

Abstract
Purpose
Sequencing-based genetic testing often identifies variants of uncertain significance (VUS) or fails to detect pathogenic variants altogether. We evaluated the utility of RNA sequencing (RNA-seq) to clarify VUS or identify missing variants in a clinical setting.
Methods
Over a 2-year period, genetics providers at a single institution referred 26 cases for clinical RNA-seq. Cases had either no candidate variant identified by prior testing or a VUS suspected to impact splicing or expression. A committee reviewed each submission to ensure it met study criteria.
Results
Among 26 cases, 8 could not be sequenced because of poor expression in an accessible tissue, 2 did not meet inclusion criteria, 3 were solved prior to collection, and 4 families declined participation or did not complete sample collection. For the 9 cases sequenced, the clinical laboratory reported two positive, four negative, and three “indeterminate.” For all three indeterminate cases, original RNA-seq data was manually evaluated and deemed explanatory.
Conclusion
Clinical RNA-seq can clarify VUS, especially splice variants, but laboratory-specific interpretation guidelines may lead to indeterminate results. Identifying individuals likely to benefit from RNA-seq and providing appropriate counseling poses unique challenges.
Keywords: RNA sequencing, variant of uncertain significance, clinical genetic testing, splice variants, germline testing
INTRODUCTION
RNA sequencing (RNA-seq) can evaluate mRNA sequence and quantity. Sequencing the transcriptome of a cell or tissue can provide a comprehensive view of gene expression, splicing, and posttranscriptional modifications.1,2 RNA-seq has recently become available in the clinical setting, after a growing number of studies demonstrated its potential to clarify variants of uncertain significance (VUS) and identify pathogenic variants missed by standard DNA sequencing.3,4 Additional applications of RNA-seq in clinical practice include cancer diagnostics,1 infectious disease pathogen identification,5 and informing disease prognosis and treatment monitoring.3
There is interest in using RNA-seq to identify or clarify pathogenic variants in suspected Mendelian conditions where existing diagnostic approaches, such as exome sequencing (ES), reveal a precise molecular diagnosis in about 30% of individuals.6 These approaches may also reveal VUS that could explain the phenotype, which RNA-seq may help clarify by identifying isoforms predicted to be deleterious (such as a skipped exon) or changes in expression that reduce the total amount of protein available.7 Such evidence may allow for variant reclassification.8
Thus far, variants predicted to contribute to disease through effects on protein function are largely limited to copy number changes, frameshift variants, start-loss variants, stop-gain or -loss variants, splice acceptor/donor variants, and missense variants with supportive functional and/or population level evidence.9 Yet, nearly 30% of pathogenic variants may occur within noncoding regions and may be difficult to detect using sequencing-based approaches.10,11 A growing number of computational predictive tools have improved our ability to discern how variants might impact splicing or expression, but their accuracy and clinical utility remains unclear.12,13 Thus, there is a need for clinical testing that can functionally validate these variants.
In the research setting, RNA-seq has been shown to improve diagnostic rates and provide insight into mechanisms leading to variant pathogenecity.14 For example, analyzing transcript sequence along with abundance may detect aberrant splicing or reduced transcript numbers to clarify the functional consequence of variants. We developed a process to identify individuals at our institution who might benefit from clinical RNA-seq to clarify VUS found by prior testing or to identify variants missed by prior clinical testing. Here, we describe our single-center experience on the use of clinical RNA-seq for cases with prior genetic testing that was either: (1) negative or nondiagnostic, but with a specific genetic condition suspected, or (2) had a VUS predicted to impact splicing or gene expression. We sought to determine the appropriate population for which RNA-seq may be most impactful, counsel individuals on its utility, and identify challenges associated with providers ordering RNA-seq.
