

Abstract
Immunotherapy has emerged as a crucial strategy to combat cancer by ‘reprogramming’ a patient’s own immune system. Although immunotherapy is typically reserved for patients possessing a high mutational burden, neoantigens produced from post-transcriptional regulation may provide an untapped reservoir of common immunogenic targets for new targeted cancer therapies. To comprehensively define tumor-specific and likely immunogenic neoantigens from patient RNA-Seq, we developed Splicing Neo Antigen Finder (SNAF), an easy-to-use and open-source computational workflow to predict splicing-derived immunogenic MHC-bound peptides (T cell antigen) and unannotated transmembrane proteins with altered extracellular epitopes (B cell antigen). This workflow employs a highly accurate deep-learning strategy for immunogenicity prediction (DeepImmuno) in conjunction with new algorithms to rank the tumor specificity of neoantigens (BayesTS) and to predict regulators of mis-splicing (RNA-SPRINT). T-cell antigens from SNAF were frequently verified as HLA-presented peptides from Mass Spectrometry (MS) and predict response to immunotherapy in melanoma. Splicing neoantigen burden was attributed to coordinated splicing factor dysregulation. Shared splicing neoantigens were found in up to 90% of patients with melanoma, correlated to overall survival in multiple cancer cohorts, induced T cell reactivity and were characterized by distinct cells of origin and amino acid preferences. In addition to T-cell neoantigens, our B-cell focused pipeline (SNAF-B) identified a new class of tumor-specific extracellular neo-epitopes which we termed ExNeoEpitopes. ExNeoEpitope full-length mRNA predictions were tumor specific and validated using long-read isoform sequencing and in vitro transmembrane localization assays. Therefore our systematic identification of splicing neoantigens revealed potential shared targets for therapy in heterogeneous cancers.
INTRODUCTION
A paramount goal for cancer treatment is standardized and accessible therapeutic strategies for shared targets that will be effective in a large percentage of patients. Tumor heterogeneity has been widely acknowledged as a hallmark of cancer, which poses challenges for developing new targeted therapies (1). Such heterogeneity is further responsible for drug resistance that leads to frequent cancer relapse. Since each tumor sample is unique with distinct mutations, the search for tumor-specific neoantigens has been considered the “final common pathway” for our immune system to fight cancer (1)(2). Focused targeting of patients with selective mutations has produced promising results in cancers with a high mutational burden, such as melanoma, non-small cell lung cancer, and Microsatellite Instability (MSI)-high (MSI-H) colorectal cancer. For example, 4-out-of-6 melanoma patients of cancer vaccinated with precision neoantigen vaccines show no evidence of relapse within 25 months post-therapy (NCT01970358) (3). Such promising clinical results have been attributed to the long-term persistence of neoantigen-specific memory T cells, illustrating the durability of neoantigen-based therapies (4). Other examples include Moderna’s mRNA-4157 combination with pembrolizumab, which achieved a 50% response rate in HPV-negative head and neck cancer compared to 14.6% for pembrolizumab monotherapy (NCT03313778) (5) and adoptive T cell transfer, in which neoantigen-reactive T cells are cultured and reinfused into the same patient, resulting in a 55% objective response and 24% complete response rate in metastatic melanoma (6).
Although immune checkpoint blockade (ICB) has become the front-line clinical treatment in patients with high mutational burden, such therapies are not used in many cancers with low mutation burden, such as glioma and leukemia (7, 8). Although historically attributed to tumor associated mutations, neoantigens can be produced from diverse post-transcriptionally regulatory mechanisms. Alternative splicing is one of the primary mechanisms used to achieve mRNA transcript and proteomic diversity in higher-order eukaryotes (9). In cancer, altered mRNA splicing can lead to aberrant protein products that promote oncogenic transformation, metastasis and confer chemotherapy resistance (10–13). Following their initial identification using proteogenomics approaches, splicing neoantigens have become increasingly recognized as a potent source of neo-peptides to potentially elicit immune response and induce cancer cell death (14).
Depending on the cancer, splicing neoantigens appear to often be the dominant source of tumor-specific peptides (15, 16). Such splicing events include intron retention, which typically results in nonsense-mediated decay, but which produce MHC-presented neopeptides that can be detected by Mass-spectrometry (MS) (17, 18). Such peptides require further experimental validation, as MHC presentation alone does not dictate the ability to mount a robust T-cell response (immunogenicity)(19). The prediction of such antigens, however, remains non-trivial, as splicing neoantigens must be degraded, bound and presented by specific cognate HLA alleles, and interact with patient-specific T-cell receptors on CD8+ T cells to induce an immune response. As such, the precise relationship between splicing neoantigen expression and patient prognosis has remained largely unknown and it is unclear whether overall splicing neoantigen burden impacts response to immunotherapy. Further, a concern for the use of splicing neoantigens as targets for therapy, is that the occurrence of a splicing event is often non-binary (changes in percent exon/intron inclusion), relative to mutations (present or absent), making it difficult to know which splicing events are truly tumor-specific. An alternative strategy to target tumor-specific splicing is to focus on events that specifically result in unannotated translated transmembrane proteins that might expose tumor-specific epitopes, bypassing the need to be presented by HLA. In principle, such peptides could be recognized by new CAR-T therapies which use B-cell receptors to bind epitopes (20) or selective monoclonal antibodies to mediate targeted tumor cell death. Although attractive, identifying such neo-isoforms requires an accurate prediction or measurement of full-length isoforms that do not undergo nonsense-mediated decay and result in properly folded protein structures that conserve the major domains of the reference protein. Given these challenges, no reusable and sufficiently comprehensive neoantigen prediction workflows exist, to unbiasedly and confidently identify splicing neoantigens that can be exploited by current immunotherapy strategies.
Here we performed a system analysis of splicing-neoantigens in cancer by creating Splicing Neo Antigen Finder (SNAF), an easy-to-use computational tool to identify, prioritize and interpret distinct classes of splicing-neoantigens. The workflow incorporates advanced deep-learning and probabilistic algorithms to discover immunogenic splicing neoantigens (SNAF-T workflow), full-length protein coding transmembrane tumor specific isoforms (SNAF-B workflow) and regulators of altered splicing (RNA-SPRINT). We demonstrate that splicing neoantigens in melanoma are frequently shared among patients, can predict survival and can be validated by multiple approaches: immunopeptidomics, targeted MS, MHC stabilization and T-cell reactivity assays, single-cell genomics, long-read isoform sequencing and neo-isoform transmembrane localization. These analyses show that splicing-neoantigens represent an untapped reservoir of shared targets for targeted cancer immunotherapy.
RESULTS
Inferring new classes of neoepitopes from RNA-Seq
To identify and characterize new forms of neoepitopes, we created two new computational workflows focused on T-cell and B-cell based therapies. T-cell based therapies include cancer vaccines, which require that target antigens are processed, presented by MHC and are immunogenic. B-cell based therapies, such as monoclonal antibodies, require the identification of transmembrane protein encoding neoantigens that will enable targeted approaches to selectively recognize cancer cells. SNAF was developed to recognize and prioritize both classes of neoantigens (SNAF-T and SNAF-B) in individual patient samples, while assessing the aggregate importance of each neoantigen at a population scale (Fig. 1A).
Figure 1. Automated discovery and confirmation of immunogenic and transmembrane splicing neoantigens with SNAF.
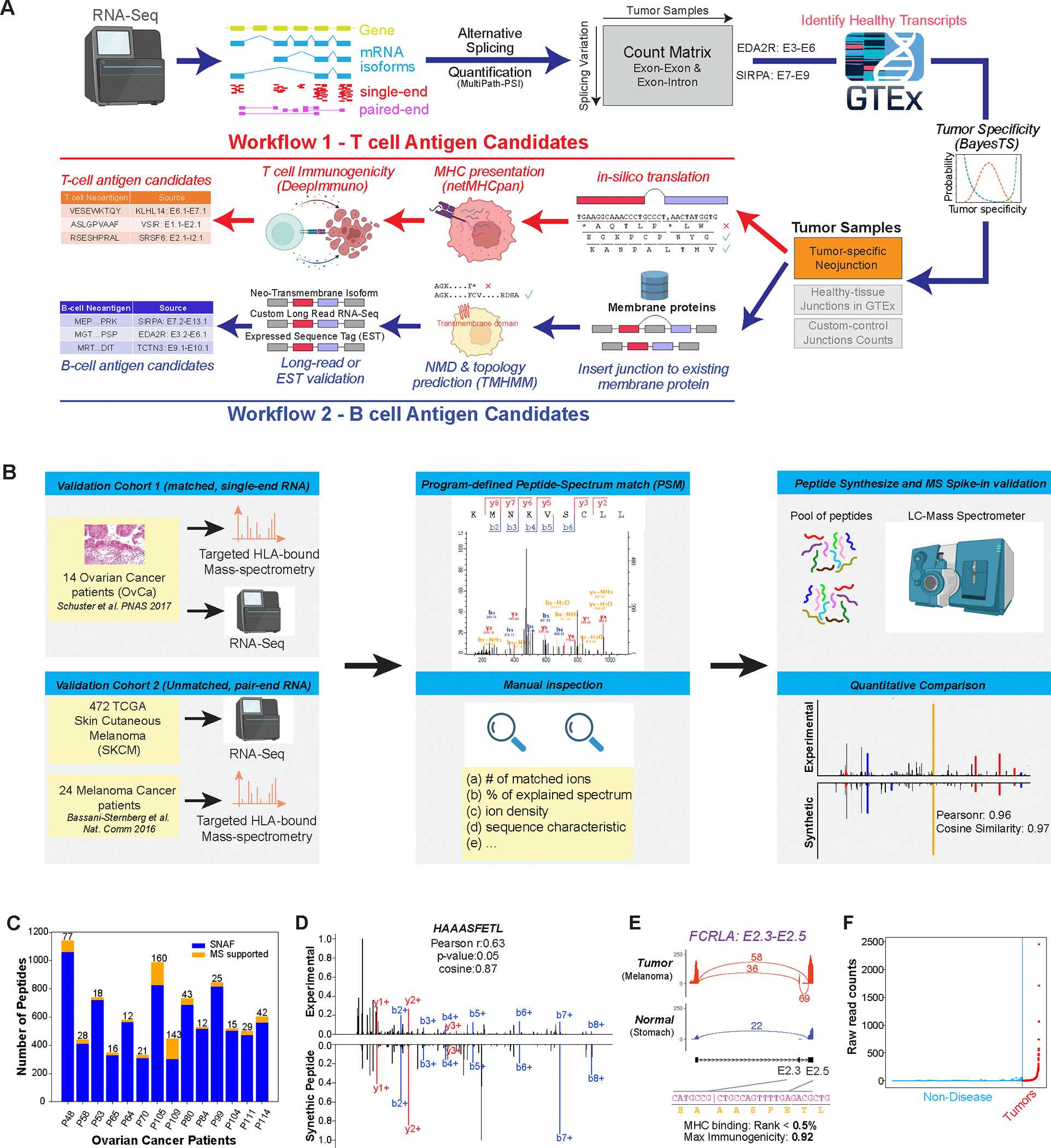
A) Outline of the two parallel workflows in the software SNAF to predict splicing neoantigens. SNAF begins with the identification and quantification of alternative splice junctions (exon-exon and exon-intron) from RNA-Seq BAM files and filters these against normal tissue reference RNA-Seq profiles (BayesTS). Retained tumor-specific splice junctions (neojunctions) are evaluated for T-cell (SNAF-T) and B-cell (SNAF-B) antigen production. SNAF-T performs in-silico translation of each junction, MHC binding affinity prediction (netMHCpan or MHCflurry) and identifies high-confidence immunogenic neoantigens through deep learning (DeepImmuno). SNAF-B predicts full-length protein coding isoforms that produce cancer-specific extracellular neo-epitopes (ExNeoEpitopes), considering existing transcript annotations and full-length isoform sequencing for targeted antibodies.
B) Validation workflow for Ovarian cancer and Melanoma immunopeptidomics with either matched or unmatched RNA-Seq. MaxQuant is applied to find Peptide-Spectrum Match (PSM), followed by quantitative and expert MS2 spectra prioritization. HPLC-MS/MS confirmation is performed on synthesized nominated neoantigens. C) Number of SNAF-T predicted neoantigens and those confirmed by immunopeptidomics across 14 of patients. D) Mirror plot of the immunopeptidomics and spike-in MS spectrum for HAAASFETL. The lines indicate mass-to-charge ratios for distinct types of fragmented ions (red/blue). E) SashimiPlot visualization of HAAASFETL, derived from an exon-exon junction in the gene FCRLA, along with the junction/peptide sequence, binding affinity and immunogenicity prediction. F) Raw read counts of the FCRLA neojunction between normal controls (blue) and TCGA melanoma cohort (red).
This workflow begins with user-supplied BAM files from tumor samples or cancer cell lines, followed by the identification and quantification of diverse classes of post-transcriptional regulation. In particular, the workflow applies a highly accurate approach for local splicing variation (MultiPath-PSI) from the AltAnalyze framework (21), to detect known and unannotated alternative splicing (cassette exon, 3′/5′ splice site exon, intron retention, alternative terminal exon, trans-splicing) and alternative promoter regulatory events, which would produce unique exon-exon or exon-intron junctions for in silico translation (Fig. S1). This approach has been benchmarked against diverse local-splicing variation (LSV) approaches, with methods to accurately quantify retained introns (22) (Fig. S2). The produced splice-junction/sample count matrix is compared against a MultiPath-PSI pre-processed database of normal human healthy tissues (GTEx and TCGA) to identify those that are tumor specific (23) (Fig. S3). Tumor-specific splice junctions can be analyzed in parallel with SNAF-T and SNAF-B. SNAF-T consists of: 1) HLA type prediction from sample FASTQ files, which are user provided, 2) in-silico translation, 3) MHC-binding prediction (NetMHCpan or embedded calls to MHCflurry) (24, 25), and 4) HLA-allele specific immunogenicity prediction (DeepImmuno) (19). SNAF-B consists of: 1) full-length isoform prediction for each tumor-specific splice-junction by augmenting existing isoform references, 2) exclusion of isoforms predicted to induce nonsense mediated decay (NMD), 3) transmembrane topology prediction, 4) long-read isoform sequence validation and augmented prediction (optional). For both workflows, a Maximum Likelihood Estimation and separate hierarchical Bayesian model (BayesTS) (26) are then applied to assess the tumor specificity of each neojunction in SNAF’s default and the optional custom healthy tissue reference RNA-Seq data with custom tissue weighting assigned by the user. Finally, to identify causal regulators of splicing neoantigen production, we developed RNA-SPRINT (RNA-based Splicing PRotein activity INference from multivariate decision Trees) to infer splicing factor activity directly from tumor RNA-Seq splicing profiles.
This workflow is unique in both its design and functionality (table 1). Unlike prior T cell based splicing neoantigen prediction approaches, SNAF is fully automated, supports any human genome version, has an embedded diverse database of healthy reference profiles (GTEx and TCGA), performs probabilistic tumor specificity modeling, quantifies splicing factor activities, identifies intron retention associated antigens and enables more accurate prediction of immunogenicity (19). SNAF-B provides independent evidence of tumor-specific transmembrane proteins (ExNeoEpitopes) that can uncover new extracellular epitopes for antibody recognition. As the program has a modular design with well-described Python classes, it can be customized to incorporate additional reference datasets for verification including control RNA-Seq, long-read sequencing and alternative algorithms such as MHC binding prediction.
Table 1.
