

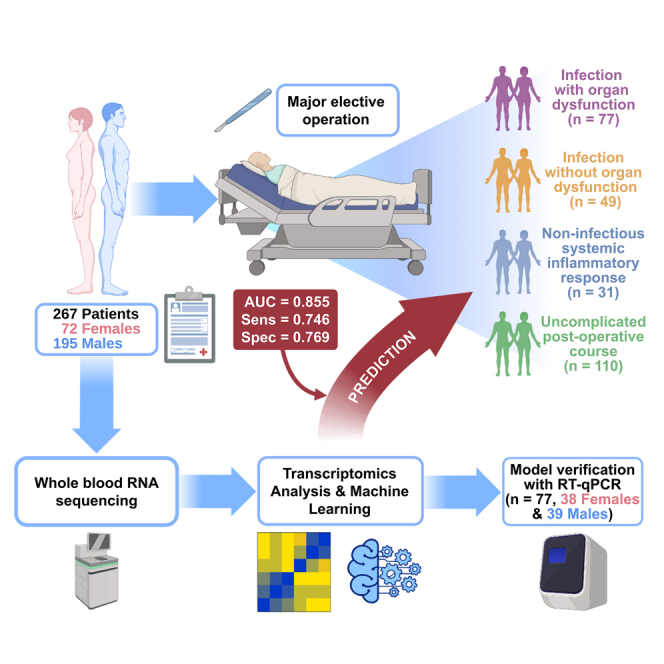
Summary
Infection is a commonplace, usually self-limiting, condition but can lead to sepsis, a severe life-threatening dysregulated host response. We investigate the individual phenotypic predisposition to developing uncomplicated infection or sepsis in a large cohort of non-infected patients undergoing major elective surgery. Whole-blood RNA sequencing analysis was performed on preoperative samples from 267 patients. These patients developed postoperative infection with (n = 77) or without (n = 49) sepsis, developed non-infectious systemic inflammatory response (n = 31), or had an uncomplicated postoperative course (n = 110). Machine learning classification models built on preoperative transcriptomic signatures predict postoperative outcomes including sepsis with an area under the curve of up to 0.910 (mean 0.855) and sensitivity/specificity up to 0.767/0.804 (mean 0.746/0.769). Our models, confirmed by quantitative reverse-transcription PCR (RT-qPCR), potentially offer a risk prediction tool for the development of postoperative sepsis with implications for patient management. They identify an individual predisposition to developing sepsis that warrants further exploration to better understand the underlying pathophysiology.
Keywords: sepsis, infectious disease, prognosis biomarkers, disease prediction, predisposition, machine learning, phenotype stratification, network analysis, RNA sequencing, RT-qPCR
Graphical abstract
Highlights
-
•
Preoperative RNA-seq data of 267 asymptomatic patients progressing to sepsis or not
-
•
776 differentially expressed genes reveal risk of complications before operation
-
•
Machine learning using preoperative samples predicts outcome with high accuracy
Garcia Lopez et al. analyze preoperative gene expression from asymptomatic patients to characterize early signals of postoperative sepsis, non-infection-associated inflammatory response, or uncomplicated outcomes. Analysis of preoperative transcriptomics shows profound changes in immune response including T cell activation and predicts postoperative patient outcome with high confidence using machine learning (ML) models.
Introduction
Sepsis represents a life-threatening organ dysfunction caused by a dysregulated host response to infection.1 It imposes a high burden on patients, families, healthcare, and social systems.2 Although outcomes are improving, mortality rates remain high (>25%), especially in low- or middle-income countries.3 Clinical risk factors for developing infection and sepsis are well recognized, including underlying malignancy, immunosuppression, diabetes, frailty, and socio-economic deprivation.4,5 Gender-specific risk factors are reported in both patients and animal models.6,7 Androgens suppress cell-mediated immune responses in males whereas female sex hormones such as estradiol possess protective effects.6 However, evidence at the genomic level for an individual intrinsic predisposition influencing susceptibility risks is conflicting.8 Studies have failed to yield any strong genomic signal in adults, and findings are often contradictory, e.g., for the tumor necrosis factor alpha (TNF-α) polymorphism −308 G/A.9
Postoperative complications following major elective surgery are common and can result in an appreciable mortality, delayed hospital discharge, and long-term morbidity. Risk factors include body mass index, age, comorbidities, serum creatinine, and prior functional status.10 Laboratory preoperative predictors of postoperative infection include serum albumin,11,12,13,14 C-reactive protein (CRP),12,13,14,15,16 hemoglobin A1c,16 interleukin (IL)-6 and alpha-1-antitrypsin,12 platelet:lymphocyte ratio,17 lymphocyte:CRP ratio,18 neutrophil:lymphocyte ratio,19 lymphopenia,20 and monocyte human leukocyte antigen-DR (HLA-DR) expression.21 Although these studies identified risk factors for postoperative complications, it remains uncertain whether these or other factors associate specifically with the risk of postoperative sepsis.
Most cases of adult sepsis present in old age22 suggesting both exogenous and endogenous non-genomic disease modifiers that influence the transcriptomic response. Here, we hypothesized that the phenotypic state of a patient undergoing elective surgery, reflected within their individual transcriptomic signature, would identify those at high risk of developing postoperative sepsis. By utilizing a unique biobank of daily samples and clinical data collected before and after elective major surgery, we identified with the aid of ML specific molecular patterns from preoperative whole-blood RNA sequencing data that could predict postoperative outcomes. This was further explored and confirmed with quantitative reverse-transcription PCR (RT-qPCR)-based ML model analysis.
Results
Preoperative RNA profiles differ between patients developing postoperative infection or not
We retrieved 267 preoperative samples from an elective surgery patient cohort who had an uncomplicated postoperative course, suffered an uncomplicated postoperative infection, or developed sepsis (STAR Methods, Figure S1).23 Demographics and preoperative clinical data between patients with different postoperative infection status were similar for all matched and most unmatched characteristics (chi-squared test, Wilcoxon rank-sum test, or Fisher’s exact test, p value > 0.05) (Tables 1 and S1). The only exceptions that were statistically significant were heart rate, CRP, and blood urea (p < 0.05).
Table 1.
