

Summary
Pancreatic ductal adenocarcinoma (PDAC) exhibits an immunosuppressive tumor microenvironment (TME) contributing to its therapeutic resistance. Following our previous studies, we report that mast cells infiltrating the PDAC TME foster this immunosuppression and desmoplasia. Mast cell infiltration correlated with human PDAC progression, and genetic or pharmacological mast cell depletion reduced tumor growth and desmoplasia while enhancing survival in mouse PDAC models. Mechanistically, mast cell-derived IL-10 promoted PDAC progression. Strikingly, combining an agonistic anti-OX40 immunotherapy with mast cell blockade synergistically elicited durable anti-tumor immunity, marked by increased infiltration of CD8+ T effector cells expressing granzyme B and dramatic survival benefit unachievable with either approach alone. An OX40-associated gene signature correlated with improved survival in human PDAC, supporting therapeutic translation. Our findings establish mast cells as promoters of the suppressive PDAC TME and rational targets for combination immunotherapy. Targeting this mast cell-mediated resistance mechanism could overcome immunotherapy failure in PDAC.
Subject areas: Molecular biology, Neuroscience, Immunology, Cancer
Graphical abstract
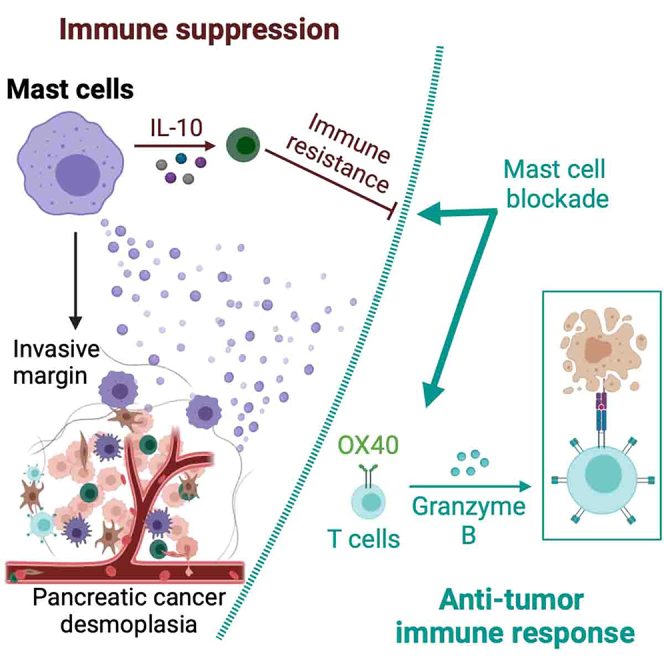
Highlights
-
•
Mast cells accumulate in PDAC, driving disease progression from precursor to tumor
-
•
Mast cell depletion suppresses tumor growth, desmoplasia, and improves survival
-
•
Mast cell-derived IL-10 drives immunosuppression, inhibiting it reduces tumors
-
•
Combining mast cell blockade and OX40 stimulation activates anti-tumor immunity
Molecular biology; Neuroscience; Immunology; Cancer
Introduction
Regardless of more than 50 years of research and therapeutic development, PDAC remains a life-threatening malignancy with a median survival of 6 months and an overall 5-year survival rate of no more than 7 percent.1 The best option for PDAC patients is surgical resection,1 however, for those who are not surgical candidates, conventional modalities, such as chemotherapy, radiotherapy, and/or immunotherapy,2 have showed little efficacy. The recent clinical successes of checkpoint control monoclonal antibody therapy have made significant strides in improving the landscape of cancer immunotherapy.3 However, PDAC is largely refractory to this treatment. Why immunotherapy, especially checkpoint control antibody therapy, is ineffective is unclear.4 The inflammatory microenvironment found in pancreatic cancer undoubtedly plays a role and the immune impairment may be presented from the start.5 Mast cells are among the first immune cells to infiltrate the site of inflammation, contributing to an immunosuppressive state in some human cancers.6
We and others have demonstrated that mast cell infiltration is an early event7 in the mouse PDAC8 tumor microenvironment (TME) and is predictive of a poor prognosis for PDAC patients.7,9,10 Inhibiting mast cell function suppresses PDAC progression.11,12 With these infrequent reports on mast cells in pancreatic cancer, we speculate that increased influx of mast cells surrounding the pancreatic parenchyma during tumor initiation may be a detrimental event whereby inflammatory cells recruited earlier into the microenvironment and continue to transform the misaligned stroma for tumor progression. The desmoplastic TME renders an important layer of temporal and spatial convergence of immune suppression and resistance to immunotherapy.
Here, we hypothesized that tumor-infiltrating mast cells mediate immunosuppression and serve as a major obstacle to effective immunotherapy. Using both human pancreas samples and complementary animal models, we report that mast cells foster an immunosuppressive and desmoplastic microenvironment, which can incapacitate endogenous anti-tumor immunity or paralyze extrinsically co-stimulatory modification. We demonstrate that mast cell blockade has a synergistic long-term durable anti-tumor effect when combined with an OX40 agonist and provide an advanced combination therapy for pancreatic cancer.
Results
Mast cells infiltrate at the invasive margin and follow the progression of tumorigenesis of human PDAC
Normal tissue was collected from the adjacent benign pancreatic tissue of patients with pancreatic endocrine tumors or solid pseudopapillary neoplasm. Samples were also collected from patients with chronic pancreatitis (CP), pancreatic intraepithelial neoplasia (PanIN), and PDAC (Figure 1). Although few mast cells were found in the human normal pancreas (NP), mast cell infiltration was noted around pancreatic intraepithelial neoplasia (PanIN) lesions and mast cells were prevalent at the tumor margin of PDAC. Although mast cells primarily accumulated at the tumor margin (Figures 1A and 1B), modest infiltration into the central region of the tumor stroma was noted (Figures 1A and 1C). Few mast cells were found in the benign pancreatic parenchyma (Figures 1A and 1D). Mast cells were rarely found in the NP (Figures 1E left and 1F), but were present in the vicinity of PanIN lesions (Figure 1E right), although their overall numbers were not different from those found in NP. Mast cell infiltration was significantly increased in PDAC (Figure 1F). These data indicate that mast cell infiltration is prominent in PDAC, especially at the tumor margins, and may be spatially and temporally associated with pancreatic cancer development.
Figure 1.

Mast cell infiltration into human pancreas
(A) Toludine blue staining (bar = 2 mm) of a tissue section from a human pancreatic cancer patient. T indicates tumor tissue, tumor area; yellow line indicates tumor invasive margin, arrowheads illustrate representative toluidine blue stained mast cells.
(B) Mast cells at the invasive margin of the tumor (T, bar = 200 μm).
(C) Mast cells at the center of the tumor.
(D) Mast cells present in the uninvolved benign tissue (bar = 200 μm).
