

Abstract
Background
Adverse drug events, encompassing both adverse drug reactions and medication errors, pose a significant threat to health, leading to illness and, in severe cases, death. Timely and voluntary reporting of adverse drug events by healthcare professionals plays a crucial role in mitigating the morbidity and mortality linked to unexpected reactions and improper medication usage.
Objectives
To assess the effectiveness of different interventions aimed at healthcare professionals to improve the reporting of adverse drug events.
Search methods
We searched CENTRAL, Embase, MEDLINE and several other electronic databases and trials registers, including ClinicalTrials.gov and WHO ICTRP, from inception until 14 October 2022. We also screened reference lists in the included studies and relevant systematic reviews.
Selection criteria
We included randomised trials, non‐randomised controlled studies, controlled before‐after studies, interrupted time series studies (ITS) and repeated measures studies, assessing the effect of any intervention aimed at healthcare professionals and designed to increase adverse drug event reporting. Eligible comparators were healthcare professionals' usual reporting practice or a different intervention or interventions designed to improve adverse drug event reporting rate. We excluded studies of interventions targeted at adverse event reporting following immunisation. Our primary outcome measures were the total number of adverse drug event reports (including both adverse drug reaction reports and medication error reports) and the number of false adverse drug event reports (encompassing both adverse drug reaction reports and medication error reports) submitted by healthcare professionals. Secondary outcomes were the number of serious, high‐causality, unexpected or previously unknown, and new drug‐related adverse drug event reports submitted by healthcare professionals. We used GRADE to assess the certainty of evidence.
Data collection and analysis
We followed standard methods recommended by Cochrane and the Cochrane Effective Practice and Organisation of Care (EPOC) Group. We extracted and reanalysed ITS study data and imputed treatment effect estimates (including standard errors or confidence intervals) for the randomised studies.
Main results
We included 15 studies (eight RCTs, six ITS, and one non‐randomised cross‐over study) with approximately 62,389 participants. All studies were conducted in high‐income countries in large tertiary care hospitals. There was a high risk of performance bias in the controlled studies due to the nature of the interventions. None of the ITS studies had a control arm, so we could not be sure of the detected effects being independent of other changes. None of the studies reported on the number of false adverse drug event reports submitted.
There is low‐certainty evidence suggesting that an education session, together with reminder card and adverse drug reaction (ADR) report form, may substantially improve the rate of ADR reporting by healthcare professionals when compared to usual practice (i.e. spontaneous reporting with or without some training provided by regional pharmacosurveillance units). These educational interventions increased the number of ADR reports in total (RR 3.00, 95% CI 1.53 to 5.90; 5 studies, 21,655 participants), serious ADR reports (RR 3.30, 95% CI 1.51 to 7.21; 5 studies, 21,655 participants), high‐causality ADR reports (RR 2.48, 95% CI 1.11 to 5.57; 5 studies, 21,655 participants), unexpected ADR reports (RR 4.72, 95% CI 1.75 to 12.76; 4 studies, 15,085 participants) and new drug‐related ADR reports (RR 8.68, 95% CI 3.40 to 22.13; 2 studies, 7884 participants).
Additionally, low‐certainty evidence suggests that, compared to usual practice (i.e. spontaneous reporting), making it easier to report ADRs by using a standardised discharge form with added ADR items may slightly improve the total number of ADR reports submitted (RR 2.06, 95% CI 1.11 to 3.83; 1 study, 5967 participants). The discharge form tested was based on the ‘Diagnosis Related Groups’ (DRG) system for recording patient diagnoses, and the medical and surgical procedures received during their hospital stay.
Due to very low‐certainty evidence, we do not know if the following interventions have any effect on the total number of adverse drug event reports (including both ADR and ME reports) submitted by healthcare professionals:
‐ sending informational letters or emails to GPs and nurses;
‐ multifaceted interventions, including financial and non‐financial incentives, fines, education and reminder cards;
‐ implementing government regulations together with financial incentives;
‐ including ADR report forms in quarterly bulletins and prescription pads;
‐ providing a hyperlink to the reporting form in hospitals' electronic patient records;
‐ improving the reporting method by re‐engineering a web‐based electronic error reporting system;
‐ the presence of a clinical pharmacist in a hospital setting actively identifying adverse drug events and advocating for the identification and reporting of adverse drug events.
Authors' conclusions
Compared to usual practice (i.e. spontaneous reporting with or without some training from regional pharmacosurveillance units), low‐certainty evidence suggests that the number of ADR reports submitted may substantially increase following an education session, paired with reminder card and ADR report form, and may slightly increase with the use of a standardised discharge form method that makes it easier for healthcare professionals to report ADRs.
The evidence for other interventions identified in this review, such as informational letters or emails and financial incentives, is uncertain.
Future studies need to assess the benefits (increase in the number of adverse drug event reports) and harms (increase in the number of false adverse drug event reports) of any intervention designed to improve healthcare professionals' reporting of adverse drug events. Interventions to increase the number of submitted adverse drug event reports that are suitable for use in low‐ and middle‐income countries should be developed and rigorously evaluated.
Keywords: Humans, Adverse Drug Reaction Reporting Systems, Adverse Drug Reaction Reporting Systems/statistics & numerical data, Bias, Controlled Before-After Studies, Drug-Related Side Effects and Adverse Reactions, Health Personnel, Interrupted Time Series Analysis, Medication Errors, Medication Errors/prevention & control, Medication Errors/statistics & numerical data, Non-Randomized Controlled Trials as Topic, Randomized Controlled Trials as Topic
Plain language summary
Improving healthcare professionals' reporting of adverse drug reactions and medication errors
Key messages
‐ Healthcare professionals have a responsibility to report unexpected and harmful responses to medicines. These responses are known as 'adverse drug events', a term that includes both adverse drug reactions (ADRs) and medication errors (MEs).
‐ An education session (outreach, in‐person workshops or via telephone), along with providing a reminder card and ADR report form, may substantially increase the number of ADR reports submitted.
‐ Using a standardised discharge form with additional ADR items that is designed to make it easier to report ADRs may slightly increase the number of ADR reports submitted.
‐ Future studies need to assess the benefit (increase in the number of adverse drug event reports submitted) and harm (increase in the number of false adverse drug event reports submitted) of any intervention designed to improve healthcase professionals' reporting of adverse drug events.
‐ Interventions suitable for use in low‐ and middle‐income countries need to be developed and rigorously evaluated.
What did we want to find out?
This Cochrane review investigated whether interventions for healthcare professionals are effective for improving at their reporting of adverse drug events. Adverse drug events include any adverse drug reaction (ADR) and any medication error (ME).
What did we do?
We looked at evidence from a range of different types of studies to find out if interventions aimed at healthcare professionals could increase the number of adverse drug event reports they make. We compared the total number of adverse drug event reports (which included both ADR and ME reports) submitted by healthcare professionals. We were also interested in the number of false adverse drug event reports they made. As well as the total number of reports, we looked separately at the number of reports submitted for adverse drug events that were categorised as serious, high‐causality (i.e. very likely to be caused by the drug), unexpected (i.e. previously unknown) or related to recent drugs (i.e. only used in the last five years).
What did we find?
This review included 15 studies (62,389 participants) that compared the effect of various interventions aimed at healthcare professionals to increase the number of adverse drug event reports they make. All the studies were carried out in high‐income countries. None of the studies looked at whether these interventions led to more false adverse drug event reports.
Compared to usual practice (spontaneous reporting and some training from regional units that monitor the safety of medicines), an education session about why and how to report adverse events, plus reminder of the session content and provision of an ADR report form, may increase the number of ADR reports made by healthcare professionals.
Compared to usual practice (spontaneous reporting), using a standardised discharge form with additional ADR items about when the ADR occurred and how it developed may also slightly improve the number of ADR reports made. The standardised form tested was based on the ‘Diagnosis Related Groups’ system for recording patient diagnoses and the medical and surgical procedures patients receive during their hospital stay.
We are very uncertain about the effectiveness of other interventions that were tested in the studies, including:
‐ sending informational letters or emails to GPs and nurses;
‐ interventions with multiple aspects, including financial and non‐financial incentives, fines, education and reminder cards;
‐ implementing government regulations together with financial incentives;
‐ including ADR report forms in quarterly bulletins and prescription pads;
‐ providing a hyperlink to the reporting form in hospitals' electronic patient records;
‐ improving the reporting method by re‐engineering the web‐based electronic error reporting system;
‐ the presence of a clinical pharmacist in hospital who actively identifies adverse drug events and encourages the identification and reporting of adverse drug events.
How up to date is this review?
The evidence in this review is based on searches up to October 2022.
Summary of findings
Summary of findings 1. Education session plus reminder card and ADR report form versus usual practice.
|
Participants: physicians and pharmacists Intervention: education session (in‐person workshop or via telephone), reminder card and ADR report form Comparator: usual practice (spontaneous reporting; briefing and standard training given by regional pharmacosurveillance unit) Setting: hospitals and outpatient centres in Northern Portugal and Spain | ||||||
| Outcomes |
Risk ratio* (95% CI) |
Illustrative comparative risks‡ (95% CI) |
Number of participants (studies) |
Certainty of the evidence (GRADE) |
Comments | |
| Assumed risk with usual practice | Corresponding risk with education session plus reminder card and report form | |||||
| Total number of ADE reports (including ADR reports and ME reports): number of ADR reports Follow‐up: 13 to 16 months |
3.00 (1.53 to 5.90) | 80 ADR reports per 1000 practitioner years | 240 ADR reports per 1000 practitioner years (122 to 472) | 21,665 (5 cRCTs)1 | Low4 | An education session, together with reminder card and ADR report form, may improve the reporting rate of ADRs. |
| Total number of false ADE reports (including false ADR reports and false ME reports) | None of the included studies reported on this outcome. | |||||
| Number of serious ADE reports (including serious ADR reports and serious ME reports) Follow‐up: 13 to 16 months |
3.30 (1.51 to 7.21) | 10 ADR reports per 1000 practitioner years | 33 ADR reports per 1000 practitioner years (15 to 72) | 21,665 (5 cRCTs)1 | Low5 | An education session, together with reminder card and ADR report form, may improve the reporting rate of serious ADRs. |
| Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports) Follow‐up: 13 to 16 months |
2.48 (1.11 to 5.57) | 20 ADR reports per 1000 practitioner years | 50 ADR reports per 1000 practitioner years (22 to 111) | 21,665 (5 cRCTs)1 | Low6 | An education session, together with reminder card and ADR report form, may improve the reporting rate of high‐causality ADRs. |
| Number of unexpected ADE reports (including unexpected ADR reports and unexpected ME reports) Follow‐up: 13 to 16 months |
4.72 (1.75 to 12.76) | 20 ADR reports per 1000 practitioner years | 94 ADR reports per 1000 practitioner years (35 to 255) | 15,085 (4 cRCTs)2 | Low7 | An education session, together with reminder card and ADR report form, may improve the reporting rate of unexpected ADRs |
| Number of new‐drug‐related ADE reports (including drug‐related ADR reports and drug‐related ME reports) Follow‐up: 13 to 16 months |
8.68 (3.40 to 22.13) | 5 ADR reports per 1000 practitioner years | 43 ADR reports per 1000 practitioner years (17 to 111) | 7884 (2 cRCTs)3 | Low8 | An education session, together with reminder card and ADR report form, may improve the reporting rate of new‐drug‐related ADRs. |
ADE: adverse drug event; ADR: adverse drug reaction; CI: confidence interval; cRCT: cluster‐randomised controlled trial; ME: medication error; vs: versus
*Risk ratios > 1 are associated with more ADRs with education session plus reminder card and ADR report form versus usual practice.
‡Illustrative comparative risks are presented as numbers of ADRs per 1000 practitioner years and are rounded to whole numbers.
1Figueiras 2006 (physicians, education group session); Herdeiro 2008 (pharmacists, education group session); Herdeiro 2012 (physicians; same intervention clusters from Herdeiro 2008 randomised a second time to telephone interview or workshop); Lopez‐Gonzalez 2015 (physicians, education group session); Ribeiro‐Vaz 2011 (pharmacists, telephone interview or workshop)
2Figueiras 2006 (physicians); Herdeiro 2008 (pharmacists); Lopez‐Gonzalez 2015 (physicians); Ribeiro‐Vaz 2011 (pharmacists)
3Figueiras 2006; Herdeiro 2008
4Downgraded once for serious risk of bias (performance bias and potential selection bias due to baseline differences in reporting rates between intervention and control group; see Figueiras 2006; Herdeiro 2012; Ribeiro‐Vaz 2011); downgraded once for serious inconsistency: I2 = 95%. The inconsistency might be explained by the mode of delivery of the education (i.e. telephone vs interactive group session vs workshop) or the different target audience (physicians vs pharmacist), but we are uncertain of this; no serious imprecision; no serious indirectness; no publication bias.
5Downgraded once for serious risk of bias (performance bias and potential selection bias due to baseline differences in reporting rates between intervention and control group; see Figueiras 2006; Herdeiro 2012; Ribeiro‐Vaz 2011); downgraded once for serious inconsistency: I2 = 96%. The inconsistency might be explained by the mode of delivery of the education (i.e. telephone vs interactive group session vs workshop) or the different target audience (physicians vs. pharmacist), or both, but we are uncertain of this; no serious imprecision; no serious indirectness; no publication bias.
6Downgraded once for serious risk of bias (performance bias and potential selection bias due to baseline differences in reporting rates between intervention and control group; see Figueiras 2006; Herdeiro 2012; Ribeiro‐Vaz 2011); downgraded once for serious inconsistency: I2 = 100%. The inconsistency might be explained by the mode of delivery of the educational outreach (i.e. telephone vs interactive group session vs workshop) or the different target audience (physicians vs pharmacist), but we are uncertain of this; no serious imprecision; no serious indirectness; no publication bias.
7Downgraded once for serious risk of bias (performance bias and potential selection bias due to baseline differences in reporting rates between intervention and control group; see Figueiras 2006; Ribeiro‐Vaz 2011); downgraded once for serious inconsistency: I2 = 64%. The inconsistency might be explained by the mode of delivery of the education (i.e. telephone vs interactive group session vs workshop) or the different target audience (physicians vs pharmacist), but we are uncertain of this; no serious imprecision; no serious indirectness; no publication bias.
8Downgraded once for serious risk of performance bias and potential selection bias due to baseline differences in reporting rates between intervention and control group (see Figueiras 2006); no serious inconsistency; downgraded once for serious imprecision (wide confidence intervals so uncertain of the true estimate of effect); no serious indirectness; no publication bias
Summary of findings 2. Informational letter or email versus usual practice.
|
Participants: general practitioners and nurses Intervention: informational letter or email Comparator: usual practice (spontaneous reporting) Setting: primary healthcare units in Sweden | ||||||
| Outcomes | Rate ratio* (95% CI) | Illustrative comparative rates‡(95% CI) | Exposure†(studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed rate with usual practice | Corresponding rate with informational letter or email | |||||
| Total number of ADE reports (including ADR reports and ME reports): number of ADR reports after one year | 1.28 (0.42 to 3.91) | 80 ADR reports per 100 practitioner years | 102 ADR reports per 100 practitioner years (34 to 313) | 268 primary healthcare unit years (2 RCTs)1 | Very low2 | We do not know if informational letters or emails to GPs and nurses increase the total number of ADR reports because the evidence is very uncertain. |
| Total number of false ADE reports (including false ADR reports and false ME reports) | None of the included studies reported on this outcome. | |||||
| Number of serious ADE reports (including serious ADR reports and serious ME reports): number of serious ADR reports after one year | 1.79 (0.69 to 4.65) | 10 ADR reports per 100 practitioner years | 18 ADR reports per 100 practitioner years (7 to 47) | 268 primary healthcare unit years (2 RCTs)1 | Very low2 | We do not know if informational letters or emails to GPs and nurses increase serious ADR reports because the evidence is very uncertain. |
| Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports) | None of the included studies reported on this outcome. | |||||
| Number of unexpected ADE reports (including unexpected ADR reports and unexpected ME reports): number of unexpected ADR reports after one year | 1.46 (0.92 to 2.30) | 20 ADR reports per 100 practitioner years | 29 ADR reports per 100 practitioner years (18 to 46) | 268 primary healthcare unit years (2 RCTs)1 | Very low2 | We do not know if informational letters or emails to GPs and nurses increase the number of unexpected ADR reports as the certainty of the evidence is very low. |
| Number of new drug‐related ADE reports (including drug‐related ADR reports and drug‐related ME reports): number of new drug‐related ADR reports after one year | 2.58 (1.12 to 5.92) | 5 ADR reports per 100 practitioner years | 13 ADR reports per 100 practitioner years (6 to 30) | 268 primary healthcare unit years (2 RCTs)1 | Very low3 | We do not know if informational letters or emails to GPs and nurses increase the total number of new drug‐related ADR reports because the evidence is very uncertain. |
ADE: adverse drug event; ADR: adverse drug reaction; CI: confidence interval; RCT: randomised controlled trials; ME: medication error; vs: versus
*Rate ratios > 1 are associated with more ADRs with informational letter or email versus usual practice.
†Unit of exposure is primary healthcare unit years.
‡Illustrative comparative rates are presented as numbers of ADR reports per 100 practitioner years and are rounded to whole numbers.
1Johansson 2009; Johansson 2011
2Downgraded once for serious risk of bias (performance bias and potential contamination bias); no serious inconsistency; downgraded twice for very serious imprecision: wide confidence intervals that cross the line of no effect (in the case of total number of ADR reports, number of serious ADR reports and number of unexpected ADR reports), small event rate (total of 242 ADR reports from 268 units in 2007 and 2008, total of 35 serious ADR reports from 268 units in 2007 and 2008, total of 85 unexpected ADR reports from 268 units in 2007 and 2008); no serious indirectness; no publication bias
3Downgraded once for serious risk of bias (performance bias and potential contamination bias); no serious inconsistency; downgraded twice for very serious imprecision: wide confidence intervals, small event rate (total of 16 new drug‐related ADRs reported from 268 units in 2007 and 2008); no serious indirectness; no publication bias
Summary of findings 3. Multifaceted interventions versus usual practice.
|
Participants: physicians and pharmacists Intervention: multifaceted intervention (including financial incentives, fines, non‐financial incentives, education, reminders) Comparator: usual practice (spontaneous reporting) Setting: hospital | ||||||
| Outcome | Relative numbers of ADES* (95% CI) | Illustrative comparative numbers of ADEs‡(95% CI) | Mean study duration (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed number with usual practice | Corresponding number with multifaceted intervention | |||||
| Total number of ADE reports (including ADR reports and ME reports): number of ADR reports after one year | 4.29 (0.32 to 56.76) | 80 ADR reports per 1000 practitioners | 343 ADR reports per 1000 practitioners (26 to 4541) | 6.5 years (2)1 | Very low2 | We do not know if multifaceted interventions increase the total number of ADR reports in physicians and pharmacists one year after implementation because the evidence is very uncertain.3 Data after two years in footnotes4 |
| Total number of false ADE reports (including false ADR reports and false ME reports) | None of the included studies reported on this outcome. | |||||
| Number of serious ADE reports (including serious ADR reports and serious ME reports): number of serious ADR reports after one year | 2.10 (0.29 to 15.20) | 10 ADR reports per 1000 practitioners | 21 ADR reports per 1000 practitioners (3 to 150) | 6.5 years (2)1 | Very low2 | We do not know if multifaceted interventions increase the total number of serious ADR reports in physicians and pharmacists one year after implementation because the evidence is very uncertain. Data after two years in footnotes5 |
| Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports) | None of the included studies reported on this outcome. | |||||
| Number of unexpected ADE reports (including unexpected ADR reports and unexpected ME reports): unexpected or previously unknown ADR reports after one year | 0.73 (0.02 to 22.75) | 20 ADR reports per 1000 practitioners | 15 ADR reports per 1000 practitioners (0 to 455) | 7.0 years (1)6 | Very low7 | We do not know if multifaceted interventions increase the total number of unexpected (previously unknown) ADR reports in physicians and pharmacists one year after implementation because the evidence is very uncertain Data after two years in footnotes8 |
| Number of new drug‐related ADE reports (including drug‐related ADR reports and drug‐related ME reports): number of new drug‐ related ADR reports after one year | 1.65 (0.20 to 13.77) | 5 ADR reports per 1000 practitioners | 8 ADR reports per 1000 practitioners (1 to 69) | 6.5 years (2)1 | Very low2 | We do not know if multifaceted interventions increase the total number of new‐drug‐related ADR reports in physicians and pharmacists one year after implementation, because the evidence is very uncertain. Data after two years in footnotes9 |
ADE: adverse drug event; ADR: adverse drug reaction; CI: confidence interval; ITS: interrupted time series; ME: medication error; vs: versus
*Relative numbers of ADRs > 1 are associated with more ADRs with multifaceted intervention versus usual practice.
‡Illustrative comparative numbers of ADRs are presented as numbers of ADRs after 1 and 2 years in a setting with 1000 practitioners.
1Meta‐analysis of data from Chang 2017: financial incentive (1% of physician salary) for spontaneous reporting of ADRs plus fine (double the amount of the incentive) for not reporting or missing an ADR (study timeline ‐ 2006 to 2009 (pre‐intervention), 2009 to 2011 (financial incentive), 2012 to 2014 (financial incentive plus government regulations for antimicrobial agents), December 2014 (last time point), total 108 observations); and Pedrós 2009: financial incentives (1% of physicians salary) for spontaneous reporting of ADRs, twice‐yearly education meeting, reminder cards and list of the most important ADRs (study timeline ‐ January 1998 (first point); December 2002 (intervention implemented); December 2005 (last time point); a total of 96 observations)
2Both studies are observational ITS studies, so GRADE starts at low; downgraded once for serious risk of bias (high risk of bias for domain: intervention independent of other changes; there are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was not influenced by other confounding variables or historic events during study period); downgraded once for serious inconsistency (I2 = 92% for year 1 and 81% for year 2, inconsistency between the studies may be explained by the fact that one study was conducted in China and the other in Spain, but not certain of this); no serious indirectness; downgraded once for serious imprecision (wide confidence intervals that cross the line of no effect); no other considerations.
3Data from Ali 2018 (intervention included implementation of financial and non‐financial incentives, i.e. employee of the month award, letters of appreciation, a day's leave, performance excellence award of extra month’s salary and a certificate) could not be included in the meta‐analysis as the length of follow‐up was much shorter (study timelines ‐ 2 years; a total of 24 observations) than Chang 2017 (8 years; 108 observations) and Pedrós 2009 (7 years; 96 observations). Data from Ali 2018 shows relative numbers of ADR reports after 1 year: 6.99, 95% CI 3.43 to 10.54; prior to intervention ‐ 80 ADR reports per 1000 practitioners, post intervention implementation ‐ 560 ADR reports per 1000 practitioners (274 to 843); very low certainty evidence as based on observational ITS study, so GRADE starts at low; downgraded once for serious risk of bias (high risk of bias for other bias ‐ seasonality not adjusted for; and intervention independent of other changes ‐ there are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was not influenced by other confounding variables or historic events during study period); no serious inconsistency; no serious indirectness; no serious imprecision; no other considerations.
4Total number of ADE reports (including ADR reports and ME reports): relative number of ADR reports after 2 years 8.11 (95% CI 0.61 to 107.93); assumed number with usual practice 160 expected ADR reports per 1000 practitioners, corresponding number with multifaceted intervention 1298 expected ADR reports per 1000 practitioners (98 to 17,269), mean study duration 6.5 years, 2 studies1 ; certainty of the evidence: very low (see footnote2)
5Relative number of serious ADR reports after 2 years: 2.57 (95% CI 0.22 to 29.93); prior to intervention ‐ 20 ADR reports per 1000 practitioners, post intervention ‐ 51 ADR reports per 1000 practitioners (4 to 599) mean study duration 6.5 years (Chang 2017; Pedrós 2009); very low certainty evidence (see footnote2)
7Observational ITS study, so GRADE starts at low; downgraded once for serious risk of bias (high risk of bias for domain: intervention independent of other changes; there are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was not influenced by other confounding variables or historic events during study period); no serious inconsistency; no serious indirectness; downgraded once for serious imprecision (wide confidence intervals that cross the line of no effect); no other considerations.
8Relative number of unexpected (previously unknown) ADR reports after 2 years: 0.67 (95% CI 0.01 to 61.55); prior to intervention ‐ 40 ADR reports per 1000 practitioners; post intervention implementation ‐ 27 ADR reports per 1000 practitioners (0 to 2462); mean study duration 7.0 years (Pedrós 2009); very low certainty evidence (see footnote7)
9Relative number of new drug‐related ADR reports after 2 years: 1.86 (95% CI 0.16 to 21.59); prior to intervention ‐ 10 ADR reports per 1000 practitioners; post intervention implementation ‐ 19 ADR reports per 1000 practitioners (2 to 216); mean study duration 6.5 years (Chang 2017; Pedrós 2009); very low certainty evidence (see footnote2)
Summary of findings 4. Government regulations plus financial incentives versus usual practice.
|
Participants: healthcare professionals Intervention: financial incentive, fines, plus government regulation, mandatory monitoring, and reporting of ADRs (timeline: 2009 to 2011 (financial incentive or fine); 2012 to 2014 (financial incentive or fine plus government regulations for antimicrobial agents) Comparator: spontaneous reporting (2006 to 2009: pre‐intervention) Setting: hospital | ||||||
| Outcome | Relative numbers of ADEs* (95% CI) | Illustrative comparative numbers of ADEs‡(95% CI) | Mean study duration (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed number with usual practice | Corresponding number with multifaceted intervention | |||||
| Total number of ADE reports (including ADR reports and ME reports): Total number of ADR reports after one year | 1.43 (0.54 to 3.79) | 80 ADR reports per 1000 practitioners | 114 ADR reports per 1000 practitioners (43 to 303) | 8.0 years (1)1 | Very low2 | We do not know if government regulations and financial incentives increase the total number of ADR reports by physicians one year after implementation of these interventions because the evidence is very uncertain. Data after two years in footnotes3 |
| Total number of false ADE reports (including false ADR reports and false ME reports) | None of the included studies reported on this outcome. | |||||
| Number of serious ADE reports (including serious ADR reports and serious ME reports) | None of the included studies reported on this outcome. | |||||
| Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports) | None of the included studies reported on this outcome. | |||||
| Number of unexpected ADE reports (including unexpected ADR reports and unexpected ME reports) | None of the included studies reported on this outcome. | |||||
| Number of new drug‐related ADE reports (including drug‐related ADR reports and drug‐related ME reports) | None of the included studies reported on this outcome. | |||||
ADE: adverse drug event; ADR: adverse drug reaction; CI: confidence interval; cRCT: cluster randomised controlled trials; ME: medication error; vs: versus
*Relative numbers of ADRs > 1 are associated with more ADRs with multifaceted intervention versus usual practice.
‡Illustrative comparative numbers of ADRs are presented as numbers of ADRs after 1 and 2 years in a setting with 1000 practitioners.
1Chang 2017: financial incentive (1% of physician salary) for spontaneous reporting of ADRs plus fine (double the amount of the incentive) for not reporting or missing an ADR plus government regulation of antimicrobial use including detailed ADR classification, mandatory monitoring, and reporting of ADRs associated with antimicrobial agents; (timeline ‐ 2006 to 2009 (pre‐intervention); 2009 to 2011 (financial incentive); 2012 to 2014 (financial incentive plus government regulations for antimicrobial agents); December 2014 (last time point); total of 108 observations)
2Observational ITS study so GRADE starts at low; downgraded by one for risk of bias (high risk of bias for domain: Intervention independent of other changes; there are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was not influenced by other confounding variables or historic events during study period); no serious inconsistency; no serious indirectness; downgraded once for serious imprecision (wide confidence intervals that cross the line of no effect); no other considerations.
3Total number of ADE reports, including ADR reports and ME reports: number of ADR reports after 2 years 1.02 (95% CI 0.24 to 4.32, mean study duration: 8 years, 1 study1; assumed number of ADR reports with usual practice 160 ADR reports per 1000 practitioners, corresponding number of ADR reports with multifaceted intervention 163 ADR reports per 1000 practitioners (38 to 346); certainty of the evidence: very low2
Summary of findings 5. Improving access to ADR report forms versus usual practice.
|
Participants: healthcare professionals Intervention: improved access to ADE reporting (standardised discharge form method; yellow card ADR report form in bulletin and prescription pad; online hyperlink to ADR report form) Comparator: spontaneous reporting Setting: hospital | ||||||
| Outcome | Relative effect (95% CI)* | Illustrative comparative rates and numbers of ADEs‡(95% CI) | Number of participants or mean study duration (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed rate or number with usual practice | Corresponding rate or number with improving access | |||||
| Total number of ADE reports, including ADR reports and ME reports Data from cRCT |
2.06 (1.11 to 3.83) | 80 ADR reports per 1000 practitioner years | 165 ADR reports per 1000 practitioner years (89 to 306) | 5967 (1)1 | Low2 | Use of a standardised discharge form (for recording patient diagnoses, medical and surgical acts received during hospital stay; based on the ‘Diagnosis Related Groups’ (DRG) system) with additional ADR items (time of occurrence and evolution) may slightly increase the number of ADR reports. |
| Total number of ADE reports, including ADR reports and ME reports Data from ITS study after one‐year follow up |
1.95 (1.33 to 2.85) | 80 ADR reports per 1000 practitioners | 156 ADR reports per 1000 practitioners (106 to 228) | 8.4 years (2)3 | Very Low4 | We do not know if including yellow card ADR report form in quarterly bulletins and prescription pads or providing a hyperlink to the ADR report form in hospitals' electronic patient records may lead to more ADRs being reported after one year because the evidence is very uncertain. Data after two years in footnote5 |
| Total number of false ADE reports (including false ADR reports and false ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of serious ADE reports (including serious ADR reports and serious ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of unexpected ADE reports (including unexpected ADR reports and unexpected ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of new‐drug‐related ADE reports (including new‐drug‐related ADR reports and new‐drug‐related ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
ADE: adverse drug event; ADR: adverse drug reaction; CI: confidence interval; cRCT: cluster randomised controlled trial; ITS: interrupted time series; ME: medication error; vs: versus
*Relative treatment effects are expressed as risk ratios and, for ITS analyses, relative expected numbers of ADRs after 1 and 2 years. Relative treatment effects > 1 are associated with more ADRs with improving access versus usual practice.
‡Illustrative comparative rates and numbers of ADRs are presented as numbers of ADRs per 1000 practitioner years (for risk ratio) and expected numbers of ADRs after 1 and 2 years in a setting with 1000 practitioners (for the ITS studies).
Serious ADRs: resulting in death; is life‐threatening; is a congenital anomaly; requires hospital admission or prolongation of stay in hospital; or results in persistent or great disability, incapacity, or both; high‐causality ADRs: ADRs with attribution of definitive or probable causality; unexpected (previously unknown) ADRs: previously unknown ADRs that are not described in the summary of product characteristics; new‐drug‐related ADRs: ADRs concerning medications that have been on the market for less than five years.
1Hanesse 1994: cluster‐RCT (with cross‐over after 8 weeks, plus 2‐week washout period); the two methods for reporting ADRs were the spontaneous reporting method (SR method; usual care) and the standardised discharge form with additional ADR items (DRG method; intervention).
2Downgraded twice for very serious risk of bias (possible contamination effect due to cross‐over design and inability to blind physicians to the intervention; also unclear if the outcome assessors were blinded); no serious inconsistency; no serious indirectness; no serious imprecision; no other considerations
3Two ITS studies; Castel 2003: combined effect of quarterly adverse drug reaction bulletin with ADR yellow card report form (introduced Sept 1985) and a ADR yellow card report form in the prescription pad (introduced January 1991 to December 1994); Ribeiro‐Vaz 2012: 2006 to 2010 ‐ hyperlinks to the ADR online reporting pharmacovigilance centre form included either in the electronic patient record or on a desktop computer.
4Both studies are observational ITS studies so GRADE starts at low; downgraded once for serious risk of bias (high risk of bias for domain: intervention independent of other changes; there are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was not influenced by other confounding variables or historic events during study period); no serious inconsistency: no serious imprecision: no serious indirectness; no other considerations.
5Total number of ADE reports, including ADR reports and ME reports: number of ADR reports after 2 years of follow‐up: RR1.80 (95% CI 1.08 to 3.01, assumed rate or number with usual practice: 160 ADR reports per 1000 practitioners; corresponding rate or number with improved access: 288 ADR reports per 1000 practitioners (173 to 482); mean study duration for 2 ITS studies3: 8.4 years; certainty of the evidence: very low4
Summary of findings 6. Improving ADE reporting method (new web‐based electronic error reporting system) versus usual practice (existing web‐based electronic error reporting system).
|
Participants: healthcare professionals Intervention: September 2010 replace existing electronic error reporting system with new web‐based electronic error reporting system (equipped with a series of standardised screens, drop‐down menu choices, and input fields designed to collect specific information and improve communication with all departments involved); post‐implementation segment (1 September 2010 to 31 October 2012) Comparator: pre‐implementation segment (1 January 2009 to 31 August 2010) ‐ web‐based electronic error reporting system Setting: hospital | ||||||
| Outcome | Relative numbers of reports (95% CI)* | Illustrative comparative numbers of reports‡ (95% CI) | Mean study duration (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed number with usual practice | Corresponding number with improved reporting system | |||||
| Total number of ADE reports: number of ME reports after one year | 1.80 (1.15 to 2.80) | 80 ME reports per 1000 practitioners | 144 ME reports per 1000 practitioners (92 to 224) | 3.75 (1)1 | Very low2 | We do not know if the re‐engineering the web‐based electronic error reporting system may have increased the number of ME reports after one year because the evidence is very uncertain. Data after two years in footnotes3 |
| Total number of false ADE reports, including false ADR reports and false ME reports | None of the included studies reported on this outcome. | |||||
| Number of serious ADE reports (including serious ADR reports and serious ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of unexpected ADE reports (including unexpected ADR reports and unexpected ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of new‐drug‐related ADE reports (including new‐drug‐related ADR reports and new‐drug‐related ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
ADE: adverse drug event; ADR: adverse drug reaction; CI: confidence interval; ITS: interrupted time series; ME: medication error; vs: versus
*Relative expected numbers of ME reports > 1 are associated with more ME reports with improving reporting practice versus usual practice.
‡Illustrative comparative rates are presented as expected numbers of ME reports after 1 and 2 years in a setting with 1000 practitioners.
1McKaig 2014: ITS; pre‐implementation segment (1 January 2009 to 31 August 2010), replace one web‐based electronic error reporting system with new web‐based electronic error reporting system (equipped with a series of standardised screens, drop‐down menu choices, and input fields designed to collect specific information and improve communication with all departments involved) implemented in September 2010, post‐implementation segment (1 September 2010 to 31 October 2012)
2Observational ITS so GRADE starts at low; downgraded once for serious risk of bias (authors do not appear to have considered seasonal effects and there is no control arm to counter this). Furthermore, there is no compelling argument that the effects of the intervention occurred independently of other changes over time; inconsistency: none; downgraded once for serious imprecision (wide confidence intervals that include little or no effect to substantial effect); indirectness: none; other: none.
