | 0 |
| Ramachandran plot | ||
| Favored (%) | 98.13 | 97.33 |
| Allowed (%) | 1.87 | 2.67 |
| Disallowed (%) | 0 | 0 |
| Rama-Z | 1.27 | 0.83 |
| CaBLAM outliers (%) | 0.68 | 0.95 |
Supplementary Material
Acknowledgements
We thank D. Toso for electron microscope operational support, J. Thorner for yeast strains and Pgk1 antibodies, N. Pfanner for Tom40 and Tom22 antibodies, and E. Craig for Pam16 and Pam18 antibodies and plasmids for photocrosslinking. We thank J. Thorner and T.A. Rapoport for critical reading of the manuscript. This work was supported by the Vallee Scholars Program (E.P.), Pew Biomedical Scholars Program (E.P.), and Hellman Fellowship (E.P.). S.I.S. was supported by an NIH training grant (5T32GM008295) and an NSF Graduate Research Fellowship (DGE 1752814). J.C.G. acknowledges support from NIH (R01 GM123169). Computational resources were provided through XSEDE (TG-MCB130173), which is supported by NSF (ACI-1548562). This work also used the Hive cluster, which is supported by the NSF (1828187) and is managed by PACE at Georgia Tech.
Footnotes
Competing interests
Authors declare no competing interests.
Supplementary information
Supplementary Information is available for this paper.
Data availability
EM maps and models are available through EM Data Bank (EMDB) and Protein Data Bank (PDB) under the following accession codes: EMD-40346 and PDB ID 8SCX for the overexpressed core TIM23 complex (Tim17/23/44 + Pam16/18); EMD-27825 and PDB ID 8E1M for the structure of the endogenous core TIM23 complex. Unprocessed gel and blot images are available in Supplementary Figure 1.
References
- 1.Wiedemann N & Pfanner N Mitochondrial Machineries for Protein Import and Assembly. Annu Rev Biochem 86, 685–714, doi: 10.1146/annurev-biochem-060815-014352 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Hansen KG & Herrmann JM Transport of Proteins into Mitochondria. Protein J 38, 330–342, doi: 10.1007/s10930-019-09819-6 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Callegari S, Cruz-Zaragoza LD & Rehling P From TOM to the TIM23 complex - handing over of a precursor. Biol Chem 401, 709–721, doi: 10.1515/hsz-2020-0101 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Schmidt O, Pfanner N & Meisinger C Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11, 655–667, doi: 10.1038/nrm2959 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Araiso Y, Imai K & Endo T Structural snapshot of the mitochondrial protein import gate. FEBS J, doi: 10.1111/febs.15661 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Kubrich M et al. The polytopic mitochondrial inner membrane proteins MIM17 and MIM23 operate at the same preprotein import site. FEBS Lett 349, 222–228, doi: 10.1016/0014-5793(94)00670-9 (1994). [DOI] [PubMed] [Google Scholar]
- 7.Lohret TA, Jensen RE & Kinnally KW Tim23, a protein import component of the mitochondrial inner membrane, is required for normal activity of the multiple conductance channel, MCC. J Cell Biol 137, 377–386, doi: 10.1083/jcb.137.2.377 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Truscott KN et al. A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat Struct Biol 8, 1074–1082, doi: 10.1038/nsb726 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Meinecke M et al. Tim50 maintains the permeability barrier of the mitochondrial inner membrane. Science 312, 1523–1526, doi: 10.1126/science.1127628 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Alder NN, Jensen RE & Johnson AE Fluorescence mapping of mitochondrial TIM23 complex reveals a water-facing, substrate-interacting helix surface. Cell 134, 439–450, doi: 10.1016/j.cell.2008.06.007 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Denkert N et al. Cation selectivity of the presequence translocase channel Tim23 is crucial for efficient protein import. Elife 6, doi: 10.7554/eLife.28324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chacinska A et al. Mitochondrial presequence translocase: switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell 120, 817–829, doi: 10.1016/j.cell.2005.01.011 (2005). [DOI] [PubMed] [Google Scholar]
- 13.van der Laan M et al. Pam17 is required for architecture and translocation activity of the mitochondrial protein import motor. Mol Cell Biol 25, 7449–7458, doi: 10.1128/MCB.25.17.7449-7458.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ieva R et al. Mgr2 functions as lateral gatekeeper for preprotein sorting in the mitochondrial inner membrane. Mol Cell 56, 641–652, doi: 10.1016/j.molcel.2014.10.010 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto H et al. Tim50 is a subunit of the TIM23 complex that links protein translocation across the outer and inner mitochondrial membranes. Cell 111, 519–528, doi: 10.1016/s0092-8674(02)01053-x (2002). [DOI] [PubMed] [Google Scholar]
