

Abstract
We have isolated 2′-Fluoro-substituted RNA aptamers that bind to streptavidin (SA) with an affinity around 7 ± 1.8 nM, comparable with that of recently described peptide aptamers. Binding to SA was not prevented by prior saturation with biotin, enabling nucleic acid aptamers to form useful ternary complexes. Mutagenesis, secondary structure analysis, ribonuclease footprinting and deletion analysis provided evidence for the essential structural features of SA-binding aptamers. In order to provide a general method for the exploitation of these aptamers, we produced derivatives in which they were fused to the naturally structured RNA elements, CopT or CopA. In parallel, we produced derivatives of CD4-binding aptamers fused to the complementary CopA or CopT elements. When mixed, these two chimeric aptamers rapidly hybridized, by virtue of CopA–CopT complementarity, to form stable, bi-functional aptamers that we called ‘adaptamers’. We show that a CD4–SA-binding adaptamer can be used to capture CD4 onto a SA-derivatized surface, illustrating their general utility as indirect affinity ligands.
INTRODUCTION
The post-genomic zeitgeist inspires the search for ways to document the activity of the proteome in experimental and diagnostic samples. Although monoclonal antibodies have provided a rich source of specific ligands for detecting the location and activity of proteins, the number of new targets outstrips the capacity of the methodology for generating and screening them. Alternative approaches to new ligand discovery involve in vitro evolution of either nucleic acids or their encoded polypeptides by selection from highly complex libraries generated by combinatorial synthesis. A number of factors will determine which of these approaches will gain widest acceptance, including the ease of use, particularly in automated systems; the physicochemical range of targets to which ligands are possible; the complexity of the library that can be screened; the chemical and structural resilience of the ligand; and the availability of downstream technologies for detection, amplification and manipulation. Nucleic acid ligands, or ‘aptamers’, have the advantages that the methods for their generation are relatively straightforward and that it is possible to screen a starting library of at least 1014 different sequences (1,2). Their potential disadvantage is their limited range of physicochemical properties, having no equivalents to the hydrophobic and basic residues of some amino acids (3). Polypeptide ligands have obvious advantages in this latter respect but phage display and similar systems for their discovery are hampered by a transfection-imposed bottleneck that limits library complexity to less than 109 and ribosome display methods have proved too fragile for general use (4,5). A recent method, called mRNA display (6), has overcome most of these difficulties through a number of elegant innovations that enable ∼1013 different, randomized, 88mer polypeptides to be screened. Recently, this approach produced peptide aptamers with 100-fold higher affinity for ATP than the best RNA aptamers (7) and with 1000-fold higher affinity for streptavidin (SA) than the best phage-display antibodies (8). Here we describe the isolation of 2′-Fluoro (2′-F)-substituted RNA aptamers that bind to SA with an affinity comparable with the peptide aptamers described above. We go on to show that our SA-binding aptamers can be used to exploit the specificity of other RNA ligands in general protein capture and detection systems, by generating hetero-bi-functional ligands through the interaction of complementary adapter sequences.
MATERIALS AND METHODS
Oligonucleotides and in vitro selection
The oligonucleotide library [5′-AATTAACCCTCACTAAAGGGAACTGTTGTGAGTCTCATGTCGAA(N)49TTGAGCGTCTAGTCTTGTCT-3′] incorporating 49 randomized nucleotides was converted into double-stranded template (9) by five rounds of polymerase chain reaction (PCR) in a 5 ml reaction volume with 5′-AATTAACCCTCACTAAAGGGAACTGTTGTGAGTCTCATGTCGAA-3′ (5′ primer) and 5′-TAATACGACTCACTATAGGGAGACAAGACTAGACGCTCAA-3′ (3′ primer). The resulting PCR products (1.8 nmol) were transcribed in a 1 ml reaction by T7 RNA polymerase in the presence of 2′-F-2′-deoxy-pyrimidine nucleoside-5′-triphosphates (TriLink BioTechnologies, Inc., San Diego, CA), together with 2′-OH-purine nucleoside-5′-triphosphates (10). After transcription, the template DNA was digested with RNase-free DNase I. The full-length 2′-F-RNA transcripts were purified by electrophoresis on 10% polyacrylamide/8 M urea gel. Affinity selection was initiated with 1.5 nmol of 2′-F-pyrimidine-containing RNA random sequence library. The RNA was incubated in water for 3 min at 95°C, cooled to room temperature before being refolded in the binding buffer (20 mM HEPES–NaOH pH 7.5, 100 mM NaCl, 50 mM KCl, 10 mM MgCl2) for 20 min at 20°C. The refolded pool of RNA was then mixed with 1 mg of Dynabeads M-280 SA (Dynal Biotech, UK) that were previously saturated with 800 pmol of a biotinylated 13-residue peptide. The first round of selection was carried out overnight at room temperature in a 500 µl volume with gentle mixing. The subsequent selection rounds 2–4 and 5–9 were scaled down to 0.6 and 0.3 mg of Dynabeads M-280 SA/biotin-saturated in 200 and 100 µl, respectively, and an incubation time of 2 h. The SA–RNA complex was separated from the unbound RNA with a Dynal magnetic particle concentrator (Dynal MPC-E) for 1 min and the supernatant removed. RNA molecules that were trapped non-specifically were removed by three washes with 200 µl of binding buffer. The bound RNA was converted to cDNA by reverse transcription with Thermus thermophilus (Tth) DNA polymerase at 70°C for 20 min following the protocol provided by the supplier (Promega, WI) followed by 15 cycles of PCR amplification (9). The resulting PCR products were used as template for in vitro transcription to produce RNA for the next round of selection. After rounds 2, 3 and 5, the enriched RNA libraries were pre-exposed to 0.3 mg of Dynabeads (without SA) in 200 µl for 1 h, to remove RNA sequences that bind to sites other than SA. Surface plasmon resonance (SPR) using BIACORE instrument (Biacore, Uppsala, Sweden) was used to assess the enrichment for SA-binders during in vitro selection.
