

Abstract
Mimivirus bradfordmassiliense (Mimivirus) is a giant virus that infects Acanthamoeba species – opportunistic human pathogens. Long- and short-read sequencing were used to generate a de novo transcriptome of the host and followed the dynamics of both host and virus transcriptomes over the course of infection. The assembled transcriptome of the host included 22,604 transcripts and 13,043 genes, with N50 = 2,372 nucleotides. Functional enrichment analysis revealed major changes in the host transcriptome, namely, enrichment in downregulated genes associated with cytoskeleton homeostasis and DNA replication, repair, and nucleotide synthesis. These modulations, together with those implicated by other enriched processes, indicate cell cycle arrest, which was demonstrated experimentally. We also observed upregulation of host genes associated with transcription, secretory pathways and, as reported here for the first time, peroxisomes and the ubiquitin-proteasome system. In Mimivirus, the early stages of infection were marked by upregulated genes related to DNA replication, transcription, translation, and nucleotide metabolism, and in later stages, enrichment in genes associated with lipid metabolism, carbohydrates, and proteases. Some of the changes observed in the amoebal transcriptome likely point to Mimivirus infection causing dismantling of host cytoskeleton and translocation of endoplasmic reticulum membranes to viral factory areas.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-76078-6.
Subject terms: Pathogens, Virology, Data acquisition, Data processing, Databases, Functional clustering, Gene ontology
Introduction
Mimivirus bradfordmassiliense (Mimivirus) is a member of the nucleo-cytoplasmatic large DNA viruses (NCLDV) clade, whose infection of Acanthamoeba spp. causes massive lysis in the amoebal population. The first giant DNA virus to be discovered, Mimivirus has a diameter of about 750 nm. Its genome, ~ 1.2 × 106 base-pair (bp) long, encodes approximately 1000 open reading frames (ORFs), many of which are orphan genes, with no detectable homologs in other species1–4.
Mimivirus’s target hosts, Acanthamoeba spp., are free-living amoebae that reside in air, soil, and water environments and are recognized as opportunistic human pathogens5–7. Indeed, Acanthamoeba polyphaga (AP) is a causative agent of several human diseases, including granulomatous amoebic encephalitis, a rare brain infection that is generally fatal, a skin ulcer in human immunodeficiency virus (HIV) patients, and Acanthamoeba keratitis—a corneal infection that can lead to loss of vision6. The latter is becoming more prevalent because of the increased use of contact lenses, which nurture amoebal growth when hygiene is inadequate.
Mimivirus enters the amoeba host cell via phagocytosis. In the phagosome, it loses its capsid and fuses with the phagosome membrane, after which the viral core enters the cytoplasm8,9. Therein, viral factories (VFs) begin to be formed and, at 5 h post infection (HPI), they fuse into a single mature VF8,10,11. Viral production occurs in the VF and, at approximately 12–14 HPI, cell lysis occurs, prompting the release of newly-formed viruses into the surrounding medium8,11.
To date, one study has described Mimivirus transcriptome dynamics during the infection cycle in Acanthamoeba castellanii (AC), another member of the Acanthamoeba genus12. However, the dynamics of the host transcriptome over the course of infection has not yet been characterized. In this study, the AP transcriptome was followed throughout the different phases of infection. Since no transcriptome was available for AP, we assembled its transcriptome de novo using PacBio long-read sequencing and Illumina sequencing, of cDNA produced from RNA isolated from uninfected AP and infected AP 1, 3, and 5 HPI corresponding to early, intermediate, and the beginning of the late stage of infection, respectively. As an example of abiotic stress, we also examined the transcriptome of AP cells 1 h after exposure to UV light. A functional enrichment analysis of AP and Mimivirus gene expression was performed at the different time points. The analysis highlights biological processes and organelles in AP whose related genes are enriched during the infection cycle, including peroxisome lipid transport, peroxisomal organization and the ubiquitin–proteasome system. Other enriched genes can be linked to the dismantling of the cytoskeleton11, translocation of ER membranes to the VF13,14, and cell-cycle arrest. The latter was observed experimentally. We also demonstrate here de novo nucleotide synthesis during Mimivirus infection, despite enrichment in host genes that downregulate nucleotide synthesis.
Results
For the de novo transcriptome assembly, we utilized PacBio Single Molecule Real-Time (SMRT) sequencing alongside Illumina next-generation sequencing (NGS). The analysis included uninfected AP samples as well as samples collected at 1, 3, and 5 h after infection. In addition, NGS sequencing was conducted on amoeba cells after exposure to UV light, allowing a comparison of the amoeba’s response to biotic and abiotic stress. AP transcriptome included 22,604 transcripts and 13,043 genes. NGS reads were employed to track transcriptome dynamics in both AP and Mimivirus functional analysis.
Functional enrichment analysis of the Mimivirus transcriptome
As infection progressed, the percentage of Mimivirus-assigned reads in the ensemble rose from 0.5 at 1 HPI, to 14.4 at 3 HPI and 23.5 at 5 HPI, while the percentage of AP assigned reads declined (Fig. 1).
Fig. 1.
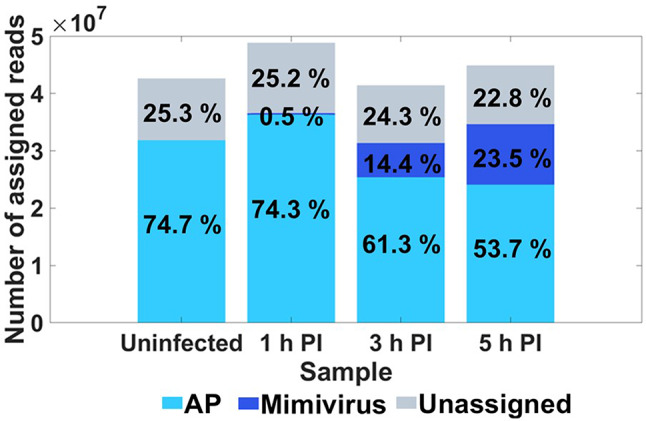
Read distribution between Acanthamoeba polyphaga (AP) and Mimivirus bradfordmassiliense (Mimivirus). Sum of total reads of AP and Mimivirus in the different samples. The percentage from total reads at a certain time point is indicated on the bars.
The Mimivirus differentially expressed genes (DEGs) are divided into three clusters (Supplementary Fig. 1). These clusters represent the expression of genes in early (208 DEGs), intermediate (274 DEGs) and late (388 DEGs) stages of infection (Fig. 2). Among the detected genes, 115 maintained approximately constant expression levels throughout the examined time points.
