

Abstract
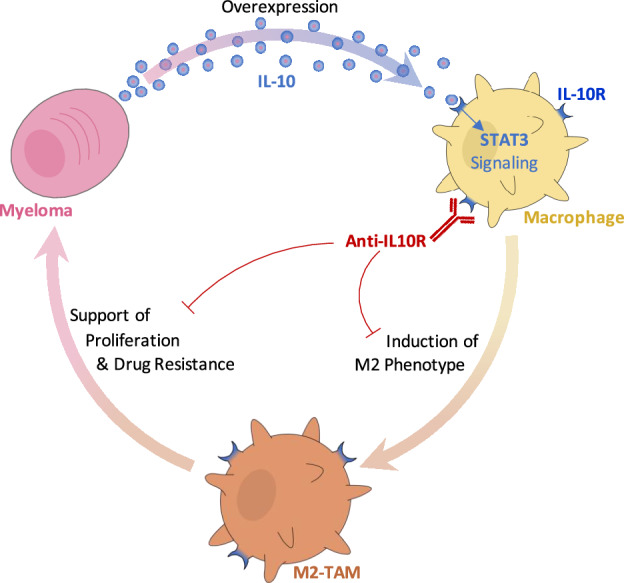
Multiple myeloma (MM) is the cancer of plasma cells within the bone marrow and remains incurable. Tumor-associated macrophages (TAMs) within the tumor microenvironment often display a pro-tumor phenotype and correlate with tumor proliferation, survival, and therapy resistance. IL-10 is a key immunosuppressive cytokine that leads to recruitment and development of TAMs. In this study, we investigated the role of IL-10 in MM TAM development as well as the therapeutic application of IL-10/IL-10R/STAT3 signaling inhibition. We demonstrated that IL-10 is overexpressed in MM BM and mediates M2-like polarization of TAMs in patient BM, 3D co-cultures in vitro, and mouse models. In turn, TAMs promote MM proliferation and drug resistance, both in vitro and in vivo. Moreover, inhibition of IL-10/IL-10R/STAT3 axis using a blocking IL-10R monoclonal antibody and STAT3 protein degrader/PROTAC prevented M2 polarization of TAMs and the consequent TAM-induced proliferation of MM, and re-sensitized MM to therapy, in vitro and in vivo. Therefore, our findings suggest that inhibition of IL-10/IL-10R/STAT3 axis is a novel therapeutic strategy with monotherapy efficacy and can be further combined with current anti-MM therapy, such as immunomodulatory drugs, to overcome drug resistance. Future investigation is warranted to evaluate the potential of such therapy in MM patients.
Subject terms: Cancer microenvironment, Myeloma
Introduction
Multiple myeloma (MM) is a cancer of plasma cells within the bone marrow (BM) and has a five-year survival of 55%, and despite development of a wide range of novel therapies, the majority of MM patients relapse, with each remission becoming shorter in duration [1]. The MM tumor microenvironment (TME) houses a complex range of elements that together support tumor progression, immunosuppression, and drug resistance [2, 3]. Thus, targeting elements in the TME has recently been a popular venue of exploration [4, 5].
Macrophages are part of the innate immune system and represent up to 30–50% of infiltrating immune cells in certain cancers [6]. Macrophages are highly plastic and polarize to different phenotypes on M1/M2 spectrum in response to environmental stimuli [7]. M1 macrophages are pro-inflammatory, participate in phagocytosis and activation of adaptive immunity, and are perceived as anti-tumor. On the other hand, M2 macrophages are anti-inflammatory, participate in wound healing and angiogenesis, and are perceived as pro-tumor. Tumor-associated macrophages (TAMs) are prominent in the TME and support tumor pathogenesis in many types of cancers, such as breast cancer, hepatocellular carcinoma, glioma, and leukemia, and present similar properties as M2 macrophages [8–11]. TAMs were shown to play a critical role in tumor growth and drug resistance[12, 13], and to be associated with negative prognosis in MM [14].
IL-10, which is a key immunosuppressive cytokine, has been found to play a critical role in polarizing macrophages to the M2 phenotype [15]. IL-10 has been reported to be highly secreted by many types of cancers including MM [16]. Macrophages have been shown to respond to IL-10 via the IL-10 receptor (IL-10R) on their surface. Activation of IL-10/IL-10R pathway leads to activation of STAT3, resulting in the transcription of M2-related genes and inhibiting M1-related genes [17, 18]. Moreover, secretion of IL-10 leads to a highly immunosuppressive feed-forward loop [19].
In this study, we aimed to elucidate the role of IL-10 in the development of TAMs in MM, as well as the therapeutic application of IL-10/IL-10R/STAT3 signaling inhibition. We hypothesized that (1) secretion of IL-10 from MM induces polarization of the TAMs to M2-like phenotype; (2) TAMs will in turn induce MM proliferation and drug resistance; and (3) inhibition of the IL-10 pathway will prevent M2-like TAMs and re-sensitize MM cells to the killing effects of therapy.
Materials and methods
Drugs
All drugs were available from commercial sources including: Phorbol 12-myristate 13-acetate (PMA) from Cayman Chemical Company (Ann Arbor, MI); recombinant human IL-10 cytokine (rhIL-10) and macrophage colony-stimulating factor (rhM-CSF) from Sino Biological (Beijing, China); Lenalidomide (LEN) and dexamethasone (DEX) from Selleck Chemicals (Houston, TX); Monoclonal Rat IgG2a Anti-human CD210 (IL-10R, clone 3F9) blocking antibody from BioLegend (San Diego, CA) (anti-IL-10R mAb). The STAT3 PROTAC was kindly provided/gifted by Professor Shaomeng Wang (University of Michigan).
Ethics statement for human tissues and animal experiments
MM patient and healthy donor BM aspirates were obtained from Siteman Cancer Center at Washington University in St. Louis School of Medicine. Informed consent was obtained from all patients with approval from the Washington University Medical School Institutional Review Board committee (protocol number: 201102270) and in accordance with the Declaration of Helsinki. All animal studies were conducted according to guidelines established and approved by the Ethical Committee for Animal Experiments at Washington University in St. Louis School of Medicine.