(E) Mast cells in normal pancreas (NP, left, bar = 200 μm). Mast cell infiltration into pancreatic intraepithelial neoplasia (PanIN, right, bar = 200 μm).
(F) Quantitative evaluation of mast cell infiltration in pancreatic tissues. One point represents one patient. Statistically significant differences were determined using a one-way ANOVA with Fisher’s least significant differences (LSD) as the post hoc multiple comparison.
Mast cells promote in vivo tumor progression and are essential for resistance to immunotherapy in mice PDAC
To determine the role of mast cells in the development of PDAC and therapeutic potential (Figures S1 and S2), mice13,14 that spontaneously develop pancreatic cancer (Pdx-1-Cre x LSL-KrasG12D; KC mice)8 were crossed with mast cell-deficient KitW-sh/W−sh mice (Figures 2A and 2B). Overall survival in mast cell-deficient KCLW (Kit W−sh/W−sh x Pdx-1-Cre x LSL-KrasG12D x LSL-Luciferase = KCLW) mice and mast cell-competent KCLB (wild-type Kit with C57BL/6 background, Pdx-1-Cre x LSL-KrasG12D x LSL-Luciferase = KCLB) mice was determined (Figure 2C). Although mast cell infiltration is an early event, mast cell activation in the pancreatic lesions occurs primarily at the PDAC stage (Figure 2D). Mast cell-deficient KCLW mice had a significantly longer lifespan, surviving 106 days (Figure 2C) and reduced desmoplasia (Figure 2E right) compared to mast cell-competent KCLB mice (Figure 2E left). This in vivo desmoplasic reaction induced by mast cells verifies our previous conclusion11,12 from in vitro investigation that mast cells aggravate the cellular and extracellular matrix dynamics of the tumor fibrotic stromal microenvironment through IL-13/TGFβ/pSmad2 signaling pathway. In addition, mast cell-competent KCLB mice at 152 days of age exhibited hunched posture, abdominal swelling, hyperpnoea, ruffled fur, emaciation, and lethargy, which were not seen in the mast cell-deficient KCLW mice. Similarly, decreased tumor growth was found when mast cell function was inhibited with masitinib, a drug reported to decrease mast cell function in vivo15 (Figure 2F), suggesting pharmacological manipulation of mast cell function in vivo is possible and will obtain similar results as genetic depletion of mast cells.
Figure 2.

Depleting mast cells suppressed PDAC growth and improved response to immunotherapy
(A and B) Breeding strategy to develop mast cell-competent and mast cell-deficient mice that spontaneously develop pancreatic cancer. (A) PDX-1Cre; KrasLSL−G12D/+(KC); LSL-Luciferase; Kitwt/wt (KCLB) mast cell-competent. (B) PDX-1Cre; KrasLSL−G12D/+(KC); LSL-Luciferase; KitW-sh/W−sh (KCLW) mast cell-deficient. Mast cell-deficiency was apparent at 3 months of age.
(C) Increased survival of KCLW mast cell-deficient mice compared to mast cell-competent KCLB mice.
(D) cDNA microarray analysis indicated the expression of mast cell proteases were primarily upregulated when the mice developed PDAC in the KPC-GEMM model.
(E) KCLW mast cell-deficient mice presented with less fibrosis in the tumor microenvironment compared to mast cell-competent KCLB mice.
(F) Blocking mast cell function with masitinib inhibited PDAC growth in the authotopic KPC model. Tumor sizes were measured by bioluminescent imaging (BLI-IVIS). Data indicate fold change in photons/sec/cm2/sr ± standard deviation, p = 0.0007 at week 5.
Mast cell-derived IL-10 represents the immune suppressive microenvironment and accelerates the tumor progression and poor prognosis
Mast cell derived cytokines, particularly interleukin (IL)-10, have been reported to affect PDAC growth16,17 and regulate immune function. To determine whether IL-10 plays a role in mast cell regulation during PDAC initiation and progression, we first examined IL-10 expression through the stages of PDAC development. Little expression of the genes encoding IL-10 and its receptors was found in the NP and during CP, but increased expression, at the protein and mRNA level was noted during PanIN and PDAC (Figures 3A–3C). We confirmed the co-localization of IL-10 and mast cells in the TME by dual-color immunofluorescence staining. Mast cell-deficient mice were reconstituted with either wild-type bone marrow-derived mast cells (BMMC) or BMMC derived from IL-10-deficient animals (Figure 3C). Mast cells were identified by tryptase staining (green) and IL-10 producing cells stained with IL-10 antibody (red). Only mice reconstituted with wild-type BMMC had IL-10 secreting mast cells in the TME. Mast cell-deficient mice were reconstituted with mast cells derived from wild-type or IL-10-deficient mice18 (Figures 3D and 3E). Two models were used: the orthotopic implantation of tumors directly into the pancreas of mice reconstituted with wild-type or IL-10-deficient mast cells, and reconstitution to PDX-1-Cre, KrasG12D mast cell-deficient mice at 100 days of age. Tumor growth was suppressed when mice were reconstituted with IL-10-deficient mast cells (Figure 3D), and survival was significantly increased when PDX-1-Cre, KrasG12D mast cell-deficient mice were reconstituted with IL-10-deficient mast cells (Figure 3E). These data indicate that mast cell-derived IL-10 is responsible for promoting PDAC growth and its depletion leads to prolonged survival in tumor-bearing hosts. No difference in tumor growth was noted when the mice were reconstituted with mast cells derived from PGE2-deficient mice (Figure S3). We found no apparent difference in the morphologic characteristics of tumors that developed in mast cell-deficient mice reconstituted with IL-10-deficient BMMC and those in mast cell-deficient mice reconstituted with wild-type BMMC (Figure 3F).
Figure 3.

Mast cell-derived IL-10 promotes tumor development
(A) cDNA microarray analysis of the expression of IL-10 and its receptors IL-10RA and IL-10RB during the development of pancreatic cancer in KPC mice.
(B) Quantitative real-time PCR confirmed microarray data. Data are represented as mean ± SEM. A one-way ANOVA, followed by the Fisher’s least significant difference (LSD) multiple comparison test, shows the significances by ∗: p < 0.05; ∗∗: p < 0.01; ∗∗∗: p < 0.001; ∗∗∗∗: p < 0.0001.
(C) IL-10 production by mast cells (bar = 50 μm). Pancreas tissues were isolated from mast cell-deficient mice that were reconstituted with normal bone marrow derived mast cells (BMMC to Kit−/−; first row) or with BMMC derived from IL-10−deficient mice (MC (IL-10−/−) to Kit−/−; second row). Pancreatic tissues were also isolated from normal mice (NP) or KPC mice during the development of pancreatic cancer (CP, PanIN, PDAC). Sections were stained with anti-tryptase (left column, green) or anti-IL-10 (middle column, red). The right column represents the merged images.
