

Abstract
Recent large-scale genomic association studies found evidence for a genetic link between increased risk of type 2 diabetes and decreased risk for adiposity-related traits, reminiscent of metabolically obese normal weight (MONW) association signatures. However, the target genes and cellular mechanisms driving such MONW associations remain to be identified. Here, we systematically identify the cellular programmes of one of the top-scoring MONW risk loci, the 2q24.3 risk locus, in subcutaneous adipocytes. We identify a causal genetic variant, rs6712203, an intronic single-nucleotide polymorphism in the COBLL1 gene, which changes the conserved transcription factor motif of POU domain, class 2, transcription factor 2, and leads to differential COBLL1 gene expression by altering the enhancer activity at the locus in subcutaneous adipocytes. We then establish the cellular programme under the genetic control of the 2q24.3 MONW risk locus and the effector gene COBLL1, which is characterized by impaired actin cytoskeleton remodelling in differentiating subcutaneous adipocytes and subsequent failure of these cells to accumulate lipids and develop into metabolically active and insulin-sensitive adipocytes. Finally, we show that perturbations of the effector gene Cobll1 in a mouse model result in organismal phenotypes matching the MONW association signature, including decreased subcutaneous body fat mass and body weight along with impaired glucose tolerance. Taken together, our results provide a mechanistic link between the genetic risk for insulin resistance and low adiposity, providing a potential therapeutic hypothesis and a framework for future identification of causal relationships between genome associations and cellular programmes in other disorders.
Obesity and type 2 diabetes (T2D)-related traits are intimately linked by both environmental and genetic factors. The global prevalence of obesity and T2D has risen dramatically over the past century and both diseases constitute a serious increasing public health concern worldwide, with T2D predicted to rise in prevalence from 451 to 693 million people between 2017 and 2045 (ref. 1). Most epidemiological and genetic studies have linked obesity to the pathogenesis of T2D through positive phenotypic correlations between adiposity and T2D. However, a small number of loci have been identified that do not follow this pattern or even correlate in the opposite phenotypic direction2. Also, up to 45% of individuals with obesity do not present with poor glycaemic or lipid profiles, commonly called the metabolically healthy obese (MHO). Concurrently, up to 30% of normal-weight individuals present with significant cardiometabolic risk factors, known as the metabolically obese normal weight (MONW)3-10.
Genome-wide association studies (GWAS) identified more than 700 genomic loci associated with glycaemic traits and T2D and more than 1,000 loci associated with adiposity-related traits, including body mass index (BMI) as a proxy of overall obesity, waist:hip ratio (WHR) as a proxy of body fat distribution, body fat percentage and direct measures of subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT) mass2,11-20. A small proportion of T2D genetic risk loci have been associated with decreased body fat percentage and a decreased SAT:VAT ratio2,21-23. Notably, these MONW/MHO genetic risk loci are characterized by distinct association signatures implying locus-specific cellular programmes2,21-23.
The metabolic risk haplotype at 2q24.3 displays cross-phenotype association signatures that are reminiscent of the MONW phenotype and are associated with an increased risk of T2D, increased homeostatic model assessment for insulin resistance, increased WHR adjusted for BMI and decreased body fat percentage, decreased estimated SAT mass and cardiometabolic trait risk2,15,24-26. Consistent with these associations, the 2q24.3 locus falls into the lipodystrophy cluster of T2D loci27, suggesting adipocytes as the mediating cell type at this locus. Notably, among the 20 loci identified in the T2D lipodystrophy process-specific cluster27, the 2q24.3 locus is the top-scoring one, suggesting the strongest contribution to a lipodystrophic-like phenotype among the T2D GWAS loci.
However, like the vast majority of genetic risk loci identified through GWAS, the mechanism of the 2q24.3 metabolic risk locus is currently unknown. There are multiple factors that still confound the goal of converting genetic associations into specific knowledge, that is, pinpointing the causal variant(s), target gene(s) and mechanism(s). First, the resolution of association mapping is inherently limited by the haplotype structure of the human genome because a common variant in the population is usually strongly genetically linked to many neighbouring variants. Second, the genetic architecture of common diseases is fundamentally different to that of rare diseases, as over 80% of associated regions do not contain any protein-altering common variants28. Importantly, genetics and descriptive catalogues of molecular activity alone are, for non-coding signals at least, insufficient to build compelling proof for a chain of causation that stretches from variant to phenotype. In this study, we set out to combine statistical and experimental methods to identify the mechanism by which 2q24.3 confers decreased adiposity and increased risk of T2D (Extended Data Fig. 1a), possibly enabling further development of therapeutic strategies to alter glycaemic control.
Results
The 2q24.3 risk locus maps to adipocyte enhancer signatures
To identify diseases and traits associated with the 2q24.3 locus tagged by the variant rs3923113, we visualized large-scale phenome-wide associations from the UK Biobank (UKB) and from a meta-analysis of a series of metabolic traits (Fig. 1a and Extended Data Fig. 1b). We observed that the 2q24.3 locus was associated with increased T2D risk, increased fasting insulin levels and a series of body fat-related traits, including increased WHR adjusted for BMI, but decreased trunk fat percentage, arm fat percentage, hip circumference and whole-body fat mass, suggesting a complex pleiotropic risk locus consistent with a MONW association signature, that is, a lean, metabolically unhealthy phenotype.
Fig. 1 ∣. The pleiotropic 2q24.3 MONW locus is associated with increased risk for T2D and decreased adiposity-related traits, and maps to sparse enhancer signatures in adipocytes.
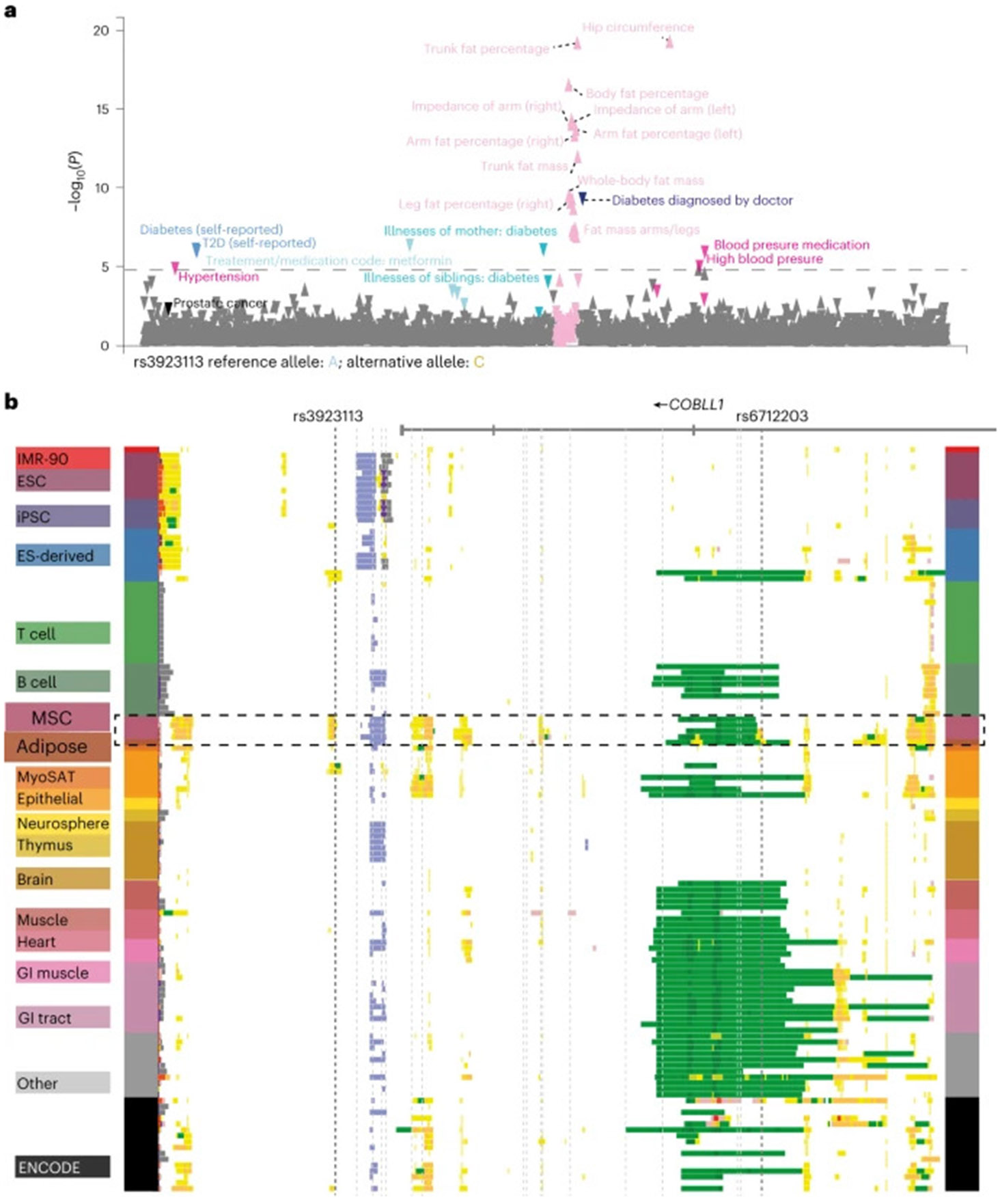
a, Phenome-wide association studies of trait associations at the rs3923113-tagged haplotype in the UKB87. The colours represent trait classes while individual rs3923113 variant association P values are shown on the y axis. The direction of effect is indicated by the orientation of the triangles: upward, increase; downward, decrease. b, Chromatin state annotations for the 55-kb-long MONW risk locus. Genomic intervals are shown across 127 human cell types and tissues reference epigenomes profiled by the Roadmap Epigenomics project, based on a 25-state chromatin state model (see Extended Data Fig. 1d for the color code of the chromatin states) learned from 12 epigenomic marks using imputed signal tracks at 25-nucleotide resolution88. Chromatin states considered included Polycomb repressed states (grey, H3K27me3), weak enhancers (yellow, H3K4me1 only), strong enhancers (orange, also H3K27ac) and transcribed enhancers (green, also H3K36me3) (https://www.freepatentsonline.com/y2022/0243178.html). ES, embryonic stem; ESC, embryonic stem cell; GI, gastrointestinal; iPSC, induced pluripotent stem cell; MSC, mesenchymal stem cell.
The 2q24.3 locus tagged by rs3923113 encompasses 55 kb, spanning from the COBLL1 intronic regions to the intergenic region between GRB14 and COBLL1 (Extended Data Fig. 1c). The MONW locus harbours 20 non-coding single0nucleotide polymorphisms (SNPs) in high linkage disequilibrium (LD) (r2 > 0.8, 1000 Genomes Project Phase 1 EUR). These single-nucleotide variants (referred to as the candidate regulatory variants) define two alternative haplotypes: the ancestral haplotype 1 (frequency 38% in European individuals), associated with a decreased risk for the MONW association pattern; and haplotype 2 (frequency 62%), associated with an increased MONW risk. To connect genetic variants at the 2q24.3 locus to relevant cell types and cell states, we examined chromatin state maps across 127 reference epigenomes from the Roadmap Epigenomics and the ENCODE consortium (Fig. 1b and Extended Data Fig. 1d). We found that the locus maps to multiple enhancer signatures, including active enhancers in mesenchymal stem cells, adipocyte progenitors and adipocytes (Fig. 1b). Several of the 20 non-coding variants map within or in the vicinity of regions with active enhancer chromatin states, suggesting that the 2q24.3 locus acts in adipocytes through gene regulatory mechanisms.
Next, we examined whether the two haplotypes (risk compared to non-risk) showed differences in chromatin structure during adipocyte differentiation. Specifically, we performed assays for enhancer activity (H3K27 acetylation (H3K27ac) chromatin immunoprecipitation followed by sequencing (ChIP–seq)) and chromatin accessibility (assay for transposase-accessible chromatin with sequencing (ATAC–seq)) on adipose-derived mesenchymal stem cells (AMSCs) from heterozygous individuals across a time course of differentiation (before induction (day 0), early differentiation (day 2), intermediate differentiation (day 6) and terminal differentiation (day 14)), and compared the numbers of reads from the two haplotypes (Extended Data Fig. 1e). The MONW risk haplotype (haplotype 2) was associated with a decrease in H3K27ac, a proxy of enhancer activity, and chromatin accessibility, with the MONW risk haplotype enriched by roughly 1.5-fold. The allele-specific difference in chromatin accessibility was reproducible across three heterozygous lines (Supplementary Tables 1 and 2), was most pronounced at day 0 of differentiation and declined after induction of differentiation (Extended Data Fig. 1f). These results indicate that haplotype 1 is associated with an active enhancer state, whereas haplotype 2 is associated with a weak enhancer state primarily in adipocyte progenitors.
The rs6712203 variant affects adipocyte COBLL1 expression
To identify which of the 20 candidate regulatory variants is likely to mediate the differential enhancer activity in adipocyte progenitors, we used two orthogonal computational approaches to prioritize variants, phylogenetic module complexity analysis (PMCA)28-30 and Bassett31 (Fig. 2a-c and Supplementary Tables 3 and 4). PMCA assesses evolutionary conservation of sequence, order and distance (in human and at least one other vertebrate species) of groups of at least three transcription factor binding motifs within a 120-bp region. Basset uses a sequence-based deep convolutional neural network (CNN) approach to predict the effects of non-coding variants on regulatory activity by training on the sequence content of a given epigenomic mark in a tissue or cell type of interest. After training on genome-wide chromatin accessibility (ATAC–seq) data in numerous cell types, including AMSC progenitors, before induction (day 0), one variant, rs6712203, stood out as consistently showing the highest score for PMCA and Bassett (Fig. 2a). Bassett predicted that the T allele on the protective haplotype increases chromatin accessibility relative to the C allele on the risk haplotype in adipocyte progenitors. These sequence-based estimates of rs6712203 C>T single-nucleotide change importance are consistent with the variant overlapping an active enhancer associated with H3K27ac and H3K4 mono-methylation in adipocyte progenitors. In line with the variant importance at rs6712203, conditional analyses of anthropometric and glycaemic traits defining MONW in the UKB confirmed an rs6712203 C/T association consistent with a primary effect in female participants for fat mass (β = 0.022, P = 4.1 × 10−08 in females and β = 0.0084, P = 0.052 in males, difference P = 0.019), hip circumference (β = 0.037, P =4.3 × 10−15 in females and β = 0.0097, P = 0.008 in males, difference P = 4.3 × 10−06) and T2D in both females and males (females β = −0.11, P = 1.4 × 10−07 and males β = −0.068, P = 2.4 × 10−05, difference P = 0.098, Supplementary Table 5 and Extended Data Figs. 1g and 2). Furthermore, we observed that the rs6712203 association with T2D was dependent on BMI (β = −0.0028, P = 0.70 in normal weight and underweight and β = −0.093, P = 2.92 × 10−06 in obese, difference P = 2.0 × 10−05, Supplementary Table 6).
Fig. 2 ∣. rs6712203 is a functional variant at the 2q24.3 MONW locus.
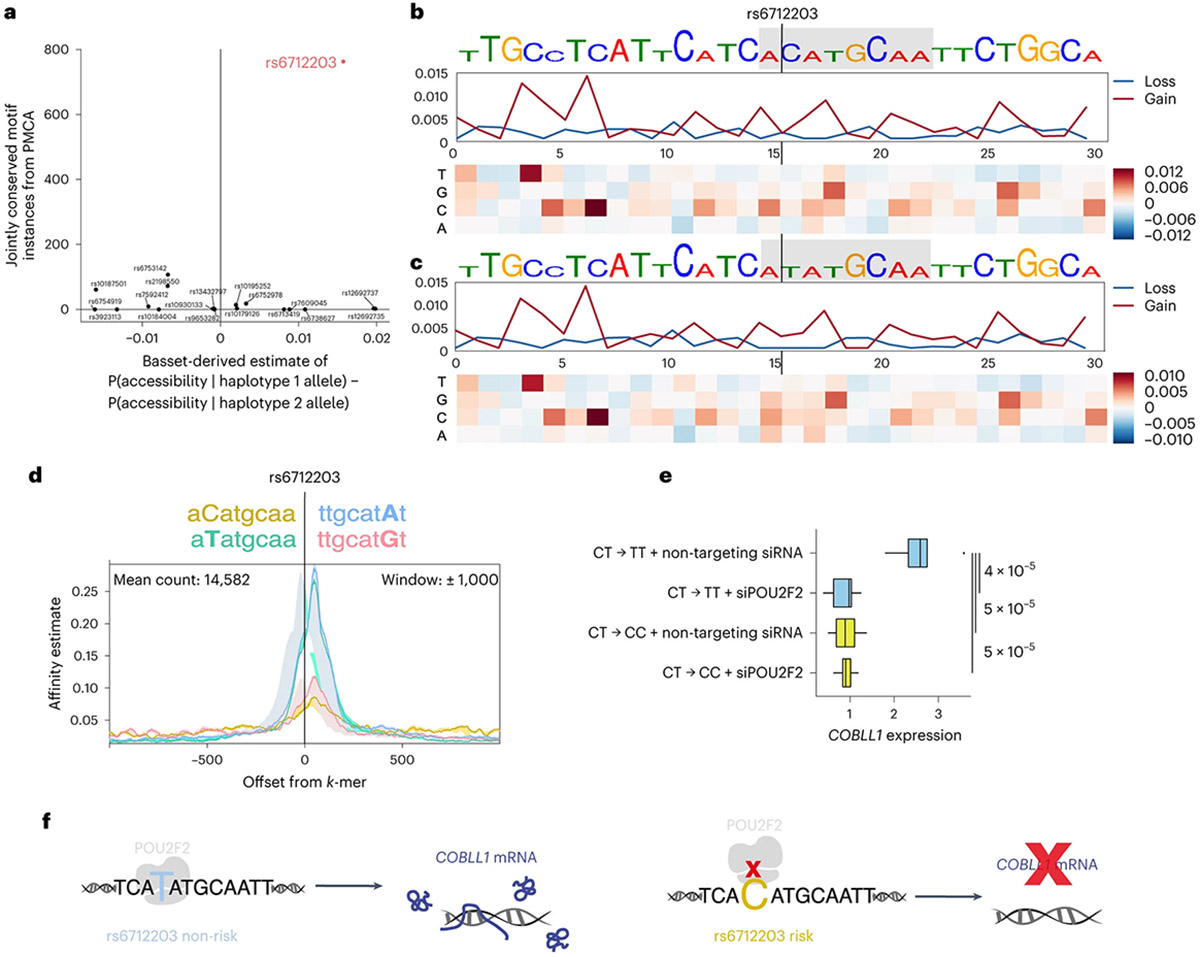
a, Phylogenetic conservation analysis and CNN-based prediction of chromatin accessibility for 20 highly linked (LD = r2 > 0.8) variants at the 2q24.3 locus (tag SNP rs3923113). Phylogenetic conservation scores of jointly conserved motifs using PMCA are shown on the x axis29. PMCA was used to identify orthologous regions in 20 vertebrates and to scan the 120-bp sequence context for groups of transcription factor binding site motifs whose sequence, order and distance range is conserved across species. Scores indicate the count of non-overlappingjointly conserved transcription factor binding site motifs whose relative positions within the window are conserved. Predicted relative change in chromatin accessibility (SNP accessibility difference SAD scores) in preadipocytes (day 0 of differentiation) for each SNP comparing alleles on haplotype 1 and haplotype 2 is shown on the y axis. A deep CNN Basset31 was trained on genome-wide ATAC–seq data assayed in preadipocytes. Alleles were assigned to each SNP in the haplotype and evaluated for predicted accessibility. b, Both PMCA and Basset predicted rs6712203 as a functional variant. For the C allele, there are no nucleotide variants that reduce binding in the rs6712203 region. Each position on the x axis represents a nucleotide and the four values in the heatmap correspond to all possible substitutions. c, For the T allele, in silico saturation mutagenesis suggests that binding loss of the POU2F motif that overlaps rs6712203, including the C allele itself, results in reduced predicted chromatin accessibility. d, Intragenomic replicates35 predict a higher binding affinity of POU2 family transcription factors for the T than C allele to both strands. Offsets from instances of the given k-mer sequence are shown on the x axis. Estimated affinity of binding (https://www.freepatentsonline.com/y2022/0243178.html) is shown on the y axis. A model with 8-mers is shown; alternatives with 6-mers through to 9-mers are shown in Extended Data Fig. 3b. e, Generation of rs6712203 CRISPR–Cas9 engineered lines starting from SGBS preadipocytes (heterozygous for rs6712203) edited to the homozygous risk (CC, yellow) and non-risk (TT, blue) alleles and qPCR-based gene expression measurement of COBLL1 conditional on the regulator POU2F2. n = 3 biologically independent experiments. f, Schematic model of the regulatory circuitry under the genetic control of rs6712203.
