

Summary
Fibrolamellar carcinoma (FLC) is a rare, lethal, early-onset liver cancer with a critical need for new therapeutics. The primary driver in FLC is the fusion oncoprotein, DNAJ-PKAc, which remains challenging to target therapeutically. It is critical, therefore, to expand understanding of the FLC molecular landscape to identify druggable pathways/targets. Here, we perform the most comprehensive integrative proteo-metabolomic analysis of FLC. We also conduct nutrient manipulation, respirometry analyses, as well as key loss-of-function assays in FLC tumor tissue slices from patients. We propose a model of cellular energetics in FLC pointing to proline anabolism being mediated by ornithine aminotransferase hyperactivity and ornithine transcarbamylase hypoactivity with serine and glutamine catabolism fueling the process. We highlight FLC’s potential dependency on voltage-dependent anion channel (VDAC), a mitochondrial gatekeeper for anions including pyruvate. The metabolic rewiring in FLC that we propose in our model, with an emphasis on mitochondria, can be exploited for therapeutic vulnerabilities.
Keywords: fibrolamellar carcinoma, proteomics, metabolomics, mitochondria, glucose, glutamine, serine, pyruvate, alpha-ketoglutarate, proline
Graphical abstract
Highlights
-
•
Proteo-metabolomics predicts SDS dependency that is validated in patient tumor slices
-
•
OAT, GLS, and ALDH18A1 form an axis central to mitochondrial rewiring in FLC
-
•
Profiling and functional studies show a lack of dependency on glycolysis
-
•
Erastin administration in tumor tissue slices demonstrates potential dependency on VDAC
Fibrolamellar carcinoma (FLC) is a rare but lethal liver cancer lacking effective therapeutics. Long, Jr. et al. provide the most comprehensive map to date of rewired metabolism in FLC. The study leverages multi-omics, nutrient manipulation, respirometry analyses, and functional studies in patient tumor tissue slices. The results reveal potential therapeutic vulnerabilities.
Introduction
Fibrolamellar carcinoma (FLC) is a devastating, early-onset, rare cancer of the liver with no standard of care.1,2,3 Surgery is the only intervention with curative intent, but many patients experience recurrence or are not eligible given the advanced stage of disease by the time of diagnosis. For patients whose tumors are unresectable, there is no standard systemic therapy, and the average survival period is 12 months.4 There is a dire need for deeper understanding of the disease toward new therapeutic options.
The most recurrent genetic lesion in FLC is a somatic, heterozygous ∼400 kb deletion on chromosome 19, which leads to an in-frame fusion of exon 1 of DNAJB1 with exons 2–12 of PRKACA.5,6 The translation of the resulting transcript leads to the chimeric protein, DNAJ-PKAc (DP). At least two studies have demonstrated that DP is sufficient to initiate tumors in mice,7,8 and even more recent work has shown that it may play a role in tumor maintenance as well.9 Although there is widespread interest in targeting DP, clinical inhibitors are not yet available. It also remains a challenge to inhibit DP without also affecting wild-type protein kinase A (PKA), which is critical for normal physiology.10,11 Therefore, it is important to investigate downstream mediators of FLC pathogenesis, as it will deepen mechanistic understanding of FLC and potentially unveil additional therapeutic targets.
Some mechanisms by which DP transforms cells and re-wires their behavior have been elucidated through a growing number of cell signaling and molecular profiling experiments.1 Genome-scale studies have brought to light potential therapeutic vulnerabilities. For example, one of our earlier studies defined FLC-specific super enhancers, which may be critical for understanding cellular rewiring.12 Other studies have identified highly dysregulated microRNAs, including those that have emerged as compelling targets in pre-clinical studies of other aggressive cancers.1 Despite these important advances, all of the “omics” landscaping has been largely focused on chromatin/transcription and gene/microRNA expression.
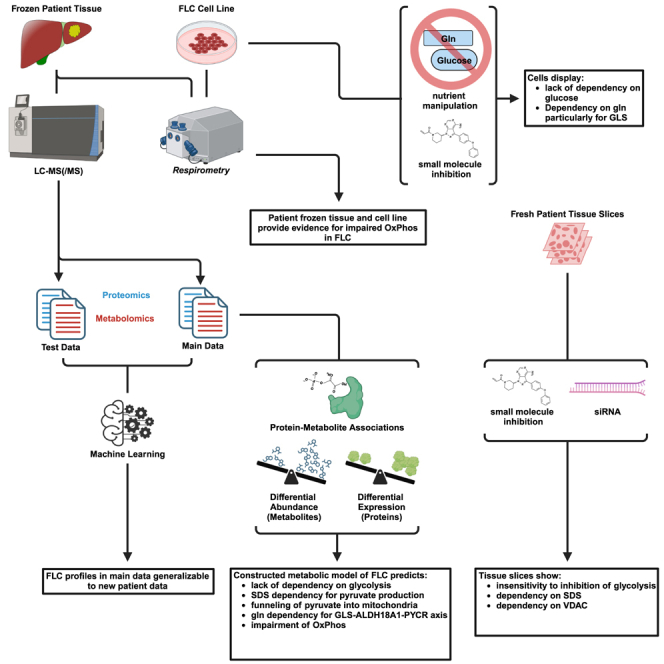
Only one prior study13 aimed to characterize the proteome and metabolome of FLC (proteomics—n = 16 patient samples; n = 4,620 proteins; metabolomics—n = 10 patient samples). Here, we have carried out the most extensive integrative proteo-metabolomic analysis of FLC (proteomics—n = 23 patient samples; n = 8,485 proteins; metabolomics—n = 26 patient samples; n = 135 metabolites). We included in the analysis previously generated transcriptomic data and performed a suite of complementary experiments and assessments including (1) respirometry on frozen, matched FLC tumors vs. non-malignant adjacent liver (NML) patient tissue, (2) nutrient manipulation and seahorse assays on a cell-based model of FLC, (3) manual curation of relevant clinical data pertaining to serum, plasma, and urine specimens, and (4) key loss-of-function experiments in FLC patient tumor slices to validate several critical components of our model.
Results
Data quality
A schematic of the proteo-metabolomics experimental design is shown in Figure S1. After applying quality control filters to the data (see STAR Methods), twenty-three (proteomics) and twenty-six samples (metabolomics) remained, of which seventeen were omics matched (present in both the proteomics and metabolomics data). Principal-component analysis (PCA) and hierarchal clustering were performed on samples with proteomic and metabolomic data (Data S1and S2). The sufficiency of our sample size was assessed by constructing an extreme gradient boost machine learning (XgBoostML) pipeline with nested cross-validation (CV) (Figures S2A and S2B; see STAR Methods) that needed to fulfill at least one of two criteria: (1) the omics-based stratification of patient cohort samples (nproteomics = 23 samples and nmetabolomics = 26 samples; used throughout this study) is comparable to that of physician diagnoses and/or (2) the omics-derived model is generalizable to a new, independent cohort of patient samples. The pipeline fulfilled both criteria for proteomics and one of the criteria for metabolomics (Figures S2C–S2F; see STAR Methods for more detail).
Amino acid transport may be favored in FLC and diverted into protein synthesis
We first assessed proteins and metabolites that comprise plasma membrane (PM) transport and metabolism of key nutrients. For glucose exchange, we focused on the SLC2A (solute carrier 2A) family given that this class of transporters specializes in the transport of monosaccharides.14,15,16,17 Seven out of the thirteen members were identified across both our proteomics and previously published transcriptomics datasets. Most notably, SLC2A2, the major glucose transporter of the liver,18,19,20 was significantly downregulated at both the protein (<1st percentile) and transcript (5th percentile) level in FLC compared to NML (FvsN), with no concomitant upregulation of any other SLC2A member (Figures 1A and S3A). Moreover, glucose was significantly depleted in FvsN (Figures 1A, S3B, and S3C).
Figure 1.

Amino acid transport may be favored in FLC and diverted into translation
(A) Descriptive model embedded with omics data depicting transport of sugars, amino acids, and fatty acids in FLC. Proteomics data—n = 23 samples; metabolomics data—n = 26 samples.
(B) Descriptive model depicting the potential relationship between amino acid transport and protein translation in FLC. (A and B) Left side of each node—gradient red is increasing log2FC (FLC vs. NML) and gradient blue is decreasing log2FC; Right side of each node—gray is not significant and yellow is significant (proteins—FDR-adj p < 0.05; metabolites—FDR-adj p < 0.1); nodes associated with edges that have open arrowheads, proteins; double-line edges, transport; nodes associated with double-line edges, metabolites; SLC, solute carrier; CEAAs, conditionally essential amino acids; EAAs, essential amino acids; NEAAs, non-essential amino acids; BCAAs, branched-chain amino acids; ECM, extracellular matrix; green traffic light, stimulation.
Amino acid (AA) transporters come from a diverse range of SLC families. We identified twelve transporters that are well known for the exchange of AAs across the PM (curated from GeneCards and Protein Atlas). Notably, six of the twelve AA transporters were significantly upregulated at the protein level (Figures 1A, 1B, and S3A). Interestingly, only four out of the nineteen AAs identified in our metabolomics analysis were significantly altered in abundance (Figures 1A, S3B, and S3C). Glutamine was the only significantly depleted AA in FLC, while proline, leucine, and valine were the 2nd, 3rd, and 7th most enriched metabolites, respectively (Figure S3B).
Enhancement of pathways for transcription and translation of mitochondrial genes
There are two main biological processes that encompass AA utilization: metabolism and translation. Regarding the latter, histological observations have shown that FLC is rich in mitochondria that are abundant in proteins.21,22,23,24,25 Therefore, we investigated whether protein synthesis is skewed toward translation of mitochondrial proteins. Indeed, the subcellular compartment with the highest increase in the median log2FC (FvsN) of its proteins compared to the median log2FC of all proteins (background set, n = 8,040) was the mitochondrion (Figure 2A). Moreover, the median log2FC of genes involved in mitochondrial translation (n = 143) as well as transcription (n = 70), but not cytoplasmic translation (n = 164) or transcription (n = 685), was significantly increased compared to the median log2FC of all genes related to the mitochondria (background set, n = 939) at the protein level (Figures 2B and 2C).
Figure 2.

Enhanced transcription and translation of mitochondrial genes
(A) Dot plot where each dot represents the difference (x axis) between the median log2FC (FLC vs. NML) of the background set and a gene set that represents a subcellular compartment (y axis). Error bars represent 95% confidence intervals.
(B–D) Comparison of log2FCs (FLC vs. NML; z-scaled) at the protein (x axis) and RNA (y axis) level. Density plots represent the distribution of the log2FCs associated with proteins (x axis) and transcripts (y axis). (E) Descriptive model of amino acid utilization in FLC. Gene sets were identified as members of (A) subcellular compartments, (B) the mitochondrial central dogma, (C) the cytoplasmic central dogma, and (D) amino acid metabolism. Background sets (bkgd) are (A and D) all genes identified in both the transcriptomic and proteomic datasets, (B) all genes related to the mitochondrion, and (C) all genes not related to the mitochondrion. (B–D) Lines associated with density plots represent the median. (A–C) MitoCarta3.0 and (A, C, and D) Protein Atlas were used to identify gene sets. (A–D) Gene sets (transcriptomic—n = 27 samples, proteomics—n = 23 samples) were compared to bkgd using Mann-Whitney (∗FDR-adj p < 0.05; ∗∗FDR-adj p < 0.01; ∗∗∗FDR-adj p < 0.001; ∗∗∗∗FDR-adj p < 0.0001). (B–D) For gene sets (excluding bkgd)—circles, genes with no significant log2FCs at either RNA or protein level; diamonds, genes with significant log2FCs at both the RNA and protein level; triangles, genes with significant log2FCs at the protein level only; squares, genes with significant log2FCs at the RNA level only; shape size, average expression values (from transcriptomic data). (A–D) Cyto, cytosol; Mito, mitochondria; DNARep&Repair, DNA replication and repair; BCAA, branched-chain amino acids; ER, endoplasmic reticulum; Lyso, lysosome; NucMem, nuclear membrane; Nucplasm, nucleoplasm; PM, plasma membrane; Perox, peroxisome.
While a few AA metabolic enzymes were upregulated in FLC, the median log2FC of genes involved in six out of nine AA metabolic pathways was significantly lower compared to the median log2FC of all genes (background set, n = 8,040) at the protein level (Figure 2D). Taken together, these results suggest that, while catabolism of some AAs may be of importance in FLC, there are likely additional purposes for the presumed elevated AA transport, including but not limited to mitochondrial translation (Figure 2E).
Suggestive enhancement of TCA cycle paired with dysfunctional ETC in FLC
We reasoned that rewiring of the network of small-molecule transporters within the mitochondrial membrane would be needed to satisfy the presumed heavy demand of AAs by the mitochondria in FLC to fuel mitochondrial translation. Therefore, we conducted a concordance analysis in FLC to assess the relationship between the abundance of small-molecule transporters (proteomics) and small molecules (metabolomics) partitioned into four groups: fatty acid (FA)-carnitine conjugates (Figure 3A), nucleotides (Figure 3B), AAs (Figure 3C), and tricarboxylic acid (TCA) cycle intermediates (Figure 3D). For each group, the number of significant metabolite-protein associations was used as a proxy for the degree of rewiring. There were no significant metabolite-protein associations for FA-carnitine conjugates. The average number of significant metabolite-protein associations per metabolite was relatively the same for nucleotides and AAs (∼2.17 and ∼1.72, respectively), while it was ∼2-fold higher for TCA cycle intermediates (∼4.5).
Figure 3.

