

Abstract
An increasing number of studies have shown that the local release of nitric oxide (NO) from hydrogels stimulates tissue regeneration by modulating cell proliferation, angiogenesis, and inflammation. The potential biomedical uses of NO-releasing hydrogels can be expanded by enabling their application in a fluid state, followed by controlled gelation triggered by an external factor. In this study, we engineered a hydrogel composed of methacrylated hyaluronic acid (HAGMA) and thiolated gelatin (GELSH) with the capacity for in situ photo-cross-linking, coupled with localized NO release. To ensure a gradual and sustained NO release, we charged the hydrogels with poly(L-lactic-co-glycolic acid) (PLGA) nanoparticles functionalized with S-nitrosoglutathione (GSNO), safeguarding SNO group integrity during photo-cross-linking. The formation of thiol–ene bonds via the reaction between GELSH’s thiol groups and HAGMA’s vinyl groups substantially accelerated gelation (by a factor of 6) and increased the elastic modulus of hydrated hydrogels (by 1.9–2.4 times). HAGMA/GELSH hydrogels consistently released NO over a 14 day duration, with the release of NO depending on the hydrogels’ equilibrium swelling degree, determined by the GELSH-to-HAGMA ratio. Biocompatibility assessments confirmed the suitability of these hydrogels for biological applications as they display low cytotoxicity and stimulated fibroblast adhesion and proliferation. In conclusion, in situ photo-crosslinkable HAGMA/GELSH hydrogels, loaded with PLGA-GSNO nanoparticles, present a promising avenue for achieving localized and sustained NO delivery in tissue regeneration applications.
Keywords: hyaluronic acid, gelatin, nitric oxide, hydrogels, in situ cross-linking, tissue regeneration
Graphical Abstract
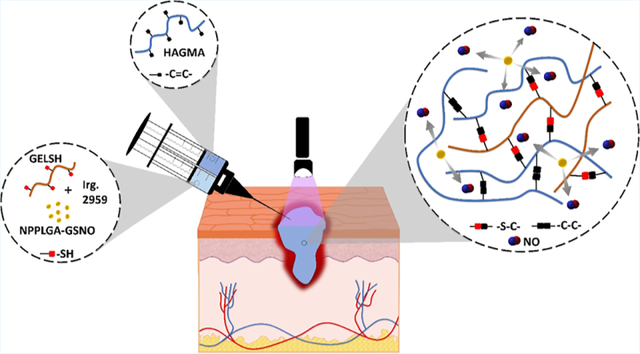
1. INTRODUCTION
Nitric oxide (NO) plays a pivotal role in tissue engineering and regenerative medicine by influencing vasodilation, inflammation, immune responses, angiogenesis, collagen production, and cell behavior. The development of hydrogels capable of releasing NO locally has been reported as an effective strategy for reducing inflammation,2 accelerating wound healing,3,4 increasing dermal vasodilation,2,5 and providing antibacterial action.6,7
Among the chosen polymers employed in developing hydrogels for biomedical applications, hyaluronic acid (HA) has emerged as an appealing choice.8 HA, an anionic biopolymer comprising repeating disaccharide units encompassing d-glucuronic acid and N-acetyl-d-glucosamine, is a key constituent in the extracellular matrix (ECM) of cartilage and skin, possessing the ability to interact with cells through specific receptors, such as CD44, thereby eliciting and regulating cell proliferation. Furthermore, HA exerts its action by diminishing the activity and downregulating the synthesis of pro-inflammatory mediators and matrix metalloproteinases.9–11 Notably, these advantageous effects of HA can be potentiated when combined with other biomacromolecules, such as gelatin (GEL), which enhances cell attachment by virtue of the arginine–glycine–aspartic acid (RGD) sequence inherited from collagen.12,13
However, it is important to acknowledge that natural biopolymers, including HA and GEL, may exhibit a limited half-life in the body due to enzymatic activity. To circumvent this constraint, one of the strategies pursued involves the functionalization of HA and GEL with reactive pendant groups, thereby facilitating their chemical cross-linking through various methodologies.14 The capacity to induce the crosslinking of hydrogels for medical applications via an external stimulus, increases their versatility and efficacy.15 This enables the hydrogel to be administered or injected in a fluid state to fill intricate cavities and subsequently undergo gelation to ensure retention at the application site. Of particular note are hydrogels capable of delivering drugs that can be spontaneously formed in situ.16,17 Among the diverse strategies available for inducing in situ cross-linking and gelation of hydrogel precursor formulations, the use of light-induced photo-cross-linking in the presence of a photoinitiator is especially noteworthy. In this context, methacrylation reactions have been frequently employed to functionalize HA and GEL with vinyl groups.18,19
Within the realm of developing hybrid photo-crosslinkable hydrogels, the strategy of functionalizing one of the formulation components with thiol (SH) groups emerges as a prominent approach. In the presence of thiolated components, the radical reactions initiated by the cleavage of the methacrylate group’s double bonds concurrently result in the formation of thiol–ene bonds, commonly referred to as click reactions, and may substantially accelerate the gelation process.20 In the present study, we developed hybrid hydrogels for in situ photo-cross-linking and localized NO release, based on mixtures of HA and GEL. Due to its cell-promoting properties, anti-inflammatory effects, ECM mimicry, formulation versatility, and its role in wound healing. HA was chosen as the main component of the hybrid hydrogels. In this sense, HA was functionalized with methacrylate groups (HAGMA) to allow it to be photo-cross-linked as a single component, compared with HAGMA hydrogels containing thiolated GEL (GELSH).
