

Abstract
Public repositories of metabolomics mass spectra encompass more than 1 billion entries. With open search, dot product or entropy similarity, comparisons of a single tandem mass spectrometry spectrum take more than 8 h. Flash entropy search speeds up calculations more than 10,000 times to query 1 billion spectra in less than 2 s, without loss in accuracy. It benefits from using multiple threads and GPU calculations. This algorithm can fully exploit large spectral libraries with little memory overhead for any mass spectrometry laboratory.
Nontargeted analyses of complex samples by mass spectrometry are used in hundreds of laboratories to enumerate exposome compounds, metabolites and lipids1,2. Thousands of tandem mass spectrometry (MS/MS) spectra are generated per sample. The complements of such spectra are compiled in public repositories such as MassIVE3 with more than 6 billion spectra and its small molecule portion, MassIVE/GNPS with around 1.2 billion spectra4. For compound identification, such experimental mass spectra must be matched against spectral libraries. We here present a Flash entropy search that enables users to query their own datasets against all publicly available spectra on personal computers, including advanced queries such as neutral loss and hybrid searches.
To comprehensively query large numbers of experimental spectra against libraries creates a large computational problem. MassBank. us lists over 2 million spectra, NIST 23 includes 2.4 million spectra and mzCloud has over 10 million spectra, plus spectra in the MetabolomicsWorkbench5 and MetaboLights6. Notably, MS/MS spectra contain information beyond direct matches (identity search, for example via MASST7), which can be revealed by open search8, neutral loss search9 or hybrid search options10,11 (Fig. 1a–d). Such searches find clusters of structurally related compounds such as illicit drug variants or modified natural products12,13. Hybrid search similarity networks are also empowered by the ‘molecular network’ using GNPS4. MSFragger software proposed omitting non-matching ions in MS/MS searches14 for open search analysis of proteomics data14; however, this software cannot be applied for metabolomics data. It cannot perform entropy similarity and it is unable to perform neutral loss and hybrid search. Current algorithms in metabolomics are far too slow to perform open similarity searches on a whole database level. For example, the classic dot-product similarity algorithm takes 25 s to perform an open search of a single MS/MS spectrum against a library of 1 million spectra15,16 (Supplementary Table 1), which extends to approximately 7 h to query 1 billion spectra. This is not a practical solution in the era of big data and machine learning.
Fig. 1 |. Overview of Flash entropy searches.
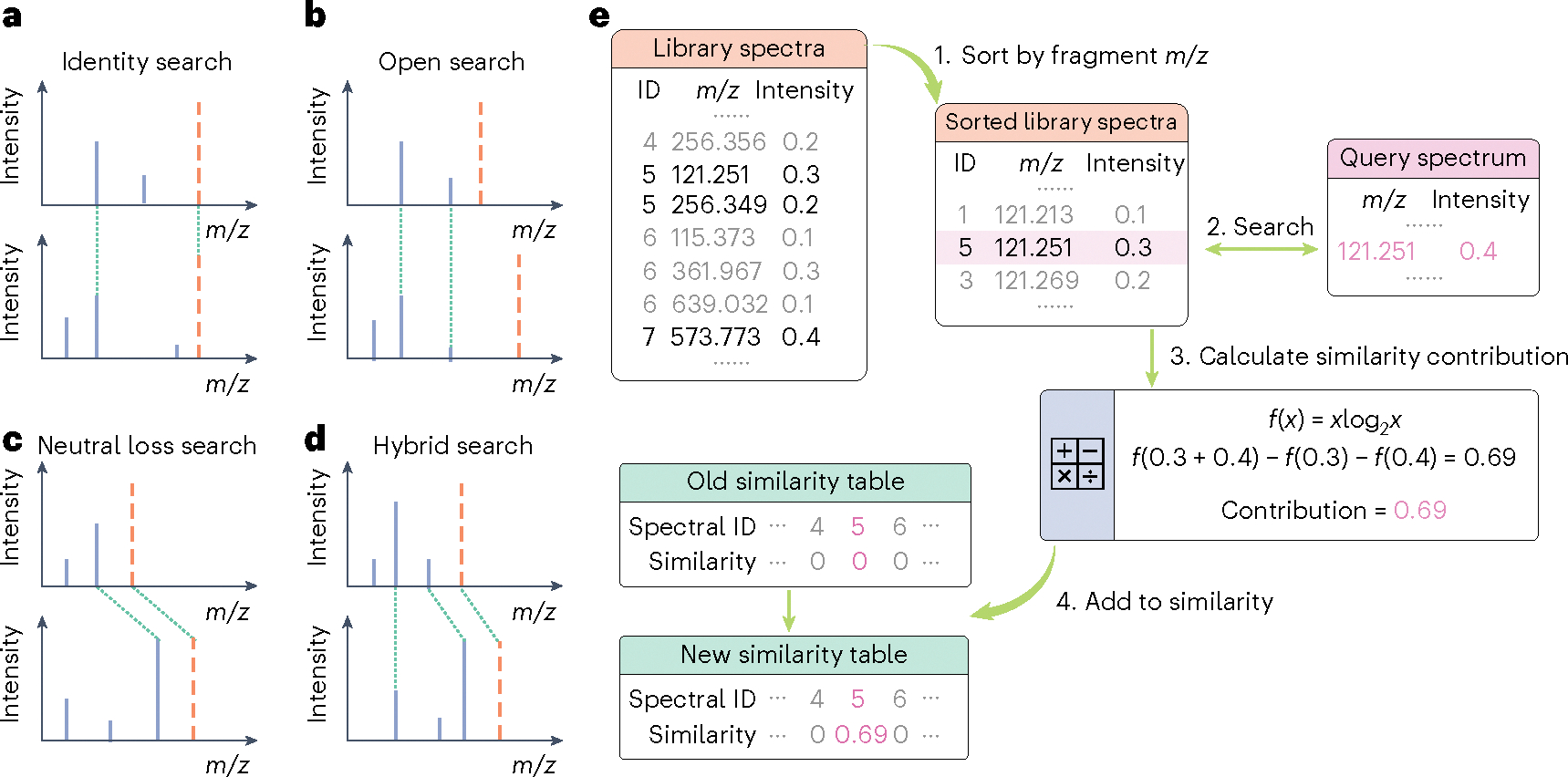
a–d, Types of MS/MS similarity searches supported by the Flash entropy search algorithm. Fragment ions are shown in blue and precursor ions are represented by orange lines. Comparisons performed by the algorithms are symbolized by dashed green lines. e, Workflow for the Flash entropy search algorithm. Spectra are cleaned and normalized to . All library fragment ions are sorted by . Query spectra are used to look up library spectra with matching fragment ions within . Subsequently, entropy similarity contributions are calculated only for these matching ions, greatly enhancing the overall search speed. Finally, this similarity contribution is added to the similarity table for each library query.
Classic dot-product similarity has recently been shown to be less accurate than entropy similarity searches17–20. This improvement in accuracy may be partly due to the additional weight for low-abundant fragment ions that are particularly important in metabolomics MS/MS spectra, as has been shown even for fragment-rich peptide spectra19,20. Unfortunately, entropy similarity-matching requires even more computational time than dot-product searches, because experimental and library spectra need to be merged for calculations. We here, speed up computations by exploiting the sparse nature of small molecule MS/MS spectra; most spectra do not have any common fragment ions. Such comparisons would therefore yield zero spectral similarity and should be avoided. We therefore propose a new formula to calculate entropy similarity (Supplementary Note 1), skipping comparisons between query spectra and library spectra that have no common fragment ions (Fig. 1e). The Flash entropy algorithm is mathematically equivalent to the original formula of the entropy algorithm but much easier to compute. First, spectra are centroided, denoised and precursor ions are removed. Normalized spectra are compiled into ion tables and sorted by . Spectra comparisons use these sorted ion tables to find matching ions. Contributions of matching ion pairs to the entropy similarity are calculated using equation (1) and added to the final similarity value. Mismatching ions contribute to final similarities only via the normalization method (Extended Data Fig. 1 and Methods).
| (1) |
To evaluate the computation time of different similarity algorithms, we randomly sampled between 100 and 1,000,000 positive and negative electrospray ionization (ESI) MS/MS spectra from Massbank.us, GNPS and public repositories. Distributions of spectral entropies show differences between these three sets of benchmark spectra with entropy interquartile ranges from 0.8–1.4 for MassBank. us to 1.2–2.7 for a combination of experimental spectra from public repositories (Extended Data Fig. 2). When we performed an open search of 100 query spectra against spectral library sized from 100 to 1 million spectra, we found that both dot-product search in MatchMS15 and the original entropy similarity search linearly increased in computation time by the size of the search libraries (Fig. 2a and Extended Data Fig. 3). Recently, an MS/MS similarity method was proposed that approximates dot-product searches (BLINK16). BLINK approximates similarities by blurring mass spectra into bins, similar to hashing strategies used in proteomics21. When implementing BLINK, we confirmed it to be about 50-times faster than MatchMS; however, while Flash entropy showed a median search time of <1 ms per spectrum when searching the MassBank.us library of 1 million entries, BLINK needed nearly 0.6 s per spectrum and MatchMS used 25 s of computation time (Fig. 2a). This comparison showed that the Flash entropy algorithm is around 500 times faster than BLINK and about 30,000 times faster than MatchMS for both entropy and dot-product similarity calculations (Extended Data Figs. 3 and 4 and Supplementary Table 1).
