

Abstract
Streptavidin, a homotetrameric protein with extremely tight biotin binding (Kd ≤ 10-14 M), has been widely used as an affinity reagent. Its utility would be increased by engineering single-chain mutants with a wide spectrum of affinities, more suitable for phage-display and chip technologies. By a circular permutation procedure, we converted streptavidin to a single-chain dimer (SCD) with two biotin-binding sites and introduced random mutations by error-prone PCR. Clones from a phagemid library, expressed as gene-3 fusion proteins on M13 bacteriophage, were panned with biotinylated beads, and SCD genes from affinity-enriched phage were subcloned to produce soluble proteins. Purification of products from the original gene and two mutants by FPLC and analysis by MALDI-TOF MS showed they exist in both dimeric (single-chain) and tetrameric (two-chain) forms, which were further characterized for their binding affinity to biotin-4-fluorescein (B4F) by fluorescence polarization and intensity measurements. Kd′ values for B4F ranged from ≈10-11 to 10-10 M, although Kd values for biotin ranged from 10-6 to 10-5 M. These results point to the possibility of combining an SCD streptavidin mutant with B4F derivatives to create a fluorescence-tagged affinity system with tight but still-reversible interaction that could be used sequentially with ordinary streptavidin–biotin for composite separation or analysis steps.
Keywords: affinity system, circular permutation, phage-display
Streptavidin, a bacterial protein, shares 33% sequence identity with its vertebrate homolog avidin. The two proteins have similar extraordinary noncovalent binding affinity for biotin (dissociation constant, Kd ≈ 10-14 M). However, its lack of glycosylation and sulfur-containing amino acids and its more favorable pI (6 rather than 10) (1) make streptavidin the preferred biotin-binding protein for technological applications. It resists high pH, high temperature, proteolytic enzymes, and organic solvents. This stability, plus the ease of incorporating biotin into biological materials, makes streptavidin a key component in many applications (2–5). For some uses, native tetrameric streptavidin has disadvantages that might be overcome by engineering dimers or monomers.** Disruption of protein quaternary structure, however, often results in lower catalytic activity, reduced binding affinity, and decreased solubility and stability. However, this paper and other work (6–10) clearly demonstrate that soluble functional dimeric and monomeric streptavidins and avidins can be engineered.
Our goal was to create a single-chain dimeric (SCD) protein, capable of subsequent variation and modification in bacteriophage display systems. Streptavidin is a tetramer comprised of two stable subunit dimers. Two monomeric subunits associate very tightly to form the primary dimer, in which the subunit β-barrels have complementary curved surfaces that interact via numerous van der Waals contacts. Two primary dimers then combine to form the tetramer via a dimer–dimer interface (6, 11–13). Our strategy was to crossconnect the two subunits of the primary dimer covalently while at the same time destabilizing the dimer–dimer interface.
The integrity of the dimer–dimer interface is essential to maintain intersubunit contacts to biotin made by W120 (6). This residue makes a major contribution to tight biotin binding (a factor of ≈10-7 in Kd) (7, 14). Replacement of streptavidin W120 by lysine led to successful expression of a two-chain dimeric protein capable of binding biotin reversibly with Kd = 6 × 10-8 M (7). Based on that result, we chose the W120K mutation to destabilize the dimer–dimer interface. By applying circular permutation to each monomer, we designed a bridge between residues 13 and 139 of core streptavidin to connect the initial N and C termini of monomers with a GGGS linker (Fig. 1 a and b). Residues 13 and 139 are located in two different antiparallel β-strands; thus, the bridge combining them forms a β turn. We broke each peptide chain to create four ends and reconnected residues G115 and S69′ to fuse the chains without major distortion of tertiary structure. The remaining two ends became the new N and C termini at E116 and G68′, respectively. The resulting covalently linked primary dimer is the concatenation of the two circularly permuted loops (Fig. 1 b and c) and has a single W120K mutation in its L domain.
Fig. 1.

Construction of circularly permuted single-chain dimeric streptavidin. Two WT genes (a) are cut, spliced, and provided with tetrapeptide linkers (b) L and R (Left and Right) designate the N- and C-terminal halves of this construct. (c) Structural representation with segments colored in order from N to C terminus, blue, green, yellow, and brown. GGGS linkers are depicted as ball-and-stick models. Residue numbering corresponds to WT streptavidin with primes added to distinguish positions in the R domain.
Streptavidin has been used for affinity purification of biotinylated proteins, but its low dissociation constant does not allow the recovery of the proteins unless very harsh conditions are used. Therefore, we engineered streptavidin mutants with lower biotin-binding affinities to allow reversible interactions with biotin and biotinylated derivatives. Discovery of the unexpectedly tight but still-reversible binding of biotin-4-fluorescein (B4F) to our mutants provides the long-sought objective of a multiflavor streptavidin system with intermediate-strength affinities (15).