Patient characteristics
| Characteristic | n | Inf+, n = 126a,b | Inf−, n = 141a,b | pc | Sepsis+, n = 77a | UInf+, n = 49a,b | SIRS+, n = 31a,b | SIRS−, n = 110a,b | pd |
|---|---|---|---|---|---|---|---|---|---|
| Gender | 267 | 0.68 | 0.11 | ||||||
| Female | 32 (25%) | 40 (28%) | 14 (18%) | 18 (37%) | 10 (32%) | 30 (27%) | |||
| Male | 94 (75%) | 101 (72%) | 63 (82%) | 31 (63%) | 21 (68%) | 80 (73%) | |||
| Age (years) | 267 | 66 (57, 73) | 66 (58, 72) | 0.97 | 68 (58, 73) | 64 (55, 74) | 66 (61, 72) | 66 (57, 72) | 0.87 |
| Ethnic origin | 262 | 0.09 | 0.23 | ||||||
| Asian or Asian British | 2 (1.6%) | 1 (0.7%) | 1 (1.3%) | 1 (2.0%) | 0 (0%) | 1 (0.9%) | |||
| Black or Black British | 2 (1.6%) | 5 (3.6%) | 2 (2.7%) | 0 (0%) | 1 (3.3%) | 4 (3.7%) | |||
| Chinese or other ethnic group | 4 (3.2%) | 0 (0%) | 4 (5.3%) | 0 (0%) | 0 (0%) | 0 (0%) | |||
| White | 116 (94%) | 132 (96%) | 68 (91%) | 48 (98%) | 29 (97%) | 103 (95%) | |||
| Temperature (0.1°C) | 221 | 36.50 (36.40, 36.70) | 36.50 (36.40, 36.70) | 0.72 | 36.50 (36.40, 36.70) | 36.50 (36.40, 36.70) | 36.40 (36.20, 36.50) | 36.50 (36.40, 36.80) | 0.04 |
| Heart rate (BPM) | 256 | 75 (67, 85) | 72 (63, 80) | 0.03 | 73 (65, 84) | 79 (71, 85) | 72 (60, 78) | 72 (63, 80) | 0.05 |
| Systolic BP (mmHg) | 255 | 129 (112, 142) | 133 (118, 146) | 0.09 | 125 (110, 142) | 130 (115, 146) | 124 (108, 146) | 135 (119, 146) | 0.10 |
| Diastolic BP (mmHg) | 255 | 75 (65, 84) | 76 (65, 85) | 0.86 | 73 (65, 84) | 78 (68, 90) | 70 (60, 79) | 77 (70, 85) | 0.02 |
| MAP | 256 | 91 (82, 101) | 93 (84, 105) | 0.42 | 90 (81, 100) | 95 (83, 110) | 86 (76, 103) | 95 (88, 105) | 0.03 |
| Total respiratory rate (bpm) | 239 | 16 (14, 17) | 15 (14, 16) | 0.13 | 15 (14, 16) | 16 (14, 17) | 16 (14, 17) | 15 (14, 16) | 0.07 |
| FiO2 | 146 | 0.21 (0.21, 0.21) | 0.21 (0.21, 0.21) | 0.12 | 0.21 (0.21, 0.21) | 0.21 (0.21, 0.21) | 0.21 (0.21, 0.21) | 0.21 (0.21, 0.21) | 0.34 |
| WBC count (×109 liter) | 184 | 7.65 (6.36, 9.87) | 7.00 (5.94, 8.54) | 0.06 | 7.40 (6.09, 9.80) | 8.03 (6.81, 9.90) | 8.14 (6.48, 9.06) | 6.84 (5.84, 8.20) | 0.03 |
| Neutrophils (×109 liter) | 114 | 4.70 (3.71, 6.10) | 4.30 (3.69, 5.90) | 0.45 | 4.26 (3.65, 5.80) | 5.46 (4.13, 6.22) | 5.10 (4.40, 6.20) | 4.10 (3.58, 5.60) | 0.07 |
| Lymphocytes (×109 liter) | 114 | 1.70 (1.33, 2.30) | 1.74 (1.20, 2.30) | 0.66 | 1.90 (1.32, 2.30) | 1.56 (1.39, 2.43) | 2.20 (1.20, 2.40) | 1.72 (1.20, 2.30) | 0.97 |
| Monocytes (×109 liter) | 114 | 0.60 (0.50, 0.70) | 0.60 (0.50, 0.70) | 0.61 | 0.60 (0.50, 0.75) | 0.60 (0.50, 0.70) | 0.60 (0.40, 0.80) | 0.59 (0.50, 0.70) | 0.96 |
| Eosinophils (×109 liter) | 113 | 0.20 (0.10, 0.30) | 0.20 (0.10, 0.26) | 0.55 | 0.20 (0.10, 0.30) | 0.20 (0.10, 0.30) | 0.10 (0.10, 0.20) | 0.20 (0.10, 0.28) | 0.73 |
| Platelets (×109 liter) | 183 | 239 (207, 294) | 244 (189, 314) | 0.40 | 231 (199, 284) | 268 (235, 320) | 235 (194, 289) | 247 (186, 315) | 0.31 |
| INR | 121 | 1.00 (0.94, 1.09) | 1.00 (0.97, 1.05) | 0.51 | 1.00 (0.94, 1.05) | 1.00 (0.94, 1.13) | 1.00 (0.98, 1.10) | 1.00 (0.97, 1.03) | 0.65 |
| APPT (seconds) | 150 | 30 (26, 32) | 31 (27, 34) | 0.31 | 30 (23, 32) | 30 (27, 32) | 30 (25, 33) | 31 (27, 34) | 0.70 |
| CRP (mg/L) | 69 | 2 (1, 7) | 1 (0, 2) | 0.02 | 2 (1, 5) | 4 (1, 12) | 1 (0, 1) | 1 (0, 4) | 0.09 |
| Blood urea (mmol/L) | 145 | 5.1 (3.9, 7.7) | 6.4 (4.8, 9.6) | 0.03 | 5.5 (4.2, 7.9) | 4.9 (3.9, 6.4) | 7.0 (5.8, 9.1) | 6.1 (4.6, 9.8) | 0.08 |
| Blood creatinine (μmol/L) | 184 | 78 (66, 92) | 80 (69, 89) | 0.78 | 78 (67, 92) | 78 (63, 93) | 82 (73, 94) | 79 (67, 88) | 0.62 |
APPT, activated partial thromboplastin time; BP, blood pressure; CRP, C-reactive protein; INR, international normalized ratio; MAP, mean arterial pressure; WBC, white blood cells.
n (%), Median (IQR).
Inf+, infection; Inf-, non-infection; UInf+, uncomplicated infection; SIRS, non-infectious systemic inflammatory response syndrome.
Fisher’s exact test, Wilcoxon rank-sum test.
Fisher’s exact test, Kruskal-Wallis rank-sum test.
Preoperative transcriptome profiles using RNA sequencing were derived for all 267 patients and yielded 776 differentially expressed genes (DEGs) (DESeq2 or edgeR, adjusted p value ≤ 0.05, STAR Methods) for patients who went on to develop postoperative infection (n = 126) compared to those who did not (n = 141) (Table S2). Preoperative samples are referred to by their respective postoperative outcome unless otherwise noted.
The large number of DEGs supported our hypothesis that a unique preoperative phenotypic state characterizing the risk of developing postoperative infection exists. Hierarchical clustering analysis revealed six clusters of differing expression patterns between the two groups (Figure 1A; Table S2). The common enriched functionality within these clusters comprised immune system-relevant categories including hapto-/hemoglobin synthesis (cluster 3), chemokine binding (cluster 5), and T cell differentiation (cluster 6) due to differential expression of immune response-relevant factors such as integrins (ITGB3, ITGB5, ITGB7), glycoproteins (CD8, CD59, CD74, CD151, CD163, CD177), and chemokines (CCR4, CCR5, CCR8) (Figure 1A).
Figure 1.

Gene expression analysis of all preoperative samples categorized by infected and non-infected postoperative outcome
(A) Heatmap and hierarchical clustering of 267 samples of both gender including 776 identified differentially expressed genes (DEGs) discriminating infected and non-infected postoperative outcome (adjusted p ≤ 0.05 according to DESeq2 or edgeR; see STAR Methods). Enriched Gene Ontology (GO) categories for six identified expression clusters are indicated. Named genes are also differentially expressed when investigating gender-specific expression.
(B) Venn Diagram of number of significant DEGs between infected and non-infected patients for each gender individually. The 72 genes are named and associated to GO categories in (A).
(C) Significant up- and down-regulated DEGs for males and females, respectively. Each panel shows purely up- or down-regulated DEGs in the upper row compared to the expression direction in the opposite gender in the lower row. Labeled genes were the top 20 up- or down-regulated DEGs per gender. Positive and negative indicate genes showing the same or opposite fold change direction, respectively, in male and female samples with infection compared to non-infection postoperative outcome. logFC = Log2 of expression fold change.
With regard to gender-specific expression signatures, we identified 72 DEGs common in both genders, opposed to 421 and 271 DEGs uniquely identified in males and females, respectively (Figure 1B; Table S2). These 72 DEGs possess immune response functionality (e.g., ITGB3, ITGB7, TNFAIP8L1) and encode for zinc-finger proteins (ZBTB47, ZCCHC3, ZNF316, ZNF580, ZNF787), transmembrane activity (TMEM185A, TMEM250), and hemoglobin (HBG1). Most of these genes showed the same fold change direction and were up-regulated in both genders. Only the adhesion-relevant gene ADGRG7 was significantly down-regulated in females opposed to significant up-regulation in males. Of note, 43 (60.5%) of the 72 common DEGs were associated with chemokine binding, while 15 (21%) are associated with T cell differentiation, a functional enrichment that was also observed when using all samples (clusters 5 and 6, Figure 1A). Only 16% of all up-regulated DEGs and 11.6% of down-regulated DEGs in males showed opposite fold changes in females, while only 1.7% up- and 8.1% down-regulated female-specific DEGs showed opposite fold changes in males (regardless of significance in the opposing gender, Figure 1C).
In summary, preoperative transcriptome profiles differed distinctly between patients who progressed to postoperative infection and those who did not. Separate gender-specific transcriptome profiles showed a primarily congruent expression direction but included differing fold changes for infection compared to an uninfected outcome. Taken together, the transcriptional phenotype appears similar between the two genders in preoperative samples with postoperative infection compared to non-infection status.
Co-expression networks reveal specific module patterns for postoperative sepsis
Co-expression network analysis offers a powerful complementary approach to reveal contextualized molecular functions.24,25,26 Using the same preoperative samples as before (developing postoperative infection or not, Figure 1), we explored co-expression networks to detect gene modules with concerted expression patterns toward further postoperative outcomes including a non-infectious systemic inflammatory response (SIRS+), uncomplicated infection (UInf+), and sepsis. Based on DEGs differentiating infection outcome using all samples or gender-specific subsets, we generated co-expression networks and identified modules by weighted gene co-expression network analysis (STAR Methods, Figure 2A; Table S3). After filtering for functionally annotated DEGs showing high correlation to at least one further annotated DEG (Spearman ρ ≥ 0.7, false discovery rate [FDR] ≤0.05), our network comprised 486 DEGs and 5,232 correlation edges (Figure 2A). Modules enriching for cellular defense and T cell activation, epithelial cell adhesion, or taste receptor activity included DEGs primarily originating from the analysis using all samples, whereas modules enriching for GTPase regulator activity, cytokinesis, or non-motile cilium assembly were dominantly differentially expressed in samples of females with differing infection status (Figure 2A).
