

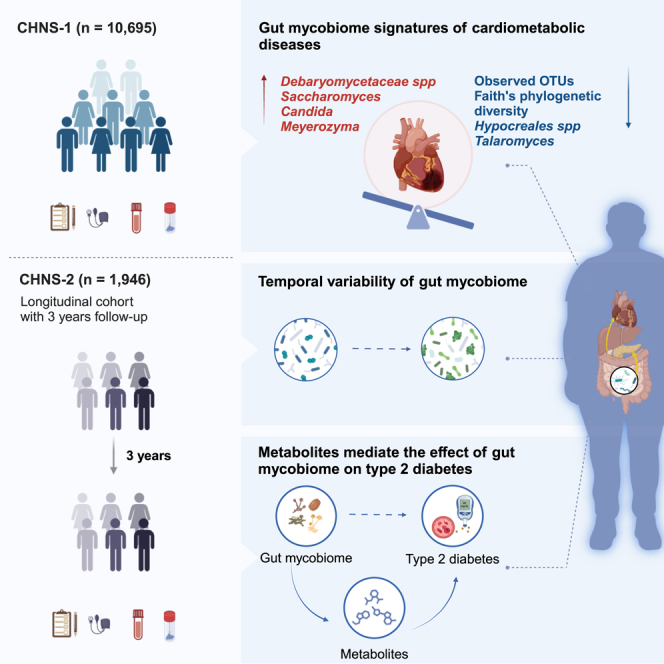
Summary
The dynamics of the gut mycobiome and its association with cardiometabolic health remain largely unexplored. Here, we employ internal transcribed spacer (ITS) sequencing to capture the gut mycobiome composition and dynamics within a nationwide human cohort of 12,641 Chinese participants, including 1,946 participants with repeated measurements across three years. We find that the gut mycobiome is associated with cardiometabolic diseases and related biomarkers in both cross-sectional and dynamic analyses. Fungal alpha diversity indices and 19 mycobiome genera are the major contributors to the mycobiome-cardiometabolic disease link. Particularly, Saccharomyces emerges as an effect modifier of traditional risk factors in promoting type 2 diabetes risk. Further integration of multi-omics data reveals key metabolites such as γ-linolenic acid and L-valine linking the gut mycobiome to type 2 diabetes. This study advances our understanding of the potential roles of the gut mycobiome in cardiometabolic health.
Keywords: gut mycobiome, cardiometabolic health, human cohort
Graphical abstract
Highlights
-
•
We characterize gut fungal composition and dynamics in 12,641 Chinese participants
-
•
Gut fungal diversity and 19 genera are associated with cardiometabolic health
-
•
The influence of gut fungi on type 2 diabetes may be mediated by serum metabolites
The association between the gut mycobiome and cardiometabolic health remains underappreciated. In a cohort of 12,641 Chinese participants, Gou et al. characterize the gut mycobiome’s composition and dynamics, demonstrating its connection to cardiometabolic health. They also identify several metabolites potentially mediating the association of the gut mycobiome with type 2 diabetes.
Introduction
The fungal microbiome, known as the mycobiome, is omnipresent in our living environment and has played a central role in the evolution of life.1 Despite fungi comprising a small proportion of the human gut microbiota, alterations in the fungal community are suggested to be correlated with various health conditions and diseases, such as host immunity,2 gastrointestinal diseases,3 and cancer.4 Yet, vast majority of the prior clinical evidence about human microbiome and health has been focused on gut bacteria but not fungi. The gut mycobiome research remains in its infancy stage, representing a rather underestimated field. Whether and how the gut mycobiome may influence the disease progression and health status in humans are largely unknown.
Cardiometabolic diseases, such as type 2 diabetes (T2D), hypertension, and dyslipidemia, are highly prevalent globally.5 Recent interest has focused on understanding the role of the gut mycobiome in these diseases.6 Studies in mice have shown that disturbances in the intestinal fungi, induced by antifungal treatments, may contribute to both intestinal and extra-intestinal pathologies.7 In humans, there are several studies examining the association of the gut mycobiome with cardiometabolic diseases, but with small sample sizes and conflicting results.8,9,10,11,12 For example, some studies report alterations in the mycobiome in individuals with T2D9,10 and hypertension11,12 while others suggest these changes may be limited.8 Furthermore, gut fungal composition varies across geographic regions.13 Previous studies, limited by geographic scope and sample size, may not have captured the full variation in fungal composition, which may confound the relationship between the gut mycobiome and cardiometabolic health. Therefore, it is essential to conduct large-scale mycobiome profiling among participants with diverse dietary and geographic backgrounds to understand the role of the gut mycobiome in cardiometabolic health.
Here, we conducted a nationwide population-based mycobiome mapping involving a deeply phenotyped cohort of 12,641 adult participants from 15 major provinces/municipalities across China. We aimed to provide a comprehensive understanding of the intricate association of the gut mycobiome with multiple host environmental phenotypes, particularly focusing on cardiometabolic health. To capture the temporal dynamics of the gut mycobiome and validate its association with cardiometabolic diseases, we further conducted a long-term follow-up analysis in a cohort of 1,946 participants over a three-year period. Furthermore, to gain potential biological insights, we integrated serum metabolome data and used mediation analysis to explore the specific metabolites that may medicate the association of the gut mycobiome with cardiometabolic diseases.
Results
Study design and cohort descriptions
In the present study, we profiled the gut mycobiome within a cohort of 12,641 Chinese adult participants (Figure 1A). These individuals were drawn from two independent sub-cohorts of the China Health and Nutrition Survey (CHNS), identified as CHNS-1 and CHNS-2. Overall characteristics of the study participants are shown in Table S1. The CHNS is a nationally representative longitudinal cohort within China, distinguished by its utilization of a meticulously designed multistage, random cluster approach, and the implementation of standardized experimental protocols.14
Figure 1.

Conceptual outline of the study and analysis workflow
(A) Overview of sampling regions and metadata. This study utilized data from the sub-cohorts of the China Health and Nutrition Survey (CHNS), referred to as CHNS-1 (n = 10,695) and CHNS-2 (n = 1,946). CHNS-1 encompassed participants from 15 provinces or municipalities across China, while CHNS-2 drew from 4 provinces within the country. Notably, there was no overlap in participants between these two cohorts. All participants contributed ITS2 sequencing data and metadata. Moreover, repeated measurements of mycobiome (n = 1,946) and metabolome (n = 996) data are accessible for CHNS-2 participants.
(B) Overview of analysis workflow. In the CHNS-1 cohort, we initially conducted a comprehensive portrayal of the inter-individual variation of the gut mycobiome and identified its associated factors. Subsequently, we delved deeper into investigating the connection between the gut mycobiome and cardiometabolic health. In the CHNS-2 cohort, our analysis focused on exploring the long-term variability of the gut mycobiome and dynamic interplay between the gut mycobiome and cardiometabolic diseases. Furthermore, we uncovered the key metabolites potential linking the gut mycobiome to cardiometabolic diseases through multi-omics integration.
See also Table S1.
The CHNS-1 cohort consisted of individuals from 15 prominent provinces or municipalities across China (Figure 1A). Within this cohort, detailed metadata and gut mycobiome profiles were available for a total of 10,695 participants (mean age [standard deviation, SD] 51.3 ± 15.5 years, 54% were women). In the CHNS-1 cohort, we provided a comprehensive depiction of the gut mycobiome’s variability and investigated the intricate associations between various host environmental phenotypes and the gut mycobiome’s variation (Figure 1B). Next, we further investigated the gut mycobiome signatures of cardiometabolic health. Subsequently, our analysis was extended to the CHNS-2 cohort, which consisted of individuals from four prominent provinces in China (Figure 1A), and featured with repeated measured clinical metadata (n = 1,946 pairs), gut mycobiome data (n = 1,946 pairs), and serum metabolome data (n = 996 pairs) spanning a median period of 3.02 years (interquartile range: 3–3.07). The inclusion of the CHNS-2 cohort facilitated an exploration of the long-term variability of the gut mycobiome and uncovered the key metabolites that may link the gut mycobiome to cardiometabolic diseases (Figure 1B).