MATERIALS AND METHODS
Study inclusion criteria
Individuals with a suspected Mendelian condition previously evaluated by a medical or biochemical genetics provider were eligible. Referrals during the study period (May 1, 2020, to May 31, 2022) were evaluated by a committee including medical geneticists, laboratory geneticists, and genetic counselors to ensure individuals met one of the inclusion criteria:
Suspected autosomal recessive (AR) condition with one pathogenic or likely pathogenic variant, and one VUS that could be clarified through RNA-seq;
Suspected AR condition with one pathogenic variant detected and a suspicious clinical or biochemical phenotype for which RNA-seq is expected to confirm the function of the other allele;
Suspected autosomal dominant (AD) condition with at least one VUS;
X-linked condition in a male with at least one VUS identified, or X-linked lethal condition in a female with one VUS;
No pathogenic or likely pathogenic variant identified on standard genetic testing but with high suspicion for a specific genetic or biochemical disease.
Clinical RNA-seq
Sequencing was performed at MNG Laboratories, a Clinical Laboratories Improvement Amendments (CLIA)-certified laboratory (CLIA ID#11D0703390), on an Illumina platform utilizing TruSeq stranded total RNA library. Sequencing data were aligned with HISAT2 and analyzed using StringTie, allowing for detection of transcript ratios and splice-site usage.15,16 Using counts corresponding to each gene, test samples were compared to tissue-specific reference data to determine a Z-score for assessing significance and relative expression. A Z-score threshold of +/− 2 was used to guide the lab for interpretation of results. The Z-score was calculated as: Z-score = (Test_sample_value - control_set_mean) / control_set_standard_deviation.
Analysis of RNA-seq data
For each sample, bam files were obtained from the clinical testing laboratory. Duplicate reads were marked using Picard Tools (http://broadinstitute.github.io/picard/), paired FASTQ files were created using bedtools17 and aligned to GRCh38 using STAR.18 Aligned sequencing data were visualized using the Integrative Genomics Viewer to evaluate transcript ratios and the impact of predicted splice variants.19
RESULTS
Study referrals and evaluation
Over a 2-year period, 26 cases were submitted to the study team for clinical RNA-seq. Of those 26 cases, 8 (31%) could not be sequenced because expression levels of the gene in one of three accessible tissues (blood, muscle biopsy, or fibroblasts) fell below cutoffs established by the clinical laboratory. Two additional cases did not meet inclusion criteria: one was a canonical splice site variant previously classified as likely pathogenic and the second was a de novo variant in a gene associated with a recessive condition that was submitted to phase the known variants.