Proteo-transcriptomic signature of mitochondrial respiration suggests an enhanced TCA cycle
(A–F) Concordance analysis in FLC patient tissue (n = 17 omics-matched sample pairs, 13 of which are FLC) between protein expression of (A–D) mitochondrial small-molecule transporters (data points) and metabolites (x axis labels) composed of (A) FA/carnitine conjugates, (B) nucleotides, (C) amino acids, and (D) TCA cycle intermediates. (A–D) Highlighted data points denote protein-metabolite pairs that are significantly (−log10(FDR-adj p) > 1.3) concordant (red) or discordant (blue) in FLC. Arrows adjacent to labels denote direction of significant alterations in the abundance of metabolites (x axis labels; FDR-adj p < 0.1) in FLC compared to NML. Gray data points indicate protein-metabolite pairs that were not statistically significant in concordance or discordance. Level of agreement between mitochondrial content (measured by Mitochondria Deep Tracker Red Fluorescent Intensity—MTDR F.I.) and protein abundance of all genes related to the mitochondria in (E) FLC (nmatchedpairs = 3; nrandompairs = 3) and (F) NML (nmatchedpairs = 4; nrandompairs = 2). Bars represent the top twenty mitochondrial pathways (x axis) with the highest percentage (y axis) of protein members whose abundance (proteomics) was significantly associated with mitochondrial content in (E) FLC and (F) NML. (E and F) Colors correspond to percentage of protein members that are significantly upregulated (sig_up, red), significantly downregulated (sig_down, blue), or unchanged (neutral, green) in FLC compared to NML. Protein_Diff_Express_Profile, protein differential expression profile; sig_up, significantly up; sig_down, significantly down; Carb, carbohydrate; BCAA, branched-chain amino acids; AA, amino acids; metab, metabolism; Cholest, cholesterol. (E and F) MitoCarta3.0 was used to identify gene sets.
Given the increased abundance of mitochondria in FvsN along with the high number of significant metabolite-protein associations between TCA cycle intermediates and small-molecule transporters, we next sought to determine if there was a strong relationship between mitochondrial abundance and TCA cycle enzymes. We directly measured mitochondrial abundance via MitoTracker Deep Red (MTDR) in FLC as well as NML patient tissue samples and assessed the degree of association with protein abundance (proteomics). Indeed, the TCA cycle contained the highest proportion of proteins that were significantly associated with mitochondrial abundance in FLC (Figure 3E). Moreover, pathways pertaining to calcium signaling and homeostasis contained the 3rd and 10th highest, respectively (Figure 3E). It is well documented that calcium modulates TCA cycle flux through regulating the activity of several TCA cycle enzymes.26 In NML, none of the three aforementioned mitochondrial pathways were among the top twenty in terms of proportion of proteins that were significantly associated with mitochondrial abundance (Figure 3F).
With the implications of an enhanced TCA cycle in FLC, we assessed if this was reflected in oxidative phosphorylation (OXPHOS)-related protein expression and activity. We detected twelve of the thirteen genes on the mitochondrial genome that encode proteins (all OXPHOS) in our proteomics dataset. Although the median log2FC of genes involved in mitochondrial translation (n = 143) showed a significant increase compared to the median log2FC of all genes localized to the mitochondria (background set, n = 939), all but one of the OXPHOS-related genes expressed and translated within the mitochondria were not significantly altered at either the protein or RNA level (Figure S4A). Additionally, there was no significant shift in the median log2FC of all genes (nuclear and mitochondrial) involved in the ETC (n = 147) compared with the median log2FC of all mitochondrial-related genes (background set, n = 939) at the protein level (Figure S4A). Intriguingly, the median log2FC of ETC assembly factors (n = 55; genes needed for the maturation, function, and stability of ETC complexes)27 displayed a significant increase compared to all mitochondrial-related genes (Figure S4A). Respirometry assays on frozen patient tissue (n = 6) revealed no significant difference in maximal respiratory capacity (MRC) between FLC and NML for all complexes that were assessed, although there was a trend toward decreased MRC in FvsN (I, II, and IV; Figure S4B). Furthermore, assessment of mitochondrial respiration in a xenograft-derived FLC cell line (n = 3 trials) showed no significant differences between basal respiration and MRC (Figure S4C), which implies low spare respiratory capacity (SRC).28 Low SRC is an indicator of high mitochondrial respiratory activity but mitigated mitochondrial fitness pertaining to aerobic respiration. Lastly, ATP was significantly depleted in FvsN although there was no significant perturbation in ATP/ADP/AMP ratios (Figure S4D). Taken together, these results suggest that high abundance of mitochondria in FLC is paralleled with high instability and turnover of ETC complexes (Figure S4E).
Glycolysis may be partially dispensable in FLC
Given the suggestive enhancement of the TCA cycle in FLC despite a dysfunctional OXPHOS profile, we investigated if glucose was used as a primary fuel. The median log2FC of genes involved in glucose production and storage (n = 45) was notably decreased compared to the median log2FC of all genes (background set, n = 8,040) at both the protein and RNA level (Figure 4A). Notably, though, the median log2FC of glycolytic genes (n = 97) also was markedly reduced compared to the median log2FC of all genes (background set, n = 8,040) at the protein level, but not at the RNA level (Figure 4A). Moreover, the significant depletion of glucose was consistent with the significant downregulation of the major glucose transporter in the liver, SLC2A2 (Figure 1). We performed a closer inspection of the glycolytic genes, separating those that are liver specific from those that are muscle specific. The median log2FC of muscle-specific glycolytic genes (n = 19) was increased compared to the median log2FC of all glycolytic genes (background set, n = 91) at the protein and RNA levels (Figure 4B). Conversely, the median log2FC of liver-specific genes (n = 26) was significantly decreased compared to the median log2FC of all glycolytic genes (background set, n = 91) at both the protein and RNA levels (Figure 4B). This preference for muscle-specific enzymes in FLC tumors, which we termed “glycolytic switch,” was not detected in a global analysis of all proteins, indicating specificity of this result to the glycolytic pathway (Figure 4C).
Figure 4.

Suggestive decoupling of glycolysis from pyruvate production
(A–C) Comparison of log2FCs (FLC vs. NML; z-scaled) at the protein (n = 23 samples) and RNA (n = 27 samples) level. Gene sets were identified that (a) participate in glycolysis as well as glucose production/storage and have (B and C) tissue specificity to liver or muscle. Background set (bkgd) is (B) all genes that participate in glycolysis and (A and C) all genes identified in both omics’ datasets. (A–C) Protein Atlas database was used to identify gene sets. Six-day glucose titration assay on FLC cell line measuring (D) ATP abundance (n = 8 samples for Ctrl 2 and zero concentration; n = 4 samples each for remaining concentrations) and (E) cell death (n = 4 samples per concentration).
(D–G) High, Normal, and Low indicate glucose concentrations above, within, and below normal physiological range, respectively. Inhibition of glycolysis via 2-deoxyglucose (2-DG) on fresh tissue slices from (F) patients afflicted with FLC (n = 4 biological replicates) or other cancers and (G) NOD scid gamma (NSG) mouse flank tumors seeded by either HepG2 or FLC primary cells. (F) BioRep, biological replicate; DMSO, control condition; CRLM, colorectal liver metastases; EHE, epithelioid hemangioendothelioma; HCC, hepatocellular carcinoma. (D–F) RQV, relative quantitative value. Mann-Whitney used for statistical assessment of (A–C) (∗FDR-adj p < 0.05; ∗∗FDR-adj p < 0.01; ∗∗∗FDR-adj p < 0.001; ∗∗∗∗FDR-adj p < 0.0001). (A–C) For gene sets (excluding bkgd), circles, genes with no significant log2FCs at either RNA or protein level; diamonds, genes with significant log2FCs at both the RNA and protein level; triangles, genes with significant log2FCs at the protein level only; squares, genes with significant log2FCs at the RNA level only; shape size, average expression values (from transcriptomic data).
We performed a six-day glucose titration assay on a xenograft-derived FLC cell line,29 and we did not observe any negative impact on ATP levels (a proxy for cell health and viability) or cell death at concentrations of glucose as low as 0.679 mM (well below the normal physiological range; Figures 4D and 4E). Even in the complete absence of glucose, cell viability was only impacted by approximately 40% to 50%. Moreover, inhibition of glycolysis via 2-deoxyglucose (2-DG) at 2.5 μM in fresh tissue slices from the liver tumors of FLC patients had no impact on cell viability (Figure 4F). This contrasts with the same dose in fresh tissue slices from tumors of other cancers known to have moderate to high glycolytic capacity (Figure 4F). The contrast remained evident when comparing the impact of 2-DG administration up to 5 mM (over three orders of magnitude higher than the previous dose) in fresh tissue slices derived from NOD scid gamma (NSG) mouse flank tumors seeded by either HepG2 or FLC primary cells (Figure 4G). While the glycolytic switch in the liver tumor to muscle-specific enzymes may be an adaptation to efficiently scavenge glucose, it appears to be dispensable as illustrated by the lack of an effect of 2-DG on the viability of fresh tissue slices derived from the tumors of patients or mouse models.
Serine may be a major contributor to pyruvate generation for the TCA cycle
As previously mentioned, the significant depletion of glucose was consistent with the significant downregulation of the major glucose transporter in the liver, SLC2A2 (Figure 1; Figure 5A). In sharp contrast, pyruvate and lactate were significantly elevated (Figure 5A), suggesting that there may be sources other than glucose for the buildup of glycolytic end products.
Figure 5.

Serine may be a major contributor to pyruvate generation for the TCA cycle
(A) Line plot of metabolomic data mapping metabolites along the trajectory of glycolysis in FLC (solid line; n = 20 samples) compared to NML (dashed line; n = 6 samples). Colored labels denote metabolites that are significantly depleted (blue) or enriched (red) in FLC. Data points and error bars represent mean abundance and standard deviation, respectively; ∗ FDR-adj p value < 0.1.
(B and C) Schematic that depicts inputs to pyruvate production. Excluding gray arrows, arrow size is directly associated with arrow color. Node size is arbitrary. Question marks refer to (C) genes that are suspected to be involved in contributing to the pyruvate pool. Background (bkgd; gray data points) is all genes identified in both omics’ datasets (transcriptomics—n = 27 samples; proteomics—n = 23 samples).
(D) Protein abundance profile of suspected contributors to pyruvate pool in FLC (n = 16 samples) compared to NML (n = 7 samples).
(E) Knockdown of SDS in fresh tissue slices derived from non-malignant liver tissue (n = 2 biological replicates) and tumors of patients afflicted with FLC (n = 2 biological replicates) or HCC (n = 1 biological replicate). HCC, hepatocellular carcinoma; BioRep, biological replicate. (C) For the highlighted genes, circles, genes with no significant log2FCs at either RNA or protein level; diamonds, genes with significant log2FCs at both the RNA and protein level; triangles, genes with significant log2FCs at the protein level only; squares, genes with significant log2FCs at the RNA level only; shape size, average expression values (from transcriptomic data). Coloration of labels correspond to genes with a significant increase (red) or decrease (blue) in log2FC at least at the protein level. A green label indicates no significant alteration at the protein level.
A thorough investigation of enzymes involved in reactions that include glucogenic AAs revealed that serine dehydratase (SDS), an enzyme that converts serine to pyruvate and ammonia, was in the 99th and 98th percentile for increased protein and RNA abundance, respectively, in FvsN (Figures 5B–5D). Importantly, knockdown of SDS in fresh, patient FLC tumor slices (n = 2 biological replicates) reduced cell viability by ∼65% (Figure 5E). Taken together these findings may be indicative of a strong reliance on SDS in FLC to sustain tumor viability via supply of pyruvate.
Rewiring of the TCA cycle is associated with urea cycle defects in FLC
A natural explanation for increased pyruvate (Figure 5A) may be to fuel the TCA cycle. Among the TCA cycle intermediates identified in our metabolomics dataset, the marked elevations of oxoglutarate (alpha-ketoglutarate [AKG]), the product of the rate-limiting step in the TCA cycle,30,31,32 was sharply contrasted with depleted malate, the intermediate positioned at the end of the TCA cycle (Figure 6A). Additionally, AKG was the fifth most enriched metabolite in the metabolomics data. Voltage-dependent anion channels (VDACs) are situated in the outer mitochondrial membrane, and they facilitate transport of negatively charged metabolites, such as pyruvate, from the cytosol to the intermembrane space.33,34,35 VDAC1 and VDAC2 were both significantly upregulated at the protein level, and most of the subunits that compose the pyruvate dehydrogenase (PDH) complex were significantly upregulated at the protein level (PDHA1, PDHB, and DLD [Figure 6B]). Erastin, a small-molecule drug, is well-known for inducing ferroptosis that is thought to be partly mediated by binding to VDAC which leads to hyperpolarization of the mitochondrial membrane and a decrease in NADH oxidation.36,37,38 Additionally, there has been some evidence for the degradation of VDAC by erastin with undetectable levels of VDAC2 after 10 h of treatment.37,38 Moreover, sensitivity to erastin has been shown to be positively correlated with VDAC2 abundance.39 A dose-response curve with administration of erastin in our FLC cell line29,40 revealed an EC50 (half maximal effective concentration) of ∼2 μM (Figure 6C), and caspase-3 activity was significantly elevated in our cell line administered 3 μM erastin compared to DMSO at every time point beyond 8 h in a 72 h trial (Figure 6D). Moreover, incubation of fresh tissue slices from the liver tumors of FLC patients with 2.5 μM of erastin over six days dramatically reduced cell viability when compared to adjacent non-malignant liver tissue (Figure 6E). This effect was not pronounced in tissue slices from patients with epithelioid hemangioendothelioma (EHE, a rare cancer that commonly originates in the liver), hepatocellular carcinoma (HCC; the most common type of liver cancer), or colorectal cancer liver metastases (CRLMs) (Figure 6E).
Figure 6.

Rewiring of the TCA cycle is associated with bottleneck of AKG in FLC
(A and B) Line plot of (A) metabolomic and (B) proteomic data mapping the trajectory of the TCA cycle in FLC (solid line; proteomics—n = 16 samples; metabolomics—n = 20 samples) compared to NML (dashed line; proteomics—n = 7 samples; metabolomics—n = 6 samples). Colored labels denote (A) metabolites and (B) proteins that are significantly decreased (blue) or increased (red) in abundance in FLC compared to NML. Data points and error bars represent log2FC and standard deviation, respectively—(A) ∗FDR-adj p value < 0.1 and (B) ∗FDR-adj p value < 0.05; ∗∗FDR-adj p value < 0.01; ∗∗∗FDR-adj p value < 0.001.
(C) Dose-response curve measuring viability of FLC cell line (n = 5) with administration of erastin. EC50 = 1.969 μM; control, DMSO. Light-shaded green area around curve represents 95% confidence interval.
(D) Caspase-3 activity in FLC cell line incubated with 3 μM of either DMSO (ctrl; n = 5) or erastin (n = 5) for 72 h. All time points were significant after 8 h. Wilcox used for statistical analysis. Data points and error bars represent mean fluorescent intensity and standard deviation, respectively.
(E) Inhibition of VDAC via erastin (six-day trial) on fresh tissue slices from patients afflicted with FLC (n = 4 biological replicates) and other liver tumors (n = 1 biological replicate for each tumor type with the exception of CRLM in which n = 2 biological replicates). EHE, epithelioid hemangioendothelioma; HCC, hepatocellular carcinoma; CRLMs, colorectal cancer liver metastases; BioRep, biological replicate; DMSO, control condition.
