


Abstract
The transcription factor CHOP/GADD153 gene is induced by cellular stress and is involved in mediating apoptosis. We report the identification of a conserved region in the promoter of the CHOP gene responsible for its inducibility by endoplasmic reticulum (ER) stress. Deletion mutants of the human CHOP promoter identify a region comprising nucleotides –75 to –104 required for both constitutive and ER-stress-inducible expression. This region of the promoter, the ER-stress element (ERSE) is sufficient to confer both increased basal activity and ER-stress inducibility to an otherwise inactive heterologous promoter. The CHOP ERSE is a novel variant of the ERSE as it contains two different functional domains, and a GA- instead of GC-rich intervening sequence. The CCAAT-box domain occupied by the constitutive transcriptional activator nuclear factor Y (NFY) is required for constitutive activation whereas the variant GCACG ‘inducible’ domain uniquely mediates ER-stress inducibility. By UV-crosslinking analysis NFY makes contact not only with the constitutive activator CCAAT box but also with the inducible GCACG domain. Deletions and nucleotide substitutions in the CCAAT box as well as its replacement by an SP1 site failed to support ER inducibility. These findings support the notion that NFY is not only required for constitutive activation of CHOP gene transcription, but is also an active and essential element for the assembly of an ER-stress-inducible enhanceosome that activates CHOP gene expression in response to cellular stress.
INTRODUCTION
In mammalian cells the endoplasmic reticulum (ER) stress response (ERSR) involves the induction of the CHOP gene (1–4). The ERSR, also known as the unfolded protein response (UPR), is a highly conserved cellular response of eukaryotic cells in both yeast and mammals that involves the induction of genes coding for ER resident proteins, the function of which is to facilitate the correct folding of secretory and transmembrane proteins. The ERSR/UPR also facilitates the degradation of the misfolded proteins by the proteosome pathway in an attempt to re-establish cellular homeostasis (5,6). Mammalian proteins induced by ER-stress such as glucose regulated proteins (GRPs) and peptide disulfide isomerase (PDI) have homologous yeast counterparts. However, the induction of CHOP by ER-stress is a relatively new event since the yeast genome does not contain a CHOP gene.
CHOP is a bZip transcription factor that belongs to the C/EBP family (7) previously cloned as a growth arrest and DNA damage-inducible gene (GADD153) (8). In yeast, Hac1p, another bZip transcription factor, is synthesized during ER-stress. The synthesis if Hac1p is regulated by Ire1p, an ER membrane-resident protein that senses unfolded proteins in the ER lumen and regulates translation of Hac1p (9–14). Hac1p then induces the expression of a set of genes involved in the UPR through binding to a specific DNA element present in their corresponding promoters (15). Because CHOP is a bZip transcription factor and is induced early during ER-stress (4), it was initially considered to be a good candidate to orchestrate the ER response in mammalian cells (3,4,16). This view was favored because of the fact that the induction of CHOP by ER-stress was shown to be dependent on the activation of the mammalian counterpart of Ire1p (17). However, more recent reports have shown that the induction of a full ERSR is preserved in primary fibroblasts obtained from chop(–/–) null mice (18). This later finding indicates that the induction of CHOP is not a requirement for the induction of the ERSR in mammalian cells. Rather CHOP seems to be part of a divergent pathway that results in the induction of further downstream genes during ER-stress (17,19). Although the data available are limited they seem to indicate that this new pathway may lead to arrest of the cell division cycle (20,21) and the active induction of cell death by apoptosis (18).
Numerous chemical agents and environmental conditions that cause cellular stress have been shown to induce CHOP expression. An important group of these compounds has been shown to generate stress in the ER, such as tunicamycin that inhibits N-linked glycosylation; thapsigargin and calcium ionophores that disrupt calcium homeostasis; and agents that disrupt protein disulphide bonds, such as cystein conjugates and dithiothreitol (4). Other agents and conditions that induce the expression of CHOP such as amino acid deprivation (22), reactive oxygen intermediates (ROI) (23) and lipopolysaccharide exposure (24) have been shown to induce CHOP expression through specific responsive elements in the CHOP promoter. In the case of oxidative stress a TRE (AP1) site located between nucleotides –250 and –225 of the CHOP promoter is required for the transcriptional activation of CHOP. A c-Jun/c-Fos heterodimeric complex was shown to mediate such activation. In fact, exposure of the cells to either oxidants or UVC stimulated the binding of c-Jun and c-Fos to the CHOP AP1 element (23). Expression of CHOP during the acute phase response (APR) was shown to be mediated by a conserved C/EBP binding site located at –339 to –320 of the CHOP promoter (24). The APR response was triggered by exposure to lipopolysaccharides resulting in an early induction of C/EBPβ, which accounted for the induction of CHOP through its interaction with the C/EBP site of the CHOP promoter. In vivo treatment with lipopolysaccharides also produced a transient protection of the AP1 site in the CHOP promoter. However, the contribution of these factors to the induction of CHOP expression was not studied in detail (24). On the other hand, induction of CHOP gene expression by amino acid deprivation was shown to require two new regions in the CHOP promoter; one located upstream of the transcription initiation site, between nucleotides –318 and –286 and the other downstream, between nucleotides +21 and +51 in the first exon. In this case, however, neither the binding proteins nor the DNA-binding elements involved in the response were well-defined (22). Induction of CHOP gene expression by ER-stress, however, has not been mapped to any particular region of its promoter and, therefore, the transcription factors involved have not been identified.
In the studies reported herein, we investigated the mechanism responsible for the transcriptional induction of the CHOP gene in response to ER-stress. We show that the induction of CHOP gene transcription in response to ER-stress is mediated via a defined DNA element in its promoter with the capacity to influence both constitutive and inducible gene expression. This element is homologous to a subgroup of ER-stress elements (ERSEs) recently identified in several promoters of the mammalian GRPs and calreticulin genes. We also identified the transcription factor NFY as being required for both constitutive and ER-stress-inducible activation of the CHOP gene through its interaction with two different domains of the CHOP ERSE.