We next used three-dimensional genome conformation data from Hi-C assays in normal human epidermal keratinocytes and human fibroblasts32 to define the physical boundaries of potential proximal and long-distant target genes. We found that the locus lies in a well-defined contact domain containing only two genes, cordon-bleu WH2 repeat protein like 1 (COBLL1) and growth factor receptor bound protein 14 (GRB14) (Extended Data Fig. 3a), without any evidence for long-range chromatin interactions. To dissect which of these two genes are targeted by rs6712203, we undertook a CRISPR interference (CRISPRi) approach by engineering immortalized human preadipocytes (in human white adipose tissue (hWAT))33 to stably express a catalytically dead Cas9 fused to a Krüppel-associated box (KRAB) domain34 (Extended Data Fig. 3b). Transduction of Cas9 Endonuclease Dead (dCas9)-hWAT preadipocytes with increasing amounts of lentiviral particles carrying single-guide RNAs (sgRNAs) targeting the transcriptional start sites (TSS) of COBLL1 and GRB14 genes led to dose-dependent downregulation of mRNA expression by 72% and 96% compared to non-targeting negative controls (Extended Data Fig. 3c). At the highest dose, COBLL1 protein was reduced by 56% without effects of sgRNAs targeting the GRB14 TSS on COBLL1 expression (Extended Data Fig. 3d). After confirming the knockdown efficiency and specificity of our system, we next designed a panel of six sgRNAs surrounding the rs6712203 variant (Extended Data Fig. 3e) and additional non-targeting and COBLL1/GRB14 TSS controls to assess its effects on COBLL1 and GRB14 mRNA expression. All TSS sgRNAs led to a reduction in COBLL1 and GRB14 mRNA content by 64% and 94% without any interference on each other (Extended Data Fig. 3f,g). More importantly, all tested sgRNAs, with exception of sgRNA 1, led to significant downregulation of COBLL1 over a range of 20–40% compared to negative controls (Extended Data Fig. 3f), while no effects were observed on GRB14 (Extended Data Fig. 3g), thus pointing to COBLL1 as the primary target of rs6712203.
We next performed in silico saturation mutagenesis to evaluate the predicted change in chromatin accessibility from mutation at every position into each alternative nucleotide within a 30-bp region surrounding rs6712203 using ATAC–seq data during AMSC differentiation. We found that the rs6712203 T allele is critical for a POU2F2 motif (Fig. 2d). The C allele of this SNP converts the chromatin in this site into a less accessible chromatin, supporting a model in which a transcription factor, possibly POU2F2, differentially binds to these allelic variants of rs6712203. To estimate the preferential binding affinity of POU2F2 to the C risk compared to the T non-risk allele, we used the intragenomic replicate (IGR) method35 on publicly available POU2F2 ChIP–seq data from the ENCODE project. By comparing the frequency of k-mers matching the rs6712203 T allele versus the C allele, we confirmed that POU2F2 preferentially binds to the T allele (9-mer change in affinity −0.38, two-tailed permutation P < 0.0034) (Fig. 2d and Extended Data Fig. 3h). These data suggest an increased POU2F2 binding to the rs6712203 T non-risk allele and suggest POU2F2 as the upstream regulator of variant action at this locus.
We next sought to establish rs6712203 causality by directly confirming that the haplotype-specific effects on enhancer activity and POU2F2 binding are mediated by rs6712203 using CRISPR-based genome editing at this SNP. We edited the human Simpson–Golabi–Behmel syndrome (SGBS) preadipocytes (n = 5) that are heterozygous at rs6712203 to create isogenic lines for the homozygous TT (non-risk genotype) and homozygous CC (risk genotype) alleles. In line with the CRISPRi rs6712203 enhancer repression experiments outlined above, we observed that cells harbouring the CC homozygous risk showed 2.4-fold lower COBLL1 expression levels compared to the TT non-risk genotype (Fig. 2e), pointing towards COBLL1 as a target gene of the rs6712203 regulatory circuitry. To test a cis/trans-conditional effect of the rs6712203 variant and the upstream regulator POU2F2 on target gene expression, we performed targeted small interfering RNA (siRNA)-mediated ablation of POU2F2 in SGBS cells and found that silencing of POU2F2 in TT allele carriers reduces COBLL1 gene expression to the level of CC allele carriers in preadipocytes (Fig. 2e), confirming POU2F2 as a functional regulator at the locus.
Together, our computational and experimental approaches support a model where the causal MONW variant rs6712203 regulates COBLL1 gene expression in a POU2F2-dependent manner (Fig. 2f).
COBLL1 affects actin remodelling in subcutaneous adipocytes
To understand the role of COBLL1 in adipocyte cellular programmes, we examined the gene expression and cellular localization of COBLL1 in differentiating adipocytes. We observed that COBLL1 is expressed at all stages of adipocyte differentiation with an increase in mRNA over the course of differentiation (Extended Data Fig. 4a). We observed consistently higher COBLL1 mRNA levels in subcutaneous compared to visceral adipocytes across the adipocyte differentiation process (Extended Data Fig. 4a). Overall, we found an enrichment of COBLL1 gene expression in adipose tissue compared to 146 other tissues and cell types36 (Extended Data Fig. 4b).
To connect the 2q24.3 locus to cellular functions in adipose tissue, we used genome-wide coexpression matrices in adipocytes matched with a series of cellular assays. We identified COBLL1 coregulated genes in genome-wide expression data from primary human AMSCs in a cohort of 12 healthy, non-obese individuals. COBLL1 coexpressed genes were highly enriched in biological processes related to regulation of actin cytoskeleton and regulation of lipolysis in adipocytes, including integrin subunit alpha M (ITGAM), phosphatidylinositol −4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), rho-associated protein kinase 2 (ROCK2), integrin alpha-1 (ITGA1), and rho guanine nucleotide exchange factor 7 (ARHGEF7), proto-oncogene, adaptor protein (CRK), fibroblast growth factor receptor 2 (FGFR2) and rho guanine nucleotide exchange factor 6 (ARHGEF6) (Fig. 3a, Extended Data Fig. 4c,d and Supplementary Tables 7-10), which are implicated in actin remodelling processes and insulin responsiveness37-40. This is consistent with recent studies showing that COBLL1 possesses a Wiskott–Aldrich syndrome protein homology 2 actin monomer-binding domain, and promotes filamentous actin (F-actin) formation in Cos-7, neuronal and prostate cancer cells41,42. Finally, we found a significantly decreased relative COBLL1 gene expression after a weight loss of 6.9 ± 1.9 kg in 18 women with obesity (BMI = 34.9 ± 3.8 kg m−2) (Extended Data Fig. 4e).
Fig. 3 ∣. The 2q24.3 effector gene COBLL1 affects actin remodelling processes in differentiating adipocytes.
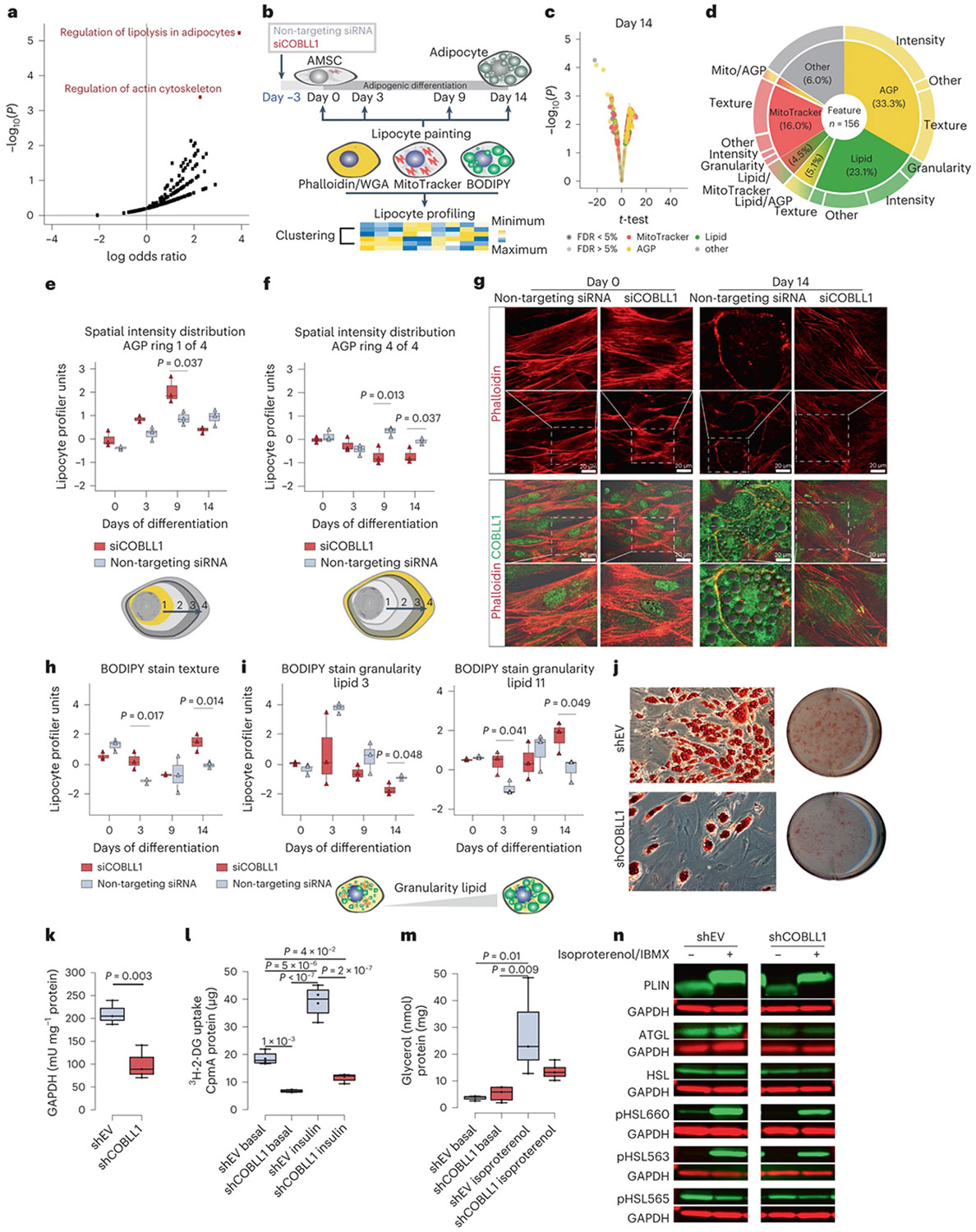
a, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of genes correlated with COBLL1 in adipocytes using Enrichr89,90. n = 30 donors, P values were derived from a hypergeometric test. b, Schematic of siCOBLL1 experiments in AMSCs across differentiation. AMSCs from a normal-weight female donor were silenced 3 d before induction and LipocyteProfiling (days 0, 3, 8 and 14 of differentiation), n = 3. c, Image-based profiles of siCOBLL1-treated compared to non-targeting siRNA-treated AMSCs on day 14; two-sided t-test, significance level FDR < 5%, n = 3. d, Pie chart illustrating differential features per channel and measurement class comparing siCOBLL1 and the non-targeting siRNA control on day 14. e,f, Spatial intensity distribution of AGP informative for the actin cytoskeleton in the centre of the cytoplasm: Cytoplasm_RadialDistribution_FracAtD_AGP_1of4 (e) and juxtaposed to the plasma membrane (Cytoplasm_RadialDistribution_RadialCV_AGP_4of4) (f). Two-sided t-test, n = 3 biologically independent experiments, median ± 95% confidence interval (CI). g, Representative images of COBLL1 knockdown and non-targeting siRNA control at days 0 and 14 of differentiation. COBLL1 staining: anti-COBLL1 primary and donkey anti-rabbit IgG H&L secondary antibodies. Actin staining: phalloidin–Atto 565. Olympus FLUOVIEW FV1000 CLSM Inverse microscope (40× magnification). Images were processed with Image J. Dotted square: image zoom-in. h,i, Texture of BODIPY stain (Cells_Texture_Correlation_Lipid_10_01) (h) and granularity measures of the BODIPY stain (Cells_Granularity_3_BODIPY) (i) in siCOBLL1 knockdown and non-targeting siRNA; two-sided t-test, n = 3 biologically independent experiments, median ± 95% CI.j, Oil Red O staining in SGBS adipocytes after stable lentiviral COBLL1 knockdown (shCOBLL1) versus empty vector control (shEV); scale bar, 15 mm. k, GPDH metabolic activity in differentiated shCOBLL1 compared to non-targeting siRNA adipocytes. Paired Student’s t-test, median ± 95% CI, n = 3. l, Basal and insulin-stimulated 3H-2-DG uptake in differentiated shCOBLL1 compared to shEV adipocytes. One-way analysis of variance (ANOVA) with Tukey’s honestly significant difference (HSD) test, median ± 95% CI, n = 3 biologically independent experiments. m, Basal and isoproterenol-stimulated lipolysis rate measured using glycerol release in differentiated shCOBLL1 compared to shEV adipocytes. One-way ANOVA with Tukey’s HSD test, median ± 95% CI, n = 3 biologically independent experiments. n, Western blots for lipolysis-relevant proteins assayed in basal or isoproterenol/IBMX-stimulated shCOBLL1 versus shEV adipocytes (n = 2).
To identify morphological and cellular traits associated with altered COBLL1 expression, we used siRNA-mediated knockdown of COBLL1 in AMSCs coupled with a high-content imaging assay that we recently developed, LipocyteProfiler43. LipocyteProfiler reports on general and adipocyte-specific cellular traits for preadipocytes before differentiation (day 0), and at three time points of adipocyte differentiation (3 d (day 3), 9 d (day 9) and 14 d (day 14) after adipogenic induction) (Fig. 3b). We examined 1,175 quantitative features, spread across two cellular compartments (whole-cell and cytoplasm only) and five dyes informative for morphological and adipocyte cellular traits (BODIPY, phalloidin, wheat germ agglutinin (WGA), SYTO14 and MitoTracker (Methods) imaged in four fluorescence channels (Fig. 3b). We observed that COBLL1 knockdown in proliferating preadipocytes with 80% knockdown efficiency (Extended Data Fig. 5a) results in changes of diverse morphological and cellular features across adipocyte differentiation with a peak at the later stages of differentiation (Fig. 3c and Extended Data Fig. 5b-d). On day 14 of differentiation, 156 features differed significantly (false discovery rate (FDR) < 5%) between COBLL1 knockdown and non-targeting control, spread across lipid (23.1%), actin, Golgi, plasma membrane (AGP)-related (33.3%) and mitochondrial (16.0%) channels (Fig. 3d and Supplementary Table 11). For AGP-related cellular processes, we observed that COBLL1 knockdown alters the spatial intensity distribution of AGP across the cytoplasm. We previously observed that these AGP-related features can be indicative of actin cytoskeleton remodelling during differentiation43. After COBLL1 silencing, we observed increased actin-associated intensity in the centre of the cell (day 9 P = 0.037; Fig. 3e) and decreased actin-associated intensity at the cell cortex (day 9 P = 0.013, day 14 P = 0.026; Fig. 3f) in differentiated subcutaneous adipocytes. This indicates that COBLL1 has a role in remodelling the actin cytoskeleton during adipocyte maturation, as reduced levels of COBLL1 disturb the disassembly of F-actin stress fibres across the cytoplasm and the reassembling to cortical F-actin (F-actin juxtaposed to the plasma membrane).
The disturbed actin remodelling process was accompanied by lowered differentiation capacity as shown by decreased lipid droplet formation. More specifically, we confirmed that COBLL1 knockdown was associated with a decreased disassembly of stiff F-actin stress fibres reaching in the middle of the cell body, at the expense of F-actin structure assembly at the cell cortex in differentiated cells (Fig. 3g and Extended Data Fig. 5e). Consistent with the notion that remodelling of F-actin stress fibres into cortical actin is linked to adipocyte differentiation, COBLL1-ablated adipocytes showed significant changes in both texture of lipid-related pixels (Cells_Texture_Correlation_Lipid; pixel intensities of BODIPY stain within the cell are more similar, day 3 P = 0.017 and day 14 P= 0.039; Fig. 3h) and lipid-related granularity measures (a class of metrics that captures the typical sizes of bright spots for the BODIPY stain) within the cell compared to adipocytes expressing COBLL1. More specifically, we observed that silencing of COBLL1 results in adipocytes with fewer smaller-sized lipid droplets (Cells_Granularity_ Lipid 3, day 14 P = 0.0483; Fig. 3i) and more medium-to-large-sized lipid droplets in mature adipocytes (Cells_Granularity_Lipid 11, day 3 P = 0.0412 and day 14 P = 0.0486; Fig. 3i), and an overall decrease in lipid droplet numbers (Cellls_Children_LargeLipidObjects_Count, P = 0.006; Extended Data Fig. 5f). These data indicate a disturbed lipid droplet formation and adipogenic differentiation in COBLL1-ablated cells, driven by an altered cytoskeleton remodelling.