The pool of RNA from the ninth round of in vitro selection was reverse transcribed and PCR amplified with primers (5′-CCGGAATTCCGGAATTAACCCTCACTAAAGGGAAC-TG-3′ and 5′-TCCCCCGGGGGATAATACGACTCACTATAGGGAGAC-3′) that introduce EcoRI and SmaI sites at the termini of the resulting DNA. The DNA was digested with EcoRI and SmaI, sub-cloned into pUC18 vector that had been previously digested with the same enzymes. Clones were sequenced by PRISM™BigDye™ cycle sequencing ready reaction kit from ABI (Perkin–Elmer).
Mutagenesis and re-selection
The nucleotide analog 6-(2-deoxy-β-d-erythropentofuranosyl)-3,4-dihydro-8H-pyrimido-[4,5C][1,2]oxazine-7-one-5′ triphosphate (dPTP) (11) was used to introduce mutations into SA19 aptamer. SA19 DNA template was amplified using 0.6 µl of Taq DNA polymerase (Promega, WI) in a 20 µl reaction containing the appropriate 5′ and 3′ primers described above at 0.5 µM, 3.5 mM MgCl2, 10 mM Tris–HCl pH 9.0, 50 mM KCl, 0.1% Triton X-100 and dATP, dCTP, dGTP, dPTP at 500 µM each. After an initial denaturation step at 93°C for 3 min, various cycles (0, 5, 10 and 15) were performed, each of which consisted of denaturation at 93°C for 50 s, annealing at 55°C for 30 s and elongation at 72°C for 1 min. The product of this first PCR was subjected to a second PCR in the presence of four natural dNTPs in order to eliminate the base analogs from the target DNA SA19 (11). The DNA from the second PCR amplification was used as template for in vitro transcription as described above. The 2′-F-pyrimidine-containing RNA transcripts from various mutagenesis cycles were analyzed by SPR to verify the abolition of the binding to SA. The pool of RNA from 15 cycles of mutagenic PCR in which SA-binders were completely lost as judged by SPR analysis was subjected to two rounds of in vitro selection as described above.
Adaptamer constructs
The CopA and/or CopT sequences (12,13) were inserted downstream of the SA19-aptamer sequence previously cloned into a pUC18 vector, using the EcoRI site. 2′-F-pyrimidine transcripts of the constructs gave aptamers with CopA or CopT at their 3′ terminus. Rat CD4 aptamers (14) were similarly engineered to contain CopA and/or CopT sequences.
Gel mobility shift assay
Dissociation constants for SA19 aptamer, chimeric SA19–CopA and the adaptamer SA19–CopA–E14–CopT binding to SA were quantified by native gel shift assays. In a typical binding experiment, 5′-32P-labeled aptamer (5000 c.p.m. Cerenkov) in 20 mM HEPES–NaOH pH 7.5, 100 mM NaCl, 50 mM KCl, 10 mM MgCl2 and 1 µg tRNA was incubated in the presence of increasing amounts of SA for 1 h at room temperature (25 µl volume). In the case of the adaptamer SA19–CopA–E14–CopT, the two 5′-end-labeled chimeric aptamers were first mixed at an equimolar ratio, heat denatured in water for 5 min at 95°C, then allowed to fold in the binding buffer for 20 min at room temperature before adding increasing concentrations of SA protein. After incubation was completed, 3 µl of 70% glycerol solution containing 0.025% (w/v) bromophenol blue was added to each binding reaction. The samples were then resolved on an 8% polyacrylamide native gel. Data were obtained from the gels using storage phosphor autoradiography and STORM phosphor imager (Molecular Dynamics). The ratio of bound to unbound RNA was quantified using ImageQuant software (Molecular Dynamics). Dissociation constants for SA19 aptamer, chimeric SA19–CopA and the adaptamer were derived from a fit by non-linear regression of GraphPad PRISM (GraphPad Software, Inc., San Diego, CA) to a hyperbolic function using the equation: Fraction bound = Bmax {[SA]/[SA] + Kd}, where Bmax represents the observed maximum fraction of aptamer bound, [SA] represents protein concentration and Kd is the dissociation constant.