MATERIALS AND METHODS
Reagents
Tissue culture media and reagents were obtained from Gibco BRL (Grand Island, NY). DNA-modifying enzymes were purchased from New England Biolabs (Beverly, MA) or Boehringer Mannheim Biochemicals (Indianapolis, IN). Radioactive compounds were obtained from Du Pont-New England Nuclear (Boston, MA). Nucleotides were purchased from Pharmacia-LKB (Piscataway, NJ). All other molecular biology grade reagents were obtained from Sigma Chemical Co. (St Louis, MO). Tunicamycin was also from Sigma and Thapsigargin from Calbiochem (San Diego, CA). Oligonucleotides were synthesized at the Molecular Biology Core Facility of the Massachusetts General Hospital. NFY A and NFY B antisera were obtained from Biodesign International (Kennebunk, ME) and NFY C and YY1 antisera were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell lines
NIH 3T3 fibroblasts were obtained from the American Type Cell Collection (ATCC, 1658-CRL). Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum in the presence of the antibiotics, penicillin (100 U/ml) and streptomycin (100 µg/ml).
Plasmid constructs
PCR amplified DNA fragments comprising nucleotides –104 to +1 and –75 to +1 were introduced into the BamHI–XbaI-digested pOCAT vector to generate the corresponding deletion reporter vectors. PCR-based mutagenesis was used to substitute the CCAA sequence of the two ERSE elements with the aacc sequence of the corresponding CCAAT sites to create the mutant construct MUT hCHOP-CAT (sequence provided in Fig. 1C). To generate the construct ERSE-TK-CAT and the MUT ERSE-TK-CAT reporters the corresponding CHOP ERSE of 39 bp oligonucleotides were inserted into the BamHI site of the minimal –41 TK-CAT vector. The sequence of the oligonucleotides MutS-ERSE and MutAS-ERSE is shown in Figure 4. To generate the constructs Mut21-ERSE, SP1-ERSE and CR-ERSE, the following double-stranded oligonucleotides were inserted into the –41 TK-CAT vector. Mut21: 5′-CCAATTGCCGGgcacCCACTTTCTGATTTGG-3′, SP1-ERSE: 5′-ggggcggggTGCCGGCGTGCCACTTTCTGAccccgcccc-3′ and for CR-ERSE: 5′-GCCGGCGTGCCACTTTCTG-3′. All recombinant DNA constructs were verified by direct sequencing.
Figure 1.
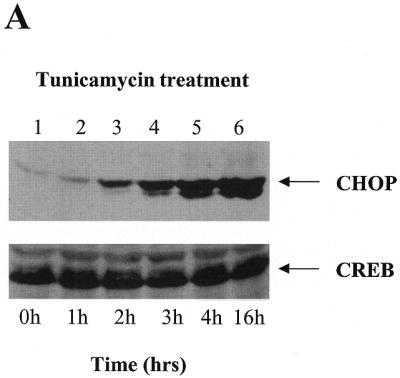


Induction of the CHOP gene expression by ER-stress maps to a promoter region homologous to the ERSE elements identified in the GRPs, PDI and calreticulin genes. (A) Tunicamycin treatment (5 µg/ml) induces a robust induction of CHOP in NIH 3T3 fibroblasts. Western blot experiments using a specific CHOP and a control CREB antisera were performed on nuclear extracts from NIH 3T3 cells treated with tunicamycin for the indicated periods of time. (B) Transcriptional activities of different CAT reporters in which activity was controlled by different promoter deletions of the human CHOP gene. Tunicamycin treatment was initiated 16 h prior to harvesting the cells. Maps of the –104 bp CHOP-CAT, –75 CHOP-CAT and the mutated form (MUT CHOP-CAT) are also shown. The reporter activity was measured and data from three independent experiments performed in duplicate were plotted. Data shown correspond to the mean ± SEM. The specific mutated sequence in the MUT CHOP-CAT construct is shown in (C). (C) Sequence analysis of the –104 –75 region of the CHOP promoter identifies two overlapping elements matching the ERSE consensus. This consensus was previously defined based on the sequence of the GRPs, PDI and calreticulin promoters (29,30).
Figure 4.




Of the two putative ERSE elements in the human CHOP promoter, only the one localized to the antisense DNA strand is functionally active. (A) Sequence comparison of the DNA sequences of the human, mouse and hamster promoters indicates conservation of the ERSE motif localized to the antisense DNA-strand but not of that localized to the sense strand. The human and hamster sequences were obtained from GenBank and the mouse sequence from our laboratory (39). (B) CAT reporter constructs were prepared by introducing the depicted oligonucleotides upstream of the TATA box in the –41 TK-CAT reporter. (C) Activity of the different constructs once transfected into NIH 3T3 cells. Tunicamycin treatment was as described in Figure 1. Results are expressed as the mean ± SEM of three independent experiments performed in duplicate. (D) The CHOP ERSE element belongs to a subgroup of ER-stress-inducible elements and differs from the reported consensus by having an AG- instead of a GC-rich spacer, and a G nucleotide substitution in the first position of the inducible domain. The domains required for constitutive activation ‘c’ and ER-stress inducibility ‘i’ were identified in our mutagenesis experiments (see also Fig. 5A).
Transfections and transactivation assays
NIH 3T3 fibroblasts were transfected by using the GenePorter system (Gene Therapy Systems, San Diego, CA). Cells were harvested 48 h after transfection. ER-stress stimulation was investigated by treating the transfected cells with tunicamycin (5 µg/ml) during a 16 h period, prior to harvesting the cells. CAT activity was measured using a non-radioactive fluorimetric assay (Fast CAT, Molecular Probes, Eugene, OR). CAT values obtained from the fluorimetric assay were expressed in arbitrary fluorimetric units per mg of protein. All values are expressed as mean ± SEM of at least three experiments carried out in duplicate. Transfections were performed with 2 µg of reporter following the manufacturer’s recommendations.
Electrophoretic mobility shift assays (EMSA)
DNA–protein-binding assays were carried out with 10 µg of nuclear extracts prepared as described earlier (25), or with purified recombinant CBF/NFY proteins, kindly provided by Dr Maity (University of Texas, TX). The CBF/NFY subunits CBF-B (60 ng) and CBF A/C (80 ng) were used at the concentrations described previously (26). Protein concentrations were determined by the Bio-Rad protein assay with bovine serum albumin as a standard. Synthetic complementary oligonucleotides with 5′-GATC overhangs were annealed and labeled by a fill-in reaction using [32P]dATP and Klenow enzyme. The sequences of the oligonucleotides used are shown in Figure 1C.
