

Abstract
The most common genetic change in aerobic organisms is a C:G to T:A mutation. C → T transitions can arise through spontaneous hydrolytic deamination of cytosine to give a miscoding uracil residue. This is also a frequent DNA lesion induced by oxidative damage, through exposure to agents such as ionizing radiation, or from endogenous sources that are implicated in the aetiology of degenerative diseases, ageing and cancer. The Ung and Smug1 enzymes excise uracil from DNA to effect repair in mammalian cells, and gene-targeted Ung−/− mice exhibit a moderate increase in genome-wide spontaneous mutagenesis. Here, we report that stable siRNA-mediated silencing of Smug1 in mouse embryo fibroblasts also generates a mutator phenotype. However, an additive 10-fold increase in spontaneous C:G to T:A transitions in cells deficient in both Smug1 and Ung demonstrates that these enzymes have distinct and nonredundant roles in suppressing C → T mutability at non-CpG sites. Such cells are also hypersensitive to ionizing radiation, and reveal a role of Smug1 in the repair of lesions generated by oxidation of cytosine.
Keywords: DNA repair, endogenous mutagenesis, oxidative DNA damage, uracil-DNA glycosylase
Introduction
Aerobic organisms must maintain the integrity of their genome in an aqueous environment, where the DNA is continuously under attack from spontaneous hydrolysis as well as active oxygen species that arise during normal cellular metabolism. Hydrolytic deamination of unmodified cytosine in DNA occurs spontaneously, generating the aberrant base uracil an estimated 500 times per proliferating human cell per day. This is one of the most abundant miscoding DNA lesions, generating C → T transition mutations upon subsequent replication of the U:G mispair, and is likely to be a major contributor, along with deamination of 5-methylcytosine (5-me-C; Millar et al, 2002) to both somatic and germline mutations. Such deamination occurs ∼100-fold more rapidly in single-stranded DNA, as would be temporarily exposed during ongoing replication and transcription, where the cytosine residues are not protected by the complementary DNA strand (Lindahl, 1993). Thus, its increased size and slower replication would make the mammalian genome a significant target for mutagenesis. Uracil may also occur in a U:A base pair through occasional use of dUTP instead of TTP during DNA replication. Multiple uracil-DNA glycosylase enzymes are widely distributed in bacteria, archaea and eukaryotes (Aravind and Koonin, 2000), signifying the importance of eliminating uracil from the genome. However, targeted enzymatic deamination of cytosine in DNA has been shown to underpin adaptive immunity and is also employed to inactivate foreign DNA (reviewed in Neuberger et al, 2003). Thus, cytosine deamination outside of the lymphoid lineage or mistargeting during antibody diversification has important implications for both oncogenesis and genome evolution.
C:G to T:A transitions are also a common genetic change induced by oxidative damage (Wang et al, 1998). Ionizing radiation can generate reactive oxygen species through radiolysis of water; the hydroxyl radical is the most biologically active, generating DNA strand breaks and base loss, but also a multitude of oxidized base lesions (reviewed in Bjelland and Seeberg, 2003). The latter damage is repaired by the DNA base-excision repair pathway, which is initiated by specific DNA glycosylases that excise the altered base (reviewed in Dizdaroglu, 2003). Further processing of the resultant abasic site by apurinic/apyrimidinic (AP) endonuclease, then polymerization and ligation of a one-nucleotide repair patch, restores the covalent DNA structure. Purine bases are sensitive to oxidation, and the abundant lesion 8-hydroxyguanine is highly mutagenic and carcinogenic in repair-deficient mammalian cells (reviewed in Barnes and Lindahl, 2004). However, oxidized pyrimidines are also formed in similar amounts and thymine derivatives such as thymine glycol are, by contrast, generally cytotoxic lesions that block replicative DNA polymerases. Thus, 8-hydroxyguanine and thymine glycol are well studied as archetypal biomarkers of oxidative stress. The identification of putative oxidation products of cytosine has been hampered due to the instability of cytosine derivatives through deamination (Bjelland and Seeberg, 2003); these lesions are thought to be largely miscoding rather than cytotoxic in vivo (Kreutzer and Essigmann, 1998).
Gene-targeted mice deficient in the ubiquitous Ung uracil-DNA glycosylase revealed a primary role of Ung during DNA replication, where slow removal of uracil from misincorporated dUMP resulted in an ∼100-fold increased steady-state level of uracil in the genome of Ung−/− mice (Nilsen et al, 2000). This is consistent with the proliferation-dependent expression of UNG in the mammalian cell nucleus (Muller-Weeks et al, 2005), its localization to replication foci during S phase (Otterlei et al, 1999), and indicates a major role of Ung in counteracting U:A base pairs (Nilsen et al, 2000). Ung was also shown to have a role in resolving U:G lesions, specifically generated by the AID enzyme (activation-induced cytidine deaminase) in immunoglobulin variable genes during somatic hypermutation and class switch recombination (Rada et al, 2002, 2004). In support of this function for Ung in the immune system, we have reported that ageing Ung−/− null mice have a greatly increased incidence of B-cell lymphomas (Nilsen et al, 2003, 2005). Ung−/− mice also show increased postischemic brain injury, which may relate to Ung's functioning in mitochondrial genome maintenance (Endres et al, 2004). In an initially surprising result, Ung-deficient mice displayed only a modest mutator phenotype (Nilsen et al, 2000), in contrast to the greatly increased frequency of spontaneous C → T mutations in Ung− mutants of bacteria and yeast (Duncan and Weiss, 1982; Impellizzeri et al, 1991). However, a second uracil-DNA glycosylase encoded by the SMUG1 gene (Haushalter et al, 1999), which is found in insects and vertebrates, was shown to account for the complementary uracil-excising activity in Ung-deficient mice (Nilsen et al, 2001).
Thus, we proposed that SMUG1 evolved as a necessary and separate antimutator acting on premutagenic U:G lesions resulting from genome-wide hydrolytic deamination of cytosine, due to the involvement of UNG in specialized roles at the replication fork and in the diversification of antibody genes. The structure of the SMUG1 active site (Wibley et al, 2003), and mutational analysis of damage recognition and catalysis (Matsubara et al, 2004), identified how SMUG1 is also able to specifically excise base derivatives from DNA that arise through oxidation of the methyl group of thymine, namely 5-hydroxymethyluracil (5-hydroxy-me-U; Boorstein et al, 2001) and 5-formyluracil (5-formyl-U; Masaoka et al, 2003), and it has been proposed that this, rather than excision of premutagenic deaminated cytosine residues, may be its main function in vivo (Matsubara et al, 2003). As 5-hydroxy-me-U could also be generated by oxidation and deamination of 5-me-C, SMUG1 may conceivably function to prevent mutagenesis at 5-me-C:G base pairs or act in a sequence of controlled oxidation, deamination and excision to effect DNA demethylation during the regulation of gene expression in higher eukaryotes (Boorstein et al, 2001). Demethylation might be achieved more directly through deamination of 5-me-C by the AID enzyme and repair of the resultant T:G mismatch (Morgan et al, 2004).
