

Abstract
Aging markedly increases cancer risk, yet our mechanistic understanding of how aging influences cancer initiation is limited. Here we demonstrate that the loss of ZNRF3, an inhibitor of Wnt signaling that is frequently mutated in adrenocortical carcinoma, leads to the induction of cellular senescence that remodels the tissue microenvironment and ultimately permits metastatic adrenal cancer in old animals. The effects are sexually dimorphic, with males exhibiting earlier senescence activation and a greater innate immune response, driven in part by androgens, resulting in high myeloid cell accumulation and lower incidence of malignancy. Conversely, females present a dampened immune response and increased susceptibility to metastatic cancer. Senescence-recruited myeloid cells become depleted as tumors progress, which is recapitulated in patients in whom a low myeloid signature is associated with worse outcomes. Our study uncovers a role for myeloid cells in restraining adrenal cancer with substantial prognostic value and provides a model for interrogating pleiotropic effects of cellular senescence in cancer.
Graphical Abstract
Aging is the most important risk factor for cancer1. Incidence rates rise steeply with age, with ~90% of newly diagnosed cancer occurring after the age of 50 years2,3. Age-driven changes in the tissue landscape cooperate with oncogenic mutations to determine overall cancer risk4. During aging, the tissue microenvironment evolves based on a multitude of physiological factors, including oxygen and nutrient availability, extracellular matrix (ECM) composition and immune cell infiltration5. These tissue-level changes influence the functional impact of oncogenic mutations3. However, despite aging being a major carcinogen, we have limited understanding of how the aged microenvironment cooperates with genetic mutations to influence tumorigenesis.
Adrenocortical carcinoma (ACC) is a highly pertinent model to study the interplay between the aging microenvironment and cell-intrinsic driver mutations. ACC is an aggressive cancer of the adrenal cortex with no effective treatments6. Moreover, peak incidence occurs at the age of 45–55 years7, which corresponds to ~12–18 months in mice8. One pathway frequently hyperactivated in ACC is Wnt–β-catenin, a highly conserved signaling cascade that mediates proper cell renewal and cell-to-cell interactions in many tissues9. Unlike most Wnt-driven tumors that are induced by activating mutations in CTNNB1 (β-catenin) or inactivation of the β-catenin destruction complex, ACC tumors frequently harbor loss-of-function (LOF) alterations in ZNRF3 (refs. 10,11), encoding an E3 ubiquitin ligase that promotes endocytic turnover of Wnt receptors12,13. Consequently, ZNRF3 loss increases receptor availability and renders cells hypersensitive to Wnt ligands within the microenvironment.
Wnt-driven ACC has been previously modeled in mice through CTNNB1 gain of function14 or APC LOF15. These approaches result in mild hyperplasia with late progression to benign adenomas or, in some rare cases, carcinomas. Combined CTNNB1 gain of function and p53 LOF shortens tumor latency but still requires 12 weeks or longer for tumors16. These studies hint at an important role for aging in the initiation and progression of adrenal tumors. However, these models are based on less frequent Wnt alterations in ACC, and the mechanism by which aging influences tumorigenesis remains unresolved.
We recently developed a model to specifically ablate Znrf3 in the adrenal cortex using steroidogenic factor 1 (SF-1)–Cre, which mediates recombination throughout the adrenal cortex beginning at embryonic day (E)9.5 (ref. 17). We initially characterized Znrf3-conditional knockout (cKO) mice between postnatal day (P)0 and adrenal maturation at 6 weeks18. We observed progressive hyperplasia that resulted in a more than eightfold increase in adrenal weight. Further characterization revealed a gradient of Wnt–β-catenin activity required for normal homeostasis with a specific role for ZNRF3 in limiting expansion of Wnt-low cells. Nevertheless, we found no evidence of neoplastic transformation during these early studies.
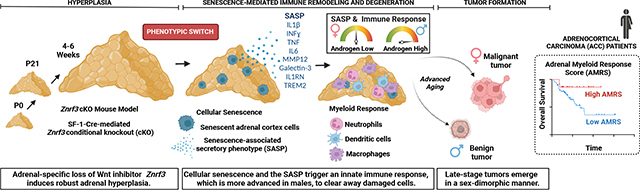
Here, we investigated the interplay between genetic Znrf3 inactivation and the aging microenvironment. We hypothesized that Znrf3-cKO mice would progress from adrenal hyperplasia to carcinoma with age. Unexpectedly, we found that Znrf3-cKO adrenals steadily regress over time. We demonstrate that this phenotypic switch from hyperplasia to regression is driven by activation of cellular senescence and a subsequent senescence-associated secretory phenotype (SASP). We characterize diverse immune infiltrates that remodel the tissue microenvironment following senescence activation, which exhibit a highly sex-dimorphic response that is enhanced in males by androgens. However, following prolonged inflammation, a large proportion of mice ultimately develop adrenal tumors. Males predominately form benign lesions, while females are significantly more prone to metastatic adrenal tumors, mirroring the higher incidence of ACC in women7. Myeloid-derived immune cells are excluded from the tissue microenvironment as tumors progress, which is recapitulated in human patients with ACC in whom a low myeloid response is associated with worse outcome. In sum, our results suggest that Znrf3 loss is permissive for ACC tumor progression with advanced age (Graphical Abstract, Supplementary Fig. 1). More broadly, our work suggests a mechanism by which extrinsic factors, including sex and the immune microenvironment, cooperate with genetic lesions to promote age-induced malignant transformation.
Results
Ultrasound imaging measures adrenal size in real time
Given the significant adrenal hyperplasia that we previously observed at 6 weeks, we hypothesized that Znrf3-cKO animals would progress to form adrenal tumors with increased age. We employed ultrasound imaging as a non-invasive technique to quantify adrenal size in real time. We first validated this approach using a mixed cohort of mice with varying adrenal sizes. We measured adrenal area and volume by ultrasound and then euthanized mice 24 h after imaging to obtain adrenal weight (Extended Data Fig. 1a). We found that adrenal weight was significantly correlated with both ultrasound area (R2 = 0.90, **P = 1.08 × 10−3) (Extended Data Fig. 1b) and volume (R2 = 0.89, **P = 1.35 × 10−3) (Extended Data Fig. 1c). These results demonstrate that ultrasound imaging provides an accurate assessment of adrenal size in real time.
Following hyperplasia, ZNRF3-deficient adrenals regress
To monitor for potential tumors, we performed weekly adrenal ultrasound imaging in Znrf3-cKO animals. We began imaging females at 4 weeks and observed an expected increase in adrenal size by 6 weeks (Fig. 1a,b). However, rather than continued growth, we unexpectedly observed a gradual decline in adrenal volume after 6 weeks (Fig. 1a,b).
Fig. 1 |. Following an initial phase of significant hyperplasia, ZNRF3-deficient adrenal glands regress over time.

a, Ultrasound imaging provides a non-invasive method for measuring adrenal size in real time. Representative three-dimensional ultrasound images from a female control and a Znrf3-cKO mouse are shown at 4 weeks (initiation of study), 6 weeks (maximum volume) and 12 weeks (end of study) of age. b, Weekly ultrasound monitoring in female control and Znrf3-cKO mice identifies a phenotypic switch from hyperplasia to regression at 6 weeks of age. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. Error bars represent mean ± s.e.m. c, Adrenal weight measurements over time confirm significant adrenal regression in female Znrf3-cKO mice beginning after 6 weeks of age. d, In males, adrenal weight peaks earlier than females at 4 weeks and progressively declines with increased age. c,d, Each dot represents an individual animal. Box-and-whisker plots indicate the median (line) within the upper (75%) and lower (25%) quartiles, and whiskers represent the range. e, Proliferation as measured by Ki67 is significantly increased in adrenals of 4- and 6-week-old female Znrf3-cKO mice compared to controls. At the onset of adrenal regression (9 weeks), proliferation is significantly reduced in ZNRF3-deficient adrenals. f, Male Znrf3-cKO mice similarly exhibit hyperproliferation at 4 weeks of age. However, proliferation returns to baseline by 6 weeks, which is earlier than in females. e,f, Each dot represents an individual animal. Error bars represent mean ± s.e.m. Statistical analysis was performed using two-way ANOVA followed by Tukey’s multiple-comparison test. Scale bars, 100 μm.
To follow up on these observations, we collected cohorts of male and female mice at 4, 6, 9, 12 and 24 weeks of age. Consistent with ultrasound measurements, we initially observed increased adrenal weight in female Znrf3-cKO animals between 4 and 6 weeks of age, followed by a progressive decline in adrenal weight (Fig. 1c). In males, adrenal weight did not continue to increase after 4 weeks and, similar to females, gradually declined with increasing age (Fig. 1d). In sum, these results indicate that ZNRF3-deficient adrenals undergo a distinct phenotypic switch from hyperplasia to regression.
Adrenal regression begins with reduced proliferation
We hypothesized that declining adrenal size in Znrf3-cKO animals was caused by enhanced cell death. To test this, we measured apoptosis, which is the dominant form of cell death attributed to homeostatic turnover of the normal adrenal cortex19. Using immunohistochemistry (IHC) for cleaved caspase 3 (CC3), we found that CC3-positive cells primarily localized at the cortical–medullary boundary in controls, as expected (Extended Data Fig. 2a,b). In Znrf3-cKO animals, apoptotic cells were scattered throughout the cortex, but there was no increase in the proportion of CC3-positive cells compared to controls (Extended Data Fig. 2a,b). We concluded that the age-dependent decrease in adrenal size in Znrf3-cKO animals was not caused by increased apoptotic cell death.
Next, we assessed proliferation in Znrf3-cKO adrenals. Previously, we observed hyperplasia at 6 weeks in females18. Consistent with these prior observations, the Ki67 index in female Znrf3-cKO animals was significantly higher than that in controls at 4 and 6 weeks (Fig. 1e). However, proliferation was reduced to baseline control levels by 9 weeks. This striking drop in proliferation was coincident with the onset of adrenal regression that we observed by adrenal ultrasound and weight. Male Znrf3-cKO animals also displayed an initial phase of hyperplasia evident by the high rate of proliferation at 4 weeks. However, the Ki67 index returned to baseline by 6 weeks (Fig. 1f). These results indicate that adrenal cortex hyperplasia in Znrf3-cKO animals is not sustained. Furthermore, the onset of regression occurs earlier in males, suggesting an underlying sexual dimorphism.
Cell cycle arrest triggers adrenal regression
To identify potential mechanisms that mediate the switch from hyperplasia to regression, we performed bulk RNA-seq on control and Znrf3-cKO adrenals. We focused our analysis on females at 6 weeks (hyperplasia), 9 weeks (regression onset) and 12 weeks (regression). Control adrenals had a highly stable expression signature, with only 23 differentially expressed genes (DEGs) between 6 and 9 weeks (17 upregulated, 6 downregulated, adjusted P value (Padj) < 0.05) (Supplementary Table 1). By contrast, we observed a striking change in gene expression in Znrf3-cKO animals, with 720 significant DEGs (383 upregulated, 337 downregulated, Padj < 0.05) (Fig. 2a).
Fig. 2 |. The switch from hyperplasia to regression is marked by activation of cellular senescence.