Comparison of features in SNAF to other published splicing neoantigen workflows.
| SNAF | IRIS | ASNEO | NeoSplice | |
|---|---|---|---|---|
| Automated Program Features | ||||
| in-silico translation | ||||
| MHC binding predictions | ||||
| Tumor-specific expression | ||||
| Immunogenicity predictions | ||||
| Surface antigen predictions | ||||
| Parallization | ||||
| Interactive web app | ||||
| Interface to proteomics analysis | ||||
| Principal tumor specificy score | ||||
| Custom normal tissue reference | ||||
| Long-read validation | ||||
| Differential gene expression | ||||
| Gene-set enrichment | ||||
| Splicing factor activity prediction | ||||
| Advanced visualization | ||||
| Integrated survival analysis | ||||
| Stand-alone python module |
Validation of predicted MHC-bound neoantigens
We recently showed that our DeepImmuno workflow can predict immunogenic tumor neoantigens with up-to a 2-fold greater sensitivity than alternative approaches(19). To determine whether SNAF-T can identify bona fide MHC-presented splicing neoantigens, we selected two prior produced cancer immunopeptidome datasets to validate its predictions. First, we evaluated bulk RNA-Seq (single-end) and matched immunopeptidome profiling data (HLA-bound peptides) from 14 patients with ovarian cancer(27). To expand these predictions, we applied SNAF-T to skin cutaneous melanoma (SKCM) biopsy RNA-Seq from The Cancer Genome Atlas (TCGA) initiative (n=472), and unmatched immunopeptidome data from 24 patients with melanoma, herein referred to as the Bassani Sternberg cohort (28) (Fig. 1B, Data S1–4). In ovarian cancer, searching for MS spectra that map directly to neoantigens SNAF identified 46 splicing neoantigens with MS support per patient sample, on average, ranging from 12–160 antigens per patient (Fig. 1C). Including the normal human proteome, a total of 41 neoantigens, on average, were still identified (Data S4) We expect this number to be an underestimate, as HLA-bound peptides arise from non-specific protease cleavage, which alters the resultant MS spectra, and hence traditional tryptic-based search engines cannot confidently recover all neoantigens (29, 30). Here, the absolute number of MS supported splicing neoantigens is higher than previously reported (average 2 peptides per sample) using untargeted proteome data (CPTAC) (14), suggesting increased sensitivity of targeted immunopeptidome for identifying valid and rare neoantigens. As MS-based predictions are subject to inherent type-1 errors (31), we validated select ovarian and melanoma SNAF-T neoantigens using targeted MS on synthesized neopeptides. Specifically, we selected 14 ovarian and 22 melanoma peptides with high-confidence spectra (32). Of the 36 tested, 27 were sufficiently detected by MS (Data S5). Comparison of the synthetic to the original mass spectra found 11 matches, of which 7 were high-scoring based on all match criteria, with varying levels of confidence (Fig. 1D and Fig. S4)..
These seven neoantigens were derived from multiple mechanisms including known alternative exons, alternative 3’ and 5’ splice sites, intron retention and undocumented cassette exons (AltAnalyze defined). For example, the shared Melanoma splicing neoantigen HAAASFETL in the gene FCRLA occurs due to a known cassette exon-exon junction in an isoform that is weakly detected in blood and spleen (average read count = 0.51, BayesTS: 0.03) (Fig. 1E). Expression of FCRLA, which is a member of the F-receptor-like immunoglobulins, is correlated with good prognosis in Melanoma (33). FCRLA gene expression is only weakly tumor-specific (BayesTS: 0.13). However, this junction was detected in >34% of TCGA patients with melanoma (162/472, Average read count = 52.91) (Fig. 1F). The resultant mass spectra of this antigen had a high-confidence match (Andromeda score: 149.06, P-value: <0.0001), with a synthetic spectrum Pearsonr similarity of 0.63 (P-value: 0.05) and cosine similarity of 0.87. This peptide was only mapped to this melanoma specific FCRLA isoform in the original mass spectrum search, using the extended human isoforms proteome database (Fig.1D). The other mass spectrum confirmed neoantigens were derived from diverse protein families, including ubiquitin protein ligase complex (FBXO7), asparagine amidase (NGLY1), cytoskeletal motor protein (DYNLT5), negative regulator of RAS signaling (RASA3) and currently uncharacterized protein coding genes (C20orf204, C6orf52) (Fig. S4). We observed lower spectrum similarity scores in the Ovarian cohort (Pearsonr 0.55) compared to the Melanoma cohort (Pearsonr: 0.84), which we attributed to differences in the MS technology applied in the synthetic peptide MS data acquisition (Ion trap MS versus Fourier transform MS) and fragmentation methods (CID versus HID).
Splicing neoantigen burden predicts overall survival and response to immunotherapy
To broadly assess the clinical relevance of splicing neoantigens in a large cancer cohort, we applied SNAF to more than 500 melanoma patient biopsies with (Van Allen cohort) (34) and without immunotherapy (TCGA-SKCM) (35). For these analyses we separately considered the number of predicted neojunctions, MHC-bound peptides, immunogenic neoantigens and overall neoantigen burden, considering RNA-Seq determined HLA alleles for each patient. In the TCGA cohort of 472 samples, we found 528 tumor-specific splice junctions (neojunctions) per patient on average, ranging from 28 to 1,549. From these neojunctions, we predicted an average of 1,090 MHC-bound peptides, ranging from 75–2,981 peptides per patient. DeepImmuno predictions reduced the number of neoantigens to 915 on average (ranging from 74–2,486/patient), filtering out 16% of potentially non-immunogenic bound peptides, which is expected as all current immunogenicity approaches suffer from low precision (Data S6) (19).
To investigate the relationship between splicing neoantigen burden and clinical outcome, we performed survival analyses on both the TCGA and Van Allen cohorts (Data S6). This analysis found that patients in TCGA with high MHC-bound neoantigen burden trended towards poor overall survival (log rank P<0.05) (Fig. 2A). Conversely, we found that patients with a high neoantigen burden that received ICB (Van Allen cohort) had improved overall survival (log rank P=0.18). These trends were also reflected in neojunction and immunogenic peptide burden (Fig. 2B). Since the bulk RNA-Seq data profiled in these cohorts were obtained prior to therapy, one plausible explanation is that patients with high neoantigen burden exhibited frequent tumor immune escape, which was overcome by CTLA-4 inhibition. Examination of differential gene expression in TCGA patients with high splicing neoantigen burden versus low found 597 up- and 227 down-regulated genes (fold>1.5 and eBayes t-test p<0.05, FDR) (Fig.2C, Data S7). The top most-differentially up-regulated genes were those previously implicated in immune evasion such as ADAM10(36), PTPN11(37), TGFBR1(38), TNPO1(39), ANKRD52(40), MIB1(41), KIF3B(42). In contrast, down-regulated genes were markers of active tumor-immune cell infiltration, in particular plasma cells and T cells (Fig. 2C) with corresponding enrichment of this immune infiltration signature, along with genes involved in antigen binding and complement activation (Fig. S5A). Gene set enrichment of high burden induced transcripts identified significant upregulation of p53 signaling, mitotic cycle-cycle, cell motility, DNA damage response, fatty-acyl-CoA metabolism, lipid biosynthesis and Epithelial-Mesenchymal Transition genes, among others (Fig. 2D, Data S8). Many of these genes are directly associated with chemotherapy or radiotherapy resistance, in particular those that mediate DNA repair including ATM, RB1, RAD21 and acyl-CoA synthesis/ferroptosis such as ACSL3, ACSL4, FASN (43–49). Combination radiotherapy and immunotherapy have been proposed as a strategy to overcome single therapy drug resistance, which aligns with our observation of elevated immune evasion genes in high-burden groups (50). Thus, high splicing neoantigen patients with melanoma (TCGA) may represent chemo- and radio-therapy resistant tumors with upregulated immune evasion capacity, representing candidates for combination therapy. In contrast, low neoantigen burden patients with melanoma show evidence of immune reactivity, which may partially explain their favorable prognosis.
Figure 2. Splicing-neoantigen burden predicts response to therapy in Melanoma.
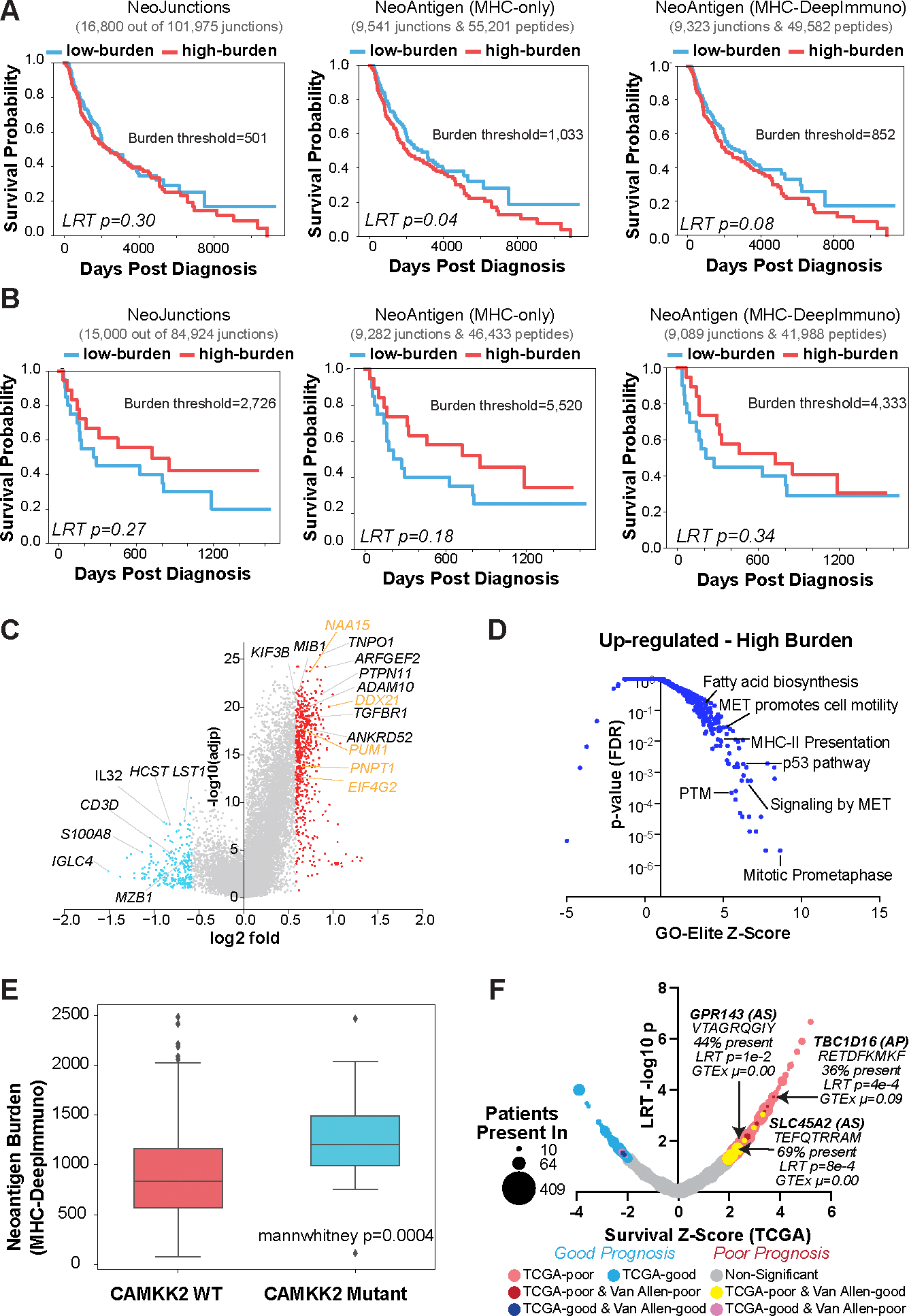
A,B) Kaplan-Meier (KM) survival plots of Melanoma patient samples stratified into low and high neoantigen burden, considering overall survival for each sequential step in SNAF for two cohorts (A) TCGA (n=472), and (B) Van Allen (n=39). These steps are: 1) tumor-specific neojunctions (left column), MHC-bound neoantigens (middle column) and immunogenic neoantigens (right column). Van Allen cohort RNA-Seq samples were subject to immune checkpoint inhibitors whereas TCGA were not. The number of neojunctions or Neoantigen peptides are shown at the top of each plot. C) Volcano plot of genes differentially expressed in patients with high versus low immunogenic splicing neoantigen burden in TCGA-SKCM, with a fold>1.5 and eBayes t-test P<0.05 (FDR corrected). Red = genes that are up-regulated in the high burden group; blue = down-regulated genes in the high burden; orange = representative RNA binding proteins. D) Gene-set enrichment with GO-Elite of ToppFun pathway gene-sets (AltAnalyze) for genes up-regulated in patients with high splicing versus low neoantigen burden(panel C). E) Immunogenic splicing neoantigen burden between patients in the TCGA Melanoma cohort with or without mutations in CAMKK2. Mann Whitney two-sided test. F) Bubble-plot of survival associated splicing neoantigens from SNAF in TCGA-SKCM. Dot size corresponds to the number of patients with melanoma that the splicing neoantigen is detected in (10–470) and are colored according to their survival significance in the TCGA-SKCM and Van Allen cohorts (LRT P<0.05 and z ≥ 1). AS = alternative splicing.
CAMMK2 mutations correlate with increased neoantigen burden in melanoma
To determine whether splicing neoantigen burden in patients with melanoma is associated with specific mutations, we compared mutations in high- versus low-burden neoantigen patients. Although no individual mutations achieved an FDR corrected P<0.05, multiple mutations trended towards significance with the most enriched mutations in the gene CAMKK2 versus wildtype (Mann-Whitney test P=0.0004, non-adjusted) (Fig. 2E, Data S9). Out of 19 patients with reported CAMKK2 mutations, 13 were found to occur in the high burden group, out of a total of 222 patients. Mutation or inhibition of CAMKK2 is known to lead to increased anti-PD1 immunotherapy efficacy (51). This effect is believed to occur as a result of CAMKK2’s ability to negatively regulate ferroptosis, a mechanism of cell death that is induced by iron-dependent lipid peroxidation, through the AMPK–NRF2 pathway(51). Our results suggest that CAMKK2 mutations may contribute to splicing neoantigen burden and immunotherapy outcome, either through direct or indirect splicing regulatory networks, in a subset of patients with melanoma.
Individual splicing neoantigens can predict response to immunotherapy
In addition to neoantigen burden, we compared the individual splicing profiles of SNAF immunogenic neoantigens with patient survival. This analysis identified 2,970 parental junctions associated with poor overall survival in TCGA (likelihood-ratio test (LRT) P<0.05 and z ≥ 1) (Fig. 2F and Data S1). Among these neoantigens, we noted that a subset (n=108 junctions) were present in over 15% of patients (shared neoantigens), suggesting these represented new potential survival biomarkers. Although a much smaller number of neoantigens were associated with survival in the Van Allen cohort due to the limited sample size, we found 1,755 poor and 227 good prognosis associated neoantigen junctions (LRT P<0.05 and z ≥ 1 or z ≤ 1, respectively). We observed 7 unique neoantigen junctions in TCGA and Van Allen associated with opposite survival associations (poor in TCGA and good in Van Allen) and present in >10 TCGA patients (Fig. 2F). For example, a splicing neoantigen in the melanin synthesis associated glucose transporter SLC45A2 (TEFQTRRAM), was detected in 212/472 patients from the TCGA SKCM cohort, was associated with poor overall survival in TCGA (Wald p <0.05, z-score>2), and was associated with good overall survival in the Van Allen cohort (Wald p<0.05, z-score<−2), suggesting that it may represent a relatively common target for therapy and prognosis (Fig. 2F). Other overlapping neoantigens produced from this same junction were present in over >64% of patients in both melanoma cohorts (FQTRRAMTL), with differences in their abundances due to varying MHC-I allele binding and immunogenicity preferences. Dozens of other shared neoantigens were associated with poor prognosis in TCGA and trending towards good prognosis in the Van Allen cohorts (Data S1). The occurrence and tumor specificity of these and other similar neojunctions were confirmed by Integrated Genome Viewer (IGV) visualization, and frequently associated with alternative splice sites in conjunction with tumor-specific gene expression (Fig. S5B). These data reveal that individual shared splicing neoantigens can predict outcomes in patients prior to and after ICB.