Luminex multiplex cytokine screen
C57BL/KaLwRij (6–11 weeks old) immunocompetent mice were injected with 5TGM1-GFP murine myeloma cells (1 × 106/mouse, i.v., n = 5), and age-matched naïve mice were used as controls (n = 5) [20]. Mice were euthanized 28 days post tumor innoculation, BM was harvested, and BM supernatants were collected. BM supernatants were analyzed with Luminex immunoassay consisted of a custom ThermoFisher Procartaplex 15-plex Panel having Th1/Th2 Cytokine 11-plex (EPX110-20820-901) beads with sRANKL (EPX01A-26037-901), IL-10 (EPX01A-20614-901), M-CSF (EPX01A-26039-901), and VEGF (EPX01A-20619-901). Samples were analyzed using a FLEXMAP3D machine (Luminex Corp, Austin, TX), and data was analyzed using Milliplex Analyst 5.1.0 software (EMD Millipore, Billerica, MA) using a 5-parameter log curve fit algorithm.
Cell culture
MM.1S, RPMI-8266, and U266 cell lines were purchased from the American Type Culture Collection (ATCC, Rockville, MD); THP-1 cell line was kindly gifted from Dr. Lori Setton’s Lab (Washington University in St. Louis); MM.1S-CBR-GFP and THP-1-CBR-GFP cell lines were kindly gifted by Dr. John DiPersio’s Lab (Washington University School of Medicine). All cell lines were cultured with RPMI-1640 media (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS; Gibco, Life Technologies, Grand Island, NY), 1% L-Glutamine, and 1% Penicillin-Streptomycin (Corning, Tewksbury, MA). Cells were cultured at 37 °C in 5% CO2 incubator (NuAire, Plymouth, MN).
Primary human samples
BM aspirates from healthy donors and MM patients were centrifuged, supernatant was collected, and cell pellets were treated with red blood cell lysis buffer (Invitrogen; Waltham, MA) to obtain BM mononuclear cells (BMMCs) [21]. Similar process was performed to obtain peripheral blood mononuclear cells (PBMCs).
Macrophage differentiation
PBMCs were culture with 25 ng/ml rhM-CSF for two days, non-adherent cells were removed, and the remaining adherent cells were used as PBMC-derived M0-macrophages. In some cases, (Figs. 1D, 3A, C, and 4B), PBMC-derived M0-macrophages were purchased from Stem Cell Technologies, Cambridge, MA. For THP-1-derived macrophages, THP-1-CBR-GFP monocyte cell line was differentiated with 50 ng/ml PMA for 2 days.
Fig. 1. MM induces M2 polarization in macrophages in patients, in vivo, and in vitro.

A Effect of MM on macrophage phenotype in human BM. Macrophage polarization, in BM macrophages in (i) healthy (n = 5) or MM (n = 20) subjects, was represented as M2/M1 ratio. (ii) Macrophage polarization stratifying for newly diagnosed MM (NDMM, n = 10) or relapsed refractory MM (RRMM, n = 10) patients. B Effect of MM on macrophage phenotype in a humanized CD34 + NCG mouse model. Macrophages polarization in naïve (n = 3) or MM-inoculated (n = 3) humanized CD34 + NCG mice. Macrophage polarization was tested by flow cytometry and represented as M2/M1 ratio. C Effect of MM co-culture on THP-1-derived macrophage M2 polarization in vitro. THP-1-derived macrophages were cultured alone, or co-cultured with MM.1S, RPMI8266, or U266 cells at 1:1 ratio in the 3D tissue-engineered bone marrow (3DTEBM) for 3 days. Macrophage polarization was tested by flow cytometry and represented as M2/M1 ratio. D Effect of MM co-culture on human primary macrophage polarization in vitro. Human PBMC-derived macrophages (from 3 independent donors) were cultured alone, or co-cultured with MM.1S, RPMI8266, or U266 cells at 1:1 ratio in the 3DTEBM for 3 days. Macrophage polarization was tested by flow cytometry and represented as M2/M1 ratio. E Effect of MM conditioned media on STAT3 signaling in macrophages. THP-1-derived macrophages were cultured for 30 min with increasing concentrations of conditioned media which was used from MM cell lines (MM.1S, RPMI8266, or U266) culture for 24 h (CM). Macrophages were lysed and levels of pSTAT3 were detected by western blotting. (Bars = Average ± SD *p < 0.05; **p < 0.01; ***p < 0.001).
Fig. 3. IL-10 induces M2 polarization in TAMs through activation of STAT3 signaling.

A Effect of IL-10 on macrophage M2 polarization in vitro. THP-1-derived and primary monocyte-derived macrophages (from 3 independent donors) were treated with 100 ng/ml rhIL-10 in the 3DTEBM for 3 days. Macrophage polarization was tested by flow cytometry and represented as M2/M1 ratio, and a fold-change of non-treated condition (NT). B Effect of IL-10 on STAT3 signaling in macrophages. THP-1-derived macrophages were treated with rhIL-10 (0, 10, 100 ng/ml) for 30 min, and STAT3 activation was measured by western blotting. C Effect of anti-IL-10R mAb on macrophage polarization in vitro. THP-1-derived and primary monocyte-derived macrophages (from 3 independent donors) treated with rhIL-10 (100 ng/ml) in combination with anti-IL-10R mAb (5 µg/ml) in the 3DTEBM for 3 days. Macrophage polarization was tested by flow cytometry and represented as M2/M1 ratio, and a fold-change of non-treated condition (NT). D Effect of anti-IL-10R inhibition on pSTAT3 activation in THP-1-derived macrophages. THP-1-derived macrophages were pretreated with or without anti-IL-10R mAb (5 µg/ml) for 30 min before addition of rhIL-10 (100 ng/ml), and STAT3 activation was measured by western blotting. E Effect of selective cellular STAT3 protein degradation on macrophage polarization. THP-1-derived macrophages were treated with the STAT3 proteolysis targeting chimera (PROTAC) molecule SD-36 (2.5uM) for 24 h. Then, rh-IL-10 (100 ng/ml) was added for 3 additional days. Macrophage polarization was tested by flow cytometry and represented as M2/M1 ratio, and a fold-change of non-treated condition (NT). (Bars = Average ± SD, **p < 0.01).
Fig. 4. IL-10R inhibition reverses MM-induced TAM M2 polarization.

