

Abstract
Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), a tissue-specific and inducible transcriptional coactivator for several nuclear receptors, plays a key role in energy metabolism. We report here that PGC-1α coactivator activity is potentiated by arginine methylation by protein arginine methyltransferase 1 (PRMT1), another nuclear receptor coactivator. Mutation of three substrate arginines in the C-terminal region of PGC-1α abolished the cooperative coactivator function of PGC-1α and PRMT1, and compromised the ability of PGC-1α to induce endogenous target genes. Finally, endogenous PRMT1 contributes to PGC-1α coactivator activity, and to the induction of genes important for mitochondrial biogenesis.
Keywords: Arginine methylation, coactivator, mitochondrial biogenesis, nuclear receptor, transcription
Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is a transcriptional coactivator for many nuclear hormone receptors (NR), such as peroxisome proliferator-activated receptor γ (PPARγ), the thyroid hormone receptors (TR), retinoic acid receptors, glucocorticoid receptor (GR), and estrogen receptors (ER); it also functions with other classes of DNA-binding transcription factors such as nuclear respiratory factor 1 (NRF-1) (Knutti and Kralli 2001; Puigserver and Spiegelman 2003). PGC-1α harbors leucine-rich motifs (LXXLL) that bind to the hormone-activated NRs. The N-terminal region contains a strong autonomous activation domain, which binds to other coactivators such as SRC-1 and CBP and is required for coactivator function (Puigserver et al. 1999). The C-terminal region of PGC-1α contains an RNA recognition motif (RRM) and an RS domain (rich in Arg and Ser residues), both of which are characteristic of proteins involved in splicing, such as hnRNP and SR proteins. Indeed, PGC-1α not only functions as a transcriptional coactivator, but can also influence splicing patterns of specific transcripts (Monsalve et al. 2000). Deletions within the C-terminal domain disrupt the ability of PGC-1α to associate with RNA polymerase II, RNA-processing factors, and the Mediator complex, and to induce target genes, suggesting that this region contains both transcriptional coactivator and splicing regulator functions (Monsalve et al. 2000; Wallberg et al. 2003).
PGC-1α mRNA is predominantly expressed in heart, brown adipose tissue, kidney, skeletal muscle, and liver, and its expression is induced in response to stimuli known to regulate metabolic activity. For example, PGC-1α expression is induced in brown adipose tissue and skeletal muscle by exposure to cold (Puigserver et al. 1998; Boss et al. 1999), in muscle in response to physical exercise, and in the liver and heart in response to short-term fasting (Knutti and Kralli 2001; Puigserver and Spiegelman 2003; Kelly and Scarpulla 2004). In accordance with the nature of the stimuli that induce its expression, PGC-1α regulates a broad range of metabolic processes, including mitochondrial biogenesis, respiration, and gluconeogenesis, and thus enables physiologic adaptation to the energy needs imposed by the stimuli. These effects of PGC-1α are mediated by several DNA-binding transcription factors. Mitochondrial biogenesis and the stimulation of oxidative phosphorylation in particular require the cooperation of PGC-1α with the nuclear respiratory factors NRF-1 and NRF-2 and with estrogen-related receptor α (ERRα), an orphan nuclear receptor (Wu et al. 1999; Mootha et al. 2004; Schreiber et al. 2004).
As the major protein arginine methyltransferase (PRMT) in mammalian cells (Tang et al. 2000), PRMT1 methylates many RNA-binding proteins (Lee et al. 2005), as well as histone H4 (Chen et al. 1999; Strahl et al. 2001). Arginine methylation has been implicated in the regulation of many cellular processes, including signal transduction, subcellular localization of proteins, protein-protein interactions, and transcriptional regulation (Lee et al. 2005). Indeed, PRMT1 serves as a coactivator for NRs (Koh et al. 2001) and other DNA-binding transcription factors, such as p53 (An et al. 2004) and YY1 (Rezai-Zadeh et al. 2003). Once recruited to the target gene promoter region through direct or indirect association with the DNA-bound transcription factor, PRMT1 cooperates with other histone-modifying enzymes to mediate chromatin remodeling and transcriptional activation (Koh et al. 2001; Metivier et al. 2003; An et al. 2004).
Since PGC-1α contains an RNA-binding domain and many RNA-binding proteins are substrates for PRMT enzymes (Stallcup 2001), we tested whether PGC-1α could be methylated on arginine residues by PRMT1 or the related PRMT family member CARM1 (Chen et al. 1999). Furthermore, because some PRMTs act as coactivators for NRs and other transcription factors, we investigated whether PGC-1α could cooperate with PRMTs as coactivators.
Results
PRMT1 and its enzymatic activity stimulate PGC-1α coactivator function
As demonstrated previously (Knutti et al. 2000; Tcherepanova et al. 2000), PGC-1α can enhance the activity of ERα on a reporter plasmid controlled by ER-responsive elements (Fig. 1). Coexpression of PRMT1 with PGC-1α dramatically increased the reporter gene activity observed with PGC-1α alone or PRMT1 alone. In contrast, coexpression of another coactivator of the PRMT class, CARM1 (Chen et al. 1999), had little or no effect on PGC-1α activity. The functional synergy between PGC-1α and PRMT1 was completely dependent on the integrity of the LXXLL motifs of PGC-1α, which are responsible for binding hormone-activated ERα, and the N-terminal activation domain of PGC-1α (Fig. 2). Thus, the effect of PRMT1 in this case depends entirely on the integrity of the coactivator functions of PGC-1α, suggesting that PRMT1 is amplifying the coactivator function of PGC-1α. PGC-1α and PRMT1 also cooperated to enhance the activity of other NRs, such as GR and TR (data not shown).
