

Abstract
Monosynaptic tracing using deletion-mutant rabies virus allows whole-brain mapping of neurons that are directly presynaptic to a targeted population of neurons. The most common and robust way of implementing it is to use Cre mouse lines in combination with Cre-dependent adeno-associated viral vectors for expression of the required genes in the targeted neurons before subsequent injection of the rabies virus. Here we present a step-by-step protocol for performing such experiments using first-generation (ΔG) rabies viral vectors.
1. Introduction
“Monosynaptic tracing” refers to the use of a deletion-mutant neurotropic virus that is complemented in vivo selectively within a targeted neuronal population of interest, so that the virus can replicate within them and spread to other neurons that are in direct synaptic contact (Wickersham et al., 2007a). In the original monosynaptic tracing system, which remains the primary version in use in neuroscience, the virus in question is a rabies virus from which the “G” gene, encoding its envelope glycoprotein, has been deleted (Wickersham et al., 2007a, b). This deletion makes the virus unable to spread beyond infected cells, unless they are engineered to express G in trans. As a second modification to the virus, it is coated with “EnvA”, the envelope protein of an avian retrovirus. This makes the rabies virus unable to infect mammalian cells unless they have been engineered to express EnvA’s receptor, “TVA” (Wickersham et al., 2007a; Young et al., 1993), a protein which is not native to mammals.
Monosynaptic tracing therefore requires engineered expression of two genes in the targeted population of starting neurons: the gene encoding the receptor TVA and the G gene encoding the rabies virus glycoprotein. The rabies virus can then infect the starting cells via the receptor TVA and, complemented by the expression of G in trans, spread beyond them. Because rabies virus naturally spreads in a retrograde direction in the central nervous system, the virus spreads to cells presynaptic to the starting cells. If the engineered G expression has been carefully restricted to the targeted postsynaptic cells and is not expressed in cells of other types that are presynaptic to them, the deletion-mutant virus cannot spread beyond them. The result is labeling of neurons that are monosynaptically connected to the targeted starting cells.
The usual implementation of all this is to use a Cre mouse line to provide genetic access to the targeted group of postsynaptic neurons in conjunction with one or two Cre-dependent “helper” adeno-associated viral vectors (AAVs) that deliver the TVA and glycoprotein genes (Ahrlund-Richter et al., 2019; Beier et al., 2015; Kohara et al., 2014; Liu et al., 2017; Szonyi et al., 2019; Wall et al., 2010; Watabe-Uchida et al., 2012; Zhang et al., 2019). The AAVs are injected in one surgery, then the rabies virus is injected some time later once the AAVs have had a chance to express the required gene. Some time after injection of the rabies virus, the mouse is perfused, imaged, etc. and the results are obtained.
Within this broad description, many variations are possible, and the devil is in the details. Many different helper AAVs have been used by different laboratories, and the degree of success varies widely. Here we present reagents and a protocol that works well for us.
The essential tension inherent in coexpressing TVA and G is this: the EnvA-TVA interaction is so sensitive (Federspiel et al., 1994; Seidler et al., 2008) that even low levels of leaky expression of TVA from the helper viruses will result in off-target infection of non-Cre-expressing neurons by the EnvA-enveloped helper virus, but a considerable amount of G expression seems to be required for labeling respectable numbers of presynaptic cells. On the other hand, too much G expression results in low efficiency of labeling as well, presumably because of toxicity due to the glycoprotein (Lavin et al., 2019; Lay et al., 2003; Sarmento et al., 2005).
Various approaches to dealing with this mismatch have been taken, including using mutated or destabilized TVA (Miyamichi et al., 2011; Sakurai et al., 2016) or use of other forms of G (Kim et al., 2016). Our preferred solution is to use a mixture of two helper AAVs (originally described in Liu et al. (2017)), carefully titrated in pilot experiments to find dilution levels for both that result in large amounts of presynaptic labeling in Cre mice and little background in wild-type controls. Both the rabies virus used in this protocol and the glycoprotein gene in the helper viruses are of the SAD B19 strain, used in the original monosynaptic tracing system (Wickersham et al., 2007a) and which remains the one most commonly used in neuroscience, although efforts to reduce toxicity and increase efficiency by using other strains of rabies virus appear promising (Reardon et al., 2016).
The first AAV in this combination, AAV1-synP-FLEX-splitTVA-EGFP-tTA, is a Cre-dependent (“FLEX” (Atasoy et al., 2008)) vector expressing TVA (actually a “split” version in which the start codon is outside the lox sites, in an effort to reduce background expression in the event of nonspecific recombination (Seidler et al., 2008)), EGFP, and the tetracycline transactivator (tTA) (Gossen and Bujard, 1992); the second AAV, AAV-TREtight-mTagBFP2-B19 G, expresses both G and the blue fluorophore mTagBFP2 (Subach et al., 2011) under the control of the tetracycline response element (“TRE-tight” variant). This combination provides Cre-dependent expression of both required genes but with the ability to dilute the TVA-expressing vector to a low enough level that the subsequently-injected rabies virus infects very few cells in wild-type mice (Lavin et al., 2019). The tTA expressed from the same vector then binds to the TRE in the second AAV and drives expression of G (and mTagBFP2) at a fairly high level, providing sufficient amplification that presynaptic cells are still labeled in large numbers in Cre mice. Note that this uses the “tet-OFF” system, so that no doxycycline (or tetracycline) is required.
As a simpler alternative, a single helper AAV, first described in Kohara et al. (2014), expressing TVA, EGFP and G can also be used, but this results in considerably more background in wild-type mice than does the two-AAV combination (Lavin et al., 2019). All three AAVs are available from Addgene packaged as serotype 1 (catalog numbers 100798-AAV1, 100799-AAV1, and 52473-AAV1), and their genome plasmids are available from Addgene as well (catalog numbers 100798, 100799, and 52473).
Using a high-quality preparation of ΔG rabies virus is key. This may or may not be available from a vector core; collaboration with a competent lab may be another option. Whatever the source, it is very important that the producers adhere to the basic principles we have emphasized previously (Wickersham et al., 2010; Wickersham and Sullivan, 2015) of keeping the passage number very low (at most 2–3 passages following the rescue step) in order not to foster the accumulation of dysfunctional mutants, which can occur rapidly and greatly degrade the efficiency of viral spread in vivo. Repeatedly passaging an inherited stock of virus obtained from another user should be avoided at all costs.
The dilutions that we recommend here for the helper viruses have been chosen based on extensive experimentation, the details of which are beyond the scope of this protocol but which we describe elsewhere (Lavin et al., 2019). We have found them to result in good transsynaptic spread in PV-Cre and DAT-Cre mice but (at least in the case of the two-AAV combination) relatively few RV-labeled cells at the injection site in wild-type mice. The optimal dilutions may be different for other Cre lines. Note that these dilutions have been determined based on a 7-day interval between the injections of AAVs and RV. We have not systematically varied the 7-day intervals between AAV and RV injections and between RV injection and perfusion. Use of a different interval between AAV and RV injections would necessitate redoing the titration to determine optimal AAV concentrations (e.g., use of a longer interval between AAV and RV injections would result in higher expression levels of TVA and G, and would therefore presumably warrant use of lower concentrations of AAVs). Use of a shorter survival time after injecting the RV (e.g., 5 days) works, and, while we assume that this yields fewer transsynaptically labeled neurons, this may be worth it if keeping cells as healthy as possible is important for the experiment.