Materials and Methods
Oligonucleotides were from Integrated DNA Technologies (Coralville, IA) or Amitof (Allston, MA). Unless otherwise noted, competent cells and PfuTurbo DNA polymerase were from Stratagene, plasmids were from Novagen, restriction enzymes and T4 DNA ligase were from New England Biolabs, and chemical reagents were from Sigma. B4F and lucifer yellow cadaverine biotin-X dipotassium salt were from Biotium (Hayward, CA).
Construction of the SCD Gene (P1234). The SCD gene was constructed by amplifying four fragments (P1–P4) of the gene for core streptavidin (16, 17) A13-S139 (Fig. 1 a and b) using PCR with appropriate primers (Fig. 5 and 6, which are published as supporting information on the PNAS web site). These also added the two linkers coding for GGGS, suitably placed initiation and termination codons, and restriction sites (Fig. 6). P1 and P2, after PCR amplification, were digested with BamHI, purified, then ligated to create the gene for the L domain (Fig. 1). This ligation product was used as a template with primers SCDf1 and SCDr2 (Fig. 5) to amplify P12. A similar procedure generated the R domain by using PstI to digest the fragments before ligation and primers SCDf3 and SCDr4 (Fig. 5) to amplify P34. The P12 and P34 genes were digested with BspeI and ligated. Direct PCR amplification of this product produced P1234, which was cloned into pET22b between NdeI and HindIII sites. To incorporate P1234 into the phagemid vector, the flanking sites were converted stepwise to SfiI and NotI, first switching P12 to SfiI and BspeI and P34 to BspeI and NotI (Table 4 and Supporting Text point 1, which are published as supporting information on the PNAS web site), then cloning these in pCR blunt vectors. P34 originally had two BspeI sites: the first at the 5′ end was left unaltered, but the second was changed from TCCGGA to TCCGGT. Modified P12 and P34 were ligated and cloned into pET22b(+), into which an SfiI site was later introduced (Supporting Text point 2).
Subcloning of SCD (SfiI/NotI) into pCANTAB 5 E. pET22b(+) carrying the 810-bp SCD gene was digested with SfiI and then with NotI. The gene was purified by electrophoresis on agarose gel, desalted with G-50, and ≈150 ng was ligated with 250 ng of pCANTAB 5 E, a 4.5-kb phagemid vector from the Amersham Pharmacia recombinant phage antibody system kit, at 16°C overnight (Supporting Text point 3). After heat inactivation at 70°C for 10 min, the reaction mixture was desalted on a Sephadex G-50 column and used to transform chemically competent ABLE K and ABLE C cells. Each phagemid clone was separately digested with BspeI (yielding the expected 5.3-kb linear product), with SfiI + NotI (yielding the 4.5-kb vector plus the 810-bp insert), and with SfiI + NotI + BspeI (yielding the vector and ≈400-bp insert fragments). All clones were confirmed by DNA sequencing.
Random Mutagenesis. The SfiI/NotI DNA fragment encoding the SCD (810-bp) gene was used as the template for error-prone PCR (Supporting Text point 4). Amplified DNA fragments were analyzed by agarose gel electrophoresis. Two more identical rounds of error-prone PCR were performed (Table 5, which is published as supporting information on the PNAS web site). The 810-bp product of the third round was digested with SfiI (50°C for 2 h) and then with NotI (37°C for 2 h), purified from a 1.4% agarose gel with a Qiagen (Chatsworth, CA) gel extraction kit, and cloned into pCANTAB 5 E with T4 DNA ligase.
Phage Display. Phagemid libraries were constructed according to the recombinant phage antibody system manual. Aliquots of ligation mixture of pCANTAB 5 E with SCD from third-round error-prone PCR (2 μl) were transformed with 50 μl of Stratagene TG1 electrocompetent cells at 1,700 volts with an ECM399 electroporator (BTX, San Diego). Fresh LB medium with 20 mM glucose (950 μl) was added into the cuvette and covered and inverted to resuspend the cells. Ten batches (≈9 ml) of electroporated cells were incubated for 1 h at 37°C with shaking and then rescued by plating. The transformation efficiency was 9 × 103 transformants per μg of DNA. The titer of this bacterial library was 162 transformants per ml. Phage rescue from 8 ml (1,296 transformants) gave 3 × 1011 phage particles used for the first round of panning.
Four rounds of affinity selection were performed with biotinylated beads prepared by immobilization of biotinylated BSA on streptavidin-coupled Dynal (Great Neck, NY) beads. Phage stocks were prepared by using M13KO7 as helper phage according to the recombinant phage antibody system manual. Phage from single infected bacterial colonies routinely yielded titers of 1010-1012 colony-forming units per ml.