Figure 1.

Specific and cooperative coactivator function between PGC-1α and PRMT1. CV-1 cells were transiently transfected with MMTV(ERE)-LUC reporter plasmid (250 ng) and expression vectors encoding ERα (0.1 ng), PGC-1α (50 ng), and PRMT1, PRMT1(E153Q) mutant or CARM1 (250 ng). Luciferase activity was measured after growth in 20 nM estradiol. (Bottom of figure) Immunoblots of extracts from COS7 cells transfected with HA-tagged PRMT1 expression vectors were performed with antibodies against HA.
Figure 2.

PGC-1α N-terminal domains required for functional synergy with PRMT1. (A) Schematic representation of PGC-1α mutants used in B. The mutant proteins are expressed at wild-type levels (data not shown). (B) CV-1 cells were transiently transfected with MMTV(ERE)-LUC reporter plasmid (250 ng) and expression vectors encoding ERα (0.1 ng) and PGC-1α wild type or mutants as indicated (50 ng) in the absence (white boxes) or presence (black boxes) of PRMT1 (200 ng). Transfected cells were grown in culture medium with 20 nM estradiol, and extracts of the harvested cells were tested for luciferase activity.
To test whether the methyltransferase activity of PRMT1 is required for coactivator synergy with PGC-1α, we used an enzymatically deficient mutant of PRMT1. In PRMT1, mutation of either Glu144 or Glu153 to Gln completely or nearly abolishes enzymatic activity, because these Glu residues are important for binding the substrate arginine residue (Zhang et al. 2000; Zhang and Cheng 2003; data not shown). The PRMT1 E153Q mutant failed to stimulate PGC-1α coactivator activity, although it was expressed at levels comparable to that of wild-type PRMT1 (Fig. 1). Thus, the specific protein arginine methyltransferase activity of PRMT1 (but not CARM1) is necessary for enhancing the coactivator function of PGC-1α. The requisite substrate that is methylated by PRMT1 to cause the enhancement of PGC-1α activity could be histone H4 (Chen et al. 1999; Strahl et al. 2001) or a component of the transcription machinery that has not been previously characterized as a PRMT1 substrate, including PGC-1α itself.
PRMT1 methylates PGC-1α
Since PGC-1α contains an RNA-binding domain and many RNA-binding proteins are known to be substrates for arginine methylation, we tested whether PGC-1α could be methylated by PRMT1. Indeed, recombinant PRMT1, but not CARM1, methylated recombinant full-length PGC-1α when incubated with [3H]AdoMet (Fig. 3A). This result correlates with the finding that PRMT1, but not CARM1, enhances PGC-1α activity.
Figure 3.

Methylation of PGC-1α by PRMT1. (A) Full-length GST-PGC-1α was incubated with GST-CARM1 or GST-PRMT1 and [3H]AdoMet for 1 h at 30°C. Products were analyzed by SDS-PAGE and fluorography. (B) Methylation of PGC-1α fragments was analyzed by SDS-PAGE (lower panels show Coomassie blue staining) and fluorography (upper panels). Diagram shows amino acid numbers. Asterisks show the stained fragments (lower panels) that were methylated (upper panels); positions of molecular weight markers are indicated beside the panels. (C) Mutant forms of the PGC-1α E fragment (sequence shown) were methylated by PRMT1 and analyzed by fluorography (left panel) and staining (right panel). In the R4K and R3K mutants, the bracketed Arg residues (R) were changed to Lys. The results presented are from a single experiment representative of three independent experiments.
Fragments of PGC-1α were used to map the region that is methylated (Fig. 3B). While N-terminal fragments containing amino acids 1-306 or 306-532 were not methylated, PRMT1 methylated two different C-terminal fragments of PGC-1α, that is, amino acids 532-640 and a subfragment of amino acids 640-798. When the C-terminal portion of PGC-1α was divided into three fragments, the acidic E region and the RS region were both substrates for PRMT1, but the RNA recognition motif (RRM) was not (Fig. 3B, right panels).
To investigate contributions of the RS and E regions to PGC-1α synergy with PRMT1, we evaluated the coactivator function of various PGC-1α deletion mutants (Fig. 4A). While PGC-1α mutants lacking various C-terminal domains were expressed at levels similar to or higher than those of wild-type PGC-1α (data not shown), they had varied abilities to cooperate synergistically with PRMT1 (Fig. 4B, black bars). Deletion of the entire C-terminal region, the E region, or the RRM domain eliminated synergy with PRMT1, but deletion of the RS region did not. Since the E region is methylated by PRMT1 and plays an important role in the stimulation of PGC-1α activity by PRMT1, we focused on identifying the methylated residues of the E region. Two groups of Arg residues within the E region were modified to Lys. A mutant GST-E(R4K) fragment with Lys substitutions at Arg residues 644, 647, 651, and 652 was methylated efficiently by PRMT1 (Fig. 3C), at levels indistinguishable from the wild-type GST-E fragment (data not shown). However, conversion of Arg residues 665, 667, and 669 to lysines in the GST-E(R3K) mutant fragment completely abolished methylation by PRMT1 (Fig. 3C), showing that these residues are important for the methylation and presumably contain the methylation site(s). Individual lysine substitutions at Arg residues 665, 667, and 669 failed to eliminate the methylation of GST-E, suggesting that more than one of these three residues are methylation sites for PRMT1 (data not shown).