Surface plasmon resonance
BIACORE 2000 was used to perform all binding studies. Research grade CM5 chips, NHS/EDC coupling reagents and ethanolamine were from BIACORE AB (Uppsala, Sweden). SA protein (Sigma) was immobilized onto sensor chip using amine-coupling chemistry. The immobilization steps were carried out at a flow rate of 5 µl/min in 20 mM HEPES–NaOH, 150 mM NaCl, 3.4 mM EDTA and 0.005% P20 surfactant. The flow cells were activated for 7 min with a mixture of NHS (0.05 M) and EDC (0.2 M). SA was injected at a concentration of 400 µg/ml in 10 mM sodium acetate pH 5.2, for 7 min. Ethanolamine (1 M, pH 8.5) was injected for 7 min to block remaining activated groups. An average of 5 kRU was immobilized on each flow cell. The assessment of enriched RNA binders to SA was done under the same running buffer that was supplemented with 50 mM KCl and 10 mM MgCl2. The RNA was refolded in the binding buffer as described above, and injected (35–60 µl) over the flow cells at 5 µl/min. Between consecutive injections, the surfaces were regenerated by long (60–120 min) washes with the running buffer. To correct for refractive index changes and instrument noise the response data from a reference surface were subtracted from the responses obtained from the reaction surface. The specificity of SA–aptamer interaction was assessed against various proteins including the soluble fraction of rat CD4, gp120, avidin and BSA. SPR analysis of adaptamers were performed in two ways: the aptamer–Cop species were either separately refolded and then injected sequentially, so that the adaptamers would form inside the flow cell, or premixed, refolded, allowed to anneal and then injected (the example shown in Figure 6B is illustrating the first case). Immediately after the RNA injections, a sample of rat CD4 was injected to test the ability of the adaptamers to simultaneously bind the two protein targets in the same flow cell.
Figure 6.

. Adaptamer formation and functional analysis. (A) Transcripts from Cop-aptamers were refolded and then loaded on a native 6% polyacrylamide gel, either separately or in pair-wise arrangement. The pair-wise combination was prepared by mixing 50 ng of each Cop–aptamer, refolding the mix to form the adaptamers, while 100 ng of each single aptamer-Cop RNA was loaded per lane. (B) SPR analysis of aptamer–Cop RNAs and adaptamers on SA-coated sensor chip. SA19–CopA (top panel) and SA19–CopT (bottom panel) were injected onto SA-coated flow cells, followed by rat CD4–Cop aptamer E14–CopT (top panel) or E14–CopA (bottom panel). Soluble rat CD4 protein was then injected (continuous lines). Control injections for specificity are shown underneath and correspond to each aptamer–Cop injection (dotted line). In particular, SA19–CopA and SA19–CopT were injected over rat CD4-coated flow cell, while E14–CopT and E14–CopA were injected over a SA-coated flow cell. Bars and arrows indicate length and end of injections, respectively.
Enzymatic probing and footprinting
SA19 aptamer was gel purified on 10% polyacrylamide/8 M urea gel, dephosphorylated and then labeled at the 5′ end with T4 polynucleotide kinase and [α-32P]ATP (15). Labeled aptamer was gel purified as above, eluted, and precipitated twice with ethanol. Before use, labeled SA19 RNA was dissolved in water, incubated at 90°C for 2 min, followed by slow cooling at 20°C in the binding buffer. Binding of 5′-end-labeled SA19 to SA protein was first allowed to form on Dynabeads M-280 SA (0.03 mg) for 1 h in the binding buffer. The unbound RNA was removed using Dynal MPC-E magnet before carrying on the experiments. As a control the RNA was incubated under identical conditions with Dynabeads lacking SA. Enzymatic hydrolysis of free or SA-bound labeled SA19 RNA was performed in 10 µl of binding buffer, in the presence of 1 µg carrier tRNA at 20°C for 10 min in presence of RNase V1 (0.07 U) or nuclease S1 (20 U). Reactions were stopped by phenol/chloroform extraction, followed by ethanol precipitation, and washing with 80% ethanol. Incubation control in the presence of Dynabeads M-280 SA but without adding any nuclease was done in parallel to detect any unspecific cleavage that may occur in the RNA. The products were then sized by electrophoresis on a 15 or 18% polyacrylamide/8 M urea gel. A partial alkaline hydrolysis ladder of the same aptamer (16) was run in parallel.
Chemical probing
Chemical probing was done as previously described (17–19). DMS (N1A, N3C), CMCT (N3U, N1G) and kethoxal (GN1, N2) modifications of 0.1 µg of gel-purified and refolded SA19 aptamer in the presence of 2 µg tRNA were carried out in 20 µl reaction volumes in 20 mM HEPES–NaOH pH 7.2 for DMS and CMCT and 50 mM sodium borate pH 8.0 for kethoxal. All buffers contained 10 mM MgCl2, 100 mM NaCl, 50 mM KCl. Reactions were performed at 20°C for 5 min in the presence of 1 µl of DMS (1:8 or 1:16 dilutions in ethanol), for 20 min in the presence of 1 µl of CMCT (40 or 20 mg/ml in water), or 5 min in the presence of 2 or 1 µl of kethoxal (20 mg/ml in 20% ethanol). After ethanol precipitation, the modified RNAs were dissolved in water. Unmodified SA19 aptamer was processed in parallel. Primer extension with 5′-32P-labeled primer (5′-AATTAACCCTCAC-3′) was used to detect the modified bases (20). Sequencing of unmodified SA19 aptamer was done as described by Sanger et al. (21).