To incorporate a NO donor into the HAGMA/GELSH hydrogels, we used S-nitrosoglutathione (GSNO), a primary S-nitrosothiol (RSNO) obtained from the S-nitrosation of glutathione.21 RSNOs act as NO donors through spontaneous dimerization reactions when in an aqueous solution.22 In addition to these reactions, RSNOs themselves, as well as free NO released from them, are also potent antioxidant agents capable of inactivating radical species.23 However, this last action, which can be very beneficial to fight cell damage in oxidative stress conditions, brings a limitation to the use of free RSNOs in photo-cross-linkable hydrogels. Since photo-crosslinking is a process that starts with the formation of free radicals from the photoinitiator and propagates through radical reactions to form the cross-linked network, the presence of free RSNOs or NO leads to the partial inactivation of these radical species, reducing the rate of the cross-linking process of the hydrogel. At the same time, such reactions consume the RSNOs incorporated in the precursor solution, reducing the amount of NO released by the hydrogel and its capability to sustain a prolonged NO release.
To overcome these limitations, we describe herein a novel strategy to prepare photo-cross-linked GSNO-containing HAGMA/GELSH hydrogels capable of releasing NO in a prolonged manner. We based our strategy on the protection of GSNO from the reactions with radical species during the photo-cross-linking process by chemically bonding GSNO to the terminal carboxyl groups of poly(L-lactic-co-glycolic acid) (PLGA) which, in turn, is precipitated as nanoparticles (NPs). In this way, most of the GSNO becomes trapped and protected in the bulk of the NPs. Upon subsequent exposure to the aqueous medium of the hydrogels, the NPs undergo hydration and hydrolysis, allowing the GSNO molecules to dimerize with NO release.
Therefore, the present hydrogels can be utilized across a spectrum of lesion types, facilitating effective coverage and filling of intricate local defects before undergoing photo-crosslinking. These versatile applications have the potential to accelerate wound healing, alleviate inflammation, or elicit microbicidal effects stemming from the biological properties of the NO released in situ. In the context of open wounds, the photo-cross-linking process can be initiated by direct irradiation of the precursor formulation applied to the wound surface. Furthermore, the composition of these hydrogels, comprising two absorbable biopolymers loaded with PLGA NPs (also absorbable), introduces the possibility of leaving them in situ for complete absorption during the tissue regeneration process. This feature may enhance the adaptability and efficacy of these hydrogels in diverse clinical scenarios.
2. MATERIALS AND METHODS
2.1. Materials.
Sodium hyaluronate (Mw 1.8 kDa and 3 MDa) was purchased from Via Farma (São Paulo, SP, Brazil). Type A gelatin, glycidyl methacrylate (GMA), PLGA—acid termination (Mw 7–17 kDa) Resomer RG 502 H, N-acetyl-homocysteine thiolactone (HCT), hyaluronidase from bovine testis type I–S (400–1000 U mg−1), sodium nitrite (NaNO2), reduced l-glutathione, 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959), L-cysteine hydrochloride monohydrate, 5,5′-dithiol-bis(2-acidonitrobenzoic) (Ellman’s Reagent), dimethyl sulfoxide (DMSO), dichloromethane (DCM), thiazolyl blue tetrazolium bromide (98%), potassium chloride (KCl), DAPI Readymade solution, and Triton X-100 were purchased from Sigma-Aldrich. N-Hydroxysuccinimide (NHS) and 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC) were purchased from Acros Organics. Ethylenediaminetetraacetic acid tetrasodium salt dihydrate (EDTA), hydrochloric acid (HCl), acetone, and ethanol were purchased from Synth. Low molar mass poly(vinyl alcohol) (PVA) (98–99% hydrolyzed) was purchased from Alfa Aesar. Calcium and magnesium-free phosphate-buffered saline (PBS) solution (1×) (CMF-PBS) was purchased from Corning Incorporated. Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and penicillin−streptomycin (P/S) were purchased from VWR. NIH/3T3 mouse fibroblast cells (ATCC 1658) for cell compatibility were obtained from American Type Culture Collection. Paraformaldehyde (4%) in PBS was purchased from Santa Cruz Biotechnology. Bovine serum albumin (BSA) was purchased from Proliant Biologicals. Alexa Fluor 488 Phalloidin was purchased from Invitrogen. All reagents were used without prior purification. The experiments were performed using Milli-Q ultrapure water (resistivity, 18.2 MΩ cm at 25 °C).
2.2. HA Methacrylation.
The functionalization of HA with GMA for the insertion of vinyl groups was based on the procedure described by Reis et al.24 Initially, 2 g of HA was solubilized in 200 mL of water at room temperature in a round-bottomed flask. The temperature of the solution was raised to 55 °C and its pH was adjusted to 3.5 by adding HCl (1 mol L−1). Then, 6.48 mL of GMA was added to the flask and the solution was kept under magnetic stirring (1100 rpm) for 24 h at 55 °C. After this period, the solution was cooled to room temperature, transferred to a cellulose acetate dialysis bag (cutoff 12–14 kDa), and dialyzed against water for 72 h, changing the water three times per day. The dialyzed solution was frozen in liquid nitrogen and lyophilized using Benchtop (Virtis) at 21 mT and −38 °C for 48 h. The methacrylated hyaluronic acid obtained was named HAGMA.