Overall gut mycobiome community profile
We profiled the gut mycobiome by sequencing the internal transcribed spacer 2 (ITS2) region. A total of 761 genera (processed via QIIME [Quantitative Insights Into Microbial Ecology] 215) were detected in the CHNS-1 cohort, and 630 were identified in the CHNS-2 cohort. After filtering out genera of low prevalence (<10% prevalence in the respective cohort), 54 genera were retained in the CHNS-1 cohort, and 75 in the CHNS-2 cohort. Among these retained genera, 53 genera were shared across both the CHNS-1 and CHNS-2 cohorts.
In the CHNS-1 cohort, the two most predominant phyla in the gut mycobiome were Ascomycota and Basidiomycota (Figure 2A), consistent with previous data from the US,16 Europe,17 and China.18 The prevalence and relative abundance of the 54 included genera are presented in Table S2. Additionally, the inter-correlation of included fungal genera is depicted in Figure S1. We identified five genera (Aspergillus, Candida, Cladosporium, Penicillium, and Saccharomyces) present in at least 50% of samples at a minimum relative abundance of 0.1%. These genera also showed enrichment in other cohorts.16,17,19 The distribution of these genera across geographic regions and urbanization gradients is depicted in the Figure 2B and S2A, respectively. We observed a noticeable increase in the relative abundance of fungal Saccharomyces with an increase in urbanization score (Figure S2A). Linear mixed-effects model revealed that the relative abundance of Saccharomyces increased by 0.029 (95% confidence interval [CI], 0.024 to 0.035) with per increase in urbanization quintile. Additionally, Saccharomyces exerted the strongest inverse association with fungal diversity (Figure S2B). Co-abundance network analysis found that Blumeria, Debaryomyces, and Candida formed central nodes in fungal networks (Figure S2C and Table S3).
Figure 2.

Landscape of the gut mycobiome and key factors explaining the gut mycobiome inter-individual variation
(A) Taxonomic tree illustrating the genera with a prevalence greater than 10% in the CHNS-1 cohort (n = 10,695). Nodes (n = 54) within the inner to outer circles depict fungal taxa from phylum to genus levels, with genus-level nodes colored based on their respective phylum assignments. Reads that could not be confidently assigned to known taxa due to insufficient matching with reference sequences were labeled with the highest taxonomic rank that could be assigned, followed by “spp.”
(B) Distribution of fungal taxa that are present in at least 50% of samples at a minimum relative abundance of 0.1%.
(C) Variance in the mycobiome composition explained by phenotype categories was assessed through multivariate PERMANOVA analysis (n = 10,695). In this analysis, 54 genera with a prevalence greater than 10% were retained. The p value was determined through 999 permutations and subsequently adjusted for multiple testing. Notably, only the statistically significant results (FDR<0.05) are depicted in the figure.
See also Figures S1 and S2 and Tables S2 and S3.
Factors explaining inter-individual gut mycobiome variation
In the CHNS-1 cohort, we used PERMANOVA (permutational multivariate analysis of variance) analysis individually to assess the contributions of each environmental and host factor to the inter-individual variability in fungal composition and found that 52 factors displayed associations with the fungal variation (false discovery rate [FDR]<0.05, Figure 2C). These factors included demography (5 factors), urbanization (2 factors), dietary and lifestyle factors (24 factors), anthropometric factors (5 factors), blood parameters (4 factors), diseases (5 factors), medications (5 factors), and other environmental factors (2 factors).
The geographic region emerged as the dominant factor associated with variations in the gut mycobiome, explaining 3.7% of the observed variability (Figure 2C). Urbanization, whether defined categorically (urban or rural) or continuously (urbanization score), along with staple foods such as rice and wheat intake, as well as exposure to smoking, ranked prominently among contributors to inter-individual variation in the gut mycobiome (Figure 2C).
Gut mycobiome signatures of cardiometabolic health
To elucidate the distinctive fungal profiles associated with cardiometabolic health, we examined the relationships between 58 fungal features (including 54 genera and 4 alpha diversity indices) and 3 cardiometabolic disorders, along with 8 clinical biomarkers, including measures of glycemic control, lipid levels, and blood pressure in the CHNS-1 cohort. The prevalence of these cardiometabolic conditions in the cohort is as follows: T2D at 11.1%, hypertension at 28%, and dyslipidemia at 32.4%. Notably, 46.6% of the T2D participants also suffer from hypertension, and 51.7% suffer from dyslipidemia. This comprehensive analysis yielded a total of 48 significant associations (FDR<0.05). Higher fungal alpha diversity indices showed protective associations with multiple cardiometabolic diseases (Figure 3A) and related biomarkers (Figure 3B). Furthermore, the abundance of eight specific genera exhibited noteworthy alterations in at least one of the cardiometabolic disease groups (Figure 3C). For instance, Saccharomyces was positively associated with T2D risk (OR [odds ration] = 1.14; 95% CI, 1.06 to 1.21). Saccharomyces was also positively associated with HbA1c (p = 0.0013), although this association did not remain significant after correcting for multiple comparisons (FDR = 0.07). Saccharomyces was found to be enriched in participants with type 1 diabetes20 and multiple sclerosis21 and has the potential to exacerbate gut inflammation as indicated by an animal study.22 Candida exhibited a positive correlation with dyslipidemia (OR = 1.08; 95% CI, 1.03 to 1.12), concurrently displaying positive association with triglycerides (TGs) and systolic blood pressure (SBP), while manifesting an inverse association with high-density lipoprotein cholesterol (HDL-C) in our study. Importantly, the inverse association of alpha diversity indices with cardiometabolic diseases persisted after further adjusting for sequencing depth (Table S4). Interestingly, 16 fungal taxa exhibited an association with TG (Figure 3B), underscoring the potential role of the mycobiome in lipid metabolism and overall cardiometabolic health.
Figure 3.

Gut mycobiome signatures associated with cardiometabolic health
(A) Boxplots illustrating observed OTUs in participants with different cardiometabolic diseases compared with those without (n = 10,695). Significance was obtained by logistic mixed-effects regression model, with adjustment for age, gender, and BMI. The logistic mixed-effects model included a random intercept and random coefficient for provinces or megacities, accounting for the variability in gut fungal composition across different regions.
(B) Associations of mycobiome with indicators of cardiometabolic diseases (n = 10,695). The analysis was conducted using linear mixed-effects regression model, with adjustment for age, gender, and BMI. The linear mixed-effects model included a random intercept and random coefficient for provinces or megacities, accounting for the variability in gut fungal composition across different regions. Associations are colored by direction of Z-scored effect (red, positive; blue, negative). Notably, only the statistically significant results (FDR < 0.05) are depicted in the figure.
(C) Associations of fungal genera (per SD unit) with cardiometabolic diseases (n = 10,695). Odds ratios and 95% CI were calculated using logistic mixed-effects regression model described above. Notably, only the statistically significant results (FDR < 0.05) are depicted in the figure.
(D) Left, boxplots illustrating Saccharomyces in participants with type 2 diabetes compared with those without. Right, interaction between Saccharomyces and traditional risk factors in relation to the risk of type 2 diabetes (n = 10,695). The traditional risk score combines multiple factors, including age, gender, waist circumference, BMI, and SBP, to estimate an individual’s overall risk of developing T2D.
See also Table S4.
Our subsequent analyses revealed compelling evidence of a synergistic interaction between the gut Saccharomyces and environmental factors on T2D risk (pinteraction = 0.012). The traditional risk score combines multiple factors, including age, gender, waist circumference, BMI, and SBP, to estimate an individual’s overall risk of developing T2D. We divided participants into four groups based on the median levels of T2D traditional risk score and Saccharomyces abundance. Comparing with participants with lower levels of both traditional risk score and Saccharomyces, the adjusted odds ratio (95% CI) for T2D was 1.30 (1.03–1.65) for those with higher Saccharomyces levels but with low traditional risk score (Figure 3D). It further increased to 4.28 (3.48–5.27) for participants exhibiting both higher levels of traditional risk scores and Saccharomyces (Figure 3D). Overall, these results suggest Saccharomyces may emerge as an effect modifier of traditional risk factors in promoting T2D risk.