Control experiments in wild-type (Cre-negative) mice should always be done, to reveal the degree to which labeling by the rabies virus at the injection site is due to leaky expression of TVA from the Cre-dependent AAV (all putatively Cre-dependent AAVs leak to some extent, it appears) and/or residual “unpseudotyped” rabies virus left over from infection of the EnvA-expressing producer cells in the last passage (Wickersham and Sullivan, 2015). While there will almost invariably be some (perhaps a few dozen, across all sections in a given mouse) RV-labeled cells at the injection site in wild-type mice due to slight leak in TVA expression, there should be no, or almost no, cells labeled by the rabies virus remote from the injection site in wild-type mice. Another valuable control condition that is possible when using the two-helper combination is to use Cre mice but to omit the second, G-encoding AAV. This is important for determining whether any putatively-transsynaptically-labeled cells might in fact just be labeled because of retrograde transduction by the TVA-encoding helper virus (some minority population of which may not be Cre-dependent, as discussed above, or the retrogradely-labeled cells could themselves express Cre, depending on the pathway and Cre line) followed by retrograde infection by the rabies virus via the TVA-expressing axons (such TVA-mediated retrograde infection being known to be perfectly possible (Huang et al., 2013)).
In addition to providing our recommendations specific to monosynaptic tracing, we have taken this opportunity to provide a detailed description of our techniques for stereotaxic injection of viruses. This of course is more broadly applicable, although the manual virus injection system is specifically designed to allow injection of small volumes at a low and carefully controlled rate and might be found tedious if used for injecting microliter-scale volumes. The core of the virus injection system that we use, namely the use of the Narishige MO-10 manual hydraulic manipulator to advance a plunger through a Drummond Wiretrol capillary, we have inherited from others; it appears to have been devised by Dr. Arturo Alvarez-Buylla and has been mentioned in print elsewhere (Agate et al., 2009; Petreanu et al., 2009; Viswanathan et al., 2015; Vogt et al., 2015; Ouzounov et al., 2017). Our implementation of this system requires custom hardware for adapting the manipulator to the stereotax, pipette and plunger. We have included machine shop drawings to allow readers to have their own made, or presumably to print them, although we have not tried this ourselves.
This protocol describes practices that have been approved by the MIT Committee for Animal Care and the MIT Environmental Health and Safety Office. The information is presented to inform readers how the authors perform these experiments, not to prescribe exactly how everyone else should. Follow your own institution’s guidelines, and common sense, for all procedures involving animals and to ensure safety of personnel. The surgery station can be set up in a biosafety cabinet if required by your institution, and this would certainly be necessary if one were working with replication-competent rabies virus rather than the deletion-mutants used for monosynaptic tracing. With regard to the specific issue of whether to vaccinate personnel working with first-generation rabies viral vectors, this is a judgement call; we have published a discussion of relevant considerations elsewhere (Wickersham et al., 2010).
2. Materials
2.1. Equipment and supplies
2.1.1. Equipment and supplies for virus storage and preparation
−80 °C freezer (e.g., Thermo Scientific, TSU series)
cryotubes, 0.5 ml with O-ring (sterile) (VWR 89004-282) for aliquoting concentrated virus. Use of tubes with O-rings, rather than, e.g, snap-cap PCR tubes, is important for preventing freeze-drying of the virus.
labels for cryotubes (Electron Microscopy Sciences 77560-Y)
freezer boxes for cryotubes (VWR 82007-162)
water bath (Southwest Science, SHW10LD), set to 37 °C for thawing viruses
vortex (Southwest Science, SBV1000), for mixing thawed virus
mini-centrifuge (Southwest Science, SC1006-M), for spinning down tubes
2.1.2. Equipment and supplies for pipette preparation
pipette puller (Sutter Instrument, P-2000)
micropipette beveler (Sutter Instrument, BV-10)
petri dish (VWR, 470210-568) for storing pipettes
dental wax (Orthomechanic, Orthowax; Amazon, B00FKDC0UU) for storing pipettes
ultraviolet sterilizer (e.g., Salon Sundry 609,728,828,431; Amazon, B007ROD0ZE)
replacement ultraviolet bulb (Sylvania, 21,061)
PCR tubes (VWR, 53509-304)
PCR tube rack (VWR, 80086-074)
2.1.3. Equipment and supplies for surgeries
stereotaxic apparatus (Stoelting Co., 51,925; includes “mouse and neonatal rat adaptor”) referred to below as “stereotax”
custom adapter parts for injection apparatus See “Equipment setup“.
thumbscrews, brass flared-collar knurled-head, 10–32 thread size, 3/4″ long (McMaster-Carr 92421A645)
thumbscrews, stainless steel, flared-collar knurled-head, 4–40 thread size, 3/8″ long (McMaster-Carr 99607A106)
washer, nylon plastic, number 4 screw size, mil. spec. (McMaster-Carr 92150A104)
micromanipulator, manual hydraulic with control unit (Narishige MO-10)
microscope, dissecting, with light source (e.g., Leica M50)
boom stand for microscope (e.g., Spot Imaging SMS6B)
glass capillaries, “Wiretrol” (Drummond # 5-000-1005; VWR, 53480-356 or Fisher, 21-175 A) Using these particular capillaries is critical, as they come with a small wire “plunger” that fits smoothly into the bore of the capillary glass and is used as an integral component of the custom injection apparatus, as described in “Equipment setup”.
oxygen tanks (Airgas, OX USPEAWBDS)
isoflurane vaporizer (Kent Scientific, VetFlo-1210S)
induction chamber (Patterson Scientific, 78933388)
hand warmers (Heat Factory, 1953)
lab timer (VWR, 62344-641)
glass bead sterilizer (Stoelting Co., 50287)
gloves, disposable, nonsterile
gloves, sterile (McKesson, 854486)
vials, sterile 30 ml (Hospira #00409581631; McKesson, #243268) for drugs
scintillation vials (Fisher, 03-337-23) for containing Nair, Betadine, and ethanol, and for disposal of used pipettes
spray bottle (e.g., Vivaplex, VPGM16-4), for 70% ethanol for de-contaminating surfaces, gloves, etc.