A Effect of anti-IL-10R mAb on patient BM macrophage polarization ex vivo. BM aspirates from 4 MM patients were cultured in the 3DTEBM model, and treated with or without anti-IL-10R mAb (5 µg/ml) for 3 days. Macrophage polarization was tested by flow cytometry and represented as M2/M1 ratio, and a fold-change of non-treated condition (NT). B Effect of anti-IL-10R mAb on macrophage polarization in vitro. THP-1-derived macrophages and primary monocyte-derived macrophages (from 3 independent donors) were co-cultured with MM1S, RPMI8266, or U266 cells (1:1 ratio) and treated with or without anti-IL-10R mAb (5µg/ml) for 3 days. Macrophage polarization was tested by flow cytometry and represented as M2/M1 ratio, and a fold-change of non-treated condition (NT). C Effect of anti-IL-10R mAb on macrophage polarization in a humanized mice model, in vivo. huCD34 NCG mice with human MM tumors were treated with (n = 3) or without (n = 3) anti-IL-10R mAb (5 mg/kg, i.p., twice a week) for 2 weeks. BM was harvested, and macrophage polarization was tested by flow cytometry and represented as M2/M1 ratio, and as fold-change of non-treated condition (NT). (Bars = Average ± SD, *p < 0.05; **p < 0.01; ***p < 0.001;).
Human IL-10 cytokine level
Determination of the IL-10 concentration in BM supernatant was performed using the MAX Human IL-10 enzyme-linked immunosorbent assay (ELISA) kit (BioLegend, San Diego, CA), according to the manufacturer’s instructions. Absorbance was read at 450 nm (signal) and 570 nm (background) with SpectraMax i3 multimode microplate spectrophotometer (Molecular Devices, San Jose, CA).
Additionally, cytokine secretion by MM.1S cells was detected by Human Cytokine Antibody Array (Abcam, Waltham, MA). Patient BM negative fraction (NF) were cultured with or without MM.1S for 2 days. The supernatants were incubated with the array membranes, washed, and analyzed for chemiluminescence, and signal intensities were analyzed by ImageJ, background corrected, and normalized against medium controls.
Patient and cell line macrophage M2 polarization
GFP + THP-1-derived macrophages co-cultured with or without MM cell lines, or primary M0-macrophages, were treated with 100 ng/ml human recombinant IL-10, with or without anti-IL-10R mAb (5 µg/ml), in 3DTEBM for 3 days, as previously described [22–25]. In some cases, cells were treated with the STAT3 degrader, SD-36 (2.5uM final concentration) for 24 h before IL-10 stimulation. 3DTEBM cultures were digested, cells were retrieved, fixed and permeablized (BD, Franklin Lakes, NJ), stained with antibodies against human or mouse CD68-FITC, CD80-APC, and CD163-BV421, and analyzed by flow cytometry. Macrophages were identified as CD68 + (or GFP+ in cell lines), M1 as CD80+ and M2 as CD163 + . Macrophage polarization was represented as ratio of the relative mean fluorescence intensities (MFI) of M2/M1in CD68+ cells.
In vivo macrophage M2 polarization
huCD34-NCG humanized mice (female, 21–31 weeks old, single donor, strain 695, Charles River Laboratories, Wilmington, MA) were inoculated with MM.1S-CBR-GFP cells (2×106/mouse, i.v., n = 6) and allowed to grow for 3 weeks until sufficient tumor burden was detected by IVIS bioluminescence imaging (BLI) system (PerkinElmer, Waltham, MA), as described before [26]. Mice were treated with anti-IL-10R mAb (5 mg/Kg, i.p., n = 3) or with PBS (i.p., n = 3), twice a week for two weeks. Naïve huCD34-NCG mice without MM inoculation were used as control (n = 3). Mice were sacrificed, BM was obtained, and macrophage polarization was determined by flow cytometry.
Western blotting
For STAT3 signaling quantification, THP-1-derived macrophages were stimulated for 30 minutes with control media, rhIL-10 (10 ng/ml or 100 ng/ml), or MM conditioned media (CM, at 50% or 100%). CM was collected from MM cell lines cultured at 1 × 106 cells/ml for 24 hours. For conditions involving IL-10R inhibition, THP-1-derived macrophages were pretreated with anti-IL-10R mAb (5 µg/ml) for 4 h before stimulation. Then, media was washed, macrophages trypsinized, lysed, and proteins collected for western blotting.
For proliferation and cell cycle signaling in MM cells, THP-1-derived macrophages were pretreated with or without anti-IL-10R (5 µg/ml) for 4 h, followed by addition of MM.1S (ratio THP-1:MM was 1:5) for 24 h. MM.1S cells were harvested for western blotting,
Blotting was performed, as previously described [27], for anti-pSTAT3 (Cat#9145) on macrophage lysate, and for anti-pAKT (Cat#4060), anti-pS6R (Cat#4858), anti-pRB (Cat #9308) for MM lysate, and anti-GAPDH (Cat#2118) in macrophages and β-Actin (Cat#8457) in MM cells, followed by anti-rabbit IgG HRP secondary antibody (Cat#7074P2) ( all from Cell Signaling Technology, Danvers, MA). Detection was performed by chemiluminescence kit (Thermo Fisher Scientific, Waltham, MA), using Biorad Gel Doc Imaging System (Biorad, Hercules, CA).
Cell survival by flow cytometry
MM.1S-CBR-GFP cells were cultured alone or co-cultured with THP-1-derived macrophages in the 3DTEBM. Cultures were treated for 3 days with anti-IL-10R mAb (5 µg/ml), lenalidomide (1 µM), or dexamethasone (1 µM). Cultures were digested and number of MM (GFP+) cells was counted and normalized to counting beads (Invitrogen, Carlsbad, CA) using flow cytometry.
In vivo efficacy
NSG-SGM3 mice (n = 28, female, 7 weeks old, Strain #013062, Jackson Laboratories, Bar Harbor, Maine) were inoculated with MM.1S-CBR-GFP cells (i.v., 2 × 106/mouse), and 3 days post-inoculation, human monocytes were injected (i.v., 1 × 106/mouse). Treatments began at 14 days post-inoculation:
For the first experiment, 2 groups (n = 7 each) were treated with PBS vehicle control, or anti-IL-10R mAb (5 mg/kg, i.p., twice/week) (Supp. Fig. S1). For the second experiment, 2 groups (n = 7 each) were treated with lenalidomide (5 mg/kg, Per Os, daily) alone, or lenalidomide and anti-IL-10R mAb (2.5 mg/kg, i.p., twice/week) (Supp. Fig. S2). Tumor burden was evaluated at days 14, 28, and 42, using BLI.