In order to assess the cellular role of SMUG1, and define the delineation or overlap of function between the Smug1 and Ung uracil-DNA glycosylases, we have employed short interfering RNA (siRNA) technology to generate stable siRNA-mediated Smug1 knockdown mouse embryo fibroblast (MEF) cell lines, in both an Ung+/+ and Ung−/− background. We report here that Smug1 knockdown cells that are also deficient in the Ung uracil-DNA glycosylase are uniquely hypersensitive to ionizing radiation, and show that SMUG1 excises two potentially cytotoxic oxidized cytosine derivatives from irradiated DNA. Importantly, analysis of spontaneous mutagenesis in Smug1 knockdown cell lines demonstrates that SMUG1 is not merely a ‘back-up' for UNG in the repair of U:G base pairs (Kavli et al, 2002; Masaoka et al, 2003). Thus, SMUG1 is identified as a novel antimutator with a unique role in suppressing the mutability of C:G base pairs in mammalian cells.
Results and discussion
Generation of stable siRNA-mediated Smug1-deficient cell lines
Stable expression of siRNA was employed to suppress or ‘knock down' Smug1 uracil-DNA glycosylase activity in both Ung+/+ and Ung−/− MEF cell lines. Ung−/− gene-targeted knockout mice and cells have been described; permanent Ung−/− and Ung+/+ cell lines were established from transformed clones arising spontaneously after repeated passage of MEFs in culture (Nilsen et al, 2000). Thus, siRNA-mediated silencing of Smug1 mRNA expression in these cell lines would not only allow analysis of the cellular role of Smug1, but also facilitate the comparison of Smug1 versus Ung function and the generation of mutant cells deficient in both these uracil-DNA glycosylases. Initially, three siRNA oligonucleotides that targeted different regions of the murine Smug1 mRNA were designed and assessed for their ability to suppress mRNA expression in transient transfection experiments. Expression of short-hairpin RNAs corresponding to the two most efficient siRNAs was achieved by insertion of appropriate sequences into expression vectors under control of an RNA pol III promoter. These two constructs gave similar results in subsequent experiments (data not shown). Following repeated passage in culture, a Smug1 knockdown in the Ung+/+ cell line (designated Smug1↓) and a Smug1 knockdown in the Ung−/− cell line (Smug1↓, Ung−/−) were isolated as clones that expressed stably suppressed levels of Smug1 mRNA. Figure 1A shows expression of Smug1 mRNA in these two cell lines relative to wild-type and Ung−/− controls, as quantified by real-time PCR of reverse-transcribed Smug1 cDNA. The Smug1↓ and Smug1↓, Ung−/− cell lines have greatly suppressed levels of Smug1 mRNA, with only ∼19 and ∼17% residual expression, respectively. This is similar to the ∼80% reduction in mRNA and protein levels reported for siRNA-mediated silencing of the Neil1 DNA glycosylase in murine cells (Carmell et al, 2003; Rosenquist et al, 2003).
Figure 1.
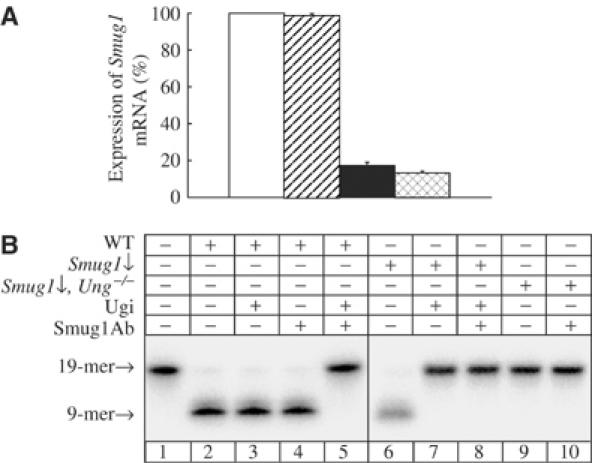
siRNA-mediated downregulation of Smug1 in MEF cell lines. (A) Smug1 mRNA level was detected by real-time PCR of reverse-transcribed cDNA from MEFs: wild-type (open bar), Ung−/− (diagonal striped bar), Smug1↓ (solid bar) and Smug1↓, Ung−/− (cross-hatched bar). Error bars show the standard error of the mean from three experiments. (B) Uracil-DNA glycosylase activity was assayed on a 19-mer double-stranded oligonucleotide with a centrally placed uracil residue in the 5′-32P-end-labelled strand (lane 1) in MEF nuclear extracts supplemented with AP endonuclease. The 9-mer radiolabelled product was resolved by denaturing PAGE and detected by phosphorimager. The substrate was incubated with wild-type extract (WT; lanes 2–5) or Smug1-deficient extracts from Smug1↓ and Smug1↓, Ung−/− cell lines (lanes 6–10), after preincubation with the UNG inhibitor Ugi and/or SMUG1 antibodies, as indicated.
The mammalian Smug1 protein is only expressed at low levels in wild-type cells and is not detected in cell-free extracts by Western blotting with SMUG1 antibodies (Nilsen et al, 2001). However, antibodies against recombinant human SMUG1 protein effectively and specifically inhibit SMUG1 uracil-DNA glycosylase activity in extracts of human and murine cells (Nilsen et al, 2001). Uracil-DNA glycosylase activity was assayed in murine nuclear extracts using a double-stranded oligonucleotide substrate containing a single, centrally placed uracil residue paired opposite guanine. Under such conditions of low substrate concentration, where the lower Km of Smug1 gives it a kinetic advantage over Ung, both enzymes contribute substantially to uracil excision and account for the detectable uracil-DNA glycosylase activity in wild-type cells, while Smug1 accounts for the residual activity in Ung−/− cells (Nilsen et al, 2001). As reported previously, addition of either the UNG inhibitor Ugi or SMUG1 antibodies alone has little effect on the uracil-DNA glycosylase activity in a wild-type extract under these experimental conditions (Figure 1B, lanes 3 and 4), where the enzyme that is not inhibited is present in saturating amounts, but coaddition of both Ugi and SMUG1 antibodies ablates all detectable activity (Figure 1B, lane 5). Similarly, no uracil-DNA glycosylase activity was detected here in a Smug1↓ extract preincubated with both SMUG1 antibodies and Ugi, or a Smug1↓, Ung−/− cell extract preincubated with SMUG1 antibodies alone (Figure 1B, lanes 8 and 10). However, there was also no detectable uracil-DNA glycosylase activity in a Smug1↓ cell extract preincubated with Ugi alone or an unsupplemented Smug1↓, Ung−/− cell extract (Figure 1B, lanes 7 and 9), even upon overexposure of the gel (data not shown). This indicates that the uracil-DNA glycosylase activity in the Smug1↓ cell line (Figure 1B, lane 6) is due almost entirely to Ung, and we estimate that there is <1% residual Smug1 activity in the Smug1↓ and Smug1↓, Ung−/− cell lines (Figure 2B, lanes 7 and 9). However, the Smug1↓ and Smug1↓, Ung−/− cell lines both still contain almost 20% residual Smug1 mRNA (Figure 1A). There is an apparent discrepancy here between levels of the Smug1 mRNA estimated by quantitative PCR of reverse-transcribed RNA and enzyme activity assayed in cell-free extracts; similarly, even transcriptionally regulated low levels of the SMUG1 mRNA are detected by Northern blot analysis, but the protein is not detected by immunoblotting of cell-free extracts with various antibodies (Nilsen et al, 2001; Elateri et al, 2003), suggesting post-transcriptional regulation of SMUG1. More importantly for the present studies, the lack of detectable Smug1 activity following siRNA-mediated Smug1 knockdown in the Smug1↓ and Smug1↓, Ung−/− cell lines means that both these cell lines may be considered functionally as Smug1-deficient mutants.