3Total number of ADE reports, including ADR reports and ME reports: Relative number of ME reports after 2 years: 2.11 (95% CI 1.03 to 4.33), assumed number with usual practice: 160 ME reports per 1000 practitioners, corresponding number with different web‐based electronic error reporting system: 338 ME reports per 1000 practitioners (165 to 693); 1 study, 3.75 years exposure to intervention; very low certainty of evidence (see footnote2)
Summary of findings 7. Case finding versus spontaneous reporting (usual practice).
|
Participants: healthcare professionals Intervention: case finding ‐ clinical pharmacist identified ADEs by joining daily hospital rounds, screening patient charts and interviewing patients, daily meetings with physicians and nurses, comprehensive review of patient charts post‐discharge using specific data extract form to identify in‐hospital ADEs; ADEs were identified by (a) spontaneous or solicited reporting by a physician, (b) spontaneous or solicited reporting by a nurse, (c) detection on regular ward rounds and (d) detection by the clinical pharmacist by chart review after hospital discharge. Comparator: usual practice (clinical pharmacist not present; ADEs identified through spontaneous reporting by nurses and physicians) Setting: hospital | ||||||
| Outcome | Relative effect (95% CI)* | Illustrative comparative numbers of ADEs‡ (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed number with usual practice | Corresponding number with case finding | |||||
| Total number of ADE reports (including ADR reports and ME reports): number of ADE reports Follow‐up: 12 months |
11.07 (6.24 to 21.38) | 1.4 per 1000 patient‐days | 15.5 (95% CI 8.74 to 29.9) per 1000 patient‐days | 1016 (1)1 | Very low2 | We do not know if having clinical pharmacists actively identifying and encouraging the identification of ADEs in a hospital setting leads to more ADEs being reported per 1000 patient‐days because the evidence is very uncertain. |
| Total number of false ADE reports (including false ADR reports and false ME reports): number of false ADE reports Follow‐up: 12 months |
None of the studies included in this comparison reported on this outcome. | |||||
| Number of serious ADE reports (including serious ADR reports and serious ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of unexpected ADE reports (including unexpected ADR reports and unexpected ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
| Number of new‐drug‐related ADE reports (including new‐drug‐related ADR reports and new‐drug‐related ME reports) | None of the studies included in this comparison reported on this outcome. | |||||
ADE: adverse drug event; ADR: adverse drug reaction; CI: confidence interval; ME: medication error; vs: versus
*Incidence rate ratio (IRR) of ADEs; IRR > 1 is associated with more ADE reports with case finding (clinical pharmacist present) versus usual practice (spontaneous reporting, no clinical pharmacist present).
‡Illustrative comparative rates are presented as expected numbers of ADEs per 1000 patient‐days.
Serious ADRs: resulting in death; is life‐threatening; is a congenital anomaly; requires hospital admission or prolongation of stay in hospital; or results in persistent or great disability, incapacity, or both); high‐causality ADRs: ADRs with attribution of definitive or probable causality; unexpected (previously unknown) ADRs: previously unknown ADRs that are not described in the summary of product characteristics; new‐drug‐related ADRs: ADRs concerning medications that have been on the market for fewer than 5 years.
1Schlienger 1999: non‐randomised cross‐over study, without a washout period. To minimise any possible learning effect, we have only included and analysed the data from the first period (1 to 12 months) of the study. In the test units: case finding ‐ clinical pharmacist identified ADEs by joining daily hospital rounds, screening patient charts and interviewing patients, daily meetings with physicians and nurses, comprehensive review of patient charts post‐discharge using specific data extract form to identify in‐hospital ADEs; ADEs were identified by (a) spontaneous or solicited reporting by a physician, (b) spontaneous or solicited reporting by a nurse, (c) detection on regular ward rounds and (d) detection by the clinical pharmacist by chart review after hospital discharge. In control units: clinical pharmacist not present; ADEs identified through spontaneous reporting by nurses and physicians.
2Because it is not a randomised study, the GRADE assessment starts at low; downgraded twice for very serious risk of bias (risk of selection bias as not randomised, risk of performance as no blinding of physicians or nurses, and risk of detection bias as no blinding of outcome assessors); no serious inconsistency: no serious indirectness; no serious imprecision; no other considerations.
Background
Description of the condition
Approximately 1.4% of the global Gross Domestic Product (US$1 trillion) is spent on medicines (World Bank). Medicines cure, arrest or prevent disease, ease symptoms or help diagnose illnesses. However, a great deal of morbidity and mortality is associated with unforeseen reactions to and inappropriate use of medicines. Adverse drug events, defined as “any untoward medical occurrence that may present during treatment with a pharmaceutical product but which does not necessarily have a causal relationship with this treatment” (Uppsala Monitoring Center), are a global public health issue.
Adverse drug events (ADEs) include all adverse drug reactions and medication errors. An adverse drug reaction is “a harmful effect suspected to be caused by a drug at doses normally used in humans for the prophylaxis, diagnosis, or therapy of disease, or for the modification of physiological function” (Uppsala Monitoring Center). A medication error is “any preventable event that may cause or lead to inappropriate medication use or patient harm while the medication is controlled by the health care professional, patient, or consumer. Such events may be related to professional practice, health care products, procedures, and systems, including prescribing, order communication, product labelling, packaging, nomenclature, compounding, dispensing, distribution, administration, education, monitoring, and use” (NCCMERP 2016).
No medicine is without ADEs. While some ADEs are detected during pre‐marketing phase clinical trials, limitations associated with the conduct of these trials make it impossible to identify all ADEs related to a product. Trial characteristics such as small sample size, relatively short follow‐up periods, close monitoring of study participants (to ensure strict adherence to study protocol), and narrowly defined characteristics of study participants and study indications (study indications for a drug are often limited to a particular disease; Gad 2009) are important for study validity and efficacy but limit the generalisability and effectiveness of the study findings. Continually monitoring the use and effects (both beneficial and harmful) of clinically approved medicines in large numbers of people is therefore important to better understand the effectiveness and safety of medication under everyday circumstances.
Pharmacovigilance, which is "the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other drug‐related problems" (WHO 2006), aims to improve patient safety related to the use of medicines (Fornasier 2018). More than 170 countries have pharmacovigilance agencies that collate and manage adverse event reporting (WHO 2016). Identifying any adverse drug events associated either with a medication or with the use of a medication, as soon as possible, prevents or minimises any potential harm. These efforts enable healthcare professionals to maximise the benefits of medicines while avoiding or minimising the risks associated with their use. Spontaneous or voluntary reporting of ADEs (i.e. case reports of ADEs that are voluntarily submitted from healthcare professionals and pharmaceutical manufacturers to the national regulatory authority (Uppsala Monitoring Center)) is the cornerstone of effective pharmacovigilance and is considered "usual practice" in most parts of the world. The level of pharmacovigilance and adverse drug event reporting differs greatly based on the regulations established by the respective regulatory agencies. Data collection also varies amongst countries. France, for example, has regional centres for collecting spontaneous reports, while Iran has a single national pharmacovigilance centre to collect data (Shalviri 2009). Although spontaneous reporting of ADEs is the most common method for collecting information on the safety of medicines during the post‐marketing phase (Figueiras 2001; Pal 2013), it is limited and is associated with gross underreporting of ADEs. According to the WHO Monitoring Center in Uppsala, annual reporting rates of over 200 adverse drug event reports per million inhabitants indicate a healthy national pharmacovigilance system (Lindquist 2008). Many countries have yet to achieve this goal. It is estimated that only 2% to 4% of non‐serious adverse drug events and 10% of serious ADEs are reported spontaneously by healthcare professionals (Hazell 2006; Moride 1997). It should be noted that reports from patients and healthcare students are also valuable contributors to drug‐related data in many countries. However, these populations are not within the scope of this review.
Description of the intervention
Although spontaneous reporting of adverse drug events is the most common method of collecting safety data associated with medications, there are other methods of collecting safety information (WHO 2006). Some countries have implemented active surveillance systems to complement spontaneous reporting, for example, the prescription event monitoring (PEM) system in New Zealand and the United Kingdom (WHO 2006). In the European Union, a set of measures called “good pharmacovigilance practices” have been drawn up to facilitate pharmacovigilance (EMA 2016). Various interventions have been used in different settings to improve healthcare professionals' spontaneous reporting of ADEs. The most commonly used interventions include the following (Gonzalez‐Gonzalez 2013; Molokhia 2009).
Educational activities such as training sessions
Reminders such as letters, emails or posters
Simplification of the adverse drug event reporting form
Increased availability of reporting forms
Modification of reporting procedures (e.g. reporting by telephone or email)
Incentives such as provision of educational credits, awards or financial motivations, or disincentives for not reporting, e.g. fines
Assistance from a colleague (e.g. a clinical pharmacist, physician or nurse) with ADE reporting
Providing feedback to reporters about adverse drug events
Use of computerised monitoring systems to signal changes in laboratory results
Some studies focus specifically on developing interventions to improve medication error reporting. For example, a study in New Zealand designed a web‐based medication error reporting programme (MERP) to supplement pharmacovigilance (Kunac 2014). Some studies choose to examine more than one intervention. For example, seven overlapping interventions were used in a study to improve ADE reporting, including a poster displaying days since the last medication error resulting in harm, a continuous slide show in the staff lounge showing performance metrics, multiple didactic curricula, unit‐wide emails providing information on medication errors, computerised physician order entry, introduction of unit‐based pharmacy technicians for medication delivery, and patient safety report form streamlining (Abstoss 2011).
How the intervention might work
Reasons for inadequate spontaneous reporting or underreporting of adverse drug events by healthcare professionals include complacency (e.g. the belief that very serious adverse drug reactions are well documented by the time a drug is marketed), insecurity (e.g. the belief that it is nearly impossible to determine whether a drug is responsible for a particular adverse reaction), diffidence (e.g. healthcare professionals are afraid of looking foolish or over‐reactive by submitting a report for an adverse event that is not severe or not obviously related to a medical product or the use of a medical product), indifference (e.g. some healthcare professionals feel that the one case they might observe could not contribute to medical knowledge), ignorance (e.g. the belief that it is only necessary to report serious or unexpected adverse drug reactions), and lack of time to complete the adverse drug event reporting procedure (Mirbaha 2015; Varallo 2014). Healthcare professionals may also fear being blamed for any adverse event they draw attention to, that acknowledging an adverse reactions may reflect negatively on their competence or put them at risk of litigation (WHO 2006).
Understanding the barriers associated with underreporting of adverse drug events guides the design of interventions to address or minimise the impact of these barriers or reasons for inadequate spontaneous reporting of ADEs. Educational interventions and informational reminders could raise awareness of the importance of reporting adverse drug events. Other interventions aim to simplify or improve the accessibility of the reporting process itself and, in this way, increase the reporting rate. Interventions that reward healthcare professionals with either financial or non‐financial incentives for reporting ADEs may also facilitate increases in the reporting rates of adverse drug events. Incentivised interventions may also lead to false reports, however, so checks and balances in the system are necessary.
Results of observational studies seem to suggest that although interventions involving educational sessions (Bäckström 2002), improving access to ADR report forms (McGettigan 1997), or financial incentives (Feely 1990) increase the reporting rate of adverse drug events, the effect of these interventions is temporary, and reporting rates decline once the intervention is removed (McGettigan 1997). The ultimate aim of interventions is to create a "culture of reporting" amongst healthcare professionals that is effective and sustained. Integrating the reporting of adverse drug events into existing hospital electronic reporting systems may be one example of a way to achieve this (Ortega 2008).
Why it is important to do this review
The World Health Organization (WHO) International Drug Monitoring Program was created in response to the lack of global harmonisation for monitoring of ADEs (WHO 2006). The programme currently includes over 170 countries as full members and associate members (WHO 2016). Despite the numerous pharmacovigilance activities undertaken in many countries, the problem of underreporting adverse drug events is still a major threat to the public's health and well‐being. Adverse drug events are a significant cause of death in many countries (Lazarou 1998; Pirmohamed 2004; Shalviri 2009; Shalviri 2012; Wester 2008), and a significant cause of hospital admissions (Al Hamid 2014; Wilson 2012). Furthermore, a substantial portion of healthcare costs are directly related to adverse drug events, with the economic burden amounting to hundreds of billions of dollars each year (Andel 2012; Classen 1997; Ernst 2001; Gyllensten 2013; Johnson 1995).
Many adverse drug events are preventable. Studies have reported that 10% to 80% of all adverse drug events can be prevented (WHO 2014). Improved spontaneous reporting of suspected adverse drug events enables early detection of any patient safety issues associated with the medication itself or with how the medication is used (Pal 2013), which can reduce drug‐related morbidity and mortality (Pal 2013). Several systematic reviews have assessed the effectiveness of interventions to enhance the reporting of adverse drug events (Gonzalez‐Gonzalez 2013; Li 2019; Pagotto 2013; Paudyal 2020). These reviews are limited in their scope in terms of intervention (educational interventions; Pagotto 2013) or outcome (ADRs; Gonzalez‐Gonzalez 2013; Li 2019; Paudyal 2020). Furthermore, none of the systematic reviews have provided an assessment of the certainty of the evidence for each of the interventions assessed (see Table 8). This Cochrane review aims to identify all interventions directed at healthcare professionals that may improve reporting of adverse drug events, including all ADRs and any MEs. Our review will also systematically assess the certainty of the evidence associated with each type of intervention.
1. Overview of published systematic reviews assessing interventions to increase ADE reporting.
| Pagotto 2013 | Gonzalez‐Gonzalez 2013 | Ribeiro‐Vaz 2016 | Li 2019 | Paudyal 2020 | Khalili 2020(scoping review) | |
| Objectives | To identify the techniques of educational intervention for promotion of pharmacovigilance by healthcare professionals and to assess their impact. | To conduct a critical review of papers that assessed the effectiveness of different strategies to increase ADR reporting, regardless of the healthcare professionals or patients included. | To describe the state of the art information systems used to promote ADR reporting. | To determine the features and successes of the various strategies undertaken to improve ADR reporting by healthcare professionals, and propose alternative initiatives that may enhance these existing methods. | To evaluate the effectiveness of interventions used for improving ADR reporting by patients and healthcare professionals. | To systematically map interventions and strategies to improve ADR reporting among health care professionals. |
| Eligible study designs | All study designs included | Pre‐post experimental design; time series; non‐randomised controlled experimental study; randomised controlled experimental study; cluster randomised controlled experimental study | Any studies describing or evaluating the use of information systems to promote adverse drug reaction reporting. Studies with data related to the number of ADRs reported before and after each intervention and the follow‐up period were included in the quantitative analysis. | RCTs, quasi‐experimental, time series studies | All forms of interventional designs were considered. Meta analysis not undertaken for non‐randomised trials | Quantitative methods focused on healthcare professionals |
| Eligible participants | Healthcare professionals | Professionals to whom the intervention for increasing ADR reporting is addressed: physicians, nurses, pharmacists, young physicians, house officers, pharmacy students, section head, ‘quality review staff’, medical students. | Healthcare professionals or patients | Healthcare professionals | Healthcare professionals and patients | Healthcare professionals |
| Eligible interventions | Educational interventions only | Educational activity, reminders, modification of reporting forms, modifciation of reporying process, incentives, assistance from another professiionl, increased availability of reporting forms, feedback on reporting | Studies describing or evaluating the use of information systems to promote adverse drug reaction reports were selected | Any intervention aimed at increasing ADR reporting | Any pharmacovigilence intervention | Any intervention or strategy (such as ones implemented by government policies, applied experimentally or non‐experimentally, or adopted in specific settings) to improve ADR reporting |
| Eligible comparison | Not stated | Not stated | Not stated | Not stated | Not stated | Not stated |
| Outcomes reported on | ADE reporting | Increase in ADR reporting | Rate of ADR reporting increase | ADR rporting | Primary outcome: quantity of ADRs reported as a result of the intervention including improvement in the number or rate of reporting. Secondary outcomes: the quality of ADR reporting including the nature of ADRs reported (e.g. serious, nonserious ADRs) and completeness of the reports. | ADR reporting rate |
| Number and type of studies | 16 met the inclusion criteria 6 RCT, 5 quasi‐experimental, 2 case‐ control studies, 2 ecological time series analysis, 1 observational analytic) | 43 studies | 33 articles were included in the analysis; these articles described 29 different projects. | 13 studies included (3 cRCTs, 1 RCT, 7 quasi‐experimental, 2 ITS) | 28 studies | 90 studies included in qualitative synthesis |
| Findings and conclusions | Pharmacovigilance‐based educational interventions showed positive impacts (quantitative and qualitative) on ADE spontaneous reporting by health professionals. Multifaceted techniques for interventions, included: lectures, placement of yellow cards, distribution of printed educational materials and giveaways, as well as the or‐ ganization of workshops | Multiple interventions have a greater impact than single. Evidence to show that, when it comes to bringing about changes in professional practice, interventions that boost the active participation of professionals (i.e. workshops) can be more effective than passive didactic sessions. Another vital factor is the duration of the effect of the intervention. It can be concluded that, as was to be expected, the longer the period from the date of the intervention, the more the latter's effect is progressively reduced. |
Most projects performed passive promotion of ADR reporting (i.e., facilitating the process). Developed in hospitals and tailored to healthcare professionals. Interventions doubled the number of ADR reports. Authors believe that it would be useful to develop systems to assist healthcare professionals with completing ADR reporting within electronic health records because this approach seems to be an efficient method to increase the ADR reporting rate. When this approach is not possible, it is essential to have a tool that is easily accessible on the web to report ADRs. This tool can be promoted by sending emails or through the inclusion of direct hyperlinks on healthcare professionals’ desktops. | Multi‐faceted approach including education, reminders, and electronic reporting would likely to be the most successful. | Limited evidence showed that active interventions involving face to face educational approaches, financial incentives, and electronic features targeted at healthcare professionals could improve ADR reporting. However, the results need to be interpreted cautiously given the short term evaluation out‐ comes, dominance of observational designs and low quality of included studies. Interventions need to be developed and tested in countries low‐and‐middle income countries. Most of the included studies included educational interventions to improve ADR reporting. A variety of educational methods were used including reminders, face to face educational sessions and newsletters. While most of these studies were reported to have improved ADR reporting, there was a lack of long‐term follow up of the outcomes. The cluster‐ randomized controlled trials included in the study reported that the impact of interventions observed by the difference in the intervention and control group in the ADR reporting rate lasted for only 12 months after which such difference was no longer significant. | Interventions aimed at enhancing ADR reporting have a good chance of producing positive results, although their effect, especially in the case of educational interventions, could be temporary. Multiple inter‐ ventions might cause greater increase in ADR reporting rates compared with single interventions. Further research is warranted to improve the methodological quality using control groups, large sample sizes, longer follow‐up periods, and adjustment for the confounders. |
| Any limits noted | Language limit: English, Portuguese, or Spanish search for publications from November 2011 to January 2012, updated in March 2013. Quality assessment of the manuscripts was not carried out. | Limit publication date: up to 2010; Language limited to English, French or Spanish | Language limited to English, Portuguese or French Excluded articles based on: (1) only focused on medication errors; (2) only focused on ADR detection; (3) studies without any information system implemented; (4) studies concerning data quality; (5) studies focused on website usability; (6) authors’ reflections on the theme; (7) studies only related to incidents that occurred in health institutions; (8) studies concerning signal detection and (9) studies concerning electronic transmission between the authority and other institutions (pharmaceutical companies or regional pharmacovigilance centres). | Search limited to studies published from 2010 to 2019; English only; NO medication error reporting | Educational research with student participants; interventions not including qualified healthcare practitioners or patients were excluded as well as the interventions related to devices and planned ADR surveillance monitoring programmes, such as those used for mass vaccinations; Abstract only publications including conference abstracts were excluded. | Search date limited from 1999 to February 2019; no language restrictions; methodological quality or risk of bias of the included articles were not appraised |
ADE: adverse drug event; ADR: adverse drug reaction; cRCT: cluster‐randomised controlled trial; ITS:interrupted time series; RCT: randomised controlled trial
Objectives
To assess the effectiveness of different interventions aimed at healthcare professionals to improve the reporting of adverse drug events.
Methods
Criteria for considering studies for this review
Types of studies
We included both individually randomised trials and cluster‐randomised trials, non‐randomised controlled studies and controlled before‐after studies. For cluster‐randomised trials, non‐randomised cluster trials and controlled before‐after studies, we included only those with at least two intervention sites and two control sites (EPOC 2013a). In addition, for controlled before‐after studies, data collection had to be contemporaneous in both the intervention and control groups during the pre‐ and post‐intervention periods, and identical measurement methods had to be used in these periods. We also included interrupted time series and repeated measures studies that had a clearly defined time point when the intervention occurred and at least three data points before and after the intervention (EPOC 2013b). We included data from both published (full‐text articles and conference abstracts) and unpublished eligible studies.
Types of participants
We included studies in which healthcare professionals (including but not limited to general practitioners, pharmacists, nurses and specialists) from any healthcare setting were the target audience of the intervention. We excluded studies aimed at patients and healthcare students (e.g. medical, nursing, pharmacy) as the target audience.
Types of interventions
We included studies assessing any intervention designed to increase adverse drug event reporting, compared with healthcare professionals' usual adverse drug event reporting practice (mainly spontaneous or voluntary reporting) or a different intervention or interventions designed to improve adverse drug event reporting. We excluded studies of interventions targeted at adverse events reporting following immunisation (AEFI) as AEFI monitoring uses different mechanisms and settings.
Types of outcome measures
An adverse drug event (ADE) is defined as "any untoward medical occurrence that may present during treatment with a pharmaceutical product but which does not necessarily have a causal relationship with this treatment" (Uppsala Monitoring Center). ADEs can include both adverse drug reactions (ADRs) and medication errors (MEs). According to the Uppsala Monitoring Center, an adverse drug reaction (ADR) is defined as "a harmful effect suspected to be caused by a drug", including "all kinds of adverse events, many of which are not 'reactions' in the strict sense and have not been subject to any assessment of causality" (Uppsala Monitoring Center). A medication error (ME) is "any preventable event that may cause or lead to inappropriate medication use or patient harm while the medication is controlled by the health care professional, patient, or consumer. Such events may be related to professional practice, health care products, procedures, and systems, including prescribing, order communication, product labelling, packaging, nomenclature, compounding, dispensing, distribution, administration, education, monitoring, and use" (NCCMERP 2016).
Primary outcomes
Total number of ADE reports, including ADR reports and ME reports, submitted by healthcare professionals
Total number of false ADE reports, including false ADR reports and false ME reports, submitted by health care professionals
Secondary outcomes
-
Number of serious ADE reports (including serious ADR reports and serious ME reports)
ADEs that result in death, are life‐threatening, are a congenital anomaly, require hospital admission or prolongation of stay in hospital, or result in persistent or significant disability or incapacity or both
-
Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports)
ADEs with attribution of definitive or probable causality
-
Number of unexpected ADE reports (including unexpected ADR reports and unexpected ME reports)
Previously unknown ADEs that are not described in the drug's summary of product characteristics
-
Number of new‐drug‐related ADE reports (including new‐drug‐related ADR reports and new‐drug‐related ME reports)
ADEs relating to medications that have been on the market for less than five years
Search methods for identification of studies
Electronic searches
An EPOC Information Specialist developed the search strategies in consultation with the review authors. We searched the following databases from inception to 14 October 2022.
Cumulative Index to Nursing and Allied Health Literature (CINAHL) via EbscoHost (1980 to 14 October 2022)
Cochrane Database of Systematic Reviews (CDSR), Database of Abstracts of Reviews of Effects (DARE), Health Technology Assessment (HTA) database, and NHS Economic Evaluation Database (NHS EED) via the Cochrane Library (Issue 10, 2022; searched on 14 October 2022)
Cochrane Central Register of Controlled Trials (CENTRAL) (includes the entirety of the EPOC Group Specialised Register) via the Cochrane Library (Issue 10, 2022; searched on 14 October 2022)
Embase via OvidSP (1974 to 14 October 2022)
Science Citation Index (SCI), Social Sciences Citation Index (SSCI) via Web of Knowledge (1975 to 14 October 2022)
Conference Proceedings Citation Index‐ Science (CPCI‐S) via Web of Science (1990 to 14 October 2022)
MEDLINE (In‐Process and other non‐indexed citations) via OvidSP (1946 to 14 October 2022)
Dissertations & Theses (COS Conference Papers Index; ProQuest Dissertations & Theses: UK & Ireland; ProQuest Dissertations & Theses Global) via ProQuest (1861 to 11 March 2021)
Virtual Health Library (VHL) Regional Portal via pesquisa.bvsalud.org/portal/advanced/?lang=en; search date: 17 October 2022
World Health Organization Library Catalogue (WHOLIS/IRIS) via https://kohahq.searo.who.int; search date: 17 October 2022
We also searched the following trial registries for potentially eligible ongoing studies on 17 October 2022.
Word Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) via www.who.int/ictrp/en;
ClinicalTrials.gov via clinicaltrials.gov.
Searches were not restricted by language, date or format of publication. The search strategies used are provided in Appendix 1.
Searching other resources
We conducted a grey literature search of the following databases using key terms "adverse drug event" OR "adverse drug reaction" OR "medication error" to identify additional potentially eligible studies.
OpenGrey via opengrey.eu (last search date 20 August 2018; database no longer updated)
Grey Literature Report, New York Academy of Medicine via www.nyam.org/library/collections-and-resources/#greylit (last search date 20 August 2018; database no longer updated)
Agency for Healthcare Research and Quality (AHRQ) via www.ahrq.gov (last search date 20 August 2018);
National Institute for Health and Clinical Excellence (NICE) via www.nice.org.uk (last search date 20 August 2018)
Bielefeld Academic Search Engine (BASE) via www.base-search.net (last search date 20 August 2018)
We also screened the reference lists of all included studies and relevant systematic reviews and primary studies. We contacted authors of relevant studies or reviews to clarify reported published information and to seek unpublished results or other data for potentially eligible studies. We contacted experts in the field for information on additional eligible ongoing or completed studies.
Data collection and analysis
Selection of studies
All references retrieved through electronic searching were downloaded into a reference management database (EndNote 2013). After removing all duplicate references, the search records were uploaded to the review management programme Covidence (Covidence). Two review authors (from GS, NM, LG, WYC) independently screened the titles and abstracts for inclusion. We obtained the full texts of all the potentially eligible studies, and two review authors (from GS, NM, LG, WYC) independently screened these for inclusion. We noted the reasons for excluding any potentially eligible full‐text studies, and these are provided in a Characteristics of excluded studies table. Any disagreement between review authors regarding study eligibility was resolved through discussion or, if required, consultation with a third author (KG). We collated multiple reports of the same study so that each study, rather than each report, was the unit of interest in the review. Details about (potentially) eligible ongoing studies are provided in a Characteristics of ongoing studies table. If we were unable to obtain the full text of a potentially eligible study and could not determine the eligibility of the study, we recorded the study details in a Characteristics of studies awaiting classification table. We presented the study selection process in a PRISMA flow diagram (Figure 1).
1.

PRISMA flow diagram
Data extraction and management
We used a standard data collection form adapted from EPOC (EPOC 2013c) to capture study characteristics and outcome data. Two review authors (GS, NM) independently extracted the following study characteristics from all included studies.
Methods: study design, number of study centres and location, study setting, date of the study, length of follow‐up
Participants: number, mean age or age range, sex, inclusion and exclusion criteria, withdrawals, loss to follow‐up, type of healthcare professionals, education
Interventions: specific components of the intervention; intensity of intervention; duration of intervention
Comparison: treatment and contact received by the control or comparison group
Outcomes: description of study outcomes reported in the study (including the number of reported ADEs, number and percentage of false reports of ADEs and number of detected ADEs), time points at which outcomes were reported, outcome data for all relevant outcomes reported
Notes: additional details about trial funding, notable author conflicts of interest, ethical approval, any outcome data reported in an unusable way and correspondence with study authors for additional data or information.
A third independent author (CR) also extracted and imputed treatment effect estimates (including standard errors or confidence intervals) for the randomised studies, and extracted and re‐analysed data from the interrupted time series (ITS) studies. For randomised studies, we either extracted published risk ratios or imputed rate ratios and exact 95% confidence intervals on the rate ratio from extracted numbers of events and exposures. Because one of the included studies was a three‐arm study in which two interventions were compared to a common comparator (Herdeiro 2012), we adjusted the standard errors for this study's comparisons using the exact adjustment method of Rücker 2017.
We extracted ITS study data from the time series graphs published in the studies using WebPlotDigitizer (accessed in March and April 2020). In some cases, there were apparent discrepancies between the dates of the interruptions shown in graphs versus those stated in the study texts. We chose to use the dates provided in the study texts where possible, which is a conservative approach that, in the case of these studies, is likely to lead to less extreme effect estimates. We re‐analysed all ITS data using piecewise linear regression, adjusted for autocorrelated disturbances and seasonality where possible, using the interrupted time series analysis add‐on command (Linden 2015) for Stata (StataCorp LLC, College Station, Texas, USA). Specifically, we estimated the pre‐interruption level and slope, post‐interruption change in level, post‐interruption slope and seasonal effects where possible. We adjusted for autocorrelated disturbances by setting the maximum lag option to a value determined by visual inspection of autocorrelation and partial correlation plots, and by using Cumby‐Huizinga general tests for autocorrelation (Cumby 1990), with a significance threshold of 0.05. We adjusted for seasonality by modelling the effect of each quarter as a fixed effect if at least two years of data were available (i.e. each quarter was observed at least twice) and if data were provided monthly or more frequently (i.e. at least three data points were available for each quarter). The included ITS studies generally reported ADR counts. We modelled ITS data on the natural logarithmic scale to constrain the error distribution to positive values (counts cannot be negative), stabilise variance, and facilitate meta‐analysis (see Measures of treatment effect). None of the included ITS studies included controls in which no intervention (or a substantively different intervention) was used in the post‐interruption period, so we could not adjust for other possible explanations for the observed changes in reporting.
At least one other review author (from LG, GS, NM) checked the extracted and imputed effect estimates. Two review authors independently compared extracted time‐series data and model fits (on the count rather than the logarithmic scale) to the graphs published in the included studies. We resolved any disagreements regarding the extracted data by consensus.
Assessment of risk of bias in included studies
Two review authors (from GS, NM, LG, WYC) independently assessed the risk of bias for each study using the Cochrane risk of bias tool (RoB 1), as outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2024), and following guidance from the EPOC group (EPOC 2015b). We resolved any disagreement by discussion or by involving a third review author (KG).
The Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2024) recommends the explicit reporting of the following individual elements for randomised controlled trials, non‐randomised controlled trials and controlled before‐after studies: random sequence generation; allocation sequence concealment; blinding (participants, personnel); blinding (outcome assessment); completeness of outcome data; selective outcome reporting; and any other sources of bias such as contamination. For each domain, we described the relevant information provided by the authors and judged each item as being at high, low or unclear risk of bias based on the criteria provided by Higgins 2024.
For cluster‐RCTs, we assessed the risk of bias associated with the following additional domains: selective recruitment of participants (recruitment bias); baseline imbalance between clusters; loss of clusters; not accounting for clustering in the analysis; and comparability with individually randomised trials to account for potential "herd effect" seen with cluster‐RCTs (Higgins 2024). We added the judgement and rationale for these domains under "Other bias" in the risk of bias tables and figures, and we report them in the Other potential sources of bias section.
For interrupted time series studies (ITS), we assessed the following additional risk of bias domains: was knowledge of the allocated interventions adequately prevented during the study; was the intervention unlikely to affect data collection; was the intervention independent of other changes; and was the shape of the intervention effect prespecified (EPOC 2013b)?
We judged each potential source of bias as high, low or unclear and provided justification for our judgement in the risk of bias table. We summarised our risk of bias judgements across different studies for each domain. We considered blinding separately for different key outcomes where necessary. Where information on the risk of bias related to unpublished data or correspondence with a trialist, we noted this in the risk of bias table. When considering treatment effects, we took into account the risk of bias in the studies that contributed to that outcome.
Measures of treatment effect
We measured relative treatment effects using risk ratios and rate ratios for the included randomised studies. For the ITS studies, we exponentiated change in level and slope (which were estimated on the logarithmic scale; see Data extraction and management) to obtain estimates of ratios of post‐ to pre‐interruption levels and slopes. These estimates describe the nature of any change in reporting. In principle, however, genuine changes in level and slope can lead to no overall change in reporting (i.e. a change in slope can effectively cancel a change in level). We, therefore, measured change in reporting as the ratio of expected numbers of ADR reports by extrapolating the pre‐interruption curve into the post‐interruption period and treating it as a counterfactual. Because this ratio is a function of time, we estimated it at one and two years post‐intervention. We excluded a study from any meta‐analysis of the data if it would be necessary to extrapolate beyond the end of follow‐up for that study. We used a consistent direction of effect for all measures of treatment effect such that a value greater than one favours the intervention over the comparator.
Unit of analysis issues
A number of eligible studies were cluster‐randomised trials. A statistician (CR) confirmed that the analyses of all these studies were appropriately adjusted for clustering. We, therefore, extracted the data as reported.
Two studies used a cross‐over design (Hanesse 1994; Schlienger 1999). After careful consideration, we decided that we would use the data from both periods for Hanesse 1994. However, in Schlienger 1999, due to limitations in the design of the study (i.e. it was non‐randomised; the same clinical pharmacologist implemented the intervention and collected the outcome; and there was no washout between study periods), we felt it prudent to include only data from the first period of the study.
Dealing with missing data
We contacted study authors to verify key study characteristics and obtain missing outcome data where possible. We noted all interactions with study authors in the Characteristics of included studies table. We did not approach the authors of ITS studies to obtain the original time series data.
Assessment of heterogeneity
When we conducted a meta‐analysis of study data, we used the I² statistic to assess heterogeneity amongst the trials in each analysis. We noted the presence of considerable heterogeneity (I2 = 75% to 100%, Higgins 2024) in the text and explored this heterogeneity through the subgroup analyses. Where there were high levels of unexplained heterogeneity, we interpreted meta‐analysis results with caution.
Assessment of reporting biases
We had planned to use funnel plots to explore possible non‐reporting biases if it were possible to pool results from more than 10 studies (Sterne 2011). We could not pool results from more than six studies, so we did not generate funnel plots.
Data synthesis
We performed random‐effects meta‐analyses following standard Cochrane methods. We used the meta‐analysis commands provided by Stata version 16.1 (StataCorp LLC, College Station, Texas, USA). We performed estimation using restricted maximum likelihood and presented results on forest plots.
Subgroup analysis and investigation of heterogeneity
Sensitivity analysis
Summary of findings and assessment of the certainty of the evidence
We summarised the findings for the main comparisons in a summary of findings table for the following primary and secondary outcomes (see Types of outcome measures).
-
Total number of ADE reports
Number of ADR reports
Number of ME reports
-
Total number of false ADE reports
Number of false ADR reports
Number of false ME reports
Number of serious ADE reports, including serious ADR reports and serious ME reports (i.e. resulting in death; is life‐threatening; is a congenital anomaly; requires hospital admission or prolongation of stay in hospital; or results in persistent or great disability, incapacity, or both).