Figure 4.

Role of PGC-1α C-terminal domains in synergy with PRMT1. (A) Schematic representation of PGC-1α deletion mutants used in B. (B) CV-1 cells were transiently transfected with MMTV(ERE)-LUC reporter plasmid (250 ng) and expression vectors encoding ERα (0.1 ng) and PGC-1α wild type or mutants as indicated (50 ng), in the absence (white boxes) or presence (black boxes) of PRMT1 (200 ng). Transfected cells were grown in culture medium with 20 nM estradiol, and extracts of the harvested cells were tested for luciferase activity.
Importance of Arg methylation in PGC-1α coactivator function
To investigate the role of the Arg methylation sites in the stimulation of PGC-1α activity by PRMT1, we constructed a plasmid expressing a full-length PGC-1α mutant with these three Arg residues (665, 667, and 669) changed to Lys. We tested the ability of this R3K mutant of full-length PGC-1α to cooperate with PRMT1 to enhance transcriptional activation by ERα. Mutant and wild-type PGC-1α were expressed at similar levels (Fig. 5A). In the absence of PRMT1, the ability of the R3K mutant of PGC-1α to enhance reporter gene activation by ERα was indistinguishable from that of wild-type PGC-1α (Fig. 5A). However, PRMT1 dramatically enhanced the coactivator function of wild-type PGC-1α but failed to stimulate the activity of the R3K mutant of PGC-1α.
Figure 5.

Role of methylated Arg residues of PGC-1α E region in coactivator function. (A) CV-1 cells were transfected with MMTV(ERE)-LUC reporter plasmid (250 ng) and plasmids encoding ERα (0.1 ng), PGC-1α wild type or R3K mutant (50 ng), and PRMT1 (200 ng) as indicated, and grown with 20 nM estradiol. (B) CV-1 cells were transfected with pERRα-Luc reporter plasmid (250 ng) and plasmids encoding PRMT1 (250 ng) and PGC-1α wild type or R3K mutant (50 ng).
To test the synergy between PGC-1α and PRMT1 and the role of arginine methylation in the context of a natural cellular target gene of PGC-1α, we compared the ability of wild-type and R3K mutant PGC-1α to induce the expression of the ERRα promoter, which is an important regulatory target of PGC-1α in the process of mitochondrial biogenesis (Mootha et al. 2004; Schreiber et al. 2004). ERRα protein binds to and activates transcription of its own gene, and PGC-1α is a required coactivator for that process. As observed previously (Laganiere et al. 2004; Mootha et al. 2004), overexpression of wild-type PGC-1α dramatically enhanced the expression of a transiently transfected reporter gene controlled by the upstream regulatory region of the ERRα promoter (Fig. 5B). In this context the R3K mutation reduced, but did not abolish, the coactivator function of PGC-1α, in the absence of exogenously expressed PRMT1. Furthermore, overexpression of PRMT1 dramatically enhanced the ability of wild-type PGC-1α to stimulate transcription from the ERRα promoter, but had little or no effect on the activity of the R3K mutant (Fig. 5B). Thus, with ERRα the inherent coactivator activity of PGC-1α depends partially, and its ability to be stimulated by PRMT1 depends entirely, on the three Arg residues that are substrates for methylation by PRMT1. These results strongly suggest that the methylation of PGC-1α by PRMT1 plays a key role in its activity and ability to function cooperatively with PRMT1.
The reduced activity of the R3K mutant on the ERRα promoter in the absence of overexpressed PRMT1 (Fig. 5B) suggests that the activity of the wild-type PGC-1α protein depends on methylation by endogenous PRMT1. To test this, we determined the effect of reducing endogenous levels of PRMT1 on the ability of overexpressed PGC-1α to stimulate expression of the transiently transfected ERRα-Luc reporter plasmid. Two different siRNAs targeting the human PRMT1 sequence efficiently reduced the level of PRMT1 protein, but not β-actin protein, in COS7 cells in a dose-dependent manner (Fig. 6, lower panels). The siRNA-treated or untreated cells were subsequently transfected with ERRα-Luc (encoding firefly luciferase), a PGC-1α expression plasmid, and a control Renilla luciferase reporter plasmid driven by a constitutive cytomegalovirus promoter. Both siRNAs against PRMT1 caused a dose-dependent decrease in expression of firefly luciferase from the ERRα promoter (Fig. 6, upper panel), which paralleled the decrease in PRMT1 protein levels (Fig. 6, lower panels). Since the firefly luciferase data are normalized against the Renilla luciferase activity, these results demonstrate that reduction in endogenous PRMT1 levels causes a gene-specific decrease in the ability of PGC-1α to stimulate expression from the transiently transfected ERRα promoter.
Figure 6.