We used ELISA to screen individual colonies obtained by plating onto SOBAG-medium plates (Supporting Text point 5). Binding of phage to biotinylated wells was detected by horseradish peroxidase/anti-M13 monoclonal conjugate (Amersham Pharmacia) reacted with 1-Step Turbo TMB-ELISA (Pierce) (18). First-round washing was done 10 times with PBS with 0.1%-Tween 20 then 10 times with PBS. Second-round washing was done 20 times the same way and third-round 30 times with 500 μl of PBS + 0.1% Tween-20 and 10 times with 500 μl of PBS to remove the detergent; the fourth round duplicated the first. Phages bound to the biotin beads were eluted by incubating with 100 mM triethylamine for 10 min (pH ≈ 6–8) for the first two rounds and increased to 20 min for the third and fourth rounds, after which the supernatant and separately the beads were immediately neutralized with 1 M Tris·HCl, pH 7.4, and used to infect Escherichia coli TG1 cells. However, because the bead eluates yielded no streptavidin-related sequences, only supernatant fractions were used for subsequent panning rounds.
Expression of SCD Mutant Proteins. The leakiness of the lac promoter and the toxicity of streptavidin prevented SCD protein expression from the phagemids in HB2151 cells. Thus we subcloned the genes into pET22b(+) vectors where the proteins were successfully expressed. E. coli strain BL21(DE3)Gold(pLysS) carrying pET22b(+) as an expression vector encoding SCD mutants under T7 promoter control was grown overnight at 37°C at 250 rpm in 50 ml of LB supplemented with 100 μg/ml ampicillin and 50 μg/ml chloramphenicol. Cultures were diluted 1:20 with fresh LB containing no antibiotics, incubated with shaking at 250 rpm at 37°C for 2 h, then induced with 1 mM isopropyl β-d-thiogalactoside to express the T7 RNA polymerase gene (controlled by the lacUV5 promoter). Cells were collected by centrifugation after incubating at 37°C for 2 h with shaking.
Isolation and Folding. We adapted methods described in Sano et al. (19) and Chu et al. (20) to isolate and fold the proteins. Cells from 1 liter of culture were washed with 200 ml of wash buffer (10 mM Tris·HCl, pH 8/100 mM NaCl/1 mM EDTA), resuspended in 20 ml of 50 mM Tris·HCl, pH 8/0.5% Triton X-100/1 mM EDTA/100 mM NaCl/0.1% NaN3, chilled on ice, and sonicated three times with a Sonifier cell disruptor (Branson). Between each round (three cycles of 30 sec on power setting 4, followed by 30 sec off), samples were centrifuged at 18,000 × g for 10 min and resuspended in the buffer. After the last round, pellets were washed with resuspension buffer minus Triton X-100, then centrifuged at 18,000 × g for 10 min, resuspended in deionized water [0.2–1 ml of H2O/(6 liters of culture)]), and homogenized to form a milky suspension to which 6 M Gu·HCl/50 mM Tris·HCl/pH 7.5 or 7 M Gu·HCl, pH 1.5 [(5.0 ml Gu·HCl/(liters of culture)] was added. The suspension was allowed to equilibrate for several hours at 4°C, then centrifuged at 18,000 × g for 10 min at 4°C to remove insoluble particles. Solubilized protein was then diluted dropwise (one drop per 5 sec) from a syringe (27 gauge, 1½ inches) into folding buffer (50 mM Tris·HCl/150 mM NaCl/5 mM EDTA/0.1 mM PMSF at pH 7.5) at 4°C while stirring forcefully. Folding buffer volume was 1/4 to 1/2 of the induced culture volume. After equilibration overnight (20), the resulting solution was centrifuged to remove insoluble material and concentrated in Millipore Centriprep and Centricon centrifugal filter devices.
Purification/Analysis by FPLC. We used Tricorn high-performance gel filtration columns (Superdex 75 10/300 GL, Amersham Pharmacia Biosciences) with an LCC-501 Plus system. Samples (80–100 μg in 200 μl of folding buffer) were injected into the column preequilibrated with the same buffer and chromatographed at 0.5 ml/min. The column was calibrated by using BSA, streptavidin, ovalbumin, chymotrypsinogen A, and ribonuclease A as standards. Rechromatography was done in the presence of biotin and B4F.
Characterization of SCD Mutants. An Applied Biosystems Voyager-DE STR in the Microchemistry Laboratory at Harvard University (Cambridge, MA) was used for MALDI-TOF MS. Semi-quantitative assay of biotin binding was done with 3H-biotin on the SCD and its mutants C2 and E2, according to ref. 6.
Fluorescence intensity and polarization were measured by using a Beacon 2000 instrument [Panvera (Madison, WI)/Millipore] at 25°C in 150 μl of 50 mM Tris·HCl (fluorescence grade)/150 mM NaCl/5 mM EDTA/0.1 mg/ml BSA [Sigma, no. A-9647; minimum 96% electrophoresis grade, to prevent non-specific binding (21)] at pH 7.5. A 0.22-μm Millipore filter was used to remove fine particulates from the buffer. Measurements were done in borosilicate culture tubes on samples prepared in microcentrifuge tubes. The order of addition was buffer, protein, ligand. Pierce streptavidin (no. 21122) was used as a control.
