

Abstract
F0F1-ATP synthases catalyze proton transport-coupled ATP synthesis in bacteria, chloroplasts, and mitochondria. In these complexes, the ɛ-subunit is involved in the catalytic reaction and the activation of the enzyme. Fluorescence-labeled F0F1 from Escherichia coli was incorporated into liposomes. Single-molecule fluorescence resonance energy transfer (FRET) revealed that the ɛ-subunit rotates stepwise showing three distinct distances to the b-subunits in the peripheral stalk. Rotation occurred in opposite directions during ATP synthesis and hydrolysis. Analysis of the dwell times of each FRET state revealed different reactivities of the three catalytic sites that depended on the relative orientation of ɛ during rotation. Proton transport through the enzyme in the absence of nucleotides led to conformational changes of ɛ. When the enzyme was inactive (i.e. in the absence of substrates or without membrane energization), three distances were found again, which differed from those of the active enzyme. The three states of the inactive enzyme were unequally populated. We conclude that the active–inactive transition was associated with a conformational change of ɛ within the central stalk.
Keywords: ɛ-subunit, F0F1-ATP synthase, fluorescence resonance energy transfer, rotation, single-molecule enzymology
Introduction
ATP is synthesized by F-type ATP synthases in the plasma membrane of bacteria, the inner membrane of mitochondria, and the thylakoid membrane of chloroplasts. These multisubunit enzymes consist of two major complexes with distinct functionalities: the hydrophobic membrane-integrated F0 part (subunits ab2c10 in Escherichia coli) is involved in proton transport across the membrane, and the hydrophilic F1 part (subunits α3β3γδɛ) contains the three catalytic nucleotide and phosphate binding sites (Yoshida et al, 2001; Capaldi and Aggeler, 2002; Weber and Senior, 2003; Kinosita et al, 2004). F0 and F1 parts are connected by a central and a peripheral stalk, as revealed by electron micrographs (Böttcher et al, 1998; Wilkens and Capaldi, 1998; Karrasch and Walker, 1999). The central stalk is built by the γ- and ɛ-subunit of F1, and parts of both subunits are interfacing the ring of c-subunits of F0. ATP synthesis takes place at the catalytic sites on the β-subunits. The Gibbs free energy derived from the electrochemical potential of the proton gradient across the membrane is transduced via conformational changes to the catalytic nucleotide binding sites (Junge, 1999; Capaldi and Aggeler, 2002; Weber and Senior, 2003), where it is used to alter the binding affinities of substrates and products as proposed by the ‘binding change mechanism' (Boyer, 1998). During ATP synthesis, translocation of protons through F0 drives rotation of the ‘rotor'-subunits γɛc10 (blue in Figure 1A) relative to the ‘stator'-subunits α3β3δab2 (orange in Figure 1A) (Zhou et al, 1997; Kaim et al, 2002; Boldt et al, 2004; Diez et al, 2004b). The actual conformational state of each of the three catalytic sites depends on the orientation of the ‘rotor' subunits relative to the αβ-pairs. Rotation of the γ-subunit has been observed with molecule ensembles (Duncan et al, 1995; Sabbert et al, 1996) and with single immobilized F1 subcomplexes (Noji et al, 1997; Yasuda et al, 2001). Rotation of the γ-subunit relative to the b-subunit (stator) has been observed with a single membrane-bound F0F1-ATP synthase in liposomes using fluorescence resonance energy transfer (FRET) (Börsch et al, 2002). Recently, a three-stepped rotation of the γ-subunit was reported during coupled catalysis with opposite directions during ATP synthesis and ATP hydrolysis (Diez et al, 2004b).
Figure 1.
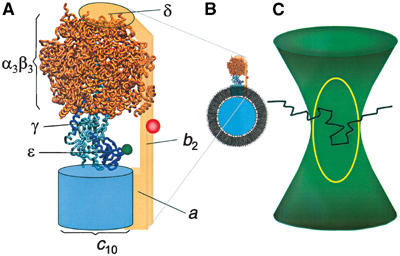
Model of the H+-ATP synthase from E. coli as derived from electron microscopic data (Böttcher et al, 2000; Rubinstein et al, 2003), the homology model of F1 (Engelbrecht and Junge, 1997), and the structure of the γɛ-complex (Rodgers and Wilce, 2000). (A) The H+-ATP synthase is labeled with the FRET donor TMR (green) at the ɛ-subunit and the FRET acceptor Cy5bis (red) at the b-subunits. ‘Rotor' subunits are depicted in blue and ‘stator' subunits are orange. (B) F0F1 incorporated into a liposome membrane. The diameter of the F1 part is 10 nm and that of the liposome is 120 nm. Liposomes used for FRET analysis contained one F0F1. (C) Photon bursts from the FRET-labeled F0F1 are observed when the liposome traverses the confocal detection volume (yellow line) within the excitation focus (green) of the laser. A schematic diffusion pathway of the liposome through the confocal volume is indicated (black line).
Not much data have been reported about movements of the ɛ-subunit. Crosslinking of the γ- and ɛ-subunits does not impair ATP hydrolysis in molecule ensembles (Tang and Capaldi, 1996; Aggeler et al, 1997; Bulygin et al, 1998), indicating that ɛ is part of the rotor. Rotation of the ɛ-subunit in single immobilized F1 parts during ATP hydrolysis was demonstrated (Häsler et al, 1998; Kato-Yamada et al, 1998). However, these experiments did not reveal a 120° stepped rotation as for the γ-subunit and give no conclusive evidence for a joint rotation of the γɛ-complex. The ɛ-subunit is also involved in the activation of the enzyme, and the active–inactive transition is a complex process that depends on several parameters including the number and type of the enzyme-bound nucleotides, energization of the membrane, and the dissociation of ɛ (Smith and Sternweis, 1977; Fischer et al, 2000). Finally, the ɛ-subunit might play an important role in switching between the ATP synthesis and ATP hydrolysis mode (Tsunoda et al, 2001). In this work, we used a single-molecule FRET approach to investigate the movements of the ɛ-subunit in the membrane-bound H+-ATP synthases from E. coli during proton transport-driven ATP synthesis and hydrolysis, and we discriminated movements during catalysis from those during activation.
Results
Labeling of F0F1 and fluorescence measurements with single enzymes
To investigate the movements of the ɛ-subunit, we prepared H+-ATP synthase from E. coli (referred to as F0F1 in the following) in freely diffusing lipid vesicles. We monitored rotation of the ɛ-subunit relative to the b-subunits during ATP synthesis and ATP hydrolysis using single-molecule FRET. The ‘rotor'-subunit ɛ of F1 was labeled with tetramethylrhodamine (TMR) as the FRET donor to yield F1-ɛH56C-TMR. A bisfunctional cyanine dye, Cy5bis (Diez et al, 2004b), was used as the FRET acceptor, which crosslinked the dimeric ‘stator'-subunits b2 of F0F1 to yield F0-bQ64C-Cy5bis-F1. The SDS gel (Figure 2) shows F0F1 before labeling (lane 1a), and F0F1 after labeling with Cy5bis (lane 2a), indicating the crosslinking of subunit b (marked b2). The fluorogram (lane 2b) reveals selective labeling of b2. Lane 3a shows F1 after labeling with TMR; the fluorogram (lane 3b) indicates selective labeling of ɛ. To obtain specifically double-labeled F0F1, the following procedure was used (for details, see Materials and methods): F0-bQ64C-Cy5bis-F1 was incorporated into liposomes, and the F1 part was removed. The liposomes with the membrane-bound Cy5bis-labeled F0 part were reassembled with F1-ɛH56C-TMR. Proteoliposomes with double-labeled F0F1 were obtained (see Figure 1B) after removal of excess F1-ɛH56C-TMR. In control experiments, we used unlabeled enzymes for incorporation into liposomes, removal of F1, and reassembling with new unlabeled F1. At each step of the preparation of both sample and control, the rates of ATP hydrolysis (vH) and ATP synthesis (vS) were measured. The results are summarized in Table I and show that the rates of ATP hydrolysis and ATP synthesis are not changed when the enzyme is labeled either with TMR at the ɛ-subunit or with Cy5bis at the b-subunit. The ATP synthesis activity of F0-bQ64C-Cy5bis-F1 is decreased by approximately 50% after removal and rebinding of F1; however, there is no difference in activity between labeled and unlabeled F1. Double-labeled enzymes show catalytic rates of vH=57±19 s−1 corresponding to a mean turnover time of tH=18±4 ms, and vS=21±4 s−1 corresponding to a mean turnover time of tS=48±8 ms. Addition of N,N′-dicyclohexylcarbodiimide (DCCD), which binds covalently to the c-subunits, inhibits both ATP synthesis and ATP hydrolysis.
