

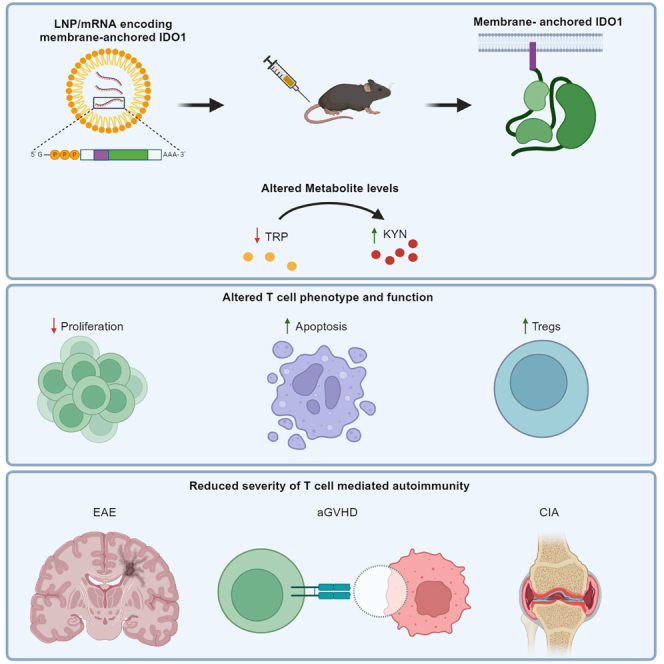
Summary
Indoleamine-2,3-dioxygenase (IDO)1 degrades tryptophan, obtained through dietary intake, into immunoregulatory metabolites of the kynurenine pathway. Deficiency or blockade of IDO1 results in the enhancement of autoimmune severity in rodent models and increased susceptibility to developing autoimmunity in humans. Despite this, therapeutic modalities that leverage IDO1 for the treatment of autoimmunity remain limited. Here, we use messenger (m)RNA formulated in lipid nanoparticles (LNPs) to deliver a human IDO1 variant containing the myristoylation site of Src to anchor the protein to the inner face of the plasma membrane. This membrane-anchored IDO1 has increased protein production, leading to increased metabolite changes, and ultimately ameliorates disease in three models of T cell-mediated autoimmunity: experimental autoimmune encephalomyelitis (EAE), rat collagen-induced arthritis (CIA), and acute graft-versus-host disease (aGVHD). The efficacy of IDO1 is correlated with hepatic expression and systemic tryptophan depletion. Thus, the delivery of membrane-anchored IDO1 by mRNA suppresses the immune response in several well-characterized models of autoimmunity.
Keywords: T cells, IDO1, tryptophan, kynurenine pathway, autoimmunity, EAE, CIA, aGVHD, mRNA, lipid nanoparticle
Graphical abstract
Highlights
-
•
Human IDO1 mRNA containing intracellular membrane anchor increases protein level
-
•
mRNA delivery of anchored IDO1 reduces disease severity in EAE, CIA, and aGVHD
-
•
Hepatic expression of anchored IDO1 is required for protection in autoimmune models
-
•
Efficacy of mRNA-delivered IDO1 is associated with reduced serum tryptophan levels
Kenney et al. explore the therapeutic potential of an mRNA-delivered IDO1 that is modified to include a myristoylation sequence, which allows the protein to insert into the membrane and extends its half-life. When used in rodent models of autoimmunity, this construct is capable of suppressing disease.
Introduction
Tryptophan (TRP) catabolism generates bioactive substances of the kynurenine (KYN) pathway, which modulate immune responses in cancer, pregnancy, viral infections, and autoimmunity. Indoleamine-2,3-dioxygenase 1 (IDO1), along with the functionally similar enzyme tryptophan-2,3-dioxygenase 2 (TDO2), catabolizes TRP to N-formylkynurenine, the rate-limiting step of the KYN pathway. N-formylkynurenine is further broken down into KYN. Even though IDO1 and TDO2 are often introduced as similar enzymes due to their role in the KYN pathway and their utilization by tumors, they differ greatly in their structure, regulation, expression profiles, and function.1,2,3 Under homeostatic conditions, TDO2, which is predominantly expressed in hepatocytes, degrades 80%–90% of dietary TRP.1,3 IDO1, on the other hand, is inducible in macrophages, dendritic cells, epithelial cells, endothelial cells, and tumors predominantly through interferon (IFN)-γ signaling.1,3,4,5
IDO1 evokes an immunoregulatory effect through the local depletion of TRP and the production of immunomodulatory KYNs. Intracellularly, the lack of TRP causes an accumulation of uncharged transfer RNAs, activating GCN2, leading to decreased translation and inactivation of mTOR, blocking effector T cell proliferation and differentiation.6 In TRP-deficient culture conditions, proliferating T cells can enter G1 but pause until TRP levels have been restored.7 KYN is a ligand for aryl hydrocarbon receptor (AhR)8; in CD4 T cells, KYN binding AhR can induce the expression of forkhead box protein P3 (FoxP3) and block retinoic-acid-receptor-related orphan receptor C, skewing CD4 T cell differentiation toward regulatory T cells (Tregs) and preventing Th17 development.9,10,11 KYN has also been found to be immunoregulatory in CD8 T cells. Within the tumor microenvironment, KYN binds to AhR in CD8 T cells and results in increased expression of PD-1, which is associated with decreased effector function.12 Furthermore, KYN and other downstream KYN metabolites have been found to induce apoptosis of activated T cells.13,14
The genetic loss of IDO1 does not result in spontaneous autoimmunity,15 but in the context of autoimmune or inflammatory models, IDO1 deficiency or blockade of IDO1 resulted in a greater severity of disease or incidence in several rodent models.9,16,17,18,19,20,21,22,23,24,25 Furthermore, endogenous IDO1 is upregulated in successful non-human primate (NHP) kidney26 and murine cardiac allografts,27 and blocking IDO1 results in allograft rejection.27,28 Conversely, overexpression of IDO1 or administration of recombinant protein reduces rodent allograft rejection29,30,31,32,33,34,35 and autoimmune disease severity, including rat collagen-induced arthritis (CIA) and murine psoriasis.36,37 In addition, T cells within IDO1-expressing allografts had reduced effector function.30 In humans, genetic variants of IDO1 correlate with susceptibility to autoimmune diseases such as systemic sclerosis38 and type 1 diabetes.39 IDO1 expression, which is found at high levels in pancreatic β cells from healthy individuals, is lost in patients with autoimmune diabetes.40 These observations suggest that elevated expression of IDO1 may exert significant clinical benefits for treating autoimmune and other inflammatory diseases.
Lipid nanoparticle (LNP)-encapsulated mRNA-based delivery systems have been used to produce vaccines and show promise in the treatment of other diseases, including use for intracellular enzyme replacement therapy.41,42,43,44,45 Enzyme replacement using mRNA therapies has been shown to be effective in preclinical models for several rare diseases, including glycogen storage disease type Ia (GSD1a) and propionic acidemia (PA),46,47,48,49,50 with ongoing clinical trials for GSD1a, PA, and methylmalonic acidemia (ClinicalTrials.gov: NCT05095727, NCT04159103, NCT05130437, NCT05295433, and NCT04899310). For autoimmunity, LNP-encapsulated mRNA has been used deliver antigens and induce immune tolerance in mouse experimental autoimmune encephalomyelitis (EAE) and a mouse model of peanut-induced anaphylaxis.51,52 Additionally, a mRNA-delivered interleukin (IL)-2 mutein has been shown to expand endogenous Tregs and demonstrated protection in both EAE and acute graft-versus-host disease (aGVHD).53
The tropisms of different LNPs is dependent on the size and charge of the particle. In general, most LNP formulations accumulate in the liver independent of mRNA cargo, allowing for the successful development of liver-specific enzyme replacement therapies.54,55 The spleen is also a prominent organ for LNP accumulation.56 Additional complexities, such as species-specific differences and inflammation, can play a role in the biodistribution of protein expression with RNA-encapsulated LNP delivery.57,58,59
In the current study, we used mRNA formulated in LNPs to overexpress a membrane-anchored version of human IDO1 in splenic myeloid cells, Kupffer cells, and hepatocytes. The anchor both increased and prolonged protein expression, leading to greater changes in KYN and TRP levels in vitro and in vivo. Delivery of IDO1 treatment was efficacious in murine EAE, rat CIA, and murine aGVHD. Furthermore, efficacy was found to be dependent on hepatic expression and systemic TRP depletion rather than elevated KYNs. Thus, delivery of IDO1 using mRNA is capable of modulating the immune response to successfully ameliorate multiple rodent models of autoimmunity.
Results
Intracellular membrane anchors improve IDO1 protein expression, leading to increased changes in metabolite levels in vitro and in vivo
Since anchoring or tethering proteins has previously been shown to increase protein stability,60,61,62,63,64,65,66,67,68 we designed human IDO1 constructs that included a membrane anchor. This included either the K-Ras tail containing a farnesylation site added to the C terminus (RAS.IDO) or the myristoylation site of Src genetically fused to the N terminus (SRC.IDO) of human IDO1 (Figure 1A). When mRNA encoding non-anchored human IDO1 (hsIDO) was transfected into RAW 264.7 cells, protein was detectable for up to 24 h (Figures 1B and 1C). To determine the functionality of the hsIDO construct, KYN and TRP were measured in the supernatants of transfected cells (Figures 1D and 1E). Supernatants from cells transfected with hsIDO had a spike in KYN and a mild reduction in TRP at 24 h. The addition of either the Ras or Src anchor extended protein expression for an additional 48 h, with protein detectable up to 72 h (Figures 1B and 1C). In the supernatants of these transfected cells, there was an increase in KYN and a decrease in TRP comparable with the non-anchored IDO1 mRNA (Figures 1D and 1E). As a negative control, an inactive IDO1 (dead IDO)69 was used, and no increase in KYN or decrease in TRP was detected with this mRNA.
Figure 1.

Insertion of intracellular membrane anchors onto mRNA-encoded IDO1 have increased and prolonged protein expression
(A) Schematic of modified IDO1 protein sequences showing location of Src or K-Ras lipid modification sites for insertion into the intracellular membrane.
(B) Western blots from RAW 264.7 cells transfected in triplicate with mRNA encoding various Src- or Ras-modified human IDO1 proteins at indicated times post-transfection.
(C) Quantification of human IDO1 normalized to HSP90 of immunoblots by using ImageJ.
(D and E) KYN (D) and TRP (E) levels were measured by ELISA from transfected cells at 24 h. Data are biological replicates and medians and representative of 3 similar studies. One-way ANOVA compared with dead SRC.IDO-treated cells to determine significance with a secondary Tukey’s multiple comparisons test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, and ∗∗∗∗p < 0.0001.
(F–H) C57BL/6 mice were injected i.v. with 1.0 mg/kg of LNP B-formulated mRNA, and at 24 h post-injection, (F) KYN and (G) TRP levels were determined in the serum by ELISA, and (H) KYN:TRP ratios were calculated. Data are individual mice and medians of n = 5 mice/group and representative of n = 2 similar experiments. Significance was determined by one-way ANOVA compared to PBS-injected mice with a secondary Dunnett’s multiple comparisons test. ∗p < 0.05, ∗∗∗p < 0.005, and ∗∗∗∗p < 0.0001.