Figure 2.

Gene co-expression networks
(A) Visualization of merged co-expression networks based on annotated differentially expressed genes (DEGs) using preoperative samples with infection or non-infection postoperative outcome. Edges refer to Spearman correlations of ≥0.7 between genes in at least one sample set (all samples, only male, or only female samples). Node color indicates for which preoperative sample set the respective gene was significantly differentially regulated. Enriched Gene Ontology (GO) terms for identified modules of the co-expression network are indicated.
(B) Visualization of expression and correlation changes given preoperative samples with sepsis, UInf+, or SIRS+ postoperative outcome compared against SIRS− postoperative outcome using all, male-, or female-associated samples. Node colors indicate the log2 fold change of mean normalized gene expression between each sub-group (sepsis, UInf+, SIRS+) against SIRS− (control). Edge colors indicate the difference in gene-gene Spearman correlations between each sub-group against SIRS−. Spearman correlation differences ≤0.3 were omitted for clarity.
To exclude base expression and correlation bias, we normalized all expression and correlation information by samples from SIRS− patients (Figure 2B; Table S3). Infected patients (sepsis and UInf+) showed significantly higher expression differences compared to SIRS+ patients irrespective of gender (p ≤ 1.7e−10 using all, only female, or only male samples; p value according to multiple test-adjusted two-tailed paired Wilcoxon test here and in the following, Table S4). Gene correlations between sepsis compared to SIRS+ samples also differed significantly (p ≤ 0.04, Table S4). Gene correlation between UInf+ and SIRS+ samples differed significantly only for male samples (p = 6.1e−04, Table S4), indicating a more pronounced expression shift in the response toward systemic inflammation in males compared to females. Three modules showed significant differences in expression pattern across different operation outcomes: cellular defense response/T cell activation, GTPase regulator activity, and non-motile cilium assembly/cytokinesis (test statistics summaries in Table S4).
Genes grouped in the module “cellular defense response/T cell activation” (Figure 2A) were more expressed in sepsis samples using all (p = 2.5e−6) or male samples (p = 1.1e−9) and significantly lower in female sepsis samples (1.6e−7) compared to UInf+ samples. Highly concerted gene expression involved immune-relevant genes that were previously identified; these included ITGB7, CCL5, and its receptor CCR5, and further targets such as SPN. SIRS+ samples showed significantly lower expression activity in this module compared to sepsis or UInf+ samples (p ≤ 6.2e−6). Of note, when comparing gender-dependent samples, females showed a significantly higher activation in this immune response-relevant module than males for UInf+ outcome (1.1e−9), but insignificant activation differences for sepsis or SIRS+.
Genes included in the module with enriched GTPase regulator activity are involved in correct actin cytoskeleton formation and are a target for bacterial manipulation to ease bacterial entry into host cells and escape phagocytosis and thus immune defense.27 The associated genes were significantly more activated in UInf+ than in sepsis or SIRS+ patients (p ≤ 5.4e−9). Irrespective of sepsis, UInf+, or SIRS+ postoperative outcomes, female-specific expression profiles yielded a significantly stronger expression response in the genes of this module compared to males (p ≤ 0.035).
Finally, genes of the “non-motile cilium assembly/cytokinesis” module were dominantly less expressed in UInf+ compared to sepsis patients irrespective of gender (p ≤ 5.2e−5), whereas UInf+ compared to SIRS+ samples were significantly less expressed only in females (p = 0.003). Cilia are associated with repeated chest infections and may thus play a role in the early defense against inhaled pathogens.28 Sepsis differentiated significantly from SIRS+ when combining samples from both genders together, in particular for male-specific gene expression in this module (p = 0.05 for samples of both genders and p = 3.7e−4 for the male subset). Gender-specific differences were present in patient samples with a UInf+ or SIRS+ outcome (p = 0.048 and p = 7.3e−7, respectively), but not in samples with a sepsis outcome.
In summary, T cell activity, GTPase, cilium assembly, and cytokinesis modules differed, and these were mostly significant between different postoperative outcomes. While the T cell activity module was highest in sepsis patients, GTPase regulator activity was most induced and cilium assembly/cytokinesis module most reduced in UInf+ patients (Figure 2B). Gender-specific outcome subsets showed primarily the same expression direction but also differed in, for example, the T cell activation module with UInf+ outcome. Taken together, both relative gene expression and co-expression correlations compared to SIRS− samples showed gender-independent as well as punctuated gender-specific characteristics for different molecular functions involved in the response to infection (sepsis and UInf+). This analysis suggests that, alongside gender-specific effects, a gender-independent phenotypic signature may be present in patients who are at risk of developing future sepsis.
Postoperative infection and sepsis can be predicted from preoperative samples
We identified minor, albeit statistically significant, differences in the patient-specific clinical factors heart rate, CRP, and blood urea levels depending on whether patients developed postoperative infection or not (Table 1). We therefore explored whether an ML classifier would be able to predict infection onset based on these preoperative clinical parameters (STAR Methods). Acquired area under the receiver operating curves (AUROC) of 0.634 (95% confidence interval [CI95] = 0.625–0.643, Table S5) suggested that the clinical characteristics of preoperative patients associated with our study cohort cannot be used for accurate risk assessment for predicting postoperative infection. When breaking down clinical outcomes into sepsis, UInf+, SIRS+, or SIRS−, the clinical parameters temperature, heart rate, diastolic blood pressure, mean arterial pressure, and white blood cell count differed significantly in at least one group (Table 1). Models of patient subsets based on these data differentiating postoperative sepsis from postoperative UInf+, SIRS+, and SIRS− achieved AUROC values of only 0.627 (CI95 = 0.614–0.641), 0.636 (CI95 = 0.619–0.653), and 0.607 (CI95 = 0.595–0.620), respectively (Table S5). As before, this exemplifies limited ML classification performance when using only clinical parameters.
Although there were only minor differences in the available preoperative clinical data between groups with differing postoperative outcomes, RNA sequencing-based expression data of preoperative samples yielded a significant number of DEGs (Table S2). By using 1,282 DEGs arising from seven pairwise comparisons as a starting point, we applied a combined feature-reduction and performance assessment procedure to develop expression-based ML prediction models (STAR Methods, Table S2). We optimized ML models with fewer genes when the increase in performance by adding more genes to the respective ML gene signatures was minimal. In contrast to the inadequate performance of ML models based on clinical data, the ML models generated on gene expression could differentiate infection and sepsis from other postoperative outcomes with convincing power. We derived gender-independent classification models with AUROC values ranging from 0.815 (CI95 = 0.810–0.821) to 0.910 (CI95 = 0.900–0.920; mean AUROC = 0.855), sensitivity ranging from 0.713 (CI95 = 0.705–0.722) to 0.767 (CI95 = 0.753–0.781; mean AUROC = 0.746), and specificity ranging from 0.743 (CI95 = 0.732–0.754) to 0.804 (CI95 = 0.786–0.821; mean AUROC = 0.769) using a balanced decision threshold (Figure 3A; Table S5). Most samples could be classified correctly, ranging from 75% for the model Inf+ vs. Inf-to 83% for the model sepsis vs. SIRS+ (Figure 3B). All tracked statistical metrics including AUROC, accuracy, sensitivity, specificity, and their respective confidence intervals as well as positive and negative predictive values as functions of assumed prevalence are shown in Table S5. Our models could predict patient outcome, even in the most challenging comparisons of sepsis vs. UInf+ and sepsis vs. SIRS+ with AUROC/sensitivity/specificity of 0.872/0.762/0.759 (CI95: 0.863–0.881/0.750–0.775/0.747–0.771) and 0.910/0.767/0.804 (CI95: 0.900–0.920/0.753–0.781/0.786–0.821), respectively. In a diagnostic context where patients need to be “ruled out” or “ruled in” for commencing antibiotic therapy on the basis of having a postoperative infection, the decision threshold for our binary classification can be optimized toward either “snout” (high sensitivity) or “spin” (high specificity) models (Table S5). Of note, using only male or female samples yielded gender-specific classification models with AUROC ≥0.8 (Table S5). Adding statistically relevant clinical features as additional features to the identified model gene signatures did not improve any of the gene expression-based model performances (Table S5).