a, Bulk RNA-seq reveals a significant change in gene expression signature during the switch from hyperplasia to regression. Heatmap of significant DEGs in adrenals from female Znrf3-cKO animals at 6, 9 and 12 weeks of age (Padj < 0.05). b, IPA identifies the most significantly altered canonical pathways in 9-versus 6-week-old female Znrf3-cKO adrenals; statistical analysis was performed using a right-tailed Fisher’s exact test (P < 0.05). AHR, aryl hydrocarbon receptor; CHK, Csk homologous kinase; GP6, Glycoprotein VI. c, DNA damage as measured by 53BP1 foci is significantly increased in female Znrf3-cKO adrenals compared to those of controls at 4 and 6 weeks of age. Quantification of 53BP1 foci was performed using QuPath digital image analysis based on the number of positive foci and normalized to total nuclei. Asterisks indicate 53BP1-positive foci. Scale bars, 10 μm. d, IPA of the most significantly activated or inhibited transcriptional regulators in adrenals of 9-versus 6-week-old female Znrf3-cKO animals. Red asterisks highlight transcriptional regulators associated with cellular senescence. p21 (e), p16INK4a (f) and SA-β-gal (g) are significantly increased in adrenals of 9-week-old female Znrf3-cKO animals compared to those of controls. Scale bars, 100 μm. Quantification of p21 and p16INK4a IHC was performed using QuPath digital image analysis based on the number of positive cells per high-powered field (HPF). Representative SA-β-gal images obtained from analysis of three independent mice are shown. h, Co-staining of p21 and GFP in 9-week-old female Znrf3-cKO mice containing the R26RmTmG lineage reporter tool confirms that p21-positive senescent cells are GFP-positive adrenal cortex cells. Representative images obtained from analysis of five female and seven male animals are shown. Scale bar, 10 μm. WT, wild type. b,d, IPA analysis includes four biological replicates per group. Statistical tests were performed using IPA (P < 0.05). c,e,f, Error bars represent mean ± s.e.m. Each dot represents an individual animal. Statistical analysis was performed using two-way ANOVA followed by Tukey’s multiple-comparison test.
Using Ingenuity Pathway Analysis (IPA)20, we analyzed DEGs to identify canonical pathways associated with the switch from hyperplasia to regression. Among the top inhibited pathways, we identified multiple pathways that regulate cell division (Fig. 2b). Conversely, among the top activated pathways, we detected several signatures associated with enhanced DNA damage and cell cycle arrest (Fig. 2b). We validated these results in situ by measuring 53BP1, which is a DNA damage-response factor recruited to DNA double-strand breaks21. Consistent with our RNA-seq data, we observed a significant increase in 53BP1 foci in Znrf3-cKO animals compared to controls (Fig. 2c and Extended Data Fig. 2c). In sum, these data suggest that ZNRF3-deficient adrenal cortex cells accumulate DNA damage and initiate cell cycle arrest near the onset of regression.
Adrenal regression is associated with senescence activation
We next performed upstream regulator analysis (URA) to identify candidate factors that could coordinate the observed change in gene expression. URA is a new IPA tool that analyzes the relationship between DEGs to identify potential upstream factors20. We focused our analysis on transcriptional regulators and identified 47 candidates (23 activated, 24 inhibited; P < 0.05). Among the most significantly activated regulators, we found TP53 (Z score = 6.20), CDKN2A (Z score = 5.68) and RB1 (Z score = 4.86) (Fig. 2d), which are central regulators of cellular senescence, one of the top activated canonical pathways (Fig. 2b).
Cellular senescence is an aging-associated stress response that causes proliferative arrest of damaged cells22. Enhanced cellular stress, which can be induced by a wide range of stimuli, activates the p53 and p16INK4a tumor-suppressor proteins, leading to cell cycle inhibition through p21 and Rb. Based on our RNA-seq analysis, in combination with observed phenotypic changes, we hypothesized that cellular senescence was activated in Znrf3-cKO animals during the transition from hyperplasia to regression. To test this hypothesis, we measured multiple hallmarks of senescence at 6 and 9 weeks. We observed increased expression of p21 (Fig. 2e and Extended Data Fig. 2d) and p16INK4a (Fig. 2f and Extended Data Fig. 2e) as well as accumulation of senescence-associated β-galactosidase (SA-β-gal) (Fig. 2g and Extended Data Fig. 2f) in adrenals from 9-week-old Znrf3-cKO animals compared to those from controls. To confirm senescence activation in adrenal cortex cells, as opposed to the stromal or immune compartment, we performed co-staining for p21 and green fluorescent protein (GFP) in adrenals isolated from mice containing the R26RmTmG lineage reporter23. In these animals, the adrenal cortex is permanently labeled with GFP through SF-1–Cre-mediated recombination. We found that all p21-positive cells were positive for GFP at 9 weeks (Fig. 2h). Collectively, these data demonstrate that cellular senescence is activated in ZNRF3-deficient adrenal cortex cells during the switch from hyperplasia to regression.
Senescent adrenal cortex cells develop a secretory phenotype
One defining feature of senescence that distinguishes it from other forms of growth arrest is the SASP24. The SASP comprises multiple families of secreted proteins including soluble signaling factors, secreted proteases and secreted insoluble proteins or ECM components. Although this phenotype is both temporally dynamic and heterogeneous across tissue types or modes of senescence induction25,26, the SASP collectively acts as a potent pro-inflammatory stimulus to facilitate tissue remodeling.
To begin to define the SASP in the context of Znrf3 loss in the adrenal gland, we performed URA for activated cytokines and growth factors in our RNA-seq dataset. During senescence activation at 9 weeks, we identified interleukin (IL)-1β (Z score = 2.96), interferon (IFN)-γ (Z score = 2.48) and tumor necrosis factor (TNF) (ligand) superfamily, member 13 (TNFSF13) (Z score = 2.00) as the most highly induced cytokines (Fig. 3a and Extended Data Fig. 3a). By 12 weeks, additional members of the IL-1 (IL-1α, Z score = 2.38), IL-6 (IL-6, Z score = 2.15 and oncostatin M (OSM), Z score = 3.15) and TNF family (TNF, Z score = 3.12 and TNFSF12, Z score = 2.37) were significantly activated (Fig. 3b and Extended Data Fig. 3b). Gene set enrichment analysis (GSEA) at both 9 weeks (Fig. 3c) and 12 weeks (Fig. 3d) further confirmed significant positive enrichment for genes associated with an inflammatory response. Growth factor analysis additionally revealed significant induction of SASP-associated factors, including transforming growth factor (TGF)-β3 (ref. 27) and brain-derived neurotrophic factor (BDNF)28 (Extended Data Fig. 3c,d).
Fig. 3 |. Senescent Znrf3-cKO adrenal glands develop a functional SASP.
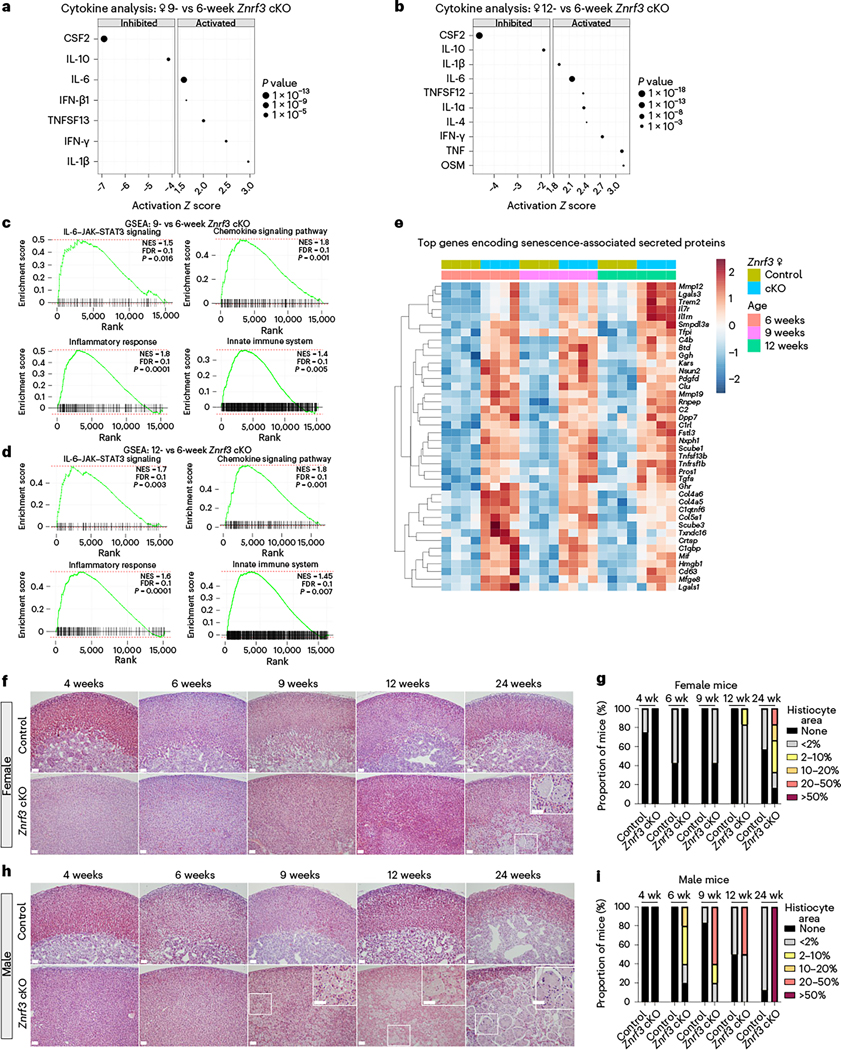
URA using bulk RNA-seq data predicts significantly activated and inhibited cytokines in adrenal tissue from Znrf3-cKO animals at 9 weeks (a) and 12 weeks (b) compared to 6 weeks. Data are representative of four biological replicates per group. Statistical analysis in IPA was performed using a right-tailed Fisher’s exact test (P < 0.05). c,d, GSEA for inflammatory signatures (IL-6–Janus kinase ( JAK)–signal transducer and activator of transcription 3 (STAT3) signaling, chemokine signaling, the inflammatory response and the innate immune system) identifies positively enriched pathways in adrenal tissue from Znrf3-cKO animals at 9 weeks (c) and 12 weeks (d) compared to 6 weeks. FDR, false discovery rate; NES, normalized enrichment score. e, Heatmap representing supervised hierarchical clustering of the 40 most significant DEGs that encode secreted proteins compared to controls; statistical analysis was performed using the Wald test (Padj < 0.05). f,g, Histological evaluation of adrenal tissue from female control and Znrf3-cKO animals based on H&E staining. Female Znrf3-cKO animals accumulate histiocytes (inset) between 12 and 24 weeks (wk) of age. h,i, Histiocytes (insets) accumulate earlier and occupy a larger proportion of the adrenal gland in male Znrf3-cKO animals than in female animals. Quantification was performed using QuPath digital analysis based on the proportion of histiocyte area normalized to the total adrenal cortex area. Scale bars, 100 μm.
Given significant activation of multiple SASP-associated cytokines and growth factors, we next sought to more broadly characterize this phenotype. We analyzed expression of genes encoding secreted proteins29 that were significantly differentially expressed in Znrf3-cKO adrenals compared to controls at 6, 9 and 12 weeks. We then prioritized top candidate SASP factors based on upregulated genes that significantly changed during the phenotypic transition from hyperplasia to senescence activation or regression. This analysis revealed a set of early secreted factors with high induction at 6 weeks and a set of later secreted factors with progressively increasing expression (Fig. 3e). We cross-referenced these early and late genes with the SASP Atlas25 and SeneQuest22 and found that >75% of the factors that we identified were established SASP components. These included the early-response gene Hmgb1, which encodes a damage-associated molecular pattern molecule actively secreted upon cellular stress25 that initiates an IL-6-dependent immune response30,31. Among our top late-response genes, we identified several central SASP components, including Mmp12 (refs. 25,32), Lgals3 (refs. 33,34) and Il1rn32,35, which play key roles in ECM remodeling and immune activation.
The SASP recruits immune cells in a sex-dimorphic manner
The SASP primarily functions to induce local inflammation and attract immune cells to clear out damaged cells36,37. Given significant SASP activity in Znrf3-cKO animals, we hypothesized that immune cells would be recruited to the adrenal gland. We first tested this hypothesis by assessing broad histological changes using hematoxylin and eosin (H&E) staining. We performed H&E at each phenotypic stage from hyperplasia (4 and 6 weeks) to senescence activation (9 weeks) and regression (12 and 24 weeks). Strikingly, we observed accumulation of large multi-nucleated cells in Znrf3-cKO adrenals over time. These highly conspicuous cells first amassed in the innermost gland and became increasingly prevalent between 12 and 24 weeks in females (Fig. 3f,g). Upon pathologic review, we identified these as potential histiocytes, which are monocyte-derived immune cells known for their phagocytic role during tissue repair38,39. Interestingly, we observed an earlier and overall greater accumulation of these cells in males than in females (Fig. 3h,i), consistent with the earlier onset of senescence and regression.
Single-cell RNA-seq reveals a diverse senescence-driven immune response
Histiocytes are complex structures derived from antigen-presenting dendritic cells (DCs) and phagocytic macrophages38,39. Moreover, histiocytes function in part to clear out neutrophils40. As a result, accumulation of histiocytes in Znrf3-cKO animals suggested that SASP-mediated tissue remodeling was likely driven by a multifaceted immune response. We performed single-cell RNA sequencing (scRNA-seq) to provide an unbiased profile of the immune microenvironment following senescence. We specifically focused on the initial switch from hyperplasia (6 weeks) to regression (9 weeks) in which we expected to identify the most proximal response. In total, we sequenced 18,294 cells from dissociated whole adrenal glands obtained from female Znrf3-cKO animals. We then used unsupervised clustering methods to define groups of cells with similar transcriptional profiles and determined the identity of each cluster based on established cell type-specific markers in conjunction with Cluster Identity Predictor41.
scRNA-seq revealed a total of 19 cell clusters (Fig. 4a,b). Expression of cell type-specific markers for each cluster is provided in Supplementary Fig. 2. Within the major adrenal clusters, we observed a massive decrease in zona fasciculata cells between 6 and 9 weeks (Fig. 4c,d and Supplementary Table 2), consistent with adrenal cortex regression. In the immune compartment, we observed increases in both myeloid and lymphoid lineages. The most striking myeloid changes included increased macrophages, conventional type 1 DCs and suppressive neutrophils. Additionally, we observed a distinct cluster of neutrophils uniquely present at 9 weeks. These cells expressed high levels of transcripts characteristic of immature neutrophils42,43, including Chil3, Ltf and Camp (Supplementary Fig. 2n). Within the lymphoid lineage, natural killer and B cells were the main increased populations. Overall, scRNA-seq uncovered a complex immune response following senescence activation with a prominent role for myeloid cells.
Fig. 4 |. scRNA-seq reveals activation of innate and adaptive immune systems in response to cellular senescence and the SASP.