Fig. 2 |. Benchmarking Flash entropy searches for speed and accuracy.
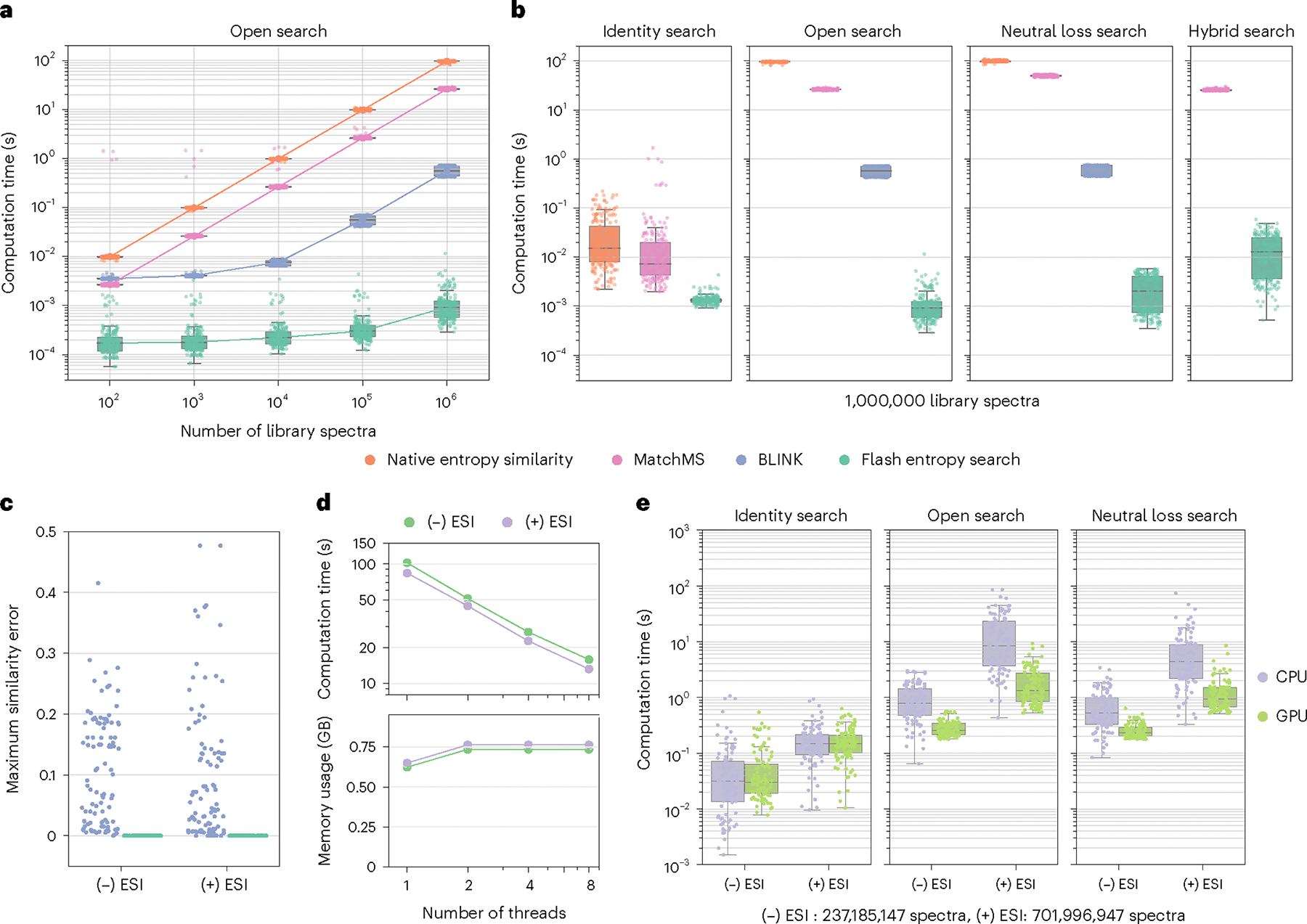
a, Calculation time to search 100 positive ESI and 100 negative ESI MS/MS spectra against randomly picked samples of the MassBank.us library. Dots represent calculation times per spectrum. b, Calculation times to perform identity, open, neutral loss and hybrid searches for 100 positive ESI and 100 negative ESI spectra against 1,000,000 MassBank.us spectra with different algorithms. c, Accuracy of MS/MS similarity results comparing Flash entropy to regular entropy searches and BLINK to MatchMS dot-product scores. d, Total computation times and memory usage for conducting an open search of 100,000 spectra against a library of 1,000,000 spectra. e, Comparison of computation times when using CPU versus GPU for Flash entropy searches. The 100 negative ESI and positive ESI spectra were searched against 237,185,147 publicly available negative ESI MS/MS spectra and 701,996,947 positive MS/MS spectra. Box plots display medians as horizontal lines inside the boxes that delineate interquartile ranges (IQRs). Whiskers extend to the lowest or highest data point within 1.5 × IQR of the 25% and 75% quartiles. n = 200 independent MS/MS spectra randomly sampled from MassBank.us library (a,b). n = 100 independent MS/MS spectra randomly sampled from public repositories (e).
Next, we compared the speed for identity search, open search, neutral loss search and hybrid search using 100 query spectra against 1 million spectra (Fig. 2b, Extended Data Figs. 5 and 6 and Supplementary Table 2). For open and neutral loss searches, the Flash entropy search was 25,000 times faster than MatchMS and 1,500 times faster for hybrid searches. Of note, the Flash entropy search can be used for hybrid searches, unlike the original entropy similarity algorithm. BLINK is not optimized for identity searches and cannot be used for hybrid searches either. Identity searches are generally faster than open, neutral loss or hybrid searches because the search space can be constrained by the precursor ions. Still, the Flash entropy search proved to be 5–10-times faster than MatchMS or the original entropy similarity tool (Fig. 2b). We then tested the calculation times to compare spectra with different complexity levels. When searching 100 spectra between entropy levels 1–4 against one million MassBank.us spectra, we confirmed that query times increased with the spectral entropy level; however, even at entropy S > 4, the Flash entropy algorithm yielded results within 10 ms. These results demonstrated the high efficiency of Flash entropy searches even when querying complex spectra against large spectral libraries (Extended Data Fig. 7).
The Flash entropy search produces highly accurate results. To compare accuracy against the BLINK fast algorithm, we used the results from the open search above and calculated the maximum errors relative to MS/MS similarity results given by the classic similarity search tools (Fig. 2c and Extended Data Fig. 8). Unlike BLINK, Flash entropy searches always generated the same result as the classical algorithm.
Next, we tested whether Flash entropy searches could be accelerated by multi-threading calculations. An open search of 100,000 spectra against 1,000,000 spectra required using an average time of 100 s on a single core (Fig. 2d). Computation times decreased almost linearly with the number of threads. With eight central processing unit (CPU) cores, the Flash entropy search needed about 15 s, with an increase in memory usage of just 17% (Fig. 2d). These results demonstrate that the Flash entropy search has excellent multi-threaded performance with minimal memory usage overhead.