Fig. 2.
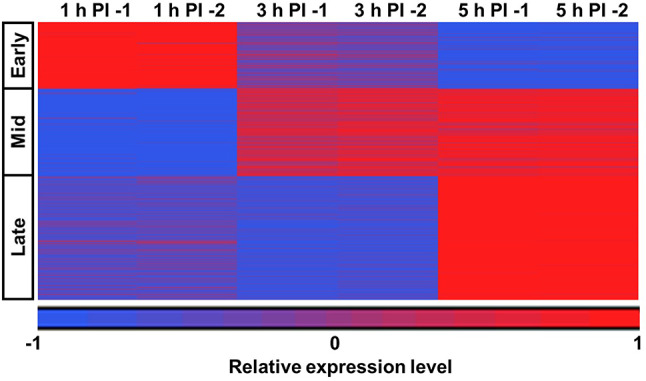
K-mean clustering of the differentially expressed genes (DEGs) of Mimivirus bradfordmassiliense (Mimivirus). Mimivirus DEGs were classified into three clusters using k-mean clustering, as was suggested by the Silhouette score (Supplementary Fig. 1). These clusters correspond to genes expressed in the early, intermediate, and late infection stages.
Since only a relatively small number of Mimivirus genes have GO annotations, we manually categorized the genes according to the top Blastx results and the literature. A statistical analysis of Mimivirus categories using a hypergeometric test indicated enriched categories in each of the time points examined (Fig. 3a).
Fig. 3.

Functional enrichment analysis. Functional enrichment analysis of Mimivirus bradfordmassiliense (Mimivirus) (a) and Acanthamoeba polyphaga (AP) (b–d). For Mimivirus, each category comprises a group of genes, with the bar size proportional to the number of genes in the category (a). For AP (b–d), the genes are annotated to each GO term, with groups of GO terms subsequently semi-automatically grouped into categories. Each category is represented by a bar whose size is proportional to the number of GO terms it includes. The functional enrichment analysis is presented with respect to the three principal GO domains: (b) biological processes; (c) molecular function; and (d) cellular components. The coloring of the bars indicates whether the category became enriched in up-regulated (red) or down-regulated (blue) genes over the course of infection. The hue represents the significance of the enrichment; in a, it is the p-value of the enriched category, and in b-d, it is based on the average p-value of the GO terms in the category.
The first cluster contained genes with the highest expression level at 1 HPI. Here, enrichment was noted for the Mimivirus ankyrin genes and TB/POZ-domain-containing proteins (Fig. 3a). The ankyrins represent the largest category in the Mimivirus transcriptome, consisting of 106 putative genes. Of these, 33 were found in the 1 HPI cluster. Ankyrin repeat-containing proteins are known to be involved in protein–protein interactions16, though no specific function of ankyrins has been demonstrated in Mimivirus.
The genes in the second cluster were highly expressed at 3 HPI, and most genes were also highly expressed at 5 HPI. Specifically, Mimivirus genes for KilA-N domain, DNA replication, transcription, nucleotide metabolism and translation were enriched (Fig. 3a), along with the corresponding AP categories (Fig. 3b–d).
The third cluster was dominated by genes that had the highest expression level at 5 HPI. These included genes encoding proteases, structural proteins, and lipid- and sugar-metabolism-related proteins (Fig. 3a). The lipid metabolism category consists of genes encoding for proteins putatively involved in lipid modification, binding, transport, and processing; the structure-related category includes, among other genes, eight putative collagen genes (collagen 1–8) present in the Mimivirus genome (L71, R196, R239, R240, R241, L668, L669 and L623).
It was reported that Mimivirus undergoes large-scale glycosylation on its fibrils17,18 and possesses various genes that encode for proteins involved in glycosylation12,18–20. In line, twenty of the 22 genes classified to the sugar metabolism category were among those assigned to the 5 HPI cluster. Many of these genes are related to glycosylation (L619, L373, R707, L140, L263, R363, L137, L138 and L193).
Functional enrichment analysis of the AP transcriptome
Illumina short reads were used for differential expression (DE) analysis. To gain insight into the dynamics of the AP transcriptome during the different Mimivirus infection stages, we selected genes that were differentially expressed at each of the time points compared to their expression in the uninfected amoeba. We then divided the DEGs into up-regulated (UR) and down-regulated (DR) genes for each time point, which yielded six gene groups (1 h UR or DR, 3 h UR or DR, and 5 h UR or DR). The different groups were analyzed using Ontologizer21 (Fig. 3b–d). We used the three primary domains of the GO acyclic tree: biological processes (Fig. 3b), molecular function (Fig. 3c) and cellular components (Fig. 3d). The GO terms belonging to each domain were grouped into categories in a semi-automatic manner.
Only 31 DEGs were enriched at 1 HPI compared to the uninfected sample. The high similarity between the two samples can also be inferred from the PCA analysis (Supplementary Fig. 2). By comparison, 3,993 DEGs were enriched at 3 HPI, and 4,025 DEGs at 5 HPI. Since most of the enriched categories were common to both 3 and 5 HPI (Fig. 3a–c), in the subsequent sections, we discuss GO terms related to these categories together.
Mitochondrial activity
The mitochondrial transport and morphogenesis category was enriched at 1 HPI. UR genes annotated to the respiratory chain complex III assembly (GO:0017062) and mitochondrial respiratory chain complex assembly (GO:0033108) GO terms were enriched at this time point. At 5 HPI, this category was enriched in DR genes, specifically with mitochondrial transmembrane transport (GO:1990542) and mitochondrion morphogenesis (GO:0070584) GO terms. These observations corroborate the finding of Legendre et al.12 of an initial increase and then, later on in the infection, a decrease in the number of reads that originate from amoebal mitochondria-related genes.
Translation regulation
Enrichment in UR genes was observed at 1 HPI for the translation regulator activity nucleic acid binding (GO:0090079), translation factor activity RNA binding (GO:0008135), and translation regulator activity (GO:0045182) GO terms. Subsequently, at 3 and 5 HPI, the categories enriched in UR genes were translation-related glutaminyl-tRNA aminoacylation (GO:0006425) (Fig. 3b) and aminoacyl-tRNA ligase activity (Fig. 3c). The tRNA-splicing ligase complex category (Fig. 3d) was enriched in UR genes at 3 HPI but not at 5 HPI. Unlike most viruses, Mimivirus encodes for translation-related proteins in its genome. The Mimivirus translation-related category was enriched at 3 HPI (Fig. 3a) with genes encoding for translation initiation factors (R458, L529, L496), tRNA ligases (L164, L124) and methionyl-tRNA synthetase (R639).