Figure 3.

RNA sequencing-based model classification performance
(A) Mean AUROC curves and performance across TPR and FPR for the indicated seven classification models.
(B) Confusion matrices for the same models described in (A). n, number of samples after ROSE-based gender balancing (STAR Methods).
(C) Upset plot of the machine learning models using all preoperative samples from seven assessed comparisons referring to different postoperative outcomes. Set size indicates the number of differentially expressed genes (DEGs) used in each comparison. Interaction size indicates the number of DEGs common across different comparisons. The interaction matrix indicates the number of shared DEGs across different classification models.
(D) Heatmap representing the top 5 most important DEGs as indicated by the highest mean importance value using the varImp function of the R caret package across all seven classification models. DEGs shared by more than one classification model are indicated in bold font. Abbreviations: TPR, true positive rate (sensitivity); FPR, false positive rate; CI95, 95% confidence interval.
Notably, all classification models required only a few genes (median 15 genes, interquartile range [IQR] 11.5–16, Figure 3C; Table S5). Of all available gene expression data, only a pool of 72 genes remained after feature elimination as the basis for all derived classification models. Forty-one of these 72 genes were also part of a highly intra-connected module of the co-expression networks, including cilium assembly/cytokinesis (e.g., TNFAIP3) and T cell activation (e.g., CCR5 and GZMH) modules but also genes such as CENPB that are present in the heterochromatin module (Figure 2A). While 13 of these genes were used in at least two ML models, the remaining 59 genes (82%) were part of individual ML models. Discriminating between different complex postoperative outcomes with high performance thus requires distinct gene signatures. By algorithmically exploring the gene importance per model (STAR Methods), we analyzed whether subsets of genes repeatedly appeared to be main drivers of classification performance. By investigating the top five most important genes per classification model (Figure 3D), EMILIN1, KLF16, and MYDGF appeared as important discriminators across multiple models; no single gene alone was sufficient to allow for a highly predictive model. Nonetheless, the achieved performance was remarkable, especially in light of using at most 17 genes as features for model prediction.
The unique property of our classification models is the ability to predict a prospective postoperative complication upon expression data before elective surgery. At the time of publication, no similar studies were available for assessing prediction of sepsis development using the transcriptional phenotype as a risk-assessment tool. Consequently, until we perform prospective validation studies, we are unable to directly test the predictive ability of our models in external cohorts but only to examine whether any of the selected genes in the ML models are still relevant after diagnosis of infection. We therefore tested whether we could correctly classify patients with septic shock as provided by an independent, publicly available dataset.29 Testing the most clear-cut models, Inf+ vs. SIRS− and sepsis vs. SIRS−, offered very high classification performance (true positive rate [c] 100% and 96%, respectively). Testing the performance of the model with the more challenging comparison, sepsis vs. SIRS+, still yielded a TPR of 75% (Table S5). Given the impact of the recent COVID-19 pandemic, we additionally performed RNA sequencing of blood samples from 61 affected COVID-19 patients (including 51 with sepsis; STAR Methods) and explored classification performance of the same models. The majority of samples (TPR 98% for mild and TPR 77% for severe, sepsis-like cases) were correctly classified for our model Inf+ vs. SIRS−, while only 31 of the 51 patients (TPR 60%) were correctly classified with our sepsis vs. SIRS− model. The model sepsis vs. SIRS+ yielded again a good TPR with 41 correctly classified samples (TPR 80%, Table S5). These results indicate that the prognostic models we developed, which are based on pre-operation samples, include in part expression information that potentially allows to also discriminate ongoing infection or sepsis.
To verify the RNA sequencing-based model classification results, we generated RT-qPCR data for all important gene features (n = 72) of the most clinically interesting preoperative classification models using 77 patients (STAR Methods, Tables S5 and S6; Figure S1). Despite the different nature of RNA sequencing and RT-qPCR techniques and the lower number of patients, using the same gene sets for new classification as with RNA sequencing-based ML models, we achieved model performances with high clinical potential as a risk-assessment tool (mean AUROC of 0.766, Table S5; Figure S2). Given the importance of predicting the risk of sepsis, we specifically tested our RT-qPCR-based classification models for identifying sepsis from other postoperative outcomes using a balanced cutoff for classification decision (Table S5). Discerning sepsis from Inf− achieved an AUROC of 0.791 (CI95 = 0.785–0.797), discerning sepsis from SIRS− an AUROC of 0.751 (CI95 = 0.739–0.763), discerning sepsis from a non-infectious inflammatory postoperative outcome (sepsis vs. SIRS+) an AUROC of 0.820 (CI95 = 0.811–0.829), and, finally, discerning sepsis from UInf+ an AUROC of 0.703 (CI95 = 0.685–0.722) using the respective RT-qPCR-based classification models (Table S5). Having achieved sufficiently high AUROC values for each of the tested models allows optimization of different class cutoffs depending on needs within a clinical context. Thus, high sensitivity at the cost of specificity (“rule-in” for treatment) or vice versa (“rule-out” for treatment) models are feasible. Taken together, our RNA sequencing-based high-performance ML models could be recapitulated using RT-qPCR data and proposed classification models based on at most 17 genes. For future perspectives, these appear technically more appropriate toward complementing clinical diagnostics than complete RNA sequencing approaches per individual.
Discussion
Many patients develop sepsis, a life-threatening complication that still carries large personal and societal burdens.2,3 Multiple studies using various omic and other approaches have identified prognostic biomarkers in septic patients.9,30,31,32,33,34 As the blood samples in these studies were collected when patients were already displaying clinical evidence of sepsis, none were predictive of the risk of developing either uncomplicated infection or sepsis. While there are well-described risk factors such as age, immune suppression, and social deprivation, all these lack sensitivity and specificity.
Preoperative blood samples show differential gene expression toward sepsis
Here, we explored the possibility of a human phenotypic predisposition at the transcriptomic level that could identify patients at risk of developing postoperative infection with or without sepsis. By analyzing the preoperative transcriptomic signatures of 267 patients undergoing elective major surgery, we detected a number of DEGs including integrins, CCR5, CD80, and VASH2 that were associated with the subsequent development of sepsis. Of note, integrins were recently proposed as novel targets for sepsis therapy.35 CCR5 is a prime example of a gene that impacts upon resistance to pathogens given that ∼1% of Caucasian humans carry a 32-bp deletion in the gene leading to an elevated resistance against HIV.36,37 CD80 may be involved in sepsis-induced acute kidney injury.38,39 We also detected a highly coordinated expression of ITGB7, CCL5/CCR5, and SPN (also termed sialophorin or CD43) by co-expression analysis. This has been associated with discordant regulation of TNF-α and IL-10 in association with mycobacteria.40 Thus our analyses suggest that differential expression predisposing the patient to developing uncomplicated infection or sepsis is already manifested in preoperative blood samples.
Despite the high number of genes that appeared in the preoperative DEG and co-expression network analysis to differentiate postoperative outcomes, a set of 72 genes was sufficient to generate classification models reaching AUROCs in the range of 0.815–0.910. Some genes, including GZMH, MYDGF, PTGDR2, and SNN, were included across different classification models. All four have been previously associated with tissue damage, infectious disease, and sepsis, implicating an elevated role in the host immune response.29,41,42,43,44 These genes also appeared in our co-expression modules (GZMH, SNN, Figure 2A) or were among the top five most important gene features in at least one ML model signature (MYDGF, PTGDR2, SNN, Figure 3D). This exemplifies their expression differences and thus potential importance for phenotypic differences in preoperative samples that could predict postoperative outcome.
Transcriptomic profiles of preoperative samples differ from established sepsis samples
Of note, the 72 genes of our ML models showed no overlap with genes of the reported gene signatures for presymptomatic infection and sepsis up to three days before diagnosis.23 This hints at remarkable differences in blood gene expression depending on whether a patient has already a confirmed infection or sepsis and may imply that early risk prediction is not achievable with gene signatures derived from patients that have, or are shortly about to have, infection or sepsis. Lastly, the biological relevance of the genes selected in specific ML models was further confirmed when classifying patients already with active infection using cohorts of 61 COVID-19 patients and 28 septic shock patients.29 These results need to be interpreted with caution as our models were trained on preoperative data of patients with future, but not ongoing, infection and sepsis. Although our models showed some capability to predict ongoing infection and sepsis, the preoperative transcriptomic expression profiles with no symptoms of infection would substantially differ from those of patients with incipient or established infection or sepsis.