Temporal variability of the gut mycobiome over time
To assess the temporal variability of the gut mycobiome, we undertook a dynamic analysis of fungal composition. This analysis focused on genera with prevalence higher than 10% in the participants with repeated mycobiome profiling data across an average period of 3.02 years (interquartile range: 3–3.07) in the CHNS-2 cohort. There was a slight increase in the inter-individual variation of the gut mycobiome over time (Figure 4A). The differences in fungal composition were more pronounced between individuals than within individuals (p < 0.0001; Figure 4A). We found that age exhibited inverse association with intra-individual variation in gut fungal composition across the baseline and follow-up (Figure 4B). Importantly, this association persisted even after adjusting for sequencing depth, as indicated by the Spearman partial correlation coefficient (corr = −0.058, p = 0.01). When considering individual fungal genera, 45.3% (34 out of 75) showed significant alterations in abundance over time (Figure S3A). Specifically, among core taxa, there was a reduction in the abundance of Aspergillus, Penicillium, and Debaryomyces, while Candida exhibited an increased over the follow-up period. Additionally, we detected an average 1.57-fold reduction in observed OTUs (operational taxonomic units) at the amplicon sequence variant (ASV) level during the follow-up compared to the baseline (Figure 4C). This observation was validated by a linear mixed model adjusted for sequencing depth (adjusted beta = −5.96, 95% CI: −8.79 to −2.40). A consistent decrease was also detected for other alpha diversity indices (Figure S3B).
Figure 4.

Dynamic interplay between the gut mycobiome and cardiometabolic diseases
(A) Comparison of inter-individual Bray-Curtis distance of fungal composition between baseline and follow-up, and the within-pair Bray-Curtis distances across the two time points (n = 1,946).
(B) Correlation between age and intra-individual variation in gut fungal composition across the baseline and follow-up (n = 1,946).
(C) Comparison of the fungal alpha diversity (observed OTUs) between baseline and follow-up. The y axis refers to the observed OTUs at the ASV level (n = 1,946).
(D) Distribution of fungal taxa that are present in at least 50% of samples at a minimum relative abundance of 0.1% across different fungal clusters (n = 1,946 pairs).
(E) Transition of all participants between the fungal clusters from the baseline to follow-up (n = 1,946).
(F) Distribution of alpha diversity across different fungal pattern transitions. The numbers of participants in each fungal pattern were marked on the top of the boxplot. Key combinations highlighted: C1-C4, C2-C4, C3-C4, and C4-C4. Others shaded in gray.
(G) Dynamic interplay between mycobiome and human cardiometabolic diseases (n = 1,946). Generalized estimating equation model was employed to assess the difference between other fungal clusters and cluster 4. The models were adjusted for age, gender, and BMI and used independent correlation structure as the generalized estimating equation model’s working correlation matrix. Additionally, the models included both a random intercept and a random coefficient for provinces or megacities.
See also Figures S3 and S4 and Table S5.
We further discerned four distinct fungal clusters by applying Dirichlet mixture modeling on the fungal composition data at both time points (Figure S4A). We found that cluster 4 (C4) was remarkably different from the remaining clusters, primarily characterized by the dominance of Saccharomyces (Figure 4D and S4B). Characteristics of the participants at baseline, stratified by fungal clusters, are presented in Table S5. Through transition trajectory analysis, we investigated the dynamic changes in participants’ fungal pattern over time and found that only a small proportion of participants exchanged within the cluster 3 and cluster 4 (Figure 4E). Those individuals who transitioned to cluster 4 during the follow-up period exhibited lower alpha diversity indices compared to those who transitioned to alternative clusters, which was found to be independent of their fungal cluster status at baseline (Figure 4F).
Dynamic interplay between the gut mycobiome, cardiometabolic diseases, and serum metabolites
To investigate the dynamic interplay between the gut mycobiome and human health in a longitudinal context, we fitted a generalized estimating equation model to account for the dynamic nature of the gut mycobiome and status of host cardiometabolic diseases in the CHNS-2 cohort. In comparison to participants assigned to fungal enterotype of cluster 4 (dominance of Saccharomyces), those assigned to cluster 3 demonstrated a significantly lower risk of cardiometabolic diseases, with odds ratio of 0.45 (0.29–0.71) for T2D and 0.67 (0.49–0.91) for hypertension (Figure 4G).
To gain a deeper understanding of the distinctive variations in serum metabolites between fungal enterotype of cluster 4 and the other clusters, we utilized the LightGBM (light gradient-boosting machine) algorithm to construct machine learning classifiers. These classifiers aimed to differentiate individuals with the fungal enterotype of cluster 4 from those with each of the other fungal enterotypes, based on their serum metabolome. Receiver operating characteristic curve analysis demonstrated that the classification for cluster 3 and cluster 4 showed the highest performance, with a mean 10-fold cross-validation area under the curve (AUC) of 0.90 (Figure 5A).
Figure 5.

Association of the gut mycobiome with serum metabolites
(A) Comparing the performance of metabolome-based classifiers (LightGBM) in categorizing participants’ fungal clusters at the baseline (n = 911). Performance of the classifiers was assessed by area under the receiver operating characteristic curve (AUC) across 10-folds, with cluster 4 as the reference group, and each of other groups and their combinations as the comparison group. Here, the numbers of participants included in the cluster 1 to 4 were 518, 227, 85, and 80, respectively.
(B) Receiver operating characteristic curve for the orthogonal projections to latent structures discriminant analysis (OPLS-DA)-selected metabolites in categorizing participants into either cluster 3 or cluster 4 groups within the discovery (n = 165) and validation cohorts (n = 138). The effectiveness of these selected metabolites in predicting fungal clusters was assessed using the machine learning model (LightGBM).
(C) Fold change of differential metabolites between the fungal cluster 3 and cluster 4 (n = 165). The identified metabolites are visualized as dots, with colors corresponding to their respective categories. The dot size indicates the importance of the metabolite in distinguishing fungal clusters during the OPLS-DA analysis.
Through further analysis using orthogonal projection to latent structures discriminant analysis (OPLS-DA), we pinpointed 212 metabolites that contributed to the differentiation of cluster 3 and cluster 4 (Table S6). These distinguished metabolites were predominantly associated with fatty acid and amino acid metabolism and their derivatives. By reconstructing a classifier using these OPLS-DA-identified metabolites at the baseline, we achieved a similar classification accuracy for cluster 3 and cluster 4 as that of the primary all-metabolites-based model (AUC = 0.90; Figure 5B). Importantly, this classifier was successfully validated with follow-up participants who were distinct from the baseline participants (AUC = 0.84; Figure 5B). These follow-up participants transitioned from cluster 1 or cluster 2 at baseline to cluster 3 or cluster 4 during the follow-up phase. Additionally, our analysis employing the Wilcoxon rank-sum test highlighted significant differences in the abundance of 132 metabolites between the two groups (Figure 5C).
Metabolites as functional link mediating the association of mycobiome with T2D
In the CHNS-2 cohort, to investigate the intricate interplay among the gut mycobiome, serum metabolites, and cardiometabolic health, we firstly performed a weighted gene co-expression network analysis. This analysis grouped the OPLS-DA-identified metabolites into 15 distinct metabolite modules based on the similarity of metabolism profiles. The distribution of identified metabolites across different metabolism modules is presented in Figure S5A. The inter-correlations among these metabolite modules ranged from weak to moderate (Figure S5B). The majority of these modules, specifically 9 out of the 15, displayed significant differences between two distinct fungal patterns, denoted as cluster 3 and cluster 4 (Figure S5C). This underscores the substantial variations in metabolic profiles associated with these fungal patterns. Additionally, four metabolite modules (M2, M9, M12, and M14) exhibited significant associations with risk of T2D or hypertension (Figure S5D).
We then hypothesized that serum metabolite modules might mediate the impact of the gut mycobiome on cardiometabolic diseases. We found that two metabolite modules (M14 and M9) emerged as mediators of the effects of the gut mycobiome on T2D, with mediated effects of 37% and 16%, respectively (Figure S6A). Importantly, we did not identify any significant inverse effect, where gut fungi might influence metabolites through their impact on the disease. Furthermore, a majority of the metabolites belonging to the M9 and M14 modules exhibited significant differences between the T2D cases and controls (Figure 6A). The inter-correlations among these metabolites ranged from weak to moderate (Figure S6B).
Figure 6.

Functional metabolites mediate the effect of the gut mycobiome on type 2 diabetes
(A) Association of serum metabolites with type 2 diabetes (n = 165). Boxplot shows the distribution of mycobiome-related metabolites abundance among the type 2 diabetes cases and healthy controls. Significance was examined by logistic regression, with adjustment for age, gender, and BMI. The names of the metabolites are color-coded based on their belonging to specific metabolite modules: purple represented M9, and yellow corresponded to M14.