To investigate if the COBLL1 effect on actin remodelling in adipocytes impacts adipocyte cellular programmes related to metabolic disease, we performed stable ablation of COBLL1 using lentivirus (shCOBLL1) in differentiating adipocytes. We observed that ablation of COBLL1 resulted in a decreased capacity to differentiate into metabolically active, round-shaped, lipid-filled mature adipocytes, as shown by decreased Oil Red O staining of accumulated triglycerides (Fig. 3j), adipocyte differentiation marker gene expression (Extended Data Fig. 5g) and glyceraldehyde-3-phosphate dehydrogenase (GPDH) activity measurements (2.1-fold, P = 3 × 10−3; Fig. 3k). We further found a correlation between the mRNA levels of COBLL1 and leptin, an adipokine produced in proportion to the size of fat depots44 in SAT (r = 0.74, P = 5 × 10−5) (Extended Data Fig. 5h,i). This effect on leptin is consistent with GWAS of serum leptin levels (rs6712203 C allele β = 0.0308, P = 9 × 10−6 and β = 0.0236, P = 1 × 10−5 (BMI-adjusted)) in Kilpeläinen et al.45; and β = 0.0285, P = 0.005889 in Folkersen et al.46. We further found a 3.4-fold (P = 2 × 10−7) decrease of insulin-responsive glucose uptake in shCOBLL1 adipocytes compared to non-targeting control, as measured by radiolabelled 3H-2-deoxyglucose (3H-2-DG) uptake assays (Fig. 3l). The failure of shCOBLL1 adipocytes to respond to insulin may have resulted from both lowered differentiation efficiency and failure of the cortical actin remodelling that mediates GLUT4 vesicle trafficking. Finally, we observed a failure of shCOBLL1 adipocytes to break down triglycerides to free fatty acids and glycerol through lipolysis after β-adrenergic stimulation using isoproterenol and the phosphodiesterase inhibitor IBMX compared to their control cells (Fig. 3m). This was accompanied with decreased protein levels of the lipolytic enzymes adipocyte triglyceride lipase, hormone-sensitive lipase (HSL), protein kinase A-dependent serine phosphorylated HSL (pHSL660, pHSL563) and on the lipid droplet-associated protein perilipin-1 (PLIN) (Fig. 3n). Notably, we did not observe an effect on cellular and morphological features when silencing COBLL1 after induction of differentiation (Extended Data Fig. 5j,k), suggesting that COBLL1 acts early in differentiation, with phenotypic effects primarily manifesting in mature adipocytes. We also did not observe an effect when COBLL1 was ablated in visceral AMSCs (Extended Data Fig. 5l), indicating that COBLL1 is critically involved in actin remodelling processes in subcutaneous adipocytes.
We additionally examined the effect of GRB14 stable knockdown in AMSCs and observed that GRB14 ablation (knockdown efficiency of 61%) did not significantly decrease in adipocyte differentiation capacity as measured by Oil Red O staining, GPDH activity (Extended Data Fig. 5m,n) or insulin-responsive glucose uptake and GLUT4 gene expression (Extended Data Fig. 5o,p), supporting COBLL1 as the effector gene at this locus.
The MONW haplotype affects the adipocyte actin cytoskeleton
To confirm that the changes on the actin cytoskeleton and subsequent effects on adipocyte functions are under the genetic control of the rs6712203 MONW risk haplotype, we used LipocyteProfiler43 to phenotypically profile primary human adipocytes differentiation throughout from individuals carrying the risk haplotype (n = 6) compared the non-risk haplotype (n = 7) (Fig. 4a). The data revealed that AGP and lipid features informative for the actin cytoskeleton and lipid accumulation differed in subcutaneous adipocytes from rs6712203 metabolic risk versus non-risk haplotype carriers (Fig. 4b,c and Extended Data Fig. 6a-c). More specifically, we found that 77 morphological features, spread across lipid (16.9%), actin-associated AGP (45.5%) and mitochondrial (26.0%) channels, significantly differed between the haplotypes on day 14 of differentiation (FDR = 5%; Supplementary Table 12). We did not observe any significant difference in visceral adipocytes (Fig. 4d and Extended Data Fig. 6d-f), which is consistent with the depot-specific effect of COBLL1 knockdown (Extended Data Fig. 5k). Notably, we found that the risk haplotype associates with increased actin-associated intensity in the centre of the cell (day 0 P = 0.018, day 3 P = 0.042, day 9 P = 0.011 day 14 P = 0.009; Fig. 4e) and decreased actin-associated intensity at the cell cortex (day 9 P = 0.024, day 14 P = 0.009; Fig. 4f). These data recapitulate our findings after COBLL1 knockdown and confirms that adipocytes from risk allele carriers are characterized by less cortical actin, which is required for insulin-stimulated glucose uptake in those cells and therefore directly relevant to fasting insulin levels and T2D. Furthermore, we observed a risk haplotype association with higher lipid object counts (day 8 P = 0.043, day 14 P = 0.034; Fig. 4g), which is representative of several lipid droplets, and higher lipid-related intensity (day 8 P = 0.001; Fig. 4h), which is indicative of dysfunctional lipid droplet formation. These genetic effects on the actin cytoskeleton dynamics and lipid accumulation in AMSCs are coherent with the effects we observed after the COBLL1 knockdown experiments (Fig. 3b-f,h,i), indicating that altered COBLL1 expression in the risk haplotype underlies the observed phenotypic effects in adipocytes. Summarized, these data show that the rs6712203 MONW risk locus, by altering COBLL1 mRNA expression levels, impacts actin remodelling in differentiating adipocytes, thereby strongly affecting fat mass-relevant and T2D-relevant cellular programmes including adipocyte differentiation, lipid droplet formation, stimulated lipolysis rate and insulin-stimulated glucose uptake.
Fig. 4 ∣. The rs6712203 MONW risk haplotype affects actin remodelling in adipocytes and adipocyte lipid storage capacity.
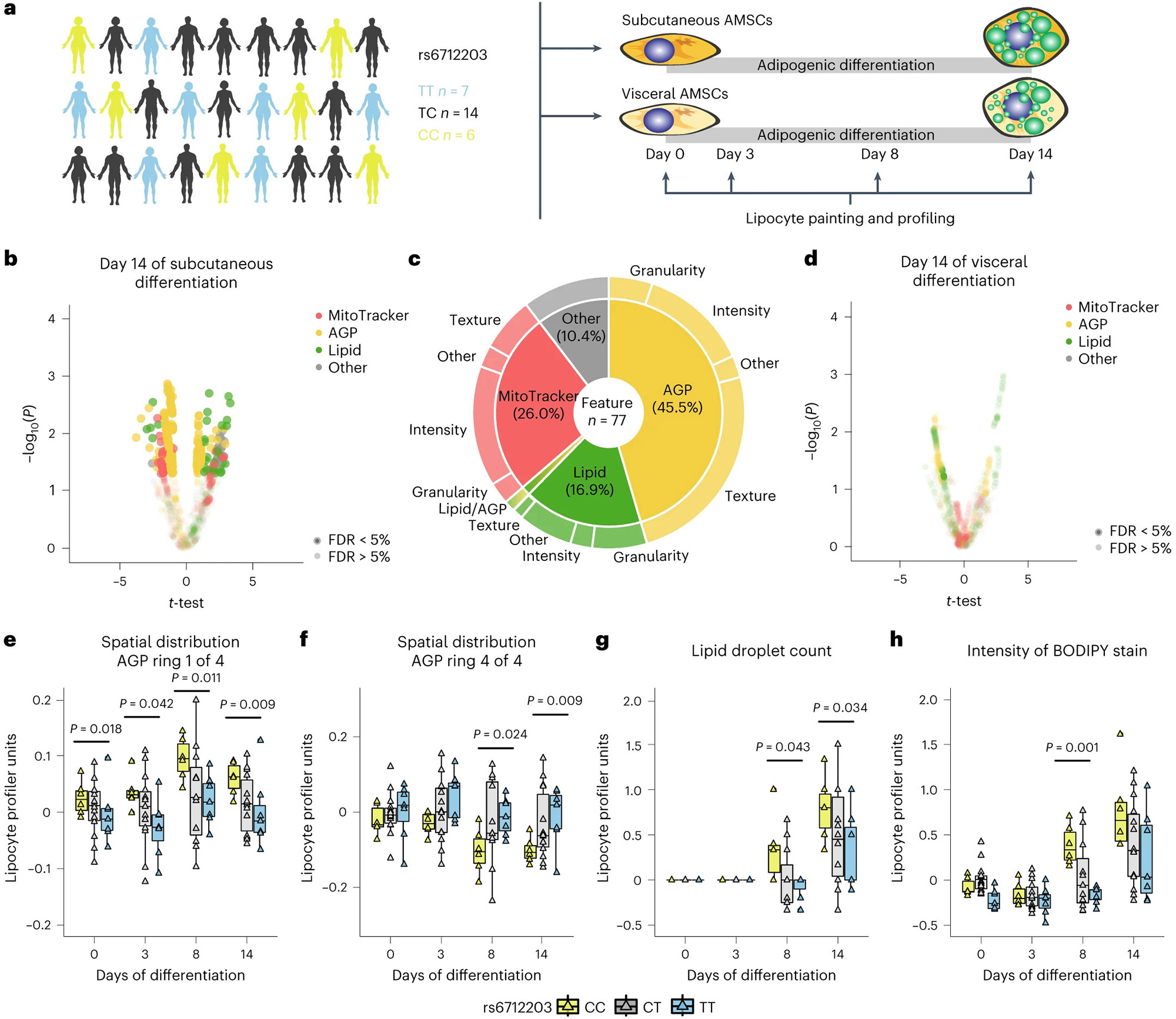
a, Schematic of adipocyte differentiation and LipocyteProfiling of subcutaneous AMSCs derived from TT (n = 7) and CC (n = 6) allele carriers of rs6712203 using LipocyteProfiler43. b,d, Differences in morphological profiles between TT (n = 7) and CC (n = 6) allele carriers at day 14 in subcutaneous (b) and visceral (d) AMSCs (multi-way ANOVA, significance level FDR < 5%). c, Pie chart illustrating non-redundant differential features per channel and class of measurement at day 14 of subcutaneous adipocyte differentiation in rs6712003 homozygous risk carriers compared to non-risk carriers. e,f, Spatial intensity distribution of AGP in the centre of the cytoplasm near the nucleus in subcutaneous adipocytes derived from TT (n = 7), TC (n = 14) and CC (n = 6) carriers of rs6712203 (Cytoplasm_RadialDistribution_ FracAtD_AGP_1of4) (e) andjuxtaposed to the plasma membrane (Cytoplasm_ RadialDistribution_RadialCV_AGP_4of4) (f) throughout differentiation. Multi-way ANOVA; data represent the median ± 95% CI. g,h, Lipid droplet count (Cells_Children_LargeLipidObjects_Count) (g) and intensity of BODIPY stain (Cells_Intensity_IntegratedIntensity_Lipid) (h) throughout differentiation. Multi-way ANOVA; data represent the median ± 95% CI.
Cobll1-deficient mice are metabolically obese normal weight
We generated a CRISPR-engineered Cobll1 knockout (Cobll1−/−) mouse model to determine a potential role for Cobll1 in the regulation of metabolic function in vivo (Extended Data Fig. 7a,b). First, we sought to assess the effect of Cobll1 knockout on morphological and cellular profiles in differentiating murine SAT AMSCs by LipocyteProfiler (days 0, 2 and 10 of differentiation; Fig. 5a). We found that mostly lipid features significantly (FDR < 5%) differed between knockout and control at day 10 of differentiation (Fig. 5b,c and Extended Data Fig. 7c-e). More specifically, the data revealed that the AMSCs of Cobll1 knockout mice showed fewer and smaller lipid droplets (Lipid object_count P = 0.0017, Lipid_Granularity size measure 3 P = 0.0003, Fig. 5d,e), lower lipid intensity (P = 0.0073; Fig. 5f) and, in line with the lipid-related observation, decreased actin cytoskeleton-related heterogeneity across the cytoplasm (Cytoplasm_Texture_Entropy_AGP, P = 0.0200; Fig. 5g). These findings indicate that Cobll1 knockout in mice affects actin cytoskeleton remodelling and lipid accumulation during in vitro adipocyte differentiation, mimicking our observations in human adipocytes. Indeed, when examining the effect of Cobll1 knockout adipocytes on lipid accumulation using Oil Red O, we observed fewer differentiated adipocytes in Cobll1−/− compared to wild-type (WT) cells (Fig. 5h). We also observed significantly lower GPDH activity, an indicator of adipocyte differentiation, in Cobll1−/− mice compared to their WT littermates (P = 0.004) (Fig. 5i), suggesting that the ablation of Cobll1−/− leads to impaired adipogenesis, supporting our finding in human adipocytes.
Fig. 5 ∣. Cobll1-deficient mice are leaner and display metabolically dysfunctional phenotypes.
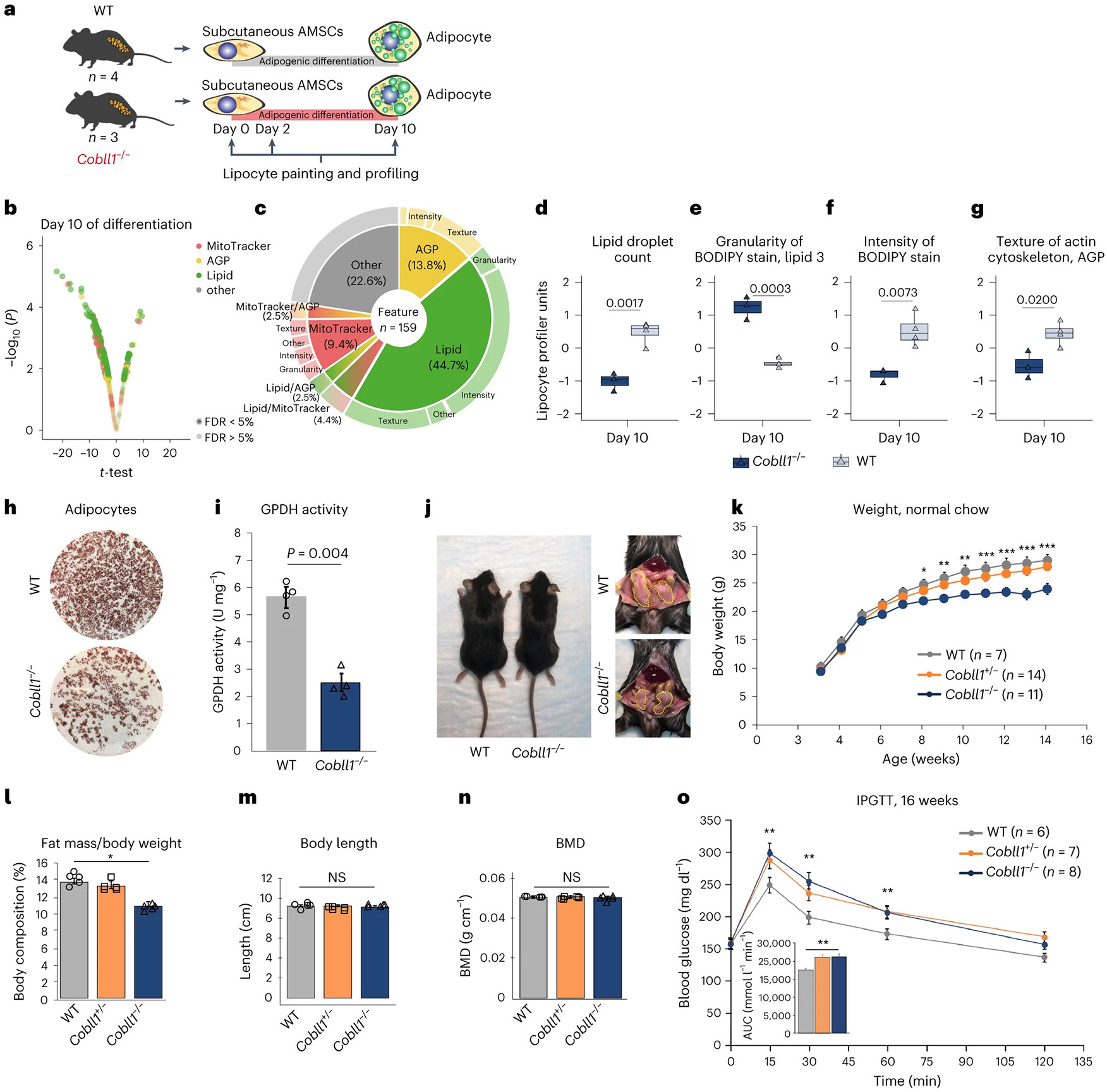
a, Schematic of differentiation and LipocyteProfiling at three time points (days 0, 2 and 10) of AMSCs derived from Cobll1−/− mice (n = 3) and WT mice (n = 4). b, Morphological profiles of the AMSCs of Cobll1−/− mice compared to the AMSCs of WT mice at day 10; two-sided t-test, significance level FDR ≤ 5%. c, Pie chart illustrating non-redundant differential features per channel and class of measurement comparing the AMSCs of Cobll1−/− and WT mice at day 10 of differentiation. d–g, Lipid droplet count (Cells_Children_LargeLipidObjects_Count) (d), granularity of BODIPY stain (Cells_Granularity_3_BODIPY) (e), intensity of BODIPY stain (Cells_Intensity_IntegratedIntensity_Lipid) (f) and texture of actin cytoskeleton (Cytoplasm_Texture_Entropy_AGP) (g) at day 10 of differentiation. Two-sided t-test; data represent the median ± 95% CI. h, Oil Red O staining of differentiated murine AMSCs. i, GPDH activity of differentiated murine AMSCs was assessed by measuring the decrease in NADH at 340 nm. Data represent the mean ± s.e.m. *P < 0.05 compared to the WT group. j, Representative photograph of 14-week-old WT and Cobll1−/− mice fed a normal chow. The yellow dotted lines delineate perigonadal WAT. k, Mouse body weight across time. Data are expressed as the mean ± s.e.m. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to the WT group. l, Body composition (fat mass/body weight). Data are expressed as the mean ± s.e.m. *P < 0.05 compared to the WT group. m, Body length measurements of WT and Cobll1−/− mice (n = 6). Data are expressed as the mean ± s.e.m. *P < 0.05 compared to the WT group. NS, not significant. n, BMD analysed by dual energy X-ray absorptiometry (DEXA). Data are expressed as the mean ± s.e.m. *P < 0.05 compared to the WT group. o, IPGTT in WT and Cobll1−/− and Cobll1+/− mice. The inset graph shows the area under the curve (AUC) of the blood glucose concentration levels measured during IPGTT. Data represent the mean ± s.e.m. **P < 0.01 compared to the WT group. In i,k–o statistical significance was determined by Student’s t-test. In l–o the experiment was repeated independently three times with similar results.
To assess the impact of the 2q24.3 MONW locus effector COBLL1 on organismal processes, we assayed for growth and body composition phenotypes in Cobll1−/− mice. At 10 weeks of age, Cobll1−/− homozygous animals displayed 20–25% less weight gain compared to the WT control and Cobll1 heterozygous (Cobll1+/−) littermates (Fig. 5j,k), reflecting a significant reduction in total fat mass percentage (3–5%), but with no difference in body length or in bone mineral density (BMD), suggesting that the phenotype of Cobll1−/− is due to reduced fat mass (Fig. 5l-n). Next, we examined glucose homeostasis by performing intraperitoneal glucose tolerance tests (IPGTTs). Cobll1−/− mice displayed impaired glucose tolerance compared to WT and heterozygous littermates (Fig. 5o). In conclusion, the phenotypic characteristics of the Cobll1 knockout mouse model recapitulate the MONW association patterns observed in humans and demonstrate how abrogation of Cobll1 links molecular and cellular phenotypes to organismal-level metabolic phenotypes associated with genetic variation in the 2q24.3 locus in humans.