(F) Comparison of log2FCs (FLC vs. NML; z-scaled) at protein (n = 23 samples) and RNA (n = 27 samples) level. Gene sets were identified that participate in alpha-ketoglutarate (AKG) consumption and production (Human Metabolome Database or HMDB used for identification). Background set (bkgd) is all genes identified in both omics’ datasets.
Investigation of enzymes involved in the TCA cycle revealed the significant upregulation of the mitochondrially located protein aconitase 2 (ACO2), while the protein abundance of cytoplasmic ACO1 was unperturbed (Figure 6B). More importantly, all three subunits of isocitrate dehydrogenase (IDH) 3, the rate-limiting enzyme of the Krebs cycle that participates in the irreversible reaction of converting isocitrate to AKG using NAD+ as a cofactor,30,31,32 were significantly increased in protein abundance (Figure 6B). IDH1 and IDH2 participate in a similar reaction to IDH3 with three key differences: (1) the reactions are reversible, (2) they use NADP+ as a cofactor rather than NAD+, and (3) IDH1 is localized to the cytoplasm.41 IDH 1 and 2 are typically not associated with the TCA cycle but rather facilitate lipogenesis, regulation of glycolysis, repair of mitochondrial oxidative damage, and defense against reactive oxygen species (ROS).41 In the context of FLC, IDH2 protein abundance was unaltered while IDH1 was significantly downregulated (Figure 6B).
To dissect the AKG finding further (Figures 6A and 6B), we performed an analysis of genes that participate in either the production or consumption of AKG (n = 52; Figure 6F). Ornithine aminotransferase (OAT) was very distinct from the remaining fifty-one genes in both its protein (99th percentile) and RNA (99th percentile) log2FC in FvsN (Figure 6F). Importantly, OAT was previously implicated as part of a sixteen-gene signature that was able to distinguish FLC from the thirty-three different cancers compiled in The Cancer Genome Atlas.42 OAT is at the nexus of the TCA cycle, urea cycle, glutamine metabolism, and proline synthesis. It commences the reversible conversion of ornithine and AKG of the TCA cycle to glutamate and pyrroline-5-carboxylate (P5C, the precursor to proline).43,44,45,46 Importantly, the second-most significant protein association with ornithine from our concordance analysis was IDH3B, a subunit of the rate-limiting enzyme from the TCA cycle that produces AKG (Figure S5A). In newborns, OAT deficiency leads to a decrease in ornithine47,48 as opposed to young children, adolescents, and adults in whom its deficiency results in an increase of ornithine and decrease in P5C.47,49,50 Therefore, we presumed that, in the context of FLC, OAT likely favors production of P5C and glutamate. This is further supported by the fact that proline, which is made from P5C, is the second-most significantly enriched metabolite in our metabolomics data (Figure S5B). Additionally, clinical metadata (Figure S5C) along with blood and urine panels (Figure S5D) from case studies of FLC patients experiencing hyperammonemia combined with our omics data suggest a rewired urea cycle that supports proline production (Figure S5E; Figure 7A).
Figure 7.

Glutamine likely serves as a critical substrate for proline production in FLC
(A–C) (A) Schematic (top) and correlation plot (bottom) depicting OAT nexus in FLC. Coloration of labels in correlation plot and nodes in schematic correspond to proteins and metabolites with a significant increase (red) or decrease (blue) in log2FC in FLC (proteomics—n = 16 samples; metabolomics—n = 20 samples) compared to NML (proteomics—n = 7 samples; metabolomics—n = 6 samples). Green coloration indicates no significant alteration. Comparison of log2FCs (FLC vs. NML; z-scaled) at protein (n = 23 samples) and RNA (n = 27 samples) level for genes that participate in the consumption or production of (B) glutamate (Glu) or (C) glutamine (Gln).
(D) Dose-response curve of FLC cell viability with varying concentrations (n = 8 replicates per concentration) of SU-1, a pan-glutaminase inhibitor. EC50 = 5.0267 μM. Light-shaded green area around curve represents 95% confidence interval.
(E) FLC cell viability under glutamine depletion and/or GLS inhibition. Mann-Whitney was used for statistical analysis. ∗ FDR-adj p < 0.05; ∗∗∗ FDR-adj p < 0.001; ∗∗∗∗ FDR-adj p < 0.0001; n.s., not significant.
(F) Serum ammonia levels in patients afflicted with FLC comparing limited (n = 10) and extensive (n = 6) tumor burden using Mann-Whitney test (∗FDR-adj p < 0.05). JHU, John Hopkins University. Dotted line indicates the upper limit for serum ammonia. (A) For schematic, arrow size is directly associated with arrow color. Node size is arbitrary. Red, increase; green, neutral; blue, decrease; gray, no data; elliptical node, protein; square node, metabolite; arrow, reaction; broken arrows, series of reactions. (B and C) Human Metabolome Database was used to identify gene sets. (B and C) Gene sets were compared to bkgd using Mann-Whitney test (∗FDR-adjusted p < 0.05; ∗∗FDRFDR-adjusted p < 0.01; ∗∗∗adjusted p < 0.001; ∗∗∗∗adjusted p < 0.0001). (A–C) Background set (bkgd) is all genes identified in both omics’ datasets. For the gene sets (excluding bkgd), circles, genes with no significant log2FCs at either RNA or protein level; diamonds, genes with significant log2FCs at both the RNA and protein level; triangles, genes with significant log2FCs at protein level; squares, genes with significant log2FCs at RNA level; shape size, average gene expression values (from transcriptomic data). (E) Bkgd, Background; RQV, relative quantitative value; Ctrl, control; FBS, fetal bovine serum; Gln, glutamine.
Mitochondrial GLS-ALDH18A1-PYCR is a critical axis in FLC
As mentioned previously, OAT connects the urea and TCA cycles with the proline synthesis pathway. Additionally, proline can be completely derived from glutamate.51 However, glutamate is a major contributor to multiple pathways that include nucleotide synthesis, glutathione synthesis, AA synthesis, TCA cycle, one-carbon metabolism, and urea cycle.51,52,53 So, we next performed an analysis on genes (n = 62) that participate in reactions involving glutamate. Strikingly, the increase in aldehyde dehydrogenase 18 family member A1 (ALDH18A1) protein and RNA expression was the first- and second-most, respectively, among all 62 enzymes (Figure 7B). Glutaminase (GLS) upregulation was the third-most at both the protein and RNA level (Figure 7B). Moreover, both genes are in the 99th percentile for top-most significantly upregulated proteins, and 94th (ALDH18A1) and 93rd (GLS) percentiles for top-most significantly upregulated RNAs, in the entire dataset (Figure 7B). ALDH18A1 is a mitochondrial enzyme that irreversibly converts glutamate into L-glutamate gamma-semialdehyde, which subsequently undergoes spontaneous degradation to produce P5C.54,55,56 GLS irreversibly converts glutamine into ammonia and glutamate. It is important to mention that there are two paralogs of GLS (both mitochondrial specific), GLS and GLS2. GLS is often implicated in cancer progression57. GLS2 expression (protein and RNA) was unperturbed.
As with glutamate, glutamine contributes to a variety of metabolic pathways. So, we next performed an analysis on genes (n = 25) that participate in reactions involving glutamine. Indeed, GLS was very distinct from the other twenty-four genes in both its protein (99th percentile) and RNA (93rd percentile) log2FC (Figure 7C). A dose-response curve of GLS inhibition in our FLC primary cell line using SU-1, a pan-GLS inhibitor,58 revealed an EC50 of ∼ 5 μM (Figure 7D). GLS inhibition using 10 μM of SU-1 negatively impacted cell viability by approximately 40% (Figure 7E), and this effect was comparable to complete glutamine depletion from media (Figure 7E). Interestingly, the combination of glutamine depletion with GLS inhibition had no further impact on cell viability than GLS inhibition alone (Figure 7E). The reactions performed by the GLS-ALDH18A1 axis are coupled with reverse reactions that are associated with ALDH4A1 and glutamate-ammonia ligase (GLUL). ALDH4A1 irreversibly converts NAD+ and P5C into glutamate and NADH. Subsequently, GLUL assimilates ammonia with glutamate and irreversibly produces glutamine. ALDH4A1 and GLUL were significantly downregulated at the protein (4th and 12th percentile, respectively) and RNA (3rd and 12th percentile, respectively) levels in FvsN (Figures 7B and 7C).
Moreover, our data indicate that, along with ornithine transcarbamylase (OTC) deficiency and OAT hyperactivity, the GLS-ALDH18A1 and ALDH4A1-GLUL axes coupled with enhanced SDS may be major contributors to the ammonia accumulation in FLC-induced hyperammonemia (HA) that we observe in patients (Figure 7F).
Potential shunting of P5C/proline into collagen production via PYCR, PRODH, and P4HA
Starting with the reactant P5C, there are three critical proteins that participate in proline metabolism: pyrroline-5-carboxylate reductase (PYCR), proline dehydrogenase (PRODH), and prolyl 4-hydroxylase subunit alpha (P4HA). There are three known paralogs of PYCR and two each for PRODH and P4HA. The proteins encoded by PYCR1, PYCR2, PRODH, and PRODH2 all reside in the mitochondria while PYCR3 resides in the cytosol.59,60,61,62,63 P4HA1 and P4HA2 reside mainly in the endoplasmic reticulum.64 PYCRs react NADH with P5C to produce proline and NAD+, which in turn could help fuel the TCA cycle and ATP production. PRODH and PRODH2 react proline and hydroxyproline, respectively, with oxidized flavin adenine dinucleotide (FAD+) to produce P5C (via PRODH), HO-P5C (via PRODH2), and reduced flavin adenine dinucleotide ([FADH2]; via PRODH & PRODH2).59,61,65,66,67 P4HAs utilize AKG to hydroxylate proline residues within procollagen chains, which results in the production of succinate and collagen with hydroxyproline residues.68,69,70
PYCR1, PYCR2, and P4HA2 were significantly upregulated at the protein (96th, 99th, and 88th percentile, respectively; Figure S6A) level, and PYCR1 and P4HA2 were also significantly upregulated at the RNA (97th and 92nd percentile, respectively; not shown) level. PRODH2 and one of the isozymes of P4HA1 were significantly downregulated at the protein (2nd and 11th percentile, respectively; Figure S6A) and RNA (12th and 11th percentile, respectively; not shown) level. PYCR3 and the other isozyme of P4HA1 were not significantly altered (Figure S6A). PRODH protein was also elevated in FLC, though barely missed significance (93rd percentile; p value = 0.061).
Given these data, we hypothesized that the rewired metabolic network relating to proline/hydroxyproline synthesis could contribute in part to enhanced production of collagen and pathways associated with extracellular matrix remodeling. We found that the median log2FC of genes related to procollagen chains (n = 55), proteoglycans (n = 32), and the lysosome (n = 93) was significantly higher relative to the median log2FC of all genes (background set, n = 8,040) at both the protein and RNA level (Figure S6B). The median log2FC of genes involved in chondroitin, heparin, and keratin sulfate synthesis (n = 28) and degradation (n = 22) was significantly higher relative to the median log2FC of all genes (background set, n = 8,040) at the protein level (Figure S6B). Additionally, the median log2FC of genes involved in protein secretion (n = 98) was significantly higher relative to the median log2FC of all genes (background set, n = 8,040) at the protein level (Figure S6B).
The overall working model of cellular energetics in FLC is shown in Figure S7.
Discussion
Our working model of FLC metabolism proposes several unique features that emerge in the cancer. First, AA import is favored in the background of impeded glucose and long-chain fatty acid (LFCA) entry (data not shown). We suspect that the metabolic switch observed in the glycolytic pathway from liver- to muscle-specific enzymes amplifies the tumor’s ability to scavenge glucose which can serve as input for pyruvate generation. However, the juxtaposition of enriched pyruvate in our FLC metabolomic profile with that of no significant impact on cell viability from administration of 2-DG in FLC patient tissue slices (same concentration of 2-DG had a negative impact on the cell viability of other cancers) suggests an alternative primary input for pyruvate production other than glucose. Fructose is another major sugar that could serve as input for the glycolytic end product, pyruvate. However, it is unlikely that fructose serves this role in FLC as ketohexokinase (KHK), the major liver-specific enzyme that allows fructose entry into the glycolytic pathway,71 is significantly downregulated at the protein level (2nd percentile). Our working model implicates SDS, the 10th most upregulated protein in FvsN, as a major contributor to enrichment of pyruvate using serine as input. Reliance on SDS for pyruvate production would likely circumvent dependence on glycolysis. Importantly, knockdown of SDS in FLC patient tissue slices, but not HCC or NML, leads to a greater than 2-fold loss in cell viability implicating SDS as a potential therapeutic vulnerability.
Our working model also suggests that combined upregulation of SLC16A3 (a major lactate transporter) and LDHB participates in the recycling of lactate into pyruvate, which is shuttled into a rewired TCA cycle. The proposed shuttling of pyruvate into the TCA cycle is supported by the apparent dependency of FLC on VDAC in patient tissue slices. Although erastin is problematic as an actual therapy, it is valuable here in elucidating the importance of VDAC function (one of which is pyruvate entry into the mitochondria) for FLC viability. This finding motivates future studies into how else this aspect of the metabolic network can be targeted in a manner that also mitigates toxicity.
The main feature of the rewired TCA cycle in FLC is a critical juncture point at AKG, one of the top-most enriched metabolites in our metabolomic data. TCA cycle-related metabolites prior to AKG display a steady increase in enrichment in FvsN, while TCA cycle-related metabolites post-AKG display diminishing enrichment to the point where malate is significantly depleted in FLC. A similar trend is evident in the proteomic data, ten proteins that are significantly upregulated pre-AKG contrasted by only one post-AKG. We suspect that the rewiring characterized by the aforementioned proteo-metabolomic profile creates a substantive bottleneck at AKG, which is subsequently provided to OAT. It may be the case that OAT, despite likely increased activity, is not increased enough to metabolize all of the available AKG, leading to an excess of AKG. Therefore, we propose that OAT is the rate-limiting step in this metabolic process.