To determine if hsIDO demonstrated function in vivo, mRNA was formulated in a murine myeloid-tropic LNP (LNP B), administered intravenously (i.v.) into C57BL/6 mice, and levels of KYN and TRP were measured in the serum at 24 h (Figures 1F and 1G). In contrast to the in vitro results, hsIDO did not increase serum KYN levels but did cause a modest 30% reduction in TRP levels. The ratio of KYN:TRP, which is commonly used to determine IDO1 activity, was not altered in hsIDO-treated mice relative to PBS controls (Figure 1H). When administered to mice, unlike the non-anchored hsIDO, RAS.IDO and SRC.IDO significantly increased the KYN:TRP ratios in the serum. RAS.IDO showed the strongest increase in KYN, and both RAS.IDO and SRC.IDO caused a similar depletion of TRP (Figures 1F–1H). Thus, the addition of a membrane anchor to IDO1 increased KYN and TRP levels in vitro and in vivo. In parallel, we also examined anchored TDO2 mRNA constructs and found that only the SRC anchor was successful at improving protein expression (data not shown). For consistency, we selected the SRC-anchored variant for further characterization.
In vivo kinetics and dose titration of SRC.IDO in mouse, rat, and NHPs
To determine the kinetics of IDO1 protein expression and activity, we ran a time course in mice injected i.v. with 0.5 mg/kg of LNP A-encapsulated mRNA (Figures 2A–2H). This LNP delivers mRNA to both the myeloid compartment and hepatocytes in mice and rats. Human IDO1 protein expression was detectable in the liver by 6 h and the spleen by 24 h post-injection (Figures 2A–2E). IDO1 expression was maintained in the liver for up to 96 h, while spleen expression was detected for up to 120 h post-injection (Figures 2A–2E).
Figure 2.

Engineered SRC.IDO treatment alters metabolite levels up to 96 h post-injection
(A–E) Naive C57BL/6 mice were injected i.v. with 0.5 mg/kg of LNP A-formulated mRNA and then monitored 6–192 h post-injection. Human IDO1 expression was determined in the (A, B, and D) liver and (A, C, and E) spleen. (A) Representative images of liver and spleen from a V5-tagged SRC.IDO-treated mouse at the indicated time stained with anti-V5 for IDO1 expression compared with PBS-treated controls. Scale bars indicate 100 μm. (B and C) IDO1 expression was determined as percentage of cells positive for V5 tag from (A) in (B) liver and (C) spleen. (D and E) Human IDO1 expression was determined in (D) liver and (E) spleen by ELISA. Dotted line indicates the levels found in PBS-treated or untreated samples.
(F–H) KYN (F) and TRP (G) levels and KYN:TRP ratios (H) were measured in serum by ELISA. Data are mean and SD of n = 5 mice/group and representative of 5 similar experiments. Significance was determined by two-way ANOVA between SRC.IDO and Dead SRC.IDO, followed by Sidak’s multiple comparisons test. ∗p < 0.05, ∗∗p < 0.005,∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
See also Figures S1–S3.
To understand the relationship between IDO1 protein expression and alteration in metabolite levels, we measured KYN and TRP levels in the serum of mice that received SRC.IDO (Figures 2F–2H). Within 6 h post-injection, when IDO1 protein is detectable in the liver, KYN levels in the serum were elevated 3-fold, and there was a concomitant increase in KYN:TRP ratios. KYN and KYN:TRP ratios peaked at 48 h and slowly declined back to control levels between 48 and 120 h (Figures 2F and 2H), mirroring the kinetics of SRC.IDO protein expression in spleen and liver (Figures 2A–2E). TRP levels were reduced by ∼50% within 6 h of treatment with SRC.IDO, and trough levels were reached by 24 h, representing a 6.7-fold reduction in TRP. This low level of TRP was maintained through 96 h and then slowly returned to baseline between 144 and 168 h (Figure 2G). Collectively, these data show that elevated KYN and TRP depletion occurs rapidly after SRC.IDO protein expression and that a single 0.5 mg/kg dose of SRC.IDO mRNA provides a window of activity of 4–6 days.
We next wanted to understand the dose range of SRC.IDO in both mice and rats where we would be able to detect changes in KYN and TRP levels in the serum. Mice were injected i.v. with either SRC.IDO or the inactive variant at 0.5, 0.25, or 0.125 mg/kg. After 72 h, there was a dose-dependent change in serum KYN and TRP levels with SRC.IDO (Figures S1A–S1C). However, KYN:TRP ratios were significantly elevated only at 0.5 and 0.25 mg/kg (Figure S1C). Similar results were found in Sprague-Dawley rats (Figures S1D–S1F). Based on these data, we selected 0.5 mg/kg as the dose for pursuing further efficacy studies using SRC.IDO mRNA.
Because of the differences in metabolic rates between rodents and primates,70 it was unclear if SRC.IDO would have similar kinetics in NHPs. Therefore, we measured metabolite levels in NHPs that were infused over 60 min with LNP B-formulated SRC.IDO or dead SRC.IDO to replicate clinical administration methods and LNP selection. KYN levels peaked at 6 h post-infusion and returned to baseline by 96 h (Figure S2A). In contrast, trough TRP levels did not occur until 24 h post-infusion, after which TRP levels returned to baseline by 96 h (Figure S2B). KYN:TRP ratios in NHPs were within a similar range to what was found in mice and rats (Figures 2H, S1C, and S1F); however, NHPs have shorter pharmacodynamics, with KYN:TRP ratios returning to baseline by 96 h (Figure S3C). Overall, SRC.IDO showed similar dose-dependent in vivo function in mice, rats, and NHPs.
In vivo function of SRC.IDO is maintained with weekly dosing
Since rodents are injected with a chimeric form of human IDO1, repeat dosing may result in a loss of in vivo function due to the development of an anti-SRC.IDO immune response. To test this, mice received 5 weekly doses of 0.5 mg/kg of SRC.IDO formulated in LNP A. At 24 h post-administration at week 1, similar to Figures 2F and 2G, SRC.IDO treatment resulted in a significant increase in KYN and a decrease in TRP compared to PBS or dead SRC.IDO treatment (Figures S3A and S3B). Similar significant differences were also observed at weeks 2–5, indicating sustained in vivo function of SRC.IDO after repeated weekly dosing. This allows us to infer the absence of efficient neutralizing anti-drug antibodies, at least within this time frame.
SRC.IDO is efficacious in multiple rodent autoimmune models
Overexpression of IDO1 previously has been shown to reduce disease score in rat CIA36 and improve allograft survival.29,30,32,33 To determine the efficacy of delivering IDO1 encoded as mRNA, we tested several rodent models of autoimmunity and inflammation: myelin oligodendrocyte glycoprotein (MOG)-induced EAE, aGVHD, and rat CIA. These models were selected because of an already established role for IDO1 in reducing disease severity.19,24,32,36,71
We tested if the overexpression of IDO1 would decrease EAE disease severity after the administration of SRC.IDO mRNA on days −1 and 6 (Figure 3A). SRC.IDO treatment significantly delayed the onset of symptoms in all mice through day 16, and only 1 out of 11 mice developed any symptoms by day 20 (Figures 3B and 3C). Moreover, SRC.IDO treatment significantly reduced EAE disease intensity and peak score (Figures 3D and 3E). Overall, these results clearly showed that administration of IDO1 mRNA can reduce the severity of disease associated with EAE.
Figure 3.

mRNA-encoded anchored IDO1 is protective in EAE
(A) Mice were induced with EAE with MOG peptide and pertussis toxin (PT) and then injected i.v. with 0.5 mg/kg of LNP A-formulated mRNA on days −1 and 6.
(B and C) Mean clinical score (B) and percentage of disease-free mice (C) were tracked over time.
(D and E) Mean disease intensity (D) and mean peak score (E) were calculated between days 10 and 21.
(B) Data are mean and SEM of 12 mice/group and representative of 4 similar experiments. Significance was determined compared to dead SRC.IDO by two-way ANOVA with a secondary Dunnett’s multiple comparisons test. ∗∗p < 0.01 and #p < 0.0001. (C) Significance was determined by log-rank (Mantel-Cox) test to dead SRC.IDO. ∗∗∗∗p < 0.0001. (D and E) Data are individual mice and median of 12 mice/group and representative of 4 similar experiments. Significance was determined by one-way ANOVA with secondary Tukey’s multiple comparisons test. ∗∗∗∗p < 0.0001.
To expand upon these results, we also examined rat CIA, a second model of CD4 T cell-mediated disease. LNP-formulated SRC.IDO or dead SRC.IDO mRNA was injected i.v. into rats treated with type II chicken collagen to elicit arthritis (Figure 4A). Dead SRC.IDO- and PBS-treated control rats developed paw swelling starting at day 10 (Figure 4B). Weekly SRC.IDO treatment starting on day 0 significantly reduced CIA disease score, disease intensity, and mean peak score (Figures 4B–4E). To further examine the benefit of IDO1 treatment after the initial priming phase in the rat CIA model, we initiated SRC.IDO treatment at various days following disease induction, i.e., day 3, 7, 10, or 14. This was followed with weekly dosing thereafter. Administration of SRC.IDO prior to the onset of symptoms (day 10) greatly attenuated disease (Figures 4F–4I) compared with PBS-treated animals. Initiating SRC.IDO treatment once disease had developed (day 14) successfully prevented the worsening of disease, resulting in reduced subsequent disease score relative to animals treated with PBS or dead SRC.IDO. Thus, SRC.IDO mRNA administration is effective in a therapeutic preclinical setting.
Figure 4.

Engineered IDO1 ameliorates rat CIA
(A) Sprague-Dawley rats were injected intradermally with type II chicken collagen/IFA on day 0 to induce collagen-induced arthritis (CIA). Rats were treated with 0.5 mg/kg LNP A-formulated mRNA i.v.
(B–E) For prophylactic dosing, administration of mRNA occurred weekly with dosing on days 0, 7, and 14.
(F–I) To dose therapeutically, rats were injected weekly starting on day 0, 3, 7, 10, or 14 as indicated in the figure legend.
(B and F) Mean clinical score and (C and G) percentage of disease-free animals were calculated. (D and H) Mean disease intensity and (E and I) mean peak score were calculated from day 10 to 21. (B and F) Data are mean and SEM of 5–8 rats/group and representative of 3 similar experiments. Significance was determined compared to dead SRC.IDO by two-way ANOVA with secondary Dunnett’s multiple comparisons tests. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005, and #p < 0.0001. (C and G) Significance was determined compared to dead SRC.IDO by log-rank (Mantel-Cox) test. ∗∗p < 0.01. (D, E, H, and I) Data are individual rats and median. Significance was determined compared to dead SRC.IDO by one-way ANOVA and secondary Dunnett’s multiple comparisons tests. ∗∗∗∗p < 0.0001.
To determine if SRC.IDO was also effective in a disease with CD8 T cell involvement, we examined SRC.IDO in the context of aGVHD. We used a non-lethal semi-allogenic model where C57BL/6 splenocytes were transferred into C57BL/6xDBA/2 F1 mice, and mice were treated on days 0 and 6 with LNP-formulated SRC.IDO mRNA (Figure 5A). In this aGVHD model, donor T cells undergo expansion with a concomitant reduction in host B cells, likely due to donor T cell-dependent killing. In SRC.IDO-treated mice, donor CD8 T cell engraftment in peripheral blood was reduced at day 7 compared with PBS- and dead SRC.IDO-treated mice; however, by day 14, splenic engraftment was similar between PBS- and SRC.IDO-treated mice (Figures 5B and 5D). Moreover, SRC.IDO treatment minimized the depletion of host B cells at both day 7 in peripheral blood and day 14 in the spleen (Figures 5C and 5E), indicating that SRC.IDO was capable of preventing one of the most significant manifestations of disease despite having similar engraftment to PBS-treated mice in this experiment.
Figure 5.

Src-anchored IDO1 delivered by mRNA protects host B cells in aGVHD
(A) C57BL/6xDBA/2 F1 recipients received C57BL/6 donor cells and were injected i.v. with 0.5 mg/kg of LNP A-formulated mRNA on days 0 and 6.