Statistical analysis
Experiments were carried out in quadruplicates and repeated at least three times. Statistical significance is determined by student’s t test, unless otherwise stated. P-value < 0.05 represents statistically significant.
Results
Induction of MM in the BM induces increased M2-like polarization of macrophages
We evaluated TAM phenotype in the BM of healthy and MM subjects by flow cytometry. We identified macrophages as CD68+ and represented TAM phenotype as M2 (CD163) versus M1 (CD80) marker ratio (Supp. Fig. S3). We demonstrated that TAMs in the BM of MM patients (n = 20) displayed elevated M2-like polarization (2.9-fold) compared to macrophages from healthy subjects (n = 5) (Fig. 1Ai). We further found that the levels of M2 polarization correlated with the stage of the disease, in which macrophages from the BM of relapsed refractory patients showed more M2 phenotype compared with these from newly diagnosed patients (Fig. 1Aii). Additionally, we examined TAM phenotype in vivo in a humanized mouse model bearing human MM. Compared to naïve mice, BM macrophages in MM-inoculated mice also displayed higher M2-like polarization (Fig. 1B).
To elucidate the effect of MM on TAMs in an in vitro/ex vivo setting, we utilized 3DTEBM to perform co-cultures of MM cells and macrophages. The 3DTEBM is a patient BM-derived 3D system that mimics the pathophysiology of the BM, and unlike traditional 2D cultures, 3DTEBM supports ex vivo primary cell proliferation and better recapitulate drug resistance, nutrient gradients, and interactions seen in patients [22, 25, 28]. Additionally, 3DTEBM allowed unique activation of macrophages compared to 2D cultures [29]. THP-1-derived macrophages (Fig. 1C) and PBMC-derived macrophages from three different healthy donors (Fig. 1D) co-cultured with three MM cell lines (MM1S, RPMI8226, and U266) exhibited about 2-fold and 2- to 6-fold, respectively, higher M2 polarization compared to cultures of macrophages alone. These results are consistent with findings from patient biopsies and mice BM.
We hypothesized that a secreted factor from myeloma cells played a role in inducing M2-phenotype in macrophage in the co-cultures. To test our hypothesis, THP-1-derived macrophages were cultured with increasing concentrations of MM conditioned media (CM) from three different MM cell lines (MM1S, RPMI8226, and U266), and the phosphorylation of key downstream signaling mediator of cytokine signaling (pSTAT3) was measured. There was a dose-dependent effect of MM conditioned media on STAT3 signaling in macrophages (Fig. 1E). These results confirm our hypothesis that a secreted factor from MM was responsible for the effect on macrophages.
Induction of MM in the BM induces increased secretion of IL-10
Next, we aimed to investigate which cytokine was responsible for the induction of the M2-polarization in the MM TME. We first examined the cytokine profile within the BM of immunocompetent MM-bearing C57BL/KaLwRij mice compared to naïve control. Multiplex immunoassay revealed that the induction of MM in mice lead to a 5-to 10-fold increase of several cytokines including IL-2, IL-5, IL-6, IL-18 and GM-CSF; however, the most significant cytokine change was observed with IL-10, in which it showed a dramatic increase of about 2,000-fold compared to naïve mice (Fig. 2A, B). Similarly, in vitro co-culture of human MM.1S cells and normal BM revealed that MM resulted in increased IL-10 production and was the most profound among the 42 cytokines tested (Supp. Fig. S4). In addition, analysis of BM supernatant from MM patients (n = 10) revealed IL-10 levels to be 3.9-fold higher than healthy subjects (n = 3) (Fig. 2C).
Fig. 2. Multiple myeloma produces high levels of IL-10.

A Cytokine screening of the BM supernatant of MM-bearing vs naïve mice. Cytokine levels in BM supernatants of naïve (n = 5) and MM-inoculated (n = 5) KaLwRij immunocompetent mice was measured by Luminex. Cytokine levels MM bearing mice are represented as fold-change of naïve mice. B Levels of IL-10 in the BM supernatant of MM-bearing vs naïve mice. IL-10 levels in BM supernatants of naïve (n = 5) and MM-inoculated (n = 5) KaLwRij immunocompetent mice was measured by Luminex. IL-10 levels we presented as pg/ml. C IL-10 concentrations in BM supernatants of healthy donors (n = 3) and MM patients (n = 10), measured using enzyme-linked immunosorbent assay (ELISA) and presented as pg/ml. (Bars: Average ± SEM, *p < 0.05).
IL-10 induced TAM M2 polarization and STAT3 phosphorylation in macrophages
To further investigate the role of IL-10 in macrophage polarization, THP-1-derived macrophages or macrophages derived from PBMCs of three different donors were cultured with or without exogenous recombinant human IL-10, and their polarization was analyzed by flow cytometry. IL-10 induced a 2-fold increase M2 polarization in THP-1-derived macrophages compared to untreated cells, and an even more robust 3–5-fold increase in PBMC-derived macrophages (Fig. 3A). Mechanistically, IL-10 induced an increase of STAT3 signaling in THP1-derived macrophages in a dose-dependent manner (Fig. 3B). Inhibition of IL-10R by a anti-IL-10R mAb reversed the IL-10-induced M2 polarization, in THP1-derived and in PBMC-derived macrophages (Fig. 3C). It is important to note that treatment with a Rat IgG2a isotype antibody did not reduce the IL-10-induced M2 polarization (Supplementary Fig. S5). Mechanistically, anti-IL-10R mAb downregulated the IL-10-induced STAT3 signaling (Fig. 3D). Furthermore, selective degradation of STAT3 protein by SD-36, a previously reported STAT3 PROTAC/degrader [30], diminished IL-10-induced M2 polarization, demonstrating that IL-10/IL-10R/STAT3 signaling axis is a key regulator of macrophage M2 polarization (Fig. 3E).
IL-10R inhibition reverses MM-induced TAM M2 polarization
With evidence showing that MM induces M2 polarization of macrophages, and that IL-10 is a highly upregulated cytokine in the MM TME with a critical role in TAMs M2 polarization, we hypothesized that inhibition of IL-10/IL-10R signaling will reverse the MM-induced M2-polarization of macrophages in the TME. To test our hypothesis, ex vivo 3DTEBM cultures derived from patient primary BM samples, with the whole BM microenvironment including MM cells and macrophages, were treated with or without an anti-IL-10R mAb. After 3 days, cultures were digested, and the polarization of macrophages was tested using flow cytometry. IL-10R inhibition significantly reduced M2 polarization in macrophages from patient-derived BM, ex vivo (Fig. 4A).