(B) Sankey diagram showing the significant mediation linkages (FDR < 0.05) where fungal genera acted as effect factors, metabolites as mediators, and type 2 diabetes as outcome (n = 165).
(C) Mediation linkages among the Blumeria, L-Valine, and type 2 diabetes (n = 165). The proportion of mediation effect is displayed at the center of the ring chart.
(D) As in (C), but for Saccharomyces, γ-linolenic acid, and type 2 diabetes (n = 165).
See also Figure S6.
For the individual core fungal taxa and metabolites belonging to the M9 and M14 modules, we uncovered three significant results in the mediation analyses, with forward mediation effects showing an FDR <0.05 and reverse mediation effects showing an FDR >0.05. These results highlight the potential role of two fungi (Saccharomyces and Blumeria) in the development of T2D via several metabolites (γ-linolenic acid, L-valine, and LPE (lysophosphatidylethanolamine) (Figure 6B). A noteworthy instance was the mediation effect of L-valine, which accounted for 45% of the total effect of Blumeria on T2D (FDRmediation = 0.02, Figure 6C). L-valine, one of the branched-chain amino acids (BCAAs), exhibited a positive correlation with Blumeria and was found to be elevated in individuals with T2D in our study (Figure 6A). Consistently, L-valine has previously been implicated in causing insulin resistance in animal studies23,24 and has shown a positive association with T2D in human cohort studies.25
Another example was γ-linolenic acid, which displayed a positive correlation with the abundance of Saccharomyces and was found to be enriched in T2D patients in our study (Figure 6A). It acted as a mediator of Saccharomyces in the progression of T2D (FDRmediation = 0.04), with a mediation effect of 16% (Figure 6D). Taken together, our findings provide potential functional insights underlying the relationships between the gut mycobiome and human cardiometabolic diseases.
Discussion
Here, we systematically characterized the gut mycobiome composition and dynamics in a large nationwide cohort of Chinese adults. Leveraging a rich array of phenotypic measurements, our research builds upon existing knowledge by uncovering robust correlations between gut mycobiome composition and cardiometabolic health. Moreover, we observed temporal variability in both individual and fungal patterns of the gut mycobiome over three years, emphasizing its inherently dynamic nature. Additionally, we found the dynamic interplay between the gut mycobiome and cardiometabolic diseases. Finally, our investigations revealed the potential functional role of metabolites as mediators in the association between the gut mycobiome and T2D. These findings provide deep insights into the intricate interactions between the gut mycobiome and the host, shedding light on its relevance to cardiometabolic health.
Several prior studies have reported associations of the gut mycobiome with geographic locations and dietary and lifestyle factors, including milk, fruit, vegetable, fish, sugar, and alcohol consumption.13,17 Our substantially expanded sample size and comprehensive phenotype data have enabled us to extend the investigation, thereby enhancing the scope of previous knowledge. Our investigation underscores the considerable impact of dietary and lifestyle factors on fungal variation, highlighting the contribution of staple food consumption (specifically wheat and rice) and physical activity. Both rice and wheat, as fundamental components of the Chinese diet, contribute substantially to caloric intake and represent distinct dietary patterns.26
Fungi are common inhabitants of the intestinal barrier surface. Animal studies have shown that the gut mycobiome is influenced by the environment and correlates with metabolic outcomes in response to diet.27 Dysbiosis in fungal communities can affect mucus production, epithelial function, and immune defense, potentially contributing to the development of metabolic diseases.6 While higher levels of gut bacterial alpha diversity have consistently been associated with improved health status,28,29 evidence for fungal diversity remains limited. In the present study, we found that fungal alpha diversity was inversely associated with multiple cardiometabolic diseases and risk factors. A recent study8 reported that fungal alpha diversity was lower in participants with T2D and hypertension based on public fecal metagenomes. However, the association was not significant, likely due to the small sample size (n < 350) and technical challenges in detecting fungi using metagenomic methods.3 We also observed that gut Saccharomyces was associated with T2D but not with dyslipidemia or hypertension, suggesting that Saccharomyces might influence T2D through mechanisms that were independent of those typically associated with dyslipidemia or hypertension. This was consistent with previous research, which also found that no single fungal genus was consistently associated with different cardiometabolic diseases.8
There was a mycobiome-dependent association between the traditional risk factors and T2D, where a stronger association was observed in individuals with enrichment of Saccharomyces. These findings suggest the importance of considering Saccharomyces as a potential effect modifier of traditional risk factors in the prevention of T2D risk. Importantly, the link between Saccharomyces and T2D is further supported by results from our independently conducted dynamic analysis. This reinforces the potential role of Saccharomyces as a predictive biomarker of T2D. It is crucial to note that these findings warrant further investigation to elucidate the underlying mechanisms and biological pathways by which Saccharomyces may influence T2D risk. Additionally, considering the potential role of Saccharomyces in modifying the impact of traditional risk factors may open avenues for personalized approaches to diabetes prevention and management.
Our mediation analysis provided evidence that several metabolites may play a mediating role in the relationship between the gut mycobiome and T2D. Of particular interest, we demonstrated that L-valine, a BCAA, may mediate the association between Blumeria and T2D. BCAAs are synthesized in bacteria, plant, and fungi and are well-known contributors to insulin resistance.23,24 Additionally, γ-linolenic, an omega-6 fatty acid, exhibited a positive association with T2D, aligning with previous findings.30 Furthermore, our observations suggest that γ-linolenic acid might mediate the impact of Saccharomyces on T2D. These associations between specific fungi and metabolites with T2D provide valuable insights into the potential mechanism by which gut mycobiota may contribute to the pathogenesis of the complex metabolic disorders. Further investigations into these specific pathways may shed light on therapeutic targets and interventions for T2D and potentially other cardiometabolic diseases. Nevertheless, additional studies are needed to validate and elucidate the underlying mechanism of these intriguing findings.
In summary, our study offers a comprehensive characterization of the gut mycobiome composition and dynamics in a nationwide Chinese cohort. By extensively mapping the gut mycobiome to environmental factors, we have created a valuable resource for unraveling the factors associated with inter-individual variation of the gut mycobiome. We reveal the inter-connections between the gut mycobiome and cardiometabolic health, underscoring gut Saccharomyces as a potential predictive biomarker of T2D risk. This study advances our understanding of the roles of the gut mycobiome in cardiometabolic health and provides potential targets for intervention and therapeutics aimed at modulating the gut mycobiome.
Limitations of the study
Our study has several limitations. Firstly, our findings regarding the associations of the gut mycobiome with cardiometabolic health are observational, without the establishment of causation. Future research is needed to confirm the potential causal relationships. Secondly, while mediation analysis has been utilized in observational microbiome studies,31,32,33 further animal studies are necessary to elucidate the underlying mechanisms and potential causal direction. Thirdly, current fungal databases are still immature, particularly for less-studied and non-culturable fungi, which results in many reads being classified as “unknown/unclassified.” Additionally, primer design can introduce biases affecting amplification efficiency.3 Future research should prioritize developing validated methodologies, including refining DNA extraction techniques, improving the quality of fungal databases, and creating more effective primers. Finally, all participants included in the present study are Chinese, so caution should be exercised in extrapolating our findings to other ethnic groups.
Resource availability
Lead contact
Further information and requests for resources should be directed to the lead contact, Prof. Ju-Sheng Zheng (zhengjusheng@westlake.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The raw ITS sequencing data have been deposited in the Genome Sequence Archive (GSA) (https://ngdc.cncb.ac.cn/gsa/) at the accession number CRA013806.
-
•
Due to informed consent regulations, phenotypic data for the cohort are available upon request from the CHNS (https://www.cpc.unc.edu/projects/china/).
-
•
The computer codes for main analyses in this study are deposited in GitHub under https://github.com/wenutrition/CHNS-fungi-cardiometabolic-diseases. The DOI at Zenodo is https://doi.org/10.5281/zenodo.13353503. The software and packages used for this analysis are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
Acknowledgments
We thank the High-Performance Computing Center and High-Throughput Core Facility at Westlake University for assistance in computing and data generation. This study was funded by the “Pioneer” and “Leading goose” R&D Program of Zhejiang (2024SSYS0032), the National Key R&D Program of China (2022YFA1303900), the National Natural Science Foundation of China (82073529, 82404242, 82103826, and U21A20427), the Research Program of Westlake Laboratory of Life Sciences and Biomedicine (202208012), and China Postdoctoral Science Foundation (2023M733177, 2022M722833). The China Health and Nutrition Survey received funding from the National Institutes of Health (NIH) (R01HD30880, R01AG065357, P30DK056350, and R01HD38700) from 1989 to 2019 and was supported by the National Institutes of Health and National Institute of Diabetes and Digestive and Kidney Diseases (R01DK104371) and the Carolina Population Center (P2CHD050924 and P30AG066615). The funders had no role in collecting data, study design, interpretation of data, or the decision to submit the manuscript for publication.