(B–E) Donor CD8 T cell engraftment (B and D) and host B cell (C and E) percentages were determined by flow cytometry on (B and C) day 7 in peripheral blood and (D and E) day 14 in the spleen.
(F and G) On day 14, splenic CD4 and CD8 T cells within the donor and host compartments were analyzed for expression of (F) active caspase-3 (apoptosis) or (G) Ki67 (proliferation).
(H) The percentages of CD4 T cells expressing FoxP3 within the spleen (top) and blood (bottom) were determined to quantify Treg populations. Data are individuals and median. Data are of n = 3–8 mice/group and representative of 3 similar experiments. Significance was determined by one-way ANOVA with a secondary Tukey’s multiple comparisons test. ∗∗p < 0.005, ∗∗∗p < 0.0005, and ∗∗∗∗p < 0.0001.
To elucidate the underlying mechanism of protection in aGVHD, we phenotyped the CD4 and CD8 T cell compartments, focusing on apoptosis and proliferation, as IDO1 has previously been found to modulate both.7,72 CD4 and CD8 T cells within both the donor and host compartments showed an increase in the apoptosis marker active caspase-3 after SRC.IDO treatment (Figure 5F) and a reduction in proliferation, as indicated by a decrease in the percentage of Ki67+ CD4 and CD8 T cells within the donor population (Figure 5G).
IDO1 previously has been shown to modulate Tregs, both in mediating differentiation and increasing suppressive function.16,17,73,74,75,76 At day 14 post-donor cell transfer, we found an increase in the percentage of both host and donor Tregs with SRC.IDO treatment (Figure 5H). These data suggest that IDO1-mediated protection in aGVHD may be due, at least in part, to reduced proliferation and increased apoptosis of effector cells and an increase in the proportion of Tregs.
Hepatic expression is required for IDO1-mediated efficacy
Most LNPs exhibit a marked tropism for hepatocytes and myeloid cells.77 In mice, the administration of LNP A i.v., which has been used for most of the experiments in this paper, results in high levels of protein in hepatocytes and Kupffer cells of the liver and the red pulp of the spleen, with little protein expressed in other organs (Figure 2; data not shown). Therefore, it was unclear whether SRC.IDO expression in the spleen, liver, or both organs was required for efficacy. We therefore utilized endogenously expressed microRNA (miRNA) to induce cell-specific degradation of our SRC.IDO mRNA. miRNAs have a sequence-specific recognition for target sequences (ts). miR122 is expressed in hepatocytes and will degrade mRNA containing the miR122 ts while miR142, which is expressed in myeloid cells, will similarly degrade mRNA containing the miR142 ts. We designed additional constructs that contained ts for either miR122 (to silence hepatocyte expression) or miR142 (to silence myeloid expression) in the 3′ UTR (Table S1), previously demonstrated to achieve programmable mRNA expression.77
The addition of the miR122 ts did not alter SRC.IDO protein expression in the spleen, as predicted; however, a 75% reduction was observed in the liver (Figures S4A, S4B, and S6A). The inverse was found with SRC.IDO_miR142ts, where there was no change in protein expression in the liver, but a significant reduction of IDO1 in the spleen was found (Figures S4A, S4B, and S6A), confirming the expected expression profiles with SRC.IDO constructs containing different miRNA ts.
We next administered the miRNA-programmable SRC.IDO mRNA into mice to understand the contribution of liver- or myeloid-derived IDO1 on metabolite levels in vivo. In mice treated with SRC.IDO_miR122ts (reduced hepatic expression), relative to PBS- or dead SRC.IDO-treated controls, there was an approximate 5- to 6-fold increase in serum KYN (Figures S4C and S6A). In contrast, mice treated with SRC.IDO_miR142ts (reduced myeloid expression) showed no increase in basal KYN levels relative to dead SRC.IDO-treated mice (Figures S4C and S6A). Collectively, these data argue that myeloid expression of IDO1 is driving the increase in serum KYN levels.
We next examined the contribution of hepatic or splenic IDO1 expression to changes in serum TRP levels. When mice were treated with SRC.IDO_miR122ts (reduced hepatic expression), a reduction in serum TRP levels from baseline was no longer observed, even though these same animals showed elevation in KYN (Figures S4C and S4D). In contrast, administration of SRC.IDO_miR142ts (reduced myeloid expression) showed a marked reduction in TRP, similar to the miRNA-less SRC.IDO construct (Figures S4D and S6A). Thus, while myeloid expression of IDO1 is necessary for regulating KYN levels, hepatic expression of SRC.IDO mRNA is needed to mediate serum TRP depletion.
To determine the role of myeloid or hepatic IDO1 expression in driving efficacy in autoimmunity, we returned to the aGVHD and EAE models. As observed previously, treatment with the miRNA-less SRC.IDO reduced the clinical score in EAE studies (Figures 3 and 6). Incorporation of the miR142 ts into the SRC.IDO mRNA reduced the severity of disease in EAE, albeit not to the same degree as miRNA-less SRC.IDO. In contrast, treatment with SRC.IDO_miR122ts failed to control disease, and the clinical score, disease intensity, mean peak score, and delay in disease onset were similar to the dead SRC.IDO_miR142ts- and PBS-treated mice (Figures 6A–6E). This indicates that hepatocyte expression of IDO1 protein is critical for controlling disease in EAE.
Figure 6.

SRC.IDO1 expression in liver is required for efficacy in autoimmune models
(A) Summary table of expression, metabolite changes, and efficacy of mRNA constructs containing miRNA ts.
(B–E) EAE-induced mice were injected i.v. with 0.5 mg/kg of LNP A-formulated mRNA on days −1 and 6. The mRNAs included either a miR122 or miR142 ts to extinguish expression in either hepatocytes or myeloid cells, respectively. (B and C) Mean clinical score (B) and percentage of disease-free mice (C) were calculated. (D and E) Mean disease intensity (D) and mean peak score (E) were calculated between days 10 and 21. Data are mean and SEM of 12 mice/group and representative of 2 similar experiments. (B) Significance was determined compared to dead SRC.IDO by one-way ANOVA with a secondary Dunnett’s multiple comparisons test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and #p < 0.0001. (C) Significance was determined by log-rank (Mantel-Cox) test to dead SRC.IDO. ∗∗p < 0.01 and ∗∗∗∗p < 0.0001.
(F–I) C57BL/6xDBA/2 F1 recipients received C57BL/6 donor cells to induce aGVHD and were injected i.v. with 0.5 mg/kg of LNP A-formulated mRNA on days 0 and 6. (F and G) On day 14, splenic (F) donor CD8 T cell engraftment and (G) host B cell percentages were determined by flow cytometry. (H and I) The percentages of CD4 T cells expressing FoxP3 within the spleen were determined in the (H) donor and (I) host compartments.
(D–I) Data are individuals and median. Data are n = 3–8 mice/group and representative of 2 similar experiments. Significance was determined by one-way ANOVA with a secondary Tukey’s multiple comparisons test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, and ∗∗∗∗p < 0.0001.
See also Figure S4.
Similar results were obtained in the aGVHD model. At day 14 post-transfer of donor cells, donor CD8 T cell engraftment was reduced with miRNA-less SRC.IDO treatment. Although SRC.IDO_miR142ts-treated mice showed a trend toward reduced engraftment, the difference was not significant (Figure 6F). Mice treated with miRNA-less SRC.IDO and SRC.IDO_miR142ts had a significant retention of the host B cell population, but this protection was not found in SRC.IDO_miR122ts-treated mice (Figure 6G), suggesting that liver targeting plays an important role. It should be noted that variability in the rate of engraftment with this model can make it challenging to determine whether SRC.IDO also impacts engraftment (Figure 5C vs. Figure 6F). Aligning with results shown in Figure 5H, there was an increase in both donor and host Treg frequencies following SRC.IDO treatment (Figures 6H and 6I). This may be driven primarily by hepatic expression of IDO1 since the increased Treg frequency is lost with the SRC.IDO_miR122ts construct, which interferes with hepatic expression of IDO1. These data further confirm the need for hepatic expression for efficacy in rodent autoimmune models.
Discussion
Overexpression of IDO1 protein has been shown previously to modulate autoimmunity and allograft rejection; however, there are still questions regarding the development of IDO1 therapeutically. We improved protein stability by engineering a Src myristoylation site that allows IDO1 to insert into the inner face of the plasma membrane. Optimized constructs were able to reduce severity and delay onset in multiple models of autoimmunity in both mice and rats.
Increased protein stability with anchor
Extracellular anchors, such as antibodies and growth factors, have been utilized in recombinant protein therapeutics to tether the protein to a targeted area.60,61,62,63,64,65 Similarly, Bracho-Sanchez et al. designed a tethered IDO1 protein using a galectin 3 anchor. While IDO1 is normally found intracellularly, after a subcutaneous injection, the anchor sequestered IDO1 to the extracellular space proximal to the injection site. This treatment reduced the severity of multiple inflammatory models where the inflammation was localized to the skin or gums. Without the anchor, there was no protection as the protein diffused out of the area.65 The mRNA approach used here allows IDO1 to be delivered to the cytoplasm, and the addition of the SRC anchor significantly prolonged protein expression, leading to changes in KYN and TRP, resulting in efficacy in systemic models of T cell-driven autoimmunity.
SRC.IDO overexpression mediates protection in models of autoimmunity and allogeneic T cell-mediated disease
Previous studies have shown that overexpression of IDO1 can reduce the rejection of allografts and be efficacious in autoimmune models.29,30,31,32,33,36,37 SRC.IDO treatment had a profound impact on delaying the onset of disease in EAE and rat CIA. In aGVHD, SRC.IDO treatment reduced hallmark indications of disease progression, such as host B cell depletion. In previous studies of lung allograft survival, IDO1 expression was required within the graft prior to transplantation.30,32 Likewise, there has been evidence of prolongation of cardiac and corneal transplant allograft survival when the graft was treated ex vivo with IDO1-containing expression vehicles prior to transplantation.31,33 These ex vivo transfection systems make it challenging to understand the therapeutic window during which IDO1 expression is required for protection. In a model of rat CIA, when IDO1 transducing agents were injected directly into the ankle joints of rats on day 7 after collagen immunization, there was a reduction in ankle swelling and disease progression.36 Similar to these results, we found that systemic administration of SRC.IDO mRNA could be withheld until past the priming phase of disease (Figure 4). In rat CIA, where disease becomes apparent starting on day 10, weekly dosing of SRC.IDO initiated on day 0, 3, 7, or 10 reduced disease score and intensity, suggesting that dosing prior to ankle swelling was sufficient for protection. Moreover, when treatment started on day 14, at which time animals had an average disease score of 9, reduced disease severity in SRC.IDO-treated rats was already evident within 24 h. Our data indicate that SRC.IDO treatment can result in the suppression of immune effector activity even when dosing occurs after initial antigen priming and activation.
The immunomodulatory function of IDO1 is multifaceted. Endogenous IDO1 expression locally depletes TRP and induces KYN and downstream KYN metabolites. which has been shown to reduce effector function of T cells and promote Treg differentiation and suppressor function.7,11,12,16,17,39,73,74,75,76 Within lung allografts from IDO1-treated donors, infiltrating host CD8 T cell proliferation and cytotoxic functions were reduced.30 Additionally, in a model of rat CIA, where IDO1 transducing agents were injected directly into the ankle joints, the reduction in ankle swelling was associated with fewer CD4 T cells within the joints and an increase in T cell apoptosis.36 We observed similar changes in a model of aGVHD. SRC.IDO treatment reduced both engraftment levels of CD8 T cells and the percentage of proliferating Ki67+ CD4 and CD8 T cells within the donor population. Effector function in this model was determined by monitoring the depletion of the host B cell population, which was reduced with SRC.IDO treatment in both the spleen and peripheral blood. This correlated with increased apoptosis of T cells as defined by active caspase-3+ CD4 and CD8 T cells following SRC.IDO treatment. These data suggest that our SRC.IDO mRNA can target effector T cells in aGVHD to reduce disease.