Limitations of the study
We recognize the need for prospective validation to confirm the predictive capability of our ML-based classification models before they are applied in a clinical setting. We show that our identified risk assessment gene signatures can also classify patients with established infection and sepsis. Nevertheless, other patient cohorts, for example cohorts over more diverse ethnicities or with a different clinical context, need to be tested to show that our identified preoperative risk signatures are generalizable. It appears reasonable to attempt deriving infecting agent-specific sepsis prediction signatures given the profound differences in impact of pathogens from different kingdoms.45,46 This question could be addressed in future, larger study cohorts.
Conclusion
We have shown here that it appears feasible to initiate counter-measures such as antimicrobial therapy, not only at the time of sepsis diagnosis but also even before any sign of infection or complications thereof. Depending on the need, diagnostic tools based on our models could be optimized toward either very high sensitivity or specificity to allow for rule-out or rule-in decision making (Table S5). If confirmed, our line of work will contribute to improved risk assessment not just in patients before elective surgery but also, potentially, more generally, in patients with elevated health risks. An example would be expedited entry of susceptible patients into vaccination programs for future pandemics.47,48,49
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Gianni Panagiotou (Gianni.Panagiotou@leibniz-hki.de).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Expression data of all preoperative patient samples were deposited in the GEO database with GEO-ID GSE208581 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208581). RNA sequencing data of COVID-19 patient samples were deposited in the GEO database with GEO-ID GSE208587 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208587). The RT-qPCR data generated in this study are included in Table S6 and are published together with this article.
-
•
The original codes used to analyze the presented data and generate machine learning models are available at zenodo (https://doi.org/10.5281/zenodo.10835070) and GitHub (https://github.com/thisisalbert/preoperative_sepsis_risk/).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank Dr. Amelia Barber, head of the junior research group Fungal Informatics at the Friedrich-Schiller University, Jena, Germany, for crossreading the manuscript. G.P. would like to thank the Area of Excellence of the Hong Kong Research Council, “Institute of Metabolic Medicine” (5/2019-4/2027), Hong Kong, China, and the Leibniz-HKI for providing institutional funding to cover all costs associated with the current study.
Author contributions
G.P. designed and conceptualized the study, administered, and acquired the funding for this study. A.G.L., B.S., M.S., and R.L. conducted the methodology. A.G.L. and T.S.-O. were in charge of software-related task. A.G.L. handled the validation process. A.G.L., B.S., and T.S.-O. performed the formal analysis. A.G.L. and S.S. carried out the investigation procedure. E.J.G.-B., G.P., M.S., R.L., and M.B. were responsible for providing the resources needed for the study. A.G.L. and S.S. took care of curating the data. A.G.L. and T.S.-O. directed the data visualization. A.G.L., G.P., and S.S. wrote the original draft. S.S. and G.P. supervised the project. All authors participated in the review and editing process.
Declaration of interests
E.J.G.-B. has received honoraria from Abbott Products Operations AG, bioMérieux, Brahms GmbH, GSK, InflaRx GmbH, Sobi, and Xbiotech Inc; independent educational grants from Abbott Products Operations, bioMérieux, Inc, InflaRx GmbH, Johnson & Johnson, MSD, UCB, and Swedish Orphan Biovitrum AB; and funding from the Horizon 2020 European Grants ImmunoSep and RISCinCOVID and the Horizon Health grant EPIC-CROWN-2 (granted to the Hellenic Institute for the Study of Sepsis). M.S. received grants from NewB, Apollo Therapeutics, and UCL Technology Fund and others from Abbott, Amormed, bioMérieux, Biotest, Deltex Medical, Fresenius, Mindray, NewB, Pfizer, Radiometer, Roche Diagnostics, Safeguard Biosystems, Shionogi, and Spiden outside of this project. M.S. is an unpaid advisor to Presymptom Health Ltd, hemotune, deepUll, and Santersus, and R.L. is a part-time advisor to Presymptom Health Ltd.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Whole blood preoperative samples | Lukaszewski et al.23 | 06/Q1702/152; IRB00001756 |
| Blood samples of patients infected with SARS-CoV-2 | ESCAPE study | 2020-001039-29; NCT04339712 |
| Chemicals, peptides, and recombinant proteins | ||
| RNAlater | ThermoFisher | Cat#AM7021 |
| Critical commercial assays | ||
| RiboPure-Blood Kit | ThermoFisher | Cat#AM1928 |
| TruSeq Stranded Total RNA with Ribo-Zero Globin kit | Illumina | Cat#20020613 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher | Cat#4368813 |
| TaqMan PreAmp Master Mix | ThermoFisher | Cat#4488593 |
| Deposited data | ||
| Raw and processed RNA sequencing data of preoperative samples | This paper | GEO: GSE208581 |
| Raw and processed RNA sequencing data of COVID-19 samples | This paper | GEO: GSE208587 |
| Human reference genome GRCh38.99 | Genome Reference Consortium | www.ensembl.org |
| Software and algorithms | ||
| Original code | This paper | Zenodo (https://doi.org/10.5281/zenodo.10835070) & GitHub (https://github.com/thisisalbert/preoperative_sepsis_risk) |
| GEO2RNAseq (v 0.100.3) | Seelbinder et al.50 | https://hub.docker.com/r/xentrics/geo2rnaseq |
| FastQC (v0.11.8) | Babraham Institute - Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimmomatic (v0.39) | The Usadel Lab | https://github.com/usadellab/Trimmomatic |
| SortMeRNA (v2.1b) | Bonsai Bioinformatics | https://bioinfo.univ-lille.fr/sortmerna/ |
| HiSAT2 (v2.1.0) | The Daehwan Kim Lab | https://daehwankimlab.github.io/hisat2/ |
| DESeq2 (v1.26) | Love et al.51 | https://bioconductor.org/packages/DESeq2/ |
| edgeR (v3.28.1) | Robinson et al.52 | https://bioconductor.org/packages/edgeR/ |
| ComplexHeatmap (v2.4.3) | Gu et al.53 | https://www.bioconductor.org/packages/ComplexHeatmap/ |
| VennDiagram (v1.6.20) | Chen et al.54 | https://cran.r-project.org/package=VennDiagram |
| org.Hs.e.g.,.db (v3.11.4) | Marc Carlson | https://bioconductor.org/packages/org.Hs.eg.db/ |
| clusterProfiler (v3.16.1) | Yu et al.55 | https://bioconductor.org/packages/clusterProfiler/ |
| Cytoscape (v3.7.0) | Shannon et al.56 | https://cytoscape.org/ |
| mice (v3.16.0) | van Buuren et al. | https://cran.r-project.org/package=mice |
| ROSE (v0.0-4) | Lunardon et al. | https://cran.r-project.org/package=ROSE |
| caret (ranger) (v6.0-94) | Max Kuhn | https://cran.r-project.org/package=caret |
| Boruta (v8.0.0) | Kursa et al.57 | https://cran.r-project.org/package=Boruta |
| R (v4.2.3) | Ross Ihaka and Robert Gentleman | https://www.r-project.org/ |
Experimental model and study participant details
The present study investigates preoperative samples from a recently reported study that demonstrated how postoperative transcriptomic changes could identify patients developing infection with or without sepsis up to three days before clinical manifestations become apparent.23 Ethical approval was granted through the Southampton and South-West Hampshire Multicentre Research Ethics Committee (ref. 06/Q1702/152) and the protocol gained US Federal Wide Assurance Independent Review Board status (IRB00001756). Patients were eligible for inclusion in the study if scheduled for “high-risk” elective surgery including extended abdominal, cardiac, gynae-urological, thoracic and vascular operations and if they gave informed consent. Preoperative blood samples from these patients were collected and RNA sequencing performed to determine whether transcriptomic signatures in these pre-surgery samples could predict postoperative outcomes, including uncomplicated infection and sepsis. Of the originally acquired 4,385 patient samples in the biobank we retrieved 267 preoperative samples of whom 126 came from patients who suffered postoperative uncomplicated infection (n = 49, “UInf+”) or sepsis (n = 77) (Figures S1A and S1B). Sepsis was identified using the Sepsis-3 criteria.1 An adjudication panel determined a high or definite likelihood of infection or sepsis from clinical and laboratory data.23 Preoperative samples were also taken from 141 patients determined as having a non-infectious post-operative course. This group was subdivided into those developing a non-infectious systemic inflammatory response (n = 31, “SIRS+”) and patients who made an uncomplicated postoperative recovery (n = 110, “SIRS-”). Patient metadata including age, gender, cause of operation and affected tissue are included in Table S1.