Figure 2.

Survival of Smug1-deficient MEF cell lines after exposure to γ-irradiation. Wild-type (filled square), Smug1↓ (open square), Ung−/− (filled triangle) and Smug1↓, Ung−/− (open triangle) MEF cell lines were treated at the doses indicated and surviving colonies scored after 10–12 days as a percentage of untreated control. All values are the mean from three experiments; error bars show the standard error of the mean.
Sensitivity of Smug1-deficient cell lines to ionizing radiation
The mammalian SMUG1 enzyme is able to excise various oxidized base lesions from DNA, and others have proposed that this (Matsubara et al, 2004) or more specifically the excision of oxidized, deaminated 5-me-C (Boorstein et al, 2001), rather than excision of premutagenic deaminated cytosine residues, may be its main function in vivo. We therefore examined whether loss of Smug1 activity would sensitize cells to ionizing radiation-induced oxidative base damage by measuring survival of the Smug1↓ and Smug1↓, Ung−/− MEF cell lines following γ-irradiation. Figure 2 shows that the sensitivity of the Smug1↓ cell line to γ-irradiation is the same as that observed for a wild-type MEF cell line, with 50% survival at 2 Gy and no significant difference in the survival curves over the 0.1–20 Gy dose range; the Ung−/− MEF cell line is also not hypersensitive to ionizing radiation in this assay. These data indicate that oxidative base lesions that are induced by ionizing radiation and are substrates of Smug1 (or Ung) are either not cytotoxic in proliferating murine cells, or else can be excised from DNA by another DNA glycosylase in vivo. However, in contrast to Smug1↓ cells, the Smug1↓, Ung−/− cell line deficient in both the Smug1 and Ung uracil-DNA glycosylases is strikingly hypersensitive to γ-irradiation (Figure 2), with a dramatic loss of survival over an ∼10-fold dose range between 0.6 and 7.5 Gy (50% survival at ∼0.8 Gy). Thus, a cytotoxic lesion is generated upon exposure to low levels of ionizing radiation that is efficiently excised by both Smug1 and Ung but not by other DNA repair enzymes in murine cells. In consequence, Smug1↓, Ung−/− cells deficient in both these uracil-DNA glycosylases are hypersensitive to killing by ionizing radiation, whereas Smug1↓ or Ung−/− cells, deficient in either Smug1 or Ung alone, show the same sensitivity as wild-type cells.
The sensitization of the Smug1↓, Ung−/− cell line to ionizing radiation is similar to that reported for siRNA-mediated suppression of the Neil1 DNA glycosylase in murine cells. Neil1 has partially overlapping substrate specificities with the Nth and Ogg1 DNA glycosylases, which excise the major oxidized base lesions thymine glycol and 8-hydroxyguanine, respectively, but candidate lesions were identified that would account for the sensitivity of Neil1 mutant cells to killing by low-level ionizing radiation (Rosenquist et al, 2003). In the case of Smug1, major substrates that are induced by ionizing radiation are the oxidized thymine derivatives 5-hydroxy-me-U and 5-formyl-U (Boorstein et al, 2001; Matsubara et al, 2003; see Figure 3). Of the other mammalian DNA glycosylases, NTH and NEIL1 are reported to have essentially negligible activity on both these lesions (Katafuchi et al, 2004), but the TDG and MBD4 DNA glycosylases, which excise thymine (resulting from deamination of 5-me-C at CpG sites) from a T:G mismatch, also excise 5-hydroxy-me-U and 5-formyl-U opposite G (Liu et al, 2003). However, 5-formyl-U is mutagenic rather than cytotoxic, promoting misincorporation of dGMP (Masaoka et al, 2001), while 5-hydroxy-me-U directs incorporation of dAMP so can also be miscoding, if generated by oxidation and deamination of 5-me-C, but not by oxidation of thymine (Boorstein et al, 2001). Moreover, mutant mammalian cells deficient in the excision of 5-hydroxy-me-U from DNA are actually resistant to its otherwise toxic effects (Boorstein et al, 1989), while the UNG enzyme has no activity on this lesion (Kavli et al, 2002). Thus, the increased ionizing radiation sensitivity of Smug1↓, Ung−/− versus Smug1↓ or Ung−/− cells would appear to be unrelated to either 5-hydroxy-me-U or 5-formyl-U; it is also not attributable to generation of 5-chlorouracil in DNA (see Supplementary data 1).
Figure 3.
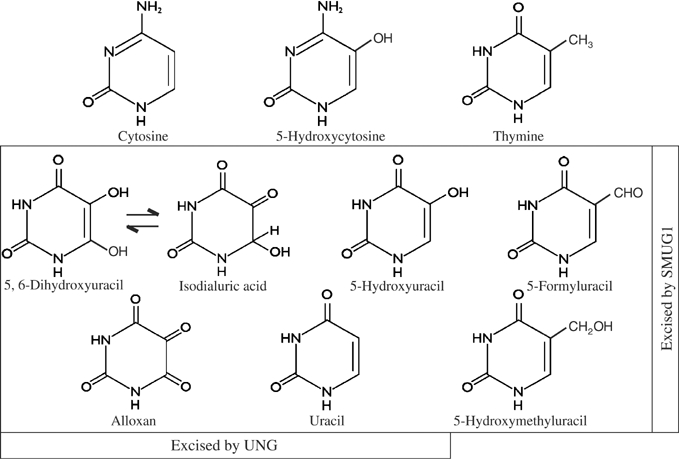
Structures of cytosine, uracil, thymine nucleobases and relevant oxidized derivatives. Lesions excised by UNG and/or SMUG1 are indicated.
Identification of cytosine-derived base lesions excised from γ-irradiated DNA by SMUG1
The SMUG1 protein differs from UNG in being able to accommodate oxidized pyrimidines with bulky substitutions at the C5 position (Wibley et al, 2003; Matsubara et al, 2004); with just a hydroxyl group at this position, both enzymes can excise 5-hydroxyuracil (5-hydroxy-U) from DNA, but not 5-hydroxycytosine (5-hydroxy-C) (Dizdaroglu et al, 1996; Masaoka et al, 2003; see Figure 3). Neither uracil-DNA glycosylase shows any activity on the normal intact cytosine and thymine bases, or on thymine derivatives arising through oxidative attack on the 5,6-double carbon bond, such as thymine glycol, which are substrates of the NTH enzyme. The 5,6-double bond of cytosine is the only primary target for oxidation of this base, converting it to a nonaromatic, nonplanar ring that perturbs normal base pairing and base stacking (Bjelland and Seeberg, 2003). UNG is able to excise certain deaminated derivatives of oxidized cytosine (Dizdaroglu et al, 1996). We have investigated the ability of SMUG1 to act on such potentially cytotoxic lesions in order to identify a common substrate for SMUG1 and UNG that would explain the marked sensitivity of the Smug1↓, Ung−/− cell line to killing by ionizing radiation.