Protein samples with final monomeric binding-site concentrations as indicated in the legend to Fig. 4 were mixed with 0.16 nM B4F. All samples were prepared in duplicate and incubated at room temperature for 30 min before polarization and intensity measurements at 25°C. K′d values were determined from the intensity vs. protein concentration curves, assuming noncooperative binding and two biotin-binding sites per SCD. For competition experiments, SCDs (9 nM monomer) were incubated with 0.16 nM B4F for 30 min at room temperature and then chased with different concentrations of biotin. B4F dissociation rates were measured by fluorescence polarization (Fig. 4 legend). In reverse-competition experiments, SCDs (9 nM monomer) were first mixed with 45 nM biotin, then chased with 0.16 nM B4F.
Fig. 4.

Fluorescence intensity and polarization behavior of B4F binding to mutant E2. Quenching curves for (a) dimer and (d) tetramer fractions. Protein samples with the final monomer concentrations shown were mixed with 0.16 nM B4F in a total volume of 150 μl. Blue and red symbols correspond to duplicate samples. Competition experiments for (b) dimer and (e) tetramer monitored by polarization. Protein samples (9 nM monomeric binding sites) were first incubated with 0.16 nM B4F and then chased with different concentrations of biotin: blue, no biotin; brown, 492 μM biotin; and green, 725.2 μM biotin. Reverse-competition experiment for (c) dimer and (f) tetramer. Protein samples (9 nM monomeric binding sites) were mixed with 45 nM biotin (red points) then chased with B4F (final concentration, 0.16 nM). Blue points, no biotin. Note the truncated y axes in c and f.
Results
Principal Genetic Constructs. Four segments (P1–P4) derived from the core streptavidin gene were combined to create the gene for a circularly permuted SCD (Materials and Methods). This construct had two GGGS linkers (Fig. 1) and a W120K mutation in P12. After ligation into a pET22b vector between the NdeI and HindIII sites, the gene was amplified and sequenced. We separately mutated the restriction sites in the two halves of the SCD gene, P12 and P34, each cloned into a PCR-blunt vector, because direct conversion of the flanking restriction sites from NdeI/HindIII to SfiI/NotI to clone the gene into pCANTAB 5 E for bacteriophage display would not work due to the repeating sequences. DNA sequencing confirmed cloning of P12 (395 bp, terminal SfiI and BspeI sites) and P34 (415 bp, terminal BspeI and NotI sites). These were purified and ligated to produce the desired P1234 (810 bp) flanked by SfiI/NotI and cloned into a pET22b(+) vector modified with an insert containing the SfiI and NotI sites. Sequencing verified that the product vector carried the intact 810-bp SCD between the SfiI and NotI sites.
The 810-bp SCD insert from the pET22b(+) vector ligated into pCANTAB 5 E was used to transform ABLE K and C cells (designed for cloning plasmids that express toxic gene products). We selected and analyzed 10 colonies from both transformations. All 20 had the expected insert and vector. DNA sequencing of two products from each cell type revealed the desired SCD gene.
Mutant-SCD Genes. To create potentially useful streptavidin variants, we subjected the SCD gene to random mutagenesis by three rounds of error-prone PCR (22). To date, there have been no reports of application of this technology to streptavidin. Display of the mutagenized SCD proteins on the surface of M13 phages was followed by four rounds of panning on biotinylated magnetic beads to capture phage encoding proteins with tighter biotin binding. The phagemid library was rescued from the transformed E. coli TG1 cells with helper phage, and rescued phage were purified by polyethylene glycol (PEG) precipitation. After each round of panning, the PEG-precipitated phage were used to infect E. coli TG1 cells, and the rescue and panning procedures were repeated. Individual colonies were screened by ELISA and mutants selected for protein expression, purification, and analysis.
Table 1 summarizes the SCD and five distinct mutant clones, two of which, C2 and E2, were analyzed in detail. They had 6 and 7 amino acid changes, respectively, compared with the SCD. The Protein Data Bank files [SCDmut.pdb (Data Set 1, which is published as supporting information on the PNAS web site), C2mut.pdb (Data Set 2, which is published as supporting information on the PNAS web site), and E2mut.pdb (Data Set 3, which is published as supporting information on the PNAS web site)] built to examine the structures use a modification of the original crystallographic structure file (1STP) to include the GGGS linkers and the chain breaks shown in Fig. 1. To maintain the original amino acid numbering scheme in these files, the interchain connection between residues G115 from the L domain (chain A) and S69′ from the R domain (chain B) was omitted. This allows programs such as rasmol (available from www.umass.edu/microbio/rasmol/getras.html) to color the two domains separately, and users can select residues of interest with the original numbers and the appropriate chain labels. The linker residues are numbered 140–143 in both domains. Fig. 1c illustrates key features of the SCD structure.