Uniform manifold approximation and projection (UMAP) plots with cluster identification of cell types in adrenal glands of female Znrf3-cKO animals at 6 weeks (9,800 cells) (a) compared to 9 weeks (8,494 cells) (b). Adrenal zG, adrenal zona glomerulosa; adrenal zF, adrenal zona fasciculata; cDC1, conventional type 1 DC; cDC2, conventional type 2 DC; MDSC, myeloid-derived suppressor cell; MoDC, monocyte-derived dendritic cell; NK, natural killer; pDC, plasmacytoid dendritic cell. c,d, Proportions of major adrenal, myeloid, lymphoid and other populations in 6-week and 9-week Znrf3-cKO samples are visualized according to the percentage of total cells. In situ validation of myeloid cell accumulation based on IHC for CD68 in control and Znrf3-cKO adrenal tissue from female (e,f) and male (g,h) cohorts. Quantification was performed using QuPath digital analysis based on the number of positive cells normalized to total nuclei. Each dot represents an individual animal. Box-and-whisker plots indicate the median (line) within the upper (75%) and lower (25%) quartiles, and whiskers represent the range. Statistical analysis was performed on log2 transformed data using two-way ANOVA followed by Tukey’s multiple-comparison test. Scale bars, 100 μm. i, Enrichment analysis using Enrichr for myeloid cell clusters (macrophages, DCs, monocytes and myeloid-derived suppressor cells) based on significant DEGs between 9- and 6-week Znrf3-cKO mice. Combined score indicates the log (P value) × z score. Padj values are indicated for the respective color groups and were calculated using Fisher’s exact test. j, Multiplex immunofluorescence images of multi-nucleated myeloid clusters based on staining for CD11c (green), F4/80 (purple), CD11b (yellow) and 4,6-diamidino-2-phenylindole (DAPI) (blue). Images shown are from a 24-week-old female Znrf3-cKO animal and are representative of clusters observed in staining of five independent 24-week-old female Znrf3-cKO animals and four 12-week-old male Znrf3-cKO mice. Quantification was performed using QuPath digital analysis on manually annotated fused cell clusters based on the percentage of total nuclei in each cluster and are visualized as parts of a whole. Scale bars, 50 μm.
Given the large proportion of immune cells detected by scRNA-seq, we next performed IHC to measure in situ changes. We chose the broad myeloid marker CD68, which was highly expressed across our DC, monocyte, neutrophil and macrophage cell clusters (Supplementary Fig. 2f). While we observed some heterogeneity between animals, CD68-positive myeloid cells progressively increased over time in female Znrf3-cKO animals compared to in controls (Fig. 4e,f). In males, we observed a significant increase in CD68-positive cells at earlier stages than in females. Moreover, CD68-positive cells occupied nearly the entire male gland by 24 weeks (Fig. 4g,h). These results suggest that cellular senescence triggers a robust immune response, which is more advanced in males, to mediate clearance of damaged adrenal cortex cells.
To further investigate the origin of this profound myeloid response, we analyzed the top DEGs within the dominant myeloid cell clusters using Enrichr44. This analysis identified multiple monocyte-derived cells among the most highly enriched cell types (Fig. 4i), suggesting a central role for bone marrow-derived immune cells. To follow up on these observations, we performed multiplex IHC for CD11b and CD11c as well as F4/80. While these markers can be expressed by multiple cell types, CD11b and CD11c are classically expressed by monocyte-derived macrophages and DCs45–48 as compared with F4/80-positive tissue-resident macrophages49. Multiplex IHC revealed a heterogeneous immune response, with contributions from both CD11c-positive as well as CD11b-positive cell populations, and involvement of resident F4/80-positive macrophages (Fig. 4j). Large multi-nucleated histiocytes were comprised of diverse cell types, including single-, double- and even some triple-positive cells. These results highlight the diversity of immune cells activated during the senescence-driven immune response and suggest a prominent role for migratory macrophages and DCs.
Males exhibit higher SASP activation
While both males and females developed an inflammatory response following senescence activation, immune cells amassed earlier and to a greater extent in males. To determine whether this differential response was due to the SASP composition and/or magnitude, we performed bulk RNA-seq on male adrenals at 4 (hyperplasia), 6 (regression onset) and 9 (regression) weeks. During the phenotypic switch from hyperplasia to regression, we found 3,161 significant DEGs (1,558 upregulated, 1,603 downregulated; Padj < 0.05) (Fig. 5a). Although we observed many sex-specific differences in gene expression (Extended Data Fig. 4), IPA identified several canonical pathways that regulate cell division as the most significantly inhibited (Fig. 5b), mirroring what we observed in females. Moreover, cellular senescence was among the top activated pathways (Fig. 5b), and the senescent mediators CDKN2A and TP53 were two of the top activated transcriptional regulators (Fig. 5c). Consistent with the strong immune phenotype and massive accumulation of phagocytic histiocytes in males, multiple immune-related signatures as well as phagosome formation were among the top activated pathways (Fig. 5b). This was similar to the later-stage response that we observed in females at 12 weeks (Extended Data Fig. 5a). When we assessed the male cytokine profile (Fig. 5d and Extended Data Fig. 5b), we found substantial overlap with females, with eight of the top ten activated cytokines shared in both sexes. However, although the profile was similar, males consistently displayed a higher magnitude of induction, including IL-1β (Z score = 5.92), IFN-γ, (Z score = 7.54) and TNF (Z score = 6.83). This was further confirmed with enhanced GSEA enrichment of immune-related signatures (Fig. 5e and Extended Data Fig. 5c). Moreover, unbiased analysis of the top DEGs that encode secreted proteins (Fig. 5f) revealed that the male SASP shared several common factors with that of females (Fig. 5g). In sum, these data suggest that both male and female Znrf3-cKO animals develop a functional SASP following senescence activation. However, males undergo the phenotypic switch earlier and display a higher magnitude of SASP induction, ultimately leading to an enhanced immune response.
Fig. 5 |. Senescence-mediated immune activation in male Znrf3-cKO mice is characterized by higher SASP induction.

a, Heatmap of the top 1,000 DEGs in adrenals from Znrf3-cKO animals at 4, 6 and 9 weeks of age (Padj < 0.05). IPA identified the top inhibited (a) and activated (b) canonical pathways, transcriptional regulators (c) and cytokines (d) in adrenals from male Znrf3-cKO animals at 6 versus 4 weeks (P < 0.05). Data are representative of four biological replicates per group, and statistical analysis in IPA was performed using a right-tailed Fisher’s exact test. BTG, B cell translocation gene; HIF-1a, hypoxia-inducible factor 1-alpha; TREM1, triggering receptor expressed on myeloid cells 1. e, GSEA for inflammatory signatures (IL-6–JAK–STAT3 signaling, chemokine signaling, the inflammatory response and the innate immune system) identifies positively enriched pathways in adrenals from male Znrf3-cKO animals at 6 versus 4 weeks. f, Heatmap representing supervised hierarchical clustering of the 40 most significant DEGs that encode secreted proteins in adrenals from male Znrf3-cKO animals compared to those of controls; statistical analysis was performed using the Wald test (Padj < 0.05). g, Comparison of the top SASP genes in males and females identified 12 shared factors induced in both sexes. h, Pharmacological inhibition of galectin 3 (encoded by Lgals3) with the selective inhibitor TD139 significantly reduced phagocytic activity in adrenal tissue from male Znrf3-cKO mice containing the R26RmTmG lineage reporter. Quantification of internalized phagocytosis beads (asterisks) was performed using QuPath digital analysis based on the number of fluorescent beads normalized to DAPI (nuclei) values. Auto-fluorescent lipid content (white) is present in large fused clusters. Three adrenal tissue slices from four independent animals per group were analyzed. i.p., intraperitoneally. Box- and-whisker plots indicate the median (line) within the upper (75%) and lower (25%) quartiles, and whiskers represent the range. Statistical analysis was performed using two-tailed Student’s t-test. Scale bars, 20 μm.
The activation of SASP factors common to both sexes suggested that these components were likely to play a key role in senescence-driven immune remodeling. To prioritize candidates to functionally interrogate, we looked for factors that (1) were expressed in the adrenal cortex in our scRNA-seq data, (2) have a known function in phagocytosis and (3) are targetable using a small-molecule inhibitor. Based on these criteria, our top candidate was Lgals3, which encodes galectin 3, a member of the β-galactoside-binding family of lectins. Moreover, galectin 3 is known to facilitate macrophage phagocytosis50 and can be selectively inhibited using TD139 (ref. 51). We administered TD139 to male Znrf3-cKO mice beginning at 6 weeks when immune cells had just begun to infiltrate. We then isolated adrenals from vehicle- and TD139-treated animals at 12 weeks and performed an ex vivo phagocytosis assay using fresh slices of adrenal tissue. This approach allowed us to preserve the native tissue architecture and avoid disruption of critical cell-to-cell interactions. We incubated adrenal tissue slices with pH-sensitive, dye-conjugated Escherichia coli particles. These particles are non-fluorescent outside the cell and only fluoresce once inside a phagosome with the shift from neutral to acidic pH52. Using this new assay system, we found a significant reduction in the number of internalized particles in adrenal slices from mice treated with TD139 as compared to those treated with vehicle control (Fig. 5h). These results demonstrate that inhibition of galectin 3 significantly impairs phagocytic activity in our model.
Androgens amplify the SASP-mediated immune response
The elevated SASP in males implicated a potential male- and/or female-specific factor capable of enhancing or repressing SASP target gene expression, respectively. However, prior studies in the normal adrenal cortex have shown that androgens suppress proliferation of stem and progenitor cells after puberty53, which is consistent with a role for androgens in promoting mechanisms related to cell cycle arrest. To functionally test whether androgens promote senescence, we castrated 4-week-old male control and Znrf3-cKO mice to lower androgen levels (Fig. 6a). At 9 weeks, we observed more than a threefold increase in adrenal weight in castrated Znrf3-cKO animals compared to that in sham controls (Fig. 6b,c). This robust increase in adrenal size was associated with both retention of proliferative adrenal cortex cells (Fig. 6d) as well as a near-complete block in the myeloid response, including neutrophils (Extended Data Fig. 6a) as well as both CD11c-positive and CD11b-positive cells (Fig. 6e). Consistent with the significant reduction in antigen-presenting cells, T cell infiltration was significantly lower in castrated animals than in sham controls (Extended Data Fig. 6b). Tissue-resident macrophages marked by F4/80 (ref. 49) were androgen responsive yet remained relatively abundant following castration (Fig. 6e). However, their distribution was more dispersed, similar to that of the normal adrenal cortex. Intriguingly, some senescent markers, including p16 and p21, were unchanged or even elevated in adrenals from castrated animals compared to those of sham controls (Fig. 6d). These results suggest that the dominant impact of androgens on senescence occurs in the later immune-mediated stages as opposed to in the initial cell cycle arrest. To further tease apart the role of androgens in early versus late senescent responses, we castrated male Znrf3-cKO mice at 6 weeks, when hyperproliferation has normally been suppressed. We analyzed mice 6 weeks later and found a significant block in the myeloid response yet no increase in proliferation (Extended Data Fig. 6c–g). These experiments suggest that androgens primarily regulate the late senescent response.
Fig. 6 |. Androgen deprivation restricts immune infiltration in male adrenals.