Discussion
The 2q24.3 locus is pleiotropic in nature and, intriguingly, is associated with increased risk of T2D and simultaneously with decreased body fat percentage, reminiscent of a MONW phenotype association signature. In this study, we applied a series of experimental and computational approaches to systematically dissect the 2q24.3 metabolic risk locus and link it to a causal variant (rs6712203), its effector gene (COBLL1), its causal cell type, cell context (developmental time point, adipose depot) and cellular mechanisms (actin remodelling). These altered cellular functions, that is, adipocyte differentiation into metabolically active subcutaneous adipocytes, lipid metabolism and insulin-responsive glucose uptake, are relevant for T2D, and body fat percentage and distribution. When ablating Cobll1 in mice, we showed a lipodystrophy-like phenotype, recapitulating the pleiotropic association with T2D and decreased body fat mass in humans. Thus, we provide genetic and mechanistic evidence that a common genetic variant limits peripheral energy storage capacity and simultaneously impairs insulin responsiveness.
The results of this study lend support to the common hypothesis that the individual risk of T2D and fasting insulin is modified by changes to the mass, distribution and function of adipose tissue23,47, and that a metabolically healthy state is largely dependent on SAT expandability. Inherited and acquired lipodystrophies, as characterized by the selective or global perturbation of adipose tissue function, mass and distribution, result in severe forms of insulin resistance and diabetes; shared molecular mechanisms between rare familial partial lipodystrophy type 1 and common forms of insulin resistance at the genetic level have been suggested previously23. Several common metabolic risk loci are characterized by a MONW and MHO association; distinct association signatures suggest that multiple mechanisms are involved, most of which are to be identified21,22,48. Previous work has convincingly implicated variants at the FAM13A locus to affect metabolic disease risk by affecting subcutaneous adipocyte differentiation48. In this work, we implicate for the first time actin cytoskeleton remodelling as a critical factor for subcutaneous adipocyte function and as causally involved in metabolic disease progression in humans, stressing the notion that MONW and MHO predisposing loci control distinct cellular programmes.
We observed evidence of sex-dimorphic effects when conditioning MONW traits on rs6712203, which is in line with a reported sexual dimorphism for WHR consistently conveying stronger effects in women15,26,49,50 and a sex-independent effect on T2D (refs. 11-14), and with a sex-dimorphic effect on gene expression for COBLL1 but not for GRB14 (ref. 19). While we did not observe a phenotypic effect of Cobll1 knockout in female mice, we cannot exclude the possibility that this is due to well-known sexual dimorphism in mouse metabolic phenotypes51.
The COBLL1 protein has been introduced as a biomarker of high prognostic value for different types of cancer, a modulator of cell morphology in prostate cancer42, and lipid metabolism and insulin signalling in adipocytes52. In this study, we established chain of causation linking the 2q24.3 locus to its functional variant, its adipocyte cell type-specific and context-specific effects, its regulatory element, its effector gene COBLL1 and finally its causal cellular function, that is, actin remodelling in differentiating adipocytes, which is under the genetic control of both locus and target gene. These findings are in line with recent reports linking actin dynamics, regulated by the F-actin:G-actin ratio, and insulin-stimulated trafficking and fusion of GLUT4 vesicles53-55. Actin remodelling also occurs in hypertrophically differentiated adipocytes55,56, an essential adaptive mechanism of post-mitotic cells, such as mature adipocytes, and was recently linked to cellular senescence in states of obesity and insulin resistance57. An unresolved question to be addressed in future work is whether COBLL1-mediated actin dynamics contributes to metabolic functions of certain adipocyte subpopulations with different degrees of insulin sensitivity as recently identified by single-cell and spatial transcriptomics58,59. Furthermore, while we did not observe a haplotype-dependent effect on actin cytoskeleton remodelling in visceral adipocytes, further studies are warranted to elucidate how actin depolymerization and repolymerization during the differentiation process may differ in subcutaneous compared to visceral adipocytes.
Besides COBLL1, the insulin receptor adaptor protein GRB14 is an intuitive effector target gene at the 2q24.3 locus60. GRB14 is a negative regulator of insulin signalling61,62. In line with our findings, physiological studies in GRB14 knockout mice show increased glucose tolerance and insulin sensitivity linked to higher IRS1 and Akt phosphorylation in liver and skeletal muscle but not adipose tissue63, which may rely more on regulation of proximal insulin signalling by GRB10 (refs. 63-65). Somewhat contradictory to these findings and to the results reported in this present work, a recent study reported that CRISPR–Cas9-mediated ablation of GRB14 in human SGBS preadipocytes resulted in decreased preadipocyte proliferation, adipocyte differentiation and adipocyte insulin responsiveness52. This contradiction might be explained by the fact that different perturbation schemes were applied, that is, small hairpin RNA (shRNA)-mediated silencing of GRB14 as described in this study compared to CRISPR–Cas9 knockout of GRB14, which may be confounded by the number of cells in a population that do not produce knockout phenotypes66 (González et al.67); also partial loss-of-function or gain-of-function phenotypes can be generated by Cas9-induced in-frame insertion and deletions, and hypomorphic alleles67, which can obscure the readout. COBLL1 as the effector gene that underlies the T2D association is further corroborated by recent exome chip and exome sequencing studies linking an rs7607980 coding variant in the COBLL1 gene to T2D and glycaemic traits (minor allele frequency (MAF) = 0.12, P = 4.7 × 10−11) (refs. 68-70). Furthermore, recent rare variant aggregation analyses at COBLL1 revealed nominal association with WHR71 but not with GRB14, which agrees with our findings that COBLL1 drives at least part of the 2q24.3 genetic risk. Our sequence-based predictive models scored rs6712203 highest across all 2q24.3 haplotype variants; our experimental data provide evidence that the SNP rs6712203 acts as a causal regulatory variant which, at least in part, drives effects at the 2q24.3 MONW locus tagged by rs3923113. Notably, our sequence-based models predict additional variants at the locus to affect regulatory activity; at least two additional variants, rs12692737 and rs10179126, are associated with allelic chromatin accessibility bias. Therefore, while beyond the scope of our study, we note that our work does not preclude multiple variants acting in concert at this locus in a cell state-dependent manner, potentially implicating GRB14 along with COBLL1 as effector genes. Furthermore, besides rs3923113, recent large-scale genetic association studies identified multiple additional signals tagged by other lead SNPs in reasonably high LD that associate with metabolic disease13,25; also, the effect of indels should be evaluated in future studies.
In conclusion, the 2q24.3 locus is a prime example of a common genetic locus that predisposes to limited peripheral adipose storage capacity and insulin resistance, driven by an impairment of dynamic actin cytoskeleton remodelling of the differentiating subcutaneous adipocyte. Our study epitomizes the potential of translating pleiotropic genetic loci into mechanistic understanding towards therapeutic hypotheses.
Methods
Human primary AMSCs isolation and needle biopsies
We obtained AMSCs from the adipose tissue of patients undergoing a range of abdominal laparoscopic surgeries (sleeve gastrectomy, fundoplication or appendectomy). VAT was derived from the proximity of the angle of His and SAT was obtained from beneath the skin at the site of surgical incision. Human liposuction material was obtained from a private plastic surgery clinic. Isolation of AMSCs was performed as described previously72,73. Abdominal specimens from SAT were obtained by needle aspiration before and 4 weeks after a very-low-calorie diet (800 kcal). Study design and the abdominal needle aspiration procedure were previously described in Ott et al.74.
Each participant gave written informed consent before inclusion and the study protocol was approved by the ethics committee of the Technical University of Munich (147 study no. 5716/13).
Differentiation of human AMSCs
For imaging, cells were seeded at 10,000 cells per well in 96-well plates (high-content imaging; CellCarrier plates, catalogue no. 6005550; PerkinElmer) or seeded at 18,000 cells per well in collagen IV-coated 8-well μ-slides (higher-resolution imaging; catalogue no. 80822, ibidi) and induced 4 d after seeding. For RNA sequencing (RNA-seq), cells were seeded at 40,000 cells per well in 12-well dishes (Corning). Before induction, cells were cultured in proliferation medium (DMEM/F-12, 1% penicillin-streptomycin, 33 μM biotin, 17 μM pantothenate supplemented with 0.13 μM insulin, 0.01 μg ml−1 epidermal growth factor, 0.001 μg ml−1 fibroblast growth factor, 2.5% FCS). Subcutaneous and visceral adipogenic differentiation was induced by changing culture medium to induction medium (basic medium supplemented with 0.861 μM insulin, 1 nM T3, 0.1 μM cortisol, 0.01 mg ml−1 transferrin, 1 μM rosiglitazone, 25 nM dexamethasone and 2.5 nM IBMX; for visceral AMSCs, 0.1 mM oleic and linoleic acid and 2% FCS were also used). On day 3 of adipogenic differentiation, culture medium was changed to differentiation medium (basic medium supplemented with 0.861 μM insulin, 1 nM T3, 0.1 μM cortisol, 0.01 mg ml−1 transferrin; for visceral AMSCs, 0.1 mM oleic and linoleic acid and 2% FCS were also used.
Genotyping
Genotyping was performed using the Illumina Global Screening bead-chip array. DNA was extracted using QIAGEN’s DNeasy Blood and Tissue Kit (catalogue no. 69504) and sent to the Oxford Genotyping Centre for genotyping on the Infinium HTS assay on Global Screening Array bead-chips (Illumina). Genotype quality control (QC) was done using GenomeStudio and genotypes were converted into PLINK format. We checked sample missingness but found no sample with missingness greater than 5%. For the remaining sample QC steps, we reduced the genotyping data down to a set of high-quality SNPs. These SNPs were: (1) common (MAF > 0%); (2) missingness < 0.1%; (3) independent, pruned at an LD (R2) threshold of 0.2; (4) autosomal only; (5) outside the lactase locus (chromosome 2), the major histocompatibility complex (chromosome 6), and outside the inversions on chromosomes 8 and 17; and (6) in Hardy–Weinberg equilibrium (P > 1 × 10−3). Using the remaining approximately 65,000 SNPs, we checked samples for inbreeding (--het in PLINK), but found no samples with excess homozygosity or heterozygosity (no sample more than 6 s.d. from the mean). We also checked for relatedness (--genome in PLINK) and found one pair of samples to be identical; we kept the sample with the higher overall genotyping rate. Finally, we performed principal component analysis using EIGENSTRAT and projected the samples onto data from HapMap 3, which includes samples from 11 global populations. Six samples had some non-European ancestral background, while most samples were of European descent. We did not remove any samples at this step, selecting to adjust for principal components (PCs) in genome-wide testing. Adjustment for PCs failed to eliminate population stratification; therefore, we restricted the process to samples of European descent only, defined as samples falling within ±10 s.d. of the first and second PCs of the CEU (Utah residents with Northern and Western European ancestry) and TSI (Tuscans in Italy) samples included in the HapMap 3 dataset. Finally, sex information was received after the initial sample QC was complete. One sample with potentially mismatching sex information (comparing genotype and phenotype information) was discovered after analyses were complete and therefore remained in the analysis.
SNP QC.
We removed monomorphic SNPs and all SNPs with missingness greater than 5% and out of Hardy–Weinberg equilibrium (P < 1 × 10−6). We set heterozygous haploid sites to missing to enable downstream imputation. The final cleaned dataset included 190 samples and approximately 700,000 SNPs.
Genotype imputation.
We performed imputation via the Michigan Imputation Server as described in Glastonbury et al.75. Briefly, we aligned SNPs to the positive strand and then uploaded the data (in VCF format) to the server. We imputed the data using the Haplotype Reference Consortium panel. We selected EAGLE as the phasing tool to phase the data. To impute chromosome X, we followed the server protocol for imputing this chromosome (including using SHAPEIT to perform the phasing step).
ATAC–seq in immortalized AMSCs
ATAC–seq was performed by adapting the protocol from Buenrostro et al.76 by adding a nucleus preparation step. Differentiating cells were lysed directly in the culture plate before adipogenesis was induced (days 0, 3, 6 and 14). Ice-cold lysis buffer was added directly onto cells grown in a 12-well plate. Plates were incubated on ice for 10 min until cells were permeabilized and nuclei released. Cells in lysis buffer were gently scraped off the well and transferred into a chilled 1.5-ml tube to create crude nuclei. Nuclei were spun down at 600g for 10 min at 4 °C. Nuclei pellets were then resuspended in 40 μl Tagmentation DNA Buffer (catalogue no. FC-121-1031, Nextera) and the quality of the nuclei was assessed using trypan blue. The volume of 50,000 nuclei was determined using a haemocytometer. The transposition reaction was performed as described previously76. All tagmented DNA was PCR-amplified for eight cycles using the following PCR conditions: 72 °C for 5 min, 98 °C for 30 s, followed by thermocycling at 98 °C for 10 s, 63 °C for 30 s and 72 °C for 1 min. The quality of the ATAC–seq libraries was assessed using a Bioanalyzer High Sensitivity Chip (Applied Biosystems). All libraries had a mean fragment size of approximately 200 bp and characteristic nucleosome patterning, indicating good quality. Libraries were pooled and sequenced on a HiSeq 4000 system (Illumina), generating 50 million 75-bp paired-end reads per sample. To reduce bias due to PCR amplification of libraries, duplicate reads were removed. Sequencing reads were aligned to the hs37d5 reference genome; the Burrows–Wheeler Aligner-MEM was used for mapping. All experiments were performed in technical duplicates.
LipocyteProfiling in human and mouse AMSCs
The full LipocyteProfiler protocol is described in Laber et al.43. Briefly, human and mouse AMSCs were plated and differentiated in 96-well CellCarrier plates for high-content imaging at days 0, 3, 8 and 14 of differentiation. On the respective day of the assay, cell culture medium was removed and replaced with 0.5 μM MitoTracker staining solution (1 mM MitoTracker Deep Red stock (catalogue no. M22426, Invitrogen) diluted in culture medium) to each well followed by 30-min incubation at 37 °C protected from light. After 30 min, the MitoTracker staining solution was removed and cells were washed twice with Dulbecco’s PBS (1×) (catalogue no. 21-030-CV, Corning) and 2.9 μM BODIPY staining solution (3.8 mM BODIPY 505/515 stock (catalogue no. D3921, Thermo Fisher Scientific) diluted in Dulbecco’s PBS) was added followed by 15-min incubation at 37 °C protected from light. Subsequently, cells were fixed by adding 16% methanol-free paraformaldehyde (PFA) (catalogue no. 15710-S, Electron Microscopy Sciences) directly to the BODIPY staining solution to a final concentration of 3.2% and incubated for 20 min at room temperature protected from light. PFA was removed and cells were washed once with Hank’s Balanced Salt Solution (HBSS) (1×) (catalogue no. 14025076, Gibco). To permeabilize cells, 0.1% Triton X-100 (catalogue no. X100, Sigma-Aldrich) was added and incubated at room temperature for 10 min protected from light. After permeabilization, multi-stain solution (10 U Alexa Fluor 568 phalloidin (catalogue no. A12380, Thermo Fisher Scientific), 0.01 mg ml−1 Hoechst 33342 (catalogue no. H3570, Invitrogen), 0.0015 mg ml−1 WGA, Alexa Fluor 555 conjugate (catalogue no. W32464, Thermo Fisher Scientific), and 3 μM SYTO 14 Green Fluorescent Nucleic Acid Stain (catalogue no. S7576, Invitrogen) diluted in HBSS) was added and cells were incubated at room temperature for 10 min protected from light. The staining solution was removed and cells were washed three times with HBSS. Cells (25 fields per well) were imaged with an Opera Phenix High-Content Screening System.
Staining and microscopy
To stain the actin cytoskeleton, COBLL1 and nuclei, cells were washed twice with ice-cold PBS and fixed with PFA Roti-Histofix 4% (Roth) for 15 min. Cells were washed twice with ice-cold PBS for 5 min and incubated with ice-cold 0.1% Triton X-100/PBS (Roth). Cells were washed again twice with PBS and incubated for 1 h at room temperature with 4% BSA, then incubated with 1:100 primary COBLL1 antibody (catalogue no. HPA053344, Atlas Antibodies, lot no. 000010746) overnight at 4 °C, followed by 1 h at room temperature (final concentration = 2 μg ml−1). Cells were washed twice with PBS and stained with 0.46% bisBenzimide H 33258 (Sigma-Aldrich), 1% phalloidin–Atto 565 (Sigma-Aldrich) and a secondary antibody against COBLL1 (1:200 Alexa Fluor 488, Abcam) at a final concentration of 0.01 mg ml−1). Cells were incubated for 1 h at room temperature in the dark. Afterwards, cells were washed twice with PBS for 5 min and kept in PBS at 4 °C until imaging. Images were acquired on a Leica DMi8 microscope (Leica Microsystems) using the HC PL APO 63×/1.40 oil objective. Images were processed with the Leica LASX software.
LipocyteProfiler
The full LipocyteProfiler protocol is described in Laber et al.43. Quantitation was performed using CellProfiler v.3.1.9. Before processing, flat-field illumination correction was performed using functions generated from the mean intensity across each plate. Nuclei were identified using with 4,6-diamidino-2-phenylindole stain and then expanded to identify whole cells using the AGP and BODIPY stains. Regions of the cytoplasm were determined by removing the nuclei from the cell segmentations. Speckles of BODIPY staining were enhanced to assist in the detection of small and large individual BODIPY objects. For each object set, measurements were collected representing size, shape, intensity, granularity, texture, colocalization and distance to neighbouring objects. After the feature extraction, data were filtered by applying automated and manual QC steps. First, fields with a total cell count less than 50 cells were removed. Second, fields that were corrupted by experimentally induced technical artefacts were removed by applying a manually defined QC mask. Furthermore, blocklisted features known to be noisy and generally unreliable were removed. After filtering, data were normalized per plate using a robust scaling approach that subtracts the median from each variable and divides it by the interquartile range. For each individual, wells were aggregated for downstream analysis by cell depot and day of differentiation. Subsequent data analyses were performed in R v.3.6.1 using base packages unless noted. For dimensionality reduction visualization, uniform manifold approximation and projection (UMAP) maps were created using the UMAP R package (https://github.com/lmcinnes/umap) with default settings.
To test for a difference in morphological profiles at any day of differentiation due to COBLL1 knockdown, individuals were analysed separately using a t-test. To test for a difference in morphological profiles at any day of differentiation between risk and non-risk haplotypes, a multi-way ANOVA was performed. Differences in morphological profiles between TT (n = 7) and CC (n = 6) allele carriers were adjusted for sex, age, BMI and batch. To overcome multiple-testing burden, P values were corrected using the FDR described in the R package qvalue (https://github.com/StoreyLab/qvalue). Features with an FDR < 5% were classified as significant and filtered based on redundancy and effect size.
COBLL1 silencing using siRNA
All silencing experiments were performed on four technical replicates. One day before silencing, AMSCs were plated into 96-well plates with 10,000 cells per well or collagen IV-coated 8-well glass μ-slides with 18,000 cells per well using growth medium. RNA-based silencing of COBLL1 was performed using the RNAiMAX Reagent (catalogue no. 13778075, Thermo Fisher Scientific) according to the manufacturer’s protocol. Briefly, Lipofectamine RNAiMAX Transfection Reagent was diluted in Opti-MEM medium (catalogue no. 11058021, Gibco). At the same time, siRNA was diluted in Opti-MEM medium. Then, diluted siRNA was added to the diluted Lipofectamine RNAiMAX reagent at a 1:1 ratio and incubated for 5 min. Coated 8-well glass μ-slides were incubated for 20 min at room temperature. The concentration of reagents per well in a 96-well plate were 0.5 μl (10 μM) of silencing oligonucleotides (catalogue no. 4392420, Ambion) or negative control duplex (catalogue no. 4390846, Ambion) and 1.5 μl Lipofectamine RNAiMAX Reagent. The plate was gently swirled and placed in a 37 °C incubator at 5% CO2 for 3 d. Cells were then induced to differentiate using a standard differentiation cocktail or collected for gene expression analysis to assess knockdown efficiency.