Method details
RNA extraction
Total RNA was extracted from whole blood preoperative samples and preserved with RNAlater (ThermoFisher). As per manufacturer’s guidelines 4 mL of vacutainer blood was added to 10.5 mL RNALater. 1.8 mL of RNAlater-preserved blood was used for manual RNA extraction using the RiboPure-Blood Kit (Thermofisher) and following the manufacturer’s instructions.
RNA-seq QC, library preparation and sequencing
RNA quantity and quality were assessed using a NanoDrop Spectrophotometer and Agilent Bioanalyzer (2100). After quality control, mRNA from organisms was enriched using oligo(dT) beads. rRNA and globin were removed using the TruSeq Stranded Total RNA with Ribo-Zero Globin kit (Illumina). Library preparation and Illumina sequencing were performed by Novogene. mRNA was randomly fragmented by adding fragmentation buffer. The cDNA was then synthesized using mRNA template and a random hexamers primer, followed by a custom second-strand synthesis buffer (Illumina), dNTPs(dUTP, dATP, dGTP and dCTP), RNase H and DNA polymerase I to initiate the second-strand synthesis. Purification followed by AMPure XP beads, terminal repair, polyadenylation, sequencing adapter ligation, size selection and degradation of second-strand U-contained cDNA by the USER enzyme. The strand-specific cDNA library was generated after final PCR enrichment. QC of the constructed library included testing library concentration preliminary with Qubit 2.0 Fluorometer (ThermoFisher), insert size with BioAnalyzer 2100 (Agilent) and qPCR to quantify effective library concentration. Qualified QC checked libraries were loaded onto a NovaSeq 6000 S4 (Illumina) for sequencing using paired end mode with 150 bp read length (2 × 150 cycles).
All preoperative samples were deposited in the GEO database with GEO-ID GSE208581 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208581).
RT-qPCR RNA QC, preprocessing and quantification
RNA transcripts to quantify with RT-qPCR were identified from all investigated classification models (n = 72, Tables S5 and S6). Extracted RNA was quantitated and quality-controlled on a QIAxpert instrument (Qiagen). An averaged concentration of RNA was calculated based on all measured samples. A fixed volume (2 μL, equal to ∼100 ng) was used for all samples as input for the subsequent cDNA synthesis, due to limited available RNA volume for some of the samples. cDNA was synthesized from 100 ng total RNA using the High-Capacity cDNA Reverse Transcription Kit (Thermofisher, cat. # 4368813) in a 20 μL reaction. Volumes of the different kit components in each reaction were: 10x RT Buffer: 2μL, 25X dNTP Mix (100 mM): 0.8μL, 10X RT Random Primers: 2 μL, MultiScribe Reverse Transcriptase: 1μL, Nuclease-Free H2O: 4.2μL, Total: 10 μL. 2 μL of total RNA and 8 μL of H2O was added to the 10 μL master-mix. An NTC (No Template Control) reaction was set up for which no RNA and 10 μL of H2O was added to the reaction mixture. A NEC (No Enzyme Control) reaction was set-up for which water was added instead of the MultiScribe Reverse Transcriptase enzyme. 2 μL of RNA was used as template for the reaction. The Standard curve was created based on a sample pool from a subset of the samples. 1000 ng of RNA from the pool was used as template in a cDNA synthesis reaction. The cycling conditions for cDNA included 25°C for 10 min, 27°C for 120 min, 85°C for 5s and 4°C otherwise. cDNA was stored at −20°C. Pre-amplification and specific target amplification (STA) was accomplished by combining a pool of each assay in a 1.5 mL tube and add 1 x TE buffer (29 samples for STA, 69 assays in in total). Assay pools were stored at −20°C. The STA mix was processed by using TaqMan PreAmp master Mix (2x) and a pooled assay mix (0.2x).
Samples were analyzed on a total number of 3 × 96.96 Fluidigm chips that were run on the Fluidigm BioMark HD instrument. The number of technical replicates was 1 for the samples and standard curve. The cycling protocol included 70 °C at 40min and 60°C for 30s for the thermal mix, 98°C 60 s for the hot start and 97°C 5 s (denaturation) and 60°C for 20 s (annealing) for the PCR cycle. The NTC and NEC samples were loaded on each of the three chips. Raw data from the Fluidigm BioMark were analyzed in the Fluidigm Real-Time PCR Analysis software v. 4.1.3 using the following settings (standard settings): Quality threshold: 0.65; Baseline correction: Linear (derivative); Ct Threshold method: User (detectors). The data from the software were then exported as Ct values to Excel spreadsheets. The assays for Otulin and RANBP3 were used to normalize the relative abundance of transcripts between samples. Missing data outside of detection limits were imputed using the R package mice (v3.16.0) with default settings.
COVID-19 sample collection
Blood was sampled from 61 patients with community-acquired pneumonia by SARS-CoV-2 (51 of those patients were classified as having sepsis because they met the Sepsis-3 criteria) of either gender within the first 24 h of hospital admission from three different study sites in Greece. RNA was preserved in RNAlater. This was a sub-study of the ESCAPE study58 which has been approved by the National Ethics Committee of Greece (approval 30/20) and by the National Organization for Medicines of Greece (approval IS 021-20). Sampling of 5 mL of whole blood was done before treatment with any biological disease-modifying drug or with corticosteroids (EudraCT number 2020-001039-29; Clinicaltrials.gov NCT04339712) and poured into PAXgene tubes. Methodological details are given in the supplementary material of the ESCAPE study publication.58
RNA sequencing was done following the protocol described above for preoperative samples and deposited in the GEO database with GEO-ID GSE208587 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208587).
RNA-seq preprocessing and analysis
RNA-seq raw data were processed following the GEO2RNA-Seq pipeline,50 an RNA-Seq pre-processing workflow and package for analyzing read files, trimming of raw reads, mapping on reference genomes, counting reads per gene and finding significant differentially expressed genes (DEGs). Quality of raw read data was checked using FastQC (v0.11.8). Reads were quality- and adapter-trimmed using Trimmomatic (v0.39). Reads were rRNA-filtered using SortMeRNA version (v2.1b) with a single rRNA database concatenated from all rRNA databases shipped with SortMeRNA. Reads were mapped against the human reference genome and corresponding annotation GRCh38.99 (obtained from www.ensembl.org) using HiSAT2 (v2.1.0). Differentially expressed genes (DEGs) were determined if significance (adjusted p ≤ 0.05) was reported by either DESeq2 (v1.26),51 or edgeR (v3.28.1)52 and a |log2(fold change)| ≥0.25. Clustering of significant DEGs was done by using K-means clustering. Heatmaps were created using the R package ComplexHeatmap (v2.4.3).53 Venn diagrams were produced using the R package VennDiagram (v1.6.20).54
Gene co-expression networks were constructed based on all significant DEGs between postoperative infection and non-infection outcome using all preoperative samples or only male or female specific preoperative samples were used. We computed Spearman correlation between all DEGs for each dataset including Sepsis, UInf+, SIRS+ and SIRS- again using all preoperative or male or female specific preoperative samples. Correlations between DEGs were included in the co-expression network if the correlation was significant (FDR ≤0.05) and high (absolute ρ ≥ 0.7). Gene co-expression networks were constructed by using weighted gene co-expression network analysis (WGCNA). GO terms for genes were obtained from Bioconductor package org.Hs.e.g.,.db (v3.11.4). Gene enrichment analysis was performed using the R package ClusterProfiler (v3.16.1).55 Networks were visualized using cytoscape (v3.7.0).56
Machine learning of classification models discriminating postoperative outcome
Classification was conducted for discriminating postoperative outcome based on preoperative samples, unless otherwise noted. Prior to classification, samples were balanced for gender if all samples were used, using down-sampling. Next, supervised random-forest using the Boruta wrapper method (up to 25 features allowed and otherwise default settings)57 to select the most relevant gene features was performed. As input for Boruta 1282 protein encoding DEGs out of a total of 1932 differentially expressed genes reported across pairwise comparisons of the infection and no infection groups with or without sepsis, UInf+, SIRS+ and SIRS- groups were used (Table S2). 10-fold cross-validation was repeated 100 times applying a random search for optimal hyperparameters. The union of all gene features from the 10 best iterations in terms of reported AUROC were determined. Based on the center-scaled protein-coding subset of these genes with non-zero variance across all considered samples recursive feature elimination was applied next to obtaining a minimum set of genes that supports AUROC≥0.8 using again random-forest based classification. Performance was identified by running 100 times 10-fold cross-validation. Mean classification performance was checked against classification using as small as possible set of genes (up to 20 features allowed) allowing for optimal AUROC values identified after 100 times repeated recursive feature elimination runs. With these, 10-fold cross-validation was repeated 100 times and mean statistical performance metrics including accuracy, AUROC, sensitivity, specificity, PPV and, NPV were tracked.