IDO1 has also been shown to promote Treg differentiation and maturation.16,17,73,75,76 In aGVHD, SRC.IDO treatment increased the percentage of Tregs relative to PBS- and dead SRC.IDO-treated animals. As expected, this was recapitulated using the protective SRC.IDO_miR142ts (reduced myeloid expression) mRNA but not with SRC.IDO_miR122ts (reduced hepatic expression) mRNA treatment. Protection in aGVHD is associated with an increased percentage of both donor and host Tregs, but it is not clear if this is due to Treg suppressive function or a lack of expansion of conventional CD4 T cells in a disease setting.
Hepatic SRC.IDO expression results in reduced serum TRP levels
In addition to driving efficacy, SRC.IDO_miR142ts mRNA, which biases SRC.IDO protein expression predominantly to the liver, also depleted serum TRP. One possible explanation for this would be the natural distribution of TRP through the body after ingestion. After absorption and concentration through the digestive system, ingested TRP travels through the portal vein to the liver prior to entering the bloodstream. Therefore, liver-expressed SRC.IDO would have direct access to ingested TRP, allowing for rapid catabolism and leading to decreased serum TRP levels. On the other hand, splenic expression of SRC.IDO requires TRP to be transported out of the bloodstream and into the spleen, which would reduce the TRP available to SRC.IDO to be degraded. These organ-specific differences potentially explain why mice treated with SRC.IDO_miR122ts mRNA did not show a reduction in systemic TRP levels.
Interestingly, even though mice treated with SRC.IDO_miR142ts mRNA (where the majority of SRC.IDO protein is expressed in the liver) have reduced TRP levels, there was no increase in serum KYN levels. Under homeostatic conditions, the liver drives 80%–90% of TRP degradation through the KYN pathway.1 Therefore, hepatocytes have basal expression of all the enzymes required to further degrade KYN into the downstream metabolites. In contrast, myeloid cells have been reported to lack the KYN enzymatic machinery.78 This would lead us to predict that in SRC.IDO_miR142ts-treated mice, where SRC.IDO is predominantly expressed in the liver, TRP was being rapidly metabolized through the entirety of the KYN pathway so that transport of KYN out of hepatocytes and into the bloodstream was not significantly changed. In SRC.IDO_miR122ts-treated mice, where SRC.IDO protein is predominantly expressed in myeloid cells, there was an accumulation of KYN in the serum, possibly due to the lack of KYN pathway enzymes in myeloid cells, allowing for KYN to be exported from the cells instead of being broken down further.1
Outside of the striking differences in the metabolite profiles between SRC.IDO_miR142ts and SRC.IDO_miR122ts, other factors may also have a role in SRC.IDO_miR142ts driving efficacy. Based on the protein expression levels (Figures 2D and 2E), the vast majority of SRC.IDO protein is produced in the liver in mice treated with miRNA-less SRC.IDO. Estimating the average liver weight at 1 g with an expression of 25 ng/mg IDO1 (25 μg total IDO1 protein) and spleen at 100 mg with an expression of 44 ng/mg (4.4 μg total IDO1 protein), approximately 85% of the SRC.IDO protein will be produced by the liver at 24 h. Therefore, the biomass of liver-derived SRC.IDO alone could be the major contributing factor to the efficacy mediated by SRC.IDO_miR142ts but not SRC.IDO_miR122ts. The addition of the miR142 ts to SRC.IDO confirmed the need for hepatic expression for efficacy, but this construct was not as protective as the miRNA-less SRC.IDO. This may be due to the additive effect of additional protein being produced within the spleen or possibly the impact of elevated KYN that was found with miRNA-less SRC.IDO treatment but not the SRC.IDO_miR142ts treatment.
TRP-reduced or -deficient diets have also been used to examine the role of systemic TRP in autoimmune disease. TRP-deficient diets have been shown to block the development of EAE in mice.79 However, mice placed on TRP-restricted diets for several weeks exhibit detrimental side effects, including weight loss and loss of muscle mass.80 Outside of the KYN pathway, TRP is also a precursor to serotonin and melatonin, so depletion of TRP may impact these downstream neurotransmitters and potentially influence mood, cognitive function, and sleep. However, 95% of serotonin is produced within the gut, maintaining intestinal homeostasis and appetite and influencing afferent nerves, while only 5% of serotonin is produced in the brain.81 Since i.v. delivery of mRNA-encoded SRC.IDO affects the catabolism of TRP after the normal uptake in the digestive tract, gut-produced serotonin may not be altered.
Potential implications of SRC.IDO as a therapeutic
This study further supports the conclusion that overexpression of IDO1 can reduce T cell-mediated autoimmunity and inflammation.36,37 Several other groups have identified a beneficial role of IDO1 in transplantation.29,30,31,32,33,34,35 Based on these data, SRC.IDO mRNA treatment could be beneficial in human disease indications with a T cell component, such as solid organ transplant or GVHD. Acute cellular rejection is mediated by the infiltration of T cells and macrophages82 and treated by a combination of calcineurin inhibitors, mTOR inhibitors, anti-proliferatives, glucocorticosteroids, and immunosuppressants.83 In GVHD, there is an activation of T cells within the graft that expand and attack tissues such as the gut, skin, and liver, and treatment relies greatly on corticosteroids.84 For treatment in humans, similar to the data shown in this study, we would predict that repeat dosing would be required to maintain immune regulation with mRNA-encoded SRC.IDO treatment.
A variety of approaches based on cell- or gene-based therapies have been developed to treat autoimmunity. Some of the earliest cell-based therapies relied on exploiting Tregs, which provide both antigen-specific as well as bystander suppression.85 While antigen-specific Tregs are more challenging to purify and expand, they show greater suppression relative to polyclonal Treg populations.86,87 Notably, the phenotype of Tregs can be plastic, with the cells losing suppressive capabilities and acquiring effector function.88,89 More recently, both CAR-T cells and CAR-Tregs have shown promise in the treatment of autoimmune disorders. Early clinical studies in patients with SLE have demonstrated a durable response, including remission, using CD19 CAR-T cells. While an exciting advance, the study sizes are currently too small, with relatively short follow-up periods, to critically evaluate the proportion of patients that will benefit from this approach or if there are unexpected side effects stemming from the continuous presence of the modified T cells. Instead of traditional DNA transfection approaches for CAR-T, the use of RNA transfections is a potential alternative.90 For example, Descartes-08, an anti-BCMA CAR-T, uses RNA to transfect T cells prior to transfer into patients with myasthenia gravis, with beneficial results to patients.91 Additional gene-based approaches such as intra-articular injection of AAV encoding soluble TNFR-Fc fusions led to reduced swelling in 2 out of 11 patients with RA, suggesting other possible therapeutic modalities that could be a significant improvement over standard of care.92 However, these approaches require substantial investments to purify and expand cells needed for modification or purifying sufficient recombinant viruses for administration in the joints.
Compared to other gene- or cell-based therapies, one of the strengths of using LNP delivery of therapeutic RNA in vivo is the lack of need to purify and expand target cells, followed by ex vivo treatment. Moreover, both the restricted tissue tropism of the LNPs and their transient expression can reduce potential target toxicities. RNA does not integrate into DNA, so there is less risk of potential genomic instability, a feature of concern associated with viral transduction approaches. This transient nature of mRNA-based therapies can also be a limitation, specifically in chronic conditions where repeated dosing with mRNA may be required. With mRNA-based therapeutics, there is the opportunity to target the intracellular space and create chimeric molecules with altered properties. This approach has exceptional advantages for treating rare diseases where protein replacement is needed. Conceptually, similar approaches for targeting the cytoplasm can be used to treat autoimmunity. Furthermore, the use of mRNA for the expression of intracellular enzymes in general may possibly reduce anti-drug antibody development compared with treatments using recombinant proteins, where antigen-presenting cells can directly interact with soluble proteins. The use of mRNA-based delivery to overexpress cytoplasmic proteins, such as IDO1, opens up additional avenues for the treatment of both autoimmunity and allograft tolerance.
Limitations of the study
While these studies represent an advancement in the therapeutic use of IDO1 in autoimmunity, there are multiple limitations. The rodent models used in these studies were selected due IDO1 previously being shown to influence disease outcome; however, these models are predominantly T cell mediated, and additional models with a stronger antibody component could be examined in the future. Additionally, the use of prophylactic dosing was only examined in context of rat CIA. To examine the potential of this dosing strategy more fully, it could be utilized in additional models.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Laurie Kenney (laurie.kenney@modernatx.com).
Materials availability
This study did not generate new unique materials or reagents.
Data and code availability
-
•
All data reported in this paper will be provided by the lead contact upon request.
-
•
The study does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Acknowledgments
The authors would like to thank Seymour de Picciotto, of Moderna, for a critical reading of the manuscript. Editorial support was provided by Hannah Lederman, of Caudex, funded by Moderna, Inc. The graphical abstract was made using a BioRender license. All experiments in this manuscript were funded by Moderna, Inc.
Author contributions
L.L.K. wrote the manuscript and designed experiments. R.S.-Y.C., M.N.D., A.W., C.H., L.S., J.J.C., K.Y., and J.D.F. executed experiments and analyzed data. K.L.J., E.H., and P.L.S. contributed to manuscript writing and interpreting results. E.H. conceived of the presented idea. All authors read and approved the final version.