DNA containing [3H]cytosine was γ-irradiated at a high cell-lethal dose in aqueous aerated solution in order to generate a spectrum of oxidized base derivatives (Dizdaroglu et al, 1996) and used in standard enzyme assays with recombinant human SMUG1 and UNG proteins. Radiolabelled ethanol-soluble products released by enzyme incubation were resolved by high-performance liquid chromatography (HPLC) (Figure 4) and identified by comparison with reference compounds (Figure 3). UNG has previously been reported to excise three of 12 base derivatives formed by γ-irradiation of DNA under similar conditions; these were 5-hydroxy-U, isodialuric acid and alloxan (Dizdaroglu et al, 1996). In the previous study, products released from unlabelled irradiated DNA were analysed by gas chromatography/mass spectrometry, during which derivatization of samples converted isodialuric acid from its keto to its enol tautomeric form detected as 5,6-dihydroxyuracil; isodialuric acid was reported to be the prevalent form in aqueous solution and in DNA (Zastawny et al, 1995). Here, we obtained analogous results for UNG by HPLC analysis, with resolution of discrete peaks of radioactivity coincident with these markers (Figure 4). Analysis of products released by SMUG1 gave a very similar HPLC profile, showing that SMUG1 can also excise 5-hydroxy-U, isodialuric acid and alloxan from γ-irradiated DNA; addition of the UNG inhibitor Ugi did not affect SMUG1 activity (Figure 4). There were no significant differences in the distribution of radiolabel in the UNG versus SMUG1 profiles, indicating that neither enzyme has a marked preference for a particular lesion. Both UNG and SMUG1 also excise uracil from the γ-irradiated DNA substrate (Figure 4); uracil was not detected in nonirradiated DNA (data not shown) and results from radiation-induced oxidation and deamination, with 5,6-dihydrocytosine as a putative reaction intermediate (Bjelland and Seeberg, 2003). Generated in DNA at elevated levels, uracil itself might be cytotoxic in irradiated Smug1↓, Ung−/− cells lacking both the Smug1 and Ung uracil-DNA glycosylases if, for example, incision but then only inefficient repair of U:G lesions by the mismatch-specific Tdg thymine-DNA glycosylase were to lead to DNA fragmentation. No other discrete products were resolved and there was negligible background due to nonenzymatic degradation of unstable intermediates, as the irradiated DNA was extensively dialysed, used immediately in enzyme assays and generated no HPLC peaks of ethanol-soluble radioactive material when incubated in reaction buffer alone (data not shown).
Figure 4.

Cytosine-derived base lesions excised from irradiated DNA by human DNA glycosylases. HPLC profiles are shown of ethanol-soluble radiolabelled material released from γ-irradiated [3H]cytosine-containing DNA by the uracil-DNA glycosylases UNG (filled triangle) and SMUG1 (open square); SMUG1 was preincubated with the UNG inhibitor Ugi. The positions of reference compounds are indicated (detected by UV absorbance).
The SMUG1 uracil-DNA glycosylase has been reported previously to act on 5-hydroxy-U, as well as other uracil derivatives with an oxidized group at C5, 5-hydroxy-me-U and 5-formyl-U (Masaoka et al, 2003), in addition to uracil itself (Haushalter et al, 1999). Here, we have extended the substrate specificity of SMUG1 to include two deaminated derivatives of oxidized cytosine resulting from saturation of the 5,6-double carbon bond, namely isodialuric acid and alloxan (Figure 3). We have shown that the hNTH DNA glycosylase does not have detectable activity on these lesions or, contrary to a number of published reports (reviewed in Dizdaroglu, 2003), on 5-hydroxy-U when assayed in the presence of the Ugi inhibitor (see Supplementary data 2). Uracil-DNA glycosylase activity has previously been spuriously assigned to several human proteins quite unrelated to DNA processing, as well as purified Escherichia coli enzymes assayed in the absence of Ugi (Bennett et al, 2004). NEIL2 has been described as a novel human DNA glycosylase for the repair of cytosine-derived lesions (Hazra et al, 2002), although it has negligible activity on 5-hydroxy-U compared to NEIL1 (Katafuchi et al, 2004), but the NEIL enzymes need to be re-evaluated in the presence of the Ugi inhibitor. Although there may currently be some disagreement over the putative activity of other DNA glycosylases on 5-hydroxy-U, we report here that SMUG1 and UNG are the only enzymes in mammalian cells that excise isodialuric acid and alloxan from γ-irradiated DNA. Whereas 5-hydroxy-U retains an intact pyrimidine ring and is a miscoding rather than cytotoxic lesion, the nonaromatic, nonplanar ring of isodialuric acid and alloxan resembles the well-characterized cytotoxic lesion thymine glycol, which is also generated by ionizing radiation (Bjelland and Seeberg, 2003). Thus, isodialuric acid and alloxan can be identified as lesions explaining the unique sensitivity of the Smug1↓, Ung−/− cell line to killing by ionizing radiation.
Spontaneous mutagenesis in Smug1-deficient cell lines
In order to investigate the effects of Smug1 deficiency on spontaneous mutagenesis, mutation frequency was determined at the endogenous X-linked hypoxanthine-guanine phosphoribosyl transferase (hprt) locus in Smug1↓ and Smug1↓, Ung−/− MEF cell lines versus a wild-type control; the Ung−/− MEF cell line was also included in this analysis. Previously, a modest (<1.5-fold) increase in in vivo spontaneous mutagenesis had been measured for a nontranscribed lacI reporter transgene in the spleen and thymus of Ung−/− mice (Nilsen et al, 2000). Mutant frequency at the hprt locus was estimated here for comparison and in the Ung−/− cell line was 5.2-fold higher than that of the wild-type Ung+/+ control (Table I). Our data are entirely consistent with previous direct comparisons between these in vivo and in vitro mutagenesis assays, where a 1.5-fold increase in mutation frequency at lacI in mouse spleen corresponded with an ∼5-fold increase at hprt in cultured splenocytes due to the higher spontaneous background at lacI; this means that mutagenic insults that produce ⩽5-fold increase in mutation frequency at an endogenous locus are difficult to detect in the lacI system (Skopek et al, 1995). The hprt mutant frequency for the control Ung+/+ cell line is correspondingly lower than that for the lacI transgene in Ung+/+ mice (Nilsen et al, 2000) but both values are within the normal range for each assay system (Skopek et al, 1995). Thus, there is no actual increase in spontaneous mutagenesis due to Ung deficiency measured at the endogenous hprt locus versus the nontranscribed lacI transgene, which might have reflected a contribution of transcription to increased deamination of cytosine and mutagenesis at C:G base pairs. Efficient and stable siRNA-mediated silencing of Smug1 expression in Ung−/− versus Ung+/+ MEFs means that an assessment of the contribution of Smug1 to spontaneous mutagenesis can now be made using the hprt system. Spontaneous mutant frequencies were estimated for the Smug1-deficient Smug1↓ and Smug1↓, Ung−/− MEF cell lines maintained in culture for a further six passages after stable siRNA suppression of Smug1 had been achieved, and were 2.4- and 9.6-fold higher than the wild-type control, respectively (Table I). Thus, estimates of hprt mutant frequency would indicate that Smug1-deficient cells, like the Ung−/− null, display a moderate mutator phenotype, but the Smug1↓, Ung−/− cell line deficient in both Smug1 and Ung shows a marked ∼10-fold increase in spontaneous mutations.