Disrupted splicing factor activity underlies high splicing neoantigen burden
The coordinated regulation of alternative splicing and alternative promoters is orchestrated by the expression or activity of conserved cis- and trans-regulatory interactions, such as splicing factor binding to RNA recognition elements. Splicing neoantigens are likely produced by modulation of such interactions, driven by direct mutations or RNA-editing that result in unique tumor specific mRNAs. The most likely immediate mediator of differential splicing neoantigen burden is a change in the expression or activity of one or more splicing factors. Examination of differential gene expression in the high versus low splicing neoantigen group found many upregulated splicing regulators (Fig. 2C, Data S3). These included 117 mRNA splicing regulators, upregulated with a fold > 1.2 and eBayes t-test p<0.05, FDR (Data S7). Patients with mutations in SF3B1 were further enriched in the high-burden group (Mann Whitney P=0.02). Given this finding, we asked whether splicing factor activity was also increased with neoantigen burden. To infer splicing factor activity we turned to existing methods for transcription regulatory network inference, which rely on a defined set of target genes or regulons (52, 53). The up- and down-regulation of these regulatory targets can indirectly provide evidence of transcription factor activity. To establish a reliable link between splicing factors and downstream splice junctions in an analogous manner, we used a large dataset of RNA-Binding Protein (RBP) knockdowns (n=191) in K562 cells from the ENCODE project. By observing the changes in splicing events upon the knockdown of a specific RBP and accounting for batch effects, we derived initial splicing regulatory targets. To refine these targets, we further incorporated evidence of direct RBP-target regulation using eCLIP sequencing (CLIP-seq) data for 120 RBPs in the K562 cell line. The resulting data were used to construct a prior network to infer splicing factor activity (Fig. 3A).
Figure 3. Regulatory networks mediating splicing neoantigen burden in Melanoma.
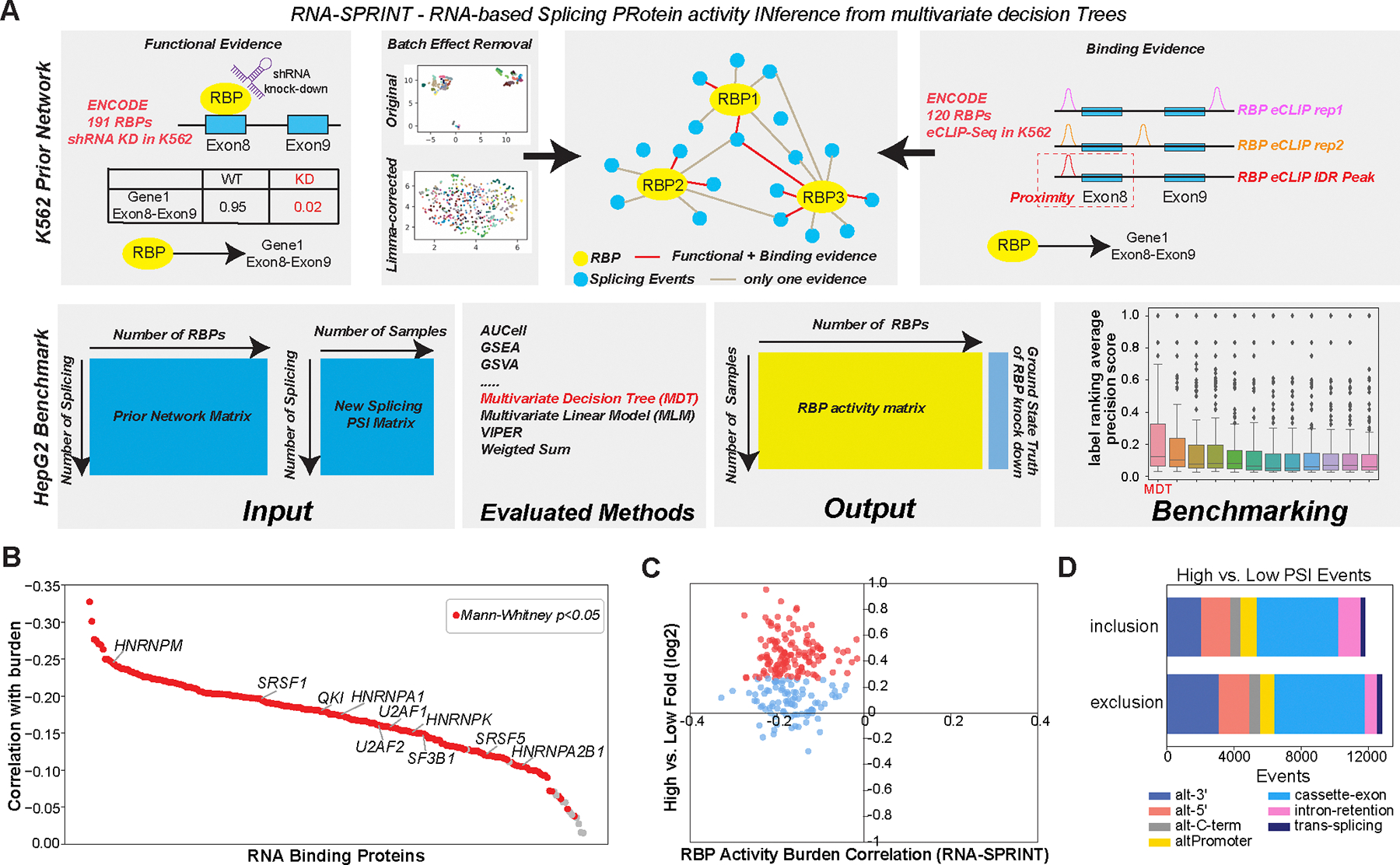
A) Schematic overview of the software RNA-SPRINT and associated benchmarking steps. The workflow involves construction of an RNA Binding Protein (RBP) prior network to predict splicing regulatory interactions. Evaluation of the method is overviewed, consisting of RNA-SPRINT benchmarking relative to 12 transcription factor (TF) activity methods in HepG2 cell line RBP knockdown RNA-Seq datasets. B) The correlation of inferred RBP activity with splicing neoantigen burden for all TCGA patients with melanoma. C) Comparison of RBP activity-burden correlations with RBP differential gene expression, for high versus low burden (TCGA SKCM). Red = upregulated genes (fold>1.2 and eBayes t-test p<0.05, FDR corrected) in high burden. D) Type of splicing events observed with exon/intron inclusion or exclusion comparing high versus low burden.
We found that a Multivariate Decision Tree (MDT)-based method produced the most accurate RBP activity predictions, compared to 12 other commonly used transcription factor (TF) activity inference methods, when considering 116 RBP knockdown splicing profiles from HepG2 cells (ENCODE) (Fig. S6A–C and Data S10). Application of this approach, which we call RNA-SPRINT, to all TCGA melanoma cohort splicing profiles, revealed that 209 out of 221 RBPs had reduced activity in the high versus low splicing neoantigen group (Mann-Whitney P<0.05) (Fig. 3B and C, Data S10). Although contrary to the observed upregulation of these RBPs, these data suggest that there exists a coordinated failure to properly splice transcripts, as previously described (54)(55). This result is further supported by an observed increase in intron-retention for patients in the high-burden group (failure to excise introns) (Fig. 3D). It is also possible that broad upregulation of splicing factors is a compensatory mechanism to counteract global splicing defects or simply a byproduct of cell-type heterogeneity within the tumor such as immune infiltration.
Shared splicing neoantigens are presented by MHC and display amino acid compositional bias
One observation from our SNAF-T analyses was the co-occurrence of splicing neoantigens among many patients. We found 940 T-cell neoantigens predicted in >15% of patients in TCGA cohort (shared) (Fig. 4A). Although a much smaller cohort, we found 439 of 8,422 shared neoantigens in the Van Allen cohort, overlapping with the 940 in TCGA. TCGA shared neoantigens were found to more frequently result from cassette exons as compared to unique neoantigens, which was only found in one patient (Fig. 4B). Genes with shared neoantigens were significantly enriched in gene sets for melanocyte biology, such as melanocyte differentiation (P = 9.3 e-4, FDR), melanin biosynthetic process (P = 3.4 e-3, FDR) and cell division (P = 1.0 e-4, FDR) (Fig. 4C, Data S11).
Figure 4. Shared splicing neoantigens are frequently detected by MS and are defined by their sequence composition in Melanoma.
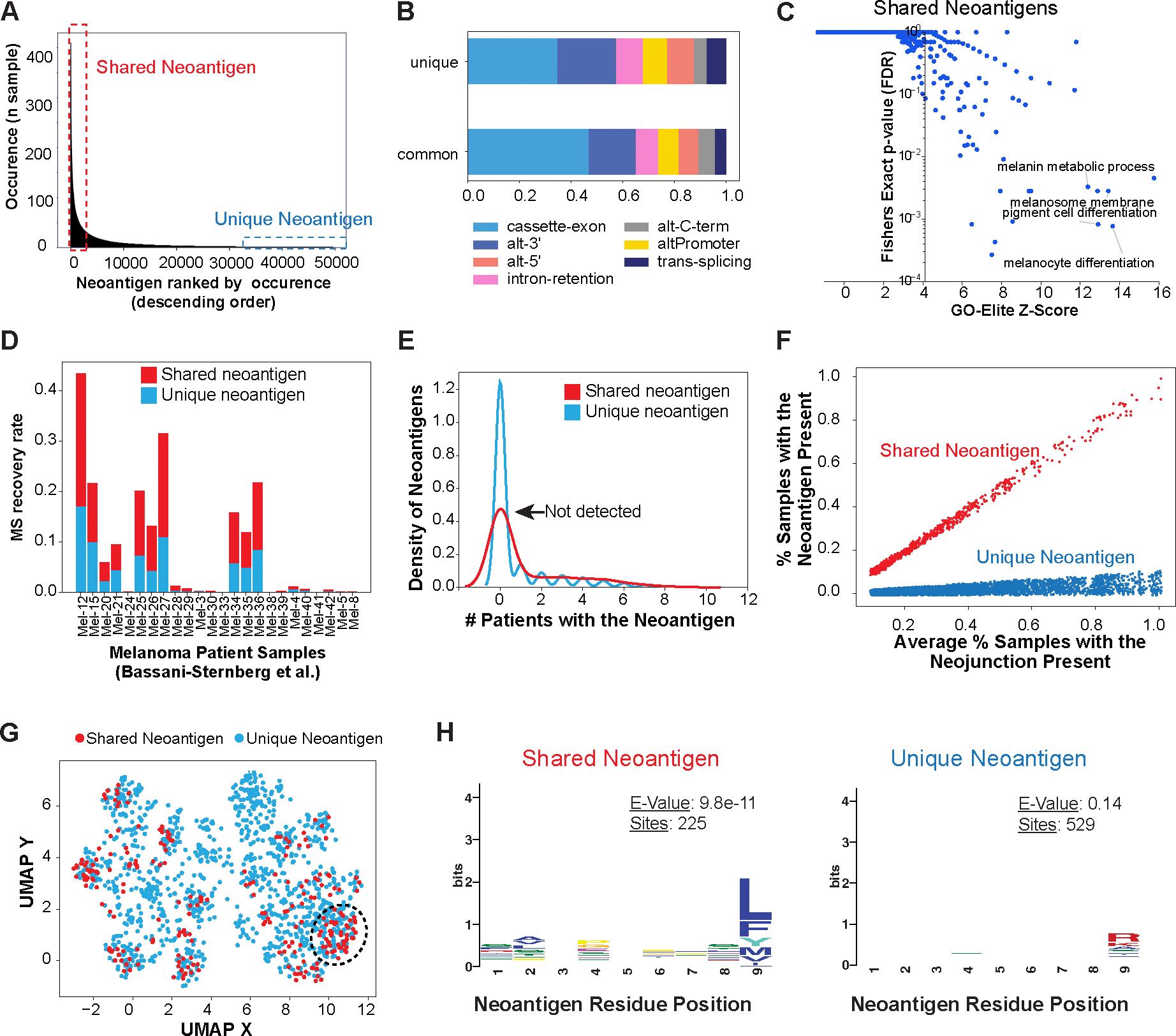
A) Identification of common (shared) and unique immunogenic splicing neoantigens in the TCGA Melanoma cohort, based on their frequency of occurrence among patients. B) Frequency of splicing-event types for shared and unique splicing neoantigen junctions in TCGA. C) Gene-set enrichment with GO-Elite of the Gene Ontology and pathways of shared neoantigens (present in >15% of patients with melanoma). D) MS recovery rate in an independent melanoma immunopeptidome dataset (Bassani-Sternberg et al.) between shared and unique neoantigens considered in the query database. E) Kernel density estimate plot comparing the observed occurrence in an independent immunopeptidomics MS experimental cohort, for all detected shared (>15% of patients with melanoma) versus unique splicing-neoantigens. F) Re-defined shared and unique neoantigens in TCGA by normalizing the occurrence of their parental splice junction, leveraging their respective observed amino acid bias. G) UMAP of splicing neoantigens based on their amino acid physiological properties in TCGA, highlighting neoantigens that cluster based on shared amino acid physicochemical features. H) Distinct enriched amino acid motifs (MEME), comparing shared versus unique neoantigens.
As shared splicing neoantigens are rare relative to all unique predicted neoantigens, we would expect such peptides to be more frequently detected using immunopeptidome profiling in an independent cohort. Hence, we again used a previously published Melanoma dataset of HLA-I bound peptides detected by MS in 24 independent patients (Bassani Sternberg cohort) (28). Considering both shared (n=613) and unique (n=16,753) peptides in our reference peptide database (combined unique reference database, excluding peptides in UniProt), we found that shared splicing neoantigens could be found at a higher rate than unique neoantigens (paired t-test, P=0.006) (Fig. 4D). The higher recovery rate was still observed even with the inclusion of the human normal proteome (Data S2). Additionally, of the 613 shared neoantigens examined, we found immunopeptidome evidence for 34% (n=210), with 98 shared neoantigens evidenced in at least 15% of patients by immunopeptidome analysis in Bassani-Sternberg cohort, as illustrated using kernel density estimates or estimator of the empirical cumulative distribution (eCDF) function (Fig. 4E, Fig. S7A). Inspection of neojunctions produced from these MHC-presented neoantigens confirmed that they were tumor-specific (Fig. S7B).