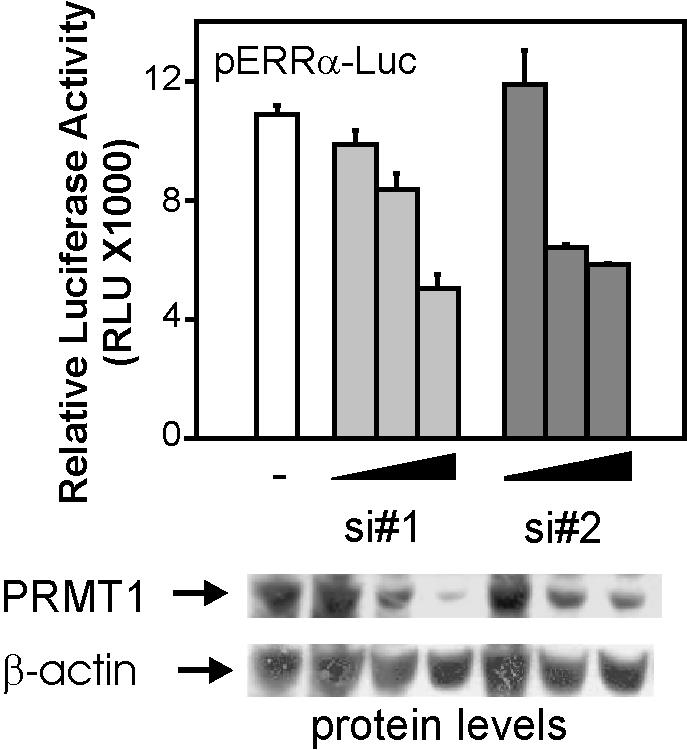
Role of endogenous PRMT1 in PGC-1α coactivator function. COS7 cells in 24-well plates were transfected with 20, 40, or 60 pmol of two different siRNAs against PRMT1 (si#1 and si#2). Two days later, cells were transfected with pERRα-Luc reporter plasmid (125 ng), a PGC-1α expression vector (25 ng), and a Renilla luciferase reporter plasmid driven by a cytomegalovirus promoter (12.5 ng). Luciferase activities were quantified 48 h later; firefly luciferase activity was normalized by Renilla luciferase activity. Protein levels were determined by immunoblots from cell extracts of a fourth siRNA-transfected well. Results shown are from a single experiment that is representative of three independent experiments.
Endogenous PRMT1 and Arg methylation of PGC-1α contribute to the induction of endogenous target genes of PGC-1α
Wild-type PGC-1α and the R3K mutant were expressed in SAOS2 osteosarcoma cells by infection with adenoviral vectors, to test their ability to stimulate expression of the endogenous ERRα and cytochrome c (cyt c) genes, which are targets of PGC-1α and important for mitochondrial biogenesis (Schreiber et al. 2003, 2004; Mootha et al. 2004). A viral vector expressing green fluorescent protein (GFP) was used as a control. Although equivalent expression of mutant and wild-type proteins was observed (Fig. 7A, inset), the PGC-1α R3K mutant was substantially less efficient than the wild-type protein at inducing the expression of ERRα and cyt c mRNA (Fig. 7A), suggesting that arginine methylation is important for PGC-1α function at endogenous target genes in their native chromatin state.
Figure 7.

Role of Arg methylation in induction of endogenous genes important for the biogenesis and function of mitochondria. (A) SAOS2 cells in six-well plates were infected with adenoviruses expressing GFP, PGC-1α wild type, or PGC-1α R3K mutant (multiplicity of infection = 40) and harvested 24 h later. mRNA levels for the ERRα and cyt c genes were determined by quantitative PCR and normalized to the level of GAPDH mRNA. (B) SAOS2 cells in six-well plates were transfected with siRNA#1 against PRMT1 (100 pmol/well) two times, separated by 72 h. Four hours after the second siRNA transfection, cells were infected with adenovirus expressing GFP or PGC-1α and harvested 24 h later. mRNA levels for the indicated genes were determined by quantitative PCR and normalized to the GAPDH mRNA level. The mean and range of variation shown are from duplicate transfections in a single experiment that is representative of five independent experiments, each with duplicate transfected cell cultures. In all five experiments, the multiplicity of infection was 10-40, and the reduction in PRMT1 mRNA levels caused by siRNA was at least 50%. A paired, two-tailed t-test performed on the values from the five experiments indicated that the siRNA against PRMT1 caused significant decreases in the PGC-1α-induced levels of Cyt c mRNA (p = 0.014) and ERRα mRNA (p = 0.008). For the five experiments, the mean decrease and 95% confidence interval was 35 ± 15% for Cyt c mRNA, 26 ± 5% for ERRα mRNA, and 75 ± 9% for PRMT1 mRNA.
To determine the role of endogenous PRMT1 in PGC-1α-dependent transcriptional programs, we next used the siRNA#1 to reduce endogenous PRMT1 mRNA levels in SAOS2 cells (Fig. 7B). The siRNA-treated or untreated SAOS2 cells were subsequently infected with adenoviral vectors encoding GFP or PGC-1α. PGC-1α expression led to increases in the levels of the endogenous cyt c and ERRα mRNAs; however, this induction by PGC-1α was partially inhibited in the cells treated with siRNA against PRMT1, compared with the cells that were not exposed to the siRNA (Fig. 7B). The effects of PGC-1α and the siRNA against PRMT1 were gene-specific, since the data shown are normalized against the expression of glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA. Thus, endogenous levels of PRMT1 contribute to the ability of PGC-1α to induce the expression of target genes that are important for mitochondrial biogenesis.