AKG feeds into a metabolic subnetwork that includes the TCA and urea cycles in which OAT is at the nexus. Reconfiguration of the urea cycle in FLC involves three important features: (1) an OTC deficiency, (2) upregulation of the cytoplasmic (SLC7A2) and mitochondrial (SLC25A15) transporters that have high specificity for ornithine,50 and (3) upregulation of the mitochondrial-specific arginase (ARG2) that participates in the penultimate step of the urea cycle converting arginine into urea and ornithine. Plasma arginine levels in patients with FLC-associated HA are at the very low end of the reference range, implying heavy demand of this AA by the tumor. Additionally, ARG2 is among the top-most significantly upregulated genes at both the protein (96th percentile) and RNA (99th percentile) level. The reconfiguration of the urea cycle results in the funneling of ornithine along with AKG into a reaction commenced by OAT that yields glutamate and P5C, the immediate precursor to proline. Additionally, the significant upregulation of 50% of the identified AA transporters in our dataset (almost exclusively at the protein level) and an enhanced GLS-ALDH18A1 axis coupled to a suppressed ALDH4A1-GLUL axis result in the shuttling of glutamine into P5C generation. We provide evidence that FLC is most dependent on GLS out of all glutamine-consuming enzymes by demonstrating that (1) the negative impact on cell viability in our FLC primary cell line with complete glutamine depletion from the media (affecting all glutamine-consuming enzymes) was comparable to inhibition of GLS with SU-1 and (2) the combination of glutamine depletion and GLS inhibition had no further impact on cell viability than GLS inhibition alone. The aforementioned coupling of the enhanced GLS-ALDH18A1 axis with that of suppressed ALDH4A1-GLUL axis also ensures that glutamate generated from OAT, GLS, other mitochondrial enzymes, or mitochondrial import, is siphoned into P5C generation.
This centralization on P5C production is leveraged by enhanced mitochondrial-specific pyrroline-5-carboxylate reductases (PYCR1 and PYCR2) that utilize NADH to convert P5C into proline and NAD+. We suspect the aforementioned reaction has three major effects in FLC: (1) generation of proline for subsequent use in producing collagen to be incorporated into strong fibrous bands, the morphological hallmark of FLC, (2) regeneration of NAD+ to fuel the TCA cycle, and (3) diversion of electrons away from the ETC. The mitochondrial proline pool is presumed to have two fates, export into the cytoplasm via SFXN4 followed by incorporation into collagen or reconversion into P5C by PRODH which requires the cofactor, FAD+. P4HA2 upregulation in FLC is consistent with what has been observed in other cancers.72 Additionally, SFXN4 and PRODH protein abundance displayed a trend toward elevation in FLC and barely missed significance (p = 0.064 and p = 0.061, respectively). PRODH is known to generate elevated levels of ROS relative to ETC complexes.73,74,75,76 Moreover, PRODH has been implicated in reducing mitochondrial respiratory fitness.76
The mitochondrial genome encodes thirteen proteins that are all involved in oxidative phosphorylation.77 We detected twelve of the thirteen in our omics datasets. While genes involved in mitochondrial-specific translation and stability of ETC complexes seemed to be enhanced in FLC, most mitochondrial-specific genes involved in OXPHOS (eleven out of the twelve) showed no significant alterations. Despite increased mitochondria in FLC,21,22,23,24,25 there was no difference in MRC of ETC complexes in FvsN, suggesting FLC-associated ETC dysfunction. ETC dysfunction was also evident in our FLC cell line, which showed no significant differences between basal respiration and MRC, which is indicative of low SRC.28 We suspect that enhanced proline metabolism in FLC compromises mitochondrial respiratory fitness by diminishing NADH and FADH2 reserves and generating ROS, which damages ETC complexes. The enhanced production of proteins involved in mitochondrial translation and stabilization of ETC subunits is likely an attempt to keep pace with rapid degradation of the electron transport chain (ETC) proteins as oxidative phosphorylation may, at least in part, still be utilized for ATP production utilizing a portion of glutamine as a substrate. The presumptive ROS generation in FLC by proline metabolism may help explain why FA-beta oxidation, a biological process that is known for copious production of ROS,78,79,80 is suppressed. FLC may be particularly susceptible to free radical damage primed by a combination of suppressed liver detoxification pathways and proline metabolism-induced ROS generation. ROS susceptibility coupled with enhanced translation in FLC is supported by a recent, major drug screen that included the use of established FLC cell lines, cell culture derived from fresh patient tissue, and patient-derived xenograft (PDX) murine models.81 Napabucasin was among the top hits, and its proposed mechanisms of action are promotion of ROS generation and inhibition of translation initiation.81 A subsequent study investigating signaling pathways in FLC associated aberrant PKA signaling with stimulation of translation initiation in FLC patient-derived and engineered cell lines.40 Moreover, cell growth was dramatically inhibited by blocking translation initiation.40
We have constructed a robust proteo-metabolomic model that proposes a centrality of AAs for energy, mitochondrial translation, and proline synthesis in FLC. The metabolic adaptations in this cancer involve dysregulation of key transporters, a glycolytic switch, rewiring of the TCA and urea cycles, upregulated OAT, revamping of the GLS-ALDH18A1/ALDH4A1-GLUL axes, and enhanced enzymes involved in the penultimate step of proline synthesis as well as proline hydroxylation. Our model serves as a metabolic roadmap for the FLC scientific community in the hopes of finding effective therapeutic vulnerabilities.
Limitations of the study
The heterogeneity across metabolomic profiles in particular is a well-documented challenge, especially with relatively small sample sizes. Future studies would benefit from the addition of a mixed sample (aggregate of all samples) to the pipeline that would serve as a representative metabolome of the patient cohort with the aim of normalizing the data. Additionally, having at least three technical replicates of each patient sample and weighting metabolite abundance in each sample by the standard deviation across replicates could serve to increase signal-to-noise ratio. In this study, to increase statistical power, we aggregated primary and metastatic samples into one group. However, we suspect that due to tissue environment there may be interesting differences between primary and metastatic categories, and in the future with increased sample sizes it would be informative to consider them separately. Finally, although we provide important functional validation in patient tissue slices for the dependency of FLC on several proposed key players in critical metabolic networks, we have not yet confirmed these findings in in vivo models of FLC. Also, the primary role of some of our metabolic enzymes of interest remains unknown, and this can be addressed at least in part with tracing studies in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Frozen patient tissue (primary, metastatic, & adjacent non-malignant) | Praveen Sethupathy (pr46@cornell.edu) | N/A |
| Patient tissue slices (primary, metastatic, & adjacent non-malignant) | Taran Gujral (tgujral@fredhutch.org) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Erastin (small molecule inhibitor that alters VDAC functionality) | Taran Gujral (tgujral@fredhutch.org) | N/A |
| SU-1 (small molecule inhibitor targeting GLS) | Rick Cerione (rac1@cornell.edu) | N/A |
| Critical commercial assays | ||
| CellTiter-Glo (FLC-H cell line) | Promega | Cat#G7570 |
| CellTox Green (FLC-H cell line) | Promega | Cat#G8741 |
| CellTiter-Glo 2.0 (FLX1 cell line) | Promega | Cat#G9241 |
| IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent (FLX1 cell line) | Sartorius | Cat#4440 |
| Incucyte ZOOM system | Sartorius | RRID: SCR_019874 |
| RealTime-Glo MT Cell Viability Assay (Tissue slices) | Promega | Cat#G9711 |
| Experimental models: Cell lines | ||
| FLC-H primary cell line (Originating as metastatic cells from ascites fluid of patient) | Praveen Sethupathy (pr46@cornell.edu) | N/A |
| FLX1 primary cell line (Originating as metastatic cells from ascites fluid of patient) | Taran Gujral (tgujral@fredhutch.org) | N/A |
| Oligonucleotides | ||
| siRNA (human) against SDS | Horizon Discovery | Cat#L-008214-01-0005 |
| siRNA (mouse) against Sds (non-targeting control) | Horizon Discovery | Cat#L-053256-01-0005 |
| Software and algorithms | ||
| Code for core analyses and generation of all main figures | Donald Long Jr (dl964@cornell.edu); Zenodo | Zenodo: 12739492; https://doi.org/10.5281/zenodo.12739492 |
| Deposited data | ||
| Raw proteomics data | ProteomeXchange Consortium | ProteomeXchange Consortium: PXD051527 |
| Raw metabolomics data | NIH Common Fund’s National Metabolomics Data Repository (NMDR) | NMDR: PR001989; https://doi.org/10.21228/M8MT61 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Praveen Sethupathy (pr46@cornell.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The mass spectrometry proteomics and metabolomics data have been deposited to the ProteomeXchange Consortium (via PRIDE82 partner repository) and NIH Common Funds National Metabolomics Data Repository (NMDR), respectively. The data is publicly available as of the date of publication. Accession numbers and DOIs are listed in the key resources table.
-
•
All code for execution of core analyses and generation of all main figures has been deposited at Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Human patients
Informed consent was obtained from all individuals and studies were performed in accordance with the protection of human subjects guidelines (U.S. Common Rule). FLC tumors (metastatic and primary) and NML samples were obtained from the Fibrolamellar Cancer Foundation (FCF) biobank in accordance with IRB protocols 1802007780, 1811008421 (Cornell University, Ithaca, NY) and 33970/1 (FCF). Both male and female subjects were included, and all samples were deidentified. Frozen patient tissue was sent to the Karolinska Institute (Stockholm, Sweden; n = 36) and Weill Cornell (NYC; n = 34) for proteomics and metabolomics, respectively. There were no metastatic and primary tissue samples in our downstream analyses that were obtained from the same patient.
Cell line
The FLC cell line used in this study was graciously donated by the Bardeesy lab at Harvard. The line was originally derived from a previously described patient-derived xenograft (PDX) murine model.25 Briefly, metastatic cells were purified from the ascites fluid of the patient, conditioned on serum-free Kubota’s medium (PhoenixSongs Biological; Branford, CT), and seeded into non-immunogenic mice. The resulting PDX tumor was dissociated from murine cells and adapted for monoculture. The FLC cell line was cultured in the following conditions: RPMI 1640 media (Gibco 11875119), 10% fetal bovine serum (Gibco 26140079, heat-treated at 56°C for 30 min, 1% penicillin-streptomycin (Gibco 15140122), and 2.5 ng/mL hepatic growth factor (Gibco PHG0321).
Method details
Nutrient manipulation assays
Cell media (stock) was generated for the FLC cell line as previously described or RPMI1640 media was exchanged for either the glucose- or glutamine-free versions (modified media). The stock was diluted with the glucose-free media to attain the following glucose concentrations (conditions): 9.77mM (Ctrl #1), 7.497mM, 5.225mM, 2.952mM, 1.815mM, 1.243mM, 0.961mM, and 0.679mM. FBS was removed from the respective modified media to generate glucose and glutamine concentrations of 0mM. DMSO was added (final concentration, 0.1%) to the 0mM concentration and 9.77mM concentration (Ctrl #2). The small molecule inhibitor targeting GLS, SU-1, was kindly donated by Rick Cerione (Cornell University College of Veterinary Medicine; Department of Molecular Medicine). FLC cells were seeded into a 96-well plate (10,000 cells/well; Corning, black with clear flat bottom, CellBIND surface) and incubated with the various conditions for six days. Subsequently, Cell-Titer Glo and CellToxGreen were used to assess cell viability and cell death, respectively, on the Gen5 Biotek Synergy 2 Microplate Reader.
Cell viability and apoptosis assays
FLX1 cells29,40 were cultured in DMEM media supplemented with 10% FBS and 1% penicillin-streptomycin. For viability measurements, 5,000 cells were seeded per well in a 96-well plate using the complete medium. The following day, Erastin was administered in a dose-dependent manner. Viability was assessed 72 h later using the CellTiter-Glo 2 reagent (Promega), following the manufacturer’s instructions. The following equation was used for curve fitting: , where hill coefficient = 1 and ec50 = 1.969 μM. For the apoptosis assays, the activity of caspase 3/7 was measured using the NucView 488 reagent (IncuCyte Caspase 3/7 Green reagent) and live-cell imaging (IncuCyte ZOOM; Essen Bioscience), following the manufacturer’s instructions. The NucView 488 reagent, at a concentration of 1 μM, was added to the cell culture medium along with either Erastin (3 μM) or a DMSO control in 96-well plates. Over a period of 72 h, apoptotic cells were identified and quantified based on the presence of green fluorescence signals (visible in the 488 nm green channel) using the IncuCyte ZOOM system.
Transcriptomics
All RNA-seq datasets analyzed in this study were generated previously.29
Proteomics
Samples were lysed by 4% SDS lysis buffer and prepared for mass spectrometry analysis using a modified version of the SP3 protein clean up and digestion protocol.83 Peptides were labeled with TMT16-plex reagent according to the manufacturer’s protocol (Thermo Scientific) and separated by immobilized pH gradient – isoelectric focusing (IPG-IEF) on 3–10 strips as described previously.84 Extracted peptide fractions from the IPG-IEF were separated using an online 3000 RSLCnano system coupled to a Thermo Scientific Q Exactive-HF. MSGF+ and Percolator in the Galaxy platform was used to match MS spectra to the Ensembl Homo sapiens (release 103 pep.all fasta) protein database.85
Metabolomics
Polar metabolite profiling was performed as previously described.86 Briefly, metabolites were extracted from cells (derived from tissue homogenate of samples) using pre-chilled 80% methanol (−80°C). The extract was dried completely with a Speedvac and redissolved in HPLC grade water prior to application on hydrophilic interaction chromatography LC-MS. Metabolites were measured on a Q Exactive Orbitrap mass spectrometer (Thermo Scientific), which was coupled to a Vanquish UPLC system (Thermo Scientific) via an Ion Max ion source with a HESI II probe (Thermo Scientific). A Sequant ZIC-pHILIC column (2.1 mm i.d. × 150 mm, particle size of 5 μm, Millipore Sigma) was used for separation of metabolites. A 2.1 × 20 mm guard column with the same packing material was used for protection of the analytical column. Flow rate was set at 150 μL/min. Buffers consisted of 100% acetonitrile for mobile phase A, and 0.1% NH 4 OH/20 mM CH 3 COONH 4 in water for mobile phase B. The chromatographic gradient ran from 85% to 30% A in 20 min followed by a wash with 30% A and re-equilibration at 85% A. The Q Exactive was operated in full scan, polarity-switching mode with the following parameters: the spray voltage 3.0 kV, the heated capillary temperature 300°C, the HESI probe temperature 350°C, the sheath gas flow 40 units, the auxiliary gas flow 15 units. MS data acquisition was performed in the m/z range of 70–1,000, with 70,000 resolution (at 200 m/z). The AGC target was 1e6 and the maximum injection time was 250 ms. The MS data was processed using Xcalibur 4.1 (Thermo Scientific) to obtain the metabolite signal intensity for relative quantitation. Metabolites were identified using an in-house library established using chemical standards. Identification required exact mass (within 5ppm) and standard retention times.