Table 1. Amino acid sequence variants (see Fig. 5 for SCD sequence).
| Sample | Differences from the SCD sequence |
|---|---|
| C6 | S45L |
| E6 | S45L |
| E9 | S45L |
| A8 | S122′P, V31′L |
| D2 | M5L, S45L |
| C2 | K134E, A138T, F29L, G68S, K80′E, G113′S |
| F7 | K134E, A138T, F29L, G68S, K80′E, G113′S |
| E2 | S136T, G142E, F29L, R103′K, G48′D, S52′T, S62′N |
Primed numbers refer to positions in the R domain.
Expression, Folding, and Purification. SCD mutants were expressed efficiently (Fig. 2) by using the tightly regulated T7 expression system (23) that allows toxic genes to be expressed successfully in E. coli strain BL21-Gold(DE3)pLysS, which also lacks the Lon and OmpT proteases that can degrade recombinant proteins. All of the SCD proteins localized to inclusion bodies. Folding was performed at pH 4.5, 6, and 7.5, and the folded proteins displayed binding to 3H-biotin at all three pHs. As monitored on SDS/PAGE, the recovered yield per liter of culture at pH 7.5 was ≈80 μg; at pH 6, 40 μg; and at pH 4.5, 20 μg. Because the initial yield of inclusion bodies was ≈1 mg/(per liter of culture), after folding, ≈90% of the protein remained aggregated even at pH 7.5.
Fig. 2.

Expression of SCD proteins in E. coli carrying pET22b(+) monitored on SDS/PAGE (4–20% VWR i-gel). (a) SCD, (b) C2, and (c) E2, as marked. Lane headings: M, protein marker (bands from top to bottom in kDa: 94, 67, 43, 30, 20, and 14.4); U, uninduced culture; I, induced culture; and 0, 2, hrs after start of induction. Expression was at 37°C and 1 mM IPTG from 500 ml of culture volume. Inclusion bodies from 500 μl of culture were loaded on the gel for SCD and from 250 μl for C2 and E2. Marker lanes contained 58 μg of protein for a and c and 29 μg for b.
Refolded proteins were purified on gel-filtration columns by FPLC (Materials and Methods). Fig. 3a shows a representative chromatogram (for mutant E2) with three principal peaks: one eluting at the void volume (≈8 ml), presumably aggregated protein; a second eluting at 10.90 ml, corresponding to Mr = ≈55–60 kDa, a tetramer of two noncovalently associated SCDs; and a third eluting at 13.25 ml, as expected for an SCD. The final peak in this chromatogram (at ≈17 ml) corresponds to an unidentified low-molecular-weight component.
Fig. 3.

FPLC of mutant E2 fractions. T and D refer to tetramer and dimer. (a) FPLC of crude E2. b and c show results of rechromatography of peaks without biotin from the crude fraction shown in a for E2T and E2D, respectively. The rechromatographed E2D peak from c was incubated with added biotin and rerun on the column, yielding only a tetramer peak (d).
Characterization of SCD, C2, and E2 by FPLC. The dimeric and tetrameric peaks from the first crude samples (Fig. 3a) were reapplied to the Superdex column in the absence and presence of excess free biotin. The tetrameric peaks reeluted as pure tetramers in both cases (Fig. 3b). The rerun dimeric peaks eluted as dimer–tetramer mixtures in the absence of biotin (Fig. 3c). However, in every case, the dimeric peaks eluted as tetramers on columns preequilibrated with buffer containing excess free biotin (Fig. 3d). To test the effect of B4F on the dimer–tetramer equilibrium (to better analyze fluorescence data; see below), we saturated samples of both dimer and tetramer fractions of mutants C2 and E2, then ran them on a column preequilibrated with 10 nM B4F. For E2, all protein from both fractions eluted in the tetramer peak. In the case of C2, the tetramer fraction gave only a tetramer peak, but the dimer fraction gave both tetramer and dimer peaks (in 2:1 ratio). These results show that binding of biotin or B4F favors tetramer formation, presumably because of the stabilization afforded by the interaction between W120 of the adjacent dimer with the bound ligand. This is consistent with other studies showing that ligand binding favors streptavidin oligomerization (10, 24–26).
MALDI-TOF MS Molecular Weights. Theoretical weights of SCD, C2, and E2 are 28,376.8, 28,434.7, and 28,499.9 Da, respectively, calculated by using protparam, http://us.expasy.org/tools/protparam.html. These masses include the N-terminal met and C-terminal (his)6 tag and matched the masses of the major MALDI peaks (Fig. 7, which is published as supporting information on the PNAS web site). All spectra also had small peaks (intensities <5–10% of the main peaks) near 57 kDa, presumably from subunits that remain noncovalently associated in MALDI-TOF MS (27, 28).
