

Abstract
Overlapping genes were thought to be essentially absent from the human genome until the discovery of abundant, frameshifted internal open reading frames (iORFs) nested within annotated protein coding sequences. However, it is currently unclear how many functional human iORFs exist and how they are expressed. We demonstrate that, in hundreds of cases, alternative transcript variants that bypass the start codon of annotated coding sequences (CDSs) can recode a human gene to express the iORF-encoded microprotein. While many human genes generate such non-coding alternative transcripts, they are poorly annotated. Here we develope a new analysis pipeline enabling the assignment of translated human iORFs to alternative transcripts, and provide long-read sequencing and molecular validation of their expression in dozens of cases. Finally, we demonstrate that a conserved DEDD2 iORF switches the function of this gene from pro- to anti-apoptotic. This work thus demonstrates that alternative transcript variants can broadly reprogram human genes to express frameshifted iORFs, revealing new levels of complexity in the human transcriptome and proteome.
The translation of thousands of noncanonical open reading frames (ncORFs) in human cells has been reported in recent years1,2. ncORFs can be translated from long non-coding RNA (lncRNA) as well as mRNAs, where ncORFs are found within the 5’ UTR (upstream or uORFs), 3’ UTR (downstream or dORFs), and nested within protein coding sequences (CDS) in alternative reading frames (internal ORFs or iORFs)3. Elucidating the expression mechanisms of these classes of ncORFs is critical to provide the conceptual foundation for their functional study. Expression of ncORFs from lncRNA is predominantly transcriptionally regulated4, while translation of uORFs has been attributed to leaky scanning and/or reinitiation5; with this understanding of their expression, examples of functional uORF-encoded microproteins have recently been demonstrated6–9. In contrast, the expression mechanisms of some other classes of ncORFs, in particular iORFs, are not established, and their biological relevance thus remains unclear.
While internal open reading frames (iORFs), a nested subclass of same-strand overlapping ORFs, have long been known in prokaryotes and viruses, they were previously thought to be rare in eukaryotes10. In this work, we focus on iORFs in alternative reading frames, which therefore encode entirely different amino acid sequences relative to the canonical proteins they overlap. Varying numbers of frameshifted human iORFs have been mapped within CDSs using ribosome profiling and proteomics platforms for unannotated protein detection, but, save a few examples, including iORFs within FUS11 and RBM1012, have remained understudied. This is because (1) iORFs are typically excluded during analysis of ribosome profiling because translation in multiple reading frames circumvents the requirement for three-nucleotide periodicity, and (2) the requirement for cap-dependent translation initiation generally means that eukaryotic mRNAs are monocistronic (Fig. 1a). It is unlikely that most iORFs mapped far downstream of CDS start sites can be translated from this context. It is therefore currently unclear whether iORFs are broadly expressed in human cells. It is also critical to exclude the hypothesis that iORFs are not independently translated, but instead arise from translational frameshifting or alternative splicing events that put them in-frame with the upstream start codon, and thus represent isoforms of canonical proteins13.
Fig. 1. Translation and global identification of anisoforms.

a, Frameshifted internal open reading frames (iORFs, green) have previously been mapped within the protein coding sequence (CDS, magenta) of annotated mRNAs (top, boxes represent exons), where their translation is likely inhibited by the upstream CDS start codon. We propose that iORFs are not translated from within annotated CDSs, but rather from alternative transcripts that lack the CDS start codon (bottom). If these alternative transcripts are currently known, they are likely annotated as non-coding variants based on the absence of the complete CDS. b, Pre-mRNA alternative splicing and alternative transcription sites (TSS) are known to generate alternative transcripts encoding protein isoforms with some constant, and some varied, domains. In contrast, alternative transcripts can also encode two entirely different proteins from the same gene (anisoforms) by including or skipping the translation initiation site of the CDS. c, Schematic of the previously reported DEDD2 gene, showing the canonical transcript and an alternatively spliced transcript. The alternative transcript lacks the second exon and the start codon for the canonical CDS, and only encodes the iORF. Transcript sequences were obtained from GENCODE. d, Schematic of the CDKN2A gene. The canonical transcript initiates from an upstream transcription start site (TSS) and encodes the INK4a protein. The alternative transcript skips the first exon, instead starting from an upstream TSS to produce the ARF protein in an alternative reading frame. e, Translation initiation (TI)-sequencing (seq) data (bottom track, blue peaks) supports expression of the previously reported DEDD2-SEP anisoform (iORF, green boxes, track 4), which overlaps the DEDD2 protein CDS (magenta boxes, track 3). A transcript variant specific for the DEDD2-SEP anisoform is currently annotated as non-coding (track 2), because it lacks exon 2 that contains the start codon of the DEDD2 CDS in the canonical transcript (track 1). f, Schematic of pipeline to identify candidate anisoforms from TI-seq data for experimental validation at the mRNA and/or protein level. The detailed workflow can be found in Method. ATs, alternative transcripts. g, Manual identification of an anisoform-encoding alternative transcript in the RUSC1 gene. Transcript 1 (C, track 1) represents the canonical transcript, while transcript 2 (Alt, track 2) represents an alternative transcript for the anisoform, both identified in GENCODE. The coding regions representing the RUSC1 CDS and the RUSC1 anisoform are schematized in track 3, magenta, and track 4, green, respectively. The mRNA-seq reads (track 5, gray) highlighted under the red bracket specifically support for the annotated alternative transcript 2. Previously published TI-seq signal (track 6, and zoom, track 8, cyan), provides evidence supporting the translation of anisoform. The green arrow indicates the peak for the start codon of anisoform in the TI-seq data. Reanalysis of previously reported Ribo-seq (tracks 7 and zoom, 9) supports low-resolution reads specific to the RUSC1 iORF reading frame (green) distinct from the CDS reading frame (magenta) within the overlapping region. Both TI-seq and Ribo-seq data were presented at the codon level.
We hypothesize that frameshifted iORFs have been mis-mapped to mRNAs that contain intact protein CDSs; while the iORF sequences are contained within canonical mRNAs, they are not translated from this context. iORFs are instead expressed from incompletely annotated, “non-coding” alternative transcript isoforms that lack the start codon of the CDS. These alternative transcripts therefore encode an entirely different protein - in contrast the well-understood role of alternative transcripts that generate isoforms of the annotated protein (Fig. 1b). There is precedent for this idea: we previously showed that the canonical transcript of the DEDD2 gene lacked the ability to translate an iORF detected via mass spectrometry14. Through RNA sequencing, we discovered an alternatively spliced transcript that lacked the second exon, which contains the start codon for the CDS, and thus exclusively encoded the iORF (Fig. 1c). Another well-known precedent is the CDKN2A transcript variants encoding the Alternative Reading Frame (ARF) and INK4A proteins, each arising from an alternative transcription start site (TSS)15 (Fig. 1d). We hypothesize that these examples are unlikely to represent isolated cases, because a majority of human protein-coding genes generate “non-coding” transcripts lacking the complete CDS16, and alternative TSS have previously been reported to increase the complexity of the human ncORFeome17. In this work, we demonstrate that alternative transcripts lacking the start codon of CDSs broadly recode human genes to express iORFs. Because frameshifted iORFs are translated from alternative transcripts of the same gene as a canonical protein, like a protein isoform, but possess entirely distinct protein sequences, we coin the term “anisoforms” to refer to them (Fig. 1b).
Global detection of anisoforms in human genes.
While iORFs have been previously reported using both ribosome profiling and mass spectrometry, no global catalog of iORF sequences and loci is currently available. We therefore first sought to generate a comprehensive list of human genes that may encode iORFs in order to both determine an upper bound on their numbers, as well as a working list of iORFs for validation and functional study. This is challenging to do with mass spectrometry because it under-detects bona fide ncORFs18. It is also difficult with ribosome profiling data generated with elongation inhibitors, because the out-of-frame signal from the CDS can obscure translation in the iORF frame4.We therefore turned to translation initiation site sequencing (TI-seq), which employs inhibitors of translation initiation such as lactimidomycin to profile start codons19,20. Although this method decreases confidence in ORF calling that arises from coverage and periodicity21, and may be subject to false positives22, it eliminates the problem of deconvoluting multiple reading frames and can be used to identify potential anisoform start sites. We thus hypothesized that reanalyzing existing TI-seq data could enable generation of a list of candidate human iORFs, with the caveat that false positives must be experimentally excluded.
We employed the PRICE23 algorithm to analyze previously published Ribo-seq and TI-seq data19 to globally identify anisoforms. As anticipated, the Ribo-seq data analysis failed to identify the anisoform within the DEDD2 gene previously reported with mass spectrometry (Supplementary Table 2). More surprisingly, however, analysis of TI-seq data also did not reveal the DEDD2 anisoform (Supplementary Table 2), despite a clear peak in intensity at its start codon (Fig. 1e). We thus developed a novel computational pipeline for the identification of anisoforms using TI-seq data (see Fig. 1f and Methods for details). Through the implementation of this pipeline, we identified a maximum of 1029 translated human anisoform candidates in HEK 293 cells, including the expected DEDD2 anisoform (Fig.1f and Supplementary Table 3). We compared the properties of iORFs identified with this pipeline to human CDSs as well as previously reported uORFs and lncRNA ORFs. Like annotated CDSs and their isoforms, the anisoform candidates show a preference for utilizing AUG within a Kozak context as the start codon, in contrast to uORFs and ncORFs (Extended Data Fig. 1a and 1b). This is consistent with cap-dependent translation24 and supports robust translational potential of iORFs. Moreover, the intensity of TI-seq peaks for anisoform candidates, when compared to uORFs, suggests that their translation initiation levels may be similar (Extended Data Fig. 1c). Strikingly, 91% of our anisoform candidates are <100 amino acids long, consistent with prior reports of ncORF detection (Extended Data Fig. 1d and 1e). These observations are consistent with anisoform translation by scanning ribosomes and support the use of TI-seq for iORF detection on a global scale.
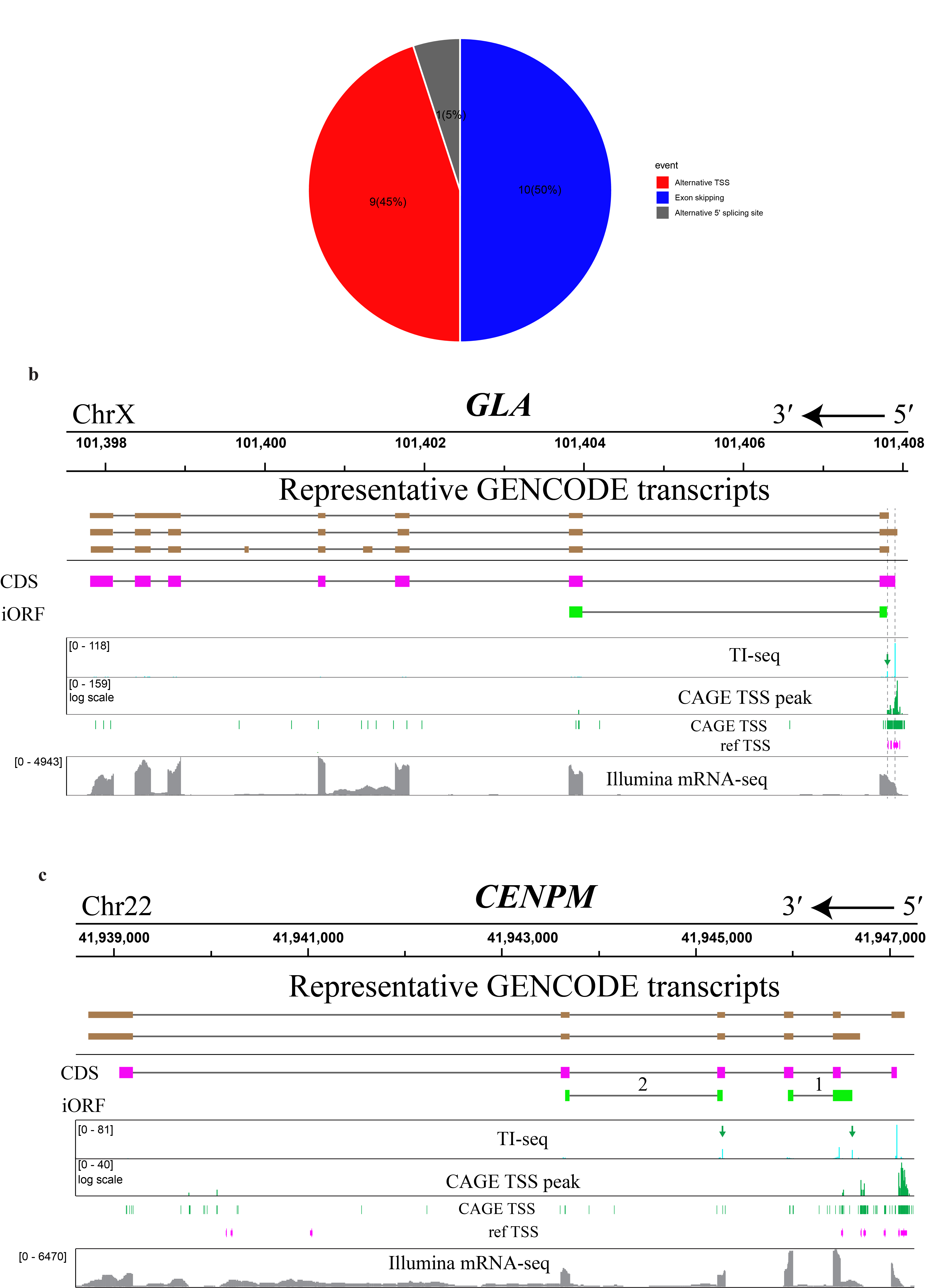
We next examined whether any alternative transcripts associated with iORF-encoding genes that have previously been deposited in GENCODE could explain iORF expression. Out of 1029 candidates, 703 could be mapped to alternative transcripts lacking the CDS start codon that were previously annotated in GENCODE with varying levels of evidence, providing strong support for our hypothesis, though these alternative transcripts are not consistently annotated in all transcriptome resources. Alternative TSS and cassette exon exclusion are the most common sources of alternative transcripts for these candidates (Fig. 2a and Extended Data Fig. 2a). The translation of the remaining 326 iORFs may speculatively be attributed to unannotated transcript isoforms25, leaky scanning, or other mechanisms. After applying additional manual filtering utilizing mRNA-seq expression support, the reference TSS database26 and Ribo-seq evidence for translation in the iORF frame19 (more details can be found in Methods), we validated 20 final high-confidence candidates within 20 genes (Supplementary Table 1). We identified examples of alternative splicing, such as the RUSC1 gene, wherein an alternative 5´ splice donor site within exon 1 can be joined directly to exon 3, excluding exon 2 which contains the CDS start codon (Fig. 1g). Among the genes utilizing alternative TSS, we observed two distinct types. The first type utilizes alternative TSSs within the same first exon but positioned after the start codon of the CDS. An example of this type of anisoform can be found in the GLA gene (Extended Data Fig. 2b). The second type involves alternative TSSs with a novel first exon that bypasses the start codon for the CDS. An illustration of this type of anisoform is evident in the CENPM gene, which may encode both a high-confidence anisoform identified by our pipeline, as well as a second candidate anisoform identified by manual inspection of an alternative transcript featuring an alternative TSS and a novel first exon – a situation mirroring ARF encoding within CDKN2A (Extended Data Fig. 2c). Overall, we conclude that our pipeline, while likely including a number of false positives, provides a preliminary genome-wide catalog of iORFs that may be translated from alternative transcripts.
Fig. 2. Cellular expression of anisoforms.

a, Schematic of iORFs and CDSs in canonical (upper panel) and alternative transcripts (lower panel) for four representative genes. b, Alternative transcripts from the 5’ UTR to the 3’ end of the iORF, which was fused with a HA-FLAG tag at the C-terminus in an expression vector. Overexpression in HEK 293T cells was followed by anti-FLAG Western blotting. The CENPM2 anisoform exhibits aberrant mobility in SDS-PAGE. c, Anisoforms overexpressed in HEK 293T from the cDNA clones described in (b) were subjected to anti-FLAG immunofluorescence (green) with DAPI counterstain (blue). Scale bars, 20 μm. d, Schematic of FLAG knock-in (KI) strategy to validate endogenous anisoform expression in cells. The HEK293T cell genome was cleaved by Cas9 and sgRNAs targeting the stop codons of two independent iORFs (DEDD2 and RUSC1). Single-stranded oligodeoxynucleotide (ssODN) templates encoding FLAG tags were then used to guide homology-directed repair (HDR). Editing in FLAG KI cells was validated with PCR (Extended Data Fig. 5c). e, Western blotting of anti-FLAG immunoprecipitates exhibited specific signal corresponding to anisoform expression in clonally selected KI cell lines relative with parental HEK 293T (WT) as a control. The DEDD2 anisoform-FLAG shows two bands, which may arise from alternative splicing.
Anisoforms are stable cellular proteins translated from alternative transcripts.