Transcription
At 3 and 5 HPI, the AP transcriptome was enriched in UR genes associated with transcription initiation, including terms related to the regulation of DNA-templated transcription initiation (GO:2000142) and regulation of transcription initiation from RNA polymerase II promoter (GO:0060260) (Fig. 3b). This was accompanied by the UR of transcription regulation activity (Fig. 3c), including DNA-binding transcription factor activity (GO:0003700) and general transcription initiation factor binding (GO:0140296) GO terms.
In the Mimivirus, the transcription category was also enriched at 3 HPI (Fig. 3a), consistent with a previous report of transcription activity in VFs at 4 HPI22. Among the genes belonging to this category were transcription regulators L544, R450, R453, R430 and R339; DNA-directed RNA polymerases R501, R867, L244, L235, R209, R470, L208, R357b and L376; the two mRNA capping enzyme-encoding genes R382 and L308; and the poly-A polymerase catalytic subunit L341 (Note that some of the Mimivirus transcription-related proteins are also packed in the virion10 and might play a role during earlier infection stages). Another pertinent Mimivirus-enriched category was proteins containing KilA N (Fig. 3a), a DNA-binding domain found in bacteria and DNA viruses23.
Peroxisomes
Found in most eukaryotic cells, peroxisomes are small organelles that carry out various oxidative-related reactions. A significant function of peroxisomes is related to lipid metabolism, including the breakdown of fatty acids and the synthesis of very-long chain fatty acids and ether lipids24. Our data reveal enrichment in UR genes related to peroxisomal transport and organization (Fig. 3b) at both 3 and 5 HPI. Go terms belonging to this category include long-chain fatty acid import into peroxisome (GO:0015910) and peroxisomal membrane transport (GO:0015919). Terms associated with peroxisome-related components (Fig. 3d) were enriched in UR genes solely at 5 HPI. These GO terms include the integral component of peroxisomal membrane (GO:0005779) and the peroxisome (GO:0005777).
The secretory pathway and the ubiquitin–proteasome system
The ER is a major component of the secretory pathway. Its membrane-bound lamellar network, which is continuous with the outer nuclear envelope, constitutes more than half the mass of the cell’s membrane24. Its primary roles are post-translational protein modification and lipid synthesis. Host GO terms related to the secretory pathway were enriched in UR genes at 3 and 5 HPI (Fig. 3b). These included ER-to-cytosol transport (GO:1903513) and the vesicle targeting and rough ER to cis-Golgi (GO:0048207) GO terms.
The proteasome is a large protein complex that degrades ubiquitin-tagged proteins. This proteolytic activity plays vital roles in various cellular processes such as transcription, cell cycle progression and cell homeostasis25. At 3 HPI, there was enrichment in UR genes related to the protein ubiquitination category (Fig. 3b), which includes protein modification by small protein conjugation (GO:0032446) and protein ubiquitination (GO:0016567) GO terms. In addition, at both 3 and 5 HPI, there was enrichment in UR genes associated with the ubiquitin–proteasome system activity category (Fig. 3c), including poly-ubiquitin modification-dependent protein binding (GO:0031593), proteasome binding (GO:0070628) and ubiquitin-protein transferase activity (GO:0004842) GO terms. Also enriched in UR genes were GO terms associated with the proteasome-related components category (Fig. 3d), including the proteasome accessory complex (GO:0022624) and the proteasome regulatory particle (GO:0005838). Another category enriched in UR genes at 3 and 5 HPI that is related to both the ubiquitin–proteasome system and ER is the cellular response to misfolded proteins category (Fig. 3b). This category includes cellular response to heat (GO:0034605), response to topologically incorrect protein (GO:0035966), and ubiquitin-dependent ERAD pathway (GO:0030433) GO terms.
Interestingly, the Mimivirus itself appears to possess genes encoding for components of the ubiquitin–proteasome system, such as putative E2/E3 enzymes. This Mimivirus gene category, however, was expressed uniformly throughout the infection cycle and was not enriched at any of the examined time points.
Cell cycle
During Mimivirus infection, AP cells stop dividing (Supplementary Video 1). This is also evidenced by the transcriptomic results, as many GO terms related to cell cycle categories were enriched in DRGs starting at 3 HPI. These terms include DNA replication origin binding, DNA repair, chromatin regulation, chromosome organization, DNA replication-related components, and anaphase-promoting complex (Fig. 3c,d). A related category, DNA replication origin binding activity (Fig. 3b), was enriched in DRGs only at 5 HPI. Some DRG GO terms associated with the two categories are shown in Table 1.
Table 1.
Host DNA replication-related categories and examples of associated GO terms that exhibit enrichment in DR genes at 3 and 5 HPI.
| AP category enriched in DR genes | Associated GO terms (examples) | |
|---|---|---|
| Name | Number | |
| At both 3 and 5 HPI | ||
| DNA-replication regulation | DNA replication-independent nucleosome organization | GO:0034724 |
| DNA strand elongation | GO:0022616 | |
| DNA-replication initiation | GO:0006270 | |
| DNA-replication-related components | MCM complex | GO:0042555 |
| Ctf18 RFC-like complex | GO:0031390 | |
| DNA polymerase complex | GO:0042575 | |
| DNA-replication preinitiation complex | GO:0031261 | |
| DNA repair | Double-strand break repair via break-induced replication | GO:0000727 |
| DNA ligation involved in DNA repair | GO:0051103 | |
| Negative regulation of DNA recombination | GO:0045910 | |
| Chromatin regulation | Chromatin silencing | GO:0006342 |
| Chromosome condensation | GO:0030261 | |
| Nucleosome assembly | GO:0006334 | |
| Chromosome organization | Anaphase-promoting complex-dependent catabolic process | GO:0031145 |
| Sister chromatid cohesion | GO:0007062 | |
| Mitotic sister chromatid separation | GO:0051306 | |
| Anaphase-promoting complex | Anaphase-promoting complex | GO:0005680 |
| Nuclear ubiquitin ligase complex | GO:0000152 | |
| Purine-nucleotide-related activity | Guanyl-nucleotide exchange factor activity | GO:0005085 |
| Carboxypeptidase activity | GO:0004180 | |
| Adenine nucleotide transmembrane transporter activity | GO:0000295 | |
| Purine ribonucleotide transmembrane transporter activity | GO:0005346 | |
| At 5 HPI only | ||
| DNA replication origin binding | DNA replication origin binding | GO:0003688 |
| Nucleotide-related processes | Adenine nucleotide transport | GO:0051503 |
| Purine ribonucleotide transport | GO:0015868 | |
In marked contrast, the viral transcriptome exhibited enrichment in UR DNA replication-related genes at 3 HPI (Fig. 3a). Amongst these were mostly genes encoding for DNA polymerases (R322, R493) and replication factors (R411, L478, L499, R395 and R510). Other UR genes associated with DNA replication were enriched at 5 HPI. These included primase polymerase (L537) and a member of DNA polymerase family X (L318). Interestingly, replication of Mimivirus DNA was observed already at 2 HPI22. This early phase is likely mediated by DNA replication-related proteins, e.g., R303, L478 R493, R771 and others, that are packed in the virion3,10 and presumably enable the commencement of replication12.