(D) 28 days post implantation, mast cell-deficient mice reconstituted with IL-10−/− BMMC developed smaller tumors and less ascites than those reconstituted with wild-type BMMC. p = 0.0253 for tumor size and 0.014 for ascites incidence.
(E) KCLW mast cell-deficient mice were reconstituted with IL-10−/− BMMC. These animals survived longer than KCLW mice reconstituted with wild-type BMMC.
(F) Morphologic comparison of tumors that develop in mast cell-deficient mice reconstituted with IL-10−/− or wild-type BMMC. H&E staining (top left, bar = 500 μm) shows tumors implanted into Kit−/− mice reconstituted with IL-10−/− mast cells. H&E staining (top right, bar = 500 μm) shows tumors implanted into Kit−/− mice reconstituted with wild-type BMMC. Toluidine blue staining (center left, bar = 500 μm) shows tumors implanted into Kit−/− mice reconstituted with IL-10−/− mast cells. Toluidine blue staining (center right, bar = 500 μm) shows tumors implanted into Kit−/− mice reconstituted with wild-type BMMC. High power magnification (bottom left and right, bar = 50 μm) of Toluidine blue staining showed mast cells density in tumors. Yellow arrowheads illustrate representative toluidine blue stained mast cells. N = benign area with normal pancreatic structure, L = peripancreatic lymphoid tissue, and T = tumor.
Mast cell deficiency is required for syngeneic allograft tumor elimination and long-term tumor free survival by OX40 agonistic modification
Because IL-10-driven immune tolerance can be blocked by an anti-OX-40 antibody,19 we tested the role of OX40 stimulation in PDAC. Tumor-bearing wild-type C57BL/6 mice (Figures 4C–4E, black sets) and age-matched mast cell-deficient KitW-sh/W−sh mice (Figures 4C–4E, green sets) were administered an agonistic anti-OX-40 antibody (Figure 4A). The growth of the highly metastatic and aggressive KPC tumor was suppressed by anti-OX40 antibody treatment (Figures 4C–4E, blue sets), and the effect was dramatically enhanced when the antibody was injected into tumor-bearing mast cell-deficient mice (Figures 4C–4E, red sets), resulting in increased survival with long-term tumor free status (Figures 4B and S2). In our more than 5 times of repeated experiments, it is worth noting that anti-OX40 antibody treatment (blue sets in Figures 4 and S2A) in wild-type C57BL/6 mice always induce partial response (2–3 responders of 5 mice), but anti-OX40 antibody treatment (red sets in Figures 4 and S2A) in mast cell-deficient mice always induce complete response (5 responders out of 5 mice). In addition, because many studies have shown that immune co-stimulatory antibodies provided potent anti-tumor activity,20,21,22,23 we also asked whether using other antibodies, such as PD-1, would have any effect in our model system. Only in mast cell-deficient tumor-bearing mice, anti-PD-1 did have minor effect (Figures S1 and S2B).
Figure 4.

Mast cell deficiency improves the anti-tumor efficacy of anti-OX40 in syngeneic KPC allograft model
(A) Experimental design and (B) survival curves in orthotopic syngeneic model. All of the anti-OX40 treated mast cell-deficient KrasG12D;P53flox/flox;PDX-1-Cre (KPC) tumor-bearing mice achieved a long-term tumor free survival, p = 0.0061.
(C) BLI-IVIS images show the tumor size before treatment at week 1 after tumor implantation (top) and after treatment at week 4 (bottom).
(D) Longitudinal tumor growths of PDAC were quantified following the treatments and presented extremely significant effect (p < 0.0001) by Two-way independent ANOVA. (Black sets in C and D) Growth of KPC tumors implanted into wild-type mice injected with saline control. (Green sets in C and D) Growth of KPC tumors implanted into mast cell-deficient mice injected with saline control. (Blue sets in C and D) Growth of KPC tumors implanted into wild-type mice injected with agonist anti-OX-40. (Red sets in C and D) Growth of KPC tumors implanted into mast cell-deficient mice injected with agonist anti-OX-40.
(E) PDAC tumor growth (tumor sizes) curves in orthotopic syngeneic model. Data indicate fold change in photons/sec/cm2/sr ± standard deviation. The Bonferroni post hoc multiple comparisons showed significance between groups over time: black vs. green = 0.014; black vs. blue <0.0001; black vs. red <0.0001; green vs. blue = 0.944; green vs. red <0.0001; blue vs. red = 0.002. This experiment has been repeated more than 5 times using 5 mice in each group.
Blocking mast cells is required to induce long-term, durable anti-tumor immunity, and survival benefit with OX40-activated microenvironment
Finally, we wanted to determine if a pharmacological approach to suppress mast cell function, coupled with immunotherapy could alter tumor growth in PDX-1Cre; KrasLSL−G12D/+; Trp53LSL−R172H/+ (KPC-GEMM) mice because it is one of the best mouse models to recapitulate human pancreatic cancer. The treatment schedule is shown in Figure 5A. Either treatment alone had a marginal effect on survival. Combining cromolyn and agonist anti-OX40 antibody resulted in a significant increase in survival (Figure 5B). Combining cromolyn and agonist anti-OX40 antibody also increased the infiltration of CD8-positive; granzyme-B positive effector cells into the pancreas (Figures 5C–5F), indicating that this treatment resulted in an influx of activated immune cells into the TME. In addition, the positive correlation between OX40 and granzyme-B expression, and the infiltration of CD8+ cells, noted in our KPC-GEMM mice, was similarly observed in 186 cases of human PDAC samples derived from TCGA database (Figure S4). Along with the influx of effector T cells, a panel of 31 genes on the pathways of Toll-like receptor24 and allergy and asthma25 were upregulated substantially in the tumor stroma of anti-OX40 treated mice (Figures S5 and S6). In order to determine if our results with experimental animals could translate to the clinic, we compared the upregulation of these pooled 30 inflammatory genes in TCGA PDAC patients. A strong correlation among the 30 OX40-related genes was noted (Figure S7), and overall survival was significantly increased when these 30 genes were upregulated (Figure 6). For discriminating the contribution of these 30 genes for survival benefit, multivariate Cox regression analysis provided unintentional selection (Table S1). The selected PDAC patients showing over-expression of the OX40-related 16-gene signature, which marked an active immune TME, presented an advantage in both overall and disease-free survival (Table S1; Figure 7). This validation (Figures S8 and S9), using the human PDAC databases, including a publicly available single-cell RNA sequencing dataset,26 dovetails well with our murine results, and indicates that OX40 agonist monoclonal antibody may provide a superior immunotherapeutic agent.
Figure 5.