Among the 16 cases approved by the committee, 9 were sequenced, 3 were withdrawn because they were solved on reanalysis of prior clinical testing data or the VUS was reclassified before a sample was collected for RNA-seq, and 4 individuals or their families could not be contacted or chose not to participate (Figure 1A). Families who declined participation cited challenges related to the need to come to a main hospital campus for Monday–Wednesday specimen collection to ensure samples reached the performing laboratory by Friday.
Figure 1. RNA sequencing workflow and examples of indeterminate variants felt to be pathogenic after re-evaluation.
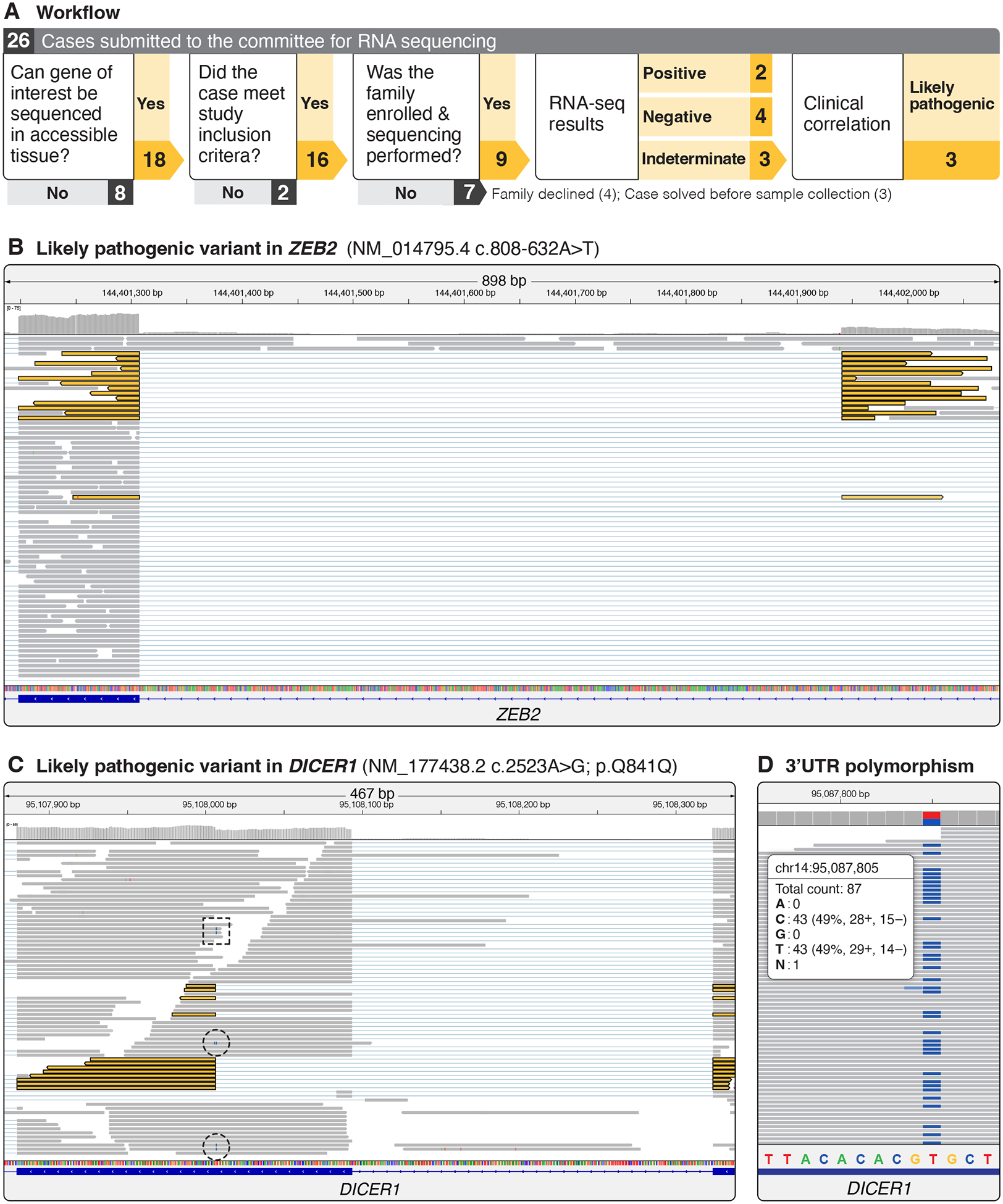
A. For each case submitted, the clinical lab was contacted to see if the specific gene could be sequenced. For those that could be sequenced, a committee reviewed the case to ensure it met the inclusion criteria. After approval, families were contacted and consented to the study. Three cases were withdrawn before the family was contacted, and 4 families either could not be reached for consent or did not complete the sample collection process. Sequencing was completed on 9 samples and original sequencing data was obtained to better understand the laboratory interpretation. Review of 3 cases reported as indeterminate by the lab resulted in the variant being treated as likely pathogenic by the clinical team. B. RNA-seq analysis for an intronic ZEB2 variant were reported as indeterminate by the clinical lab. Review of the data revealed evidence of altered splicing in approximately 25% of reads that were split between an exon and the position of the intronic variant (split reads are colored yellow). C. A synonymous variant in DICER1 predicted to alter splicing and result in a 29-amino acid in-frame deletion within exon 16 was reported as indeterminate by the clinical lab. Review of the RNA sequencing data revealed that of the 61 reads mapping to the c.2523A>G polymorphism 12 did not contain the first 29 amino acids (highlighted in yellow), suggesting altered splicing. A further 3 reads contained the c.2523A>G polymorphism, suggesting they were processed normally (dashed circle); 2 reads contained the c.2523A>G, but at the third-to-last base pair position of the read (dashed box), which is also contained on the neighboring exon, thus it is unclear if these reads should be split or if this is evidence of normal processing of the transcript. D. To assess if nonsense-mediated decay resulted in underrepresentation of abnormally spliced transcripts in the DICER1 case, we identified a heterozygous polymorphism in the 3’UTR that was present in a 50/50 ratio in the RNA-seq data. This suggests that both alleles were equally represented in the data but that difficulties with mapping short reads masked the true impact of the splice variant.