Binding reactions were carried out in the presence of 0.1 µg of poly(dI·dC), using nuclear extracts (10 µg of protein). Incubations were performed in the presence of 20 000 c.p.m. of radiolabeled probe (~6–10 fmol) in a total volume of 20 µl containing 20 mM potassium phosphate (pH 7.9), 70 mM KCl, 1 mM DTT, 0.3 mM EDTA and 10% glycerol.
UV cross-linking analysis
For in situ DNA–protein crosslinking, polyacrylamide slabs from EMSA were placed under a 300 nm UV light source (Fotodyne 7000 mW/cm2) and irradiated during 20 min in the cold room as described previously (27). The DNA–protein complexes were excised and analyzed on a 10% SDS–PAGE under reducing conditions.
Western immunoblots
Equivalent amounts of the cellular extracts used for determinations of CAT activity (30 µg) were subjected to SDS–PAGE after the addition of an equivalent volume of Laemmli buffer. After overnight transfer to a nitrocellulose membrane, filters were blocked and incubated for 1 h with a specific CHOP antiserum (Santa Cruz Biotechnology). Immune complexes were detected with the ECL system (Amersham Pharmacia Biotech, Arlington Heights, IL) after incubation with a horseradish peroxidase-conjugated secondary antibody.
RESULTS
Inducibility of CHOP gene expression by ER-stress maps to a promoter region with homology to the recently identified ERSE element in the GRPs and calreticulin promoters. To study the mechanism by which ER-stress induces the expression of the CHOP gene we chose to perform our experiments in NIH 3T3 cells since they respond to ER-stress agents such as tunicamycin with a robust induction of CHOP (Fig. 1A). Numerous reports (1–3) and our own work have shown that this response is mediated by transcriptional activation of the CHOP promoter. Our previous results and those obtained by other investigators (28) indicate that a reporter driven by a minimal CHOP promoter retains full ER-stress inducibility. We now report additional experiments performed with CAT reporters driven by shorter deletion mutants of the human CHOP promoter in an attempt to identify the element responsive to ER-stress. As shown in Figure 1B, the –104 CHOP-CAT reporter was fully inducible by tunicamycin, whereas the –75 CHOP-CAT reporter was not inducible. This latter reporter construct, still containing the TATA box and a SP1 site, also showed a decreased basal activity compared to the –104 CHOP-CAT reporter. These data indicated that ER-stress-responsiveness of the CHOP gene required an element residing in the –104 to –75 bp region of its promoter. Sequence analysis of this region identified two overlapping regions with homology to the newly identified ERSEs of the GRPs, PDI and calreticulin genes (Fig. 1C) (29,30). Nucleotide substitutions in the CAAT boxes of these two elements (Fig. 1C) rendered the –104 CHOP reporter insensitive to ER-stress (Fig. 1B); however, it also decreased its basal activity to the level detected in the –75 CHOP-CAT construct. The low basal promoter activity of the CAAT sequence with the nucleotide substitutions makes the effects in response to tunicamycin induction difficult to interpret. However, this marked loss of basal activity clearly indicates the importance of the CAAT sequences in contributing to the transcriptional activity of the ERSE.
The CHOP ERSE motif is in itself sufficient to confer constitutive activation and ER-stress inducibility to a heterologous promoter. In the experiments described above ER-stress inducibility and basal activation were abolished both by deletion of the –104 –75 region of the promoter and by introducing nucleotide substitutions in the CAAT boxes of the identified ERSE homologous sequences. To investigate whether the new identified element was not only required but also sufficient to mediate ER-stress inducibility of the CHOP gene we performed additional functional studies. In these studies, we placed the identified CHOP ERSE in the context of a heterologous promoter that by itself was not responsive to ER-stress. Both the wild-type and mutant forms of the CHOP ERSE comprising nucleotides –104 to –75 were introduced upstream of the minimal –41 TK-CAT reporter and their response to ER-stress agents was tested in NIH 3T3 cells after transfection. The data presented in Figure 2A show that the identified CHOP ERSE motif contains the information required to mediate ER-stress inducibility since it conferred tunicamycin inducibility to the otherwise insensitive –41 TK-CAT reporter. This response was consistently reproduced with other agents known to induce ER-stress such as thapsigargin and dithiothreitol, which induce CHOP expression by a different mechanism (28). Nucleotide substitutions in the corresponding CAAT boxes abolished both ER-stress inducibility and basal constitutive activity. That tunicamycin treatment was equally effective in all the experimental conditions was demonstrated by western blot detection of endogenous CHOP induction (Fig. 2A). We also observed that the magnitude of the CHOP ERSE response to tunicamycin is in fact larger in the heterologous construct than in its natural context in the CHOP promoter (Fig. 2B) (6- versus 3-fold, compared to their corresponding basal activity). This circumstance may indicate the presence of some negative regulatory influences in its natural context. As it occurred for ER-stress inducibility, the effect on basal constitutive activity of the CHOP ERSE element was also transferred to the heterologous promoter (Fig. 2A and B). This finding indicates that the same element is responsive for both constitutive and inducible activation. Whether these two functions can be separated and ascribed to different domains of the CHOP ERSE element or whether they are mediated by a unique mechanism was then investigated.
Figure 2.


The CHOP ERSE element contains in itself the information required for ER-stress inducibility and constitutive activation. (A) CAT activity assayed in cellular extracts from NIH 3T3 cells transfected with the corresponding reporter constructs and treated with tunicamycin as indicated in Figure 1. ‘Empty reporter’ is the –41 TK-CAT alone. The wild-type (WT ERSE-CAT) and mutant (MUT ERSE-CAT) constructs contain inserts of the CHOP ERSE and CHOP ERSE MUT (sequences shown in Fig. 1C) upstream of the –41 TK-CAT. Each experiment was performed in duplicate. Tunicamycin-inducibility of the endogenous CHOP gene was determined by western blot assay of the same cell extracts in which the CAT activities were determined thereby serving as an internal control for ER-stress induction. (B) The level of CAT activity of each of the reporter constructs was calculated based on three independent experiments performed in duplicate and the results were plotted as the mean ± SEM.