Blocking mast cell function improves the anti-tumor efficacy of anti-OX40 in both syngeneic allograft and autochthonous KPC model
(A) Experimental design and (B) survival curves of KPC-GEMM mice. Combination therapy with anti-OX40 and cromolyn significantly extended survival in KPC mice compared with control (p = 0.0007); anti-OX40 alone had no survival benefit (p = 0.1318); cromolyn significantly increased survival (p = 0.0388); and combination therapy with anti-OX40 and cromolyn resulted in longer survival than did cromolyn alone (p = 0.0435).
(C–F) Immunohistochemical analysis of CD8 infiltration (C and D) and granzyme B expression (E and F) showed little infiltration of effector T cells into the tumor microenvironment in control mice (black sets in C–F) and after treatment with cromolyn (green sets in C–F), slightly increased with the treatment with anti-OX40 (blue sets in C–F), however, a dramatic increase was found in mice treated with agonist anti-OX40 and cromolyn (red sets in C–F).
(D and F) Quantification of CD8 and granzyme B positive cells—average from 5 representative high-power fields. A one-way ANOVA, followed by the Fisher’s least significant difference (LSD) multiple comparison test, shows the significances by ∗: p < 0.05; ∗∗: p < 0.01; ∗∗∗: p < 0.001; ∗∗∗∗: p < 0.0001.
Figure 6.

TCGA survival analysis of patients in which 30 inflammatory genes were upregulated by OX40
(A) OncoPrint of 31 genes and percentage of alteration from 185 patients (MUC5AC data were not available in TCGA database) showed that 108 patients (red) had alterations in these 30 genes and 77 patients (blue) did not have alterations in these 30 genes; overall survival months were presented on the top of gene list.
(B) The median overall survival increased for 4.81 months (p = 0.0198) in patients where the mRNA for these 30 genes was upregulated.
(C) Disease free survival was presented; differences were not statistically significant.
Figure 7.

TCGA survival validation of OX40 upregulated 16-gene signature after Cox regression
(A) OncoPrint of the 16-gene signature and percentage of alteration from 184 patients showed that 67 patients (red) had alterations in these 16-gene signature and 117 patients (blue) did not have alterations in these 16-gene signature; overall survival months were presented on the top of gene list.
(B) The median overall survival increased for 25.74 months (p = 1.317e-5) in patients with these 16-gene signature mRNA upregulation.
(C) The median disease-free survival increased for 12.85 months (p = 0.00825) in patients with these 16-gene signature mRNA up regulation.
Discussion
As PDAC is an immunological privileged tumor aggregated with endogenous stromal reaction, we evaluated the contribution of mast cells to tumorigenesis and combination regimens in multiple immunocompetent mouse models: two autochthonous spontaneous PDAC models (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre mice, “KPC” mice, and p53 wild type Pdx-1-Cre x LSL-KrasG12D, “KC” mice), and two syngeneic orthotopic allografts (KPC and Panc-02). Mast cell-deficient mice (KitW-sh/W−sh) were clear of mast cells at 3 months of age.27 Because KPC-GEMM developed CP and PanIN before 3 months of age, this line was not suitable to monitor the role of mast cells during the development of pancreatic cancer. However, KC mice with a normal copy of p53 (Pdx-1-Cre x LSL-KrasG12D) mice remained histologically normal until 3 months old and served as an appropriate model to cross with Kit W−sh/W−sh mice, in order to develop mast cell-deficient mice that spontaneously developed pancreatic cancer (Kit W−sh/W−sh x Pdx-1-Cre x LSL-KrasG12D x LSL-Luciferase = KCLW). Their mast cell-competent littermates (wild-type Kit with C57BL/6 background, Pdx-1-Cre x LSL-KrasG12D x LSL-Luciferase = KCLB) were used as controls. Hence, KCLW becomes a valid tool in studying mast cell function during pancreatic tumorigenesis for this work and more applications to future study, which is required to dissect the precise regulatory mechanism of mast cell cytokine release and degranulation in PDAC TME.
By converging on the role of mast cells and overcoming the off-target effects derived from any individual model system, we employed either mast cell-deficient (Kit W−sh/W−sh) mice or those compounds that well accepted as mast cell blockades (cromolyn and masitinib) to validate our results. Considering some controversial effects28 of cromolyn and KitW-sh/W−sh mice was discussed previously, we11 and others29 have confirmed that cromolyn treatment inhibits mast cell degranulation in the TME of mouse models, as well as mast cell deficiency30 in KitW-sh/W−sh mice. Likewise, masitinib, a c-Kit antagonist, has been recognized as mast cell inhibitor in canine mast cell tumors and rheumatoid arthritis in humans by robustly blocking the proliferation, differentiation, and degranulation of mast cells.15,31 Thus, it is worth noting that our genetic and pharmacological approaches provide complementary justification on mast cell function and its synergistic effect with OX40 agonist in PDAC.
In this study, we utilized two distinct mast cell inhibitors—masitinib and cromolyn—to assess their effects on PDAC progression and response to immunotherapy. Masitinib is a tyrosine kinase inhibitor that specifically targets the c-Kit receptor, which is critical for mast cell differentiation and survival. In contrast, cromolyn11,12 is a mast cell stabilizer that inhibits the degranulation and activation of mature mast cells. The decision to employ these two different pharmacological agents was based on their distinct mechanisms of action and potential applications in the PDAC setting. Masitinib32,33 was used in the syngeneic allograft model to acutely deplete mast cells and evaluate their impact on tumor growth, while cromolyn was utilized in the autochthonous KPC-GEMM to chronically suppress mast cell function during the natural progression of pancreatic cancer. The complementary use of these two mast cell-targeting drugs allowed us to more comprehensively dissect the role of mast cells at different stages of PDAC development and their influence on the response to immunotherapies like anti-OX40. This multi-pronged approach strengthens the translational relevance of our findings and highlights the potential clinical utility of mast cell-directed therapies, either as single agents or in combination with immune checkpoint blockade, for the treatment of this highly aggressive malignancy.
Incorporating our animal results and human data (Figure S8), our findings suggest that the activation of the OX40 downstream signatures upon OX40 stimulation may exert a significant influence on the survival outcomes of tumor-bearing hosts. Furthermore, analysis of TCGA patients’ data (Figures 6 and 7) hints at the possibility of potential endogenous drivers bolstering the OX40 pathway, thereby conferring a survival advantage to those patients through the overexpression of OX40 signatures. However, it is imperative to acknowledge the limitations inherent in analyzing bulk RNA sequencing data from large-scale datasets like TCGA, as such findings may not definitively establish the causal relationship between OX40 pathway activation and survival outcomes. Therefore, while our results offer intriguing insights, they underscore the necessity for further investigation with multiple single-cell sequencing datasets into the large-scale mechanism underlying OX40 pathway modulation and its impact on anti-tumor immunity. Future studies should aim to explore not only OX40 upstream immune stimulation but also the potential rewiring of anti-tumor immunity through OX40 activity, thereby elucidating its precise role in tumor progression and therapeutic response.