2.3. Gelatin Thiolation.
Gelatin thiolation was performed according to the procedure described by Vlierberghe et al.25 Initially, 5 g of gelatin and 50 mL of carbonate buffer solution (pH 10) were added to a round-bottomed flask and left overnight at room temperature for the hydration of gelatin. After this time, the temperature was raised to 40 °C for complete solubilization of the gelatin. Next, 0.015 g of EDTA and 3.81 g of HCT were added. The reaction mixture was kept at 40 °C for 3 h under vigorous magnetic stirring and performed under a nitrogen atmosphere. After this time, the reaction mixture was diluted with the addition of 100 mL of water, dialyzed against water for 48 h at 40 °C, changing the water three times per day, and lyophilized. The thiolated gelatin obtained was named GELSH.
2.4. S-Nitrosoglutathione Synthesis.
S-Nitrosoglutathione (GSNO) was synthesized through the S-nitrosation of glutathione (GSH) with sodium nitrite in an acidic medium according to Vercelino et al.2 Initially, 4.5 g of GSH and 1 g of NaNO2 were solubilized in 23 mL of water containing 1 mL of HCl (37 wt %). The reaction mixture was kept under stirring in an ice bath for 90 s, protected from room light, and then precipitated through the addition of 30 mL of cold acetone. After 5 min, the precipitate was vacuum filtered, washed with 15 mL of cold acetone, and lyophilized for 24 h. The solid dry GSNO was stored at −20 °C in a vial protected from light.
2.5. Functionalization of PLGA with GSNO.
Coupling of PLGA with GSNO was performed according to the procedure described by Lee et al.26 Initially, 2 g of PLGA, 0.452 g of EDC, and 0.268 g of NHS were dissolved in 12 mL of DMSO under magnetic stirring (500 rpm) for 24 h at room temperature. These amounts correspond to a molar excess of NHS over PLGA of 8.6 times. The product formed (PLGA-NHS) was precipitated in 100 mL of water, separated by centrifugation (10,000 rpm), frozen in liquid nitrogen, and lyophilized for 24 h. Next, 1 g of PLGA-NHS and 0.4 g of GSNO were dissolved in 6 mL of DMSO under stirring (500 rpm) for 24 h at room temperature. These amounts correspond to a molar excess of GSNO over PLGA-NHS of 8.8 times. The product formed (PLGA-GSNO) was precipitated in 100 mL of cold water and separated by centrifugation (10,000 rpm) at 10 °C. Excess-free GSNO was removed by washing the solid product twice with cold water during the centrifugation step. Subsequently, the solid PLGA-GSNO was frozen in liquid nitrogen, lyophilized for 24 h, and stored at −20 °C. All the above steps involving GSNO were performed with the reaction flasks protected from room light with aluminum foil.
2.6. Preparation of the PLGA-GSNO Nanoparticles.
The PLGA-GSNO NPs were prepared by the emulsion solvent evaporation method according to Lee et al.26 Initially, 100 mg of PLGA-GSNO was dissolved in 4 mL of DCM and poured into 20 mL of cold 1 wt % aqueous PVA solution. The mixture was sonicated for 90 s using a Sonics Vibra-cell Sonicator VCX 500 instrument at 40% of the maximum amplitude. The dispersion was kept under stirring (600 rpm) in an ice bath under a gentle N2 flow for solvent evaporation. Subsequently, the PLGA-GSNO NP dispersion was centrifuged (10,000 rpm) twice at 10 °C, with the addition of cold water, frozen in liquid nitrogen, and lyophilized. The obtained NPs were named NPPLGA-GSNO.
2.7. Measurement of the GSNO Loading.
The GSNO loading of the NPPLGA-GSNO [L(GSNO)] was calculated by measuring the absorbance of the NPPLGA-GSNO solution at 336 nm, which is the maximum of the characteristic UV absorption band of GSNO in a diode-array UV–vis spectrophotometer (Agilent, model 8453) in triplicate using a 1 cm path-length quartz cuvette. Solutions of NPPLGA-GSNO were prepared by dissolving 5.0 mg of the dry NPPLGA-GSNO in 2 mL of DMSO. The calibration curve was obtained with standard GSNO solutions in DMSO in the concentration range of 0.1–0.7 mmol L−1.
2.8. Preparation of the Photo-Cross-Linked Hydrogels.
To prepare the photo-cross-linkable HA hydrogel, HAGMA (1.5% w/v) was dissolved in a PBS solution (pH 7.4) containing 1 mmol L−1 of EDTA. Then, 200 μL of an ethanolic solution of the photoinitiator Irgacure 2959 was added to the solution to reach a final concentration of 0.5 w/v %. The HAGMA solution was poured into a cylindrical polypropylene mold that was 2 cm high and 1 cm in diameter. It was then irradiated from the top for 10 min using a 3 mW cm−2 power UV source emitting at 365 nm (Lightning Enterprises UV LED Spot Cure System). The resulting photo-cross-linked HA hydrogel was named HHA.
For the preparation of photo-cross-linkable HA hydrogels containing GELSH, aqueous buffered solutions of GELSH (0.5 and 1.0 w/v %) were prepared by dissolving them at 40 °C in a PBS solution containing 1 mmol L−1 of EDTA (pH 7.4). These GELSH solutions were then cooled to room temperature, and HAGMA was added to reach a final concentration of 1.5% w/v. The mixture was stirred magnetically until complete homogenization. Then, 200 μL of an ethanolic solution of the photoinitiator Irgacure 2959 was added to the solution to reach a final concentration of 0.5 w/v %. Subsequently, the combined HAGMA/GELSH solutions were poured into cylindrical polypropylene molds that were 2 cm high and 1 cm in diameter. They were irradiated from the top for 10 min using the previously mentioned light source. The HA photo-cross-linked in the presence of GELSH (0.5 and 1.0 w/v %) was named HAG0.5 and HAG1, respectively.