Figure 2.
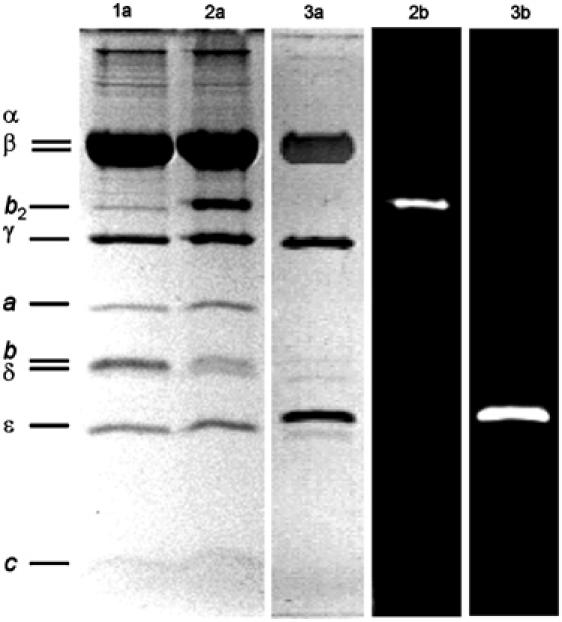
SDS–PAGE (13%) of F1 and F0F1. Lanes marked with (a) show the protein stained with Coomassie blue and those marked with (b) show the corresponding fluorograms. Lane 1: F0-bQ64C-F1; lane 2: F0-bQ64C-Cy5bis-F1 showing crosslinking of subunits b by Cy5bis; lane 3: F1-ɛH56C-TMR.
Table 1.
Rates of ATP hydrolysis (vH) and ATP synthesis (vS) of liposome-bound F0F1 at 23°C, measured at different states during the preparation procedure
| Enzymes |
vH (s−1) |
vS (s−1) |
||
|---|---|---|---|---|
| +DCCD | +DCCD | |||
| F0F1 ‘wild type' | 91±2 | ND | 87±11 | ND |
| F0-bQ64C-F1 | 71±4 | ND | 60±1 | ND |
| F0-bQ64C-Cy5bis-F1 | 75±9 | ND | 48±1 | ND |
| F0-bQ64C (after removal of F1) | 0 | ND | 0 | ND |
| F0-bQ64C-Cy5bis (after removal of F1) | 0 | ND | 0 | ND |
| F0-bQ64C-F1, after removal of F1 and reassembling with F1-ɛH56C | 55±8 | 4±2 | 22±3 | 4±3 |
| F0-bQ64C-F1, after removal of F1 and reassembling with F1-ɛH56C-TMR | 55±9 | 5±1 | 21±3 | 4±2 |
| F0-bQ64C-Cy5bis after removal of F1 and reassembling with F1-ɛH56C | 52±8 | 5±1 | 23±3 | 5±2 |
| F0-bQ64C-Cy5bis after removal of F1 and reassembling with F1-ɛH56C-TMR | 57±19 | 5±2 | 21±4 | 4±0 |
| ND: not determined. | ||||
Single-molecule FRET measurements of the membrane-integrated H+-ATP synthases with selectively attached fluorophores were carried out in a confocal setup with two-channel detection (Börsch et al, 2002). TMR was excited at 532 nm, and the emission of both fluorophores was simultaneously recorded. Fluorescence of TMR was detected in the spectral range between 545 and 610 nm (FD), and of Cy5bis above 660 nm (FA). Well-separated photon bursts with count rates up to 100 photons/ms were observed when a single liposome containing one double-labeled F0F1 diffused through the confocal detection volume as shown schematically in Figure 1C. Time-dependent fluctuations of the fluorescence intensities in both channels reflect the stochastic pathway of the liposome through the confocal volume. This volume can be approximated by a three-dimensional Gaussian excitation intensity distribution (green in Figure 1C) and a confocal detection volume of the fluorescence emission (yellow in Figure 1C). The FRET efficiency EFRET=FA/(FA+FD) calculated from the ratio of corrected fluorescence intensities in the donor channel (FD) and the acceptor channel (FA) is independent of the localization within the confocal volume. Therefore, a constant ratio of the fluorescence intensities throughout a photon burst indicated a constant EFRET that corresponds to a fixed distance between the two fluorophores. The distance can be calculated according to the Förster theory of FRET (Förster, 1948; Van der Meer et al, 1994).
Fluorescence trajectories and dynamics of ɛ-subunit movement during catalysis
In the presence of 1 mM AMPPNP (an ATP analog that is bound at the catalytic sites but is not hydrolyzed), the photon bursts simultaneously exhibited large changes in fluorescence intensities of FRET donor and acceptor due to the diffusion through the confocal volume as shown in Figure 3A (FD, green trajectories; FA, red trajectories). The FRET efficiency EFRET calculated from these trajectories (blue trace) remained unchanged within the bursts, indicating a constant distance of the ɛ-subunit with respect to the b-subunits during the observation time. We observed three different FRET states, one with low FRET efficiency (L), one with medium FRET efficiency (M), and one with high FRET efficiency (H). In addition, Figure 3A shows a photon burst resulting from an enzyme carrying only the donor fluorophore (D). In all trajectories, the mean background counts were subtracted in each channel, and, therefore, the acceptor channel count rate of a donor-only-labeled enzyme is zero as well as the FRET efficiency.
Figure 3.
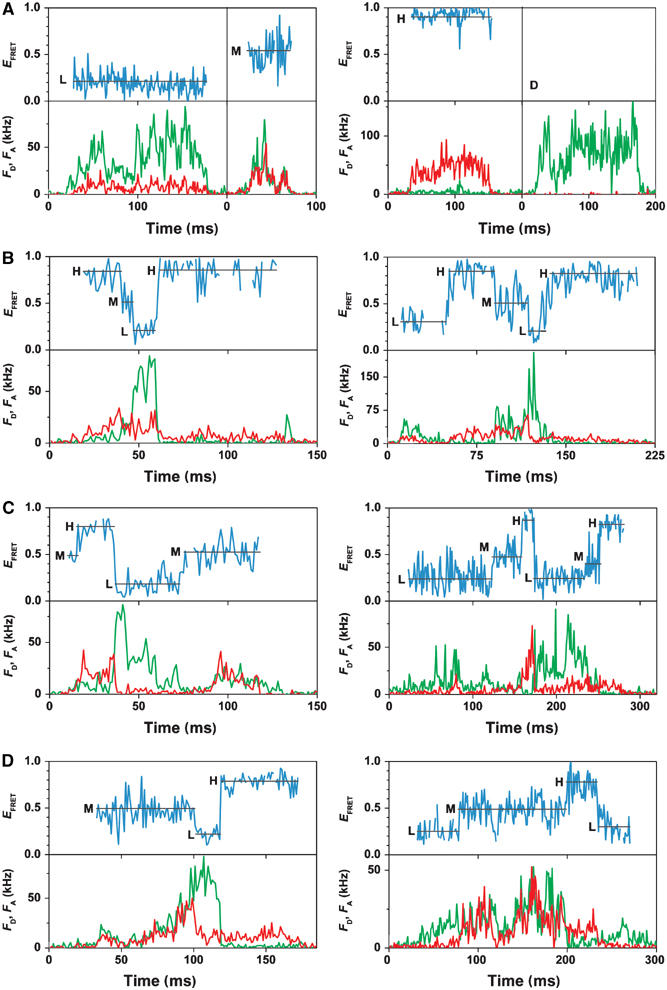
Photon bursts of FRET-labeled F0F1 in liposomes. Corrected fluorescence intensity traces of the donor, FD, and acceptor, FA, are green and red, respectively. The FRET efficiencies (EFRET) calculated from these traces are shown as blue traces. For each FRET level, the arithmetic mean value is calculated (black line). The three FRET states are labeled L, M, and H. (A) Traces in the presence of AMPPNP showing constant FRET efficiencies and a donor-only-labeled F0F1 in a liposome (D). (B) Traces during ATP hydrolysis showing stepwise changes of FRET levels in the sequence → L → H → M → L. (C) Traces during ATP synthesis showing stepwise changes of FRET levels in the sequence → L → M → H → L. (D) Traces during proton transport showing both sequences.