The secondary structure model of SA19 aptamer was deduced from STAR software package (22,23) using stochastic and genetic folding algorithms. The predictions were constrained by imposing the data from solution probing.
RESULTS
Isolation of streptavidin-binding aptamers
A DNA library was synthesized, having a 49 nt randomized region flanked by constant regions that incorporate T7 and T3 RNA polymerase promoters for positive and negative strand transcription, respectively. Approximately 1014 different 2′-F-pyrimidine-substituted RNAs were synthesized by T7 RNA polymerase and those binding SA were selected using Dynabeads M-280 SA complexed to a biotinylated peptide. SA-bound aptamers were eluted and amplified by PCR to generate a library enriched for SA-binding RNA sequences. Progress of selection was monitored using a SPR biosensor to which SA had been covalently coupled (Fig. 1). The results show that RNA species with SA-binding properties become a significant component of the mixture by round 4 and the dominant component by round 8. There was no significant enrichment of SA-binders after round 9, consequently the aptamers were cloned and sequenced at this stage. The enriched RNA pool from round 9 did not show any binding to BSA (Fig. 1), indicating the specificity of the interaction with SA protein.
Figure 1.

Overlay of sensorgrams from SPR analysis showing enrichment for SA-aptamer during in vitro selection. Pool of 2′-F-RNA transcripts from rounds 2, 4, 5, 6, 8 and 9 were injected (∼75 nM) at a flow rate of 5 µl/min over a sensor chip pre-coated with 4.5 kRU SA. The specificity of the enriched RNA pool from round 9 was assessed against immobilized BSA (4.2 kRU). The arrow indicates the end of injections and start of buffer chase.
Binding characteristics of streptavidin aptamers
Fifty aptamer clones were sequenced. The alignment revealed that only 12 clones were distinct and fell into one sequence group with considerable sequence similarities (Table 1). The ability of these clones to bind SA protein was qualitatively assessed by SPR. All of them except clone SA27 were found to bind SA. We chose SA19 clone as the representative for further analysis. To determine the binding affinity of SA for this aptamer we used gel mobility shift assay. In our experimental conditions the interaction between SA19 aptamer and SA produced a single, saturable complex (Fig. 2A). Titration of SA protein to SA19 aptamer at a concentration of ∼3 nM yielded a complex of slower electrophoretic mobility than the free aptamer as shown in Figure 2A. To determine the apparent dissociation constant for this protein–aptamer interaction, the amount of 32P present in the free and bound aptamer bands was quantified and the binding data were fit to an equation that describes a simple bimolecular equilibrium (Fig. 2B). The result indicated a Kd of 7.0 ± 1.8 nM. SPR analysis was also used to obtain an independent value for the apparent dissociation constant, which was calculated to be similar to that obtained by gel mobility shift assay (data not shown).
Table 1. Sequence alignment of 2′-F-pyrimidine-containing RNA aptamers derived from affinity selection on SA.

Only the random region is shown. Aptamers derived from the parental SA19 by random mutagenesis followed by two rounds of in vitro re-selection are also aligned. The alignment was obtained with Clustal X program (version 1.64B). The cross symbol indicates non-binder aptamers and the diamond and asterisk indicate aptamers with slow on-rate and fast off-rate, respectively. Nucleotides that are variants between clones are shown in italic, those that cause loss of binding to SA, when mutated, are underlined and in bold, whereas the ones that are just underlined seem not to be essential for binding.
Figure 2.

Native gel mobility shift assay for SA binding to SA19 aptamer. (A) Storage phosphor autoradiogram of a representative gel used to separate free aptamer SA19 from SA19–SA complex using a range of increased protein concentrations from 1.8 to 447 nM. (B) Representative plot of fraction of aptamer bound by SA as a function of protein concentration. The data were fitted to a hyperbolic function by non-linear curve fitting method of GraphPad PRISM. This titration yielded an equilibrium dissociation constant (Kd) of 7 ± 1.8 nM.
The in vitro selection process was designed in order to isolate aptamers that would not compete for the biotin-binding site on the SA protein, which was achieved by pre-saturating SA with a biotinylated peptide. We used SPR analysis to test whether the aptamers were capable of binding SA protein that was pre-saturated with biotin. Three adjacent flow cells were coated with SA protein and one flow cell (number 2) was pre-saturated with biotin. The remaining flow cell (number 4) was used as a reference surface to correct for refractive index changes and instrument noise. SA19 aptamer was then injected over all flow cells and it was able to bind biotin-saturated SA on flow cell 2. The amount of aptamer bound was, however, approximately half of that on flow cells 1 and 3 (Fig. 3A). The kinetics of the interaction between the aptamer and the SA protein were not affected by the presence of biotin (data not shown). Interestingly, SA19 aptamer did not interact with the functionally related avidin. The specificity of the interaction was also assessed against other proteins, including gp120 and CD4, none of which was recognized by the aptamer (Fig. 3B).