The CAAT box binding factor CBF also known as nuclear factor Y (NFY) represents the major protein complex binding to the CHOP ERSE element. Once we had defined the DNA-responsive element required for ER-stress inducibility of the CHOP gene, we attempted to identify transcription factors that bind the ERSE. These factors are expected to be involved in both constitutive activation and ER-stress inducibility. DNA–protein binding EMSA experiments were performed with nuclear extracts from both control and tunicamycin-treated cells. Two major retarded complexes (A and B) were identified (Fig. 3A). However, no tunicamycin-inducible DNA-binding complexes were detected in these experiments. This finding was not surprising since no inducible DNA-binding complexes have been detected for other ERSE motifs in the GRP78 gene by using conventional gel retardation assays (29,31–33). A recent report by Roy and Lee (30) indicated the appearance of an ER-stress inducible complex (ERSF) on the rat GRP78 ERSE-98 and the mouse GRP72 ERSE-194 only after modifying the EMSA binding conditions. This inducible complex binds, however, to a GGC motif present in the 9 nt intervening domain that Yoshida et al. (29) reported as being irrelevant for ER-stress inducibility. We used these modified EMSA conditions and still were unable to find an inducible complex with the CHOP ERSE motif. This finding is consistent with fact that the CHOP ERSE lacks a GGC sequence in its intervening domain. Extensive comparison between the different ERSE sequences of the CHOP, GRPs, PDI and calreticulin promoters indicate that the CHOP ERSE element belongs to a characteristic subgroup of ERSE that lack a GC-rich intervening sequence (Fig. 4D).
Figure 3.


Identification of NFY as a transcription factor that binds the CHOP ERSE element in both basal and ER-stress stimulated conditions. (A) EMSA experiments performed using nuclear extracts obtained from control (–) and tunicamycin (+) treated NIH 3T3 cells. Nuclear extracts were incubated with the CHOP ERSE in the conditions described in the Materials and Methods section. Results show a similar DNA–protein-binding pattern in both control and tunicamycin-treated conditions. The most prominent complexes are called A and B, respectively. Super-shift experiments using specific antibodies for candidate factors identify the transcription factors NFY-A and NFY-B as the major component of A and B complexes (arrows). Nuclear extracts were incubated in the presence of specific antisera for 15 min prior to adding the DNA probe. (B) Competition experiments using increasing amounts of unlabeled CHOP ERSE (ERSE) and its corresponding mutant ERSE (MUT) oligonucleotides (the sequence of the ERSE and MUT is depicted in Fig. 1C). Unlabeled competitor oligonucleotides were used at 10- to 100-fold molar excess. Extracts are from cells treated with tunicamycin.
To identify the transcription factors involved in the formation of the A and B complexes we attempted super-shift experiments in which antibodies against several candidate factors were used. The results shown in Figure 3A indicate that the histone-fold motif and CCAAT box binding factor CBF/NFY is the major component of both complexes. NFY-A seems to be part of both A and B complexes, NFY-B super-shifted only complex B, and NFY-C could not be detected. Since the three subunits of NFY have been reported to be required for in vitro DNA binding, it is not clear if these differences are real or if they depend on the position of the different epitopes in the final complex. Competition experiments with the wild-type and the previously described CCAAT box CHOP ERSE mutant (Fig. 1C) indicate that complex A is formed by NFY binding to the CCAAT boxes (Fig. 3B). Complex B, also containing NFY, is competed by the CHOP ERSE mutant oligonucleotide and therefore it must bind outside the CCAAT boxes. Since this mutant is functionally inactive, it is suggested that only complexes A and not B are required for constitutive activation and ER-stress inducibility of the CHOP gene.
Only the highly conserved CHOP ERSE motif located in the antisense DNA strand of the human CHOP promoter is functionally active. Our initial analysis of the –104 to –75 region of the human CHOP promoter indicated the existence of two different elements highly homologous to the reported ERSE (Fig. 1C). One of these elements is located in the sense orientation and the other in the antisense orientation with some overlap between the two. Analysis of the corresponding sequences of the mouse and hamster promoters indicated that the sense element is not conserved in these other species (Fig. 4A). Since all of these promoters are strongly induced by ER-stress, we investigated whether both elements of the human gene were required for ER-stress inducibility and whether there was any synergistic interaction between the two. This is important since Roy and Lee (30) reported that a unique ERSE element was insufficient to confer inducibility of a heterologous promoter and that at least two elements were required to mediate an ER-stress response. Therefore, we studied the functional activity of mutants in which the sense or the antisense elements had been individually inactivated by nucleotide substitutions of their corresponding CCAAT boxes (Fig. 4B). Results obtained with these constructs (Fig. 4C) indicate that only the element in the antisense orientation is responsible for ER-stress inducibility. Interestingly, this element was also the one required to support constitutive activation of the promoter. The reason why the element placed in the sense strand is functionally inactive is unknown. This finding is of interest because the sequence of the element is closer to the published ERSE consensus than the one in the antisense orientation. A single nucleotide change (T to G) in the last position of the inducible domain (Fig. 4D) does not explain this difference since a similar substitution is found in some other functional ERSE elements, such as the ERSE-194 of the mouse GRP72 gene and the ERSE 136 of the Caenorhabditis elegans GRP78 gene. To determine whether the orientation of the element was the reason for such functional differences we placed the CHOP ERSE element in the reverse orientation (Fig. 4B) in the same minimal reporter. This reporter construct is inactive (Fig. 4C), indicating that the sense element cannot substitute for the antisense element even when placed in the same position. This experiment also indicates that the antisense element positioned in the sense orientation was inactive as well. Therefore, we can conclude that both the nucleotide sequence and the spatial location of the CHOP ERSE are critical factors for ER-stress inducibility and constitutive activation of the CHOP gene.
Constitutive activation and ER-stress inducibility can be ascribed to different domains of the CHOP ERSE motif. We turned our attention to the fact that all the mutations generated to eliminate ER-stress inducibility also caused a decrease in basal activity. It is possible that an alteration in the basal transcription machinery could affect, in a non-specific manner, the inducibility of the reporter construct to ER-stress. We then performed additional mutagenesis of the CHOP ERSE and found that a 4 nt substitution (gtgc instead of cacg) in the inducible domain (ERSE-Mut-21) eliminated tunicamycin inducibility without affecting constitutive activation (Fig. 5A). EMSA experiments performed with this mutant oligonucleotide showed a retardation pattern identical to the one obtained with the wild-type oligonucleotide (Fig. 5B). These findings are consistent with the previous results in which DNA binding did not correlate with ER-stress inducibility. Again, nucleotide substitutions of the inverted CCAAT box resulted in both a decrease of basal activity and abrogation of ER-stress inducibility (Fig. 5A). These results have important implications. First, the fact that the Mut-21 construct could not be induced further by tunicamycin treatment indicates that its basal activity was not due to a basal level of ER-stress due to the culture conditions of the cells. Therefore, two different mechanisms of transcriptional activation appear to channel through the newly identified element, one responsible for constitutive activation and the other for ER-stress inducibility. Second, the finding that NFY still binds to the non-inducible Mut-21 rules out the possibility that a post-translational modification of NFY is the mechanism responsible for ER-stress induction of the CHOP gene. Induction of an ER-stress factor with a capacity to interact weakly with the inducible domain of the CHOP ERSE motif seems to be more likely. DNA binding of this hypothetical factor would not be stable enough to be detected in gel-shift assays. However, in vivo, it could be stabilized by interactions with NFY and perhaps by other proteins already associated with the promoter.