To provide molecular evidence that the anisoforms we prioritized with our TI-seq analysis pipeline can be translated to produce detectable proteins, we selected 20 genes (encoding 21 putative anisoforms) as described above as a first-round test set of genes with evidence for anisoform-encoding alternative transcripts in GENCODE or NCBI for experimental validation (see Supplementary Table 1). First, we examined OpenProt27, an open access database incorporating experimental evidence for ncORF expression. 12 of our 21 iORF candidates exhibited previously reported proteomic evidence supporting protein expression. Next, we utilized translation reporters: a portion of each alternative transcript inclusive of the 5´ UTR through the stop codon of the iORF with an epitope tag fused to the 3´ end of the anisoform was cloned into a mammalian expression vector. Transient over-expression followed by Western blotting and/or immunofluorescence provided support for 15 anisoforms mapping to 14 genes (Fig. 2b, 2c, Extended Data Fig. 3a,3b and summarized in Supplementary Table 1). This represents an accumulated positivity rate of 70%. Finally, we sought to detect endogenous expression of anisoform proteins from the corresponding genomic loci. Given that antibodies against anisoforms do not exist, we turned to CRISPR/Cas9-mediated homologous recombination to append two iORFs, RUSC1 and DEDD2, with epitope tags for detection. These two genes were selected because the TI-seq signal for these iORFs were among the top 4 candidates prioritized by our pipeline, and also because they are non-essential28, so disruption of the CDS by inserting the epitope tag in the iORF reading frame should not affect cell viability. Immunoprecipitation-Western blotting (IP-WB) validated endogenous expression of the RUSC1 and DEDD2 anisoforms from the assigned genomic iORF sequences (Fig. 2d–e and Extended Data Fig. 3c). These results collectively support the ability of our anisoform identification pipeline to successfully identify stable and detectable anisoform proteins expressed from genes with previously reported “non-coding” transcripts.
We next tested the hypothesis that anisoforms are exclusively translated from alternative transcripts that lack the CDS start codon, rather than from canonical transcripts with an intact CDS. For the four candidates described above, we cloned the iORFs along with their 5’ UTRs from both the canonical transcripts and alternative transcripts into an overexpression vector, incorporating an epitope tag reporter at the 3´ end of the anisoform in both sequence contexts. Transient transfection followed by Western blotting confirmed that the anisoforms of DEDD2, CENPM, and GLA were exclusively translated by alternative transcripts (Fig. 3a). Even when the canonical transcript supported detectable anisoform expression, as for RUSC1, the alternative transcript produced higher anisoform levels. Consistent with the scanning model, when we mutated the CDS start codons within the canonical transcripts for RUSC1 and GLA (selected because only one AUG start codon is present before the iORF in both of these transcripts), anisoform translation became detectable (Fig. 3b). We also confirmed that the CDS can be expressed from the canonical transcripts, as expected (Fig. 3c). We conclude that anisoform-encoding alternative transcripts are required to bypass iORF translational repression by the start codon of the CDS and enable expression of anisoforms.
Fig. 3. Canonical and alternative transcripts specify CDS or iORF expression, respectively.

a, GENCODE canonical (C) and alternative (Alt) transcripts from four representative genes were cloned from HEK 293 cell cDNA from the 5’ end through the iORF stop codon, with an HA-FLAG epitope tag appended to the 3’ end of the iORF. Anti-FLAG Western blotting was performed after transient transfection to probe iORF expression from each context. b, The start codon of CDSs was mutated to TAA within canonical transcripts to test its suppression of iORF expression. Western blotting for an epitope tag appended to the 3’ end of the iORF was performed after transient transfection.c, GENCODE C and Alt transcripts were cloned from HEK 293 cDNA from the 5’ end through the CDS stop codon, immediately before which an HA epitope tag was encoded. Western blotting from transiently transfected cells was performed to examine CDS expression from each context.
Detection and quantitation of anisoform-encoding alternative transcripts.
Anisoforms are often mapped to overlapping iORFs deep within protein-coding sequences because, while some “non-coding”, anisoform-encoding alternative transcripts have been reported, many are not annotated consistently across transcriptome databases. It is also possible that the full-length sequences of some anisoform-encoding alternative transcripts are incompletely or incorrectly annotated. It is furthermore necessary to validate and quantify alternative transcript expression. Therefore, our next step involved validating known alternative transcripts and discovering novel ones for a total of 70 genes, comprising the prior set of 20 high-confidence candidates associated with annotated alternative transcripts, as well as 50 additional candidates that lack strong Illumina sequencing evidence or existing annotation for alternative transcripts (Supplementary Table 1). Specifically, we included 18 candidates that lack evidence for anisoform-encoding alternative transcripts in NCBI release 110 and GENCODE V38 to investigate whether unannotated transcripts could account for their translation. Since short-read RNA-seq exhibits limited capacity to assemble RNA splicing isoforms29, we employed third-generation sequencing of full-length transcript isoforms30. We elected to increase the depth of sequencing at our desired loci via enrichment due to the high complexity of the human transcriptome. This allowed us to survey our target genes of interest due to their particular complexity, as 98.5%, 80.0%, 57.1%, and 45.7% are already annotated as complex loci exhibiting alternative TTS, exon skipping, alternative 5´ and 3´ splice donor/acceptor sites, and intron retention, respectively (Extended Data Fig. 4a). To this end, we employed fragmented, biotinylated oligonucleotide probes to selectively capture the cDNA corresponding to the genes of interest followed by PacBio sequencing (Fig. 4a)31. The resulting HiFi reads were demultiplexed and refined to discard non-full-length reads. The clustered high-quality reads were then mapped to the genome and collapsed into unique transcript isoforms. We then employed an in-house script for de novo assignment of coding potential (e.g., anisoform, CDS, CDS isoform, or other) to each transcript isoform. Finally, we utilized prior Cap Analysis of Gene Expression (CAGE) sequencing32 and RNA polymerase II chromatin immunoprecipitation (ChIP) sequencing data33 to provide orthogonal evidence for the authenticity of these alternative TSSs and mRNA 5´ ends. Due to the failure to generate biotinylated oligonucleotide probes for one target, the final analysis includes 69 genes.
Fig. 4. ORF-capture and long-read sequencing to confirm, reveal and quantify anisoform alternative transcripts.

a, Schematic showing the workflow of ORF-capture full-length sequencing and data analysis. Full-length first strand cDNA was synthesized with poly(T) reverse transcription primers from the 3’ end and further extended over the 5’ end by template switching. Simultaneously, plasmids containing cloned cDNA of target genes were PCR-amplified with biotin-dUTP to generate biotinylated cDNA, which was then fragmented to create biotinylated probes. The cDNA of targeted genes was enriched using biotinylated probes and streptavidin beads. Following amplification and library preparation, the cDNA was sequenced using PacBio Sequel II. The resulting HiFi reads were processed to retain only high-quality full-length reads. Finally, the processed reads were mapped to the genome to obtain transcript information. b, Evidence for alternative transcripts encoding the C7orf50 anisoform. Top, representative transcripts deposited in GENCODE. Below, PacBio full-length transcripts by read count. Taupe boxes represent exons. The positions of the C7orf50 CDS (magenta) and anisoform-encoding iORF (green) are represented below the transcript diagrams. Reanalysis of TI-seq (cyan) and CAGE TSS (FANTOM5, green, and NCBI reference TSS, pink) from HEK293 cells, as well as RNA PolII CHIP-seq33 (purple), reveals peaks (red arrows) supporting the internal TSS governing anisoform expression. More detailed views can be found in Extended Data Fig. 5. c, Pie plot shows the distribution of the strongest evidence for each anisoform transcript, categorized into “Top 10 & >1% isoform,” “Top 10 isoform,” “>1% isoform,” “Detectable isoform,” “Read support,” and “Other transcript”. Genes from the test set (42/69) lacking anisoform-encoding transcript evidence in NCBI were selected for analysis. PacBio full-length sequencing data for each gene were inspected for the presence and evidence level for anisoform transcripts. “Top10&>1% isoform” signifies isoforms ranking in the top 10 by read count for the gene, with read percentages exceeding 1%. “>1% isoform” refers to isoforms where read percentages exceed 1% of the gene’s total. “Top10 isoform” denotes isoforms within the top 10 by read count. “Detectable isoform” categorizes those isoforms not in the top 10 and with read counts not surpassing 1%. “Other transcript” refers to transcript isoforms that do not encode the iORF. d, Alluvial plot illustrating the distribution and transitions of PacBio evidence levels for transcript isoforms across four categories: “PacBio All” (all detected isoforms), “PacBio Only” (isoforms not shown in previous annotations), “GENCODE”, and “NCBI”. The plot visualizes the flow of transcript variants from those detected by PacBio (first column), to those uniquely identified by PacBio sequencing (second column), and then compared against existing annotations in the “GENCODE” and “NCBI” databases (third and fourth columns respectively). Each stream represents transitions across different evidence levels, including “Top 10 & > 1% isoform”, “Top10 isoform”, “>1% isoform”, “detectable isoforms”, “read support isoform”, and “other transcript”. The “read support isoform” refers to instances where Isoseq 3 did not call the isoform, but raw reads clearly indicate its existence. Each stream represents a unique gene, illustrating the alignment or discrepancy between PacBio findings and established genomic annotations. e, Violin plot illustrating the relative abundances of iORF vs. CDS-encoding transcripts detected by PacBio. The coding ability was assigned based on the first start codon within the mRNA sequence. The left violin plot represents the full-length read count percentage for CDS-encoding transcripts, while the right plot illustrates the full-length read count percentage for iORF-coding alternative transcripts. Median and mean values are indicated for each group. Genes with more than 50% read count for an anisoform-encoding alternative transcript are labeled.
As anticipated, full-length sequencing not only validated the existing annotation but also unveiled novel alternative transcripts selectively encoding anisoforms. These variable alternative transcription events include alternative TSSs, exon skipping, and alternative 5´ splice donor sites. For instance, PacBio data, along with CAGE-Seq and ChIP-Seq data, confirmed an anisoform-encoding alternative transcript previously deposited in GENCODE that uses a downstream alternative TSS in the gene C7orf50 (Fig. 4b, and Extended Data Fig. 5), and revealed a novel anisoform transcript that uses a downstream TSS in the gene RPS8 (Extended Data Fig. 6). Moreover, 17 out of 18 candidates lacking annotated anisoform-encoding alternative transcripts indeed exhibited novel transcripts that selectively encode anisoforms (Supplementary Table 1).
Further analysis showed that 97% of the target genes produce unannotated transcript isoforms (of any type, not just those that encode anisoforms) that rank in the top 10 by isoform count and accounting for more than 1% of the total transcript count for the gene (Top 10&>1%, Extended Data Fig. 4b). Furthermore, 71% of the genes exhibited alternative events absent from current annotations at Top 10& >1% isoform level. While most genes exhibited 100–300 detectable transcript variants, only 1–3 isoforms emerged as major products. In contrast, certain genes such as CREM, DMKN, and RUSC1 displayed a multitude of transcript variants, none of which constituted a majority of reads (Extended Data Fig. 4c and 4d). In brief, this data confirms the diversity of the human transcriptome and the incompleteness of existing annotations.
While we initially chose these genes based on existing annotations to elucidate the expression of anisoforms, our findings revealed limited support for previously annotated transcript isoforms in the cells under study (HEK 293). For example, while we confirmed the alternative TSS for anisoform encoding transcripts in the gene C7orf50 (Fig. 4b), the most abundant alternative transcripts in our dataset have different exon combinations at the 3’ end compared to the annotated variants. Furthermore, 40 out of 42 analyzed genes that lack NCBI-annotated anisoform-encoding isoforms were found to produce other anisoform-encoding transcripts (Fig. 4c), demonstrating that our method can successfully identify anisoform-encoding isoforms in these complex genes. Despite the fact that anisoform-encoding isoforms have been deposited in NCBI and GENCODE for 27 and 50, respectively, of the 69 genes analyzed in this work, only 7/27 and 9/50 of these show PacBio evidence at Top 10& >1% isoform level to substantiate these annotations (Fig. 4d). Despite their inconsistent annotation status, our PacBio data suggest that anisoform-encoding alternative transcripts reach more than 1% of transcript isoforms, and rank in the top 10 transcript isoforms, by read count for 37 of the genes studied; 33 of these anisoform encoding transcripts are previously unannotated. Finally, PacBio data indicates that anisoforms can, in certain instances, emerge as the predominant products of their parent genes, rather than the annotated CDS (Fig. 4e). Calculated TPM values demonstrate a high expression level for anisoform-encoding transcripts relative to CDS-encoding transcripts from these genes (Extended Data Fig. 4f), suggesting their biological significance. We conclude that anisoform-encoding alternative transcripts can be abundant, and existing annotations are insufficient to fully elucidate the protein-coding capacity of some genes.
Anisoforms have distinct functions from the CDSs they overlap.
Because anisoforms have distinct amino acid sequences from the proteins they overlap, yet arise from the same gene, it is important to determine if their functions are related, or different. We first probed the interactomes of our four lead anisoform candidates (DEDD2, CENPM, GLA and RUSC1) using co-immunoprecipitation mass spectrometry (Fig. 5a). None interacted directly with the canonical proteins they overlap, but each significantly enriched a unique set of proteins or complexes. Next, we performed immunofluorescence to investigate the subcellular localization of overexpressed anisoforms and CDSs. As expected, the anisoforms and CDSs from the same genes localized to different regions of the cell (Fig. 5b). Taken together, these results confirm that anisoforms undergo specific protein-protein interactions and localize to subcellular regions consistent with biological roles distinct from the overlapping proteins expressed from the same genes.
Fig. 5. Anisoforms have biological functions distinct from the proteins they overlap.
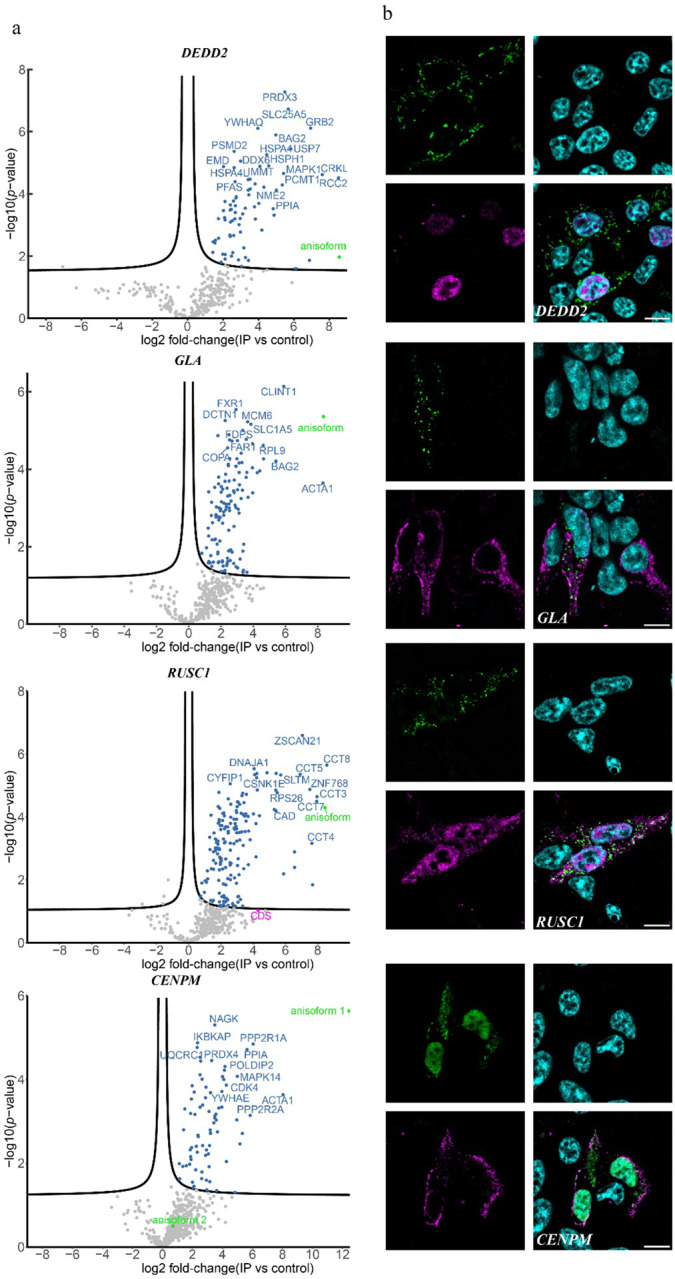
a, Volcano plots for proteins enriched by anisoform co-immunoprecipitation and label-free proteomics (co-IP-MS). The anisoforms were overexpressed in HEK293T cells using plasmids shown in Fig. 3a and subjected to anti-FLAG co-IP, with (empty vector/untransfected cells) as the negative control, in biological triplicate. The green dots represent the anisoform baits. The threshold line indicates a 5% FDR (false discovery rate). b, Immunofluorescence (IF) showing the distinct cellular locations of anisoforms and their corresponding overlapping proteins. The anisoforms and canonical proteins were tagged with FLAG (green) and HA (magenta), respectively; DAPI counterstain is shown in cyan. Scale bars, 10 μm.
DEDD2 anisoform switches gene function.