lab coat, fluid-resistant (Bioexpress, L-1305-M)
surgical masks (McKesson, 91-2003)
hair covers (McKesson, 37311100)
pipette, 0.1–2.5 μl (e.g. Eppendorf 022470001)
pipette tips, filtered, 0.1–10 μl (VWR, 89174–520)
balance (Smart weigh, TOP500) for weighing mouse
syringes, 1 ml (McKesson, 26049)
needles, 27 gauge (McKesson, 1031799)
clippers (Wahl Professional, 8685; Amazon, B00011K2BA)
cotton tip applicators (McKesson Brand 24–106; McKesson, 508716)
coverslips, square (VWR, 10118-641)
autoclave pouch (Fisher, 01-812-54)
sterilized cling film (Glad Press’n Seal; Sai Infusion Technologies, PSS-70)
scissors, fine (Fine Science Tools, 14040-10)
forceps, straight (Fine Science Tools, 11251-20)
forceps, curved, 2 pairs (Fine Science Tools, 11651-10)
drill (Vogue Professional, 6000 or similar)
burrs for drill, 0.5 mm diameter (Fine Science Tools, 19007-05)
surgical eye spears (Beaver-Visitec International, 0008685; McKesson, 406875)
needle holder (Fine Science Tools, 12002-12)
sutures, nylon, 6-0 (McKesson Brand, S1698GX; McKesson, 1034505)
warm water heating pad (Gaymar Stryker, TP22C; Amazon, B002TI26IK)
warm water pump (Gaymar Stryker, TP 700 T/Pump; Amazon, B00VQOF6J4)
2.1.4. Equipment and supplies for perfusion and histology
rotating shakers, 2 (Southwest Science SBT300) one in a cold room for postfixing brains before sectioning and for overnight incubation of sections in secondary antibody, the other at room temperature for other immunostaining steps
vibrating microtome (Leica VT1000S)
12-well plates (e.g., Corning, 353043)
“Netwell” net inserts (Corning, 3478)
syringe filters, 0.2 μm (VWR 28145-477) for filtering primary and secondary antibody solutions
slides, coated (Superfrost Plus, VWR 48311-703)
cover glass, 24 × 60 mm (VWR 16004-312)
2.2. Reagents
2.2.1. Mice, viruses, and associated reagents
Cre mouse (e.g., PV-Cre, Jackson 017320, or DAT-Cre, Jackson 020080)
helper adeno-associated viruses:
either:
AAV1-synP-FLEX-splitTVA-EGFP-B19 G (Addgene, 52473-AAV1)
or both of:
AAV1-syn-FLEX-splitTVA-EGFP-tTA (Addgene, 100798-AAV1)
AAV1-TREtight-mTagBFP2-B19 G (Addgene, 100799-AAV1)
These viruses should be diluted, stored, and mixed as described below under Reagent Setup.
Dulbecco’s phosphate-buffered saline (DPBS) (Fisher, 14-190-250)
rabies virus, ΔG and EnvA-enveloped (e.g., RVΔG-4mCherry(EnvA) (Weible et al., 2010)). Either made in-house or obtained from a core facility or by collaboration.
2.2.2. Reagents for surgeries
95% ethanol (e.g. Pharmco-AAPER 111000190)
bleach (e.g., VWR 76245-190)
isoflurane (Piramal, NDC 66794-0017)
ketamine, 100 mg/ml (Vedco, NDC 50989-996-06)
xylazine, 100 mg/ml (Patterson, NDC 59399-111-50)
buprenorphine, slow-release, 0.5 mg/ml (compounded)(Zoopharm, IZ-74-000-18) This must not be diluted, otherwise the slow release quality will be negated.
meloxicam (Patterson, NDC 14043-909-20)
sterile normal saline (Nurse Assist, 6240)
bottles, sterile, 30 ml (McKesson, 243268) for mixing and storing drugs
lactated Ringer’s solution (Westnet, R8306)
Nair (Nair, 794628234946 741655452645 524883464495 885857837111)
mineral oil (Sigma, M8410-100ML)
eye ointment (Puralube, FBA_2680; Amazon, B00HGMZ7RQ)
povidone-iodine swabs (McKesson, 854753)
swab (McKesson, 24-106)
2.2.3. Reagents for perfusion and histology
sodium pentobarbital solution (Fatal Plus, Vortech, NDC 0298-9373-68)
phosphate buffer, 10x (Sigma P3619-1GA)
phosphate-buffered saline (PBS), 10X (Sigma, P5493-1 L)
16% paraformaldehyde ampules (Electron Microscopy Sciences, 15710)
sucrose (Sigma, S1888-500 G)
ethylene glycol (Sigma, 324558-1 L)
polyvinylpyrrolidone (Sigma, PVP40-50 G)
Triton-X 10% solution in water (Sigma 93443-100ML)
normal donkey serum (Jackson Immuno 017-000-121)
chicken anti-GFP (Aves Labs GFP-1020)
primary antibody for Cre-defined starting cells (e.g., for PV-Cre mice, guinea pig anti-parvalbumin (Synaptic Systems 195004); for DAT-Cre mice, sheep anti-tyrosine hydroxylase (Millipore AB1542))
donkey anti-chicken Alexa Fluor 488 (Jackson Immuno 703-545-155)
secondary antibody for Cre-defined starting cells (e.g., donkey anti-guinea pig, AlexaFluor 647 conjugated (Jackson Immuno 706-605-148) or donkey anti-sheep, AlexaFluor 647 conjugated (Jackson Immuno 713-605-147))
water, sterile (e.g., Ambion AM9937) for reconstituting lyophilized serum and antibodies
mounting medium (Prolong Diamond Antifade, Thermo Fisher P36970)
2.3. Equipment setup
2.3.1. Injection apparatus assembly
First, have the four custom stereotax adapter parts fabricated by a machine shop according to the precision drawings provided as Supplementary File 1. Note: these parts could presumably be 3D-printed much more cheaply, but we have not tried this.
Assemble the injection apparatus using these four fabricated parts and the MO-10 hydraulic micromanipulator, using the thumb screws and nylon washer listed above, as shown in Fig. 1 and following the instructions given in the legend of that figure.
The plunger that comes with each package of capillary glass comes with a plastic handle attached; remove this handle by using wire cutters to clip the metal shaft of the plunger just where it connects to the base. Using needle-nose pliers, grasp the last ~3 mm of the cut end of the plunger and bend into a right angle with the rest of the shaft, making an “L” shape. Make sure that you are bending the end that has been cut and not the other end, which must be unperturbed in order to fit into the bores of the pipettes. During surgeries, this little bent-over leg will rest on the top of the plunger-clamping block (Fig. 1).
Fig. 1. Custom injection apparatus assembly.

The injection apparatus consists of a manual hydraulic manipulator that advances a plunger through the bore of a pulled glass pipette. Three of the four custom parts (see Supplementary File 1 for fabrication information) are mounted on the vertical rod that comes with the stereotax and hold both the micromanipulator and the pipette; the fourth is mounted on the moveable shaft of the manipulator and holds the plunger that moves within the bore of the pipette to expel the virus. For best results, adhere to the positioning of the components shown here. The vertical metal rod extends approximately 10.5 cm below the metal clamp that holds the vertical rod to the stereotax arm, and the bottom of the pipette-clamping block is attached to the vertical rod (with a 4–40 screw) and flush with the end of it. The two symmetrical micromanipulator-clamping blocks are clamped (with two brass 10–32 screws) to both the vertical rod and the micromanipulator; the blocks should be flush against the metal clamp, and the end of the micromanipulator shaft is ~1.8 cm below the bottom edge of the clamping blocks. The plunger-clamping block is attached (with a 4–40 screw) to the piston of the micromanipulator, with the grooves of the pipette-clamping block and those of the plunger-clamping block aligned so that the plunger is in line with the pipette. The two 4–40 thumbscrews that pin the pipette and its plunger into the grooves in their respective blocks have nylon washers to provide grip and prevent breakage of the glass. While the exact positioning of the custom parts on the stereotax rod is not critical, the positions shown here work well, and major deviation from them could result in the inability to raise the pipette high enough to allow the stereotax arm to be swung out of the way when inserting and removing the mouse or, conversely, the inability to lower the pipette to the desired depth in the brain.
2.3.2. Pipette preparation
Place a strip of dental wax along the bottom of a petri dish so that pulled pipettes can be stuck to it for storage.