Figure 5.

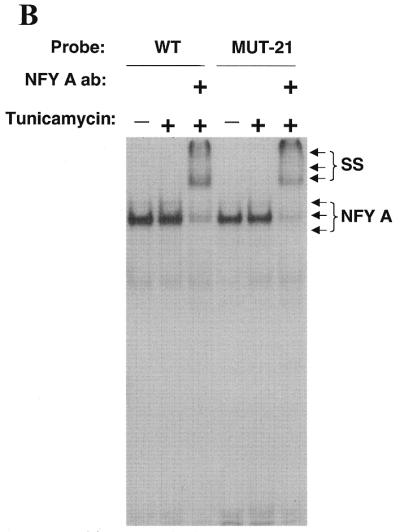
Two different domains are identified in the CHOP ERSE element. One is required for ER-stress inducibility and the other for constitutive activation. (A) Schematic representation of the constructs transfected into NIH 3T3 cells and CAT activity determined for each of the constructs. The Mut21 contains a gcac instead of CGTG substitution in its inducible domain. The MutAS has a ggtt instead of TTGG substitution in the functional inverted CCAAT motif. (B) EMSA experiments in which both the wild-type CHOP ERSE (WT) and the Mut21 (MUT21) oligonucleotides were labeled and incubated in the presence of nuclear extracts from control and tunicamycin-treated cells. Arrows indicate the CBF/NFY-containing band (NFY) and the band super-shifted by the NFY-A antibody (SS).
NFY binding to the CAAT box is also required for ER-stress inducibility and cannot be substituted by another constitutive activator such as SP1. Our results indicate that the ER-stress inducibility of the CHOP gene was mediated by a particular region of the ERSE motif that is different from the CCAAT box. However, the nucleotide substitutions in the CCAAT box abrogated not only constitutive activation but also ER-stress induciblity, indicating that this domain may also be required for ER inducibility. We then examined the role played by NFY and the CCAAT domain of the CHOP ERSE motif in ER-stress inducibility. We created additional CAT reporter constructs. In one of them, the central region of the CHOP ERSE containing the inducible domain but not the CCAAT boxes was placed into the –41 TK-CAT reporter to create the CR-ERSE-TK-CAT. In another construct, five copies of the CR-ERSE were multimerized (5×-CR-ERSE) and placed in the same reporter to create the 5×(CR-ERSE) TK-CAT. In an additional construct, the constitutive CCAAT domains of the CHOP ERSE were substituted by equivalent SP1 elements (SP1-ERSE) (Fig. 6). The CR-ERSE and the 5×-CR-ERSE constructs showed residual activity similar to that of the empty vector. Only the SP1-ERSE displayed some increase in basal activity that did not reach the level of the wild-type CHOP ERSE construct. None of these reporters showed ER-stress inducibility (Fig. 6). These findings and those presented above indicate that the CCAAT domain of the CHOP ERSE is required not only for constitutive activation but also for ER-stress inducibility. The findings also indicate that NFY is an absolute requirement for both constitutive activation and ER-stress inducible expression and that it cannot be substituted by another constitutive activator.
Figure 6.

CBF/NFY binding to the CHOP ERSE element is required for ER-stress inducibility and is not functionally substituted by another constitutive activator such as SP1. Schematic representation of the constructs used in these experiments and CAT activity determined in three independent experiments. Data are expressed as the mean ± SEM. Each experiment was performed in duplicate. The full sequence of the different oligonucleotides is described in the Materials and Methods. CR-ERSE contains the inducible domain and all the flanking sequence with the exception of the CCAAT boxes of the human CHOP ERSE. The same sequence is repeated five times in the 5×-CR-ERSE. In the SP1-ERSE the flanking CCAAT boxes are substituted by respective SP1 binding sites. SP1 binding was tested in gel shift experiments (not shown).
UV-crosslinking experiments indicate that NFY also establishes contact with the inducible domain of the CHOP ERSE. To further understand the mechanism by which NFY binding to the CCAAT motif can influence ER-stress inducibility we considered some additional possibilities. Taking into consideration the way NFY interacts with DNA, one possibility was that once bound to the CCAAT motif NFY can interact with the inducible domain located 9 nt apart. In this manner, both NFY and the specific DNA sequence of the inducible domain could contribute to the creation of a new interacting surface for an ER-stress inducible factor. This hypothetical factor would represent the mammalian counterpart of the yeast Hac1p. If this hypothesis is correct the inducible factor may not require strong affinity for the inducible DNA sequence since its binding would be stabilized by a protein–protein interaction with NFY. This hypothesis is based on published observations concerning the nature of the NFY interaction with its DNA-binding element in the collagen promoter (26,34). In the context of the collagen promoter, NFY acts as a constitutive activator. In these studies (26,34) different approaches including UV-crosslinking, hydroxyl radical footprinting and methylation interference showed that NFY interacts with three different regions of its DNA-binding element. Interaction with the domain II, containing the CCAAT box was sequence-specific, whereas interaction with the other two domains was non-specific and did not require a particular DNA-sequence. We observed that the relative location of the inducible ‘i’ domain and the CCAAT box in the CHOP promoter is equivalent to the position of the domains I and II (CCAAT box) in the α1-collagen promoter (Fig. 8). We then experimentally tested the possibility of a direct interaction between NFY and the inducible ‘i’ domain of the CHOP ERSE. To this end, a CHOP ERSE element with a BrdU substitution in the inducible domain and an element with unsubstituted control nucleotides were synthesized (Fig. 7A). These oligonucleotides were incubated with NIH 3T3 nuclear extracts or purified reconstituted NFY/CBF and subjected to non-denaturing gel electrophoresis. A similar gel-shifting pattern was observed with both nuclear extracts and with purified recombinant NFY/CBF (Fig. 7B). DNA–protein complex ‘A’ was also generated with the recombinant NFY/CBF (Fig. 7B); complex B, however, was only observed with nuclear extracts. After in situ UV-crosslinking complex A was excised, denatured and its composition determined by SDS–PAGE. Three different proteins corresponding to the three subunits of the NFY/CBF complex were identified (Fig. 7C). A similar pattern was observed with both the purified NFY/CBF and the nuclear extracts. No crosslinked complexes were observed with the unsubstituted oligonucleotide (probe 2). The sizes of the different DNA–protein complexes, 60, 40 and 32 kDa (Fig. 7C), correspond to those previously described for crosslinked NFY/CBF subunits (26). These data indicate a direct and specific interaction between the three subunits of NFY with the inducible domain of the CHOP ERSE. These findings indicate that interactions of NFY with the specific sequence of the inducible ‘i’ domain may form a new contact surface for a putative ER-stress inducible factor (Fig. 8). The mutant-21 of the CHOP ERSE would function in a manner similar to the collagen element and supports only constitutive activation. This model also may explain some other findings of our studies as well as reported observations such as: (i) the fact that NFY is essential not only for constitutive activation but also for ER-stress inducibility; (ii) the lack of detection of specific ER-stress inducible binding to the ERSE by EMSA; and (iii) why the distance between the inducible domain and the CCAAT box is critical for inducibility.