We selected the DEDD2 anisoform for functional characterization because it is conserved in higher mammals. The presence of the DEDD2 anisoform was observed in DEDD2 genes from Boreoeutheria (Fig. 6a and 6b), but attempts to identify a syntenic start codon in the DEDD2 anisoform reading frame in Laurasiatheria species such as elephant, manatee, and aardvark were unsuccessful. This suggests that the anisoform of DEDD2 may have emerged 77.4–73.2 million years ago, coinciding with the divergence of Placentalia into Boreoeutheria34. Interestingly, the overlapping region shared by the DEDD2 CDS and anisoform is variable among Boreoeutheria sequences, the rest of the CDS shows a greater degree of sequence conservation (Extended Data Fig. 7), potentially indicating that the anisoform emerged after DEDD2 and is under selection35.
Fig. 6. Anisoform of DEDD2 is conserved and regulated by SRSF2.

a, Multiple sequence alignment of the DEDD2 anisoform sequence across twelve placental mammals, performed using seeded guide trees and HMM profile-profile techniques. Gaps in the alignment are marked with dashes. b, Phylogenetic tree constructed from the aligned DEDD2 anisoform sequences in a. Branch lengths indicate evolutionary distances (scale bar, arbitrary unit of evolutionary distance based on sequence divergence). c, Violin plots illustrating the percentage of DEDD2 exon 2 exclusion observed in RNA-seq data from ENCODE. The median values are indicated by horizontal blue lines. d, RNA-seq (ENCODE) read depth within exons 1 through 3 and counts of reads spanning splice junctions (exon 1–2, 2–3, and 1–3) in the DEDD2 gene, from three normal human tissues. e, Diagram illustrating requirement of SRSF2 binding for exon 2 inclusion in the DEDD2 mRNA. f and g. qRT-PCR measurement of the relative amounts of transcript variant 1 (exon 2 included) and 2 (exon 2 excluded) of the DEDD2 gene upon f, overexpression of SRSF 2 or the unrelated eGFP protein as a control, or g, shRNA-mediated silencing of SRSF2 vs a non-targeting shRNA control. p values, Welch two Sample t-test. Error bars represent the standard error of the mean (SEM) for n = 3.
While we caried out anisoform discovery in cell lines, we rationalized that detecting expression of the DEDD2 anisoform in vivo would better support its biological importance. We first analyzed RNA sequencing data from 20 cell lines and 20 normal tissues from ENCODE36. The splice junction unique to the alternative transcript encoding the DEDD2 anisoform is elevated in cancer cell lines, and exhibits lower but detectable expression in normal tissues (Fig. 6c and 6d). This supports expression of the DEDD2 alternative transcript and, correspondingly, the anisoform, in human.
We also noticed that the expression ratio of the canonical and alternative DEDD2 transcript isoforms is variable, and thus hypothesized that DEDD2 alternative splicing may be regulated. We used an alternative splicing site predictor37 to analyze the DEDD2 pre-mRNA and found that the 5’ splice site of exon 2 is a weak acceptor with a low score. Additionally, the RNA-binding protein database38 predicted an SRSF2 binding motif in exon 2. With these clues, we tested the hypothesis that SRSF2 binds to the exonic splicing enhancer (ESE)39 in exon 2 of DEDD2 pre-mRNA to promote the inclusion of exon 2 and thus canonical DEDD2 protein production; in the absence of SRSF2, exon 2 would be excluded, leading to production of the alternative transcript and anisoform (Fig. 6e, and Extended Data Fig. 8a). Consistent with our hypothesis, overexpression of SRSF2 increases the ratio of canonical transcript isoform 1 that encodes the CDS to alternative transcript isoform 2 (Fig. 6f), while knockdown of SRSF2 decreases the relative abundance of the canonical transcript (Fig. 6g). A previous RNA-seq dataset in K562 cells wherein SRSF2 expression was silenced with siRNA was consistent with our data (Extended Data Fig. 8b)40. It is noteworthy that the ESE interacting domain of SRSF2 is frequently mutated in myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML) and other myeloid malignancies41. To determine whether these SRSF2 mutations affect DEDD2 alternative splicing, we reanalyzed RNA-seq datasets collected from K562 cells overexpressing the MDS-associated SRSF2 P95H, P95L and P95R mutations40 or K562 cells engineered to bear a genomic SRSF2 P95L mutation42; we observed upregulation of the anisoform-encoding alternative transcript in these cells relative to wild-type SRSF2 controls (Extended Data Fig. 8c–d). DEDD2 pre-mRNA alternative splicing is thus controlled by SRSF2 and the anisoform is upregulated with cancer-associated mutations.
Given its conservation and regulation, we reasoned that the DEDD2 anisoform was likely to be biologically functional. The canonical, annotated DEDD2 protein was identified based on sequence homology and has been reported to exhibit a weak pro-apoptotic phenotype upon overexpression and anti Fas treatment43. To examine the cellular function of the anisoform, we required two knockout (KO) cell lines: (1) abrogating the CDS alone and (2) abrogating both the CDS along with anisoform (Fig. 7a and Extended Data Fig. 9a and 9b). Because the DEDD2 anisoform locates in the mitochondrion14 and is upregulated in cancer, we hypothesized that it may function in apoptosis, so we examined cleavage of poly (ADP-ribose) polymerase (PARP) in our cell lines under treatment with recombinant soluble human Fas ligand (FasL)44,45. While the loss of the CDS alone had no significant effect on the cell’s response to FasL, the loss of both the CDS and the anisoform greatly increased the level of cleaved PARP (Fig. 7b–d and Extended Data Fig. 9c), consistent with increased apoptosis. Moreover, the crystal violet assay revealed that the dual KO, but not the CDS-only KO, significantly slowed cell proliferation (Extended Data Fig. 9d). Finally, rescue with the DEDD2 CDS via lentivirus slightly increased the level of cleaved PARP in two dual KO clonal cell lines, consistent with previous reports that overexpressing the CDS of DEDD2 promotes apoptosis43,46 (Fig. 7e and 7f). In contrast, rescue with the DEDD2 anisoform decreased the level of cleaved PARP by 41.2% and 29.6% in the two dual KO cell lines, respectively. The DEDD2 anisoform is thus anti-apoptotic and opposes the function of DEDD2 in FasL-induced apoptosis.
Fig. 7. DEDD2 anisoform is antiapoptotic.

a, Schematic representing the CRISPR/Cas9 editing strategy used to disrupt the DEDD2 gene to differentiate the expression and function of canonical CDS and anisoform. “CDS KO1” and “CDS KO2” used different sgRNA combinations to delete exon 2, which contains the start codon of the CDS and thus only deletes the DEDD2 CDS while leaving anisoform expression unchanged. “dual KO1” and “dual KO2” used different sgRNA combinations to delete exon 2, which contains the start codon of the CDS, and exon 3, which contains the start codon for the anisoform, thus abrogating expression of both DEDD2 and the anisoform. The gray squares represent UTRs, magenta bars denote the canonical CDS, and the green bar indicates the anisoform. KO validation can be found in Extended Data Fig. 9a and 9b. b, Western blot analysis of cleaved PARP (Asp214), an apoptosis marker, in wild-type, dual KO strains 1 and 2, and CDS-only KO 1 and 2. In all apoptosis assays, equal numbers of cells were seeded for each cell line, and apoptosis was induced with 200 ng/ml FasL for 4 hours followed by lysis and Western blotting. Beta-actin was a loading control. Results for additional independent clonal lines derived from each KO can be found in Extended Data Fig. 9c. c, RT-PCR and agarose gel analysis to detect altered transcript isoform production as a result of DEDD2 gene editing. RT-PCR of beta-actin was a loading control. d, Quantitation of normalized PARP cleavage with representative data shown in b. For all Western blot quantitation, cleaved PARP intensities were first normalized by the sum of replicates for each condition and then further normalized against actin levels to control for variations in protein loading and transfer across samples. Individual data points on the bars represent measurements from separate experimental replicates. All p values by two-sided, two-sample Student’s t-test. Error bars represent the standard error of the mean (SEM). n = 4. e, Western blot analysis of apoptosis marker cleaved PARP after rescue of dual knockout (KO) cell lines with either DEDD2 canonical protein or anisoform. Each clonal dual KO cell line was stably transfected via lentivirus with constructs encoding either the anisoform (FLAG-tagged), the canonical CDS (HA-tagged), or eGFP as a control. Beta-actin served as the loading control to ensure equal protein loading across samples. f, Quantitation of apoptosis marker in various cell lines (representative data in e) across four biological replicates as described in d.
Discussion
With the prevailing paradigm that same-strand, frameshifted, overlapping ORFs are unique to viruses, human iORFs have generally been overlooked. As a result, while multiple reports have identified large numbers of translated, frameshifted iORFs, the numbers and biological significance of this class has remained uncertain14,47. In this work, we showed that, due to the incomplete annotation of transcript isoform diversity, iORFs have been mis-mapped within protein coding sequences by both ribosome profiling and proteomics, since both of these methods rely on reference annotations. By developing a new pipeline to reanalyze TI-seq data, we identified >1000 putative iORFs in a human cell line, >700 of which are likely to be encoded in previously reported but inconsistently annotated alternative transcripts that lack the start codon of the annotated, overlapping CDS. We note that these numbers represent upper bounds for the total numbers of human iORFs and likely include false positives. We validated alternative transcripts from 70 target genes, chosen on the basis of strong TI-seq signal, refTSS signal, and/or the presence of alternative transcripts in public databases. Furthermore, we specifically queried whether any candidates the 328 genes exhibiting internal TI-seq signals but no known alternative transcripts may express iORFs via un-annotated alternative transcripts. We identified unannotated alternative transcripts that specifically encode anisoforms in 17 out of the 18 selected genes. Although these unannotated transcripts are expressed at low levels, future studies could investigate whether these low-abundance alternative transcripts are regulated or specifically expressed in certain cell clusters due to transcriptome heterogeneity48. While the total number of iORF-encoded anisoforms with cellular functions remains uncertain, our experimental results support expression of nearly six dozen human anisoforms from alternative transcripts, suggesting that this may be a general phenomenon. More anisoforms will likely be found in different cell lines and tissues, considering that TSS and alternative splicing vary among different cell lines49,50.
It is important to note that our data analysis, long-read sequencing, and experimental pipelines broadly exclude the competing hypothesis that putative iORFs are translated in-frame with the CDS start codon via translational frameshifting or alternative splicing, because we computationally assign and exclude transcripts containing the CDS start codon. In the genes considered in this study, anisoforms are therefore translated from bona fide, generally short ORFs within alternative transcripts lacking the CDS start codon. In this light, our study suggests that dozens, and potentially as many as hundreds, of human genes are completely recoded to produce two totally different proteins – an annotated protein and an anisoform – that overlap in genomic sequence but are translated from mutually exclusive alternative transcripts.
Dual coding genes producing two distinct proteins have already been recognized in the case of microprotein-encoding uORFs, such as MIEF16, ASDURF7, oSCRIB/EMBOW9 and alt-RPL368. It is of interest to consider how the mechanism of expression of each class of dual coding genes – e.g., uORFs vs. iORFs – may influence the functional relationship between the ncORF and associated CDS. In the case of uORFs and a subset of iORFs that can be co-expressed from the same transcript as the CDS via leaky scanning and/or reinitiation, several studies have suggested that the uORF-encoded protein and downstream protein have related functions or phenotypes, and in some cases may even directly interact47. In contrast, iORF expression requires the generation of alternative transcripts from mutually exclusive TSS or alternative splicing events relative to the CDS. CDS and iORF expression thus occur in opposition to each other at the mRNA level, despite arising from the same gene locus and pre-mRNA. This opens the possibility that the protein and anisoform can have different, or even opposing, functions – as in DEDD2 – associated with the cellular conditions under which each transcript is produced. To determine whether this hypothesis generalizes to more overlapping genes will require further experimental investigation of iORF functions. It will also be of interest to determine if some uORFs or dORFs are expressed from dedicated transcripts that lack the CDS, instead of via translational reinitiation, and if so, to determine whether the same principle of distinct functions holds for these classes of dual-coding genes as well.
An important unanswered question raised by the existence of iORFs is the mechanism by which their overlapping sequences arise in evolution. While we do not attempt to address this question in our study, we can consider whether and how our results comport with models of de novo gene origination and overprinting, since understanding iORF molecular evolution will be just as critical as elucidating their expression mechanisms to support their biological reality and functionality. While overprinting of frameshifted iORFs on viral genes is known, it is nonetheless somewhat surprising that this process might occur in human genes, as mutations required to create a start codon in the iORF reading frame could be deleterious in the CDS reading frame. De novo gene origination from non-coding loci51,52, is better understood and is not subject to this limitation. How, then, might iORF overprinting hypothetically occur? First, we observed, to our surprise, that the overlapping region shared by the DEDD2 CDS and iORF exhibits lower sequence conservation in the CDS reading frame than the rest of the protein. This contrasted our naïve expectation that the shared region might be more evolutionarily constrained than the rest of the CDS by the presence of functional coding sequences in two reading frames. One speculative explanation could be that the anisoform has been overprinted on a region of the DEDD2 coding sequence that tolerates mutations, and is currently under selection. An alternative hypothesis could be that the canonical DEDD2 protein is not essential, or, at an extreme, may be undergoing pseudogenization; a mutation creating the DEDD2 anisoform would then be more analogous to de novo gene origination within a non-coding RNA. Interestingly, whereas de novo gene origination from non-coding loci require acquisition of transcription, polyadenylation, and nuclear export53 of the RNA before novel ORFs can be accessed by ribosomes, non-coding transcript variants processed from coding loci already meet these requirements. More rigorous evolutionary analysis of a larger set of anisoform-coding genes would be required to test these hypotheses or posit alternative explanations.
Regardless of their origins, the existence of overlapping iORFs and anisoforms has substantial implications for understanding and treating disease, as has previously been noted11. Disease-associated mutations mapping to genomic regions encoding overlaps, especially for the genes identified in this study,54,55 should be reconsidered to determine if the iORF contributes to the disease phenotype, as has previously been suggested for the alt-FUS and alt-RPL36 overlapping proteins. The presence of anisoforms should also be carefully considered when designing siRNA therapies, because iORF-encoding alternative transcripts share most of their sequence with canonical transcript targets.
Methods
Antibodies
The following primary antibodies were used for immunoblotting: anti-FLAG (1:1000, Sigma, F1804), anti-HA (1:3,000, Invitrogen, 71–5500), anti-β-actin (1:3,000, Invitrogen, MA5–15739), anti-Histone-H3 (1:2000, Cell Signaling, 4499), anti-Cleaved-PARP (Asp214) (1:1000, Cell Signaling, 5625). The following secondary antibodies were used for immunoblotting: goat anti-rabbit IgG horseradish peroxidase conjugate (1:4,000, Rockland, 611–1302) and goat anti-mouse IgG horseradish peroxidase conjugate (1:4,000, Rockland, 610–1319). Primary antibodies for immunostaining were mouse anti-FLAG (1:1000, Sigma, F1804) and rabbit anti-HA (1:500, Invitrogen, 71–5500). Secondary antibodies for immunostaining were goat anti-rabbit IgG Alexa Fluor 568 (1:500, Invitrogen, A11011), goat anti-rabbit IgG Alexa Fluor Plus 555 (1:500, Invitrogen, A32732), goat anti-mouse IgG Alexa Fluor 647 (1:500, Invitrogen, A21235), and goat anti-mouse IgG Alexa Fluor Plus 647 (1:500, Invitrogen, A32728).