Number of high‐causality ADE reports, including high‐causality ADR reports and high‐causality ME reports (i.e. ADEs with attribution of definitive or probable causality)
Number of unexpected ADE reports, including unexpected ADR reports and unexpected ME reports (i.e. previously unknown ADEs that are not described in the summary of product characteristics)
Number of new drug‐related ADE reports, including new drug‐related ADR reports and new drug‐related ME reports (i.e. ADEs concerning medications that have been on the market for less than five years)
We re‐expressed meta‐analytical risk and rate ratio estimates as assumed and corresponding numbers of ADE reports per 1000 practitioner years. We used data from study control arms to estimate the "assumed" rate of ADE reports, which necessarily differs with respect to the type of ADE. For example, the total number of ADR reports is larger than that for serious ADRs. We then rounded these estimates to the nearest 10 to aid reasoning. We then calculated the "corresponding" numbers of ADE reports under the intervention by multiplying the assumed rate by the relative treatment estimate and the bounds on its 95% CI. A similar approach was used to re‐express ratios of numbers of ADE reports at one and two years as assumed and corresponding numbers of ADE reports in a setting with 1000 practitioners (for simplicity, the assumed number of ADE reports at two years was taken to be twice the number of ADE reports at one year).
Two review authors (from GS, NM, LG, WYC) independently assessed the certainty of the body of evidence (i.e. high, moderate, low and very low) as it relates to these outcomes (GRADEpro GDT 2015; Guyatt 2008), using the five GRADE considerations (risk of bias, consistency of effect, imprecision, indirectness and publication bias). We resolved disagreements on certainty ratings by discussion. We provided justification for decisions to downgrade or upgrade the ratings using footnotes in the table, and we added comments to aid readers' understanding of the review where necessary. We used plain language statements to report these findings in the review (EPOC 2013d).
We considered whether there was any additional outcome information that could not be incorporated in our meta‐analyses. We noted this in the comments and stated if it supported or contradicted the information from the meta‐analyses. If it was not possible to meta‐analyse the data, we narratively summarised the results in the text.
Results
Description of studies
Our study selection process is outlined in a PRISMA flow diagram (Liberati 2009) ‐ see Figure 1.
Results of the search
A comprehensive search of electronic databases from inception to 14 October 2022 retrieved 17,705 records. We also screened the reference lists of six recently published relevant systematic reviews (see Table 8 for details). Following de‐duplication, we screened the titles and abstracts of the remaining 11,520 records and retrieved 99 full‐text reports (including trial registrations) to assess for eligibility. Of these 99, 15 studies (17 references) met the review inclusion criteria (Included studies), and we excluded 69 studies (70 references), documenting our reasons ‐ see Excluded studies. We have eight studies awaiting classification, and three studies are ongoing (Hutchinson 2020; Kiguba 2022; NCT05402254). We identified an errata paper for one of the ongoing studies during our preparation of the review (Kiguba 2022).
Study design
There were two individually randomised controlled trials (Johansson 2009; Johansson 2011), five cluster‐randomised controlled trials (Figueiras 2006; Herdeiro 2008; Herdeiro 2012; Lopez‐Gonzalez 2015; Ribeiro‐Vaz 2011) and one cluster‐randomised cross‐over trial (Hanesse 1994). Six were interrupted time series studies (Ali 2018; Castel 2003; Chang 2017; McKaig 2014; Pedrós 2009; Ribeiro‐Vaz 2012) and one was a non‐randomised cross‐over study (Schlienger 1999).
Four of the studies were connected (Figueiras 2006; Herdeiro 2008; Herdeiro 2012; Ribeiro‐Vaz 2011). Figueiras 2006 and Herdeiro 2008 were conducted in the same 15 Portuguese clusters (which consisted of one reference hospital plus the outpatient centre and any other hospital in the catchment area). Figueiras 2006 targeted the physicians in these clusters and Herdeiro 2008 targeted the pharmacists. A second randomisation of the four intervention clusters from Figueiras 2006 and Herdeiro 2008 was performed, with two clusters receiving the workshop intervention and two clusters receiving the telephone‐interview intervention, and 11 clusters remaining assigned to the control arm. Herdeiro 2012 targeted the physicians and Ribeiro‐Vaz 2011 targeted the pharmacists in the newly randomised clusters.
Study setting
Five of the studies were conducted in Portugal (Figueiras 2006; Herdeiro 2008; Herdeiro 2012; Ribeiro‐Vaz 2011; Ribeiro‐Vaz 2012), three in Spain (Castel 2003; Lopez‐Gonzalez 2015; Pedrós 2009), two in Sweden (Johansson 2009; Johansson 2011) and one in each of the following countries: France (Hanesse 1994), Switzerland (Schlienger 1999), USA (McKaig 2014), China (Chang 2017) and Saudi Arabia (Ali 2018). Most of the studies were conducted in large tertiary care hospitals.
Participants
Approximately 62,389 participants were enroled in the 15 included studies. While some studies reported approximate participant numbers, four (all ITS studies) did not report the number of healthcare professionals exposed to the intervention. In seven studies, the intervention targeted physicians (Castel 2003; Chang 2017; Figueiras 2006; Hanesse 1994; Herdeiro 2012; Lopez‐Gonzalez 2015; Pedrós 2009). In three studies, the intervention targeted hospital pharmacists (Ali 2018; Herdeiro 2008; Ribeiro‐Vaz 2011). The intervention in Schlienger 1999 targeted physicians and nurses. The intervention in Johansson 2009 and Johansson 2011 targeted hospital GPs and nurses. In McKaig 2014 and Ribeiro‐Vaz 2012, the intervention targeted all hospital medical staff (physicians, nurses, and pharmacists).
Interventions
Based on the characteristics of the various interventions, the main focus of the interventions and usual practices implemented in the eligible studies, we devised the following overarching categories and comparisons.
Educating and informing
Comparison 1. Education session plus reminder card and ADR report form versus usual practice (Figueiras 2006; Herdeiro 2008; Herdeiro 2012; Lopez‐Gonzalez 2015; Ribeiro‐Vaz 2011)
Comparison 2. Informational letter and email versus usual practice (Johansson 2009; Johansson 2011)
Multifaceted intervention
Comparison 3. Multifaceted intervention (financial and non‐financial incentives, fines, education, reminders) versus usual practice (Ali 2018; Chang 2017; Pedrós 2009)
Comparison 4. Government regulations plus financial incentives versus usual practice (Chang 2017)
Process improvement
Comparison 5. Improving access to ADR report forms versus usual practice (Castel 2003; Hanesse 1994; Ribeiro‐Vaz 2012)
Comparison 6. Improving the reporting method versus usual practice (McKaig 2014)
Adverse drug event champion
Comparison 7. Case finding versus spontaneous reporting (Schlienger 1999)
Outcomes
See Table 9 for a summary of the outcomes measured and reported on in each study. In terms of our first primary outcome, all but two studies reported the number of ADR reports submitted (McKaig 2014; Schlienger 1999). McKaig 2014 (comparison 6 ‐ improving ME reporting method versus usual practice) reported the mean number of monthly medication error reports submitted pre‐ and post‐intervention implementation. Schlienger 1999 (comparison 7 ‐ case finding versus spontaneous reporting (usual practice) reported the number of ADE reports per 1000 patient‐days. None of the included studies provided data on our other primary outcome, the total number of false ADE reports (including ADR and ME) submitted by healthcare professionals.
2. Outcomes reported by the included studies.
| Total number of ADE reports (including ADR reports and ME reports) | Total number of false ADE reports (including false ADR reports and false ME reports) | Number of serious ADE reports (including serious ADR reports and serious ME reports) | Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports) | Number of unexpected (previously unknown) ADE reports (including unexpected ADR reports and unexpected ME reports) | Number of new drug‐related ADE reports (including drug‐related ADR reports and drug‐related ME reports) |
| Comparison 1. Education session plus reminder card and report form versus usual practice | |||||
| Figueiras 2006; Herdeiro 2008; Herdeiro 2012; Lopez‐Gonzalez 2015; Ribeiro‐Vaz 2011 (ADR reports) | Figueiras 2006; Herdeiro 2008; Herdeiro 2012; Lopez‐Gonzalez 2015; Ribeiro‐Vaz 2011 (ADR reports) | Figueiras 2006; Herdeiro 2008; Herdeiro 2012; Lopez‐Gonzalez 2015; Ribeiro‐Vaz 2011 (ADR reports) | Figueiras 2006; Herdeiro 2008; Lopez‐Gonzalez 2015; Ribeiro‐Vaz 2011 (ADR reports) | Figueiras 2006; Herdeiro 2008 (ADR reports) | |
| Comparison 2. Informational letter or email versus usual practice | |||||
| Johansson 2009; Johansson 2011 (ADR reports) | Johansson 2009; Johansson 2011 (ADR reports) | Johansson 2009; Johansson 2011 (ADR reports) | Johansson 2009; Johansson 2011 (ADR reports) | ||
| Comparison 3. Multifaceted intervention versus usual practice | |||||
| Ali 2018; Chang 2017; Pedrós 2009 (ADR reports) | Chang 2017; Pedrós 2009 (ADR reports) | Pedrós 2009 (ADR reports) | Chang 2017; Pedrós 2009 (ADR reports) | ||
| Comparison 4. Government regulations and financial incentives versus usual practice | |||||
| Chang 2017 (ADR reports) | |||||
| Comparison 5. Improving access to ADR report form versus usual practice | |||||
| Castel 2003; Ribeiro‐Vaz 2012; Hanesse 1994 (ADR reports) | |||||
| Comparison 6. Improving reporting method versus usual practice | |||||
| McKaig 2014 (mean number of monthly ME reports pre‐ and post‐intervention) | |||||
| Comparison 7. Case finding versus spontaneous reporting (usual practice) | |||||
| Schlienger 1999 (number of ADE reports per 1000 patient‐days) | |||||
- Serious ADRs: adverse drug reactions resulting in death; is life‐threatening; is a congenital anomaly; requires hospital admission or prolongation of stay in hospital; results in persistent or great disability, incapacity or both
- High‐causality ADRs: adverse drug reactions with attribution of definitive or probable causality; unexpected (previously unknown)
- ADRs: unknown adverse drug reactions that are not described in the drug's summary of product characteristics
- New‐drug‐related ADRs: adverse drug reactions relating to medications that have been on the market for less than five years
- ADE: adverse drug event
- ME: medication error
Excluded studies
We excluded 69 full‐text articles (70 studies). The most common reason for exclusion was ineligible study design. For more details, see Characteristics of excluded studies.
Risk of bias in included studies
For a summary of our assessment of the studies' risk of bias, see Figure 2 (controlled studies) and Figure 3 (ITS studies).
2.

Risk of bias summary for the controlled trials
3.
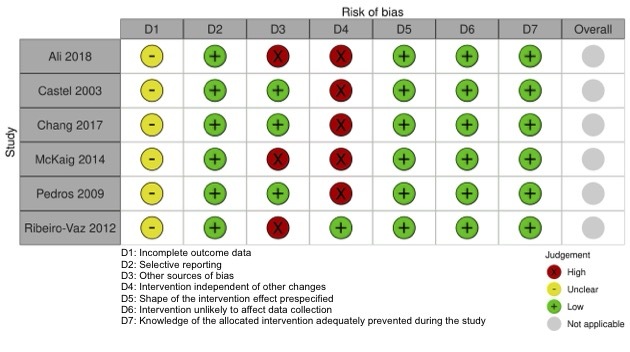
Risk of bias summary for interrupted time series (ITS) studies
Allocation
We judged Schlienger 1999 to have a high risk of selection bias as it was a non‐randomised study. Johansson 2009 and Johansson 2011 both have an unclear risk of selection bias as they did not describe how the random sequence was generated. However, both studies stated that "a person not involved in the study and without knowledge about the study protocol performed the randomisation procedure". Assuming that the randomisation procedure includes the allocation procedure, it is likely that overall, the risk of selection bias was low in both these studies. We judged Hanesse 1994 to have an unclear risk of selection bias. The study authors state that reporting methods were randomly assigned; however, they did not describe how the random sequence was generated or provide details regarding allocation concealment. We judged the remainder of the randomised studies to have a low risk of selection bias (Figueiras 2006; Herdeiro 2008; Herdeiro 2012; Lopez‐Gonzalez 2015; Ribeiro‐Vaz 2011).
Performance bias
We judged all the studies to have a high risk of performance bias as blinding of the participant or intervention targets was not possible due to the nature of the various interventions implemented.
Detection bias
We judged Schlienger 1999 to have a high risk of detection bias as the outcome assessor was not blinded to the group allocation. The risk of detection bias was judged unclear in Hanesse 1994 as the data on adverse drug reactions were extracted from patients' medical files in the study, and it was unclear who extracted them and if this person was blinded. Although blinding of the outcome assessors was not clearly or explicitly described in most of the studies, we judged the remaining studies to have a low risk of detection bias as the person who extracted the data was likely to be unaware of the intervention allocation.
Incomplete outcome data
We judged Schlienger 1999 as unclear risk of attrition bias as neither the flow of participants nor the retention of healthcare professionals was described in the published study. We judged all remaining randomised trials as having a low risk of attrition bias. There was no loss of clusters in the cluster‐randomised studies, and even though retention of healthcare professionals (which might affect the number of adverse drug event reports submitted) was not clearly reported, we assumed that any loss or addition of healthcare professionals was just as likely to occur in the intervention and control arms.
Selective reporting
We judged all the studies to have a low risk of reporting bias as, even though the protocol was not available for all the studies, all the outcomes mentioned in the methods section were reported on in the results section. Furthermore, all expected outcomes were reported in the studies. Only three studies were registered in a trial registry (Herdeiro 2008; Herdeiro 2012; Lopez‐Gonzalez 2015).
Other potential sources of bias
We judged Herdeiro 2012 and Ribeiro‐Vaz 2011 as having a high risk of other bias due to a baseline imbalance in adverse drug reaction reporting rate between the intervention and control groups. We judged Johansson 2009 as having a high risk of other bias due to potential contamination bias. The authors stated: "The intervention may also have spilled over to the control units. Doctors may work in more than one primary health care unit, i.e. both in the intervention group and in the control group. Also, the units all belong to the same organisation, and information may easily be passed on from one unit to another." We judged Schlienger 1999 as having an unclear risk of other bias due to an imbalance in the number of males and females in the intervention and control arms, and the effect of this imbalance on the number of reported adverse drug events being unclear. Schlienger 1999 is a non‐randomised cross‐over study; to mitigate the potential risk of bias due to the carry‐over effect, we included only data from the first phase of the study prior to cross‐over.
Interrupted time‐series (ITS) studies
Incomplete outcome data (attrition bias)
We judged all the ITS studies to have an unclear risk of attrition bias. Most of the studies reported the number of centres and hospitals exposed to the intervention and the number included in the analysis, but they did not report the number of healthcare professionals serving these hospitals or centres or the retention rate of healthcare professionals in the study.
Selective reporting (reporting bias)
We judged all the ITS studies to have a low risk of reporting bias. Even though we did not have access to the study protocols, all relevant outcomes in the method section were reported in the results section of the published studies.
Other potential sources of bias
We judged Ali 2018, McKaig 2014 and Ribeiro‐Vaz 2012 to have a high risk of other bias as none of these studies appeared to adjust for seasonality. Furthermore, there is a probable clustering effect in Ribeiro‐Vaz 2012 (there appears to be a hierarchy of hospitals within centres), which does not seem to be modelled in the analyses.
Intervention independent of other changes
We judged Ali 2018, Castel 2003, Chang 2017, McKaig 2014 and Pedrós 2009 to have a high risk of bias in this domain as the study authors did not provide any compelling argument that the intervention occurred independently of other changes over time and that the outcome was not influenced by other confounding variables or historic events during the study period. Ribeiro‐Vaz 2012 was the only study to explicitly address this issue: "From the initial 18 centres (31 hospitals), we excluded four hospitals that established other cooperation protocols with UFN to avoid a possible confounder bias. For the other 16 centres, we believe that there were no external interventions that could potentially explain the observed results.”
Shape of the intervention effect prespecified
We judged all the ITS studies to have a low risk of bias in this domain as the point of analysis matched the point of intervention and was clearly described in all the studies.
Intervention unlikely to affect data collection
We judged all the ITS studies to have a low risk of bias in this domain because we thought that the intervention itself was unlikely to affect data collection as sources and data collection methods were the same before and after the intervention in all studies.
Knowledge of the allocated interventions adequately prevented during the study
We judged all the ITS studies to have a low risk of bias in this domain because the outcome is objective. In all the studies, the number of ADE reports were retrieved from central or pharmacovigilance databases.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7
For a summary of the key comparisons and outcomes, see Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7.
None of the included studies reported on our primary outcome 'total number of false ADE reports' (see Primary outcomes).
Comparison 1. Education session plus reminder card and report form versus usual practice
Data for this comparison came from five cluster‐RCTs (Figueiras 2006; Herdeiro 2008; Herdeiro 2012; Lopez‐Gonzalez 2015; Ribeiro‐Vaz 2011; see Table 1). All trials assessed the effect of an education session (consisting of 30‐ to 60‐minute long face‐to‐face group workshops or telephone interviews, or both), plus a reminder card (similar to the ADR report form summarising the main points from the education session) and an ADR report form on the number of ADR reports submitted. The effect of the intervention was measured amongst participants (i.e. physicians and pharmacists).
At 13 to 16 months after the implementation of the intervention, compared to usual practice, education sessions together with reminder cards and ADR report forms may substantially increase the number of submitted ADR reports (risk ratio 3.00, 95% CI 1.53 to 5.90; Figure 4), serious ADR reports (risk ratio 3.30, 95% CI 1.51 to 7.21; Figure 5), high‐causality ADR reports (risk ratio 2.48, 95% CI 1.11 to 5.57; Figure 6), unexpected or previously unknown ADR reports (risk ratio 4.72, 95% CI 1.75 to 12.76; Figure 7) and new‐drug‐related ADR reports (risk ratio 8.68, 95% CI 3.40 to 22.13; Figure 8). The certainty of the evidence was low for all these findings.
4.
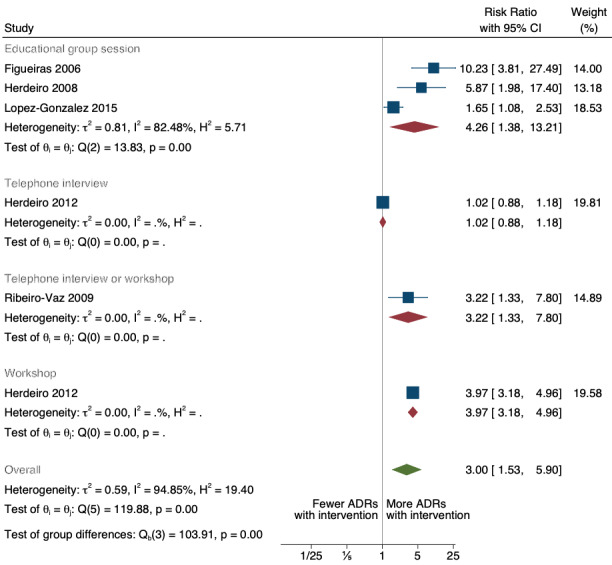
Comparison 1. Education session plus reminder card and report form versus usual practice. Outcome: total number of adverse drug reaction reports submitted. Education delivered in group sessions, workshops or via telephone. Meta‐analysis of five cluster‐randomised controlled studies.
5.
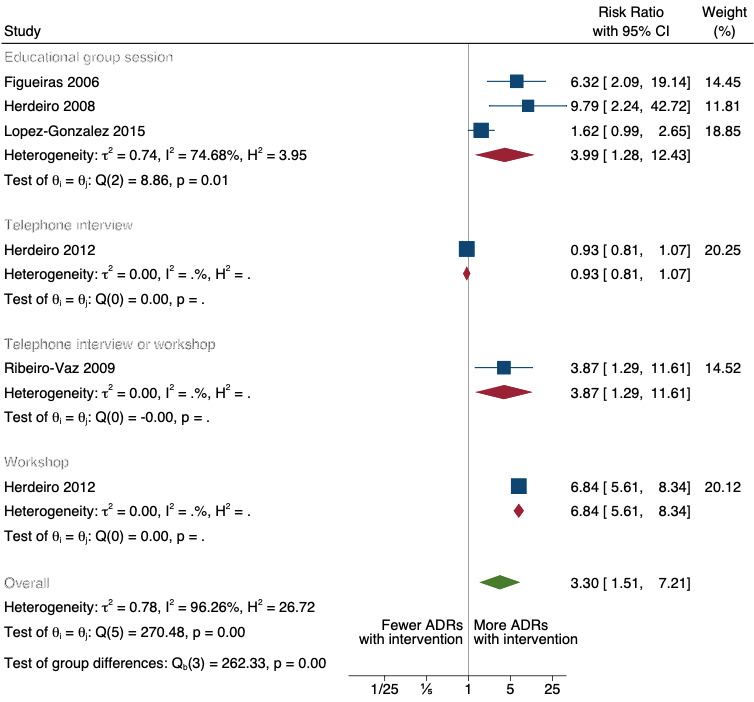
Comparison 1. Education session plus reminder card and report form versus usual practice. Outcome: number of serious adverse drug reaction reports submitted
6.
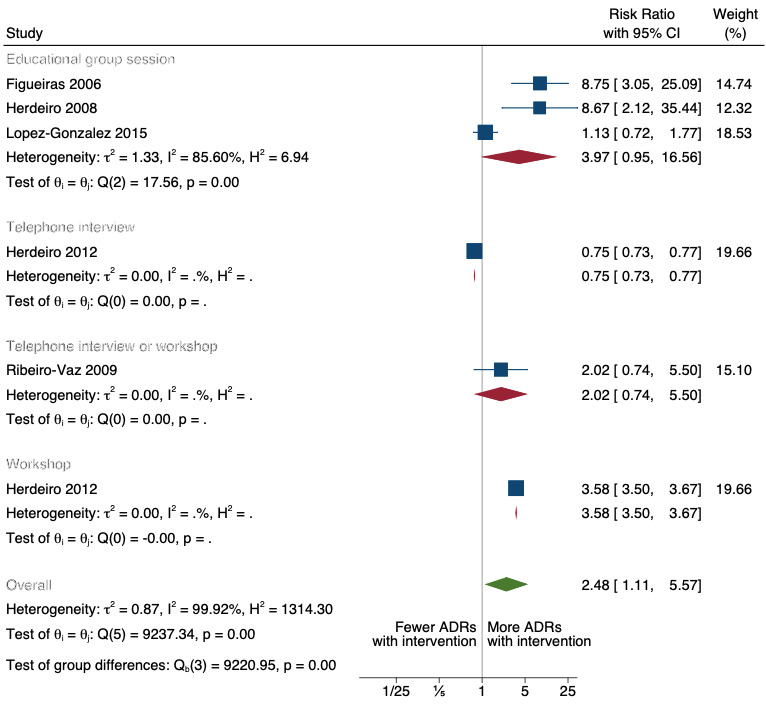
Comparison 1. Education session plus reminder card and report form versus usual practice. Outcome: high‐causality adverse drug reaction reports submitted
7.
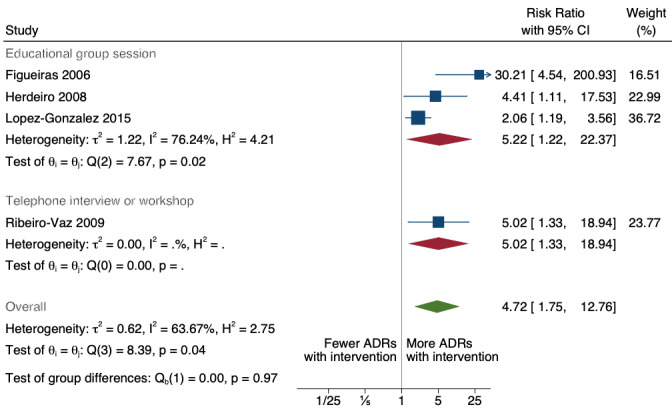
Comparison 1. Education session plus reminder card and report form versus usual practice. Outcome: unexpected adverse drug reaction reports
8.
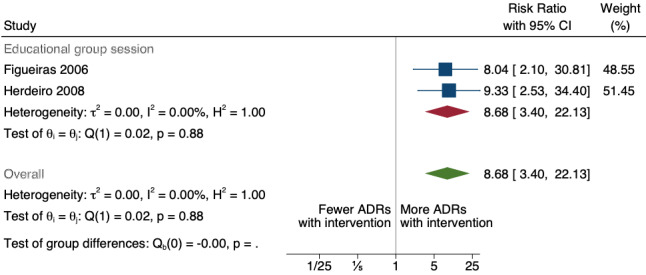
Comparison 1. Education session plus reminder card and report form versus usual practice. Outcome: number of new drug‐related adverse drug reaction reports submitted
Comparison 2. Informational letter or email versus usual practice
Two RCTs provided data for this comparison (Johansson 2009; Johansson 2011; see Table 2). The two studies assessed the effectiveness of distributing a series of informative emails or letters to GPs and nurses in primary healthcare units describing the importance of reporting ADRs and instructions on how to do so. The effect of the intervention was measured in primary healthcare unit years of exposure.
At the one‐year follow‐up time point following the implementation of the intervention, compared to usual practice, we do not know if sending informational letters or emails to GPs and nurses increases the number of total ADR reports because the certainty of the evidence is very low (rate ratio 1.28, 95% CI 0.42 to 3.91; very low certainty evidence; Figure 9). Similarly, we do not know if sending informational letters or emails to GPs and nurses increases the number of serious ADR reports (rate ratio 1.79, 95% CI 0.69 to 4.65; very low certainty evidence; Figure 10), new‐drug‐related ADR reports (rate ratio 2.58, 95% CI 1.12 to 5.92; very low certainty evidence; Figure 11) or unexpected or previously unknown ADR reports (rate ratio 1.46, 95% CI 0.92 to 2.30; very low certainty evidence; Analysis 1.1). This is because the certainty of the evidence for these outcomes is also very low.
9.

Comparison 2. Informational letter or email vs usual practice. Outcome: total number of adverse drug reaction reports submittedd
10.

Comparison 2. Informational letter or email versus usual practice. Outcome: number of serious adverse drug reaction reports submitted
11.

Comparison 2. Informational letter or email versus usual practice. Outcome: number of new drug‐related adverse drug reaction reports submitted
1.1. Analysis.

Comparison 1: Comparison 2: Informational letter or email versus usual practice, Outcome 1: Number of unexpected (previously unknown) adverse drug reaction reports
Comparison 3. Multifaceted interventions (financial incentives, fines, non‐financial incentives, education, reminder) versus usual practice
Three ITS studies assessed the effectiveness of various interventions that included financial and non‐financial (i.e. letters of appreciation, employee of the month award, certificate and a day's leave) incentives for spontaneous reporting of ADRs, fines for not reporting or missing an ADR, education workshops, and periodic reminders to report ADRs (Ali 2018; Chang 2017; Pedrós 2009). The data were presented only in the form of published figures, and thus we extracted and re‐analysed these data (see Figure 12; Figure 13; Figure 14; Figure 15; Figure 16; Figure 17; Figure 18; Figure 19). The periods over which the studies were conducted and the lengths of follow‐up differed across the three studies in this comparison as detailed below.
12.
Re‐analysis of data from published graph. Ali 2018: Comparison 3. Multifaceted interventions versus usual practice. Outcome: total number of adverse drug reaction reports submitted
13.
Re‐analysis of data from published graph. Chang 2017 Comparison 3. Multifaceted intervention versus usual practice. Outcome: total number of adverse drug reaction reports submitted
14.
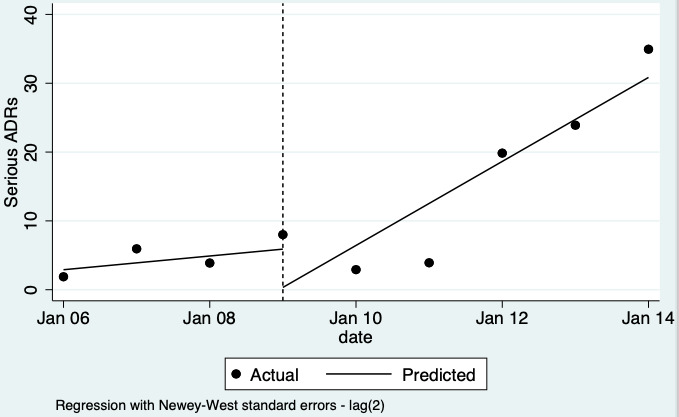
Re‐analysis of data from published graph. Chang 2017 Comparison 3. Multifaceted intervention versus usual practice. Outcome: number of serious adverse drug reaction reports submitted
15.
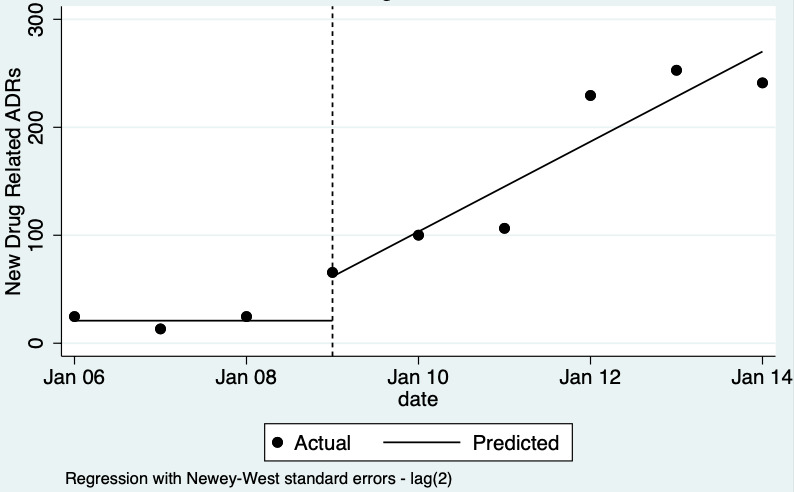
Re‐analysis of data from published graph. Chang 2017 Comparison 3. Multifaceted intervention versus usual practice. Outcome: number of new drug‐related adverse drug reaction reports submitted
16.
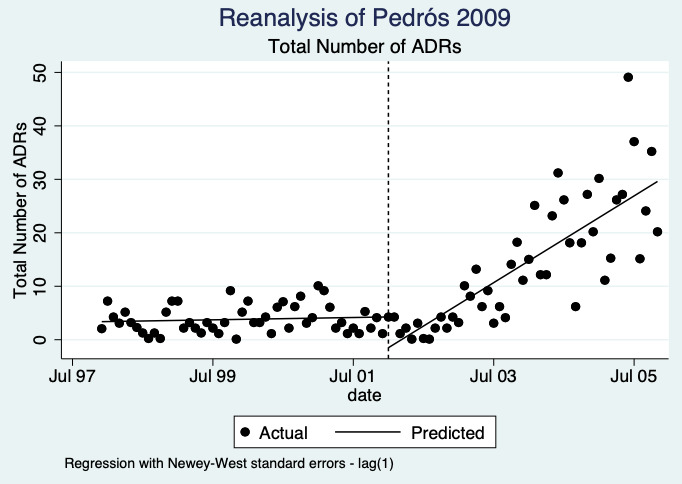
Pedrós 2009: re‐analysis of total number of adverse drug reaction reports submitted
17.
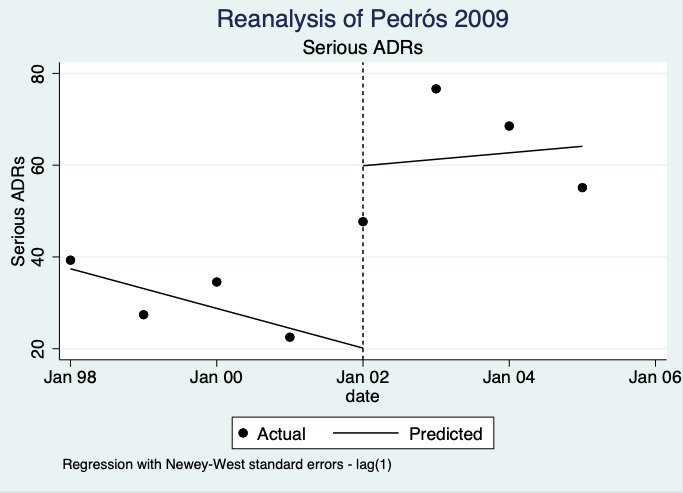
Pedrós 2009: re‐analysis of number of serious adverse drug reaction reports submitted
18.
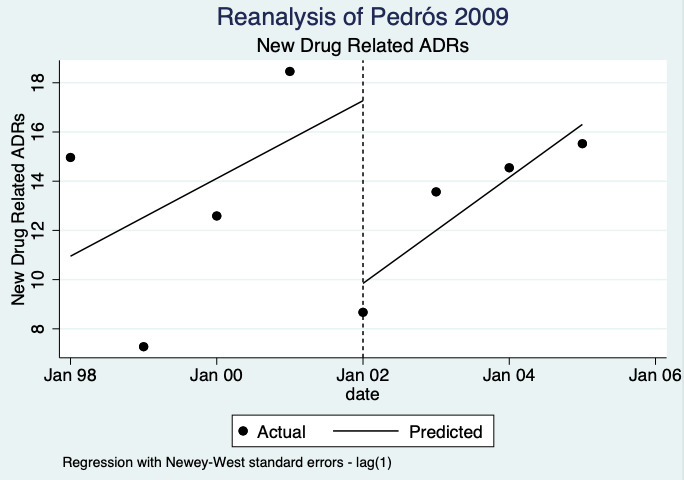
Pedrós 2009: re‐analysis of number of new‐drug‐related adverse drug reaction reports submitted
19.
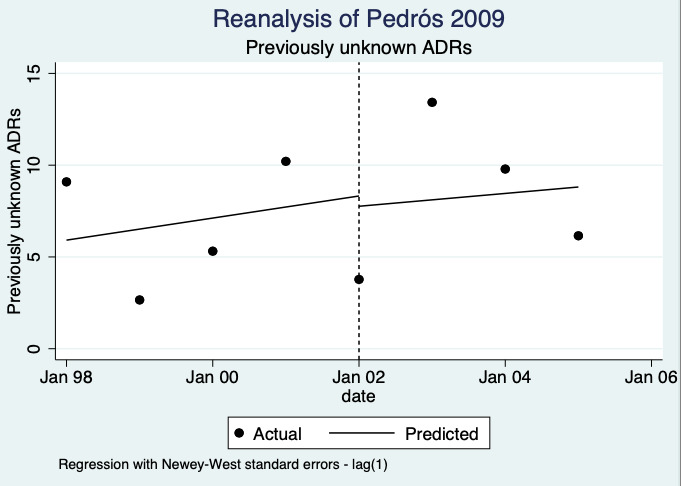
Pedrós 2009: re‐analysis of number of previously unknown adverse drug reaction reports submitted
Ali 2018: December 2015 (first time point); January 2016 (intervention implemented); November 2016 (last time point); a total of 24 observations
Chang 2017: 2006 to 2009 (pre‐intervention); 2009 to 2011 (financial incentive); 2012 to 2014 (financial incentive plus government regulations for antimicrobial agents); December 2014 (last time point); total 108 observations
Pedrós 2009: January 1998 (first point); December 2002 (intervention implemented); December 2005 (last time point); a total of 96 observations
Table 3 presents the meta‐analysed data from Chang 2017a and Pedrós 2009. Ali 2018 data is presented separately (see Table 10), as the follow‐up period after the implementation of the intervention was too short to be included in the meta‐analysis. The data for this comparison is generated through ITS studies, in which it is often impossible to know how many people are exposed to the intervention. Therefore, the effectiveness of the intervention is assessed over the mean duration of the contributing studies (i.e. the length of time participants were exposed to the intervention) instead of the number of participants.