Figure 8.

Model depicting the differential interaction of NFY with the α1 collagen promoter in which it serves only as a constitutive activator, and with the CHOP promoter in which it is required not only for constitutive activation but also ER-stress inducibility. It is proposed that the ER-stress inducible ‘i’ domain locates right to a DNA region in which NFY makes contact. Both the specific sequence of the ‘i’ domain and the region of NFY that makes contact with such a domain contribute to the creation of a new interaction surface for a putative ERSF, equivalent to the yeast Hac1p.
Figure 7.

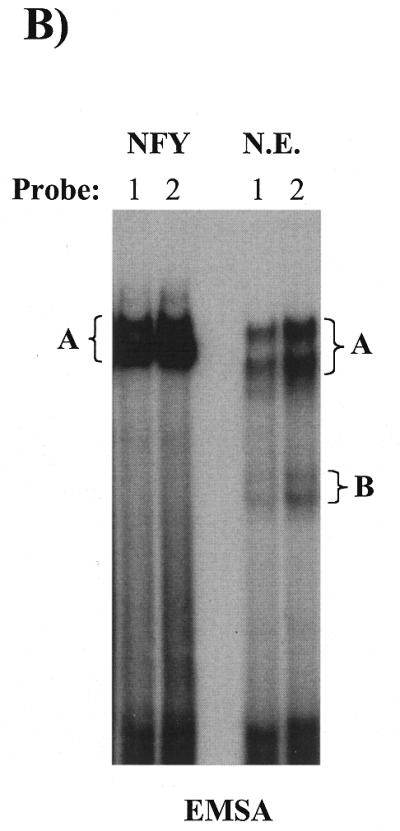
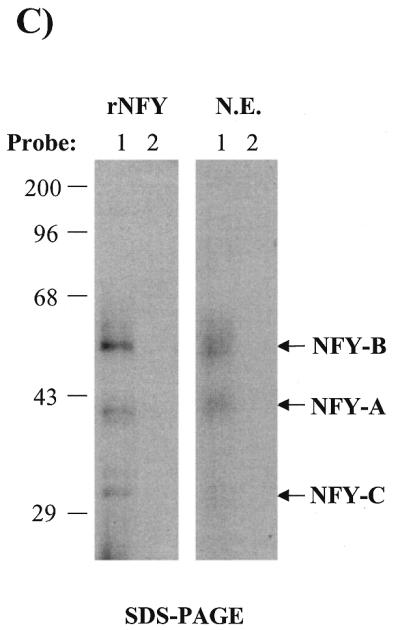
The transcription factor CBF/NFY also interacts with the inducible domain of the CHOP ERSE. (A) A CHOP ERSE nucleotide containing a BrdU substitution in its inducible (‘i’) domain and an unsubstituted control oligonucleotide were used in these experiments. (B) Purified recombinant NFY/CBF generates a major EMSA shifting complex in both the presence of the BrdU substituted and unsubstituted probes. This complex is equivalent to the major A complex in NIH 3T3 nuclear extracts containing NFY as indicated by its specific recognition by an NFY antiserum (see Figs 3A and 5B). (C) After UV-crosslinking, the A complex was excised and its components were separated on a denaturing polyacrylamide gel (SDS–PAGE). Three different proteins crosslink specifically to the BrdU substituted probe (1) and not to the unsubstituted control probe (2). Both nuclear extracts (N.E.) and recombinant purified NFY/CBF (rNFY) show a similar crosslinking pattern that corresponds to the three NFY/CBF subunits with approximate molecular weights of 60 (NFY-B), 40 (NFY-A) and 32 kDa (NFY-C).
DISCUSSION
Numerous reports have shown that CHOP/GADD153 is induced by a variety of agents that produce ER-stress mediated by an increased rate of transcription (1–3). However, the mechanism by which transcription of the CHOP gene responds to ER-stress signals remains largely unknown. Some progress has been made recently by the identification of an ER membrane resident protein, Ire1p, as an upstream activator of chop gene transcription during ER-stress (35). However, the promoter region and the DNA-binding factor(s) required for such a response remained to be identified. In this manuscript, we report the identification of a short region in the CHOP promoter required for its inducibility by ER-stress. This region of the human CHOP promoter contains two overlapping domains that share sequence homology with the recently identified ERSEs in the GRP78, GRP94, PDI and calreticulin promoters (29,30). However, our studies show that only one of the elements, the one localized in the antisense DNA strand is functionally active. This finding is in agreement with the fact that only this element is conserved between the human, mouse and hamster promoters of the corresponding CHOP genes. Also of interest is the fact that the sense element in the human CHOP promoter is inactive, in spite of having closer overall homology to the reported ERSE elements. This circumstance may be related to the fact that the different ERSE elements present considerable heterogeneity with respect to their DNA binding and functional activity (36). For instance, of the three functional ERSE motifs of the rat GRP78 promoter (ERSE-163, ERSE-131 and ERSE-98), only one (ERSE-163) is responsive to the yeast Hac1p (36). Our results show that the functional CHOP ERSE element differs from the consensus ERSE sequence (Fig. 4D) reported previously (30). By comparing the CHOP sequence with those of other similar elements present in the PDI, ERP72 and calreticulin genes, we were able to identify a new subgroup of ERSEs. The consensus sequence for this subgroup of ERSEs differs from the previous consensus in its 9 nt spacer (Fig. 4D) containing an AG- instead of a GC-rich sequence. It also contains a nucleotide substitution in the first position of the inducible domain, a G instead of C. The functional consequences of these differences remain to be elucidated. A higher potency of activation could be a possible difference between the two ERSE subtypes since only one copy of the CHOP ERSE is sufficient to confer ER-stress inducibility to a heterologous promoter. At least two copies of the ERSE element containing a GC-rich spacer were reported to be required to confer inducibility to a heterologous promoter (30).