Declaration of interests
L.L.K., R.S.-Y.C., M.N.D., A.W., C.H., L.S., J.J.C., J.D.F., K.L.J., P.L.S., and E.H currently are or have previously been employees of Moderna, Inc., and may hold shares and/or stock options in the company. L.L.K., E.H., P.L.S., and M.N.D. are authors of patents associated with this work (WO2023015261A1 and WO202115567A2).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-IDO1 (D727U) rabbit mAb | Cell Signaling Technology | Cat#68572 RRID:AB_2799750 |
| anti-HSP90 (C45G5) | Cell Signaling Technology | Cat#4877 RRID:AB_2233307 |
| IRDYE 800 CW goat anti-rabbit | LI-COR Biosciences | Cat#926-32211 RRID:AB_621843 |
| TruStain FcX (anti-mouse CD16/32) antibody | Biolegend | Cat#101320 RRID:AB_1574975 |
| BV805 Hamster anti-mouse CD3 (clone 145-2C11) | BD Biosciences | Cat#749276 RRID:AB_2873651 |
| Brilliant Violet 785 Rat anti-mouse CD4 (clone GK1.5) | Biolegend | Cat#100453 RRID:AB_2565843 |
| BD Horizon BUV737 Rat anti-mouse CD8a (clone 53–6.7) | BD Biosciences | Cat#612759 RRID:AB_2870090 |
| Alexa Fluor 700 Rat anti-mouse CD19 (clone 6D5) | Biolegend | Cat#115528 RRID:AB_493735 |
| PE MHC class I (H-2Kd) anti-mouse (clone SF1-1.1.1) | Invitrogen | Cat#12-5957-82 RRID:AB_2043875 |
| Brilliant Violet 421 (H-2Kb) anti-mouse (clone AF6-88.5) | Biolegend | Cat#116525 RRID:AB_2876430 |
| Alexa Fluor 647 anti-mouse/rat/human FoxP3 (clone 150D) | Biolegend | Cat#320014 RRID:AB_439750 |
| Brilliant Violet 605 Rat anti-mouse Ki-67 (clone 16A8) | Biolegend | Cat#652413 RRID:AB_2562664 |
| FitC Rabbit anti-Active Caspase-3 | BD Biosciences | Cat#559341 RRID:AB_397234 |
| Biological samples | ||
| Heat Inactivated fetal bovine serum certified one shot | Gibco | Cat#A38400-01 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbeco’s Modified Media (DMEM) | Gibco | Cat#11965-092 |
| MOG35-55 | AnaSpec | Cat#AS-60130-1 |
| CFA with 2.5 mg/mL Mycobacterium tuberculosis H37Ra | BD Biosciences | Cat#211372 |
| Pertussis toxin | List Labs | Cat#180 |
| Chicken collagen type II | Sigma | Cat#C9301 |
| IFA | BD Biosciences | Cat#210486 |
| RIPA lysis buffer | Thermo Fisher Scientific | Cat#89901 |
| NuPage 4–12% Bis-Tris Midi gels | Invitrogen | Cat#WG1403BX10 |
| iBlot 2 nitrocellulose regular stacks membranes | Invitrogen | Cat#IB23001 |
| Odyssey LI-COR Blocking Buffer | Li-Corbio | Cat#927-60001 |
| 10x lysis buffer | Cell Signaling Technology | Cat#9803 |
| 7mm stainless steel bead | Qiagen | Cat#69990 |
| 70-μm cell strainer | Celltreat | Cat#229484 |
| K2 EDTA-coated tubes | Griener Bio One | Cat#450532 |
| ACK lysis buffer | Gibco | Cat#A1049201 |
| PBS pH 7.4 (1x) | Gibco | Cat#10010-023 |
| Invitrogen eBioscience Foxp3/Transcription Factor Staining Buffer Set | Invitrogen | Cat#05-5523-00 |
| Leica Bond Epitope retrieval solution 1 | Leica Biosystems | Cat#AR0086 |
| Rabbit monoclonal anti-V5 antibody | Cell Signaling Technology | Cat#13202 |
| Bond Polymer Refine Detection system | Lecia Biosystems | Cat#DS9914 |
| Invitrogen eBioscience Fixable Viability Dye eFluor 780 | Invitrogen | Cat#65086514 |
| 20mM Tris Buffer with 8% sucrose, pH 7.4 | Teknova | Cat#3T6100 |
| Critical commercial assays | ||
| Pierce BCA kit | Thermo Fisher Scientific | Cat#23227 |
| Tryptophan Competitive Enzyme Immunoassay | IBL-America | Cat#IB89127R |
| Kynurenine Competitive Enzyme Immunoassay | IBL-America | Cat#IB89190R |
| Human IDO ELISA Kit | Abcam | Cat#ab245710 |
| Experimental models: Cell lines | ||
| Raw 264.7 | ATCC | Cat# TIB-71 RRID:CVCL_0493 |
| Experimental models: Organisms/strains | ||
| C57Bl/6 (female) | Charles River Laboratories | Cat#027 RRID:IMSR_CRL:027 |
| B6D2F1 (female) | Charles River Laboratories | Cat#099 RRID:IMSR_CRL:099 |
| Sprague-Dawley rats (male) | Charles River Laboratories | Cat#001 RRID:RGD_734476 |
| Software and algorithms | ||
| FlowJo | BD Life Science | https://www.flowjo.com/ |
| Graphpad Prism 10.0 | Graphpad | https://www.graphpad.com/ |
| ImageJ | ImageJ | https://imagej.net/ij/ |
| HALO image analysis software | Indica Labs | https://indicalab.com/ |
Experimental model and study participant details
Cells
Raw 264.7 cells were obtained from ATCC (TIB-71) and maintained in DMEM (Gibco, catalog no. 11965-092) with 10% heat inactivated fetal bovine serum (Gibco, catalog no. A38400-01) at 37C and 5% CO2.
Mice
Female 8-week-old C57BL/6 and C57BL/6 x DBA/2 F1 (B6D2F1) were purchased from Charles River Laboratories (#027 and #099) and maintained at Moderna in Cambridge, MA. Mice housed in groups of four or five in specific pathogen-free housing in accordance with Moderna Institutional Animal Care and Use Committee at Moderna (IACUC). All animal experiments were performed in accordance with the federal, state, local, and institutional IACUC polices. These policies are determined by the United States Department of Agriculture and are further enforced by the Massachusetts Society for the Prevention of Cruelty to Animals and Cambridge Commission. Moderna is regularly monitored to ensure compliance with these rules.
To run the EAE model 12-week-old C57BL/6 female mice were injected on day 0 at two sites on the rear flanks with a total of 100 μL of emulsion containing 100 μg of myelin oligodendrocyte glycoprotein peptide (MOG)35-55 peptide emulsified in CFA with 2.5 mg/mL Mycobacterium tuberculosis H37Ra. Mice were also injected with 100 μL of 6 μg/mL Pertussis toxin i.v. on days 0 and 2. Formulated mRNA was administered i.v. to mice on day −1 and day 6. Starting on day 10, mice were scored and weighed daily. The scoring system was as follows: 0 – Normal, no overt signs of disease, 1 tail paresis, 2 – righting reflex impaired, 3 – partial hindlimb paralysis, 4 – complete hindlimb paralysis or absence of ambulation, 5 – complete hindlimb paralysis with front limb paresis. Mean disease intensity was calculated as the area under the curve for the clinical score for each individual animal and the mean peak score was determined as the mean of the peak scores of individuals within each group. Experiments contained 12 mice/group.
For the aGVHD model spleen and lymph nodes were isolated from 8-week-old female C57BL/6J mice (Charles River) and processed to a single-cell suspension by passing through a sterile 70-μm cell strainer (Celltreat, Catalog No. 229484) using the end of a sterile syringe plunger. Single-cell suspensions were then lysed in ACK lysis buffer (Gibco, Catalog No. A1049201), counted and resuspended in PBS (Gibco, catalog no. 10010-023) at 500 × 106 cells/mL. Recipient female C57BL/6 x DBA/2 F1 (B6D2F1) mice (Charles River) were injected i.v. with 50 × 106 C57BL/6 cells in 100ul. Mice were injected with formulated mRNA at 0.5 mg/kg i.v. on days 0 and 6. Peripheral blood was isolated on day 7. Mice were euthanized and spleen and peripheral blood were isolated on day 14. Experiments were performed with 3–8 mice per group.
Rats
Male Sprague-Dawley rats were purchased from Charles River Labs (#001) and housed in groups of two or three in specific pathogen-free housing in accordance with Pharma Models IACUC. All animal experiments were performed in accordance with the federal, state, local, and institutional IACUC polices. These policies are determined by the United States Department of Agriculture and are further enforced by the Massachusetts Society for the Prevention of Cruelty to Animals and Cambridge Commission. Moderna is regularly monitored to ensure compliance with these rules.
To run the CIA model 8-week-old male Sprague-Dawley rats were injected intradermally with chicken collagen type II (Sigma, Catalog no. C9301) in IFA on day 0. Formulated mRNA was administered i.v. at the timepoints indicated in Figure 4. After injection, animals were scored daily from day 10 to day 21. Evaluation of disease involved scoring each paw on the following scale: 0 – no swelling or erythema, 1 – slight swelling and/or erythema, 2 – low to moderate swelling, 3 – pronounced swelling with limited joint usage, 4 – excessive swelling with joint rigidity. Each animal had a possible total disease score of up to 16. Mean disease intensity was calculated as the area under the curve for the clinical score for each individual animal and the mean peak score was determined as the mean of the peak scores of individuals within each group. Experiments contained 5–8 rats per group.
Non-human primates
Male cynomolgus monkeys were between the ages of 2 and 4 years and were housed at a Charles River facility. The animal facility is specific pathogen-free and the control animals were co-housed or held in the same holding room. NHP are group house with up to 3 animals of the same sex and same dosing group housed together. Housing set-up is as specified in the USDA Animal Welfare Act (9 CFR, Parts 1, 2 and 3) and as described in the Guide for the Care and Use of Laboratory Animals. All animal experiments were performed in accordance with the federal, state, local, and institutional IACUC polices. These policies are determined by the United States Department of Agriculture and are further enforced by the Massachusetts Society for the Prevention of Cruelty to Animals and Cambridge Commission. Moderna is regularly monitored to ensure compliance with these rules.
Method details
mRNA production and formulation
hsIDO, SRC.IDO, and RAS.IDO mRNA encoding the H. sapiens IDO1 wild-type or a chimeric molecule was fused to a lipidation signal to facilitate protein anchoring to the cytoplasmic side of the cell membrane. This included either the 21 amino acid K-Ras tail containing a farnesylation site added to the C-terminus (RAS.IDO) or the 14 amino acids of SRC containing a myristoylation site added to the N-terminus (SRC.IDO) of human IDO1. A catalytically inactive IDO1 (Dead IDO) construct was also designed and tested. The Dead IDO1 protein is not enzymatically active due to a single phenylalanine-to-alanine point mutation at position 226 (F226A).69 mRNA containing target sequences (ts) for miR122 or miR142 included 3x miR ts in the 3′ untranslated region (UTR).77 Inclusion of the microRNA ts allowed for silencing expression in hepatocytes or myeloid cells, respectively. For immunohistochemistry, a C-terminal V5-tagged version of SRC.IDO was used (Figure 2A). The complete sequence of the mRNA constructs can be found in Table S1.
The resulting mRNA constructs were synthesized by in vitro transcription. A plasmid encoding the T7 RNA polymerase promoter followed by 5′UTR, open reading frame, 3′UTR, and polyA tail was overexpressed in E. coli, linearized, and purified to homogeneity. The mRNA was synthesized by using a modified T7 RNA polymerase for in vitro transcription, where uridine triphosphate was substituted completely with N1-methyl-pseudouridine triphosphate.93 A donor methyl group S-adenosylmethionine was added to the methylated capped RNA (Cap 0) resulting in a Cap 1 structure that was utilized to improve translation efficiency. After the transcription reaction, mRNAs were purified, buffer exchanged into sodium citrate, and stored at −20°C until use. Further enhancements included codon optimization and addition of inverted deoxythymidine to interfere with de-adenylation of the mRNA and prolong mRNA stability.
Formulation of mRNA was performed through modified ethanol-drop nanoprecipitation process as described previously.94 Ionizable, structural, helper and polyethylene glycol lipids were mixed with mRNA. The mixture was neutralized with Tris-Cl pH7.5 and filter sterilized. An analytical characterization assay, which included particle size and polydispersity, encapsulation, and endotoxin content, had to meet predefined specifications before the material was deemed acceptable for in vivo study use. Final particle size was between 50 nm and 120 nm, encapsulation was greater than 80%, and endotoxin content <10 EU/ml.