Pull pipettes using a Sutter P-2000 puller with the following settings: HEAT 475, FIL 4, VEL 50, DEL 140, PUL 45. If necessary, adjust puller settings according to manufacturer’s instructions in order to produce long (> 10 mm), skinny pipettes.
Under dissecting microscope and holding each pipette alongside of a metric ruler, use straight forceps to break tips to a length of ~8 mm from the “shoulder” of the pipette.
Use a micropipette beveler to bevel the ends of the snipped injection pipettes at 35 degrees from the longitudinal axis of the pipette.
The day before surgery, place beveled pipettes, blunt end down, in PCR tubes filled with mineral oil to backload mineral oil and sterilize overnight in ultraviolet sterilizer.
2.3.3. Autoclaving tools and coverslips
Take an empty pipette tip box and remove the rack that held the tips.
Place an aluminum foil square on the bottom and a layer of coverslips on top of that. Layer and repeat for as many coverslips as desired. Close box with a piece of autoclave indicator tape.
Place surgical tools in an autoclavable pouch.
Autoclave all on a dry cycle and check indicators to ensure sterilization has occurred.
2.4. Reagent setup
2.4.1. 70% ethanol
734 ml 95% ethanol +263 ml deionized water. Can be stored in sealed glass bottles indefinitely.
2.4.2. 1:10 bleach solution
10 ml bleach +90 ml water (deionized or otherwise). Can be stored in sealed glass bottles indefinitely. Fill two scintillation vials with 20 ml 1:10 bleach for decontamination of coverslips and pipettes.
2.4.3. Helper viruses
These come from Addgene in 100 μl tubes. For initial aliquoting of undiluted viruses: thaw each tube in a 37 °C water bath, then vortex briefly (perhaps two seconds) to mix, then spin down briefly (another few seconds) on the mini-centrifuge to consolidate all the liquid in the bottom of the tube. Make 5 μl aliquots in cryotubes, label and refreeze. For preparing aliquots of diluted viruses: thaw one 5 μl aliquot in a water bath as above (note that thawing this smaller volume takes only a few seconds), briefly vortex and briefly spin down, then dilute as described below. Note that we have found these dilutions to work well in PV-Cre and DAT-Cre mice; the optimal dilutions may be different for other Cre lines because of AAV1′s tropism.
If using AAV1-synP-FLEX-splitTVA-EGFP-B19G: dilute to 2.4 × 10^12 gc (genome count) per ml (this is a 1:10 dilution of Addgene lot# v14715, with undiluted titer of 2.4 × 10^13 gc/ml, which we have tested in our lab; if using this lot, therefore, add 45 μl DPBS to the 5 μl aliquot), then recap, vortex, and spin down as above, then divide this diluted stock into 5 μl aliquots in new cryotubes, label appropriately and refreeze at −80 °C.
If using AAV1-syn-FLEX-splitTVA-EGFP-tTA and AAV1-TREtight-mTagBFP2-B19G: dilute AAV1-syn-FLEX-splitTVA-EGFP-tTA to 8.5 × 10^10 gc/ml (1:200 dilution of Addgene lot # v15287, with undiluted titer of 1.7 × 10^13 gc/ml), and dilute AAV1-TREtight-mTagBFP2-B19 G to 1.6 × 10^12 GC/ml (1:20 dilution of Addgene lot # v14716, undiluted titer 3.2 × 10^13 gc/ml). Divide the diluted stocks into 5 μl aliquots in new cryotubes, label appropriately and refreeze at −80 °C.
On the day of the first surgery (day 0, below), thaw either one tube of diluted AAV1-synP-FLEX-splitTVA-EGFP-B19 G or one tube each of diluted AAV1-syn-FLEX-splitTVA-EGFP-tTA and AAV1-TREtight-mTagBFP2-B19 G, as applicable, in a 37 °C water bath, vortex, and spin as above. If using the two-AAV combination, combine the diluted AAV1-syn-FLEX-splitTVA-EGFP-tTA and AAV1-TREtight-mTagBFP2-B19 G 50/50 by volume in one of the tubes, then vortex to mix and spin down. Keep all tubes of thawed viruses on wet ice when not working with them.
2.4.4. Anesthetic cocktail
Mix26.7 ml sterile normal saline +3 ml ketamine (100 mg/ml) +0.3 ml xylazine (100 mg/ml) in a sterile 30 ml vial. Store at room temperature until expiration date of any component. Use of controlled substances such as ketamine, buprenorphine, and sodium pentobarbital must be logged according to your institution’s guidelines.
2.4.5. Diluted meloxicam
Mix28.8 ml sterile normal saline +1.2 ml meloxicam (5 mg/ml) in a sterile 30 ml vial. Store at room temperature until expiration date of any component.
2.4.6. 4% paraformaldehyde in PB
Mix10 ml 16% PFA, 4 ml 10X PB and 26 ml distilled water, per mouse. May be store at 4 °C for up to one week. Additional volumes of 4% paraformaldehyde should be made so that the brain will be completely submerged in fixative postoperatively.
2.4.7. Cryoprotectant
Mix500 ml DPBS +300 g sucrose +300 ml ethylene glycol +10 g polyvinylpyrrolidone; add distilled water to a final volume of 1 L. Shake vigorously until dissolved (this may take up to a day).
2.4.8. PBS-0.5% Triton
100ml 10X PB +50 ml 10% Triton-X +850 ml double-distilled water.
3. Procedure
3.1. First surgery: Injection of helper AAVs (Day 0)
Prepare equipment, supplies, and reagents as described above, and select a Cre mouse of the desired age and genotype (e.g., PV-Cre).
Determine the desired total displacement of the pipette plunger, given the desired injectate volume. To prevent overshoots caused by operator inattention, stick a small sliver of tape at the appropriate reading on the edge of the manipulator control unit drum, assuming the injection will begin at a reading of zero. For the capillaries described here (internal diameter = 0.494 mm), each tick of the manipulator drum (2 μm each) injects 0.3833 nl. Injecting 100 nl therefore entails advancing 521.7 μm (just over one 500 μm revolution of the manipulator drum).
Put on personal protective equipment for surgery (e.g., gloves, lab coat, hair cover, surgical mask), following your institution’s guidelines.
Turn on warm water heating pad and place a new cage on it to warm up. Place a new burr in the drill; turn on drill, stereotax digital display, and glass bead sterilizer. Remove a sterile drape section from its pouch and set on bench next to stereotax. Slide sterile surgical tools out of autoclave pouch onto it without touching tools. The glass bead sterilizer is for disinfecting surgical tools in between mice when doing multiple surgeries on the same day.
Record relevant information about the mouse and surgical experiment (animal number, injection date, start time, target region, virus, species/strain, sex, age, atlas coordinates, and any additional information specific to your experiment).
Weigh mouse and record weight. A large plastic measuring cup, or an empty pipette tip box, can be used to contain the mouse on the balance.
- Draw up drugs for mouse:
- Anesthetic cocktail (ketamine/xylazine cocktail): 0.1 ml cocktail / 10 g mouse body weight. This corresponds to a dose of 100 mg/kg ketamine, 10 mg/kg xylazine.