Interpretation of RNA-seq test reports
A positive result was returned in 2 of 9 cases (22%), a negative result in 4 of 9 cases (44%), and an indeterminate result in 3 of 9 cases (33%) (Figure 1A, Table 1). For the positive result pertaining to the CTC1 (HGNC:26169, NM_025099.6) c.2385G>A variant, RNA sequencing demonstrated aberrant splicing consisting of two types of aberrations: 1) intron 13 inclusion as the predominant aberration and 2) creation of a novel donor splice site at NC_000017.10:g.8135340, resulting in an in-frame deletion of the last 40 amino acids of exon 13. For the RPL30 (HGNC:10333, NM_000989.4) c.167+769C>T variant, RNA sequencing demonstrated that an alternate GT acceptor splice site was used which resulted in a novel coding region from NC_000008.10:g.99056404–99056469. This splicing alteration results in a shift in the reading frame that is predicted to lead to a change in amino acid sequence at position 56 from an arginine to a serine with a stop codon 21 amino acids after this change and a truncation of the final 39 amino acids of the protein. While these positive results displayed clear aberrant splicing and negative results showed no aberrant splicing or changes to expression, lab reports for participants with indeterminate results all commented on some degree of altered splicing or reduced gene expression. To better understand why these were indeterminate, original sequencing data were obtained and reviewed.
Table 1. Summary of RNA-seq results from 9 study participants.
A specific variant was evaluated in 6 of 9 cases, 5 of which were predicted to affect splicing (the impact of the TP53 intronic insertion was uncertain). The clinical laboratory returned a positive result in 2 of 6 cases, an indeterminate result in 3 of 6 cases, and a negative result in 1 case. After evaluation of the original RNA-seq data for the 3 indeterminate results, each participant’s clinical team felt that the testing supported the pathogenicity of the suspected variant. In 3 of 9 cases in which a missing variant was suspected the clinical interpretation was negative and subsequent reevaluation of the RNA sequencing data by the study team did not identify a candidate variant.
| Gene | Inclusion Criteria | Suspected Condition | Variant Coding Sequence Change | Variant Genomic Change | Observed Variant RNA Change | Predicted Variant Amino Acid Change | RNA source | RNA Sequencing Result | Clinical Interpretation |
|---|---|---|---|---|---|---|---|---|---|
| CTC1 | Evaluate VUS predicted to affect splicing | Dyskeratosis congenita | NM_025099.6:c.2385G>A | NC_000017.10:g.8135221C>T | NM_025099.6:r.2385g>a | NP_079375.3:p.(Lys795=) | Blood | Positive | Positive |
| DICER1 | Evaluate VUS predicted to affect splicing | DICER1 tumor predisposition | NM_001195573.1:c.2523A>G | NC_000014.8:g.95574344T>C | NM_001195573.1:r.2523a>g | NP_001182502.1:p.(Gln841=) | Blood | Indeterminate | Positive |
| RASA1 | Evaluate VUS identified by clinical testing predicted to affect splicing | Capillary malformation-arteriovenous malformation | NM_002890.3:c.2011+6T>A | NC_000005.9:g.86670739T>A | N/A | N/A | Blood | Negative | Negative |
| RPL30 | Evaluate deep intronic variant found by research testing predicted to affect splicing | Diamond-Blackfan anemia | NM_000989.4:c.167+769C>T | NC_000008.10:g.99056402G>A | NM_000989.4: r.167_168ins167+767_167+702 | NP_000980.1:p.(Arg56Serfs*21) | Blood | Positive | Positive |