RNA preparation and quantitative PCR
Total RNA was extracted with TRIzol (catalogue no. 15596026, Ambion) and the Direct-zol RNA MiniPrep Kit (catalogue no. R2052, Zymo Research) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized with the High-Capacity cDNA Reverse Transcription Kit (catalogue no. 4368814, Applied Biosystems) according to the manufacturer’s instructions. Quantitative PCR (qPCR) was performed using the PCR Master Mix (catalogue no. K0172, Thermo Fisher Scientific) and TaqMan probes for the target gene COBLL1 (catalogue no. 4448892, ID: Hs01117513_m1, Thermo Fisher Scientific) and the housekeeping gene CANX (catalogue no. 4448892, ID Hs01558409_m1, Thermo Fisher Scientific). Relative gene expression was calculated by the 2–ΔΔCt method. Target gene expression was normalized to the expression of CANX.
RNA-seq and splicing analysis
RNA-seq reads were trimmed using SeqPurge with the following command: SeqPurge -a1 CTGTCTCTTATACACATCTCCGAGCCCACGAGAC-a2 CTGTCTCTTATACACATCTGACGCTGCCGACGA.
For the transcript-level quantification, trimmed reads were analysed with Kallisto (with 25 bootstraps) and the transcripts per million (TPM) estimates were log-transformed; the top 10 PCs were computed. Next, reads were summed across all transcripts of a given gene to obtain gene-level estimates of the expression in each sample.
For the splicing analysis with Leafcutter, reads were mapped with STAR using the following parameters: STAR--twopassMode Basic--outSAMstrandField intronMotif--readFilesCommand zcat-outSAMtype BAM Unsorted.
They were then processed using Samtools and RegTools to convert to a JUNC file with the following command: regtools junctions extract -s 1 -a 8 -m 50 -M 500000.
Finally, reads were clustered into splicing events with the following command from the Leafcutter project: leafcutter cluster regtools.py -j <files> -m 50 −1500000. These clusters were then converted to TPM and modelled as a function of the rs6712203 genotype.
RNA pathway enrichment analysis
Transcript-level (log) RNA expression was compared between COBLL1 and all other quantified genes using linear regression. The effect of COBLL1 on other genes was compared and adjusted for the expression PCs, sample depot source, cell line and day of differentiation. This resulted in effect sizes of individual genes in terms of how similar they were to COBLL1; those with estimates that had a Bonferroni-adjusted P > 1 × 10−13 and an absolute effect size less than 0.1 or greater than 10 were excluded. Similarly expressed genes with strong association with COBLL1 were uploaded to Enrichr and analysed as a gene list against the KEGG, and the WikiPathways and HCI pathways. The full set of tests is available at https://maayanlab.cloud/Enrichr/enrich?dataset=1a9a07019bfd8bbddc6eb6c26641bfef; the sensitivity evaluation in which very-lowly-expressed and highly-expressed genes were not excluded are available at https://maayanlab.cloud/Enrichr/enrich?dataset=231b12708d04818007d93364e489fab7.
PMCA variant conservation analysis
The PMCA results were replicated from Claussnitzer et al.29. Briefly, transcription factor binding sites and their cooccurrence across species were tallied and classified into complex and non-complex regions. Complex regions were counted on the basis of motifs aligned across species and were then plotted against Basset scores to discover putative causal variants.
Basset variant effect prediction analysis
Basset models were trained and evaluated as in Sobreira et al.77. Models were trained to capture chromatin regulation relevant to adipocyte differentiation and a number of other metabolic tissues; these effects were estimated by determining the difference in effect between alleles at each variant. The variants with the largest effect on accessibility were considered the most important and most likely to be causal.
Allele-specific accessibility analysis
Allele-specific analyses were performed as in Sinnott-Armstrong et al.78. Briefly, reads were aligned from a heterozygous individual on the basis of the variant; the number of reads supporting each allele were tallied at each time point and across variants on the haplotype.
Conditional and BMI-dependent variant association analysis
Variants (n = 6,167) within 100 kb of rs6712203 were included in the analysis. White British individuals in the UKB were analysed with phenotype type 2 diabetes (as described in Eastwood et al.79), log WHR adjusted for BMI, hip circumference and whole-body fat mass. Individuals were stratified on the basis of reported sex and filtered to White British unrelated individuals as described in Sinnott-Armstrong et al.80. Conditional analyses and all associations were performed using PLINK 2.0.
POU2F2 affinity modelling using the IGR method
The IGR method was used for POU2F2 affinity modelling using the POU2F2 ChIP–seq data35. To correct for systematic bias in the sequencing depth around particular k-mers, all scores were offset by a baseline value, defined as the average signal between the forward and reverse complement instances of the k-mer between −200 and −195 and between 195 and 199 bases away from the k-mer centre. To include only large-effect binding differences, the prominence was defined as the maximum score across any point in the context of either the forward or reverse complement version of the k-mer for both alleles and the maximum difference as the maximum absolute difference in scores between the two alleles at any point in the window. The baseline ratio was defined as the ratio of the maximum difference to the prominence, which varied between 0 (if the two alleles were equal at all points) and 2 (if they were perfectly complementary at their highest absolute point).
To map high-quality putative disrupted binding sites, the k-mer sequence that gave the highest affinity under the germline was recorded as reference and the k-mer sequence that gave the highest affinity under the somatic variant as alternate. The quality of a given k-mer was defined as the correlation between the average context plot forward and the reverse of the average context plot of the reverse complement; the symmetry of a given k-mer was the correlation between the average context plot forward and the average context plot reverse. Quality was high when the antiparallel binding was preserved, and symmetry was high when the peak signal was centred with respect to the variant. The results were included as passed when the Bonferroni-corrected P value for the comparison was less than 0.05, the baseline ratio was greater than 0.5, quality and symmetry were both greater than 0.85 for one of the alleles, and quality and symmetry were both greater than 0.5 for the other allele.
Microarray expression data
A global gene expression measurement was performed, using Illumina HumanRef-8 v.3 BeadChip microarrays from whole abdominal SAT. Signal intensities were quantile-normalized before the correlation analysis.
SGBS genome editing
To edit the rs6712203 heterozygous allele in SGBS preadipocytes to the homozygous risk (CC) and non-risk (TT) alleles, we applied the CRISPR–Cas9 homology-directed repair genome editing approach. The hCas9 vector was purchased from Addgene (plasmid no. 41815). The guide sequence was selected using the design tool (Zhang Lab, MIT) with a predicted number of 228 potential off-target sites, located 211 bp upstream of rs6712203. It was cloned in front of the U6 promoter into the BbsI cloning site of the sgRNA expression vector (R. Kühn, Helmholtz Zentrum München), using the double-stranded oligonucleotides 5′-CACCGACTCTCCACTACCATTGCCA-3′ and 5′-AAACTGGCAATGGTAGTGGAGAGTC-3′. To amplify the 2,009-bp homology region with the risk or non-risk allele of rs6712203 at mid-position, the genomic DNA (gDNA) of SGBS cells was amplified with the primers 5′-GGTGGTCCCATTAAAAAGAAAGAAGCTTGG-3′ and 5′-CTTCTCTTTTACCCTGCTGGCTACTGGTTG-3′ using High-Fidelity Q5 DNA polymerase (New England Biolabs). The gel-purified PCR product was cloned into the blunt end pjet1.2 vector using the CloneJET PCR Cloning Kit (Fermentas). A clone with the rs6712203 C allele was selected and the corresponding T allele vector was generated using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) with the primers 5′-TCATTCATCATATGCAATTCTGG-3′ and 5′-GGCAAATTAATATTTAGGATTATATC-3′. To avoid Cas9 reactivity after genome editing, the NGG guide target sequence was mutated to NCG in both homology vectors with the primers 5′-CCATTGCCAACGGCTGAGTCAG-3′ and 5′-TAGTGGAGAGTTCTCACAAAAC-3′. SGBS cells were cotransfected with green fluorescent protein (Lonza), hCas9, the respective sgRNA and pMACS v.4.1 (Miltenyi Biotec) plasmids using the Amaxa Nucleofector device (programme U-033) (Lonza). Cells were sorted using the MACSelect Transfected Cell Selection Kit (Miltenyi Biotec). The integrity of each edited vector construct and the SGBS cell nucleotide exchange was confirmed by DNA sequencing (Eurofins).
Lentiviral SGBS cell transduction
The MISSON Lentiviral Packaging Mix (Sigma-Aldrich) was used according to the manufacturer’s instructions. Packaging HEK 293T cells were grown in a low-antibiotic growth medium (DMEM, 10% FCS, 0.1% penicillin-streptomycin). When cells were approximately 70% confluent, they were cotransfected using X-treme GENE HP (Roche), with the packaging plasmid pCMVdeltaR8.91, the envelope plasmid pMD2.G and the pLKO-based plasmid containing shRNA against the human target gene COBLL1 (NM_014900.2-3071s1c1), COBLL1 (NM_014900.2-4440s1c1), GRB14 (NM_004490.1-1243s1c1) or empty vector MISSION TRC2 pLKO.5-puro plasmid (Sigma-Aldrich). Cells were incubated for 24 h, and the medium was discarded and replaced with a serum-rich medium (30% FCS). The supernatant containing the viable virus particles was collected 48 and 72 h after transfection, centrifuged to remove cellular debris and stored at −80 °C.
SGBS cells were seeded at a concentration of 2.6 × 104 cells per 6-well plate and grown in normal growth medium. After 24 h the medium was replaced and supplemented with 8 μg ml−1 Polybrene (Sigma-Aldrich) and virus supernatant with a multiplicity of infection of 2. On consecutive days, cells were washed with PBS and medium was replaced to remove the virus. The medium was supplemented with 0.5 μg ml−1 puromycin 96 h after infection to select stable clones. When cells were grown confluent, puromycin was removed from the medium and cells were differentiated until day 16. Target gene silencing was confirmed by qPCR with reverse transcription (RT–qPCR).
Generation of dCas-hWAT preadipocyte cell line
We generated an hWAT cell line, an established white adipocyte line33, stably expressing dCas9-KRAB. Lentivirus was delivered to preadipocytes using spinfection. For single-copy integration, the virus titre was optimized to reach an integration efficiency of 30-40%. Four days after infection, we positively selected for cells expressing the integrated transgene using blasticidin. We validated the generated line by assessing dCas9-KRAB activity delivering a sgRNA targeting CD81, stably expressed in adipocytes, and measuring CD81 gene expression.
CRISPRi
sgRNAs targeting TSS were designed using CRISPick (https://portals.broadinstitute.org/gppx/crispick/public) and sgRNAs targeting non-coding regions were designed with assistance from the Genetic Perturbation Platform at the Broad Institute. Guide sequences are as follows: non-targeting control 1: AAAAAGCTTCCGCCTGATGG; non-targeting control 2: CGTCCCTTCGTCTCTGCTTA; non-targeting control 3: TCACCTCCGAACGAACACCT; non-targeting control 4: GGACGC ACCATTCCGGGTGA; COBLL1_1: AGCTCGCCTGTTCTCCCTCG; COB LL1_2: GAGTAGGAGAGGAAGCCGCG; COBLL1_3: AGAGCCGAGCGGC AAGAGCG; COBLL1_4: GAGCCGAGCGGCAAGAGCGC; GRB14_1: AC CACACCTGCAGAGCGCTC; GRB14_2: GCCCGAGCGCTCTGCAGGTG; GRB14_3: CGAGCGCTCTGCAGGTGTGG; GRB14_4: CACCACACCTGC AGAGCGCT; RS4: AGAGTTCTCACAAAACTCCA; RS9: CTCTCCACTA CCATTGCCAA; RS20: CCATTGCCAAGGGCTGAGTC; RS1: CATCACA TGCAATTCTGGCA; RS8: AATACAAATAGAGCAAGTAT; RS11: TTGT AAGTGGAAAAAAAGCT. sgRNAs were cloned into the BsmBI site of a lentiviral pXPR_050 vector (plasmid no. 96925, Addgene)81 and verified using Sanger sequencing.
Lentiviral particles were generated by transfecting HEK 293T cells (8 × 105 per well of a 6-well plate seeded the day before transfection) with 1,250 ng sgRNA, 1,250 ng psPAX2 and 250 ng vesicular stomatitis virus G vectors using a TransIT-LT1 transfection reagent at 8.5 μl per well (Mirus Bio). Viral particles were collected 72 h after transfection, passed through a 0.45-μm filter, aliquoted and stored at −80 °C until use. For lentiviral transduction by spinfection, cells were seeded at 1 × 105 per well of 12-well plates in the presence of 50, 250 or 500 μl viral supernatant and Polybrene (4 μg ml−1) and spun at 930 g for 2 h. Cells were selected with 1 μg ml−1 puromycin for 3 d, split once and collected 6 d after transduction for RNA extraction and gene expression by qPCR. Cas9 expression and COBLL1 knockdown were verified by western blot using specific antibodies (Cas9: catalogue no. C15310258, Diagenode; COBLL1: catalogue no. HPA053344, Sigma-Aldrich). Ponceau-S staining or β-actin (catalogue no. 4970, Cell Signaling Technology).
Measurement of GPDH activity
Cells were grown to confluence and differentiated until day 16 in 6-well plates. Cells were collected in a GPDH buffer with 0.05 M Tris-HCl (pH 7.4), 1 mM EDTA and 1 mM mercaptoethanol before they were stored at −80 °C until further use. Samples were gently defrosted at 4 °C and sonified for 7 s at 29%, and centrifuged for 10 min at 10,000g at 4 °C. GPDH activity was measured as described previously82. Briefly, GPDH activity was assessed, measuring the conversion of dihydroxyacetone phosphate (Sigma-Aldrich) in the presence of the coenzyme nicotinamide adenine dinucleotide (NADH) (Omnilab) at a wavelength of 340 nm, using the Infinite 200 (Tecan). Protein concentrations were assessed using the BCA-reducing agent compatible protein assay kit (Thermo Fisher Scientific) with BSA standard samples in GPDH buffer for quantification. The value for each condition was calculated using the ratio between GPDH activity and protein concentration.
Glucose uptake, lipolysis and western blot analysis
For glucose, glycerol and western blot analysis, shRNA COBLL1, shRNA empty vector and shRNA GRB14 SGBS cells were differentiated until day 16 in 6-well plates.
The insulin-stimulated 2-DG uptake experiment was performed as described previously83. Briefly, cells were incubated in glucose-free DMEM and F-12 (1:1) containing 1% penicillin-streptomycin, 16 μM biotin, 36 μM pantothenic acid, 14.3 mM NaHCO3 and 0.5 mM sodium pyruvate(Sigma-Aldrich) for 12 h. The medium was replaced with 118 mM NaCl, 1.2 mM KH2PO4, 4.8 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 10 mM HEPES, 2.5 mM sodium pyruvate (Sigma-Aldrich) and 0.5% BSA (Sigma-Aldrich), pH 7,35. After 1.5 h the same buffer was added fresh either without supplement or with 1 μM insulin for 30 min. Radioactive uptake was started by adding KRH 3H-2-DG at an activity of 1 μCi ml−1 and 50 μM 2-DG. Cells were incubated for 30 min and then washed with PBS. Cells were scraped off after adding 200 μl IGEPAL and 150 μM phloretin. Radioactivity was measured using liquid scintillation counting with an external standard. Protein concentrations to normalize 2-DG uptake were measured.
To measure glycerol release, cells were washed with PBS and incubated for 3 h in phenol red-free DMEM containing 2% free fatty acid-free BSA (Roth). The medium was changed and cells were incubated for 1 h without supplement for basal lipolysis or the addition of 10 μM isoproterenol (Sigma-Aldrich) and 0.5 mM IBMX for stimulated lipolysis. The supernatant was collected for spectrophotometric glycerol measurement in a Sirius tube luminometer (Berthold Technologies) using glycerol kinase (Sigma-Aldrich) and the ATP Kit SL (BioThema). Remaining cells were collected for protein quantification and western blot analysis in radioimmunoprecipitation assay buffer containing 50 mM Tris-HCl (pH 8), 150 mM NaCl, 0.2% SDS, 1% NP-40, 0.5% deoxycholate, 1 mM phenylmethylsulfonyl fluoride, phosphatase and protease inhibitors. Western blot analysis was performed using a mouse anti-human GPDH IgG (Ambion) and the Lipolysis Activation Antibody Sampler Kit (catalogue no. 8334, Cell Signaling Technology) according to the manufacturer’s protocol. Secondary IRDye IgG (LI-COR) was used to generate the fluorescence, detected by the Odyssey scanner (LI-COR).
Relative gene expression RT–qPCR
Primer pairs were designed using published nucleotide sequences from the human genome (GenBank NCBI/UCSC) and Ensembl, primer3input84, net primer (PREMIER Biosoft) and primer blast (NCBI GenBank)85. Primers against the human target genes COBLL1 (forward TGATCCTGACTCAGCCCTT, reverse CAACTACCTTCTTTCCATTGC) LEFTIN (forward TGGGAAGGAAAATGCATTGGG, reverse ATAAGGT CAGGATGGGGTGG) and GLUT4 (forward CTGTGCCATCCTGATGACTG, reverse CCAGGGCCAATCTCAAAA) and the reference genes HPRT1 (forward TGAAAAGGACCCCACGAAG, reverse AAGCAGATGGCCACAGAACTAG), PPIA (forward TGGTTCCCAGTTTTTCATC, reverse CGAGTTGTCCACAGTCAGC) and IPO8 (forward CGGATTATAGTCTCTGACCATGTG, reverse TGTGTCACCATGTTCTTCAGG) were synthesized by Eurofins.
Total RNA was extracted using the RNeasy Mini Kit (QIAGEN) and 0.5 μg was reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit. RT–qPCR was performed using 96-well plates (black frame, white wells) with heat-sealing films, fixed by the 4s2 Automated Heat Sealer (all from 4titude). The Maxima SYBR Green Mix (Thermo Fisher Scientific) was used for amplification in a RT–qPCR Mastercycler ep realplex (Eppendorf), with a denaturation step of 95 °C for 10 min and 40 cycles of 95 °C for 15 s and 60 °C for 40 s, followed by a melting curve. Relative gene expression was calculated by the 2–ΔΔCt method86 with a reference gene index of HPRT, PPIA and IPO8 or the reference gene HPRT (weight loss study).
Mice
All mice (C57BL/6J) were originally obtained from the Charles River Laboratories. To genetically engineer a Cobll1 whole-body knockout (Cobll1−/−) model, we used CRISPR–Cas9 genome editing system. Male mice were weaned at 4 weeks of age and body weight was measured every week from 4 to 14 weeks of age. All mice were housed in a temperature-controlled room (22 °C with approximately 50–65% humidity) on a 12-h light–dark cycle with ad libitum access to food (normal diet: 14% fat, 64.8% carbohydrate and 21.2% protein, catalogue no. 2920X, Harlan Teklad) and under pathogen-free conditions. To analyse body fat mass (%), body length (cm) and BMD (g cm−2), we used the DEXA scan. Before scanning, animals were anaesthetized with ketamine. All procedures were conducted with approval from the Institutional Animal Care and Use Committee of the University of Chicago (no. ACUP-71656; IBC0934).