a, Male control or Znrf3-cKO mice were castrated at 4 weeks of age to lower androgen levels. b,c, At 9 weeks of age, adrenal glands from castrated mice were significantly larger than those from sham-operated controls. b, Representative gross histology images are shown. Scale bars, 1 mm. c, Normalized adrenal weight. Each dot represents an individual animal. Box-and-whisker plots indicate the median (line) within the upper (75%) and lower (25%) quartiles, and whiskers represent the range. Relative fold change is indicated below each group. Senescence markers (Ki67, p16 and p21) (d) and myeloid immune markers (CD68, CD11c, CD11b and F4/80) (e) in adrenals from sham-operated versus castrated animals. Representative images are shown for each group. Scale bars, 20 μm. Quantification was performed using QuPath digital image analysis based on the number of positive cells normalized to total nuclei. Each dot represents an individual animal. Box-and-whisker plots represent mean with variance across quartiles. Statistical analysis was performed using two-tailed Student’s t-test (c,d) or two-way ANOVA (e) followed by Tukey’s multiple-comparison test. IPA of RNA-seq data from adrenals of 9-week-old males identified the top inhibited and activated canonical pathways (f) and cytokines (g) in adrenals from castrated versus sham-operated Znrf3-cKO animals; statistical analysis in IPA was performed using a right-tailed Fishers exact test (P < 0.05). CREB, cAMP response element-binding protein; FAK, focal adhesion kinase; LXR, liver X receptor; RXR, retinoid X receptor. h, Heatmap representing supervised hierarchical clustering of the 40 most significant DEGs that encode secreted proteins in adrenals from castrated compared to sham-operated Znrf3-cKO mice; statistical analysis was performed using the Wald test (Padj < 0.05). Asterisks indicate genes previously identified as shared SASP components common to both male and female Znrf3-cKO animals. Data are representative of four biological replicates per group, and statistical tests were performed using IPA.
To further examine the mechanism by which androgens regulate senescence, we performed bulk RNA-seq on whole adrenals from 9-week-old castrated and sham-operated Znrf3-cKO animals. We found that castration reversed many of the top canonical pathways (Fig. 6f), cytokines (Fig. 6g) and genes encoding secreted proteins (Fig. 6h) that were previously enhanced in males compared with females. In sum, these experiments demonstrate that androgens amplify the SASP-mediated immune response.
Senescence-driven tissue remodeling leads to adrenal tumors
Based on high levels of immune infiltration and adrenal degeneration, we predicted that Znrf3-cKO animals would succumb to adrenal insufficiency. To test this hypothesis, we generated late-stage cohorts at 44, 52 (1 year) and 78 weeks (18 months) (Fig. 7a,b). Surprisingly, rather than adrenal failure, a large proportion of Znrf3-cKO animals ultimately developed adrenal tumors (Fig. 7c). Notably, tumor development and progression were highly sex dimorphic, with females exhibiting increased malignancy.
Fig. 7 |. Following senescence-mediated remodeling of the tissue microenvironment, metastatic ACC tumors arise in a sex-dimorphic manner.