| TP53 | Evaluate impact of intronic alpha-satellite insertion found by research testing on expression | Li-Fraumeni syndrome | NM_000546.6:c.919+15_919+17delinsTGGAAACGAATGGAATCATCATCGAATGGAAATGAAAGGAGTCATCATCTAATGGAATTGCATGGAATCATCATAAAATGGAATCGAATGGAATCAACATCAAATGGAATCAAATGGAATCATTGAACGGAATTGAATGGAATCGTCATCGAATGAATTGACTGCAATCATCGAATGGTCTCGAATGGAATCATCTTCAAATGGAATGGAATGGAATCATCGCATAGAATCGAATGGAATTATCATCGAATGGAATCGAATGGAATCAACATCAAACGGAAAAAAACGGAATTATCGAATGGAATCGAAGAGAATCATC | NC_000017.10:g.7577002_7577004delinsGATGATTCTCTTCGATTCCATTCGATAATTCCGTTTTTTTCCGTTTGATGTTGATTCCATTCGATTCCATTCGATGATAATTCCATTCGATTCTATGCGATGATTCCATTCCATTCCATTTGAAGATGATTCCATTCGAGACCATTCGATGATTGCAGTCAATTCATTCGATGACGATTCCATTCAATTCCGTTCAATGATTCCATTTGATTCCATTTGATGTTGATTCCATTCGATTCCATTTTATGATGATTCCATGCAATTCCATTAGATGATGACTCCTTTCATTTCCATTCGATGATGATTCCATTCGTTTCCA | N/A | None | Blood | Indeterminate | Positive |
| ZEB2 | Evaluate deep intronic variant found by research testing predicted to affect splicing | Mowat-Wilson syndrome | NM_014795.4:c.808–632T>A | NC_000002.11:g.145159506T>A | NM_014795.4: r.807_808ins808–632_808–1 | NP_055610.1: p.(269_270ins X[292]) | Blood | Indeterminate | Positive |
| DMD | X-linked condition; no variant identified by prior clinical testing | Dystrophinopathy (Becker muscular dystrophy) | N/A | N/A | N/A | N/A | Muscle biopsy | Negative | Negative |
| NGLY1 | Single hit in recessive condition; no 2nd variant identified by clinical testing | Disorder of N-linked glycosylation | N/A | N/A | N/A | N/A | Blood | Negative | Negative |
| PRKDC | Single hit in recessive condition; no 2nd variant identified by clinical testing | Severe Combined Immunodeficiency (SCID) | N/A | N/A | N/A | N/A | Blood | Negative | Negative |
In a child with suspected Mowat-Wilson syndrome based on exam, but negative clinical testing that included ES, research long-read genome sequencing on the Nanopore platform identified a deep intronic variant in ZEB2 (HGNC:14881, NM_014795.4) c.808–632A>T predicted to alter splicing (SpliceAI score: 0.97 for acceptor gain).12 RNA sequencing confirmed that this variant created a novel splice junction utilizing an aberrant GT donor site at position NC_000002.11:g.145159506–145159507 within intron 6 that extended to the canonical exon 7 acceptor site, but the clinical lab reported the result as indeterminate because only 25% of transcripts contained evidence of altered splicing (Figure 1B). We visually inspected the RNA-seq data and confirmed that 25% (15/61) of transcripts appeared aberrantly spliced, indicating that functional ZEB2 mRNA levels would be reduced relative to controls. Because the phenotype was classic for Mowat-Wilson syndrome, this was interpreted by the clinical team as a likely pathogenic change.
A second indeterminate result was reported in a participant with a pleuropulmonary blastoma (PPB) and a synonymous germline DICER1 (HGNC:17098, NM_001195573.1) c.2523A>G identified by clinical testing and classified as a VUS.20 Splicing prediction software predicted a high likelihood of altered splicing (SpliceAI score: 0.99 for acceptor gain), thus the clinical team felt it was likely explanatory given the phenotype.12 Visual inspection of the RNA-seq data revealed that 12/70 reads at the DICER1 c.2523 position were discernably transcribed from the DNA strand containing this variant (Figure 1C). Of these 12 reads, 9 exhibited aberrant splicing, indicating at least 12% of total reads were aberrantly spliced. This is likely an underrepresentation of the number of mis-spliced reads given that a minority of copies (12/70) came from the affected haplotype. That not all transcripts are captured or aligned properly is supported by the observation of a 3’UTR polymorphism that was present at 50% allele frequency in the RNA-seq data (Figure 1D). Given the strong association of PPB with DICER1 syndrome, the presence of mis-spliced reads, and that transcripts with altered splicing were likely missing, this change was clinically interpreted as likely pathogenic.