Whereas HLA genes are highly polymorphic and have different binding preferences for neoantigens, the existence of shared splicing neoantigens suggests that diverse HLA genotypes may bind in a more promiscuous manner to these peptides versus those present only in one or few individuals. To evaluate the potential recurrence of amino acid sequences, we first redefined the shared and unique neoantigens by normalizing the frequency of their parental splice junctions (Fig. 4F). Since 9-mer neoantigens had a balanced number of shared and unique neoantigens, we focused specifically on these peptides. To derive a physicochemical profile of the amino acids associated with each, we encoded each 9-mer as a numerical vector based on all amino acid physicochemical parameters in the AAIndex database (566 parameters) and projected each neoantigen vector as a point in UMAP space (Fig. 4G). Whereas the majority of shared and unique splicing neoantigens have similar physicochemical characteristics, a few empirically observed clusters were enriched in shared versus unique neoantigens (circled regions, Fig. 4G). To find preferentially detected amino acids present in the shared and unique neoantigens, we performed motif analysis with MEME(56) in these two sets. MEME finds that shared neoantigens frequently end in Lysine and Phenylalanine (e-value =9.8e-14), whereas the most dominant motif found in unique neoantigens disproportionately end in Arginines (e-value=0.14), the less significant E-value also suggested the more diverse binding modes for unique neoantigen (Fig. 4H). We observed near identical motif enrichments for shared versus unique splicing neoantigens in the Van Allen cohort (Fig. S7C). These data suggest that amino acid sequence bias could be used to find shared neoantigens that are bound by most MHC alleles.
Splicing neoantigens stabilize MHC and elicit T cell responses
To demonstrate that such splicing neoantigens represent viable targets for broadly applicable immunotherapies, we experimentally verified their ability to directly bind MHC and induce T-cell reactivity. For validation, we selected 5 shared splicing neoantigens derived from three neojunctions in PMEL, SLC45A2 and CDH19. Although PMEL is an existing target for immunotherapy in melanoma (NCT00509496), the predicted splicing neoantigen has not been previously described (unannotated exon-exon junction). These neoantigens were selected based on availability of HLA matched donor cells, high neoantigen frequency in melanoma and tumor specificity. A MHC stabilization assay using TAP deficient HLA-A*02 containing T2 cells, confirmed the binding of two neoantigens to HLA-A*02 (Fig. 5A–B). To assess immunogenicity, we leveraged HLA-typed healthy blood peripheral blood mononuclear cells (PBMC) to prime T cells and tested IFNγ responses upon pMHC stimulation (Fig. S8). Frozen PBMC were primed 3 times with autologous peptide loaded Dendritic Cells (DCs) and thereafter tested for their IFNγ response against single HLA expressing 721.221 (221) cells, to ensure immunogenicity was specific for a single HLA genotype. The 5 neoantigen peptides loaded on 221 cells expressing their respective allele, generated similar IFNγ responses compared to known immunogenic FLU and/or HCMV peptide antigens in 3 donors (Fig. 5C–D). Only weak IFNγ responses were detected in unstimulated T cells and in response to 221 without peptide, showing peptide specificity. Thus, these analyses provide strong evidence of T-cell immunogenicity by all 5 shared splicing neoantigens tested. These results are in contrast to previously described neoantigen prediction workflows that reported far lower validation accuracies (57, 58).
Figure 5. Shared splicing neoantigens bind HLA and induce T-cell reactivity.
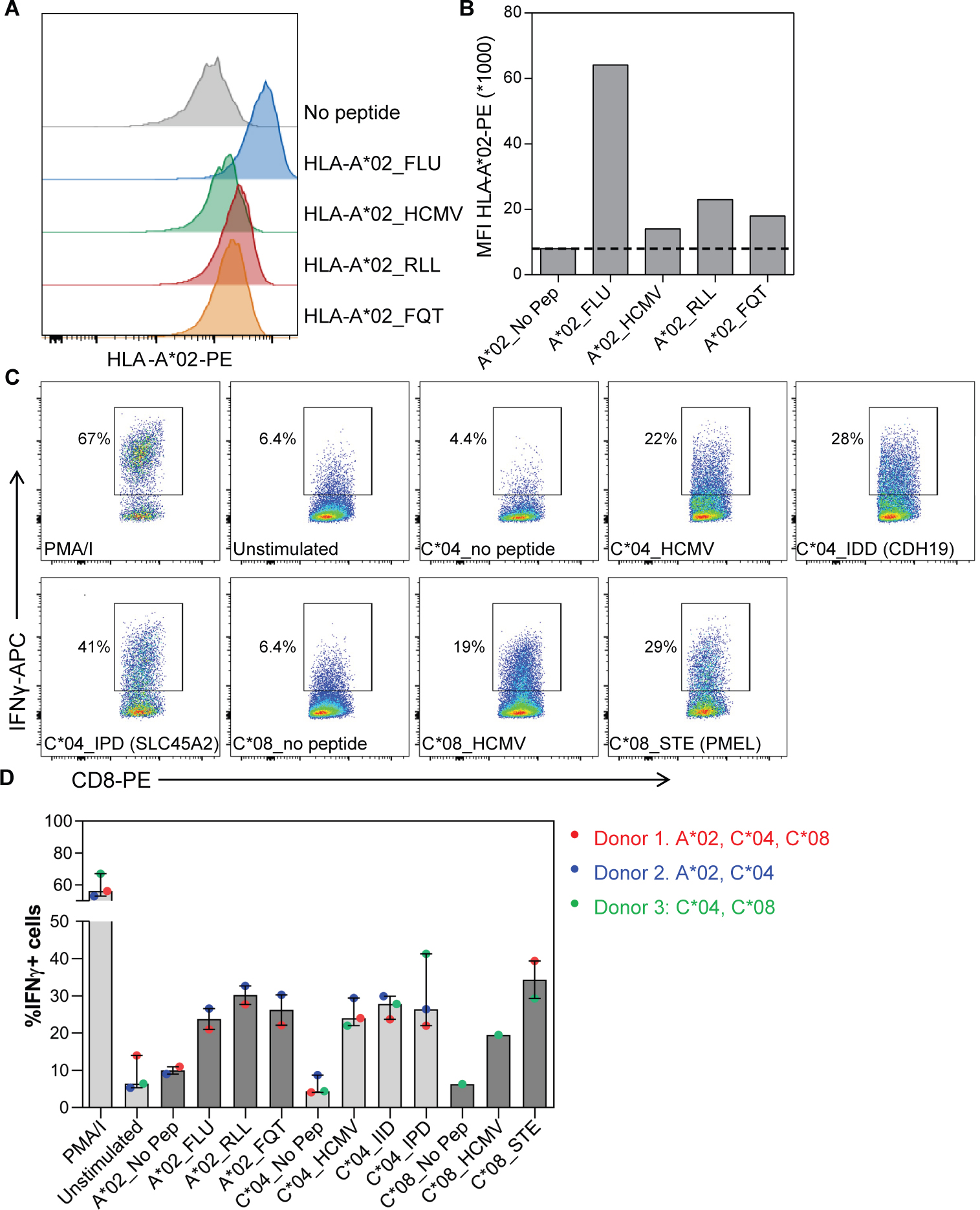
(A) Histograms and (B) graph show HLA-A*02-PE staining on HLA-A*02 containing TAP deficient T2 cells without peptide (no pep), loaded with FLU and HCMV control peptides and RLLGTEFQT (RLL) and FQTTRRAMTL (FQT) peptide neoantigens. MFI = median fluorescence intensity. PE = Phycoerythrin conjugated antibodies. C) Dot plots and (D) graph show the percentage of Interferon gamma-positive (IFNγ+) CD8+ T-cells in response to 5 melanoma shared splicing antigens compared to negative (unstimulated, no pep) or positive (PMA/I, FLU, HMCV) controls. CD8+ T cells were primed using peptide loaded monocyte derived dendritic cells and thereafter tested against 721.221 cells selectively expressing the indicated HLA allele with and without peptide loading. Bars indicate median of 2–3 donors and lines interquartile range.
Shared splicing neoantigens derive from tumor cells rather than the tumor microenvironment
Although bulk tumor RNA-Seq enables the detection of splicing neoantigens it cannot clarify the precise cellular source. Emerging data suggest that neoantigens can also be derived from the tumor microenvironment including immune cells (59). To determine which cell types splicing neoantigens derive from, we re-analyzed single-cell (sc)-RNA-Seq from tumor biopsies from 17 patients with melanoma (4,454 cells) (60). As these data were profiled using SmartSeq2 chemistry, we were able to detect over 340,000 exon-exon and exon-intron junctions present throughout the gene body, for 7 prior annotated cell-types (tumor, endothelial, cancer associated fibroblasts, B cell, T cell, natural killer cells and macrophages). This analysis found only a small proportion of scRNA-Seq detected junctions were enriched in tumor cells >2 fold (n=30,523) versus those enriched in immune cells (n=236,954) (Fig. 6A). Considering all TCGA predicted melanoma neojunctions in this dataset, we find roughly equal immune and tumor enriched neojunctions (1,289 and 979, respectively). However, restricting this analysis to shared splicing neoantigens in >15% of patients, we found these neojunctions were almost entirely derived from tumor cells (n=195) versus immune cells (n=10) (Fig. 6B, Data S12). Tumor specific neojunctions included experimentally confirmed splicing antigens (SLC45A2, CDH19, FCRLA) (Fig.6C). Thus, the tumor microenvironment appears to be a contributor to splicing neoantigen burden, but not a primary source for shared neoantigens.
Figure 6. Splicing-neoantigen cell of origin is dependent on its mechanism of regulation.
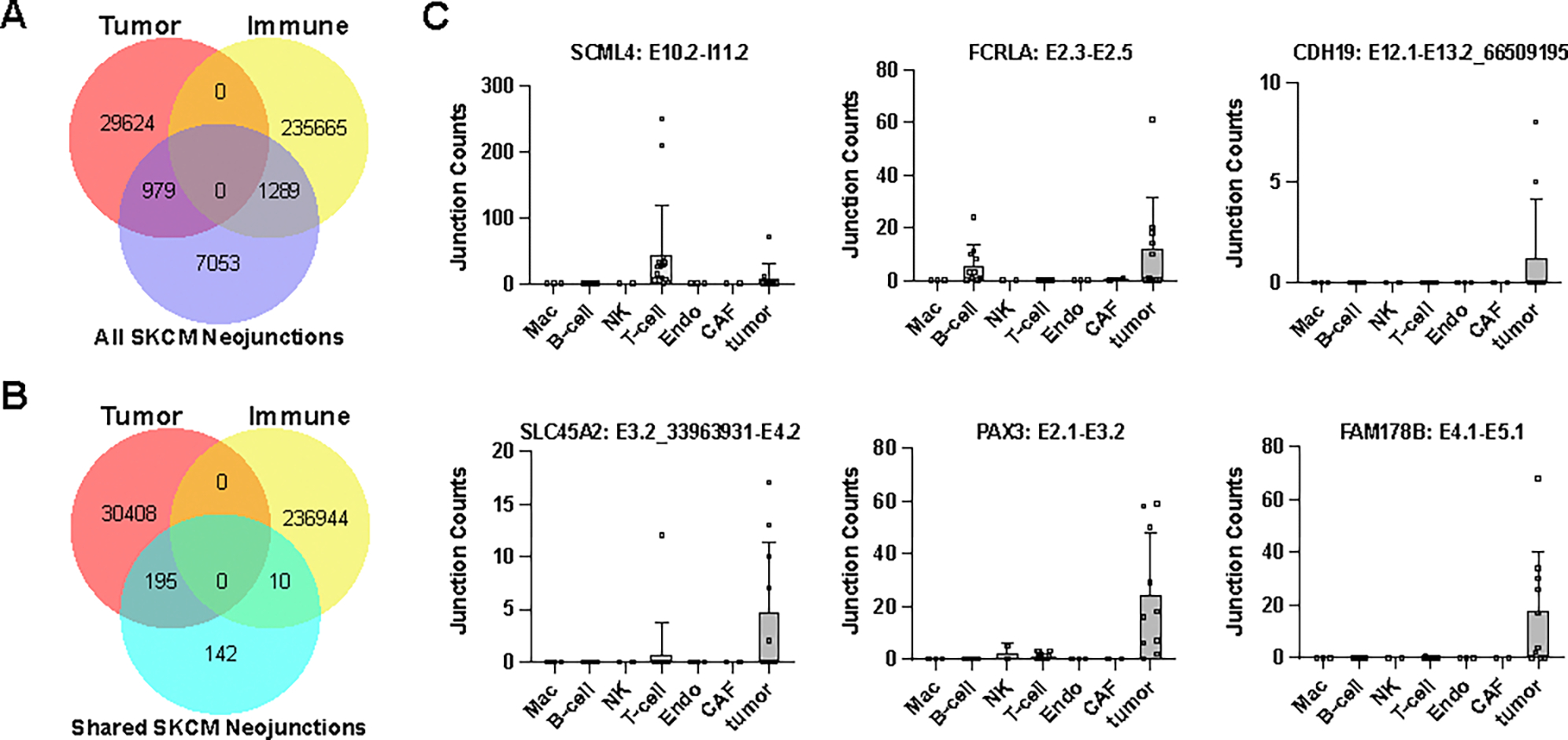
A,B) Venn diagrams comparing the number of parental neojunctions for TCGA SKCM splicing neoantigens unique to a single-patient (A) or shared in >15% of patients (B) to the specific cell-types they derive from in independent melanoma tumor biopsies by single-cell RNA-Seq analysis. Neojunctions are defined as tumor or immune if they are >2 fold enriched in either cell-population (absolute number of reads in all patients and cells for each lineage). C) Neojunction expression in individual cell populations for select shared splicing neoantigens. Each dot denotes the combined neojunction read counts in a single patient (n=19) with melanoma, separately per cell annotated cell population.
To ensure that such neoantigens are not only a byproduct of a normal cellular proliferative program, which would limit their therapeutic by presenting a potential side-effect, we analyzed multiple in vitro bulk RNA-Seq datasets, in which human epidermal cells were induced to proliferate or do so naturally (embryonic). We found proliferation associated junctions only accounted for a small percentage (~6%) of all TCGA melanoma identified neojunctions (Fig S9A–C). Neojunctions from shared splicing neoantigens were only frequently observed in one out of 170 proliferative samples, specifically from 16-week fetal fibroblasts, but not from other fetal fibroblasts profiled (61) (Data S13). Furthermore, these overlapping melanoma neojunctions were only weakly detected (1–50 reads) in any cultured epidermal sample and were associated with cell cycle regulation (Fig. S9D and Data S14). Thus, non-malignant melanocyte proliferation is not a dominant source of splicing neoantigen production.
SNAF accurately predicts full-length mRNAs and stable proteoforms
Several SNAF-T splicing neoantigens were those that occur in transmembrane proteins, such as GPR143 and SLC45A2, which occur due to undocumented in-frame alternative splice-sites (5′ or 3′). In principle, such splicing events could result in undescribed cell-surface expressed proteins. As an alternative source of neoantigens that do not require degradation, MHC presentation, and T-cell receptor recognition, we applied SNAF-B to the same TCGA melanoma cohort. The SNAF-B workflow can be used to identify ExNeoEpitopes that have retained transmembrane domains, but altered N-terminal or other extracellular sequences that could serve as new epitopes for specifically designed monoclonal antibodies. To predict full length isoforms, unannotated exon-exon or exon-intron junctions (not in the Ensembl or UCSC mRNA database), are inserted into the best matching isoform models based on exon composition, followed by in silico translation (Fig. 7A). This workflow can optionally exclude predicted mRNAs expected to result in NMD and selectively include those that have high-confidence transmembrane domains based on a prior published Hidden Markov Model based topology prediction approach (TMHMM) (62). Putative ExNeoEpitopes that result in deleted or new extracellular polypeptides can be assessed using the SNAF-B interactive viewer.
Figure 7. SNAF-B finds full-length mRNAs and stable-proteoforms for targeted therapies.