Last, we demonstrated that Flash entropy is suitable for searching against all publicly available mass spectra, even for open and neutral loss search tests. We tested Flash entropy speed for 100 negative ESI and 100 positive ESI spectra against >938 million spectra accumulated from public repositories. Without compression, such a library is >30 TB. After compacting, the library size was 318 GB, which is too much memory space for a personal computer. We therefore stored this library on a hard disk, limiting memory usage to 4–16 GB. The Flash entropy search had a medium time of <1 s per spectrum for searching the negative ESI library and <10 s for searching the larger positive ESI library (Fig. 2e). This result shows that the Flash entropy search does not require loading the entire dataset into memory. In fact, the Flash entropy search can be further accelerated by using graphic processing units (GPUs) instead of CPUs. GPUs finished searches against the full negative ESI and positive ESI libraries 3.6–6.7-times faster than CPUs, with a median time of 0.25 s per negative ESI spectra and <1.5 s for positive ESI spectra (Fig. 2e). In comparison, MSFragger cannot be boosted by GPUs.
We developed, implemented and evaluated Flash entropy to calculate similarity-matching of millions of accurate mass MS/MS spectra within less than 10 ms (or a billion spectra in <2 s), using classic low-memory personal computers. Flash entropy presents ultrafast computing on a big-data scale to every laboratory. It extends similarity-matching from simple identity searches to include open, neutral loss and hybrid searches. This method has five benefits over alternative approaches: (1) It greatly improves computation efficiency when comparing large spectral libraries; (2) it does not require binning of the product ions and does not alter the accuracy of similarity results; (3) it can be run in parallel using multiple cores with minimal overhead; (4) it retains high performance when analyzing spectral libraries that are too large to be entirely loaded into the memory; and (5) its speed can be boosted by GPUs (Supplementary Note 2). As a cautionary remark, best practice in compound annotations requires additional data complementing MS, such as chromatographic retention time or collision cross section.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41592-023-02012-9.
Methods
Entropy similarity
The entropy similarity17 between two spectra and is defined as:
| (2) |
and represent the spectral entropy of spectra and . is the spectral entropy of the 1:1 mixed spectrum and . Ion intensities are weighted by equation (3) with as the intensity of each ion.
| (3) |
Equation (2) can be transformed as given in the Supplementary Note 1. If spectra A and B are normalized to give a , only the intensities of matched peaks are needed to calculate entropy similarity. Entropy similarity can then be calculated as:
| (4) |
Calculations are slightly faster if the spectra are normalized to , then using entropy similarity as:
| (5) |
Here, is the intensity of peak in spectrum is the intensity of peak in spectrum and represents the mass/charge ratio of product ions.
MS/MS spectra search
MS/MS spectra can be compared against spectra libraries by four different methods: identity search, open search, neutral loss search and hybrid search. For identity searches, scientists seek to find the direct hit that identifies a query spectrum against a library spectrum. This search is usually the fastest search because library entries can be constrained to those spectra that match the precursor ion of the query spectrum within the mass accuracy range of a mass spectrometer. Although modern high resolution mass spectrometers today typically yield a mass error of < 2 mDa, we here selected a wide search range of ±10 mDa to simulate cases where experimental mass accuracies might be compromised by local noise ions or by low-ion statistics for low-abundant molecules.
The ‘open search’ mode, compares all MS/MS spectra without constraining precursor ion masses. This search mode is used to match experimental spectra to any other molecule that shares critical substructures, such as aglycones and aglycones-glycosides. ‘Open search’ in small molecule MS/MS similarity is slightly different from the ‘open search’ method used in proteomics field, as proteomics still uses precursor ion constraints, but at wide ranges. Next, ‘neutral loss’ searches are performed in a similar way as open searches, but transform all search spectra by subtracting the fragment ion from the precursor ion values. In effect, neutral loss searches require similar search times as open searches. For ‘hybrid search’ (also called ‘modified cosine search’), every ion in the query spectrum must match either the identical fragment ions or a corresponding neutral loss ions. In this sense, hybrid searches are a mixture of open searches and neutral loss searches. Therefore, hybrid searches require the largest computational time. For Flash entropy hybrid searches, we clarified the algorithm by mandating that each query ion can only be used to match either an open search fragment ion or a neutral loss ion, but cannot be used for both matches. We further prioritize matching fragment ions over matching neutral loss ions.
Flash entropy algorithm
Both library spectra and query spectra are normalized before a Flash entropy search. First, the non-fragmented portion of precursor ions and any ion larger than the precursor ion are removed from MS/MS spectra by filtering out > (precursor − 1.6). The accurate mass of the precursor ion is known for each MS/MS spectrum and can be used for searches, but its abundance is not useful in MS/MS matching. Removing the precursor ions improves the performance of library searching17 and we here used the 1.6 Da window settings as implemented in the NIST MS search v.3.0 software. MS/MS spectra may also contain varying abundance and number of noise ions, depending on the abundance of the precursor ion, the complexity of co-eluting ions within the ion isolation window during precursor ion selection, tuning, parameters and operation of the mass spectrometer itself. While noise is therefore hard to define, possible fragment ions at <1% the maximal fragment ion abundance carry a high probability to stem from other sources than the precursor ion and are removed. Notably, the fragment intensities are normalized by applying the entropy weights according to equation (3) and afterwards, MS/MS spectra are normalized to 0.5 for the sum of all peak intensities. Spectra are listed consecutively for library spectra entries so that all fragment ions are represented as a tuple: (spectrum identifier by continuous numbering from zero, fragment ion , ion intensity) in a large list. Next, all ion tuples in the library spectra are sorted by the fragment ion (step 1 in Fig. 1). To speed up the lookup processes, we created an index of all fragment ion values. This step can be omitted if the library is small, but for very large libraries with billions of fragment ions, indexing is advantageous.
For querying spectra in an ‘open search’ against library spectra (ignoring precursor ions), we first initiated zero similarity for all library spectra. We then found all matching fragment ions within the library spectra within a defined ± mass tolerance (step 2). As maximum mass errors may occur for low-abundant ions at 10-mDa difference, we select a generous 20-mDa-wide window for fragment ion-matching to also account for possible measurement errors in library spectra. Lowering this window may speed up calculations even further. Then, for each matching ion pair an entropy similarity value contribution is added, as per equation (6).
| (6) |
This process is given as step 3 in Fig. 1. Here is query spectrum ion intensity and is the library spectrum ion intensity. When all ions in the query spectrum are fully queried against the library, the similarity score calculation is completed.
Variations of Flash entropy similarity searches
The Flash entropy search can be easily adopted for other types of MS/MS spectral searches. For ‘identity searches’, library spectra are restricted to hits within user-defined mass accuracy windows of the precursor ion accurate mass. For the benchmark tests, we used a wide search window with up to 10 mDa error. Here, all library spectra are sorted by their precursor and then indexed as given above. Because the library spectra are sorted, the continuous spectral library numbers give the search range within ±10 mDa. Only spectra within the search range are calculated with equation (5) to yield spectral similarities. Hence, only spectra within the search range have nonzero spectral similarities. For ‘neutral loss’ searches, the fragment ion values are replaced by the mass of molecular neutral losses. These are calculated as precursor ion minus fragment ion . All other steps including index and search steps are the same as given in ‘open search’. For ‘hybrid search’ we transform search libraries in the following way: first, all fragment ions in the spectral library are represented as a tuple: (spectral number, fragment ion , neutral loss mass and ion intensity). Then all entries are sorted from the lowest fragment ion to the highest fragment ion value. Entries are copied and recorded by the simplified tuple (spectral number, fragment ion and peak intensity). This is the fragment ion table. This list is only used for fragment lookups. All ions are given an ion continuous number, to reflect the sorted list. Next, the original tuple list is sorted by neutral loss masses, generating a new list with a tuple (spectral number, neutral loss mass, ion intensity and ion number). This is called the neutral loss table. Now, all ions from the query spectrum are first compared to the ions in the fragment ion table. Matching ion pairs are recorded by their original ion continuous number. Subsequently, the query fragment ions are compared to the neutral loss table. If a query ion/neutral loss pair is already present in the matching fragment ion table, this ion will be ignored in the neutral loss match. Finally, all matching fragment ions and all matching query ion/neutral loss matches are calculated to yield entropy similarity using equation (6).