NPPLGA-GSNO were incorporated into the photo-cross-linkable HHA, HAGE0.5, and HAGE1 hydrogels at a concentration of 20 wt % relative to the dry mass of HAGMA. To achieve this, NPPLGA-GSNO were initially dispersed in the PBS solutions used for dissolving HAGMA and GELSH, as described above. To ensure homogeneous dispersion of NPPLGA-GSNO in the PBS solutions, the solutions were sonicated in an ultrasonic bath for 4 min at room temperature. The resulting HHA, HAG0.5, and HAG1 hydrogels containing NPPLGA-GSNO were named NPHHA, NPHAG0.5, and NPHAG1, respectively.
2.9. Nuclear Magnetic Resonance.
The functionalization of HA was confirmed by 1H NMR. For the NMR analysis, 15 mg of pure and modified polymers were solubilized, separately, in 500 μL of D2O and the spectra were obtained in a Bruker AVANCE 250 spectrometer operating at a frequency of 300.059 MHz. The methacrylation degree (MD) of HAGMA was estimated through integration of the 1H NMR peaks of the vinylic hydrogens of the methacrylate group at 6.17 and 5.74 ppm (I6.17 and I5.74, respectively) and the peaks of the methyl hydrogens of the n-acetyl group of HA at 2.0 ppm (I2.0), and the calculation of their relative intensity was according to eq 1.
| (1) |
2.10. Infrared Spectroscopy.
Attenuated total reflectance Fourier-transformed infrared spectroscopy (ATR-FTIR) was used to confirm the formation of HAGMA, PLGA-GSNO, and GELSH. All the materials were analyzed as dry powders after lyophilization. The ATR-FTIR spectra were acquired in an Agilent Cary 660 spectrometer, in the spectral range of 4000–400 cm−1 with a total of 64 scans/spectrum and 4 cm−1 resolution.
2.11. Raman Spectroscopy.
The presence of thiol groups in the GELSH, as well as the formation of photo-cross-linked HAGMA/GELSH hydrogels, was confirmed by Raman spectroscopy using a Horiba XploRA One 785 nm spectrometer in the spectral range of 4000–400 cm−1 using a 785 nm laser. Each spectrum was obtained with an exposure time of 60 and 2 scans.
2.12. Thiol Quantification.
Ellman’s assay was used to quantify the thiol groups in GELSH. A stock solution was prepared by dissolving 4 mg of Ellman’s reagent into 1 mL of PBS solution (pH 8.0) containing 1 mmol L−1 of EDTA. GELSH solutions were prepared through the dissolution of 30 mg of solid dry GELSH into 30 mL of water at 40 °C under magnetic stirring for 2 h. For the reaction with the Ellman’s reagent, 250 μL of the GELSH solution was mixed with 2.5 mL of PBS solution (pH 8.0) and 50 μL of the Ellman’s stock solution and kept at room temperature for 15 min. After this time, the solution was transferred to a 1 cm path-length quartz cuvette and the absorbance was measured at 412 nm using the above-described UV–vis spectrophotometer, in triplicate. The calibration curve was obtained following a similar procedure, using a buffered cysteine hydrochloride monohydrate solution in the range 1 × 10−4 to 1 × 10−5 mol L−1 as standard.
2.13. Characterization of Nanoparticles.
The morphology of the NPPLGA-GSNO prepared was analyzed by transmission electron cryomicroscopy (cryo-TEM) performed in a Talos Arctica microscope (ThermoFisher) operating at 200 kV. Dispersions of NPs in water containing 5 mg mL−1 were prepared using an ultrasonic bath (Fisherbrand, model CPX1800) for 5 min. The mean diameter of NPPLGA-GSNO was obtained by analyzing 320 NPs in the cryo-TEM micrographs with the Size Meter software. The OriginPro 2018 software was used to plot a histogram of the NP size distribution. The size distribution of PLGA NPs was also analyzed by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS particle size analyzer. The analysis was performed in triplicate at 25 °C with an equilibrium time of 120 s. The average particle size and the polydispersity index were determined simultaneously. The surface charge of the NPPLGA-GSNO was determined by measuring the zeta potential of the particles using the above-described particle size analyzer. For this analysis, 0.4 mg mL−1 of lyophilized NPPLGA-GSNO was dispersed in a 1 mmol L−1 KCl solution by using an ultrasonic bath. The pH of the dispersions was adjusted in the range of 2–10 using HCl and NaOH solutions. Measurements were done in triplicate at 25 °C.
2.14. Rheological Measurements.
The gelation time of the hydrogels was investigated by rheology using a Thermo Scientific HAAKE MARS 40 rheometer equipped with a Thermo Scientific A25 thermostatic bath. A plate–plate (P35) geometry with a 1 mm gap was employed. The tests were carried out at a frequency of 1 Hz and at 37 °C to obtain G′ and G″ as a function of time. The solutions were irradiated through the gap between the parallel plates of the cell with a 3 mW cm−2 power UV source emitting at 365 nm (Lightning Enterprises UV LED Spot Cure System). The gelation time was defined by the crossover of G′ and G″.