Discussion
As an important controller of energy homeostasis, PGC-1α expression is highly regulated at the transcription level, in a tissue-specific manner and in response to several physiologic signals (Knutti and Kralli 2001; Puigserver and Spiegelman 2003; Kelly and Scarpulla 2004). In addition, the important roles of PGC-1α in metabolic regulation also appear to be modulated by a variety of post-translational modifications and interactions with other proteins. The docking of PPARγ to PGC-1α induces an apparent conformational change that allows binding of the collaborating coactivators SRC-1 and CBP/p300 to the N-terminal activation domain of PGC-1α (Puigserver et al. 1999). Moreover, there is evidence that a repressor protein competes with NRs for binding to PGC-1α, and this competition appears to be regulated by phosphorylation of the PGC-1α suppression domain by MAPK p38, which inhibits binding of the repressor protein (Knutti et al. 2001; Puigserver et al. 2001; Fan et al. 2004). Our data add arginine methylation to the list of post-translational regulatory mechanisms that govern PGC-1α activity and, in addition, implicate PRMT1 as an important component cooperating with PGC-1α in metabolic processes such as mitochondrial biogenesis. Although reductions in the level of PRMT1 caused relatively modest but significant decreases in the coactivator function of PGC-1α (Figs. 6, 7), it is noteworthy that the decreases in PGC-1α levels and the activity of PGC-1α-dependent pathways that have been correlated to insulin resistance in humans are very modest—∼35% decrease in PGC-1α expression and ∼20% decrease in expression of genes encoding oxidative phosphorylation components such as cyt c (Mootha et al. 2003; Patti et al. 2003). These findings are indicative of the fact that small changes in pathways important for energy metabolism can have potent consequences over time, and suggest that modest effects on PGC-1α activity can be crucial for human health. Understanding the signals and mechanisms that enable PRMT1 to modify PGC-1α and modulate its activity may lead to ways to enhance PGC-1α activity and intervene in such diseases.
Our evidence indicates that methylation by PRMT1 of two or three of the Arg residues in an RERQR sequence, found within the C-terminal Glu-rich E region of PGC-1α (Fig. 3), plays a critical role in the coactivator function of PGC-1α and its ability to function synergistically with PRMT1 (Figs. 5, 6, 7). Detecting methylation of specific proteins in vivo is still a developing art, as witnessed by a variety of recent methodological papers (Boisvert et al. 2003; Rappsilber et al. 2003). We have been unable to demonstrate methylation of the PGC-1α E region in vivo by mass spectrometry, labeling of cells with methyl-labeled methionine, or antibodies with supposedly general specificity toward proteins containing methyl-arginine. Our experience suggests that success with these approaches depends on the specific protein. Methylation of PGC-1α by PRMT1 may be regulated, and may occur in vivo only, for example, when they are both recruited into a transcription complex and PGC-1α adopts the appropriate conformation. In support of this idea, the conformation of PGC-1α is regulated by protein-protein interactions (Puigserver et al. 1999).
In spite of our lack of direct evidence for in vivo methylation, our data provide strong indirect evidence that methylation of PGC-1α by PRMT1 does occur in vivo and plays an important role in facilitating PGC-1α coactivator function: (1) PGC-1α and its E region are good substrates for PRMT1 but not CARM1. (2) PRMT1, but not CARM1, acts synergistically with PGC-1α as a coactivator, and this synergy depends on the methyltransferase activity of PRMT1 and the E region of PGC-1α. (3) The Arg residues that are methylated by PRMT1 are important for the synergy between PGC-1α and PRMT1 and for the ability of PGC-1α to stimulate expression of transiently transfected and endogenous target genes. (4) Endogenous PRMT1 is important for the coactivator function of PGC-1α in the induction of transiently transfected and endogenous target genes.
While PRMT1 and its methyltransferase activity clearly affect the coactivator function of PGC-1α, considerable PGC-1α coactivator activity remains when PRMT1 levels are severely reduced (Figs. 6, 7B), when the Arg methylation sites in the E region are mutated (Figs. 5, 7A), or even when the entire C-terminal region (RS, E, and RRM domains) is deleted (Fig. 4). Thus PGC-1α coactivator function has two components, one independent of PRMT1 and one dependent on PRMT1. The strong N-terminal activation domain appears to be important for both of these activities (Fig. 2), but the specific downstream mechanisms that mediate these two components of PGC-1α activity remain to be defined.
Arginine methylation of PGC-1α occurs in the E region, which contains a subnuclear localization signal (Monsalve et al. 2000), and thus could modulate the subnuclear distribution of PGC-1α, as has been shown for the methylation of some RNA-binding proteins (Lee et al. 2005). If so, such an effect would be subtle, because no differences in the nuclear distribution of wild-type or R3K PGC-1α protein, in the presence or absence of overexpressed PRMT1, were evident by immunofluorescence (data not shown). A PGC-1α mutant lacking the C-terminal region exhibited reduced coactivator function (Fig. 4) and was deficient in binding to RNA polymerase II and some SR-type splicing factors (Monsalve et al. 2000), suggesting that this region is important for both transcriptional activation and regulation of mRNA splicing by PGC-1α. The positive effect of PRMT1-mediated methylation suggests that methylation may cause an activating conformational change in PGC-1α, enhance binding of positively acting proteins such as TRAP220 (Wallberg et al. 2003), and/or inhibit binding of repressor proteins. Further studies will be required to elucidate the downstream effects of PGC-1α methylation and how it contributes to enhanced transcriptional activation.