Table 1.
Increased spontaneous mutation frequencies at the hprt locus in MEF cell lines deficient in Smug1 and/or Ung
| Genotypea | Cells screened (× 106) | Mutants | Mutant frequency (× 10−6) | Increase (× control) |
|---|---|---|---|---|
| Control (Ung+/+) | 25 | 36 | 1.44 | — |
| Ung−/− | 22.65 | 170 | 7.51 | 5.2 |
| Smug↓ | 20.35 | 70 | 3.44 | 2.4 |
|
Smug↓, Ung−/− |
18.65 |
257 |
13.78 |
9.6 |
| Ung+/+ and Ung−/− MEFs were assayed at passage 28 and used to generate the Smug1↓ mutant cell lines; stable Smug1 suppression was achieved within eight passages and cells were assayed after a further six passages in culture. | ||||
Results from assays of both mutation frequency (Table I) and also mutation rate (see Supplementary data 3) clearly demonstrate that Smug1 as well as Ung is an antimutator in mammalian cells. In order to analyse the specific contributions of the Smug1 and Ung uracil-DNA glycosylases to preventing spontaneous mutagenesis in mammalian cells, individual hprt mutant clones present in nonselected cultures (Table I) were sequenced from Smug1↓ and Ung−/− versus Smug1↓, Ung−/− MEF cell lines; the mutational spectra are shown in Figure 5. The most prevalent mutational change in wild-type cells was C:G to T:A transition, but the number of mutations in this class was markedly elevated in uracil-DNA glycosylase-deficient cells and would appear to correlate with the 5.2-fold (Ung−/−), 2.4-fold (Smug1↓) and 9.6-fold (Smug1↓, Ung−/−) increase in spontaneous mutants measured at the hprt locus in these cell lines (Table I). There was no increase in transversions at C:G base pairs. Furthermore, while ∼60% of C:G to T:A transition mutations in wild-type cells occurred as expected at CpG dinucleotides due to deamination of 5-me-C to thymine (Millar et al, 2002), >90% of C:G to T:A transitions in Ung−/− cells, Smug1↓ and Smug1↓, Ung−/− cells occurred at non-CpG sequences, consistent with replication of premutagenic U:G mispairs as would result from spontaneous deamination of cytosine. The increased frequency of C:G to T:A mutations at the hprt locus in Ung−/− MEFs is in good agreement with the small increase in C → T mutagenesis of a lacI transgene in Ung−/− null mice (Nilsen et al, 2000). Here, deficiency in the Smug1 enzyme similarly leads to a moderately increased frequency of spontaneous transition mutations at C:G base pairs in Smug1↓ cells. The apparently lower level of mutagenesis due to Smug1 versus Ung deficiency may be due to some residual Smug1 activity in the siRNA-mediated Smug1↓ knockdown compared with the ablation of Ung activity in the Ung−/− gene-targeted knockout, or the greater activity of Ung in proliferating cells (Nilsen et al, 2000, 2001); thus, the contribution of Smug1 to the prevention of spontaneous mutagenesis in vivo may be underestimated here.
Figure 5.
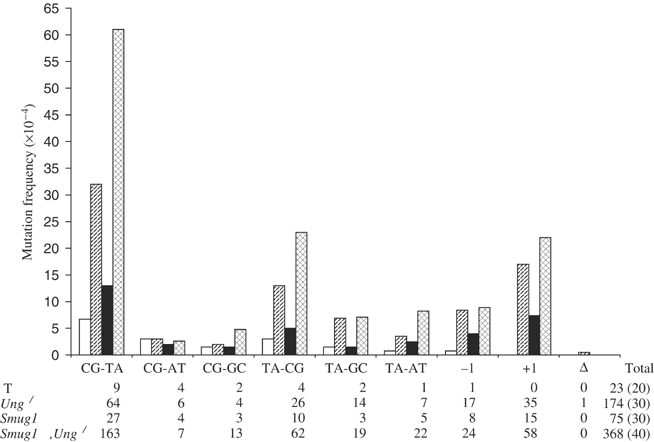
Mutational spectra in MEF cell lines deficient in Smug1 and/or Ung. The hprt open reading frame (672 bp) was sequenced in individual mutant colonies (as in Table I) from wild-type (WT; open bar), Ung−/− (diagonal striped bar), Smug1↓ (solid bar) and Smug1↓, Ung−/− (cross-hatched bar) MEF cell lines. Data are presented for the different classes of mutation (as indicated on the x-axis). The number of individual mutations of each class (N) and the total number of mutations are shown below for each cell line; the number of mutant clones sequenced (M) is given in parentheses. The frequency of each class of mutation (y-axis) is calculated as N/(672M).
Compared with cells deficient in either Smug1 or Ung alone, Smug1↓, Ung−/− cells deficient in both uracil-DNA glycosylases have a marked mutator phenotype with an ∼10-fold increase in C:G to T:A transition mutations (Figure 5). The apparently additive effect of combined Smug and Ung deficiency indicates that they have nonredundant roles in preventing mutagenesis at C:G base pairs. This does not appear to be due to different activities of Smug1 and Ung in terms of the spontaneous lesions excised by each enzyme (Figure 3), as Smug1-deficient cells do not show an increase in C → T transitions at CpG or of T:A → C:G mutations that would be the hallmarks of mutagenesis due to the Smug1-specific substrates 5-hydroxy-me-U (Boorstein et al, 2001) and 5-formyl-U (Masaoka et al, 2001), respectively. Rather, the mutational spectra are entirely consistent with both Ung and Smug1 acting on deaminated cytosine, and their nonredundancy may reflect differences in subnuclear compartmentalization and cell cycle regulation of the two enzymes. Smug1 appears to be constitutively expressed (Nilsen et al, 2001), whereas Ung is localized to replication foci (Otterlei et al, 1999). Consistent with a function before or coincident with the onset of DNA replication, the nuclear isoform of UNG is degraded during S phase and the protein does not reappear until early G1 (Fischer et al, 2004). Clearly, not all U:G lesions occurring outside of replication foci are corrected by Ung prior to replication as has been proposed (Kavli et al, 2002), and some become fixed during replication in Smug1↓ cells, while, conversely, some U:G lesions are apparently not accessible to Smug1, possibly arising in temporarily exposed single-stranded DNA within replication foci, and so become fixed in Ung−/− cells; these effects are then additive in the Smug1↓, Ung−/− cell line deficient in both enzymes. There is also a marked increase here in frameshift mutations, especially +1 insertions, that is predominantly due to Ung deficiency (Figure 5) and was noted previously in proliferating Ung−/− mouse tissue (Nilsen et al, 2000); although the mechanism is presently unclear, it appears to be largely due to polymerase error at -CC-, or -GG-, dinucleotides (data not shown). This would be consistent with a role of Ung at replication forks, although +1 frameshifts are also increased in the Smug1↓ cell line. Interestingly, mutations at T:A base pairs of all classes were slightly elevated in mutant cells, and again predominantly but not exclusively due to Ung deficiency (Figure 5). U:A mispairs due to misincorporation of dUMP that persist in Ung−/− cells (Nilsen et al, 2000) may become targets for mutagenic misinsertions opposite the U by error-prone DNA polymerases such as pol iota at the replication fork (Vaisman and Woodgate, 2001; Kannouche et al, 2002).