During ATP hydrolysis in the presence of 1 mM ATP, large fluctuations in fluorescence intensities of donor and acceptor were observed within single photon bursts (Figure 3B). Three different FRET levels were found with similar FRET efficiencies as in the presence of AMPPNP, which were called also L, M, and H. However, we detected often more than one FRET level per photon burst. The time traces of the FRET efficiency (blue traces) show distinct FRET levels (between 5 and 150 ms) but in contrast to the AMPPNP case, there were rapid changes to the next FRET level. The stepping occurred faster than the time resolution of these measurements (1 ms). We analyzed photon bursts that showed at least one FRET transition (two FRET levels). In 454 photon bursts, we found FRET level transitions with the sequence → H → M → L → H… for more than 81% of the bursts, and 19% showed apparently the wrong sequence (details of the statistics are provided as Supplementary data). From these data, it can be concluded that the distances between the ɛ-subunit and the b-subunits change sequentially in one direction, and this implies a three-stepped rotary movement of ɛ relative to b.
To investigate ATP synthesis, a pH difference plus an additional electric potential difference was generated across the liposome membrane in the presence of 100 μM Mg-ADP and 5 mM phosphate (see Materials and methods). We measured the rate of ATP synthesis with luciferin/luciferase under these conditions in ensemble experiments, and found ATP synthesis activity up to 3 min. Therefore, the single-molecule data were collected for 3 min after energization. During ATP synthesis, again three different FRET states were observed at similar levels as during ATP hydrolysis, and with rapid changes between them (Figure 3C). The sequence → L → M → H → L… of FRET states was found for 84% of 386 analyzed bursts with at least one FRET transition. These data indicate unidirectional sequential changes in the distances between the two fluorophores, that is, rotation of the ɛ-subunit relative to the b-subunits in three steps during ATP synthesis, and in the opposite direction as observed during ATP hydrolysis.
To investigate the movements of the ɛ-subunit during proton transport without ATP synthesis or ATP hydrolysis, the proteoliposomes were energized by ΔpH and Δφ in the absence of nucleotides. Figure 3D shows two examples of photon bursts under these conditions. Three similar FRET efficiencies were discriminated as during ATP hydrolysis and ATP synthesis, which changed rapidly. However, the sequence in synthesis direction was found for 66% of 140 analyzed bursts with at least two FRET levels, and 34% rotated in hydrolysis direction, with a mean dwell time of tE=38±8 ms.
Donor–acceptor distances and populations of the three FRET states
For a statistical evaluation of all photon bursts, the mean EFRET value of each FRET level during ATP hydrolysis, ATP synthesis, proton transport, and AMPPNP binding was calculated and is shown as black lines in Figure 3. The resulting FRET level histograms are depicted in Figure 4. All histograms exhibited three peaks, and each peak was fitted with a Gaussian distribution (for details, see Supplementary data). From the maxima of each peak, the most probable donor–acceptor distance (rDA) was calculated using EFRET=(R0)6/[(R0)6+(rDA)6] and a Förster radius R0=6.4 nm for the pair TMR at ɛ and Cy5 at b2 (see Materials and methods). The maxima of the three FRET levels were found at approximately the same FRET efficiencies during ATP hydrolysis, ATP synthesis, and proton transport (Figure 4B–D). These efficiencies corresponded to distances between donor and acceptor of 7.8 nm (L), 6.3 nm (M), and 4.6 nm (H).
Figure 4.
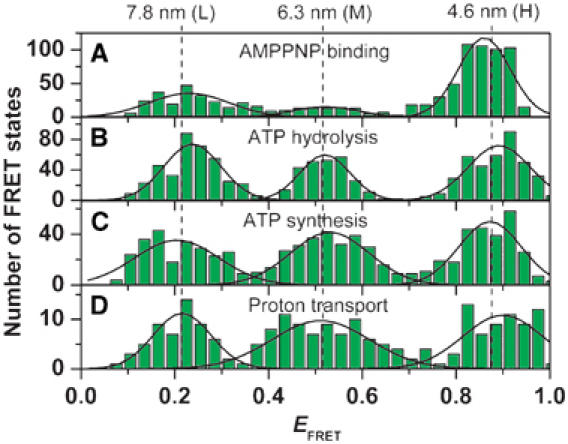
Histograms of the FRET efficiencies for the active F0F1. The mean FRET efficiency was calculated for each observed FRET level (see black lines in Figure 3) and the number of observations was plotted as a function of EFRET. (A) In the presence of AMPPNP (data from 888 photon bursts). (B) ATP hydrolysis (454 photon bursts). (C) ATP synthesis (368 photon bursts). (D) proton transport (140 photon bursts). The peaks were fitted by Gaussian distributions. For all conditions, the maxima of the distributions are observed at the same FRET efficiency (black lines). The calculated distances are shown at the top.
In the presence of AMPPNP, no catalysis takes place, and, correspondingly, no changes of FRET levels during the photon bursts were observed. Still the maxima of the Gaussian distributions give similar distances as during catalysis (Figure 4A). Obviously, the three different FRET distances during catalysis are frozen by AMPPNP binding.
The turnover time of catalysis at the different αβ-pairs
The dwell time of a FRET state is the duration of a constant ɛ conformation in F0F1 during catalysis, and, therefore, it represents the docking time of the γɛ-complex to one specific αβ-pair. In the presence of high nucleotide concentrations (ATP, or ADP plus Pi), this time interval represents, presumably, the time of synthesis or hydrolysis of one ATP at one catalytic site. Switching of the γɛ-complex to the next αβ-pair occurs faster than 1 ms. In Figure 5A the histogram of all dwell times during ATP hydrolysis is shown, and in Figure 5B that of all dwell times during ATP synthesis. Since the dwell times of the first and the last FRET level in a photon burst cannot be determined (because the enzyme in a liposome enters or leaves the confocal volume), only photon bursts with at least two FRET level transitions were used for this analysis. Fitting the distributions during ATP synthesis and hydrolysis monoexponentially yielded mean dwell times tS=17.7±0.9 ms and tH=14.3±0.7 ms, which are in good agreement with the ensemble turnover times.
Figure 5.
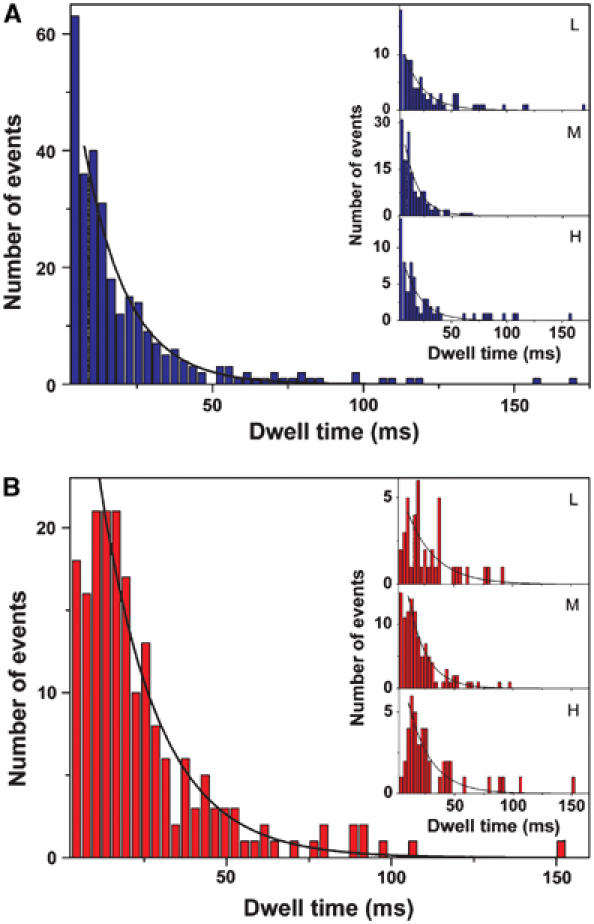
Histograms of dwell times of all FRET levels (A) during ATP hydrolysis (292 dwells) and (B) during ATP synthesis (193 dwells). Monoexponential fits (black lines) yield average dwell times of tH=14.3±0.7 ms (mean±standard deviation of the mean) during ATP hydrolysis and tS=17.7±0.9 ms during ATP synthesis. Monoexponential fits of the dwell times of each FRET level separately (see insets) give t=17.6±1.4 ms (L, 89 dwells), t=12.7±1.0 ms (M, 139 dwells), and t=15.8±1.7 ms (H, 64 dwells) for ATP hydrolysis, and t=24.0±4.4 ms (L, 41 dwells), t=15.4±1.0 ms (M, 108 dwells), and t=19.5±2.2 ms (H, 44 dwells) for ATP synthesis.