3. Re‐analysis of data in three included studies (Ali 2018, Chang 2017, Pedros 2009).
| Study | Outcome | Relative treatment effect | ITS parameter | 95% CI |
| Ali 2018 | Total number of ADR reports | Relative change in level | 2.533 | 1.658 to 3.408 |
| Relative change in slope | 0.308 | 0.073 to 0.543 | ||
| Relative expected adverse drug reaction reports1 | 6.99 | 3.43 to 10.54 | ||
|
Chang 2017 Financial incentives |
Total number of ADR reports | Relative change in level | 2.084 | 1.490 to 2.678 |
| Relative change in slope | 0.061 | ‐0.005 to 0.126 | ||
|
Chang 2017 Government regulations |
Total number of ADR reports | Relative change in level | 0.695 | 0.129 to 1.261 |
| Relative change in slope | ‐0.028 | ‐0.069 to 0.014 | ||
| Pedrós 2009 | Total number of ADR reports | Relative change in level | ‐0.538 | ‐1.142 to 0.065 |
| Relative change in slope | 0.059 | 0.026 to 0.092 |
1See Figure 22 for data source
ADR: adverse drug reactions; CI: confidence interval; ITS: interrupted time series
We do not know if multifaceted interventions (including incentives, fines, education meetings, and reminder cards) increase the total number of ADE reports by physicians and pharmacists at the one‐ and two‐year time points following intervention implementation. This is because the certainty of the evidence is very low (relative number of ADR reports after one year: 4.29, 95% CI 0.32 to 56.76; Figure 20; after two years: 8.11, 95% CI 0.61 to 107.93; Figure 21; very low certainty evidence; see Table 3. Although Ali 2018 showed an increase in the total number of ADR reports (relative number of ADR reports after one year: 6.99, 95% CI: 3.43 to 10.54; Figure 22 and Table 10) following the implementation of incentives (both non‐financial and financial) targeting the hospital’s clinical pharmacists, the certainty of this evidence is very low, so we do not know if this intervention is truly effective or not.
20.
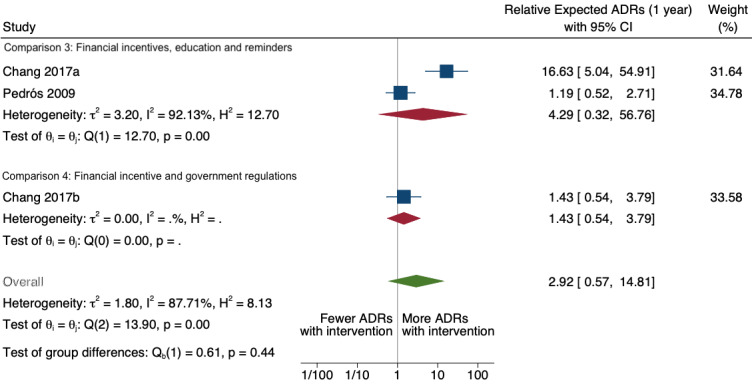
Comparison 3 and 4. Outcome: total number of ADR reports submitted after 1 year
21.
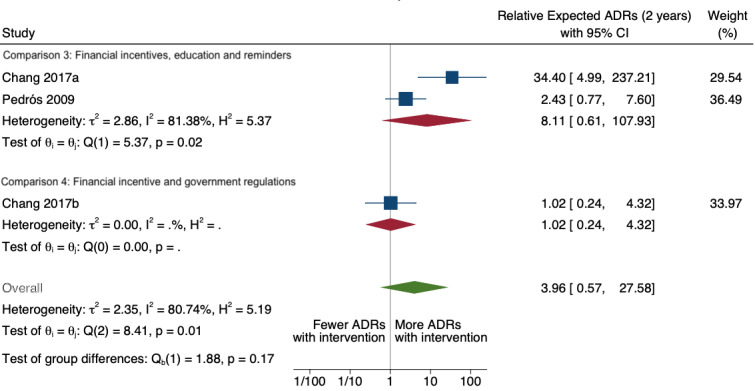
Comparison 3 and Comparison 4. Outcome: total number of ADR reports after 2 years
22.
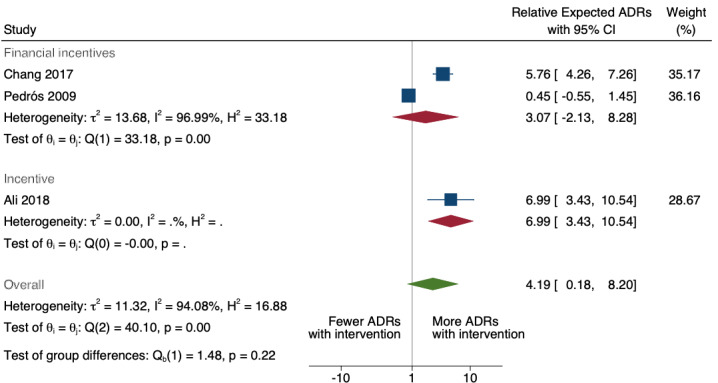
Multifaceted interventions: total number of adverse drug reaction reports submitted. This was the first draft of the analyses. We subsequently took Ali 2018 out of the meta‐analysis as the follow‐up time was too short compared to Chang 2017 and Pedrós 2009.
Similarly, we do not know if multifaceted interventions directed at physicians and pharmacists increase the total number of serious ADR reports (relative number of ADR reports after one year: 2.10, 95% CI 0.29 to 15.20; Figure 23; after two years: 2.57, 95% CI 0.22 to 29.93; Figure 24; very low certainty evidence), the total number of new drug‐related ADR reports (after one year: 1.65, 95% CI 0.20 to 13.77; Figure 25; after two years: 1.86, 95% CI 0.16 to 21.59; Figure 26; very low certainty evidence), or the total number of unexpected or previously unknown ADR reports (after one year 0.73, 95% CI 0.02 to 22.75; Figure 27; after two years: 0.67, 95% CI 0.01 to 61.55; Figure 28; very low certainty evidence). This is because the certainty of the evidence is very low. The 95% CIs on the relative treatment effect, and the corresponding illustrative comparative numbers of ADEs, likely reflect (a) the limitations of re‐analysis of uncontrolled ITS studies that often reported relatively few data points; (b) highly heterogeneous study results and the effect of the random‐effects assumption; and (c) extrapolation using a relatively simple model.
23.
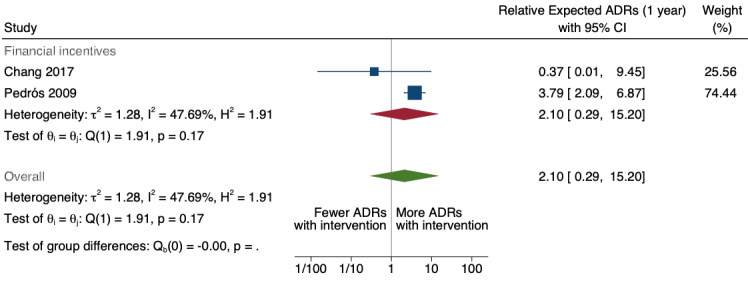
Comparison 3. Multifaceted interventions. Outcome: serious adverse drug reaction reports submitted after 1 year
24.
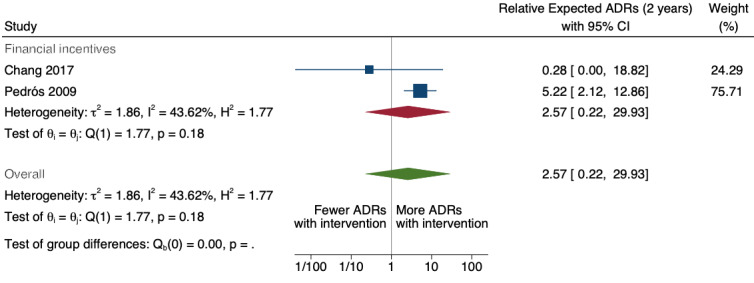
Comparison 3. Multifaceted interventions. Outcome: serious adverse drug reaction reports submitted after 2 years
25.
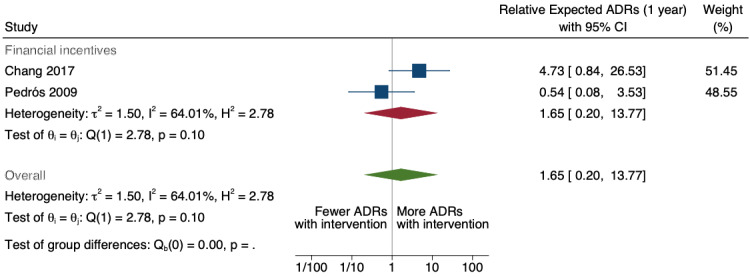
Comparison 3. Multifaceted interventions. Outcome: new‐drug‐related adverse drug reaction reports submitted after 1 year
26.
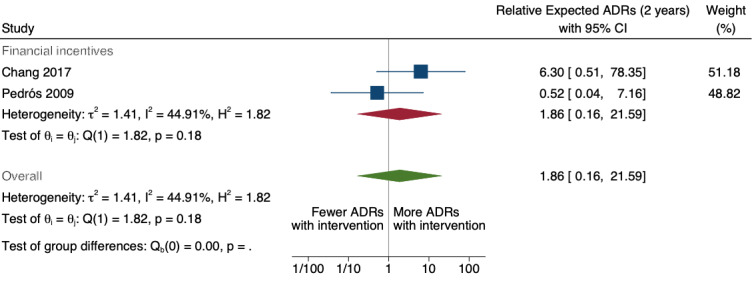
Comparison 3. Multifaceted interventions. Outcome: new‐drug‐related adverse drug reaction reports submitted after 2 years
27.
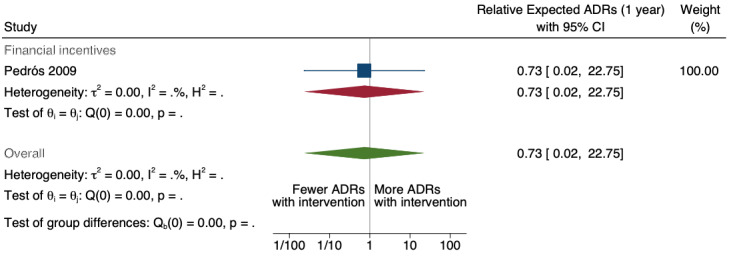
Comparison 3. Multifaceted interventions. Outcome: unexpected (previously unknown) adverse drug reaction reports submitted after 1 year
28.
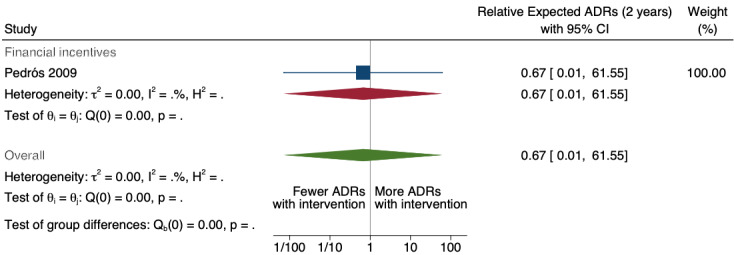
Comparison 3. Multifaceted interventions. Outcome: unexpected (previously unknown) adverse drug reaction reports submitted after 2 years
Comparison 4: Government regulations plus financial incentives versus usual practice
Chang 2017 assessed the effect of implementing financial incentives for ADR reporting together with government regulations on the clinical use of antimicrobial agents, which included detailed reporting on the total number of ADR reports by physicians (see Figure 20; Figure 21; Table 4). As the data were generated by an ITS study, the effectiveness of the intervention is assessed over the mean duration of the contributing study (i.e. length of time exposed to the intervention) instead of the number of participants.
We do not know if government regulations and financial incentives increase the total number of ADR reports by physicians one or two years after the implementation of these interventions as the certainty of the evidence is very low (after one year: 1.43, 95% CI 0.54 to 3.79; Figure 20; after two years: 1.02, 95% CI 0.24 to 4.32; Figure 21; very low certainty evidence). Chang 2017 did not report on the number of serious ADR reports, new drug‐related ADR reports or unexpected or previously unknown ADR reports.
Comparison 5: Improving access to ADE report forms versus usual practice
One randomised cross‐over study (Hanesse 1994) and two ITS studies (Castel 2003; Ribeiro‐Vaz 2012) provided data for this comparison. We extracted the data from the ITS studies from published figures and re‐analysed them (see Figure 29 and Figure 30).
29.
Re‐analysis of data from published graph. Castel 2003 Comparison 4. Improving access to adverse drug reaction report form versus usual practice. Outcome: total number of adverse drug reaction reports submitted
30.
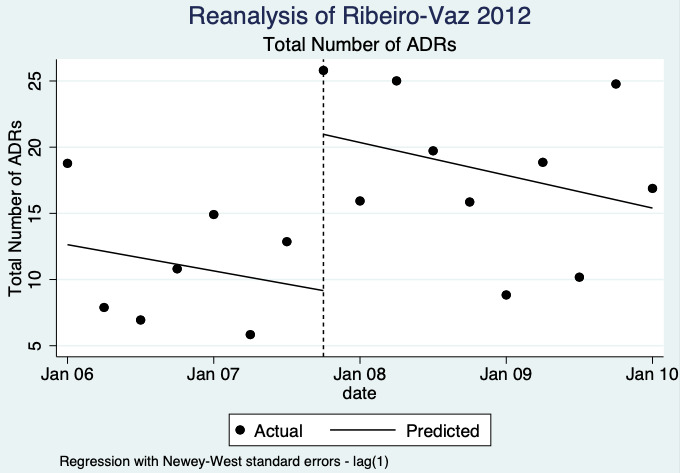
Ribeiro‐Vaz 2012: re‐analysis of total number of adverse drug reaction reports submitted
Data from Hanesse 1994 suggests that the use of a standardised discharge form (based on the ‘Diagnosis Related Groups’ (DRG) system and used by physicians for recording patient diagnoses, and medical and surgical acts received during hospital stay) with additional ADR items (addressing time of occurrence and evolution) may slightly improve the number of ADR reports (risk ratio 2.06, 95% CI 1.11 to 3.83; low‐certainty evidence). See Table 5 and Table 11 for more details).
4. Comparison 5. Improving access to ADR report forms (Hanesse 1994).
| Study | Intervention | Outcome | Risk ratio | 95% CI |
| Hanesse 1994 | Improving access vs usual practice | Total number of ADR reports | 2.06 | 1.11 to 3.83 |
ADR: adverse drug reactions; CI: confidence interval; vs: versus
We do not know if making it easier to report ADEs by including ADR yellow card report forms in quarterly bulletins and prescription pads or by providing a hyperlink to the reporting form in hospitals' electronic patient records leads to more ADRs being reported because the certainty of this evidence, based on combined data from Castel 2003 and Ribeiro‐Vaz 2012, is very low (after one year: 1.95, 95% CI 1.33 to 2.85; Figure 31; after two years: 1.80, 95% CI 1.08 to 3.01; Figure 32; very low certainty evidence). See Table 5 and Table 12 for more details.
31.
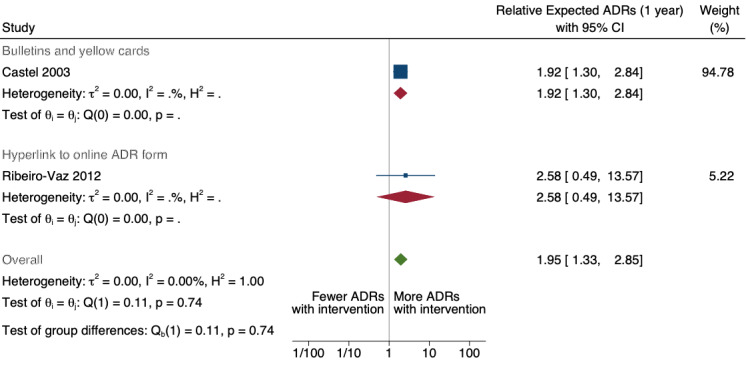
Comparison 5. Improving access to adverse drug reaction report form versus usual practice. Outcome: total number of adverse drug reaction reports submitted after 1 year
32.
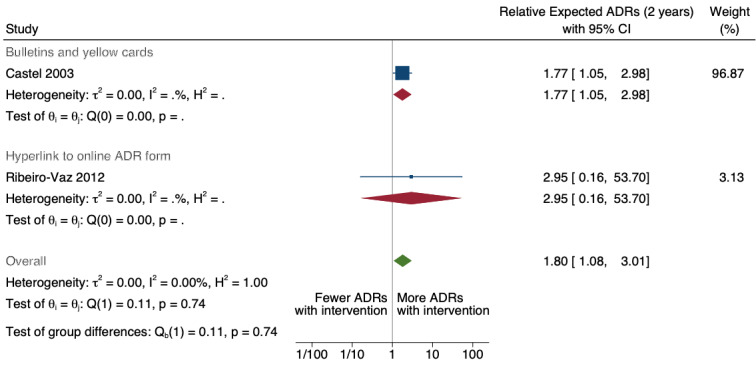
Comparision 5. Improving access to adverse drug reaction report form versus usual practice. Outcome: total number of adverse drug reaction reports submitted after 2 years
5. Comparison 5. Improving access to ADR report forms (Castel 2003 and Ribeiro‐Vaz 2012).
| Study ID | Intervention | Outcome | Measure | ITS parameter | 95% CI |
| Castel 2003 | Bulletins and ADR report forms | Total number of ADR reports | Relative change in level | 0.734 | 0.422 to 1.047 |
| Relative change in slope | ‐0.007 | ‐0.022 to 0.008 | |||
| Relative expected ADRs (1 year) | 0.653 | 0.263 to 1.043 | |||
| Relative expected ADRs (2 years) | 0.572 | 0.050 to 1.093 | |||
| Ribeiro‐Vaz 2012 | Hyperlink to online ADR form | Total number of ADR reports | Relative change in level | 0.813 | 0.302 to 1.324 |
| Relative change in slope | 0.034 | ‐0.281 to 0.348 | |||
| Relative expected ADRs (1 year) | 0.947 | ‐0.714 to 2.608 | |||
| Relative expected ADRs (2 years) | 1.081 | ‐1.822 to 3.983 |
ADR: adverse drug reaction; CI: confidence interval; ITS: interrupted time series
Comparison 6. Improving medication error reporting method versus usual practice
One ITS study assessed the effect on the number of ME reports of re‐engineering the web‐based electronic error reporting system (i.e. incorporating a series of standardised screens, drop‐down menu choices, and input fields designed to collect specific information and improve communication with all departments involved) (McKaig 2014). We extracted the data from a published figure and re‐analysed them (see Table 13 for a detailed re‐analysis of the data). As the data were from an ITS study, the effectiveness of the intervention is assessed over the mean duration of the contributing study (i.e. length of time exposed to the intervention) instead of the number of participants.
6. Comparison 6. Improving reporting practice vs usual practice (McKaig 2014).
| Study | Outcome | Relative treatment effect | ITS parameter | 95% CI |
| McKaig 2014 | Total number of ME reports | Relative change in level | 0.426 | 0.229 to 0.624 |
| Relative change in slope | 0.013 | ‐0.010 to 0.037 | ||
| Relative expected ME reports (1 year) | 0.586 | 0.142 to 1.031 | ||
| Relative expected ME reports (2 years) | 0.746 | 0.026 to 1.466 |
CI: confidence interval; ITS: interrupted time series; ME: medication error
We do not know if the number of ME reports are greater one year or two years after re‐engineering the web‐based electronic error reporting system as the certainty of the evidence is very low for both time points (after one year: 1.80, 95% CI 1.15 to 2.80; Figure 33; after two years: 2.11, 95% CI 1.03 to 4.33; Figure 34). See Table 6.
33.
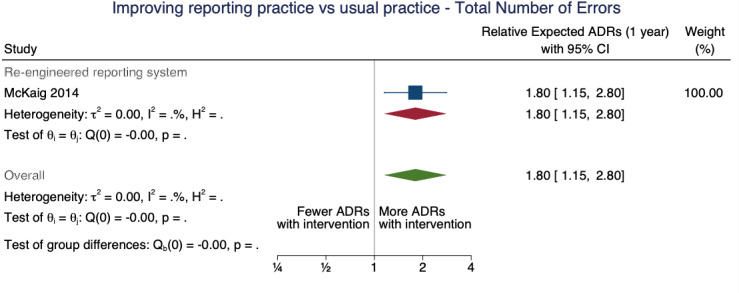
Comparison 6. Improving usability of reporting form versus usual practice. Outcome: total number of medication error reports submitted after 1 year
34.
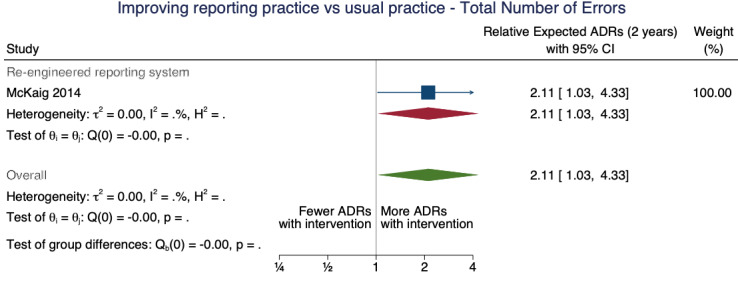
Comparison 6. Improving usability of reporting form versus usual practice. Outcome: total number of medication error reports submitted after 2 years
Comparison 7. Case finding versus spontaneous reporting
A non‐randomised, prospective cross‐over study evaluated the effectiveness of employing a clinical pharmacist to encourage physicians and nurses to identify and report ADEs in hospitalised patients (Schlienger 1999). As Schlienger 1999 is a cross‐over study without a washout period, we used only the data from the first period of the study to minimise any possible "learning effect" on the findings. Using data from Schlienger 1999 (from Table 3 in the full‐text study report), we imputed the incidence rates and 95% CIs; then we computed the incidence rate ratios (IRRs) to compare the ADE reporting rate with and without the presence of a clinical pharmacist (see Table 14).
7. Comparison 7. Case finding versus spontaneous reporting (usual practice) (Schlienger 1999).
| Study | Incidence rate (95% CI) of ADE reports: period 1 (12 months) | Incidence rate ratio | |
| With clinical pharmacist | Without clinical pharmacist | ||
| Schlienger 1999 | 15.5 (95% CI 12.87 to 18.51) ADEs per 1000 patient‐days |
1.4 (95% CI 0.75 to 2.39) ADEs per 1000 patient‐days |
11.07 (95% CI 6.24 to 21.38) more ADEs per 1000 patient‐days with clinical pharmacist than without |
ADE: adverse drug event; CI: confidence interval
As Schlienger 1999 is a cross‐over study without a washout period, we only used the data from the first period of the study, prior to the treatment allocation cross‐over. Using data from Table 3 in the full‐text study report (first study period), we imputed the incidence rates and 95% CI; then we computed the incidence rate ratios to compare the rate of ADE reporting rate with and without the presence of a clinical pharmacist actively checking for and submitting reports on any ADEs.
We do not know if the presence of a clinical pharmacist actively identifying and encouraging the identification and reporting of ADEs in a hospital setting increases the number of ADE reports as the certainty of the evidence is very low (IRR 11.07, 95% CI 6.24 to 21.38). See Table 7.
Discussion
A comprehensive search for published and unpublished evidence identified 15 studies of various designs (enroling approximately 62,389 participants) that addressed the review question.
Educating and informing
Comparison 2. Informational letter or email versus usual practice
Fairly robust evidence from five cluster‐randomised trials suggests that, compared to usual practice, education sessions along with reminder cards, and provision of ADR report forms may increase the number of adverse drug reactions reported (see Table 1). The inconsistency in the effect of educational interventions may be explained by the mode of delivery. Based on the limited data available, outreach group education sessions delivered in‐person, appeared to be more effective in increasing the number of ADR reports submitted compared with delivering similar information in a 3‐ to 8‐minute telephone interview. The different target audience (physicians versus pharmacists) in the studies may also explain the inconsistency in the effect of the education intervention on the number of ADR reports submitted. Participation rates varied between studies and modes of delivering the education intervention. For example, participation rates in the telephone interviews ranged from 7.9% to 36%, whereas participation rates in the education workshops ranged from 27% to 81%. Almost all the studies reported that the effect of the educational interventions lasted no longer than six months, suggesting that education sessions might have to be conducted on a continuous 6‐monthly basis to maintain any benefits. The benefits of the intervention would have to be balanced against the time and human resource costs required to implement the education sessions, which may limit the scalability of the intervention.
In contrast, we do not know if sending informational letters or emails to GPs and nurses increases the quantity of ADR reports as the certainty of this evidence is very low (see Table 2). A major limitation of this intervention is not knowing if the informational email or letter reached its target audience. It would be beneficial to know the true effect of this intervention as it is cheap and easy to implement with broad‐reaching potential.
Multifaceted interventions, including educating, informing, incentivising (financial and non‐financial), and reprimanding
Comparison 4. Government regulations plus financial incentives versus usual practice
Based on very low certainty evidence from three ITS studies, we do not know if multifaceted interventions, including incentives, fines, educational meetings, and reminder cards, increase the number of adverse drug reaction reports submitted by physicians and pharmacists one year and two years after the implementation of the intervention (see Table 3). While in most cases, quite dramatic increases in adverse drug reaction reports were noted, wide confidence intervals around the summary estimates of effect, and the corresponding illustrative comparative numbers of adverse drug reactions, reflected the uncertainty in the evidence and the possibility of the results being compatible with both an increase or a decrease in the number of reports, or no difference between them. We do not know if implementing government regulations together with financial incentives increases the number of adverse drug reaction reports submitted by physicians one year or two years after implementing these interventions, as the certainty of the evidence is also very low (see Table 4). Financially and non‐financially incentivising or punishing healthcare professionals based on the number of adverse drug reaction reports submitted may increase the risk of false adverse drug event reports, as healthcare professionals' better judgement might be clouded by greed or fear. Unfortunately, none of the studies included in this review reported the effect of any interventions on the number of false reports of adverse drug events submitted.
Process improvement
Comparison 6. Improving medication error reporting method versus usual practice
While it may seem evident that improving the reporting process should result in an increase in adverse drug event reports being submitted, the certainty of the available evidence is low or very low. Making it easier to report adverse drug reactions by implementing a standardised discharge form (based on the ‘Diagnosis Related Groups’ (DRG) system for recording patient diagnoses, medical and surgical acts received during hospital stay) with additional ADR items (i.e. time of occurrence and evolution) may slightly improve the number of adverse drug reaction reports (Hanesse 1994). However, we do not know if including ADR report forms in quarterly bulletins and prescription pads or providing a hyperlink to the reporting form in hospitals' electronic patient records to improve the ease of adverse drug reaction reporting leads to more adverse drug reaction reports being submitted because the certainty of this evidence is very low (Castel 2003; Ribeiro‐Vaz 2012; see Table 5). We also do not know if improving the reporting process by re‐engineering the web‐based electronic error reporting system (i.e. incorporating standardised screens, drop‐down menu choices, and input fields to collect specific information and improve communication with all departments involved) will increase the expected number of medication error reports one or two years after implementing the changes as the certainty of the evidence is very low (see Table 6).
Adverse drug event champion
Comparison 7. Case finding versus spontaneous reporting
Finally, we do not know if the presence of a clinical pharmacist actively identifying and encouraging the identification and reporting of adverse drug events in a hospital setting increases the number of adverse drug event reports as the certainty of the evidence is very low (see Table 7).
Overall completeness and applicability of evidence
Although we identified 15 eligible studies investigating the effectiveness of various interventions on the reporting rate of adverse drug events, important gaps in the evidence base remain.
All 15 studies included in this review were conducted in high‐income countries with relatively well‐established pharmacovigilance systems. This may limit the applicability of the evidence to countries without established functioning pharmacovigilance systems. For the most part, interventions to improve reporting of adverse drug events (including adverse drug reactions and medication errors) were targeted at hospital physicians. General practitioners, nurses and pharmacists were targeted to a lesser extent. As a result, we are not sure of the transferability of the effectiveness of the various interventions to all health professionals.
Most studies assessed the effectiveness of interventions to increase the number of adverse drug reaction reports. Only one study provided data on ways to increase reporting of medication errors (McKaig 2014), and one study provided data on ways to increase adverse drug event reporting (Schlienger 1999). Importantly, none of the studies included in the review investigated the impact of the tested interventions on the number of false adverse event reports submitted. This outcome is of particular concern with interventions offering financial or non‐financial incentives or punishment based on the number of adverse drug reaction reports submitted.
Quality of the evidence
Overall, the certainty of the evidence related to the effectiveness of interventions to improve the reporting of adverse drug events is low to very low. Of the 15 included studies, eight studies incorporated randomisation and a control group. Most of the remaining studies were interrupted time series studies without control populations. Our certainty in the evidence from randomised studies was reduced due to possible risk of bias, inconsistency of effects and imprecision around the effect estimates. We further downgraded the low certainty of the evidence of the observational studies because lack of a control group made it impossible to rule out confounding variables or events as the cause of any effects observed. The inconsistency of the effects between the studies may be explained, but we cannot be certain of our explanations. Lastly, the evidence provided by observational studies was imprecise, including both benefits and harms.
Potential biases in the review process
We used a comprehensive method to identify all eligible studies investigating interventions aimed at healthcare professionals with the intention of increasing the reporting rate of adverse drug events (including adverse drug reactions and medication errors). We used a sensitive search strategy, without date or language limits, to conduct a comprehensive search of a number of electronic databases for both published and unpublished studies. We supplemented our search of electronic databases by hand‐searching the reference lists of relevant systematic reviews and eligible studies for additional eligible studies. We also searched clinical trial registry sites for ongoing studies.
Throughout the review process, every effort was made to reduce any potential risk of bias in this review. We consistently adhered to our published review protocol. We have detailed any deviations we made from the published protocol in the Differences between protocol and review section of this review. At least two review authors independently screened identified records for eligibility and extracted data from eligible studies. At least two review authors independently assessed the certainty of the evidence using GRADE. Any disagreements regarding eligibility, extracted data, or certainty of the evidence were discussed or referred to a third review author for resolution. We could not assess publication bias in our meta‐analyses using funnel plots as we did not have enough studies to do so reliably.
Agreements and disagreements with other studies or reviews
A number of systematic reviews have examined this question in whole or in part. We briefly describe these reviews in Table 8. We have included all relevant studies from these reviews in this Cochrane review. The findings of this Cochrane review closely mirror those of the other systematic reviews, which agree that educational sessions, particularly outreach, in‐person group workshops (see Table 1), may improve the number of adverse drug reaction reports by healthcare professionals. This finding is further supported by the findings of a Cochrane review looking at the effectiveness of educational outreach visits on healthcare professionals' practice (O'Brien 2007). Based on data from 69 studies, educational outreach improved the care delivered to patients through small to moderate changes in practice (O'Brien 2007).
Another key finding of the systematic reviews was that multifaceted interventions are likely to be more effective than single interventions. Our Cochrane review was not designed to test this (i.e. single versus multifaceted interventions), so we cannot make such claims. Most of the interventions assessed in our Cochrane Review were multifaceted, including some form of educational input (even when it was not the focus of the intervention), reminders and ADR report forms.
The other systematic reviews also drew attention to the limitations of the current body of evidence; while uncontrolled, observational studies are pragmatic, they do not provide robust data. The evidence is limited to high‐income countries with relatively good pharmacovigilance systems, and there is no long‐term follow‐up data on the degree of the longevity of any intervention effect.
A major difference between our Cochrane Review and previous systematic reviews is that we looked for evidence of harm (i.e. an increase in the number of false adverse drug event reports) as a result of the interventions. Unfortunately, we did not find any data on this outcome, but it remains an important aspect for consideration going forward.
Authors' conclusions
Implications for practice.
Low‐certainty evidence suggests that two of the interventions tested in the studies may be useful for increasing the number of ADR reports submitted by healthcare professionals: an education session together with reminder card and adverse drug reaction (ADR) report form may substantially increase the number of ADR reports, and making it easier to report ADRs by using a modified standard discharge form may slightly increase ADR reports. The local applicability of these interventions and how the interventions might be implemented in different settings needs to be assessed. All other interventions identified in the eligible studies, such as informational letters or emails, government regulations and financial incentives, and multifacted interventions warrant further rigorous evaluation in real‐world practice settings as the certainty of the evidence for these interventions is very low.
Implications for research.
Although randomised controlled trials provide the most robust evidence of effectiveness, it may be more feasible and pragmatic to assess the effectiveness of interventions to improve adverse drug event (ADE) reporting using carefully controlled observational study designs. For example, the effectiveness of a clinical pharmacist actively identifying and encouraging the identification and reporting of ADEs in a hospital setting (the intervention) could be assessed using an interrupted time series study design. Two or more prespecified geographic regions, where data from routine systems is readily available and where interventions can be implemented across a very wide jurisdiction, would be identified. The intervention could be implemented in some hospitals in the regions and not in others (these would serve as the control group). Routine data from all the hospitals would be collected at three or more time points prior to the implementation of the intervention and at three or more time points after the intervention. The type of intervention being assessed would determine the study setting and number of sites to be included.
Most of the studies in the current body of evidence are focused on physicians. While future studies should continue to focus on testing the effectiveness of the various interventions for physicians, studies should also focus on other healthcare professionals, such as nurses and general practitioners.
Studies might also consider investigating the effectiveness of these interventions in both inpatient and outpatient settings.
Studies should assess any intervention in terms of both the benefit (increase in the number of ADE reports) and potential harm (increase in the number of false ADE reports).
Countries around the world have different pharmacovigilance infrastructures and capacities, different disease burdens and distribution, different medical care cultures, different medical education programmes, and vastly different economic status, so interventions to increase the reporting rate of ADEs need to be developed and tested in more countries, particularly low‐ and middle‐income countries.
This review focused on interventions to enhance ADE reporting in healthcare providers only. However, other relevant populations (e.g. patients and students) are also significant contributors to this information, and reviews of interventions targeted at enhancing their reporting rates are also valuable.
Healthcare professional students (e.g. medical, nursing and pharmacy students) are a crucial target population for interventions that enhance ADE reporting. Identifying effective educational interventions is an important area for medical education research to help inform future curriculum development.
We excluded many studies, with relevant interventions targeted at relevant participants (healthcare professionals), from this review due to having an ineligible study design. Depending on the availability and integrity of the data, it may be possible to re‐analyse the data from uncontrolled before‐after studies and retrospective observational studies using interrupted time series or repeated‐measures analyses.
History
Protocol first published: Issue 3, 2017
Acknowledgements
The methods section of this review is based on a standard template used by Cochrane Effective Practice and Organisation of Care (EPOC) (EPOC 2013a). This article is a part of the first author’s PhD dissertation sponsored by Tehran University of Medical Sciences’ Deputy of Research under project number 18360‐102‐02‐91.
Cochrane Effective Practice and Organisation of Care (EPOC) supported the authors in the development of this review. The authors thank Julia Worswick who provided comments to improve the review, and Paul Miller for developing the search strategy and conducting the electronic database searches. LG and CR were members of Cochrane EPOC but were not involved in the editorial process or decision‐making for this review. National Institute for Health Research funded Cochrane EPOC via Cochrane Infrastructure Funding prior to its closure in March 2023. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health and Social Care.