Finally, in a participant with early-onset embryonal rhabdomyosarcoma of the neck and a family history of early-onset cancer, targeted long-read sequencing identified an intronic 319-bp alpha-satellite insertion in TP53 (HGNC:11998, NM_000546.6) c.919+15_919+17delins319. This variant was not readily analyzed via splicing prediction software. No mis-spliced reads were observed, but a statistically significant decrease in expression of TP53 transcripts compared to controls (Z-score: −2.3) was above the laboratory cutoff of −2.5 and was therefore interpreted as indeterminate. Examination of sequencing data confirmed reduced expression of transcripts from one haplotype. Given the phenotype’s association with Li-Fraumeni syndrome, this decrease in expression was considered significant and likely explanatory by the clinical team. A clinical testing lab later updated the variant classification to likely pathogenic after an unrelated family with a similar insertion and phenotype was identified.
DISCUSSION
While RNA-seq is extensively used in research, it has only recently become available in the clinical setting for diagnostic purposes. Complexities to RNA-seq exist and include the need for special handling and extraction protocols to ensure sample integrity; the inability to sequence all genes because of tissue-specific expression; batch effects related to extraction, library preparation, and sequencing methods; and challenges identifying significant changes in splicing or gene expression. The goal of our study was to understand how these complexities as well as limited provider experience with this technology might impact use in clinical practice. Demonstrating clinical utility of RNA-seq may increase coverage by payers and improve overall access to this new testing modality.
While one-third (3/9) of results were indeterminate, manual review allowed us to use clinical judgement to establish the pathogenicity of each variant. This guided screening for the participants and their family members with the TP53 or DICER1 variants. Manual review of the sequencing data from indeterminate cases may limit the use of RNA-seq by providers who lack this expertise, although we expect this to improve as experience and guidelines for RNA-seq interpretation improve. Clinical interpretation of the indeterminate cases was strengthened by clear and specific clinical presentations: a clear Mowat-Wilson phenotype in a participant with a ZEB2 splice variant; a pleuropulmonary blastoma in a participant with a predicted splice variant in DICER1; and an early-onset embryonal rhabdomyosarcoma in a participant with a TP53 variant. It may be difficult to make similar determinations in cases where the phenotype is less clear or where the phenotype may evolve over time. Inhibiting cellular processes such as nonsense-mediated decay could have reduced the number of indeterminate reports by increasing the number of mis-spliced transcripts for analysis, although this may increase test complexity without a clear improvement to clinical utility.
Our study highlights other challenges with the use of RNA-seq in clinical practice. Approximately one-third (8/26) of submissions could not be sequenced because of poor expression in accessible tissues, some families declined participation because of stricter collection requirements to ensure sample stability, and significant local expertise was needed to properly interpret indeterminate results. Our sample size was not large enough to evaluate the utility of RNA-seq in identifying variants missed by prior clinical testing. Cases referred for testing had previous genetic testing at our institution, so our findings may not be relevant to other RNA-seq use cases.
All families that consented to the study demonstrated understanding about the use of the test to clarify the impact of a VUS or identify a missing variant. Families expressed interest in understanding how mRNA sequencing could show differences in gene expression or splicing. Because of this, counseling took longer than other tests, such as exome sequencing.
Despite challenges, clinical RNA-seq was useful in clarifying uncertain results in about one-third (9/26) of cases. Variants reclassified as benign may guide additional genetic testing recommendations; those reclassified as pathogenic may resolve outstanding diagnostic questions for an individual or family, guiding management, reducing additional genetic testing, and ending their diagnostic odyssey. RNA-seq may be used in place of cascade testing to resolve a VUS, potentially streamlining variant resolution as testing may not need to be coordinated among multiple family members.