Other host categories enriched in DRGs were nucleotide-related processes, at 5 HPI (Fig. 3b), and purine nucleotide-related activity, at both 3 and 5 HPI (Fig. 3c) (Table 1). Unlike AP, the Mimivirus category of nucleotide metabolism was enriched at 3 HPI (Fig. 1A). To identify the source of nucleotides used for the replication of the viral genome, the host cells were treated with a nucleoside analog of thymidine prior to infection. The analog, 5-ethynyl-2´-deoxyuridine (EdU), can be fluorescently labeled after fixation and permeabilization of the cells. EdU was incorporated only into the host genome, thus enabling to distinguish between existing and de novo-synthetized nucleotides. While we observed prominent staining by the DNA dye 4′,6-diamidino-2-phenylindole (DAPI) in the VFs following infection, it was not accompanied by EdU staining (Fig. 4). Thus, nucleotides used during Mimivirus genome replication originate primarily or solely from de novo synthesis, where viral proteins are likely to play a major role.
Fig. 4.

Nuclear DNA staining with EDU-labeled nucleotides. AP cells were grown in PYG medium supplemented with EDU-labeled nucleotides for approximately 24 h prior to infection. Prior to infection, the medium was replaced with fresh PYG. AP cells were infected for the indicated time periods, after which cells were fixed and stained. Green—EDU labeling (nuclear DNA). Blue—DAPI staining. White line—cell contour. Scale bar: 10µm.
Cytoskeleton
Similarly to other eukaryotes, AP possesses an extensive cytoskeletal network that confers mechanical stability to the cells and plays important roles in cellular transport and locomotion5. Mimivirus infection induces the rearrangement of tubulin and actin networks within AP cells11 and causes them to lose their asymmetrical shape with pseudopodia. As a result, the cells of the amoeba round, with their actin fibers retracting to form shell-like structures at the cell periphery. Consistent with these observations, we found that categories and associated GO terms related to cytoskeletal components were enriched in DRGs upon infection. Specifically, both at 3 and 5 HPI, there was enrichment in DRGs in the microtubule nucleation (Fig. 3b), tubulin binding activity (Fig. 3c), and microtubule (Fig. 3d) categories. Likewise, actin-related processes (Fig. 3b), actin binding (Fig. 3c) and actin-related cellular components (Fig. 3d) all showed enrichment in DRGs. The myosin binding (Fig. 3c) and myosin complex (Fig. 3d) categories were also enriched in DRGs at 3 and 5 HPI. Examples for cytoskeleton related categories and GO terms are described in Table 2.
Table 2.
Host cytoskeleton-related categories and examples of associated GO terms that exhibit enrichment in down regulated (DR) genes 3 and 5 HPI.
| AP category enriched in DR genes | Associated GO terms (examples) | |
|---|---|---|
| Name | Number | |
| Microtubule nucleation | Microtubule nucleation | GO:0007020 |
| Tubulin binding activity | Tubulin binding | GO:0015631 |
| Microtubule | Microtubule | GO:0005874 |
| Actin-related processes | Negative regulation of cytoskeleton organization | GO:0051494 |
| Actin crosslink formation | GO:0051764 | |
| Actin binding | Actin filament binding | GO:0051015 |
| Actin-related cellular components | Cortical cytoskeleton | GO:0030863 |
| Lamellipodium | GO:0030027 | |
| Cortical actin cytoskeleton | GO:0030864 | |
| Myosin binding | Myosin binding | GO:0017022 |
| Myosin complex | Actomyosin | GO:0042641 |
| Myosin I complex | GO:0045160 | |
Discussion
In this work, we generated the transcriptome of the free-living amoeba Acanthamoeba polyphaga and analyzed its dynamics during Mimivirus infection, along with the corresponding changes in the viral transcriptome.
A summary of the key findings of the AP and Mimivirus functional enrichment analyses, as well as the state of the infected cells along the infection, is presented in Fig. 5. The transcriptome dynamics of Mimivirus observed in this study are largely consistent with those reported by Legendre et al.12. Both studies highlight genes involved in DNA replication, nucleotide synthesis, and translation during the intermediate phase, as well as structural proteins during the late phase. We have, however, assigned some of the changes in viral gene expression to a different stage of infection (early, intermediate, or late) as compared to Legendre et al.12. In part, these discrepancies may be due to differences between the amoeba hosts—Acanthamoeba castellanii and Acanthamoeba polyphaga 26. Although these Acanthamoeba species are closely related, differences in the infectivity of Mimivirus strains toward AC and AP have been observed27. Other causes may be related to variations in multiplicity of infection (MOI), sampling times, sample preparation, sequencing technology, and other experimental variations.
Fig. 5.

Summary of the Acanthamoeba polyphaga (AP) and Mimivirus bradfordmassiliense (Mimivirus) transcriptome enrichment analysis. Columns represent time points post-infection. Upper panel: cell state illustration showing the nucleus (dark purple), viral factory (red), and Mimivirus (blue pentagon in a white circle).  —Nucleus,
—Nucleus,  —Viral factory. Lower panel: transmission light microscopy with DAPI labeling. Scale bar: 10 µm. Table at bottom: Summary of the processes that were upregulated in AP
—Viral factory. Lower panel: transmission light microscopy with DAPI labeling. Scale bar: 10 µm. Table at bottom: Summary of the processes that were upregulated in AP  , downregulated in AP
, downregulated in AP  , and upregulated in Mimivirus
, and upregulated in Mimivirus  .
.