Frozen tissue respirometry
The frozen patient tissue from each sample (2 x ∼100 mg chunks per sample) was thawed and homogenized in 500uL of MAS buffer (70 mM sucrose, 220 mM mannitol, 5 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2 mM HEPES pH 7.4) with protease inhibitor cocktail (Roche). For each sample, the set of homogenates was centrifuged at 1,160×g for 10 min at 4°C, and the resulting supernatant (liver homogenate) was collected. Subsequently, one of the homogenates was taken through another round of centrifugation at 10,000xg to attain mitochondrial isolates (pellet). The pellet was washed and resuspended in MAS buffer. Protein concentrations were determined by Bradford colorimetric assay (Thermo Fisher). Liver homogenates (15 μg/well) and mitochondrial isolates (15ug/well or 6ug/well) were loaded into Seahorse XF96 microplate in MAS buffer (20 μL/well) and centrifuged at 2,000×g for 5 min at 4°C. After centrifugation the volume was increased to 150μL by adding 130μL MAS with cytochrome c (10 μg/mL) void (wells with mitochondrial isolates) or present (wells with liver homogenate). At port A, substrates of NADH (1 mM) were injected to determine the respiratory capacity of Complex I; succinate (5 mM) + rotenone (2 μM) were injected to determine the respiratory capacity of Complex II. Subsequently, the following compounds were injected so that final concentrations were: port B—rotenone (2 μM) + antimycin (4 μM); port C—TMPD (0.5 mM) + ascorbic acid (1 mM); port D: azide (50 mM). Port C injection determined the respiratory capacity of Complex IV. OCR rates were measured using Seahorse XF96 Extracellular Flux Analyzer (Agilent Technologies) and normalized to either 1) protein (liver homogenates) or mitochondrial content (mitochondrial isolates) quantified by MTDR. Isolates were stained with 500 nM MTDR for 10 min followed by two wash steps to remove the dye (Thermo Fisher). MTDR fluorescence was read on a Tecan Spark plate reader (Excitation: 633 nm; Emission: 678 nm).
Tissue slice preparation and drug treatments
The process for preparing tumor slices was carried out as previously described.87,88 In summary, tumor tissues were sliced into 400-micrometer sections using a Leica VT1200S vibratome (Leica Biosystems), employing HBSS as the slicing medium. These slices were further processed into 400-micrometer cuboids using the McIlwain tissue chopper (Ted Pella) as described in.89 These cuboids were immediately placed into 96-well ultralow-attachment plates (Corning) in Williams’ medium supplementing with 12 mM nicotinamide, 150 nM ascorbic acid, 2.25 mg/mL sodium bicarbonate, 20 mM HEPES, an additional 50 mg/mL glucose, 1 mM sodium pyruvate, 2 mM L-glutamine, 1% (v/v) ITS, 20 ng/mL EGF, 40 IU/mL penicillin, and 40 μg/mL streptomycin. The RealTime Glo reagent (Promega) was added to the incubation media as per the guidelines provided by the manufacturer. The baseline cell viability in the cuboids was determined after 24 h using RealTime Glo bioluminescence, measured with a Synergy H4 instrument (Biotek). The cuboids were subjected to either DMSO as a control or various experimental drugs as indicated. Measurements of the overall viability of the tumor tissues were taken daily for up to 7 days following the treatment.
Gene knockdown experiments in tissue slices
The preparation of tissue slices followed the method as described above. We obtained small interfering RNA (siRNA) targeting SDS, or non-targeting control from Horizon Discovery. To evaluate changes in aggregate viability, we performed siRNA transfections in 96-well plates using Lipofectamine RNAiMax (provided by Invitrogen), following the instructions of the manufacturer. Each transfection process involved a minimum of two wells, with each well containing 4 to 6 3D cuboids. We monitored the viability changes using the Synergy H4 instrument from Biotek. To assess the overall changes in the viability of the tumor tissues, we carried measurements for a period of up to 7 days after the treatment.
FLC cell line respirometry
Oxygen consumption rate (OCR) was measured by the Agilent seahorse xFe 24 Bioanalyzer. FLC cells were plated on xFe24 cell culture plates at a seeding density of 60,000 cells per well and incubated for 48-h at 37°C with 5% CO2. Following incubation, cells were washed in PBS and culture media was replaced with unbuffered Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 4.5 g/L glucose, 4 mM glutamine, and 1 mM pyruvate. The compound concentrations for the mitochondrial stress test were as follows (final concentration): Port A: Oligomycin (1 μM), Port B: Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) (1 μM), Port C: Rotenone/Antimycin A (2 μM each). The OCR was normalized to the total cell number using the Celigo image cytometer. Respirometry data were collected using Agilent Wave v2.4 software and were expressed as the mean ± SEM.
Quantification and statistical analysis
Proteomics
For proteomics, samples were run through LC MS/MS in two consecutive batches (16 samples per batch). Protein quantification by TMT16plex reporter ions was calculated using TMT PSM ratios to the entire sample set (all 16 TMT-channels) and normalized to the sample median. The median PSM TMT reporter ratio from peptides unique to a gene symbol was used for quantification. Protein false discovery rates were calculated using the picked-FDR method using gene symbols as protein groups and limited to 1% FDR.90 Quantified proteins (n = 10065) displayed 1) values for all samples across both batches, 2) values missing for all samples in one of the batches, or 3) values missing for all samples across both batches. For the full and omics-matched datasets, only proteins containing values for all samples across both batches were retained (n = 8485). Covariate adjustment using R was performed to remove batch effect. Python and R were used to generate hierarchal clustering with heatmap and PCA-based clustering, respectively. Data profiles from metastatic (MET) and primary (PRI) tissue samples were aggregated and deduplicated (random removal of either MET or PRI that came from the same patient) to create the FLC cohort. DeqMS91 was used for differential protein expression analysis.
Metabolomics
For metabolomics, observations that were either blank or displayed a zero value were considered missing.92,93 Subsequently, metabolites (n = 180) were filtered using the modified 80% rule where any metabolite that contained less than or equal to 20% missing values in at least one of the cohorts (NML or tumor) were retained (nfull_dataset = 135; nomics-matched_dataset = 119). At this point, approximately 9% of observations were missing values. The following five different imputation methods were assessed for optimally imputing missing values in our dataset: imputeBasic, multivariate imputation by chained equations (MICE),94 missMDA,95 missForest,96 and softImpute.97 A test dataset (n = 76) was derived from the metabolomics dataset where the modified 80% rule had been applied. The test dataset was void of any missing values. For each imputation method (except for MICE, a very computationally intensive platform) one-hundred simulations were run generating three different missing data patterns (missing completely at random or MCAR, missing at random or MAR, and missing not at random or MNAR)98 at 3%, 10%, 30%, or 50% of the data missing. Thirty simulations were run for MICE. In total, 1200 simulations each (100 simulations x 3 missing data patterns x 4 different degrees of data missing) were run for imputeBasic, missMDA, missForest, and sofImpute, while 360 simulations (30 simulations x 3 missing data patterns x 4 different degrees of data missing) were run for MICE. Five quality metrics generated from the simulations were used to compare and contrast the imputation methods—raw bias (difference between predicted value of point estimate and it’s true value from the test dataset), percent bias (raw bias expressed as a percent), coverage rate (proportion of confidence intervals generated from simulations that contain the true value), average width (average width of confidence interval), and root mean squared error (represents compromise between bias and variance or accuracy and precision). Given the distribution of missing values in our metabolomics dataset and our unawareness of any bias that would influence the distribution, we presumed the missing values were MCAR. At a missing value rate of 10%, missForest displayed the best combination of quality metrics, and it was used for imputing missing values (∼9%) in our full and omics-matched metabolomic datasets. Samples were run through LC/MS in four consecutive batches. In the resulting peak intensity file, MET and PRI tissue samples were aggregated and deduplicated (random removal of either MET or PRI that came from the same patient) to create the FLC cohort. Subsequently, the peak intensity files from the full and omics-matched data was processed in the following manner after imputation: (1) batch median normalization (each observation of a metabolite divided by its batch median), (2) log2 transformation, (3) co-variate adjustment for batch, (4) reverse-log2 transformation, (5) metabolite median normalization (each metabolite within a sample divided by the median of all metabolites within that sample), (6) log2 transformation and (7) co-variate adjustment for injection order. Differential abundance analysis was performed using MetaboAnalyst99,100 where Empirical Bayes Analysis of Microarrays (EBAM) was used for statistical testing.
Full datasets vs. omics-matched profiles
Pearson was utilized to assess the correlation between log2FCs of significantly altered features (proteins and metabolites) in either our full dataset (nproteomics = 23; nmetablomics = 26) or omics-matched dataset (nmetabolomics-matched = 17; nproteomics-matched = 26) but present in both. The overlap of differentially abundant features between the full sample set and omics-matched sample set were 53.6% and 27.8% for proteomics and metabolomics, respectively (Data S3 and S4). However, the effect sizes between the sample sets were comparable in magnitude and direction (Data S3 and S4). Quantification of the relationship of effect sizes via Pearson between the sample sets showed a strong, positive correlation (R ≥0.98, p < 2.2e−16; data not shown). The use of Pearson was justified by the Central Limit Theorem, which states that a sample size of greater than or equal to thirty approximates a normal distribution.
Concordance analysis
Proteomics & metabolomics
For concordance analysis between our proteomics and metabolomics data, omics-matched tumor samples (patient tumor samples that were present in both the proteomics and metabolomics datasets; n = 13 out of the 17 omics-matched samples) were used as input for pipeline adapted from Benedetti et al. 2023101 to assess level of agreement between proteomic and metabolomic abundance in FLC. Importantly, concordance assumes no underlying distribution. We leveraged the ‘concordance’ function from the ‘survival’ package in R to perform the computations (n = 1,009,715 measured associations).
Proteomics & mitochondrial abundance
For concordance analysis between mitochondrial abundance measured via MitoTracker Deep Red (MTDR) and our proteomics data, proteins specific to the mitochondria (identified by MitoCarta3.0) were retained for downstream analysis (n = 1003). Four of the six NML samples measured via MTDR were patient-matched with samples in the mitochondria-specific, omics (MSO) dataset. The two unmatched samples measured via MTDR were randomly paired with two samples from the remaining MSO sample set (n = 3). For FLC, three of the six samples measured via MTDR were patient-matched with samples in the MSO dataset. The three unmatched samples measured via MTDR were randomly paired with three samples from the remaining MSO sample set (n = 13).
The library of mitochondrial pathways contained in MitoCarta3.0 (n = 150) were filtered by the following criteria: (1) The sub-pathways of generalized over-arching pathways were retained to avoid redundancy of protein members. For example, “Gluconeogenesis” was retained, but its overarching pathway, “Carbohydrate Metabolism”, was removed. (2) Retained pathways had at least one protein member identified in the MSO dataset that was significantly concordant or discordant with mitochondrial abundance. (3) Retained pathways had at least ten protein members. Subsequently, mitochondrial pathways were ranked by (1) highest proportion of pathway members that were significantly associated with mitochondrial abundance (pathway members identified in MSO dataset that were significantly associated with mitochondrial abundance divided by all pathway members identified in MSO dataset), (2) highest proportion of pathway members identified in MSO dataset, and (3) absolute value of scaled concordance (2 ∗ concordance – 1). Finally, a threshold was set for attaining the top twenty mitochondrial pathways where at least 10% of a pathway’s protein members that were identified in the MSO dataset were significantly associated with mitochondrial content.
To address the potential issue of reproducibility associated with random pairing, we performed Monte Carlo simulations with the aforementioned pipeline for identifying all possible sets of top twenty mitochondrial pathways from all possible sample combinations of NML (4 matched samples + [3choose2] = 3 possible combinations) or FLC (3 + [13choose3] = 286 possible combinations). Subsequently, we attained the top twenty mitochondrial pathways with the greatest hits. A hit was considered a gene that was significantly associated with mitochondrial abundance in at least one of the simulations. Cumulative distribution function (CDF) was used for statistical testing.
Machine learning
Sufficient sample size was assessed by the fulfillment of at least one of two criteria: (1) the omics-based classification of the patient cohort samples (nproteomics = 23 samples and nmetabolomics = 26 samples; used throughout this study) was in alignment with the physician-based classifications and/or (2) the omics-derived profiles were generalizable to a new, independent cohort of patient samples. An extreme gradient boost machine learning (XgBoostML) pipeline with nested cross-validation (CV) was constructed (Figures S2A and S2B) and trained on the twenty-three (proteomics) and twenty-six (metabolomics) samples used throughout this study. Subsequently, the trained model was tested on thirteen (proteomics) and ten (metabolomics) independent samples (these samples were used exclusively for machine learning and not any other downstream analyses). The ML pipeline consistently performed significantly better than random chance at classifying patient samples across both the training (fulfillment of first criteria) and testing (fulfillment of second criteria) phases for proteomics (Figures S2C and S2D). For the metabolomics data (nmetabolomics = 26 samples), the ML pipeline fulfilled the first criteria but failed to meet the second criteria (Figures S2E and S2F). This is consistent with the greater inter-individual heterogeneity associated with metabolomics, which has been observed previously.102,103,104
Data processing was performed independently on the training and hold-out sets to minimize data leakage,105 with the exception of imputation for metabolomics which was performed on all the samples combined. Training data went through two (proteomics) to three (metabolomics) iterations of inner-loop train/test splitting (ninner-loop train_proteomics = 13 samples and ninner-loop test_proteomics = 10 samples; ninner-loop train_metabolomics = 18 samples and ninner-loop test_metabolomics = 8 samples). With each iteration, the inner training (IT) data was subsetted for the most relevant features using a PCA-based method106,107 that involved the following steps: (1) performing PCA with centering (proteomics, metabolomics) and scaling (metabolomics), (2) selecting principal components that explained greater than 10% of the variance in the data, (3) performing varimax rotation on raw loadings, and (4) isolating features that were greater than or equal to an absolute value of 0.5 in any of the selected principal components. The subsetted IT data was subsequently used for stratified 2-fold,3-repeat CV where each fold (resample) contained at least six samples (nNML_proteomics_resample = 2 and nFLC_proteomics_resample ≥ 4; nNML_metabolomics_resample = 2 and nFLC_metabolomics_resample = 5) within each partition for training and testing. Hyperparameter tuning (described in more detail below) was incorporated into the 2-fold, 3-repeat CV. The best model from the inner training was assessed for performance against the inner test data (ninner-loop test_proteomics = 10 samples; ninner-loop train_metabolomics = 8 samples). Overall evaluation of training performance was attained by averaging of the two (proteomics) to three (metabolomics) training assessments from the two (proteomics) to three (metabolomics) iterations of inner training. The union of the feature subsets from the two (proteomics) to three (metabolomics) iterations was used to subset the whole training dataset (ntraining_proteomics = 23 and ntraining_metabolomics = 26), and 2-fold,3-repeat CV with hyperparameter tuning was performed. A final evaluation was executed by assessing the performance of the trained model from the whole training dataset against the hold-out set (nholdout_proteomics = 13 and nholdout_metabolomics = 10). Performance metrics of interest were balanced accuracy (the accuracy of the training model taking class imbalance of the testing set into account), precision-recall area under curve (similar to ROC AUC but takes class imbalance of the testing set into account), Kappa (the performance of the classifier compared to classification at random according to class frequency), and learning rate (lower values indicate better learning).