We have shown that Smug1 acts as an antimutator in mammalian cells. While deficiency of either Ung or Smug1 alone confers a moderate spontaneous mutator phenotype, cells lacking both enzymes have a greatly elevated level of C:G to T:A transition mutations, indicating that these are the only enzymes that effectively repair deaminated cytosine in mammalian cells, and confirming that they have separable, nonredundant roles in this regard. The highly efficient siRNA-mediated suppression of Smug1 means that we can further exploit this approach to achieve stable germline transmission of RNA interference in the mouse (Carmell et al, 2003; Kunath et al, 2003) and so evaluate the role of Smug1 in vivo. Furthermore, we have demonstrated that SMUG1 and UNG, but not NTH, excise several cytosine-derived products of oxidative damage, including the cytotoxic lesions isodialuric acid and alloxan, such that cells deficient in both the Smug1 and Ung uracil-DNA glycosylases are hypersensitive to ionizing radiation. As cells are sensitized to killing by low levels of radiation in the therapeutic range, suppression of these DNA repair enzymes may be of relevance to the efficacy of cancer radiotherapy.
Materials and methods
siRNA-mediated suppression of Smug1 in MEFs
Gene-targeted Ung−/− mice and permanent Ung−/− and Ung+/+ MEF cell lines have been described previously (Nilsen et al, 2000). Cells were cultured in Dulbecco's MEM/Ham's F12 (3:1) with 10% fetal bovine serum. siRNA oligonucleotides targeted to the Smug1 mRNA were designed based on the murine Smug1 cDNA (NM_027885) and their specificity to the target sequences confirmed by BLAST analysis (www.dharmacon.com). Three optimum 19-mer oligonucleotides that targeted different regions of the Smug1 open reading frame were synthesized (Dharmacon RNA Technologies), and these corresponded to nucleotides 205–223, 522–540 and 800–818 of the 837-nucleotide Smug1 open reading frame. Oligonucleotides were transfected into wild-type (Ung+/+) MEFs using Oligofectamine™ Reagent (Invitrogen Life Technologies), and the efficiency of siRNA-mediated silencing of Smug1 mRNA expression evaluated by real-time PCR 48 h post-transfection (see below). Then, 64-mer sense and antisense DNA oligonucleotides (Sigma Genosys) were annealed, phosphorylated and ligated into the pSUPER.retro plasmid (OligoEngine) to encode small hairpin RNAs corresponding to two of these siRNA oligonucleotides, which targeted the sequences -AACTATGTGACTCGCTACT- (nucleotides 205–223) and -TGCCTCTGCTAACGGATGA- (nucleotides 800–818). Expression vectors were transfected into the Ung−/− and Ung+/+ MEF cell lines at passage 28 using TransIT Transfection Reagent (Mirus Corp.). Puromycin-resistant colonies were isolated following culture in 5 μg ml−1 puromycin (Sigma), and those showing optimal silencing of Smug1 expression were again identified by real-time PCR (see below). Stable siRNA-mediated Smug1 knockdown cell lines were isolated following repeated cloning of appropriate colonies for up to eight passages in selective medium (2 μg ml−1 puromycin), with re-evaluation of Smug1 mRNA expression at each stage. All data presented here are for cells expressing the former construct, which targeted nucleotides 205–223 of the murine Smug1 open reading frame.
Real-time PCR
Total RNA was isolated from cell pellets and cDNA prepared using the Cells-to-cDNA II kit (Ambion) as recommended by the manufacturer. Smug1 mRNA was quantified by real-time PCR using the ABI PRISM 7900HT Detection System (Applied Biosystems). PCR primers and fluorescent probes were obtained from Applied Biosciences; reactions were performed and results analysed according to the standard protocol using the TaqMan Universal PCR Master Mix (Applied Biosystems).
γ-Irradiation cell survival assays
Cells were harvested and resuspended in phosphate-buffered saline (PBS) at a concentration of 106 cells ml−1. Cells were exposed to a dose range of 0–20 Gy in an IBL437C 137Cs irradiator (CIS bio international; dose rate 2.4 Gy min−1). Cells were plated in triplicate for each dose (100 cells per 100 mm dish at 0–2 Gy; 500 cells per 100 mm dish at ⩾5 Gy). Surviving colonies were stained with Giemsa and counted after 10–12 days in culture. Survival at each dose was calculated as a percentage of the untreated control after adjustment for the actual plating efficiency of each cell line, which was ∼50%.
Assay of uracil-DNA glycosylase activity in cell-free extracts
Cell-free extracts were prepared from MEF cell lines, using the PARIS kit (Ambion). Uracil-DNA glycosylase activity was assayed as described previously (Nilsen et al, 2001). Briefly, 5 μg of extract was preincubated on ice for 10 min with either 10 μg SMUG1 antibodies (Nilsen et al, 2001), 4 U Ugi (Bennett et al, 1993), or SMUG1 antibodies and Ugi together, then incubated at 37°C for 1 h in 20 mM Tris–HCl pH 8.0, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, 100 μg ml−1 BSA, with 1 ng AP endonuclease and 0.24 pmol of a 19-mer double-stranded oligonucleotide substrate, containing a centrally placed uracil residue in the 5′-32P-end-labelled strand. Reactions were stopped by addition of 1 M piperidine, AP sites cleaved by incubation at 90°C for 20 min and samples dried under vacuum, resuspended in 95% formamide/dyes and analysed by gel electrophoresis and phosphorimager, as described. His-tagged recombinant human AP endonuclease was a gift from I Hickson.