Author contributions
J.-S.Z., B.Z., and H.W. conceived the study. B.Z. and H.W. established the human cohorts. J.-S.Z. and W.G. designed and performed the statistical analyses. H.W., J.Z., W.D., X.J., and C.S. collected the data. W.G., C.S., W.D., J.Z., K.Z., X.W., Z.M., C.G., and Y.F. contributed to data curation. W.G. and J.-S.Z. wrote the first draft of the manuscript. W.G., H.W., C.S., and Y.F. contributed equally to the work. All authors contributed to the discussion and critical revision of the manuscript and approved the final draft.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human feces | This paper | N/A |
| Critical commercial assays | ||
| E.Z.N.A.® Soil DNA Kit | Omega Bio-tek, Norcross, GA, U.S. | Cat# D5625-01 |
| Deposited data | ||
| ITS2 rRNA sequencing data | This paper | Genome Sequence Archive (GSA): CRA013806 |
| Metadata | This paper | https://www.cpc.unc.edu/projects/china/ |
| Oligonucleotides | ||
| ITS2 rRNA Forward Primer GCATCGATGAAGAACGCAGC |
Op De et al.34 | ITS3F |
| ITS2 rRNA Reverse Primer TCCTCCGCTTATTGATATGC |
Op De et al.34 | ITS4R |
| Software and algorithms | ||
| Stata package v15.0 | StataCorp LLC, TX, USA | https://www.stata.com |
| NetCoMi R package v.1.0.2 | Peschel et al.35 | https://github.com/stefpeschel/NetCoMi?tab=readme-ov-file |
| vegan R package v.2.5.7 | CRAN | https://cran.r-project.org/web/packages/vegan/index.html |
| microbiome R package v.1.19.1 | bioconductor | https://www.bioconductor.org/packages/release/bioc/html/microbiome.html |
| lightgbm Python package v.3.2.1 | sklearn | https://lightgbm.readthedocs.io/en/stable/ |
| ropls R package v.1.26.4 | bioconductor | https://bioconductor.org/packages/release/bioc/html/ropls.html |
| WGCNA R package v.1.70.3 | CRAN | https://cran.r-project.org/web/packages/WGCNA/index.html |
| mediation R package v.4.5.0 | CRAN | https://cran.r-project.org/web/packages/mediation/index.html |
| QIIME 2 | Bolyen et al.15 | https://qiime2.org/ |
| The computer code for main analyses | This paper |
https://github.com/wenutrition/CHNS-fungi-cardiometabolic-diseases Zenodo: https://doi.org/10.5281/zenodo.13353503 |
Experimental model and study participant details
Study cohorts
The present study was based on data from the China Health and Nutrition Survey (CHNS). CHNS is nationwide longitudinal cohort within China, distinguished by its utilization of a multistage, random cluster design.14 This intricate design ensures the comprehensive coverage of a diverse range of critical public health risk factors, health outcomes, dietary and lifestyle components, as well as demographic and economic factors. The CHNS rounds have been completed in 1989, 1991, 1993, 1997, 2000, 2004, 2006, 2009, 2011, 2015 and 2018. Notably, during the 2015 and 2018 rounds of the CHNS, stool samples were collected for the measurement of the gut mycobiome. In this study, we included two sub-cohorts of the CHNS, identified as CHNS-1 and CHNS-2. In the CHNS-1 cohort, a total of 10,865 participants from 12 provinces (Guangxi, Guizhou, Heilongjiang, Henan, Hubei, Hunan, Jiangsu, Liaoning, Shandong, Shanxi, Yunnan, Zhejiang) and 3 municipalities (Beijing, Chongqing, Shanghai) provided stool samples for the measurement of ITS rRNA. After excluding participants with low depth of sequencing reads (<20,000, n = 32) and those without detailed metadata (n = 138), 10,695 participants (mean age 51.3 ± 15.5 years, 54% were women) were included in the CHNS-1 cohort. They were utilized to comprehensively profile the variations in the mycobiome and establish associations between the gut mycobiome and cardiometabolic health.
After adopting the same inclusion and exclusion criteria as used in the CHNS-1 cohort, we included an additional sub-cohort of independent participants (CHNS-2) from four prominent provinces in China (Liaoning, Guangxi, Jiangsu, Hubei), with repeatedly collected metadata (n = 1,946 pairs, mean age 56.5 ± 13.7 years, 55% were women), mycobiome data (1,946 pairs), and metabolome data (n = 996 pairs) over a period of 3 years. This sub-cohort was utilized to explore the inherent long-term variability of the gut mycobiome and to provide a deeper biological insight into the intricate interplay between the gut mycobiome, metabolome and cardiometabolic diseases.
Ethical approval
The CHNS protocol was approved by the Institutional Review Boards of the Chinese Center for Disease Control and Prevention (No. 201524), the Westlake University (No. 20220429ZJS002), the University of North Carolina at Chapel Hill, USA, and the US National Institute for Nutrition and Health (No. 07–1963). Informed consent was obtained from all participants.
Method details
Metadata collection
We collected phenotypes across a diverse range of categories, including demographic factors, dietary and lifestyle components, anthropometric factors, chronic diseases, medications, blood parameters, urbanization-related indices, and various other environmental factors. Demographic, medication, lifestyle, and dietary data were collected through standard questionnaires during home visits over three consecutive days. Habitual dietary intakes were evaluated using three successive 24-h dietary recalls, including two on weekdays and one on a weekend day. Trained investigators guided participants in reporting the specific types and quantities of all foods consumed within the preceding 24 h.36 Physical activity was assessed as a total metabolic equivalent (MET) for task hours per week.37 Anthropometric factors were measured on-site by trained staff.
We utilized the urbanization index as a comprehensive measure to assess the degree of urbanization within each community. This index comprises 12 community indicators, encompassing factors such as population density, economic activity, presence of traditional and modern markets, transportation and health infrastructure, sanitation facilities, communication access, availability of social services, cultural diversity, and housing conditions. Each of these 12 components was computed based on the presence of relevant infrastructure, the percentage of households utilizing these services within the community, and assigned a maximum score of 10 (ranging from 0 to 10). The methodology for scoring algorithms, threshold values are elaborated elsewhere.38
Following an overnight fasting, blood samples were collected via venipuncture. All samples were analyzed in a national central lab in Beijing (medical laboratory accreditation certificate ISO 15189:2007) with strict quality control. Blood glucose levels were measured using a glucose oxidase phenol 4-aminoantipyrine peroxidase kit (Randox, Crumlin, UK) and a Hitachi 7,600 Analyzer (Hitachi, Tokyo, Japan). Glycated hemoglobin (HbA1c) was measured via high-performance liquid chromatography system (model HLC-723 G7; Tosoh Corporation, Tokyo, Japan). All blood lipid measures were on the Hitachi 7,600 automated analyzer (Hitachi Inc., Tokyo, Japan).
Assessments of cardiometabolic diseases
Type 2 diabetes (T2D) cases were ascertained based on fasting blood glucose ≥7.0 mmol/L or HbA1c ≥ 47.5 mmol/mol (6.5%), or being currently under medical treatment for T2D during the collection of stool samples, according to the American Diabetes Association criteria for the diagnosis of diabetes.39 Dyslipidemia cases were ascertained based on TC ≥ 240 mg/dL (6.2 mmol/L) or TG ≥ 200 mg/dL (2.3 mmol/L) or LDL-C ≥ 160 mg/dL (4.1 mmol/L) or HDL-C as <40 mg/dL (1.0 mmol/L), according to the guidelines for the prevention and treatment of dyslipidemia in Chinese adults.40 Hypertension was defined as systolic blood pressure over 140 mmHg, diastolic blood pressure over 90 mmHg, or taking antihypertensive medicine.41
Fecal sample collection and gut mycobiome analyses
Stool samples were collected by the participants themselves, who received instructions for the collection and storage process, and were immediately frozen at −20°C refrigerators after collection. All stool samples were transported through a cold chain to the central laboratory within 1–2 days and stored at −40°C until processing.