Materials and methods
Plasmids
The following plasmids were previously described: pcDNA3-HA-PGC-1α, encoding full-length human PGC-1α, PGC-1α.L2A, PGC-1α.L2/3A (PGC-1α bearing point mutations within the Leu-rich or LXXLL motifs, as indicated), and PGC-1α.91C (PGC-1α lacking the N-terminal activation domain) (Knutti et al. 2000); pSG5.HA-PRMT1, encoding HA epitope-tagged PRMT1 (Koh et al. 2001); GST-PRMT1, encoding a GST-PRMT1 fusion protein (Lin et al. 1996); pSG5.HA-CARM1, encoding HA-CARM1; pGEX4T1.CARM1, encoding GST-CARM1 (Chen et al. 1999); pHE0, encoding human estrogen receptor α (Green et al. 1988); and the luciferase reporter plasmid for ER, MMTV(ERE)-LUC (Umesono and Evans 1989).
Construction of plasmids encoding fragments of PGC-1α fused to GST: PCR-amplified cDNA fragments encoding PGC-1α fragments that include amino acids 1-306, 306-532, 532-640, and 640-798, were inserted into the BamHI and XhoI sites of pGEX4T1; pGEX4T3-RS, encoding PGC-1α(566-632); pGEX4T3-E, encoding PGC-1α(632-676); and pGEX4T3-RRM, encoding PGC-1α(676-755), were constructed by inserting the appropriate cDNA fragments, amplified from pcDNA3-HA-PGC-1α as a template and with primers containing BamHI and SalI sites, into the BamHI and SalI sites of pGEX4T3. The following mutations were generated with the Quickchange site-directed mutagenesis kit (Stratagene), using the corresponding wild-type plasmids as templates: pSG5.HA-PRMT1(E153Q); pGEX4T3-E(R4K), containing R644K, R647K, R651K, and R652K mutations; pGEX4T3-E(R3K), containing R665K, R667K, and R669K mutations; pcDNA3-HA-PGC-1α(R3K), pcDNA3-HA-PGC-1α.ΔC (Δ566-755), pcDNA3-HA-PGC-1α.ΔRS (Δ566-632), pcDNA3-HA-PGC-1α.ΔE (Δ632-676), and pcDNA3-HA-PGC-1α.ΔRRM (Δ676-755). To construct the luciferase reporter pERRα-Luc, the ERRα-responsive sequences of the ESRRA promoter (nucleotides -537 to -829, relative to the transcription initiation site) were amplified by PCR and subcloned upstream of the minimal alcohol dehydrogenase promoter of pΔLuc (Iñiguez-Lluhí et al. 1997).
Cell culture and transfection
CV-1 and COS7 cells were transiently transfected in 12-well dishes as described previously (Ma et al. 1999). Where indicated, medium contained 20 nM estradiol during the last 30 h of growth. Luciferase activity results shown (mean and range of variation of two transfected wells) are from a single experiment, representative of three to five independent experiments. pSG5.HA and pcDNA3.1 empty vectors were added to all transfections to balance total DNA. Luciferase activity was determined with the Promega Luciferase Assay Kit according to the manufacturer's protocols. Because some coactivators enhance the activities of so-called constitutive promoters, constitutive reporter plasmids were not used routinely to normalize the luciferase activities but were used strategically to monitor transfection efficiency.
Adenovirus infections and RNA analysis
SAOS2 cells were infected with adenoviral vectors expressing GFP and PGC-1α as described previously (Schreiber et al. 2003). Adenoviruses expressing PGC-1α R3K were constructed by using the insert of pcDNA3-HA-PGC-1α R3K. Total RNA was isolated using the TRIzol reagent (Invitrogen), cDNA was synthesized with the SuperScript Choice system (Invitrogen), and specific transcripts were quantified by quantitative real-time PCR using the SYBR Green PCR system (Applied Biosystems) and the Chromophore detection system (MJ Research) (Fig. 7A) or the Stratagene Mx3000P System (Fig. 7B). Gene-specific primers were used for human ERRα (Schreiber et al. 2003), human cyt c (Schreiber et al. 2004), and human GAPDH (5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGATGGTGATGGGATTTC-3′). ERRα and cyt c mRNA levels were normalized to GAPDH mRNA levels and estimated using standard curves (Fig. 7A; Schreiber et al. 2004) or the ΔΔCt method cited in ABI User Bulletin #2 (Fig. 7B).
Methylation assays
Methylation assays (Chen et al. 1999) were performed with 1 μg each of GST-fused enzyme and substrate.