Importantly, by blocking mast cells to abolish the desmoplastic and immunosuppressive TME, this work visualized that OX40 agonist can promote enhanced effector cell accumulation to eradicate PDAC when the local cytokine milieu is suitable, a similar precondition to the OX40 signaling in autoimmunity disease model,34 regardless of the perceived dichotomy in OX40 effects on different biologic events. Although more mechanistic pathways remain to be illustrated to understand the contribution of mast cells and OX40 in the immune microenvironment of pancreatic cancer, this work demonstrated that suppressing mast cell function is required for OX40 immunotherapy to be effective as quantified by granzyme B expression and survival validation.
Taken together, our data shed new light on the mechanisms regulating immune resistance in PDAC. We provide evidence that intratumoral mast cells cf. immune suppression in the pancreatic cancer microenvironment. This finding is of great clinical relevance since interfering with mast cell function may provide a regimen to increase the efficacy of immunotherapy in pancreatic cancer. We saw the most dramatic effect when anti-OX40 was used to stimulate the immune system, but only when this therapy was used in combination with mast cell depletion. Coupled with increased survival and tumor destruction was an influx of activated effector cells into the TME and the upregulation of inflammatory gene signature. Whether this approach works with other immunotherapy reagents remains to be seen. Our work supports a multimodality immunotherapy (blocking mast cell-derived immunosuppression coupled with stimulating immunity) that offers a favorable prognosis for hosts with pancreatic cancer.
Limitations of the study
While our findings provide compelling evidence for the role of mast cells in promoting an immunosuppressive PDAC microenvironment, certain limitations should be noted. First, the reliance on mouse models, including genetically engineered and syngeneic allograft systems, may not fully recapitulate the complex biology and heterogeneity of human PDAC. Differences in the TME, immune landscapes, and mast cell biology between mice and humans could limit the direct translational applicability of these findings. Additionally, the Kit mutant mice used for genetic mast cell depletion could potentially have developmental defects impacting other cell types, though the corroborating pharmacological data mitigates this concern. Our study also did not exhaustively evaluate the effects of targeting mast cells on all potential immunotherapies beyond OX40 agonism and PD-1 antagonism. Another potential limitation is the pharmacological agents used to target mast cells, such as cromolyn and masitinib. While these compounds have been widely used to inhibit mast cell function, they may have off-target effects that could confound the interpretation of the results. The development of more selective and potent mast cell-targeted therapies could help address this limitation. Furthermore, the mechanisms underpinning the synergy between mast cell inhibition and OX40 activation require deeper interrogation. Additional mechanistic investigations, potentially leveraging emerging single-cell sequencing and multi-omics technologies, will be needed to better understand the complex interplay between mast cells, the TME, and anti-tumor immunity. Finally, while our human data analyses support therapeutic translation, clinical trials will be needed to validate the efficacy and safety of this combinatorial approach in PDAC patients.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ying Ma (yingma_maying@hotmail.com or yingma@tmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Publicly available human TCGA dataset were analyzed. Further details are provided in the method details.
-
•
The human TCGA dataset and single cell RNA sequencing data that support the findings of this study are openly available. DOIs are listed in the key resources table.
-
•
All original code is publicly available. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
The authors thank Drs. Stephen E. Ullrich, Cassian Yee, and Anirban Maitra for insightful discussions and comments on the manuscript, technical support from Nasser Kazimi for animal maintenance. The graphical abstract was created with BioRender.
Grant support: this work was supported by The National Natural Science Foundation of China (NSFC) grants, grant number 82072691 and 82272767, and a Hirshberg Foundation for pancreatic cancer research seed grant to Y.M. This work was supported by the NIH grant CA131207, Cancer Prevention & Research Institute of Texas Grant RP120777, and a G.S. Hogan Gastrointestinal Cancer Research Fund. The Animal Facilities and the Characterized Cell Line Core were supported in part by a National Cancer Institute Core grant, CA16672.
Author contributions
Conceptualization, Y.M.; methodology, Y.M.; software, Y.M., L.H., and J.F.; formal analysis, Y.M., S.Q., and H.W.; investigation, Y.M.; resources, B.J., L.H., P.H., and C.D.L.; data curation, Y.M.; writing—original draft, Y.M.; writing—review and editing, Y.M. and X.Z.; visualization, Y.M.; funding acquisition, Y.M.; supervision, Y.M. and H.W.
All authors have read and agreed to the published version of the manuscript.
Declaration of interests
The authors disclose no potential conflicts of interest.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors used ChatGPT and Claude-3 (poe.com) in order to improve readability and language. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMAb anti-mouse OX40 (CD134) | Bio X cell | Cat#BE0031 |
| InVivoMAb anti-mouse PD-1 (CD279) | Bio X cell | Cat#BE0146 |
| Purified Rat Anti-Mouse CD8a | BD PharmingenTM | Cat#553027; RRID: AB_394565 |
| Biotin Rat Anti-Mouse Granzyme B Monoclonal Antibody | eBioscience | Cat#13-8822-82; RRID: AB_1603282 |
| Mast Cell Tryptase (G-12) | Santa Cruz | Cat#Sc-32474; RRID: AB_2206484 |
| Purified Rat Anti-Mouse IL-10 | BD Pharmingen | Cat#554464; RRID: AB_398557 |
| Biological samples | ||
| Pancreatic disease patients samples | UT MD Anderson Cancer Center | www.mdanderson.org |
| Chemicals, peptides, and recombinant proteins | ||
| cromolyn | Sigma | Cat#C0399 |
| masitinib | Sigma | Cat#SML3594 |
| D-Luciferin | Caliper Life Sciences | Cat#122796 |
| Toluidine blue | VWR | Cat#0672-25G |
| Critical commercial assays | ||
| RT2 SYBR@ Green Master Mixes | SABiosciences | Cat#330502 |
| First Strand Kits | SABiosciences | Cat#330401 |
| RT2 ProfilerTM PCR Array Mouse Allergy & Asthma | SABiosciences | Cat#PAMM-067ZD-12 |
| RT2 ProfilerTM PCR Array Mouse Toll-Like Receptor | SABiosciences | Cat#PAMM-018ZD-12 |
| Trichome-Masson Kit | Sigma-Aldrich | Cat#HT15 |
| Experimental models: Cell lines | ||
| Panc02 mouse pancreatic cancer cell line | https://doi.org/10.1158/0008-5472.CAN-12-4479 | NA |
| KPC mouse pancreatic cancer cell line | https://doi.org/10.1158/0008-5472.CAN-12-4479 | NA |
| Experimental models: Organisms/strains | ||