Adrenal weight measurements in cohorts of female (a) and male (b) control and Znrf3-cKO mice, including at 44, 52 and 78 weeks of age. After 52 weeks, adrenal tumors of varying sizes begin to form. Each dot represents an individual animal. The black line indicates the mean. c, Representative images of the gross histology (left) and H&E staining (middle and right) from each pathology observed at 78 weeks of age. Blinded samples were scored as atrophic, benign or malignant. The respective proportion of each pathology in male compared to female cohorts is indicated. Scale bars, 1 mm (left), 200 μm (middle);, 500 μm (right). d,e, Proliferation as measured by the Ki67 index is significantly higher, with progression to benign and malignant tumors in Znrf3-cKO mice. Each symbol represents an individual animal. The black line indicates the mean. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. f, Malignant tumors metastasize to distant sites, including the liver. Representative images are shown from mice expressing the R26RmTmG lineage reporter, which permanently labels adrenal cortex cells with GFP. Scale bars, 100 μm. g,h, Tissue weight is significantly higher, with progression to benign and malignant tumors in Znrf3-cKO mice. Each symbol represents an individual animal. The black line indicates the mean. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. i, Representative H&E (left), CD68 (middle) and Ki67 (right) staining in atrophic, benign and malignant adrenals from Znrf3-cKO 78-week-old mice. Scale bars, 500 μm (whole tissue), 100 μm (high-magnification inset). j, In Znrf3-cKO adrenals, benign nodules have a significantly higher Ki67 index and lower CD68 index than the background gland. Each dot represents an individual nodule. Box-and-whisker plots indicate the median (line) within the upper (75%) and lower (25%) quartiles, and whiskers represent the range. Statistical analysis was performed using two-tailed Student’s t-test. k, In malignant tumors, there is a significant inverse correlation between the Ki67 index and the CD68 index. Each dot represents a distinct tumor region. Colors represent distinct regions from the same tumor. Statistical analysis was performed using simple linear regression (n = 23 tumor regions from six animals).
To evaluate tumors without bias, we performed H&E and Ki67 staining. Blinded samples were then pathologically reviewed based on criteria derived from the Weiss system conventionally used to evaluate adrenal cortical neoplasms in adult patients54,55. This analysis revealed three main phenotypes in Znrf3-cKO adrenals: atrophic glands, benign lesions and malignant tumors (Fig. 7c). Atrophic glands were characterized by high histiocyte accumulation (Fig. 7c) and an overall Ki67 index at or below that of age-matched controls (Fig. 7d,e). This pathology was observed in 14.3% of females (three of 21 mice) and 25% of males (three of 12 mice). Benign tumors were most prevalent in males (66.7%, eight of 12 mice). These lesions were characterized by formation of discrete nodules contained within the encapsulated adrenal cortex (Fig. 7c) with a significantly higher Ki67 index than that of matched controls (Fig. 7d,e). Finally, malignant tumors were most prevalent in females (61.9%, 13 of 21 mice). We conservatively classified tumors as malignant strictly based on the presence of metastases, which occurred to clinically relevant sites including the lung and the liver (Fig. 7f). However, malignant tumors also had a significantly higher Ki67 index (>10%) (Fig. 7d,e) and were significantly larger in size (Fig. 7g,h). Overall, these findings demonstrate that Znrf3 loss is permissive for ACC in a sex-dependent manner with advanced aging.
Reduced myeloid cells are predictive of poor patient outcome
Our Znrf3-cKO model presented two striking sexual dimorphisms. Males exhibited an earlier, more robust immune response following senescence and were less likely to develop malignant tumors. Conversely, females displayed a dampened immune response and were significantly more likely to develop metastatic tumors. To further examine the relationship between the immune microenvironment and tumor progression, we quantified CD68-positive myeloid cells in aged animals at 44 and 52 weeks of age before the onset of tumors and in atrophic, benign and malignant tumors at 78 weeks. With increased aging, female Znrf3-cKO animals continued to accumulate histiocytes (Extended Data Fig. 7a,b) yet retained a greater proportion of adrenal cortex cells than males (Extended Data Fig. 7c,d). Control adrenals also displayed an age- and sex-dependent accumulation of CD68-positive cells at the cortical–medullary boundary (Extended Data Fig. 7e–h), which is where the longest-lived adrenal cortex cells reside. These results are consistent with myeloid cell recruitment in the adrenal gland being driven by age-related mechanisms. At 78 weeks, we observed a heterogeneous immune landscape, dependent on the tumor stage. Atrophic glands exhibited a high CD68 index, consistent with the accumulation of histiocytes noted in H&E staining (Fig. 7i and Extended Data Fig. 7i). In benign cases, there was an inverse correlation between proliferation and myeloid cell infiltration. Nodules displayed a significantly higher Ki67 index and lower CD68 index than those of the background gland (Fig. 7i,j and Extended Data Fig. 7j). This apparent exclusion of myeloid cells in more proliferative regions was also evident in malignant tumors. Here, we took advantage of high intertumoral and intratumoral heterogeneity and analyzed CD68-positive myeloid cells across different tumors as well as subpopulations. We found that regions with the highest Ki67 index had the lowest CD68 index (R2 = 0.54, ****P = 6.73 × 10−5) (Fig. 7i,k). In sum, these results suggest that, as adrenal tumors become more aggressive, the immune microenvironment is remodeled to exclude tumor-suppressive myeloid cells.
Given these observations, we next sought to assess myeloid cell infiltration in ACC patient tumors. A myeloid response score (MRS) was recently established in hepatocellular carcinoma that predicts patient prognosis and therapy response56. We evaluated expression of each hepatocellular carcinoma MRS gene in our scRNA-seq dataset and identified four genes (Cd33, Cd68, Itgam, Msr1) that were myeloid specific (Fig. 8a–d) and for which the combined expression represented the full adrenal myeloid compartment (Fig. 8e). We then evaluated the adrenal MRS (AMRS) in ACC tumors from The Cancer Genome Atlas (TCGA) (TCGA-ACC) in conjunction with established markers of poor prognosis (high MKI67 (ref. 57) and low G0S2 (ref. 58); Fig. 8f). Consistent with our Znrf3-cKO model, females had a significantly lower AMRS than males (Fig. 8g). Moreover, low AMRS was associated with significantly reduced overall (Fig. 8h) and progression-free survival (Fig. 8i), even in females alone (Extended Data Fig. 8). To compare the AMRS to that of individual immune cell populations, we derived signatures for populations of interest based on established cell type-specific markers that were robustly expressed in TCGA-ACC data. We found that signatures of individual myeloid cell populations, including macrophages, DCs and neutrophils, failed to predict both overall and progression-free survival (Supplementary Table 3). By contrast, a signature of monocyte-derived cells was strongly predictive of patient outcome and was significantly higher in males than in females, consistent with our AMRS and the collective importance of multiple recruited myeloid cell populations.
Fig. 8 |. A low myeloid response is associated with worse patient outcome in ACC.
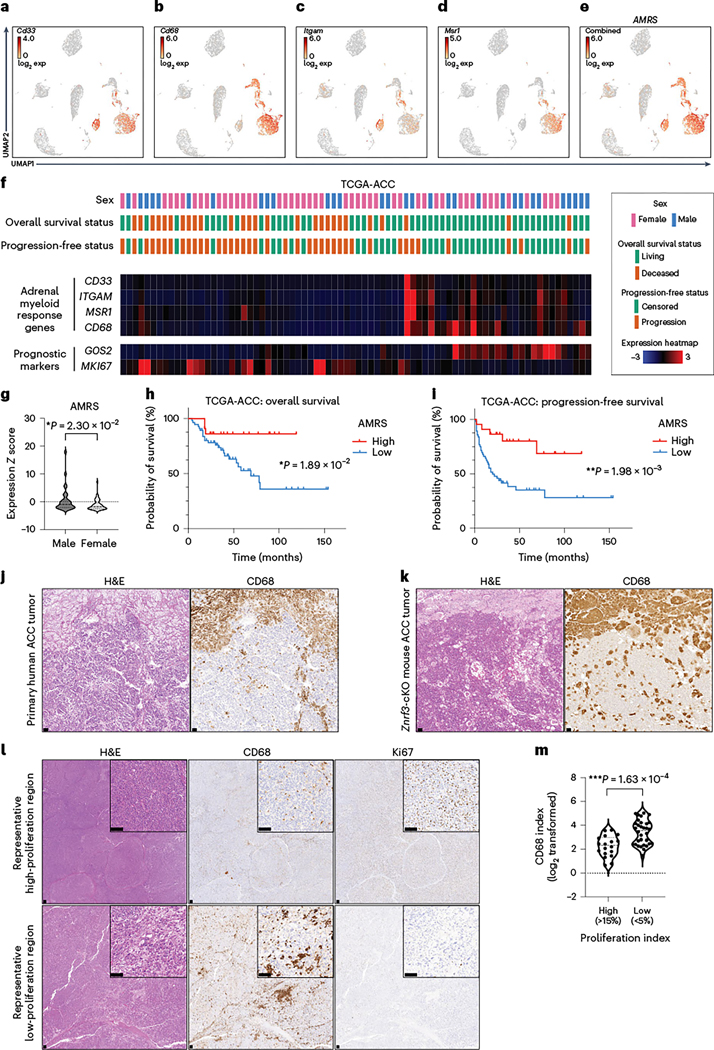
UMAP of scRNA-seq data from adrenal tissue of female Znrf3-cKO animals. Expression of individual myeloid cell markers Cd33 (a), Cd68 (b), Itgam (also known as CD11b) (c) and Msr1 (also known as CD204) (d) is shown. e, The combined expression of these four markers was used to generate an AMRS. Exp, expression. f, Analysis of TCGA-ACC data, including patient demographics and outcome, in accordance with mRNA expression of AMRS genes (CD33, CD68, ITGAM, MSR1) and established prognostic markers (high MKI67 (proliferation) and low G0S2 (recurrent disease)). AMRS is significantly lower in female than in male TCGA patients with ACC (g), and low AMRS is associated with shorter overall (h) and progression-free (i) survival. j, In primary human ACC tumors, CD68+ histiocytes are found in a subset of cases. k, A representative example region is shown relative to a Znrf3-cKO mouse adrenal tumor. l,m, The infiltration of CD68+ myeloid cells in primary human ACC tumors is heterogeneous. Regions with a high Ki67 index (>15%) have a significantly lower CD68 index than regions of low proliferation (Ki67 < 5%). IHC quantification was performed using QuPath digital analysis based on the number of positive cells normalized to total nuclei. Tumor-associated stroma and highly necrotic regions were excluded from analysis. Each dot represents a distinct tumor region (n = 50 tumor regions from 38 patients). Statistical analysis was performed using two-tailed Mann–Whitney test (g), log-rank Mantel–Cox test (h,i) or two-tailed Student’s t-test (m). Scale bars, 100 μm.
Our results implicated myeloid cells as a key component of the ACC tumor microenvironment, yet ACC tumors are classically described as immune poor59. However, this notion is primarily based on evaluation of lymphoid cells60, and myeloid cells have thus far remained unexplored. To extend the translational relevance of our findings, we obtained formalin-fixed, paraffin-embedded (FFPE) primary tumor samples from 38 patients with ACC. We found that all primary tumor specimens contained CD68-positive myeloid cells, with an average overall index of 11.52% (Supplementary Table 4). Moreover, a subset of cases (seven of 38 tumors or 18.4%) contained distinct histiocytes, similar to what we observed in tumors from Znrf3-cKO mice (Fig. 8j,k). In addition, we found that tumor regions with a high Ki67 index (>15%) had a significantly lower CD68 index than regions of low proliferation (Ki67 < 5%) (Fig. 8l,m), mirroring what we observed in our mouse model. In sum, our results reveal an important role for myeloid cells in restraining ACC tumor progression with substantial prognostic value.
Discussion
Our data demonstrate that loss of the Wnt inhibitor ZNRF3 is permissive for metastatic adrenal cancer with advanced aging. However, genetic Znrf3 loss is not completely sufficient and depends on additional extrinsic factors, including sex and the aged immune microenvironment. These factors are closely linked to cellular senescence, an age-associated stress response activated in ZNRF3-deficient adrenals. Our in vivo characterization of senescence-induced tissue remodeling reveals a prominent role for myeloid-derived immune cells during the phenotypic switch from hyperplasia to regression. Males activate senescence earlier and mount a greater immune response, mediated in part by androgens, which ultimately results in fewer metastatic tumors. We translate these findings to human patients with ACC, in whom a high myeloid response is associated with better outcome. Collectively, our model recapitulates the age and sex dependence of ACC and reveals a new role for myeloid cells in restraining adrenal cancer progression.
Sex-specific differences in stem cell renewal, the tissue microenvironment and innate and adaptive immune responses have been increasingly acknowledged as key factors in both aging61 and cancer62, including immunotherapy response63. Our model, in which males exhibit an advanced senescent response and subsequently become more protected from malignancy, provides a system to interrogate sex-dependent mechanisms of immunosurveillance. While there are multiple underlying differences between males and females, we demonstrate a specific role for male androgens in amplifying the SASP to enhance recruitment and activation of monocyte-derived macrophages and DCs. These results are consistent with prior work showing that androgens activate cellular senescence in vitro64,65 and recently published observations that androgens enhance the phagocytic activity of macrophages66. In our model, we found that androgen deprivation had no effect on the initial cell cycle arrest but instead blocked manifestation of a full senescent phenotype by suppressing the SASP. This not only reduced macrophages but also DCs and other myeloid cells. The adaptive immune response and T cell infiltration were also impaired, likely due to the loss of antigen-presenting cells, and consistent with recent reports of senescence promoting anti-tumor immunity in other cancers67,68.
Following extensive immune remodeling and advanced aging, Znrf3-cKO animals ultimately develop adrenal tumors at 12–18 months. This corresponds to peak ACC incidence at an age of 45–55 years7,8. Moreover, metastatic tumors in our model were more common in females, which is analogous to the higher prevalence of ACC in women7. In Znrf3-cKO animals, tumor development requires escape from senescence-mediated tissue degeneration. While our studies focus on ZNRF3 as a common ACC tumor suppressor, we expect that tumorigenesis is influenced by additional genetic mutations, particularly within genes that bypass cellular senescence and are recurrently altered in patients (for example, TP53, CDKN2A and RB1 (refs. 10,11)). However, in addition to mutational events, we also expect that age-dependent changes in the tissue microenvironment substantially impact the functional consequence of acquired mutations. In particular, we observe a striking exclusion of myeloid cells with advanced tumor progression, suggesting an age-dependent conversion of the SASP from an anti-tumorigenic to a pro-tumorigenic process. These findings translate to human patients and align with the immune-poor nature of ACC59 as well as the modest efficacy of immunotherapy thus far69. Given that a strong intratumor IFN-γ signature correlates with better immunotherapy response in other cancers70–72, exploiting underlying mechanisms that regulate immune cell recruitment in our model may ultimately help enhance the efficacy of immune-based therapy for ACC and other immune-cold tumors.
Overall, our Znrf3-cKO model is a highly unique in vivo system that highlights the pleiotropic effects of cellular senescence in cancer. Traditionally, cellular senescence has been studied as a tumor-suppressive mechanism that halts proliferation of damaged cells at risk for neoplastic transformation73. However, more recent studies reveal that some senescent cells may be released from their growth arrest and actually promote more aggressive phenotypes in the long term74,75. In our model, tumors may potentially arise from cells that evaded senescence or from cells released from senescence. Newly developed lineage-tracing tools to track senescent cells will provide further insight on the origin of these tumors as well as metastatic outgrowths. This has important clinical implications given that conventional chemotherapy agents, including EDP (etoposide, doxorubicin and cisplatin), frequently used as standard of care for advanced ACC76, are known to act in part by triggering ‘therapy-induced senescence’ (ref. 77). Consequently, EDP therapy may favor long-term maintenance of senescent tumor cells primed for late-stage recurrence78. Senolytic agents that selectively kill senescent cells may be a promising strategy to eliminate tumor cells following therapy-induced senescence, which is an active area of ongoing clinical investigation79 and future study.
Methods
Mice
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Michigan (protocol 00010217) and the University of Utah (protocol 21–02009). Mouse strains used in this study have been previously described: SF1Cre-high17, Znrf3-floxed13, R26RmTmG (ref. 23). All animals were maintained on the C57Bl/6J background with a 12-h light–12-h dark cycle and ad libitum access to food and water. Littermate control animals were used in all experiments. Tumor-bearing mice were monitored based on body weight and condition. Mice that exhibited pain, loss of appetite, distress, abnormal behavior or loss of condition were evaluated by a veterinarian and euthanized accordingly.
Ultrasound
Mice were anesthetized using 3% isoflurane in O2 (1 l min−1) in an induction chamber until reaching the surgical plane. Following induction, mice were weighed and moved to a heated platform in the prone position in a modified Trendelenburg position. To maintain a surgical plane of anesthesia, 1–1.5% isoflurane in O2 was supplied via a nose cone. Eye lubricant was applied to prevent corneal damage during prolonged anesthesia. ECG and respiration were monitored via non-invasive resting ECG electrodes. Hair was removed from the left dorsal area with depilatory cream. Two-dimensional (B-mode) and three-dimensional ultrasound images were recorded using a VisualSonics Vevo 2100 in vivo micro-imaging system with an MS-550D transducer, which has a center frequency of 40 MHz and a bandwidth of 22–55 MHz.
Surgical castration