2.15. Compression Tests.
Uniaxial compression tests were carried out on a TA.XT plusC texture analyzer (TA Instruments), which was outfitted with a 5 kgf load cell and a 15 mm diameter circular Delrin probe (P/0.5). The analyzer was programed to deform 30% of the hydrogel samples at a speed of 1 mm s−1. Cylinders of hydrogels 1 cm in diameter and 1 cm high photo-cross-linked for 10 min of irradiation were used as specimens for the compression tests. All of the specimens were previously swollen for 24 h in a PBS solution (pH 7.4). The compression tests were performed in triplicate. The elastic modulus of compression was obtained through the slope of the stress–strain curve, according to eq 2 (Young’s equation), where σ is the stress in MPa, is the elastic modulus, and is the strain in absolute value.
| (2) |
2.16. Swelling Measurements.
The water absorption profiles of the photo-cross-linked hydrogels were characterized gravimetrically during their immersion in PBS solution (pH 7.40) at 37 °C in a thermostatic bath until they reached the swelling equilibria. Cylinders of hydrogels 1 cm in diameter and 1 cm high photo-cross-linked for 10 min of irradiation were used as specimens for the swelling measurements. To monitor the water absorption, we removed the hydrogel samples from the PBS solution at predetermined time points over 48 h. Each sample was carefully dried with filter paper to remove excess surface water before weighing. Measurements were performed in quadruplicate. The swelling degree was expressed as the percentage of mass increase according to eq 3, where is the initial mass of the sample and is the mass of the completely swollen sample.
| (3) |
2.17. Enzymatic Degradation.
The degradation of hydrogels was characterized gravimetrically during the 14 days of immersion in PBS solution (pH 7.40) at 37 °C in a thermostatic bath in the presence of hyaluronidase (10 U mL−1). Cylinders of hydrogels 1 cm in diameter and 1 cm high photo-cross-linked for 10 min of irradiation were used as specimens for the degradation measurements. To monitor the mass loss, the freeze-dried hydrogel samples were initially weighted and then immersed in the PBS solution. The samples were removed from the PBS solution at predetermined time points, freeze-dried, and weighed again. Measurements were performed in quadruplicate. The mass loss (ML) was expressed as the percentage of mass loss according to eq 4, where is the initial mass of the dry sample and is the mass of the degraded dry sample at each time .
| (4) |
2.18. Scanning Electron Microscopy.
The morphology of the hydrogels was characterized by SEM using a Quanta 450 FEG (Thermo Fischer Scientific) microscope operating at an acceleration voltage of 10 kV. The hydrogels were frozen in liquid nitrogen and freeze-dried for 24 h before being cryogenically fractured in liquid nitrogen.
2.19. Real-Time NO Release.
The profiles of real-time NO release of the NPPLGA-GSNO were characterized by ozone-based chemiluminescence using a nitric oxide analyzer (Sievers 208i, GE Analytical Instruments) operating at a 6.0 psig oxygen pressure and a cell pressure of 8.0 Torr. For the measurements, 10 mg of NPPLGA-GSNO, were introduced into the reaction flask of the instrument, containing PBS solution (pH 7.4) and 1 mmol L−1 of EDTA, protected from light and maintained at 37 °C.
2.20. Long-Term NO Release.
The long-term NO release profiles from NPHHA, NPHAG0.5, and NPHAG1 were characterized over a 14 day monitoring period under incubation in a PBS solution (pH 7.4) at 37 °C by quantifying the released NO by chemiluminescence. In this case, since NO released in the PBS solution can be oxidized to nitrite and nitrate over extended incubation times, the vanadium chloride (VCl3) method,27 which quantitatively reduces nitrite and nitrate back to NO, was used for NO quantification, using the same above-described NO analyzer (NOA). A saturated solution of VCl3 in 1.0 mol L−1 HCl was used, 5 mL of which was transferred to the NOA reaction flask and maintained at a temperature of 90 °C, with a cell pressure of 7.0 Torr. For incubation, specimens of the hydrogels were immersed in 20 mL of PBS solution contained in a Schott flask kept closed, protected from ambient light with aluminum foil, and immersed in a thermostated bath at 37 °C. At time intervals between 1 and 14 days, aliquots of 100 μL of the PBS solution were removed from the Schott flask and injected into the NOA for triplicate quantification of the cumulative NO released. After each aliquot extraction, the total extracted volume was replaced with fresh PBS solution (pH 7.4) to keep the solution volume of the Schott flask constant. Pure PBS was analyzed as a control. A calibration curve was obtained in the range of 12.5–500 mmol L−1 using a standard sodium nitrite solution.
2.21. 3T3 Mouse Fibroblast Cell Culturing.
Cell cytocompatibility was assessed for the hydrogels using NIH 3T3 mouse fibroblast cells per the ISO 10993–5 standard.28 The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was employed by using the indirect testing method. The cells were cultured on a T75 (75 cm2) flask with complete DMEM (DMEM supplemented with 10% FBS and 1% P/S) at 37 °C and 5% CO2. When the cells were 70% confluent, they were seeded on sterile 96-well plates at a cell seeding density of 5 × 103 cells well−1 to adhere.
2.22. Leachate Preparation.
The hydrogels were previously prepared with the same volume of polymeric solution (250 μL) according to the procedure described in Section 2.8. The control and test groups (n = 4) were first disinfected with 70% ethanol and UV-sterilized for 30 min. The hydrogels were then submerged in complete DMEM for 24 h at 37 °C and 5% CO2 to prepare the leachates. After 24 h, the hydrogels were centrifuged at 500 g for 10 min. The hydrogels were then removed, and the leachates were diluted (10%) and used for cytocompatibility studies.