We further confirmed these findings in 3DTEBM co-cultures of macrophages with MM cell lines. THP-1-derived and PBMC-derived macrophages were co-cultured with different MM cell lines (MM1S, RPMI8226, and UT66) in the 3DTEBM model, with or without of anti-IL-10R mAb. Inhibition of IL-10/IL-10R significantly decreased M2 polarization compared to untreated control (Fig. 4B). It is important to note that an isotype control antibody did not reduce the MM-induced M2 polarization (Supp. Fig. S5), and it is worth noting that treatment with anti-IL-10R mAb did not result in any toxicity toward macrophages (Supp. Fig. S6).
We then examined the effect of IL-10/IL-10R axis inhibition on the polarization of TAMs in vivo. Humanized NCG mice were inoculated with human MM and treated with or without anti-IL-10R mAb twice a week for two weeks. The BM was then extracted, and TAM polarization was analyzed by flow cytometry. Similar to the observation in primary MM and in co-cultures of macrophages with MM cell lines, TAMs from MM-bearing mice treated with anti-IL-10R mAb displayed a decrease in M2 polarization compared to macrophages from untreated mice (Fig. 4C).
IL-10R inhibition reverses TAM-induced proliferation of MM
After characterizing the effect of MM on TAM polarization in vitro and in vivo, in the next part of this study, we aimed to investigate the effect of TAMs on MM proliferation and drug resistance. Co-culturing of MM cells with macrophages in the 3DTEBM elevated proliferation of MM cells compared to MM cell monoculture, by 1.4- and 2.0-fold, at day 3 and day 7, respectively (Fig. 5A). Mechanistically, co-culture of MM cells with macrophages increased MM cell cycle and proliferation cell signaling, as demonstrated with increased pRb expression for G1 to S phase cell cycle transition, as well as pAKT and pS6R for proliferation. Moreover, inhibition of IL-10/IL10R by anti-IL-10R mAb reversed macrophage-induced overexpression of pRb, pAKT and pS6R in MM cells (Fig. 5B). Furthermore, treatment with anti-IL-10R mAb reversed the macrophage-induced MM proliferation advantage, but did not exert a change in MM cell proliferation when cultured alone (Fig. 5C). It is important to note that an isotype control antibody did not reduce the macrophage-induced proliferation of MM (Supp. Fig. 7).
Fig. 5. IL-10R inhibition reverse TAM-induced MM proliferation.

A Effect of macrophages on MM proliferation. MM.1S-CBR-GFP cells were cultured alone or co-cultured at 1:1 ratio with THP-1-derived macrophages for 3 or 7 days in 3DTEBM. MM proliferation is determined by GFP cell count, presented as % of MM alone. Statistical significance is compared between the co-culture condition and the respective monoculture condition of each day. B Effect of IL-10R inhibition on TAM-induced MM proliferation and cell cycle signaling. THP-1-derived macrophages were pretreated with or without of anti-IL-10R mAb (5µg/ml) for 4 h, and MM.1S cells were cultured alone or with macrophages at 1:1 ratio for 24 h. MM.1S cells were then detached from macrophages, lyzed, and proteins were analyzed by western for proliferation (pAKT and S6R) and cell cycle signaling (pRB). Actin was blotted as the loading control. C Effect of IL10R inhibition on TAM-induced MM proliferation. MM.1S-CBR-GFP cells were cultured alone or co-cultured at 1:1 ratio with THP-1-derived macrophages in 3DTEBM and treated with or without anti-IL-10R mAb (5 µg/ml) for 3 days. MM survival is determined by GFP cell count by flow cytometry, presented as % of MM monoculture NT condition. Statistical significance is compared between anti-IL-10R mAb and NT. D Effect of anti-IL-10R mAb on in vivo MM tumor progression. NSG mice (n = 14) were inoculated with MM.1S-CBR-GFP and injected with human PBMC-derived macrophages. Treatment started at day 14 post-inoculation, mice were randomly divided into two groups which were treated with vehicle control (n = 7), or anti-IL-10R mAb (5 mg/kg, i.p, twice/week, n = 7). Tumor progression was measured at days 14, 28, and 42 by BLI, and represented as fold of day 14. Statistical significance is compared between anti-IL-10R mAb and NT. (Bars = Average ± SD, **p < 0.01; ***p < 0.001).
Then, we explored the effect of anti-IL-10R mAb as monotherapy on MM proliferation in vivo. NCG mice were inoculated with human MM and PBMC-derived macrophages, and 14 days later, treated with vehicle control or anti-IL-10R mAb, and tumor burden was measured by BLI. Treatment with anti-IL-10R mAb dramatically reduced MM tumor progression in mice compared to vehicle control (Fig. 5D).
IL-10R inhibition reverses TAM-induced drug resistance in MM
Next, we investigated the effect of macrophages and the inhibition IL-10/IL-10R axis on MM drug resistance to frontline anti-MM therapies, in vitro and in vivo. In MM monocultures in the 3DTEBM, lenalidomide induced 60% killing of MM cells, and the combination of lenalidomide with anti-IL-10R mAb did not change the sensitivity of MM cells to lenalidomide (Fig. 6A). On the other hand, co-culture of MM cells with macrophages in the 3DTEBM resulted in significant resistance to lenalidomide therapy, in which lenalidomide induced only 20% of MM killing (compared to 60% killing in monocultures). Moreover, inhibition of the IL-10/IL-10R axis with anti-IL-10R mAb re-sensitized MM cells to lenalidomide, in which the combination of anti-IL-10R mAb and lenalidomide synergized and induced killing of 83% of MM cells, reversing TAM-induced drug resistance in MM in vitro (Fig. 6A). Similarly, anti-IL-10R mAb reversed TAM-induced drug resistance to dexamethasone in MM cells but had no effect on drug sensitivity in MM monocultures without the presence of macrophages (Supp. Fig. S8).
Fig. 6. Anti-IL10R mAb reverses MM drug resistance to lenalidomide in vitro and in vivo.