Total fungal genomic DNA was extracted using the E.Z.N.A. soil DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) according to manufacturer’s instructions. The quality and concentration of extracted DNA were determined using 1.0% agarose gel electrophoresis and a NanoDrop ND-2000 spectrophotometer (Thermo Scientific Inc., USA). The hypervariable region of ITS2 was amplified using the primer pairs ITS3F (GCATCGATGAAGAACGCAGC) and ITS4R (TCCTCCGCTTATTGATATGC)34 by an ABI GeneAmp 9700 PCR thermocycler (ABI, CA, USA). The PCR reaction mixture including 4 μL of 5 × Fast Pfu buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of forward and reverse primer (5 μM), 0.4 μL of Fast Pfu polymerase, and 10 ng of template DNA. The volume was then brought up to 20 μL with ddH2O. The PCR was carried out under the following conditions: initial denaturation for 3 min at 95°C, followed by 35 cycles of denaturing for 30 s at 95°C, annealing for 30 s at 55°C, and extension for 45 s at 72°C, and single extension at 72°C for 10 min, and finally hold at 10°C. The PCR product was extracted from a 2% agarose gel and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to manufacturer’s instructions, and quantified using Quantus Fluorometer (Promega, USA).
The gut mycobiome was profiled by sequencing of Internal Transcribed Spacer (ITS). The hypervariable region of ITS2 was amplified using the primer pairs ITS3F (GCATCGATGAAGAACGCAGC) and ITS4R (TCCTCCGCTTATTGATATGC) by an ABI GeneAmp 9700 PCR thermocycler (ABI, CA, USA). Purified amplicons were pooled in equimolar amounts and paired-end sequenced on NovaSeq 6000 platform (Illumina, San Diego,USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). We obtained an average of 145,675 ± 65,187 reads/sample (mean ± SD) in the CHNS-1 cohort and 226,503 ± 65,272 reads in the CHNS-2 cohort.
The raw sequencing reads were further processed with the Quantitative Insights Into Microbial Ecology 2 platform (QIIME 2).15 In summary, DADA242 was used to filter sequencing reads with quality score Q < 25 and to denoise reads into amplicon sequence variants (ASVs), resulting in feature tables and representative sequences. The ASV features that were presented in only one sample were excluded before conducting the taxonomy analysis. Taxonomy was assigned to ASVs based on the UNITE (version 8.2, 99%) database using the VSEARCH tool wrapped in QIIME2. Reads that could not be confidently assigned to known taxa due to insufficient matching with reference sequences were labeled with the highest taxonomic rank that could be assigned, followed by ‘spp’. ASVs that were not filtered were rarefied at 20,000 reads to calculate alpha diversity. The measures of alpha diversity at the ASV level include observed OTUs, Shannon’s diversity index, Faith’s phylogenetic diversity, and Pielou’s evenness.
Serum metabolome profiling
The serum samples were stored at −80°C in a refrigerator and thawed on ice before being vortexed for 10 s. For metabolomic analysis, 50 μL of the sample and 300 μL of the extraction solution (ACN:Methanol = 1:4, V/V) containing internal standards were added to a 2 mL microcentrifuge tube. The sample was vortexed for 3 min and then centrifuged at 12,000 rpm for 10 min at 4°C. Subsequently, 200 μL of the supernatant was collected and placed at −20°C for 30 min, followed by another centrifugation at 12,000 rpm for 3 min at 4°C. An aliquot of 180 μL of the supernatant was transferred for LC-ESI-MS/MS analysis.
The sample extracts were analyzed using an LC-ESI-MS/MS system (UPLC, ExionLC AD, https://sciex.com.cn/; MS, QTRAP System, https://sciex.com/) following standard protocols. The triple quadrupole-linear ion trap mass spectrometer (QTRAP) was used to perform LIT and triple quadrupole (QQQ) scans, operated and controlled by Analyst 1.6.3 software (Sciex) with standard parameters. The source temperature 500°C; ion spray voltage (IS) was 5500 V (positive) and −4500 V (negative); ion source gas I (GSI), gas II (GSII), and curtain gas (CUR) were set at 55, 60, and 25.0 psi, respectively; the collision gas (CAD) was set to high. Instrument tuning and mass calibration were performed with10 and 100 μmol/L polypropylene glycol solutions in QQQ and LIT modes, respectively. A specific set of MRM transitions were monitored for each period according to the metabolites eluted within this period.
Quantification and statistical analysis
The gut mycobiome taxa data were analyzed at the genus level. To address the issue of sparse fungal data in subsequent analyses, taxa present in less than 10% of the population were excluded. For the mycobiome-phenotype association analysis, the taxa data were transformed using the centered log-ratio (CLR) method to address the compositional nature of the mycobiome data. Prior to performing the CLR transformation, multiplicative imputation was employed to handle zero values.43 As for blood metabolome data, a natural logarithmic (loge) transformation was applied. All statistical tests used were two sided. To account for potential false positive discoveries, multiple-testing correction was implemented using the Benjamini–Hochberg method. An adjusted p value of less than 0.05 was considered indicative of statistical significance. All regression analyses were performed using Stata (v15.0).
Core taxa identification
Core fungal taxa were identified as those appearing in at least 50% of samples with a minimum relative abundance of 0.1%, or forming central nodes in fungal network. NetCoMi package 43 was used to perform fungal co-abundance networks analyses. We used SparCC analysis to estimate the fungal correlation matrix. Correlations with FDR adjusted p values <0.05 and with a magnitude above 0.2 were selected for further visualization and network analysis. Here, we used eigenvector centrality to define the central node in the co-abundance network (node with a centrality value above the empirical 95% quantile in the network).
Estimation of the effect of environmental factors on fungal variability
We used the vegdist function in the R package vegan (version 2.5.7) to calculate the gut fungal Bray-Curtis dissimilarity matrix. The variations of Bray-Curtis dissimilarity matrix explained by each of the environmental variables was determined by PERMANOVA analysis using the function adonis2 in vegan. The p value was determined through 999 permutations.
Correlation between fungal genera and observed OTUs
Spearman correlation analysis was used to examine the correlation between measured fungal genera and observed OTUs.
Association of the gut mycobiome with human cardiometabolic diseases
We used logistic mixed-effects regression and linear mixed-effects regression, respectively, to independently evaluate the associations of each fungal features (per SD) with human cardiometabolic diseases and their indicative factors (fasting glucose, HbA1c, HDL, LDL, TC, TG, SBP and DBP). The models were adjusted for age, gender, and BMI and included a random intercept and random coefficient for provinces or megacities, accounting for the variability in gut fungal composition across different regions.
For the fungus that was significantly associated with T2D, we further evaluated the interaction between the fungus and T2D traditional risk score for the risk of T2D using a logistic mixed-effects regression model. The model simultaneously included the two main effects, as well as the product of the two main effects, along with random intercepts and random coefficients to account for the variability across provinces or megacities. The risk score utilized in the analysis was developed based on the standards of medical care for T2D in China. It encompassed factors such as age, gender, waist circumference, BMI, and systolic blood pressure.44 Furthermore, for the fungal taxa that showed a significant interaction with risk score for T2D, we performed a subgroup analysis stratified by the median level of the corresponding factor.
Temporal variability of gut mycobiome over time
We conducted dynamic analyses on participants with paired mycobiome data from the 2015 and 2018 surveys. To evaluate differences in the gut mycobiome between the two time points, we performed a principal component analysis (PCA) based on between-samples Aitchison distance, obtained from CLR-transformed abundance data. We performed paired two-sided Wilcoxon-signed rank tests on the first two axes of principal components to assess the variation between the two time points. To compare differences in overall fungal composition between and within individuals, we employed the Wilcoxon rank-sum test on the fungal Bray-Curtis dissimilarity. Furthermore, we utilized paired two-sided Wilcoxon-signed rank test to evaluate temporal changes in both alpha diversity and the relative abundance of gut fungi. For the clustering of samples across the two time points, we adopted the R package microbiome (version 1.19.1) to perform Dirichlet Multinomial Mixture (DMM) analysis.45 The optimal number of clusters was identified based on the local minimum of the Laplace approximation score. To ensure the robustness of this cluster count, we used additional alternative criteria, such as the Bayesian Information Criterion (BIC) and Akaike Information Criterion (AIC). We quantified the contribution of each fungal taxa to the fungal clusters, considering taxa with contribution values exceeding the 99th percentile of the overall contribution values as the primary contributor to the fungal cluster. We then analyzed the transition of all participants between the DMM clusters from the baseline to follow-up. Subsequently, we analyzed the transitions of all participants between the DMM clusters from baseline to follow-up.