Hyperparameter tuning was performed with the ‘bayesOpt’ function of the ‘ParBayesianOptimization’ package in R.108 The following tuning parameters were considered: max_depth (maximal depth of decision tree), eta (learning rate, lower rates allow for better granularity of the model), gamma (modulator for tree complexity), colsample_bytree (the proportion of features used for each tree), min_child_weight (additional modulator for tree complexity), nrounds (number of boosting rounds), and subsample (proportion of samples used for each tree). A grid design containing fifty different combinations of tuning parameters was used to initialize the search (generated by using the ‘maximinLHS’ function from the ‘LHS’ package in R). After initialization, the optimization algorithm was programmed to perform ten optimization steps with ten samplings of the search space (optimums) at each step. The acquisition function type used to assess performance was excepted improvement (ei) accompanied by the tunable parameter, epilson, which balances exploitation of the search space with exploration. Other important parameters considered were the following: convergence threshold (maximum acceptable difference in ei between successive optimization steps), utility (score that represents the potential to find a better parameter set than the previous), acquisition threshold (percentage of global optimal utility required for a local optimum to be included as a candidate parameter set in the successive optimization step), and time limit on whole optimization process. Additionally, we utilized the upsampling method, SMOTE (Synthetic minority over-sampling technique),109 during resampling to account for class imbalances (FLC samples ≫ NML samples).
Eight datasets were used for XgBoostML nested CV. Background datasets (noise) were created by permutating the original datasets (proteomics and metabolomics) before data processing (normalization, transformation, or co-variate adjustment). A second set of background datasets (adjusted noise) were generated by processing the former datasets (noise). The original datasets (both preprocessed and postprocessed) were the remaining four inputs. Each input went through ten consecutive rounds of nested CV, and the following comparisons were made with each performance metric: noise-to-adjusted noise, noise-to-original preprocessed, adjusted noise-to-original postprocessed, and original preprocessed-to-original postprocessed. Mann Whitney was used for statistical testing.
Pathway enrichment
For Qiagen Ingenuity Pathway Analysis (IPA), differential abundance profiles of mitochondrial-specific proteins (n = 1004) and the metabolome (n = 135) from our proteomics and metabolomics data, respectively, were used as initial input. Qiagen IPA required a threshold to determine what it called “analysis-ready” molecules. We used an FDR-adjusted p-value cut-off of 0.1 as the threshold, which filtered the initial input to 617 analysis-ready molecules. Pathway enrichment was determined by comparing the number of analysis-ready molecules from our differential abundance profiles that overlapped with molecule sets from Qiagen pathways to the overlap of 617 randomly chosen molecules from the initial input with molecule sets from Qiagen pathways. A right-tailed Fisher’s Exact test was used for statistical analysis, and p-values were adjusted using Benjamini-Hochberg. For each pathway, Qiagen IPA used a Z score algorithm on the analysis-ready molecules to compute a Z score that represented pathway activity. This pathway activity score was statistically compared to Qiagen IPA’s canonical patterns of activity for a given pathway. A pathway with an activity score greater than 2 or less than −2 was predicted to be activated or suppressed, respectively.
Retrospective cohort studies
Serum ammonia & tumor burden
A retrospective cohort study was performed on patients with FLC using the Johns Hopkins Liver Cancer Database. Only patients with: (i) a confirmed pathologic diagnosis, (ii) imaging of the chest, abdomen, and pelvis sufficient for evaluation of FLC tumor burden, and (iii) serum ammonia measurement ordered as standard of care, were eligible for this analysis. Ammonia was considered to be elevated above 32 mmol/dL for adults (age ≥18) and above 55 mmol/dL for children (age 12–18). If patients had multiple assessments of serum ammonia, the serum ammonia level obtained at the time when FLC tumor burden was greatest was reported. FLC tumor burden was assessed by adding the sum of the diameter of up to 5 measurable tumor lesions using RECIST 1.1 criteria.
Examination of case studies from literature
Case studies of patients with FLC were collected by literature search. “FLC”, “fibrolamellar carcinoma”, “hyperammonemia”, and “case study” were the combination of search terms in PubMed and Google Scholar. A total of eighteen case studies from the literature were identified with an additional two cases generously donated from the John Hopkins University (JHU) medical center cleared via IRB approval. Cases from the literature were split evenly by gender (nine male and nine female), while the gender of the two cases from JHU were undisclosed. The median age of the cohort was 23. These demographics are in alignment with data from the Surveillance, Epidemiology, and End Results (SEER) program. Data was collected on levels of serum ammonia, plasma AAs, urinary orotic acid, urinary creatinine, and circulating liver enzymes. The time at which the aforementioned measurements were performed (pre- or post-treatment of HA) was noted and information on drugs used for treatment was also documented. In some cases, the following additional parameters were noted: suspected OTC deficiency, OTC mutation status, patient responsiveness to arginine supplementation, viral infection status, cirrhosis, chronic liver disease, drug-induced HA encephalopathy, portosystemic shunting, portal hypertension, liver failure, and idiopathic etiology.
All data visualization was generated using a combination of R, Python, and BioRender.
Acknowledgments
We are grateful for the support and comments provided by the members of the Sethupathy lab, especially Michael T. Shanahan, who offered rich intellectual discourse on this project and beyond. We also thank the Proteomics Core Facility at the Karolinska Institute as well as the Weill Cornell Metabolomics Facility for their service. We are particularly grateful to the FLC patient donors whose contributions made this work possible. Some figure schematics were created with BioRender.com. This work was supported by funding from the National Science Foundation Graduate Research Fellowship (awarded to D.L.,Jr.), the Howard Hughes Medical Institute Gilliam Fellows Program (awarded to D.L.,Jr. and P.S.), the National Cancer Institute (R01CA273081, awarded to T.S.G.), and the Fibrolamellar Cancer Foundation Research Grant (two grants awarded respectively to P.S. and T.S.G.). Data presented in Figures 4M and 6E were funded by a grant from the U.S. Food and Drug Administration (U01FD007925, awarded to T.S.G.). We are indebted to the foundation for their tireless efforts to promote FLC research, coordinate tissue banks and other collaborative efforts, and ultimately serve patients and caregivers.
Author contributions
Project lead: D.L.,Jr. Conception and design: D.L.,Jr. and P.S. Acquisition of data: D.L.,Jr., T.S.G., M.H., P.-Y.T., R.K.M., A.B.F., M.Y., L.M.O., and N.M.V. Analysis and interpretation of data: primarily D.L.,Jr. but also M.H., P.-Y.T., R.K.M., J.B., D.B.S., M.Y., Z.K., L.M.O., N.M.V., M.J.M., and P.S. Writing – original draft: D.L.,Jr. and P.S. Writing – editing: D.L.,Jr., M.H., M.Y., L.M.O., J.B., N.M.V., and P.S. Writing – final review: all authors. Acquisition of funding: P.S. Study supervision: P.S.
Declaration of interests
The authors declare no competing interests.
Published: August 28, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101699.
Contributor Information
Donald Long, Jr., Email: dl964@cornell.edu.
Praveen Sethupathy, Email: pr46@cornell.edu.
Supplemental information
Output data from PCA analysis of (a) total patient cohort or (b) omics-matched cohort. Output data that serve as input for hierarchical clustering and heatmap encompassing (c) total patient cohort or (d) omics-matched cohort. (a–d) MET, metastatic tissue (ntotal = 9, nomics_matched = 9); PRI, primary tissue (ntotal = 7, nomics_matched = 4); NML, non-malignant tissue (ntotal = 7, nomics_matched = 4). Omics-matched refers to patient samples that were present in both the proteomic and metabolomic datasets.
Output data from PCA analysis of (a) total patient cohort or (b) omics-matched cohort. Output data that serve as input for hierarchical clustering and heatmap encompassing (c) total patient cohort or (d) omics-matched cohort. (a–d) MET, metastatic tissue (ntotal = 12, nomics_matched = 10); PRI, primary tissue (ntotal = 8, nomics_matched = 3); NML, non-malignant tissue (ntotal = 6, nomics_matched = 4). Omics-matched refers to patient samples that were present in both the metabolomic and proteomic datasets.
(a) Data related to the degree of overlap between differentially expressed proteins that are associated with either the full (n = 23 patient samples) or omics-matched (n = 17 patient samples) dataset. Data that serves as input for MA plots that depict protein expression from the (b) full and (c) omics-matched datasets. (b and c) The top-ten most highly expressed proteins that overlap between the two datasets are indicated by the last column.
(a) Data related to the degree of overlap between differentially abundant metabolites that are associated with either the full (n = 26 patient samples; 135 metabolites) or omics-matched (n = 17 patient samples; 119 metabolites) dataset. Data that serve as input for MA plots that depict protein expression from the (b) full and (c) omics-matched datasets.
References
- 1.Dinh T.A., Utria A.F., Barry K.C., Ma R., Abou-Alfa G.K., Gordan J.D., Jaffee E.M., ’Sco’t J.D., Zucman-Rossi J., O'Neill A.F., et al. A framework for fibrolamellar carcinoma research and clinical trials. Nat. Rev. Gastroenterol. Hepatol. 2022;19:328–342. doi: 10.1038/s41575-022-00580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lalazar G., Simon S.M. Fibrolamellar Carcinoma: Recent Advances and Unresolved Questions on the Molecular Mechanisms. Semin. Liver Dis. 2018;38:51–59. doi: 10.1055/s-0037-1621710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Neill A.F., Church A.J., Perez-Atayde A.R., Shaikh R., Marcus K.J., Vakili K. Fibrolamellar carcinoma: An entity all its own. Curr. Probl. Cancer. 2021;45:100770. doi: 10.1016/j.currproblcancer.2021.100770. [DOI] [PubMed] [Google Scholar]
- 4.Stipa F., Yoon S.S., Liau K.’H., Fong Y., Jarnagin W.R., D'Angelica M., Abou-Alfa G., Blumgart L.H., DeMatteo R.P. Outcome of patients with fibrolamellar hepatocellular carcinoma. Cancer. 2006;106:1331–1338. doi: 10.1002/cncr.21703. [DOI] [PubMed] [Google Scholar]
- 5.Ramai D., Ofosu A., Lai J.K., Gao Z.H., Adler D.G. Fibrolamellar Hepatocellular Carcinoma: A Population-Based Observational Study. Dig. Dis. Sci. 2021;66:308–314. doi: 10.1007/s10620-020-06135-3. [DOI] [PubMed] [Google Scholar]
- 6.Honeyman J.N., Simon E.P., Robine N., Chiaroni-Clarke R., Darcy D.G., Lim I.I.P., Gleason C.E., Murphy J.M., Rosenberg B.R., Teegan L., et al. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science (New York, N.Y.) 2014;343:1010–1014. doi: 10.1126/science.1249484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelholm L.H., Riaz A., Serra D., Dagnæs-Hansen F., Johansen J.v., Santoni-Rugiu E., Hansen S.H., Niola F., Frödin M. CRISPR/Cas9 Engineering of Adult Mouse Liver Demonstrates That the Dnajb1–Prkaca Gene Fusion Is Sufficient to Induce Tumors Resembling Fibrolamellar Hepatocellular Carcinoma. Gastroenterology. 2017;153:1662–1673.e10. doi: 10.1053/J.GASTRO.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kastenhuber E.R., Lalazar G., Houlihan S.L., Tschaharganeh D.F., Baslan T., Chen C.C., Requena D., Tian S., Bosbach B., Wilkinson J.E., et al. DNAJB1–PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA. 2017;114:13076–13084. doi: 10.1073/pnas.1716483114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumayer C., Ng D., Jiang C.S., Qureshi A., Lalazar G., Vaughan R., Simon S.M. Oncogenic Addiction of Fibrolamellar Hepatocellular Carcinoma to the Fusion Kinase DNAJB1-PRKACA. Clin. Cancer Res. 2023;29:271–278. doi: 10.1158/1078-0432.CCR-22-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schalm S.S., O’Hearn E., Wilson K., LaBranche T.P., Silva G., Zhang Z., DiPietro L., Bifulco N., Woessner R., Stransky N., et al. Evaluation of Protein Kinase cAMP-Activated Catalytic Subunit Alpha as a Therapeutic Target for Fibrolamellar Carcinoma. Gastro Hep Advances. 2023;2:307–321. doi: 10.1016/j.gastha.2022.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Averill A.M., Rehman H.tul, Charles J.W., Dinh T.A., Danyal K., Verschraegen C.F., Stein G.S., Dostmann W.R., Ramsey J.E. Inhibition of the chimeric DnaJ-PKAc enzyme by endogenous inhibitor proteins. J. Cell. Biochem. 2019;120:13783–13791. doi: 10.1002/jcb.28651. [DOI] [PubMed] [Google Scholar]