RNA interference and immunoblots
The following siRNA oligonucleotides for human PRMT1 were designed using the Target Finder program (Ambion), and chemically synthesized by the USC Norris Comprehensive Cancer Center Microchemical Core: siRNA#1 sense, 5′-AGAUUACUACUUUGACUCCdTdT-3′; siRNA#1 antisense, 5′-GGAGUCAAAGUAGUAAUCUdTdT-3′; siRNA#2 sense, 5′-CUUAUGUUUUUAUAUGGUUdTdT-3′; siRNA#2 antisense, 5′-AACCAUAUAAAAACAUAAGdTdT-3′. Annealed siRNAs were transfected with Lipofectamine 2000 (Invitrogen) into COS7 cells in 24-well plates 48 h before DNA transfection. Luciferase assays were performed 48 h after DNA transfection. Parallel cell extracts were prepared in SDS sample buffer, separated by 12% SDS-PAGE, and transferred onto PVDF membrane. Immunoblots used antibodies against PRMT1 (Upstate Biotechnology) and, after stripping, β-actin (Santa Cruz). SAOS2 cells in six-well plates were transfected with siRNA#1 against PRMT1 (100 pmol/well). After 72 h, cells were transfected again with siRNA#1 against PRMT1 (100 pmol/well), medium was changed after 4 h, and then cells were infected with adenovirus expressing GFP or PGC-1α and harvested 24 h later. mRNA levels for the indicated genes were determined by quantitative PCR and normalized to the GAPDH mRNA level.
Acknowledgments
We thank Mr. Dan Gerke (University of Southern California) and Ms. Jessica Cardenas and Dr. M. Benjamin Hock (Scripps Research Institute) for technical help and advice, and Ms. Catherine Yang (University of Southern California) for assistance with statistical analyses. This work was supported by Grants DK55274 (to M.R.S.) and DK064951 (to A.K.) from the National Institutes of Health.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1295005.
Corresponding authors.
References
- An W., Kim, J., and Roeder, R.G. 2004. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117: 735-748. [DOI] [PubMed] [Google Scholar]
- Boisvert F.M., Cote, J., Boulanger, M.C., and Richard, S. 2003. A proteomic analysis of arginine-methylated protein complexes. Mol. Cell Proteomics 2: 1319-1330. [DOI] [PubMed] [Google Scholar]
- Boss O., Bachman, E., Vidal-Puig, A., Zhang, C.Y., Peroni, O., and Lowell, B.B. 1999. Role of the β(3)-adrenergic receptor and/or a putative β(4)-adrenergic receptor on the expression of uncoupling proteins and peroxisome proliferator-activated receptor-γ coactivator-1. Biochem. Biophys. Res. Commun. 261: 870-876. [DOI] [PubMed] [Google Scholar]
- Chen D., Ma, H., Hong, H., Koh, S.S., Huang, S.-M., Schurter, B.T., Aswad, D.W., and Stallcup, M.R. 1999. Regulation of transcription by a protein methyltransferase. Science 284: 2174-2177. [DOI] [PubMed] [Google Scholar]
- Fan M., Rhee, J., St Pierre, J., Handschin, C., Puigserver, P., Lin, J., Jaeger, S., Erdjument-Bromage, H., Tempst, P., and Spiegelman, B.M. 2004. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1α: Modulation by p38 MAPK. Genes & Dev. 18: 278-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S., Issemann, I., and Sheer, E. 1988. A versatile in vivo and in vitro eukaryotic expression vector for protein engineering. Nucleic Acids Res. 16: 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iñiguez-Lluhí J.A., Lou, D.Y., and Yamamoto, K.R. 1997. Three amino acid substitutions selectively disrupt the activation but not the repression function of the glucocorticoid receptor N terminus. J. Biol. Chem. 272: 4149-4156. [DOI] [PubMed] [Google Scholar]
- Kelly D.P. and Scarpulla, R.C. 2004. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes & Dev. 18: 357-368. [DOI] [PubMed] [Google Scholar]
- Knutti D. and Kralli, A. 2001. PGC-1, a versatile coactivator. Trends Endocrinol. Metab. 12: 360-365. [DOI] [PubMed] [Google Scholar]
- Knutti D., Kaul, A., and Kralli, A. 2000. A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol. Cell. Biol. 20: 2411-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutti D., Kressler, D., and Kralli, A. 2001. Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proc. Natl. Acad. Sci. 98: 9713-9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S.S., Chen, D., Lee, Y.-H., and Stallcup, M.R. 2001. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J. Biol. Chem. 276: 1089-1098. [DOI] [PubMed] [Google Scholar]
- Laganiere J., Tremblay, G.B., Dufour, C.R., Giroux, S., Rousseau, F., and Giguere, V. 2004. A polymorphic autoregulatory hormone response element in the human estrogen-related receptor alpha (ERRα) promoter dictates peroxisome proliferator-activated receptor γ coactivator-1α control of ERRα expression. J. Biol. Chem. 279: 18504-18510. [DOI] [PubMed] [Google Scholar]
- Lee D.Y., Teyssier, C., Strahl, B.D., and Stallcup, M.R. 2005. Role of protein methylation in regulation of transcription. Endocr. Rev. 26: 147-170. [DOI] [PubMed] [Google Scholar]
- Lin W.-J., Gary, J.D., Yang, M.C., Clarke, S., and Herschman, H.R. 1996. The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J. Biol. Chem. 271: 15034-15044. [DOI] [PubMed] [Google Scholar]