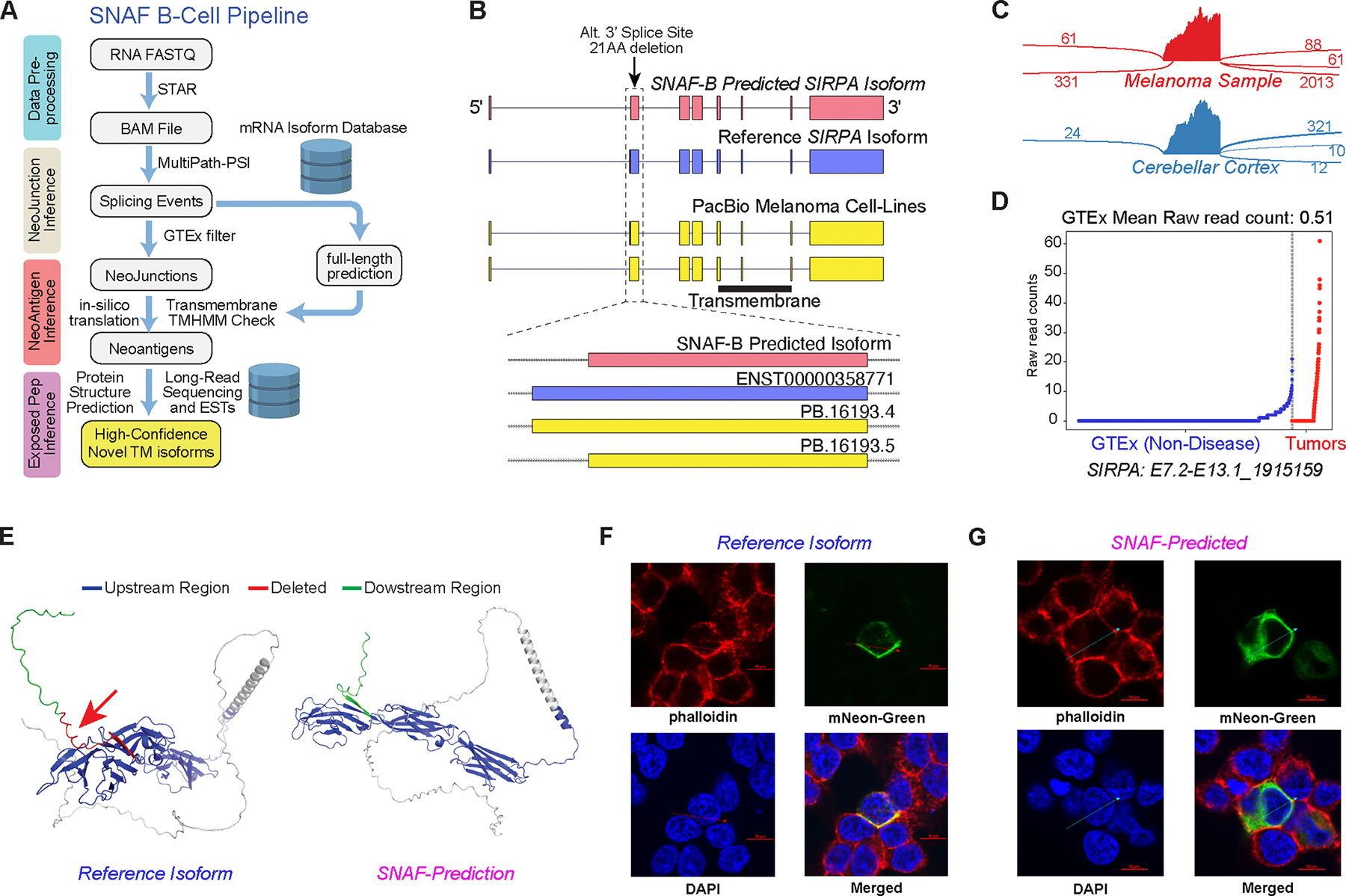
A) Overview of the SNAF-B prediction workflow to define ExNeoEpitopes. The workflow begins with bulk RNA-Seq datasets and optional long-read sequencing data integration to produce results with multiple levels of in silico evidence. B) Comparison of a SNAF-B predicted full-length isoform in the transmembrane protein SIRPA to documented mRNA isoforms and those predicted from PacBio long-read IsoSeq of melanoma cell lines. C) SashimiPlot of alternative 3’ splice site selection in Melanoma and Brain RNA-Seq for SIRPA. D) Specificity of the indicated SIRPA ExNeoEpitope for TCGA melanoma samples versus an integrated healthy controls tissue database (GTEx + TCGA). E) Alphafold2 3D modeling of the reference isoform and the long-read verified ExNeoEpitope. Arrow denotes the deleted region in the alternative isoform. F,G) Co-localization of the SIRPA reference (F) or Melanoma-specific (G) splice isoform by confocal microscopy with a cell surface stain (phalloidin). The arrow indicates the cross-section used to quantify fluorophore spatial coincidence.
This analysis found 378 initial ExNeoEpitopes in the TCGA melanoma cohort, using prior well annotated transcripts as reference models (Ensembl, UCSC mRNAs) (Data S15). To initially assess the validity of such ExNeoEpitopes, we performed long-read RNA isoform sequencing (“Iso-Seq”) in four commonly used melanoma cell lines using the PacBio Sequel II platform. This Iso-Seq analysis found 17 of our predicted ExNeoEpitopes isoforms that perfectly matched our in silico predictions and an additional 20 that partially matched (overlapping neojunction) (Data S15). One example was an undocumented alternative 3′ site in Signal Regulatory Protein Alpha (SIRPA), which resulted in an in-frame deletion of the 5′ end of exon 13, missing 21AA. The predicted full-length isoform was directly evidenced by Iso-Seq (Fig. 7B).
In addition to predicting full-length isoforms SNAF-B can match short-read identified junctions to other supplied isoform models including long-read or Expressed sequence tags (EST). Matching exon-exon and exon-intron junctions from the TCGA melanoma cohort to a publicly available PacBio Iso-Seq dataset of 10 commonly used cancer cell lines (Universal Human Reference), we found 1207 additional full length neo-isoforms associated with junctions not detected in GTEx (Data S15). This included the same SLC45A2 neojunction, predicted by SNAF-T to be associated with poor survival, shared in 69% of patients with melanoma, found to be MHC-presented (MS) and only expressed in tumors (Fig. S10A,B). This alternative 5′ donor site resulted in the deletion of 80AA which disrupted the 6th transmembrane domain. The deleted region (AA 215–295) appeared to be composed of a transmembrane segment (AA 215–237) and a cytoplasmic segment (AA 237–295) in the reference protein (Fig. S10C,D). As a result of the deletion, a region of polypeptide sequence normally positioned at the cytoplasmic face of the membrane was now predicted to reside in the extracellular domain, representing a potential new neoepitope for CAR-T therapy.
Considering all SNAF-B short- and long-read predictions from 10 tumor and 5 melanoma cell lines together, we ran SNAF-B to identify a total of 514 unique ExNeoEpitope proteins (Data S15). We filtered these to 187 predictions in which the neojunction overlapped with the extracellular domain (UniProt) and was not contained within any other Ensembl or UCSC protein isoforms. In addition to proteoforms with missing polypeptide sequences, we identified 12 initial candidate long-read supported isoforms with high GTEx evidence of tumor-specificity that result in the inclusion of undocumented alternative first or cassette exons. These were initially identified through manual inspection of BLAT sequence matches to the human genome and mRNA transcript databases (UCSC and Ensembl) and biased to ExNeoEpitopes detected in >15% of patients with melanoma. Visualization in the SNAF-B viewer found that 5 out of these 12 ExNeoEpitopes result in new inserted polypeptide sequences that impact a cytoplasmic region of the protein (OCA2, SLC2A10, TMEM9, IL13RA1, ATP13A1), one occurring within a transmembrane domain (ANO10) and 5 predicted to alter the extracellular region of the protein and result in stable transmembrane predictions (DCBLD2, NALCN, MET, SEMA6A, IGSF11). Although orthogonal long-read RNA sequencing indicated the validity of these transcripts from neojunction inference, it is possible such isoforms are not properly folded or inserted into the cell membrane. First, to initially show the ability of these isoforms to produce functional transmembrane proteins, we predicted 2D and 3D protein structures for the tumor specific and reference mRNA isoforms using Protter (63) and Alphafold2 (64), respectively. In each of the examples, we observed high-confident 2D and 3D structures of both the tumor and reference isoforms, suggesting that the deleted or inserted polypeptide sequence selectively impacted the extracellular portion of these proteins (Fig. 7 and Fig. S10–12). Finally, we tested the ability of ExNeoEpitopes with distinct in silico evidence to traffic to the plasma membrane. Specifically, we synthesized and transfected C-terminal fluorescent tagged cDNAs for three ExNeoEpitopes that gained new peptide sequences (SEMA6A, ANO10, IGSF11, DCBLD2) and three isoforms with deletions (SIRPA, MET, SLC45A2), along with the reference isoform for each. The synthesized reference isoforms for 5 out of the 7 ExNeoEpitopes (SEMA6A, SIRPA, MET, SLC45A2, DCBLD2) were expressed and found to co-localize to the transmembrane using a selective cell surface stain in HEK-293 cells. Confirming their expression and trafficking, we could also observe partial transmembrane localization in 4 of 7 of the evaluated ExNeoEpitopes predicted by SNAF-B (SEMA6A, SIRPA, MET, SLC45A2) (Fig. 7F,G and Fig. 12B–F). These data illustrate the potential of tumor-specific splice isoforms as higher precision candidates for CAR-T or monoclonal antibodies over existing targets, which include conformational epitopes that impact protein structure (65, 66).
Interactive Neoantigen Web Explorer
To facilitate the exploration and prioritization of the predicted neoantigens from SNAF, we developed two interactive web applications to visualize both T-cell and B-cell neoantigens (Figure S13). The SNAF-T viewer allows users to explore different global neoantigen features, including amino acid length, and frequency within a cohort, in a projected 2D UMAP space. Here, each neoantigen is embedded based on its physicochemical properties. Users can manually select clusters to identify enriched web logo AA motifs and explore individual neojunctions and neoantigens (Supplementary Movie). The SNAF-B viewer can be used to interactively visualize GTEx and tumor counts for neojunctions, generate protein sequence alignments between a putative ExNeoEpitope and selected reference proteins, compare protein feature composition, secondary structure and solvability prediction(67), and perform topology modeling (62). Hence, these tools can be used to select optimal targets for experimental validation.
DISCUSSION
The identification and prioritization of shared neoantigens within and across cancers provides the potential to lead to new targeted immunotherapies. However, the development of existing targeted neoantigen immunotherapies is time-consuming and costly, as they must exploit specific MHC-presented mutations that are evidenced by precision proteomics and immunogenicity assays (68). Our study provides evidence that aberrant splicing in melanoma frequently results in shared MHC-presented neoantigens, which can be confirmed in different patient cohorts and used to predict survival and response to immunotherapy. Patients with high splice neoantigen burden skew towards poor outcomes and associate with genes important to block immune-tumor recruitment. Further, Although prior computational strategies for splicing neoantigen discovery have been proposed, SNAF is unique in its inclusion of probabilistic modeling to quantify immunogenicity and tumor specificity, interactive exploratory methods, quantification of splicing factor activities and interfaces for long-read and immunopeptidomics analysis. We attribute these new methods to our high validation rate. These analyses establish the broad existence of highly shared splicing neoantigens in melanoma and nominate coordinated splicing failure as a broad mediator of mis-splicing. Shared versus patient specific splicing neoantigens were found to have distinct physicochemical characteristics and cells of origins, suggesting distinct mechanisms of regulation. From these analyses, one prediction, SLC45A2 stands out due to its: 1) prognostic indication for immunotherapy outcomes, 2) specificity for tumor versus immune cells, 3) MHC-I stabilization, 4) high immunogenicity, 5) support from long-read sequencing, and 6) localization to the cell surface, making it an immensely promising target for future therapies.
A key challenge faced by SNAF and other tools is the identification of optimal targets for experimental validation. Improving such predictions in the future will depend on well-designed prospective studies, experimentally validating a range of predicted immunogenic and non-immunogenic peptides (MHC-presentation, immunogenicity) from mutations and splicing that are shared or unique and derived from different tools and statistical cutoffs. Similarly, the validation of ExNeoEpitopes will rely on new proteogenomics approaches that leverage targeted long sequencing isoform sequencing and proteomics along with antibodies that target specific conformational epitopes. Such antibodies could represent powerful new molecular reagents for CAR-T or monoclonal antibody strategies, for shared and patient-specific neoantigens. Although our current pipeline enables the identification of likely ExNeoEpitopes and deep visual interrogation of the impact and position of residues in undocumented cancer protein isoforms, ultimately improved automated bioinformatics methods are needed to determine which introduced or removed residues will result specifically in new extracellular or transmembrane sequences that retain the conformational integrity of the new protein isoforms. Moreover, transposable elements have been reported to contribute to a subset of alternative splicing events (74) and give rise to neoantigens (75–78), including endogenous retrovirus (79). Although we do not observe such overlapping elements in our validated shared splicing neoantigens (UCSC genome browser), these warrant more systemic analysis to assess the potential convergence between these two types of neoantigen and their contribution, respectively.
A final important consideration is the tumor specificity of such neoantigens. As antigen assays do not provide information on tumor specificity, there is a need for more comprehensive normal tissue references. Most conventional RNA-Seq studies are on a limited set of adult human tissues, without considering rare cell-types or fetal developmental isoforms. It is likely that an expanded atlas of normal tissues, with extremely high sequencing depths (>100 million reads) and longer reads (>100nt) are needed, which may be aided by newly reported cheaper and longer sequencing approaches (80) and ideally single-cell resolution for hundreds of cell types.
Finally, it is important to note that our study has several limitations and outstanding questions. It has been suggested that cancer cells maintain a delicate balance between mutations in oncogenes and suppression of their presentation by MHC (69). Whether a similar mechanism exists for splicing neoantigens could further inform the type of therapy administered. Second, although current neoantigen predictions focus on HLA-I presentation and CD8 T-cell function, HLA-II and CD4 T -cells have also been reported to play an essential role in enhancing anti-tumor activities, together with other major immune cell types such as neutrophils, dendritic cells (70). As current bioinformatics pipelines do not consider the activities of these other HLA mechanisms and T-cell subsets, future methods may need to incorporate additional T-cell and antigen presentation mechanisms. Further, to confirm SNAF observations, we leveraged existing immunopeptidomics and synthetic peptide MS data. Although it represents an important high-throughput validation, we note that MS-based neoantigen validations suffer from both false positives and false negatives due to the non-tryptic nature of immunopeptides and the complexed peptide search space considering combinatorial Post-Translational Modifications. The incomplete nature of the MS2 spectrum necessitates orthogonal assays and manual annotation to confirm individual splicing neoantigen predictions, which accounts for low prior reported MS/MS identification rates (5%) compared to normal proteome (50%) (29, 30, 71–73).
Given its flexibility, SNAF can be easily extended to new datasets, sequencing technologies, and neoantigen prediction libraries which can be deployed in a modular manner in custom bioinformatics pipelines. Applied broadly to new cancers and distinct forms of malignancy, we believe SNAF could be used to identify splicing neoantigens that are unique and shared across malignancies and discover new sequence motif preferences that expand the repertoire of targets for precision cancer therapy.