- Meloxicam: 0.1 ml / 10 g mouse body weight. This corresponds to a dose of 2 mg/kg,
- Slow release buprenorphine: 0.04 ml / 10 g mouse body weight. This corresponds to a dose of 2 mg/kg,
Create a platform for loading the virus into the pipette tip by cutting a ~1 inch square of parafilm and placing it on top of the stereotax’s ear bars so that it forms an inverted U shape. Placed in this way, the parafilm square forms a springy platform for the coverslip so that the pipette tip will not break if it contacts the coverslip. Loading the pipette from a droplet on the coverslip, rather than simply lowering the pipette tip into the tube of virus, is necessitated because the pipettes are not long enough to reach to the bottoms of typical O-ring cryotubes.
Spray off your gloves with 70% ethanol, then use sterile forceps to place an autoclaved coverslip on the parafilm.
Remove beveled pipette from the mineral oil, keeping the pipette tip pointed up. Carefully insert the end of the plunger into the back of the pipette, avoiding any trapping of air bubbles. When plunger is successfully inserted, and keeping the pipette tip pointed up, push in plunger until the oil has displaced all air from the pipette. Invert the pipette and plunger so that the pipette points down, and secure it to the track of the lower injection head with the thumbscrew. Ensure that the metal plunger is resting in the groove of the upper injection head. Gently push down on the plunger until the small bent end is resting on the top of the upper injection head, then secure it with its thumbscrew. Blot away the oil droplet with an eye spear. Avoiding the introduction of any air into the system is critical.
Pipette 1–1.5 μl of virus solution onto the coverslip. Expel a small amount of mineral oil from the pipette onto another section of the coverslip in order to displace any air pocket that may have formed. Using a sterile eye spear, dab any remaining mineral oil off the pipette tip, then move the tip into the pool of virus. Use the manipulator drum to retract the plunger to draw up a volume of virus well in excess of the desired amount of virus solution to be injected. In a later step, the drum will be advanced to the 0 mark before the desired volume of virus is injected, so the amount loaded in the pipette should be sufficient to allow this.
Still observing through the microscope, raise pipette out of the virus solution and advance the manipulator drum by a small amount to expel a drop of solution, confirming that there is no problem with virus expulsion. Blot drop with eye spear.
Move microscope out of the way. Raise pipette to the maximum height possible and rotate stereotax arm back somewhat more than 90 degrees so that the pipette is well out of the way. Pick up parafilm with coverslip and remaining virus, and discard in scintillation vial filled with 1:10 bleach that, when full, should be placed in a sharps container to be autoclaved subsequently.
Place mouse in isoflurane induction chamber and turn on oxygen (~1 L per minute) and isoflurane (4%). Monitor the mouse closely while anesthesia takes effect, to prevent overdose. The induction chamber should be suitably ventilated either by a snorkel, fume hood, or otherwise, to prevent release of isoflurane into the room air.
When mouse’s breathing has slowed to approximately one breath per second, turn off flow to induction chamber and inject the ketamine/xylazine cocktail intraperitoneally. Record time of administration.
Place mouse into an enclosed container or clean cage without any other animals while waiting for injectable anesthetic to take effect.
Prepare 1 ml syringe of sterile normal saline or lactated Ringer’s solution. Set aside in a safe location, to be used later to maintain hydration of the tissue.
When mouse appears anesthetized, confirm unresponsiveness with foot pinch.
Lift the skin on the back of the neck and inject the slow release buprenorphine and meloxicam subcutaneously.
Shave scalp with clippers and wipe away cut hair with a Kimwipe or paper towel.
Apply ophthalmic ointment liberally to protect eyes from Nair, ethanol, and Betadine and to prevent their drying out.
Apply Nair to scalp in the area of the planned incision with a cotton-tipped applicator. After one minute, begin wiping off shed hair with a paper towel, leaving a denuded scalp. Nair removes all hair and stubble remaining after shaving. The shaving step is still necessary, however, to allow the Nair to contact the scalp readily.
Place mouse in ear bars. Begin with the left ear bar fixed and the right one withdrawn to make room, then place the mouse’s left ear hole onto ear bar and carefully insert the right one until resistance is encountered. Affix the right ear bar. Check for symmetry by eye and use curved forceps to test whether head can rotate freely in vertical plane only. Mounting the mouse into the ear bars is usually the most difficult step for new surgeons to master.
Center head by holding one ear bar while loosening its screw, then applying extremely light pressure to it while very slightly loosening the other bar’s screw, so that the other bar is pushed back while being controlled by a finger to avoid overshoot. When the bars are at equal readings, affix with screws.
Lift up snout with curved forceps and slide bite bar forward under nose, opening mouth with curved forceps as necessary to allow the bar into the mouse. When incisors seem to drop down into hole in bar, withdraw bar exceedingly gently to make sure that resistance is encountered, confirming incisors are indeed in hole, then very slightly bring the bar forward once again a tiny amount so that no tension is being applied to teeth in either direction. Put snout clamp on snout and tighten, making that the clamp it situated high on the snout so as not to restrict the nasal passages and cause suffocation. Ensure that head is even in the stereotax and otherwise looks right; return to previous step if necessary. Typical parameters on Stoelting Mouse & Neonatal Rat adaptor: snout clamp height 9.8–10.0 mm, ear bar height 15 mm, ear bars 2.7–3.4 mm.
Place a freshly-opened hand warmer under the mouse, with a paper towel between the mouse and the hand warmer. This helps prevent hypothermia, which can be fatal.
Sanitize scalp: proceeding from center to periphery with an out-ward-spiraling motion, perform 3 scrubs with povidone-iodine swabs followed by 3 scrubs with 70% ethanol-soaked swabs.
Spray off gloves with 70% ethanol and place a sterile drape over the mouse’s body if required by your institution. Alternatively, sterilized clear consumer quality wrap can be used so that respiration can be observed during surgery. Also if required by your institution, don sterile gloves and spray or wipe down microscope controls and other non-sterile things that will be handled.
Using clean curved forceps, tent the skin away from the skull, and then use scissors to create an ~1 cm long cut midsagittally down the scalp starting from the occipital plate. This should allow both bregma and lambda landmarks to be visualized. Scissors work well for making the scalp incision on a mouse and do not damage the skull and cause bleeding, unlike a scalpel blade.
Use curved forceps to pull the skin flaps laterally to expose the skull, ensuring bregma and lambda points are clearly visible and that the vicinity of the drilling location is well exposed.
Rotate the stereotax arm back to the working orientation (perpendicular to the anterior-posterior axis) and secure in place. Use stereotax control knobs to maneuver pipette tip to roughly above the bregma point and lower to within 1–2 cm of the skull surface. Move microscope head to focus on skull surface at low magnification.
Move the pipette tip within the field of view of the microscope, then move it towards the bregma point, increasing the magnification as it approaches so that both the tip and the skull surface remain visible as the tip moves until the desired working magnification is reached.
Move the pipette tip until it is just above the bregma point. Pushing down very gently with curved forceps on the skull plates assists with locating the skull sutures. Reset the X and Y coordinates to zero. Bregma is defined as the intersection point of the skull sutures while lambda is defined as the intersections of lines tangent to the skull sutures, rather than the intersection of the sutures themselves. See Paxinos atlas (Paxinos and Franklin, 2013) for further discussion.
Expel a small droplet of virus solution from the pipette tip to ensure that it is not clogged. Blot droplet away with a spear tip and retain this spear, setting it down so as to keep its tip from touching any surfaces.