- Ma H., Hong, H., Huang, S.-M., Irvine, R.A., Webb, P., Kushner, P.J., Coetzee, G.A., and Stallcup, M.R. 1999. Multiple signal input and output domains of the 160-kDa nuclear receptor coactivator proteins. Mol. Cell. Biol. 19: 6164-6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metivier R., Penot, G., Hubner, M.R., Reid, G., Brand, H., Kos, M., and Gannon, F. 2003. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115: 751-763. [DOI] [PubMed] [Google Scholar]
- Monsalve M., Wu, Z., Adelmant, G., Puigserver, P., Fan, M., and Spiegelman, B.M. 2000. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol. Cell 6: 307-316. [DOI] [PubMed] [Google Scholar]
- Mootha V.K., Lindgren, C.M., Eriksson, K.F., Subramanian, A., Sihag, S., Lehar, J., Puigserver, P., Carlsson, E., Ridderstrale, M., Laurila, E., et al. 2003. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately down-regulated in human diabetes. Nat. Genet. 34: 267-273. [DOI] [PubMed] [Google Scholar]
- Mootha V.K., Handschin, C., Arlow, D., Xie, X., St Pierre, J., Sihag, S., Yang, W., Altshuler, D., Puigserver, P., Patterson, N., et al. 2004. Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. 101: 6570-6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti M.E., Butte, A.J., Crunkhorn, S., Cusi, K., Berria, R., Kashyap, S., Miyazaki, Y., Kohane, I., Costello, M., Saccone, R., et al. 2003. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. 100: 8466-8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P. and Spiegelman, B.M. 2003. Peroxisome proliferator-activated receptor-γ coactivator 1 α (PGC-1α): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 24: 78-90. [DOI] [PubMed] [Google Scholar]
- Puigserver P., Wu, Z., Park, C.W., Graves, R., Wright, M., and Spiegelman, B.M. 1998. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92: 829-839. [DOI] [PubMed] [Google Scholar]
- Puigserver P., Adelmant, G., Wu, Z., Fan, M., Xu, J., O'Malley, B., and Spiegelman, B.M. 1999. Activation of PPARγ coactivator-1 through transcription factor docking. Science 286: 1368-1371. [DOI] [PubMed] [Google Scholar]
- Puigserver P., Rhee, J., Lin, J., Wu, Z., Yoon, J.C., Zhang, C.Y., Krauss, S., Mootha, V.K., Lowell, B.B., and Spiegelman, B.M. 2001. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 8: 971-982. [DOI] [PubMed] [Google Scholar]
- Rappsilber J., Friesen, W.J., Paushkin, S., Dreyfuss, G., and Mann, M. 2003. Detection of arginine dimethylated peptides by parallel precursor ion scanning mass spectrometry in positive ion mode. Anal. Chem. 75: 3107-3114. [DOI] [PubMed] [Google Scholar]
- Rezai-Zadeh N., Zhang, X., Namour, F., Fejer, G., Wen, Y.D., Yao, Y.L., Gyory, I., Wright, K., and Seto, E. 2003. Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes & Dev. 17: 1019-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S.N., Knutti, D., Brogli, K., Uhlmann, T., and Kralli, A. 2003. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor α (ERRα). J. Biol. Chem. 278: 9013-9018. [DOI] [PubMed] [Google Scholar]
- Schreiber S.N., Emter, R., Hock, M.B., Knutti, D., Cardenas, J., Podvinec, M., Oakeley, E.J., and Kralli, A. 2004. The estrogen-related receptor α (ERRα) functions in PPARγ coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. 101: 6472-6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallcup M.R. 2001. Role of protein methylation in chromatin remodeling and transcriptional regulation. Oncogene 20: 3014-3020. [DOI] [PubMed] [Google Scholar]
- Strahl B.D., Briggs, S.D., Brame, C.J., Caldwell, J.A., Koh, S.S., Ma, H., Cook, R.G., Shabanowitz, J., Hunt, D.F., Stallcup, M.R., et al. 2001. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol. 11: 996-1000. [DOI] [PubMed] [Google Scholar]
- Tang J., Frankel, A., Cook, R.J., Kim, S., Paik, W.K., Williams, K.R., Clarke, S., and Herschman, H.R. 2000. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 275: 7723-7730. [DOI] [PubMed] [Google Scholar]
- Tcherepanova I., Puigserver, P., Norris, J.D., Spiegelman, B.M., and McDonnell, D.P. 2000. Modulation of estrogen receptor-alpha transcriptional activity by the coactivator PGC-1. J. Biol. Chem. 275: 16302-16308. [DOI] [PubMed] [Google Scholar]
- Umesono K. and Evans, R.M. 1989. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell 57: 1139-1146. [DOI] [PubMed] [Google Scholar]
- Wallberg A.E., Yamamura, S., Malik, S., Spiegelman, B.M., and Roeder, R.G. 2003. Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1α. Mol. Cell 12: 1137-1149. [DOI] [PubMed] [Google Scholar]
- Wu Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., Troy, A., Cinti, S., Lowell, B., Scarpulla, R.C., et al. 1999. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115-124. [DOI] [PubMed] [Google Scholar]
- Zhang X. and Cheng, X.D. 2003. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 11: 509-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhou, L., and Cheng, X. 2000. Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 19: 3509-3519. [DOI] [PMC free article] [PubMed] [Google Scholar]