MATERIALS AND METHODS
Study Design
The objective of this study is to comprehensively define tumor-specific and potentially immunogenic neoantigens produced from post-transcriptional regulation, particularly through alternative splicing. A systematic pipeline for the identification of splicing neoantigens in heterogeneous cancers is presented as a strategy to reveal new shared targets for therapy. To broadly assess the presence and specificity of splicing neoantigens shared among patients with melanoma and ovarian cancer, our described bioinformatics workflow was applied to existing well-curated cancer molecular omics datasets. These cancers and datasets were selected as they possess matched or unmatched multiomic measurements (immunopeptidome, RNA-Seq), clinical outcomes and diverse therapy regimens. Bulk long-read RNA-sequencing was applied in melanoma cell lines (1 library replicate per cell line) to capture a sufficient diversity of full-length mRNA isoforms, not necessarily present in other included cancer cell line long-read datasets. As the majority of analyses in the study are retrospective, sample size for these bulk RNA-Seq, immunoproteomics and single-cell RNA-Seq datasets is dependent on the original study design. For in vitro functional validation, neoantigen-MHC binding was confirmed using the TAP deficient T2 cell line’s capacity to stabilize HLA-A*02 upon the binding of candidate peptides. The immunogenicity and T cell reactivity of neoantigens were evaluated using peripheral blood collected from a minimum of three healthy donors. The selection of donors was contingent on the availability of MHC-I matches predicted by DeepImmuno and NetMHCpan SNAF. The experiment was conducted three times to ensure the reliability of the results and each replication followed the same protocol and conditions to minimize variability and enhance the robustness of the findings. The analyses were blinded to the study participants by the clinical study coordinators.
Statistics
Replicate sample genomic comparative analyses employed a two-sided empirical Bayes moderated t-test (p<0.05) for all bulk RNA-Seq gene expression and alternative splicing analyses. All analyses in which greater than 30 measurements were obtained, were subject to false discovery testing procedure (Benjamini Hochberg). Associations for individual neojunctions or neoantigens with patient survival were derived using a univariate Cox Regression analysis, with positive and negative associations results reported for all results and significant results reported for a Coxph < 0.05.
SNAF Architecture
SNAF was designed as a modular python package to automate splicing neoantigen identification, using a series of embedded workflows. This workflow consists of distinct steps (see below) which are divided into functionally distinct modules which can be called on a single SNAF python object or independently produced data files. These functions can be mixed and matched to identify T-cell or B-cell neoantigens or perform orthogonal such as survival, MS proteomics, long-read analysis. Additional documentation and tutorials are provided from the GitHub repository (https://github.com/frankligy/SNAF). Specific SNAF algorithm details are provided in Supplementary Materials and Methods.
–Bulk and single-cell melanoma splicing evaluation datasets
To evaluate shared and unique splicing neoantigens identified by SNAF, we reanalyzed prior reported bulk and single-cell RNA-Seq datasets through SNAF, using the same genome alignment and splicing quantification workflows applied to TCGA samples. To assess the association of melanoma splicing neoantigens with non-cancerous proliferative skin cell splicing events, we reanalyzed five proliferative melanocyte RNA-Seq datasets in the GEO database (GSE102983, GSE111786, GSE149189, GSE197471, GSE202700) (Fig. S9). To determine the cell of origin for melanoma splicing antigens, we obtained access to the controlled access raw sequencing data (DUOS-000002). For 4,645 individual cell transcriptomes corresponding to 19 patients with melanoma (GSE72056). Only 3,877 with cell annotations were retained for further analysis. These individual cell-level FASTQ files were re-analyzed in STAR and AltAnalyze to produce aggregate junction read counts for each patient and author annotated cell-populations. These junction read counts were summed per cell-population to identify tumor versus immune neojunction enrichments (fold>2 enriched). These analyses are biased towards immune cells, as twice as many immune cells (n=2,605) versus tumor (n=1,174) were present.
I–Peptide synthesis and MS spike-in validation
36 peptide candidate splicing neoantigens were synthesized (GenScript, Piscataway, NJ) at minimum of 70% purity with an average yield of 0.2–0.5 mg. Peptides were reconstituted with water to a final stock concentration of 1 pmol/μL. Peptides were pooled (except for LELLVKGTV and STLEFGLRV, which did not solubilize sufficiently) at a concentration of 1 pmol/μL and then diluted 1:10 for a 100 fmol/μL working solution. LC-MS analysis was performed on a 50 fmol injection of pooled peptides using a Ultimate 3000 nanoflow HPLC (Dionex) and Orbitrap Eclipse Tribrid mass spectrometer (ThermoFisher Scientific) as described below. Injections were loaded onto an Acclaim PepMap 100 trap column (300 μm x 5 mm x 5 μm C18) and gradient-eluted from an Acclaim PepMap 100 analytical column (75 μm x 25 cm, 3 μm C18) equilibrated in 96% solvent A (0.1% formic acid in water) and 4% solvent B (80% acetonitrile in 0.1% formic acid). The peptides were eluted at 300 nL/min using the following gradient: 4% B from 0–5 min, 4 to 10% B from 5–10 min, 10–35% B from 10–60 min, 35–55% B from 60–70 min, 55–90% B from 70–71 min, 90% B from 71–73 min, 90–4%B from 73–74 min and 4% B from 74–90 min. The Orbitrap Eclipse was operated in positive ion mode with 2.0 kV at the spray source, RF lens at 30% and data dependent MS/MS acquisition with XCalibur version 4.3.73.11. Positive ion Full MS scans were acquired in the Orbitrap from 375–1500 m/z with 120,000 resolution. Data dependent selection of precursor ions was performed in Cycle Time mode, with 3 seconds in between Master Scans, using an intensity threshold of 2 × 104 ion counts and applying dynamic exclusion (n=1 scans within 30 seconds for an exclusion duration of 60 seconds and +/− 10 ppm mass tolerance). Monoisotopic peak determination was applied and charge states 2–6 were included for HCD MS2 scans (quadrupole isolation mode; 1.6 m/z isolation window, Normalized collision energy at 30%). The resulting fragments were detected in the Orbitrap at 15,000 resolution with Standard AGC target and Dynamic maximum injection time mode.
IValidation of peptide-MHC binding by MHC stabilization assay
To test MHC-I binding of synthesized neoantigen peptides, we used TAP deficient T2 cells that are defective in transporters required for endogenous peptide loading (104, 105). T2 cells were obtained from ATCC and grown at 37°C, 5% CO2 in ‘scove’s Modified Du’becco’s Medium supplemented with 20% FBS and pen/strep. 1×105 T2 cells were used without and loaded with 100ug/ml peptides and thereafter incubated overnight. All T2 cells were harvested and stained with HLA-A2-PE antibody (clone BB7.2; Biolegend) for 30 mins on ice. Cells were washed once with cell culture medium and acquired on a Fortessa II flow cytometer. Median fluorescence intensity was determined using FlowJo software.
Immunogenicity assay
Immunogenicity of predicted splicing neoantigen peptides was determined as described previously (16, 77). In short, HLA-typed PBMCs from leukocyte reduction system (LRS) (Cincinnati Hoxworth Blood Center) chambers were isolated using Ficoll hypaque density gradient centrifugation, aliquoted in 20×106 cells per vial and frozen in liquid nitrogen until use. At day 0, PBMC were thawed and used to set up monocyte derived dendritic cells by plating 4×106 PBMC in a 24 well. Cells were incubated at 37°C, 5% CO2 in DC medium (RPMI 1640 supplemented with 10% FBS, 1% L-Glutamine (200mM) + IL-4 (1000 U/ml) and GM-CSF (800U/ml). After 4h, the non-adherent fraction was removed by rinsing the wells twice with PBS. Adherent cells were cultured for 7 days in DC medium. On day 7, DCs were loaded with 10ug/ml peptides dissolved in DC medium and incubated at 37°C, 5% CO2. After 4h, 1.5ml DC maturation medium was added (RPMI 1640 supplemented with 10% FBS, 1% L-Glutamine, IL-4 (1000U/ml), GM-CSF (800U/ml), IL-1β (10ng/ml), IL-6 (10ng/ml), TNF-α (10ng/ml) and LPS (30ng/ml). After 16h of DC maturation, peptide loaded DCs were used to stimulate autologous PBMC, by adding 1×106 PBMC of the same donor to the DC cultures. DC and PBMC co-cultures were grown in T cell medium (60% RPMI 1640, 40% Click’s medium supplemented with 10% FBS, 1% L- glutamine, IL-6 (100ng/ml), IL-7 (10ng/ml), IL-12 (10ng/ml), and IL-15 (5ng/ml). Medium was changed on day 3 and day 6 based on medium color change. On day 14 and 21 T cells were harvested and stimulated with new autologous peptide loaded DCs. After three rounds of T cell priming, at day 28, T cells were harvested and tested for their IFNγ response to peptide loaded 721.221 (221) single HLA antigen target cells. Fist the target cells 221.A*02:01, 221.C*04:01, and 221.C*08:01 were loaded with relevant peptides at 10μg/ml in separate wells. 221 and peptides were incubated for 4h at 37°C, 5% CO2 in RPMI 1640 supplemented with 10% FBS. Peptide loaded 221 target cells with and without peptides were co-cultured with primed T cells at 3:1 ratio in the presence of 50 ng/ml PMA for 6h at 37°C, 5% CO2 in RPMI 1640 supplemented with 10% FBS and monensin. PMA and ionomycin stimulation (each at 1μg/ml) was used at positive control. Thereafter the cells were harvested and stained for extracellular CD45-BV786 (Biolegend), CD14-PerCP (Biolegend) and CD8-PE (biolegend) for 30 mins on ice. Cells were fixed and permeabilized using the CytoFix/CytoPerm kit (BD) according to manufacturers instructions and thereafter stained for intracellular IFNγ-APC expression (clone 4S.B3; Biolegend) for 20 mins on ice and directly analyzed on a BD Fortessa flow cytometer. Analysis of CD45+CD14-CD8+ IFNγ+ cells was determined using FlowJo software.
Validation of ExNeoEpitope localization
To confirm the expression and cell membrane localization of SNAF-B neo-isoforms, we synthesized the long-read sequencing evidenced alternative isoforms and their annotated reference isoforms as C-terminal tagged cDNAs with either mNeon-Green or eGFP on a plasmid vector (VectorBuilder, USA). Streak LB agar plates with 100 μg/mL Ampicillin were made for each isoform. A single colony was picked from each plate and expanded in 1 mL of LB broth for 8 hours at 37 Celsius respectively. 20 μL of the pre-expansion broth was then taken and pipetted into a Elenmyer flask with 50 mL of LB broth. The competent cells were expanded overnight at 37 Celsius. Medi-preps were performed for each construct with the ZymoPure II plasmid midiprep kit. On an Ibidi 4-well chamber μ-slide, HEK-293T cells were seeded in prior with a concentration of 0.15 M/mL. When HEK-293T cells reached 60% of confluency, these constructs (CMV promoter) were transfected into the cells separately with 1 μg of plasmid (99% pUC19 negative control plasmid+1% engineered plasmid) with TransIT-LT1 (Mirus) following the manufacturer’s protocol.
The cells were fixed and permeabilized with 4% PFA 24 hours post-transfection. After two rounds of washing, the cells were treated with a warm 1x citrate buffer (diluted from 10X stock; Sigma-Aldrich) to break the protein cross-links. The cells were then washed once with PBS again and a membrane actin stain was performed with 1x Phalloidlin 647 in PBS with 1% BSA (Abcam) at room temperature for an hour. To stain the nuclei of the fixed cells, the cells were washed with PBS and stained with DAPI (Thermo Scientific) (1:4000 diluted in PBS with 1% BSA) at room temperature for 5 minutes. The stained cells were immediately washed with PBS, and the PBS removed by vacuum. Prolong gold mounting media (Thermo Fisher Scientific) was evenly applied to the fixed cells surface. 24 hours post transfection, the expression and co-localization of the reference and alternative isoforms were assessed by a confocal spinning disk microscopy (Yokogawa SoRa W1 dual camera system) using the Nikon Elements software. The confocal raw imaging files with all z-stacks are provided in Synapse (https://www.synapse.org/#!Synapse:syn52063953).
Melanoma Cell-Line Long-Read Isoform Sequencing
For long-read mRNA isoform sequencing, cells were grown and isolated from five independent Melanoma cell lines: A375, SKMEL, MeWo, UACC62, and UACC257 (ATCC). Total RNA was isolated (Trizol) and analyzed on a Thermo Nanodrop UV-Vis and an Agilent Bioanalyzer to confirm the nominal concentration and ensure RNA integrity. From the RNA, cDNA was synthesized using the Clontech SMARTr cDNA Synthesis Kit, in which a barcode was added to the oligo-dT at the 3′ end. Each melanoma cell line cDNA was pooled and then converted into a SMRTbell library using the Iso-Seq Express Kit SMRT Bell Express Template prep kit 2.0 (Pacific Biosciences). We sequenced each library on a SMRT cell on the Sequel II system using a 30 hour movie collection time. The “ccs” command from the PacBio SMRTLink suite (SMRTLink version 9) was used to convert Raw reads into Circular Consensus Sequence (CCS) reads. The resulting data was analyzed in SQUANTI to assign reads to full-length collapsed reference or neo-isoforms. The isoform GTF files, barcode sequences and raw data are available in Synapse (https://www.synapse.org/#!Synapse:syn32785802). (table S1).
Melanoma RNA-Seq Analyses
RNA-Seq paired-end BAM files from 472 patients with melanoma collected by TCGA (SKCM) were obtained from the GDC portal following dbGAP approval (phs000178.v10.p8). A second collection of 40 RNA-Seq FASTQ files from patients who underwent immunotherapy (archival formalin-fixed, paraffin-embedded) in Van Allen cohort (34) files were obtained from the dbGAP database (phs000452.v2.p1). These FASTQ files were aligned to the reference human genome (hg38) and transcriptome (Ensembl 91) using STAR. Among these 40 Van Allen RNA-Seq samples, patient 41 was excluded (only partial sequencing data available), as previously reported (106). The HLA genotype of each patient sample was determined from the RNA-Seq FASTQ files using the software Optitype 1.3.3 (107). We chose Optitype based on its superior performance in calling HLA-I alleles (over 99% accuracy) from RNA-Seq data based on a recent large-scale benchmarking study, evaluated on “gold-standard” HLA genotyping data (108)(109). AltAnalyze v 2.1.4 was used to quantify splicing independently in these two cohorts using the Ensembl version 91 database. MultiPath-PSI identified splicing events were used as inputs for SNAF. The TCGA survival and mutation data were downloaded from Xena Browser (110). Survival analysis was performed using the snaf.survival_analysis function with stratification argument n=2 (high burden is equivalent to greater than the median burden, low burden is equivalent to less than the median burden, with the outliers excluded). Mutation analysis was conducted using snaf.mutation_analysis function. To identify individual neojunctions or Neoantigens associated with survival, an univariate Cox Regression analysis was used to identify events/antigens with a parental PSI value that are positively or negatively associated with patient outcome (Wald test p-value and z-score). Here, the neojunction and its parental PSI value is ignored if the neoantigen was not predicted to be presented in that sample, resulting in different survival associations for different neoantigens produced from the same neojunction. For analysis of Melanoma RNA-Seq TCGA samples in the SNAF-B workflow, long-read Iso-Seq cDNA sequences were obtained from pan-cancer cell line sequencing, using the PacBio provided isoform GTF file (https://downloads.pacbcloud.com/public/dataset/UHRRisoseq2021/Final-MappedTranscripts/).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Daniel J. Schnell for his valuable feedback on statistical analysis recommendations and Dr. J. Matthew Kofron and the Confocal Imaging Core for assistance with the imaging analyses.
Funding
This work was partially supported by the Cincinnati Children’s Hospital Research Foundation, the University of Cincinnati Cancer Center, the Melanoma Research Foundation (Career Development Award to G.S.), an American Cancer Society Institutional Research Grant to G.S., and the National Institutes of Health (R01CA226802 to N.S, RC2DK122376 to H.L.G. and N.S., and R01HG013328 to M.T.W.)