The following people conducted the editorial process for this review:
Editorial and peer‐reviewer contributions
Sign‐off Editor (final editorial decision): Yoon Loke, University of East Anglia;
Managing Editor (provided editorial guidance to authors, edited the article): Joey Kwong, Cochrane Central Editorial Service;
Editorial Assistant (conducted editorial policy checks, selected peer reviewers, collated peer‐reviewer comments and supported editorial team): Sara Hales‐Brittain, Cochrane Central Editorial Service;
Copy Editor (copy editing and production): Laura MacDonald. Cochrane Central Production Service;
Peer reviewers (provided comments and recommended an editorial decision): MO Reumerman, Amsterdam UMC, location VUmc, RECIPE AMSTERDAM (clinical/content review); Cheryl Sumner (consumer review); Jennifer Hilgart, Cochrane Evidence Production & Methods Directorate (methods review); Steve McDonald, Cochrane Australia and Jo Platt, Cochrane Evidence Production & Methods Directorate (search reviews). One additional peer reviewer provided clinical/content peer review but chose not to be publicly acknowledged.
Appendices
Appendix 1. Search strategies
Science Citation Index
Science Citation Index Expanded (SCI‐EXPANDED), 1945 to present
Conference Proceedings Citation Index ‐ Science (CPCI‐S), 1990 to 14 October 2022
| No. | Search terms |
| # 01 | TS=(adverse NEAR/2 drug* NEAR/2 report*) |
| # 02 | TS=((pharmacovigilan* near/2 surveillance) OR (pharmacovigilan* near/2 monitor*) OR (pharmacovigilan* near/2 report*)) |
| # 03 | #2 OR #1 |
| # 04 | TS=(physician* OR clinician* OR nurs* OR provider* OR family practice* OR general practice* OR clinical practice* OR doctor* OR caregiver* OR therap* OR physiotherapy* OR pharmac* OR professional* OR personnel OR practitioner* OR staff) |
| # 05 | #4 AND #3 |
| # 06 | TS=((increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*) near/5 report*) |
| # 07 | #6 AND #5 |
| # 08 | TS=(randomis* OR randomiz* OR randomly OR groups) |
| # 09 | TS=(trial OR multicenter OR "multi center" OR multicentre OR "multi centre") |
| # 10 | TS=(intervention* OR effect* OR impact* OR controlled OR "control group*" OR (before near/5 after) OR (pre near/5 post) OR ((pretest OR "pre test") AND (posttest OR "post test")) OR quasiexperiment* OR "quasi experiment*" OR "pseudo experiment*" OR pseudoexperiment* OR evaluat* OR time series OR "time point*" OR "repeated measur*") |
| # 11 | #10 OR #9 OR #8 |
| # 12 | #11 AND #7 |
CINAHL (EbscoHost)
1980 to 14 October 2022
| No. | Search terms |
| S1 | (MH "Pharmacovigilance") |
| S2 | ((pharmacovigilan* or medication error? or ADE? or adverse drug reaction?) N2 (surveillance or monitor* or report*)) |
| S3 | yellow card? |
| S4 | (MH "Adverse Drug Event+") |
| S5 | (MH "Voluntary Reporting") |
| S6 | S4 AND S5 |
| S7 | S1 OR S2 OR S3 OR S6 |
| S8 | (increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*) |
| S9 | S7 AND S8 |
| S10 | ((increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*) N5 ((unexpected or undesirable or harm* or serious or toxic* or adverse) N2 (drug* or medicine? or medication? or pharmaceutical?) N2 (effect? or reaction? or event? or outcome?))) |
| S11 | (surveillance or monitor* or report*) |
| S12 | S10 AND S11 |
| S13 | ((increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*) N5 ((unexpected or undesirable or harm* or serious or toxic* or adverse) N2 (effect? or reaction? or event? or outcome?)) N2 (surveillance or monitor* or report*)) |
| S14 | (drug* or medicine? or medication? or pharmaceutical?) |
| S15 | S13 AND S14 |
| S16 | TI ((unexpected or undesirable or harm* or serious or toxic* or adverse) and (drug* or medicine? or medication? or pharmaceutical?) and (effect? or reaction? or event? or outcome?) and (surveillance or monitor* or report*)) |
| S17 | S9 OR S12 OR S15 OR S16 |
| S18 | PT randomised controlled trial |
| S19 | PT clinical trial |
| S20 | PT research |
| S21 | (MH "Randomized Controlled Trials") |
| S22 | (MH "Clinical Trials") |
| S23 | (MH "Intervention Trials") |
| S24 | (MH "Nonrandomized Trials") |
| S25 | (MH "Experimental Studies") |
| S26 | (MH "Pretest‐Posttest Design+") |
| S27 | (MH "Quasi‐Experimental Studies+") |
| S28 | (MH "Multicenter Studies") |
| S29 | (MH "Health Services Research") |
| S30 | TI ( randomis* or randomiz* or randomly) OR AB ( randomis* or randomiz* or randomly) |
| S31 | TI (trial or effect* or impact* or intervention* or before N5 after or pre N5 post or ((pretest or "pre test") and (posttest or "post test")) or quasiexperiment* or quasi W0 experiment* or pseudo experiment* or pseudoexperiment* or evaluat* or "time series" or time W0 point* or repeated W0 measur*) OR AB (trial or effect* or impact* or intervention* or before N5 after or pre N5 post or ((pretest or "pre test") and (posttest or "post test")) or quasiexperiment* or quasi W0 experiment* or pseudo experiment* or pseudoexperiment* or evaluat* or "time series" or time W0 point* or repeated W0 measur*) |
| S32 | S18 OR S19 OR S20 OR S21 OR S22 OR S23 OR S24 OR S25 OR S26 OR S27 OR S28 OR S29 OR S30 OR S31 |
| S33 | S17 AND S32 |
| S34 | S17 AND S32 Limiters ‐ Exclude MEDLINE records |
Cochrane Library (Wiley)
Databases: CDSR, DARE, CENTRAL, HTA, NHS EED
Issue/year searched: 5/2022 (CDSR), 2/2015 (DARE), 4/2022 (CENTRAL), 10/2022 (Cochrane Library)
| No. | Search terms |
| #1 | ((pharmacovigilan* or medication error? or ADE? or adverse drug reaction?) near/2 (surveillance or monitor* or report*)):ti,ab |
| #2 | yellow card?:ti,ab |
| #3 | [mh "drug‐related side effects and adverse reactions"] |
| #4 | [mh "product surveillance, postmarketing"] |
| #5 | #3 and #4 |
| #6 | {or #1‐#2, #5} |
| #7 | (increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*):ti,ab |
| #8 | #6 and #7 |
| #9 | ((increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*) near/5 ((unexpected or undesirable or harm* or serious or toxic* or adverse) near/2 (drug* or medicine? or medication? or pharmaceutical?) near/2 (effect? or reaction? or event? or outcome?))):ti,ab |
| #10 | (surveillance or monitor* or report*):ti,ab |
| #11 | #9 and #10 |
| #12 | ((increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*) near/5 ((unexpected or undesirable or harm* or serious or toxic* or adverse) near/2 (effect? or reaction? or event? or outcome?)) near/2 (surveillance or monitor* or report*)):ti,ab |
| #13 | (drug* or medicine? or medication? or pharmaceutical?):ti,ab |
| #14 | #12 and #13 |
| #15 | ((unexpected or undesirable or harm* or serious or toxic* or adverse) and (drug* or medicine? or medication? or pharmaceutical?) and (effect? or reaction? or event? or outcome?) and (surveillance or monitor* or report*)):ti |
| #16 | [mh "Adverse Drug Reaction Reporting Systems"] |
| #17 | {or #8, #11, #14‐#16} |
Embase (OvidSP)
1974 to 2022 October 14
| No. | Search terms |
| 1 | ((pharmacovigilan* or medication error? or ADE? or adverse drug reaction?) adj2 (surveillance or monitor* or report*)).ti,ab. |
| 2 | yellow card?.ti,ab. |
| 3 | *adverse drug reaction/ |
| 4 | exp *postmarketing surveillance/ |
| 5 | *drug surveillance program/ |
| 6 | 3 and (4 or 5) |
| 7 | (increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*).ti,ab. |
| 8 | or/1‐2,6 |
| 9 | 7 and 8 |
| 10 | ((increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*) adj5 ((unexpected or undesirable or harm* or serious or toxic* or adverse) adj2 (drug* or medicine? or medication? or pharmaceutical?) adj2 (effect? or reaction? or event? or outcome?))).ti,ab. |
| 11 | (surveillance or monitor* or report*).ti,ab. |
| 12 | 10 and 11 |
| 13 | ((increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz* or manag* or enhanc*) adj5 ((unexpected or undesirable or harm* or serious or toxic* or adverse) adj2 (effect? or reaction? or event? or outcome?)) adj2 (surveillance or monitor* or report*)).ti,ab. |
| 14 | (drug* or medicine? or medication? or pharmaceutical?).ti,ab. |
| 15 | 13 and 14 |
| 16 | ((unexpected or undesirable or harm* or serious or toxic* or adverse) and (drug* or medicine? or medication? or pharmaceutical?) and (effect? or reaction? or event? or outcome?) and (surveillance or monitor* or report*)).ti. |
| 17 | or/9,12,15‐16 |
| 18 | (physician* or clinician* or nurs* or provider* or family practice? or general practice? or clinical practice? or doctor? or caregiver? or GP? or therap* or physiotherapy* or pharmac* or (health* adj2 (professional? or personnel)) or practitioner? or staff).ti,ab. |
| 19 | exp health care personnel/ |
| 20 | or/18‐19 |
| 21 | 17 and 20 |
| 22 | randomised controlled trial/ |
| 23 | controlled clinical trial/ |
| 24 | quasi experimental study/ |
| 25 | pretest posttest control group design/ |
| 26 | time series analysis/ |
| 27 | experimental design/ |
| 28 | multicenter study/ |
| 29 | (randomis* or randomiz* or randomly).ti,ab. |
| 30 | groups.ab. |
| 31 | (trial or multicentre or multicenter or multi centre or multi center).ti. |
| 32 | (intervention? or effect? or impact? or controlled or control group? or (before adj5 after) or (pre adj5 post) or ((pretest or pre test) and (posttest or post test)) or quasiexperiment* or quasi experiment* or pseudo experiment* or pseudoexperiment* or evaluat* or time series or time point? or repeated measur*).ti,ab. |
| 33 | or/22‐32 |
| 34 | (systematic review or literature review).ti. |
| 35 | "cochrane database of systematic reviews".jn. |
| 36 | exp animals/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ |
| 37 | human/ or normal human/ or human cell/ |
| 38 | 36 not (36 and 37) |
| 39 | 34 or 35 or 38 |
| 40 | 33 not 39 |
| 41 | 21 and 40 |
ClinicalTrials.gov
| No. | Search terms |
| adverse drug reaction AND report [intervention field] + Interventional Studies | |
| pharmacovigilance [intervention field] + Interventional Studies | |
| adverse drug AND surveillance [intervention field] + Interventional Studies |
WHO ICTRP
| No. | Search terms |
| adverse* AND drug* AND report* | |
| pharmacovigilan* | |
| adverse* AND drug* AND surveillance* |
MEDLINE (OvidSP)
Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE Daily and Ovid MEDLINE
1946 to present
| No. | Search terms |
| 1 | "Adverse Drug Reaction Reporting Systems"/ |
| 2 | ((pharmacovigilan* or medication error? or ADE? or adverse drug reaction?) adj2 (surveillance or monitor* or report*)).ti,ab. |
| 3 | yellow card?.ti,ab. |
| 4 | ((unexpected or undesirable or harm* or serious or toxic* or adverse) adj2 drug* adj2 (effect? or reaction? or event? or outcome?) adj3 (surveillance or monitor* or report*)).ti,ab. |
| 5 | "drug‐related side effects and adverse reactions"/ |
| 6 | product surveillance, postmarketing/ |
| 7 | 5 and 6 |
| 8 | or/1‐4,7 |
| 9 | (increas* or improv* or motivat* or decreas* or chang* or volunteer* or voluntary or spontaneous or encourag* or promot* or alter or alters or altered or influenc* or willing* or stimulat* or prompt* or program* or system* or organis* or organiz? or manag* or enhanc*).ti,ab. |
| 10 | 8 and 9 |
| 11 | ((unexpected or undesirable or harm* or serious or toxic* or adverse) and (drug* or medicine? or medication? or pharmaceutical?) and (effect? or reaction? or event? or outcome?) and (surveillance or monitor* or report*)).ti. |
| 12 | *"Adverse Drug Reaction Reporting Systems"/ |
| 13 | or/10‐12 |
| 14 | randomised controlled trial.pt. |
| 15 | controlled clinical trial.pt. |
| 16 | multicenter study.pt. |
| 17 | pragmatic clinical trial.pt. |
| 18 | (randomis* or randomiz* or randomly).ti,ab. |
| 19 | groups.ab. |
| 20 | (trial or multicenter or multi center or multicentre or multi centre).ti. |
| 21 | (intervention? or effect? or impact? or controlled or control group? or (before adj5 after) or (pre adj5 post) or ((pretest or pre test) and (posttest or post test)) or quasiexperiment* or quasi experiment* or pseudo experiment* or pseudoexperiment* or evaluat* or time series or time point? or repeated measur*).ti,ab. |
| 22 | non‐randomized controlled trials as topic/ |
| 23 | interrupted time series analysis/ |
| 24 | controlled before‐after studies/ |
| 25 | or/14‐24 |
| 26 | exp animals/ |
| 27 | humans/ |
| 28 | 26 not (26 and 27) |
| 29 | review.pt. |
| 30 | meta analysis.pt. |
| 31 | news.pt. |
| 32 | comment.pt. |
| 33 | editorial.pt. |
| 34 | cochrane database of systematic reviews.jn. |
| 35 | comment on.cm. |
| 36 | (systematic review or literature review).ti. |
| 37 | or/28‐36 |
| 38 | 25 not 37 |
| 39 | 13 and 38 |
ProQuest Dissertations & Theses Global
| No. | Search terms |
| (TI, AB(adverse AND drug AND report) OR TI,AB(pharmacovigilan*)) AND (SU(health*) OR TI(effect OR effects OR impact OR influenc* OR random* OR study OR controlled OR trial OR effectiveness) OR ALL(random* OR intervention OR collaborat* OR team* OR multidisciplin* OR multi‐disciplin* OR crossdisciplin* OR cross‐disciplin* OR interdisciplin* OR community OR quasi*) OR ALL(before NEAR/10 after) OR ALL(before NEAR/10 during) OR ALL("time series" OR timeseries) OR ALL((control* NEAR/2 group) OR (control NEAR/2 study) OR (control NEAR/2 cohort))) |
Other sources
Based on recommendations from EPOC information specialist, we used key terms only (in titles/abstracts) of "adverse drug event", "adverse drug reaction", "medication error" for VHL/IRIS/WHOLIS and reviewed the following number of extracted items, retrieved on 17 October 2022.
Virtual Health Library (VHL) (http://pesquisa.bvsalud.org/portal/advanced/?lang=en): 197 items
World Health Organization IRIS (http://apps.who.int/iris/simple‐search?query=): 285 items
WHO Library Information System (WHOLIS) (https://kohahq.searo.who.int/): 43 items
We also searched the below sources on 20 August 2018.
OpenGrey: 42 items
Grey Literature Report (New York Academy of Medicine): 43 items
Agency for Healthcare Research and Quality (AHRQ): 97 items
National Institute for Health and Clinical Excellence (NICE): 82 items
Data and analyses
Comparison 1. Comparison 2: Informational letter or email versus usual practice.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Number of unexpected (previously unknown) adverse drug reaction reports | 2 | 268 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.92, 2.30] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ali 2018.
| Study characteristics | ||
| Methods |
Study design: ITS Study setting: Riyadh, Saudi Arabia (large hospital, 1400 ward beds; estimated annual outpatient visit of about 300,000) Duration of follow‐up: December 2015 to November 2016; total of 24 observations |
|
| Participants |
Participants: clinical pharmacists Number of participants: not reported Mean age: not reported Sex: not reported |
|
| Interventions |
Comparison 3: multifaceted intervention (financial and non‐financial incentives) vs usual practice Intervention: in January 2016, pharmacy leadership of the hospital introduced incentives to encourage ADR reporting amongst the hospital’s pharmacy staff. A member of staff with the highest number of ADR reports, who has also excelled in other aspects of their job including ME reporting and participation in ongoing research projects, was nominated every month for the employee of the month award. Letters of appreciation were given to the awardees; their names were posted on the hospital’s notice board, and they were entitled to a full day’s leave. Becoming the employee of the month also increased their chances of nomination for the annual performance excellence award (PEA). PEA awardees were eligible for an extra month’s salary and a certificate. December 2015 (first time point); January 2016 (intervention implemented); November 2016 (last time point); a total of 24 observations Comparison: usual practice |
|
| Outcomes |
|
|
| Notes | Funding: this research received no specific grant from any funding agency in the public, commercial or not for‐profit sectors. Conflict of interest: the authors declare that there is no conflict of interest. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study authors do not provide information on the number of clinical pharmacists working in this hospital before or after the implementation of the intervention, so it is not possible to assess completeness of follow‐up. |
| Selective reporting (reporting bias) | Low risk | No access to the study protocol; however, all relevant outcomes in the methods section are reported in the results section. |
| Other bias | High risk | Seasonality was not adjusted for. Study authors say: "A limitation of this study is the few timepoints before and after intervention, hence we were unable to evaluate the reports for seasonal variations." |
| Intervention independent of other changes | High risk | There are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was not influenced by other confounding variables or historic events during study period. |
| Shape of the intervention effect pre‐specified | Low risk | Point of analysis is the point of intervention (i.e. the point in time at which the intervention was implemented). "The change in the level of average monthly adverse drug reaction reporting between 2015 and 2016 was determined using segmented regression analysis of an interrupted time series, with 24 observation points." |
| Intervention unlikely to affect data collection | Low risk | The intervention itself was unlikely to affect data collection: sources and methods of data collection were the same before and after the intervention. |
| Knowledge of the allocated interventions adequately prevented during the study | Low risk | The primary outcome (number of adverse drug reaction reports noted in the pharmacovigilance database) is objective. |
Castel 2003.
| Study characteristics | ||
| Methods |
Study design: ITS Study setting: Catalan Centre of Pharmacovigilance; Barcelona, Spain Duration of follow‐up: 1983 to 1995 |
|
| Participants |
Participants: physicians Number of participants: approximately 30,000 physicians Mean age: not reported Sex: not reported |
|
| Interventions |
Comparison 5: improving access to reporting form vs usual practice Intervention 1982 ‐ Catalan Centre of Pharmacovigilance began its activities at the end of 1982. 1985 to 1991: quarterly adverse drug reaction bulletin (Adverse Drug Reaction (Butlletí Groc) ‐ ADRB) with ADR yellow card report form enclosed, mailed to all physicians (approximately 30,000) in catchment area. 1991 to 1994: quarterly adverse drug reaction bulletin with ADR yellow card report form, plus ADR yellow card report form included in the prescription pads of the main provider organisation for the Catalan Health Service (Institut Catala de la Salut, ICS), with approximately 6500 prescribers. Comparison: usual practice |
|
| Outcomes | Primary outcome: number of spontaneous ADR reports received at the Catalan Center per month | |
| Notes |
Funding: Servei Català de la spontan‐Salut. Departament de Sanitat i Seguretat Social. Generalitat eous de Catalunya Conflict of interest: the authors have no conflicts of interest relevant to the contents of this study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | We know that approximately 30,000 physicians and 6500 prescribers were potentially exposed to the interventions. It is unclear how these numbers changed over the time period of the study, so we cannot be sure how this might affect the primary outcome: number of spontaneously reported ADRs per month. |
| Selective reporting (reporting bias) | Low risk | Although we do not have a copy of the study protocol, all relevant outcomes in the methods section are reported in the results section. |
| Other bias | Low risk | Analyses appropriate; seasonality considered |
| Intervention independent of other changes | High risk | The duration of the study was long (from 1983 to 1995), so the number of adverse drug reaction reports might be influenced by factors other than the intervention. There are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was not influenced by other confounding variables or historic events during study period. |
| Shape of the intervention effect pre‐specified | Low risk | Point of analysis is the point of intervention (i.e. the point in time at which the intervention was implemented). |
| Intervention unlikely to affect data collection | Low risk | The intervention itself was unlikely to affect data collection: sources and methods of data collection were the same before and after the intervention. |
| Knowledge of the allocated interventions adequately prevented during the study | Low risk | The primary outcome (number of adverse drug reaction reports per month received by the centre) is objective. |
Chang 2017.
| Study characteristics | ||
| Methods |
Study design: ITS Study setting: First Affiliated Hospital of Zhengzhou University (Henan Province), a Chinese tertiary care university hospital Duration of follow‐up: 2006 to 2014 |
|
| Participants |
Participants: physicians Number of participants: unclear, although it states that, "The general ward of the hospital has approximately 5000 beds, with more than 6624 staff physicians and pharmacists." Mean age: not reported Sex: not reported |
|
| Interventions |
Comparison 3: multifaceted intervention vs usual practice Comparison 4: financial incentives plus government regulations vs usual practice Intervention: financial arrangements (incentives, fines), government regulations 2006: multifaceted intervention aimed at improving pharmacovigilance in hospital initiated; included implementation of the ADR database. 2009: financial incentives for spontaneous ADR reporting initiated (economic incentives for ADR reporting were integrated in clinical activity at 2 levels: medical staff and ward; included a bonus of 20 RMB for a spontaneous ADR report and a fine of 50 RMB (2.5 times the bonus) for a withheld report (missed ADRs found by routine retrospective review of the medical charts by dedicated pharmacists; if ADR in the chart or if the patient was rehospitalised because of an ADR and that the ADR had not been reported by the physician, then the fine was charged). The financial incentive was fixed and represented less than 1% of the physician's salary. 2012: government‐enacted strict regulations on the clinical use of antimicrobial agents, including detailed ADR classification and mandatory monitoring and reporting of ADRs associated with antimicrobial agents. Comparison: usual practice |
|
| Outcomes | Primary outcome: spontaneous reports of ADRs (per month; general, serious or new) | |
| Notes | Funding: authors report that there was no funding for this study. Study on the optimal model of health management for elderly chronic diseases based on the theory of social network (grant no. 71673298) Conflict of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The monthly number of spontaneous ADR reports received is the primary outcome. We know that the general ward of the hospital was served by 6624 physicians and pharmacists, but it is not clearly described how this number changed over time. |
| Selective reporting (reporting bias) | Low risk | Although we do not have access to the study protocol, all relevant outcomes in the methods section are reported in the results section. |
| Other bias | Low risk | Analyses appropriate and seasonality considered |
| Intervention independent of other changes | High risk | There are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was not influenced by other confounding variables or historic events during study period. |
| Shape of the intervention effect pre‐specified | Low risk | Point of analysis is the point of intervention (i.e. the point in time at which the intervention was implemented). |
| Intervention unlikely to affect data collection | Low risk | The intervention itself was unlikely to affect data collection: sources and methods of data collection were the same before and after the intervention. |
| Knowledge of the allocated interventions adequately prevented during the study | Low risk | The primary outcome (number of ADR reports noted in the pharmacovigilance database) is objective. |
Figueiras 2006.
| Study characteristics | ||
| Methods |
Study design: cluster‐RCT (4 intervention clusters; 11 control clusters; each cluster consisted of 1 reference hospital plus the outpatient centre and any other hospital in its catchment area). See comments on Herdeiro 2012 Study setting: Northern Region of Portugal; 104 outpatient centres, 25 hospitals (15 general medical hospitals; 5 small satellite hospitals; 5 speciality hospitals (e.g. cancer, maternity, paediatric) Duration of intervention: March to July 2004 Duration of follow‐up: 13 to 16 months |
|
| Participants | All National Health System physicians in the north of Portugal Number of participants: 6451 (4 intervention clusters: N = 1388; 11 control clusters: N = 5063) Mean age: 45.1 years (control); 43.5 years (intervention) Sex (% females): 52 (control); 51 (intervention) |
|
| Interventions |
Comparison 1: education session plus reminder card and report form vs usual practice Intervention
A reminder card similar to the report form and containing the principal messages of the presentation was distributed to approximately 90% of physicians attending the sessions. Intervention delivered March 2004 to July 2004 Intervention fidelity: only 655/1388 intervention physicians attended the education sessions. Comparison: usual practice Authors do not specify what the control group received or what usual practice entailed. |
|
| Outcomes |
|
|
| Notes |
Study funding: PRODEP, the Portuguese Educational Development Program; Dr Figueiras was in part funded by Health Research Fund (Fondo de Investigación Sanitaria ‐ FIS) grant PI021512 from the Spanish Ministry of Health. Financial disclosures: none reported Role of the sponsor: sponsors had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; or preparation, review or approval of the manuscript. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated procedure was used to generate the randomisation sequence. |
| Allocation concealment (selection bias) | Low risk | A computer‐generated procedure was used to generate the randomisation sequence and assign the clusters to intervention and control groups. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Due to the type of intervention, participants could not be blinded. Study personnel delivering the intervention would also not have been blinded to the group allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The Pharmacosurveillance Unit expert responsible for codifying adverse reactions was blinded to the physician study group assignment. Confidentiality was maintained, with data only being available for study purposes under a code number assigned to each physician that precluded any further identification. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ADR reporting is a passive process; every report that is generated is received by the Northern Pharmacosurveillance Unit then given to the researchers. Because of this, there was 100% assessment of ADR outcomes in the study population, and effectively no loss to follow‐up. Potential source of error: if physicians in the study left clinical practice or died, it could distort the per‐physician rates but would not affect the accuracy of the number of ADR reports. |
| Selective reporting (reporting bias) | Low risk | No access to study protocol; however, the published report includes all expected outcomes as well as those those specified in the methods section. |
| Other bias | High risk | Statistically significant differences between groups at baseline with respect to age, specialities and work setting; baseline rates of reporting for all outcomes were lower in the intervention group, but not statistically significantly so, therefore we judged it to be high risk of other bias. Additional risk of bias domains pertaining to cluster‐randomised trials listed below.
|
Hanesse 1994.
| Study characteristics | ||
| Methods |
Study design: cluster‐RCT (with cross‐over after 8 weeks, plus 2‐week washout period) 10 participating departments were pair‐matched according to the medical or surgical nature of their activities resulting in five strata: internal medicine, otorhinolaryngology, ophthalmology, rheumatology, cardiology. 1st 8 weeks ‐ reporting methods randomly assigned to one of the departments within each stratum 2‐week washout 2nd 8 weeks ‐ attribution of reporting methods reversed within each stratum Study setting: Centre Hospitalier et Universitaire de Nancy, France; 50 departments receiving about 100,000 patients a year; six medical departments and four surgical departments participated in the trial, including two of each of the following specialities: internal medicine, rheumatology, cardiology, otorhinolaryngology and ophthalmology Duration of follow‐up: start date ‐ early 1993; plus 18‐week study duration |
|
| Participants | Intervention was aimed at physicians working in 1 of 10 participating departments; participating physicians were aware of the aims of the study. Outcomes were measured in all patients discharged from a participating department during one of the two 8‐week periods of the study (N = 5967 participants; 1st 8 weeks N = 3106 spontaneous reporting, N = 2861 standardised form); 44% of 5967 participants were female. *Study authors did not report any details regarding the physicians. |
|
| Interventions |
Comparison 5: improving access to ADR report form vs spontaneous ADR reporting Intervention:standardised discharge form used by physicians for recording patient diagnoses by ICD‐9th classification (required to determine Diagnosis Related Group (DRG) system (DRG method)), and medical or surgical acts received during hospital stay with additional items addressing time of ADR occurrence (i.e. whether it was the cause of admission or occurred during hospital stay) and its evolution; on discharge from hospital, physician completes standardised outcome summary form for each patient; each completed questionnaire is transmitted to the centre; then medical data about ADRs are extracted from patients’ medical files; during this time no weekly meeting between medical student and physician to summarise and file ADRs and no spontaneous reports to ‘Centre Regional de Pharmacovigilance de Lorraine’. Comparison: spontaneous reporting method. Medical students meet weekly with hospital physicians in their department. They record ADR physicians’ reports and refer them to the ‘Centre Regional de Pharmacovigilance de Lorraine’. Usual practice for the past 16 years. |
|
| Outcomes |
|
|
| Notes | Study funding: no information provided Conflict of interest: no information provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The authors state: "For the first eight‐week period, reporting methods were randomly assigned to one of the departments within each stratum. After a two‐week ‘wash‐out’ period, the attribution of methods was reversed within each stratum in a second eight‐week period", but do not describe how the randomisation sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | Study authors do not decribe who allocated the departments to the experimental groups and whether or not this person knew to which group the departments were being allocated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The authors state participating physicians were aware of the study aims, which suggests that they were not blinded. Authors do not describe blinding study personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Data on ADRs were extracted from patient medical files ‐ it is not clear who extracted this data or if this person was blinded to the group allocations. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ADRs were collected either spontaneously on a weekly basis or on discharge of patients. Patient attrition is not an issue in this instance. |
| Selective reporting (reporting bias) | Low risk | We do not have access to the study protocol. The methods do not clearly outline which outcomes will be assessed and reported on; however, all expected outcomes appear to be reported on. |
| Other bias | Low risk | 10 participating departments were pair‐matched according to the medical or surgical nature of their activities resulting in five strata: (1) internal medicine, (2) otorhinolaryngology, (3) ophthalmology, (4) rheumatology, (5) cardiology. Departments within each strata = clusters; reduced risk of contamination Data on prior reporting of ADRs per department not provided Although patients in the standardised form group appear to be older and might therefore present with more ADRs, it seems that the difference was not statistically significant. Recruitment bias: low risk; as clusters assigned and intervention implemented; correct analyses: outcomes adequately adjusted for clustering; no reported loss of clusters |
Herdeiro 2008.
| Study characteristics | ||
| Methods |
Study design: cluster‐RCT (4 intervention clusters; 11 control clusters; each spatial‐cluster consisted of one reference hospital plus the community pharmacist and any other hospital in catchment area). See comments on Ribeiro‐Vaz 2011 Study setting: Portugal’s Northern Region Health Authority (104 outpatient centres, 25 hospitals (5 speciality hospitals) and 761 community pharmacies) Duration of intervention: March 2004 to June 2004 Duration of follow‐up: July 2004 to June 2005 |
|
| Participants | All pharmacists employed in hospital and community pharmacies across Portugal’s Northern Regional Health Authority Number of participants: 1433 (control N = 1091; intervention: N = 342) Mean age: control: 37.5 years, intervention: 38.2 years Sex (% female): 79.7 (control); 79.5 (intervention) |
|
| Interventions |
Comparison 1: education session plus reminder card and report form versus usual practice Intervention: 60‐minute education session Active (group‐session slide presentation) and passive approaches (distribution of informational leaflets):
Intervention fidelity: 276/342 attended the intervention Comparison: both groups received briefing and standard training provided by Portugal's Northern Pharmacovigilance Unit. |
|
| Outcomes |
|
|
| Notes | Study funding: Program for the Educational Development in Portugal; Professor Dr Adolfo Figueiras’ work on this project was in part funded by Health Research Fund (Fondo de Investigacio´n Sanitaria) grants 99/1189 from the Spanish Ministry of Health, and by CIBER of Epidemiology and Public Health. Conflict of interest: the authors declare that they have no conflicts of interest that are directly relevant to the content of this study. Trial registration ID: ISRCTN45894687 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated procedure was used to generate the randomisation sequence. |
| Allocation concealment (selection bias) | Low risk | A computer‐generated procedure was used to generate the randomisation sequence and assign four clusters to the intervention group and 11 to the control group. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Due to the type of intervention, participants could not be blinded. Study personnel delivering the intervention would also not have been blinded to the group allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The Pharmacosurveillance Unit expert (Jorge Polonia) responsible for codifying adverse reactions was blinded as regards the study group to which the reporting pharmacist belonged. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All these data came from the Northern Pharmacosurveillance Unit part of the Portuguese Health Authority. |
| Selective reporting (reporting bias) | Low risk | The study protocol is available (ISRCTN45894687) and all of the study’s prespecified (primary and secondary) outcomes including rate of total notifications per month, rate of serious, unexpected, high‐causality, and new‐drug‐related ADRs per month) that are of interest in the review have been reported in the prespecified way. |
| Other bias | Low risk | Low risk of recruitment bias, as clusters assigned and intervention implemented; outcomes adequately adjusted for clustering; no reported loss of clusters; allocation was by cluster, which reduced the risk of contamination between experimental groups; baseline characteristics of pharmacists and ADR reporting rates were similar between the intervention and control groups. |
Herdeiro 2012.
| Study characteristics | ||
| Methods |
Study design: cluster‐RCT Each cluster ‐ one reference hospital, small hospitals in the selected geographic area, plus the outpatient centre. This study builds on previous studies: Figueiras 2006 targeted physicians in the 15 clusters randomised to receive 1‐hour education session, tailored to training needs or no intervention; Herdeiro 2008 targeted pharmacists in the 15 clusters randomised to receive 1‐hour education sessions, tailored to training needs or no intervention. A second randomisation of the four intervention clusters (from Figueiras 2006 and Herdeiro 2008) was performed, and two clusters received the workshop intervention and two clusters received the telephone‐interview intervention, with 11 clusters remaining assigned to the control arm. Herdeiro 2012 targeted the physicians and Ribeiro‐Vaz 2011 targeted the pharmacists in the newly randomised clusters. Study setting: National Health System in the northern region of Portugal (25 hospitals and their respective outpatient centres); 15 of the hospitals were general medical hospitals, which cover a designated geographic catchment area; five were small satellite hospitals of general hospitals; and five were speciality hospitals (e.g. cancer, maternity, paediatrics) Duration of follow‐up: 2008 to 2009 (20 months) |
|
| Participants |
Participants: clinically active National Health System physicians based in northern Portugal Number of participants: 438 physicians (workshop); 1034 physicians (telephone interview); 5107 physicians (control) Mean age: control: 48.55 years, intervention (workshop): 49.31 years, intervention (telephone): 46.29 years Sex: control: 52.1% female, intervention (workshop): 42.9% female, intervention (telephone): 54.6% female |
|
| Interventions |
Comparison 1: education session plus reminder card and report form versus usual practice Intervention: of the 4 intervention clusters (from 2004 study), 2 clusters received the workshop intervention and 2 clusters the telephone‐interview intervention Telephone interviews ‐ telephone conversation (pre‐established script; 3 to 8 minutes); physicians asked if (i) they had ever had any suspicion of ADRs; (ii) they had experienced any difficulty in reporting; (iii) they remembered the different methods that could be used for reporting purposes (telephone, fax, email or internet); (iv) they attached importance to the individual physician’s role in reporting; (v) they remembered any cases of an alert in which reporting had played a vital role; and finally (vi) they had any questions concerning the reporting system; following telephone interview participant sent (via post) support material (letter of acknowledgement, ADR spontaneous report form and one NPC presentation folder); 3 attempts made to contact physician, after which deemed impossible to contact. Workshops ‐ brief presentation; approximately 1 hour (definitions of pharmacovigilance, ADRs and their impact on public health, plus more in‐depth approach to spontaneous ADR reporting and physicians’ attitudes to and knowledge of the practice); clinical case presented, and each physician invited to discuss it, an ADR form completed with the case data and the support of the SPC; attendance certificate, NPC presentation folder, ADR report form sent to attending physicians. Intervention delivered in 2008 Intervention fidelity: 118/438 attended workshop (26.9%); 82/1034 telephone interview (7.9%) Comparison: NPC and Portuguese National Medicine and Health Product Authority‐run awareness campaigns |
|
| Outcomes |
Rate data calculated as follows: (no. of reports of a study group during a specific follow‐up period (months))/(no. of physicians belonging to the study group during that follow‐up period)*(no. of months in that follow‐up period)*1000*12 |
|
| Notes | Funding: Prof Figueiras ‐ Consortium for Biomedical Research in Epidemiology and Public Health (CIBER en Epidemiologıa y Salud Publica [CIBERESP]) [AC08_008]; Marıa Pineiro (CIBERESP); Professor Maria Teresa Herdeiro ‐ Science and Technology Fund (Fundação para a Ciencia e Tecnologia) grant SFRH/BPD/35746/2007 from the Portuguese Ministry of Science, Technology and Higher Education Conflict of interest: the authors declare that they have no conflicts of interest directly relevant to the contents of this study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Study authors do not describe how the random sequence was generated; however, they state that randomisation was performed. |
| Allocation concealment (selection bias) | Low risk | Study authors refer to Figueiras 2006, for methodological details. In Figueiras 2006, a computer‐generated procedure was used to generate the randomisation sequence and assign the clusters to intervention and control group. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Study authors do not discuss participant blinding; it was not possible to blind participants to the intervention due to the type of intervention. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All data was sourced from the Portuguese Health Authority (NPC). A code number was attributed to the physicians so as to prevent further identification and ensure confidentiality. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ADRs are either reported or not, so attrition bias is not relevant in this instance. There was no loss of clusters. |
| Selective reporting (reporting bias) | Low risk | The study protocol is available (ISRCTN45894687) and all of the study’s prespecified primary and secondary outcomes have been reported. |
| Other bias | High risk | Low risk of recruitment bias; clusters assigned then intervention implemented; outcome data appropriately adjusted for clustering; no reported loss of clusters; allocation by clusters reducing risk of contamination; baseline characteristics different between groups (workshop group higher % males and general medicine; telephone group higher % work in hospital), but unclear if any differences are statistically significant; baseline reporting rates different between groups (telephone group had higher baseline reporting rates for total, serious and high‐causality ADRs compared to the control group; workshop group had lower reporting rates for serious and high‐causality ADRs compared with the control group; both workshop and telephone groups had higher rate of unexpected ADRs compared to the control group), but unclear if any of these differences were statistically significant. |
Johansson 2009.