Dynamic interplay between gut mycobiome and human cardiometabolic diseases
We fitted a generalized estimating equation (GEE) model to examine the association of dynamic changes in gut mycobiome clusters over time with cardiometabolic diseases. The models were adjusted for age, gender, and BMI and used independent correlation structure as the GEE model’s working correlation matrix. Additionally, the models included both a random intercept and a random coefficient for provinces or megacities.
Associations of gut mycobiome with human serum metabolites
Among the participants with measured mycobiome and metabolome data in the 2015 survey, we constructed metabolome-based machine learning classifiers (LightGBM)46 to classify the fungal clusters. Performance of the classifiers was assessed by area under the Receiver-Operating Characteristic curve (AUC) across 10-folds. The reference group was cluster 4, and each of other groups and their combinations as the comparison group.
We performed orthogonal projections to latent structures discriminant analysis (OPLS-DA) to investigate the differential metabolites between fungal cluster 3 and cluster 4. This analysis was performed in the R language using the ‘ropls (version 1.26.4). Metabolites with a variable importance in projection (VIP) score exceeding 1 were selected for further investigation. We evaluated the chosen metabolites using the machine learning model (LightGBM). This analysis was conducted using the Python package lightgbm (version 3.2.1). The model was trained and initially validated using 10-fold cross-validation with the 2015 dataset. Subsequently, the model trained on the 2015 data (discovery model) was directly applied and validated on the 2018 dataset, which did not include any participants from the training dataset. Additionally, we employed the Wilcoxon rank-sum test to calculate the fold change of selected metabolites between the two groups.
Mediation analysis
We utilized weighted correlation network analysis (WGCNA, version 1.70.3)47 to detect co-expressed metabolite modules based on the metabolites selected from OPLS-DA. To assess the associations between fungal clusters and diseases, as well as between fungal clusters (cluster 3 and cluster 4) and metabolite modules, logistic mixed-effects and linear mixed-effects regression analyses were conducted, respectively. Furthermore, logistic mixed-effects regression was used to evaluate the association between metabolite modules and diseases. All models were adjusted for age, gender, and BMI and included a random intercept and random coefficient for provinces or megacities, accounting for the variability in gut fungal composition across different regions. For metabolite modules that exhibited significant associations with both fungal partitions and diseases, we conducted mediation analysis using the R package mediation (v4.5.0) to investigate the potential mediation effect of metabolites on the link between mycobiome and cardiometabolic diseases. In addition, we also performed mediation analysis to explore whether disease status could mediate the association between mycobiome and the metabolites.
Published: October 4, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101775.
Contributor Information
Bing Zhang, Email: zhangbing@chinacdc.cn.
Ju-Sheng Zheng, Email: zhengjusheng@westlake.edu.cn.
Supplemental information
References
- 1.Peay K.G., Kennedy P.G., Talbot J.M. Dimensions of biodiversity in the Earth mycobiome. Nat. Rev. Microbiol. 2016;14:434–447. doi: 10.1038/nrmicro.2016.59. [DOI] [PubMed] [Google Scholar]
- 2.Iliev I.D., Leonardi I. Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat. Rev. Immunol. 2017;17:635–646. doi: 10.1038/nri.2017.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richard M.L., Sokol H. The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 2019;16:331–345. doi: 10.1038/s41575-019-0121-2. [DOI] [PubMed] [Google Scholar]
- 4.Saftien A., Puschhof J., Elinav E. Fungi and cancer. Gut. 2023;72:1410–1425. doi: 10.1136/gutjnl-2022-327952. [DOI] [PubMed] [Google Scholar]
- 5.Stefan N., Schulze M.B. Metabolic health and cardiometabolic risk clusters: implications for prediction, prevention, and treatment. Lancet Diabetes Endocrinol. 2023;11:426–440. doi: 10.1016/S2213-8587(23)00086-4. [DOI] [PubMed] [Google Scholar]
- 6.Wang L., Zhang K., Zeng Y., Luo Y., Peng J., Zhang J., Kuang T., Fan G. Gut mycobiome and metabolic diseases: The known, the unknown, and the future. Pharmacol. Res. 2023;193:106807. doi: 10.1016/j.phrs.2023.106807. [DOI] [PubMed] [Google Scholar]
- 7.Wheeler M.L., Limon J.J., Bar A.S., Leal C.A., Gargus M., Tang J., Brown J., Funari V.A., Wang H.L., Crother T.R., et al. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe. 2016;19:865–873. doi: 10.1016/j.chom.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan Q., Li S., Yan Q., Huo X., Wang C., Wang X., Sun Y., Zhao W., Yu Z., Zhang Y., et al. A genomic compendium of cultivated human gut fungi characterizes the gut mycobiome and its relevance to common diseases. Cell. 2024;187:2969–2989.e24. doi: 10.1016/j.cell.2024.04.043. [DOI] [PubMed] [Google Scholar]
- 9.Van Syoc E., Nixon M.P., Silverman J.D., Luo Y., Gonzalez F.J., Elbere I., Klovins J., Patterson A.D., Rogers C.J., Ganda E. Changes in the type 2 diabetes gut mycobiome associate with metformin treatment across populations. mBio. 2024;15:e0016924. doi: 10.1128/mbio.00169-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayasudha R., Das T., Kalyana Chakravarthy S., Sai Prashanthi G., Bhargava A., Tyagi M., Rani P.K., Pappuru R.R., Shivaji S. Gut mycobiomes are altered in people with type 2 Diabetes Mellitus and Diabetic Retinopathy. PLoS One. 2020;15:e0243077. doi: 10.1371/journal.pone.0243077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu J., Zhao L., Cheng Y., Chen Q., Xu Y., Lu Y., Gao J., Lei W., Yan C., Ling Z., Wu S. Exploring the gut mycobiome: differential composition and clinical associations in hypertension, chronic kidney disease, and their comorbidity. Front. Immunol. 2023;14:1317809. doi: 10.3389/fimmu.2023.1317809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou Y., Ge A., Lydia B., Huang C., Wang Q., Yu Y. Gut mycobiome dysbiosis contributes to the development of hypertension and its response to immunoglobulin light chains. Front. Immunol. 2022;13:1089295. doi: 10.3389/fimmu.2022.1089295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Y., Zuo T., Cheung C.P., Gu W., Wan Y., Zhang F., Chen N., Zhan H., Yeoh Y.K., Niu J., et al. Population-Level Configurations of Gut Mycobiome Across 6 Ethnicities in Urban and Rural China. Gastroenterology. 2021;160:272–286.e11. doi: 10.1053/j.gastro.2020.09.014. [DOI] [PubMed] [Google Scholar]
- 14.Popkin B.M., Du S., Zhai F., Zhang B. Cohort Profile: The China Health and Nutrition Survey--monitoring and understanding socio-economic and health change in China, 1989-2011. Int. J. Epidemiol. 2010;39:1435–1440. doi: 10.1093/ije/dyp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F., et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nash A.K., Auchtung T.A., Wong M.C., Smith D.P., Gesell J.R., Ross M.C., Stewart C.J., Metcalf G.A., Muzny D.M., Gibbs R.A., et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome. 2017;5:153. doi: 10.1186/s40168-017-0373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szóstak N., Handschuh L., Samelak-Czajka A., Tomela K., Schmidt M., Pruss Ł., Milanowska-Zabel K., Kozlowski P., Philips A. Host Factors Associated with Gut Mycobiome Structure. mSystems. 2023;8:e0098622. doi: 10.1128/msystems.00986-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shuai M., Fu Y., Zhong H.-L., Gou W., Jiang Z., Liang Y., Miao Z., Xu J.J., Huynh T., Wahlqvist M.L., et al. Mapping the human gut mycobiome in middle-aged and elderly adults: multiomics insights and implications for host metabolic health. Gut. 2022;71:1812–1820. doi: 10.1136/gutjnl-2021-326298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmad H.F., Castro Mejia J.L., Krych L., Khakimov B., Kot W., Bechshøft R.L., Reitelseder S., Højfeldt G.W., Engelsen S.B., Holm L., Nielsen D.S. Gut Mycobiome dysbiosis is linked to hypertriglyceridemia among home Dwelling elderly Danes. bioRxiv. 2020 doi: 10.1101/2020.04.16.044693. Preprint at. [DOI] [Google Scholar]