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Splicing Neo Antigen Finder (SNAF) identifies shared immunogenic MHC-presented splicing neoantigens and tumor-specific transmembrane isoforms.
Data Availability
The SNAF application is available as a Python3 package (https://pypi.org/project/SNAF/). The source code is available at (https://github.com/frankligy/SNAF) and has been deposited to Zenodo (https://zenodo.org/records/10252900). The scripts and data for reproducing the results are available at (https://github.com/frankligy/SNAF/tree/main/reproduce) along with the raw and processed SNAF results (https://www.synapse.org/#!Synapse:syn32057176/files/).
REFERENCES
- 1.Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Tran E, Robbins PF, Rosenberg SA, “Final common pathway” of human cancer immunotherapy: targeting random somatic mutations. Nat. Immunol. 18, 255–262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ, An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu Z, Leet DE, Allesøe RL, Oliveira G, Li S, Luoma AM, Liu J, Forman J, Huang T, Iorgulescu JB, Holden R, Sarkizova S, Gohil SH, Redd RA, Sun J, Elagina L, Giobbie-Hurder A, Zhang W, Peter L, Ciantra Z, Rodig S, Olive O, Shetty K, Pyrdol J, Uduman M, Lee PC, Bachireddy P, Buchbinder EI, Yoon CH, Neuberg D, Pentelute BL, Hacohen N, Livak KJ, Shukla SA, Olsen LR, Barouch DH, Wucherpfennig KW, Fritsch EF, Keskin DB, Wu CJ, Ott PA, Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 27, 515–525 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bauman J, Burris H, Clarke J, Patel M, Cho D, Gutierrez M, Julian R, Scott A, Cohen P, Frederick J, Robert-Tissot C, Zhou H, Mody K, Keating K, Meehan R, Gainor J, 798 Safety, tolerability, and immunogenicity of mRNA-4157 in combination with pembrolizumab in subjects with unresectable solid tumors (KEYNOTE-603): an update. J Immunother Cancer 8 (2020), doi: 10.1136/jitc-2020-SITC2020.0798. [DOI] [Google Scholar]
- 6.Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US, Klebanoff CA, Hughes MS, Restifo NP, Langhan MM, Shelton TE, Lu L, Kwong MLM, Ilyas S, Klemen ND, Payabyab EC, Morton KE, Toomey MA, Steinberg SM, White DE, Rosenberg SA, Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J. Clin. Oncol. 34, 2389–2397 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortés ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau D-A, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CWM, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G, Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yarchoan M, Hopkins A, Jaffee EM, Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 377, 2500–2501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nilsen TW, Graveley BR, Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park SM, Ou J, Chamberlain L, Simone TM, Yang H, Virbasius C-M, Ali AM, Zhu LJ, Mukherjee S, Raza A, Green MR, U2AF35(S34F) Promotes Transformation by Directing Aberrant ATG7 Pre-mRNA 3’ End Formation. Mol. Cell 62, 479–490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calabretta S, Bielli P, Passacantilli I, Pilozzi E, Fendrich V, Capurso G, Fave GD, Sette C, Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 35, 2031–2039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S-J, Rampal R, Manshouri T, Patel J, Mensah N, Kayserian A, Hricik T, Heguy A, Hedvat C, Gönen M, Kantarjian H, Levine RL, Abdel-Wahab O, Verstovsek S, Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 119, 4480–4485 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oudin MJ, Hughes SK, Rohani N, Moufarrej MN, Jones JG, Condeelis JS, Lauffenburger DA, Gertler FB, Characterization of the expression of the pro-metastatic Mena(INV) isoform during breast tumor progression. Clin. Exp. Metastasis 33, 249–261 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahles A, Lehmann KV, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Rätsch G, Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 34 (2018), doi: 10.1016/j.ccell.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rivera OD, Mallory MJ, Quesnel-Vallières M, Chatrikhi R, Schultz DC, Carroll M, Barash Y, Cherry S, Lynch KW, Alternative splicing redefines landscape of commonly mutated genes in acute myeloid leukemia. Proc. Natl. Acad. Sci. U. S. A. 118 (2021), doi: 10.1073/pnas.2014967118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rivero-Hinojosa S, Grant M, Panigrahi A, Zhang H, Caisova V, Bollard CM, Rood BR, Proteogenomic discovery of neoantigens facilitates personalized multi-antigen targeted T cell immunotherapy for brain tumors. Nat. Commun. 12, 6689 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, Fugmann T, Wong K-K, Van Allen EM, Intron retention is a source of neoepitopes in cancer. Nat. Biotechnol. 36, 1056–1058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehx G, Larouche J-D, Durette C, Laverdure J-P, Hesnard L, Vincent K, Hardy M-P, Thériault C, Rulleau C, Lanoix J, Bonneil E, Feghaly A, Apavaloaei A, Noronha N, Laumont CM, Delisle J-S, Vago L, Hébert J, Sauvageau G, Lemieux S, Thibault P, Perreault C, Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity 54, 737–752.e10 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Li G, Iyer B, Prasath VBS, Ni Y, Salomonis N, DeepImmuno: deep learning-empowered prediction and generation of immunogenic peptides for T-cell immunity. Brief. Bioinform. 22 (2021), doi: 10.1093/bib/bbab160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narayan V, Barber-Rotenberg JS, Jung IY, Lacey SF, Rech AJ, Davis MM, Hwang WT, Lal P, Carpenter EL, Maude SL, Plesa G, Vapiwala N, Chew A, Moniak M, Sebro RA, Farwell A. Marshall, Gilmore J, Lledo L, Dengel K, Church SE, Hether TD, Xu J, Gohil M, Buckingham TH, Yee SS, Gonzalez VE, Kulikovskaya I, Chen F, Tian L, Tien K, Gladney W, Nobles CL, Raymond HE, Hexner EO, Siegel DL, Bushman FD, June CH, Fraietta JA, Haas NB, PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat. Med. 28 (2022), doi: 10.1038/s41591-022-01726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenix AM, Miyaoka Y, Bertero A, Blue SM, Spindler MJ, Tan KKB, Perez-Bermejo JA, Chan AH, Mayerl SJ, Nguyen TD, Russell CR, Lizarraga PP, Truong A, So P-L, Kulkarni A, Chetal K, Sathe S, Sniadecki NJ, Yeo GW, Murry CE, Conklin BR, Salomonis N, Gain-of-function cardiomyopathic mutations in RBM20 rewire splicing regulation and re-distribute ribonucleoprotein granules within processing bodies. Nat. Commun. 12, 6324 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itskovich SS, Gurunathan A, Clark J, Burwinkel M, Wunderlich M, Berger MR, Kulkarni A, Chetal K, Venkatasubramanian M, Salomonis N, Kumar AR, Lee LH, MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia. Nat. Commun. 11, 2369 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369 (2020), doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M, NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data Nucleic Acids Research 48, W449–W454 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Systems 11, 42–48.e7 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Li G, Bhattacharjee A, Salomonis N, Quantifying tumor specificity using Bayesian probabilistic modeling for drug target discovery and prioritization bioRxiv, 2023.03.03.530994 (2023). [Google Scholar]
- 27.Schuster H, Peper JK, Bösmüller H-C, Röhle K, Backert L, Bilich T, Ney B, Löffler MW, Kowalewski DJ, Trautwein N, Rabsteyn A, Engler T, Braun S, Haen SP, Walz JS, Schmid-Horch B, Brucker SY, Wallwiener D, Kohlbacher O, Fend F, Rammensee H-G, Stevanović S, Staebler A, Wagner P, The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl. Acad. Sci. U. S. A. 114, E9942–E9951 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, Martignoni ME, Werner A, Hein R, Busch DH, Peschel C, Rad R, Cox J, Mann M, Krackhardt AM, Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 7, 1–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilhelm M, Zolg DP, Graber M, Gessulat S, Schmidt T, Schnatbaum K, Schwencke-Westphal C, Seifert P, de Andrade Krätzig N, Zerweck J, Knaute T, Bräunlein E, Samaras P, Lautenbacher L, Klaeger S, Wenschuh H, Rad R, Delanghe B, Huhmer A, Carr SA, Clauser KR, Krackhardt AM, Reimer U, Kuster B, Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat. Commun. 12, 3346 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Declercq A, Bouwmeester R, Hirschler A, Carapito C, Degroeve S, Martens L, Gabriels R, MSRescore: Data-Driven Rescoring Dramatically Boosts Immunopeptide Identification Rates. Mol. Cell. Proteomics 21, 100266 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen C, Sheng Q, Dai J, Li Y, Zeng R, Tang H, On the estimation of false positives in peptide identifications using decoy search strategy. Proteomics 9, 194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller RM, Jordan BT, Mehlferber MM, Jeffery ED, Chatzipantsiou C, Kaur S, Millikin RJ, Dai Y, Tiberi S, Castaldi PJ, Shortreed MR, Luckey CJ, Conesa A, Smith LM, Deslattes Mays A, Sheynkman GM, Enhanced protein isoform characterization through long-read proteogenomics. Genome Biol. 23, 1–28 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Chen Y, Hu X, Meng J, Li X, Development and Validation of the B Cell-Associated Fc Receptor-like Molecule-Based Prognostic Signature in Skin Cutaneous Melanoma. Biomed Res. Int. 2020 (2020), doi: 10.1155/2020/8509805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D, Garraway LA, Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350 (2015), doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas Network, Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith TM Jr, Tharakan A, Martin RK, Targeting ADAM10 in Cancer and Autoimmunity. Front. Immunol. 11, 499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Mohseni M, Grauel A, Diez JE, Guan W, Liang S, Choi JE, Pu M, Chen D, Laszewski T, Schwartz S, Gu J, Mansur L, Burks T, Brodeur L, Velazquez R, Kovats S, Pant B, Buruzula G, Deng E, Chen JT, Sari-Sarraf F, Dornelas C, Varadarajan M, Yu H, Liu C, Lim J, Hao H-X, Jiang X, Malamas A, LaMarche MJ, Geyer FC, McLaughlin M, Costa C, Wagner J, Ruddy D, Jayaraman P, Kirkpatrick ND, Zhang P, Iartchouk O, Aardalen K, Cremasco V, Dranoff G, Engelman JA, Silver S, Wang H, Hastings WD, Goldoni S, SHP2 blockade enhances anti-tumor immunity via tumor cell intrinsic and extrinsic mechanisms. Sci. Rep. 11, 1399 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Batlle E, Massagué J, Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 50, 924–940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang B, Chen J, Teng Y, TNPO1-Mediated Nuclear Import of FUBP1 Contributes to Tumor Immune Evasion by Increasing NRP1 Expression in Cervical Cancer. Journal of Immunology Research 2021 (2021), doi: 10.1155/2021/9994004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song T-Y, Long M, Zhao H-X, Zou M-W, Fan H-J, Liu Y, Geng C-L, Song M-F, Liu Y-F, Chen J-Y, Yang Y-L, Zhou W-R, Huang D-W, Peng B, Peng Z-G, Cang Y, Tumor evolution selectively inactivates the core microRNA machinery for immune evasion. Nat. Commun. 12, 7003 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vlaykova T, Talve L, Hahka-Kemppinen M, Hernberg M, Muhonen T, Franssila K, Collan Y, Pyrhönen S, MIB-1 immunoreactivity correlates with blood vessel density and survival in disseminated malignant melanoma. Oncology 57, 242–252 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Gao Y, Zheng H, Li L, Zhou C, Chen X, Zhou X, Cao Y, KIF3C Promotes Proliferation, Migration, and Invasion of Glioma Cells by Activating the PI3K/AKT Pathway and Inducing EMT. Biomed Res. Int. 2020 (2020), doi: 10.1155/2020/6349312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang L, Wei F, Wu Y, He Y, Shi L, Xiong F, Gong Z, Guo C, Li X, Deng H, Cao K, Zhou M, Xiang B, Li X, Li Y, Li G, Xiong W, Zeng Z, Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 37, 1–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaal EA, Berkers CR, The Influence of Metabolism on Drug Response in Cancer. Front. Oncol. 8 (2018), doi: 10.3389/fonc.2018.00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dick FA, Goodrich DW, Sage J, Dyson NJ, Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 18, 442–451 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding Q, Xu Z, Chen Y, Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J. Exp. Clin. Cancer Res. 38, 149 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng P, Wang Z, Chen J, Liu S, Yao X, Liu S, Liu L, Yu Z, Huang Y, Xiong Z, Xiao R, Gao J, Liang W, Chen J, Liu H, Hong JH, Chan JY, Guan P, Chen J, Wang Y, Yin J, Li J, Zheng M, Zhang C, Zhou P, Kang T, Teh BT, Yu Q, Zuo Z, Jiang Q, Liu J, Xiong Y, Xia X, Tan J, RAD21 amplification epigenetically suppresses interferon signaling to promote immune evasion in ovarian cancer. J. Clin. Invest. 132 (2022), doi: 10.1172/JCI159628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rossi Sebastiano M, Konstantinidou G, Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. Int. J. Mol. Sci. 20 (2019), doi: 10.3390/ijms20153624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Germain N, Dhayer M, Boileau M, Fovez Q, Kluza J, Marchetti P, Lipid Metabolism and Resistance to Anticancer Treatment. Biology 9 (2020), doi: 10.3390/biology9120474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li G, Jiang Y, Li G, Qiao Q, Comprehensive analysis of radiosensitivity in head and neck squamous cell carcinoma. Radiother. Oncol. 159 (2021), doi: 10.1016/j.radonc.2021.03.017. [DOI] [PubMed] [Google Scholar]
- 51.Wang S, Yi X, Wu Z, Guo S, Dai W, Wang H, Shi Q, Zeng K, Guo W, Li C, CAMKK2 Defines Ferroptosis Sensitivity of Melanoma Cells by Regulating AMPK–NRF2 Pathway. J. Invest. Dermatol. 142 (2022), doi: 10.1016/j.jid.2021.05.025. [DOI] [PubMed] [Google Scholar]
- 52.Miraldi ER, Pokrovskii M, Watters A, Castro DM, De Veaux N, Hall JA, Lee JY, Ciofani M, Madar A, Carriero N, Littman DR, Bonneau R, Leveraging chromatin accessibility for transcriptional regulatory network inference in T Helper 17 Cells. Genome Res. 29 (2019), doi: 10.1101/gr.238253.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P, Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS One 5, e12776 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas JD, Lu SX, De Neef E, Sabio E, Rousseau B, Gigoux M, Knorr DA, Greenbaum B, Elhanati Y, Hogg SJ, Chow A, Ghosh A, Xie A, Zamarin D, Cui D, Erickson C, Singer M, Cho H, Wang E, Lu B, Durham BH, Shah H, Chowell D, Gabel AM, Shen Y, Liu J, Jin J, Rhodes MC, Taylor RE, Molina H, Wolchok JD, Merghoub T, Jr LAD, Abdel-Wahab O, Bradley RK, Abstract 5742: Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cancer Res. 83, 5742–5742 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wan L, Lin KT, Rahman MA, Ishigami Y, Wang Z, Jensen MA, Wilkinson JE, Park Y, Tuveson DA, Krainer AR, Splicing Factor SRSF1 Promotes Pancreatitis and KRASG12D-Mediated Pancreatic Cancer. Cancer Discov. (2023), doi: 10.1158/2159-8290.CD-22-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bailey TL, Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Bipolymers (1994). [PubMed] [Google Scholar]