Figure 3.

Overlay of sensorgrams showing the effect of biotin saturated-SA on the binding of aptamer and its specificity. (A) Flow cells 1–3 were pre-coated with 5.7, 4.9 and 4.8 kRU SA, respectively, and flow cell 4 was left as a blank control. Flow cell 2 was saturated with 0.113 kRU of biotin before injecting 200 nM of SA19 aptamer over flow cells 1–4 in series. (B) SA19 aptamer was injected at ∼50 nM over flow cells 1–4 previously coated with BaLgp120 (9 kRU), rat sCD4 (6 kRU), avidin (4.9 kRU) and SA (4.8 kRU), respectively. The arrow indicates the end of injections and start of buffer chase.
Streptavidin binds to a defined region of the aptamer
Clone SA19 was subjected to mutagenic PCR using the nucleotide analog dPTP and subsequent de novo selection in order to (i) identify mutants with improved binding properties; and (ii) map the positions of nucleotides that are involved in the interaction with the target protein.
The DNA from various mutagenic PCR cycles (see Materials and Methods) was used as template for in vitro transcription to generate 2′-F-transcripts. The resulting pools of RNAs were analyzed by SPR to examine the effects of the mutagenesis on the binding to SA protein (Fig. 4). By comparison with the control (0 cycle), the binding to SA was significantly reduced after five cycles of mutagenic PCR, almost abolished by cycle 10 and not detected by cycle 15 (Fig. 4). The pool of 2′-F-transcripts corresponding to 15 cycles of mutagenic PCR was the starting material from which the rare remaining SA ligands were rescued. This was achieved by two rounds of de novo selection on SA-coated magnetic beads followed by RT–PCR, cloning and sequencing. The resulting aptamers were designated SA19Mxx, where SA19M refers to the fact that each clone is a mutant form of the SA-binding aptamer SA19 and xx is an arbitrary two digit number referring to the clone (Table 1). Thirty aptamer clones were sequenced. Sequence comparison and alignment showed that nine clones were distinct and that the mutant aptamers were very similar to the parental sequence (Table 1). SPR analysis of the mutant aptamers showed that three clones (SA19M21, SA19M24 and SA19M27) had lost the ability to bind SA, while mutant SA19M15 had slower on-rate (4.7 × 103 s–1 M–1) as compared with the parental SA19 (2.5 × 104 s–1 M–1), whereas the off-rate remained the same. On the contrary, aptamer SA19M22 had a faster off-rate (6.0 × 10–3 s–1) as compared with SA19 (3.5 × 10–4 s–1), and an increased on-rate (4.4 × 104 s–1 M–1). The overall binding characteristics of the remaining mutants were comparable with those of the parental SA19 aptamer. Analysis of the primary sequence of the mutant SA19M15 showed four mutations (A49G, A56G, A58G and C59U). Since these mutations affect the association rate of the interaction, they are likely to be important in the initial binding events.
Figure 4.

Overlay of sensorgrams showing the effect of mutagenic PCR on binding of SA19 aptamer to SA. Template DNA from various mutagenesis cycles was used to produce 2′-F-RNA. Transcripts (∼65 nM) were injected over sensor chip pre-coated with 4.2 kRU of immobilized SA. The binding to SA was significantly reduced after five cycles of mutagenesis, almost abolished by cycle 10 and completely lost by cycle 15, as compared with the control (0 cycle).
The analysis of the three mutants, SA19M21, SA19M24 and SA19M27, as well as clone SA27 from the first selection, which had lost the ability to bind SA protein, allowed us to determine key nucleotides that are involved in the interaction. Two mutations, U48C and U57C, in SA27 and SA19M2, respectively, were sufficient to abolish the binding to SA protein. Additional nucleotide differences were also observed between non-binder SA27 and other binders, namely the A at position 32, G at position 68 and the U at position 71, none of which seemed to affect the binding to SA when mutated to G, A and G/A, respectively. This indicated a critical role of the 2′-F-UTP at position 48 in the binding to SA. Sequence analysis of SA19M21 and SA19M27 revealed that the presence of 2′-F-UTP at positions 25 and 57 was essential in the binding to SA. Nucleotides A38, U46 and G51 are mutated in the non-binding SA19M24, which also carries the C25U mutation, which is sufficient to abolish activity alone.