Out of the 31 AP DEGs identified at 1 HPI, 29 were upregulated. These included genes associated with translation regulation and mitochondrial transport and morphogenesis (Fig. 3b and c). Twenty-eight of the 31 genes were also differentially expressed and upregulated 1 h after UV stress. This might suggest that the amoeba detects the virus early on during the infection and launches a general stress response. AC was reported to possess genes that encode for putative pathogen recognition receptors such as LBP/BPI and c-lectin5, which may be involved in Mimivirus detection and the subsequent stress response.
Major changes in AP gene transcription patterns begin at 3 HPI (Fig. 3b–d). Based on our analysis, we suggest that the infection interferes with cell cycle progression by obstructing transition into the synthesis (S) and mitotic (M) phases. During the S phase, the cell is engaged in the execution of DNA replication28,29. However, during infection, we observed a broad downregulation of host genes involved in DNA replication, DNA repair, and nucleotide metabolism. Our analysis similarly indicates enrichment in DRGs related to various mitotic processes, including chromosome organization and segregation, chromatin regulation and the formation of the anaphase-promoting complex (Fig. 3a-d). By contrast, Mimivirus categories of DNA replication and nucleotide metabolism were enriched at 3 HPI (Fig. 3a). In addition, we show that nucleotides used during replication of the viral genome originate primarily from de novo synthesis and not from the breakdown of the host DNA or cellular reservoirs. Modulation of the cell cycle is by no means unique to the Mimivirus; infection by herpes simplex virus-1 (HSP1), Epstein-Barr virus (EBV)30, both double-stranded DNA viruses, as well as RNA viruses like influenza A virus30,31, HIV, and Severe acute respiratory syndrome coronavirus (SARS CoV)30, also result in cell cycle arrest, typically at the G2/M phase. This may be an example of the advantage of the Mimivirus’s elaborate DNA replication system, which endows the virus with more autonomy in cases where genes associated with similar host tasks are downregulated.
As an example of the amoeba cells’ response to abiotic stress, we analyzed the AP transcriptome after exposure to UV light. Following the treatment, AP DRGs associated with cell cycle-related categories were also enriched, including cell cycle (GO:0007049), DNA replication (GO:0006260), sister chromatid segregation (GO:0000070), and anaphase-promoting complex (GO:0005680). This suggests that cell cycle arrest is part of a general cellular stress response of the host. Nonetheless, the arrest might be beneficial for the Mimivirus infection cycle, as it leaves metabolic resources, such as nucleotide precursors and amino acids, available for the virus.
The enrichment in DRGs related to the cytoskeleton at 3 and 5 HPI is consistent with microscopic observations of alterations in actin and tubulin structures during Mimivirus infection11. The rounding of cells and loss of the pseudopodia during infection are aligned with this notion and can be attributed to the downregulation of actin, myosin and tubulin-related processes (Fig. 3b-d). However, actin is required for the proper fusion of VFs11. Thus, although actin-related processes are enriched in DRGs, actin is still required and is, therefore, likely present in amounts and forms that enable completion of the infection cycle.
It has been proposed that the Mimivirus membrane originates from the ER13,14. We suggest that the mechanism by which Mimivirus induces the uptake of ER membranes relies on enhanced activation of the secretory pathway, as there is enrichment in UR genes related to that pathway. Viruses are known to utilize the ER for various tasks, such as entry into the proper cellular compartment, exiting the cell, viral replication and assembly, synthesis of glycoprotein32,33, and membrane assembly13,14,32.
Our analysis also reveals enrichment in host UR genes associated with the ubiquitin–proteasome system, commencing at 3 HPI. The Mimivirus may utilize this system similarly to other viruses, such as HIV, influenza A virus (RNA virus), human papillomavirus (HPV) and adenovirus (double-stranded DNA virus), which use it to degrade host proteins, or human cytomegalovirus (HCMV) (double-stranded DNA virus), which exploits the ERAD pathway34,35. Notably, the Mimivirus also possesses genes encoding proteins belonging to the ubiquitin–proteasome system, suggesting viral use of the system.
Finally, our analysis highlights changes in peroxisomal activities during infection. Other viruses were reported to modulate peroxisome numbers and activity upon infection36,37. These include influenza A virus, which increases lipid metabolism in the peroxisome, and HCMV, Herpes simplex virus 1 (HSV-1) and Kaposi’s sarcoma-associated herpesvirus (KSHV), all three are double-stranded DNA viruses, which were associated with an increase in the number of peroxisomes in the host cells36.
In conclusion, we propose that the amoeba host detects and responds to the Mimivirus early on during the infection; a result of this response may be cell cycle arrest observed during Mimivirus infection. In addition, we suggest that infection is accompanied by proteasome and peroxisome involvement in Mimivirus infection, increase in secretory pathway activity and the downregulation of actin and tubulin polymerization-related genes. The modulation in the latter probably underlines the massive alterations in the cytoskeleton of host cells during infection.
Potential implications
Our work makes the Acanthamoeba polyphaga transcriptome publicly available. In addition, it provides leads for future research into the specific roles of host cellular components, such as the peroxisome and ubiquitin–proteasome system, during Mimivirus infection. Future work should also focus on the sensing mechanisms used by the host to detect the Mimivirus at early stages of infection and on the molecular events that lead to cell cycle arrest of the host during infection, as indicated by our analysis.
Methods
Mimivirus purification
AP cells were plated in 175 cm2 flasks and infected by exposure to the virus for 48 h at 30°C. The content of the flasks was then transferred to 50 mL tubes and centrifuged (100 g, 20 min, 4°C). The supernatant was transferred to a new 50 mL tube and centrifuged (10,000 g, 30 min, 4°C). The supernatant was discarded and virus pellets were washed twice with phosphate-buffered saline (PBS). The pellets were resuspended in PBS, filtered with 1.4-µm filter, and kanamycin (0.5 mg/mL) and ampicillin (1 mg/mL) were added. Viruses were stored at 4°C.
Host cell growth and infection
AP cells were grown in peptone-yeast-glucose (PYG) in the presence of kanamycin (0.05 mg/mL) and ampicillin (0.1 mg/mL) in 9-cm untreated cell-culture plates to 70–80% confluency at 30 °C. AP cells were infected with Mimivirus using a multiplicity of infection (MOI) of 10. At 0.5 HPI, plates were washed with fresh medium and incubated for the desired infection time. Cells were then scraped from plates and frozen in liquid nitrogen until the RNA purification step. For the UV-stress experiment, cells were plated in a similar manner. Medium was replaced with Hanks′ Balanced Salt solution (Merck H9269) and exposed to 50 J/m2 UV light. After 1 h, cells were scraped and frozen.