- 12.Dinh T.A., Sritharan R., Smith F.D., Francisco A.B., Ma R.K., Bunaciu R.P., Kanke M., Danko C.G., Massa A.P., Scott J.D., Sethupathy P. Hotspots of Aberrant Enhancer Activity in Fibrolamellar Carcinoma Reveal Candidate Oncogenic Pathways and Therapeutic Vulnerabilities. Cell Rep. 2020;31:107509. doi: 10.1016/j.celrep.2020.03.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solomon L.N., Tomasini M.D., Knox J., Shirani M., Shebl B., Requena D., Clark J., Heissel S., Alwaseem H., Surjan R., et al. Disruption of proteome by an oncogenic fusion kinase alters metabolism in fibrolamellar hepatocellular carcinoma. Sci. Adv. 2023;9:eadg7038. doi: 10.1126/sciadv.adg7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navale A.M., Paranjape A.N. Glucose transporters: physiological and pathological roles. Biophys. Rev. 2016;8:5–9. doi: 10.1007/s12551-015-0186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joost H.G., Bell G.I., Best J.D., Birnbaum M.J., Charron M.J., Chen Y.T., Doege H., James D.E., Lodish H.F., Moley K.H., et al. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am. J. Physiol. Endocrinol. Metab. 2002;282:E974–E976. doi: 10.1152/ajpendo.00407.2001. [DOI] [PubMed] [Google Scholar]
- 16.Thorens B., Mueckler M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010;298:E141–E145. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uldry M., Thorens B. The SLC2 family of facilitated hexose and polyol transporters. Pflugers Arch. 2004;447:480–489. doi: 10.1007/s00424-003-1085-0. [DOI] [PubMed] [Google Scholar]
- 18.Waeber G., Thompson N., Haefliger J.A., Nicod P. Characterization of the murine high Km glucose transporter GLUT2 gene and its transcriptional regulation by glucose in a differentiated insulin-secreting cell line. J. Biol. Chem. 1994;269:26912–26919. [PubMed] [Google Scholar]
- 19.Thorens B., Cheng Z.Q., Brown D., Lodish H.F. Liver glucose transporter: a basolateral protein in hepatocytes and intestine and kidney cells. Am. J. Physiol. 1990;259:C279–C285. doi: 10.1152/ajpcell.1990.259.2.C279. [DOI] [PubMed] [Google Scholar]
- 20.Mueckler M., Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 2013;34:121–138. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kersten C.A., Sloey E.N., Zhou E., Peng Y., Torbenson M.S., Guo Y. Fibrolamellar hepatocellular carcinoma: Exploring molecular mechanisms and differentiation pathways to better understand disease outcomes and prognosis. Liver Research. 2017;1:187–192. doi: 10.1016/j.livres.2017.12.003. [DOI] [Google Scholar]
- 22.Vivekanandan P., Daniel H., Yeh M.M., Torbenson M. Mitochondrial Mutations in Hepatocellular Carcinomas and Fibrolamellar Carcinomas. Mod. Pathol. 2010;23:790–798. doi: 10.1038/2Fmodpathol.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Craig J.R., Peters R.L., Edmondson H.A., Omata M. Fibrolamellar carcinoma of the liver: A tumor of adolescents and young adults with distinctive clinico-pathologic features. Cancer. 1980;46:372–379. doi: 10.1002/1097-0142(19800715)46:2/3C372::AID-CNCR2820460227/3E3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 24.Payne C.M., Nagle R.B., Paplanus S.H., Graham A.R. Fibrolamellar carcinoma of liver: a primary malignant oncocytic carcinoid? Ultrastruct. Pathol. 1986;10:539–552. doi: 10.3109/01913128609007211. [DOI] [PubMed] [Google Scholar]
- 25.Oikawa T., Wauthier E., Dinh T.A., Selitsky S.R., Reyna-Neyra A., Carpino G., Levine R., Cardinale V., Klimstra D., Gaudio E., et al. Model of fibrolamellar hepatocellular carcinomas reveals striking enrichment in cancer stem cells. Nat. Commun. 2015;6:8070. doi: 10.1038/ncomms9070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S.H., Duron H.E., Chaudhuri D. Beyond the TCA cycle: new insights into mitochondrial calcium regulation of oxidative phosphorylation. Biochem. Soc. Trans. 2023;51:1661–1673. doi: 10.1042/2FBST20230012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cogliati S., Lorenzi I., Rigoni G., Caicci F., Soriano M.E. Regulation of Mitochondrial Electron Transport Chain Assembly. J. Mol. Biol. 2018;430:4849–4873. doi: 10.1016/j.jmb.2018.09.016. [DOI] [PubMed] [Google Scholar]
- 28.Marchetti P., Fovez Q., Germain N., Khamari R., Kluza J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. Faseb. J. : official publication of the Federation of American Societies for Experimental Biology. 2020;34:13106–13124. doi: 10.1096/fj.202000767R. [DOI] [PubMed] [Google Scholar]
- 29.Francisco A.B., Kanke M., Massa A.P., Dinh T.A., Sritharan R., Vakili K., Bardeesy N., Sethupathy P. Multiomic analysis of microRNA-mediated regulation reveals a proliferative axis involving miR-10b in fibrolamellar carcinoma. JCI insight. 2022;7:e154743. doi: 10.1172/jci.insight.154743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X., Qiao Y., Ting X., Si W. Isocitrate dehydrogenase 3A, a rate-limiting enzyme of the TCA cycle, promotes hepatocellular carcinoma migration and invasion through regulation of MTA1, a core component of the NuRD complex. Am. J. Cancer Res. 2020;10:3212–3229. https://pubmed.ncbi.nlm.nih.gov/33163266 [PMC free article] [PubMed] [Google Scholar]
- 31.May J.L., Kouri F.M., Hurley L.A., Liu J., Tommasini-Ghelfi S., Ji Y., Gao P., Calvert A.E., Lee A., Chandel N.S., et al. IDH3α regulates one-carbon metabolism in glioblastoma. Sci. Adv. 2019;5:eaat0456. doi: 10.1126/sciadv.aat0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du B., Sun T., Li X., Diao Y., Li Y. Effect of IDH3a on glucose uptake in lung adenocarcinoma: A pilot study based on [18 F]FDG. Cancer Med. 2019;8:5341–5351. doi: 10.1002/cam4.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemasters J.J., Holmuhamedov E. Voltage-dependent anion channel —DA— as mitochondrial governator--thinking outside the box. Biochim. Biophys. Acta. 2006;1762:181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Fang D., Maldonado E.N. VDAC Regulation: A Mitochondrial Target to Stop Cell Proliferation. Adv. Cancer Res. 2018;138:41–69. doi: 10.1016/bs.acr.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCommis K.S., Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem. J. 2015;466:443–454. doi: 10.1042/BJ20141171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang D., Chen X., Kang R., Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–125. doi: 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y., Li Y., Zhang R., Wang F., Wang T., Jiao Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. OncoTargets Ther. 2020;13:5429–5441. doi: 10.2147/2FOTT.S254995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yagoda N., von Rechenberg M., Stockwell B.R., Zaganjor E., Bauer A.J., Yang W.S., Fridman D.J., Wolpaw A.J., Smukste I., Peltier J.M., et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/2Fnature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Y., Luo M., Zhang K., Zhang J., Gao T., Connell D.O.’, Yao F., Mu C., Cai B., Shang Y., Chen W. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020;11:433. doi: 10.1038/s41467-020-14324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan G.K.L., Maisel S., Hwang Y.C., Pascual B.C., Wolber R.R.B., Vu P., Patra K.C., Bouhaddou M., Kenerson H.L., Lim H.C., et al. Oncogenic PKA signaling increases c-MYC protein expression through multiple targetable mechanisms. Elife. 2023;12:e69521. doi: 10.7554/eLife.69521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Khallaf H. Isocitrate dehydrogenases in physiology and cancer: biochemical and molecular insight. Cell Biosci. 2017;7:37. doi: 10.1186/s13578-017-0165-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dinh T.A., Vitucci E.C.M., Wauthier E., Graham R.P., Pitman W.A., Oikawa T., Chen M., Silva G.O., Greene K.G., Torbenson M.S., et al. Comprehensive analysis of The Cancer Genome Atlas reveals a unique gene and non-coding RNA signature of fibrolamellar carcinoma. Sci. Rep. 2017;7:44653. doi: 10.1038/srep44653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrett D.J., Bateman J.B., Sparkes R.S., Mohandas T., Klisak I., Inana G. Chromosomal localization of human ornithine aminotransferase gene sequences to 10q26 and Xp11.2. Invest. Ophthalmol. Vis. Sci. 1987;28:1037–1042. [PubMed] [Google Scholar]
- 44.Inana G., Totsuka S., Redmond M., Dougherty T., Nagle J., Shiono T., Ohura T., Kominami E., Katunuma N. Molecular cloning of human ornithine aminotransferase mRNA. Proc. Natl. Acad. Sci. USA. 1986;83:1203–1207. doi: 10.1073/pnas.83.5.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuzawa T., Katsunuma T., Katunuma N. Crystallization of ornithine transaminase and its properties. Biochem. Biophys. Res. Commun. 1968;32:161–166. doi: 10.1016/0006-291x(68)90363-x. [DOI] [PubMed] [Google Scholar]
- 46.Storici P., Capitani G., Müller R., Schirmer T., Jansonius J.N. Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine. J. Mol. Biol. 1999;285:297–309. doi: 10.1006/jmbi.1998.2289. [DOI] [PubMed] [Google Scholar]
- 47.Boffa I., Polishchuk E., De Stefano L., Dell'Aquila F., Nusco E., Marrocco E., Audano M., Pedretti S., Caterino M., Bellezza I., et al. Liver-directed gene therapy for ornithine aminotransferase deficiency. EMBO Mol. Med. 2023;15:e17033. doi: 10.15252/emmm.202217033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cleary M.A., Dorland L., de Koning T.J., Poll-The B.T., Duran M., Mandell R., Shih V.E., Berger R., Olpin S.E., Besley G.T.N. Ornithine aminotransferase deficiency: diagnostic difficulties in neonatal presentation. J. Inherit. Metab. Dis. 2005;28:673–679. doi: 10.1007/s10545-005-0074-1. [DOI] [PubMed] [Google Scholar]
- 49.Wang T., Steel G., Milam A.H., Valle D. Correction of ornithine accumulation prevents retinal degeneration in a mouse model of gyrate atrophy of the choroid and retina. Proc. Natl. Acad. Sci. USA. 2000;97:1224–1229. doi: 10.1073/pnas.97.3.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monné M., Miniero D.V., Daddabbo L., Robinson A.J., Kunji E.R.S., Palmieri F. Substrate specificity of the two mitochondrial ornithine carriers can be swapped by single mutation in substrate binding site. J. Biol. Chem. 2012;287:7925–7934. doi: 10.1074/jbc.M111.324855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walker M.C., van der Donk W.A. The many roles of glutamate in metabolism. J. Ind. Microbiol. Biotechnol. 2016;43:419–430. doi: 10.1007/s10295-015-1665-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nissen J.D., Pajęcka K., Stridh M.H., Skytt D.M., Waagepetersen H.S. Dysfunctional TCA-Cycle Metabolism in Glutamate Dehydrogenase Deficient Astrocytes. Glia. 2015;63:2313–2326. doi: 10.1002/glia.22895. [DOI] [PubMed] [Google Scholar]
- 53.Chaudhry F.A., Reimer R.J., Edwards R.H. The glutamine commute: take the N line and transfer to the A. J. Cell Biol. 2002;157:349–355. doi: 10.1083/jcb.200201070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aral B., Schlenzig J.S., Liu G., Kamoun P. Database cloning human delta 1-pyrroline-5-carboxylate synthetase (P5CS) cDNA: a bifunctional enzyme catalyzing the first 2 steps in proline biosynthesis. C. R. Acad. Sci. III. 1996;319:171–178. https://pubmed.ncbi.nlm.nih.gov/8761662/ [PubMed] [Google Scholar]
- 55.Baumgartner M.R., Hu C.A., Almashanu S., Steel G., Obie C., Aral B., Rabier D., Kamoun P., Saudubray J.M., Valle D. Hyperammonemia with reduced ornithine, citrulline, arginine and proline: a new inborn error caused by a mutation in the gene encoding delta(1)-pyrroline-5-carboxylate synthase. Hum. Mol. Genet. 2000;9:2853–2858. doi: 10.1093/hmg/9.19.2853. [DOI] [PubMed] [Google Scholar]
- 56.Bicknell L.S., Pitt J., Aftimos S., Ramadas R., Maw M.A., Robertson S.P. A missense mutation in ALDH18A1, encoding Delta1-pyrroline-5-carboxylate synthase (P5CS), causes an autosomal recessive neurocutaneous syndrome. Eur. J. Hum. Genet. 2008;16:1176–1186. doi: 10.1038/ejhg.2008.91. [DOI] [PubMed] [Google Scholar]
- 57.Masisi B.K., El Ansari R., Alfarsi L., Rakha E.A., Green A.R., Craze M.L. The role of glutaminase in cancer. Histopathology. 2020;76:498–508. doi: 10.1111/his.14014. [DOI] [PubMed] [Google Scholar]
- 58.Lukey M.J., Cluntun A.A., Katt W.P., Lin M.C.J., Druso J.E., Ramachandran S., Erickson J.W., Le H.H., Wang Z.E., Blank B., et al. Liver-Type Glutaminase GLS2 Is a Druggable Metabolic Node in Luminal-Subtype Breast Cancer. Cell Rep. 2019;29:76–88.e7. doi: 10.1016/j.celrep.2019.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reversade B., Escande-Beillard N., Dimopoulou A., Fischer B., Chng S.C., Li Y., Shboul M., Tham P.Y., Kayserili H., Al-Gazali L., et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat. Genet. 2009;41:1016–1021. doi: 10.1038/ng.413. [DOI] [PubMed] [Google Scholar]
- 60.De Ingeniis J., Ratnikov B., Richardson A.D., Scott D.A., Aza-Blanc P., De S.K., Kazanov M., Pellecchia M., Ronai Z., Osterman A.L., Smith J.W. Functional specialization in proline biosynthesis of melanoma. PLoS One. 2012;7:e45190. doi: 10.1371/journal.pone.0045190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakayama T., Al-Maawali A., El-Quessny M., Rajab A., Khalil S., Stoler J.M., Tan W.H., Nasir R., Schmitz-Abe K., Hill R.S., et al. Mutations in PYCR2, Encoding Pyrroline-5-Carboxylate Reductase 2, Cause Microcephaly and Hypomyelination. Am. J. Hum. Genet. 2015;96:709–719. doi: 10.1016/j.ajhg.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maynard T.M., Meechan D.W., Dudevoir M.L., Gopalakrishna D., Peters A.Z., Heindel C.C., Sugimoto T.J., Wu Y., Lieberman J.A., Lamantia A.S. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol. Cell. Neurosci. 2008;39:439–451. doi: 10.1016/j.mcn.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Phang J.M. The regulatory mechanisms of proline and hydroxyproline metabolism: Recent advances in perspective. Front. Oncol. 2022;12:1118675. doi: 10.3389/fonc.2022.1118675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Myllyharju J. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann. Med. 2008;40:402–417. doi: 10.1080/07853890801986594. [DOI] [PubMed] [Google Scholar]