Identification of anisoforms
To identify candidate anisoforms, lactimidomycin (LTM)-based translation initiation site sequencing (TI-seq) data (SRA: SRR618772 and SRR618773) generated from HEK293 cells were utilized. Adaptor trimming was performed using Cutadapt (version 4.4), and transfer RNA and ribosomal RNA were filtered out with STAR (version 2.7.11a). The remaining reads were aligned to the hg38 reference genome with the guidance of GENCODE Human Release v38 using STAR. Subsequently, the mapped reads were analyzed using PRICE (Gedi version 1.0.2) with the assistance of Ensembl annotation version 104 to identify codons. The resulting cit file was then converted to bedgraph format using the ViewCIT command. A python script (TI_Seq_ORF.py, available at https://github.com/slavofflab/TI-seq-ORF) was used to process the bedgraph files. A peak, defined at the codon level, was called when satisfied following conditions: (i) The peak is among the top 5 highest signals in the gene. (ii) The peak intensity reached 5 observed reads calculated by PRICE. (iii) The peak intensity is greater than 20% of the highest intensity in the gene. (iv) The position must be a local maximum within a span of seven nucleotides. We subsequently assigned peaks to ORF types based on their positions within GENCODE V38 transcripts. The types include ncORF (ORF within non-coding RNA), CDS (annotated coding sequence), N-extension (unannotated N-terminal extension in-frame with annotated protein CDS), uORF (ORF upstream of an annotated CDS), uoORF (ORF that initiates upstream and partially overlaps an annotated CDS in an alternative reading frame), ooORF(outside overlapping ORF that initiates before, ends after, and entirely encompasses the annotated CDS in an alternative reading frame), N-deletion (N-terminal truncation/internal, in-frame translation initiation site of an annotated CDS), dORF (ORF downstream of an annotated protein CDS), doORF (ORF that initiates within CDS, and partially overlaps an annotated CDS in an alternative reading frame), and iORF (internal, frameshifted, overlapping ORF within an annotated CDS). We then implemented a computational procedure to identify candidate iORFs that may be expressed from either known or unannotated anisoform alternative transcripts – in other words, anisoforms. First, we rigorously identified a training set of iORFs with evidence for alternative transcripts in GENCODE V38. If a peak is designated as an iORF or doORF in any transcript isoform and is not assigned to CDS or any of their isoforms in any transcript isoform the peak will be considered as a candidate for follow up study. Only iORFs have AUG start codon were selected for validation. The first round of anisoform candidates were then manually checked against the following criteria with the order from highest to lowest intensity: (1) expression of the annotated alternative transcript for anisoform that cased by exon skipping, alternative 5’ splicing site and alternative first exon should be supported by mRNA-Seq data. Specifically, exon skipping must be evidenced by reads spanning the splice junctions. Similarly, the alternative 5’ splice site and the alternative first exon should have reads mapped to their respective specific regions. The mRNA-Seq data (SRA: SRR24971804, SRR24971805, SRR24971806, SRR24971813, SRR24971814, SRR24971815) were aligned to the hg38 reference genome using the GENCODE Human Release v38 annotation as a guide, employing STAR after trimming and filtering out non-coding RNA reads. The resulted bam files were merged by samtools (version 1.18) to get the final single bam file. (2) The anisoform should exhibit other in-frame Ribo-Seq peaks. The Ribo-Seq (SRR618771) data underwent identical preprocessing steps for TI-Seq analysis. The Ribo-Seq data (SRR618771) underwent identical preprocessing steps as those used for TI-Seq analysis. No ORF calling was applied. Instead, the observed reads, calculated by PRICE, were used to manually check for in-frame peaks. The in-frame peaks of the iORF should appear at a comparable level (>10% of adjacent in-frame peaks of the CDS), and the in-frame peaks of the third ORF should be absent or present at a much lower frequency. This helps to ensure that the in-frame peaks of the iORF are not produced by non-specific signals. (3) If the annotated alternative transcript for anisoform uses an alternative transcription start site (TSS), it should be supported by the refTSS database26 or CAGE-Seq or RNA Pol II-Seq. The CAGE-Seq results were downloaded directly from FANTOM5 with FF ontology id 10450–106F9. The RNA Pol II ChIP-Seq data (GSE152062) were aligned to human hg38 genome with STAR after trimming. Next, we identified a second set of experimental iORFs for further validation at the mRNA level. All selection criteria were followed, except for the requirement of existing annotation.
Cloning
The constructs pcDNA3.1-HA, pcDNA3.1-FLAG, and pcDNA3.1-HA-FLAG were created by introducing HA, FLAG, and HA-FLAG tags into pcDNA3.1, respectively, using EcoRI and XbaI restriction enzymes. To validate each anisoform, total RNA was extracted from HEK293T cells using TRIzol (Invitrogen, 15596026), followed by cDNA synthesis with the cDNA Synthesis Kit (NEB, E6300S). Target alternative transcript isoforms (iORF) and canonical transcripts isoforms (CDS) from the were PCR amplified form from the 5’ UTR to the stop codon of ORFs. Restriction sites were introduced using gene-specific primers and Q5 polymerase (NEB, M0491S) with High GC Enhancer. The PCR program ran for 35 cycles, each cycle comprising denaturation at 98°C for 10s, annealing at 62°C for 15s, and extension at 72°C for 30s (with 2 mins final extension). The PCR products were purified via agarose gel electrophoresis, digested with corresponding restriction enzymes, and then ligated into pcDNA3.1 vectors containing different tags for various purposes. Following transformation into DH5-alpha cells, single colonies were selected and confirmed by Sanger sequencing to ensure the correct transcript isoform (from 5’ UTR to stop codon of selected ORF) was obtained. The C7orf50 5’ UTR-iORF-HA-FLAG construct was synthesized by GenScript and inserted into pcDNA3.1.
For shRNA lentivirus, the pLKO.1-TRC cloning vector (Plasmid #10878) was digested using EcoRI and AgeI restriction enzymes, followed by purification via agarose gel electrophoresis. shRNA oligos (obtained from Sigma) were annealed in T4 ligase buffer and then inserted into pLKO.1 using T4 ligase. For stable expression, cDNAs were inserted into pLJM1-eGFP (a gift from David Sabatini, addgene plasmid #19319) vectors modified with FLAG/HA tags utilizing EcoRI and AgeI restriction sites. The lentivirus plasmids were transformed into NEB® Stable Competent E. coli. Single colonies were selected and confirmed by Sanger sequencing to ensure the correct construction was obtained.
Cell culture
HEK 293 (CRL-1573) and HEK 293T cells (CRL-3216) were purchased from ATCC, and early-passage stocks were established to ensure cell line identity; cells were maintained up to no more than 10 passages. Cells were cultured in DMEM (Corning, 10–013-CV) with 10% FBS (Sigma, F0392) and 1% penicillin-streptomycin (Gibco, 15140122) in a 5% CO2 atmosphere at 37 °C.
Immunofluorescence
One day before transfection, HEK293T cells were seeded in a 24-well plate with antibiotic-free DMEM with 10% FBS. The cells were 70–90% confluent at the time of transfection. Plasmids were transfected using Lipofectamine 2000 (Invitrogen, 11668019) or Lipofectamine 3000 (Invitrogen, L3000015) according to the manufacturer’s instructions. After 6–8 hours, the transfected cells were reseeded onto poly-L-lysine (Sigma, P4832) coated glass coverslips in a 12-well plate in complete media. After allowing the cells to attach overnight, the cells were fixed with 10% formalin (Fisher, SF100–4), quenched with 10 mM glycine in PBST (phosphate-buffered saline with 0.1 % Tween-), and permeabilized with 0.25% Triton X-100 in PBST. After rinsing 3 times with PBST for 5 minutes each, the cells were blocked with 1% BSA in PBST at room temperature for 1 hour. Then, the cells were incubated with primary antibodies (1:300 dilution) in PBST with 1% BSA at 4°C overnight. After rinsing 3 times with PBST for 5 minutes each, the cells were incubated with fluorochrome-conjugated secondary antibodies (1:300 dilution) and DAPI in PBST with 1% BSA for 1 hour, protected from light. After rinsing 3 times with PBST for 5 minutes each, the glass coverslips were mounted onto microscope slides. Confocal imaging was performed on a Leica SP8 LS confocal microscope with a 63× oil immersion objective. The images were processed with ImageJ (version 1.54g) using the DeconvolutionLab2 plugin.
Immunoprecipitation
For IP-WB of transfected cells and stable expression cells, the HEK293T cells were grown in T25 flasks. For IP-WB of FLAG KI cells and IP-MS, the HEK293T cells were grown in 15 cm dishes. Plasmids were transfected using Lipofectamine 2000, Lipofectamine 3000, or polyethyleneimine (Polysciences, 23966) according to the manufacturer’s instructions. The cells were lysed with RIPA buffer in the presence of a protease inhibitor cocktail (Roche, 11836170001). After centrifuging at 14,800 rpm for 30 minutes, the supernatant of the lysate was incubated with 15 μl (for T25 flasks) or 30 μl (for 15 cm dishes) of ANTI-FLAG® M2 affinity gel (Sigma, A2220) at 4 °C overnight. After removing the supernatant, the beads were washed with RIPA buffer 3 times. Elution was in 30 μl of 3× FLAG peptide (Sigma, F4799) at a final concentration of 100 μg ml−1 in RIPA buffer at 4 °C for 2 h. Eluted proteins were subjected to SDS–PAGE or Tricine–SDS-PAGE for WB or LC-MS/MS.
Proteomics
Gel slices (protein bands or entire lanes) were digested with trypsin (Promega, V5111) at 37 °C overnight. After reduction by DTT (Sigma, D0632) and alkylation by IAA (TCI, 144-48-9), the resulting peptide mixtures were extracted from the gel and dried. Residual detergent was removed with ethyl acetate followed by desalting with a peptide cleanup C18 spin column (Pierce, 89870). Peptides were resuspended in 35 μl of 0.1% formic acid (FA), followed by centrifugation at 14,800 rpm at 4 °C for 30 min. Five microliters of each sample was injected on a C18 nano column (CoAnn, HEB05005001718I) attached to an EASY-nLC (Thermo) in-line with a Thermo Scientific Q Exactive Plus Hybrid Quadrupole Orbitrap mass spectrometer. A 95-minute gradient was used to further separate the peptide mixtures as follows (solvent A: 0.1% FA; solvent B: 80% acetonitrile (ACN) with 0.1% FA) at a flow rate of 0.1 μl/min: The initial condition was 5% solvent B. Solvent B was then increased to 45% over 60 minutes. The gradient increased to 85% B over the next 1 minute. Solvent B was held at 85% for 10 minutes. Finally, the gradient returned to 5% B over 23 minutes until the end of the run. The full MS spectrum was collected over the mass range of 300–1,700 m/z with a resolution of 70,000, and the automatic gain control target was set at 3 × 106. MS/MS data were collected using a top 10 high-collision energy dissociation method in data-dependent mode with a normalized collision energy of 27.0 eV and a 1.6-m/z isolation window. MS/MS resolution was 17,500, and dynamic exclusion was 90 s.
The proteomic data were analyzed using MaxQuant (version 2.0.3.0). Oxidation of methionine and protein N-terminal acetylation were set as variable modifications. Carbamidomethyl of cystine was set as fix modification. The data were searched against the human UniProt protein database (version 2021) plus the corresponding anisoform sequence. For all analyses, a mass deviation of 20 ppm was set for MS1 peaks, MS/MS tolerance was 0.6 Da, and a maximum of two missed cleavages were permitted. Maximum false discovery rates were set to 1% both on peptide and protein levels. The minimum required peptide length was five amino acids. Protein quantitation was calculated by the label-free quantification and setting the LFQ min ratio count to 2. Data normalization, filtering, and imputation were performed using Perseus (version 2.0.3.0), and the enrichment analysis was performed using the volcano plot with FDR set to 0.05 and S0 set to 0.1. Missing values were imputed from a normal distribution with a downshift of 1.8 and a width of 0.15.
Construction of CRISPR-Cas9 mediated knock-in and knock-out cell lines
Guid RNAs (gRNAs) were designed with CRISPOR56 and listed in Supplementary table 5. The HDR templates for KI were using single-stranded DNA oligonucleotides (ssODN)57 and listed in Supplementary table 5. Double-stranded DNA oligonucleotides corresponding to the gRNAs were inserted into pSpCas9(BB)-2A-GFP vector (a gift from Feng Zhang, addgene plasmids #48138).
For generation of KO cells, 625 ng of each two gRNA plasmids were co-transfected with 2.5 μg P3000 and 3.75 μg Lipofectamine 3000 (Invitrogen, L3000015) according to the manufacturer’s instructions into HEK293 cells in 12-well plate. The top 5% GFP-positive cells were sorted using a BD Aria flow cytometer and seeded into a 96-well plate at a density of !1 cell per well. The genomic DNA from clonal strains was isolated using 0.05% SDS and 16 units/mL of proteinase K (NEB, P8107S) in 10 mM Tris-HCl, pH 8 at 37°C for 2 hours. Subsequently, deactivation was carried out at 80°C for 30 minutes. Target loci were amplified using Q5 polymerase (NEB, M0491S) with GC enhancer and analyzed by agarose gel electrophoresis. PCR products from correctly cloned single cells were purified using spin columns, followed by re-amplification using OneTaq polymerase (NEB, M0480S) with GC Reaction Buffer. Subsequently, they were subcloned using the TOPO™ TA Cloning™ Kit (Invitrogen, 450641) and validated by Sanger sequencing. The generation of knock-in (KI) cells followed the same protocol, except for transfection: 1 μg of gRNA plasmid and 2 μL of 10 μM ssODN were transfected into the cells.
Generating stable cell lines
HEK293T cells were seeded in a 6-well plate and transfected with 500 ng of either the shRNA vector or stable expression vector, along with 375 ng of psPAX2 (a gift from Didier Trono, addgene plasmid #12260) and 125 ng of pMD2.G (a gift from Didier Trono, addgene plasmid #12259) using Lipofectamine 3000. The resulting lentivirus was harvested at 48 and 72 hours post the transfection. The combining viral solution was filtered using a 0.45 μm syringe filter (Millipore, SLHA033SS) and used to infect cells in the presence of 8 μg/mL polybrene (Sigma, H8268). Two days post-infection, the cells were subjected to selection with 4 μg/mL Puromycin (Sigma, P8833). Subsequently, the cells were maintained in culture with 2 μg/mL Puromycin.
Generating ORFeome and biotin-labeling probes
A fragment between 600 bp to 1.5 kb was amplified from the target genes from cDNA using specific primers and Taq polymerase (NEB, M0480S) with GC Reaction Buffer. The program was run for 45 cycles, with each cycle consisting of denaturation at 94°C for 30s, annealing at 52°C for 15s, and extension at 68°C for 2 min. The PCR products were subcloned directly with TOPO™ TA Cloning™ Kit (Invitrogen™, 450641). Then, biotinylated cDNAs for each clone were synthesized by PCR with universal primers for pCR®2.1-TOPO® vector using a modified dNTP mixture containing 33% biotin-dUTP (prepared by mixing 5 μL of dATP, dCTP, dGTP, 3.35 μL of dTTP (NEB, N0447S) and 33 μL of biotin-16-Aminoallyl-2′-dUTP (Trilink, N-5001)). After purifying with a mini spin column (Econospin, 1910–250), the biotin-labeled cDNAs were randomly fragmented in microTUBEs (Covaris, 520052) on a Covaris S220 sonicator. The sonication method parameters are as follows: peak power of 175 W, duty cycle of 10%, 200 cycles per burst, and duration of 430 s. The fragmented products were dried in a SpeedVac and resuspended in ddH2O to get a high-concentration solution. Finally, the fragmented biotin-labeled cDNAs were mixed to generate the biotin-labeling probe sets at 1 ng/ μL.
ORF-capture sequencing
The total RNA isolated from HEK293 cells with TRIzol (Invitrogen, 15596026) was sent to Yale Center for Genome Analysis (YCGA) for library construction utilizing the “Preparing Iso-Seq libraries using SMRTbell prep kit 3.0” (PacBio, PN 102-396-000 REV02 APR 2022) protocol. During the processes, the full-length cDNA was taken back after Step 2.4.16. The cDNA was then amplified using NEBNext® High-Fidelity 2X PCR Master Mix (NEB, M0541S) and cDNA primers bc1001-F/R in a 100 μL reaction. The PCR program was run for 14 cycles after initial heating at 98°C for 45 seconds. Each cycle consisted of denaturation at 98°C for 10 seconds, annealing at 62°C for 15 seconds, and extension at 72°C for 3 minutes, with a final extension step at 72°C for 5 minutes. The PCR products were cleaned using 1.8X AMPure XP beads (BECKMAN COULTER, A63880) according to manufacturer’s protocol and eluted with 50 μL nuclease-free water.
The amplified cDNA was subjected to target transcript enrichment using the “cDNA Capture Using IDT xGen® Lockdown® Probes” protocol (PacBio, PN 101-604-300 Version 01 June 2018) by using with modifications. Gen Hybridization™ and Wash Kit (IDT, 413096243) were utilized for this purpose. 1 μg of purified cDNA was combined with 1 nmol of PCR primer bc1001-F/R (Sigama) and 1 nmol of PolyT blocker Oligo (IDT) in a 1.5 mL LoBind tube (Eppendorf, 022431021). After drying in a SpeedVac, 8.5 μL of 2X Hybridization Buffer, 2.7 μL of Hybridization Buffer Enhancer and 1.8 μL of nuclease-free water were added to resuspend the DNA. The resulting solution was transferred to a PCR tube and cDNA was denatured by placing it in a 95°C Thermal Cycler (Bio-Rad, C1000) for 10 minutes. The tube was quickly spun down after heating. Once the solution cooled to room temperature, 4 μL of biotin-labeling probe sets (4 ng) were added, mixed thoroughly, and then spun down. The mixture was incubated at 65°C for 4 hours. Subsequently,100 μL of M-270 streptavidin beads (Invitrogen) was added and a series of washes were performed according to the protocol. After adding 50 μL nuclease-free water, on-bead PCR was performed in a total volume of 300 μL with the same program used previously, except the cycle number was increased to 30. The PCR product was cleaned using 1.8X AMPure XP according to manufacturer’s protocol and eluted with 50 μL nuclease-free water. The cDNA was sent back to YCGA, and the process continued at step 4.1. The final library was sequenced on a PacBio Sequel II.