Illumina short read sequencing. RNA was purified from (1) uninfected AP samples, (2) AP samples at 1, 3, 5 HPI, and (3) AP samples 1 h after UV exposure, using RNeasy Mini Kit (Qiagen catalog number: 74104). RNA integrity was assessed using the 4200 TapeStation System. For library preparation, the poly-A fraction of mRNA was purified from 500 ng total RNA, followed by heat fragmentation and production of double-stranded cDNA. Thereafter, Agencourt Ampure XP bead cleanup (Beckman Coulter), end repair, A base addition, adapter ligation, and PCR amplification steps were performed. Libraries were quantified by Qubit (Thermo Fisher Scientific) and TapeStation (Agilent). Sequencing was performed on a Hiseq2500 instrument (Illumina) using a PE125 V4 kit (paired end sequencing).
PacBio single-molecule real-time (SMRT) sequencing of long reads
RNA was purified from (1) uninfected AP samples, (2) AP samples at 1, 3, 5 HPI, and (3) AP samples 1 h after UV exposure, using RNeasy Mini Kit (Qiagen catalog number: 74104), and mixed. SMARTer® PCR cDNA synthesis kit (TaKaRa,cat 634926) and PrimeSTAR® GXL DNA polymerase (TaKaRa, cat R050B) were used for cDNA preparation. Libraries were prepared using the SMRTbell® Express Template Prep Kit 2.0. Sequencing was then performed using a SMRT cell 1M v3 on the Sequel instrument, using Sequel sequencing plate 3.0.
Transcriptome assembly
Raw data obtained using PacBio were subjected to IsoSeq3 (version 8) for raw reads’ processing. Illumina reads of AP samples after 1, 3, and 5 h of infection with Mimivirus, as well as of uninfected AP cells and AP cells 1 h after UV treatment were mapped to PacBio reads using Bowtie2 (Version 2.3.5.1)38. Unmapped reads were utilized using two independent approaches. The first was genome-based de novo assembly using StringTie software,version 2.1.4 (https://github.com/gpertea/stringtie)39 and the publicly-available genome assembly of AP (INSDC: CDFK CDFK00000000.1). We only used transcripts that originated from DNA contigs longer than 2000 nucleotides that were at least 10 nucleotides away from each end of the contig. The second approach involved de novo assembly without utilizing a reference genome, using rnaSPAdes (Version 3.14.0)40 (The names of transcripts originated from IsoSeq3, StringTie and rnaSPAdes begin with “UnnamedSample_HQ_transcript_”, “new.strg.” and “NODE_”, respectively). Transcripts from the three assemblies were integrated and EvidentialGene, tr2aacds41 was used to remove redundancy. The transcriptome was run against the non-redundant protein database (NCBI) using Blastx (Blast + version 2.5.0), for 20 hits, and the best hit results were used for further analysis. The NCBI Taxonomy browser42 was used to analyze the organisms from which the annotation was borrowed, based on the Blastx results. In order to remove transcripts originating from contaminants by other organisms that may reside in the amoeba43 or may have been introduced during sample preparation, we filtered Blastx results for bacteria and removed those that had a hit of > 85% identity and a coverage > 50%. Transcripts that aligned with viruses were likewise filtered out. The Cogent—collapse_by_cogent_category.py script (version 6.0.0. Python 3.7 version, https://github.com/Magdoll/Cogent) was later used to define genes, and the longest transcript was chosen as the family representative. A figure describing the transcriptome assembly workflow can be found in supplementary (Supplementary Fig. 3). The assembled AP transcriptome yielded 22,604 transcripts (N50 = 2,372 bp) (Supplementary File 1), and 13,043 genes (Supplementary File 2). Histograms describing transcripts length, and number of isoforms per gene can be found in supplementary Fig. 4A and B respectively.
BUSCO analysis
BUSCO (Version 3, https://gitlab.com/ezlab/busco)44,45 was used to evaluate the completeness and duplication of the transcriptome. In order to assess the quality of the assembly, we compared our de novo AP transcriptome BUSCO results with those of its closest relative with a known transcriptome, namely, AC (Acanthamoeba castellanii Acastellanii.str NEFF_v1 ,GCA_000313135.1, https://protists.ensembl.org/Acanthamoeba_castellanii_str_neff_gca_000313135/Info/Index)46. We used both eukaryota_odb9 and protists_ensembl databases for the comparison (supplementary Table 1).
Sanger sequencing
The KAPA Taq PCR Kit (Sigma-Aldrich KR0352_S) was used to amplify the reads from a cDNA mixture of uninfected samples and 1, 3 and 5 HPI samples. Primers were designed using the Primer3web software, version 4.1.0 (https://primer3.ut.ee/)47,48 (Supplementary Table 2). The forward primers were used in the sequencing (3730 DNA Analyzer, ABI). DEGs with Blastx annotations from each assembly tool were selected randomly. Sanger results are detailed in supplementary Table 3.
Mapping and gene quantification
The RSEM tool49 was used to map Illumina reads from each sample to a unified transcriptome that included the Mimivirus and AP (with isoforms). Differential expression analysis was applied at the gene level by summing the number of reads assigned to all isoforms of the same gene. The fold-change value and the p-value between each two time points were calculated using the DESeq2 package in R50. The criteria for DEGs were ≥ twofold change and a false discovery rate (FDR) (adjusted p-value) < 0.05 in at least one comparison between the samples. Mimivirus differentially expressed genes can be found at Supplementary File 3. Acanthamoeba polyphaga differentially expressed genes can be found at Supplementary File 4, and the data 1 h after UV in Supplementary File.
Data clustering and PCA
DESeq2 output of normalized rlog transformed (rld) values were used for principal component analysis (PCA), performed with Partek Genomics Suite software, version 6.5 (https://www.partek.com/partek-flow/), (Partek Inc., St. Louis, MO). K-means clustering was performed using standardized log2 DEG expression values as input (rld). A Silhouette plot was used to determine the optimal number of clusters for the Mimivirus data (Matlab) (Supplementary Fig. 1), and clustering was then performed using the Partek Genomics Suite. Mimivirus gene clusters can be found at Supplementary File 6.
Gene annotation by GO
The Blast2Go tool15 was used to find GO-terms associated with the hit sequences obtained by Blastx (against the non-redundant protein database (July 2020, ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz). Top Blast hits can be found in Supplementary File 7. The output was analyzed by Ontologizer (Supplementary File 8). Ontologizer assignment of genes to GO terms can be found in Supplementary File 9.