2.23. Cytocompatibility of the Hydrogels.
MTT stock reagent (5 mg mL−1) was prepared by dissolving and sterile filtering thiazolyl blue tetrazolium bromide in CMF-PBS. After 24 h of cell exposure to the leachate dilutions, the leachates were removed, and diluted MTT reagent (0.5 mg mL−1) was exposed to the cells for 2.5 h at 37 °C and 5% CO2. After incubation, the MTT reagent was removed from the wells; the formazan crystals present were redissolved in DMSO. The absorbance was read with a BioTek (Winooski, VT) plate reader at 570 and 690 nm and translated into a relative percent viability of the fibroblast cells (eq 5).
| (5) |
2.24. Cell Adhesion.
The hydrogels’ ability to promote cell adhesion was assessed by direct cell seeding onto the hydrogels. The materials, described in Section 2.8, were formed (n = 4) directly on 8-well culture sides, using the same volume (150 μL), and irradiated for 5 min. NIH 3T3 mouse fibroblast cells were initially cultured on a T75 (75 cm2) flask with complete DMEM (supplemented with 10% FBS and 1% P/S) at 37 °C and 5% CO2. When the cells were 70% confluent, they were harvested and seeded on UV-sterilized (30 min) hydrogels at a cell seeding density of 2 × 104 cells well−1 to adhere. The seeded hydrogels were incubated at 37 °C and 5% CO2 for 24 h. After allowing cells to adhere to the hydrogels for 24 h, the remaining cells in suspension were counted, and the number of cells adhered to the hydrogels was calculated. The cell suspension from each hydrogel was transferred from the well into a microcentrifuge tube and centrifuged at 500g for 5 min. The supernatant was then removed, and the cell pellet was resuspended in CMF-PBS. Equal parts of the cell suspension and 0.4 % Trypan blue were mixed and counted on a hemocytometer. The number of cells adhered to the hydrogels was calculated according to eq 6.
| (6) |
2.25. Staining and Fluorescent Imaging of Adhered Cells.
After the initial 24 h of incubation, the hydrogels with cells adhered were incubated with fresh complete DMEM for another 48 h to allow for the cells to proliferate and spread. At 72 h, the medium was removed, and the cells were fixed with 4% paraformaldehyde for 1 h. Once the cells were fixed and rinsed with CMF-PBS, they were permeabilized with 0.1% (v/v) Triton X-100, followed by treatment with 10% (w/v) BSA to prevent indiscriminate staining of hydrogel components in succession. Then, the cell cytoskeleton (F-actin) was stained with 0.165 μM Alexa Fluor 488 conjugated Phalloidin, while the cell nucleus was stained with 1μg/mL DAPI, in succession, and protected from light. The cells were imaged with an Advanced Microscopy Group’s EVOS FL Fluorescence Imaging Microscope (AMG, Mill Creek, WA).
2.26. Statistical Analysis.
Analyses were performed in triplicate, except for the swelling and in vitro degradation tests which were performed in quadruplicate and are expressed such as mean ± standard deviation. The calculated standard deviation is represented by error bars. A standard Student’s t-test was used to determine statistical significance—*P < 0.05.
3. RESULTS AND DISCUSSION
3.1. HA Methacrylation.
Figures 1a and S1 show the 1H NMR spectra of HA and HAGMA. The HA spectrum shows a signal at 2.0 ppm, assigned to the proton of the methyl group of the N-acetylglucosamine unit, and an envelope of signals between 3.3 and 5.4 ppm, which are assigned to the hydrogens of the saccharide units of HA.29 The HAGMA spectrum shows additional signals at 5.74 and 6.17 ppm (inset) characteristic of vinylic hydrogens, as well as at 1.93 ppm (proton of the methyl group of GMA), and between 3.5 and 4.3 ppm (protons of the methylene group of the glycidyl spacer).30 The presence of these additional peaks confirms the methacrylation of HA through the ring-opening reaction of GMA, according to the reaction shown in Figure S2a.31 Under the experimental conditions used, an estimated MD of HA of 75% was obtained (eq 1). Additional evidence of HA methacrylation is shown in the comparison among the FTIR spectra of HA, GMA, and HAGMA (Figure S3a,b).
Figure 1.