Lower the tip until it is just touching the skull surface at the bregma point. Zero the Z coordinate.
Immediately raise the tip away from the skull and expel another droplet of solution to prevent clogging. Remove this droplet with a spear.
Move the pipette tip to just above the lambda point and record X and Y coordinates.
Expel a small droplet of virus solution from the pipette tip to ensure not clogged and blot with a spear.
Lower the tip until it is just touching the skull surface at the lambda point. Record the Z coordinate. The Z coordinate of lambda should be very close (within<0.1 mm) to that of bregma; if not (and if the snout clamp and ear bar heights are set as recommended in step 25), this is usually an indication that the mouse’s head has been improperly placed in the stereotax. The snout clamp height can be adjusted to attempt to correct for this, but the mouse’s head may need to be removed from the stereotax and placed in it again.
Immediately raise the tip away from the skull and expel another droplet of solution to prevent clogging, blotting with a spear.
Determine the target coordinate values: for each of the three coordinate axes, scale the coordinates according to the measured bregma-distance (scaled coordinate = atlas coordinate * BLMeasured/BLatlas, where BLMeasured = measured bregma-lambda anterior-posterior distance and BLatlas = atlas bregma-lambda anterior-posterior distance = 4.2 mm for Paxinos atlas).
Withdraw the pipette along the Z axis until it is well clear of the mouse’s head, then move to the scaled target X and Y values.
Advance the pipette along the Z axis until it is almost, but not quite, touching the skull surface.
Using a 27 gauge needle, scratch an identifying mark on the skull surface just under the pipette tip to indicate the desired location of the craniotomy.
Withdraw pipette along Z axis until it is safely out of the way. Note: follow your institutional safety guidelines, and common sense, to make sure that you do not stick yourself with the virus-containing pipette. Readers are encouraged to develop whatever additional safety procedures are necessary to prevent needle sticks or other accidents.
Use the drill to carefully thin the skull in a circle around the center of the mark, leaving a thick central region. Use the non-dominant hand to stabilize the hand holding the drill to minimize motion if needed. Move the pipette tip back down to ensure that the circle is properly placed before moving back to the previous position and adjusting the circle if necessary.
Thin the circular perimeter until it is thin enough to be punctured by the needle in the next step, using translucency as a visual indication of thickness. Leave one side (the left side, for right-handed surgeons) slightly more firmly attached than the rest of the circumference, as a “hinge”. Small cracks should appear at the lowest points in the resulting groove when thinned enough.
When the thinning is complete, press the tip of the needle into the right (for right-handed surgeons) side of the thick central region and gently lift the disc of skull away from the brain on the side opposite the “hinge”. When right side of the skull disc is lifted away from the brain, put away needle and use forceps to remove disc completely and discard. No bleeding should occur if performed correctly.
To keep tissue hydrated, dribble sterile saline or lactated Ringer’s onto the skull surface, forming a mound surrounded by the scalp, then blot excess using an eye spear, leaving a thin layer of solution in the surface.
Advance the manipulator drum to the nearest whole millimeter (i.e. so that the 0 on the drum is aligned with the line) and use a sterile spear to dab off any expelled virus.
Looking through microscope at the craniotomy, advance tip along Z axis and to the surface of brain. If a blood vessel is directly over the injection site, adjust the X and Y coordinates to avoid the blood vessel and record the new coordinates and reason for the change in your notes. While initially observing through microscope to ensure that the tip advances into the brain uneventfully, advance the tip until it. has penetrated the dura, then monitor readings of stereotax positional display while continuing to advance to the desired Z coordinate, being careful not to overshoot.
Starting a timer to maintain an even pace (e.g., 2 μm (one tick of manipulator drum), or 0.3833 nl, per second), advance the injection manipulator to inject 200 nl. Injecting slowly and steadily minimizes flux back up along the pipette track.
When the injection is complete, leave the pipette in place for 5 min before withdrawal. This is also to minimize flux along the pipette track.
While waiting, fill out cage cards and begin cleaning up or preparing for next surgery, if applicable.
Withdraw the pipette from the brain; raise stereotax arm to maximum extent and rotate stereotax arm out of the way.
Blot away any liquid from the skull surface using a spear.
Remove the mouse from the stereotax, place it on the drape, and close the incision with sutures.
Place the mouse in a clean cage on a heating pad. Mice should be housed one per cage to recover from anesthesia before returning to cagemates.
Used pipettes can be placed in a scintillation vial filled with 10% bleach that, when full, should be disposed of in a sharps container to be autoclaved.
Return mouse to vivarium when recovered from anesthesia.
Check mouse on the first, second, and third days following surgery to ensure recovering well. Administer 2 mg/kg meloxicam subcutaneously on postoperative day 1 and further analgesic on following days if symptoms of distress are noted.
3.2. Second surgery: rabies virus injection (day 7)
The second surgery is very similar to the first surgery, with the following exceptions:
The rabies virus, rather than the helper AAVs, is being injected. Store, thaw, vortex, and spin the rabies virus as for the AAVs, and again keep thawed virus on wet ice when not actively working with the tubes. The virus can be injected at the same rate as the AAVs. The injection volume can vary depending on the structure: we have used 100 nl for cortex (in PV-Cre mice) and larger volumes for subcortical structures (e.g., substantia nigra, pars compacta in DAT-Cre mice) in which hitting the site previously injected with AAV is less certain.
The hair on the mouse may not have grown back, therefore the hair removal steps may be skipped.
Once the mouse is anesthetized and mounted in the stereotax, sterilize the scalp as before, then use a pair of forceps and small pair of sterile scissors to remove the sutures and make an incision along the line of the previous incision.
The bregma-lambda distance does not need to be remeasured: after bregma has been located and the coordinates zeroed, the previous injection site coordinates can be used without relocating lambda.
Drilling is unnecessary, as the injection site is exposed from the previous surgery. The use of the same craniotomy also helps to ensure that the second injection is at the same site as the first one.
Be careful when pulling the skin laterally to expose the previous injection site. A film may have formed above the injection site, and quickly pulling the skin could pull on it and damage any blood vessels.
3.3. Perfusion and histology (day 14+)
We usually stain tissue both for EGFP (without which the EGFP signal from the helper AAV is not usually visible) and for a marker of the Cre-defined population (e.g., parvalbumin in PV-Cre mice). Staining for mCherry or other transgene expressed by the rabies virus is not necessary, and the mTagBFP2 expressed from the second helper AAV is typically clearly visible as well. Cell labeled with EGFP, mTagBFP2, and mCherry may be assumed to be “starter cells”, although in fact it is impossible to say based on postmortem inspection alone which cells were the initially-infected ones, because some could have died prior to perfusion.
Seven days after the second surgery, deeply anesthetize the mouse with sodium pentobarbital and perfuse transcardially with 40 ml of PBS to flush followed by 40 ml of 4% paraformaldehyde, then remove the brain. Place the brain in 4% paraformaldehyde and shake overnight at approximately 1 Hz at 4 °C.
The next morning, set up 6 Eppendorf tubes per brain, suitably labeled with mouse identification number and series number (1–6), in a tube rack. Add 1 ml cryoprotectant to each tube.
On a vibrating microtome, section the brain coronally into 50 μm thick sections. Use a small paintbrush to place each section into the appropriate tube, proceeding through the set of six tubes repeatedly so that each tube contains a series of every sixth section through the brain.