According to the binding change mechanism (Boyer, 1993), all three αβ-pairs carry out consecutively the same reaction steps in a strictly cooperative manner. Thus, it is expected that during steady-state catalysis, each αβ-pair equally accounts for the catalytic rate. Our data allow for the analysis of the dwell times for each of the three αβ-pairs separately, that is, the three FRET levels (see insets in Figure 5). During ATP hydrolysis, the M-level has an average dwell time of 12.7±1.0 ms, the L-level 17.6±1.4 ms, and the H-level 15.8±1.7 ms. During ATP synthesis, the M-level has an average dwell time of 15.4±1.0 ms, the L-level 24.0±4.4 ms, and the H-level 19.5±2.2 ms. In both directions of catalysis, the M-level exhibits the shortest dwell time.
The docking time of the γɛ-complex to one αβ-pair (that with the medium distance of the label at the ɛ-subunit to the label at the b-subunit) is shorter than the other two. Intrinsically, all αβ-pairs are identical and they carry out the same catalytic reaction. However, the peripheral stalk with subunits δ and b2 is attached to one pair (McLachlin et al, 2000). Possibly, the conformation of this αβ-pair differs slightly from the two other pairs, and, accordingly, might effect the required conformational changes for catalysis. This could lead to subtle changes in the Gibbs free energy and the activation barriers for the docking/undocking steps.
Conformational changes of ɛ and the active–inactive transition
Finally, we investigated the enzyme under conditions where it does not carry out catalysis. Different noncatalytic conditions were analyzed and, as in the case of AMPPNP addition (see Figure 3A), photon bursts with a single constant FRET state were observed. We calculated the mean EFRET value for each FRET level, and collected them in histograms for the different experimental conditions (Figure 6). Again, three FRET states were found for F0F1 in buffer at pH 8 without substrates, in the presence of ADP or Pi, and in the presence of both ADP and Pi. Since energization of the membrane is achieved by an acid–base transition starting at pH 4.7 (see Materials and methods), we investigated also the enzyme at pH 4.7 (Figure 6F). In addition, FRET level distributions were analyzed after ATP synthesis (Figure 6G) and after dissipation of ΔpH in the absence of substrates (Figure 6F), that is, 5 min after the acid–base transition. The maxima for all ‘inactive' conditions (with the exception of those in the presence of AMPPNP) were found at different FRET efficiencies compared to those of the catalytically active enzyme. Therefore, we called the three FRET states of the inactive enzymes L*, M*, and H* (for details, see Supplementary data). From the FRET efficiencies of the maxima, donor–acceptor distances of 8.3 nm (L*), 6.4 nm (M*), and 4.0 nm (H*) were calculated. The population of the three inactive FRET states was not equal and differed markedly from that of active enzymes: without nucleotides as well as in the presence of ADP or Pi (Figure 6B–F), approximately 50% of the enzymes were found in the L*-state, 15% in the M*-state, and 35% in the H*-state. After ATP synthesis (Figure 6E), after proton transport (Figure 6F), and in the presence of AMPPNP (Figure 6A), 30% are in the L-state, 15% are in the M-state, and 55% are in the H-state. In all cases, the M*-state is only weakly populated. This implies that the free energy of this state is higher than that of the two others. The population distribution in the presence of AMPPNP is peculiar (28% L, 10% M, 62% H): the FRET levels did not change within the photon bursts, indicating that the enzyme did not carry out catalysis; however, the FRET level maxima corresponded to those of a catalytically active enzyme (see Figures 4 and 6). Therefore, AMPPNP binding traps the enzyme in a ‘frozen catalytic conformation', which differs from the inactive state.
Figure 6.
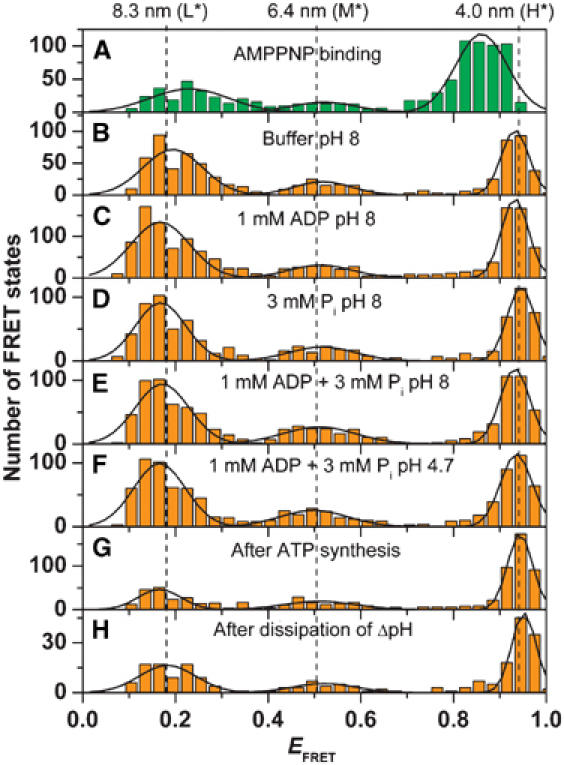
Histograms of the FRET efficiencies for the inactive F0F1. The mean FRET efficiency was calculated for each observed FRET state and the number of observations was plotted as a function of EFRET. For all conditions, the FRET state remained constant during the burst (see Figure 3A). (A) In the presence of AMPPNP (same data as in Figure 4). (B) Buffer, pH 8 (761 photon bursts). (C) Buffer, pH 8, in the presence of ADP (1428 photon bursts). (D) Buffer, pH 8, in the presence of phosphate (869 photon bursts). (E) Buffer, pH 8, in the presence of ADP and phosphate (960 photon bursts). (F) Buffer, pH 4.7, in the presence of ADP and phosphate (985 photon bursts). (G) 5 min after ATP synthesis (755 photon bursts). (H) 5 min after energization with ΔpH and Δφ (230 photon bursts). The peaks were fitted by Gaussian distributions. For all conditions, the same maxima of the distributions are observed (black lines) except for AMPPNP. The calculated distances are shown at the top.
Discussion
The ɛ-subunit during catalysis
In this work, intramolecular single-molecule FRET was used to investigate the movements of the ɛ-subunit during catalysis in membrane-integrated F0F1. During catalysis, we found characteristic fluctuations of the FRET efficiencies within photon bursts of a single enzyme. The FRET efficiencies remained constant for short time intervals (with dwell times of about 5–150 ms), and then rapidly changed to the next FRET level within 1 ms. A statistical evaluation of the data revealed FRET level histograms with three distinct maxima of the distribution, which were defined the H-, the M-, and the L-level. During ATP hydrolysis and ATP synthesis, these maxima were observed at similar FRET efficiencies; however, the sequence of FRET levels was opposite for both directions of the reaction.
From the mean FRET efficiencies of each level, the distances between the two fluorophores were calculated according to the Förster theory. We combined the three ɛ56–b64 distances using the program ‘FRETsg' (Schröder and Grubmüller, 2003), assuming that the three positions of ɛ56 were allocated on a circle around the axis of rotation. The positions were spaced by 120° rotational steps, and are indicated by green spheres in Figure 7A. Thereby, the position of the second label on the b-subunits is fixed in three dimensions by the three FRET distances 4.6 nm (H), 6.3 nm (M), and 7.8 nm (L), and is visualized as a red sphere in Figure 7A. We fitted these positions into a homology model of the E. coli H+-ATP synthase (Engelbrecht and Junge, 1997) with the aligned structure of the γɛ-complex (Rodgers and Wilce, 2000). We conclude that the dynamics of FRET level changes indicate a rotational movement of the ɛ-subunit relative to the b-subunit, presumably in 120° steps. In accordance with earlier measurements using the actin filament method (Noji et al, 1997), the FRET level sequence (→ H → M → L → H) during ATP hydrolysis corresponds to a counterclockwise rotation of the ɛ-subunit in the model, when viewed from F0 (blue arrow in Figure 7A). Consequently, clockwise rotation corresponds to ATP synthesis (yellow arrow in Figure 7A). The fact that not all bursts exhibited the same sequence for ATP hydrolysis (only 81%) and for ATP synthesis (only 84%) is due to the kinetics of catalysis. In our analysis, only levels with more than 4 ms were identified as a separate level. Assuming an exponential distribution of the dwell times, we calculated from the average dwell times (see Figure 5) 76% (ATP hydrolysis) and 80% (ATP synthesis) FRET levels with a duration longer than 4 ms. Therefore, apparently wrong sequences could be observed because short levels were not recognized.
Figure 7.