Ligand Binding. B4F detects streptavidin concentrations as low as 200 pM accurately with fluorescence polarization (21). We used both intensity and polarization data from B4F to study its interactions with streptavidin and the SCD mutants. Competition experiments were used to determine the binding affinity of biotin. All of the SCD proteins partially quench B4F fluorescence (Table 2).
Table 2. The change in the total fluorescence intensities of B4F upon addition of excess SCD, C2 dimer, C2 tetramer, and E2 tetramer.
| Protein | Total intensity at 0.18 nM protein (monomer) | Total intensity at 0.25 nM protein (monomer) |
|---|---|---|
| SCD | 259 | 198 |
| C2D | 317 | 272 |
| C2T | 260 | 208 |
| E2T | 325 | 250 |
| No protein | 425 | 425 |
The E2 dimer was not tested with 0.18 and 0.25 nM, but with 1 nM monomer (total intensity = 290); it also showed quenching.
Fig. 4 a and d show binding curves obtained by fluorescence intensity titration of B4F with dimeric and tetrameric E2. Dissociation constants (Table 3) for B4F (Kd′) were estimated assuming a simple one-step binding reaction. When the fluorescence intensity is half maximally quenched, the dye is half-bound:
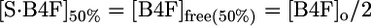 |
Thus,
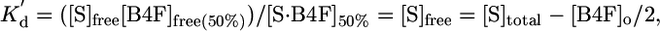 |
where [S·B4F]50% = molarity of bound B4F at half-saturation; [B4F]free(50%) = molarity of free B4F at half-saturation; [B4F]o = molarity of total B4F; [S]free = molarity of unoccupied biotin-binding sites; and [S]total = total protein monomer concentration.
Table 3. Dissociation constants for ligand binding to streptavidin, SCD, and mutants C2 and E2.
| Mutant | K′d, M | Kd, M | Kd/K′d |
|---|---|---|---|
| streptavidin | 1.4 × 10-10 | 4 × 10-14* | 2.86 × 10-4 |
| SCD | 1.2 × 10-10 | 6.72 × 10-6 | 5.60 × 104 |
| C2 dimer | 1.6 × 10-10 | 1.33 × 10-5 | 8.31 × 104 |
| C2 tetramer | 8 × 10-11 | 3.3 × 10-6 | 4.13 × 104 |
| E2 dimer | 3 × 10-11 | 1.5 × 10-6 | 5.00 × 104 |
| E2 tetramer | 8 × 10-11 | 1.28 × 10-5 | 1.60 × 105 |
K′d is the dissociation constant for B4F, and Kd is the dissociation constant for biotin (as estimated from competition experiments).
Data from ref. 1
Fig. 4 b and e show competition experiments for dimeric and tetrameric E2, and Fig. 4 c and f show the corresponding reverse-competition experiments. (Fig. 8, which is published as supporting information on the PNAS web site, shows additional reverse-competition experiments).
Competition experiments use previously determined Kd′ values for B4F (Table 3 and Fig. 4 a and d) to measure the dissociation constants of biotin for SCD, C2, and E2. SCD proteins were first incubated with B4F and then chased with biotin. In reverse-competition experiments, protein samples were first mixed with biotin and then chased with B4F. These confirmed the reversibility of biotin binding and the stronger affinity of the SCD proteins for B4F. Note the distinctly slower kinetics for displacement of biotin from the E2 tetramer (Fig. 4 f vs. c).
Parallel experiments with another biotin-dye conjugate, lucifer yellow cadaverine biotin-X dipotassium salt (LYB), showed similar behavior to that observed with B4F, although in this case quenching was much less. The Kd′ values for LYB with E2 dimer and E2 tetramer are both ≈10-8 M, i.e., >100-fold weaker than for B4F.
Discussion
SCD has been successfully engineered and expressed in E. coli. It has a dissociation constant of ≈10-5 M for biotin and ≈10-10 Mfor B4F. Random mutation via error-prone PCR produces mutants with higher affinity for biotin or its derivatives that can be selected by phage display.†† Attempts to express SCD and its mutants by conventional methods from phagemids grown in HB2151 cells failed, most likely because of streptavidin toxicity. Subcloning of SCD and its mutants in a system with tight expression control, however, allowed protein production (18). As seen from the FPLC experiments, SCD mostly tetramerizes; the mutant proteins exist as mixtures of dimers and tetramers, but upon saturation with biotin, they also tetramerize.
The binding results are totally unexpected. While streptavidin has ≈104-fold preference for biotin over B4F, the SCD and all of the mutant forms tested bind B4F 104-105 times stronger than biotin (Table 3). Thus, the relative preference for biotin and B4F changes by >108. This has enormous practical consequences for the utility of the new derivatives. Note that some of the SCD constructs bind B4F even more strongly than streptavidin does.