| Study characteristics | ||
| Methods |
Study design: RCT Setting: primary healthcare units in the Region Västra Götaland, Sweden. Duration of intervention: three emails January, May and September 2007 Duration of follow‐up: 1 year (2007) In addition, a small number of the units were participating in a contemporary educational intervention during the study period. Consequently, the allocation was stratified according to number of ADR reports in 2006 and whether or not the unit had received the educational intervention. |
|
| Participants |
Target audience:doctors employed in primary healthcare units Number of primary healthcare units: 117 (58 control; 59 intervention) Baseline ADR reporting rate (2006): "total of 89 reports from the primary health care units were registered, with the number of reports per unit ranging from zero to 12 (intervention group) and zero to five (control group)" |
|
| Interventions |
Comparison 2: informational email or letter vs usual practice Intervention: three emails with attachments sent in January, May and September 2007. Emails followed the established system for emails on drug news and included one page with (1) the heading “Every adverse drug reaction report is important", (2) a current case report of an ADR and (3) instructions on how to report. Each email included a new current case report of an ADR. Comparison: usual practice (no informative email) |
|
| Outcomes |
|
|
| Notes | Funding: Swedish Foundation for Strategic Research and The Swedish Society of Medicine Conflict of interest: "No conflict of interest exists" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study authors do not describe how the random sequence was generated. Study authors state that the primary healthcare units were randomised in a 1:1 ratio to intervention and control, and stratification of primary healthcare units is also discussed. |
| Allocation concealment (selection bias) | Low risk | "A person not involved in the study and without any knowledge of the study protocol performed the randomization procedure". Assume that "randomisation procedure" includes the allocation of primary healthcare units. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Due to the nature of the intervention, it would not have been possible to blind the participants or study personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study authors only say, "The number of reports from each primary care unit run by the same head was registered", and thus it is not clear if the outcome assessor was blinded to the allocation of the primary healthcare unit, but it is unlikely that this would affect the number of reports. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ADR reports were sought from all primary healthcare units randomised to intervention or control, so no loss to follow‐up. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but the published report includes all expected outcomes. |
| Other bias | High risk | Possible high risk of contamination bias: "The intervention may also have spilled over to the control units. Doctors may work in more than one primary health care unit, i.e. both in the intervention group and in the control group. Also, the units all belong to the same organization, and information may easily be passed on from one unit to another." Not enough information is provided to determine whether or not there were significant differences in primary healthcare unit characteristics or ADR reporting rate ("total of 89 reports from the primary health care units were registered, with the number of reports per unit ranging from zero to 12 (intervention group) and zero to five (control group) between groups at baseline"). |
Johansson 2011.
| Study characteristics | ||
| Methods |
Study design: RCT (151 PHCs randomly allocated) Setting: PHCs in the Region Västra Götaland, Sweden Duration of intervention: letters sent in January, May and September 2008 Duration of follow‐up: 1 year |
|
| Participants |
Target audience: physicians and nurses working in the PHCs Number of PHCs: 77 intervention units, 74 control units Total number of physicians and nurses: 845 physicians, 1423 nurses Median (IQR) physicians and nurses per PHC unit: intervention = 6 (4 to 7) physicians and 9 (7 to 13) nurses; control = 5 (4 to 7) physicians and 9 (6 to 13) nurses Baseline ADR reporting rate (2007): 62 reports from 32 (42%) intervention units, and 55 reports from 31 (42%) control units (mean number of reports per unit ± standard deviation: 0.8 ± 1.4 vs 0.74 ± 1.1, P = 0.93) |
|
| Interventions |
Comparison 2: informational email or letter vs usual practice Intervention: a one‐page ADR information letter was sent to the secretary of each PHC unit with instructions to distribute the ADR information letter to all physicians and nurses at the PHC unit. The ADR information letter contained (i) the heading “adverse drug reaction Information Letter”, (ii) a current case report of an ADR and (iii) instructions on what and how to report. The information letters were sent out 3 times in January, May and September 2008. *Intervention fidelity assessed through questionnaire: 57% response rate (1292/2268 questionnaires returned); 300 respondents reported having received at least one ADR information letter during 2008 (23%), and 362 (28%) had read at least one ADR information letter during the year. More people in the intervention group than in the control group had received (29% vs 19%, P < 0.001) and read (31% vs 26%, P < 0.001) an ADR information letter during 2008. In the intervention group, more physicians than nurses had received (36% vs 28%, P < 0.015) but not read (36% vs 37%, P = 0.89) the ADR information letter. Comparison: usual practice (no informative letter) |
|
| Outcomes |
PHC adverse drug reaction reports extracted from the SWEdish Drug Information System. |
|
| Notes | Research grants from the Swedish Foundation for Strategic Research. The authors’ work was independent of the funders. The authors declare that they have no competing interests. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | How the randomisation sequence was generated is not described. Study stated that the primary healthcare units were randomised 1:1 to intervention and control groups. Allocation was stratified according to number of ADR reports in 2007 and whether or not the unit had received the repeated drug safety e‐mails (see Johansson 2009). |
| Allocation concealment (selection bias) | Low risk | Statement in study: "a person not involved in the study and without knowledge about the study protocol performed the randomisation procedure. Assume that randomisation procedure includes the allocation procedure |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Due to the nature of the intervention, it would not be possible to blind participants and personnel. In the discussion, the study authors believe that the intervention spilled over to the control units as questionnaire responders in the control units reported having received and read the ADR information letter. Physicians and nurses may work in more than one primary healthcare unit, so they may have been exposed to both the intervention and control settings. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessors is not described. Primary healthcare units' ADR reports were extracted from the SWEdish Drug Information System (SWEDIS), so it is unlikely that lack of blinding would have affected the outcome. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ADR reports from the included primary healthcare units were extracted from the SWEdish Drug information System (SWEDIS), the Swedish ADR database where all ADR reports are registered, after being assessed for, e.g. causality and seriousness, according to the definitions by the WHO. There is no reported loss of PHC units. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available but the published report includes all expected outcomes. |
| Other bias | Low risk | Possible high risk of contamination: Based on the questionnaire 19% of respondents in the control group had received and 26% had read an adverse drug reaction information letter during 2008. There does not appear to be any significant differences in PHC unit characteristics or adverse drug reaction reporting rate between groups at baseline |
Lopez‐Gonzalez 2015.
| Study characteristics | ||
| Methods |
Study design: cluster‐RCT Each cluster made up of referral hospital, small hospitals and primary healthcare centres in the respective catchment area. Randomisation was stratified by geographical, social and economic differences between coastal and inland areas. Setting: Galician, in the northwest of Spain; local public health system provides hospitalised care at 13 general hospitals (seven referral and six small) and primary care at 405 healthcare centres distributed throughout the territory. Duration of follow‐up: 8 months |
|
| Participants |
Participants: all physicians actively engaged in clinical practice in the Galician public health system Number of participants: 5734 (control N = 3614; intervention: N = 2120) Mean age: data not provided Sex: data not provided |
|
| Interventions |
Comparison 1: education session plus reminder card and report form vs usual practice Intervention: education intervention programme with active component (group sessions) and passive component (educational material) Active: 20‐ to 25‐minute presentation, dynamic, objective and brief; importance of reporting ADRs, expressed in terms of morbidity, mortality and cost; limitations of clinical trials for the detection of adverse reactions and the advantages of a spontaneous voluntary reporting scheme; under‐reporting highlighted as one of the main disadvantages of this scheme; messages were reinforced to modify attitudes of complacency, insecurity, lack of self‐confidence (diffidence), indifference and ignorance, with additional stress being laid on the fact that only a few minutes were needed to complete an ADR report form; procedures for reporting to the Galician Pharmacovigilance Center were explained. Passive: a specimen ADR yellow card report form was handed out to each of those attending the educational session. Who delivered the intervention: 4 pharmacy researchers trained in pharmacovigilance; to ensure that the sessions would be as uniform as possible, all researchers met beforehand to agree on the use of common criteria. Intervention delivered from November 2007 to December 2008 Intervention fidelity: 57.2% of the physicians in the intervention clusters attended the presentations. Comparison: both groups received the continuing education course imparted by the Galician Pharmacovigilance Center. |
|
| Outcomes |
|
|
| Notes | Funding: Health Research Fund (Fondo de Investigacion Sanitaria) grants PI 081239, PI09/90609 from the Spanish Ministry of Health. Conflict of interest: study authors report no conflict of interest directly relevant to the content of this study. Clinical trial registration number: ISRCTN91140684 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Study authors do not describe how the random sequence was generated; however, based on the description of how clusters were defined and how randomisation was stratified, we feel that the randomisation sequence was probably generated appropriately. |
| Allocation concealment (selection bias) | Low risk | Study authors do not describe who allocated the clusters to the experimental groups and whether the person was blinded to the group allocation; however, selection bias was unlikely because in forming the clusters the particularities of the public healthcare system were taken into account (i.e. each cluster consisted of a referral hospital, small hospitals and primary healthcare centres in the respective health catchment areas), and randomisation was stratified by geographical, social and economic differences between Galicia’s coastal and inland areas. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The nature of the intervention does not allow for blinding of study participants or study personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded to the assignment of the regions: “adverse drug reaction reports were assessed at the Galician Pharmacovigilance Center. To eliminate subjectivity, assessors were kept ignorant of which geographical areas pertained to the control and intervention clusters.” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ADRs were either reported or not, so attrition bias is not relevant. There was a well‐established system for collecting ADR reports (Galician Pharmacovigilance Center). |
| Selective reporting (reporting bias) | Low risk | Based on the registered study protocol (ISRCTN91140684), all measured outcomes are reported in the published paper. |
| Other bias | Low risk | Clusters were assigned and then the intervention was implemented; no reported loss of clusters; outcome data appropriately adjusted for clustering; allocation by clusters thus reducing the risk of contamination; baseline ADR reporting rates for all adverse drug reaction categories studied were higher, albeit not statistically significantly so, in the control group than in the intervention group; no information available on baseline characteristics |
McKaig 2014.
| Study characteristics | ||
| Methods |
Study design: ITS Setting: a 719‐bed multidisciplinary urban medical centre, USA Duration of follow‐up: 2009 to 2012 |
|
| Participants |
Participants: nurses, physicians, pharmacists Number of participants: unclear Mean age: unclear Sex: unclear |
|
| Interventions |
Comparison 6: improving reporting method vs usual practice Intervention Pre‐implementation: 1 January 2009 to 31 August 2010 Post‐implementation: 1 September 2010 to 31October 2012 September 2010: web‐based electronic error‐reporting system replaced previous electronic error‐reporting system Previous electronic error‐reporting system: limited number of entry fields and drop‐down menu choices, reducing the quantity and quality of event‐specific information that can be entered at the time of reporting. Reports were directly routed to risk managers, who would identify stakeholders, such as floor nurse managers, and contact them for additional details regarding the event. This was performed using standard e‐mail communication, which resulted in potential delays in gathering event‐specific details. The risk manager entered the final event report through the electronic error‐reporting system. On a monthly basis, a system‐wide report was generated and provided to the institution wide Patient Safety and Quality Improvement Committees. The lack of access of the previous electronic error‐reporting system to staff or clinical managers limited the ability to add new information to the event or to document actions taken or improvements that had been made as a result of the report. Web‐based electronic error‐reporting system: staff access web‐based electronic error‐reporting system using secure web‐based portal from any computer in the hospital through the hospital intranet; reporting system is equipped with a series of standardised screens, drop‐down menu choices, and input fields designed to collect specific information regarding the nature and specific type of the event, time and location of event occurrence and discovery, involved personnel, impact on patient care, and subsequent patient outcomes. Events are routed by the system in real time for peer review to managers in the patient care area where the error has occurred, as well as to other departments identified by the reporter as involved in the event (e.g. pharmacy, laboratory). The system generates an e‐mail notification to alert managers that an event has been reported in the area and that it has been added to the task list for review. For all types of events, area managers perform a “first‐level” review to validate the accuracy of the information included in the report. Manager is also responsible for:
The Quality Management Department manages the overall hospital‐wide reporting process, providing high‐level review of the documented responses to events; performs general trending of event reporting; and follows up with managers who have not responded to outstanding events in a timely manner. The department is responsible for marking event status as “closed” when appropriate corrective action and follow‐up has been documented by the manager. Reported events that are identified by the reporter as resulting in patient harm are immediately routed by the system to senior hospital leadership and risk management for appropriate corrective action and for event reporting to appropriate outside agencies, such as the Massachusetts Department of Health. These reports receive an expedited formal review that uses the hospital’s root cause analysis (RCA) process, whereby an appropriate corrective action plan is developed. The action plan is entered into the RCA module within the electronic error‐reporting system for evaluation of the action plan and tracking of follow‐up actions. |
|
| Outcomes |
|
|
| Notes |
Study funding: not reported Conflict of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study authors do not clearly descibe the number of nurses, physicians or pharmacists exposed to the intervention or how these numbers changed over the duration of the study. |
| Selective reporting (reporting bias) | Low risk | Although we do not have access to the study protocol, all relevant outcomes in the methods section are reported in the results section. |
| Other bias | High risk | Authors did not appear to consider seasonal effects and there is no control arm. |
| Intervention independent of other changes | High risk | Comment: there are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was influenced by other confounding variables or historic events during study period. Comment: “A limitation of our study design is that we could not prospectively quantify nor isolate the impact of human factors and culture change that may have accompanied the launch of the current electronic error‐reporting system. Therefore, we are unable to attribute the impact of changing the electronic error‐reporting system on error‐reporting rates in the absence of any other extraneous influence”. |
| Shape of the intervention effect pre‐specified | Low risk | Point of analysis is the point of intervention (i.e. the point in time at which the intervention was implemented). |
| Intervention unlikely to affect data collection | Low risk | The intervention itself was unlikely to affect data collection: sources and methods of data collection were the same before and after the intervention. |
| Knowledge of the allocated interventions adequately prevented during the study | Low risk | The primary outcome (number of ME reports in the hospital‐wide medication‐error‐reporting programme) is objective. |
Pedrós 2009.
| Study characteristics | ||
| Methods |
Study design: ITS Study setting: Spain, Vall d’Hebron Hospital, tertiary care teaching hospital; general area of the hospital: 700 beds Duration of follow‐up: 1998 to 2005 (intervention implemented in 2002); total of 96 observations First period prior to the intervention: 1998 to 2002); intervention implemented in December 2002; second period post implementation of intervention: 2003 to 2005 |
|
| Participants |
Participants: physicians Number of participants: approximately 500 staff physicians and pharmacists; 200 physicians and pharmacists in training. Mean age: not reported Sex: not reported |
|
| Interventions |
Comparison 3: multifaceted intervention vs usual practice Intervention (financial and education) Implemented in December 2002: a multifaceted intervention based on healthcare management agreements between hospital managers and clinical services implemented. Physicians offered economic incentives with aim of increasing number of spontaneous reports of ADRs. The economic incentives for ADR reporting were integrated with other clinical objectives at three levels: (i) institution or whole hospital; (ii) clinical department or clinical team; and (iii) physician. The financial incentive was variable according to the objectives achieved, and was approximately 5 to 7% of the physician’s salary. The size of the financial payment to physicians for ADR reporting was not fixed, instead being variable depending on the prioritisation of other commitments, and it accounted for less than 10% of the total of agreed incentives. Therefore, the financial incentive obtained for reporting was, on average, less than 1% of the physician’s salary. Twice‐yearly education meetings (45 to 60 minutes long) ‐ in each clinical service, an initial meeting between physicians and the hospital pharmacovigilance team was held with the objective of discussing spontaneous reporting of ADRs, a summary of the hospital’s pharmacovigilance activities, the way to report adverse drug reactions, and the changes in the pharmacovigilance legal rules recently established in the EU and Spain were presented. In addition, reminder cards containing the contact telephone number of the pharmacovigilance team in charge of the hospital pharmacovigilance programme and a list of the most important ADRs to be reported (serious, unexpected and those associated with new drugs) were distributed to the hospital wards. Comparison: usual practice |
|
| Outcomes |
|
|
| Notes |
Study funding: no sources of funding were used to assist in the development of this study. Conflict of interest: the authors have no conflicts of interest that are directly relevant to the content of this study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | While we know that the general area of the hospital was served by approximately 500 staff physicians and pharmacists; 200 physicians and pharmacists in training, it is not clearly described how this number changed over the duration of the study. |
| Selective reporting (reporting bias) | Low risk | No access to study protocol. However, there is no evidence that outcomes were selectively reported (all relevant outcomes in the methods section are reported in the results section). |
| Other bias | Low risk | Study authors used an autoregressive integrated moving average (ARIMA) model that accounted for seasonality. |
| Intervention independent of other changes | High risk | There are no compelling arguments that the intervention occurred independently of other changes over time and the outcome was not influenced by other confounding variables or historic events during study period. |
| Shape of the intervention effect pre‐specified | Low risk | Point of analysis is the point of intervention (i.e. the point in time at which the intervention was implemented). |
| Intervention unlikely to affect data collection | Low risk | The intervention itself was unlikely to affect data collection: sources and methods of data collection were the same before and after the intervention. |
| Knowledge of the allocated interventions adequately prevented during the study | Low risk | The primary outcome (number of adverse drug reaction reports noted in the pharmacovigilance database) is objective. |
Ribeiro‐Vaz 2011.
| Study characteristics | ||
| Methods |
Study design: cluster‐RCT Geospacial clusters: pharmacists working in a referral hospital and in community pharmacies in the geographic area; 4 intervention clusters (2 education workshop, 2 telephone interview), 11 control clusters. This study builds on previous studies: Figueiras 2006 targeted physicians in the 15 clusters randomised to receive 1‐hour education session, tailored to training needs or no intervention; Herdeiro 2008 targeted pharmacists in the 15 clusters randomised to receive 1‐hour education sessions, tailored to training needs or no intervention. A second randomisation of the four intervention clusters (from Figueiras 2006 and Herdeiro 2008) was performed, and two clusters received the workshop intervention and two clusters received the telephone‐interview intervention, with 11 clusters remaining assigned to the control arm. Herdeiro 2012 targeted the physicians and Ribeiro‐Vaz 2011 targeted the pharmacists in the newly randomised clusters. Study setting: 20 hospitals in the northern region of Portugal Duration of intervention: May to June 2007 Duration of follow‐up: 20 months |
|
| Participants | Clinically active pharmacists based in the northern region of Portugal (Herdeiro 2012 reports on the physicians in these clusters) Number of participants: 1467 (control: N = 1103; intervention: N = 364 including 261 pharmacists in telephone group and 103 pharmacists in workshop group) Mean age: unclear Sex: 79% female |
|
| Interventions |
Comparison 1: education session plus reminder card and report form versus usual practice Intervention: of the 4 intervention clusters, 2 received educational workshops and 2 received telephone interviews. Workshop ‐ 60‐minute sessions; problem of ADR and its impact on public health; approach to spontaneous ADR notification; attitudes and knowledge that pharmacists have on the subject; guidance on how to complete the spontaneous ADR report form; discussion of a practical case and completion of the spontaneous notification of adverse drug reaction with data from the case study; after session, each participant provided with support material for filing and facilitating the process: a copy of the practical case, a copy of a spontaneous notification of ADR to be filled in with the practical case, of presentation of the Northern Pharmacovigilance Unit and certificate of attendance. Telephone ‐ telephone conversation (pre‐established script); 4 to 12 minutes of fluid conversation; 3 attempts made to contact participants, after which deemed impossible to contact; following telephone interview participant sent (via post) support material (thank‐you letter, spontaneous notification of adverse drug reaction report form and presentation of the Northern Pharmacovigilance Unit). Intervention delivered from 29 May to 26 June 2007 (workshop) and 2 to 20 July 2007 (telephone) Intervention fidelity: workshop ‐ attended by 46% of pharmacists; telephone interview ‐ 36% of all individuals contacted Comparison: usual practice; no workshop or telephone interview |
|
| Outcomes | 1. Total number of ADR reports (assessed monthly; raw data; per 1000 pharmacist‐month) 2. Number of serious ADRs (one that results in death; is life‐threatening; is a congenital anomaly; requires hospital admission or prolongation of stay in hospital; or results in persistent or great disability, incapacity, or both) 3. Number of ADRs with attribution of definitive or probable causality 4. Number of unexpected ADRs (unknown ADRs that are not described in the SPC) |
|
| Notes | Study funding: no information provided Conflict of interest: no statement provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Study authors do not describe how the random sequence was generated; however, they do state that the intervention and control clusters were assigned in a ratio of 1:3, so it is likely that a legitimate sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | Study authors do not describe how the clusters were allocated to the experimental groups. The risk of bias is therefore unclear. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Due to the nature of the intervention, it is not possible for the participants to be blinded. It is not clear to what extent the study personnel were blinded to the group allocation of the pharmacist. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The outcome assessor was blinded to the study group of each pharmacist and data on notifers was transformed into a numeric code. "The expert responsible for assessing adverse reactions was unknown to the study group every pharmacist belonged. Confidence was throughout the study and data on notifiers have been transformed into a numeric code rich.” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ADRs are spontaneously reported or not reported. Therefore, attrition bias is not applicable in this instance. All ADR data was obtained from the Northern Pharmacovigilance Unit. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available; however, all the outcomes described in the methods are reported in the results. |
| Other bias | High risk | Low risk of recruitment bias as clusters were allocated and then intervention was implemented; outcome data appropriately adjusted for clustering; no reported loss of clusters; baseline ADR reporting rate in all the categories was higher in the workshop group, but it was unclear if this difference was statistically significant; no information regarding participants' baseline characteristics; allocation by clustering thus low risk of contamination |
Ribeiro‐Vaz 2012.
| Study characteristics | ||
| Methods |
Study design: ITS Setting: hospitals in Northern Portugal Duration of study: 2006 to 2010 Duration of intervention: implemented over 6 months in 2007 to 2008 Duration of follow‐up: 31 months |
|
| Participants |
Target population: healthcare professionals (i.e. physicians, pharmacists, nurses) working in hospitals in northern Portugal Number of centres (hospitals) in the area: 18 centres (31 hospitals) Number of centres (hospitals) excluded: 2 centres (3 hospitals) plus 1 other hospital Number of centres (hospitals) included: 16 centres (27 hospitals) Number of centres (hospitals) included in analysis: 11 centres (18 hospitals) ‐ 5 centres (9 hospitals) did not include the hyperlink intervention Study does not report the number of healthcare professionals serving these centres and hospitals. |
|
| Interventions |
Comparison 4: improving access to ADR report form vs usual practice Intervention: hyperlinks to the ADR online reporting UFN (Northern Pharmacovigilance Center) form were proposed to 18 northern Portugal hospitals. The hyperlinks can be included either in healthcare professional‐specific software (typically electronic patient records or pharmacy‐specific applications used by doctors, nurses and pharmacists) or on hospital computer desktops. October 2007: letter was sent to the chief physicians of these 18 northern Portuguese hospitals regarding inclusion of the hyperlink. If no response within 2 weeks, clinical administration boards were reminded by telephone. Five of the centres failed to respond by the end of 2010. December 2007: five hospital centres implemented the hyperlink. Another six implemented it over the course of the next 5 months. Hyperlink in the electronic patient record: 8 centres (12 hospitals) Hyperlink in the desktop: 2 centres (5 hospitals) Hyperlink in both the desktop and in the electronic patient record: 1 centre (1 hospital) Comparison: usual practice |
|
| Outcomes |
23 months before intervention and 31 months after the intervention in each hospital |
|
| Notes |
Study funding: not reported Conflict of interest: no statement provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | We know the number of centres and hospitals offered the intervention (16 centres; 27 hospitals) and the number included in the analysis (11 centres; 18 hospitals ‐ 5 centres; 9 hospitals did not implement the hyperlink), but the study does not report the number of health professionals serving these hospitals. |
| Selective reporting (reporting bias) | Low risk | Although we do not have access to the study protocol, there is no evidence that outcomes were selectively reported (all relevant outcomes in the methods section are reported in the results section). |
| Other bias | High risk | There is a probable clustering effect in Ribeiro‐Vaz 2012 (there appears to be a hierarchy of hospitals within centres), which does not seem to be modelled. The study covers a period of about 4 years, but there does not seem to be any consideration of seasonality effects. |
| Intervention independent of other changes | Low risk | "From the initial 18 centres (31 hospitals) we excluded four hospitals that established other cooperation protocols with UFN to avoid a possible confounder bias. For the other 16 centres, we believe that there were no external interventions that could potentially explain the observed results.” |
| Shape of the intervention effect pre‐specified | Low risk | Point of analysis is the point of intervention (i.e. the point in time at which the intervention was implemented). |
| Intervention unlikely to affect data collection | Low risk | The intervention itself was unlikely to affect data collection: sources and methods of data collection were the same before and after the intervention. |
| Knowledge of the allocated interventions adequately prevented during the study | Low risk | The primary outcome (number of adverse drug reaction reports noted in the pharmacovigilance database) is objective. |
Schlienger 1999.
| Study characteristics | ||
| Methods |
Study design: a prospective, comparative, open, cross‐over study Cross‐over study: we decided to only use data from the first year of the study (first period data) in order to reduce the risk of contamination bias and any carry‐over effect inherent in a cross‐over study. We judged the risk of bias of the study based on the first period data only. Setting: Department of Medicine of the University Hospital of Basel, Switzerland; 900‐bed teaching hospital providing primary and tertiary care and tertiary care referral centre for the north west of Switzerland; the ward specialised in the care of patients with infectious diseases, cardiovascular, haematological, oncological and peripheral vascular disorders; it included four adjacent 13‐bed nursing units located on the same floor. Duration of follow‐up: 24 months (12 months, intervention and control units crossed over, 12 months) |
|
| Participants |
Target of the intervention:physicians, nurses, pharmacists Each unit was staffed with one medical intern (second or third year of postgraduate training on a 3‐ to 6‐month rotation scheme). The four interns were supervised by a full‐time chief resident board‐certified in internal medicine. The outcome was assessed per participant. Consecutive patients were included who were admitted to one of the units of the General Medical Service included in the study, and were prescribed at least one pharmacologically active drug. First study period (months 1 to 12) Number of participants: 507 (intervention); 509 (control) #Mean age: 63 ± 17 years (intervention); 63 ± 17 (control) #Sex (% male): 66 (intervention); 53 (control) #combined data for both 12‐month periods; as data not presented separately for each 12‐month period. |
|
| Interventions |
Comparison 7: case finding (pharmacists) vs usual practice (spontaneous reporting) No structured system for reporting ADEs was in place in the institution. Clinical pharmacist responsible for reporting and identification of ADEs; graduated from the School of Pharmacy and a full‐time member of the staff of the hospital’s Division of Clinical Pharmacology. Intervention in the test units consisted of the following three components.
Comparison: information about ADEs from control units was based only on spontaneous voluntary reports from physicians and nurses; the clinical pharmacist did not visit the patients or the medical and nursing staff. |
|
| Outcomes |
Primary outcome
*The definition of ADEs in this study is a little bit unclear. Firstly, the study authors defined ADEs based on a modified WHO definition: an ADE is an event that is "noxious and unintended and occurs at doses used in humans for prophylaxis, diagnosis, therapy, or modification of physiologic functions. Furthermore, for the purpose of this investigation, this definition included injuries resulting from inappropriate use of a drug as well as errors in drug administration, whereas therapeutic failures, poisonings, and intentional overdoses were excluded." Secondly, the authors go on to say that ADEs were classified as having "definite, probable, or possible causal relationship with a drug", which would usually be ADRs. |
|
| Notes | Financial support: study supported in part by grants from the Swiss Society of Hospital Pharmacists (GSASA) and from Sintetica SA, Mendrisio Switzerland. Conflict of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | No randomisation was performed. |
| Allocation concealment (selection bias) | High risk | "At the beginning of the study and after 12 months ‐ with the change from test to control unit status ‐ physicians and the nursing teams of study and control units were informed about the study design and goals during a one‐hour oral presentation by the clinical pharmacist. In addition, at the beginning of each rotation period, interns joining the teams were also informed about the intervention." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The outcome assessor (clinical pharmacist) was not blinded to the group allocation. There were four ways by which ADEs were identified in test unit patients: (a) spontaneous or solicited reporting by a physician; (b) spontaneous or solicited reporting by a nurse; (c) detection on regular ward rounds; and (d) detection by the clinical pharmacist by chart review after hospital discharge. In control unit patients, spontaneous reporting by members of the medical and nursing team was the only method used. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The flow of patients through the study is not clearly described. "Consecutive patients were included who were admitted to one of the units of the General Medical Service included in the study, and were prescribed at least one pharmacologically active drug." |
| Selective reporting (reporting bias) | Low risk | No access to study protocol. There is no evidence that outcomes were selectively reported (all relevant outcomes in the methods section are reported in the results section). |
| Other bias | Unclear risk | Baseline characteristics: "Patients in test and control units were similar in age, length of stay and average number of med‐ ications, whereas the gender distribution differed significantly (Table 1)" The effect of this imbalance on the number of reported AREs is unclear. As this is a cross‐over study, it is also unclear how any potential carry‐over effect might bias the outcome measured. |
ADE: adverse drug event; ADR: adverse drug reaction; IQR: interquartile range; IRR: incidence rate ratio; ITS: interrupted time series; ME: medication error; N: number of participants; NPC: Northern Pharmacovigilance Centre; P: probability; PHC: primary healthcare unit; RCT: randomised controlled trial; RMB: renminbi, Chinese currency; SPC: summary of product characteristics; vs: versus; WHO: World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aldeyab 2016 | Ineligible participants: intervention targets patients not health professionals |
| Anbalagan 2019 | Ineligible study design: uncontrolled before‐after study |
| Åserød 2017 | Ineligible intervention: description of mobile application design |
| Aspinall 2002 | Ineligible study design: descriptive observational study |
| Avong 2018 | Ineligible study design: no control/comparison and not ITS |
| Bäckström 2002 | Ineligible study design: CBA study with only one intervention and one control group |
| Bäckström 2006 | Ineligible study design: CBA study with only one intervention and one control group |
| Belhekar 2022 | Ineligible study design: CBA study with only one intervention and one control group |
| Bracchi 2005 | Ineligible study design: CBA study with only one intervention and one control group and not enough data points to re‐analyse as ITS |
| Candore 2022 | Ineligible study design: retrospective observational study (data includes adverse reactions reported by patients and HCPs) |
| Cano‐Sandoval 2020 | Ineligible study design: descriptive observational study |
| Capucho 2008 | Ineligible study design: not enough data points post intervention for ITS analysis |
| Chatas 1990 | Ineligible study design: uncontrolled cohort and not enough data pre intervention to analyse as ITS/repeated measures |
| Clarkson 2001 | Ineligible study design: only one intervention region or cluster |
| Colodny 1999 | Ineligible study design: ITS ‐ monthly ADR reporting rates in 1996 (prior to programme implementation) and 1997 (post implementation) provided, but apart from "early 1997", there is not a clear‐cut point in time when the programme/policy was implemented. |
| Correa 2019 | Ineligible study design: uncontrolled before‐after study and unable to re‐analyse as ITS |
| Costello 2007 | Ineligible study design: not enough data collection timepoints for ITS analysis |
| Deslandes 2022 | Ineligible study design: not enough data points pre and post intervention to analyse as ITS |
| Fang 2017 | Ineligible study design: retrospective, uncontrolled before‐after study; not enough data point provided to analyse as ITS/repeated measures |
| Fracchiolla 2018 | Ineligible study design: observational cohort study |
| Gony 2010 | Ineligible study design: controlled before‐after study with only one control group |
| Haramburu 1988 | Ineligible study design: retrospective observational study |
| Haw 2011 | Ineligible study design: ITS but no data pre‐intervention, only data for post‐intervention |
| He 2018 | Ineligible study design: uncontrolled before‐after study |
| Iessa 2021 | Ineligible study design: descriptive observational study |
| Jacqout 2012 | Ineligible study design: uncontrolled before‐after study |
| Jha 2017 | Ineligible study design: descriptive observational study |
| Kronenfeld 2019 | Ineligible study design: historical control and not possible to re‐analyse as ITS/repeated measures |
| Lata 2004 | Ineligible study design: uncontrolled before‐after study |
| Lee 2004 | Ineligible study design: ITS but only one time point post intervention |
| Li 2020 | Ineligible participants: intervention directed to both consumers and HCPs and data not reported separately |
| Linder 2010 | Ineligible intervention: no health professional involved |
| Lynn 2010 | Ineligible study design: ITS but no pre intervention data |
| Marquez 2016 | Ineligible study design: controlled post‐intervention study with no pre‐intervention data collection; has only two clusters |
| Massah 2021 | Ineligible study design: uncontrolled before‐after study, not enough data points for ITS |
| McGettigan 1997 | Ineligible study design: historical controls, not enough data points for ITS analysis |
| Menat 2021 | Ineligible study design: only 2 medicine units involved in the study, no control group and multiple time points not collected so cannot re‐analyse as ITS; study compared if more ADRs reported by spontaneous reporting or triggers (e.g. red flags on the computerised medical records) method. |
| Morales 2016 | Ineligible study design: uncontrolled before‐after study |
| Morgan 1990 | Ineligible study design: uncontrolled before‐after study, not enough data to re‐analyse as ITS |
| Mwamwitwa 2022 | Ineligible study design: uncontrolled before‐after study but not enough data to re‐analyse as ITS |
| NCT02087293 | Ineligible participants: intervention targeted patient reporting of ADEs |
| Ng 2020 | Ineligible intervention: ITS study, intervention aimed at reducing MEs, not aimed at improving the reporting of MEs |
| Opadeyi 2019 | Ineligible study design: focus of the study was to improve knowldege, attitude and process outcomes |
| Ortega 2008 | Ineligible study design: not enough timepoints pre and post implementation of the intervention provided |
| Potlog 2020 | Ineligible study design: only 1 intervention cluster and 1 control cluster (made up of B and C) |
| Praveen 2021 | Ineligible study design: cross‐sectional observational study assessing knowledge, attitude and practice/process outcomes |
| Reumerman 2021a | Ineligible study design: although this is a CBA design, our review inclusion criteria stipulated that CBA should have at least two intervention sites and two control sites to be eligible. This study has one intervention site and two control sites. |
| Reumerman 2021b | Ineligible study design: no control group or baseline data; ADRs were reported by medical students not qualified healthcare professionals (so wrong participants) |
| Rogers 1989 | Ineligible study design: CBA study with only one intervention group |
| Roy 2018 | Ineligible study design: pre and post survey |
| Rubin 2019 | Ineligible study design: retrospective observational study |
| Salcedo de Diego 2015 | Ineligible study design: uncontrolled before‐and‐after study; monthly data are not available, which would allow re‐analysis as an ITS |
| Sánchez 2014 | Ineligible study design: no control group and no mention of baseline data |
| Sanko 2020 | Ineligible intervention: ADR reporting was from a simulation programme |
| Schindler 2019 | Ineligible study design: not interventional, just discusses how adverse drug reactions might be reported |
| Segec 2021 | Ineligible participants: patients/consumers were intervention target population |
| Sonowa 2020 | Ineligible study design: before‐after study without a control group; only one time point assessed pre‐intervention so cannot be re‐analysed as ITS |
| Srikanth 2019 | Ineligible study design: before‐after study without a control group |
| Tabali 2009 | Ineligible study design: uncontrolled before‐after study |
| Terblanche 2018 | Ineligible study design: before‐after study with no control group and not enough time points pre and post intervention to analyse as ITS/repeated measures |
| Touchette 2012 | Ineligible participants: intervention targeted patient reporting of ADEs |
| Varallo 2015 | Ineligible study design: uncontrolled before‐after study |
| Weant 2010 | Ineligible study design: retrospective cohort review |
| Welsh 1996 | Ineligible study design: uncontrolled before‐after study (not enough data points before and after intervention for ITS re‐analysis) |
| Wengrove 2021 | Ineligible study design: uncontrolled before‐after study |
| Wentzell 2017 | Ineligible study design: only one intervention and one control site |
| Winstanley 1989 | Ineligible study design: ITS, but only 2 data timepoints pre‐intervention provided |
| Xu 2014 | Ineligible intervention: intervention aimed at improving patient safety and reducing MEs |
| Yen 2010 | Ineligible study design: uncontrolled before‐after study (data collected 39 months before and after the intervention of the electronic system ‐ the (monthly) data is not provided so we cannot reanalyse as an ITS/repeated measures study) |
ADE: adverse drug events; ADR: adverse drug reaction; CBA: controlled before‐and‐after study; HCP: healthcare professional; ITS: interrupted time series; ME: medication error
Characteristics of studies awaiting classification [ordered by study ID]
Biagi 2013.