The newly identified CHOP ERSE is a highly specific sensor for ER-stress. This element was induced not only by tunicamycin but also by other agents known for their capacity to induce ER-stress such as thapsigargin and dithiothreitol. However, it did not respond to other agents or conditions such as deprivation of amino acids or glucose, also known to cause a robust induction of the CHOP gene (data not shown). In this study we also identified the histone-fold motif transcription factor NFY/CBF (NFY) as the major DNA-binding protein associated with the CHOP ERSE. Stress-induced binding of NFY to the CHOP ERSE appears not to be a mechanism by which ER-stress stimulates CHOP expression since our data did not show an inducible protein–DNA complex after tunicamycin treatment; therefore, we considered other possibilities. One of these involved post-translational modifications of NFY that could affect its transactivation activity without affecting its DNA-binding capacity. This possibility is ruled out by the fact that our mutant (MUT-21) lost specifically ER-stress inducibility while maintaining full binding activity to NFY. Another possibility we considered involves the synthesis of an inducible factor similar to the yeast Hac1p. Although several candidate factors have been proposed (29,37), so far no true mammalian counterpart of Hac1p has been identified. An ortholog of Hac1p function must meet three requirements: (i) its functional activity has to be regulated by the activated mammalian Ire1 proteins. Since the RNase L domain of Ire1p has been conserved in the mammalian Ire1p, a mechanism involving RNA processing is likely. (ii) It has to be able to translocate to the nucleus when stimulated with ER-stress; and (iii) it must serve as a transcriptional regulator of the ERSE-mediated transcriptional activation. Our data support the notion introduced by Yoshida et al. (29) in which the mammalian homolog of Hac1p may not necessarily require direct DNA binding to the ERSE motif. A mechanism involving tethering to a pre-existing protein–DNA complex is proposed. This model involves a critical role of relatively weak protein–protein and protein–DNA interactions and favors a more flexible and versatile mechanism. This mechanism would allow for a fine regulation of a larger set of genes by the same pathway via different stress-inducible regulators of the ERSE-mediated transcription. This situation is especially important when taking into consideration the diversity of cell types present in higher organisms and the requirement for conservation of the ERSR. The difficulty in attempting to identify the mammalian counterpart of the yeast Hac1p supports this possibility. This view is also favored by the existence of more than one putative factor with the capacity to induce the UPR in mammals, such as ATF6 (29) and YY1 (36,37). Induction of GRP78 expression by the zinc-finger transcription factor YY1, in response to ER-stress, was reported previously to be mediated by an ERSE element (37). This induction required YY1 binding to the DNA ‘core’ region in the GRP78 promoter that encompasses the inducible CCACG domain of the ERSE element (36,37). However, YY1 is constitutively expressed and so far no induction or processing by ER-stress has been reported. Therefore, the possibility of YY1 being the mammalian counterpart of the yeast hac1p seems unlikely. Even if YY1 is not the mammalian counterpart of the yeast Hac1p, its functional interaction with the yeast Hac1p (36) suggests it could still play an important role as a co-activator of the ER-stress response in mammalian cells.
On the other hand, ATF6 was originally identified as an activator of ERSE-mediated transcription by using the yeast one-hybrid screening technique (29). However, so far no direct or indirect binding of ATF6 to the ERSE element has been reported. Furthermore, no Ire1p-dependent processing of its mRNA was observed (29). These data suggest that ATF6 is not the mammalian counterpart of the yeast Hac1p. Interestingly, the ATF6 protein was shown to be specifically cleaved during ER-stress (29,38). While the full-length 90 kDa ATF6 protein resides in the ER membrane, the cleaved 50 kDa form of ATF6 concentrates in the nucleus where it activates the expression of the genes involved in the mammalian UPR. If eventually it is proven that ATF6 is not the real mammalian counterpart of the yeast Hac1p it seems at least likely that it remains an important modulator of the mammalian transcriptional response to ER-stress.
In summary, we report the identification of a new promoter DNA element in the CHOP gene responsible for both ER-stress inducibility and constitutive activation. The new element is homologous to the recently identified ERSE in the GRPs, calreticulin and PDI genes. However, the CHOP element differs from the consensus ERSE in its potency since one single copy is sufficient to provide a heterologous promoter with ER-stress inducibility. It also differs in its sequence, since it has an AG-rich spacer instead of a GC-rich one, apart from a nucleotide (G instead of C) substitution in the first position of its inducible domain. In this manner, the CHOP ERSE may define a new subgroup of ERSE elements also present in the GRP72, calreticulin and PDI genes. Two different domains in the CHOP ERSE were identified, a CCAAT box required for constitutive activation and a GCACG motif required for ER-stress inducibility. Our data also show that the constitutive expressed CCAAT box binding transcription factor NFY/CBF plays a critical role for both ER-stress inducibility and constitutive activation of the CHOP gene. CBF/NFY binds to the CCAAT box in a sequence-specific manner and controls constitutive activation. NFY also interacts with the inducible domain and affects its interaction with a putative mammalian homolog of the yeast Hac1p to support ER-stress inducibility. Therefore, our data not only explain the mechanism by which the expression of the CHOP gene is regulated in conditions of ER-stress but they may also have important implications for understanding the mechanism by which other mammalian ERSE elements work.
Acknowledgments
ACKNOWLEDGEMENTS
We thank L. Fucci for expert experimental assistance, M. Matzilevich for critical reading of the manuscript and T. Budde and R. Larraga for help in the preparation of the manuscript. J.F.H. is an Investigator with the Howard Hughes Medical Institute.