Biological pathways analysis
GO functional enrichment analysis for the annotated genes in the different clusters was performed using Ontologizer (version 2.1, http://ontologizer.de/.)21 and the Obo library (http://purl.obolibrary.org/obo/go/go-basic.obo). The topology weighted algorithm51 was used and the threshold p-value was 0.005. GO terms in each domain were grouped into categories in a semi-automatic manner, using a combination of Revigo52 and manual classification, in order to simplify data interpretation. Manual classification was based on GO hierarchical classification and common genes between GO terms. A full list of GO terms assigned to categories and time points can be found in Supplementary File 10. Enrichment analysis of AP genes 1 h post UV treatment can be found in Supplementary File 11.
Mimivirus enrichment analysis
Mimivirus genes were categorized manually, based on the top blast annotations and the literature (Supplementary File 6). Category counts were performed using an in-house script (10.6084/m9.figshare.20154740). The enrichment analysis was performed using hypergeometric test, in R. Enrichment was considered statistically significant with a p-value < 0.05 (Supplementary File 12).
Fluorescence microscopy
Uninfected AP cells were plated on 3.5-cm glass-bottomed plates (µ-Slide, 8-well, tissue culture-treated (Ibidi IBD-80826 i)). Cells were infected with Mimivirus at MOI ~ 10 for 1, 3 or 5 h. The plates were then washed and incubated with PFA 4% in PBS for 20 min at room temperature (RT), washed three times with PBS for 5 min each, and incubated for an additional 10 min at RT with triton X100 0.1% in PBS. Cells were washed three times with PBS for 5 min each and incubated with a 1:1000 dilution of DAPI (5 µg/mL) in PBS for ~ 1 h, and then finally washed three times with PBS. Samples were imaged using an Eclipse TI-E Nikon inverted microscope (Nikon Instruments Inc., Melville, NY) with a Plan Apo 100X/1.4 NA lens. Images were acquired with a cooled electron-multiplying charge-coupled device (EMCCD) camera (IXON ULTRA 888; Andor).
Amoebal genomic DNA staining by EdU
We used the Click-iT® EdU Cell Proliferation Kit for Imaging (Thermo Fischer, catalog number: C10337). AP cells (5–10% confluency) were grown in PYG in a 3.5-cm petri dish containing EdU nucleotides (20 μM; 1:500), for 24 h at 30°C. Cells were transferred to 8-well plates (Ibidi 80826), to achieve 50–70% confluency, for 10 min, and then washed thrice with fresh PYG and left in the incubator for 1 h, followed by Mimivirus infection at MOI ~10 for 4, 5, 6, 8, and 10 h. Infection was terminated by washing the cells with PBS and incubating them in formaldehyde (3.7% in PBS) for 15 min, followed by three washes with PBS. Triton® X-100 (300 μL, 0.1% in PBS) was added, and the cells were then incubated for additional 10 min at RT, after which they were washed thrice with PBS. We then followed the manufacturer’s instructions for using the kit. Briefly, 300 μL of Click-iT® reaction mixture were added to each well and the cells were incubated for 30 min at RT, protected from light. The reaction mixture was then removed, and the cells were washed three times with 3% BSA in PBS and stained with DAPI 1:1000.
Time-lapse imaging during Mimivirus infection
AP cells were grown on 35 mm2 glass-bottom plates (Mat‐Tek corp. P35G‐1.5‐14‐C), treated with 1 mg/mL poly-L lysine for 1 h, and then infected with Mimivirus at MOI ~ 50, at 30 °C. Widefield images were acquired using a Deltavision microscope (Applied Precision) equipped with a 60X UPlanSApo NA 1.40 objective and a photometrics coolSNAP HQ2 CCD (Roper Scientific, Tucson, AZ). Images were captured every 60 s from 250 min post-infection onward.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We dedicate this paper to Professor Avi Minsky, who passed away this year. We thank Drs. Merav Kedmi and David Pilzer, for their support in SMRT sequencing sample preparation; Maor Knafo, for his assistance with the SMRT sequencing and data analysis; Dr. Liran Ben Yaakov for establishing the UV protocol; and Drs. Ruti Kapon, Shifra Ben Dor, and Inbal Neta-Sharir, for their helpful advice and for reviewing the manuscript.
Abbreviations
- AC
Acanthamoeba castellanii
- AP
Acanthamoeba polyphaga
- bp
Base pairs
- DEGs
Differentially expressed genes
- DR
Downregulated
- ER
Endoplasmic reticulum
- ERAD
ER-associated degradation
- FDR
False discovery rate
- GO
Gene Ontology
- HIV
Human immunodeficiency virus
- HPV
Human papilloma virus
- HCMV
Human cytomegalovirus
- Mimivirus
Mimivirus bradfordmassiliense
- NCLDV
Nucleo-cytoplasmatic large DNA virus
- NGS
Next-generation sequencing
- ORF
Open reading frame
- PCA
Principal component analysis
- HPI
Hours post infection
- RT
Room temperature
- SMRT
Single-molecule, real-time
- SR
Short reads
- URG
Upregulated genes
- VF
Viral factory
Author contributions
Conception and design of the research -RN, YFS, EF. Experiment execution -RN, YFS. Data analysis and interpretation -RN , EF, YFS, Reinat Nevo, HKS. Writing the manuscript- RN, EF, YFS. Critical revision of the article-RN, ZR, AM, Reinat Nevo, HKS, EF, YFS. Visualization- RN, Reinat Nevo. Supervision- AM and ZR. Funding acquisition-AM and ZR.
Data availability
The data sets supporting the results of this article are available in the Figshare repository, https://figshare.com/s/2e61150ec8bea07a5e13Raw data have been deposited in the NCBI BioProject database, BioProject accession number PRJNA720295 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA720295).
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Reut Bickels Nuri, Email: reutnuri@gmail.com.
Ziv Reich, Email: ziv.reich@weizmann.ac.il.