- 16.Chacinska A et al. Mitochondrial translocation contact sites: separation of dynamic and stabilizing elements in formation of a TOM-TIM-preprotein supercomplex. EMBO J 22, 5370–5381, doi: 10.1093/emboj/cdg532 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mokranjac D, Bourenkov G, Hell K, Neupert W & Groll M Structure and function of Tim14 and Tim16, the J and J-like components of the mitochondrial protein import motor. EMBO J 25, 4675–4685, doi: 10.1038/sj.emboj.7601334 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Silva PR, Schilke B, Hayashi M & Craig EA Interaction of the J-protein heterodimer Pam18/Pam16 of the mitochondrial import motor with the translocon of the inner membrane. Mol Biol Cell 19, 424–432, doi: 10.1091/mbc.e07-08-0748 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mokranjac D et al. Role of Tim50 in the transfer of precursor proteins from the outer to the inner membrane of mitochondria. Mol Biol Cell 20, 1400–1407, doi: 10.1091/mbc.E08-09-0934 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Josyula R, Jin Z, Fu Z & Sha B Crystal structure of yeast mitochondrial peripheral membrane protein Tim44p C-terminal domain. J Mol Biol 359, 798–804, doi: 10.1016/j.jmb.2006.04.020 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Sim SI, Chen Y & Park E Structural basis of mitochondrial protein import by the TIM complex. bioRxiv (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demishtein-Zohary K et al. Role of Tim17 in coupling the import motor to the translocation channel of the mitochondrial presequence translocase. Elife 6, doi: 10.7554/eLife.22696 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demishtein-Zohary K, Marom M, Neupert W, Mokranjac D & Azem A GxxxG motifs hold the TIM23 complex together. FEBS J 282, 2178–2186, doi: 10.1111/febs.13266 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Ting SY, Schilke BA, Hayashi M & Craig EA Architecture of the TIM23 inner mitochondrial translocon and interactions with the matrix import motor. J Biol Chem 289, 28689–28696, doi: 10.1074/jbc.M114.588152 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ting SY, Yan NL, Schilke BA & Craig EA Dual interaction of scaffold protein Tim44 of mitochondrial import motor with channel-forming translocase subunit Tim23. Elife 6, doi: 10.7554/eLife.23609 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiller D, Cheng YC, Liu Q, Walter W & Craig EA Residues of Tim44 involved in both association with the translocon of the inner mitochondrial membrane and regulation of mitochondrial Hsp70 tethering. Mol Cell Biol 28, 4424–4433, doi: 10.1128/MCB.00007-08 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schilke BA, Hayashi M & Craig EA Genetic analysis of complex interactions among components of the mitochondrial import motor and translocon in Saccharomyces cerevisiae. Genetics 190, 1341–1353, doi: 10.1534/genetics.112.138743 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiss C et al. Domain structure and lipid interaction of recombinant yeast Tim44. Proc Natl Acad Sci U S A 96, 8890–8894, doi: 10.1073/pnas.96.16.8890 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamura Y et al. Identification of Tam41 maintaining integrity of the TIM23 protein translocator complex in mitochondria. J Cell Biol 174, 631–637, doi: 10.1083/jcb.200603087 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kutik S et al. The translocator maintenance protein Tam41 is required for mitochondrial cardiolipin biosynthesis. J Cell Biol 183, 1213–1221, doi: 10.1083/jcb.200806048 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qi L et al. Cryo-EM structure of the human mitochondrial translocase TIM22 complex. Cell Res 31, 369–372, doi: 10.1038/s41422-020-00400-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y et al. Structure of the mitochondrial TIM22 complex from yeast. Cell Res 31, 366–368, doi: 10.1038/s41422-020-00399-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dekker PJ et al. The Tim core complex defines the number of mitochondrial translocation contact sites and can hold arrested preproteins in the absence of matrix Hsp70-Tim44. EMBO J 16, 5408–5419, doi: 10.1093/emboj/16.17.5408 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589, doi: 10.1038/s41586-021-03819-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Humphreys IR et al. Computed structures of core eukaryotic protein complexes. Science 374, eabm4805, doi: 10.1126/science.abm4805 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S et al. The Mgr2 subunit of the TIM23 complex regulates membrane insertion of marginal stop-transfer signals in the mitochondrial inner membrane. FEBS Lett 594, 1081–1087, doi: 10.1002/1873-3468.13692 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Gebert M et al. Mgr2 promotes coupling of the mitochondrial presequence translocase to partner complexes. J Cell Biol 197, 595–604, doi: 