- 65.Meng Z., Lou Z., Liu Z., Li M., Zhao X., Bartlam M., Rao Z. Crystal structure of human pyrroline-5-carboxylate reductase. J. Mol. Biol. 2006;359:1364–1377. doi: 10.1016/j.jmb.2006.04.053. [DOI] [PubMed] [Google Scholar]
- 66.Bender H.U., Almashanu S., Steel G., Hu C.A., Lin W.W., Willis A., Pulver A., Valle D. Functional consequences of PRODH missense mutations. Am. J. Hum. Genet. 2005;76:409–420. doi: 10.1086/428142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim S.Z., Varvogli L., Waisbren S.E., Levy H.L. Hydroxyprolinemia: comparison of a patient and her unaffected twin sister. J. Pediatr. 1997;130:437–441. doi: 10.1016/s0022-3476(97)70207-5. [DOI] [PubMed] [Google Scholar]
- 68.Halme J., Kivirikko K.I., Simons K. Isolation and partial characterization of highly purified protocollagen proline hydroxylase. Biochim. Biophys. Acta. 1970;198:460–470. doi: 10.1016/0005-2744(70)90124-5. [DOI] [PubMed] [Google Scholar]
- 69.Lamberg A., Pihlajaniemi T., Kivirikko K.I. Site-directed mutagenesis of the alpha subunit of human prolyl 4-hydroxylase. Identification of three histidine residues critical for catalytic activity. J. Biol. Chem. 1995;270:9926–9931. doi: 10.1074/jbc.270.17.9926. [DOI] [PubMed] [Google Scholar]
- 70.Annunen P., Helaakoski T., Myllyharju J., Veijola J., Pihlajaniemi T., Kivirikko K.I. Cloning of the human prolyl 4-hydroxylase alpha subunit isoform alpha(II) and characterization of the type II enzyme tetramer. The alpha(I) and alpha(II) subunits do not form a mixed alpha(I)alpha(II)beta2 tetramer. J. Biol. Chem. 1997;272:17342–17348. doi: 10.1074/jbc.272.28.17342. [DOI] [PubMed] [Google Scholar]
- 71.Cirillo P., Gersch M.S., Mu W., Scherer P.M., Kim K.M., Gesualdo L., Henderson G.N., Johnson R.J., Sautin Y.Y. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J. Am. Soc. Nephrol. 2009;20:545–553. doi: 10.1681/ASN.2008060576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xiong G., Deng L., Zhu J., Rychahou P.G., Xu R. Prolyl-4-hydroxylase α subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer. 2014;14:1. doi: 10.1186/1471-2407-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu Y., Borchert G.L., Donald S.P., Surazynski A., Hu C.A., Weydert C.J., Oberley L.W., Phang J.M. MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis. 2005;26:1335–1342. doi: 10.1093/carcin/bgi083. [DOI] [PubMed] [Google Scholar]
- 74.Liu Y., Borchert G.L., Surazynski A., Hu C.A., Phang J.M. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene. 2006;25:5640–5647. doi: 10.1038/sj.onc.1209564. [DOI] [PubMed] [Google Scholar]
- 75.Cooper S.K., Pandhare J., Donald S.P., Phang J.M. A novel function for hydroxyproline oxidase in apoptosis through generation of reactive oxygen species. J. Biol. Chem. 2008;283:10485–10492. doi: 10.1074/jbc.M702181200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hancock C.N., Liu W., Alvord W.G., Phang J.M. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino acids. 2016;48:859–872. doi: 10.1007/s00726-015-2134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taanman J.W. The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta. 1999;1410:103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 78.Schönfeld P., Wieckowski M.R., Lebiedzińska M., Wojtczak L. Mitochondrial fatty acid oxidation and oxidative stress: lack of reverse electron transfer-associated production of reactive oxygen species. Biochim. Biophys. Acta. 2010;1797:929–938. doi: 10.1016/j.bbabio.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 79.Rosca M.G., Vazquez E.J., Chen Q., Kerner J., Kern T.S., Hoppel C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes. 2012;61:2074–2083. doi: 10.2337/db11-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Panov A., Mayorov V.I., Dikalov S. Metabolic Syndrome and β-Oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria. Int. J. Mol. Sci. 2022;23:4047. doi: 10.3390/ijms23074047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lalazar G., Requena D., Ramos-Espiritu L., Ng D., Bhola P.D., de Jong Y.P., Wang R., Narayan N.J.C., Shebl B., Levin S., et al. Identification of Novel Therapeutic Targets for Fibrolamellar Carcinoma Using Patient-Derived Xenografts and Direct-from-Patient Screening. Cancer Discov. 2021;11:2544–2563. doi: 10.1158/2159-8290.CD-20-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., García-Seisdedos D., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moggridge S., Sorensen P.H., Morin G.B., Hughes C.S. Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics. J. Proteome Res. 2018;17:1730–1740. doi: 10.1021/acs.jproteome.7b00913. [DOI] [PubMed] [Google Scholar]
- 84.Branca R.M.M., Orre L.M., Johansson H.J., Granholm V., Huss M., Pérez-Bercoff Å., Forshed J., Käll L., Lehtiö J. HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods. 2014;11:59–62. doi: 10.1038/nmeth.2732. [DOI] [PubMed] [Google Scholar]
- 85.Boekel J., Chilton J.M., Cooke I.R., Horvatovich P.L., Jagtap P.D., Käll L., Lehtiö J., Lukasse P., Moerland P.D., Griffin T.J. Multi-omic data analysis using Galaxy. Nat. Biotechnol. 2015;33:137–139. doi: 10.1038/nbt.3134. [DOI] [PubMed] [Google Scholar]
- 86.Chen W.W., Freinkman E., Wang T., Birsoy K., Sabatini D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell. 2016;166:1324–1337.e11. doi: 10.1016/j.cell.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nishida-Aoki N., Bondesson A.J., Gujral T.S. Measuring Real-time Drug Response in Organotypic Tumor Tissue Slices. J. Vis. Exp. 2020 doi: 10.3791/61036. [DOI] [PubMed] [Google Scholar]
- 88.Sivakumar R., Chan M., Shin J.S., Nishida-Aoki N., Kenerson H.L., Elemento O., Beltran H., Yeung R., Gujral T.S. Organotypic tumor slice cultures provide a versatile platform for immuno-oncology and drug discovery. OncoImmunology. 2019;8:e1670019. doi: 10.1080/2F2162402X.2019.1670019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Horowitz L.F., Rodriguez A.D., Au-Yeung A., Bishop K.W., Barner L.A., Mishra G., Raman A., Delgado P., Liu J.T.C., Gujral T.S., et al. Microdissected "cuboids" for microfluidic drug testing of intact tissues. Lab Chip. 2021;21:122–142. doi: 10.1039/D0LC00801J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Savitski M.M., Wilhelm M., Hahne H., Kuster B., Bantscheff M. A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol. Cell. Proteomics. 2015;14:2394–2404. doi: 10.1074/mcp.M114.046995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu Y., Orre L.M., Zhou Tran Y., Mermelekas G., Johansson H.J., Malyutina A., Anders S., Lehtiö J. DEqMS: A Method for Accurate Variance Estimation in Differential Protein Expression Analysis. Mol. Cell. Proteomics. 2020;19:1047–1057. doi: 10.1074/mcp.TIR119.001646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Do K.T., Wahl S., Raffler J., Molnos S., Laimighofer M., Adamski J., Suhre K., Strauch K., Peters A., Gieger C., et al. Characterization of missing values in untargeted MS-based metabolomics data and evaluation of missing data handling strategies. Metabolomics. 2018;14:128. doi: 10.1007/s11306-018-1420-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang J., Zhao X., Lu X., Lin X., Xu G. A data preprocessing strategy for metabolomics to reduce the mask effect in data analysis. Front. Mol. Biosci. 2015;2:4. doi: 10.3389/fmolb.2015.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mera-Gaona M., Neumann U., Vargas-Canas R., López D.M. Evaluating the impact of multivariate imputation by MICE in feature selection. PLoS One. 2021;16:e0254720. doi: 10.1371/journal.pone.0254720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Josse J., Husson F. missMDA: A Package for Handling Missing Values in Multivariate Data Analysis. J. Stat. Softw. 2016;70:1–31. doi: 10.18637/jss.v070.i01. [DOI] [Google Scholar]
- 96.Stekhoven D.J., Bühlmann P. MissForest--non-parametric missing value imputation for mixed-type data. Bioinformatics. 2012;28:112–118. doi: 10.1093/bioinformatics/btr597. [DOI] [PubMed] [Google Scholar]
- 97.Mazumder R., Hastie T., Tibshirani R. Spectral Regularization Algorithms for Learning Large Incomplete Matrices. J. Mach. Learn. Res. 2010;11:2287–2322. [PMC free article] [PubMed] [Google Scholar]
- 98.Cornelisz I., Cuijpers P., Donker T., van Klaveren C. Addressing missing data in randomized clinical trials: A causal inference perspective. PLoS One. 2020;15:e0234349. doi: 10.1371/journal.pone.0234349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chong J., Wishart D.S., Xia J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinformatics. 2019;68:e86. doi: 10.1002/cpbi.86. [DOI] [PubMed] [Google Scholar]
- 100.Pang Z., Chong J., Zhou G., de Lima Morais D.A., Chang L., Barrette M., Gauthier C., Jacques P.É., Li S., Xia J. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021;49:W388–W396. doi: 10.1093/nar/gkab382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Benedetti E., Liu E.M., Tang C., Kuo F., Buyukozkan M., Park T., Park J., Correa F., Hakimi A.A., Intlekofer A.M., et al. A multimodal atlas of tumour metabolism reveals the architecture of gene–metabolite covariation. Nat. Metab. 2023;5:1029–1044. doi: 10.1038/s42255-023-00817-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.González-Domínguez Á., Estanyol-Torres N., Brunius C., Landberg R., González-Domínguez R. QComics: Recommendations and Guidelines for Robust, Easily Implementable and Reportable Quality Control of Metabolomics Data. Anal. Chem. 2024;96:1064–1072. doi: 10.1021/acs.analchem.3c03660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wanichthanarak K., Jeamsripong S., Pornputtapong N., Khoomrung S. Accounting for biological variation with linear mixed-effects modelling improves the quality of clinical metabolomics data. Comput. Struct. Biotechnol. J. 2019;17:611–618. doi: 10.1016/j.csbj.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.González-Domínguez Á., González-Domínguez R. How far are we from reliable metabolomics-based biomarkers? The often-overlooked importance of addressing inter-individual variability factors. Biochim. Biophys. Acta, Mol. Basis Dis. 2024;1870:166910. doi: 10.1016/j.bbadis.2023.166910. [DOI] [PubMed] [Google Scholar]
- 105.Whalen S., Schreiber J., Noble W.S., Pollard K.S. Navigating the pitfalls of applying machine learning in genomics. Nat. Rev. Genet. 2022;23:169–181. doi: 10.1038/s41576-021-00434-9. [DOI] [PubMed] [Google Scholar]
- 106.Tsai C.-F., Sung Y.-T. Ensemble feature selection in high dimension, low sample size datasets: Parallel and serial combination approaches. Knowl. Base Syst. 2020;203:106097. doi: 10.1016/j.knosys.2020.106097. [DOI] [Google Scholar]
- 107.Tsai C.-F., Hsiao Y.C., Yu-Chieh Combining multiple feature selection methods for stock prediction: Union, intersection, and multi-intersection approaches. Decis. Support Syst. 2010;501:258–269. doi: 10.1016/j.dss.2010.08.028. [DOI] [Google Scholar]
- 108.Snoek J., Larochelle H., Adams R.P. Practical Bayesian Optimization of Machine Learning Algorithms. Neural Information Processing Systems. 2012 doi: 10.5555/2999325.2999464. [DOI] [Google Scholar]
- 109.Chawla N.V., Bowyer K.W., Hall L.O., Kegelmeyer W.P. Smote: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002;16:321–357. doi: 10.1613/jair.953. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Output data from PCA analysis of (a) total patient cohort or (b) omics-matched cohort. Output data that serve as input for hierarchical clustering and heatmap encompassing (c) total patient cohort or (d) omics-matched cohort. (a–d) MET, metastatic tissue (ntotal = 9, nomics_matched = 9); PRI, primary tissue (ntotal = 7, nomics_matched = 4); NML, non-malignant tissue (ntotal = 7, nomics_matched = 4). Omics-matched refers to patient samples that were present in both the proteomic and metabolomic datasets.
Output data from PCA analysis of (a) total patient cohort or (b) omics-matched cohort. Output data that serve as input for hierarchical clustering and heatmap encompassing (c) total patient cohort or (d) omics-matched cohort. (a–d) MET, metastatic tissue (ntotal = 12, nomics_matched = 10); PRI, primary tissue (ntotal = 8, nomics_matched = 3); NML, non-malignant tissue (ntotal = 6, nomics_matched = 4). Omics-matched refers to patient samples that were present in both the metabolomic and proteomic datasets.
(a) Data related to the degree of overlap between differentially expressed proteins that are associated with either the full (n = 23 patient samples) or omics-matched (n = 17 patient samples) dataset. Data that serves as input for MA plots that depict protein expression from the (b) full and (c) omics-matched datasets. (b and c) The top-ten most highly expressed proteins that overlap between the two datasets are indicated by the last column.
(a) Data related to the degree of overlap between differentially abundant metabolites that are associated with either the full (n = 26 patient samples; 135 metabolites) or omics-matched (n = 17 patient samples; 119 metabolites) dataset. Data that serve as input for MA plots that depict protein expression from the (b) full and (c) omics-matched datasets.
Data Availability Statement
-
•
The mass spectrometry proteomics and metabolomics data have been deposited to the ProteomeXchange Consortium (via PRIDE82 partner repository) and NIH Common Funds National Metabolomics Data Repository (NMDR), respectively. The data is publicly available as of the date of publication. Accession numbers and DOIs are listed in the key resources table.
-
•
All code for execution of core analyses and generation of all main figures has been deposited at Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.