CRISPR–Cas9-mediated generation of a Cobll1 knockout mouse model.
To confirm directly that ablation of Cobll1 affects T2D-related phenotypes in vivo, we applied the CRISPR–Cas9 system to genetically engineer a Cobll1 whole-body knockout (Cobll1−/−) model. Using specific sgRNAs, we targeted the Cobll1 gene in the C57BL/6 genetic background. We used guides with the following sequences: gRNA (exon 2) 5′-TTGCTCACTAGTGGGGTCGCAGG-3′ and gRNA (exon 6) 5′-CTTCCTCCGGCCGAGACGAAGGG-3′.
Genotyping.
The genotypes of Cobll1 mutant mice were determined by PCR amplification of gDNA extracted from tails. PCR was performed for 30 cycles at 95 °C for 30 s, 60 °C for 15 s and 72 °C for 30 s, with a final extension at 72 °C for 5 min. PCR amplification was performed using the following primer sets: forward 5′-AAAAGTTTCCTGATGTGAAAGTCA-3′ and reverse 5′-AAAAACAGATGCTCCCCAGA-3′. The PCR products were size-separated by electrophoresis on a 4% agarose gel for 1 h.
In vivo glucose tolerance test.
At 16 weeks old, mice were tested for glucose sensitivity by IPGTT. Before IPGTT, mice were fasted for 4 h and an initial blood glucose reading was taken. This fast was followed by intraperitoneal injection of 2 mg kg−1 dextrose (catalogue no. CAS 50-99-7, Merck Millipore) and subsequent blood glucose checks using an Accu-Chek Aviva glucometer (Roche). Blood glucose readings were taken at 15, 30, 60 and 120 min after dextrose injection. After IPGTT, mice resumed a high-fat diet. An unpaired, two-sided Student’s t-test was used to test for significance.
Real-time qPCR.
After establishment of stable Cobll1 knockout mice, ablation of Cobll1 expression was confirmed by RT–qPCR in relevant tissues, which showed significant decrease in the mRNA fold change of Cobll1 knockout mice compared to WT and heterozygous littermates. Total RNA was isolated from the inguinal white fat pad, kidney and liver using the RNA extraction reagent RNeasy Mini Kit (catalogue no. 74104, QIAGEN). cDNA synthesis was performed using the Superscript III First-Strand Synthesis System (catalogue no. 18080-044, Thermo Fisher Scientific). Real-time qPCR reactions were performed using the SsoAdvanced Universal SYBR Green Supermix (catalogue no. 1725270, Bio-Rad Laboratories). Real-time qPCR amplification was performed using the primer sets: qPcrF 5′-CGTCACAGAGCAACAAGACA-3′ and qPcrR5′-ACTGAGCACAGAGGAACACG-3′.
Isolation, culture and differentiation of mouse preadipocytes.
Primary adipocytes were isolated from dissected SAT of 6-week-old mice and digested in 1 g ml−1 type I collagenase solution (catalogue no. LS004174, Worthington Biochemical Corporation) solution (containing 3.5% BSA v/v (catalogue no. A9418, Merck Millipore)) in a 37 °C water bath with shaking at 120 rpm for 45 min. The suspension was centrifuged at 250g for 5 min; then, the cell pellet was resuspended in culture medium (high-glucose DMEM, catalogue no. D5796, Merck Millipore), 20% FCS (catalogue no. A31605-01, Thermo Fisher Scientific), 100 U ml−1 penicillin and 0.1 mg ml−1 streptomycin (catalogue no. 15140-122, Thermo Fisher Scientific), was filtered through a 45-μm strainer and was seeded in 25-cm2 flasks. Confluent preadipocytes were induced for 2 d with an adipogenic medium (high-glucose DMEM, 10% FCS, penicillin-streptomycin (10,000 U ml−1, 10,000 μg ml−1), 850 nM insulin (catalogue no. 12585014, Thermo Fisher Scientific), 1 nM T3 (catalogue no. T6397, Merck Millipore), 500 μM IBMX (catalogue no. 15879, Merck Millipore), 1 μM dexamethasone (catalogue no. D4902, Merck Millipore), 125 μM indomethacin (catalogue no. 70270, Cayman Chemical) and 1 μM rosiglitazone (catalogue no. 71740, Cayman Chemical), and then switched to differentiation medium (adipogenic medium without IBMX, dexamethasone and indomethacin). Cells were collected on the eight day of differentiation and used for further analysis.
Oil Red O and GPDH assay of mouse preadipocytes.
Oil Red O staining was used to assess the presence of lipids in mature adipocytes. For Oil Red O staining, cells were washed with PBS (catalogue no. P38135, Merck Millipore) and fixed with 4% PFA (catalogue no. P6148, Merck Millipore). Fixed cells were then covered with 3 mg ml−1 Oil Red O (catalogue no. O0625, Merck Millipore) dissolved in 60% isopropanol (v/v) for 20 min; then the dye was washed away with H2O. To determine GPDH activity, we used a commercially available kit from Takara Bio (catalogue no. MK426), by monitoring the dihydroxyacetone phosphate-dependent oxidation of NADH at 340 nm. Enzyme activity was calculated by the formula described in the manufacturer’s protocol; GPDH activity was expressed as U mg−1 protein.
Statistics and reproducibility
Statistical analyses were performed using a two-tailed Student’s t-test or ANOVA to compare the means of two or multiple groups, respectively. No data were excluded from the analyses.
Extended Data
Extended data Fig. 1 ∣. The pleiotropic 2q24.3 MONW locus is associated with increased risk for type 2 diabetes and decreased adiposity related traits and maps to sparse enhancer signatures in adipocytes.
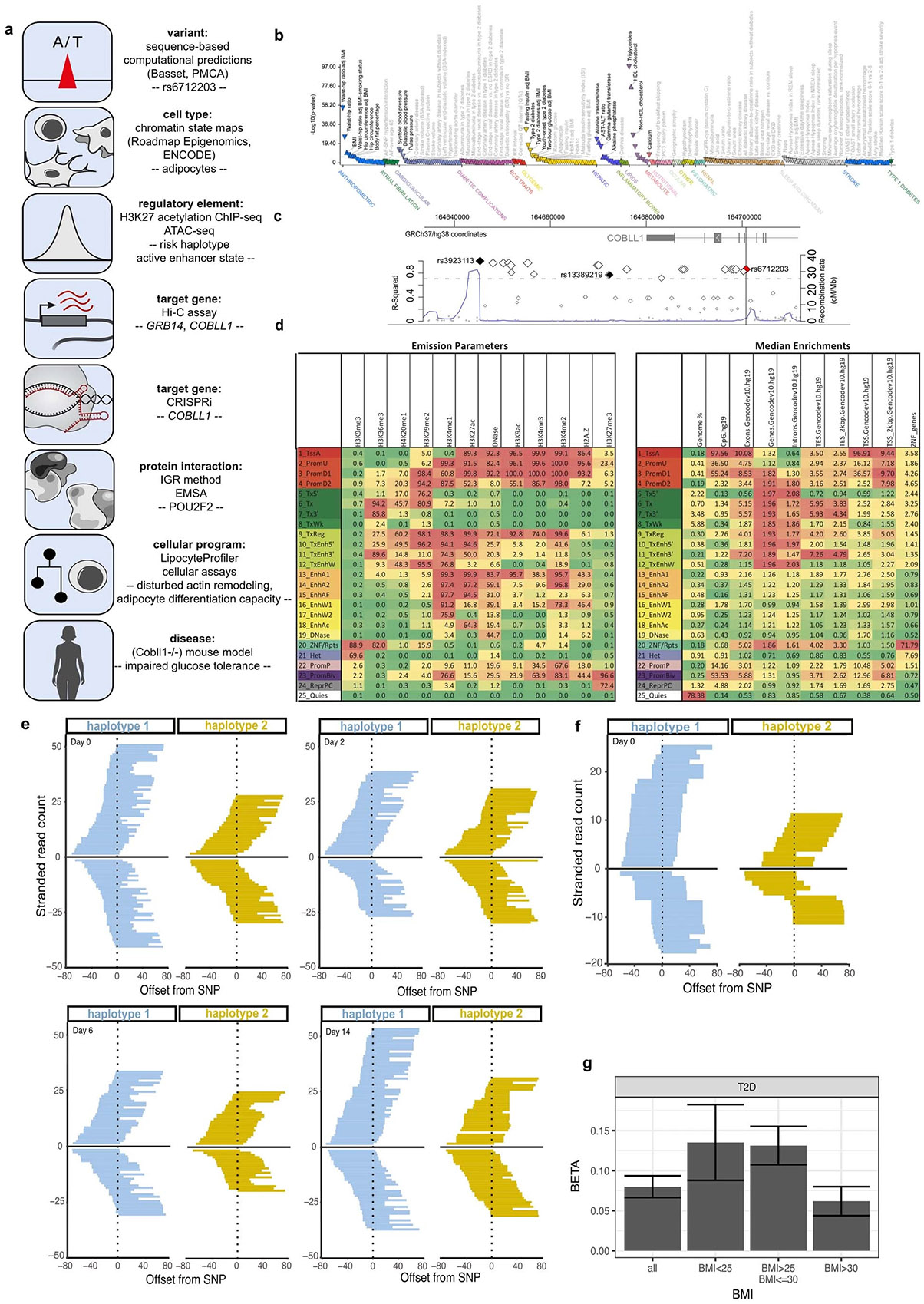
(a) Schematic overview for the 2q24.3 metabolic risk locus dissection. Aim of step (top, bold); methods/experiments used (middle); key finding/result of each step (bottom). (b) PheWAS of trait associations at the rs3923113-tagged haplotype of a meta-analysis https://t2d.hugeamp.org/. Colors represent trait classes while individual rs3923113 variant association p-values are shown on the Y axis. Direction of effect is indicated by orientation of triangles, upward: increase, downward: decrease (c) The 2q24.3 MONW locus spans 19 non-coding SNPs in high linkage disequilibrium with rs3923113 (LD r2 > 0.8). The region of association localizes to a >55 kb interval in an intergenic region between COBLL1 and GRB14. (d) Annotation panel and color key for the twenty-five state chromatin model70. Rows represent chromatin states abbreviations, columns are emission parameters, corresponding to the frequency with which each mark is expected in each state (left table) and genome coverage and median enrichments of relevant genomic annotations (right panel). TssA: Active TSS, TssAFlnk: Flanking Active TSS, TxFlnk: Transcription at gene 5’ and 3’, Tx: Strong Transcription, TxWk: Weak Transcription, EnhG: Genic enhancers: Enh: Enhancers, ZNF/Rpts: ZNF genes & repeats, Het: Heterochromatin, TssBiv: Bivalent/Poised TSS, BivFlnk: Flanking Bivalent TSS/Enhancer, EnhBiv: Bivalent Enhancer, ReprPC: Repressed Polycomb, ReprPCWk: Weak Repressed Polycomb, Quies: Quiescent/Low. (e) Stranded allele-specific chromatin accessibility measures at the haplotype using ATAC-seq data in differentiating adipocytes from a heterozygous individual. For each day of differentiation of an individual heterozygous, the number of reads overlapping with 20 non-coding SNPs in the haplotype, ordered by their start position and strand relative to the position of the variant, are shown. More reads indicate higher activity in haplotype 1 (non-risk, blue) compared to haplotype 2 (risk yellow). x-axis: offset from SNP position (bp), y-axis: stranded read count. (f) Replication of the effect at time 0 (mesenchymal stem cells) with ATAC-seq. (g) BMI-dependent variant association analysis. Bar plots represent the beta of the rs6712203 association with type 2 diabetes following BMI stratification. The cohort analysed is the UK Biobank self-identified white British individuals (total N = 327,960; N = 109198 with BMI < 25, N = 140539 with BMI between 25 and 30, and N = 78223 with BMI > = 30), and overlay of data points is not practical. Betas and 95% confidence intervals are shown, derived from a two-sided generalized linear model on outcome adjusted for demographic covariates (age, sex, genotyping array, 40 PCs).
Extended data Fig. 2 ∣. Conditional ana- lyses implicating rs6712203 in the genetic control of anthropometric traits and type 2 diabetes.
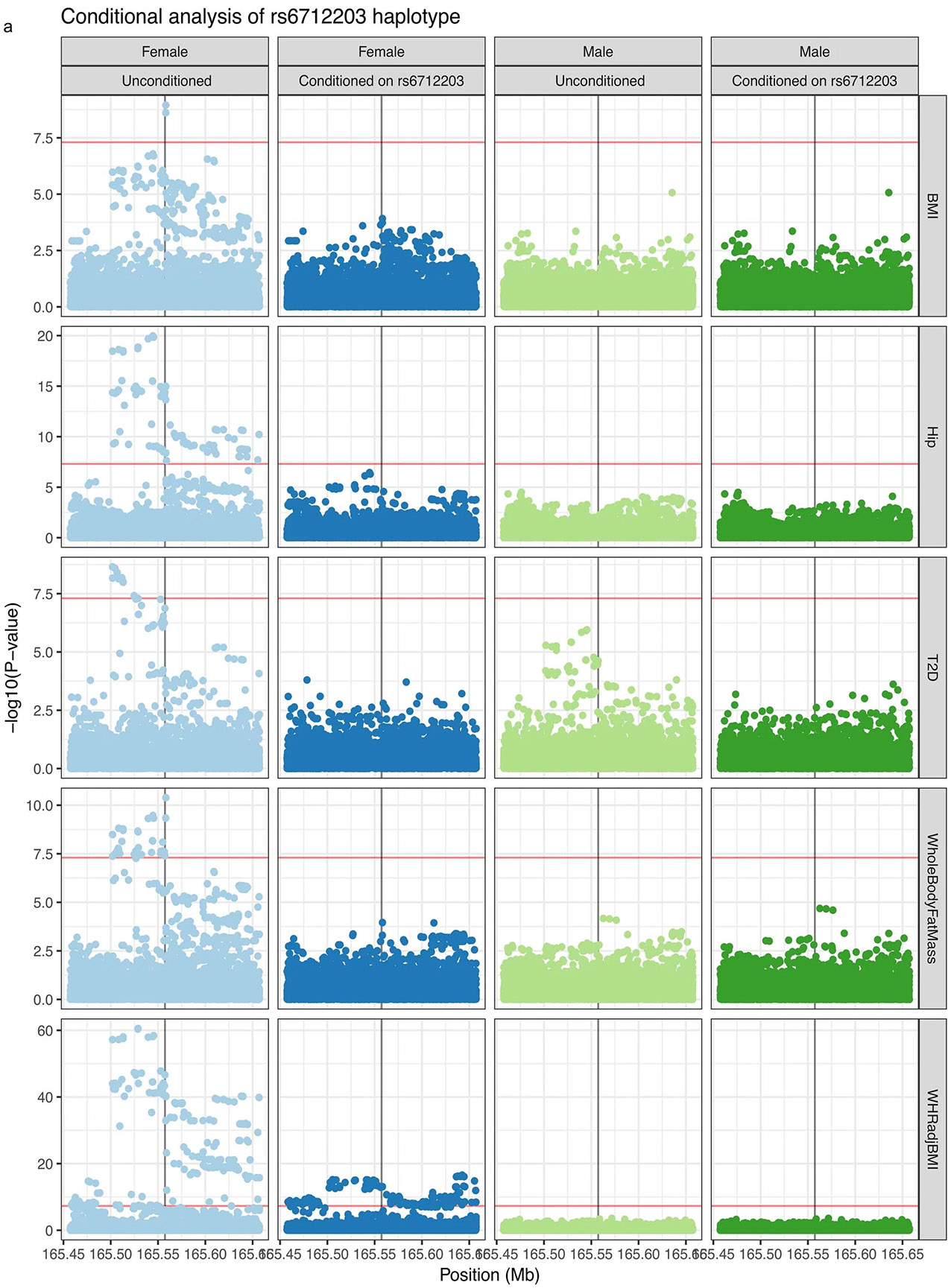
a, Conditional analyses implicating rs6712203 in the genetic control of anthropometric traits and type 2 diabetes. Each panel represents a different trait/sex/conditional analysis window, and all panels have an X axis corresponding to 100 kb on either side of the rs6712203 variant. The Y axis shows, for each variant in the window, the association strength for the given trait conditioned on the variants noted in White British participants in UK Biobank with the sex shown, and red lines indicate the significance threshold 5 × 10-8). −log10 p-values are shown, derived from a two-sided generalized linear model on outcome adjusted for demographic covariates (age, sex, genotyping array, 40 PCs).
Extended data Fig. 3 ∣. Chromatin inter- actions and CRISPRi of 2q24.3 locus identify COBLL1 as target genes.