Male control or Znrf3-cKO mice at 4 or 6 weeks of age were anesthetized using 3% isoflurane in O2 (1 l min−1) in an induction chamber until reaching the surgical plane. To maintain a surgical plane of anesthesia, 1–1.5% isoflurane in O2 was supplied via a nose cone. Eye lubricant was applied to prevent corneal damage during prolonged anesthesia. A single incision was made at each scrotum, and testes were exposed and removed. Pain relief was administered using bupivacaine (2 mg ml−1) subcutaneously and lidocaine (4 mg per kg) at the surgical site. Sham operations followed the same protocol, without exposing and removing the testes. Mice were housed singly after the operation and monitored in accordance with Institutional Animal Care and Use Committee protocols. Mice were euthanized at 9 or 12 weeks, respectively, and prostate regression was visually inspected to confirm complete castration.
TD139 study
TD139 (15 mg ml−1) or DMSO was administered intraperitoneally to 6-week-old male Znrf3-cKO mice every 3 d for 6 weeks. Upon termination of the study, adrenals were removed, cleaned of excess fat, embedded in 4% low-melt agarose (Lonza, 50101) and cut into 150-μm tissue sections using a Leica VT1200S vibratome. Tissue slices were cleaned of excess agarose and incubated in complete medium (DMEM/F12, 1× GlutaMAX, 1× ITSX and 1% penicillin–streptomycin) at 37 °C with 0% CO. Phagocytosis beads (pHrodo Deep Red E. coli BioParticles; Thermo Fisher, P35360) reconstituted at 1 mg ml−1 in sterile 1× PBS were added to adrenal slices (5 μg ml−1) and incubated for 90 min with agitation. Upon assay termination, slices were washed with 1× ice-cold PBS and fixed for 15 min using 4% paraformaldehyde (Fisher Scientific, NC1537886). Slices were then stained with DAPI (Thermo Fisher, D21490) and mounted using a glycerol-based mounting medium. Images were acquired using a Leica SP8 laser scanning confocal microscope with a ×40, 1.10 water-immersion objective (HC PL APO CS2). Volume acquisition was performed in unidirectional xyz scan mode to obtain z stacks using a slice interval of 1 μm and a resolution of 0.284 μm per voxel. The pinhole was set to 1 Airy unit. Serial acquisition was used to minimize cross-talk for 405-, 488-, 561-and 633-nm lasers. The number of phagocytosis beads per image was quantified using QuPath digital image-analysis software and normalized to DAPI values.
Adrenal weight
Adrenals were cleaned of excess fat, weighed and snap frozen in liquid nitrogen or fixed as described for histology. In control animals, we observed an age-dependent increase in adrenal weight across our time series that significantly correlated with an age-dependent increase in body weight (Supplementary Fig. 3a–c). To account for these normal aging differences, adrenal weights in control and Znrf3-cKO mice were compared based on the adrenal-to-body weight ratio calculated as the sum of both adrenal glands (mg) normalized to body weight (g). Raw adrenal weight data are provided for reference in Supplementary Fig. 3d–g.
Mouse adrenal histology
Adrenals were fixed in 10% normal buffered formalin (Fisher Scientific, 23–427098) for 24 h at room temperature, embedded in paraffin and cut into 5-μm sections. Antibody information and staining conditions are listed in Supplementary Table 5. Endogenous peroxidase activity was blocked with 0.3% H2O2 for 30 min at room temperature. For brightfield IHC, primary antibodies were detected with HRP polymer solution (Vector Laboratories, MP-7401 (anti-rabbit), MP-7402 (anti-mouse) or MP-7405 (anti-goat)) and DAB EqV peroxidase substrate (Vector Laboratories, SK-4103–100), nuclei were counterstained with hematoxylin (Sigma, GHS132), and slides were mounted using VectaMount (Vector Laboratories, H-5000–60). For immunofluorescence, primary antibodies were detected with HRP polymer solution and Alexa Fluor tyramide reagent (Thermo Fisher, B40953 (488), B40955 (555) or B40958 (647)), nuclei were counterstained with Hoechst (Thermo Fisher, H3570), and slides were mounted using ProLong Gold (Thermo Fisher, P36930). For multiplexed immunofluorescence, consecutive staining of primary antibodies and the corresponding HRP-conjugated polymers followed by signal detection with Alexa Fluor tyramide reagents was performed using the same conditions as described for brightfield IHC. Antibody stripping was performed after detection of each primary antibody by boiling in the antigen-retrieval buffer of the next primary antibody. Single-stain and fluorescence-minus-one controls were run to confirm complete stripping. Imaging was performed on a Zeiss Apotome microscope with an AxioCam MRm camera, a Pannoramic MIDI II (3DHISTECH) digital slide scanner or a Zeiss Axio Scan.Z1 digital slide scanner. Quantification was performed using QuPath digital pathology software. Multiplex immunofluorescence quantification was performed using QuPath on manually annotated fused cell clusters.
Senescence-associated β-galactosidase staining
Freshly isolated adrenal tissue was cryopreserved in optimal cutting temperature compound. Cryosections (10 μm) were stained using the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology, 9860) according to the manufacturer’s protocol with minor modifications. Briefly, cryosections were air dried at room temperature for 30 min, fixed for 15 min and washed with PBS. Sections were stained inside a sealed, humidified chamber at 37 °C overnight. Sections were then counterstained with eosin, dehydrated and mounted for imaging.
RNA isolation
Adrenals isolated from mice at the indicated time points were cleaned of excess fat, weighed, snap frozen in liquid nitrogen and stored at −80 °C. For homogenization, RLT lysis buffer (Qiagen, 74104) containing 1% β-mercaptoethanol was added to thawed adrenal tissue in high-impact zirconium 1.5-mm bead tubes (Benchmark Scientific, D1032–15). Samples were homogenized for 30 s using the BeadBug 3 homogenizer (Benchmark Scientific, D1030), returned to ice and homogenized for an additional 30 s. Total RNA isolation was completed using the RNeasy Kit (Qiagen, 74104) according to manufacturer guidelines. DNases were removed using the RNase-Free DNase Set (Qiagen, 79254) and subsequently purified using the RNeasy MinElute Cleanup Kit (Qiagen, 74204) according to manufacturer guidelines.
Bulk RNA sequencing
Following RNA isolation and DNase treatment, RNA quality was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies). All samples used for sequencing had an RNA-integrity number value >8.0. Libraries were prepared using the NEBNext Ultra II Directional RNA Library Prep Kit with the poly(A) mRNA isolation protocol and sequenced using the NovaSeq Reagent Kit version 1.5 150 × 150-bp sequencing protocol. Libraries were sequenced on an Illumina NovaSeq 6000. The mouse GRCm38 genome and gene-feature files were downloaded from Ensembl release 102, and a reference database was created using STAR version 2.7.6a with splice junctions optimized for 150-bp reads80. Optical duplicates were removed from the paired-end FASTQ files using clumpify version 38.34, and reads were trimmed of adaptors using cutadapt 1.16 (ref. 81). The trimmed reads were aligned to the reference database using STAR in two-pass mode to output a BAM file sorted by coordinates. Mapped reads were assigned to annotated genes using featureCounts version 1.6.3. Expected gene counts were filtered to remove features with zero counts and features with fewer than ten reads in any sample. The age and genotype were combined into a single column in the design formula, and DEGs were identified using a 5% false discovery rate with DESeq2 version 1.30.1 (ref. 82). Male and female datasets were analyzed in separate DESeq2 models. Pathways were analyzed using the fast gene set enrichment package83 and IPA20. Z scores reported from IPA represent the activation Z score, which was used to infer the activation states of pathways and upstream regulators.
Adrenal gland dissociation
Adrenal glands were obtained from 6- or 9-week-old mice following rapid decapitation to minimize stress-induced transcriptional changes. Adrenals were placed immediately into ice-cold 1× Hank’s Balanced Salt Solution (HBSS) containing calcium and magnesium (Thermo Fisher, 14025134). Tissues were then finely chopped and digested enzymatically using enzymes and reagents from the Neural Tissue Dissociation Kit (Miltenyi Biotec, 130–092-628). All steps were carried out at 4 °C, including centrifugation. All tubes and pipette tips used to handle cell suspensions were pre-coated with 3% BSA in HBSS to prevent cell loss. During the dissociation, the cell suspension was gently agitated with mechanical pipetting every 10 min and visually assessed under a stereomicroscope until the tissue was fully digested. The suspension was then filtered through 70-μm filters to obtain a single-cell suspension, and enzymes were neutralized using HBSS containing 10% FBS. Red blood cells were removed using Red Blood Cell Lysis buffer (Roche, 11814389001) according to manufacturer guidelines, and the cells were washed twice with HBSS containing 2% FBS before counting on a Countess Automated Cell Counter (Thermo Fisher).
Single-cell RNA sequencing
Immediately following dissociation, cells were stained with 0.1 μg ml−1 DAPI and 5 μM Vybrant DyeCycle Ruby (Invitrogen, V10309). Viable Ruby-positive, DAPI-negative single cells were sorted based on fluorescent properties using the BD FACSAria III Cell Sorter at the University of Utah Flow Cytometry Shared Resource. Cells were collected in 1× PBS (free of calcium and magnesium) containing 0.04% BSA and kept on ice. Single-cell droplets with a target capture of 10,000 cells were immediately prepared on the 10x Chromium system at the Huntsman Cancer Institute High-Throughput Genomics Shared Resource (Supplementary Fig. 4 Gating Strategy). Single-cell libraries were prepared using the Chromium Next GEM Single Cell 3′ Gene Expression Library Construction Kit version 3.1 according to manufacturer instructions. Sequencing was performed on an Illumina NovaSeq 6000 instrument using an S4 flow cell generating 150-bp paired-end reads at a target depth of 300 million reads per sample. Raw data were converted to demultiplexed FASTQ files with 10x Cellranger mkfastq. Reads were mapped to the prebuilt mm10 reference distributed by 10x Genomics (version mm10–2020-A), and genes were counted with 10x Cellranger count. Samples were also aggregated into a single Loupe file with 10x Cellranger aggr. Filtering parameters for gene-count range (>200 and <5,500), unique molecular identified counts (>200 and <30,000) and mitochondrial transcript fraction (<15%) were used to remove low-quality cells.
scRNA-seq analysis and cluster identification
For analysis of scRNA-seq clusters, major cell types were distinguished using graph-based and k-mean methods using the Loupe Cell Browser Interface. For major cell clusters, core cell type-specific genes were used to validate initial clustering methods: adrenal cortex (Nr5a1, also known as Sf1), immune (Ptprc), endothelial (Plvap) and medulla (Th) (Supplementary Fig. 2). Validation of subclusters was completed by identifying defined transcriptional signatures from previously annotated mouse scRNA-seq datasets84–86 and established identification markers (Supplementary Fig. 2). Both inter-cluster and intra-cluster comparisons were performed for cells of similar lineage. Final clustering was based on both gene expression patterns and the proportion of significant DEGs between respective clusters. Validation of immune-based clusters was completed using Cluster Identity Predictor correlation analysis to compare new clusters with the established ImmGen reference dataset41. Enrichment analysis was performed using Enrichr with which DEGs in the dominant myeloid clusters (macrophages, monocytes, DCs and MDSCs) were compared between 9 and 6 weeks. Combined scores were used to indicate statistical significance (P) and ranking (x) based on c = log (P) × x (ref. 87).
TCGA-ACC immune signatures
The relative AMRS in each tumor from TCGA-ACC data was calculated using mRNA expression Z scores obtained from cBioPortal (https://www.cbioportal.org, PanCancer Atlas). A composite enrichment score was calculated based on the average expression of CD33, CD68, ITGAM (also known as CD11b) and MSR1 (also known as CD204). TCGA-ACC tumors without mRNA expression data were excluded (n = 14), resulting in final analysis of 31 male and 47 female tumors. A score cutoff above or below the median, respectively, was used to classify AMRS-high versus AMRS-low tumors. Individual immune cell scores were derived using the same approach for macrophages (C1QA, C1QB, APOE, MERTK), DCs (CD209, ITGAE, IRF8, TLR3), neutrophils (S100A8, S100A9, MMP9, CSF3R), B cells (CD79A, CD79B, RAG1, CD22), natural killer cells (GZMA, GZMB, CCL5, NKG7), T cells (CD3E, CD3D, CCR7, CTLA4) and monocyte-derived cells (ITGAX, CD68, CCR2, ITGAM). Gene markers for each cell population were chosen based on established cell type-specific genes that were also robustly expressed across the TCGA-ACC cohort. MKI67, which is an established prognostic marker in ACC57, as well as a published gene set for phagocytic macrophages66 (CD68, TREM2, TYROBP) were included for comparison.
Human ACC tumor histology
Primary ACC FFPE tumor samples were obtained from the Huntsman Cancer Institute and the University of Colorado School of Medicine at Colorado Anschutz Medical Campus in accordance with study protocols approved by the Institutional Review Board at each institution. Surgical specimens from each case were reviewed by a clinical pathologist to select representative FFPE blocks enriched for viable tumor tissue. H&E and immunostaining were performed by the ARUP Research Histology Lab and the Huntsman Cancer Institute Biorepository and Molecular Pathology Shared Resource, respectively, on 5-μm serial sections. Immunostaining was performed on the Leica Bond automated staining platform (Leica Biosystems). Antigen retrieval was performed with Epitope Retrieval Solution 2 (Leica Biosystems) for 20 min (CD68) or 30 min (Ki67) at 95 °C, followed by primary antibody incubation (anti-CD68, Leica, PA0273, ready to use; anti-Ki67, Abcam, ab1667, 1:400) for 15 min at room temperature and the Bond Polymer Refine Detection kit (Leica Biosystems) for 8 min at room temperature. Slides were imaged on a Zeiss Axio Scan.Z1 slide scanner, and automated quantification was performed using QuPath digital pathology software. Surrounding fat and stroma as well as necrotic tumor regions were excluded from analysis. Tumors with substantial intratumor heterogeneity with respect to the Ki67 proliferation index were scored based on region.
Statistics and reproducibility
Sample size was determined based on power calculations performed using G*Power to obtain >80% power with 5% type 1 error. Statistical analysis was performed using R or GraphPad Prism 9. For comparison of two groups, a two-tailed Student’s t-test was performed. For more than two groups, one-way ANOVA followed by Tukey’s post hoc test was performed. When analyzing two independent variables, two-way ANOVA followed by Tukey’s post hoc test was used. Normal distribution was tested with an F-test (two groups) or the Brown–Forsythe test (ANOVA). If normal distribution could not be assumed, a non-parametric analysis was used or data were log2 transformed before statistical analysis. Specific statistical tests used for each analysis are indicated in the figure legends. A P value <0.05 was considered significant, and actual P values for each test are indicated in each figure. Samples were blinded before analysis whenever possible. This included the pathological review of tumors performed on samples from 78-week-old animals. In many experiments, blinding was not feasible, as the genotype was evident from the image data based on differences in adrenal size. Results from the adrenal ultrasound were not replicated. Rather, we reproduced these finding using an expanded orthogonal approach based on adrenal weight with increased numbers and in both sexes. Results from bulk RNA-seq and scRNA-seq were confirmed using orthogonal approaches. All other laboratory experiments were replicated successfully, and no data were excluded. For further information on experimental numbers for in vivo studies, see the Supplementary Information. For TCGA-ACC analysis using cBioPortal, patients without mRNA expression data were excluded. For RNA-seq experiments, mice were randomly chosen from the respective cohorts. For the TD139 drug study, mice were randomly assigned vehicle control (DMSO) or drug treatment. For castration studies, male mice were randomly assigned castration or sham surgical procedure. For all other experiments, mice were assigned to control or Znrf3-cKO groups based on genotype.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequencing datasets have been deposited to the NCBI Gene Omnibus Database (accession code GSE201127 (GSE201125, bulk RNA-seq; GSE201126, scRNA-seq). Patient data analyzed from TCGA-ACC are publicly available using https://www.cbioportal.org/study/summary?id=acc_tcga_pan_can_atlas_2018. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
Code availability
Code used for bulk RNA-seq and scRNA-seq analyses is freely available at https://github.com/HuntsmanCancerInstitute. Additional scRNA-seq packages used for analysis are available at https://github.com/atakanekiz/CIPR-Package.
Extended Data
Extended Data Fig. 1 |. Ultrasound imaging provides an accurate measure of adrenal size in real-time.