To gain a structural insight into the significance of these nucleotide substitutions, the secondary structure of SA19 RNA was probed using a combination of chemical and enzymatic probes. The footprint with V1 and S1 nucleases allowed us to delineate the binding site of SA on SA19 aptamer (Fig. 5A and B). The predicted secondary structure of the representative SA19 aptamer (Fig. 5A) can be divided into three domains. Domain I, from nucleotide 1 to 31, for which no chemical probing data were available, is predicted to fold into a stem, a symmetrical internal loop and a hairpin loop. Domain II, from nucleotide 32 to 75, and for which most of the nucleotides have been probed, presented a reactivity pattern that correlated well with the presence of two stem–loops linked by a stretch of 4 nt. Although there was a good correlation between the secondary structure predicted by STAR software and the solution probing data, some nucleotides such as 44, 45 and 47 in the first loop of domain II as well as nucleotides 96–98 in the loop of domain III were not reactive toward DMS. Similarly, there were nucleotides in the second stem of domain II that showed a degree of reactivity toward CMCT (Fig. 5A). These discrepancies could be explained by the presence of alternative conformers and/or the existence of tertiary structures that cannot be probed under the experimental conditions used for this study. Binding of SA induced several protections against nuclease S1 and RNase V1 hydrolysis in this domain. The major protections were located in a region encompassing residues 50–62, well correlated with the mutagenesis data showing that the modified U57 is essential for binding. Domain III, which contains the remainder of the aptamer sequence, was predicted to fold into a hairpin loop that is flanked by two single-stranded regions and was confirmed by the solution probing data. Deletion of this domain did not affect the binding to SA protein (data not shown).
Figure 5.

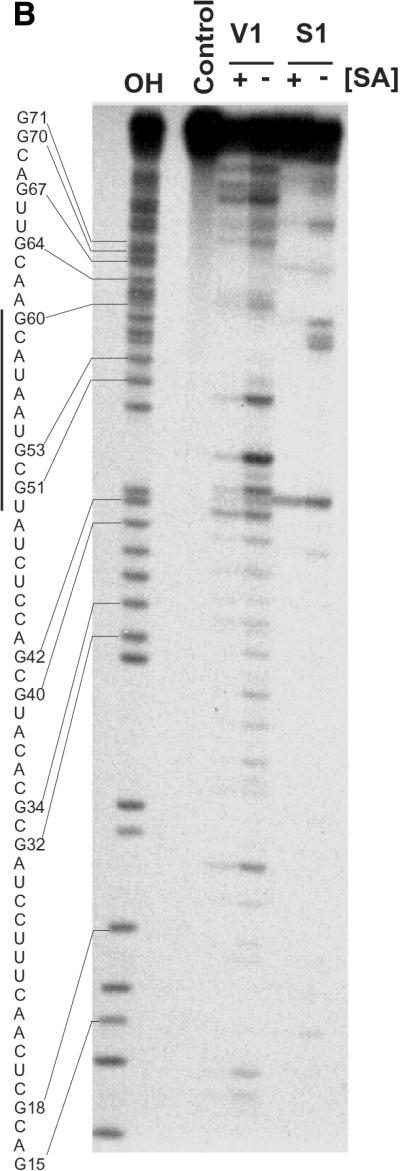
Solution structure of SA19 aptamer and SA footprinting. (A) Proposed secondary structure of SA19 aptamer as deduced from solution probing data and predictions with STAR software package. Reactive nucleotides towards DMS (C at N3), CMCT (U at N3, G at N1) and kethoxal (G at N1, N2) are encircled, and non-reactive nucleotides are boxed. No symbol is for not determined. The protected area is delimitated by black dots. (B) Autoradiogram of a 18% polyacrylamide/8 M urea gel, showing digestion products of 5′-end-labeled SA19 with RNase V1 and nuclease S1 in the presence (+) or absence (–) of SA; the major protected area is shown by a vertical line. Lane OH is the ladder from partially alkaline hydrolyzed SA19. The control lane corresponds to the 5′-end-labeled SA19 incubated in the presence of SA but in the absence of any nucleases. The gaps in the OH ladder are indicative of 2′-F-pyrimidines.
Formation of streptavidin–CD4 adaptamers
In order to develop a general method for forming aptamers that simultaneously bind SA and a second target protein, we fused the gene encoding SA19 with that encoding CopA or CopT structured RNAs from Escherichia coli plasmid R1 (13). In parallel, we also fused the gene encoding a rat CD4-binding aptamer, E14 (14), with that encoding the complementary CopT or CopA sequence. The addition of ∼100 nt that formed the Cop sequence at the 3′ of SA19 aptamer resulted in a chimeric aptamer that we designated SA19–CopA/T. When this chimeric aptamer was mixed with a similarly engineered CD4 chimeric aptamer (E14–CopA/T) the resulting molecule was designated adaptamer. Transcripts from sequences containing Cop–aptamers were refolded and then analyzed by native PAGE, either separately or in pair-wise arrangements to monitor the formation of the hybrid molecules (adaptamers), which formed efficiently (Fig. 6A). The ability of aptamer–Cop RNAs and their derived adaptamers to bind to SA and CD4, was analyzed by SPR on SA-coated sensor chip (Fig. 6B). SA–Cop aptamers SA19–CopT and SA19–CopA were injected onto SA-coated flow cells, followed by CD4–Cop aptamers E14–CopT and E14–CopA, respectively. The rapid rise in response in each case demonstrates the formation of adaptamers through the CopA–CopT interaction. Recombinant rat CD4 was injected subsequently and a further substantial response was seen, showing that the adaptamers could bind simultaneously to both SA and CD4.