| C57BL/6 wild type mice | The Jackson Laboratory | Cat#000664 |
| C57BL/6 albino mice (B6(Cg)-Tyrc-2J/J) | The Jackson Laboratory | Cat#000058 |
| mast cell-deficient mice (KitW-sh/W-sh) | The Jackson Laboratory | Cat#012861 |
| IL-10–deficient mice (B6.129P2-IL10tmiCgn/J) | The Jackson Laboratory | Cat#002251 |
| PGE2-deficient mice (B6.129[FVB]-Ptgs2tm2.1(ptgs1)Fn/J) | The Jackson Laboratory | Cat#008104 |
| cLGL-KRasG12V/Ela-CreER mice | https://doi.org/10.1053/j.gastro.2009.05.052 | NA |
| KPC-GEMM (KrasG12D/+; Trp53R172H/+; Pdx-1-Cre) | Frederick National Laboratory for Cancer Research (NCI, Maryland, USA https://doi.org/10.1016/j.ccr.2005.04.023 |
NA |
| KC-GEMM (KrasG12D/+; Pdx-1-Cre) | Frederick National Laboratory for Cancer Research (NCI, Maryland, USA https://doi.org/10.1016/S1535-6108(03)00309-X |
NA |
| Software and algorithms | ||
| ImageJ | https://wsr.imagej.net/notes.html | Version 1.52p 22 June 2019 |
| Living Image | IVIS Spectrum in https://store.caliperls.com | 3.1 Software |
| TCGA cBioPortal | http://www.cbioportal.org/index.do | NA |
| Prism | https://www.graphpad.com | NA |
| RT2 Profiler PCR Array Data Analysis | http://pcrdataanalysis.sabiosciences.com | NA |
| scTIME Portal | http://sctime.sklehabc.com | NA |
| Single cell RNA sequencing data | http://sctime.sklehabc.com/#/visual/11/CRA001160?species=%5B%22Homo%20sapiens%22%5D&addpath&compare=trueA | NA |
Experimental model and study participant details
Human tissue
We searched the record database at The University of Texas MD Anderson Cancer Center for patients who had undergone pancreaticoduodenectomy and had not received any form of preoperative chemotherapy or radiotherapy. We identified 67 samples that met those criteria: 16 cases of normal pancreas, 26 cases of PanIN lesions, and 25 cases of PDAC. The Institutional Review Board of MD Anderson Cancer Center approved this study. The study approval number is Protocol: Lab05-0854. This protocol has a waiver for informed consent. The clinical and demographic details (age, sex/gender, ancestry, race, and ethnicity) of participants are not available due to strict regulations of this IRB on patient’s Protected Health Information (PHI). Archival tissue blocks and their matching hematoxylin and eosin (H&E)-stained slides were retrieved, reviewed, and screened by two gastrointestinal pathologists (H. Wang & S. Qiu) to identify representative tumor regions and nonneoplastic pancreatic parenchyma.
Mice
The mice were maintained in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care International, in accordance with current regulations and standards of the United States Department of Agriculture, Department of Health and Human Services, and National Institute of Health. All animal procedures, along with justification of selected strains with experimental arms, and statistical justification of the number of mice to be used and randomized, were reviewed and approved by the MD Anderson Cancer Center Animal Care and Use Committee. The study was approved by the University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee (IACUC) under protocol A3343-01. For tumor implantation procedures, male mice and male-derived cell lines were used to ensure immune compatibility, while both male and female subjects were utilized for spontaneous tumor development experiments and breeding. In tumor implantation experiments, we used 5–10-week-old mice. For spontaneous tumor development, animals were monitored from birth until euthanasia when they became ill. Although our tumor implantation studies primarily used male mice, we validated these results using spontaneous models that included both sexes, allowing us to account for potential sex-based differences in our findings.
Eight-to 10-week-old C57BL/6 wild type or albino mice, mast cell-deficient mice (KitW-sh/W−sh), IL-10–deficient mice (B6.129P2-IL10tmiCgn/J), and PGE2-deficient mice (B6.129 [FVB]-Ptgs2tm2.1(ptgs1)Fn/J) on the C57BL/6 background were obtained from The Jackson Laboratory (Bar Harbor, ME).
Genetically engineered mouse models of chronic pancreatitis (CP), PanIN, and PDAC were developed by crossing cLGL-KRasG12V with Bac-Ela-CreER mice.35 KPC-GEMM13,14 mice were used for treatment evaluation using the previous described breeding strategy to yield mice that possessed conditional p53 mutation and endogenous levels of mutant KRasG12D and the luciferase-reporter expressed by pancreatic tissue.36 When PDAC was diagnosed by amplified luciferase signal in the pancreas, mice were blindly allocated to the groups by random assignment and then treatments were started. The breeder mice for the genetically engineered mouse models were obtained from the Mouse Repository at the Frederick National Laboratory for Cancer Research (NCI, Maryland, USA).
Method details
Orthotopic implantation of PDAC
Surgical implantation of male mouse PDAC cells to male mouse pancreas was performed as orthotopic injection.7 Briefly, 10-week-old male mice were anesthetized with isoflurane and a 1 cm incision in the left subcostal region was made. Murine PDAC cells were injected into the caudal pancreas. The peritoneum and skin were closed with the EZ Clip wound closing kit (Stoelting Co., Wood Dale, IL). Analgesia (Buprenorphine; 0.5 mg/kg) was administered post-operatively to minimize pain.
Treatments with antibodies or cromolyn
Mice received cromolyn (10 mg/kg body weight)11 or masitinib (100 mg/kg body weight)15 into the peritoneal cavity. Treatments were administered daily for one month. Anti-PD-1 (Clone RMP1-14) or Anti-OX40 (Clone OX-86) treated mice received 4 mg/kg body weight of the antibody 6 times at days 1, 4, 7, 13, 16, 19 ip. Phosphate-buffered saline was used as a control.
Readout of tumor growth and sample collection
The implanted tumor cells were transfected with enhanced firefly luciferase,11 and at various times post implantation the mice were injected with D-Luciferin (150 μg/mouse; Caliper Life Sciences, Hopkinton, MA.). Bioluminescence (BLI) was measured with the IVIS imaging system (Caliper Life Sciences). Tumor volume was measured once a week until termination of the experiment using the formula: (π x long axis x short axis x short axis)/6. After the one-month treatment period, KPC-GEMM mice were monitored daily. When the mice presented with obvious morbidity, they were euthanized, necropsy performed and survival date was recorded. Pancreas, tumor, and spleen were harvested for sectioning or RNA precipitation.
Cell culture and mast cell reconstitution
Murine PDAC cells11 (Panc-02, or K-rasG12Dp53−/−), authenticated by genotyping, were cultured in RPMI 1640 (Invitrogen) containing 10% FCS with penicillin and streptomycin (Invitrogen) at 37°C in a humidified atmosphere of 5% CO2.
Mast cell-deficient mice were reconstituted with bone marrow derived mast cells (BMMCs) by intraperitoneal injection.7 A total of 1 × 106 BMMCs were injected into the peritoneal cavity of mast cell-deficient mice. Mice were used six weeks post reconstitution.