Memory and time usage of the Flash entropy algorithm
During library searching, the Flash entropy search uses very little memory, with a minimum requirement of , where s is the total number of spectra in the library and is the maximum number of matched peaks. This process enables the algorithm to run on low configuration computers, even when processing large spectral libraries. The Flash entropy search has a time complexity of , where is the total number of peaks in the query spectra, is the total number of peaks in the library spectra and is the matching tolerance. The more accurate MS measurements become, the smaller the user-defined matching tolerance gets. Smaller mass windows result in correspondingly shorter run times for the Flash entropy search algorithm.
Benchmark
Spectra from MassBank.us and GNPS were downloaded on 3 March 2023. Additional MS/MS spectra from public repositories were downloaded from the MassIVE/GNPS, MetabolomicsWorkbench. org, MetaboLights and the West Coast Metabolomics Center in December 2022. In total, more than 939 million spectra were available (237,185,147 negative ESI and 701,996,947 positive ESI MS/MS spectra). As library and query spectra, between 100 to 1,000,000 spectra were randomly sampled from those repositories using the function ‘numpy.random.choice’ from Numpy package, v.1.23.5 (ref. 22). All spectra were centroided, summarizing ion intensities with < 50 mDa. Spectra were denoised by removing ion intensities at <1% of the most abundant fragment ion. Ion intensities were normalized to a sum of 0.5. Spectra were indexed before testing for computational times.
The spectral similarity calculation time is measured as wall clock time. The algorithm is implemented in Python and tested on major CPU architectures such as x86_64 and ARM from Intel, AMD and Apple. It was also verified in different operating systems, including Linux, Windows and MacOS. All benchmark tests were performed on a personal computer with an AMD Ryzen 9 3900 × 12-Core Processor, 64 GB memory, Nvidia Geforce RTX 2060 Super GPU and 2 TB WD_BLACK SN850X NVMe SSD, installed with a KDE neon 5.26 operation system and Python v.3.9. To benchmark the accuracy of similarity queries, the Flash entropy search was compared to the original implementation of entropy similarity. The native entropy similarity is calculated using the code downloaded from GitHub at https://github.com/YuanyueLi/SpectralEntropy, v.1.0.0. The BLINK package is downloaded from https://github.com/biorack/blink on 9 February 2023. Dot-product score results obtained by the BLINK algorithm were compared to the CosineGreedy function in the MatchMS package, v.0.18.0. We used the recommended bin size for BLINK at 1 mDa. Precursor ion matching tolerances were set to <10 mDa and the MS/MS ion matching tolerance was set to <20 mDa. Memory usage was measured using the command ‘/usr/bin/time -v’ and limited memory with the command ‘systemd-run–scope -p MemoryMax = 4G (16G)’.
Extended Data
Extended Data Fig. 1 |. Examples for calculating Flash entropy similarity.
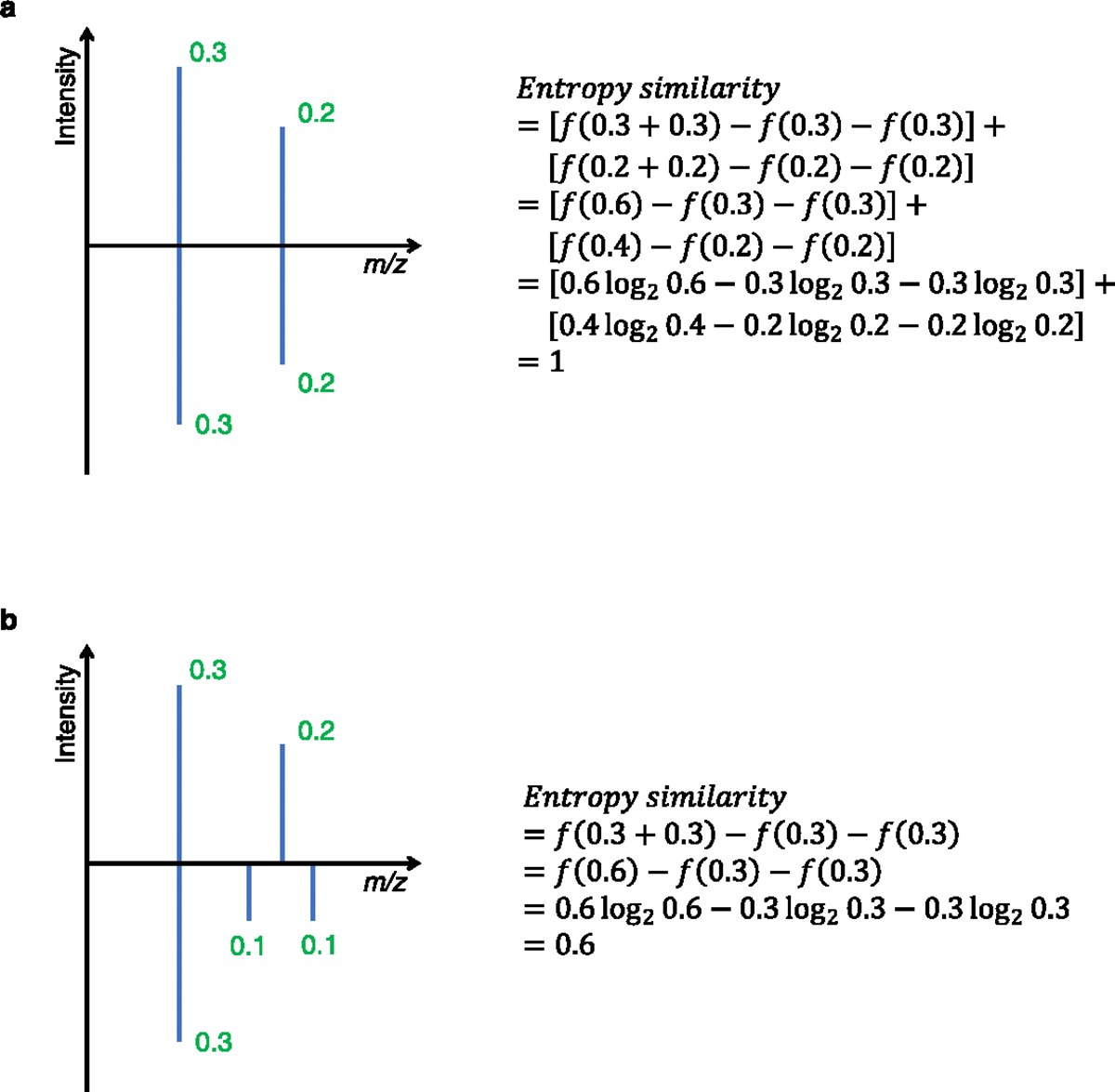
(a) Example when all ions match between query spectrum (top) and library spectrum (bottom). in the two spectra are matched. (b) Example when only one pair of ions matches between query and library spectra. Note that the sum intensities of ion abundances in each spectrum are normalized to equal 0.5 (see Supplementary Note 1 for equations). Hence, mismatched ions do not contribute themselves into the calculations, but are considered during the normalization process.
Extended Data Fig. 2 |. Distributions of spectral entropies when sampling spectra from different MS/MS repositories for benchmarking studies.