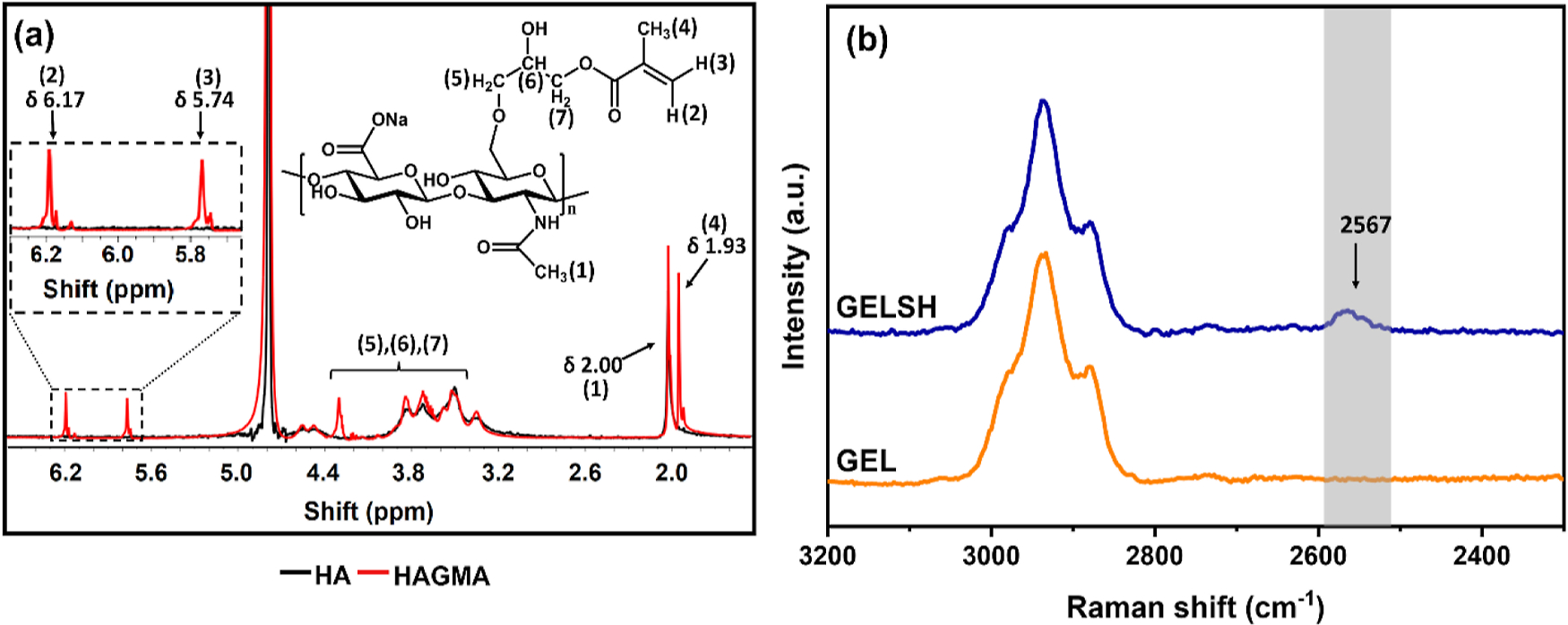
(a) 1H NMR spectra of HA and HAGMA in D2O. (b) Raman spectra of GEL and GELSH.
3.2. Gelatin Thiolation.
The functionalization of GEL through the formation of a secondary amide generated by the reaction of primary amines of the GEL and the carboxylic acid of HCT,25 led to the formation of GELSH, according to the reaction shown in Figure S2b, which was characterized by the Ellman assay. Quantification of the thiol groups by this method allowed estimating an extent of thiolation of the GEL of 24 wt %. Although the comparison between the FTIR-ATR spectra of GEL and GELSH shows no changes that can be attributed to the functionalization of GEL with HCT (the characteristic −SH band at 2567 cm−1 is too weak to be detected) (Figure S3c), the formation of GELSH could be confirmed by Raman spectroscopy through the detection of the characteristic −SH group band at 2567 cm−1 in the spectrum of GELSH (Figure 1b).32
3.3. PLGA-GSNO Nanoparticles.
GSNO was coupled to PLGA through the formation of an amide bond between the primary amine of GSNO and the terminal carboxyl of PLGA in a carbodiimide-mediated reaction (Figure 2a I and II). Figure 2b shows the ATR-FTIR spectra of pure PLGA and PLGA-GSNO. Both spectra display the characteristic bands of PLGA at 2996 and 2949 cm−1 (C−H stretching of CH2 and CH3, respectively); 1748 cm−1 (CO stretching of esters) and 1168 and 1083 cm−1 (axial and angular C−O deformation).32 The new weak band that appeared at 1532 cm−1 in the spectrum of PLGA-GSNO can be assigned to the angular deformation of secondary amides,32 confirming the formation of PLGA-GSNO.
Figure 2.
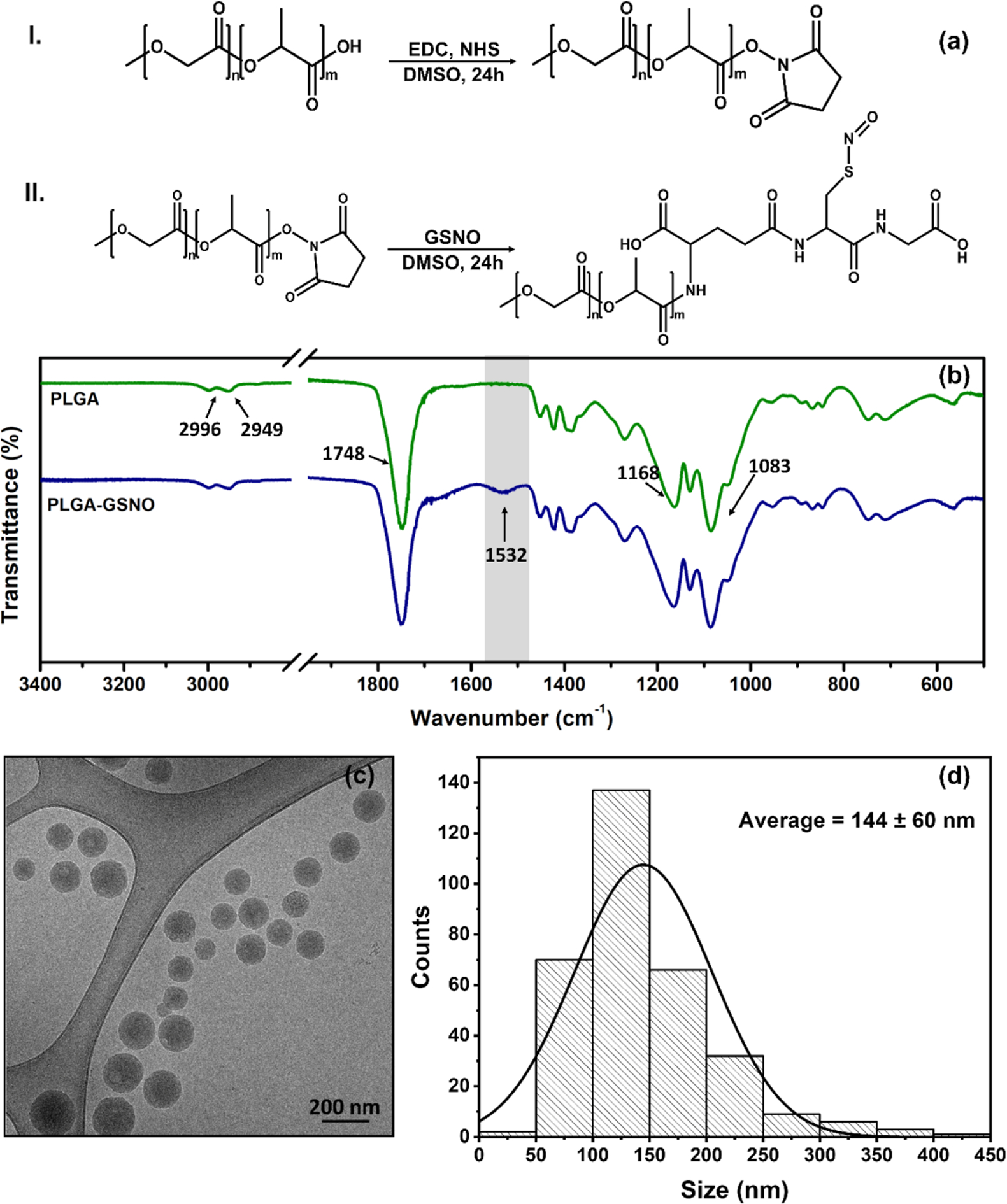
(a) Scheme of the coupling reaction of PLGA with GSNO mediated by EDC. The carbonyl is activated in step I, and the intermediate species is coupled with GSNO in step II. (b) FTIR-ATR spectra of PLGA and PLGA-GSNO. (c) Representative cryo-TEM image of PLGA-GSNO NPs dispersed in ultrapure water. (d) Size distribution of the PLGA-GSNO NPs based on cryo-TEM images.
PLGA-GSNO nanoparticles (NPPLGA-GSNO) were prepared by emulsion formation followed by solvent evaporation. As shown in the cryo-TEM image of Figure 2c, the NPPLGA-GSNO are spherical and well dispersed in water. The size distribution of NPPLGA-GSNO is shown in the bar graph of Figure 2d, from which an average diameter of 144 ± 60 nm can be estimated. This estimate is in agreement with the order of magnitude of the average hydrodynamic diameter of the NPPLGA-GSNO (230 ± 89 nm) determined by DLS measurements (Figure S4a). It should be noted that the PLGA used in this study has terminal carboxylic groups. Assuming a reaction