Long-read data processing
The analysis was following the PacBio Iso-Seq3 (https://github.com/ylipacbio/IsoSeq3) workflow. The HiFi reads were initially processed using lima (version 2.7.1) to demultiplex, remove barcode sequences, adjust orientation, and eliminate unwanted primer combinations. Subsequently, the resulting BAM file underwent further refinement with isoseq3 refine (version 4.0.0) to ensure the presence of PolyA and to remove concatemers. Following this, the refined full-length reads were clustered using isoseq3 cluster (version 4.0.0). After filtering out low-quality reads, the clustered reads were aligned to the hg38 reference genome using pbmm2 (version 1.13.0). Then, isoseq3 collapse (version 4.0.0) was employed to annotate the transcriptome. The annotated transcripts were sorted using pigeon sort (version 1.1.0), and pigeon classify (version 1.1.0) was utilized to classify and quantify transcripts, guided by GENCODE V38 annotation. Finally, the PacBio transcript isoforms of target genes were classified based on their potential coding abilities with a python script (coding_assign.py, available at https://github.com/slavofflab/Codingtype_assign) based on the first start codon with the mRNA sequence. The RNA-seq datasets mentioned above were aligned to the hg38 genome using Salmon (version 1.10.2). The quantified data was then subjected to further analysis with DESeq2 (version 1.42.1) to obtain the Transcripts Per Million (TPM) values for each gene. The TPM values of transcript isoforms were calculated by multiplying the TPM of the gene with the transcript percentage derived from the long-read sequencing data.
RT-PCR
Total RNA was extracted from cells using TRIzol (Invitrogen, 15596026), followed by cDNA synthesis using the iScript™ cDNA Synthesis Kit (BIO-RAD, 1708891). Subsequently, RT-PCR was performed with Luna® Universal qPCR Master Mix (NEB, M3003L) in CFX96 Touch (Bio-Rad, 1854095). The RT-PCR program was initiated with an initial heating step at 95°C for 60 seconds, followed by 45 cycles. Each cycle comprised denaturation at 95°C for 15 seconds and extension at 60°C for 1 minute, along with plate reading. Finally, a melt curve step was conducted from 60–95°C over 30 minutes.
Conservation and evolution analysis
Sequences of DEDD2 and DEDD2-anisoform were obtained from NM_001270614.2 (human, Homo sapiens), XM_024351836.3 (Chimpanzee, Pan troglodytes), XM_015124083.2 (rhesus monkey, Macaca mulatta), XM_008567239.1 (colugo, Galeopterus variegatus,), NM_207677.3 (house mouse, Mus musculus), XM_003748789.4 (brown rat, Rattus norvegicus), NM_001076017.1 (cattle, Bos taurus), XM_004015286.5 (sheep, Ovis aries), XM_023651436.1 (horse, Equus caballus), XM_014867556.2 (donkey, Equus asinus), XM_019819482.3 (cat, Felis catus), XM_002923941.4 (giant panda, Ailuropoda melanoleuca), XM_023544014.1 (elephant, Loxodonta africana), XM_023742697.1 (manatee, Trichechus manatus), XM_007942940.1 (aardvark, Orycteropus afer). Multiple sequence alignment was performed with Clustal Omega (https://www.ebi.ac.uk/jdispatcher/msa/clustalo).
Statistical analysis
Statistical analyses were performed using two-sided, two-sample Student’s t-test with Microsoft Excel (version 2404) or Welch two Sample t-test with R (version 4.3.3). Error bars represent the mean ± SEM. Further statistical details of experiments are reported in the figure legends. No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Splicing variation analysis
For cancer cell lines and tissue samples, the processed RNA-seq data (BAM files) were downloaded from ENCODE and listed in Supplementary table 4. The analysis included data from 20 cancer cell lines and 20 tissue samples. For siRNA knock down and SRSF2 mutation samples, the raw data were downloaded from GEO (GSE65349 and GSE164666) and mapped to hg38 using STAR. The resulted BAM files were used for downstream analysis. SGSeq (version 1.36.0) was utilized to identify and quantify splice variants. The splice graph analysis was based on de novo prediction.
Cell proliferation assay
At day 0 in the afternoon, cells were treated with trypsin (Sigma, T4090) for digestion. Subsequently, 10 μL of cell suspension was mixed with one part trypan blue (Sigma, T8154) and counted using a cell counter (Bio-Rad, Tc20). Following counting, the cells were seeded onto poly-lysine-coated (Sigma, P4707) 12-well plates at a concentration of 104 cells per well. After reaching the desired time point, the medium was aspirated, and the cells were fixed with 10% formalin (SF100–4) for 5 minutes after being washed with 1 mL of PBS. Subsequently, the cells were incubated with 0.5 mL of 0.1% crystal violet (Sigma, C0775) in 20% methanol for 15 minutes. Following the incubation, the cells were washed three times with PBS. Finally, 1 mL of 10% acetic acid was added to each well to dissolve the crystal violet. Subsequently, 100 μL of the dissolved solution was transferred to a 96-well plate, and the absorbance at 590 nm was measured using a BioTek Synergy 4 spectrophotometer.
Apoptosis assay
After trypsinization (Sigma, T4090), 10 μL of cell suspension was mixed with one part trypan blue (Sigma, T8154) and counted using a cell counter (Bio-Rad, Tc20). Following counting, the cells were seeded onto 12-well plates at a concentration of 5×105 cells per well and cultured overnight. The growth medium was replaced with 1 mL of fresh medium containing 200 ng/mL Fas Ligand (PeproTech, 310–03H) for 4 hours prior to lysis and Western blotting.
Supplementary Material
Extended Data Fig. 1 TI-seq unveiled the presence and properties of anisoforms. a, Start codon identifies and translated ORFs were defined in individual transcripts based on TI-seq data and assigned using a script (TI_Seq_ORF.py) to ORF types, including CDS (annotated protein coding sequence or any ORF that contains a portion/isoform of the annotated CDS), iORF (internal, frameshifted ORF), dORF (downstream of CDS in any reading frame), uORF (upstream of CDS in any reading frame), uoORF (upstream and partially overlapping CDS in alternative reading frame), overprinted ORF (ORF that initiates before, ends after, and entirely encompasses the annotated CDS in an alternative reading frame), doORF (ORF that initiates within CDS, and partially overlaps an annotated CDS in an alternative reading frame), and ncORF (ORF within a “non-coding” RNA, inclusive of pseudogenes, long non-coding RNA, and other non-coding classes), based on their positions and properties. The start codon composition was then quantified by ORF type, represented as a bar plot. AUG variants means a TI-seq putative start codon exhibiting any single-nucleotide difference from AUG, excluding CUG, which is considered separately; purple bars denote putative ORF start codons that are neither AUG nor its near-cognate variants. b, Bar plot illustrating the distribution of Kozak sequence contexts around the start codons of TI-seq-detected ORFs according to ORF class. Red indicates strong AUG Kozak sequences (RNNAUGG)58, green denotes intermediate AUG Kozak sequences (YNNAUGG or RNNAUGH), cyan represents weak AUG Kozak sequences (YNNAUGH), and purple signifies ORFs that start with a non-AUG codon, which were not subjected to further sequence context analysis. ‘R’ represents A or G, ‘Y’ stands for C or T, and ‘H’ indicates A, C, or T. c, Violin and box plot illustrating the start codon peak intensity distribution of TI-seq-detected ORFs across different categories. Mean and median are highlighted for each category. The p-value was calculated using Welch’s two-sample t-test. Red dots represent outliers, and blue dots represent the mean intensity. Error bars represent the standard error of the mean (SEM). d, Cumulative frequency plot illustrating the length distribution of different ORF types detected using TI-seq, measured in codons (not including stop codon). e, Histogram depicting the frequency distribution of iORF (anisoform) lengths detected via TI-seq. The apparent peak at >100 aa is not an increase at larger sizes but an artifact of the larger “bin” encompassing all longer anisoforms.
{kind=link}
Extended Data Fig. 2 Alternative transcription start sites (TSS) and alternative pre-mRNA splicing generate iORF-encoding alternative transcripts. a, Pie chart representing the proportion of each event type that generates iORF-encoding alternative transcripts, relative to the corresponding canonical transcript assigned to the same gene: alternative TSS (red), cassette exon exclusion (blue), or alternative 5’ splice site usage (gray). b, GLA gene showcasing an example of an anisoform-encoding alternative transcript that initiates at an alternative TSS within the same first exon, yet positioned after the start codon of the canonical coding sequence (CDS). The top tracks represent GENCODE v38 representative transcripts for GLA, indicating the chromosomal location and the orientation from 3’ to 5’; taupe boxes, exons; lines, introns. Below, the positions of the CDS (magenta boxes) and the anisoform (green boxes) are presented, and start codons are indicated by dashed lines. Green arrows point to the peaks of the start codon for the anisoform in the TI-seq data (cyan track). A broad CAGE TSS peak (green track), and split mRNA-seq intensity within the first exon (gray track, bottom), may also be consistent with alternative TSS usage. c, Analysis of the CENPM gene provides an example of an anisoform utilizing an alternative TSS within a novel 5’ extended exon that bypasses the annotated first exon and start codon for the CDS. Top, GENCODE v38 representative transcripts for CENPM, indicating the chromosomal location and the orientation from 3’ to 5’. Taupe boxes, exons. Below, the positions of the multiple protein products are indicated in magenta for the CDS and green for two putative iORFs whose start codons were identified via TI-seq (cyan, green arrows). Transcription from the novel TSS is supported by CAGE-seq and mRNA-seq (green and gray, respectively).
{kind=link}
Extended Data Fig. 3 Anisoforms can be translated from alternative transcripts. a, Anisoform-encoding transcripts encompassing the 5’UTR through the stop codon of the iORF were cloned into pcDNA3.1 and fused with HA-FLAG tags at the C-terminus. The resulting plasmids were transfected into HEK293T cells. Anti-FLAG Western blotting and anti-FLAG immunoprecipitation followed by anti-FLAG Western blotting were used to validate their translation. b, Epitope-tagged anisoform expression plasmids generated as described in (a) were transfected into HEK293T cells to validate anisoform translation and subcellular localization with confocal microscopy. After fixation and permeabilization, cells were subjected to anti-FLAG immunofluorescence (green), with DAPI counterstain (cyan) for nuclei. Scale bars, 10 μm. c, PCR validation of DEDD2 and RUSC1 anisoform-FLAG knock-in (KI) cells, related to Fig. 2. Left panel, schematic showing the primer positions. The primer set “FI” and “RI” is used to confirm the insertion in the target gene locus. The primer set “FT” and “RT” is used to confirm that the tag is inserted into the target gene locus. Right panel, agarose gel analysis of PCR products from genomic DNA generated with the specified primer pairs.
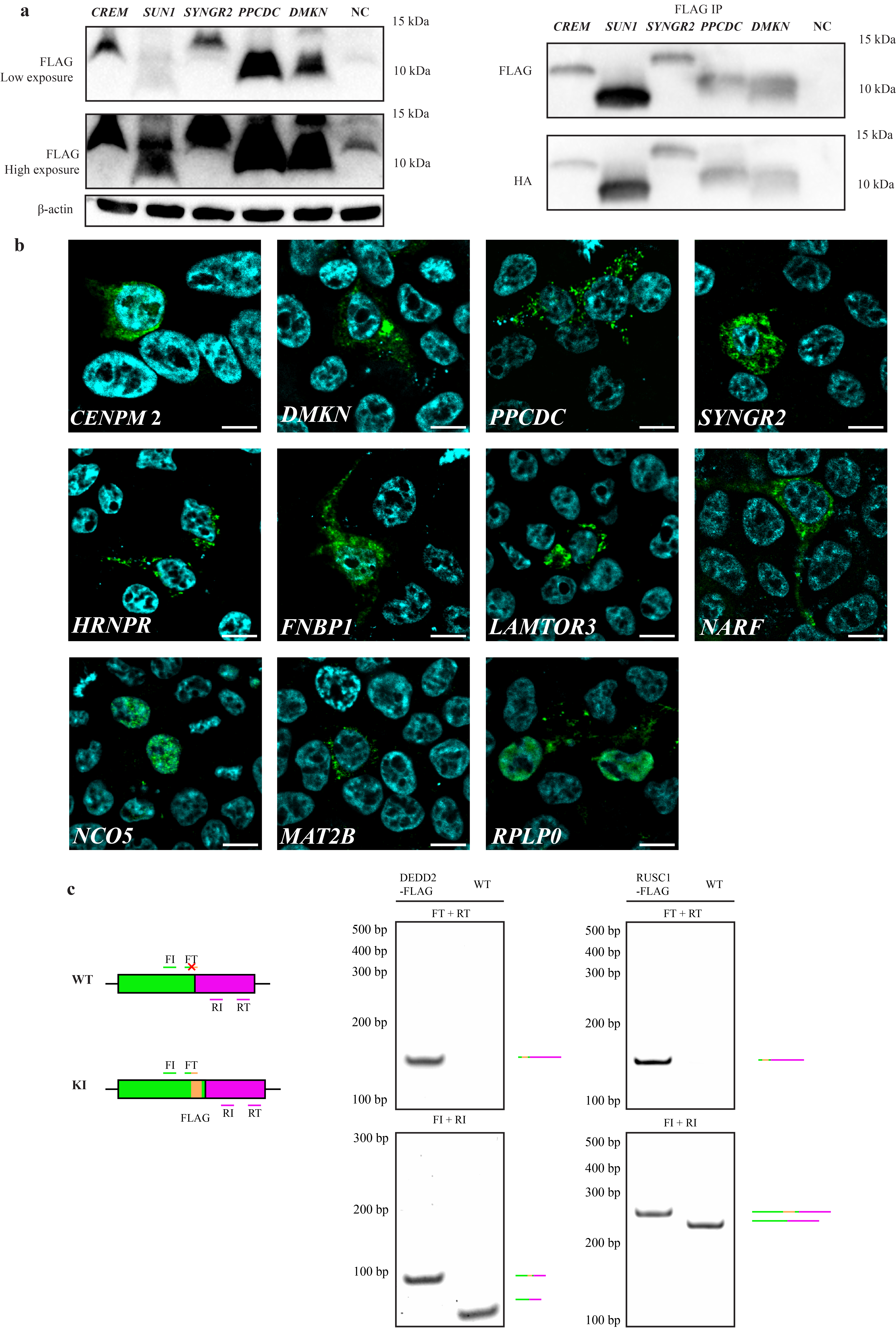{kind=link}
Extended Data Fig. 4 Comprehensive analysis of transcript diversity for target genes using PacBio full-length sequencing. a, Overview of all alternative transcript and mRNA processing events previously deposited in GENCODE V38 and NCBI release 110 within the 69 iORF-encoding target genes considered in this study, representing previously known transcript diversity generated from these loci. b, Overview of transcript isoforms and events identified through ORF capture and PacBio full-length sequencing of 69 target genes. The bar plot categorizes all alternative transcript-generating events that not include in GENCODE v38 and NCBI release 110, such as alternative transcript isoforms, alternative TSS, cassette exon exclusion, alternative 5’ or 3’ splice donors, unannotated exon inclusion, intron retention, and alternative processing events combination, which include all event above. The colors represent the levels of evidence from PacBio sequencing: soft coral red for transcripts in the top 10 and top 1% by read count, golden olive for variants present above 1% (but not top 10), bright green for transcripts in the top 10 (but not above 1%), teal blue for variants that are detected at any level, cornflower blue for transcripts with read support, and orchid pink for no evidence. c, Histogram depicting the distribution of detected transcript variant numbers within target genes. The x-axis represents the isoform number, and the y-axis represents the count of genes with a specific number of detected transcript variants. d, Tile plot illustrating the distribution of the top 10 transcript isoforms for each gene and the total number of isoforms detected. Each row represents a gene, and the tiles are color-coded to show the read count percentage corresponding to each of the top 10 isoforms. The gradient bar on the right indicates the isoform percentage (0–100%), while the additional color scale represents the total number of isoforms detected per gene, ranging from 100 to 500. e, Tile plot illustrating the distribution of transcripts for anisoforms within the top 10 transcript isoforms. Each row represents a gene, with the tiles color-coded to reflect the percentage of transcripts corresponding to anisoforms. The gradient scale on the right indicates the percentage values (0–100%) for each isoform. Gray tiles marked “NA” represent cases where the transcript does not encode the anisoform within the top 10 isoforms for the corresponding gene. f, Tile plot illustrating the calculated TPM (Transcripts Per Million) values of anisoforms within the top 10 transcript isoforms and the total TPM of the parent gene. The TPM values were computed by multiplying the total percentage of transcripts for anisoforms with the TPM values of the respective parent genes. The color gradient, presented in log scale, represents the TPM values, ranging from low (light colors) to high (dark colors). Gray tiles marked “NA” indicate cases where the transcript does not encode an anisoform within the top 10 isoforms for the corresponding gene.