Interpretation of our binding results requires caution. In addition to the dimer–tetramer equilibrium, this system is complicated by the fundamental asymmetry of the SCD (Supporting Text point 6 and Fig. 9, which is published as supporting information on the PNAS web site). In solutions containing SCDs and ligand, there can be up to 24 protein species, 21 of which have bound ligand (Supporting Text point 6 and Figs. 10–12, which are published as supporting information on the PNAS web site). The apparent Kd values in Table 3 ignore this complex set of multiple equilibria. The L and R domains of the SCD (Fig. 1) are nonequivalent, and the use of a single W120K mutation to destabilize the dimer–dimer interface increases the basic asymmetry (Figs. 10 and 11 and Supporting Text point 6). The mutations (Table 1) further increase these differences. Tetramerization introduces an additional binding pocket asymmetry in the SCDs, because each dimer has only a single W120 that can insert into a binding site across the dimer–dimer interface and contribute to ligand stabilization. SCD tetramers will have two distinct types of biotin-binding sites; each with a characteristic Kd value. In experiments where 9 nM C2 dimer was mixed with 0.16 nM B4F and then chased with 10–1 mM biotin (data not shown), some of the fluorescence polarization data appear to demonstrate the presence of strong and weak sites, because the polarization decreases in two steps, at ≈ 10 μM and at ≈1 mM biotin concentrations. However, because of the quenching and potential energy transfer between fluoresceins bound to the same dimer or tetramer, a detailed interpretation of the fluorescence polarization binding data is not achievable (Supporting Text point 7).
To optimize SCDs for applications, additional modifications are desirable. For example, the tendency for tetramerization might be eliminated by making changes to key residues at the dimer–dimer interface, as we have done in a related project to create a soluble two-chain dimeric streptavidin (18). From the structure of C2 and E2 (Fig. 9) we can propose mutations that can be accepted by an SCD, allowing it to fold, to have reasonable binding affinity, and to be soluble. The monomer–monomer interface must apparently remain intact; no mutations occurred there. This also correlates with experience with the two-chain dimer Stv-43 (6). The dimer–dimer interface also seems resistant to change (only one peripheral mutation occurred there, S62′N in E2), suggesting that it has a role in determining which mutants are successfully expressed, folded, and purified. As expected, changes rarely occur in or near the biotin-binding site. Most of the mutations that we have isolated occur on the original molecular surface of the tetramer, suggesting that such sites could easily tolerate additional engineered changes.
The unexpected tight binding of B4F to the SCDs begs for an explanation. In preliminary computational docking studies to a WT monomer (data not shown), we found that when the biotin moiety is inserted into its normal location in the binding pocket, the fluorescein group of B4F can sit directly above the biotin in a position analogous to the location of the W120 side chain in the normal wt tetramer. Thus, B4F may be a ligand that brings part of its binding pocket along with it. In our dimeric SCDs, both binding sites lack the W120, so that such a binding mode is feasible. As tetramers form, presumably half of the sites acquire the W120, so a more complex situation must prevail. Interestingly, B4F does not destabilize tetramers.
Supplementary Material
Acknowledgments
We thank Dr. Sandor Vajda for initial suggestions and Dr. Zhiping Weng for the final design of the SCD, including its Protein Data Bank structure; Paola Favaretto for modifying the structure for C2 and E2; and Taner Kaya for performing ligand-docking calculations. This work was supported by grants from Defense Advanced Research Planning Agency and Sequenom, Inc.
Author contributions: F.M.A., Y.Y., S.C.M., and C.R.C. designed research; F.M.A. and Y.Y. performed research; F.M.A., Y.Y., S.C.M., and C.R.C. analyzed data; and F.M.A., S.C.M., and C.R.C. wrote the paper.
Abbreviations: SCD, single-chain dimer; B4F, biotin-4-fluorescein.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AY884152–AY884154).
Footnotes
We note, however, that extremely high-affinity mutants might not be recovered in this procedure due to near-irreversible binding of biotin from the culture medium.
Throughout this paper, we use the terms monomer(ic), dimer(ic), and tetramer(ic) to refer to the number of biotin-binding domains present in a molecule, regardless of the number of polypeptide chains it has.