A Effect of IL-10R inhibition on TAM-induced MM drug resistance to lenalidomide (LEN), in vitro. MM.1S-CBR-GFP cells were cultured alone or at 1:1 ratio with THP-1-derived macrophages in 3DTEBM and treated with or without LEN (1 uM), with or without anti-IL-10R mAb (5 µg/ml) for 3 days. MM survival was determined by GFP cell count by flow cytometry, represented as % of untreated MM respective mono- or co-culture. Statistical significance is compared between the combination and LEN-only conditions. B Effect of IL-10R inhibition on TAM-induced MM drug resistance to lenalidomide (LEN), in vivo. NSG mice (n = 14) were inoculated with MM.1S-CBR-GFP and injected with human PBMC-derived macrophages. Treatment started at day 14 post-inoculation) for all cohort (n = 14), mice were randomly divided into two groups which were treated with LEN alone (4mg/kg, Per Os, Daily, n = 7), or a combination of LEN with anti-IL10R mAb (2.5 mg/kg, i.p., twice/week, n = 7). Tumor progression was measured at days 14, 28 and 42 by BLI, and represented as fold of day 14. Statistical significance is compared between LEN alone and the combination of LEN + anti-IL-10R mAb. (Bars= Average ±SD,*: p < 0.05; ***: p < 0.001).
Lastly, we investigated the effect IL-10/IL-10R axis inhibition on resistance to lenalidomide, in vivo. NCG mice were inoculated with human MM and PBMC-derived macrophages, and 14 days later, treated with lenalidomide alone or lenalidomide in combination with anti-IL-10R mAb, and tumor burden was measured by BLI. In our previous in vivo experiment, mice treated with 5 mg/kg α-IL-10R mAbs exhibited slow tumor growth, therefore we decreased the dose to 2.5 mg/kg in this experiment to explore the combination effect. Consistent with our in vitro observation, treatment with anti-IL-10R mAb sensitized MM tumors to lenalidomide and enhanced its effect compared to lenalidomide monotherapy (Fig. 6B).
Discussion
Immunotherapeutic strategies are expanding to better elicit activation of various components of the immune system [31]. Many studies has been focused on engagement and activation of the adaptive immune system [32–34]. Recently, attention has been shifted to potentiate activity of the innate immune system in general [35] and macrophages in particular [36]. In the context of cancer, TAMs are a prominent population within the TME [37], especially in MM, TAMs constitute around 10% of the BM, and correlate negatively with patient survival [14, 38]. Several strategies have been explored to target TAMs in a variety of cancers, including reducing TAMs, reprogramming TAMs, inhibiting the CD47/SIRPα checkpoint, and overcoming immunosuppression [13]. IL-10 was previously shown to have an immunosuppressive role in the TME, especially in TAM polarization to M2 [39–42]; however, IL-10/IL-10R pathway has not been explored as an immunotherapeutic target in the field of TAMs, and limited data is available regarding IL-10/STAT3 in the biology of MM. One study showed that high levels of IL-10 in serum correlated with poor prognosis in newly diagnosed MM patients [16]. To our knowledge, this is the first study to investigate the biological role of IL-10 in the progression of MM, and the first to target the IL-10/IL-10R/STAT3 as a therapeutic strategy for the reprograming of macrophages in MM or any other type of cancer.
The results demonstrate an increased prevalence of M2-like TAMs in the MM TME, a phenomenon we observed in BM samples of MM patients compared to normal subjects, which correlated with the progression of the disease in patients, as well as in vivo and in vitro models of MM. M2-like TAM phenotype was a direct result of introducing MM to the BM microenvironment, including the induction of this phenotype by MM-conditioned media. This suggested that the effect was driven by a secreted cytokine. Therefore, we investigated the changes in secreted cytokines in the BM of MM-bearing mice and found a dramatic 2000-fold increase of the anti-inflammatory cytokine IL-10. We further confirmed that IL-10 levels were also increased in the BM supernatants of MM patients compared to normal subjects. Then, we investigated the role of IL-10 in TAMs in the MM TME. The results showed that IL-10 induced an M2-like phenotype in macrophages through STAT3 signaling and that the inhibition of the IL-10/IL-10R axis using an anti-IL-10R mAb or a STAT3 protein degrader, significantly reduced the IL-10 driven M2 polarization. Anti-IL-10R mAb reversed M2 polarization in primary MM patient cultures ex vivo, in MM models in vivo, and in MM cell line-based models in vitro. These results provide direct evidence that IL-10 is overexpressed in the TME due to MM progression, and that it induces an M2-like phenotype in TAMs, which emphasizes that targeting and inhibiting IL-10/IL10R/STAT3 axis can be used to reprogram TAMs, as a potential novel immuno-therapeutic strategy.
We have previously demonstrated that MM progression and drug resistance is heavily dependent on the interaction with different compartments of the TME [43] including stromal cells [2, 44–46], endothelial cells [44, 47, 48], and others[49–52]. We have also shown that inhibition of the interaction of MM with the TME increases the sensitivity of MM cells to different therapies, including proteasome inhibitors[2, 25, 27, 44, 48], immunomodulators[23, 25, 47], and others[23, 25, 49, 51–53]. This study demonstrates that macrophages induce MM proliferation through activation of proliferative cell signaling in MM cells, including the PI3K pathway and cell cycle. Importantly, inhibition of the IL-10/IL-10R axis reversed the macrophage-induced MM proliferation in vitro, while it had no direct effect on MM cells themselves, without the presence of macrophages. Furthermore, inhibition of IL-10/IL-10R dramatically reduced the rate of progression of MM, in vitro and in vivo. It also demonstrates that macrophages induced resistance to immunomodulatory drugs (lenalidomide and dexamethasone) in MM, which is in agreement with previous reports showing that macrophages drive resistance to dexamethasone in MM [54]. Most importantly, the inhibition of IL-10/IL-10R axis significantly restored the sensitivity of MM cells to lenalidomide in vitro and in vivo (and dexamethasone in vitro). Interestingly, BM macrophages from relapsed/ refractory MM patients were found to be significantly more skewed to the M2 phenotype compared to newly diagnosed MM, which presents additional evidence for the tumor-supportive and resistance-promoting role of MM-associated TAMs, and warrants further studies to target MM-TAM interactions in drug resistant MM.
Our findings present a novel strategy to reprogram TAMs, inhibiting a tumor-supporting property to harness their killing capabilities to supplement current MM treatment. Additionally, macrophage- infiltrating solid tumors, such as breast cancer, have also been shown to be a source of IL-10 production, which is associated with drug resistance [55]. Thus, therapeutic blockade of the IL-10/IL-10R/STAT3 axis could be a promising strategy beyond MM.