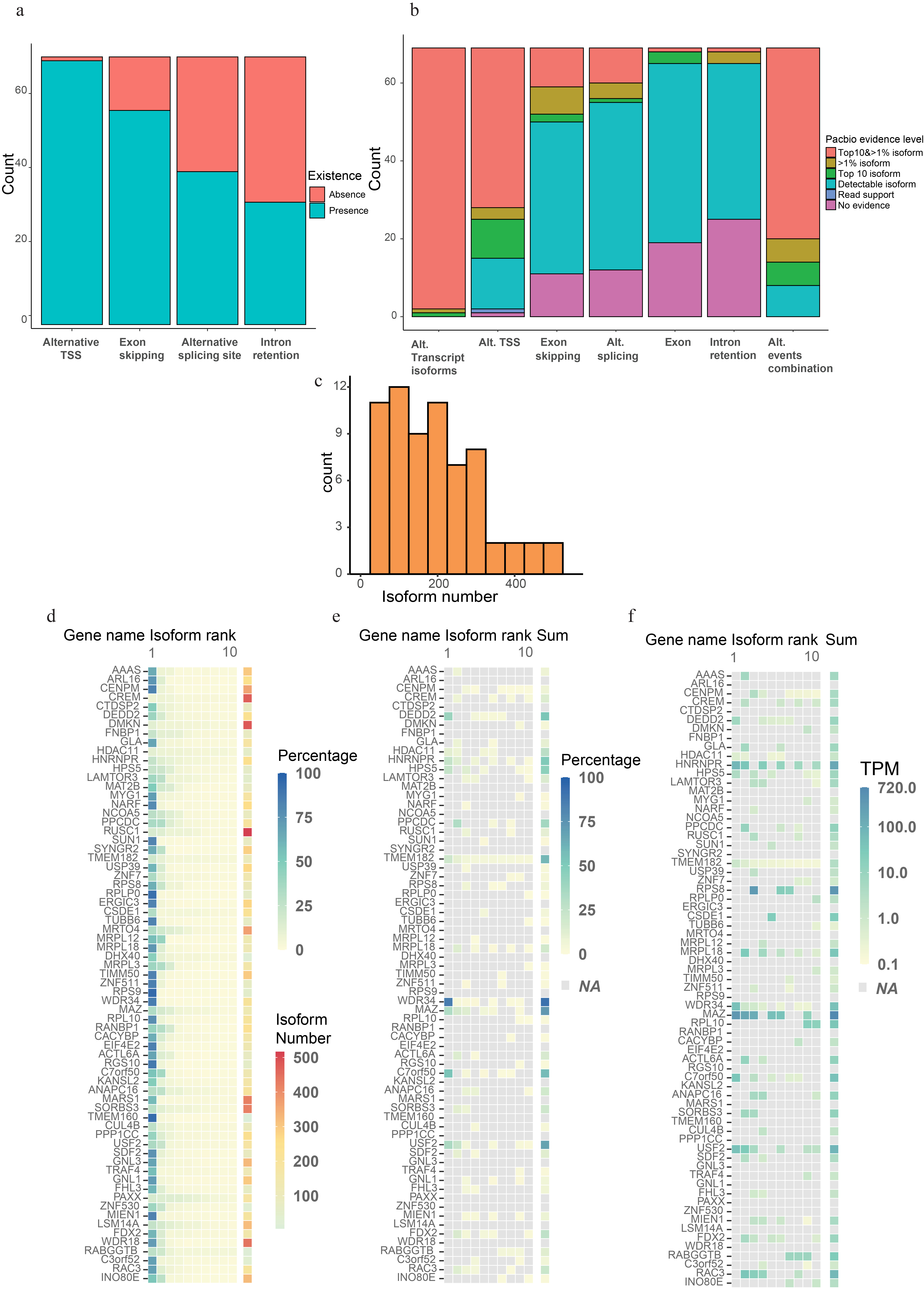{kind=link}
Extended Data Fig. 5 PacBio full-length sequencing validation of the C7orf50 alternative transcript and anisoform. a, Genome view of C7orf50. Representative transcript variants obtained from GENCODE (tracks 1–3; taupe, exons) encode either the annotated protein (tracks 1 and 2) or the anisoform (track 3). The most abundant transcript detected by PacBio long-read sequencing (track 4; other PacBio transcripts, tracks 5 and 6) matches the anisoform-encoding transcript variant found in GENCODE. The positions of the CDS (magenta) and iORF (green) within these transcripts are represented below. TI-seq (cyan) supports the use of multiple start codons, one of which is consistent with the downstream CAGE-seq (green), reference TSS (pink), PolII ChIP-seq (purple), mRNA-seq (gray) and PacBio (orange) data supporting transcription of the anisoform-encoding transcript from an internal TSS (red arrows). In addition to the TI-seq signal, further support for translation in the anisoform reading frame specifically within the region of the iORF is provided by reanalyzing Ribo-seq data (bottom zoom; pink, reads mapping to CDS reading frame; green, reads mapping to iORF reading frame). b, Analysis of translation and stability of the C7orf50 anisoform. The most abundant PacBio-detected anisoform transcript mapping to C7orf50 was cloned from the 5’ end to the stop codon of the iORF into pcDNA3.1 by gene synthesis, and fused with a dual HA-FLAG tag at the C-terminus of the iORF. The resulting plasmid was transfected into HEK293T cells, followed by lysis and anti-FLAG Western blotting, to validate expression. Beta actin served as a loading control, and untransfected cells as a negative control.
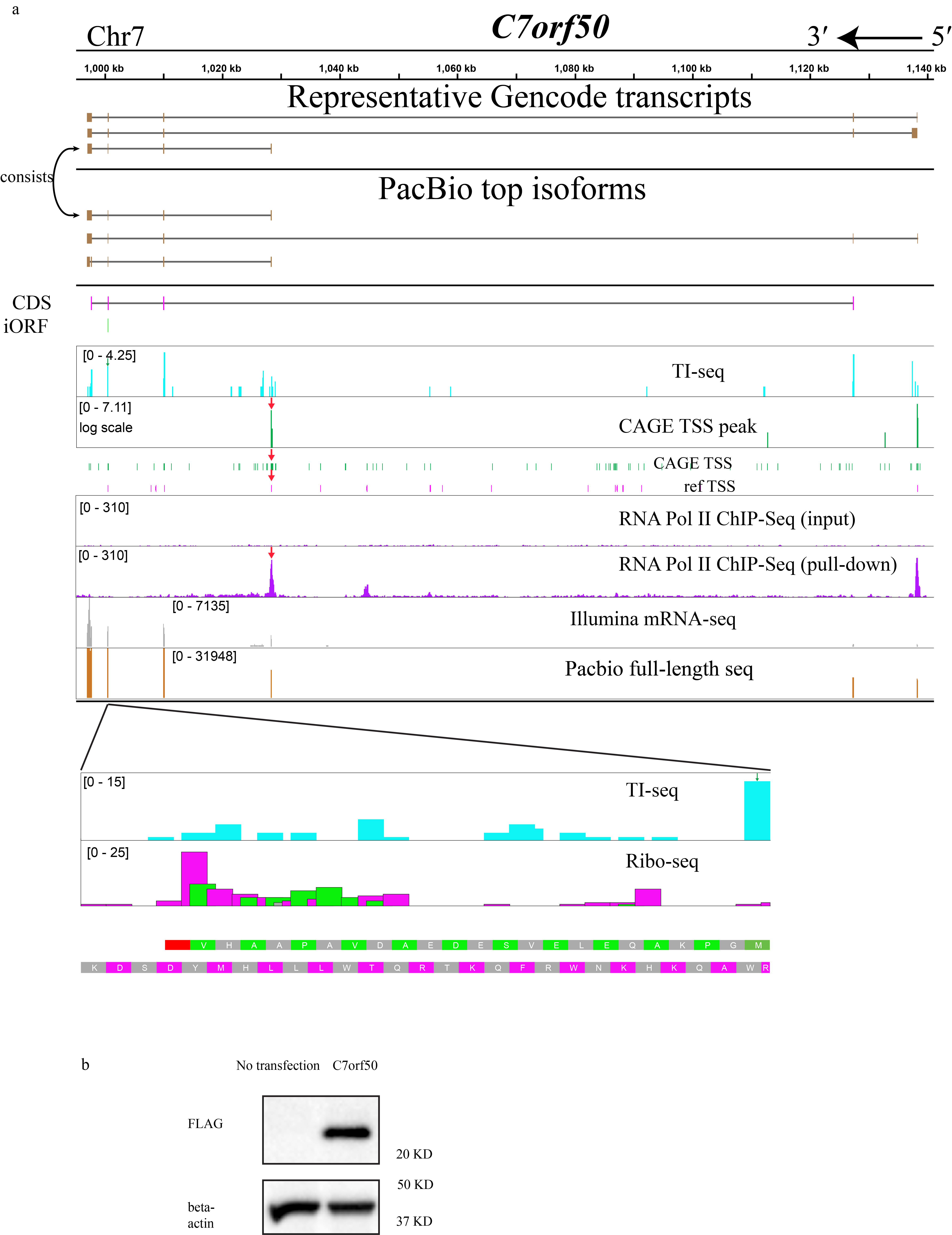{kind=link}
Extended Data Fig. 6 PacBio full-length sequencing validation of the RPS8 alternative transcript and anisoform. a, Top, genome view of RPS8. Tracks 1–10, RPS8 transcript isoforms detected using PacBio and ranked by read count, representing various putative processing and alternative TSS events. Taupe boxes, exons. The third-ranked transcript encodes the anisoform with a unique 5’ end that may utilize a novel TSS. All transcripts from NCBI release 110 and GENCODE v38 are shown for reference. The positions of the CDS (magenta boxes) and iORF (green boxes) are represented below the transcript diagrams, and evidence for translation initiation at the anisoform start codon is provided by TI-seq (cyan track, start codon indicated with green arrow). Ribo-seq reads (orange tracks) are also observed in the anisoform reading frame and specific to the positions of the iORF (bottom, zoom; magenta signal indicates Ribo-seq reads in the CDS reading frame, and green for the iORF reading frame). The position of the candidate alternative TSS is indicated by red arrows overlaid on CAGE-seq, CAGE-TSS (both green) refTSS (magenta) and PolII CHIP-seq (purple) data. Illumina mRNA-seq data (gray) and PacBio long-read sequencing data (dark orange) provide support for expression. b, The third most abundant transcript, which encodes the anisoform, was cloned from the 5’ end to the stop codon of the anisoform into pcDNA3.1 and fused with dual HA-FLAG tag at the C-terminus. The resulting plasmids were transiently transfected into HEK293T cells followed by lysis and anti-FLAG Western blotting. c, The construct described in b was transiently transfected into HEK 293T cells, followed by fixation and anti-FLAG immunofluorescence, with DAPI as a counterstain.
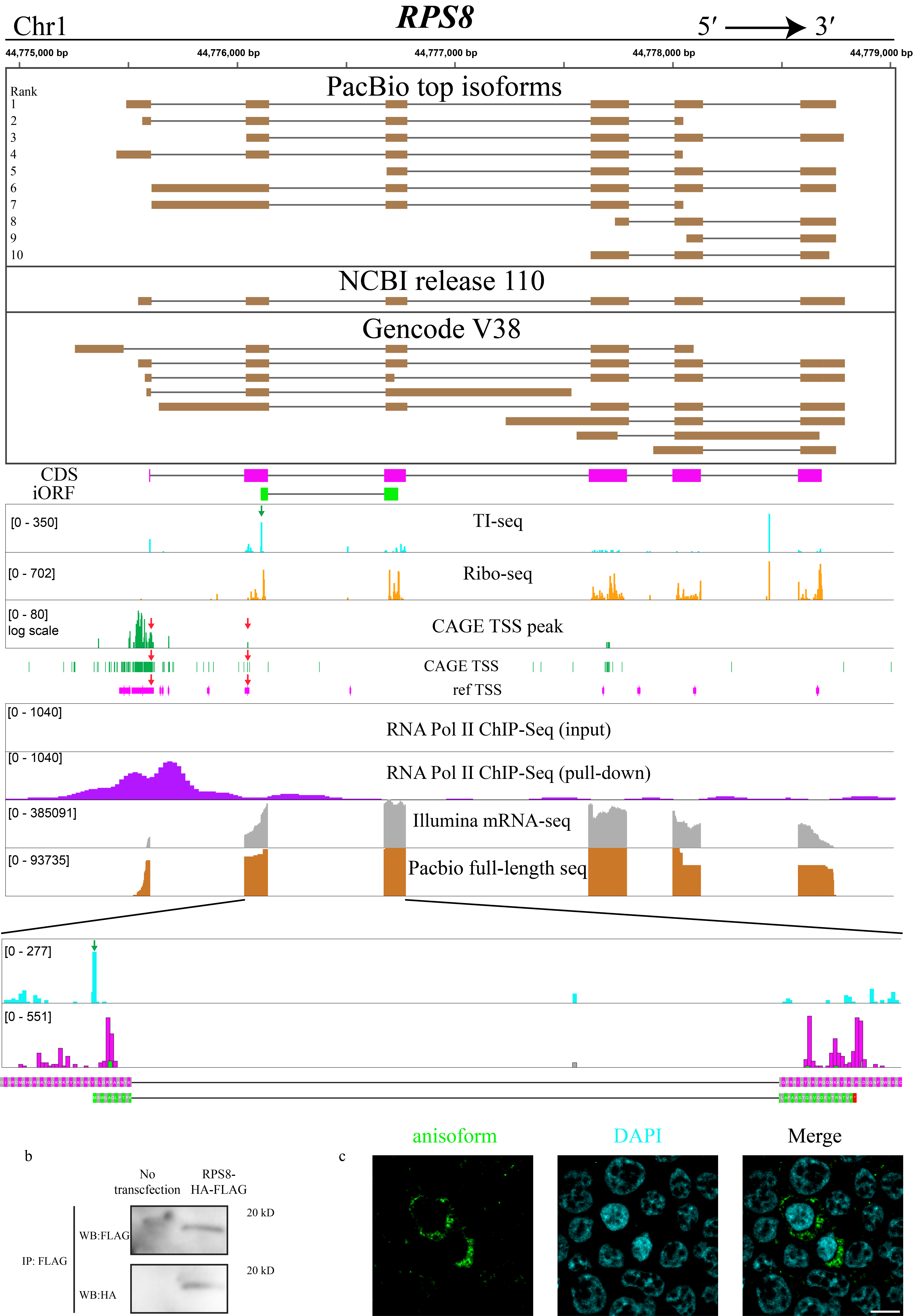{kind=link}
Extended Data Fig. 7 Conservation analysis of DEDD2 CDS. a, Multiple sequence alignment of DEDD2 among 12 species. The yellow box indicates the region that the anisoform overlaps in an alternative reading frame. b and c, Phylogenetic trees constructed from the entire CDS and the region overlapping with the anisoform, respectively. Branch lengths represent evolutionary distances.
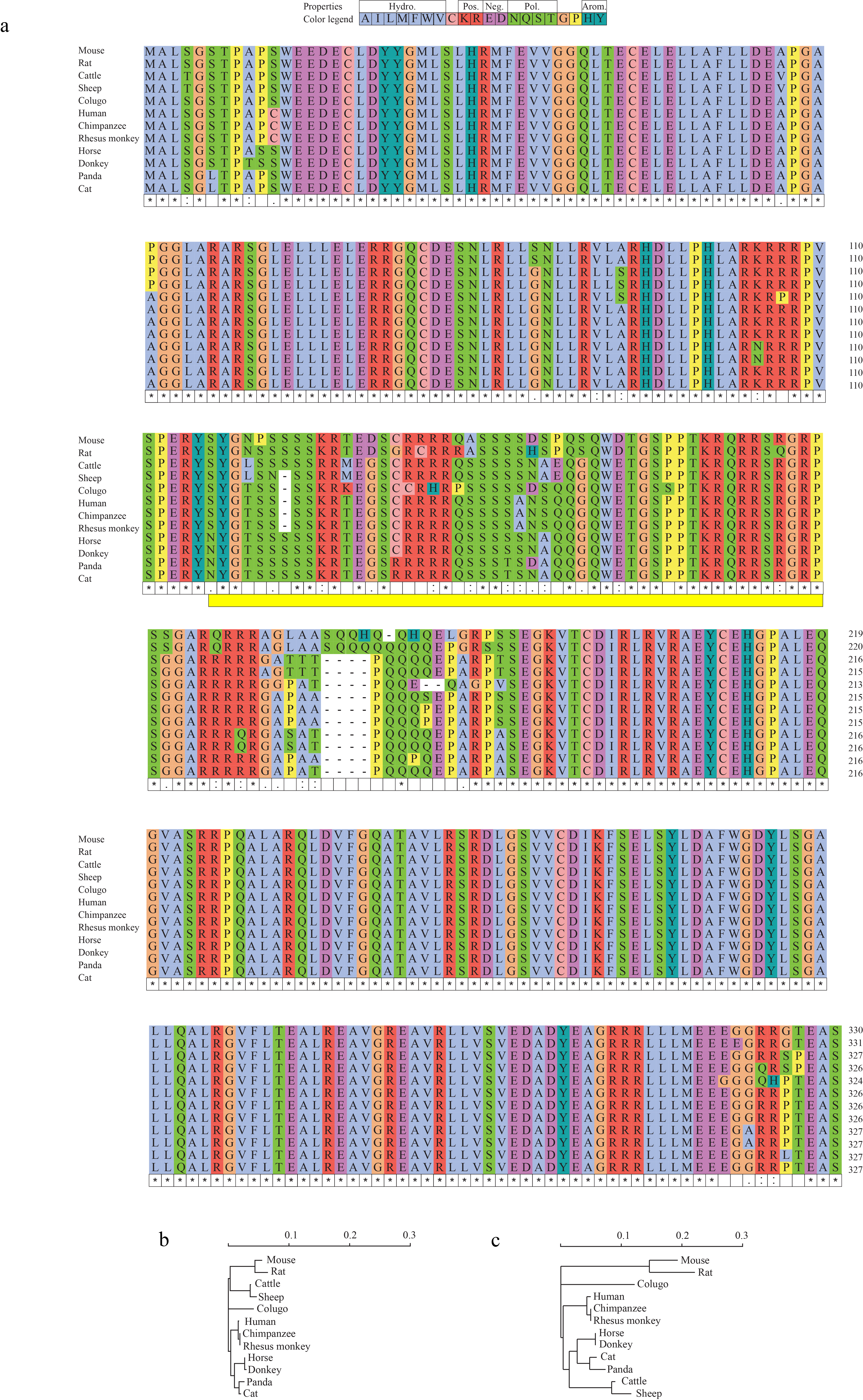{kind=link}
Extended Data Fig. 8 SRSF2 regulates the splicing of DEDD2. a, Schematic representing a portion of the pre-mRNA of DEDD2 up to position 2867 inclusive of exons 1–3. Boxes, exons; lines, introns. The magenta region represents the annotated CDS, while the green region encompasses the region where the frameshifted iORF overlaps the CDS. The predicted SRSF2 binding site is indicated with an arrow. b, c, and d Reanalysis of RNA-seq read depth across DEDD2 exons 1 through 3 and read counts spanning splice junctions between exons 1–2 and 2–3 (specific to transcript variant 1 encoding the CDS) and exons 1–3 (specific to transcript variant 2 encoding the iORF) in various cells and models, including b, TF-1 cells treated with SRSF2 siRNA or control siRNA; e, K562 cells over expressing various point mutants or wild-type SRSF2; and c, K562 cells expressing knock-in P95L or wild-type SRSF2. d, Bar plot showing the percentage of exon 2 exclusion in K562 cells expressing SRSF2 WT and SRSF2 P95L.All p values by Welch two Sample t-test. Error bars represent the standard error of the mean (SEM). Sample sizes are as follows: n = 3 in panels b and c, n = 6 in the SRSF2 WT K562 group in panel g, and n = 4 in the SRSF2 P95L K562 group in panel g.