10.1083/jcb.201110047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura Y et al. Tim23-Tim50 pair coordinates functions of translocators and motor proteins in mitochondrial protein import. J Cell Biol 184, 129–141, doi: 10.1083/jcb.200808068 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geissler A et al. The mitochondrial presequence translocase: an essential role of Tim50 in directing preproteins to the import channel. Cell 111, 507–518, doi: 10.1016/s0092-8674(02)01073-5 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Gevorkyan-Airapetov L et al. Interaction of Tim23 with Tim50 Is essential for protein translocation by the mitochondrial TIM23 complex. J Biol Chem 284, 4865–4872, doi: 10.1074/jbc.M807041200 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Dayan D et al. A mutagenesis analysis of Tim50, the major receptor of the TIM23 complex, identifies regions that affect its interaction with Tim23. Sci Rep 9, 2012, doi: 10.1038/s41598-018-38353-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singha UK et al. Protein translocase of mitochondrial inner membrane in Trypanosoma brucei. J Biol Chem 287, 14480–14493, doi: 10.1074/jbc.M111.322925 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pyrihova E et al. A Single Tim Translocase in the Mitosomes of Giardia intestinalis Illustrates Convergence of Protein Import Machines in Anaerobic Eukaryotes. Genome Biol Evol 10, 2813–2822, doi: 10.1093/gbe/evy215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaudhuri M et al. Tim17 Updates: A Comprehensive Review of an Ancient Mitochondrial Protein Translocator. Biomolecules 10, doi: 10.3390/biom10121643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schneider A Mitochondrial protein import in trypanosomatids: Variations on a theme or fundamentally different? PLoS Pathog 14, e1007351, doi: 10.1371/journal.ppat.1007351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu X & Rapoport TA Translocation of Proteins through a Distorted Lipid Bilayer. Trends Cell Biol 31, 473–484, doi: 10.1016/j.tcb.2021.01.002 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramesh A et al. A disulfide bond in the TIM23 complex is crucial for voltage gating and mitochondrial protein import. J Cell Biol 214, 417–431, doi: 10.1083/jcb.201602074 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin J, Mahlke K & Pfanner N Role of an energized inner membrane in mitochondrial protein import. Delta psi drives the movement of presequences. J Biol Chem 266, 18051–18057 (1991). [PubMed] [Google Scholar]
- 49.Turakhiya U et al. Protein Import by the Mitochondrial Presequence Translocase in the Absence of a Membrane Potential. J Mol Biol 428, 1041–1052, doi: 10.1016/j.jmb.2016.01.020 (2016). [DOI] [PubMed] [Google Scholar]
Methods-only references
- 50.McIsaac RS et al. Fast-acting and nearly gratuitous induction of gene expression and protein depletion in Saccharomyces cerevisiae. Mol Biol Cell 22, 4447–4459, doi: 10.1091/mbc.E11-05-0466 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee ME, DeLoache WC, Cervantes B & Dueber JE A Highly Characterized Yeast Toolkit for Modular, Multipart Assembly. ACS Synth Biol 4, 975–986, doi: 10.1021/sb500366v (2015). [DOI] [PubMed] [Google Scholar]
- 52.Lin A et al. Utilization of a Strongly Inducible DDI2 Promoter to Control Gene Expression in Saccharomyces cerevisiae. Front Microbiol 9, 2736, doi: 10.3389/fmicb.2018.02736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meisinger C, Pfanner N & Truscott KN Isolation of yeast mitochondria. Methods Mol Biol 313, 33–39, doi: 10.1385/1-59259-958-3:033 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Meyer L et al. A simplified workflow for monoclonal antibody sequencing. PLoS One 14, e0218717, doi: 10.1371/journal.pone.0218717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51, doi: 10.1016/j.jsb.2005.07.007 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Tegunov D & Cramer P Real-time cryo-electron microscopy data preprocessing with Warp. Nat Methods 16, 1146–1152, doi: 10.1038/s41592-019-0580-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296, doi: 10.1038/nmeth.4169 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501, doi: 10.1107/S0907444910007493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Afonine PV et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544, doi: 10.1107/S2059798318006551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Terwilliger TC, Sobolev OV, Afonine PV & Adams PD Automated map sharpening by maximization of detail and connectivity. Acta Crystallogr D Struct Biol 74, 545–559, doi: 10.1107/S2059798318004655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21, doi: 10.1107/S0907444909042073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baker NA, Sept D, Joseph S, Holst MJ & McCammon JA Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98, 10037–10041, doi: 10.1073/pnas.181342398 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612, doi: 10.1002/jcc.20084 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Goddard TD et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25, doi: 10.1002/pro.3235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krishnamurthy M et al. Caught in the act: covalent cross-linking captures activator-coactivator interactions in vivo. ACS Chem Biol 6, 1321–1326, doi: 10.1021/cb200308e (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Varadi M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50, D439–D444, doi: 10.1093/nar/gkab1061 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schneiter R et al. Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J Cell Biol 146, 741–754, doi: 10.1083/jcb.146.4.741 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zinser E et al. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol 173, 2026–2034, doi: 10.1128/jb.173.6.2026-2034.1991 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schlame M, Ren M, Xu Y, Greenberg ML & Haller I Molecular symmetry in mitochondrial cardiolipins. Chem Phys Lipids 138, 38–49, doi: 10.1016/j.chemphyslip.2005.08.002 (2005). [DOI] [PubMed] [Google Scholar]
- 70.van Meer G, Voelker DR & Feigenson GW Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9, 112–124, doi: 10.1038/nrm2330 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu EL et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comput Chem 35, 1997–2004, doi: 10.1002/jcc.23702 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW & Klein ML Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 79, 926–935, doi: 10.1063/1.445869 (1983). [DOI] [Google Scholar]
- 73.Horvath SE & Daum G Lipids of mitochondria. Prog Lipid Res 52, 590–614, doi: 10.1016/j.plipres.2013.07.002 (2013). [DOI] [PubMed] [Google Scholar]
- 74.Huang J et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 14, 71–73, doi: 10.1038/nmeth.4067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klauda JB et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843, doi: 10.1021/jp101759q (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Phillips JC et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys 153, 044130, doi: 10.1063/5.0014475 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Darden T, York D & Pedersen L Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. The Journal of Chemical Physics 98, 10089–10092, doi: 10.1063/1.464397 (1993). [DOI] [Google Scholar]
- 78.Balusek C et al. Accelerating Membrane Simulations with Hydrogen Mass Repartitioning. J Chem Theory Comput 15, 4673–4686, doi: 10.1021/acs.jctc.9b00160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zorova LD et al. Mitochondrial membrane potential. Anal Biochem 552, 50–59, doi: 10.1016/j.ab.2017.07.009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38, doi: 10.1016/0263-7855(96)00018-5 (1996). [DOI] [PubMed] [Google Scholar]
- 81.Romo TD, Leioatts N & Grossfield A Lightweight object oriented structure analysis: tools for building tools to analyze molecular dynamics simulations. J Comput Chem 35, 2305–2318, doi: 10.1002/jcc.23753 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Romo TD & Grossfield A LOOS: an extensible platform for the structural analysis of simulations. Annu Int Conf IEEE Eng Med Biol Soc 2009, 2332–2335, doi: 10.1109/IEMBS.2009.5335065 (2009). [DOI] [PubMed] [Google Scholar]
- 83.Katoh K, Misawa K, Kuma K & Miyata T MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066, doi: 10.1093/nar/gkf436 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van der Laan M et al. A role for Tim21 in membrane-potential-dependent preprotein sorting in mitochondria. Curr Biol 16, 2271–2276, doi: 10.1016/j.cub.2006.10.025 (2006). [DOI] [PubMed] [Google Scholar]
- 85.Tucker K & Park E Cryo-EM structure of the mitochondrial protein-import channel TOM complex at near-atomic resolution. Nat Struct Mol Biol 26, 1158–1166, doi: 10.1038/s41594-019-0339-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ieva R et al. Mitochondrial inner membrane protease promotes assembly of presequence translocase by removing a carboxy-terminal targeting sequence. Nat Commun 4, 2853, doi: 10.1038/ncomms3853 (2013). [DOI] [PubMed] [Google Scholar]
- 87.Gotzke H et al. The ALFA-tag is a highly versatile tool for nanobody-based bioscience applications. Nat Commun 10, 4403, doi: 10.1038/s41467-019-12301-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
EM maps and models are available through EM Data Bank (EMDB) and Protein Data Bank (PDB) under the following accession codes: EMD-40346 and PDB ID 8SCX for the overexpressed core TIM23 complex (Tim17/23/44 + Pam16/18); EMD-27825 and PDB ID 8E1M for the structure of the endogenous core TIM23 complex. Unprocessed gel and blot images are available in Supplementary Figure 1.