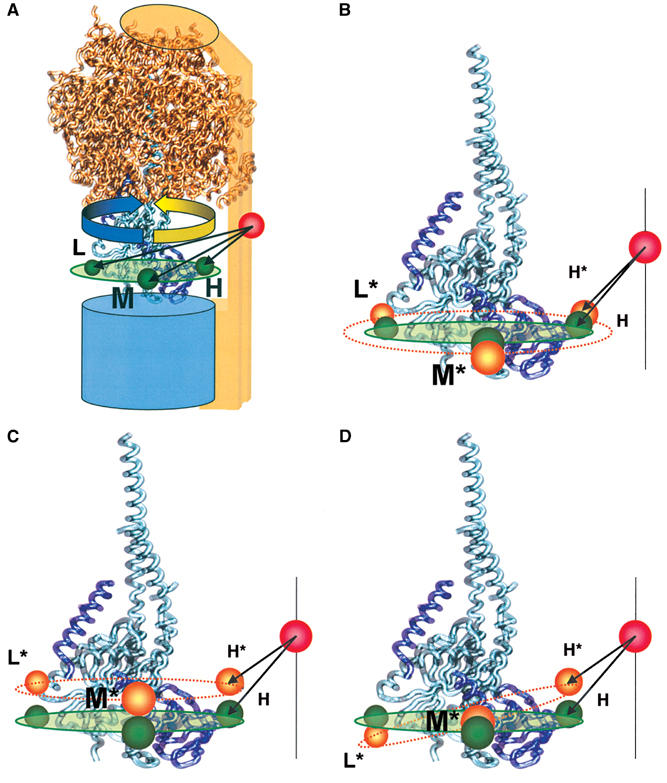
Visualization of the FRET distances in the F0F1 model. (A) The three donor–acceptor distances calculated from the FRET efficiencies obtained during catalysis are shown in green and labeled H, M, and L. The ATP synthesis direction is shown by an yellow arrow and ATP hydrolysis direction by a blue arrow. (B–D) The three donor–acceptor distances calculated from the FRET efficiencies of the inactive enzyme are shown in orange and labeled H*, M*, and L*, in addition to the active positions in green. The different active–inactive distance changes in the H-, M-, and L-state by the same conformational change in the γɛ-complex. (B) Distance changes by expanding the radius of rotation at ɛ56. (C) Distance change by shifting perpendicular to the membrane plane. (D) Distance changes by tilting with respect to the membrane plane.
Earlier single-molecule investigations of ɛ-movements in immobilized F1 parts reported a complex behavior. With CF1, an irregular stepwise motion of ɛ was observed (Häsler et al, 1998). With F1 from PS3, the rotation of the ɛ-subunit was demonstrated using the fluorescent actin filament as a marker; however, the rate of rotation was a factor 2 lower than that of the γ-subunit under the comparable conditions (Kato-Yamada et al, 1998). Presumably, it is complicated to separate the ɛ-movements resulting from catalysis and those from activation–inactivation of ɛ in immobilized F1 parts. In single membrane-bound F0F1, a three-stepped rotational movement of ɛ was observed with the same rate as reported earlier for the γ-subunit (Diez et al, 2004b), supporting the joint movement of the γɛ-complex as expected from ensemble measurements (Aggeler et al, 1997).
The same three maxima in the FRET level histograms were found for all catalytic conditions. This indicates that the three resting positions of ɛ relative to the b-subunit are not changed when the enzyme is switched from ATP synthesis to ATP hydrolysis. Two different structures of the isolated ɛ-subunit of F0F1 from E. coli have been reported (Uhlin et al, 1997; Rodgers and Wilce, 2000), and it has been speculated whether these two conformations might be related to the function of ɛ as a regulatory switch between the directions of catalysis. Several models including up-and-down movements of the C-terminal part of ɛ (Tsunoda et al, 2001; Suzuki et al, 2003; Bulygin et al, 2004), a ratchet-like mechanism with the C-terminus sticking at different angles between α- and β-subunits (Cipriano et al, 2002), or an additional ATP binding site (Kato-Yamada and Yoshida, 2003) have been proposed. In our measurements with the catalytically active holoenzyme, the three distances found during ATP synthesis, ATP hydrolysis, and proton transport without nucleotides are very similar. Thus, our data give no evidence for an up-and-down rearrangement of ɛ in the ɛ56-region relative to the b-subunit, when the enzyme switches between ATP synthesis and ATP hydrolysis. Of course, we cannot exclude that switching between both modes of catalysis might be connected with movement of other parts of the ɛ-subunit, which does not change the distance between the b-subunit and the ɛ56-region.
The movement of the ɛ-subunit during proton translocation in the absence of nucleotides is surprising. We conclude that this might reflect the presence of a ‘proton slip' (Feniouk et al, 2005). So far, we have detected only a small number of photon bursts with two or more FRET level transitions, which showed significantly increased dwell times of a FRET level as compared to ATP synthesis. However, in contrast to our conclusion, crosslinking data during energization have been interpreted to indicate no rotation of the γ-subunit during energization in the absence of nucleotides (Zhou et al, 1997).
AMPPNP inhibits ATP synthesis and hydrolysis, and photon bursts without FRET level changes were observed within the photon bursts (Figure 3A–C). The FRET states exhibited the same FRET efficiencies as during catalysis. We conclude from these data that binding of AMPPNP does not lead to an inactive conformation but it ‘freezes' the enzyme in a state with the same ɛ–b distances as during catalysis.
The ɛ-subunit during active–inactive transition
The ɛ-subunit has a dual role in the F0F1-ATP synthase. First, as a part of the rotor, it rotates in 120° steps relative to the b-subunits in the α3β3-barrel, and second, it regulates the enzyme activity (Sternweis and Smith, 1980; Fischer et al, 2000). We analyzed the ɛ–b distances also under conditions where the enzyme did not carry out catalysis. Three FRET states (called H*, M*, and L*) were observed, and the FRET levels did not change during the photon burst, that is, the distances remained constant. However, the mean distances differed from those of the active enzyme. Despite the fact that the corresponding active and inactive FRET levels showed a small overlap of the standard deviations of the Gaussian fits (see Supplementary data), the maxima of the H, M, and L levels for all active conditions, as well as the maxima of the H*, M*, and L* states for the seven inactive conditions, were highly reproducible. Therefore, we conclude that (i) the enzyme has different conformations of ɛ depending on active or inactive conditions and that (ii) the active–inactive transition is connected with a conformational change of ɛ within the central stalk. Three possibilities for the conformational changes associated with the active–inactive transition have to be considered (Figure 7B–D). Assuming that the acceptor position at b64 is the same in the active and the inactive state, Figure 7 shows the structural models with the three FRET distances of the essential γ–ɛ–b section of F0F1. The stopping positions of the ɛ56-region as observed in the active state are shown by green spheres, and those observed in the inactive state are shown by orange spheres. The high FRET distance decreased from 4.6 to 4.0 nm, the medium FRET distance remained almost constant (6.3–6.4 nm), and the low FRET distance increased from 7.8 to 8.3 nm. This could be caused by an increase in the radius of rotation for ɛ56 (Figure 7B). Alternatively, an up-shift of the three ɛ positions during the active–inactive transition including a small inward movement of the b64 position (Figure 7C), or a tilting of the plane of rotation for ɛ56 (Figure 7D), could explain the measured FRET distance changes. Each model could explain the observed changes, or the conformational shift of the ɛ56-region has components of all models. It should be mentioned that the conformational changes might lead to differences in mobility or orientation of the transition moments of the fluorophores, which would result also in changes of the energy transfer efficiency. The distances given above do not account for such an effect and should be taken as a first-order approximation. However, the lack of high-resolution structural data for the complete F0F1 makes it difficult to describe in detail the conformational changes of ɛ monitoring only one position at ɛ56 with respect to one position at b64.
Functional asymmetry of F0F1
According to the binding change theory, each catalytic site adopts sequentially three conformations and works strictly in a cooperative way (Boyer, 1993). This implies that catalysis occurs with the same turnover time at each αβ-pair. The dwell time represents the docking time of the γɛ-complex to one specific αβ-pair, and we assume that it represents the catalytic turnover time at this αβ-pair. A detailed analysis of the dwell times of each FRET level separately (see Figure 5, insets) showed that during ATP synthesis as well as during ATP hydrolysis, the dwell time of the M-level is significantly shorter than that of L- and H-level. Correspondingly, the catalytic turnover is higher (between 20 and 30%) than in the two other states. We assume that the relative movement of the subunits is not influenced by the fluorophores since both probes are small. However, we cannot rigorously exclude that there might be an orientation-specific steric hindrance of the rotor movement by the attached TMR.