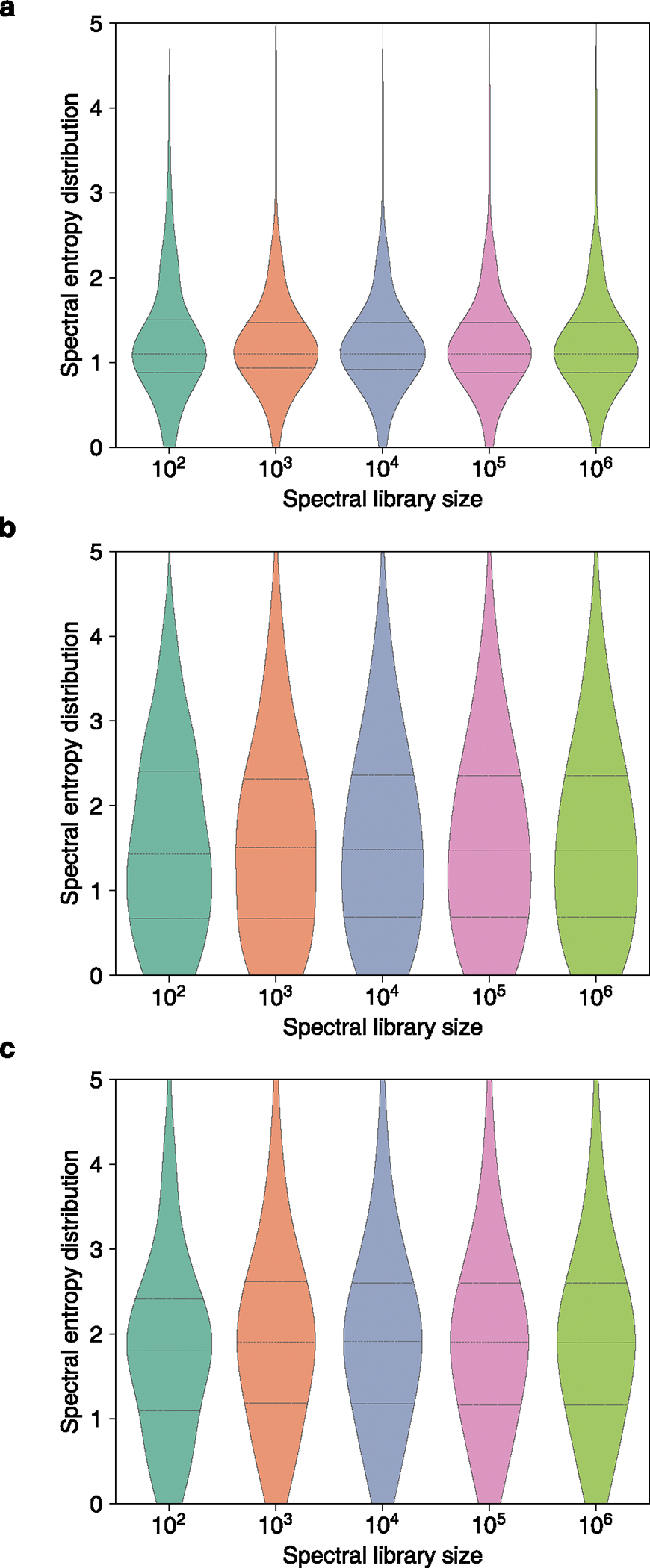
(a) MassBank.us, (b) GNPS for annotated compounds (library), (c) all combined experimental public MS/MS repositories including MassIVE/GNPS, MetaboLights, MetabolomicsWorkbench and West Coast Metabolomics Center.
Extended Data Fig. 3 |. Computation time required to perform ‘open search’ queries using entropy similarity for 100 positive ESI and 100 negative ESI mass spectra against spectral libraries of different sizes.
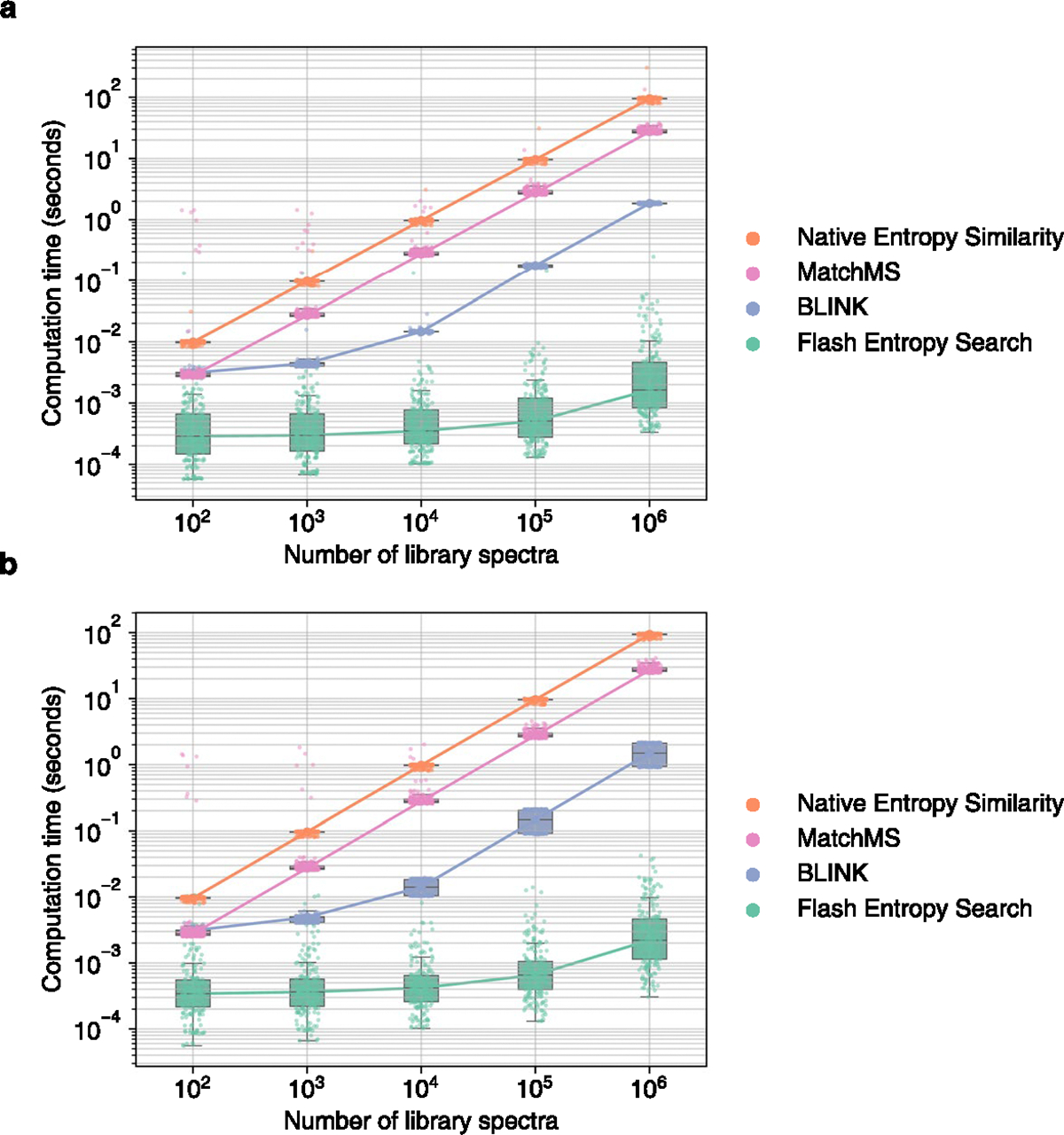
MS/MS spectra were sampled from (a) GNPS (b) public repositories. Box plots display medians as horizontal lines inside the boxes that delineate interquartile ranges (IQR). Whiskers extend to the lowest or highest data point within 1.5x IQR of the 25% and 75% quartiles. N = 200 independent MS/MS spectra randomly sampled from (a) GNPS (b) public.
Extended Data Fig. 4 |. Computation time required to perform ‘open search’ queries using dot product similarity for 100 positive ESI and 100 negative ESI mass spectra against spectral libraries of different sizes.