| Methods | Objective: assessing the knowledge, attitudes, and behavior of GPs regarding ADR reporting; evaluating whether a monthly e‐mail‐based newsletter on drug safety could affect the rate or quality of the ADR reports submitted by these GPs Design: controlled before‐after study (questionnaire study) Setting: Italy (three local health authorities) Duration: 3 years. Was divided into: (1) identification of the reasons leading to underreporting through a questionnaire (Phase I); (2) the intervention, i.e. sending a newsletter for a 10‐month period (Phase II); (3) evaluation of the intervention outcomes during the 10 months following the period in which the newsletter had been received (Phase III) |
| Participants | All GPs (N = 737) associated with these three local health authorities were recruited. |
| Interventions | GPs associated with three local health authorities exposed to the intervention; study authors report: "The pooled number of ADR reports sent by GPs in the remaining seven LHAs of the region was used as controls". Seven local health authorities of the region not exposed to intervention were used as controls; however, it is unclear if these controls were considered historical controls. We have contacted the study authors for additional information. |
| Outcomes | Pooled number of ADR reports sent by GPs; number of GP reports per 100,000 inhabitants |
| Notes | The design of this study is not clear; if it is to be considered a CBA study, we need to be sure that the seven unexposed local health authorities are not historical controls. We do not have sufficient data from the study to re‐analyse as an ITS study. |
Kim 2012.
| Methods | Objective: to examine the impact of the implementation of an electronic‐based reporting system on the occurrence of ME reports in the hospice setting. Method: an electronic ME reporting system was developed and an inservice was provided to participants. |
| Participants | Healthcare workers in hospice setting |
| Interventions | Pre‐intervention data was collected for a two‐year period and consisted of ME reports from the hospice's paper‐based system. Post‐intervention data was collected for a 120‐day period and consisted of reports from the newly electronic‐based system. |
| Outcomes | ME reports collected and analysed comparing two specific time periods. |
| Notes | Potentially eligible ‐ poster abstract only; probably an eligible ITS study |
Kumar 2021.
| Methods | The design of this ongoing study is not clearly described: "Method of generating randomization sequence: Random Number Table Method of allocation concealment: Sequentially numbered, sealed, opaque envelopes Blinding and masking: Not Applicable". It looks like an RCT, but trial registration entry goes on to say the following about the control group: "Standard ADR reporting before our training on mobile app ADR reporting: The ADR reporting done before mobile app intervention will be the control arm" |
| Participants | Medical officers (target sample size: 50) |
| Interventions | Intervention: ADR reporting by mobile app: medical officers will be trained on reporting ADR using mobile app namely Pharmacovigilance Program of India (PvPI) ADR app developed by Government of India, and will be asked to use it for ADR reporting when encountered while treating patients with tuberculosis. Control: standard ADR reporting before our training on mobile app ADR reporting: The ADR reporting done before mobile app intervention will be the control arm |
| Outcomes |
Time points: baseline, 3rd, 6th, 9th and 12th months |
| Notes | It is unclear how the data will be collected; while the participants and intervention are appropriate for our review, it appears that the control data might be historical data, so it is not clear if the study is a controlled before‐after study. Recruitment status: open to recruitment Emailed authors on 9 March 2023 for additional study information (Dr S Ramesh Kumar: drrameshskumar@yahoo.co.in) |
Lan 2022.
| Methods | Conducted in China (a large general hospital). Authors retrospectively analysed the number of inpatient ADRs submitted to the Center for Advanced Drug Monitoring per month, the number of unreported ADRs per month, and the standardised ADRs reporting rate per month in 2010 to 2019 to investigate the immediate and long‐term efects of clinical pharmacists intervention on ADRs reporting. The intervention was implemented on 1 June 2015, and a segmented regression model was used to analyse the data from 1 January 2010 to 31 December 2019. |
| Participants | Clinical pharmacists |
| Interventions | A team of clinical pharmacists detected ADRs among hospital patients and promptly reminded clinicians to report them. In cases of potential ADRs, clinical pharmacists assessed causality, verifed the event with clinicians, and reminded the clinicians to report any missed ADRs. In addition, clinical pharmacists reviewed ADRs reports for missing items, measures taken to treat the ADRs, improvement or resolution of the ADRs after drug discontinuation or dose reduction, recurrence of ADRs after the resumption of the suspected drug, and causality, to reduce potential under‐reporting and omission and promote the standardisation of ADRs reporting. |
| Outcomes | Number of inpatient ADRs reports submitted to the Center for Advanced Drug Monitoring Number of unreported ADRs Number of standardised ADRs reporting rate per month |
| Notes | Study identified in the updated search conducted just prior to submitting the review for peer review; study to be assessed for inclusion in the updated review; retrospective observational study, however, data analysed as ITS |
Lander 2013.
| Methods | Study design: unclear (retrospective CBA study) or repeated measures study (although the described analyses does not support this) Setting: Denmark "The number of ADEs reported during the study period (1 October 2010–30 September 2011) was compared with the number of reports between 1 October 2009 and 30 September 2010, which furthermore was compared with the number of reports between 1 October 2008 and 30 September 2009", so there appear to be repeated measures data. "The total number of ADE reports from all hospitals in the Capital Region of Denmark was provided by the DMA", so there appears to be a control or comparison group. |
| Participants | Two junior doctors and one resident from the Department of Clinical Pharmacology took on the role of the ADE manager and were responsible for reporting the ADEs. |
| Interventions | Intervention: 1 hospital ADE manager introduced at a university hospital (five medical wards) in Denmark in the period October 2010 to September 2011. Function of the ADE manager was to complete the ADE report, whenever a physician required assistance. ADE manager used electronic health records to collect the necessary information about the ADE and complete a report to the DMA, now part of Danish Health and Medicines Authority. Comparison: 11 hospitals; study authors do not describe ADE reporting usual practice in the other hospitals. |
| Outcomes | Number of ADE reports per month |
| Notes | Study is presented as a descriptive observational study or a CBA study; if we assume it is a CBA, it would be ineligible as there is only one intervention site (1 hospital); it may be possible to re‐analyse the data using ITS analysis; closer inspection of Figure 1 data ‐ no data points post the intervention implementation period (the study states that in the period between October 2010 and September 2011, an ADE manager was introduced at Bispebjerg University Hospital, and Figure 1 only provides data up to September 2011, so we do not have our required 3 data points post intervention needed for ITS analysis). This study was identified in the search update and we did not have the resources to re‐analyse the data at that stage. |
Marquez 2015.
| Methods | The aim of this study is to evaluate the quantitative and qualitative increase of ADR reports by nurses after an educational intervention; quasi‐experimental study; 113 nurses in the intervention group; 590 nurses in the control group; January 2013 to September 2014. |
| Participants | Nurses working in primary care (Portugal) |
| Interventions | Two educational interventions (no further information provided) |
| Outcomes | Quantitative and qualitative increase of ADR reports |
| Notes | Awaiting classification ‐ abstract only, and not enough information; non‐randomised controlled trial of primary care nurses, so there may be more than one site and might possibly be eligible; co‐authors include Herdeiro and Ribeiro‐Vaz (authors of currently included studies) |
Opadeyi 2021.
| Methods | This study evaluated the impact of an educational lecture followed by repeated text messages via the Short Messaging System on ADR reporting. Six teaching hospitals in the South‐South zone of Nigeria were randomised in 1:1 ratio into intervention and non‐intervention hospitals. |
| Participants | We assume healthcare professionals were the intervention target (4912 healthcare professionals were working in the 6 hospitals at the time of the study; 3099 in the intervention and 1813 in the control) |
| Interventions | The intervention hospitals received an educational lecture followed by monthly SMS reinforcements over 12 months. Educational intervention conducted between January and March 2016. No further information provided in the abstract. |
| Outcomes | The number and quality of ADR reports from the local pharmacovigilance centers of each teaching hospital over the 12 months before and after the intervention were described |
| Notes | Study identified in updated search; we have not been able to access a copy of the full text and there is not enough information in the abstract to determine study eligibility. We have contacted the study authors for a copy of the full text and for more information and data. |
Sawangjit 2022.
| Methods | Cluster‐RCT (based on the study title) |
| Participants | HCPs including doctors and pharmacists work at public tertiary hospital in Lao People's Democratic Republic, who have a role in reporting ADRs in hospital. |
| Interventions | Education‐related ADR report for 2 hour 3 time plus using TaWai mobile system as a tool to report ADR in hospital for 3 months Education‐related ADR report for 2 hour 3 time plus using classical (usual) form as a tool to report ADR in hospital for 3 months |
| Outcomes | Total number of ADR reported in 3 months Satisfaction of reporters: mean of satisfaction score Quality of ADR report during 3 months: number of ADR reports with good quality Rate of ADR report 1, 2 and 3 months: the number of ADR reports/measurement time |
| Notes | No full text or data published for this study; need more information on the study design hence awaiting classification; emailed authors on 9 March for additional study information (Ratree Sawangjit: ratree.m@msu.ac.th); copy of PhD dissertation (completed after submission of our review) found online on 27 January 2024 ‐ to be reviewed and assessed for future review update |
ADR: adverse drug reaction; ADE: adverse drug event; CBA: controlled before and after; DMA: Danish Medicines Agency; GP: general practitioner; HCP: healthcare professional; ITS: interrupted time series; RCT: randomised controlled trial
Characteristics of ongoing studies [ordered by study ID]
Hutchinson 2020.
| Study name | Use of an audit with feedback implementation strategy to promote medication error reporting by nurses |
| Methods |
Study design: quasi‐experimental design (possible re‐analysis as controlled before‐after study or cluster RCT if additional data provided) Study setting: study was undertaken at one large, private, not‐for‐profit hospital in Melbourne, Australia (hospital selected for convenience; associated pilot research conducted at this hospital see Hutchinson 2015). Sampling of wards was purposive, with ward selection and matching guided by an initial assessment of the clinical case mix (diagnostic group, average length of stay, occupied beds) of all acute care wards at the participating hospital. Duration of follow up: intervention implemented over 12 months then data collected during 6 months post implementation |
| Participants |
Participants: nurses; medical wards included a neurology or stroke ward and a general medical or aged care assessment ward; surgical wards included a cardiothoracic ward and a plastics or general surgery ward. All participating wards were located in close proximity to each other and comprised teams of medical doctors that worked across multiple wards. All managers (n = 4) and full‐ and part‐time nurses (both registered and enrolled nurses; n = 162) working in the participating wards were considered eligible for inclusion. Casual nurse bank or agency nurses and nursing students were excluded due to their inconsistent presence on wards. Number of participants: 162 nurses, whether full‐time or part‐time, and whether registered or enrolled nurses, in all 4 wards |
| Interventions |
Comparison 8: audit and feedback of analysed audit data (intervention) versus audit without feedback (control) Intervention: nurses within intervention wards received audit with feedback on a quarterly basis over a 12‐month implementation period (i.e. four times in total). Feedback report incorporated a brief educational component, but mainly consisted of a coloured, one‐page infographic poster, with content based on medication error data obtained from audits and the hospitals’ risk management system (RiskMan). Feedback posters were placed in the intervention wards by a member of the research team in locations deemed appropriate by the stakeholder group and nurse unit managers of participating wards (i.e. medication rooms, the mirror next to hand basin in staff bathrooms, on the back of toilet doors and on the walls above toilet roll holders, on tables in tea rooms, in staff communication books and on the wall near staff lockers). Feedback posters were also sent electronically via email by the senior nurse or nurses to nurses in the intervention wards. The feedback report was unique to each intervention ward and specifically related to processes of care. The content of the posters was sourced from the following.
Comparison: nurses in control wards underwent quarterly point‐prevalence audits of medication documentation in patients’ medical records. No feedback provided. |
| Outcomes |
Primary outcome: rate of ME reports per month (models considered number of reported medication errors in study wards as the outcome, offset by the average number of occupied beds in the wards per month; i.e. modelling rate of medication errors per ward per month) Rate of ME reports per month determined in both groups at pre‐implementation (12‐month period prior to the implementation), implementation (12‐month period of the implementation) and post‐implementation phases (6‐month period following the implementation phase). The outcome data was collected retrospectively: "all medication‐related RiskMan data were retrospectively extracted for all four wards by a staff member of the Clinical Governance Unit at the participating hospital, who was blinded to ward allocation. Relevant RiskMan data were extracted for the following periods: a 12‐month period prior to the implementation phase (pre‐implementation); the 12‐month period of the implementation phase (implementation); and a 6‐month period following the implementation phase (postimplementation). The purpose of extracting data over the three specified periods was to establish a trend in medication error reporting over pre‐implementation, implementation and post‐implementation periods" |
| Starting date | Study dates and phases: March 2014 to March 2015 (pre‐implementation); March 2015 to March 2016 (implementation phase); April to September 2016 (post implementation) |
| Contact information | Alison M Hutchinson, School of Nursing & Midwifery, Centre for Quality & Patient Safety Research, Institute for Health Transformation, Faculty of Health, Deakin University, Geelong, VIC 3220, Australia. Email: alison.hutchinson@deakin.edu.au |
| Notes | The study meets the eligibility criteria for this review but the published paper only provides the rate of ME reports per month for the intervention wards and the control wards, without any indication of variation around the mean. We have emailed the study authors for the required information. Trial registration: not registered Funding: this work was supported by the Australian Government through the Australian Research Council (project number LP120200197). The funding source of the study had no role in study design, data collection, data analysis, data interpretation, writing of the report or decision to submit for publication. Conflict of interest: the authors have no conflicts of interest to declare. |
Kiguba 2022.
| Study name | Effectiveness of the Med Safety mobile application in improving adverse drug reaction reporting by healthcare professionals in Uganda |
| Methods | A pragmatic cluster‐RCT will be implemented over 30 months at 191 intervention and 191 comparison cART sites to evaluate the Med Safety app. |
| Participants | Healthcare professionals |
| Interventions | The Med Safety mobile application (developed through the European Union’s Innovative Medicines Initiative WEB‐Recognising Adverse Drug Reactions project) to promote digital pharmacovigilance. Healthcare professionals enrolled in the intervention arm will be trained in the use of mobile‐based, paper‐based and web‐based reporting, while those in the comparison arm will be trained in paper‐based and web‐based reporting only. |
| Outcomes | Primary outcome: number of HCP‐reported ADRs per 100,000 person‐months of treated people living with HIV per study arm. Secondary outcomes: number of app ADR reports per 1000 app downloads per month of follow‐up; causality (by Naranjo Scale and Liverpool Causality Assessment Tool); seriousness as per the WHO definitions (threatens life, i.e. leads to or prolongs hospitalisation, causes incapacitation or death); ADR outcome; cost per ADR report; cost per additional ADR report; and cost per additional avoidable serious ADR report. |
| Starting date | 1 July 2020 (anticipated stated date on registration site) |
| Contact information |
Primary Sponsor: Medical Research Council, London, United Kingdom Principal Investigator: Dr. Ronald Kiguba, Mulago Hill Road, Kampala, Uganda |
| Notes | Eligible ongoing study; protocol stage; trial registration number PACTR202009822379650 |
NCT05402254.
| Study name | Impact of a pharmacovigilance program led by advanced practice nursing (IMPACTO) |
| Methods | Hypothesis ‐ an advanced practice nursing intervention in the area of pharmacovigilance performed on patients and professionals improves the identification and reporting of suspected ADRs and improves the overall experience of hospitalised patients. |
| Participants | Based on hypothesis, intervention is aimed at patients and healthcare professionals |
| Interventions | Pharmacovigilance Program ‐ intensive nursing intervention for the identification and notification of ADEs; knowledge of the risks in the use of the drug, identification and notification of ADEs. |
| Outcomes | Number of identified ADEs |
| Starting date | 1 May 2022; estimated primary study completion date: 1 June 2023 |
| Contact information | Natalia Rodriguez, Hospital San Carlos, Madrid, Spain, 28040 |
| Notes |
ADE: adverse drug event; ADR: adverse drug reactions; cART: combination antiretroviral therapy; HCP: healthcare professionals; ME: medication error; RCT: randomised controlled trials; WHO: World Health Organization
Differences between protocol and review
Wording of the objective
In the published protocol for our review (Shalviri 2017), the review objective was "to assess the effects of different interventions implemented for improving adverse drug event reporting by health professionals". While the objective of the review has not changed, the wording of the objective has been amended slightly to comply with Cochrane Review style guidance. The objective is now "to assess the effectiveness of different interventions aimed at healthcare professionals to improve the reporting of adverse drug events".
Authorship
Liesl Nicol, Christopher Rose, and Weng Chin were added to the author team. Liesl and Weng brought Cochrane review experience to the team, and Christopher Rose brought statistical expertise to the team.
Search update
In the protocol, we stated we would search the following databases from inception:
Embase, 1974 to present, OvidSP;
Dissertations and Theses Database, 1861 to present, ProQuest;
Science Citation Index and Social Sciences Citation Index, 1975 to present, ISI Web of Knowledge;
Web of Science, Conference Proceedings Citation IndexScience, 1990 to present, (ISI Web of Knowledge);
World Health Organization Library Information System (WHOLIS/IRIS);
Virtual Health Library (VHL);
CINAHL (Cumulative Index to Nursing and Allied Health Literature), 1980 to present, EbscoHost.
In the most recent search update, we did not search AMED, CRD, Psychinfo, Agency for Healthcare Research and Quality (AHRQ), National Institute for Health and Clinical Excellence (NICE) and BASE (https://www.base-search.net/) as we did not consider them a priority in relation to the review question. A check of all of the included studies we had identified (15 at 19 October 2022) indicated that the studies are all indexed in either MEDLINE or CENTRAL: Ali 2018; Castel 2003; Chang 2017; Figueiras 2006; Hanesse 1994; Herdeiro 2008, Herdeiro 2012; Johansson 2009; Johansson 2011; Lopez‐Gonzalez 2015; McKaig 2014; Pedrós 2009; Ribeiro‐Vaz 2011; Ribeiro‐Vaz 2012; Schlienger 1999.
All included are indexed in MEDLINE, Ovid except for Hanesse 1994.
Hanesse 1994 is indexed in CENTRAL, Cochrane Library.
Additional databases to MEDLINE and CENTRAL have so far not identified unique eligible studies for inclusion.
Types of studies
In our review, we followed the updated guidance provided by the EPOC group and included all of the different types of studies prespecified in the protocol but with some additional caveats to reduce inherent bias associated with the observational studies: "For cluster‐randomised trials, non‐randomised cluster trials, and controlled before‐after studies, we only included those with at least two intervention sites and two control sites (EPOC 2013a). In addition, for controlled before‐after studies, data collection had to be contemporaneous in both the intervention and control groups during the pre‐ and post‐intervention periods, and identical measurement methods had to be used in these periods. We also included interrupted time series and repeated measures studies that had a clearly defined time point when the intervention occurred and at least three data points before and after the intervention (EPOC 2013b)."
Types of interventions
In the protocol we stated that we would classify interventions according to the following categories:
delivery arrangements;
financial arrangements;
governance arrangements; and
implementation strategies.
While it seemed to make sense to do this at the protocol stage, it did not make sense to do this at the review stage because it was not possible to cleanly classify the interventions assessed in the included studies in this way since various aspects of the assessed interventions fit into some or all of the prespecified categories.
Outcome measures
In the protocol, the primary outcomes of interest were:
number of ADE reports, including ADR reports and ME reports, submitted by health care professionals;
number of false ADE reports, including false ADR reports and false ME reports, submitted by health care professionals
The secondary outcome was:
number of detected adverse drug events by health professionals.
During the review development phase, we noted that for this clinical question there are other important outcomes, which we did not consider at the protocol stage and which were also reported by the eligible studies.
-
Number of serious ADE reports (including serious ADR reports and serious ME reports)
ADEs resulting in death, are life‐threatening, are a congenital anomaly, require hospital admission or prolongation of stay in hospital, or result in persistent or significant disability or incapacity or both
-
Number of high‐causality ADE reports (including high‐causality ADR reports and high‐causality ME reports)
ADEs with attribution of definitive or probable causality
-
Number of unexpected ADE reports (including unexpected ADR reports and unexpected ME reports)
Previously‐unknown ADEs that are not described in the summary of product characteristics
-
Number of new drug‐related ADE reports (including drug‐related ADR reports and drug‐related ME reports)
ADEs concerning medications that have been on the market for less than five years
For optimal clinical relevance and applicability, we made a post hoc decision to include the above outcomes as secondary outcomes in the review. See Secondary outcomes and Summary of findings and assessment of the certainty of the evidence.
Data collection and analysis
In the protocol we stated we would use Endnote to screen records. In the review we uploaded all records into Covidence (Covidence) and used this platform to screen records identified by the comprehensive searches.
Subgroup analysis
In the protocol we planned to conduct subgroup analyses based on the different categories of interventions (e.g. delivery arrangements, governance arrangements). In the review we separated the different categories or types of interventions into stand‐alone comparisons, for each section (see Types of interventions for details of how this was presented). We were unable to perform any of the other subgroup analyses as planned as there were not enough data in each of the comparisons.
In the protocol we also said that we would conduct subgroup analyses based on the type of ADE reported (i.e. adverse drug reaction or medication error). In the review, we separated the type of ADEs and did not attempt to combine or subgroup the data for these different types of ADEs.
Sensitivity analysis
We were unable to perform the sensitivity analyses we had planned in the protocol as there were not enough studies in any of the comparisons to justify this.
Summary of findings and assessment of the certainty of the evidence
In the protocol we planned to include the following outcomes in the SOF tables:
number of ADE reports (including ADR reports and ME reports) submitted by health professionals;
number of false adverse event reports (including false ADR reports and false ME reports) submitted by health professionals.
In the review, for optimal clinical relevance and applicability, we made a post hoc decision to assess the certainty of the evidence, and we included the following outcomes in 'Summary of findings' tables (see Summary of findings and assessment of the certainty of the evidence):
total number of ADE reports submitted, including number of ADR reports and number of ME reports;
total number of false ADE reports submitted, including number of false ADR reports and number of false ME reports;
number of serious ADE reports submitted, including serious ADR reports and serious ME reports (i.e. ADEs resulting in death; is life‐threatening; is a congenital anomaly; requires hospital admission or prolongation of stay in hospital; or results in persistent or great disability, incapacity, or both);
number of high‐causality ADE reports submitted, including high‐causality ADR reports and high‐causality ME reports (i.e. ADEs with attribution of definitive or probable causality);
number of unexpected ADE reports submitted, including unexpected ADR reports and unexpected ME reports (i.e. previously unknown ADEs that are not described in the summary of product characteristics);
number of new drug‐related ADE reports submitted, including new drug‐related ADR reports and new drug‐related ME reports (i.e. ADEs concerning medications that have been on the market for fewer than five years).
Contributions of authors
Screened titles/abstracts and full texts; extracted data and assessed risk of bias/GRADE: GS, NM, LG, WYC, KG Co‐ordinated the review development process: NM, GS, LG, RM, BY, KG, FM Wrote the review: GS, LG, CR, NM Provided general content/methods advice: RM, FM, BY, KG Analysed data: CR
GS is the guarantor of the review.
Sources of support
Internal sources
-
Tehran University of Medical Sciences’ Deputy of Research, Iran
This article is a part of the first author’s PhD dissertation sponsored by Tehran University of Medical Sciences’ Deputy of Research under project number 18360‐102‐02‐91.
External sources
-
National Institute for Health Research, UK
National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to Cochrane Effective Practice and Organisation of Care (EPOC) (which closed in March 2023). The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR or the Department of Health & Social Care.
Declarations of interest
Gloria Shalviri: none known Niayesh Mohebbi: none known Fariba Mirbaha: none known Reza Majdzadeh: none known Bahareh Yazdizadeh: none known Kheirollah Gholami: none known Liesl Grobler: NHMRC, South African Medical Research Council (employment); Associate Editor of Cochrane Effective Practice and Organisation of Care (closed March 2023) but not involved in the editorial process for this review. Chris Rose: no relevant interests; Statistical Editor of Cochrane Effective Practice and Organization of Care (closed March 2023) but not involved in the editorial process for this review. Weng Yee Chin: Norwegian Institute of Public Health (independent contractor)
New
References
References to studies included in this review
Ali 2018 {published data only}
- Ali S, Egunsola O, Al-Dossari DS, Al-Zaagi IA. Adverse drug reaction reporting in a large tertiary hospital in Saudi Arabia: results of an incentive strategy. Therapeutic Advances in Drug Safety 2018;9(10):585-90. [DOI: 10.1177/2042098618790209] [DOI] [PMC free article] [PubMed] [Google Scholar]
Castel 2003 {published data only}
- Castel JM, Figueras A, Pedros C, Laporte JR, Capella D. Stimulating adverse drug reaction reporting: effect of a drug safety bulletin and of including yellow cards in prescription pads. Drug Safety 2003;25(14):1049-55. [DOI] [PubMed] [Google Scholar]
Chang 2017 {published data only}
- Chang F, Xi Y, Zhao J, Zhang X, Lu Y. A time series analysis of the effects of financial incentives and mandatory clinical applications as interventions to improve spontaneous adverse drug reaction reporting by hospital medical staff in China. Journal of Evaluation Clinical Practice 2017;23(6):1316-21. [DOI] [PubMed] [Google Scholar]
Figueiras 2006 {published data only}
- Figueiras A, Herdeiro MT, Polonia J, Gestal-Otero JJ. An educational intervention to improve physician reporting of adverse drug reactions: a cluster-randomized controlled trial. Journal of the American Medical Association 2006;296(9):1086-93. [DOI: 10.1001/jama.296.9.1086] [DOI] [PubMed] [Google Scholar]
Hanesse 1994 {published data only}
- Hanesse B, Legras B, Royer RJ, Guillemin F, Briancon S. Adverse drug reactions: comparison of two report methods. Pharmacoepidemiology and Drug Safety 1994;3(4):223-9. [Google Scholar]
Herdeiro 2008 {published data only}
- Herdeiro MT, Polonia J, Gestal-Otero JJ, Figueiras A. Improving the reporting of adverse drug reactions: a cluster-randomized trial among pharmacists in Portugal. Drug Safety 2008;31(4):335-44. [DOI] [PubMed] [Google Scholar]
- ISRCTN45894687. An educational intervention to improve adverse drug reactions reporting: a cluster-randomised trial among Portuguese physicians and pharmacists. https://www.isrctn.com/ISRCTN45894687 (first received 7 June 2006).
Herdeiro 2012 {published data only}
- Herdeiro MT, Ribeiro-Vaz I, Ferreira M, Polonia J, Falcao A, Figueiras A. Workshop- and telephone-based interventions to improve adverse drug reaction reporting: a cluster-randomized trial in Portugal. Drug Safety 2012;35(8):655-65. [DOI] [PubMed] [Google Scholar]
Johansson 2009 {published data only}
- Johansson ML, Brunlof G, Edward C, Wallerstedt SM. Effects of e-mails containing ADR information and a current case report on ADR reporting rate and quality of reports. European Journal of Clinical Pharmacology 2009;65(5):511-4. [DOI] [PubMed] [Google Scholar]
Johansson 2011 {published data only}
- Johansson ML, Hagg S, Wallerstedt SM. Impact of information letters on the reporting rate of adverse drug reactions and the quality of the reports: a randomized controlled study. BMC Clinical Pharmacology 2011;11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lopez‐Gonzalez 2015 {published data only}
- ISRCTN91140684. An educational intervention to improve adverse drug reactions reporting among Galician physicians. https://www.isrctn.com/ISRCTN91140684 (first received 7 May 2009).