Three independent results revealed an asymmetry in the function of the enzyme. (i) In the M-level, the catalytic turnover is faster than in the L- and H-level. (ii) Binding of AMPPNP traps the enzyme preferentially in the H-state. (iii) Without catalytic turnover, the enzyme is found preferentially in the L*-state (or H*-state after ATP synthesis or energization) as the inactive states. The reason for this heterogeneity and its significance for the mechanism were not investigated in this work. The interactions of the γɛ-complex with the three αβ-pairs during rotation induce the conformational changes proposed by the binding change theory. One might speculate that the binding of the peripheral stalk b2δ to one αβ-pair leads to some asymmetry in the otherwise identical αβ-pairs and this gives rise to the observed functional heterogeneities. The b-dimer interacts strongly with one αβ-pair (Diez et al, 2004a; Weber et al, 2004), leading to a slightly different conformation of the tagged αβ-pair (Kersten et al, 2000). This αβ-pair might have smaller activation barriers (M → L for hydrolysis and M → H for synthesis) or a higher Gibbs free energy. It is expected that in the absence of the peripheral stalk, for example, in isolated F1 parts, this asymmetry is not observed.
In ensemble measurements, neither static nor dynamic heterogeneities within a population of molecules can be detected. In single-molecule enzymology, functional single holoenzymes are investigated selectively, and mechanisms are derived from a statistical analysis. This general approach is applicable to a broad variety of biological systems for monitoring structural dynamics, binding and unbinding, oligomerization, and catalytic processes.
Materials and methods
Purification and labeling of F1 and F0F1-ATP synthase from E. coli
The plasmid pRAP100 carrying the ɛH56C mutation was constructed from pRA100 (Aggeler and Capaldi, 1992) by Medigenomix (Germany) and expressed in strain RA1. F1 was isolated as described (Gogol et al, 1989). Preparation of F0F1 from pRR76, containing the mutation bQ64C expressed in strain RA1, was carried out as described (Börsch et al, 2002). The F0F1 preparation before labeling contained 1.5 ATP/F0F1 and 0.9 ADP/F0F1. Protein and fluorophore concentrations were determined by UV absorption. F1-ɛH56C (20 μM) was labeled with tetramethylrhodamine-C5-maleimide (TMR; Molecular Probes) with a molar ratio of 1:0.9 in 50 mM MOPS–NaOH (pH 7.0) and 100 μM MgCl2 at 0°C for 4 min. The molar ratio of bound fluorophore to protein (54%) was calculated from absorption spectra with correction of the absorption of the fluorophore at 280 nm. The following absorption coefficients were used: ɛ(555 nm)=95 000 M−1 cm−1, ɛ(280 nm)=19 000 M−1 cm−1 for TMR (Molecular Probes), and ɛ(280 nm)=191 000 M−1 cm−1 for F1 (Gill and von Hippel, 1989). The two b-subunits of F0-bQ64C-F1 (20 μM) were crosslinked with Cy5bis-C5-maleimide (Cy5bis; Diez et al, 2004b) with a molar ratio of 1:1 in the same buffer as described for F1 labeling in the presence of 0.1% n-dodecylmaltoside (Glycon) at 0°C for 90 min. The labeling efficiency was 94%, as determined from the absorption spectra using ɛ(650 nm)=250 000 M−1 cm−1 and ɛ(280 nm)=5200 M−1 cm−1 for Cy5bis (Diez et al, 2004b), and ɛ(280 nm)=340 000 M−1 cm−1 for F0F1 (Gill and von Hippel, 1989). Selectivity of labeling of F1 and F0F1 was checked by measuring the fluorograms after gel electrophoresis. Approximately 90% of the b-subunits were crosslinked by Cy5bis according to SDS–PAGE. The rate of ATP hydrolysis (vH) of F1 was measured with an ATP regenerating system (Fischer et al, 2000) at 37°C in a buffer containing 100 mM Tris–HCl (pH 8), 25 mM KCl, 4 mM MgCl2, 2.5 mM phosphoenolpyruvate, 18 U/ml pyruvate kinase, 16 U/ml lactate dehydrogenase, and 0.2 mM NADH.
Preparation of F0F1 in a liposome
Labeled (or unlabeled) F0F1 was reconstituted into liposomes as described (Fischer and Gräber, 1999). The number of F0F1 per liposome was adjusted as follows. The diameter of the liposomes was determined by light scattering yielding 120±10 nm. The sum of inner and outer surfaces of the liposome was 7.9 × 104 nm2. Using 0.6 nm2 (Nagle et al, 1996) for the average area of one lipid, the number of lipid molecules was 1.32 × 105. The lipid concentration during reconstitution was 7.5 g/l or 10 mM. Using 1.32 × 105 lipids per liposome, the liposome (or vesicle) concentration was 75 nM. Addition of 15 nM F0F1 resulted in a ratio of five liposomes per F0F1. If the incorporation of F0F1 into liposomes occurred statistically, about 80% of the vesicles contained no enzyme, and the number of liposomes containing more than one was negligible. F0-liposomes were prepared by stripping the F1 part (Lötscher et al, 1984) as follows: F0F1-liposomes were centrifuged (90 min at 264 000 g, room temperature) and resuspended in 1 mM tricine–NaOH (pH 8.0), 1 mM DTT, 0.5 mM EDTA, and 4% (v/v) glycerol. The resuspended liposomes were incubated for 90 min at room temperature, centrifuged, and resuspended twice. Finally, the stripped vesicles were resuspended in the same buffer to yield an F0 concentration of 60 nM. The F0-liposome suspension (less than one F0 per liposome) was incubated with labeled (or unlabeled) F1 at a molar excess of two F1 per F0 in the presence of 2.5 mM MgCl2 and 50 mM NaCl, first for 45 min at 37°C and then for 90 min at 0°C. The excess of unbound F1 was removed by three subsequent ultracentrifugation steps (90 min, 264 000 g, 4°C) and by resuspending the pellet in 20 mM tricine–NaOH (pH 8.0), 20 mM succinic acid, 0.6 mM KCl, and 4% (v/v) glycerol. The final concentration of F0F1 was adjusted to 40–50 nM.
The rates of ATP hydrolysis (vH) and ATP synthesis (vS) were measured at 23°C to control the efficiency of reconstitution. All activities were calculated on the basis of the F0 concentration. ATP hydrolysis was carried out with an ATP regeneration system (see above) after preincubation of the F0F1-liposomes with 25 mM KCl for 24 h. ATP synthesis was measured after an acid–base transition in the presence of a K+/valinomycin diffusion potential. F0F1-liposomes (30 μl) were mixed with 120 μl of a buffer with 20 mM succinic acid–NaOH (pH 4.7), 5 mM NaH2PO4, 0.6 mM KOH, 2.5 mM MgCl2, 100 μM ADP, and 20 μM valinomycin. After 3 min incubation, 150 μl of the acidic suspension was injected into 850 μl of 200 mM tricine–NaOH (pH 8.8), 5 mM NaH2PO4, 160 mM KOH, 2.5 mM MgCl2, and 100 μM ADP. The generated ATP was monitored by the luciferin/luciferase assay as described (Turina et al, 2003). For specific inhibition of ATP hydrolysis, the proteoliposomes were incubated with 100 μM DCCD (60 min at 30°C), and with 60 μM DCCD (60 min at 23°C) for inhibition of ATP synthesis.
Single-molecule fluorescence measurements
Single-molecule fluorescence measurements were performed at room temperature using a home-built two-channel confocal microscope setup (Börsch et al, 2002). The intensity of the laser beam (Nd:YAG, 532 nm, Coherent) was attenuated to 105 μW and fluorescence from FRET-labeled F0F1-liposomes at a final concentration of 50–90 pM was recorded by a multichannel scaler PC-card (PMS 300, Becker&Hickl) using 1 ms binning time. Measurements under various conditions (no substrate, 3 mM phosphate, 1 mM ADP, 1 mM AMPPNP, 1 mM ATP) were carried out in a buffer containing 20 mM tricine–NaOH (pH 8.0), 20 mM succinic acid, 2.5 mM MgCl2, 0.6 mM KCl, and 4% (v/v) glycerol. Measurements at pH 4.7 were carried out in a buffer containing 20 mM succinic acid–NaOH (pH 4.7), 0.6 mM KOH, 2.5 mM MgCl2, 3 mM phosphate, and 1 mM ADP. Fluorescence measurements during ATP synthesis were carried out after energization of the F0F1-liposomes (generation of a ΔpH plus Δφ by mixing the same two buffers as described above for bulk measurements of ATP synthesis with two syringes) in a microscopic flow chamber, which was implemented in the confocal setup. Data were recorded for 3 min after mixing. The concentrations of AMPPNP, ADP, and ATP were determined by UV absorption using ɛ(260 nm)=15 400 M−1 cm−1.