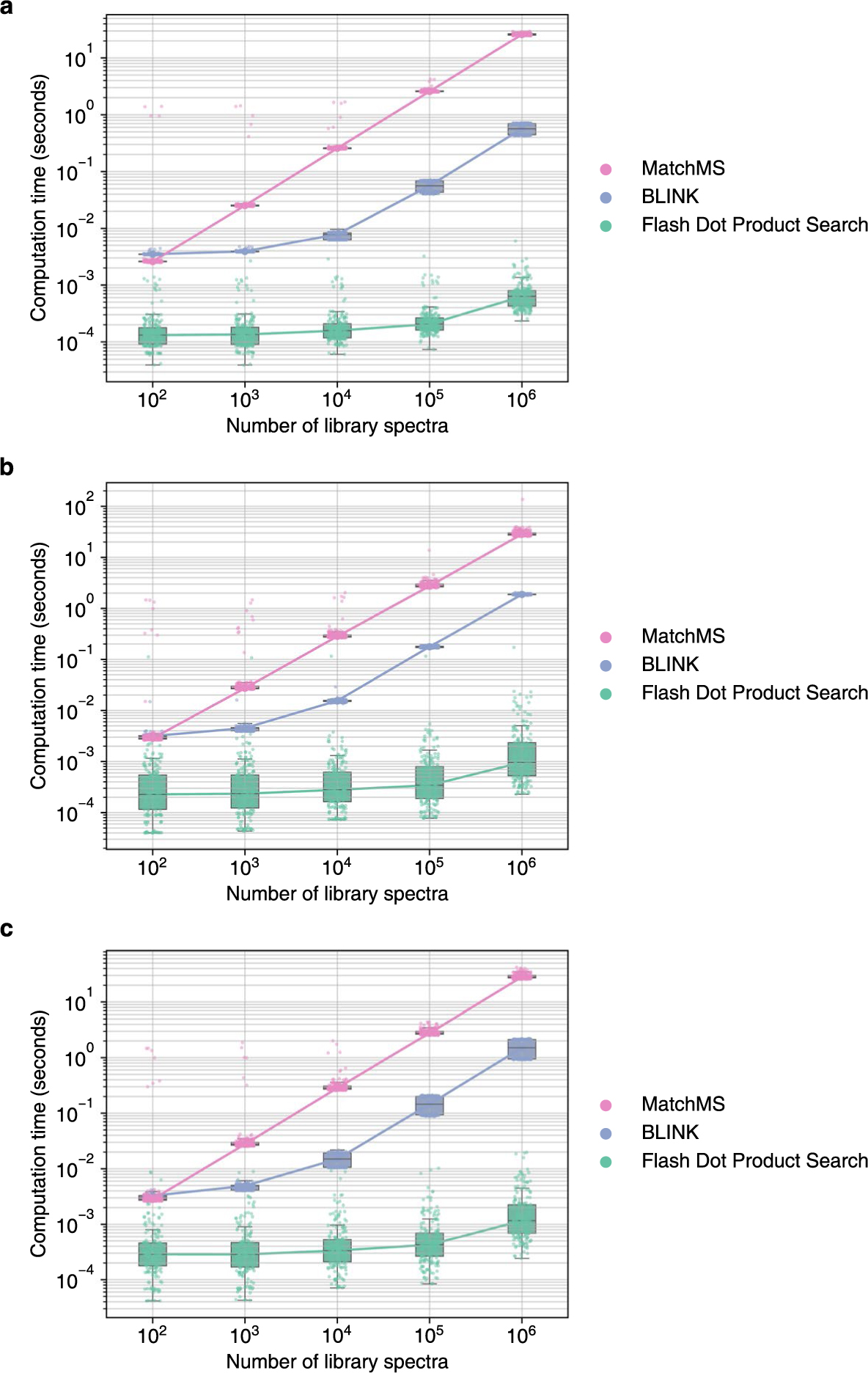
MS/MS spectra were sampled from (a) MassBank.us, (b) GNPS, (c) public repositories. Box plots display medians as horizontal lines inside the boxes that delineate interquartile ranges (IQR). Whiskers extend to the lowest or highest data point within 1.5x IQR of the 25% and 75% quartiles. N = 200 independent MS/MS spectra randomly sampled from (a) MassBank.us, (b) GNPS, (c) public repositories.
Extended Data Fig. 5 |. Computation time required to perform ‘neutral loss’ searches with entropy similarity for 100 positive ESI and 100 negative ESI mass spectra against spectral libraries of different sizes.
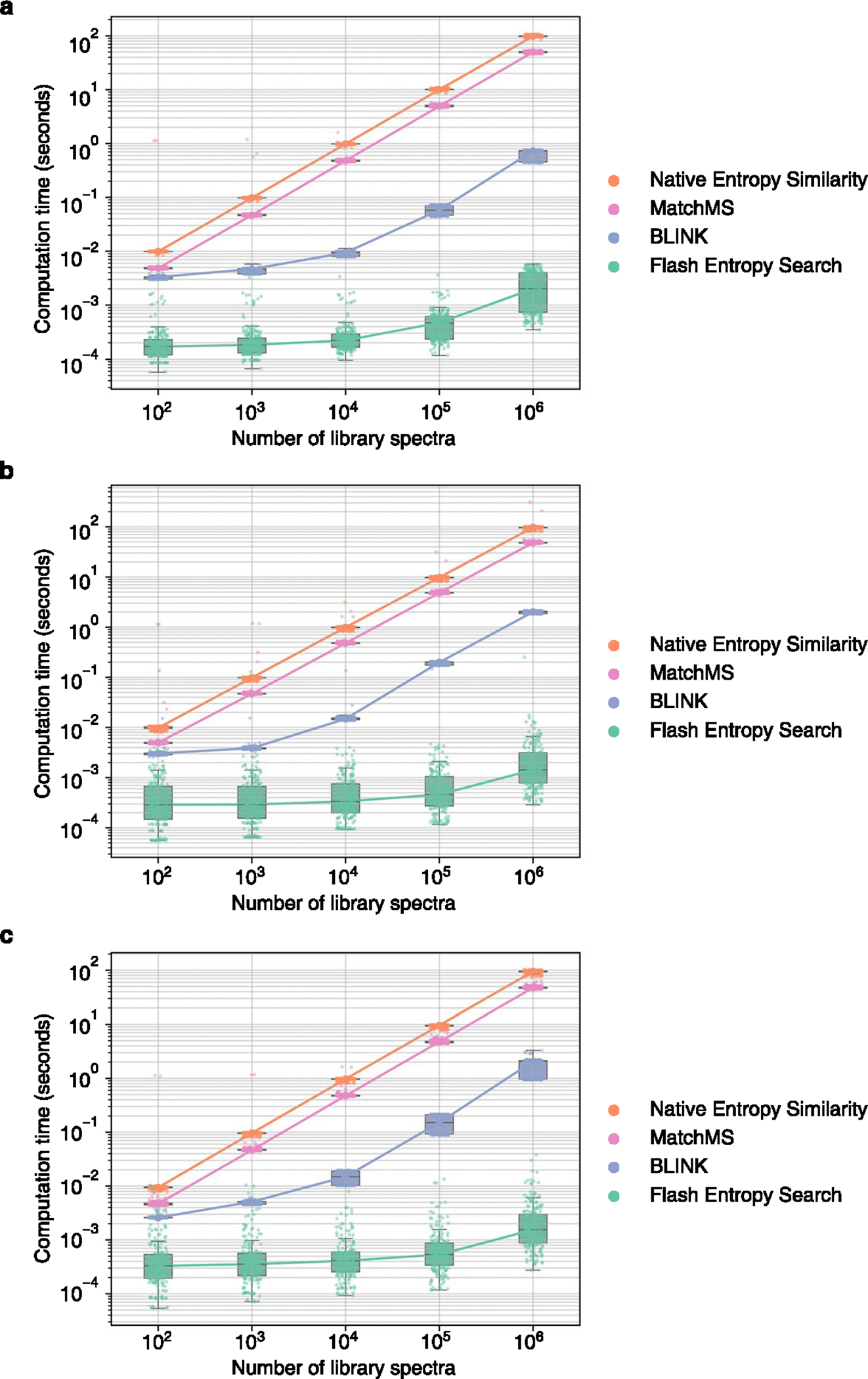
MS/MS spectra were sampled from (a) MassBank.us, (b) GNPS, (c) public repositories. Box plots display medians as horizontal lines inside the boxes that delineate interquartile ranges (IQR). Whiskers extend to the lowest or highest data point within 1.5x IQR of the 25% and 75% quartiles. N = 200 independent MS/MS spectra randomly sampled from (a) MassBank.us, (b) GNPS, (c) public repositories.
Extended Data Fig. 6 |. Computation time required to perform ‘hybrid searches’ with entropy similarity for 100 positive ESI and 100 negative ESI mass spectra against spectral libraries of different sizes.