Quantification and statistical analysis
All statistical analyses were processed in the R programming environment (v4.0.5). Differentially expressed genes were identified by DESeq2 (v1.22.1),51 or edgeR (v3.24.3)52 as detailed out above. Significance for co-expression networks were calculated by one-tailed Fisher exact test and using higher expression or correlation differences between the indicated groups as input for the required 2 × 2 tables.
Published: September 3, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101712.
Supplemental information
References
- 1.Singer M., Deutschman C.S., Seymour C.W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G.R., Chiche J.D., Coopersmith C.M., et al. The third international consensus definitions for sepsis and septic shock (sepsis-3) JAMA, J. Am. Med. Assoc. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paoli C.J., Reynolds M.A., Sinha M., Gitlin M., Crouser E. Epidemiology and costs of sepsis in the United States-an analysis based on timing of diagnosis and severity level. Crit. Care Med. 2018;46:1889–1897. doi: 10.1097/CCM.0000000000003342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fleischmann-Struzek C., Mellhammar L., Rose N., Cassini A., Rudd K.E., Schlattmann P., Allegranzi B., Reinhart K. Incidence and mortality of hospital- and ICU-treated sepsis: results from an updated and expanded systematic review and meta-analysis. Intensive Care Med. 2020;46:1552–1562. doi: 10.1007/s00134-020-06151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.López-Mestanza C., Andaluz-Ojeda D., Gómez-López J.R., Bermejo-Martín J.F. Clinical factors influencing mortality risk in hospital-acquired sepsis. J. Hosp. Infect. 2018;98:194–201. doi: 10.1016/j.jhin.2017.08.022. [DOI] [PubMed] [Google Scholar]
- 5.Donnelly J.P., Lakkur S., Judd S.E., Levitan E.B., Griffin R., Howard G., Safford M.M., Wang H.E. Association of Neighborhood Socioeconomic Status With Risk of Infection and Sepsis. Clin. Infect. Dis. 2018;66:1940–1947. doi: 10.1093/CID/CIX1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angele M.K., Pratschke S., Hubbard W.J., Chaudry I.H. Gender differences in sepsis. Virulence. 2014;5:12–19. doi: 10.4161/viru.26982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sunden-Cullberg J., Nilsson A., Inghammar M. Sex-based differences in ED management of critically ill patients with sepsis: a nationwide cohort study. Intensive Care Med. 2020;46:727–736. doi: 10.1007/s00134-019-05910-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu H., Wen D., Wang X., Gan L., Du J., Sun J., Zeng L., Jiang J., Zhang A. Host genetic variants in sepsis risk: a field synopsis and meta-analysis. Crit. Care. 2019;23:26. doi: 10.1186/S13054-019-2313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Georgescu A.M., Banescu C., Azamfirei R., Hutanu A., Moldovan V., Badea I., Voidazan S., Dobreanu M., Chirtes I.R., Azamfirei L. Evaluation of TNF-α genetic polymorphisms as predictors for sepsis susceptibility and progression. BMC Infect. Dis. 2020;20:221. doi: 10.1186/S12879-020-4910-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Visser A., Geboers B., Gouma D.J., Goslings J.C., Ubbink D.T. Predictors of surgical complications: A systematic review. Surgery (United States) 2015;158:58–65. doi: 10.1016/j.surg.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Lin M.Y.C., Liu W.Y., Tolan A.M., Aboulian A., Petrie B.A., Stabile B.E. Preoperative serum albumin but not prealbumin is an excellent predictor of postoperative complications and mortality in patients with gastrointestinal cancer. Am. Surg. 2011;77:1286–1289. doi: 10.1177/000313481107701002. [DOI] [PubMed] [Google Scholar]
- 12.Haupt W., Hohenberger W., Mueller R., Klein P., Christou N.V. Association between preoperative acute phase response and postoperative complications. European Journal of Surgery, Acta Chirurgica. 1997;163:39–44. [PubMed] [Google Scholar]
- 13.Moyes L.H., Leitch E.F., McKee R.F., Anderson J.H., Horgan P.G., McMillan D.C. Preoperative systemic inflammation predicts postoperative infectious complications in patients undergoing curative resection for colorectal cancer. Br. J. Cancer. 2009;100:1236–1239. doi: 10.1038/sj.bjc.6604997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohri Y., Miki C., Kobayashi M., Okita Y., Inoue M., Uchida K., Tanaka K., Inoue Y., Kusunoki M. Correlation between preoperative systemic inflammation and postoperative infection in patients with gastrointestinal cancer: A multicenter study. Surg. Today. 2014;44:859–867. doi: 10.1007/s00595-013-0622-5. [DOI] [PubMed] [Google Scholar]
- 15.Fransen E.J., Maessen J.G., Elenbaas T.W., van Aarnhem E.E., Van Dieijen-Visser M.P. Enhanced preoperative C-reactive protein plasma levels as a risk factor for postoperative infections after cardiac surgery. Ann. Thorac. Surg. 1999;67:134–138. doi: 10.1016/S0003-4975(98)00973-4. [DOI] [PubMed] [Google Scholar]
- 16.Gustafsson U.O., Thorell A., Soop M., Ljungqvist O., Nygren J. Haemoglobin A1c as a predictor of postoperative hyperglycaemia and complications after major colorectal surgery. Br. J. Surg. 2009;96:1358–1364. doi: 10.1002/bjs.6724. [DOI] [PubMed] [Google Scholar]
- 17.Inaoka K., Kanda M., Uda H., Tanaka Y., Tanaka C., Kobayashi D., Takami H., Iwata N., Hayashi M., Niwa Y., et al. Clinical utility of the platelet-lymphocyte ratio as a predictor of postoperative complications after radical gastrectomy for clinical T2-4 gastric cancer. World J. Gastroenterol. 2017;23:2519–2526. doi: 10.3748/wjg.v23.i14.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okugawa Y., Toiyama Y., Yamamoto A., Shigemori T., Ide S., Kitajima T., Fujikawa H., Yasuda H., Hiro J., Yoshiyama S., et al. Lymphocyte-C-reactive Protein Ratio as Promising New Marker for Predicting Surgical and Oncological Outcomes in Colorectal Cancer. Ann. Surg. 2020;272:342–351. doi: 10.1097/SLA.0000000000003239. [DOI] [PubMed] [Google Scholar]
- 19.Mohri Y., Tanaka K., Toiyama Y., Ohi M., Yasuda H., Inoue Y., Kusunoki M. Impact of preoperative neutrophil to lymphocyte ratio and postoperative infectious complications on survival after curative gastrectomy for gastric cancer: A single institutional cohort study. Medicine (United States) 2016;95 doi: 10.1097/MD.0000000000003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schroth J., Weber V., Jones T.F., Del Arroyo A.G., Henson S.M., Ackland G.L. Preoperative lymphopaenia, mortality, and morbidity after elective surgery: systematic review and meta-analysis. Br. J. Anaesth. 2021;127:32–40. doi: 10.1016/j.bja.2021.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheadle W.G., Hershman M.J., Wellhausen S.R., Polk H.C. HLA-DR antigen expression on peripheral blood monocytes correlates with surgical infection. Am. J. Surg. 1991;161:639–645. doi: 10.1016/0002-9610(91)91247-G. [DOI] [PubMed] [Google Scholar]
- 22.Martin G.S., Mannino D.M., Moss M. The effect of age on the development and outcome of adult sepsis. Crit. Care Med. 2006;34:15–21. doi: 10.1097/01.ccm.0000194535.82812.ba. [DOI] [PubMed] [Google Scholar]
- 23.Lukaszewski R.A., Jones H.E., Gersuk V.H., Russell P., Simpson A., Brealey D., Walker J., Thomas M., Whitehouse T., Ostermann M., et al. Presymptomatic diagnosis of postoperative infection and sepsis using gene expression signatures. Intensive Care Med. 2022;48:1133–1143. doi: 10.1007/s00134-022-06769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Dam S., Võsa U., van der Graaf A., Franke L., de Magalhães J.P. Gene co-expression analysis for functional classification and gene–disease predictions. Brief. Bioinform. 2018;19:575–592. doi: 10.1093/BIB/BBW139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y., Han L., Yuan Y., Li J., Hei N., Liang H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014;5:3231. doi: 10.1038/ncomms4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ulas T., Pirr S., Fehlhaber B., Bickes M.S., Loof T.G., Vogl T., Mellinger L., Heinemann A.S., Burgmann J., Schöning J., et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat. Immunol. 2017;18:622–632. doi: 10.1038/ni.3745. [DOI] [PubMed] [Google Scholar]