- 20.Kowalewska B., Zorena K., Szmigiero-Kawko M., Wąż P., Myśliwiec M. Higher diversity in fungal species discriminates children with type 1 diabetes mellitus from healthy control. Patient Prefer. Adherence. 2016;10:591–599. doi: 10.2147/PPA.S97852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shah S., Locca A., Dorsett Y., Cantoni C., Ghezzi L., Lin Q., Bokoliya S., Panier H., Suther C., Gormley M., et al. Alterations of the gut mycobiome in patients with MS. EBioMedicine. 2021;71:103557. doi: 10.1016/j.ebiom.2021.103557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiaro T.R., Soto R., Zac Stephens W., Kubinak J.L., Petersen C., Gogokhia L., Bell R., Delgado J.C., Cox J., Voth W., et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci. Transl. Med. 2017;9:eaaf9044. doi: 10.1126/scitranslmed.aaf9044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pedersen H.K., Gudmundsdottir V., Nielsen H.B., Hyotylainen T., Nielsen T., Jensen B.A.H., Forslund K., Hildebrand F., Prifti E., Falony G., et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535:376–381. doi: 10.1038/nature18646. [DOI] [PubMed] [Google Scholar]
- 24.Neinast M., Murashige D., Arany Z. Branched Chain Amino Acids. Annu. Rev. Physiol. 2019;81:139–164. doi: 10.1146/annurev-physiol-020518-114455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morze J., Wittenbecher C., Schwingshackl L., Danielewicz A., Rynkiewicz A., Hu F.B., Guasch-Ferré M. Metabolomics and Type 2 Diabetes Risk: An Updated Systematic Review and Meta-analysis of Prospective Cohort Studies. Diabetes Care. 2022;45:1013–1024. doi: 10.2337/dc21-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu K., Zhang B., Liu Y., Mi B., Wang Y., Shen Y., Shi G., Dang S., Liu X., Yan H. Staple Food Preference and Obesity Phenotypes: The Regional Ethnic Cohort Study in Northwest China. Nutrients. 2022;14:5243. doi: 10.3390/nu14245243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mims T.S., Abdallah Q.A., Stewart J.D., Watts S.P., White C.T., Rousselle T.V., Gosain A., Bajwa A., Han J.C., Willis K.A., Pierre J.F. The gut mycobiome of healthy mice is shaped by the environment and correlates with metabolic outcomes in response to diet. Commun. Biol. 2021;4:281. doi: 10.1038/s42003-021-01820-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z., Radjabzadeh D., Chen L., Kurilshikov A., Kavousi M., Ahmadizar F., Ikram M.A., Uitterlinden A.G., Zhernakova A., Fu J., et al. Association of Insulin Resistance and Type 2 Diabetes With Gut Microbial Diversity: A Microbiome-Wide Analysis From Population Studies. JAMA Netw. Open. 2021;4:e2118811. doi: 10.1001/jamanetworkopen.2021.18811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aasmets O., Krigul K.L., Lüll K., Metspalu A., Org E. Gut metagenome associations with extensive digital health data in a volunteer-based Estonian microbiome cohort. Nat. Commun. 2022;13:869. doi: 10.1038/s41467-022-28464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miao Z., Lin J.S., Mao Y., Chen G.D., Zeng F.F., Dong H.L., Jiang Z., Wang J., Xiao C., Shuai M., et al. Erythrocyte n-6 polyunsaturated fatty acids, gut microbiota and incident type 2 diabetes: a prospective cohort study. Diabetes Care. 2020;43:2435–2443. doi: 10.1101/2020.03.29.20039693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin L., Yi X., Liu H., Meng R., Li S., Liu X., Yang J., Xu Y., Li C., Wang Y., et al. The airway microbiome mediates the interaction between environmental exposure and respiratory health in humans. Nat. Med. 2023;29:1750–1759. doi: 10.1038/s41591-023-02424-2. [DOI] [PubMed] [Google Scholar]
- 32.Chen L., Wang D., Garmaeva S., Kurilshikov A., Vich Vila A., Gacesa R., Sinha T., Lifelines Cohort Study. Segal E., Weersma R.K., et al. The long-term genetic stability and individual specificity of the human gut microbiome. Cell. 2021;184:2302–2315.e12. doi: 10.1016/j.cell.2021.03.024. [DOI] [PubMed] [Google Scholar]
- 33.Chen L., Zhernakova D.V., Kurilshikov A., Andreu-Sánchez S., Wang D., Augustijn H.E., Vich Vila A., Lifelines Cohort Study. Weersma R.K., Medema M.H., et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat. Med. 2022;28:2333–2343. doi: 10.1038/s41591-022-02014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Op De Beeck M., Lievens B., Busschaert P., Declerck S., Vangronsveld J., Colpaert J.V. Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS One. 2014;9:e97629. doi: 10.1371/journal.pone.0097629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peschel S., Müller C.L., von Mutius E., Boulesteix A.L., Depner M. NetCoMi: network construction and comparison for microbiome data in R. Brief. Bioinform. 2021;22:bbaa290. doi: 10.1093/bib/bbaa290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X., Zhang J., Du W., Su C., Ouyang Y., Huang F., Jia X., Li L., Bai J., Zhang B., et al. Multi-Trajectories of Macronutrient Intake and Their Associations with Obesity among Chinese Adults from 1991 to 2018: A Prospective Study. Nutrients. 2021;14:13. doi: 10.3390/nu14010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng S.W., Howard A.-G., Wang H.J., Su C., Zhang B. The physical activity transition among adults in China: 1991-2011. Obes. Rev. 2014;15:27–36. doi: 10.1111/obr.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones-Smith J.C., Popkin B.M. Understanding community context and adult health changes in China: development of an urbanicity scale. Soc. Sci. Med. 2010;71:1436–1446. doi: 10.1016/j.socscimed.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.American Diabetes Association Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 2013;36:S67–S74. doi: 10.2337/dc13-S067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.2016 Chinese guidelines for the management of dyslipidemia in adults Journal of geriatric cardiology. 2018;15:1–29. doi: 10.11909/j.issn.1671-5411.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mills K.T., Stefanescu A., He J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020;16:223–237. doi: 10.1038/s41581-019-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J.A., Holmes S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palarea-Albaladejo J., Martín-Fernández J.A. zCompositions—R package for multivariate imputation of left-censored data under a compositional approach. Chemometr. Intell. Lab. Syst. 2015;143:85–96. doi: 10.1016/j.chemolab.2015.02.019. [DOI] [Google Scholar]
- 44.Zhou X., Qiao Q., Ji L., Ning F., Yang W., Weng J., Shan Z., Tian H., Ji Q., Lin L., et al. Nonlaboratory-based risk assessment algorithm for undiagnosed type 2 diabetes developed on a nation-wide diabetes survey. Diabetes Care. 2013;36:3944–3952. doi: 10.2337/dc13-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmes I., Harris K., Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One. 2012;7:e30126. doi: 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ke G., Meng Q., Finley T., Wang T., Chen W., Ma W., Ye Q., Liu T.Y. NIPS; 2017. LightGBM: A Highly Efficient Gradient Boosting Decision Tree.https://proceedings.neurips.cc/paper_files/paper/2017/file/6449f44a102fde848669bdd9eb6b76fa-Paper.pdf [Google Scholar]
- 47.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The raw ITS sequencing data have been deposited in the Genome Sequence Archive (GSA) (https://ngdc.cncb.ac.cn/gsa/) at the accession number CRA013806.
-
•
Due to informed consent regulations, phenotypic data for the cohort are available upon request from the CHNS (https://www.cpc.unc.edu/projects/china/).
-
•
The computer codes for main analyses in this study are deposited in GitHub under https://github.com/wenutrition/CHNS-fungi-cardiometabolic-diseases. The DOI at Zenodo is https://doi.org/10.5281/zenodo.13353503. The software and packages used for this analysis are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.