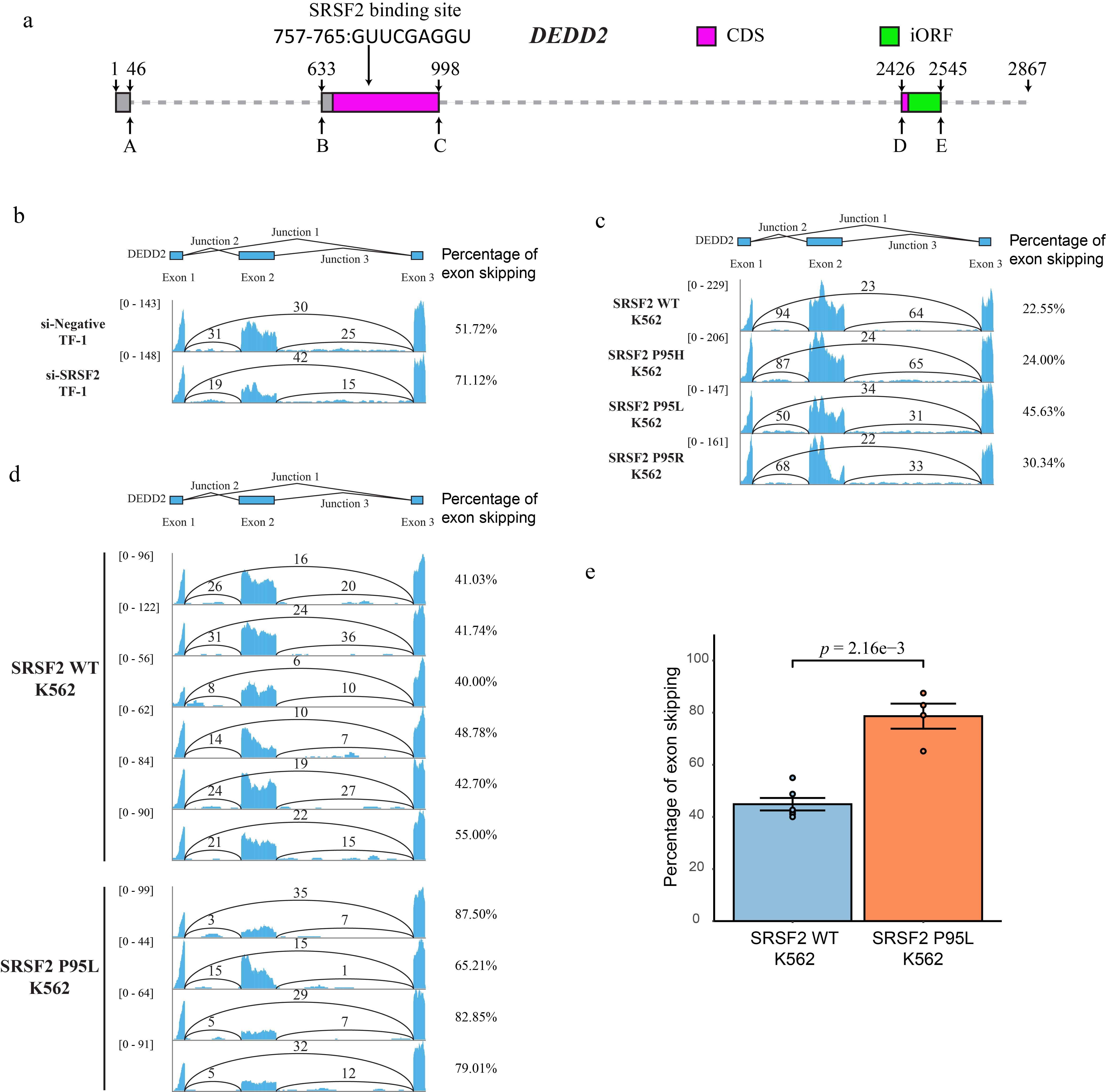{kind=link}
Extended Data Fig. 9 Validation of knock-out cell lines. a, Sequencing validation of DEDD2 CDS knockout (CDS KO) cell lines. The top panel shows the genome reference sequence for DEDD2 with the sgRNA2, sgRNA5, and sgRNA6 binding sites highlighted. Below, the sequencing chromatograms for CDS KO1 and CDS KO2 are shown, confirming the successful deletion at these sites. b, Sequencing validation of DEDD2 dual KO cell lines. The top panel shows the genome reference sequence for DEDD2 with the sgRNA1, sgRNA3, and sgRNA4 binding sites highlighted. Below, the sequencing chromatograms for dual KO1 and dual KO2 are shown, confirming the successful deletion at these sites. The figure includes the positions of the forward primer (FP) and reverse primer (RP) used for genomic DNA PCR. c Western blot analysis, corresponding to Fig. 7b, showing the levels of cleaved PARP (Asp214), an apoptosis marker, across additional independent clonal CDS KO and dual dual KO cell lines. Clone numbers are indicated above each lane. Beta-actin was used as a loading control. d Cell proliferation for two independent, clonal DEDD2 CDS KO cell lines (CDS-KO-1 and CDS-KO-2) and two dual KO cell lines (Dual-KO-1 and Dual-KO-2), compared to parental (wild-type, WT) HEK 293T cells. Equal numbers of cells from each line were seeded and grown for six days, then cell numbers were measured with crystal violet staining. The y-axis represents the optical density at 590 nm (OD 590). Error bars represent the standard error of the mean (SEM) from four independent experiments. Statistical significance is indicated by p-values for comparisons on Day 6. All p values by Welch two Sample t-test. Error bars represent the standard error of the mean (SEM).
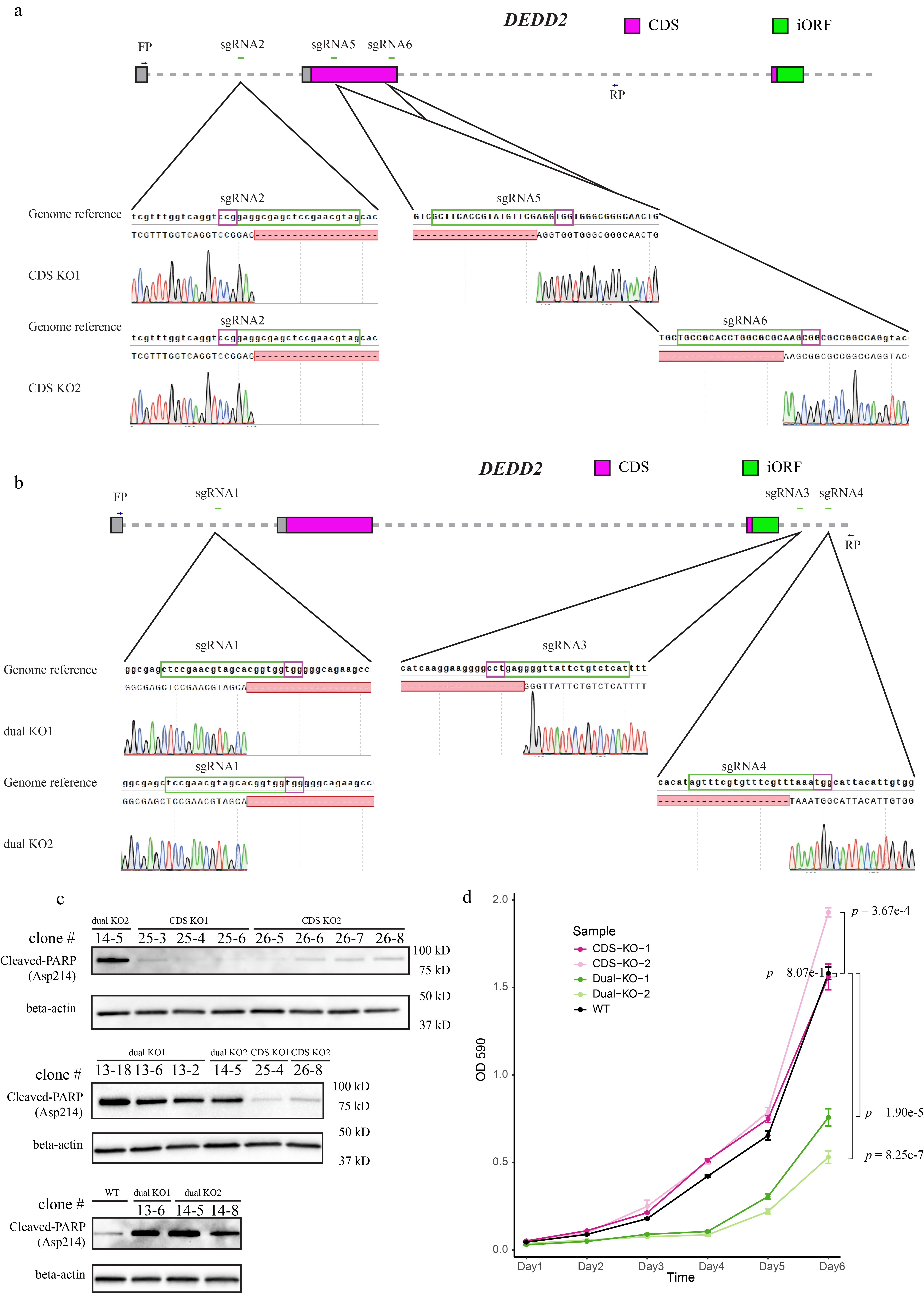{kind=link}
Acknowledgments
This work was supported in part by the National Institutes of Health (1R01GM155404), an Emerging Leader Award from the Mark Foundation for Cancer Research, a Distinguished Investigator Award from the Paul G. Allen Frontiers Group, and a Sloan Research Fellowship (FG-2022-18417), to S.A.S. We thank members of the Slavoff and Loh labs for helpful discussions. We also thank the Yale West Campus Analytical Core (WCAC) for their assistance with LCMS. We thank Yale Center for Genome Analysis and Keck Microarray Shared Resource at Yale University for providing PacBio sequencing and high-performance computing services, which are funded in part by the National Institutes of Health instrument grants 1S10OD028669-01 and 1S10OD030363-01A1, respectively. We thank the Yale West Campus Imaging Core for providing confocal microscopy.
Footnotes
Code availability
Python scripts for TI-Seq analysis is available at https://github.com/slavofflab/TI-seq-ORF. Other analysis scripts/codes are available upon request.