In conclusion, we have shown that MM cells release high levels of IL-10, which induces polarization of macrophages to a pro-tumor M2-like phenotype. In turn, macrophages support proliferation and drug resistance in MM cells, creating a vicious cycle of tumor progression that leads to immuno-supression in the form of M2-like macrophage polarization that leads to more tumor progression. Targeting the IL-10/IL-10R/STAT3 axis with an anti-IL-10R mAb reversed MM-induced M2-polarization of TAM, which further inhibited TAM-induced proliferation and drug resistance in MM. We therefore suggest targeting IL-10/IL-10R/STAT3 as a novel therapeutic strategy in MM, and beyond.
Supplementary information
Acknowledgements
We thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis, MO., for the use of the Bursky Center Immunmonitoring Laboratory which provided the Luminex immunoassay service. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant #P30 CA091842. The Shokeen Lab thanks Drs. Hathi, Cho and Ghai for help with sample preparation and data organization.
Author contributions
J.S. conceived the idea, designed and conducted experiments, analyzed the data, and wrote the manuscript. F.A., S.C., B.M., K.M., M.M., C.D., O.A., K.A., C.P., B.L., Y.C., O.A., H.B., D.E.B. and H.Z. conducted experiments and analyzed data. S.K. and M.F. provided clinical samples. R.V. designed experiments and provided clinical samples. M.S., M.T.S.W. and S.W. designed experiments and analyzed data. A.K.A. conceived the idea, designed experiments, analyzed the data, wrote the manuscript, and supervised the study. All authors reviewed and approved the manuscript.
Funding
Dr. Azab received funding for this study from the Paula C. and Rodger O. Riney Blood Cancer Research Initiative Fund, and from the Cancer Prevention & Research Institute of Texas (CPRIT # RR220088). Jennifer Sun was supported by the Spencer T. and Ann W. Olin Fellowship for Women in Graduate Study at the Washington University in St. Louis.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
Competing interests
Dr. Azab is the founder and owner of Cellatrix LLC that has an exclusive license for the 3DTEBM technology described in part of the experiments in this paper; however, Cellatrix has no contribution to this study. Other authors state no conflicts of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41375-024-02391-8.
References
- 1.van de Donk NWCJ, Pawlyn C, Yong KL. Multiple myeloma. Lancet. 2021;397:410–27. [DOI] [PubMed] [Google Scholar]
- 2.Azab AK, Runnels JM, Pitsillides C, Moreau A-S, Azab F, Leleu X, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawano Y, Moschetta M, Manier S, Glavey S, Görgün GT, Roccaro AM, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunological Rev. 2015;263:160–72. [DOI] [PubMed] [Google Scholar]
- 4.Uckun FM. Dual targeting of multiple myeloma stem cells and myeloid-derived suppressor cells for treatment of chemotherapy-resistant multiple myeloma. Front Oncol. 2021;11. [DOI] [PMC free article] [PubMed]
- 5.Shah UA, Mailankody S. Emerging immunotherapies in multiple myeloma. Bmj. 2020. [DOI] [PubMed]
- 6.Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-associated macrophages: recent insights and therapies. Front Oncol. 2020;10. [DOI] [PMC free article] [PubMed]
- 7.Zhang C, Yang M, Ericsson AC. Function of macrophages in disease: current understanding on molecular mechanisms. Front Immunol. 2021;12. [DOI] [PMC free article] [PubMed]
- 8.Komohara Y, Niino D, Saito Y, Ohnishi K, Horlad H, Ohshima K, et al. Clinical significance of CD163+ tumor‐associated macrophages in patients with adult T‐cell leukemia/lymphoma. Cancer Sci. 2013;104:945–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong N, He Z, Ma Y, Wang Z, Huang Z, Cao H, et al. Tumor associated macrophages, as the dominant immune cells, are an indispensable target for immunologically cold tumor—glioma therapy? Front Cell Develop Biol. 2021;9. [DOI] [PMC free article] [PubMed]
- 10.Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H, et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/β-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018;9. [DOI] [PMC free article] [PubMed]
- 11.Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Science. 2019;26. [DOI] [PMC free article] [PubMed]
- 12.Cencini E, Fabbri A, Sicuranza A, Gozzetti A, Bocchia M. The role of tumor-associated macrophages in hematologic malignancies. Cancers. 2021;13. [DOI] [PMC free article] [PubMed]
- 13.Sun J, Park C, Guenthner N, Gurley S, Zhang L, Lubben B, et al. Tumor-associated macrophages in multiple myeloma: advances in biology and therapy. J Immunother Cancer. 2022;10. [DOI] [PMC free article] [PubMed]
- 14.Wang H, Hu W-m, Xia Z-j, Liang Y, Lu Y, Lin S-x, et al. High numbers of CD163+ tumor-associated macrophages correlate with poor prognosis in multiple myeloma patients receiving bortezomib-based regimens. J Cancer. 2019;10:3239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopes RL, Borges TJ, Zanin RF, Bonorino C. IL-10 is required for polarization of macrophages to M2-like phenotype by mycobacterial DnaK (heat shock protein 70). Cytokine. 2016;85:123–9. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Wang L, Chi P-d, Wang W-d, Chen X-q, Geng Q-r, et al. High level of interleukin-10 in serum predicts poor prognosis in multiple myeloma. Br J Cancer. 2016;114:463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, et al. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Advances in Immunology2014. p. 177-210. [DOI] [PMC free article] [PubMed]
- 18.Chen L, Shi Y, Zhu X, Guo W, Zhang M, Che Y, et al. IL‑10 secreted by cancer‑associated macrophages regulates proliferation and invasion in gastric cancer cells via c‑Met/STAT3 signaling. Oncology Reports. 2019. [DOI] [PMC free article] [PubMed]
- 19.Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–81. [DOI] [PubMed] [Google Scholar]
- 20.Maes K, Boeckx B, Vlummens P, De Veirman K, Menu E, Vanderkerken K, et al. The genetic landscape of 5T models for multiple myeloma. Sci Rep. 2018;8:15030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun J, Muz B, Alhallak K, Markovic M, Gurley S, Wang Z, et al. Targeting CD47 as a novel immunotherapy for multiple myeloma. Cancers. 2020;12. [DOI] [PMC free article] [PubMed]
- 22.Alhallak K, de la Puente P, Jeske A, Sun J, Muz B, Rettig MP, et al. 3D tissue engineered plasma cultures support leukemic proliferation and induces drug resistance. Leuk Lymphoma. 2021;62:2457–65. [DOI] [PubMed] [Google Scholar]