Release of cytosine base derivatives from irradiated DNA by recombinant enzymes
A [3H]cytosine containing DNA substrate was prepared by nick translation of calf thymus DNA by DNA polymerase I in the presence of [3H]deoxycytidine 5′-triphosphate (60 Ci mmol−1, labelled at multiple sites with a hydrogen transfer catalyst; Amersham Biosciences). DNA was precipitated with ethanol, redissolved in 10 mM phosphate buffer (pH 7.4) at a concentration of 50 μg ml−1 and irradiated in an IBL437C 137Cs irradiator (CIS bio international) at a dose of 227 Gy (dose rate 2.4 Gy min−1). Such treatment generates <5% S1 nuclease-sensitive material, indicating that the double-stranded DNA structure is largely preserved (Breimer and Lindahl, 1985). Following irradiation, the DNA was dialysed extensively (20 h) against 10 mM phosphate buffer (pH 7.4) and 10 mM NaCl and then a further 4 h against enzyme reaction buffer (20 mM Tris–HCl pH 8.0, 50 mM NaCl, 1 mM EDTA, 1 mM DTT). DNA was used immediately in enzyme assays, concentrated enzymes being added directly to aliquots of the DNA solution (1.5 μg). Purified recombinant human SMUG1 (Nilsen et al, 2001) has been described; recombinant human UNG protein lacking the N-terminal 84 amino acids (UNGΔ84) was expressed and purified as described by Slupphaug et al (1995). All reactions contained 0.1 μg AP endonuclease (as above), and either 7.5 μg SMUG1 or 0.4 μg UNG. Where applicable, enzymes were incubated with 4 U Ugi (as above) at 0°C for 10 min before addition to the DNA. Reactions were incubated at 37°C for 1 h. After incubation, DNA was precipitated with ethanol and the supernatant fractions recovered. Reference compounds (1–2 μg each isodialuric acid, alloxan, 5-hydroxy-U, uracil and cytosine) were added to each supernatant fraction, which was then dried under vacuum and resuspended in 10 μl water. All chemicals were purchased from Sigma, except isodialuric acid, which was a gift from M Dizdaroglu.
Derivatives of 3H-labelled cytosine in supernatant fractions were analysed by HPLC, on a Phenomenex Synergi Hydro reverse phase column (250 × 4.6 mm) in water, at a flow rate of 0.5 ml min−1. After 10 min, a linear gradient of 0–50% methanol was applied over 25 min. Reference compounds (as above) were detected by UV absorbance. The ring-saturated markers isodialuric acid and alloxan were detected by their weak end absorption in the near-UV region. Fractions were collected at 0.5 min intervals and radioactivity quantified by scintillation counting.
hprt mutation assays
To calculate spontaneous mutant frequencies at the hprt locus, ∼2 × 107 plated cells were screened in selective medium without hypoxanthine containing 6-thioguanine (6-TG; 40 μM) in 150 cm plates and 6-TG-resistant mutants scored after 10–12 days incubation. 6-TG-resistant colonies were isolated from 150 cm plates and direct sequencing of cDNA was performed essentially as described (Yang et al, 1989). A total of 20–40 colonies were analysed for each cell line. cDNA was prepared using the Cells-to-cDNA II kit (Ambion). The hprt open reading frame (657 bp) was amplified using the Expand High Fidelity PCR System (Roche) with annealing temperature at 50°C (sense PCR primer: 5′-GTCATGCCGACCCGCAG-3′ (−3 to +14); antisense PCR primer 5′-CTTGCGCTCATCTTAGG-3′ (+653–669)) and sequenced after purification (QIAquick PCR Purification Kit, Qiagen) with the antisense PCR primer using the Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Where necessary, putative mutations were confirmed by sequencing with an appropriate internal sense primer. The sequences were analysed using Sequencher software, version 4.1.2, and mutations were scored as sequences deviating from consensus nucleotide database annotations.
Supplementary Material
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3
Acknowledgments
We thank Pertti Koivisto for help with HPLC analysis, Miral Dizdaroglu (National Institute of Standards and Technology, Gaithersburg, USA) for the kind gift of isodialuric acid and Ian Hickson (Cancer Research UK, Oxford) for providing AP endonuclease. This work was supported by Cancer Research UK.
References
- Aravind L, Koonin EV (2000) The alpha/beta fold uracil DNA glycosylases: a common origin with diverse fates. Genome Biol 1: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Lindahl T (2004) Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet 38: 445–476 [DOI] [PubMed] [Google Scholar]
- Bennett SE, Chen C-Y, Mosbaugh DW (2004) Escherichia coli nucleoside diphosphate kinase does not act as a uracil-processing DNA repair nuclease. Proc Natl Acad Sci USA 101: 6391–6396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett SE, Schimerlik MI, Mosbaugh DW (1993) Kinetics of the uracil-DNA glycosylase/inhibitor protein association. J Biol Chem 268: 26879–26885 [PubMed] [Google Scholar]
- Bjelland S, Seeberg E (2003) Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res 531: 37–80 [DOI] [PubMed] [Google Scholar]
- Boorstein RJ, Chiu L-N, Teebor GW (1989) A mammalian cell line deficient in activity of the DNA repair enzyme 5-hydroxymethyluracil-DNA glycosylase is resistant to the toxic effects of the thymidine analog 5-hydroxymethyl-2′-deoxyuridine. Mol Cell Biol 12: 5536–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorstein RJ, Cummings A Jr, Marenstein DR, Chan MK, Ma Y, Neubert TA, Brown SM, Teebor GW (2001) Definitive identification of mammalian 5-hydroxymethyluracil DNA N-glycosylase activity as SMUG1. J Biol Chem 276: 41991–41997 [DOI] [PubMed] [Google Scholar]
- Breimer LH, Lindahl T (1985) Thymine lesions produced by ionizing radiation in double-stranded DNA. Biochemistry 24: 4018–4022 [DOI] [PubMed] [Google Scholar]
- Carmell MA, Zhang L, Conklin DS, Hannon GJ, Rosenquist TA (2003) Germline transmission of RNAi in mice. Nat Struct Biol 10: 91–92 [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M (2003) Substrate specificities and excision kinetics of DNA glycosylases involved in base-excision repair of oxidative DNA damage. Mutat Res 531: 109–126 [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M, Karakaya A, Jaruga P, Slupphaug G, Krokan HE (1996) Novel activities of human uracil DNA N-glycosylase for cytosine-derived products of oxidative DNA damage. Nucleic Acids Res 24: 418–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan BK, Weiss B (1982) Specific mutator effects of ung (uracil-DNA glycosylase) mutations in Escherichia coli. J Bacteriol 151: 750–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elateri I, Muller-Weeks S, Caradonna S (2003) The transcription factor, NFI/CTF plays a positive regulatory role in expression of the hSMUG1 gene. DNA Repair 2: 1371–1385 [DOI] [PubMed] [Google Scholar]