- Lopez-Gonzalez E, Herdeiro MT, Pineiro-Lamas M, Figueiras A, Grephepi group. Effect of an educational intervention to improve adverse drug reaction reporting in physicians: a cluster randomized controlled trial. Drug Safety 2015;38(2):189-96. [DOI] [PubMed] [Google Scholar]
McKaig 2014 {published data only}
- McKaig D, Collins C, Elsaid KA. Impact of a reengineered electronic error-reporting system on medication event reporting and care process improvements at an urban medical center. Joint Commission Journal on Quality & Patient Safety 2014;40(9):398-407. [DOI] [PubMed] [Google Scholar]
Pedrós 2009 {published data only}
- Pedrós C, Vallano A, Cereza G, Mendoza-Aran G, Agusti A, Aguilera C, et al. An intervention to improve spontaneous adverse drug reaction reporting by hospital physicians: a time series analysis in Spain. Drug Safety 2009;32(1):77-83. [DOI] [PubMed] [Google Scholar]
Ribeiro‐Vaz 2011 {published data only}
- Ribeiro-Vaz I, Herdeiro MT, Polonia J, Figueiras A. Strategies to increase the sensitivity of pharmacovigilance in Portugal [Estratégias para aumentar a sensibilidade da farmacovigilância em Portugal]. Revista Saude Publica 2011;45(1):129-35. [DOI] [PubMed] [Google Scholar]
Ribeiro‐Vaz 2012 {published data only}
- Ribeiro-Vaz I, Santos Cda, Costa-Pereira A, Cruz-Correia R. Promoting spontaneous adverse drug reaction reporting in hospitals using a hyperlink to the online reporting form: an ecological study in Portugal. Drug Safety 2012;33(5):387-94. [DOI] [PubMed] [Google Scholar]
Schlienger 1999 {published data only}
- Schlienger RG, Luscher TF, Schoenenberger RA, Haefeli WE. Academic detailing improves identification and reporting of adverse drug events. Pharmacy World & Science 1999;21(3):110-5. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Aldeyab 2016 {published data only}
- Aldeyab MA, Noble SC, Cuthbert M, Maxwell S, Dear J, Boyter A. Assessment of the impact of the Scottish public health campaign on patient reporting of adverse drug reactions. Drugs and Therapy Perspectives 2016;32(5):209-18. [Google Scholar]
Anbalagan 2019 {published data only}
- Anbalagan K, Shanmugasundaram P. A non-randomized interventional study to promote the knowledge, attitude and practices of community pharmacists towards pharmacovigilance. International Journal of Research in Pharmaceutical Sciences 2019;10(4):3286-92. [Google Scholar]
Åserød 2017 {published data only}
- Åserød H, Babic A. Designing a mobile system for safety reporting of arthroplasty adverse events. In: In: Eskola H, Väisänen O, Viik J, Hyttinen J (eds) EMBEC & NBC 2017. EMBEC NBC 2017 2017. IFMBE Proceedings. Vol. 65. Singapore: Springer, 2018:571-4. [DOI: ]
Aspinall 2002 {published data only}
- Aspinall MB, Whittle J, Aspinall SL, Maher Jr RL, Good CB. Improving adverse-drug-reaction reporting in ambulatory care clinics at a Veterans Affairs Hospital. American Journal of Health-System Pharmacy 2002;59(9):841-5. [DOI] [PubMed] [Google Scholar]
Avong 2018 {published data only}
- Avong YK, Jatau B, Gurumnaan R, Danat N, Okuma J, Usman I, et al. Addressing the under-reporting of adverse drug reactions in public health programs controlling HIV/AIDS, Tuberculosis and Malaria: a prospective cohort study. PLoS One 2018;13(8):e0200810. [DOI: doi: 10.1371/journal.pone.0200810. PMID: 30133453; PMCID: PMC6104922.] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bäckström 2002 {published data only}
- Bäckström M, Mjorndal T, Dahlqvist R. Spontaneous reporting of adverse drug reactions by nurses. Pharmacoepidemiology and Drug Safety 2002;11(8):647-50. [DOI] [PubMed] [Google Scholar]
Bäckström 2006 {published data only}
- Bäckström M, Mjorndal T. A small economic inducement to stimulate increased reporting of adverse drug reactions--a way of dealing with an old problem? European Journal of Clinical Pharmacology 2006;62(5):381-5. [DOI: ] [DOI] [PubMed] [Google Scholar]
Belhekar 2022 {published data only}
- Belhekar MN, Dhorajiwala SS. A prospective comparative study to evaluate the impact of educational interventions on knowledge, attitude, and practice about pharmacovigilance among nurses. National Journal of Physiology, Pharmacy and Pharmacology 2022;12(8):1164-70. [Google Scholar]
Bracchi 2005 {published data only}
- Bracchi RC, Houghton J, Woods FJ, Thomas S, Smail SA, Routledge PA. A distance-learning programme in pharmacovigilance linked to educational credits is associated with improved reporting of suspected adverse drug reactions via the UK yellow card scheme. British Journal of Clinical Pharmacology 2005;60(2):221-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Candore 2022 {published data only}
- Candore G, Monzon S, Slattery J, Piccolo L, Postigo R, Xurz X, Strauss S, Arlett P. The impact of mandatory reporting of non-serious safety reports to EudraVigilance on the detection of adverse reactions. Drug Safety 2022;45(1):83-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cano‐Sandoval 2020 {published data only}
- Cano-Sandoval MA, Lopez-Armas GC, Perfecto-Avalos Y, Vazquez-Alvarez AO, Brennan-Bourdon LM. Opportunities to improve the electronic reporting system for adverse drug reactions in Mexico: a comparative evaluation with the United States of America and the European Union. Pharmacoepidemiology and Drug Safety 2020;29(11):1523-6. [DOI] [PubMed] [Google Scholar]
Capucho 2008 {published data only}
- Capucho H, Reis L, Siqueira S, Pereira L. Strategies impact to increase the reporting of pharmacovigilance in a Brazilian university hospital. Drug Safety 2008;31(10):885. [Google Scholar]
Chatas 1990 {published data only}
- Chatas CA, Vinson B. Program for improving adverse drug reaction reporting. American Journal of Hospital Pharmacy 1990;47:155-7. [PubMed] [Google Scholar]
Clarkson 2001 {published data only}
- Clarkson A, Ingleby E, Choonara I, Bryan P, Arlett P. A novel scheme for the reporting of adverse drug reactions. Archives of Disease in Childhood 2001;84:337-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Colodny 1999 {published data only}
- Colodny L, Spillane J. Toward increased reporting of adverse drug reactions. Hospital Pharmacy 1999;34(10):1179-85. [Google Scholar]
Correa 2019 {published data only}
- Correa E, Jauregui OI, Frangella J, Peuchot V, Otero C, Luna D. Adverse drug event reporting rates after the implementation of an EHR integrated reporting system. Studies in Health Technology and Informatics 2019;21(264):1652-3. [DOI] [PubMed] [Google Scholar]
Costello 2007 {published data only}
- Costello JL, Torowicz DL, Yeh TS. Effects of a pharmacist-led pediatrics medication safety team on medication-error reporting. American Journal of Health-System Pharmacy 2007;64(13):1422-6. [DOI] [PubMed] [Google Scholar]
Deslandes 2022 {published data only}
- Deslandes PN, Bracchi R, Jones K, Haines KE, Carey E, Adams A, et al. Changes in suspected adverse drug reaction reporting via the yellow card scheme in Wales following the introduction of a National Reporting Indicator. British Journal of Clinical Pharmacology 2022;88(8):3829-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fang 2017 {published data only}
- Fang H, Lin X, Zhang J, Hong Z, Sugiyama K, Nozaki T, et al. Multifaceted interventions for improving spontaneous reporting of adverse drug reactions in a general hospital in China. BMC Pharmacology and Toxicology 2017;18(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fracchiolla 2018 {published data only}
- Fracchiolla NS, Artuso S, Cortelezzi A, Pelizzari AM, Tozzi P, Bonfichi M, et al. FarmaREL: an Italian pharmacovigilance project to monitor and evaluate adverse drug reactions in haematologic patients. Hematological Oncology 2018;36(1):299-306. [DOI] [PubMed] [Google Scholar]
Gony 2010 {published data only}
- Gony M, Badie K, Sommet A, Jacquot J, Baudrin D, Gauthier P, etal. Improving adverse drug reaction reporting in hospitals: results of the French Pharmacovigilance in Midi-Pyrenees region (PharmacoMIP) network 2-year pilot study. Drug Safety 2010;33(5):409-16. [DOI] [PubMed] [Google Scholar]
Haramburu 1988 {published data only}
- Haramburu F, Pere JC, Begaud B, Albin H. Has mandatory reporting changed the rate of adverse drug reaction spontaneous reports? [Declaration obligatoire des effets indesirables des medicaments: le decret a-t-il modifie la notification spontanee?]. Therapie 1988;43(6):493-6. [PubMed] [Google Scholar]
Haw 2011 {published data only}
- Haw C, Cahill C. A computerized system for reporting medication events in psychiatry: the first two years of operation. Journal of Psychiatric and Mental Health Nursing 2011;18(4):308-15. [DOI] [PubMed] [Google Scholar]
He 2018 {published data only}
- He W, Yao D, Hu Y, Dai H. Analysis of a pharmacist-led adverse drug event management model for pharmacovigilance in an academic medical center hospital in China. Therapeutics and Clinical Risk Management 2018;30(14):2139-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Iessa 2021 {published data only}
- Iessa N, Macolic Sarinic V, Ghazaryan L, Romanova N, Alemu A, Rungapiromnan W, et al. Smart Safety Surveillance (3S): multi-country experience of implementing the 3S concepts and principles. Drug Safety 2021;44(10):1085-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jacqout 2012 {published data only}
- Jacquot J, Gony M, Baudrin D, Chastel X, Montastruc JL, Bagheri H. Could we improve notification of adverse drugs reactions in hospital? Assessment of 5 years of network PharmacoMIP's activities [Peut-on ameliorer la notification des effets indesirables dans les etablissements hospitaliers? Bilan de 5 ans d'activite du reseau PharmacoMIP]. Therapie 2012;67(3):231-6. [DOI] [PubMed] [Google Scholar]
Jha 2017 {published data only}
- Jha N, Rathore DS, Shankar PR, Bhandary S, Alshakka M, Gyawali S. Knowledge, attitude and practice regarding pharmacovigilance and consumer pharmacovigilance among consumers at Lalitpur District, Nepal. Journal of Nepal Health Research Council 2017;15(1):31-7. [DOI] [PubMed] [Google Scholar]
Kronenfeld 2019 {published data only}
- Kronenfeld, N, Gamsu, S, Keidar, R, Livneh, A, Berkovitch, M, Goldman, M, Bahat, H, Shchory-Potlog, M, Ziv-Baran, T, Abu-Kishk, I. The Role of an Interventional Program for Improving Pharmacovigilance at a Pediatric Facility. Front Pharmacol 2019;10:1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lata 2004 {published data only}
- Lata PF, Mainhardt M, Johnson CA. Impact of nurse case manager-pharmacist collaboration on adverse-drug-event reporting. American Journal of Health-System Pharmacy 2004;61(5):483-7. [DOI] [PubMed] [Google Scholar]
Lee 2004 {published data only}
- Lee, S B, Schepers, G P, Goldberg, K L. Electronic adverse drug reaction reporting program. Am J Health Syst Pharm 2004;61(12):1230-1233. [DOI] [PubMed] [Google Scholar]
Li 2020 {published data only}
- Li, R, Curtis, K, Zaidi, S T R, Van, C, Castelino, R. Effect of the black triangle scheme and its online educational campaign on the quantity and quality of adverse drug event reporting in Australia: a time series analysis. Expert Opin Drug Saf 2020;19(6):747-753. [DOI] [PubMed] [Google Scholar]
Linder 2010 {published data only}
- Linder, J A, Haas, J S, Iyer, A, Labuzetta, M A, Ibara, M, Celeste, M, Getty, G, Bates, D W. Secondary use of electronic health record data: spontaneous triggered adverse drug event reporting. Pharmacoepidemiol Drug Saf 2010;19(12):1211-1215. [DOI] [PubMed] [Google Scholar]
Lynn 2010 {published data only}
- Lynn, R M, Riding, K, McIntosh, N. The use of electronic reporting to aid surveillance of ADRs in children: a proof of concept study. Arch Dis Child 2010;95(4):262-265. [DOI] [PubMed] [Google Scholar]
Marquez 2016 {published data only}
- Marquez ST, Herdeiro MT, Vaz IR. An educational intervention to improve nurses reporting of adverse drug reactions. Pharmacoepidemiology and Drug Safety 2016;25:403. [DOI: ] [Google Scholar]
- Marquez, S, Herdeiro, MT, Ribeiro-Vaz, I. An Educational Intervention to improve nurses reporting of Adverse drug reactions. Clinical Therapeutics 2015;37:e57. [Google Scholar]
Massah 2021 {published data only}
- Massah L, Mohammadi R, Namnabati M. Improvement of medication error reporting: An applied motivation program in pediatric units. J 2021;10(1):189. [DOI] [PMC free article] [PubMed] [Google Scholar]
McGettigan 1997 {published data only}
- McGettigan P, Golden J, Conroy RM, Arthur N, Feely J. Reporting of adverse drug reactions by hospital doctors and the response to intervention. British Journal of Clinical Pharmacology 1997;44(1):98-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Menat 2021 {published data only}
- Menat, U, Desai, C K, Panchal, J R, Shah, A N. An evaluation of trigger tool method for adverse drug reaction monitoring at a tertiary care teaching hospital. Perspectives in Clinical Research 2021;12(1):33-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Morales 2016 {published data only}
- Morales Rios O, Jasso Gutierrez L, Talavera JO, Tellez-Rojo MM, Olivar Lopez V, Garduno Espinosa J, etal. A comprehensive intervention for adverse drug reactions identification and reporting in a Pediatric Emergency Department. International Journal of Clinical Pharmacy 2016;38(1):80-7. [DOI] [PubMed] [Google Scholar]
Morgan 1990 {published data only}
- Morgan SA, Frank JT. Development of a videotape on adverse drug reactions. American Journal of Hospital Pharmacy 1990;47(6):1340-1342. [PubMed] [Google Scholar]
Mwamwitwa 2022 {published data only}
- Mwamwitwa KW, Fimbo AM, Bukundi EM, Nkayamba AF, Buma D, Muro EP, Maganda BA, Shewiyo DH, Shearer MC, Smith AD, Kaale EA. Effectiveness of a structured stimulated spontaneous safety monitoring of medicines reporting program in strengthening pharmacovigilance system in Tanzania. Sci 2022;12(1):16131. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT02087293 {published data only}
- NCT02087293. e-Pharmacovigilance II - surveillance for safety and effectiveness - Calling for Earlier Detection of Adverse Reactions (CEDAR). https://ClinicalTrials.gov/show/NCT02087293 (first received 11 March 2014).
Ng 2020 {published data only}
- Ng TM, Teo CJ, Heng ST, Chen YR, Lim WP, Teng CB. Impact of round-the-clock pharmacist inpatient medication chart review on medication errors. JACCP Journal of the American College of Clinical Pharmacy 2020;3(8):1437-43. [Google Scholar]
Opadeyi 2019 {published data only}
- Opadeyi AO, Fourrier-Reglat A, Isah AO. Educational intervention to improve the knowledge, attitude and practice of healthcare professionals regarding pharmacovigilance in South-South Nigeria. Therapeutic Advances in Drug Safety 2019;10:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ortega 2008 {published data only}
- Ortega A, Aguinagalde A, Lacasa C, Aquerreta I, Fernández-Benítez M, Fernández LM. Efficacy of an adverse drug reaction electronic reporting system integrated into a hospital information system. Annals of Pharmacotherapy 2008;42:1491-6. [DOI] [PubMed] [Google Scholar]
Potlog 2020 {published data only}
- Potlog Shchory M, Goldstein LH, Arcavi L, Shihmanter R, Berkovitch M, Levy A. Increasing adverse drug reaction reporting-how can we do better? PLoS ONE 2020;15(8):e0235591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Praveen 2021 {published data only}
- Praveen KS, Ramesh B, Sachin PR, Mubashir A. Impact of an awareness program on adverse drug reaction reporting among community pharmacist. European Journal of Clinical Pharmacy 2021;23(2):133-8. [Google Scholar]
Reumerman 2021a {published data only}
- Reumerman M, Tichelaar J, Van Eekeren R, Van Puijenbroek EP, Richir MC, Van Agtmael MA. The potential of training specialist oncology nurses in real-life reporting of adverse drug reactions. European Journal of Clinical Pharmacology 2021;77(10):1531-42. [DOI: 10.1007/s00228-021-03138-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Reumerman 2021b {published data only}
- Reumerman M, Tichelaar J, Richir MC, Van Agtmael MA. Medical students as adverse drug event managers, learning about side effects while improving their reporting in clinical practice. Naunyn-Schmiedeberg's Archives of Pharmacology 2021;394:1467–6. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rogers 1989 {published data only}
- Rogers SA. Strategies to promote physician reporting of adverse drug events based on the experience of an FDA-sponsored project in Maryland. Journal of Clinical Research and Drug Development 1989;3(1):29-37. [Google Scholar]
Roy 2018 {published data only}
- Roy R, Ma J. Impact of a policy change on pharmacists' reporting of adverse drug reactions. Canadian Journal of Hospital Pharmacy 2018;71(4):227-33. [PMC free article] [PubMed] [Google Scholar]
Rubin 2019 {published data only}
- Rubin DS, Pesyna C, Jakubczyk S, Liao C, Tung A. Introduction of a mobile adverse event reporting system Is associated with participation in adverse event reporting. American Journal of Medical Quality 2019;34(1):30-5. [DOI] [PubMed] [Google Scholar]
Salcedo de Diego 2015 {published data only}
- Salcedo de Diego I Serrano Gallardo P, Ruiz Antorán B, De Andrés Gimeno B, Revuelta Zamorano M, Avendaño Solá C. Impact of a multicomponent intervention on registered nurses to increase reporting of adverse drug reactions and medication errors. Clinical Therapeutics 2015;37(8):e153. [Google Scholar]
Sánchez 2014 {published data only}
- Sánchez I, Amador C, Plaza JC, Correa G, Amador R. Assessment of an active pharmacovigilance system carried out by a pharmacist. Revista Médica de Chile 2014;142(8):998-1005. [DOI] [PubMed] [Google Scholar]
Sanko 2020 {published data only (unpublished sought but not used)}
- Sanko JS, McKay M. Participation in a system-thinking simulation experience changes adverse event reporting. Simulation in Healthcare 2020;15(3):167-71. [DOI] [PubMed] [Google Scholar]
Schindler 2019 {published data only}
- Schindler TM, Bridge H. For safety’s sake part 2: reporting of adverse event data. AMWA Journal 2019;34(1):32-7. [Google Scholar]
Segec 2021 {published data only}
- Segec A, Slattery J, Morales DR, Januskiene J, Kurz X, Arlett P. Does additional monitoring status increase the reporting of adverse drug reactions? An interrupted time series analysis of EudraVigilance data. Pharmacoepidemiology and Drug Safety 2021;30:350– 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sonowa 2020 {published data only}
- Sonowa S, Desai CK, Panchal JR. Impact of certain educational interventions on adverse drug reaction reporting by nursing health professionals at a tertiary care hospital. Asian Journal of Pharmaceutical and Clinical Research 2020;13(6):175-80. [Google Scholar]
Srikanth 2019 {published data only}
- Srikanth MS, Adepu R. Assessment of structured educational intervention and barriers of community pharmacists towards adverse drug reaction reporting and monitoring in South Indian district. Drug Invention Today 2019;12:131-41. [Google Scholar]
Tabali 2009 {published data only}
- Tabali M, Jeschke E, Bockelbrink A, Witt CM, Willich SN, Ostermann T, et al. Educational intervention to improve physician reporting of adverse drug reactions (ADRs) in a primary care setting in complementary and alternative medicine. BMC Public Health 2009;9(1):274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Terblanche 2018 {published data only}
- Terblanche A, Meyer JC, Godman B, Summers RS. Impact of a pharmacist-driven pharmacovigilance system in a secondary hospital in the Gauteng Province of South Africa. Hospital Practice (1995) 2018;46(4):221-8. [DOI] [PubMed] [Google Scholar]
Touchette 2012 {published data only}
- Touchette DR, Masica AL, Dolor RJ, Schumock GT, Choi YK, Kim Y. Safety-focused medication therapy management: a randomized controlled trial. Journal of the American Pharmacists Association: JAPhA 2012;52(5):603-12. [DOI] [PubMed] [Google Scholar]
Varallo 2015 {published data only}
- Varallo FR. Educational intervention in pharmacovigilance for hospital health professionals. https://ClinicalTrials.gov/show/NCT02134587 (first received 5 May 2014). [NCT02134587]
Weant 2010 {published data only}
- Weant KA, Humphries RL, Hite K, Armitstead JA. Effect of emergency medicine pharmacists on medication-error reporting in an emergency department. American Journal of Health-System Pharmacy 2010;67(21):1851-5. [DOI] [PubMed] [Google Scholar]
Welsh 1996 {published data only}
- Welsh CH, Pedot R, Anderson RJ. Use of morning report to enhance adverse event detection. Journal of General Internal Medicine 1996;11(8):454-60. [DOI] [PubMed] [Google Scholar]
Wengrove 2021 {published data only}
- Wengrove McCracken A, Grimes M, Sherman B, Decker K, Hirsch D, Pham A, et al. Implementation of standard nursing workflow for adverse drug reaction reporting within health system ambulatory infusion services. Infusion 2021;27(1):12-7. [Google Scholar]
Wentzell 2017 {published data only}
- Wentzell J, Nguyen T, Bui S, MacDonald E. Pharmacy student facilitation of reporting of adverse drug reactions in a hospital. Canadian Journal of Hospital Pharmacy 2017;70(4):276-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Winstanley 1989 {published data only}
- Winstanley PA, Irvin LE, Smith JC, Orme ML, Breckenridge AM. Adverse drug reactions: a hospital pharmacy-based reporting scheme. British Journal of Clinical Pharmacology 1989;28(1):113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Xu 2014 {published data only}
- Xu C, Li G, Ye N, Lu Y. An intervention to improve inpatient medication management: a before and after study. Journal of Nursing Management 2014;22(3):286-94. [DOI] [PubMed] [Google Scholar]
Yen 2010 {published data only}
- Yen Y-H, Kuo L-N, Hsu M-H, Li Y-C, Cheng K-J. Evaluation of the electronic adverse drug event management system. Journal of Experimental & Clinical Medicine 2010;2(6):287-91. [Google Scholar]
References to studies awaiting assessment
Biagi 2013 {published data only}
- Biagi C, Montanaro N, Buccellato E, Roberto G, Vaccheri A, Motola D. Underreporting in pharmacovigilance: an intervention for Italian GPs (Emilia-Romagna region). European Journal of Clinical Pharmacology 2013;69(2):237-44. [DOI: 10.1007/s00228-012-1321-7] [DOI] [PubMed] [Google Scholar]
Kim 2012 {published data only}
- Kim M, McPherson ML, Smith SW, Kopochis E. Impact of an electronic medication error reporting system in a hospice organization. Journal of Pain and Symptom Management 2012;43(2):449. [DOI: ] [Google Scholar]
Kumar 2021 {published data only}
- Acceptability and feasibility of mobile app in adverse drug reactions (ADRs) reporting in Revised National Tuberculosis Control Program (RNTCP) centres. https://trialsearch.who.int/Trial2.aspx?TrialID=CTRI/2021/07/035163 (first received 27 July 2021).
Lan 2022 {published data only}
- Lan T, Wang H, Li X, Yin H, Shao D, Jiang Y, Yu Q. The effect of clinical pharmacists' intervention in adverse drug reaction reporting: a retrospective analysis with a 9-year interrupted time series. BMC Health Services Research 2022;22(1):925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lander 2013 {published data only}
- Lander AR, Blicher TM, Jimenez-Solem E, Jespersen M, Kampmann JP, Christensen HR. Introducing an adverse drug event manager. European Journal of Hospital Pharmacy: Science and Practice 2013;20:78-81. [Google Scholar]
Marquez 2015 {published data only}
- Marquez S, Herdeiro MT, Ribeiro-Vaz I. An educational intervention to improve nurses reporting of adverse drug reactions. Clinical Therapeutics 2015;37(8):e57. [Google Scholar]
Opadeyi 2021 {published data only}
- Opadeyi AO, Fourrier-Reglat A, Isah AO. Impact of an educational intervention on adverse drug reaction reporting in tertiary hospitals in South-South Nigeria. West African Journal of Medicine 2021;38(7):634-45. [PubMed] [Google Scholar]
Sawangjit 2022 {published data only}
- Effectiveness of TaWai mobile system in Lao version: a cluster randomized controlled trial in Lao, PDR. https://trialsearch.who.int/Trial2.aspx?TrialID=TCTR20220607002 (first received 7 June 2022).
References to ongoing studies
Hutchinson 2020 {published data only}
- Hutchinson AM, Brotto V, Chapman A, Sales AE, Mohebbi M, Bucknall TK. Use of an audit with feedback implementation strategy to promote medication error reporting by nurses. Journal of Clinical Nursing 2020;29(21-2):4180-93. [DOI] [PubMed] [Google Scholar]
- Hutchinson AM, Sales AE, Brotto V, Bucknall TK. Implementation of an audit with feedback knowledge translation intervention to promote medication error reporting in health care: a protocol. Implementation Science 2015;10:70. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kiguba 2022 {published data only}
- Correction: Effectiveness of the med Safety mobile application in improving adverse drug reaction reporting by healthcare professionals in Uganda: a protocol for a pragmatic cluster-randomised controlled trial. BMJ Open 2024;14:e061725corr1. [DOI: 10.1136/bmjopen-2022-061725corr1] [DOI] [PMC free article] [PubMed]
- Kiguba R, Mwebaza N, Ssenyonga R, Ndagije HB, Nambasa V, Katureebe C, et al. Effectiveness of the Med Safety mobile application in improving adverse drug reaction reporting by healthcare professionals in Uganda: a protocol for a pragmatic cluster-randomised controlled trial. BMJ Open 2022;12(7):e061725. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT05402254 {published data only}
- NCT05402254. Impact of a pharmacovigilance program led by advanced practice nursing (IMPACTO). https://www.clinicaltrials.gov/study/NCT05402254 (first received 23 May 2022).
Additional references
Abstoss 2011
- Abstoss KM, Shaw BE, Owens TA, Juno JL, Commiskey EL, Niedner MF. Increasing medication error reporting rates while reducing harm through simultaneous cultural and system-level interventions in an intensive care unit. BMJ Quality and Safety 2011;20(11):914-22. [DOI] [PubMed] [Google Scholar]
Al Hamid 2014
- Al Hamid A, Ghaleb M, Aljadhey H, Aslanpour Z. A systematic review of hospitalization resulting from medicine-related problems in adult patients. British Journal of Clinical Pharmacology 2014;78(2):202-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andel 2012
- Andel C, Davidow SL, Hollander M, Moreno DA. The economics of health care quality and medical errors. Journal of Health Care Finance 2012;39(1):39. [PubMed] [Google Scholar]
Classen 1997
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. Journal of the American Medical Association 1997;277(4):301-6. [PubMed] [Google Scholar]
Covidence [Computer program]
- Covidence. Version accessed prior to 22 October 2024. Melbourne, Australia: Veritas Health Innovation, 2024. Available at https://www.covidence.org.
Cumby 1990
- Cumby RE, Huizinga J. Testing the autocorrelation structure of disturbances in ordinary least squares and instrumental variables regressions. Econometrica 1992;60(1):185-95. [Google Scholar]
EMA 2016
- European Medicines Agency (EMA). Good pharmacovigilance practices. www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000345.jsp. Accessed 2 October 2016.
EndNote 2013 [Computer program]
- EndNote. Version X4. Thomson Reuters, 2013.
EPOC 2013a
- Effective Practice and Organisation of Care (EPOC). What study designs should be included in an EPOC review? EPOC Resources for review authors. Oslo: Norwegian Knowledge Centre for the Health Services; 2013. epoc.cochrane.org/epoc-specific-resources-review-authors Accessed prior to 28 February 2017.
EPOC 2013b
- Effective Practice and Organisation of Care (EPOC). Interrupted time series (ITS) analyses. EPOC Resources for review authors. Oslo: Norwegian Knowledge Centre for the Health Services; 2013. epoc.cochrane.org/epoc-specific-resources-review-authors Accessed prior to 28 February 2017.
EPOC 2013c
- Effective Practice and Organisation of Care (EPOC). Data collection form. EPOC resources for review authors. Oslo: Norwegian Knowledge Center for the Health Services; 2013. epoc.cochrane.org/epoc-specific-resources-review-authors Accessed prior to 28 February 2017.
EPOC 2013d
- Effective Practice and Organisation of Care (EPOC). EPOC worksheets for preparing a Summary of Findings (SoF) table using GRADE. EPOC Resources for review authors. Oslo: Norwegian Knowledge Centre for the Health Services; 2013. epoc.cochrane.org/epoc-specific-resources-review-authors Accessed prior to 28 February 2017.
EPOC 2015b
- Effective Practice and Organisation of Care (EPOC). Suggested risk of bias criteria for EPOC reviews. EPOC Resources for review authors. Oslo: Norwegian Knowledge Centre for the Health Services; 2015. epoc.cochrane.org/epoc-specific-resources-review-authors Accessed prior to 28 February 2017.
Ernst 2001
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. Journal of the American Pharmaceutical Association 2001;41(2):192-199. [DOI] [PubMed] [Google Scholar]
Feely 1990
- Feely J, Moriarty S, O’Connor P. Stimulating reporting of adverse drug reactions by using a fee. British Medical Journal 1990;300(6716):22-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Figueiras 2001
- Figueiras A, Tato F, Fontaiñas J, Takkouche B, Gestal-Otero JJ. Physicians’ attitudes towards voluntary reporting of adverse drug events. Journal of Evaluation in Clinical Practice 2001;7(4):347-54. [DOI] [PubMed] [Google Scholar]
Fornasier 2018
- Fornasier G, Francescon S, Leone R, Baldo P. An historical overview over Pharmacovigilance. International Journal of Clinical Pharmacy 2018;40:744–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gad 2009
- Gad SC. Postmarketing safety evaluation: monitoring, assessing, and reporting of adverse drug responses. In: Drug Safety Evaluation. Second edition. Hoboken, New Jersey: John Wiley & Sons, 2009:943-44. [Google Scholar]
Gonzalez‐Gonzalez 2013
- Gonzalez-Gonzalez C, Lopez-Gonzalez E, Herdeiro MT, Figueiras A. Strategies to improve adverse drug reactions reporting: a critical and systematic review. Drug Safety 2013;36(5):317-28. [DOI] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- GRADEpro GDT. Version Accessed prior to 28 February 2017. Hamilton (ON): GRADE Working Group, McMaster University, 2015.
Guyatt 2008
- Guyatt GH, Oxman AD, Vist G, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al GRADE Working Group. Rating quality of evidence and strength of recommendations GRADE: An emerging consensus on rating quality of evidence and strength of recommendations. British Medical Journal 2008;336(7650):924-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gyllensten 2013
- Gyllensten H, Rehnberg C, Jönsson AK, Petzold M, Carlsten A, Andersson Sundell K. Cost-of-illness of patient-reported adverse drug events – a population-based cross-sectional survey. BMJ Open 2013;3(6):e002574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hazell 2006
- Hazell L, Shakir SAW. Under reporting of adverse drug reactions: a systematic review. Drug Safety 2006;29(5):385-96. [DOI] [PubMed] [Google Scholar]
Higgins 2024
- Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.5 (updated August 2024). Cochrane 2024:Available from www.training.cochrane.org/handbook.
Johnson 1995
- Johnson JA, Bootman JL. Drug-related morbidity and mortality. A cost-of-illness model. Archives of Internal Medicine 1995;155(18):1949-56. [PubMed] [Google Scholar]
Khalili 2020
- Khalili M, Mesgarpour B, Sharifi H, Daneshvar Dehnavi S, Haghdoost AA. Interventions to improve adverse drug reaction reporting: a scoping review. Pharmacoepidemiology and Drug Safety 2020;29(9):965-92. [DOI] [PubMed] [Google Scholar]
Kunac 2014
- Kunac DL, Tatley MV, Seddon ME. A new web-based Medication Error Reporting Programme (MERP) to supplement pharmacovigilance in New Zealand--findings from a pilot study in primary care. New Zealand Medical Journal 2014;127(1401):69-81. [PubMed] [Google Scholar]
Lazarou 1998
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients. Journal of the American Medical Association 1998;279(15):1200-5. [DOI] [PubMed] [Google Scholar]
Li 2019
- Li R, Zaidi STR, Chen T, Castelino R. Effectiveness of interventions to improve adverse drug reaction reporting by healthcare professionals over the last decade: a systematic review. Pharmacoepidemiology and Drug Safety 2019;29:1-8. [DOI] [PubMed] [Google Scholar]
Liberati 2009
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Medicine 2009;6:e1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Linden 2015
- Linden A. Conducting interrupted time-series analysis for single- and multiple-group comparisons. Stata Journal 2015;15(2):480-500. [Google Scholar]
Lindquist 2008
- Lindquist M. The WHO Global ICSR database system: basic facts. Drug Information Journal 2008;42:409-19. [Google Scholar]
McGettigan 1997
- McGettigan P, Golden J, Conroy RM, Arthur N, Feely J. Reporting of adverse drug reactions by hospital doctors and the response to intervention. British Journal of Clinical Pharmacology 1997;44(1):98-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mirbaha 2015
- Mirbaha F, Shalviri G, Yazdizadeh B, Gholami K, Majdzadeh R. Perceived barriers to reporting adverse drug events in hospitals: a qualitative study using theoretical domains framework approach. Implementation Science 2015;10:110. [DOI: 10.1186/s13012-015-0302-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Molokhia 2009
- Molokhia M, Tanna S, Bell D. Improving reporting of adverse drug reactions: systematic review. Clinical Epidemiology 2009;1:75-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moride 1997
- Moride Y, Haramburu F, Requejo A, Bégaud B. Under-reporting of adverse drug reactions in general practice. British Journal of Clinical Pharmacology 1997;43(2):177-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCCMERP 2016
- National Coordinating Council for Medication Error Reporting and Prevention (NCCMERP). What is a medication error? www.nccmerp.org. Accessed 28 February 2024.
O'Brien 2007
- O'Brien MA, Rogers S, Jamtvedt G, Oxman AD, Odgaard‐Jensen J, Kristoffersen DT, et al. Educational outreach visits: effects on professional practice and health care outcomes. Cochrane Database of Systematic Reviews 2007, Issue 4. Art. No: CD000409. [DOI: 10.1002/14651858.CD000409.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ortega 2008
- Ortega A, Aguinagalde A, Lacasa C, Aquerreta I, Fernández-Benítez M, Fernández LM. Efficacy of an adverse drug reaction electronic reporting system integrated into a hospital information system. Annals of Pharmacotherapy 2008;42(10):1491-6. [DOI] [PubMed] [Google Scholar]
Pagotto 2013
- Pagotto C, Varallo F, Mastroianni F. Impact of educational interventions on adverse drug events reporting. International Journal of Technology Assessment in Health Care 2013;29(4):410-17. [DOI] [PubMed] [Google Scholar]
Pal 2013
- Pal S, Duncombe C, Falzon D, Olsson S. WHO strategy for collecting safety data in public health programmes: complementing spontaneous reporting systems. Drug Safety 2013;36(2):75-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Paudyal 2020
- Paudyal V, Al-Hamida A, Bowena M, Abdul Hadi M, Hasan SS, Jalala Z, Stewart D. Interventions to improve spontaneous adverse drug reaction reporting by healthcare professionals and patients: systematic review and meta-analysis. Expert Opinion on Drug Safety 2020;19(9):1173–91. [DOI] [PubMed] [Google Scholar]
Pirmohamed 2004
- Pirmohamed M, James S, Meakin S, Green C, Scott AK, Walley TJ, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18,820 patients. British Medical Journal 2004;329(7456):15-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ribeiro‐Vaz 2016
- Ribeiro-Vaz I, Silva AM, Costa Santos C, Cruz-Correia R. How to promote adverse drug reaction reports using information systems - a systematic review and meta-analysis. BMC Medical Informatics and Decision Making 2016;16(27):1-10. [DOI: 10.1186/s12911-016-0265-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rücker 2017
- Rücker G, Cates CJ, G Schwarzer. Methods for including information from multi-arm trials in pairwise meta-analysis. Research Synthesis Methods 2017;8(4):392-403. [DOI] [PubMed] [Google Scholar]
Shalviri 2009
- Shalviri G, Valadkhani M, Dinarvand R. Ten years pharmacovigilance activities in Iran. Iranian Journal of Public Health 2009;38(Suppl 1):162-5. [Google Scholar]
Shalviri 2012
- Shalviri G, Yousefian S, Gholami K. Adverse events induced by ceftriaxone: a 10-year review of reported cases to Iranian Pharmacovigilance Centre. Journal of Clinical Pharmacy and Therapeutics 2012;37(4):448-51. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Sutton AJ, Ioannidis JP, Terrin N, Jones DR, Lau J, et al. Recommendations for examining and interpreting funnel plot asymmetry in meta-analyses of randomised controlled trials. British Medical Journal 2011;343:d4002. [DOI: 10.1136/bmj.d4002] [DOI] [PubMed] [Google Scholar]
Uppsala Monitoring Center
- Uppsala Monitoring Center. Glossary of terms used in pharmacovigilance. www.who-umc.org. Accessed 28 January 2024.
Varallo 2014
- Varallo FR, Guimarães S de O, Abjaude SA, Mastroianni P de C. Causes for the underreporting of adverse drug events by health professionals: a systematic review. Revista da Escola de Enfermagem da USP 2014;48(4):739-47. [DOI] [PubMed] [Google Scholar]
Wester 2008
- Wester K, Jönsson AK, Spigset O, Druid H, Hägg S. Incidence of fatal adverse drug reactions: a population based study. British Journal of Clinical Pharmacology 2008;65(4):573-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2006
- World Health Organization. The safety of medicines in public health programmes:pharmacovigilance an essential tool. www.who.int/medicines/areas/quality_safety/safety_efficacy/Pharmacovigilance_B.pdf?ua=1 Accessed 2 October 2016. [ISBN 92 4 159391 1]
WHO 2014
- World Health Organization. Reporting and learning systems for medication errors: the role of pharmacovigilance centres. apps.who.int/iris/bitstream/10665/137036/1/9789241507943_eng.pdf?ua=1 Accessed prior to 28 February 2017. [ISBN 978 92 4 150794 3]
WHO 2016
- World Health Organization. Annual meeting of representatives of National Pharmacovigilance Centres participating in the WHO Programme for International Drug Monitoring. www.who.int//medicines/regulation/medicines-safety/npvc-meeting/en/ Accessed 2 October 2016.
Wilson 2012
- Wilson RM, Michel P, Olsen S, Gibberd RW, Vincent C, El-Assady R, et al. Patient safety in developing countries: retrospective estimation of scale and nature of harm to patients in hospital. British Medical Journal 2012;344:e832. [DOI: ] [DOI] [PubMed] [Google Scholar]
World Bank
- World Bank. World Development Indicators (GDP). http://data.worldbank.org/indicator/NY.GDP.MKTP.CD 2014.
References to other published versions of this review
Shalviri 2017
- Shalviri G, Mohebbi N, Mirbaha F, Majdzadeh R, Yazdizadeh B, Gholami K. Improving adverse drug event reporting by health care professionals. Cochrane Database of Systematic Reviews 2017, Issue 3. Art. No: CD012594. [DOI: 10.1002/14651858.CD012594] [DOI] [PMC free article] [PubMed] [Google Scholar]