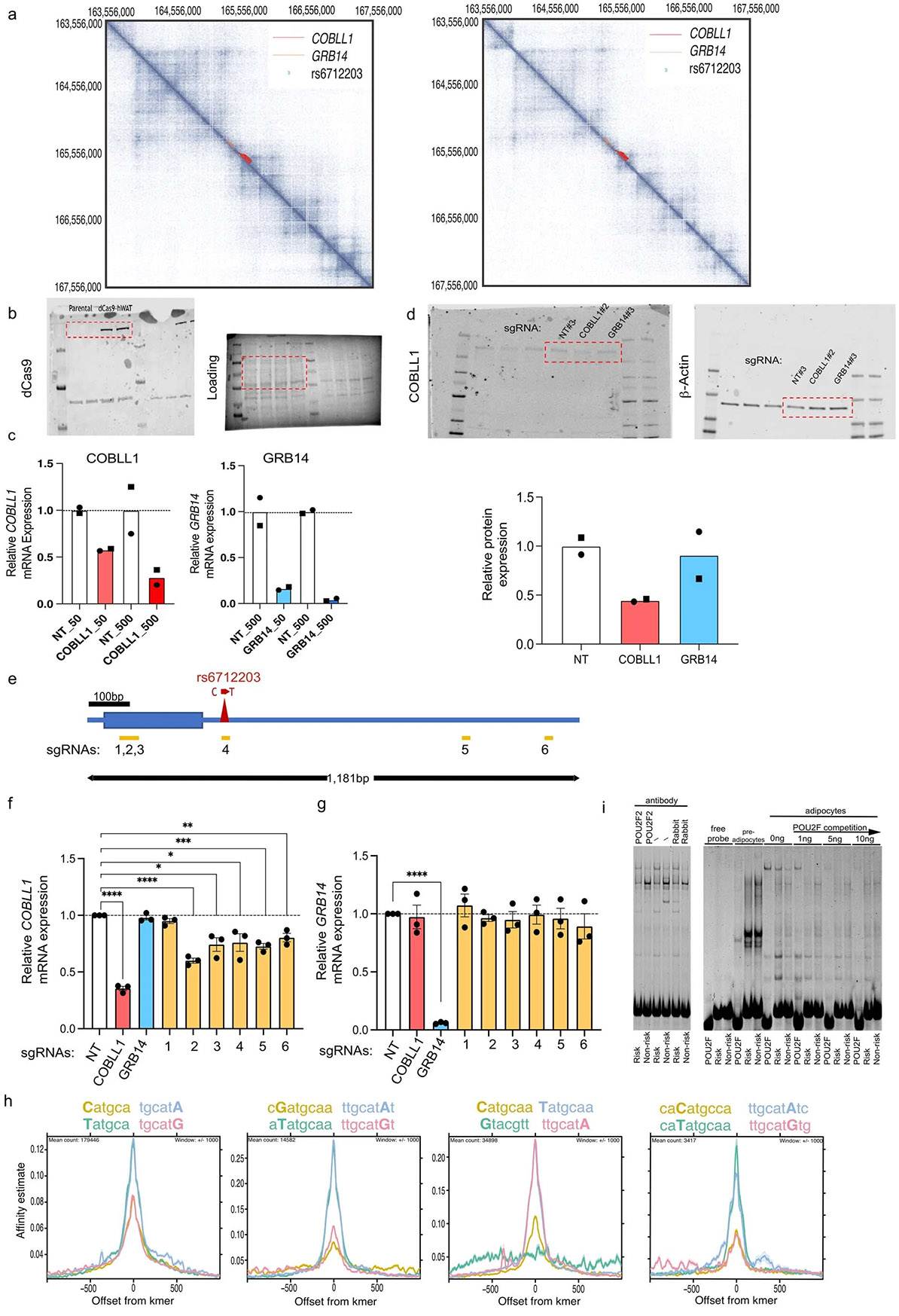
(a) Cross-cell type conserved genome-wide higher order chromatin interactions for the 2q24.3 locus analyzed by Hi-C assays in human fibroblasts (left) and NHEK primary normal human epidermal keratinocytes (right), chr2: 163,556,000 - 167,558,000 (hg19), binned at 2 kb resolution. (b) Cas9 protein expression in dCas9 hWAT compared to the parental hWAT cell line. (c) mRNA expression of COBLL1 and GRB14 in response to increasing amounts of lentiviral sgRNA vectors (2 sgRNAs, virus volume 50 μl and 500 μl) targeting TSS regions of each gene compared to non-targeting controls (NT, 2 sgRNAs). Columns are means of individual sgRNAs indicated by different symbols. (d) COBLL1 protein expression normalized to b-actin in dCas9 hWATs transduced with sgRNAs targeting COBLL1 or GRB14 compared to controls. Top panel: Image of gel of representative sgRNA targeting NT, COBLL1 or GRB14. Bottom panel: plot of protein expression; 2 sgRNA for each target in 2 replicates. (e) Representation of 1,181 bp region flanking the COBLL1 intronic variant rs6712203 at the 2q24.3 MONW locus showing individual sgRNAs (n = 6) targeting the rs6712203 flanking regulatory region used in the CRISPRi experiments. (f, g) mRNA expression of (f) COBLL1 and (g) GRB14 in undifferentiated dCas9-hWAT preadipocytes at 6 days post lentiviral transduction with sgRNAs targeting TSS regions (red: COBLL1 TSS; blue GRB14 TSS) and the rs6712203-flanking regulatory element at position 1 to 6 as depicted in (e). Data are mean +/− SEM of 3 independent experiments. **** P < 0.0001, *** P = 0.0004, ** P = 0.006, * P = 0.013 – 0.036, two-tailed Student’s t test. (h) Predicted binding of POU2F2 between the two alleles using the Intragenomic Replicate Method (Cowper-Sal lari et al. 2012). As in Fig. 2d with different kmer counts.
Extended data Fig. 4 ∣. COBLL1 regulates actin cytoskeleton remodeling.
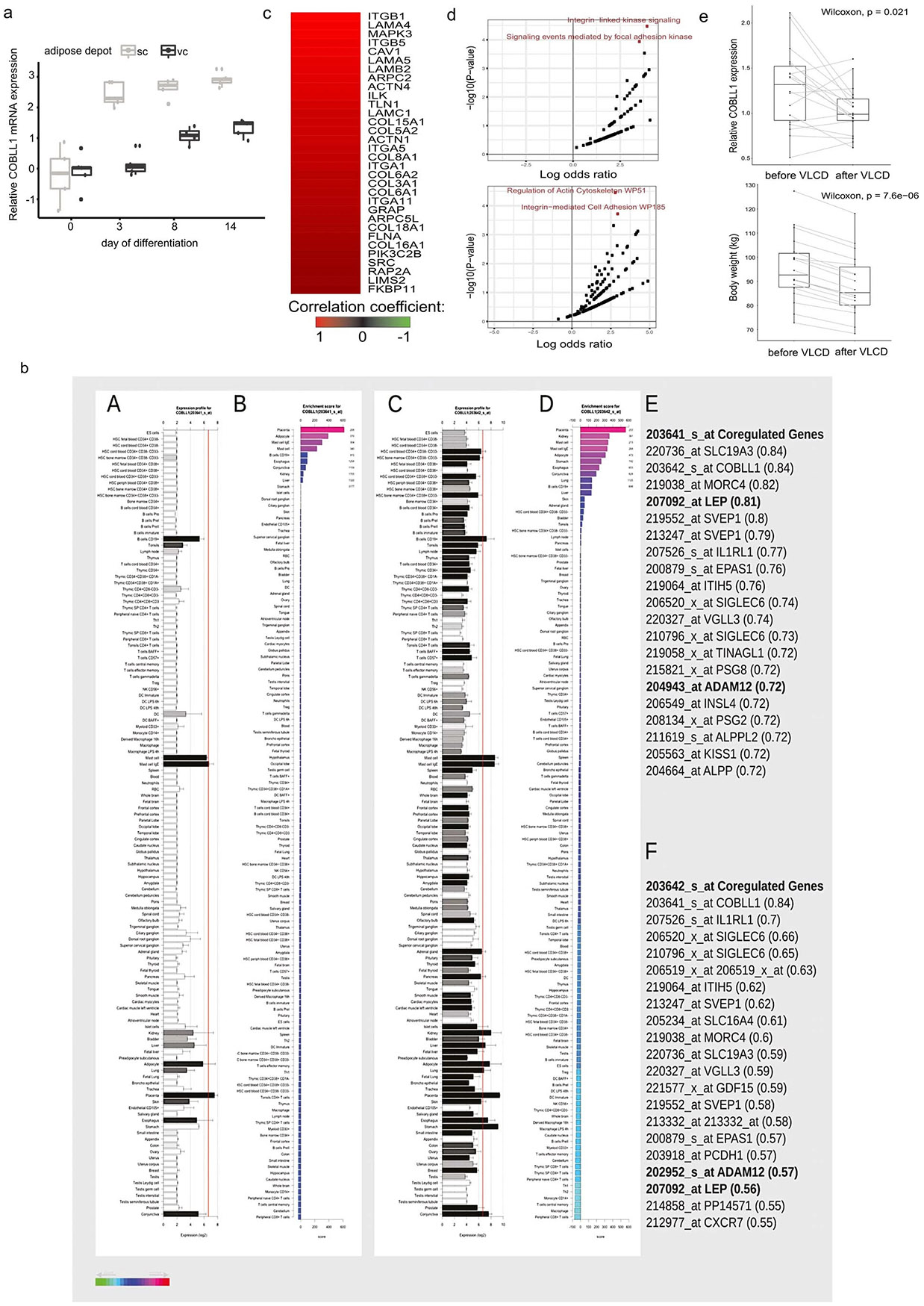
(a) COBLL1 expression in subcutaneous and visceral AMSCs throughout adipogenic differentiation, N = 4 biologically independent experiments, t-test two-sided, data represent median + 95% CI. (b) COBLL1 gene expression enrichment across 142 tissues (A-D) from enrichment profiler36. COBLL1 probes 203641_s_at and 203642_s_at were used for coregulation analysis (E-F). (c) Correlation with COBLL1 probe ILMN_1761260 using microarray data from lean and individuals with obesity. (d) Enrichment of pathways in the HCI (upper panel) and WikiPathways (lower panel) gene set lists from Enrichr, plotted as in Fig. 3A (KEGG), with p-value thresholds corresponding to the FDR cutoffs in those data. p-values are derived from a hypergeometric test. (e) COBLL1 expression in subcutaneous adipose tissue before and after a very low caloric diet (VLCD, upper panel, n = 18), corresponding body weight (lower panel), Wilcoxon signed-rank test.
Extended data Fig. 5 ∣. Knockdown of COBLL1 affects actin remodeling processes in differentiating adipocytes along with adipocyte differentiation insulin sensitivity and lipolysis rate.
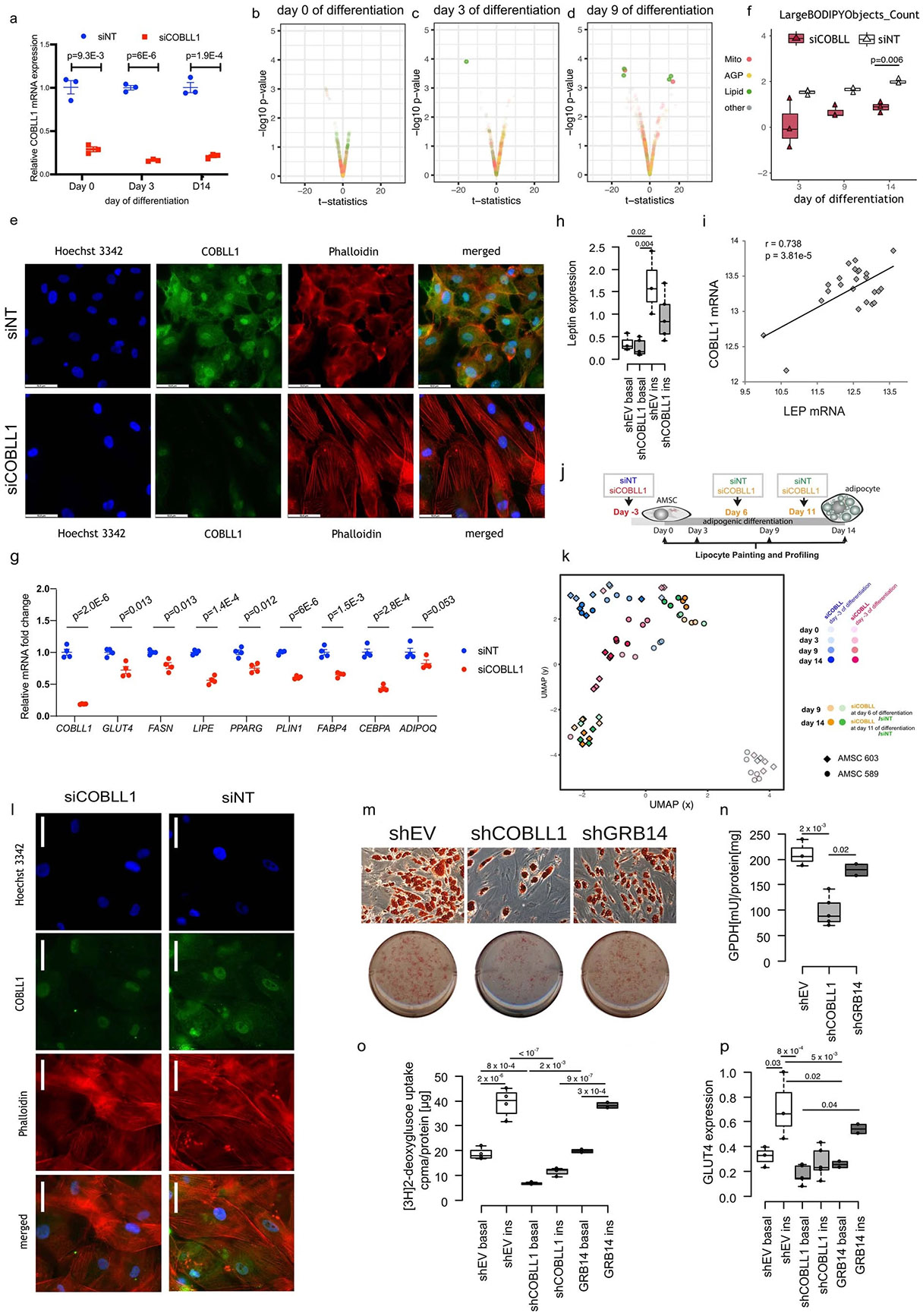
(a) COBLL1 expression in siCOBLL1 and siNT at day 0, 3 and 14 of differentiation, N = 3 biologically independent experiments, t-test two-sided. knock-down efficiency 80%, mean values + SEM. (b–d) Morphological profiles of siCOBLL compared to siNT AMSCs at day 0 (b) day 3 (c) and day 9 (d) of differentiation, t-test two-sided, significance level < 5%FDR. (e) Actin and COBLL1 staining in siCOBLL1 compared to siNT subcutaneous adipocytes at day 9 using phalloidin and COBLL1 antibody staining (HPA053344, Alexa-Fluor 488), magnification x63/oil. Scale bar = 52.8 um. Representative results from N = 3 independent experiments. (f) Cells_Children_ LargeBODIPY_objects_count in siCOBLL1- and siNT AMSCs at day 3, 9, 14, N = 3 biologically independent experiments, t-test two-sided, significance level < 5% FDR. (G) qPCR-based gene expression of COBLL1 and adipocyte marker genes GLUT4, FASN, LIPE, PPARG, PLIN1, FABP4, CEBPA, ADIPOQ in siCOBLL1 and siNT AMSCs at day 14 of differentiation, t-test two-sided, N = 4 biologically independent experiments, mean values +/− SEM. (h) qPCR-based leptin gene expression in shCOBLL1 compared to shEV adipocytes. Data are represented as median + 95% CI, one-way ANOVA with Tukey’s HSD test, N = 4 biologically independent experiments (i) Correlation of COBLL1 mRNA with LEP mRNA in subcutaneous adipose tissue from 24 lean individuals measured by Illumina microarrays. The pearson’s correlation coefficient r and p-value are depicted (j) Schematic of siCOBLL1 KD and AMSCs differentiation. (k) UMAP-based dimensionality reduction of LipocyteProfiler features in siCOBLL1 and siNT AMSCs. (l) Actin and COBLL1 staining in siCOBLL1 and siNT visceral adipocytes at day 14 using phalloidin and COBLL1 antibody staining (HPA053344, Alexa-Fluor 488), magnification x63/oil. Representative result from N = 2 independent experiments, scale bar = 52,8um (m) Representative Oil-Red-O lipid staining in SGBS adipocytes following lentiviral COBLL1 knock-down (shCOBLL1, knock-down efficiency 69%) and GRB14 (shGRB14, knock-down efficiency 61%) compared to empty vector control (shEV), scale bar = 15 mm. (n) GPDH metabolic activity in shCOBLL1, shGRB14 and shEV SGBS adipocytes, one-way ANOVA with Tukey’s HSD test, mean + 95% CI, N = 4 biologically independent experiments (o) Basal and insulin-stimulated 3H-2-deoxyglucose uptake in shCOBLL1, shGRB14 and shEV SGBS adipocytes, one-way ANOVA with Tukey’s HSD test, mean + 95% CI, N = 4 biologically independent experiments, 1st and 3rd quartiles (box) and median (middle line) are indicated, p = 4.3 × 10-8. (p) qPCR-based GLUT4 gene expression in shCOBLL1, shGRB14 and shEV adipocytes, one-way ANOVA with Tukey’s HSD test, mean + 95% CI, N = 4 biologically independent experiments.
Extended data Fig. 6 ∣. Lipocyte profiles of risk versus non-risk haplotype carriers.
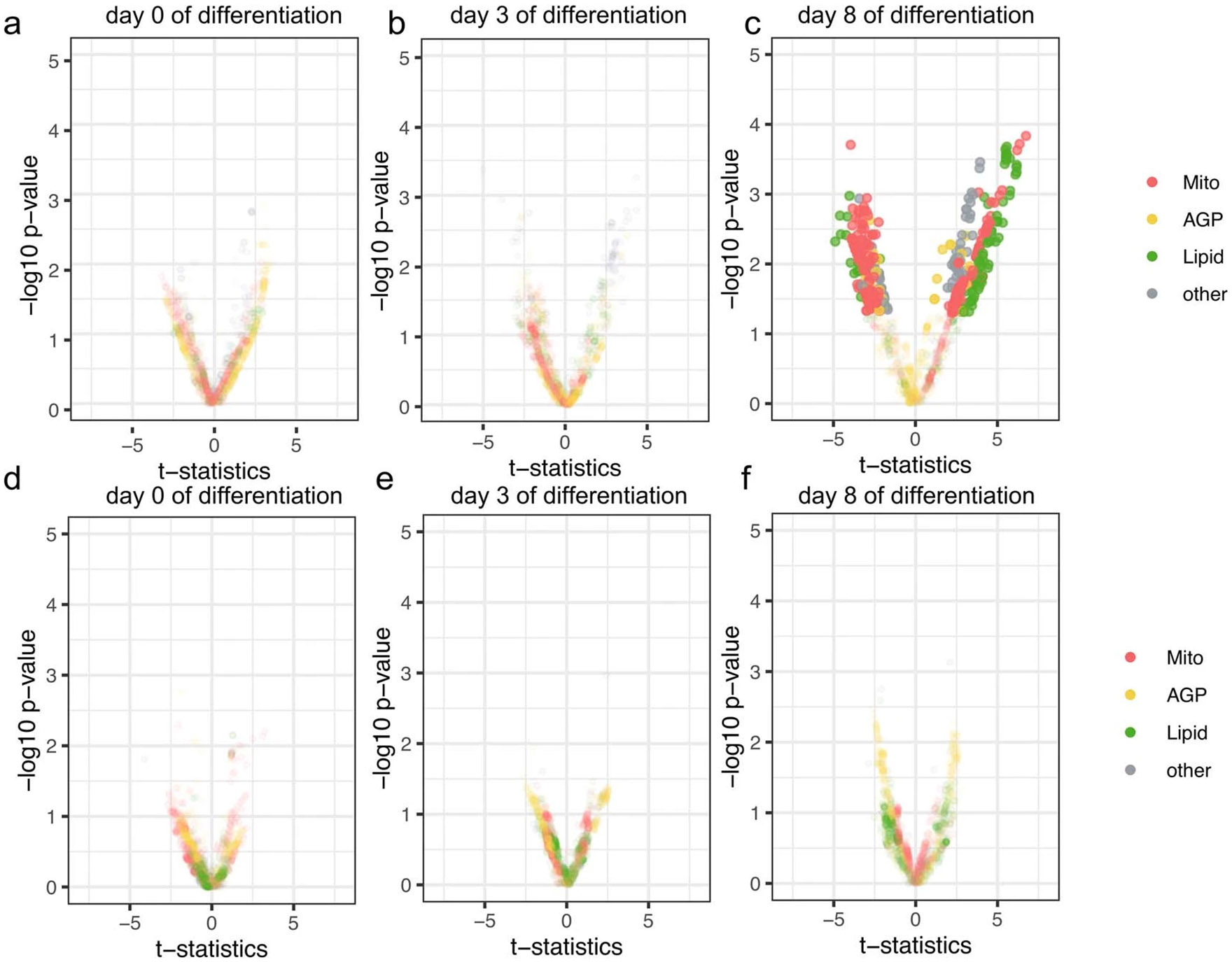
(a–c) Differences in morphological profiles between TT (n = 7) and CC (n = 6) allele carriers at day 0 (a), day 3 (b) and day 8 (c) in subcutaneous AMSCs (multi-way ANOVA, significance level < 5% FDR). (d–f) Differences in morphological profiles between TT (n = 7) and CC (n = 6) allele carriers at (d) day 0, (e) day 3 and (f) day 8 in visceral AMSCs (multi-way ANOVA, significance level < 5% FDR).