- Endres M, Biniszkiewicz D, Sobol RW, Harms C, Ahmadi M, Lipski A, Katchanov J, Mergenthaler P, Dirnagl U, Wilson SH, Meisel A, Jaenisch R (2004) Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J Clin Invest 113: 1711–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer JA, Muller-Weeks S, Caradonna S (2004) Proteolytic degradation of the nuclear isoform of uracil-DNA glycosylase occurs during S phase of the cell cycle. DNA Repair 3: 505–513 [DOI] [PubMed] [Google Scholar]
- Haushalter KA, Todd Stukenberg MW, Kirschner MW, Verdine GL (1999) Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr Biol 9: 174–185 [DOI] [PubMed] [Google Scholar]
- Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldagh I, Mokkapati SK, Mitra S, Izumi T (2002) Identification of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem 277: 30417–30420 [DOI] [PubMed] [Google Scholar]
- Impellizzeri KJ, Anderson B, Burgers PM (1991) The spectrum of spontaneous mutations in a Saccharomyces cerevisiae uracil-DNA-glycosylase mutant limits the function of this enzyme to cytosine deamination repair. J Bacteriol 173: 6807–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannouche P, de Henestrosa ARF, Coull B, Vidal AE, Gray C, Zicha D, Woodgate R, Lehmann AR (2002) Localisation of DNA polymerases eta and iota to the replication machinery is tightly co-ordinated in human cells. EMBO J 21: 6246–6256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katafuchi A, Nakano T, Masaoka A, Terato H, Iwai S, Hanaoka F, Ide H (2004) Differential specificity of human and Escherichia coli endonuclease III and VIII homologues for oxidative base lesions. J Biol Chem 279: 14464–14471 [DOI] [PubMed] [Google Scholar]
- Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G (2002) hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem 277: 39926–39936 [DOI] [PubMed] [Google Scholar]
- Kreutzer DA, Essigmann JM (1998) Oxidized, deaminated cytosines are a source of C --> T transitions in vivo. Proc Natl Acad Sci USA 95: 3578–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath T, Gash G, Lickert H, Jones N, Pawson T, Rossant J (2003) Transgenic RNA interference in ES cell-derived embryos recapitulates a genetic null phenotype. Nat Biotech 21: 559–561 [DOI] [PubMed] [Google Scholar]
- Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362: 709–715 [DOI] [PubMed] [Google Scholar]
- Liu P, Burdzy A, Sowers LC (2003) Repair of the mutagenic DNA oxidation product, 5-formyluracil. DNA Repair 2: 199–210 [DOI] [PubMed] [Google Scholar]
- Masaoka A, Matsubara M, Hasegawa R, Tanaka T, Kurisu S, Terato H, Ohyama Y, Karino N, Matsuda A, Ide H (2003) Mammalian 5-formyluracil-DNA glycosylase. 2. Role of SMUG1 uracil-DNA glycosylase in repair of 5-formyluracil and other oxidized and deaminated base lesions. Biochemistry 42: 5003–5012 [DOI] [PubMed] [Google Scholar]
- Masaoka A, Terato H, Kobayashi M, Ohyama Y, Ide H (2001) Oxidation of thymine to 5-formyluracil in DNA promotes misincorporation of dGMP and subsequent elongation of a mismatched primer terminus by DNA polymerase. J Biol Chem 276: 16501–16510 [DOI] [PubMed] [Google Scholar]
- Matsubara M, Masaoka A, Tanaka T, Miyano T, Kato N, Terato H, Ohyama Y, Iwai S, Ide H (2003) Mammalian 5-formyluracil-DNA glycosylase. 1. Identification and characterization of a novel activity that releases 5-formyluracil from DNA. Biochemistry 42: 4993–5002 [DOI] [PubMed] [Google Scholar]
- Matsubara M, Tanaka T, Terato H, Ohmae E, Izumi S, Katayanagi K, Ide H (2004) Mutational analysis of the damage-recognition and catalytic mechanism of human SMUG1 DNA glycosylase. Nucleic Acids Res 32: 5291–5302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A (2002) Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 297: 403–405 [DOI] [PubMed] [Google Scholar]
- Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK (2004) Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues. J Biol Chem 279: 52353–52360 [DOI] [PubMed] [Google Scholar]
- Muller-Weeks S, Balzer RJ, Anderson R, Caradonna S (2005) Proliferation-dependent expression of nuclear uracil-DNA glycosylase is mediated in part by E2F-4. DNA Repair 4: 183–190 [DOI] [PubMed] [Google Scholar]
- Neuberger MS, Harris RS, di Noia J, Petersen-Mahrt SK (2003) Immunity through DNA deamination. Trends Biochem Sci 28: 305–312 [DOI] [PubMed] [Google Scholar]
- Nilsen H, An Q, Lindahl T (2005) Mutation frequencies and AID activation state in B-cell lymphomas from UNG-deficient mice. Oncogene 24: 3063–3066 [DOI] [PubMed] [Google Scholar]
- Nilsen H, Haushalter KA, Robins P, Barnes DE, Verdine GL, Lindahl T (2001) Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J 20: 4278–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE (2000) Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell 5: 1059–1065 [DOI] [PubMed] [Google Scholar]
- Nilsen H, Stamp G, Andersen S, Hrivnak G, Krokan HE, Lindahl T, Barnes DE (2003) Gene-targeted mice lacking the Ung uracil-DNA glycosylase develop B-cell lymphomas. Oncogene 22: 5381–5386 [DOI] [PubMed] [Google Scholar]
- Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakke O, Krokan HE (1999. Post-replicative base excision repair in replication foci. EMBO J 18: 3834–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Di Noia JM, Neuberger MS (2004) Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic hypermutation. Mol Cell 16: 163–171 [DOI] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS (2002) Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol 12: 1748–1755 [DOI] [PubMed] [Google Scholar]
- Rosenquist TA, Zaika E, Fernandes AS, Zharkov DO, Miller H, Grollman AP (2003) The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death. DNA Repair 147: 1–11 [DOI] [PubMed] [Google Scholar]
- Skopek TR, Kort KL, Marino DR (1995) Relative sensitivity of the endogenous hprt gene and lacI transgene in ENU-treated Big Blue B6C3F1™ mice. Environ Mol Mut 26: 9–15 [DOI] [PubMed] [Google Scholar]
- Slupphaug G, Eftedal I, Kavli B, Bharati S, Helle NM, Haug T, Levine DW, Krokan HE (1995) Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry 34: 128–138 [DOI] [PubMed] [Google Scholar]
- Vaisman A, Woodgate R (2001) Unique misinsertion specificity of polι may decrease the mutagenic potential of deaminated cytosines. EMBO J 20: 6520–6529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Kreutzer DA, Essigmann JM (1998) Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat Res 400: 99–115 [DOI] [PubMed] [Google Scholar]
- Wibley JE, Waters TR, Haushalter K, Verdine GL, Pearl LH (2003) Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol Cell 11: 1647–1659 [DOI] [PubMed] [Google Scholar]
- Yang JL, Maher VM, McCormick JJ (1989) Amplification and direct nucleotide sequencing of cDNA from the lysate of low numbers of diploid human cells. Gene 83: 347–354 [DOI] [PubMed] [Google Scholar]
- Zastawny TH, Doetsch PW, Dizdaroglu M (1995) A novel activity of E. coli uracil-DNA N-glycosylase. Excision of isodialuric acid (5, 6-dihydroxyuracil), a major product of oxidative DNA damage, from DNA. FEBS Lett 364: 255–258 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3