When finished sectioning, add 300 μl more cryoprotectant to each tube, cap tubes (taking care that no sections are sticking out of the cryoprotectant and able to be damaged by the cap when closing), and store in −20 °C freezer until needed.
- Series can then be immunostained individually, so that every sixth section is stained for the same proteins. In brief, immunostain as follows:
- Transfer sections into a Netwell insert in a 12-well plate well.
- Rinse sections with 4 quick washes in PB followed by 4 5-minute washes 0.5% Triton in PB (5 ml per wash). During the 5-minute incubations, prepare blocking/dilution buffer: 10% normal donkey serum + 0.5% Triton in PBS.
- Incubate sections in 2 ml blocking buffer per well for 2 h on shaker at room temperature. During the incubation, prepare primary antibody solution, diluting the antibodies in blocking/dilution buffer for a total volume of 2.5 ml per well, with the following dilutions:
- chicken anti-GFP 1:1000 (note: because this antibody comes diluted 50/50 in glycerol, use 1:500 of the stock)
- antibody to marker of starting cells (e.g., guinea pig anti-parvalbumin or sheep anti-tyrosine hydroxylase, both 1:1000)
- Filter primary antibody solution through 0.2 μm filter to remove antibody aggregates immediately before adding to tissue.
- Incubate sections in primary antibody solution (2.5 ml per well) overnight on shaker at 4 °C, with plate covered with lid and wrapped with plastic wrap to minimize evaporation.
- The following morning, rinse sections with 4 quick washes in 0.5% Triton in PB followed by 4 10-minute washes 0.5% Triton in PB (5 ml per wash). During the washes, prepare secondary antibody solution, diluting the antibodies in blocking/dilution buffer for a total volume of 2 ml per well, with the following dilutions:
- donkey anti-chicken Alexa Fluor 4881:200
- secondary antibody to antibody to marker of starting cells (e.g., donkey anti-guinea pig Alexa Fluor 647 or donkey anti-sheep Alexa Fluor 647, both 1:200)
- Filter secondary antibody solution through 0.2 μm filter to remove antibody aggregates immediately before adding to tissue.
- Incubate sections in secondary antibody solution (2 ml per well) for two hours on shaker at room temperature. A smaller volume (2 ml vs 2.5 ml) is used for this incubation, which is shorter and for which evaporation is therefore less of a factor.
- Rinse sections with 4 quick washes in PB followed by 5 10-minute washes in 0.5% Triton in PB, followed by 1 5-minute wash in PB (5 ml per wash).
Mount brain sections onto coated slides, allow to dry (slides either flat or tilted), covered to protect from light, at room temperature until all liquid droplets have evaporated (30–45 minutes).
Coverslip with mounting medium, allow to dry flat at room temperature overnight, then image using a confocal or epifluorescence microscope. Example results are shown in Fig. 2.
Fig. 2. Typical monosynaptic tracing results: inputs to parvalbumin-positive cells in somatosensory cortex.
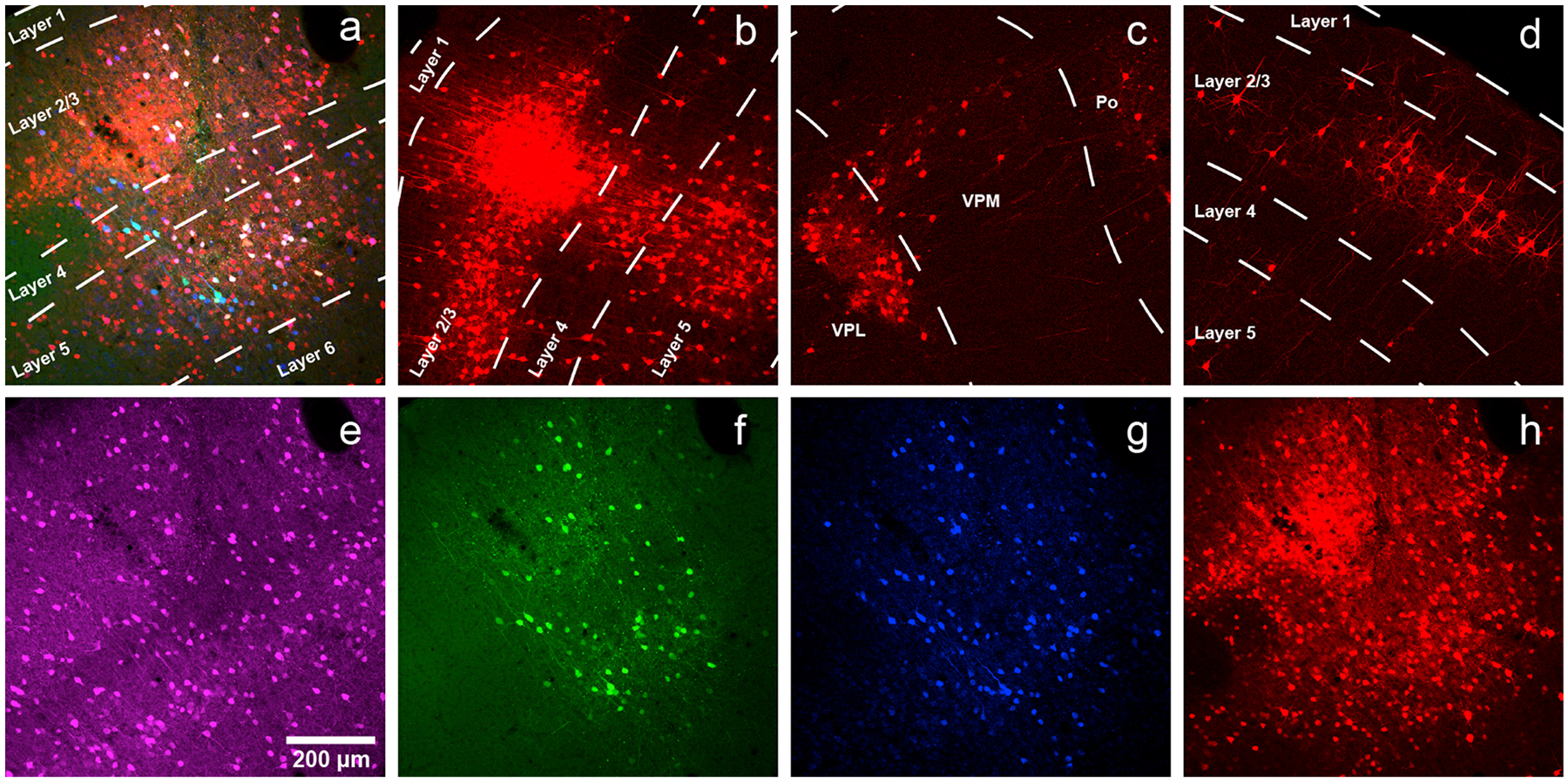
a) Injection site in primary somatosensory cortex (coordinates: AP= −0.58 mm with respect to bregma, LM = 3.00 mm with respect to bregma, DV = −1.00 mm with respect to the surface of the brain) of a PV-Cre mouse injected with a mixture of AAV1-syn-FLEX-splitTVA-EGFP-tTA and AAV1-TREtight-mTagBFP2-B19 G, followed by RVΔG-4mCherry(EnvA) a week later. Green = anti-EGFP staining, blue = mTagBFP2, red = mCherry. Individual channels from this field are shown in panels e–h. Scale bar: 200 μm, applies to all panels. b) Labeled presynaptic neurons in ipsilateral secondary somatosensory cortex. The dense central region is not an injection site but instead just a concentrated group of transsynaptically-labeled neurons. c) Labeled presynaptic neurons in ipsilateral thalamus (VPL, VPM and Po). d) Labeled presynaptic neurons in contralateral S1. e–h) Individual channels from the field shown in panel a. e) anti-parvalbumin staining (not shown in panel a). f) anti-EGFP staining, indicating expression from the first, Cre-dependent AAV (note that the EGFP signal is not usually visible when the helper viruses are used at the recommended dilutions). g) mTagBFP2, indicating expression from the second, tTA-dependent AAV. h) mCherry, marking activity of the rabies virus.