Affinity characterization of SA19–Cop and adaptamer SA interactions
It was necessary to verify that the affinity of the chimeric aptamer as well as the adaptamer was not altered by these modifications. For this we have used gel mobility shift assay as for the unmodified parental aptamer. Titration of SA protein to the chimeric aptamer (Fig. 7A) as well as to the adaptamer (Fig. 7B) at a concentration of ∼3 nM yielded a complex of slower electrophoretic mobility compared with the free chimeric or free adaptamer, as shown in Figure 7A and B, respectively. The apparent dissociation constants for these interactions were calculated as before (Fig. 7C), and yielded values of 18 ± 2.7 and 24 ± 4.2 nM for the chimeric aptamer and the adaptamer, respectively. It appeared from these results that the affinities of the newly engineered chimeric aptamer and the adaptamer have dropped only by ∼2–3-fold in comparison with the parental aptamer.
Figure 7.

Native gel mobility shift assay of SA binding to chimeric SA19 and adaptamer. (A) Storage phosphor autoradiogram of a representative gel used to separate free chimeric aptamer SA19–CopA from SA19–CopA/SA complex, using a range of increasing protein concentrations from 0.45 to 3584 nM. (B) The same analysis as described in (A) but with the adaptamer SA19–CopA–E14–CopT. (C) Representative plots of fraction of the chimeric SA19–CopA (continuous line) and the adaptamer SA19–CopA–E14–CopT (dotted line) bound by SA as a function of protein concentration. The data were fitted to a hyperbolic function by non-linear curve fitting method of GraphPad PRISM. These titrations yielded an equilibrium dissociation constant of ∼18 and 24 nM for the chimeric SA19–CopA and the adaptamer SA19–CopA–E14–CopT, respectively.
DISCUSSION
We describe here the isolation of nucleic acid ligands for SA with potentially general utility as reagents in analytical and diagnostic applications. The relatively straightforward method used to isolate these SA-binding aptamers contrasts with the elaborate, if elegant, approach for producing SA-binding peptide aptamers recently described (8). The affinity of the nucleic acid aptamers described here is comparable with that of the peptide aptamers. Moreover, because of their 2′-F-chemistry the modified ligands are relatively nuclease resistant and are intrinsically not susceptible to proteases.
Sequence comparison of the isolated aptamers revealed that they all belong to one family with probably a unique motif. Mutagenesis and de novo selection confirmed that this motif represented the highest-affinity aptamer accessible within the sequence space of the library used.
A recently published paper (24) has described the isolation of SA aptamers with interesting properties as affinity tags to study RNAs and ribonucleoproteins. We compared the sequences of our aptamers with the published S1 and S12 aptamers (24) and found no significant similarities. The affinity of our SA19 aptamer toward SA was 10-fold higher than the one reported for S1 aptamer (24). The aptamers here described contain 2′-F-pyrimidines while the published aptamers were synthesized with canonical nucleotides. Not surprisingly, when we substituted the 2′-F-pyrimidines of our SA aptamers with canonical nucleotides, binding to SA protein was completely lost. This loss of binding in the unmodified RNA was not due to its degradation (unpublished data). This feature could provide a suitable model to investigate the impact of the presence of a fluorine atom at the 2′ position of the sugar on the folding of aptamers and on their functional properties.
Usefully, the binding of aptamer SA19 to SA was not prevented by saturation of SA with biotin, in contrast with the previously published aptamers (24). We found that the affinity was slightly reduced and the stoichiometry of interaction changed in a way that suggests non-competitive inhibition. This reinforces the idea that the biotin-binding site and the aptamer-binding site on SA are distinct as expected from the design of the selection strategy. Equally important, the 2′-F-aptamers did not bind to avidin, which, together with the non-competitive binding with biotin, would make them very useful in multiplex detection and diagnostic platforms.
It occurred to us that SA-binding aptamers could be useful as tools for linking other aptamers to biotin-dependent detection systems of the sort frequently employed in immunoassays, confocal microscopy and so on. When we attempted to produce chimeric aptamers by direct fusion of the gene encoding SA19 with that encoding an aptamer to a second protein, we found that in most cases the resultant RNA transcript had lost the ability to bind one or both of its target molecules (data not shown). This most probably results from inter-domain interactions that prevent the formation of the parental folds. Accordingly, we developed a general method for forming bi-functional aptamers, in which the gene encoding one aptamer is fused with that encoding the CopA structured RNA from E.coli plasmid R1 (13), and the gene for a second aptamer is fused with that encoding the complementary CopT sequence. We call these bi-functional ligands adaptamers. Because the two Cop elements are highly stable structures, they fold independently of any RNA to which they are fused (25). Any CopA-bearing aptamer will pair with high efficiency with any CopT-bearing aptamer to generate a bi-functional adaptamer. Adaptamers bearing ligands for both SA and rat CD4 were constructed and were successfully used to capture soluble CD4 in a sandwich biosensor assay.