FRET data analysis
The recorded fluorescence intensities were corrected as follows: mean background count rates (usually between 0.5 and 2 kHz), which were obtained from measurements of pure buffer solutions, were subtracted. Additionally, a small leak of donor signal into the acceptor channel (‘crosstalk') was measured and taken into account to yield the corrected fluorescence intensities of donor (FD) and acceptor (FA). Mean diffusion times, τD, through the confocal detection volume of approximately 7 fl were measured by fluorescence correlation spectroscopy (Börsch et al, 1998) for the unbound dye (τD=0.4 ms), donor-labeled F1 (not bound to F0, τD=3.6 ms), and labeled F0F1-liposomes (τD=25 ms). Photon bursts with fluorescence intensities above the background were selected by the following criteria: (i) minimal duration longer than 20 ms, and (ii) at least 400 photons as a sum of photon counts in both detection channels.
For each photon burst, the FRET efficiencies were calculated in 1 ms steps when the sum of corrected count rates in both channels was greater than 10 kHz. The FRET efficiency was calculated according to EFRET=FA/(FA+γFD) with a correction factor γ=ηAΦA/ηDΦD where ηD and ηA are the overall detection efficiencies of the donor and the acceptor channel. ΦD and ΦA are the fluorescence quantum yields of the enzyme-bound donor and acceptor. ηD=0.35 and ηA=0.35 were calculated by comparing the corrected emission spectra of donor and acceptor with the transmission characteristics of the filters and the spectral sensitivities of the detectors of the single-molecule setup. The quantum yield ΦD=0.37 of TMR at ɛ56 was measured relative to sulforhodamine 101 as a standard (Karstens and Kobs, 1980). For the FRET acceptor Cy5bis, the quantum yield ΦA=0.32 was reported previously (Diez et al, 2004b). With these data, we obtain γ=0.87.
For photon bursts with changes of the FRET efficiency, a FRET level was recognized when this level remains constant for longer than 4 ms. The mean EFRET values for all levels within all bursts were calculated and were collected in histograms for the different experimental conditions. The peaks in the histograms were approximated by Gaussian distributions, and the maxima were used to calculate the mean distances between the two fluorophores. The relative population of the different FRET states was calculated by integration of the Gaussian fits. When several FRET level were observed within one photon burst (during ATP hydrolysis, during ATP synthesis, and during energization without ADP), the dwell times of the FRET levels were also measured. The first and the last FRET levels were omitted in the dwell time histograms because their duration before entering and after leaving the confocal volume remained unknown. The dwell time histograms were fitted by an exponential function to obtain the average dwell times. Donor-labeled proteoliposomes (about 60% of all bursts) were identified as bursts without photons in the acceptor channel, and were omitted from further data analysis. Liposomes with two labeled F0F1 were expected to have a doubled fluorescence intensity. Such bursts (less than 1%) were excluded.
Calculation of intramolecular distances from single-molecule FRET data
Distances (rDA) between the FRET donor and acceptor were calculated according to rDA=R0[(1/EFRET)−1]1/6 (Förster, 1948). FRET efficiencies EFRET were taken from the maxima of Gaussian fits of the FRET level histograms. The Förster radius R0=64 Å for the pair TMR/Cy5 was determined from R0=0.2111[J(λ)n−4κ2ΦD]1/6 as follows: the overlap integral, J(λ), was calculated from the emission spectrum of F1-ɛH56C-TMR and the absorption spectrum of F0-bQ64C-Cy5bis-F1 to yield J(λ)=9.90 × 10−13 M−1 cm3. The donor quantum yield ΦD=0.37 was determined as described above. For the index of refraction, n=1.33 was used, and for the orientation factor, κ2=2/3 was assumed. This value is in the range of possible κ2 values for the case of linear and planar (due to rotation) transition moments of donor and acceptor (Van der Meer et al, 1994). The anisotropies r of the fluorophores bound to the protein were measured for F1-ɛH56C-TMR and F0-bQ64C-Cy5bis-F1. The data indicate that TMR at ɛ56 (rTMR=0.06) can move unrestricted, whereas the mobility of Cy5bis at b64 (rCy5bis=0.25) is more restricted.
Supplementary Material
Supplementary Information
Acknowledgments
We thank CAM Seidel for the gift of the Cy5 dye, W Wangler for excellent technical assistance, A Aird for software solutions, P Turina for inspiring discussions, J Wrachtrup and DJ Cipriano for critical reading of the manuscript, and A Börsch-Haubold for editorial suggestions. The financial support by the Landesstiftung Baden-Württemberg in the network of competence ‘functional nanodevices' is gratefully acknowledged.
References
- Aggeler R, Capaldi RA (1992) Cross-linking of the γ subunit of the Escherichia coli ATPase (ECF1) via cysteines introduced by site-directed mutagenesis. J Biol Chem 267: 21355–21359 [PubMed] [Google Scholar]
- Aggeler R, Ogilvie I, Capaldi RA (1997) Rotation of a γ–ɛ subunit domain in the Escherichia coli F1F0-ATP synthase complex. The γ–ɛ subunits are essentially randomly distributed relative to the α3β3δ domain in the intact complex. J Biol Chem 272: 19621–19624 [DOI] [PubMed] [Google Scholar]
- Boldt F-M, Heinze J, Diez M, Petersen J, Börsch M (2004) Real-time pH microscopy down to the molecular level by combined scanning electrochemical microscopy/single molecule fluorescence spectroscopy. Anal Chem 76: 3473–3481 [DOI] [PubMed] [Google Scholar]
- Börsch M, Diez M, Zimmermann B, Reuter R, Gräber P (2002) Stepwise rotation of the γ-subunit of EF0F1-ATP synthase observed by intramolecular single-molecule fluorescence resonance energy transfer. FEBS Lett 527: 147–152 [DOI] [PubMed] [Google Scholar]
- Börsch M, Turina P, Eggeling C, Fries JR, Seidel CA, Labahn A, Gräber P (1998) Conformational changes of the H+-ATPase from Escherichia coli upon nucleotide binding detected by single molecule fluorescence. FEBS Lett 437: 251–254 [DOI] [PubMed] [Google Scholar]
- Böttcher B, Bertsche I, Reuter R, Gräber P (2000) Direct visualisation of conformational changes in EF0F1 by electron microscopy. J Mol Biol 296: 449–457 [DOI] [PubMed] [Google Scholar]
- Böttcher B, Schwarz L, Gräber P (1998) Direct indication for the existence of a double stalk in CF0F1. J Mol Biol 281: 757–762 [DOI] [PubMed] [Google Scholar]
- Boyer PD (1993) The binding change mechanism for ATP synthase—some probabilities and possibilities. Biochim Biophys Acta 1140: 215–250 [DOI] [PubMed] [Google Scholar]
- Boyer PD (1998) ATP synthase—past and future. Biochim Biophys Acta 1365: 3–9 [DOI] [PubMed] [Google Scholar]
- Bulygin VV, Duncan TM, Cross RL (1998) Rotation of the ɛ subunit during catalysis by Escherichia coli FOF1-ATP synthase. J Biol Chem 273: 31765–31769 [DOI] [PubMed] [Google Scholar]
- Bulygin VV, Duncan TM, Cross RL (2004) Rotor/Stator interactions of the ɛ subunit in Escherichia coli ATP synthase and implications for enzyme regulation. J Biol Chem 279: 35616–35621 [DOI] [PubMed] [Google Scholar]
- Capaldi RA, Aggeler R (2002) Mechanism of the F1F0-type ATP synthase, a biological rotary motor. Trends Biochem Sci 27: 154–160 [DOI] [PubMed] [Google Scholar]
- Cipriano DJ, Bi Y, Dunn SD (2002) Genetic fusions of globular proteins to the ɛ subunit of the Escherichia coli ATP synthase: implications for in vivo rotational catalysis and epsilon subunit function. J Biol Chem 277: 16782–16790 [DOI] [PubMed] [Google Scholar]
- Diez M, Börsch M, Zimmermann B, Turina P, Dunn SD, Gräber P (2004a) Binding of the b-subunit in the ATP synthase from Escherichia coli. Biochemistry 43: 1054–1064 [DOI] [PubMed] [Google Scholar]