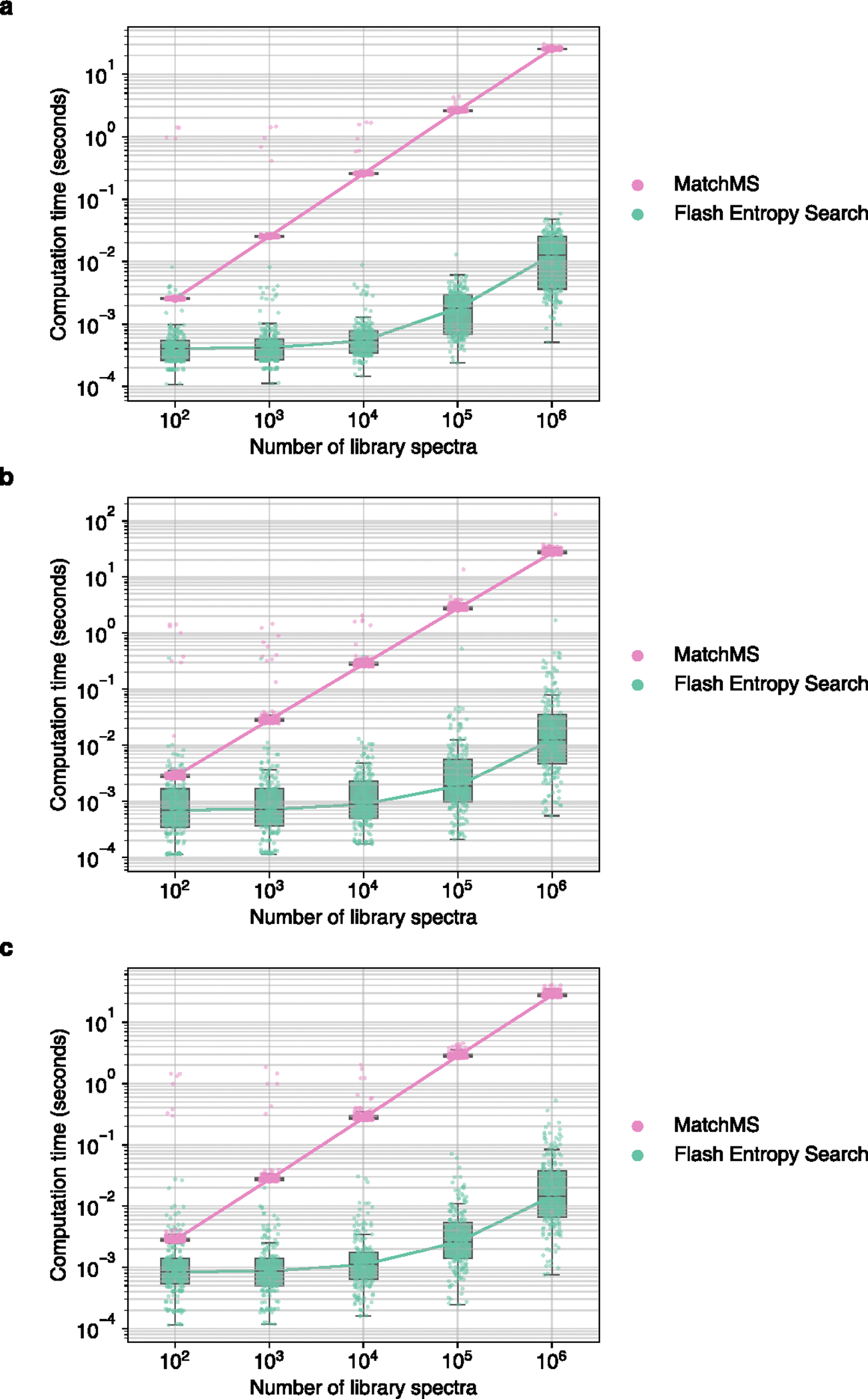
MS/MS spectra were sampled from (a) MassBank.us, (b) GNPS, (c) public repositories. Box plots display medians as horizontal lines inside the boxes that delineate interquartile ranges (IQR). Whiskers extend to the lowest or highest data point within 1.5x IQR of the 25% and 75% quartiles. N = 200 independent MS/MS spectra randomly sampled from (a) MassBank.us, (b) GNPS, (c) public repositories.
Extended Data Fig. 7 |. Calculation time to open search 100 positive ESI and 100 negative ESI MS/MS spectra at different spectral entropy levels against randomly picked samples from the MassBank.us library.
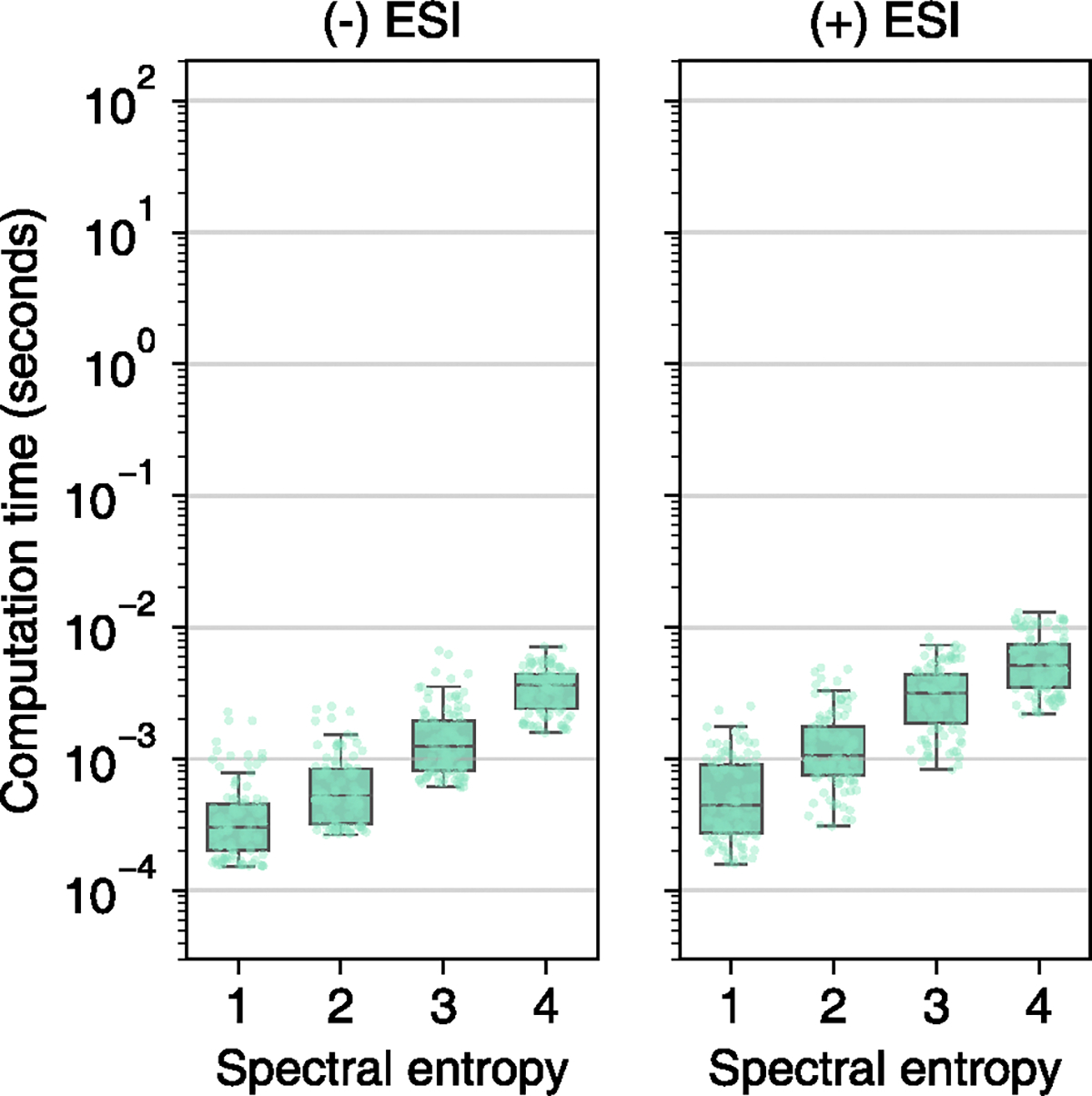
Box plots display medians as horizontal lines inside the boxes that delineate interquartile ranges (IQR). Whiskers extend to the lowest or highest data point within 1.5x IQR of the 25% and 75% quartiles. N = 100 independent MS/MS spectra randomly sampled from MassBank.us.
Extended Data Fig. 8 |. Comparison of the accuracy of similarity query results between Flash entropy search and BLINK.