- 23.Alhallak K, Jeske A, de la Puente P, Sun J, Fiala M, Azab F, et al. A pilot study of 3D tissue-engineered bone marrow culture as a tool to predict patient response to therapy in multiple myeloma. Sci Rep. 2021;11:19343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Puente P, Azab AK. 3D tissue-engineered bone marrow: what does this mean for the treatment of multiple myeloma? Future Oncol. 2016;12:1545–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de la Puente P, Muz B, Gilson RC, Azab F, Luderer M, King J, et al. 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials. 2015;73:70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alhallak K, Sun J, Wasden K, Guenthner N, O’Neal J, Muz B, et al. Nanoparticle T-cell engagers as a modular platform for cancer immunotherapy. Leukemia. 2021;35:2346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de la Puente P, Luderer MJ, Federico C, Jin A, Gilson RC, Egbulefu C, et al. Enhancing proteasome-inhibitory activity and specificity of bortezomib by CD38 targeted nanoparticles in multiple myeloma. J Control Release. 2018;270:158–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alhallak K, Jeske A, de la Puente P, Sun J, Fiala M, Azab F, et al. A pilot study of 3D tissue-engineered bone marrow culture as a tool to predict patient response to therapy in multiple myeloma. Scientific Reports. 2021;11. [DOI] [PMC free article] [PubMed]
- 29.Sun J, Chen Y, Lubben B, Adebayo O, Muz B, Azab AK. CD47-targeting antibodies as a novel therapeutic strategy in hematologic malignancies. Leuk Res Rep. 2021;16:100268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36:498–511.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30:507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN Guidelines with the level of evidence. Cancers. 2020;12. [DOI] [PMC free article] [PubMed]
- 33.Alhallak K, Sun J, Jeske A, Park C, Yavner J, Bash H, et al. Bispecific T cell engagers for the treatment of multiple myeloma: achievements and challenges. Cancers. 2021;13. [DOI] [PMC free article] [PubMed]
- 34.van de Donk NWCJ, Usmani SZ, Yong K. CAR T-cell therapy for multiple myeloma: state of the art and prospects. Lancet Haematol. 2021;8:e446–e61. [DOI] [PubMed] [Google Scholar]
- 35.Rothlin CV, Ghosh S. Lifting the innate immune barriers to antitumor immunity. J Immunother Cancer. 2020;8. [DOI] [PMC free article] [PubMed]
- 36.Hirayama D, Iida T, Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Inter J Mol Sciences. 2017;19. [DOI] [PMC free article] [PubMed]
- 37.Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Targeted Ther. 2021;6. [DOI] [PMC free article] [PubMed]
- 38.Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood. 2009;114:3625–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boehler RM, Kuo R, Shin S, Goodman AG, Pilecki MA, Gower RM, et al. Lentivirus delivery of IL-10 to promote and sustain macrophage polarization towards an anti-inflammatory phenotype. Biotechnol Bioeng. 2014;111:1210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dominguez-Soto A, Sierra-Filardi E, Puig-Kroger A, Perez-Maceda B, Gomez-Aguado F, Corcuera MT, et al. Dendritic cell-specific ICAM-3-grabbing nonintegrin expression on M2-polarized and tumor-associated macrophages is macrophage-CSF dependent and enhanced by tumor-derived IL-6 and IL-10. J Immunol. 2011;186:2192–200. [DOI] [PubMed] [Google Scholar]
- 41.Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, et al. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. 2017;112:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mia S, Warnecke A, Zhang XM, Malmstrom V, Harris RA. An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-beta yields a dominant immunosuppressive phenotype. Scand J Immunol. 2014;79:305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de la Puente P, Azab AK. Contemporary drug therapies for multiple myeloma. Drugs Today (Barc). 2013;49:563–73. [DOI] [PubMed] [Google Scholar]
- 44.Federico C, Alhallak K, Sun J, Duncan K, Azab F, Sudlow GP, et al. Tumor microenvironment-targeted nanoparticles loaded with bortezomib and ROCK inhibitor improve efficacy in multiple myeloma. Nat Commun. 2020;11:6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de la Puente P, Quan N, Hoo RS, Muz B, Gilson RC, Luderer M, et al. Newly established myeloma-derived stromal cell line MSP-1 supports multiple myeloma proliferation, migration, and adhesion and induces drug resistance more than normal-derived stroma. Haematologica. 2016;101:e307–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muz B, Azab F, Fiala M, King J, Kohnen D, Fogler WE, et al. Inhibition of E-Selectin (GMI-1271) or E-selectin together with CXCR4 (GMI-1359) re-sensitizes multiple myeloma to therapy. Blood Cancer J. 2019;9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Azab AK, Quang P, Azab F, Pitsillides C, Thompson B, Chonghaile T, et al. P-selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment. Blood. 2012;119:1468–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muz B, Buggio M, Azab F, de la Puente P, Fiala M, Padval MV, et al. PYK2/FAK inhibitors reverse hypoxia-induced drug resistance in multiple myeloma. Haematologica. 2019;104:e310–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muz B, Kusdono HD, Azab F, de la Puente P, Federico C, Fiala M, et al. Tariquidar sensitizes multiple myeloma cells to proteasome inhibitors via reduction of hypoxia-induced P-gp-mediated drug resistance. Leuk Lymphoma. 2017;58:2916–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muz B, Azab F, de la Puente P, Landesman Y, Azab AK. Selinexor Overcomes Hypoxia-Induced Drug Resistance in Multiple Myeloma. Transl Oncol. 2017;10:632–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de la Puente P, Muz B, Jin A, Azab F, Luderer M, Salama NN, et al. MEK inhibitor, TAK-733 reduces proliferation, affects cell cycle and apoptosis, and synergizes with other targeted therapies in multiple myeloma. Blood Cancer J. 2016;6:e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Azab F, Vali S, Abraham J, Potter N, Muz B, de la Puente P, et al. PI3KCA plays a major role in multiple myeloma and its inhibition with BYL719 decreases proliferation, synergizes with other therapies and overcomes stroma-induced resistance. Br J Haematol. 2014;165:89–101. [DOI] [PubMed] [Google Scholar]
- 54.Chen X, Chen J, Zhang W, Sun R, Liu T, Zheng Y, et al. Prognostic value of diametrically polarized tumor-associated macrophages in multiple myeloma. Oncotarget. 2017;8:112685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26:623–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.