References
- 1.La Scola, B. et al. A giant virus in amoebae. Science299(5615), 2033 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Raoult, D. et al. The 12-megabase genome sequence of Mimivirus. Science306(5700), 1344–1350 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Renesto, P. et al. Mimivirus giant particles incorporate a large fraction of anonymous and unique gene products. J. Virol.80(23), 11678–11685 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Legendre, M. et al. Breaking the 1000-gene barrier for Mimivirus using ultra-deep genome and transcriptome sequencing. Virol J8, 99 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan, N. A. Acanthamoeba: Biology and Pathogenesis 2nd edn, 334 (Caister Academic Press, 2015). [Google Scholar]
- 6.Lorenzo-Morales, J., Khan, N. A. & Walochnik, J. An update on Acanthamoeba keratitis: Diagnosis, pathogenesis and treatment. Parasite22, 10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marciano-Cabral, F. & Cabral, G. Acanthamoeba spp. as agents of disease in humans. Clin. Microbiol. Rev.16(2), 273–307 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrade, A. C. D. S. et al. Filling knowledge gaps for mimivirus entry, uncoating, and morphogenesis. J. Virol.10.1128/JVI.01335-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zauberman, N. et al. Distinct DNA exit and packaging portals in the virus Acanthamoeba polyphaga mimivirus. PLoS Biol.6(5), e114 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fridmann-Sirkis, Y. et al. Efficiency in complexity: Composition and dynamic nature of mimivirus replication factories. J. Virol.90(21), 10039–10047 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yaakov, L. B. et al. Kinetics of mimivirus infection stages quantified using image flow cytometry. Cytometry A95(5), 534–548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legendre, M. et al. mRNA deep sequencing reveals 75 new genes and a complex transcriptional landscape in Mimivirus. Genome Res.20(5), 664–674 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mutsafi, Y. et al. Membrane assembly during the infection cycle of the giant Mimivirus. PLoS Pathog.9(5), e1003367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suárez, C. et al. Open membranes are the precursors for assembly of large DNA viruses. Cell Microbiol.15(11), 1883–1895 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conesa, A. et al. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics21(18), 3674–3676 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Sedgwick, S. G. & Smerdon, S. J. The ankyrin repeat: A diversity of interactions on a common structural framework. Trends Biochem. Sci.24(8), 311–316 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Notaro, A. et al. Expanding the occurrence of polysaccharides to the viral world: The case of mimivirus. Angew. Chem. Int. Ed. Engl.60(36), 19897–19904 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piacente, F. et al. The rare sugar N-acetylated viosamine is a major component of Mimivirus fibers. J. Biol. Chem.292(18), 7385–7394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piacente, F. et al. The autonomous glycosylation of large DNA viruses. Int. J. Mol. Sci.16(12), 29315–29328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parakkottil Chothi, M. et al. Identification of an L-rhamnose synthetic pathway in two nucleocytoplasmic large DNA viruses. J. Virol.84(17), 8829–8838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauer, S. et al. Ontologizer 2.0—A multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics24(14), 1650–1651 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Mutsafi, Y. et al. Vaccinia-like cytoplasmic replication of the giant Mimivirus. Proc. Natl. Acad. Sci. U.S.A107(13), 5978–5982 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iyer, L. M., Koonin, E. V. & Aravind, L. Extensive domain shuffling in transcription regulators of DNA viruses and implications for the origin of fungal APSES transcription factors. Genome Biol.3(3), 1. 10.1186/gb-2002-3-3-research0012 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alberts, B. Molecular Biology of the Cell 5th edn. (Garland Science, 2008). [Google Scholar]
- 25.Nandi, D. et al. The ubiquitin-proteasome system. J. Biosci.31(1), 137–155 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Chelkha, N. et al. A Phylogenomic study of acanthamoeba polyphaga draft genome sequences suggests genetic exchanges with giant viruses. Front. Microbiol.9, 2098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dornas, F. P. et al. Isolation of new Brazilian giant viruses from environmental samples using a panel of protozoa. Front. Microbiol.10.3389/fmicb.2015.01086 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lane, A. N. & Fan, T. W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucl. Acids Res.43(4), 2466–2485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desany, B. A. et al. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev.12(18), 2956–2970 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshizawa-Sugata, N. & Masai, H. Cell cycle synchronization and flow cytometry analysis of mammalian cells. In Cell Cycle Control. Methods in Molecular Biology (Methods and Protocols) Vol. 1170 (eds Noguchi, E. & Gadaleta, M.) (Humana Press, 2014). 10.1007/978-1-4939-0888-2_13. [DOI] [PubMed] [Google Scholar]
- 31.He, Y. et al. Influenza A virus replication induces cell cycle arrest in G0/G1 phase. J. Virol.84(24), 12832–12840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ravindran, M. S. et al. Opportunistic intruders: How viruses orchestrate ER functions to infect cells. Nat. Rev. Microbiol.14(7), 407–420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inoue, T. & Tsai, B. How viruses use the endoplasmic reticulum for entry, replication, and assembly. Cold Spring Harb. Perspect. Biol.5(1), a013250 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Randow, F. & Lehner, P. J. Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol.11(5), 527–534 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Pizzorno, A. et al. Influenza A viruses alter the stability and antiviral contribution of host E3-ubiquitin ligase Mdm2 during the time-course of infection. Sci. Rep.8(1), 3746 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cook, K. C. et al. Peroxisome plasticity at the virus-host interface. Trends Microbiol.27(11), 906–914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jean Beltran, P. M. et al. Infection-induced peroxisome biogenesis is a metabolic strategy for herpesvirus replication. Cell Host Microbe24(4), 526-541.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods9(4), 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol.33(3), 290–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bushmanova, E. et al. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience10.1093/gigascience/giz100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbert, D. Gene-omes built from mRNA seq not genome DNA. In 7th Annual Arthropod Genomics Symposium.
- 42.Schoch, C.L., et al., NCBI Taxonomy: A Comprehensive Update on Curation, Resources and Tools. Database (Oxford) (2020). [DOI] [PMC free article] [PubMed]
- 43.Rayamajhee, B. et al. A Systematic review of intracellular microorganisms within acanthamoeba to understand potential impact for infection. Pathogens10(2), 225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.BUSCO. Version 3 (2019). https://gitlab.com/ezlab/busco.
- 45.Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics31(19), 3210–3212 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Acanthamoeba castellanii str. Neff (GCA_000313135.1) (Acastellanii.strNEFF_v1). https://protists.ensembl.org/Acanthamoeba_castellanii_str_neff_gca_000313135/Info/Index.
- 47.Koressaar, T. & Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics23(10), 1289–1291 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Untergasser, A. et al. Primer3–new capabilities and interfaces. Nucl. Acids Res40(15), e115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li, B. & Dewey, C. N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform.12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol.15(12), 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexa, A., Rahnenführer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics22(13), 1600–1607 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Supek, F. et al. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE6(7), e21800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets supporting the results of this article are available in the Figshare repository, https://figshare.com/s/2e61150ec8bea07a5e13Raw data have been deposited in the NCBI BioProject database, BioProject accession number PRJNA720295 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA720295).