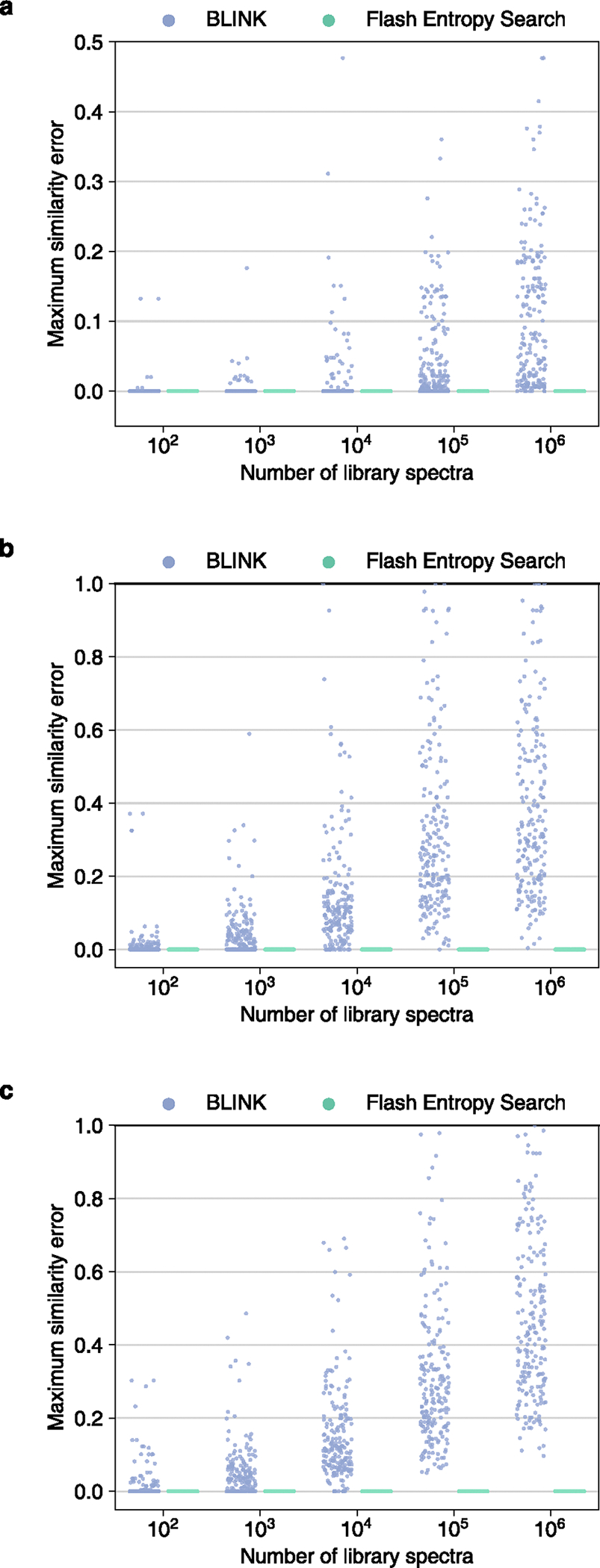
Each dot shows the maximum similarity difference between the fast algorithms and their classic algorithm counterparts. 100 positive ESI and 100 negative ESI MS/MS spectra were sampled from (a) MassBank.us, (b) GNPS, (c) public repositories.
Supplementary Material
Acknowledgements
This study was funded by National Institutes of Health grants U2C ES030158 and R03 OD034497 (to O.F.).
Footnotes
Competing interests
The authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
The original source code and benchmark data for the Flash entropy search are available under the Apache License 2.0 on GitHub (https://github.com/YuanyueLi/FlashEntropySearch) and Zenodo (https://doi.org/10.5281/zenodo.7972082), as well as on CodeOcean (https://doi.org/10.24433/CO.8809500.v1). The GUI can be downloaded from the GitHub repository: https://github.com/YuanyueLi/EntropySearch. Flash entropy search is also integrated into the ‘MSEntropy’ package, available for download from https://github.com/YuanyueLi/MSEntropy. Comprehensive documentation for the ‘MSEntropy’ package can be found at https://msentropy.readthedocs.io.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41592-023-02012-9.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41592-023-02012-9.
Peer review information Nature Methods thanks Xusheng Wang and Jianguo Xia for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Arunima Singh, in collaboration with the Nature Methods team.
Data availability
All spectra from MassBank.us (https://massbank.us/) and GNPS (https://gnps-external.ucsd.edu/gnpslibrary/ALL_GNPS.mgf) were downloaded on 3 March 2023. Additional MS/MS spectra from public repositories were downloaded from the MassIVE/GNPS (https://gnps.ucsd.edu/ProteoSAFe/datasets.jsp#%7B%22query%22%3A%7B%7D%2C%22title_input%22%3A%22GNPS%22%7D), MetabolomicWorkbench. org (https://www.metabolomicsworkbench.org/) and MetaboLights (https://www.ebi.ac.uk/metabolights/) in December 2022. In total, more than 939 million spectra were available (237,185,147 negative ESI and 701,996,947 positive ESI MS/MS spectra). All the spectra from those sources were used in this study. Source data are provided with this paper.
References
- 1.Liang L et al. Metabolic dynamics and prediction of gestational age and time to delivery in pregnant women. Cell 181, 1680–1692 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li D & Gaquerel E Next-generation mass spectrometry metabolomics revives the functional analysis of plant metabolic diversity. Annu. Rev. Plant Biol. 72, 867–891 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Choi M et al. MassIVE.quant: a community resource of quantitative mass spectrometry–based proteomics datasets. Nat. Methods 17, 981–984 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang M et al. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 34, 828–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sud M et al. Metabolomics Workbench: an international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 44, D463–D470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haug K et al. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 48, D440–D444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M et al. Mass spectrometry searches using MASST. Nat. Biotechnol. 38, 23–26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chick JM et al. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat. Biotechnol. 33, 743–749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aisporna A et al. Neutral loss mass spectral data enhances molecular similarity analysis in METLIN. J. Am. Soc. Mass. Spectrom. 33, 530–534 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watrous J et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl Acad. Sci. USA 109, E1743–E1752 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burke MC et al. The hybrid search: a mass spectral library search method for discovery of modifications in proteomics. J. Proteome Res. 16, 1924–1935 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Moorthy AS, Wallace WE, Kearsley AJ, Tchekhovskoi DV & Stein SE Combining fragment-ion and neutral-loss matching during mass spectral library searching: a new general purpose algorithm applicable to illicit drug identification. Anal. Chem. 89, 13261–13268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bittremieux W et al. Comparison of cosine, modified cosine, and neutral loss based spectrum alignment for discovery of structurally related molecules. J. Am. Soc. Mass. Spectrom. 33, 1733–1744 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D & Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 14, 513–520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber F et al. matchms - processing and similarity evaluation of mass spectrometry data. J. Open Source Softw. 5, 2411 (2020). [Google Scholar]
- 16.Harwood T et al. BLINK: Ultrafast tandem mass spectrometry cosine similarity scoring. Sci. Rep. 13, 13462 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y et al. Spectral entropy outperforms MS/MS dot product similarity for small-molecule compound identification. Nat. Methods 18, 1524–1531 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King E, Overstreet R, Nguyen J & Ciesielski D Augmentation of MS/MS libraries with spectral interpolation for improved identification. J. Chem. Inf. Model. 62, 3724–3733 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang KL et al. MSBooster: improving peptide identification rates using deep learning-based features. Nat. Commun. 14, 4539 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi X et al. Deep learning prediction boosts phosphoproteomics-based discoveries through improved phosphopeptide identification. Preprint at bioRxiv 10.1101/2023.01.11.523329 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bittremieux W, Laukens K & Noble WS Extremely fast and accurate open modification spectral library searching of high-resolution mass spectra using feature hashing and graphics processing units. J. Proteome Res. 18, 3792–3799 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harris CR et al. Array programming with NumPy. Nature 585, 357–362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All spectra from MassBank.us (https://massbank.us/) and GNPS (https://gnps-external.ucsd.edu/gnpslibrary/ALL_GNPS.mgf) were downloaded on 3 March 2023. Additional MS/MS spectra from public repositories were downloaded from the MassIVE/GNPS (https://gnps.ucsd.edu/ProteoSAFe/datasets.jsp#%7B%22query%22%3A%7B%7D%2C%22title_input%22%3A%22GNPS%22%7D), MetabolomicWorkbench. org (https://www.metabolomicsworkbench.org/) and MetaboLights (https://www.ebi.ac.uk/metabolights/) in December 2022. In total, more than 939 million spectra were available (237,185,147 negative ESI and 701,996,947 positive ESI MS/MS spectra). All the spectra from those sources were used in this study. Source data are provided with this paper.