All of the cell lines were tested to be free of mycoplasma contamination before being injected into animals.
Quantitative real-time PCR array and cDNA microarray analysis
Total RNA of mouse pancreatic cancer tissue was extracted with Trizol (Invitrogen) and purified further by using the RNeasy RNA cleanup protocol (Qiagen, Germantown, MD, USA). cDNA was reverse-transcribed from total RNA using a high-capacity cDNA RT kit (RT2 SYBR@ Green Master Mixes 330502 & First Strand Kits 330401, SABiosciences, Frederick, MD, USA). cDNA (25 ng) was subjected to Illumina Microarray (San Diego, CA, USA) and real-time RT-PCR array (RT2 Profiler PCR Array Mouse Allergy & Asthma, PAMM-067ZD-12; RT2 Profiler PCR Array Mouse Toll-Like Receptor Signaling Pathway, PAMM-018ZD-12, SABiosciences, Frederick, MD, USA) using a sequence detector (Model BioRad CFX96).
Quantification and statistical analysis
Histo-pathology analysis
Immunohistochemistry was followed by the routine immunohistochemical staining protocol. Antibodies specific for CD8 (Cat. 553027, clone 53-6.7, BD Pharmingen, San Jose, CA, USA) and granzyme B (Cat. 13–8822, clone 16G6, eBioscience, San Jose, CA, USA).
Immunofluorescence analysis: Sections were stained with the antibodies against: mast cell tryptase (Goat polyclonal IgG anti-mouse mast cell tryptase antibody, Cat. Sc-32474, Santa Cruz, CA, USA) and IL-10 (Rat IgG2b anti-mouse IL-10 antibody, Cat. 554464, clone JES5-16E3, BD Pharmingen, San Jose, CA, USA).
Histology staining with Trichome-Masson Kit (HT15, Sigma-Aldrich, St. Louis, MO, USA) was used to characterize the fibrosis.
Mast cell infiltration into the pancreas was measured using Toluidine blue staining.11
Images were obtained with an Olympus DP70 microscope (Olympus, Melville, NY). All the histo-pathology staining images were analyzed on 5 high-power fields on each slides using ImageJ software (NIH, Bethesda, MD).
TCGA
The results here are in whole or part based upon data generated by the TCGA Research Network, which are available in a public repository from the TCGA cBioPortal (http://www.cbioportal.org/index.do).37,38 Data are analyzed and downloaded on Select Cancer Study from 186 samples of Pancreatic Adenocarcinoma (TCGA, Provisional) and Select Genomic Profiles from mRNA Expression z-Scores (RNA Seq V2 RSEM).
Single cell sequencing analysis
The correlation results are generated by the publicly available single-cell RNA sequencing datasets of PDAC26 in the public scTIME Portal: (http://sctime.sklehabc.com/#/visual/11/CRA001160?species=%5B%22Homo%20sapiens%22%5D&addpath&compare=trueA). The mean expressions (TPM) of signature genes in each sample or each cell type are presented in the heatmap. T cell proportion, the percentage of T cells in all cells in each sample, scores the immune infiltration in the patient.
Statistical analysis
Statistical differences between groups were determined using the unpaired, two-tailed, Student’s t test; a one-way ANOVA, followed by the Fisher’s least significant difference (LSD) multiple comparison test; or two-way ANOVA. Normality tests along with the variations within groups and between groups were performed before the significance differences can be evaluated. The relationship of genes expression was determined by Pearson correlation and the contributions of genes to survival were determined by multivariate Cox regression. Scatterplots and Clustergrams of quantitative real-time PCR array were generated from RT2 Profiler PCR Array Data Analysis version 3.5 (http://pcrdataanalysis.sabiosciences.com, SABiosciences, QIAGEN). Survival curves were constructed using the Kaplan-Meier method, and statistical significance was determined using the Log Rank test. p values less than 0.05 were considered statistically significant. Representative experiments are shown; each experiment was repeated independently at least 3 times.
Published: September 30, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111085.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2015. CA A Cancer J. Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Wagner M., Redaelli C., Lietz M., Seiler C.A., Friess H., Büchler M.W. Curative resection is the single most important factor determining outcome in patients with pancreatic adenocarcinoma. Br. J. Surg. 2004;91:586–594. doi: 10.1002/bjs.4484. [DOI] [PubMed] [Google Scholar]
- 3.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 4.Le D.T., Lutz E., Uram J.N., Sugar E.A., Onners B., Solt S., Zheng L., Diaz L.A., Jr., Donehower R.C., Jaffee E.M., Laheru D.A. Evaluation of Ipilimumab in Combination With Allogeneic Pancreatic Tumor Cells Transfected With a GM-CSF Gene in Previously Treated Pancreatic Cancer. J. Immunother. 2013;36:382–389. doi: 10.1097/CJI.0b013e31829fb7a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark C.E., Hingorani S.R., Mick R., Combs C., Tuveson D.A., Vonderheide R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472. [DOI] [PubMed] [Google Scholar]
- 6.Dalton D.K., Noelle R.J. The roles of mast cells in anticancer immunity. Cancer Immunol. Immunother. 2012;61:1511–1520. doi: 10.1007/s00262-012-1246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang D.Z., Ma Y., Ji B., Wang H., Deng D., Liu Y., Abbruzzese J.L., Liu Y.j., Logsdon C.D., Hwu P. Mast Cells in Tumor Microenvironment Promotes the in vivo Growth of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2011;17:7015–7023. doi: 10.1158/1078-0432.CCR-11-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris J.P., Cano D.A., Sekine S., Wang S.C., Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J. Clin. Invest. 2010;120:508–520. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai S.W., Yang S.Z., Gao J., Pan K., Chen J.Y., Wang Y.L., Wei L.X., Dong J.H. Prognostic significance of mast cell count following curative resection for pancreatic ductal adenocarcinoma. Surgery. 2011;149:576–584. doi: 10.1016/j.surg.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Strouch M.J., Cheon E.C., Salabat M.R., Krantz S.B., Gounaris E., Melstrom L.G., Dangi-Garimella S., Wang E., Munshi H.G., Khazaie K., Bentrem D.J. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin. Cancer Res. 2010;16:2257–2265. doi: 10.1158/1078-0432.CCR-09-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Y., Hwang R.F., Logsdon C.D., Ullrich S.E. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 2013;73:3927–3937. doi: 10.1158/0008-5472.CAN-12-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma Y., Ullrich S.E. Intratumoral mast cells promote the growth of pancreatic cancer. OncoImmunology. 2013;2 doi: 10.4161/onci.25964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hingorani S.R., Petricoin E.F., Maitra A., Rajapakse V., King C., Jacobetz M.A., Ross S., Conrads T.P., Veenstra T.D., Hitt B.A., et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 14.Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H., Rustgi A.K., Chang S., Tuveson D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 15.Humbert M., Castéran N., Letard S., Hanssens K., Iovanna J., Finetti P., Bertucci F., Bader T., Mansfield C.D., Moussy A., et al. Masitinib combined with standard gemcitabine chemotherapy: in vitro and in vivo studies in human pancreatic tumour cell lines and ectopic mouse model. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alam M.S., Gaida M.M., Bergmann F., Lasitschka F., Giese T., Giese N.A., Hackert T., Hinz U., Hussain S.P., Kozlov S.V., Ashwell J.D. Selective inhibition of the p38 alternative activation pathway in infiltrating T cells inhibits pancreatic cancer progression. Nat. Med. 2015;21:1337–1343. doi: 10.1038/nm.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasan S., Satake M., Dawson D.W., Funahashi H., Angst E., Go V.L.W., Reber H.A., Hines O.J., Eibl G. Expression analysis of the prostaglandin E2 production pathway in human pancreatic cancers. Pancreas. 2008;37:121–127. doi: 10.1097/MPA.0b013e31816618ba. [DOI] [PubMed] [Google Scholar]
- 18.Grimbaldeston M.A., Nakae S., Kalesnikoff J., Tsai M., Galli S.J. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat. Immunol. 2007;8:1095–1104. doi: 10.1038/ni1503. [DOI] [PubMed] [Google Scholar]
- 19.Ito T., Wang Y.H., Duramad O., Hanabuchi S., Perng O.A., Gilliet M., Qin F.X.F., Liu Y.J. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc. Natl. Acad. Sci. USA. 2006;103:13138–13143. doi: 10.1073/pnas.0603107103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinberg A.D., Rivera M.M., Prell R., Morris A., Ramstad T., Vetto J.T., Urba W.J., Alvord G., Bunce C., Shields J. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J. Immunol. 2000;164:2160–2169. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- 21.Petty J.K., He K., Corless C.L., Vetto J.T., Weinberg A.D. Survival in human colorectal cancer correlates with expression of the T-cell costimulatory molecule OX-40 (CD134) Am. J. Surg. 2002;183:512–518. doi: 10.1016/s0002-9610(02)00831-0. [DOI] [PubMed] [Google Scholar]
- 22.Weinberg A.D. OX40: targeted immunotherapy--implications for tempering autoimmunity and enhancing vaccines. Trends Immunol. 2002;23:102–109. doi: 10.1016/s1471-4906(01)02127-5. [DOI] [PubMed] [Google Scholar]
- 23.Weinberg A.D., Morris N.P., Kovacsovics-Bankowski M., Urba W.J., Curti B.D. Science gone translational: the OX40 agonist story. Immunol. Rev. 2011;244:218–231. doi: 10.1111/j.1600-065X.2011.01069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voo K.S., Foglietta M., Percivalle E., Chu F., Nattamai D., Harline M., Lee S.T., Bover L., Lin H.Y., Baladandayuthapani V., et al. Selective targeting of Toll-like receptors and OX40 inhibit regulatory T-cell function in follicular lymphoma. Int. J. Cancer. 2014;135:2834–2846. doi: 10.1002/ijc.28937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jember A.G., Zuberi R., Liu F.T., Croft M. Development of allergic inflammation in a murine model of asthma is dependent on the costimulatory receptor OX40. J. Exp. Med. 2001;193:387–392. doi: 10.1084/jem.193.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng J., Sun B.F., Chen C.Y., Zhou J.Y., Chen Y.S., Chen H., Liu L., Huang D., Jiang J., Cui G.S., et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019;29:725–738. doi: 10.1038/s41422-019-0195-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grimbaldeston M.A., Chen C.C., Piliponsky A.M., Tsai M., Tam S.Y., Galli S.J. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 2005;167:835–848. doi: 10.1016/S0002-9440(10)62055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oka T., Kalesnikoff J., Starkl P., Tsai M., Galli S.J. Evidence questioning cromolyn's effectiveness and selectivity as a 'mast cell stabilizer' in mice. Lab. Invest. 2012;92:1472–1482. doi: 10.1038/labinvest.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soucek L., Lawlor E.R., Soto D., Shchors K., Swigart L.B., Evan G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007;13:1211–1218. doi: 10.1038/nm1649. [DOI] [PubMed] [Google Scholar]
- 30.Chacón-Salinas R., Chen L., Chavez-Blanco A.D., Limon-Flores A.Y., Ma Y., Ullrich S.E. An essential role for platelet-activating factor in activating mast cell migration following ultraviolet irradiation. J. Leukoc. Biol. 2014;95:139–148. doi: 10.1189/jlb.0811409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deplanque G., Demarchi M., Hebbar M., Flynn P., Melichar B., Atkins J., Nowara E., Moyé L., Piquemal D., Ritter D., et al. A randomized, placebo-controlled phase III trial of masitinib plus gemcitabine in the treatment of advanced pancreatic cancer. Ann. Oncol. 2015;26:1194–1200. doi: 10.1093/annonc/mdv133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drayman N., DeMarco J.K., Jones K.A., Azizi S.-A., Froggatt H.M., Tan K., Maltseva N.I., Chen S., Nicolaescu V., Dvorkin S., et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science (New York, N.Y.) 2021;373:931–936. doi: 10.1126/science.abg5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lortholary O., Chandesris M.O., Bulai Livideanu C., Paul C., Guillet G., Jassem E., Niedoszytko M., Barete S., Verstovsek S., Grattan C., et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet. 2017;389:612–620. doi: 10.1016/S0140-6736(16)31403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruby C.E., Yates M.A., Hirschhorn-Cymerman D., Chlebeck P., Wolchok J.D., Houghton A.N., Offner H., Weinberg A.D. Cutting Edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J. Immunol. 2009;183:4853–4857. doi: 10.4049/jimmunol.0901112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji B., Tsou L., Wang H., Gaiser S., Chang D.Z., Daniluk J., Bi Y., Grote T., Longnecker D.S., Logsdon C.D. Ras activity levels control the development of pancreatic diseases. Gastroenterology. 2009;137:1072–1082. doi: 10.1053/j.gastro.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Safran M., Kim W.Y., Kung A.L., Horner J.W., DePinho R.A., Kaelin W.G., Jr. Mouse reporter strain for noninvasive bioluminescent imaging of cells that have undergone Cre-mediated recombination. Mol. Imag. 2003;2:297–302. doi: 10.1162/15353500200303154. [DOI] [PubMed] [Google Scholar]
- 37.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6 doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Publicly available human TCGA dataset were analyzed. Further details are provided in the method details.
-
•
The human TCGA dataset and single cell RNA sequencing data that support the findings of this study are openly available. DOIs are listed in the key resources table.
-
•
All original code is publicly available. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.