- 57.Yang W, Lee K-W, Srivastava RM, Kuo F, Krishna C, Chowell D, Makarov V, Hoen D, Dalin MG, Wexler L, Ghossein R, Katabi N, Nadeem Z, Cohen MA, Tian SK, Robine N, Arora K, Geiger H, Agius P, Bouvier N, Huberman K, Vanness K, Havel JJ, Sims JS, Samstein RM, Mandal R, Tepe J, Ganly I, Ho AL, Riaz N, Wong RJ, Shukla N, Chan TA, Morris LGT, Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 25, 767–775 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu SX, De Neef E, Thomas JD, Sabio E, Rousseau B, Gigoux M, Knorr DA, Greenbaum B, Elhanati Y, Hogg SJ, Chow A, Ghosh A, Xie A, Zamarin D, Cui D, Erickson C, Singer M, Cho H, Wang E, Lu B, Durham BH, Shah H, Chowell D, Gabel AM, Shen Y, Liu J, Jin J, Rhodes MC, Taylor RE, Molina H, Wolchok JD, Merghoub T, Diaz LA Jr, Abdel-Wahab O, Bradley RK, Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell 184, 4032–4047.e31 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savanur MA, Weinstein-Marom H, Gross G, Implementing Logic Gates for Safer Immunotherapy of Cancer. Front. Immunol. 12, 780399 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin J-R, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CGK, Kazer SW, Gaillard A, Kolb KE, Villani A-C, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jané-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA, Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Endicott JL, Nolte PA, Shen H, Laird PW, Cell division drives DNA methylation loss in late-replicating domains in primary human cells. Nat. Commun. 13, 6659 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krogh A, Larsson B, von Heijne G, Sonnhammer EL, Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001). [DOI] [PubMed] [Google Scholar]
- 63.Omasits U, Ahrens CH, Müller S, Wollscheid B, Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang C-Y, Cheng I-C, Chang Y-C, Tsai P-S, Lai S-Y, Huang Y-L, Jeng C-R, Pang VF, Chang H-W, Identification of Neutralizing Monoclonal Antibodies Targeting Novel Conformational Epitopes of the Porcine Epidemic Diarrhoea Virus Spike Protein. Sci. Rep. 9, 2529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dreyer AM, Beauchamp J, Matile H, Pluschke G, An efficient system to generate monoclonal antibodies against membrane-associated proteins by immunisation with antigen-expressing mammalian cells. BMC Biotechnol. 10, 1–14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adamczak R, Porollo A, Meller J, Combining prediction of secondary structure and solvent accessibility in proteins. Proteins 59, 467–475 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Schumacher TN, Schreiber RD, Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Hoyos D, Zappasodi R, Schulze I, Sethna Z, de Andrade KC, Bajorin DF, Bandlamudi C, Callahan MK, Funt SA, Hadrup SR, Holm JS, Rosenberg JE, Shah SP, Vázquez-García I, Weigelt B, Wu M, Zamarin D, Campitelli LF, Osborne EJ, Klinger M, Robins HS, Khincha PP, Savage SA, Balachandran VP, Wolchok JD, Hellmann MD, Merghoub T, Levine AJ, Łuksza M, Greenbaum BD, Fundamental immune-oncogenicity trade-offs define driver mutation fitness. Nature 606, 172–179 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brightman SE, Naradikian MS, Miller AM, Schoenberger SP, Harnessing neoantigen specific CD4 T cells for cancer immunotherapy. J. Leukoc. Biol. 107, 625–633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M, Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10 (2011), doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 72.Purcell AW, Is the Immunopeptidome Getting Darker?: A Commentary on the Discussion around Mishto et al., 2019. Front. Immunol. 12, 720811 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ouspenskaia T, Law T, Clauser KR, Klaeger S, Sarkizova S, Aguet F, Li B, Christian E, Knisbacher BA, Le PM, Hartigan CR, Keshishian H, Apffel A, Oliveira G, Zhang W, Chen S, Chow YT, Ji Z, Jungreis I, Shukla SA, Justesen S, Bachireddy P, Kellis M, Getz G, Hacohen N, Keskin DB, Carr SA, Wu CJ, Regev A, Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat. Biotechnol. 40 (2022), doi: 10.1038/s41587-021-01021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clayton EA, Rishishwar L, Huang T-C, Gulati S, Ban D, McDonald JF, Jordan IK, An atlas of transposable element-derived alternative splicing in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 375, 20190342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shah NM, Jang HJ, Liang Y, Maeng JH, Tzeng S-C, Wu A, Basri NL, Qu X, Fan C, Li A, Katz B, Li D, Xing X, Evans BS, Wang T, Pan-cancer analysis identifies tumor-specific antigens derived from transposable elements. Nat. Genet. 55, 631–639 (2023). [DOI] [PubMed] [Google Scholar]
- 76.Burbage M, Rocañín-Arjó A, Baudon B, Arribas YA, Merlotti A, Rookhuizen DC, Heurtebise-Chrétien S, Ye M, Houy A, Burgdorf N, Suarez G, Gros M, Sadacca B, Carrascal M, Garmilla A, Bohec M, Baulande S, Lombard B, Loew D, Waterfall JJ, Stern M-H, Goudot C, Amigorena S, Epigenetically controlled tumor antigens derived from splice junctions between exons and transposable elements. Sci Immunol 8, eabm6360 (2023). [DOI] [PubMed] [Google Scholar]
- 77.Merlotti A, Sadacca B, Arribas YA, Ngoma M, Burbage M, Goudot C, Houy A, Rocañín-Arjó A, Lalanne A, Seguin-Givelet A, Lefevre M, Heurtebise-Chrétien S, Baudon B, Oliveira G, Loew D, Carrascal M, Wu CJ, Lantz O, Stern M-H, Girard N, Waterfall JJ, Amigorena S, Noncanonical splicing junctions between exons and transposable elements represent a source of immunogenic recurrent neo-antigens in patients with lung cancer. Sci Immunol 8, eabm6359 (2023). [DOI] [PubMed] [Google Scholar]
- 78.Laumont CM, Vincent K, Hesnard L, Audemard É, Bonneil É, Laverdure J-P, Gendron P, Courcelles M, Hardy M-P, Côté C, Durette C, St-Pierre C, Benhammadi M, Lanoix J, Vobecky S, Haddad E, Lemieux S, Thibault P, Perreault C, Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 10 (2018), doi: 10.1126/scitranslmed.aau5516. [DOI] [PubMed] [Google Scholar]
- 79.Ng KW, Boumelha J, Enfield KSS, Almagro J, Cha H, Pich O, Karasaki T, Moore DA, Salgado R, Sivakumar M, Young G, Molina-Arcas M, de Carné Trécesson S, Anastasiou P, Fendler A, Au L, Shepherd STC, Martínez-Ruiz C, Puttick C, Black JRM, Watkins TBK, Kim H, Shim S, Faulkner N, Attig J, Veeriah S, Magno N, Ward S, Frankell AM, Al Bakir M, Lim EL, Hill MS, Wilson GA, Cook DE, Birkbak NJ, Behrens A, Yousaf N, Popat S, Hackshaw A, Hiley CT, Litchfield K, McGranahan N, Jamal-Hanjani M, Larkin J, Lee S-H, Turajlic S, Swanton C, Downward J, Kassiotis G, Antibodies against endogenous retroviruses promote lung cancer immunotherapy. Nature 616, 563–573 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Almogy G, Pratt M, Oberstrass F, Lee L, Mazur D, Beckett N, Barad O, Soifer I, Perelman E, Etzioni Y, Sosa M, Jung A, Clark T, Lithwick-Yanai G, Pollock S, Hornung G, Levy M, Coole M, Howd T, Shand M, Farjoun Y, Emery J, Hall G, Lee S, Sato T, Magner R, Low S, Bernier A, Gandi B, Stohlman J, Nolet C, Donovan S, Blumenstiel B, Cipicchio M, Dodge S, Banks E, Lennon N, Gabriel S, Lipson D, Cost-efficient whole genome-sequencing using novel mostly natural sequencing-by-synthesis chemistry and open fluidics platform bioRxiv, 2022.05.29.493900 (2022). [Google Scholar]
- 81.Emig D, Salomonis N, Baumbach J, Lengauer T, Conklin BR, Albrecht M, AltAnalyze and DomainGraph: analyzing and visualizing exon expression data. Nucleic Acids Res. 38, W755–62 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dong Y, Liu X, Jiang B, Wei S, Xiang B, Liao R, Wang Q, He X, A Genome-Wide Investigation of Effects of Aberrant DNA Methylation on the Usage of Alternative Promoters in Hepatocellular Carcinoma. Front. Oncol. 11, 780266 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB, Rech AJ, Zaretsky JM, Comin-Anduix B, Ng AHC, Chour W, Yu TV, Rizvi H, Chen JM, Manning P, Steiner GM, Doan XC, Merghoub T, Guinney J, Kolom A, Selinsky C, Ribas A, Hellmann N. Hacohen, Sette A, Heath JR, Bhardwaj N, Ramsdell F, Schreiber RD, Schumacher TN, Kvistborg P, Defranoux NA, Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 183 (2020), doi: 10.1016/j.cell.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Paul S, Croft NP, Purcell AW, Tscharke DC, Sette A, Nielsen M, Peters B, Benchmarking predictions of MHC class I restricted T cell epitopes in a comprehensively studied model system. PLoS Comput. Biol. 16, e1007757 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nibeyro G, Girotti R, Prato L, Moron G, Luján HD, Fernandez EA, MHC-I binding affinity derived metrics fail to predict tumor specific neoantigen immunogenicity, doi: 10.1101/2022.03.14.484285. [DOI] [Google Scholar]
- 86.Salomonis N, Nelson B, Vranizan K, Pico AR, Hanspers K, Kuchinsky A, Ta L, Mercola M, Conklin BR, Alternative Splicing in the Differentiation of Human Embryonic Stem Cells into Cardiac Precursors. PLoS Comput. Biol. 5 (2009), doi: 10.1371/journal.pcbi.1000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Salomonis N, Schlieve CR, Pereira L, Wahlquist C, Colas A, Zambon AC, Vranizan K, Spindler MJ, Pico AR, Cline MS, Clark TA, Williams A, Blume JE, Samal E, Mercola M, Merrill BJ, Conklin BR, Alternative splicing regulates mouse embryonic stem cell pluripotency and differentiation. Proc. Natl. Acad. Sci. U. S. A. 107, 10514 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Behura SK, Severson DW, Codon usage bias: causative factors, quantification methods and genome-wide patterns: with emphasis on insect genomes. Biol. Rev. Camb. Philos. Soc. 88, 49–61 (2013). [DOI] [PubMed] [Google Scholar]
- 89.Bowman MJ, Pulman JA, Liu TL, Childs KL, A modified GC-specific MAKER gene annotation method reveals improved and novel gene predictions of high and low GC content in Oryza sativa BMC Bioinformatics 18 (2017), doi: 10.1186/s12859-017-1942-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brogna S, Wen J, Nonsense-mediated mRNA decay (NMD) mechanisms Nature Structural & Molecular Biology 16, 107–113 (2009). [DOI] [PubMed] [Google Scholar]
- 91.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L, Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Frazee AC, Jaffe AE, Langmead B, Leek JT, Polyester: simulating RNA-seq datasets with differential transcript expression. Bioinformatics 31, 2778–2784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wolf FA, Angerer P, Theis FJ, SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bingham E, Chen JP, Jankowiak M, Obermeyer F, Pradhan N, Karaletsos T, Singh R, Szerlip P, Horsfall P, Goodman ND, Pyro: Deep Universal Probabilistic Programming (2018) (available at http://arxiv.org/abs/1810.09538). [Google Scholar]
- 95.Slaff B, Radens CM, Jewell P, Jha A, Lahens NF, Grant GR, Thomas-Tikhonenko A, Lynch KW, Barash Y, MOCCASIN: a method for correcting for known and unknown confounders in RNA splicing analysis. Nat. Commun. 12, 1–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, Halow J, Van Nostrand EL, Freese P, Gorkin DU, Shen Y, He Y, Mackiewicz M, Pauli-Behn F, Williams BA, Mortazavi A, Keller CA, Zhang X-O, Elhajjajy SI, Huey J, Dickel DE, Snetkova V, Wei X, Wang X, Rivera-Mulia JC, Rozowsky J, Zhang J, Chhetri SB, Zhang J, Victorsen A, White KP, Visel A, Yeo GW, Burge CB, Lécuyer E, Gilbert DM, Dekker J, Rinn J, Mendenhall EM, Ecker JR, Kellis M, Klein RJ, Noble WS, Kundaje A, Guigó R, Farnham PJ, Cherry JM, Myers RM, Ren B, Graveley BR, Gerstein MB, Pennacchio LA, Snyder MP, Bernstein BE, Wold B, Hardison RC, Gingeras TR, Stamatoyannopoulos JA, Weng Z, Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, van den Oord J, Atak ZK, Wouters J, Aerts S, SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102 (2005), doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hänzelmann S, Castelo R, Guinney J, GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 1–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Belinda Ding B, Hilda Ye B, Califano A, Network-based inference of protein activity helps functionalize the genetic landscape of cancer. Nat. Genet. 48, 838 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cox J, Mann M, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification Nature Biotechnology 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 103.Chen Y, Kwon SW, Kim SC, Zhao Y, Integrated approach for manual evaluation of peptides identified by searching protein sequence databases with tandem mass spectra. J. Proteome Res. 4, 998–1005 (2005). [DOI] [PubMed] [Google Scholar]
- 104.Stuber G, Leder GH, Storkus WT, Lotze MT, Modrow S, Székely L, Wolf H, Klein E, Kärre K, Klein G, Identification of wild-type and mutant p53 peptides binding to HLA-A2 assessed by a peptide loading-deficient cell line assay and a novel major histocompatibility complex class I peptide binding assay. Eur. J. Immunol. 24 (1994), doi: 10.1002/eji.1830240341. [DOI] [PubMed] [Google Scholar]
- 105.Grommé M, Neefjes J, Antigen degradation or presentation by MHC class I molecules via classical and non-classical pathways. Mol. Immunol. 39 (2002), doi: 10.1016/s0161-5890(02)00101-3. [DOI] [PubMed] [Google Scholar]
- 106.Zhang Z, Zhou C, Tang L, Gong Y, Wei Z, Zhang G, Wang F, Liu Q, Yu J, ASNEO: Identification of personalized alternative splicing based neoantigens with RNA-seq. Aging 12 (2020), doi: 10.18632/aging.103516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O, OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Orenbuch R, Filip I, Comito D, Shaman J, Pe’er I, Rabadan R, arcasHLA: high-resolution HLA typing from RNAseq. Bioinformatics 36, 33–40 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Buchkovich ML, Brown CC, Robasky K, Chai S, Westfall S, Vincent BG, Weimer ET, Powers JG, HLAProfiler utilizes k-mer profiles to improve HLA calling accuracy for rare and common alleles in RNA-seq data. Genome Med. 9, 1–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, Zhu J, Haussler D, Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kawashima S, Kanehisa M, AAindex: Amino Acid index database. Nucleic Acids Res. 28, 374–374 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Crooks GE, Hon G, Chandonia J-M, Brenner SE, WebLogo: A Sequence Logo Generator. Genome Res. 14, 1188 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The SNAF application is available as a Python3 package (https://pypi.org/project/SNAF/). The source code is available at (https://github.com/frankligy/SNAF) and has been deposited to Zenodo (https://zenodo.org/records/10252900). The scripts and data for reproducing the results are available at (https://github.com/frankligy/SNAF/tree/main/reproduce) along with the raw and processed SNAF results (https://www.synapse.org/#!Synapse:syn32057176/files/).