- 27.Popoff M.R. Bacterial factors exploit eukaryotic Rho GTPase signaling cascades to promote invasion and proliferation within their host. Small GTPases. 2014;5 doi: 10.4161/sgtp.28209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tilley A.E., Walters M.S., Shaykhiev R., Crystal R.G. Cilia dysfunction in lung disease. Annu. Rev. Physiol. 2015;77:379–406. doi: 10.1146/annurev-physiol-021014-071931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barcella M., Bollen Pinto B., Braga D., D’Avila F., Tagliaferri F., Cazalis M.A., Monneret G., Herpain A., Bendjelid K., Barlassina C. Identification of a transcriptome profile associated with improvement of organ function in septic shock patients after early supportive therapy. Crit. Care. 2018;22:312–313. doi: 10.1186/S13054-018-2242-3/FIGURES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almansa R., Heredia-Rodríguez M., Gomez-Sanchez E., Andaluz-Ojeda D., Iglesias V., Rico L., Ortega A., Gomez-Pesquera E., Liu P., Aragón M., et al. Transcriptomic correlates of organ failure extent in sepsis. J. Infect. 2015;70:445–456. doi: 10.1016/j.jinf.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Giamarellos-Bourboulis E.J., Apostolidou E., Lada M., Perdios I., Gatselis N.K., Tsangaris I., Georgitsi M., Bristianou M., Kanni T., Sereti K., et al. Kinetics of circulating immunoglobulin M in sepsis: Relationship with final outcome. Crit. Care. 2013;17 doi: 10.1186/cc13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sweeney T.E., Perumal T.M., Henao R., Nichols M., Howrylak J.A., Choi A.M., Bermejo-Martin J.F., Almansa R., Tamayo E., Davenport E.E., et al. A community approach to mortality prediction in sepsis via gene expression analysis. Nat. Commun. 2018;9:694. doi: 10.1038/s41467-018-03078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsalik E.L., Langley R.J., Dinwiddie D.L., Miller N.A., Yoo B., van Velkinburgh J.C., Smith L.D., Thiffault I., Jaehne A.K., Valente A.M., et al. An integrated transcriptome and expressed variant analysis of sepsis survival and death. Genome Med. 2014;6:111. doi: 10.1186/s13073-014-0111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reyes M., Filbin M.R., Bhattacharyya R.P., Billman K., Eisenhaure T., Hung D.T., Levy B.D., Baron R.M., Blainey P.C., Goldberg M.B., Hacohen N. An immune-cell signature of bacterial sepsis. Nat. Med. 2020;26:333–340. doi: 10.1038/s41591-020-0752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nader D., Curley G.F., Kerrigan S.W. A new perspective in sepsis treatment: could RGD-dependent integrins be novel targets? Drug Discov. Today. 2020;25:2317–2325. doi: 10.1016/J.DRUDIS.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samson M., Libert F., Doranz B.J., Rucker J., Liesnard C., Farber C.-M., Saragosti S., Lapoumeroulie C., Cognaux J., Forceille C., et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 37.Proudfoot A.E.I. Chemokine receptors: multifaceted therapeutic targets. Nat. Rev. Immunol. 2002;2:106–115. doi: 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Netti G.S., Sangregorio F., Spadaccino F., Staffieri F., Crovace A., Infante B., Maiorano A., Godeas G., Castellano G., Di Palma A.M., et al. LPS removal reduces CD80-mediated albuminuria in critically ill patients with Gram-negative sepsis. Am. J. Physiol. Renal Physiol. 2019;316:F723–F731. doi: 10.1152/AJPRENAL.00491.2018. [DOI] [PubMed] [Google Scholar]
- 39.Miyake H., Tanabe K., Tanimura S., Nakashima Y., Morioka T., Masuda K., Sugiyama H., Sato Y., Wada J. Genetic Deletion of Vasohibin-2 Exacerbates Ischemia-Reperfusion-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2020;21:4545. doi: 10.3390/IJMS21124545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fratazzi C., Manjunath N., Arbeit R.D., Carini C., Gerken T.A., Ardman B., Remold-O’Donnell E., Remold H.G. A macrophage invasion mechanism for mycobacteria implicating the extracellular domain of CD43. J. Exp. Med. 2000;192:183–192. doi: 10.1084/jem.192.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voskoboinik I., Whisstock J.C., Trapani J.A. Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 2015;15:388–400. doi: 10.1038/nri3839. [DOI] [PubMed] [Google Scholar]
- 42.Snyder A., Jedreski K., Fitch J., Wijeratne S., Wetzel A., Hensley J., Flowers M., Bline K., Hall M.W., Muszynski J.A. Transcriptomic Profiles in Children With Septic Shock With or Without Immunoparalysis. Front. Immunol. 2021;12 doi: 10.3389/FIMMU.2021.733834/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Houseright R.A., Miskolci V., Mulvaney O., Bortnov V., Mosher D.F., Rindy J., Bennin D.A., Huttenlocher A. Myeloid-derived growth factor regulates neutrophil motility in interstitial tissue damage. J. Cell Biol. 2021;220 doi: 10.1083/JCB.202103054/212198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burnham K.L., Davenport E.E., Radhakrishnan J., Humburg P., Gordon A.C., Hutton P., Svoren-Jabalera E., Garrard C., Hill A.V.S., Hinds C.J., Knight J.C. Shared and Distinct Aspects of the Sepsis Transcriptomic Response to Fecal Peritonitis and Pneumonia. Am. J. Respir. Crit. Care Med. 2017;196:328–339. doi: 10.1164/rccm.201608-1685OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsalik E.L., Henao R., Nichols M., Burke T., Ko E.R., McClain M.T., Hudson L.L., Mazur A., Freeman D.H., Veldman T., et al. Host gene expression classifiers diagnose acute respiratory illness etiology. Sci. Transl. Med. 2016;8:322ra11. doi: 10.1126/SCITRANSLMED.AAD6873/SUPPL_FILE/8-322RA11_SM.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalantar K.L., Neyton L., Abdelghany M., Mick E., Jauregui A., Caldera S., Serpa P.H., Ghale R., Albright J., Sarma A., et al. Integrated host-microbe plasma metagenomics for sepsis diagnosis in a prospective cohort of critically ill adults. Nat. Microbiol. 2022;7:1805–1816. doi: 10.1038/s41564-022-01237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Magouras I., Brookes V.J., Jori F., Martin A., Pfeiffer D.U., Dürr S. Emerging Zoonotic Diseases: Should We Rethink the Animal–Human Interface? Front. Vet. Sci. 2020;7 doi: 10.3389/fvets.2020.582743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White R.J., Razgour O. Emerging zoonotic diseases originating in mammals: a systematic review of effects of anthropogenic land-use change. Mamm Rev. 2020;50:336–352. doi: 10.1111/mam.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabozzi G., Bonizzi L., Crespi E., Somaruga C., Sokooti M., Tabibi R., Vellere F., Brambilla G., Colosio C. Emerging zoonoses: The “one health approach.”. Saf. Health Work. 2012;3:77–83. doi: 10.5491/SHAW.2012.3.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seelbinder B., Wolf T., Priebe S., McNamara S., Gerber S., Guthke R., Linde J. GEO2RNAseq: An easy-to-use R pipeline for complete pre-processing of RNA-seq data. bioRxiv. 2019 doi: 10.1101/771063. [DOI] [Google Scholar]
- 51.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 54.Chen H., Boutros P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinf. 2011;12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu G., Wang L.G., Han Y., He Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/GR.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kursa M.B., Rudnicki W.R. Feature selection with the boruta package. J. Stat. Softw. 2010;36:1–13. doi: 10.18637/jss.v036.i11. [DOI] [Google Scholar]
- 58.Karakike E., Dalekos G.N., Koutsodimitropoulos I., Saridaki M., Pourzitaki C., Papathanakos G., Kotsaki A., Chalvatzis S., Dimakopoulou V., Vechlidis N., et al. ESCAPE: An Open-Label Trial of Personalized Immunotherapy in Critically lll COVID-19 Patients. J. Innate Immun. 2022;14:218–228. doi: 10.1159/000519090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Expression data of all preoperative patient samples were deposited in the GEO database with GEO-ID GSE208581 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208581). RNA sequencing data of COVID-19 patient samples were deposited in the GEO database with GEO-ID GSE208587 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208587). The RT-qPCR data generated in this study are included in Table S6 and are published together with this article.
-
•
The original codes used to analyze the presented data and generate machine learning models are available at zenodo (https://doi.org/10.5281/zenodo.10835070) and GitHub (https://github.com/thisisalbert/preoperative_sepsis_risk/).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.