REFERENCES
- 1.Bartlett J., Luethy,J., Carlson,S., Sollott,S. and Holbrook,N.J. (1992) J. Biol. Chem., 267, 20465–20470. [PubMed] [Google Scholar]
- 2.Chen Q., Yu,K., Holbrook,N.J. and Stevens,J.L. (1992) J. Biol. Chem., 267, 8207–8212. [PubMed] [Google Scholar]
- 3.Price B. and Calderwood,S. (1992) Cancer Res., 52, 3814–3817. [PubMed] [Google Scholar]
- 4.Wang X.-Z., Lawson,B., Brewer,J., Zinszner,H., Sanjay,A., Mi,L., Boorstein,R., Kreibich,G., Hendershot,L. and Ron,D. (1996) Mol. Cell. Biol., 16, 4273–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufman R.J. (1999) Genes Dev., 13, 1211–1233. [DOI] [PubMed] [Google Scholar]
- 6.Mori K. (2000) Cell, 101, 451–454. [DOI] [PubMed] [Google Scholar]
- 7.Ron D. and Habener,J.F. (1992) Genes Dev., 6, 439–453. [DOI] [PubMed] [Google Scholar]
- 8.Fornace A.J., Neibert,D.W., Holander,M.C., Luethy,J.D., Papathanasiou,M., Fragoli,J. and Holbrook,N.J. (1989) Mol. Cell. Biol., 9, 4196–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bork P. and Sander,C. (1993) FEBS Lett., 334, 149–152. [DOI] [PubMed] [Google Scholar]
- 10.Cox J.S., Shamu,C.E. and Walter,P. (1993) Cell, 73, 1197–1206. [DOI] [PubMed] [Google Scholar]
- 11.Mori K., Ma,W., Gething,M.J. and Sambrook,J. (1993) Cell, 74, 743–756. [DOI] [PubMed] [Google Scholar]
- 12.Sidrauski C. and Walter,P. (1997) Cell, 90, 405–413. [DOI] [PubMed] [Google Scholar]
- 13.Shamu C.E. and Alter,P. (1996) EMBO J., 15, 3028–3039. [PMC free article] [PubMed] [Google Scholar]
- 14.Welihinda A.A. and Kaufman,R.J. (1996) J. Biol. Chem., 271, 18181–18187. [DOI] [PubMed] [Google Scholar]
- 15.Mori K., Ogawa,N., Kawahara,T., Yagani,H. and Yura,T. (1998) J. Biol. Chem., 273, 9912–9920. [DOI] [PubMed] [Google Scholar]
- 16.Carlson S.G., Fawcett,T.W., Bartlett,J.D., Bernier,M. and Holbrook,N.J. (1993) Mol. Cell. Biol., 13, 4736–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X.Z., Kuroda,M., Sok,J., Batchvarova,N., Kimmel,R., Chung,P., Zinszner,H. and Ron,D. (1988) EMBO J., 17, 3619–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zinszner H., Kuroda,M., Wang,X., Batchvarova,N., Lightfoot,R.T., Remotti,H., Stevens,J.L. and Ron,D. (1998) Genes Dev., 12, 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sok J., Wang,X.Z., Batchvarova,N., Kuroda,M., Harding,H. and Ron,D. (1999) Mol. Cell. Biol., 19, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barone M.V., Crozat,A.Y., Tabaee,A., Philipson,L. and Ron,D. (1994) Genes Dev., 8, 453–464. [DOI] [PubMed] [Google Scholar]
- 21.Zhan Q., Liebermann,D.A., Alamo,I., Hollander,M.C. and Ron,D. (1994) Mol. Cell. Biol., 14, 2361–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bruhat A., Jousse,C. and Fafournoux,P. (1999) Proc. Nutr. Soc., 58, 625–632. [DOI] [PubMed] [Google Scholar]
- 23.Guyton K.Z., Xu,Q. and Holbrook,N.J. (1996) Biochem. J., 314, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sylvester S.L., Rhys,C.M.J., Luethy-Martindale,J.D. and Holbrook,N.J. (1994) J. Biol. Chem., 269, 20119–20125. [PubMed] [Google Scholar]
- 25.Ubeda M., Wang,X.-Z., Zinszner,H., Wu,I., Habener,J.F. and Ron,D. (1996) Mol. Cell. Biol., 16, 1479–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang S.G. and Maity,S.N. (1998) J. Biol. Chem., 273, 31590–31598. [DOI] [PubMed] [Google Scholar]
- 27.Molitor J.A., Walker,W.A., Doerre,S., Ballard,D.W. and Greene,W.C. (1990) Proc. Natl Acad. Sci. USA, 87, 10028–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jousse C., Bruhat,A., Harding,H.P., Ferrara,M., Ron,D. and Fafournoux,P. (1999) FEBS Lett., 448, 211–216. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida H., Haze,K., Yanagi,H., Yura,T. and Mori,K. (1998) J. Biol. Chem., 273, 33741–33749. [DOI] [PubMed] [Google Scholar]
- 30.Roy B. and Lee,A.S. (1999) Nucleic Acids Res., 27, 1437–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy B. and Lee,A.S. (1995) Mol. Cell. Biol., 15, 2263–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W.W., Sistonen,L., Morimoto,R.I. and Lee,A.S. (1994) Mol. Cell. Biol., 14, 5533–5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy B., Li,W.W. and Lee,A.S. (1996) J. Biol. Chem., 271, 28995–29002. [DOI] [PubMed] [Google Scholar]
- 34.Bi W., Wu,L., Coustry,F., Crombrugghe,B. and Maity,S.N. (1997) J. Biol. Chem., 272, 26562–26572. [DOI] [PubMed] [Google Scholar]
- 35.Wang X.Z., Harding,H.P., Zhang,Y., Jolicoeur,E.M., Kuroda,M. and Ron,D. (1998) EMBO J., 17, 5708–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foti D.M., Welihinda,A., Kaufman,R.J. and Lee,A.S. (1999) J. Biol. Chem., 274, 30402–30409. [DOI] [PubMed] [Google Scholar]
- 37.Li W.W., Hsiung,Y., Zhou,Y., Roy,B. and Lee,A.S. (1997) Mol. Cell. Biol., 17, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haze K., Yoshida,H., Yanagi,H., Yura,T. and Mori,K. (1999) Mol. Biol. Cell, 10, 3787–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitt-Ney M. and Habener,J.F. (2000) J. Biol. Chem., in press. [Google Scholar]