References
- 1.Green, N. M. (1990) Methods Enzymol. 184, 51-67. [DOI] [PubMed] [Google Scholar]
- 2.Wilchek, M. & Bayer, E. A. (1990) Methods Enyzmol. 184, 5-45. [DOI] [PubMed] [Google Scholar]
- 3.Wilbur, D., Pathare, P., Hamlin, D., Stayton, P., To, R., Klumb, L., Buhler, K. & Vessella, R. (1999) Biomol. Eng. 16, 113-118. [DOI] [PubMed] [Google Scholar]
- 4.Hamblett, K. J., Kegley, B. B., Hamlin, D. K., Chyan, M. K., Hyre, D. E., Press, O. W., Wilbur, D. S. & Stayton, P. S. (2002) Bioconjug. Chem. 13, 588-598. [DOI] [PubMed] [Google Scholar]
- 5.Demidov, V. V., Bukanov, N. O. & Frank-Kamenetskii, D. (2000) Curr. Issues Mol. Biol. 2, 31-35. [PubMed] [Google Scholar]
- 6.Sano, T., Vajda, S., Smith, C. L. & Cantor, C. R. (1997) Proc. Natl. Acad. Sci. USA 94, 6153-6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laitinen, O. H., Airenne, K. J., Marttila, A. T., Kulik, T., Porkka, E., Bayer, E. A., Wilchek, M. & Kulomaa, M. S. (1999) FEBS Lett. 461, 52-58. [DOI] [PubMed] [Google Scholar]
- 8.Qureshi, M. H., Yeung, J. C., Wu, S. C. & Wong, S. L. (2001) J. Biol. Chem. 276, 46422-46428. [DOI] [PubMed] [Google Scholar]
- 9.Qureshi, M. & Wong, S. (2002) Protein Expr. Purif. 25, 409. [DOI] [PubMed] [Google Scholar]
- 10.Laitinen, O. H., Nordlund, H. R., Hytonen, V. P., Uotila, S. T., Marttila, A. T., Savolainen, J., Airenne, K. J., Livnah, O., Bayer, E. A., Wilchek, M., et al. (2003) J. Biol. Chem. 278, 4010-4014. [DOI] [PubMed] [Google Scholar]
- 11.Sano, T., Vajda, S., Reznik, G. O., Smith, C. L. & Cantor, C. R. (1996) Ann. N.Y. Acad. Sci. 799, 383-390. [DOI] [PubMed] [Google Scholar]
- 12.Weber, P. C., Ohlendorf, D. H., Wendoloski, J. J. & Salemme, F. R. (1989) Science 243, 85-88. [DOI] [PubMed] [Google Scholar]
- 13.Hendrickson, W. A., Pahler, A., Smith, J. L., Satow, Y., Merritt, E. A. & Phizackerley, R. P. (1989) Proc. Natl. Acad. Sci. USA 86, 2190-2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sano, T. & Cantor, C. R. (1995) Proc. Natl. Acad. Sci. USA 92, 3180-3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reznik, G. O., Vajda, S., Sano, T. & Cantor, C. R. (1998) Proc. Natl. Acad. Sci. USA 95, 13525-13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sano, T., Pandori, M. W., Chen, X., Smith, C. L. & Cantor, C. R. (1995) J. Biol. Chem. 270, 28204-28209. [DOI] [PubMed] [Google Scholar]
- 17.Pahler, A., Hendrickson, W. A., Kolks, M. A., Argarana, C. E. & Cantor, C. R. (1987) J. Biol. Chem. 262, 13933-13937. [PubMed] [Google Scholar]
- 18.Aslan, F. M. (2005) Ph.D. thesis (Boston University, Boston).
- 19.Sano, T., Smith, C. L. & Cantor, C. R. (1997) Methods Mol. Biol. 63, 119-128. [DOI] [PubMed] [Google Scholar]
- 20.Chu, V., Freitag, S., Le Trog, I., Stenkamp E., R. & Stayton, P. (1998) Protein Sci. 7, 848-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kada, G., Kaiser, K., Falk, H. & Gruber, H. J. (1999) Biochim. Biophys. Acta 1427, 44-48. [DOI] [PubMed] [Google Scholar]
- 22.Cirino, P., Mayer, K. & Umeno, D. (2003) Methods Mol. Biol. 231, 3-10. [DOI] [PubMed] [Google Scholar]
- 23.Studier, F. W., Rosenberg, A. H., Dunn, J. J. & Dubendorff, J. W. (1990) Methods Enzymol. 185, 60-89. [DOI] [PubMed] [Google Scholar]
- 24.Pazy, Y., Eisenberg-Domovich, Y., Laitinen, O. H., Kulomaa, M. S., Bayer, E. A., Wilchek, M. & Livnah, O. (2003) J. Bacteriol. 185, 4050-4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laitinen, O. H., Marttila, A. T., Airenne, K. J., Kulik, T., Livnah, O., Bayer, E. A., Wilchek, M. & Kulomaa, M. S. (2001) J. Biol. Chem. 276, 8219-8224. [DOI] [PubMed] [Google Scholar]
- 26.Nordlund, H. R., Laitinen, O. H., Hytonen, V. P., Uotila, S. T., Porkka, E. & Kulomaa, M. S. (2004) J. Biol. Chem. 279, 36715-36719. [DOI] [PubMed] [Google Scholar]
- 27.Juhasz, P. & Biemann, K. (1994) Proc. Natl. Acad. Sci. USA 91, 4333-4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bordini, E. & Hamdan, M. (1999) Rapid Commun. Mass Spectrom. 13, 1143-1151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.