Although larger studies are needed, our results suggest that the higher number of indeterminate results should be communicated during pretest counseling. Providers may require guidance on limitations of the technology and awareness that manual review of original data may be necessary. Because of difficulties interpreting transcripts generated using short-read-based approaches, we hypothesize that emerging technologies like long-read RNA-seq, which can sequence complete isoforms and potentially simplify interpretation, may supersede short-read approaches.
ACKNOWLEDGEMENTS
We thank the families who participated in this study. We thank Angela Miller for editorial assistance and figure preparation.
FUNDING STATEMENT
MRT is supported by the American Cancer Society (ACS) and the Andy Hill Washington State Cancer Research Endowment. CL is supported by the National Institute of Neurological Diseases and Stroke (NINDS), the National Center for Advancing Translational Sciences (NCATS), and the Rare Diseases Clinical Research Network (RDCRN) at the National Institutes of Health through the Frontiers in Congenital Disorders of Glycosylation Grant 1U54NS115198-01. GM is supported Jordan’s Guardian Angels and the Sunderland Foundation. JTB is supported by the National Institutes of Health through HD104435. DEM is supported by the National Institutes of Health through the NIH Director’s Early Independence Award DP5OD033357. This work was supported in part by the Seattle Children’s Foundation (Elizabeth and George Smith Endowment in Molecular Diagnostics and the Laboratory Test Development Fund) and by the Seattle Children’s Focus on Kids Laboratory Guild.
CONFLICT OF INTEREST
DEM holds stock options in MyOme, is on a scientific advisory board at Oxford Nanopore Technologies (ONT), is engaged in a research agreement with ONT, and has received travel support from ONT.
Footnotes
ETHICS DECLARATION
The study was approved by the Seattle Children’s Hospital IRB, study #00002289. All participants or their legal guardian provided written consent for this study.
DATA AVAILABILITY
Clinical RNA sequencing data cannot be made available as part of this study. Long-read sequencing data described in this manuscript will be deposited in AnVIL.
REFERENCES
- 1.Ergin S, Kherad N, Alagoz M. RNA sequencing and its applications in cancer and rare diseases. Mol Biol Rep. 2022;49(3):2325–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ketkar S, Burrage LC, Lee B. RNA Sequencing as a Diagnostic Tool. JAMA. 2023;329(1):85–86. [DOI] [PubMed] [Google Scholar]
- 3.Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, Craig DW. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet. 2016;17(5):257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yepez VA, Gusic M, Kopajtich R, et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 2022;14(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liebhoff AM, Menden K, Laschtowitz A, Franke A, Schramm C, Bonn S. Pathogen detection in RNA-seq data with Pathonoia. BMC Bioinformatics. 2023;24(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wojcik MH, Reuter CM, Marwaha S, et al. Beyond the exome: What’s next in diagnostic testing for Mendelian conditions. Am J Hum Genet. 2023;110(8):1229–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma M, Ru Y, Chuang LS, et al. Disease-associated variants in different categories of disease located in distinct regulatory elements. BMC Genomics. 2015;16 Suppl 8(Suppl 8):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(6):665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176(3):535–548 e524. [DOI] [PubMed] [Google Scholar]
- 13.Scalzitti N, Jeannin-Girardon A, Collet P, Poch O, Thompson JD. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genomics. 2020;21(1):293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marco-Puche G, Lois S, Benitez J, Trivino JC. RNA-Seq Perspectives to Improve Clinical Diagnosis. Front Genet. 2019;10:1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyle ANJ, Ohlsen TJD, Miller DE, et al. Congenital pleuropulmonary blastoma in a newborn with a variant of uncertain significance in DICER1 evaluated by RNA-sequencing. Matern Health Neonatol Perinatol. 2023;9(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Clinical RNA sequencing data cannot be made available as part of this study. Long-read sequencing data described in this manuscript will be deposited in AnVIL.