Extended data Fig. 7 ∣. Generation of COBLL1 mutant mice using CRISPR/Cas9 editing.
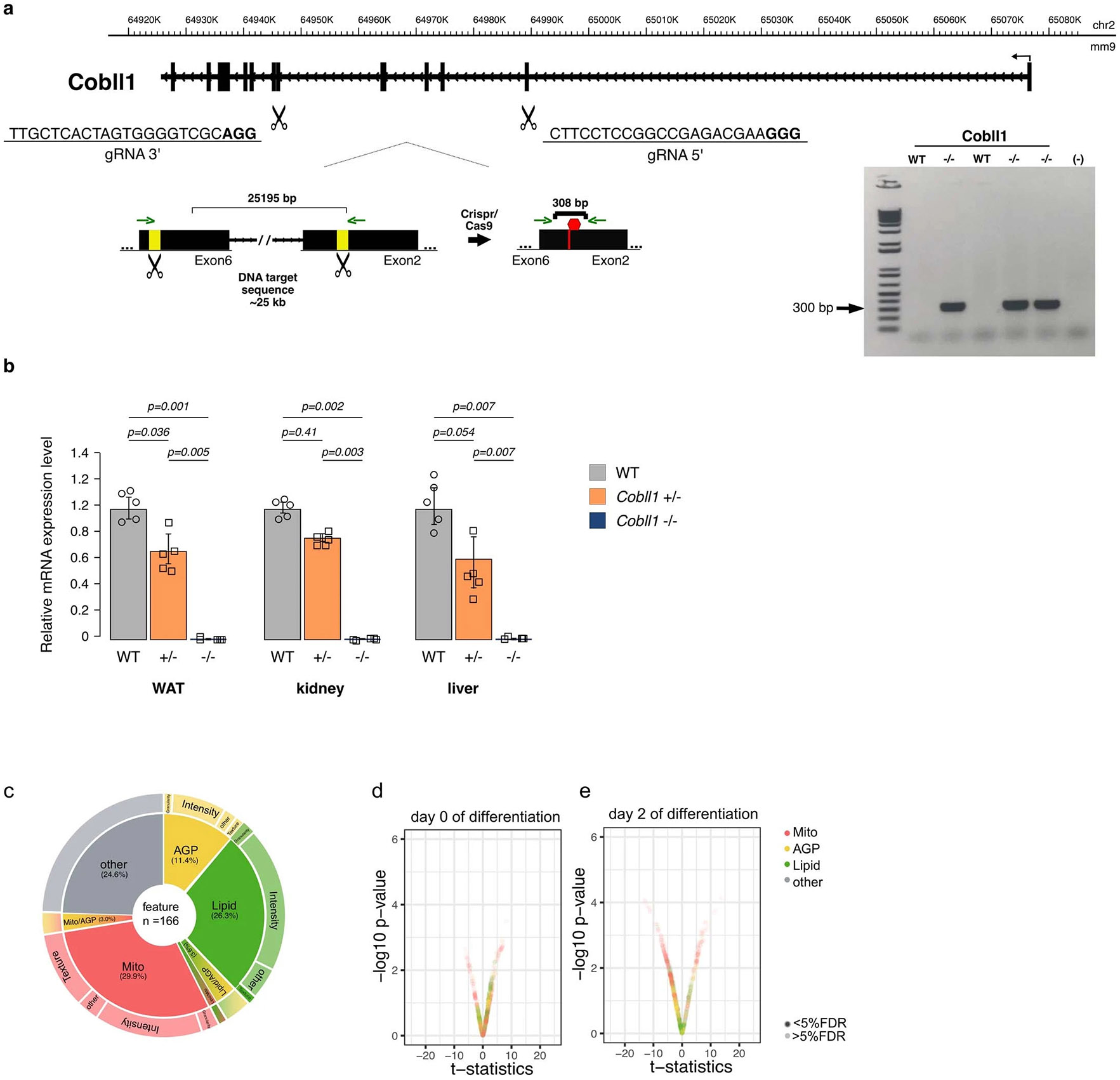
(a) Overview of the CRISPR/− Cas9 strategy to delete ~20 kb of the Cobll1 gene. The gRNA-targeting sequences (gRNAs) are underlined, and the PAM sequences are indicated in bold. Exons are represented as thick black boxes introns are indicated as black lines with arrows, and the yellow boxes indicate the DNA-targeting region. Red hexagon indicates a stop codon gen- erating a Cobll1 truncated protein. Agarose gel showing the PCR products generated from DNA containing success- fully targeted Cobll1 from F0 mouse tail genomic DNA. The 308 bp band corresponds to the genomic deletion. (b) A real-time quantitative PCR of levels of Cobll1 mRNA in white adipose tissue (WAT), liver and kidney of Cobll1 WT, Cobll1 heterozygous (+/−) and null knockout Cobll1 (−/−) animals to confirm the Cobll1 ablation in knockout animals. Each group was analyzed using 5 different mice and the values were expressed as the mean ± s.e.m and P values by Student’s t-test the experiment was repeated independently two times with similar results. (c) Pie chart illustrating non-redundant differential features per channel and class of measurement at day 8 of subcutaneous adipocyte differentiation in rs6712003 homozygous risk compared to non-risk carriers. (d, e) Differences in morphological profiles between AMSCs from Cobll1−/− mice (n = 3) and WT (n = 4) at (h) day 0 (i) day 2 (t-test two-sided, significance level < 5%FDR).
Supplementary Material
Acknowledgements
Work in the Claussnitzer lab is supported by the Foundation for the National Institutes of Health (NIH) (AMP-T2D RFB8b), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (UM1 DK126185 and NIDDK DK102173), the Novo Nordisk Foundation (NNF21SA0072102) and by a Next Generation Award from the Broad Institute of MIT and Harvard. We acknowledge support from the Center for Advanced Light Microscopy of the Technical University of Munich School of Life Sciences, T. Kufer at the University of Hohenheim for access to microscopes, the Clinical Cooperation Group ‘Nutrigenomics and Type 2 Diabetes’ from the Helmholtz Center Munich and the German Center for Diabetes Research, the Else Kröner-Fresenius-Foundation, a Stanford Graduate Fellowship, a Center for Computational, Evolutionary and Human Genomics Graduate Fellowship, the Novo Nordisk Foundation Challenge Grant (no. NNF18OC0033754), NIH grant nos. R01HL128075 and P30DK020595, the Federal Ministry of Education and Research (award no. 0315674), the German Research Foundation (award no. 338582098) and the Federal Ministry of Education and Research (grant no. 0315674). We thank A. Flaccus, A. Kozza, D. Mvondo, M. Hubersberger, E. Hofmair, J. Rüttgen, C. Deuschle, A. Saadat and S. Harken for technical support.
Footnotes
Competing interests
M.C. holds equity in Waypoint Bio, consults for Pfizer, and is a member of the Nestle scientific advisory board. The authors have filed a provisional patent application (63/218,656).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended data is available for this paper at https://doi.org/10.1038/s42255-023-00807-w.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s42255-023-00807-w.
Data availability
The ATAC–seq data for the immortalized human adipocytes are deposited in the database of Genotypes and Phenotypes under accession no. PRJNA539992. The high-content imaging data are available at the Cell Painting Gallery on the Registry of Open Data on Amazon Web Services (https://registry.opendata.aws/cellpainting-gallery/) under accession no. cpg0011. Source data are provided with this paper.
Code availability
The code used is publicly available on GitHub (https://github.com/ClaussnitzerLab/). The GitHub LP-2q24.3-metabolic-risk-locus repository containing the code and files used to generate LipocyteProfiles can be accessed at https://github.com/sophiestrobel/LP-2q24.3-metabolic-risk-locus.git.
References
- 1.Cho NH et al. IDF Diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract 138, 271–281 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Lu Y. et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat. Commun 7, 10495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hosseinpanah F, Barzin M, Sheikholeslami F & Azizi F Effect of different obesity phenotypes on cardiovascular events in Tehran Lipid and Glucose Study (TLGS). Am. J. Cardiol 107, 412–416 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Caleyachetty R. et al. Metabolically healthy obese and incident cardiovascular disease events among 3.5 million men and women. J. Am. Coll. Cardiol 70, 1429–1437 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Arnlöv J, Ingelsson E, Sundström J & Lind L Impact of body mass index and the metabolic syndrome on the risk of cardiovascular disease and death in middle-aged men. Circulation 121, 230–236 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Aung K, Lorenzo C, Hinojosa MA & Haffner SM Risk of developing diabetes and cardiovascular disease in metabolically unhealthy normal-weight and metabolically healthy obese individuals. J. Clin. Endocrinol. Metab 99, 462–468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calori G. et al. Prevalence, metabolic features, and prognosis of metabolically healthy obese Italian individuals: the Cremona Study. Diabetes Care 34, 210–215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wildman RP et al. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch. Intern. Med 168, 1617–1624 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Primeau V. et al. Characterizing the profile of obese patients who are metabolically healthy. Int. J. Obes 35, 971–981 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Yaghootkar H. et al. Genetic evidence for a normal-weight ‘metabolically obese’ phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 63, 4369–4377 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vujkovic M. et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet 52, 680–691 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spracklen CN et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 582, 240–245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shungin D. et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fox CS et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 8, e1002695 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris AP et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet 44, 981–990 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu AY et al. Multiethnic genome-wide meta-analysis of ectopic fat depots identifies loci associated with adipocyte development and differentiation. Nat. Genet 49, 125–130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H. et al. Genotype-by-environment interactions inferred from genetic effects on phenotypic variability in the UK Biobank. Sci. Adv 5, eaaw3538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao C. et al. Genome-wide study of subcutaneous and visceral adipose tissue reveals novel sex-specific adiposity loci in Mexican Americans. Obesity 26, 202–212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lagou V. et al. Sex-dimorphic genetic effects and novel loci for fasting glucose and insulin variability. Nat. Commun 12, 24 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J. et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet 53, 840–860 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loos RJF & Kilpeläinen TO Genes that make you fat, but keep you healthy. J. Intern. Med 284, 450–463 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kilpeläinen TO et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat. Genet 43, 753–760 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lotta LA et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet 49, 17–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kooner JS et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat. Genet 43, 984–989 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet 46, 234–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heid IM et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat. Genet 42, 949–960 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Udler MS et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med. 15, e1002654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hindorff LA et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA 106, 9362–9367 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Claussnitzer M. et al. Leveraging cross-species transcription factor binding site patterns: from diabetes risk loci to disease mechanisms. Cell 156, 343–358 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Claussnitzer M. et al. FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med 373, 895–907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelley DR, Snoek J & Rinn JL Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 26, 990–999 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixon JR et al. Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xue R. et al. Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat. Med 21, 760–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilbert LA et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cowper-Sal lari R. et al. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet 44, 1191–1198 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benita Y et al. Gene enrichment profiles reveal T-cell development, differentiation, and lineage-specific transcription factors including ZBTB25 as a novel NF-AT repressor. Blood 115, 5376–5384 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawaguchi N. et al. ADAM12 induces actin cytoskeleton and extracellular matrix reorganization during early adipocyte differentiation by regulating β1 integrin function. J. Cell Sci 116, 3893–3904 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Qian Y et al. PI3K induced actin filament remodeling through Akt and p70S6K1: implication of essential role in cell migration. Am. J. Physiol. Cell Physiol 286, C153–C163 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Morandi EM et al. ITGAV and ITGA5 diversely regulate proliferation and adipogenic differentiation of human adipose derived stem cells. Sci. Rep 6, 28889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Truebestein L, Elsner DJ, Fuchs E & Leonard TA A molecular ruler regulates cytoskeletal remodelling by the Rho kinases. Nat. Commun 6, 10029 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Izadi M. et al. Cobl-like promotes actin filament formation and dendritic branching using only a single WH2 domain. J. Cell Biol 217, 211–230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takayama K-I, Suzuki T, Fujimura T, Takahashi S & Inoue S COBLL1 modulates cell morphology and facilitates androgen receptor genomic binding in advanced prostate cancer. Proc. Natl Acad. Sci. USA 115, 4975–4980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laber S. et al. Discovering cellular programs of intrinsic and extrinsic drivers of metabolic traits using LipocyteProfiler. Preprint at bioRxiv 10.1101/2021.07.17.452050 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris RBS Direct and indirect effects of leptin on adipocyte metabolism. Biochim. Biophys. Acta 1842, 414–423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kilpeläinen TO et al. Genome-wide meta-analysis uncovers novel loci influencing circulating leptin levels. Nat. Commun 7, 10494 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Folkersen L. et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat. Metab 2, 1135–1148 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Small KS et al. Regulatory variants at KLF14 influence type 2 diabetes risk via a female-specific effect on adipocyte size and body composition. Nat. Genet 50, 572–580 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fathzadeh M. et al. FAM13A affects body fat distribution and adipocyte function. Nat. Commun 11, 1465 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Randall JC et al. Sex-stratified genome-wide association studies including 270,000 individuals show sexual dimorphism in genetic loci for anthropometric traits. PLoS Genet. 9, e1003500 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sung YJ et al. Genome-wide association studies suggest sex-specific loci associated with abdominal and visceral fat. Int. J. Obes 40, 662–674 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palmer BF & Clegg DJ The sexual dimorphism of obesity. Mol. Cell. Endocrinol 402, 113–119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z. et al. Functional screening of candidate causal genes for insulin resistance in human preadipocytes and adipocytes. Circ. Res 126, 330–346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L, Hu H, Qiu W, Shi K & Kassem M Actin depolymerization enhances adipogenic differentiation in human stromal stem cells. Stem Cell Res. 29, 76–83 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Kanzaki M & Pessin JE Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J. Biol. Chem 276, 42436–42444 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Kim JI et al. During adipocyte remodeling, lipid droplet configurations regulate insulin sensitivity through F-actin and G-actin reorganization. Mol. Cell. Biol 39, e00210–e00219 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hansson B. et al. Adipose cell size changes are associated with a drastic actin remodeling. Sci. Rep 9, 12941 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Q. et al. Obesity and hyperinsulinemia drive adipocytes to activate a cell cycle program and senesce. Nat. Med 27, 1941–1953 (2021). [DOI] [PubMed] [Google Scholar]
- 58.Bäckdahl J. et al. Spatial mapping reveals human adipocyte subpopulations with distinct sensitivities to insulin. Cell Metab. 33, 1869–1882 (2021). [DOI] [PubMed] [Google Scholar]
- 59.Emont MP et al. A single-cell atlas of human and mouse white adipose tissue. Nature 603, 926–933 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cariou B. et al. Increased adipose tissue expression of Grb14 in several models of insulin resistance. FASEB J. 18, 965–967 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Béréziat V. et al. Inhibition of insulin receptor catalytic activity by the molecular adapter Grb14. J. Biol. Chem 277, 4845–4852 (2002). [DOI] [PubMed] [Google Scholar]
- 62.Depetris RS et al. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol. Cell 20, 325–333 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cooney GJ et al. Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J. 23, 582–593 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang L. et al. Peripheral disruption of the Grb10 gene enhances insulin signaling and sensitivity in vivo. Mol. Cell. Biol 27, 6497–6505 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holt LJ et al. Dual ablation of Grb10 and Grb14 in mice reveals their combined role in regulation of insulin signaling and glucose homeostasis. Mol. Endocrinol 23, 1406–1414 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi J. et al. Discovery of cancer drug targets by CRISPR–Cas9 screening of protein domains. Nat. Biotechnol 33, 661–667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.González F. et al. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell 15, 215–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fuchsberger C. et al. The genetic architecture of type 2 diabetes. Nature 536, 41–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Albrechtsen A. et al. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia 56, 298–310 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wessel J. et al. Low-frequency and rare exome chip variants associate with fasting glucose and type 2 diabetes susceptibility. Nat. Commun 6, 5897 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kan M. et al. Rare variant associations with waist-to-hip ratio in European-American and African-American women from the NHLBI-Exome Sequencing Project. Eur. J. Hum. Genet 24, 1181–1187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Claussnitzer MC et al. Leveraging transcription factor binding site patterns: from diabetes risk loci to disease mechanisms. Cell 156, 343–358 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hauner H, Skurk T & Wabitsch M Cultures of human adipose precursor cells. Methods Mol. Biol 155, 239–247 (2001). [DOI] [PubMed] [Google Scholar]
- 74.Ott B. et al. Effect of caloric restriction on gut permeability, inflammation markers, and fecal microbiota in obese women. Sci. Rep 7, 11955 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Glastonbury CA et al. Machine learning based histology phenotyping to investigate the epidemiologic and genetic basis of adipocyte morphology and cardiometabolic traits. PLoS Comput. Biol 16, e1008044 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sobreira DR et al. Extensive pleiotropism and allelic heterogeneity mediate metabolic effects of IRX3 and IRX5. Science 372, 1085–1091 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sinnott-Armstrong N. et al. A regulatory variant at 3q21.1 confers an increased pleiotropic risk for hyperglycemia and altered bone mineral density. Cell Metab. 33, 615–628 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eastwood SV et al. Algorithms for the capture and adjudication of prevalent and incident diabetes in UK Biobank. PLoS ONE 11, e0162388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sinnott-Armstrong N. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet 53, 185–194 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanson KR et al. Optimized libraries for CRISPR–Cas9 genetic screens with multiple modalities. Nat. Commun 9, 5416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pairault J & Green H A study of the adipose conversion of suspended 3T3 cells by using glycerophosphate dehydrogenase as differentiation marker. Proc. Natl Acad. Sci. USA 76, 5138–5142 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Claussnitzer M, Skurk T, Hauner H, Daniel H & Rist MJ Effect of flavonoids on basal and insulin-stimulated 2-deoxyglucose uptake in adipocytes. Mol. Nutr. Food Res 55, S26–S34 (2011). [DOI] [PubMed] [Google Scholar]
- 84.Untergasser A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ye J. et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pfaffl MW A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 29, e45 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Elliott L. et al. The genetic basis of human brain structure and function: 1,262 genome-wide associations found from 3,144 GWAS of multimodal brain imaging phenotypes from 9,707 UK Biobank participants. Preprint at bioRxiv 10.1101/178806 (2017). [DOI] [Google Scholar]
- 88.Kundaje A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ATAC–seq data for the immortalized human adipocytes are deposited in the database of Genotypes and Phenotypes under accession no. PRJNA539992. The high-content imaging data are available at the Cell Painting Gallery on the Registry of Open Data on Amazon Web Services (https://registry.opendata.aws/cellpainting-gallery/) under accession no. cpg0011. Source data are provided with this paper.
The code used is publicly available on GitHub (https://github.com/ClaussnitzerLab/). The GitHub LP-2q24.3-metabolic-risk-locus repository containing the code and files used to generate LipocyteProfiles can be accessed at https://github.com/sophiestrobel/LP-2q24.3-metabolic-risk-locus.git.