(a) Images of adrenal ultrasound area, ultrasound volume, and gross histology from representative animals with adrenals of varying size. Scale bars, 1 mm. (b) Adrenal area and (c) adrenal volume are significantly correlated with adrenal weight. Ultrasound imaging was performed 24-hours prior to necropsy. All data shown is from female mice. Each dot represents an individual animal. Statistical analysis was performed using simple linear regression.
Extended Data Fig. 2 |. Znrf3 cKO adrenals activate cellular senescence during adrenal regression.

(a, b) Apoptotic cell death as measured by cleaved caspase 3 (CC3) is not significantly increased in female or male Znrf3 cKO adrenals compared to controls during the initial phase of tissue regression. Quantification of CC3-positive cells was performed using QuPath digital image analysis based on the number of positive cells and normalized to total adrenal cortex nuclei. Arrows indicate CC3-positive cells. Dashed line indicates histological boundary between adrenal cortex and medulla. Scale bars, 100 μm. (c) DNA damage as measured by 53BP1 foci is significantly increased in male Znrf3 cKO adrenals compared to controls at 4- and 6-weeks of age. Quantification of 53BP1 foci was performed using QuPath digital image analysis based on the number of positive foci and normalized to total nuclei. Asterisks indicate 53BP1-positive foci. Scale bars, 10 μm. (d) p21, (e) p16INK4a, and (f) senescence-associated beta-galactosidase (SA-β-gal) are significantly increased in 9-week male Znrf3 cKO adrenals compared to controls. Scale bars, 100 μm. Quantification of p21 and p16INK4a IHC was performed using QuPath digital image analysis based on the number of positive cells per high powered field (HPF). Representative SA-β-gal images obtained from analysis of 3 independent mice is shown. Each dot represents an individual animal. Error bars represent mean ± s.e.m. Statistical analysis was performed using two-way ANOVA followed by Tukey’s multiple comparison’s test.
Extended Data Fig. 3 |. Senescent female Znrf3 cKO adrenal glands activate production of cytokines and growth factors.

(a) URA using RNA-seq data predicts significantly activated and inhibited cytokines in 9-week and (b) week female Znrf3 cKO adrenal tissue compared to 6-week, p < 0.05. (c) URA predicts significantly activated and inhibited growth factors in 9-week and (d) 12-week female Znrf3 cKO adrenal tissue compared to 6-week, p < 0.05. Data is representative of 4 biological replicates per group, statistical analysis in IPA was performed using a right-tailed fishers exact test, p < 0.05.
Extended Data Fig. 4 |. Sex- and stage-specific differences in gene expression in control and Znrf3 cKO adrenals.

Bulk RNA-seq analysis reveals the top DEGs based on (a-c) sex or (d-f) phenotypic stage in control and Znrf3 cKO adrenals. Heatmaps of the top 50 DEGs are shown, statistical analysis was perform using the Wald test, p adj <0.05. Known sex-linked genes were excluded from the analysis.
Extended Data Fig. 5 |. Inflammation and cytokine production in male senescent Znrf3 cKO adrenal glands is further enhanced at 9-weeks.

(a) IPA identifies the most significantly altered canonical pathways in 12- vs 6-week female Znrf3 cKO adrenals. Similar pathways are altered earlier in male Znrf3 cKOs, consistent with the more accelerated phenotype in males. (b) IPA identified the top activated and inhibited cytokines in 9- vs 4-week male Znrf3 cKO adrenals, p < 0.05. (c) GSEA for inflammatory signatures (IL6/JAK/STAT3 signaling, chemokine signaling, the inflammatory response, and the innate immune system) identifies positively enriched genes in 9- vs 4-week male Znrf3 cKO mice. Data is representative of 4 biological replicates per group and, statistical analysis in IPA was performed using a right-tailed fishers exact test, p < 0.05.
Extended Data Fig. 6 |. Androgen deprivation restricts immune infiltration in early and late models of castration in male adrenals.

Markers of (a) neutrophils (Ly6g) and (b) T cells (CD3e) are reduced in castrated male Znrf3 cKO mice compared to sham controls. Scale bars 20μM. Castration was performed at 4-weeks of age and analysis was performed at 9-weeks of age. To further tease apart a role for androgens in the early (cell cycle arrest) versus late (immune) senescent response, (c) male Znrf3 cKO mice were castrated at 6-weeks of age when hyperproliferation has normally already been suppressed. (d-e) At 12-weeks of age, adrenal glands from castrated mice were significantly larger than sham-operated controls. (d) Representative gross histology images. Scale bars 1 mm. (e) Normalized adrenal weight. Relative fold change is indicated below each group. (f) Senescence markers (Ki67, p16, and p21) and (g) myeloid immune markers (CD68, CD11c, CD11b, and F4/80) in adrenals from sham versus castrated animals. Representative images are shown for each group. Scale bars 20 μM. Quantification was performed using QuPath digital image analysis based on the number of positive cells normalized to total nuclei. Each dot represents an individual animal. Box and whisker plots indicate the median (line) within the upper (75%) and lower (25%) quartiles, and whiskers represent the range. Statistical analysis was performed using two-tailed Student’s t-test.
Extended Data Fig. 7 |. Immune cell recruitment in the adrenal gland is sex- and age-dependent.

(a, b) Histological evaluation of female control and Znrf3 cKO adrenal tissue based on H&E. Female Znrf3 cKOs continue to accumulate histiocytes with advanced aging at 44- and 52-weeks of age. (c-d) Male Znrf3 cKOs sustain high levels of histiocytes previously observed as early as 24-weeks of age. Quantification was performed using QuPath digital analysis based on the proportion of histiocyte area normalized to total adrenal cortex area. Scale bars, 100 μm. Data from 4- to 24-weeks of age was previously shown in Fig. 3f-i, and is included as reference. (e-f) In situ validation of myeloid cell accumulation based on IHC for CD68 in control and Znrf3 cKO adrenal tissue from female and (g-h) male cohorts at 44- and 52-weeks of age. Data from 4- to 24-weeks of age was previously shown in Fig. 4e-h, and is included as reference. Quantification was performed using QuPath digital analysis based on the number of positive cells normalized to total nuclei. Each dot represents an individual animal. Box and whisker plots indicate the median (line) within the upper (75%) and lower (25%) quartiles, and whiskers represent the range. Statistical analysis was performed on log2 transformed data using two-way ANOVA followed by Tukey’s multiple comparison’s test. Scale bars, 100 μm. (i) At 78-weeks of age, atrophic Znrf3 cKO adrenal glands have a significantly lower Ki67-index and higher CD68-index compared to age-matched controls. (j) In 78-week-old benign Znrf3 cKO adrenals, nodules have a significantly higher Ki67-index and lower CD68-index compared to the background gland. Each dot represents an individual animal. Box and whisker plots represent mean with variance across quartiles. Statistical analysis was performed using two-tailed Student’s t-test.
Extended Data Fig. 8 |. A low myeloid response score (MRS) is associated with worse patient outcome in female ACC.