Protein expression by Western blot
Cell pellets from RAW 264.7 cells (mouse macrophage) transfected with IDO1 LNPs were lysed in 1× RIPA lysis buffer (ThermoFisherScientific, Catalog No. 89901) on ice for 10 min. Lysates were cleared by centrifugation, and supernatant was collected. Protein was quantified using the Pierce BCA kit (ThermoFisher Scientific, Catalog No. 23227). 20ug of protein was run per sample. Samples were separated by NuPage 4–12% Bis-Tris Midi gels (Invitrogen, Catalog No. WG1403BX10) and transferred to iBlot 2 nitrocellulose regular stacks membranes (Invitrogen, Catalog No. IB23001). Nonspecific binding was prevented by incubation with Odyssey LI-COR Blocking Buffer. Primary antibodies were incubated for a minimum of 18 h at 4°C. Primary anti–mouse antibodies used for immunoblot analysis included: anti-IDO1 (D727U) rabbit mAb (Cell Signaling Technology, Catalog No. 68572) and anti-HSP90 (C45G5) (Cell Signaling Technology, Catalog No. 4877). Subsequent incubation with the secondary antibody IRDYE 800 CW goat anti-rabbit (Li-Cor, Catalog No. 926–32211) at 1:20,000 in 1xTBST for 30 min at RT. Membranes were imaged with Odyssey CLx Infrared Imaging System (LI-COR Biotechnology). Immunoblot quantification was performed using ImageJ software. For quantification, IDO1 protein levels were normalized to normalized HSP90. HSP90 protein levels for individual samples were normalized to the median HSP90 protein level for all samples on a single membrane.
LNP administration and selection
Mice and rats were injected with LNP diluted to in 20mM Tris Buffer with 8% sucrose, pH 7.4 (Teknova, catalog no. 3T6100) or PBS pH 7.4 (Gibco, catalog no. 10010-023) through tail vein bolus intravenous injections. NHP were infused through a peripheral vein with LNP diluted in Tris sucrose at a dose volume of 10 mL/kg over a period of 60 min. The infusion used for NHP was utilized to replicate the route of administration that would be used in the clinic. The LNP, either LNP B or LNP A was selected for each experiment. In initial mouse experiments we used myeloid-tropic LNP B (Figure 1). We determined we had better systemic changes in TRP and KYN with the use of LNP A, which delivers mRNA to both myeloid cells and hepatocytes so we switched LNP for the remainder of the rodent studies (Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, S1, and S3). For LNP biodistribution there are species specific differences. While LNP B does not deliver well the mouse liver in NHP there is delivery to both spleen and liver. Additionally, LNP B is a clinically relevant LNP. For these reasons we selected LNP B for the NHP studies (Figure S2).
Assessment of KYN and TRP levels by ELISA
KYN and TRP levels were determined from transfected cell supernatant at 24 h and plasma or serum collected from individual mice within each study using the Tryptophan Competitive Enzyme Immunoassay (IBL-America, Catalog No. IB89127R) or the Kynurenine Competitive Enzyme Immunoassay (IBL-America, Catalog No. IB89190R) according to the manufacturer’s instructions.
Assessment of IDO1 protein by ELISA
Spleen and liver homogenates were prepared from weighed portions of each organ. Organ pieces were placed into 2 mL Eppendorf tubes containing 0.5–1.0 mL 1X lysis buffer (Cell Signaling Technology, Catalog no. 9803) and a single 7mm stainless steel bead (Qiagen, Catalog No. 69990). Samples were homogenized using the Qiagen TissueLyser II (Catalog No. 85300). Samples were stored at −80°C prior to use. Human IDO1 enzyme was determined by Human IDO ELISA Kit (Abcam, Catalog No. ab245710) according to the manufacturer’s instructions.
Flow cytometry for aGVHD
To prepare single-cell suspensions, spleens were collected and passed through a 70-μm cell strainer (Celltreat, Catalog No. 229484) using the end of a sterile syringe plunger. Whole blood was collected by cardiac puncture into K2 EDTA-coated tubes (Griener Bio One, Catalog No. 450532). Single-cell suspensions were then lysed in ACK lysis buffer (Gibco, Catalog No. A1049201). For cell surface staining, cells were pre-incubated with TruStain FcX (anti-mouse CD16/32) antibody (Biolegend, Catalog No. 101320, clone 93), diluted in FACS buffer (PBS +2% FBS), for 5 min at room temperature. Surface staining was performed with fluorochrome-conjugated antibodies diluted in FACS buffer for 30–60 min at 4°C. For intracellular staining, cells were washed with FACS buffer, and fixed and permeabilized with the Invitrogen eBioscience Foxp3/Transcription Factor Staining Buffer Set (Invitrogen, Catalog No. 05-5523-00) for 30 min at 4°C. Cells were then washed with 1x permeabilization buffer (Invitrogen, Catalog No. 05-5523-00) and stained with intracellular antibodies diluted in permeabilization buffer overnight at 4°C. Flow cytometry data were acquired on a Cytek Aurora cytometer (Cytek Biosciences) and analyzed using FlowJo software (BD Life Sciences). Staining was performed using the antibodies shown in Table S2.
Immunohistochemistry for Ido1
Liver and spleen tissues were collected at 6, 24, 48, 72, 96, 120, 144, 168, and 192 h post dosing and fixed in 10% neutral-buffered formalin for 48 h. Tissues went through routine paraffin processing and embedded into paraffin blocks. Tissue blocks were then cut into sections of 5-micron thickness and mounted onto charged glass slides.
Immunohistochemistry was performed for the detection of the expression of SRC.IDO. The immunohistochemistry detection system is a biotin-free, polymeric horseradish peroxidase (HRP)-linker antibody conjugate system run on the Leica Bond RX autostainer. Slides were baked, dewaxed with subsequent antigen retrieval performed for 20 min using Leica Bond Epitope retrieval solution 1 (Leica Biosystems, Catalog no. AR0086). Using a Rabbit monoclonal anti-V5 antibody (Cell Signaling Technology, Catalog no. 13202) at a 1:400 dilution along with the Bond Polymer Refine Detection system (Lecia Biosystems, Catalog no. DS9914), followed by hematoxylin and bluing reagent counterstain (Leica Microsystems). Slides were imaged using 3D Histech Panoramic 250 whole-slide scanner and image analysis was completed with HALO image analysis software.
Quantification and statistical analysis
Data was analyzed by one-way ANOVA, two-way ANOVA with either secondary Dunnet’s or Tukey’s multiple comparisons tests as indicated in the figure legend or by log rank (Mantel-Cox) test compared to Dead SRC.IDO. All statistical analysis was completed in Graphpad prism. In vitro studies were run in triplicate and in vivo studies examined between 3 and 12 animals per group.
Published: September 6, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101717.
Supplemental information
References
- 1.Badawy A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017;10 doi: 10.1177/1178646917691938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ye Z., Yue L., Shi J., Shao M., Wu T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J. Cancer. 2019;10:2771–2782. doi: 10.7150/jca.31727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.2015. Targeting the Broadly Pathogenic Kynurenine Pathway. [DOI] [Google Scholar]
- 4.Pallotta M.T., Rossini S., Suvieri C., Coletti A., Orabona C., Macchiarulo A., Volpi C., Grohmann U. Indoleamine 2,3-dioxygenase 1 (IDO1): an up-to-date overview of an eclectic immunoregulatory enzyme. FEBS J. 2022;289:6099–6118. doi: 10.1111/febs.16086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones S.P., Guillemin G.J., Brew B.J. 2015. Targeting the Broadly Pathogenic Kynurenine Pathway; pp. 257–272. [DOI] [Google Scholar]
- 6.Munn D.H., Sharma M.D., Baban B., Harding H.P., Zhang Y., Ron D., Mellor A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity. 2005;22:633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 7.Munn D.H., Shafizadeh E., Attwood J.T., Bondarev I., Pashine A., Mellor A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Opitz C.A., Litzenburger U.M., Sahm F., Ott M., Tritschler I., Trump S., Schumacher T., Jestaedt L., Schrenk D., Weller M., et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 9.Yan Y., Zhang G.-X., Gran B., Fallarino F., Yu S., Li H., Cullimore M.L., Rostami A., Xu H. IDO Upregulates Regulatory T Cells via Tryptophan Catabolite and Suppresses Encephalitogenic T Cell Responses in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2010;185:5953–5961. doi: 10.4049/jimmunol.1001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mezrich J.D., Fechner J.H., Zhang X., Johnson B.P., Burlingham W.J., Bradfield C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010;185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munn D.H., Sharma M.D., Johnson T.S., Rodriguez P. IDO, PTEN-expressing Tregs and control of antigen-presentation in the murine tumor microenvironment. Cancer Immunol. Immunother. 2017;66:1049–1058. doi: 10.1007/s00262-017-2010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amobi-McCloud A., Muthuswamy R., Battaglia S., Yu H., Liu T., Wang J., Putluri V., Singh P.K., Qian F., Huang R.-Y., et al. IDO1 Expression in Ovarian Cancer Induces PD-1 in T Cells via Aryl Hydrocarbon Receptor Activation. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.678999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terness P., Bauer T.M., Röse L., Dufter C., Watzlik A., Simon H., Opelz G. Inhibition of Allogeneic T Cell Proliferation by Indoleamine 2,3-Dioxygenase–expressing Dendritic Cells. J. Exp. Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dagenais-Lussier X., Aounallah M., Mehraj V., El-Far M., Tremblay C., Sekaly R.-P., Routy J.-P., van Grevenynghe J. Kynurenine Reduces Memory CD4 T-Cell Survival by Interfering with Interleukin-2 Signaling Early during HIV-1 Infection. J. Virol. 2016;90:7967–7979. doi: 10.1128/jvi.00994-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mellor A.L., Baban B., Chandler P., Marshall B., Jhaver K., Hansen A., Koni P.A., Iwashima M., Munn D.H. Cutting Edge: Induced Indoleamine 2,3 Dioxygenase Expression in Dendritic Cell Subsets Suppresses T Cell Clonal Expansion. J. Immunol. 2003;171:1652–1655. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 16.van der Marel A.P.J., Samsom J.N., Greuter M., van Berkel L.A., O’Toole T., Kraal G., Mebius R.E. Blockade of IDO Inhibits Nasal Tolerance Induction. J. Immunol. 2007;179:894–900. doi: 10.4049/jimmunol.179.2.894. [DOI] [PubMed] [Google Scholar]
- 17.Matteoli G., Mazzini E., Iliev I.D., Mileti E., Fallarino F., Puccetti P., Chieppa M., Rescigno M. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut. 2010;59:595–604. doi: 10.1136/gut.2009.185108. [DOI] [PubMed] [Google Scholar]
- 18.Ravishankar B., Liu H., Shinde R., Chandler P., Baban B., Tanaka M., Munn D.H., Mellor A.L., Karlsson M.C.I., McGaha T.L. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc. Natl. Acad. Sci. USA. 2012;109:3909–3914. doi: 10.1073/pnas.1117736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakurai K., Zou J.-P., Tschetter J.R., Ward J.M., Shearer G.M. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002;129:186–196. doi: 10.1016/s0165-5728(02)00176-5. [DOI] [PubMed] [Google Scholar]