Supplementary Material
Acknowledgements
We thank Carlos Lois for first showing us an injection apparatus based on the MO-10/Wiretrol combination. We thank Andrew Gallant of the MIT Central Machine Shop for fabricating the stereotax adapter parts and for producing the detailed drawings of them. Research reported in this publication was supported by BRAIN Initiative awards U01MH106018, U01MH114829, and U19MH114830 from the National Institute of Mental Health.
Footnotes
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.jchemneu.2019.101661.
References
- Agate RJ, Scott BB, Haripal B, Lois C, Nottebohm F, 2009. Transgenic songbirds offer an opportunity to develop a genetic model for vocal learning. Proc. Natl. Acad. Sci. U. S. A 106, 17963–17967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrlund-Richter S, Xuan Y, van Lunteren JA, Kim H, Ortiz C, Pollak Dorocic I, Meletis K, Carlen M, 2019. A whole-brain atlas of monosynaptic input targeting four different cell types in the medial prefrontal cortex of the mouse. Nat. Neurosci 22, 657–668. [DOI] [PubMed] [Google Scholar]
- Atasoy D, Aponte Y, Su HH, Sternson SM, 2008. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J. Neurosci 28, 7025–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, Gao XJ, Kremer EJ, Malenka RC, Luo L, 2015. Circuit architecture of VTA dopamine neurons revealed by systematic input-output mapping. Cell 162, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federspiel MJ, Bates P, Young JA, Varmus HE, Hughes SH, 1994. A system for tissue-specific gene targeting: transgenic mice susceptible to subgroup A avian leukosis virus-based retroviral vectors. Proc. Natl. Acad. Sci. U. S. A 91, 11241–11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Bujard H, 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U. S. A 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, et al. , 2013. Convergence of pontine and proprioceptive streams onto multimodal cerebellar granule cells. eLife 2, e00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Jacobs MW, Ito-Cole T, Callaway EM, 2016. Improved monosynaptic neural circuit tracing using engineered rabies virus glycoproteins. Cell Rep. 15, 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, et al. , 2014. Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nat. Neurosci 17, 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin TK, Jin L, Lea NL, Wickersham IR, 2019. Monosynaptic tracing success depends critically on helper virus concentrations. bioRxiv. 10.1101/736017.736017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay S, Prehaud C, Dietzschold B, Lafon M, 2003. Glycoprotein of nonpathogenic rabies viruses is a major inducer of apoptosis in human jurkat T cells. Ann. N. Y. Acad. Sci 1010, 577–581. [DOI] [PubMed] [Google Scholar]
- Liu K, et al. , 2017. Lhx6-positive GABA-releasing neurons of the zona incerta promote sleep. Nature 548, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamichi K, et al. , 2011. Cortical representations of olfactory input by trans-synaptic tracing. Nature 472, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzounov DG, et al. , 2017. In vivo three-photon imaging of activity of GCaMP6-labeled neurons deep in intact mouse brain. Nat. Methods 14, 388–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ, 2013. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates. Elsevier/AP, Amsterdam. [Google Scholar]
- Petreanu L, Mao T, Sternson SM, Svoboda K, 2009. The subcellular organization of neocortical excitatory connections. Nature 457, 1142–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon TR, et al. , 2016. Rabies Virus CVS-N2c(DeltaG) Strain Enhances Retrograde Synaptic Transfer and Neuronal Viability. Neuron 89, 711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai K, et al. , 2016. Capturing and Manipulating Activated Neuronal Ensembles with CANE Delineates a Hypothalamic Social-Fear Circuit. Neuron 92, 739–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmento L, Li XQ, Howerth E, Jackson AC, Fu ZF, 2005. Glycoprotein-mediated induction of apoptosis limits the spread of attenuated rabies viruses in the central nervous system of mice. J. Neurovirol 11, 571–581. [DOI] [PubMed] [Google Scholar]
- Seidler B, Schmidt A, Mayr U, Nakhai H, Schmid RM, Schneider G, Saur D, 2008. A Cre-loxP-based mouse model for conditional somatic gene expression and knock-down in vivo by using avian retroviral vectors. Proc. Natl. Acad. Sci. U. S. A 105, 10137–10142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach OM, Cranfill PJ, Davidson MW, Verkhusha VV, 2011. An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS One 6, e28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szonyi A, Sos KE, Nyilas R, Schlingloff D, Domonkos A, Takacs VT, Posfai B, Hegedus P, Priestley JB, Gundlach AL, et al. , 2019. Brainstem nucleus incertus controls contextual memory formation. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan S, et al. , 2015. High-performance probes for light and electron microscopy. Nat. Methods 12, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt D, et al. , 2015. Viral-mediated labeling and transplantation of medial ganglionic eminence (MGE) cells for in vivo studies. J. Vis. Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall NR, Wickersham IR, Cetin A, De La Parra M, Callaway EM, 2010. Monosynaptic circuit tracing in vivo through Cre-dependent targeting and complementation of modified rabies virus. Proc. Natl. Acad. Sci U. S. A 107, 21848–21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N, 2012. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74, 858–873. [DOI] [PubMed] [Google Scholar]
- Weible AP, et al. , 2010. Transgenic targeting of recombinant rabies virus reveals monosynaptic connectivity of specific neurons. J. Neurosci 30, 16509–16513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR, Sullivan HA, 2015. Rabies viral vectors for monosynaptic tracing and targeted transgene expression in neurons. Cold Spring Harb. Protoc 2015, 375–385. [DOI] [PubMed] [Google Scholar]
- Wickersham IR, et al. , 2007a. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 53, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR, Finke S, Conzelmann KK, Callaway EM, 2007b. Retrograde neuronal tracing with a deletion-mutant rabies virus. Nat. Methods 4, 47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR, Sullivan HA, Seung HS, 2010. Production of glycoprotein-deleted rabies viruses for monosynaptic tracing and high-level gene expression in neurons. Nat. Protoc 5, 595–606. [DOI] [PubMed] [Google Scholar]
- Young JA, Bates P, Varmus HE, 1993. Isolation of a chicken gene that confers susceptibility to infection by subgroup A avian leukosis and sarcoma viruses. J. Virol 67, 1811–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhong P, Hu F, Barger Z, Ren Y, Ding X, Li S, Weber F, Chung S, Palmiter RD, et al. , 2019. An excitatory circuit in the perioculomotor midbrain for non-REM sleep control. Cell 177 1293–1307 e1216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.