(a) Low adrenal myeloid response score (AMRS) is associated with shorter progression-free survival in female TCGA-ACC patients. Statistical analysis was performed using Log-rank Mantel-Cox test.
Supplementary Material
Acknowledgements
We thank H. Clevers and B.-K. Koo for providing Znrf3-floxed mice and the late K. Parker for providing SF1Cre transgenic mice. Research reported in this publication used the High-Throughput Genomics and Bioinformatic Analysis Shared Resource and the Biorepository and Molecular Pathology Shared Resource at the Huntsman Cancer Institute at the University of Utah and was supported by the National Cancer Institute of the National Institutes of Health under award P30CA042014. We also used the University of Utah Flow Cytometry Shared Resource supported by the Office of the Director of the NIH (award S10OD026959 and NCI award 5P30CA042014-24) and the Cell Imaging Core at the University of Utah. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We especially thank J. Marvin, O. Allen and M. Bridge for respective technical assistance with flow cytometry, 10x Genomics library preparation and confocal imaging. We also thank J. Gertz, B. Myers and S. Holmen for helpful scientific discussions and comments on the manuscript. This work was supported by funding from a Cancer Center Support Grant (P30CA040214, K.J.B.), the V Foundation (V2021-021, K.J.B.) and 5 For The Fight (K.J.B.).
Footnotes
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s43587-023-00420-2.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43587-023-00420-2.
References
- 1.National Cancer Institute. Age and Cancer Risk https://www.cancer.gov/about-cancer/causes-prevention/risk/age (2021).
- 2.Siegel RL, Miller KD & Jemal A. Cancer statistics, 2018. CA Cancer J. Clin 68, 7–30 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Rozhok A. & DeGregori J. A generalized theory of age-dependent carcinogenesis. eLife 8, e39950 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laconi E, Marongiu F. & DeGregori J. Cancer as a disease of old age: changing mutational and microenvironmental landscapes. Br. J. Cancer 122, 943–952 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillip JM, Aifuwa I, Walston J. & Wirtz D. The mechanobiology of aging. Annu. Rev. Biomed. Eng 17, 113–141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crona J. & Beuschlein F. Adrenocortical carcinoma—towards genomics guided clinical care. Nat. Rev. Endocrinol 15, 548–560 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Else T. et al. Adrenocortical carcinoma. Endocr. Rev 35, 282–326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flurkey K, Mcurrer J. & Harrison D. Mouse models in aging research. In The Mouse in Biomedical Research Vol. 3, 637–672 (Elsevier, 2007). [Google Scholar]
- 9.Nusse R. & Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Assie G. et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet 46, 607–612 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Zheng S. et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 29, 723–736 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hao HX et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 485, 195–200 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Koo BK et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488, 665–669 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Berthon A. et al. Constitutive β-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum. Mol. Genet 19, 1561–1576 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Heaton JH et al. Progression to adrenocortical tumorigenesis in mice and humans through insulin-like growth factor 2 and β-catenin. Am. J. Pathol 181, 1017–1033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borges KS et al. Wnt/β-catenin activation cooperates with loss of p53 to cause adrenocortical carcinoma in mice. Oncogene 39, 5282–5291 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bingham NC, Verma-Kurvari S, Parada LF & Parker KL Development of a steroidogenic factor 1/Cre transgenic mouse line. Genesis 44, 419–424 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Basham KJ et al. A ZNRF3-dependent Wnt/β-catenin signaling gradient is required for adrenal homeostasis. Genes Dev. 33, 209–220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitani F, Mukai K, Miyamoto H, Suematsu M. & Ishimura Y. Development of functional zonation in the rat adrenal cortex. Endocrinology 140, 3342–3353 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Krämer A, Green J, Pollard J. & Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panier S. & Boulton SJ Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol 15, 7–18 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Gorgoulis V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Muzumdar MD, Tasic B, Miyamichi K, Li L. & Luo L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Coppé J-P et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, e301 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basisty N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiley CD et al. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 16, 1043–1050 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim S-J et al. Endothelial Toll-like receptor 4 maintains lung integrity via epigenetic suppression of p16INK4a. Aging Cell 18, e12914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Limbad C. et al. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 15, e0227887 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jochems F. et al. The Cancer SENESCopedia: a delineation of cancer cell senescence. Cell Rep. 36, 109441 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schiraldi M. et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med 209, 551–563 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lotze MT & Tracey KJ High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol 5, 331–342 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Lau L, Porciuncula A, Yu A, Iwakura Y. & David G. Uncoupling the senescence-associated secretory phenotype from cell cycle exit via interleukin-1 inactivation unveils its protumorigenic role. Mol. Cell. Biol 39, e00586–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galanos P. et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol. 19, 37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y. et al. Senescent mesenchymal stem cells promote colorectal cancer cells growth via galectin-3 expression. Cell Biosci. 5, 21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mosteiro L, Pantoja C, de Martino A. & Serrano M. Senescence promotes in vivo reprogramming through p16INK4a and IL-6. Aging Cell 17, e12711 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coppé J-P, Desprez P-Y, Krtolica A. & Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kale A, Sharma A, Stolzing A, Desprez P-Y & Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 17, 16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naeim F. Histiocytic and dendritic cell disorders. In Hematopathology (eds Naeim F, Nagesh Rao P. & Grody WW) 489–512 (Elsevier, 2008). [Google Scholar]
- 39.Picarsic JL & Chikwava K. Disorders of histiocytes. In Hematopathology 3rd edn (ed. Hsi ED) 567–616 (Elsevier, 2018). [Google Scholar]
- 40.Dale DC, Boxer L. & Liles WC The phagocytes: neutrophils and monocytes. Blood 112, 935–945 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Ekiz HA, Conley CJ, Stephens WZ & O’Connell RM CIPR: a web-based R/shiny app and R package to annotate cell clusters in single cell RNA sequencing experiments. BMC Bioinformatics 21, 191 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evrard M. et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity 48, 364–379 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Volberding PJ et al. Suppressive neutrophils require PIM1 for metabolic fitness and survival during chronic viral infection. Cell Rep. 35, 109160 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC & Schultze JL The myeloid cell compartment—cell by cell. Annu. Rev. Immunol 37, 269–293 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Guilliams M. et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity 45, 669–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H. et al. Role of bone marrow-derived CD11c+ dendritic cells in systolic overload-induced left ventricular inflammation, fibrosis and hypertrophy. Basic Res. Cardiol 112, 25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song P. et al. Hepatic recruitment of CD11b+Ly6C+ inflammatory monocytes promotes hepatic ischemia/reperfusion injury. Int. J. Mol. Med 41, 935–945 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dolfi B. et al. Unravelling the sex-specific diversity and functions of adrenal gland macrophages. Cell Rep. 39, 110949 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sano H. et al. Critical role of galectin-3 in phagocytosis by macrophages. J. Clin. Invest 112, 389–397 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirani N. et al. Target inhibition of galectin-3 by inhaled TD139 in patients with idiopathic pulmonary fibrosis. Eur. Respir. J 57, 2002559 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lindner B, Burkard T. & Schuler M. Phagocytosis assays with different pH-sensitive fluorescent particles and various readouts. BioTechniques 68, 245–250 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Grabek A. et al. The adult adrenal cortex undergoes rapid tissue renewal in a sex-specific manner. Cell Stem Cell 25, 290–296 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Weiss LM Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am. J. Surg. Pathol 8, 163–169 (1984). [DOI] [PubMed] [Google Scholar]
- 55.Weiss LM, Medeiros LJ & Vickery AL Pathologic features of prognostic significance in adrenocortical carcinoma. Am. J. Surg. Pathol 13, 202–206 (1989). [DOI] [PubMed] [Google Scholar]
- 56.Wu C. et al. Myeloid signature reveals immune contexture and predicts the prognosis of hepatocellular carcinoma. J. Clin. Invest 130, 4679–4693 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beuschlein F. et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J. Clin. Endocrinol. Metab 100, 841–849 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Mohan DR et al. Targeted assessment of G0S2 methylation identifies a rapidly recurrent, routinely fatal molecular subtype of adrenocortical carcinoma. Clin. Cancer Res 25, 3276–3288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thorsson V. et al. The immune landscape of cancer. Immunity 48, 812–830 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Landwehr L-S et al. Interplay between glucocorticoids and tumor-infiltrating lymphocytes on the prognosis of adrenocortical carcinoma. J. Immunother. Cancer 8, e000469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hägg S. & Jylhävä J. Sex differences in biological aging with a focus on human studies. eLife 10, e63425 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clocchiatti A, Cora E, Zhang Y. & Dotto GP Sexual dimorphism in cancer. Nat. Rev. Cancer 16, 330–339 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Guan X. et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature 606, 791–796 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roediger J. et al. Supraphysiological androgen levels induce cellular senescence in human prostate cancer cells through the Src–Akt pathway. Mol. Cancer 13, 214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mirzakhani K. et al. The androgen receptor–lncRNA"SAT1–AKT–p15 axis mediates androgen-induced cellular senescence in prostate cancer cells. Oncogene 41, 943–959 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilmouth JJ et al. Sexually dimorphic activation of innate antitumor immunity prevents adrenocortical carcinoma development. Sci. Adv 8, eadd0422 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen H-A et al. Senescence rewires microenvironment sensing to facilitate antitumor immunity. Cancer Discov. 13, 432–453 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marin I. et al. Cellular senescence is immunogenic and promotes antitumor immunity. Cancer Discov. 13, 410–431 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jimenez C. et al. Endocrine and neuroendocrine tumors special issue—checkpoint inhibitors for adrenocortical carcinoma and metastatic pheochromocytoma and paraganglioma: do they work? Cancers 14, 467 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ayers M. et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest 127, 2930–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prat A. et al. Immune-related gene expression profiling after PD-1 blockade in non-small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res. 77, 3540–3550 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Riaz N. et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 11, S27–S31 (2001). [DOI] [PubMed] [Google Scholar]
- 74.Roberson RS, Kussick SJ, Vallieres E, Chen S-YJ & Wu DY Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 65, 2795–2803 (2005). [DOI] [PubMed] [Google Scholar]
- 75.Milanovic M. et al. Senescence-associated reprogramming promotes cancer stemness. Nature 553, 96–100 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Fassnacht M. et al. Combination chemotherapy in advanced adrenocortical carcinoma. N. Engl. J. Med 366, 2189–2197 (2012). [DOI] [PubMed] [Google Scholar]
- 77.Prasanna PG et al. Therapy-induced senescence: opportunities to improve anti-cancer therapy. J. Natl Cancer Inst 113, 1285–1298 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saleh T. et al. Therapy-induced senescence: an ‘old’ friend becomes the enemy. Cancers 12, 822 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Myrianthopoulos V. et al. Senescence and senotherapeutics: a new field in cancer therapy. Pharmacol. Ther 193, 31–49 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Dobin A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 17, 10–12 (2011). [Google Scholar]
- 82.Love MI, Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Korotkevich G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv 10.1101/060012 (2016). [DOI] [Google Scholar]
- 84.Consortium ImmGen et al. The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nat. Commun 12, 2856 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Han X. et al. Construction of a human cell landscape at single-cell level. Nature 581, 303–309 (2020). [DOI] [PubMed] [Google Scholar]
- 86.Zilionis R. et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 50, 1317–1334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing datasets have been deposited to the NCBI Gene Omnibus Database (accession code GSE201127 (GSE201125, bulk RNA-seq; GSE201126, scRNA-seq). Patient data analyzed from TCGA-ACC are publicly available using https://www.cbioportal.org/study/summary?id=acc_tcga_pan_can_atlas_2018. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.