- 20.Wetzel L.A., Hurtado M., Kaswan Z.A.M., McCusker R.H., Steelman A.J. Deletion of indoleamine 2,3 dioxygenase (Ido)1 but not Ido2 exacerbates disease symptoms of MOG35-55-induced experimental autoimmune encephalomyelitis. Brain. Behav., Immun. - Heal. 2020;7 doi: 10.1016/j.bbih.2020.100116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takamatsu M., Hirata A., Ohtaki H., Hoshi M., Hatano Y., Tomita H., Kuno T., Saito K., Hara A. IDO1 Plays an Immunosuppressive Role in 2,4,6-Trinitrobenzene Sulfate–Induced Colitis in Mice. J. Immunol. 2013;191:3057–3064. doi: 10.4049/jimmunol.1203306. [DOI] [PubMed] [Google Scholar]
- 22.Jasperson L.K., Bucher C., Panoskaltsis-Mortari A., Mellor A.L., Munn D.H., Blazar B.R. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood. 2009;114:5062–5070. doi: 10.1182/blood-2009-06-227587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jasperson L.K., Bucher C., Panoskaltsis-Mortari A., Taylor P.A., Mellor A.L., Munn D.H., Blazar B.R. Indoleamine 2,3-dioxygenase is a critical regulator of acute graft-versus-host disease lethality. Blood. 2008;111:3257–3265. doi: 10.1182/blood-2007-06-096081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Criado G., Šimelyte E., Inglis J.J., Essex D., Williams R.O. Indoleamine 2,3 dioxygenase–mediated tryptophan catabolism regulates accumulation of Th1/Th17 cells in the joint in collagen-induced arthritis. Arthritis Rheum. 2009;60:1342–1351. doi: 10.1002/art.24446. [DOI] [PubMed] [Google Scholar]
- 25.Ju J.-M., Nam G., Lee Y.-K., Jung M., Chang H., Kim W., Shon W.J., Lim J.Y., Kim J.Y., Chang J., et al. IDO1 scavenges reactive oxygen species in myeloid-derived suppressor cells to prevent graft-versus-host disease. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2011170118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poirier N., Azimzadeh A.M., Zhang T., Dilek N., Mary C., Nguyen B., Tillou X., Wu G., Reneaudin K., Hervouet J., et al. Inducing CTLA-4–Dependent Immune Regulation by Selective CD28 Blockade Promotes Regulatory T Cells in Organ Transplantation. Sci. Transl. Med. 2010;2 doi: 10.1126/scitranslmed.3000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sucher R., Fischler K., Oberhuber R., Kronberger I., Margreiter C., Ollinger R., Schneeberger S., Fuchs D., Werner E.R., Watschinger K., et al. IDO and Regulatory T Cell Support Are Critical for Cytotoxic T Lymphocyte-Associated Ag-4 Ig-Mediated Long-Term Solid Organ Allograft Survival. J. Immunol. 2012;188:37–46. doi: 10.4049/jimmunol.1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guillonneau C., Hill M., Hubert F.-X., Chiffoleau E., Hervé C., Li X.-L., Heslan M., Usal C., Tesson L., Ménoret S., et al. CD40Ig treatment results in allograft acceptance mediated by CD8+CD45RClow T cells, IFN-γ, and indoleamine 2,3-dioxygenase. J. Clin. Invest. 2007;117:1096–1106. doi: 10.1172/jci28801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu H., Liu L., Fletcher B.S., Visner G.A. Novel Action of Indoleamine 2,3-Dioxygenase Attenuating Acute Lung Allograft Injury. Am. J. Respir. Crit. Care Med. 2006;173:566–572. doi: 10.1164/rccm.200509-1413oc. [DOI] [PubMed] [Google Scholar]
- 30.Liu H., Liu L., Liu K., Bizargity P., Hancock W.W., Visner G.A. Reduced Cytotoxic Function of Effector CD8+ T Cells Is Responsible for Indoleamine 2,3-Dioxygenase-Dependent Immune Suppression. J. Immunol. 2009;183:1022–1031. doi: 10.4049/jimmunol.0900408. [DOI] [PubMed] [Google Scholar]
- 31.Li J., Meinhardt A., Roehrich M.-E., Golshayan D., Dudler J., Pagnotta M., Trucco M., Vassalli G. Indoleamine 2,3-dioxygenase gene transfer prolongs cardiac allograft survival. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H3415–H3423. doi: 10.1152/ajpheart.00532.2007. [DOI] [PubMed] [Google Scholar]
- 32.Swanson K.A., Zheng Y., Heidler K.M., Mizobuchi T., Wilkes D.S. CDllc+ Cells Modulate Pulmonary Immune Responses by Production of Indoleamine 2,3-Dioxygenase. Am. J. Respir. Cell Mol. Biol. 2004;30:311–318. doi: 10.1165/rcmb.2003-0268oc. [DOI] [PubMed] [Google Scholar]
- 33.Beutelspacher S.C., Pillai R., Watson M.P., Tan P.H., Tsang J., McClure M.O., George A.J.T., Larkin D.F.P. Function of indoleamine 2,3-dioxygenase in corneal allograft rejection and prolongation of allograft survival by over-expression. Eur. J. Immunol. 2006;36:690–700. doi: 10.1002/eji.200535238. [DOI] [PubMed] [Google Scholar]
- 34.Yu G., Dai H., Chen J., Duan L., Gong M., Liu L., Xiong P., Wang C.Y., Fang M., Gong F. Gene delivery of indoleamine 2,3-dioxygenase prolongs cardiac allograft survival by shaping the types of T-cell responses. J. Gene Med. 2008;10:754–761. doi: 10.1002/jgm.1201. [DOI] [PubMed] [Google Scholar]
- 35.Yu G., Fang M., Gong M., Liu L., Zhong J., Feng W., Xiong P., Wang C.-Y., Gong F. Steady state dendritic cells with forced IDO expression induce skin allograft tolerance by upregulation of regulatory T cells. Transpl. Immunol. 2008;18:208–219. doi: 10.1016/j.trim.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 36.Chen S.-Y., Wu C.-L., Lai M.-D., Lin C.-C., Yo Y.-T., Jou I.-M., Lee C.-H., Weng C.-T., Shiau A.-L., Wang C.-R. Amelioration of Rat Collagen-Induced Arthritis Through CD4+ T Cells Apoptosis and Synovial Interleukin-17 Reduction by Indoleamine 2,3-Dioxygenase Gene Therapy. Hum. Gene Ther. 2011;22:145–154. doi: 10.1089/hum.2009.217. [DOI] [PubMed] [Google Scholar]
- 37.Elizei S.S., Pakyari M., Ghoreishi M., Kilani R., Mahmoudi S., Ghahary A. IDO-expressing Fibroblasts Suppress the Development of Imiquimod-induced Psoriasis-like Dermatitis. Cell Transplant. 2018;27:557–570. doi: 10.1177/0963689718757482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tardito S., Negrini S., Conteduca G., Ferrera F., Parodi A., Battaglia F., Kalli F., Fenoglio D., Cutolo M., Filaci G. Indoleamine 2,3 dioxygenase gene polymorphisms correlate with CD8+ Treg impairment in systemic sclerosis. Hum. Immunol. 2013;74:166–169. doi: 10.1016/j.humimm.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 39.Pallotta M.T., Orabona C., Volpi C., Vacca C., Belladonna M.L., Bianchi R., Servillo G., Brunacci C., Calvitti M., Bicciato S., et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011;12:870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 40.Anquetil F., Mondanelli G., Gonzalez N., Rodriguez Calvo T., Zapardiel Gonzalo J., Krogvold L., Dahl-Jørgensen K., Van den Eynde B., Orabona C., Grohmann U., von Herrath M.G. Loss of IDO1 Expression From Human Pancreatic β-Cells Precedes Their Destruction During the Development of Type 1 Diabetes. Diabetes. 2018;67:1858–1866. doi: 10.2337/db17-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jackson L.A., Anderson E.J., Rouphael N.G., Roberts P.C., Makhene M., Coler R.N., McCullough M.P., Chappell J.D., Denison M.R., Stevens L.J., et al. An mRNA Vaccine against SARS-CoV-2 — Preliminary Report. N. Engl. J. Med. 2020;383:1920–1931. doi: 10.1056/nejmoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mulligan M.J., Lyke K.E., Kitchin N., Absalon J., Gurtman A., Lockhart S., Neuzil K., Raabe V., Bailey R., Swanson K.A., et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature. 2020;586:589–593. doi: 10.1038/s41586-020-2639-4. [DOI] [PubMed] [Google Scholar]
- 43.Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Pérez Marc G., Moreira E.D., Zerbini C., et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/nejmoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aliprantis A.O., Shaw C.A., Griffin P., Farinola N., Railkar R.A., Cao X., Liu W., Sachs J.R., Swenson C.J., Lee H., et al. A phase 1, randomized, placebo-controlled study to evaluate the safety and immunogenicity of an mRNA-based RSV prefusion F protein vaccine in healthy younger and older adults. Hum. Vaccines Immunother. 2021;17:1248–1261. doi: 10.1080/21645515.2020.1829899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cafri G., Gartner J.J., Zaks T., Hopson K., Levin N., Paria B.C., Parkhurst M.R., Yossef R., Lowery F.J., Jafferji M.S., et al. mRNA vaccine–induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Invest. 2020;130:5976–5988. doi: 10.1172/jci134915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao J., Choi M., Guadagnin E., Soty M., Silva M., Verzieux V., Weisser E., Markel A., Zhuo J., Liang S., et al. mRNA therapy restores euglycemia and prevents liver tumors in murine model of glycogen storage disease. Nat. Commun. 2021;12:3090. doi: 10.1038/s41467-021-23318-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei G., Cao J., Huang P., An P., Badlani D., Vaid K.A., Zhao S., Wang D.Q.-H., Zhuo J., Yin L., et al. Synthetic human ABCB4 mRNA therapy rescues severe liver disease phenotype in a BALB/c.Abcb4−/− mouse model of PFIC3. J. Hepatol. 2021;74:1416–1428. doi: 10.1016/j.jhep.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.An D., Frassetto A., Jacquinet E., Eybye M., Milano J., DeAntonis C., Nguyen V., Laureano R., Milton J., Sabnis S., et al. Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. EBioMedicine. 2019;45:519–528. doi: 10.1016/j.ebiom.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu X., Yin L., Theisen M., Zhuo J., Siddiqui S., Levy B., Presnyak V., Frassetto A., Milton J., Salerno T., et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019;104:625–637. doi: 10.1016/j.ajhg.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang L., Park J.-S., Yin L., Laureano R., Jacquinet E., Yang J., Liang S., Frassetto A., Zhuo J., Yan X., et al. Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic acidemia. Nat. Commun. 2020;11:5339. doi: 10.1038/s41467-020-19156-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu X., Wang X., Liao Y.-P., Luo L., Xia T., Nel A.E. Use of a Liver-Targeting Immune-Tolerogenic mRNA Lipid Nanoparticle Platform to Treat Peanut-Induced Anaphylaxis by Single- and Multiple-Epitope Nucleotide Sequence Delivery. ACS Nano. 2023;17:4942–4957. doi: 10.1021/acsnano.2c12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krienke C., Kolb L., Diken E., Streuber M., Kirchhoff S., Bukur T., Akilli-Öztürk Ö., Kranz L.M., Berger H., Petschenka J., et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science. 2021;371:145–153. doi: 10.1126/science.aay3638. [DOI] [PubMed] [Google Scholar]
- 53.de Picciotto S., DeVita N., Hsiao C.J., Honan C., Tse S.-W., Nguyen M., Ferrari J.D., Zheng W., Wipke B.T., Huang E. Selective activation and expansion of regulatory T cells using lipid encapsulated mRNA encoding a long-acting IL-2 mutein. Nat. Commun. 2022;13:3866. doi: 10.1038/s41467-022-31130-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki Y., Katsurada Y., Hyodo K. Differences and Similarities of the Intravenously Administered Lipid Nanoparticles in Three Clinical Trials: Potential Linkage between Lipid Nanoparticles and Extracellular Vesicles. Mol. Pharm. 2023;20:4883–4892. doi: 10.1021/acs.molpharmaceut.3c00547. [DOI] [PubMed] [Google Scholar]