The availability of SA-binding aptamers, together with the adaptamer approach, opens the possibility of applying the wide range of SA–biotin-based detection systems—currently used in conjunction with antibody ligands—to the analysis of molecules to which nucleic acid aptamers have been isolated.
REFERENCES
- 1.Ellington A.D. and Szostak,J.W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 2.Tuerk C. and Gold,L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 3.Joyce G.F. (1998) Nucleic acid enzymes: playing with a fuller deck. Proc. Natl Acad. Sci. USA, 95, 5845–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanes J., Jermutus,L., Weber-Bornhauser,S., Bosshard,H.R. and Pluckthun,A. (1998) Ribosome display efficiently selects and evolves high-affinity antibodies in vitro from immune libraries. Proc. Natl Acad. Sci. USA, 95, 14130–14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanes J. and Pluckthun,A. (1997) In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl Acad. Sci. USA, 94, 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts R.W. and Szostak,J.W. (1997) RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl Acad. Sci. USA, 94, 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keefe A.D. and Szostak,J.W. (2001) Functional proteins from a random-sequence library. Nature, 410, 715–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson D.S., Keefe,A.D. and Szostak,J.W. (2001) The use of mRNA display to select high-affinity protein-binding peptides. Proc. Natl Acad. Sci. USA, 98, 3750–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuerk C. (1997) Using the SELEX combinatorial chemistry process to find high affinity nucleic acid ligands to target molecules. Methods Mol. Biol., 67, 219–230. [DOI] [PubMed] [Google Scholar]
- 10.Heidenreich O., Kang,S.H., Brown,D.A., Xu,X., Swiderski,P., Rossi,J.J., Eckstein,F. and Nerenberg,M. (1995) Ribozyme-mediated RNA degradation in nuclei suspension. Nucleic Acids Res., 23, 2223–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaccolo M., Williams,D.M., Brown,D.M. and Gherardi,E. (1996) An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol., 255, 589–603. [DOI] [PubMed] [Google Scholar]
- 12.Nordstrom K., Molin,S. and Light,J. (1984) Control of replication of bacterial plasmids: genetics, molecular biology and physiology of the plasmid R1 system. Plasmid, 12, 71–90. [DOI] [PubMed]
- 13.Persson C., Wagner,E.G. and Nordstrom,K. (1988) Control of replication of plasmid R1: kinetics of in vitro interaction between the antisense RNA, CopA and its target, CopT. EMBO J., 7, 3279–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraus E., James,W. and Barclay,A.N. (1998) Novel RNA ligands able to bind CD4 antigen and inhibit CD4+ T lymphocyte function. J. Immunol., 160, 5209–5212. [PubMed] [Google Scholar]
- 15.Maniatis T., Fritsch,E.F. and Sambrook,J. (1982) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 16.Aurup H., Williams,D.M. and Eckstein,F. (1992) 2′-Fluoro- and 2′-amino-2′-deoxynucleoside 5′-triphosphates as substrates for T7 RNA polymerase. Biochemistry, 31, 9636–9641. [DOI] [PubMed] [Google Scholar]
- 17.Ehresmann C., Baudin,F., Mougel,M., Romby,P., Ebel,J.P. and Ehresmann,B. (1987) Probing the structure of RNAs in solution. Nucleic Acids Res., 15, 9109–9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peattie D.A. and Gilbert,W. (1980) Chemical probes for higher-order structure in RNA. Proc. Natl Acad. Sci. USA, 77, 4679–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stern S., Moazed,D. and Noller,H.F. (1988) Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Methods Enzymol., 164, 481–489. [DOI] [PubMed] [Google Scholar]
- 20.Felden B., Florentz,C., Giege,R. and Westhof,E. (1994) Solution structure of the 3′-end of brome mosaic virus genomic RNAs. Conformational mimicry with canonical tRNAs. J. Mol. Biol., 235, 508–531. [DOI] [PubMed] [Google Scholar]
- 21.Sanger F., Nicklen,S. and Coulson,A.R. (1977) DNA sequencing with chain-terminating inhibitors. Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Batenburg F.H., Gultyaev,A.P. and Pleij,C.W. (1995) An APL-programmed genetic algorithm for the prediction of RNA secondary structure. J. Theor. Biol., 174, 269–280. [DOI] [PubMed] [Google Scholar]
- 23.Gultyaev A.P. (1991) The computer simulation of RNA folding involving pseudoknot formation. Nucleic Acids Res., 19, 2489–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srisawat C. and Engelke,D.R. (2001) Streptavidin aptamers: affinity tags for the study of RNAs and ribonucleoproteins. RNA, 7, 632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malmgren C., Wagner,E.G., Ehresmann,C., Ehresmann,B. and Romby,P. (1997) Antisense RNA control of plasmid R1 replication. The dominant product of the antisense RNA-mRNA binding is not a full RNA duplex. J. Biol. Chem., 272, 12508–12512. [DOI] [PubMed] [Google Scholar]