- Diez M, Zimmermann B, Börsch M, König M, Schweinberger E, Steigmiller S, Reuter R, Felekyan S, Kudryavtsev V, Seidel CA, Gräber P (2004b) Proton-powered subunit rotation in single membrane-bound F0F1-ATP synthase. Nat Struct Mol Biol 11: 135–141 [DOI] [PubMed] [Google Scholar]
- Duncan TM, Bulygin VV, Zhou Y, Hutcheon ML, Cross RL (1995) Rotation of subunits during catalysis by Escherichia coli F1-ATPase. Proc Natl Acad Sci USA 92: 10964–10968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelbrecht S, Junge W (1997) ATP synthase: a tentative structural model. FEBS Lett 414: 485–491 [DOI] [PubMed] [Google Scholar]
- Feniouk BA, Mulkidjanian AY, Junge W (2005) Proton slip in the ATP synthase of Rhodobacter capsulatus: induction proton conduction and nucleotide dependence. Biochim Biophys Acta 1706: 184–194 [DOI] [PubMed] [Google Scholar]
- Fischer S, Gräber P, Turina P (2000) The activity of the ATP synthase from Escherichia coli is regulated by the transmembrane proton motive force. J Biol Chem 275: 30157–30162 [DOI] [PubMed] [Google Scholar]
- Fischer S, Gräber P (1999) Comparison of ΔpH- and Δφ-driven ATP synthesis catalyzed by the H+-ATPases from Escherichia coli or chloroplasts reconstituted into liposomes. FEBS Lett 457: 327–332 [DOI] [PubMed] [Google Scholar]
- Förster T (1948) Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann Phys 2: 55–70 [Google Scholar]
- Gill SC, von Hippel PH (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 182: 319–326 [DOI] [PubMed] [Google Scholar]
- Gogol EP, Lucken U, Bork T, Capaldi RA (1989) Molecular architecture of Escherichia coli F1 adenosinetriphosphatase. Biochemistry 28: 4709–4716 [DOI] [PubMed] [Google Scholar]
- Häsler K, Engelbrecht S, Junge W (1998) Three-stepped rotation of subunits γ and ɛ in single molecules of F-ATPase as revealed by polarized, confocal fluorometry. FEBS Lett 426: 301–304 [DOI] [PubMed] [Google Scholar]
- Junge W (1999) ATP synthase and other motor proteins. Proc Natl Acad Sci USA 96: 4735–4737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaim G, Prummer M, Sick B, Zumofen G, Renn A, Wild UP, Dimroth P (2002) Coupled rotation within single F0F1 enzyme complexes during ATP synthesis or hydrolysis. FEBS Lett 525: 156–163 [DOI] [PubMed] [Google Scholar]
- Karrasch S, Walker JE (1999) Novel features in the structure of bovine ATP synthase. J Mol Biol 290: 379–384 [DOI] [PubMed] [Google Scholar]
- Karstens T, Kobs K (1980) Rhodamine B and rhodamine 101 as reference substances for fluorescence quantum yield measurements. J Phys Chem 84: 1871–1872 [Google Scholar]
- Kato-Yamada Y, Noji H, Yasuda R, Kinosita K Jr, Yoshida M (1998) Direct observation of the rotation of ɛ subunit in F1-ATPase. J Biol Chem 273: 19375–19377 [DOI] [PubMed] [Google Scholar]
- Kato-Yamada Y, Yoshida M (2003) Isolated ɛ subunit of thermophilic F1-ATPase binds ATP. J Biol Chem 278: 36013–36016 [DOI] [PubMed] [Google Scholar]
- Kersten MV, Dunn SD, Wise JG, Vogel PD (2000) Site-directed spin-labeling of the catalytic sites yields insight into structural changes within the F0F1-ATP synthase of Escherichia coli. Biochemistry 39: 3856–3860 [DOI] [PubMed] [Google Scholar]
- Kinosita K Jr, Adachi K, Itoh H (2004) Rotation of F1-ATPase: how an ATP-driven molecular machine may work. Annu Rev Biophys Biomol Struct 33: 245–268 [DOI] [PubMed] [Google Scholar]
- Lötscher HR, deJong C, Capaldi RA (1984) Modification of the F0 portion of the H+-translocating adenosinetriphosphatase complex of Escherichia coli by the water- soluble carbodiimide 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide and effect on the proton channeling function. Biochemistry 23: 4128–4134 [DOI] [PubMed] [Google Scholar]
- McLachlin DT, Coveny AM, Clark SM, Dunn SD (2000) Site-directed cross-linking of b to α, β, and a subunits of the Escherichia coli ATP synthase. J Biol Chem 275: 17571–17577 [DOI] [PubMed] [Google Scholar]
- Nagle JF, Zhang R, Tristram-Nagle S, Sun W, Petrache HI, Suter RM (1996) X-ray structure determination of fully hydrated L alpha phase dipalmitoylphosphatidylcholine bilayers. Biophys J 70: 1419–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noji H, Yasuda R, Yoshida M, Kinosita K Jr (1997) Direct observation of the rotation of F1-ATPase. Nature 386: 299–302 [DOI] [PubMed] [Google Scholar]
- Rodgers AJ, Wilce MC (2000) Structure of the γ/ɛ complex of ATP synthase. Nat Struct Biol 7: 1051–1054 [DOI] [PubMed] [Google Scholar]
- Rubinstein JL, Walker JE, Henderson R (2003) Structure of the mitochondrial ATP synthase by electron cryomicroscopy. EMBO J 22: 6182–6192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbert D, Engelbrecht S, Junge W (1996) Intersubunit rotation in active F-ATPase. Nature 381: 623–625 [DOI] [PubMed] [Google Scholar]
- Schröder GF, Grubmüller H (2003) FRETsg: biomolecular structure model building from multiple FRET experiments. Comp Phys Comm 158: 150–157 [Google Scholar]
- Smith JB, Sternweis PC (1977) Purification of membrane attachment and inhibitory subunits of the proton translocating adenosine triphosphatase from Escherichia coli. Biochemistry 16: 306–311 [DOI] [PubMed] [Google Scholar]
- Sternweis PC, Smith JB (1980) Characterization of the inhibitory (ɛ) subunit of the proton-translocating adenosine triphosphatase from Escherichia coli. Biochemistry 19: 526–531 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Murakami T, Iino R, Suzuki J, Ono S, Shirakihara Y, Yoshida M (2003) F0F1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of ɛ subunit in response to proton motive force and ADP/ATP balance. J Biol Chem 278: 46840–46846 [DOI] [PubMed] [Google Scholar]
- Tang C, Capaldi RA (1996) Characterization of the interface between γ and ɛ subunits of Escherichia coli F1-ATPase. J Biol Chem 271: 3018–3024 [DOI] [PubMed] [Google Scholar]
- Tsunoda SP, Rodgers AJ, Aggeler R, Wilce MC, Yoshida M, Capaldi RA (2001) Large conformational changes of the ɛ subunit in the bacterial F1F0 ATP synthase provide a ratchet action to regulate this rotary motor enzyme. Proc Natl Acad Sci USA 98: 6560–6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turina P, Samoray D, Gräber P (2003) H+/ATP ratio of proton transport-coupled ATP synthesis and hydrolysis catalysed by CF0F1-liposomes. EMBO J 22: 418–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlin U, Cox GB, Guss JM (1997) Crystal structure of the ɛ subunit of the proton-translocating ATP synthase from Escherichia coli. Structure 5: 1219–1230 [DOI] [PubMed] [Google Scholar]
- Van der Meer BW, Cooker G, Chen SS-Y (1994) Resonance Energy Transfer: Theory and Data. New York: VHC [Google Scholar]
- Weber J, Senior AE (2003) ATP synthesis driven by proton transport in F1F0-ATP synthase. FEBS Lett 545: 61–70 [DOI] [PubMed] [Google Scholar]
- Weber J, Wilke-Mounts S, Nadanaciva S, Senior AE (2004) Quantitative determination of direct binding of b subunit to F1 in Escherichia coli F1F0-ATP synthase. J Biol Chem 279: 11253–11258 [DOI] [PubMed] [Google Scholar]
- Wilkens S, Capaldi RA (1998) ATP synthase's second stalk comes into focus. Nature 393: 29. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Noji H, Yoshida M, Kinosita K Jr, Itoh H (2001) Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase. Nature 410: 898–904 [DOI] [PubMed] [Google Scholar]
- Yoshida M, Muneyuki E, Hisabori T (2001) ATP synthase—a marvellous rotary engine of the cell. Nat Rev Mol Cell Biol 2: 669–677 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Duncan TM, Cross RL (1997) Subunit rotation in Escherichia coli FoF1-ATP synthase during oxidative phosphorylation. Proc Natl Acad Sci USA 94: 10583–10587 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information