- 55.LoPresti S.T., Arral M.L., Chaudhary N., Whitehead K.A. The replacement of helper lipids with charged alternatives in lipid nanoparticles facilitates targeted mRNA delivery to the spleen and lungs. J. Contr. Release. 2022;345:819–831. doi: 10.1016/j.jconrel.2022.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki Y., Ishihara H. Structure, activity and uptake mechanism of siRNA-lipid nanoparticles with an asymmetric ionizable lipid. Int. J. Pharm. 2016;510:350–358. doi: 10.1016/j.ijpharm.2016.06.124. [DOI] [PubMed] [Google Scholar]
- 57.Lokugamage M.P., Gan Z., Zurla C., Levin J., Islam F.Z., Kalathoor S., Sato M., Sago C.D., Santangelo P.J., Dahlman J.E. Mild Innate Immune Activation Overrides Efficient Nanoparticle-Mediated RNA Delivery. Adv. Mater. 2020;32 doi: 10.1002/adma.201904905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parhiz H., Brenner J.S., Patel P.N., Papp T.E., Shahnawaz H., Li Q., Shi R., Zamora M.E., Yadegari A., Marcos-Contreras O.A., et al. Added to pre-existing inflammation, mRNA-lipid nanoparticles induce inflammation exacerbation (IE) J. Contr. Release. 2022;344:50–61. doi: 10.1016/j.jconrel.2021.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen K.-H., Lundy D.J., Toh E.K.-W., Chen C.-H., Shih C., Chen P., Chang H.-C., Lai J.J., Stayton P.S., Hoffman A.S., Hsieh P.C.H. Nanoparticle distribution during systemic inflammation is size-dependent and organ-specific. Nanoscale. 2015;7:15863–15872. doi: 10.1039/c5nr03626g. [DOI] [PubMed] [Google Scholar]
- 60.Ishihara J., Fukunaga K., Ishihara A., Larsson H.M., Potin L., Hosseinchi P., Galliverti G., Swartz M.A., Hubbell J.A. Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aan0401. [DOI] [PubMed] [Google Scholar]
- 61.Ishihara J., Ishihara A., Potin L., Hosseinchi P., Fukunaga K., Damo M., Gajewski T.F., Swartz M.A., Hubbell J.A. Improving Efficacy and Safety of Agonistic Anti-CD40 Antibody Through Extracellular Matrix Affinity. Mol. Cancer Therapeut. 2018;17:2399–2411. doi: 10.1158/1535-7163.mct-18-0091. [DOI] [PubMed] [Google Scholar]
- 62.Kitajima T., Terai H., Ito Y. A fusion protein of hepatocyte growth factor for immobilization to collagen. Biomaterials. 2007;28:1989–1997. doi: 10.1016/j.biomaterials.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 63.Martino M.M., Briquez P.S., Güç E., Tortelli F., Kilarski W.W., Metzger S., Rice J.J., Kuhn G.A., Müller R., Swartz M.A., Hubbell J.A. Growth Factors Engineered for Super-Affinity to the Extracellular Matrix Enhance Tissue Healing. Science. 2014;343:885–888. doi: 10.1126/science.1247663. [DOI] [PubMed] [Google Scholar]
- 64.Kitajima T., Sakuragi M., Hasuda H., Ozu T., Ito Y. A chimeric epidermal growth factor with fibrin affinity promotes repair of injured keratinocyte sheets. Acta Biomater. 2009;5:2623–2632. doi: 10.1016/j.actbio.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 65.Bracho-Sanchez E., Rocha F.G., Bedingfield S.K., Partain B.D., Macias S.L., Brusko M.A., Colazo J.M., Fettis M.M., Farhadi S.A., Helm E.Y., et al. Suppression of local inflammation via galectin-anchored indoleamine 2,3-dioxygenase. Nat. Biomed. Eng. 2023;7:1156–1169. doi: 10.1038/s41551-023-01025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wright M.H., Heal W.P., Mann D.J., Tate E.W. Protein myristoylation in health and disease. J. Chem. Biol. 2010;3:19–35. doi: 10.1007/s12154-009-0032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Resh M.D. Palmitoylation of Ligands, Receptors, and Intracellular Signaling Molecules. Sci. STKE. 2006;2006 doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- 68.Chang Y.-H. Impact of Protein Nα-Modifications on Cellular Functions and Human Health. Life. 2023;13:1613. doi: 10.3390/life13071613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sugimoto H., Oda S.i., Otsuki T., Hino T., Yoshida T., Shiro Y. Crystal structure of human indoleamine 2,3-dioxygenase: Catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. USA. 2006;103:2611–2616. doi: 10.1073/pnas.0508996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Elgar M.A., Harvey P.H. Basal Metabolic Rates in Mammals: Allometry, Phylogeny and Ecology. Funct. Ecol. 1987;1:25. doi: 10.2307/2389354. [DOI] [Google Scholar]
- 71.Lemos H., Huang L., Chandler P.R., Mohamed E., Souza G.R., Li L., Pacholczyk G., Barber G.N., Hayakawa Y., Munn D.H., Mellor A.L. Activation of the STING Adaptor Attenuates Experimental Autoimmune Encephalitis. J. Immunol. 2014;192:5571–5578. doi: 10.4049/jimmunol.1303258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fallarino F., Grohmann U., Vacca C., Bianchi R., Orabona C., Spreca A., Fioretti M.C., Puccetti P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- 73.Chen W., Liang X., Peterson A.J., Munn D.H., Blazar B.R. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J. Immunol. 2008;181:5396–5404. doi: 10.4049/jimmunol.181.8.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fallarino F., Grohmann U., You S., McGrath B.C., Cavener D.R., Vacca C., Orabona C., Bianchi R., Belladonna M.L., Volpi C., et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006;176:6752–6761. doi: 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- 75.Sharma M.D., Baban B., Chandler P., Hou D.-Y., Singh N., Yagita H., Azuma M., Blazar B.R., Mellor A.L., Munn D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest. 2007;117:2570–2582. doi: 10.1172/jci31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baban B., Chandler P.R., Sharma M.D., Pihkala J., Koni P.A., Munn D.H., Mellor A.L. IDO Activates Regulatory T Cells and Blocks Their Conversion into Th17-Like T Cells. J. Immunol. 2009;183:2475–2483. doi: 10.4049/jimmunol.0900986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jain R., Frederick J.P., Huang E.Y., Burke K.E., Mauger D.M., Andrianova E.A., Farlow S.J., Siddiqui S., Pimentel J., Cheung-Ong K., et al. MicroRNAs Enable mRNA Therapeutics to Selectively Program Cancer Cells to Self-Destruct. Nucleic Acid Therapeut. 2018;28:285–296. doi: 10.1089/nat.2018.0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schramme F., Crosignani S., Frederix K., Hoffmann D., Pilotte L., Stroobant V., Preillon J., Driessens G., Van den Eynde B.J. Inhibition of Tryptophan-Dioxygenase Activity Increases the Antitumor Efficacy of Immune Checkpoint Inhibitors. Cancer Immunol. Res. 2020;8:32–45. doi: 10.1158/2326-6066.cir-19-0041. [DOI] [PubMed] [Google Scholar]
- 79.Sonner J.K., Keil M., Falk-Paulsen M., Mishra N., Rehman A., Kramer M., Deumelandt K., Röwe J., Sanghvi K., Wolf L., et al. Dietary tryptophan links encephalogenicity of autoreactive T cells with gut microbial ecology. Nat. Commun. 2019;10:4877. doi: 10.1038/s41467-019-12776-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ninomiya S., Nakamura N., Nakamura H., Mizutani T., Kaneda Y., Yamaguchi K., Matsumoto T., Kitagawa J., Kanemura N., Shiraki M., et al. Low Levels of Serum Tryptophan Underlie Skeletal Muscle Atrophy. Nutrients. 2020;12:978. doi: 10.3390/nu12040978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Floc’h N.L., Otten W., Merlot E. Tryptophan metabolism, from nutrition to potential therapeutic applications. Amino Acids. 2011;41:1195–1205. doi: 10.1007/s00726-010-0752-7. [DOI] [PubMed] [Google Scholar]
- 82.Hart A., Lentine K.L., Smith J.M., Miller J.M., Skeans M.A., Prentice M., Robinson A., Foutz J., Booker S.E., Israni A.K., et al. OPTN/SRTR 2019 Annual Data Report: Kidney. Am. J. Transplant. 2021;21:21–137. doi: 10.1111/ajt.16502. [DOI] [PubMed] [Google Scholar]
- 83.Szumilas K., Wilk A., Wiśniewski P., Gimpel A., Dziedziejko V., Kipp M., Pawlik A. Current Status Regarding Immunosuppressive Treatment in Patients after Renal Transplantation. Int. J. Mol. Sci. 2023;24 doi: 10.3390/ijms241210301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Holtan S.G., Yu J., Choe H.K., Paranagama D., Tang J., Naim A., Galvin J., Joachim Deeg H. Disease progression, treatments, hospitalization, and clinical outcomes in acute GVHD: a multicenter chart review. Bone Marrow Transplant. 2022;57:1581–1585. doi: 10.1038/s41409-022-01764-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raffin C., Vo L.T., Bluestone J.A. Treg cell-based therapies: challenges and perspectives. Nat. Rev. Immunol. 2020;20:158–172. doi: 10.1038/s41577-019-0232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tang Q., Henriksen K.J., Bi M., Finger E.B., Szot G., Ye J., Masteller E.L., McDevitt H., Bonyhadi M., Bluestone J.A. In Vitro–expanded Antigen-specific Regulatory T Cells Suppress Autoimmune Diabetes. J. Exp. Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tarbell K.V., Yamazaki S., Olson K., Toy P., Steinman R.M. CD25+ CD4+ T Cells, Expanded with Dendritic Cells Presenting a Single Autoantigenic Peptide, Suppress Autoimmune Diabetes. J. Exp. Med. 2004;199:1467–1477. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhou X., Bailey-Bucktrout S.L., Jeker L.T., Penaranda C., Martínez-Llordella M., Ashby M., Nakayama M., Rosenthal W., Bluestone J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bailey-Bucktrout S.L., Martinez-Llordella M., Zhou X., Anthony B., Rosenthal W., Luche H., Fehling H.J., Bluestone J.A. Self-antigen-Driven Activation Induces Instability of Regulatory T Cells during an Inflammatory Autoimmune Response. Immunity. 2013;39:949–962. doi: 10.1016/j.immuni.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Janssens I., Campillo Davó D., Van den Bos J., De Reu H., Berneman Z.N., Wens I., Cools N. Engineering of regulatory T cells by means of mRNA electroporation in a GMP-compliant manner. Cytotherapy. 2022;24:659–672. doi: 10.1016/j.jcyt.2022.01.001. [DOI] [PubMed] [Google Scholar]
- 91.Granit V., Benatar M., Kurtoglu M., Miljković M.D., Chahin N., Sahagian G., Feinberg M.H., Slansky A., Vu T., Jewell C.M., et al. Safety and clinical activity of autologous RNA chimeric antigen receptor T-cell therapy in myasthenia gravis (MG-001): a prospective, multicentre, open-label, non-randomised phase 1b/2a study. Lancet Neurol. 2023;22:578–590. doi: 10.1016/s1474-4422(23)00194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mease P.J., Hobbs K., Chalmers A., El-Gabalawy H., Bookman A., Keystone E., Furst D.E., Anklesaria P., Heald A.E. Local delivery of a recombinant adenoassociated vector containing a tumour necrosis factor α antagonist gene in inflammatory arthritis: a phase 1 dose-escalation safety and tolerability study. Ann. Rheum. Dis. 2009;68:1247–1254. doi: 10.1136/ard.2008.089375. [DOI] [PubMed] [Google Scholar]
- 93.Nelson J., Sorensen E.W., Mintri S., Rabideau A.E., Zheng W., Besin G., Khatwani N., Su S.V., Miracco E.J., Issa W.J., et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aaz6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sabnis S., Kumarasinghe E.S., Salerno T., Mihai C., Ketova T., Senn J.J., Lynn A., Bulychev A., McFadyen I., Chan J., et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 2018;26:1509–1519. doi: 10.1016/j.ymthe.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be provided by the lead contact upon request.
-
•
The study does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.