Data availability
The PacBio sequencing data are deposited in the Sequence Read Archive with accession number PRJNA1130103. MS-based proteomics data are available via PRIDE with accession number PXD053420. During review, log in to the PRIDE website using the following details:
Username: reviewer_pxd053420@ebi.ac.uk
Password: xOKh0QreU491
References
- 1.van Heesch S. et al. The Translational Landscape of the Human Heart. Cell 178, 242–260 e229 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Chothani S. P. et al. A high-resolution map of human RNA translation. Mol Cell 82, 2885–2899 e2888 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Mohsen J. J., Martel A. A. & Slavoff S. A. Microproteins-Discovery, structure, and function. Proteomics 23, e2100211 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez T. F. et al. Accurate annotation of human protein-coding small open reading frames. Nat Chem Biol 16, 458–468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gunisova S., Hronova V., Mohammad M. P., Hinnebusch A. G. & Valasek L. S. Please do not recycle! Translation reinitiation in microbes and higher eukaryotes. FEMS Microbiol Rev 42, 165–192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rathore A. et al. MIEF1 Microprotein Regulates Mitochondrial Translation. Biochemistry 57, 5564–5575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cloutier P. et al. Upstream ORF-Encoded ASDURF Is a Novel Prefoldin-like Subunit of the PAQosome. J Proteome Res 19, 18–27 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Cao X. et al. Alt-RPL36 downregulates the PI3K-AKT-mTOR signaling pathway by interacting with TMEM24. Nat Commun 12, 508 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y. et al. Unannotated microprotein EMBOW regulates the interactome and chromatin and mitotic functions of WDR5. Cell Rep 42, 113145 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright B. W., Molloy M. P. & Jaschke P. R. Overlapping genes in natural and engineered genomes. Nat Rev Genet 23, 154–168 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunet M. A. et al. The FUS gene is dual-coding with both proteins contributing to FUS-mediated toxicity. EMBO reports 22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao X. et al. Nascent alt-protein chemoproteomics reveals a pre-60S assembly checkpoint inhibitor. Nat Chem Biol 18, 643–651 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren G. et al. Ribosomal frameshifting at normal codon repeats recodes functional chimeric proteins in human. Nucleic Acids Res 52, 2463–2479 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slavoff S. A. et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol 9, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quelle D. E., Zindy F., Ashmun R. A. & Sherr C. J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 83, 993–1000 (1995). [DOI] [PubMed] [Google Scholar]
- 16.Dhamija S. & Menon M. B. Non-coding transcript variants of protein-coding genes - what are they good for? RNA Biol 15, 1025–1031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oyama M. et al. Diversity of translation start sites may define increased complexity of the human short ORFeome. Mol Cell Proteomics 6, 1000–1006 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Wacholder A. & Carvunis A. R. Biological factors and statistical limitations prevent detection of most noncanonical proteins by mass spectrometry. PLoS Biol 21, e3002409 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S. et al. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci U S A 109, E2424–2432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao X. et al. Quantitative profiling of initiating ribosomes in vivo. Nat Methods 12, 147–153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bazzini A. A. et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J 33, 981–993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji Z., Song R., Huang H., Regev A. & Struhl K. Transcriptome-scale RNase-footprinting of RNA-protein complexes. Nat Biotechnol 34, 410–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erhard F. et al. Improved Ribo-seq enables identification of cryptic translation events. Nat Methods 15, 363–366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinnebusch A. G. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem 83, 779–812 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Pardo-Palacios F. J. et al. SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms. Nat Methods 21, 793–797 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abugessaisa I. et al. refTSS: A Reference Data Set for Human and Mouse Transcription Start Sites. J Mol Biol 431, 2407–2422 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Brunet M. A. et al. OpenProt 2021: deeper functional annotation of the coding potential of eukaryotic genomes. Nucleic Acids Res 49, D380–D388 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo H. et al. DEG 15, an update of the Database of Essential Genes that includes built-in analysis tools. Nucleic Acids Res 49, D677–D686 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X. et al. Full-length transcriptome reconstruction reveals a large diversity of RNA and protein isoforms in rat hippocampus. Nat Commun 10, 5009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mardis E. R. DNA sequencing technologies: 2006–2016. Nat Protoc 12, 213–218 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Sheynkman G. M. et al. ORF Capture-Seq as a versatile method for targeted identification of full-length isoforms. Nat Commun 11, 2326 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noguchi S. et al. FANTOM5 CAGE profiles of human and mouse samples. Sci Data 4, 170112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosa-Mercado N. A. et al. Hyperosmotic stress alters the RNA polymerase II interactome and induces readthrough transcription despite widespread transcriptional repression. Mol Cell 81, 502–513 e504 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alvarez-Carretero S. et al. A species-level timeline of mammal evolution integrating phylogenomic data. Nature 602, 263–267 (2022). [DOI] [PubMed] [Google Scholar]
- 35.Pal C., Papp B. & Lercher M. J. An integrated view of protein evolution. Nat Rev Genet 7, 337–348 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Zhang J. et al. An integrative ENCODE resource for cancer genomics. Nat Commun 11, 3696 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang M. & Marin A. Characterization and prediction of alternative splice sites. Gene 366, 219–227 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Cook K. B., Kazan H., Zuberi K., Morris Q. & Hughes T. R. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res 39, D301–308 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keren H., Lev-Maor G. & Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11, 345–355 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Kim E. et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 27, 617–630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshida K. & Ogawa S. Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA 5, 445–459 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Wheeler E. C. et al. Integrative RNA-omics Discovers GNAS Alternative Splicing as a Phenotypic Driver of Splicing Factor-Mutant Neoplasms. Cancer Discov 12, 836–855 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roth W., Stenner-Liewen F., Pawlowski K., Godzik A. & Reed J. C. Identification and characterization of DEDD2, a death effector domain-containing protein. J Biol Chem 277, 7501–7508 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Strasser A., Jost P. J. & Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity 30, 180–192 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuda I. et al. The C-terminal domain of the long form of cellular FLICE-inhibitory protein (c-FLIPL) inhibits the interaction of the caspase 8 prodomain with the receptor-interacting protein 1 (RIP1) death domain and regulates caspase 8-dependent nuclear factor kappaB (NF-kappaB) activation. J Biol Chem 289, 3876–3887 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alcivar A., Hu S., Tang J. & Yang X. DEDD and DEDD2 associate with caspase-8/10 and signal cell death. Oncogene 22, 291–297 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Chen J. et al. Pervasive functional translation of noncanonical human open reading frames. Science 367, 1140–1146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian L. et al. Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing. Genome Biol 22, 310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Djebali S. et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Klerk E. & t Hoen P. A. Alternative mRNA transcription, processing, and translation: insights from RNA sequencing. Trends Genet 31, 128–139 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Vakirlis N., Vance Z., Duggan K. M. & McLysaght A. De novo birth of functional microproteins in the human lineage. Cell Rep 41, 111808 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carvunis A. R. et al. Proto-genes and de novo gene birth. Nature 487, 370–374 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Broeils L. A., Ruiz-Orera J., Snel B., Hubner N. & van Heesch S. Evolution and implications of de novo genes in humans. Nat Ecol Evol 7, 804–815 (2023). [DOI] [PubMed] [Google Scholar]
- 54.Liu Y., Yu W., Ren P. & Zhang T. Upregulation of centromere protein M promotes tumorigenesis: A potential predictive target for cancer in humans. Mol Med Rep 22, 3922–3934 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schafer E. et al. Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum Mutat 25, 412 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Concordet J. P. & Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 46, W242–W245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Z. Generation of epitope tag knock-in mice with CRISPR-Cas9 to study the function of endogenous proteins. STAR Protoc 4, 102518 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hernandez G., Osnaya V. G. & Perez-Martinez X. Conservation and Variability of the AUG Initiation Codon Context in Eukaryotes. Trends Biochem Sci 44, 1009–1021 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended Data Fig. 1 TI-seq unveiled the presence and properties of anisoforms. a, Start codon identifies and translated ORFs were defined in individual transcripts based on TI-seq data and assigned using a script (TI_Seq_ORF.py) to ORF types, including CDS (annotated protein coding sequence or any ORF that contains a portion/isoform of the annotated CDS), iORF (internal, frameshifted ORF), dORF (downstream of CDS in any reading frame), uORF (upstream of CDS in any reading frame), uoORF (upstream and partially overlapping CDS in alternative reading frame), overprinted ORF (ORF that initiates before, ends after, and entirely encompasses the annotated CDS in an alternative reading frame), doORF (ORF that initiates within CDS, and partially overlaps an annotated CDS in an alternative reading frame), and ncORF (ORF within a “non-coding” RNA, inclusive of pseudogenes, long non-coding RNA, and other non-coding classes), based on their positions and properties. The start codon composition was then quantified by ORF type, represented as a bar plot. AUG variants means a TI-seq putative start codon exhibiting any single-nucleotide difference from AUG, excluding CUG, which is considered separately; purple bars denote putative ORF start codons that are neither AUG nor its near-cognate variants. b, Bar plot illustrating the distribution of Kozak sequence contexts around the start codons of TI-seq-detected ORFs according to ORF class. Red indicates strong AUG Kozak sequences (RNNAUGG)58, green denotes intermediate AUG Kozak sequences (YNNAUGG or RNNAUGH), cyan represents weak AUG Kozak sequences (YNNAUGH), and purple signifies ORFs that start with a non-AUG codon, which were not subjected to further sequence context analysis. ‘R’ represents A or G, ‘Y’ stands for C or T, and ‘H’ indicates A, C, or T. c, Violin and box plot illustrating the start codon peak intensity distribution of TI-seq-detected ORFs across different categories. Mean and median are highlighted for each category. The p-value was calculated using Welch’s two-sample t-test. Red dots represent outliers, and blue dots represent the mean intensity. Error bars represent the standard error of the mean (SEM). d, Cumulative frequency plot illustrating the length distribution of different ORF types detected using TI-seq, measured in codons (not including stop codon). e, Histogram depicting the frequency distribution of iORF (anisoform) lengths detected via TI-seq. The apparent peak at >100 aa is not an increase at larger sizes but an artifact of the larger “bin” encompassing all longer anisoforms.
Extended Data Fig. 2 Alternative transcription start sites (TSS) and alternative pre-mRNA splicing generate iORF-encoding alternative transcripts. a, Pie chart representing the proportion of each event type that generates iORF-encoding alternative transcripts, relative to the corresponding canonical transcript assigned to the same gene: alternative TSS (red), cassette exon exclusion (blue), or alternative 5’ splice site usage (gray). b, GLA gene showcasing an example of an anisoform-encoding alternative transcript that initiates at an alternative TSS within the same first exon, yet positioned after the start codon of the canonical coding sequence (CDS). The top tracks represent GENCODE v38 representative transcripts for GLA, indicating the chromosomal location and the orientation from 3’ to 5’; taupe boxes, exons; lines, introns. Below, the positions of the CDS (magenta boxes) and the anisoform (green boxes) are presented, and start codons are indicated by dashed lines. Green arrows point to the peaks of the start codon for the anisoform in the TI-seq data (cyan track). A broad CAGE TSS peak (green track), and split mRNA-seq intensity within the first exon (gray track, bottom), may also be consistent with alternative TSS usage. c, Analysis of the CENPM gene provides an example of an anisoform utilizing an alternative TSS within a novel 5’ extended exon that bypasses the annotated first exon and start codon for the CDS. Top, GENCODE v38 representative transcripts for CENPM, indicating the chromosomal location and the orientation from 3’ to 5’. Taupe boxes, exons. Below, the positions of the multiple protein products are indicated in magenta for the CDS and green for two putative iORFs whose start codons were identified via TI-seq (cyan, green arrows). Transcription from the novel TSS is supported by CAGE-seq and mRNA-seq (green and gray, respectively).
Extended Data Fig. 3 Anisoforms can be translated from alternative transcripts. a, Anisoform-encoding transcripts encompassing the 5’UTR through the stop codon of the iORF were cloned into pcDNA3.1 and fused with HA-FLAG tags at the C-terminus. The resulting plasmids were transfected into HEK293T cells. Anti-FLAG Western blotting and anti-FLAG immunoprecipitation followed by anti-FLAG Western blotting were used to validate their translation. b, Epitope-tagged anisoform expression plasmids generated as described in (a) were transfected into HEK293T cells to validate anisoform translation and subcellular localization with confocal microscopy. After fixation and permeabilization, cells were subjected to anti-FLAG immunofluorescence (green), with DAPI counterstain (cyan) for nuclei. Scale bars, 10 μm. c, PCR validation of DEDD2 and RUSC1 anisoform-FLAG knock-in (KI) cells, related to Fig. 2. Left panel, schematic showing the primer positions. The primer set “FI” and “RI” is used to confirm the insertion in the target gene locus. The primer set “FT” and “RT” is used to confirm that the tag is inserted into the target gene locus. Right panel, agarose gel analysis of PCR products from genomic DNA generated with the specified primer pairs.
Extended Data Fig. 4 Comprehensive analysis of transcript diversity for target genes using PacBio full-length sequencing. a, Overview of all alternative transcript and mRNA processing events previously deposited in GENCODE V38 and NCBI release 110 within the 69 iORF-encoding target genes considered in this study, representing previously known transcript diversity generated from these loci. b, Overview of transcript isoforms and events identified through ORF capture and PacBio full-length sequencing of 69 target genes. The bar plot categorizes all alternative transcript-generating events that not include in GENCODE v38 and NCBI release 110, such as alternative transcript isoforms, alternative TSS, cassette exon exclusion, alternative 5’ or 3’ splice donors, unannotated exon inclusion, intron retention, and alternative processing events combination, which include all event above. The colors represent the levels of evidence from PacBio sequencing: soft coral red for transcripts in the top 10 and top 1% by read count, golden olive for variants present above 1% (but not top 10), bright green for transcripts in the top 10 (but not above 1%), teal blue for variants that are detected at any level, cornflower blue for transcripts with read support, and orchid pink for no evidence. c, Histogram depicting the distribution of detected transcript variant numbers within target genes. The x-axis represents the isoform number, and the y-axis represents the count of genes with a specific number of detected transcript variants. d, Tile plot illustrating the distribution of the top 10 transcript isoforms for each gene and the total number of isoforms detected. Each row represents a gene, and the tiles are color-coded to show the read count percentage corresponding to each of the top 10 isoforms. The gradient bar on the right indicates the isoform percentage (0–100%), while the additional color scale represents the total number of isoforms detected per gene, ranging from 100 to 500. e, Tile plot illustrating the distribution of transcripts for anisoforms within the top 10 transcript isoforms. Each row represents a gene, with the tiles color-coded to reflect the percentage of transcripts corresponding to anisoforms. The gradient scale on the right indicates the percentage values (0–100%) for each isoform. Gray tiles marked “NA” represent cases where the transcript does not encode the anisoform within the top 10 isoforms for the corresponding gene. f, Tile plot illustrating the calculated TPM (Transcripts Per Million) values of anisoforms within the top 10 transcript isoforms and the total TPM of the parent gene. The TPM values were computed by multiplying the total percentage of transcripts for anisoforms with the TPM values of the respective parent genes. The color gradient, presented in log scale, represents the TPM values, ranging from low (light colors) to high (dark colors). Gray tiles marked “NA” indicate cases where the transcript does not encode an anisoform within the top 10 isoforms for the corresponding gene.
Extended Data Fig. 5 PacBio full-length sequencing validation of the C7orf50 alternative transcript and anisoform. a, Genome view of C7orf50. Representative transcript variants obtained from GENCODE (tracks 1–3; taupe, exons) encode either the annotated protein (tracks 1 and 2) or the anisoform (track 3). The most abundant transcript detected by PacBio long-read sequencing (track 4; other PacBio transcripts, tracks 5 and 6) matches the anisoform-encoding transcript variant found in GENCODE. The positions of the CDS (magenta) and iORF (green) within these transcripts are represented below. TI-seq (cyan) supports the use of multiple start codons, one of which is consistent with the downstream CAGE-seq (green), reference TSS (pink), PolII ChIP-seq (purple), mRNA-seq (gray) and PacBio (orange) data supporting transcription of the anisoform-encoding transcript from an internal TSS (red arrows). In addition to the TI-seq signal, further support for translation in the anisoform reading frame specifically within the region of the iORF is provided by reanalyzing Ribo-seq data (bottom zoom; pink, reads mapping to CDS reading frame; green, reads mapping to iORF reading frame). b, Analysis of translation and stability of the C7orf50 anisoform. The most abundant PacBio-detected anisoform transcript mapping to C7orf50 was cloned from the 5’ end to the stop codon of the iORF into pcDNA3.1 by gene synthesis, and fused with a dual HA-FLAG tag at the C-terminus of the iORF. The resulting plasmid was transfected into HEK293T cells, followed by lysis and anti-FLAG Western blotting, to validate expression. Beta actin served as a loading control, and untransfected cells as a negative control.
Extended Data Fig. 6 PacBio full-length sequencing validation of the RPS8 alternative transcript and anisoform. a, Top, genome view of RPS8. Tracks 1–10, RPS8 transcript isoforms detected using PacBio and ranked by read count, representing various putative processing and alternative TSS events. Taupe boxes, exons. The third-ranked transcript encodes the anisoform with a unique 5’ end that may utilize a novel TSS. All transcripts from NCBI release 110 and GENCODE v38 are shown for reference. The positions of the CDS (magenta boxes) and iORF (green boxes) are represented below the transcript diagrams, and evidence for translation initiation at the anisoform start codon is provided by TI-seq (cyan track, start codon indicated with green arrow). Ribo-seq reads (orange tracks) are also observed in the anisoform reading frame and specific to the positions of the iORF (bottom, zoom; magenta signal indicates Ribo-seq reads in the CDS reading frame, and green for the iORF reading frame). The position of the candidate alternative TSS is indicated by red arrows overlaid on CAGE-seq, CAGE-TSS (both green) refTSS (magenta) and PolII CHIP-seq (purple) data. Illumina mRNA-seq data (gray) and PacBio long-read sequencing data (dark orange) provide support for expression. b, The third most abundant transcript, which encodes the anisoform, was cloned from the 5’ end to the stop codon of the anisoform into pcDNA3.1 and fused with dual HA-FLAG tag at the C-terminus. The resulting plasmids were transiently transfected into HEK293T cells followed by lysis and anti-FLAG Western blotting. c, The construct described in b was transiently transfected into HEK 293T cells, followed by fixation and anti-FLAG immunofluorescence, with DAPI as a counterstain.
Extended Data Fig. 7 Conservation analysis of DEDD2 CDS. a, Multiple sequence alignment of DEDD2 among 12 species. The yellow box indicates the region that the anisoform overlaps in an alternative reading frame. b and c, Phylogenetic trees constructed from the entire CDS and the region overlapping with the anisoform, respectively. Branch lengths represent evolutionary distances.
Extended Data Fig. 8 SRSF2 regulates the splicing of DEDD2. a, Schematic representing a portion of the pre-mRNA of DEDD2 up to position 2867 inclusive of exons 1–3. Boxes, exons; lines, introns. The magenta region represents the annotated CDS, while the green region encompasses the region where the frameshifted iORF overlaps the CDS. The predicted SRSF2 binding site is indicated with an arrow. b, c, and d Reanalysis of RNA-seq read depth across DEDD2 exons 1 through 3 and read counts spanning splice junctions between exons 1–2 and 2–3 (specific to transcript variant 1 encoding the CDS) and exons 1–3 (specific to transcript variant 2 encoding the iORF) in various cells and models, including b, TF-1 cells treated with SRSF2 siRNA or control siRNA; e, K562 cells over expressing various point mutants or wild-type SRSF2; and c, K562 cells expressing knock-in P95L or wild-type SRSF2. d, Bar plot showing the percentage of exon 2 exclusion in K562 cells expressing SRSF2 WT and SRSF2 P95L.All p values by Welch two Sample t-test. Error bars represent the standard error of the mean (SEM). Sample sizes are as follows: n = 3 in panels b and c, n = 6 in the SRSF2 WT K562 group in panel g, and n = 4 in the SRSF2 P95L K562 group in panel g.
Extended Data Fig. 9 Validation of knock-out cell lines. a, Sequencing validation of DEDD2 CDS knockout (CDS KO) cell lines. The top panel shows the genome reference sequence for DEDD2 with the sgRNA2, sgRNA5, and sgRNA6 binding sites highlighted. Below, the sequencing chromatograms for CDS KO1 and CDS KO2 are shown, confirming the successful deletion at these sites. b, Sequencing validation of DEDD2 dual KO cell lines. The top panel shows the genome reference sequence for DEDD2 with the sgRNA1, sgRNA3, and sgRNA4 binding sites highlighted. Below, the sequencing chromatograms for dual KO1 and dual KO2 are shown, confirming the successful deletion at these sites. The figure includes the positions of the forward primer (FP) and reverse primer (RP) used for genomic DNA PCR. c Western blot analysis, corresponding to Fig. 7b, showing the levels of cleaved PARP (Asp214), an apoptosis marker, across additional independent clonal CDS KO and dual dual KO cell lines. Clone numbers are indicated above each lane. Beta-actin was used as a loading control. d Cell proliferation for two independent, clonal DEDD2 CDS KO cell lines (CDS-KO-1 and CDS-KO-2) and two dual KO cell lines (Dual-KO-1 and Dual-KO-2), compared to parental (wild-type, WT) HEK 293T cells. Equal numbers of cells from each line were seeded and grown for six days, then cell numbers were measured with crystal violet staining. The y-axis represents the optical density at 590 nm (OD 590). Error bars represent the standard error of the mean (SEM) from four independent experiments. Statistical significance is indicated by p-values for comparisons on Day 6. All p values by Welch two Sample t-test. Error bars represent the standard error of the mean (SEM).
Data Availability Statement
The PacBio sequencing data are deposited in the Sequence Read Archive with accession number PRJNA1130103. MS-based proteomics data are available via PRIDE with accession number PXD053420. During review, log in to the PRIDE website using the following details:
Username: reviewer_pxd053420@ebi.ac.uk
Password: xOKh0QreU491