under stoichiometric conditions, these groups (that participate in the protonation–deprotonation equilibrium, affecting the charge of the nanoparticles), are completely consumed in the GSNO functionalization, according to the reactions schematized in Figure 2a I and II. The remaining protonable groups, which then determine the net electric charge that the NPPLGA-GSNO can assume, are the carboxylate groups present on the glutamate residue (pKa = 2.1) and glycine residue (pKa = 3.5) of GSNO.2 Therefore, it can be expected that below pH 2.1, NPPLGA-GSNO should show a net charge close to zero.
With increasing pH, the gradual deprotonation of the two – COOH groups from the glutamate and glycine residues starts to impart a negative net charge to the NPPLGA-GSNO. This prediction agrees with the results of Figure S4b, which show that, starting at pH ca. 3.0, the NPPLGA-GSNO show a negative zeta potential, which stabilizes above pH 6.0, indicating the presence of four negative charges per molecule of PLGA-GSNO (two at each end of the chain). Therefore, the good dispersion of the NPPLGA-GSNO, with no aggregation after their redispersion in water, as shown in Figure 2c, can be attributed to the negative surface charge. Using spectrophotometric measurements based on the absorption band of GSNO at 336 nm, the molar amount of GSNO ( transition)33 coupled to the PLGA nanoparticles was estimated to be 0.104 mol mg−1 (Figure S4c,d).
3.4. Hydrogel Formation.
HHA, HAG0.5, HAG1, NPHHA, NPHAG0.5, and NPHAG1 were formed through photo-cross-linking reactions under irradiation in the presence of the photoinitiator. In the case of HHA, only the photo-cross-linking reaction between carbon radicals formed in the cleavage of the C═C bonds of HAGMA is involved (Figure S5a), while in the case of HAG0.5 and HAG1, a parallel reaction between the carbon radicals formed in the cleavage of the C═C bonds and thiol groups takes place and leads to the formation of thiol–ene bonds (Figure S5b).34,35 In the last case, hydrogen abstraction from the thiol group by a radical species leads to the formation of a thiyl radical (RS•) that attacks the double bond (−C═C−), establishing a C−S bond. The thiol–ene reactions are known as click reactions and have the advantages of being considerably faster than the reaction between two carbon-centered radicals, and less susceptible to inhibition by oxygen (O2).36 These two parallel reactions also take place in the presence of NPPLGA-GSNO. The two reactions involved in the formation of the hydrogels, along with photographs of the photo-cross-linked hydrogels after their removal from a cylindrical mold, are shown in Figure 3a–c.
Figure 3.
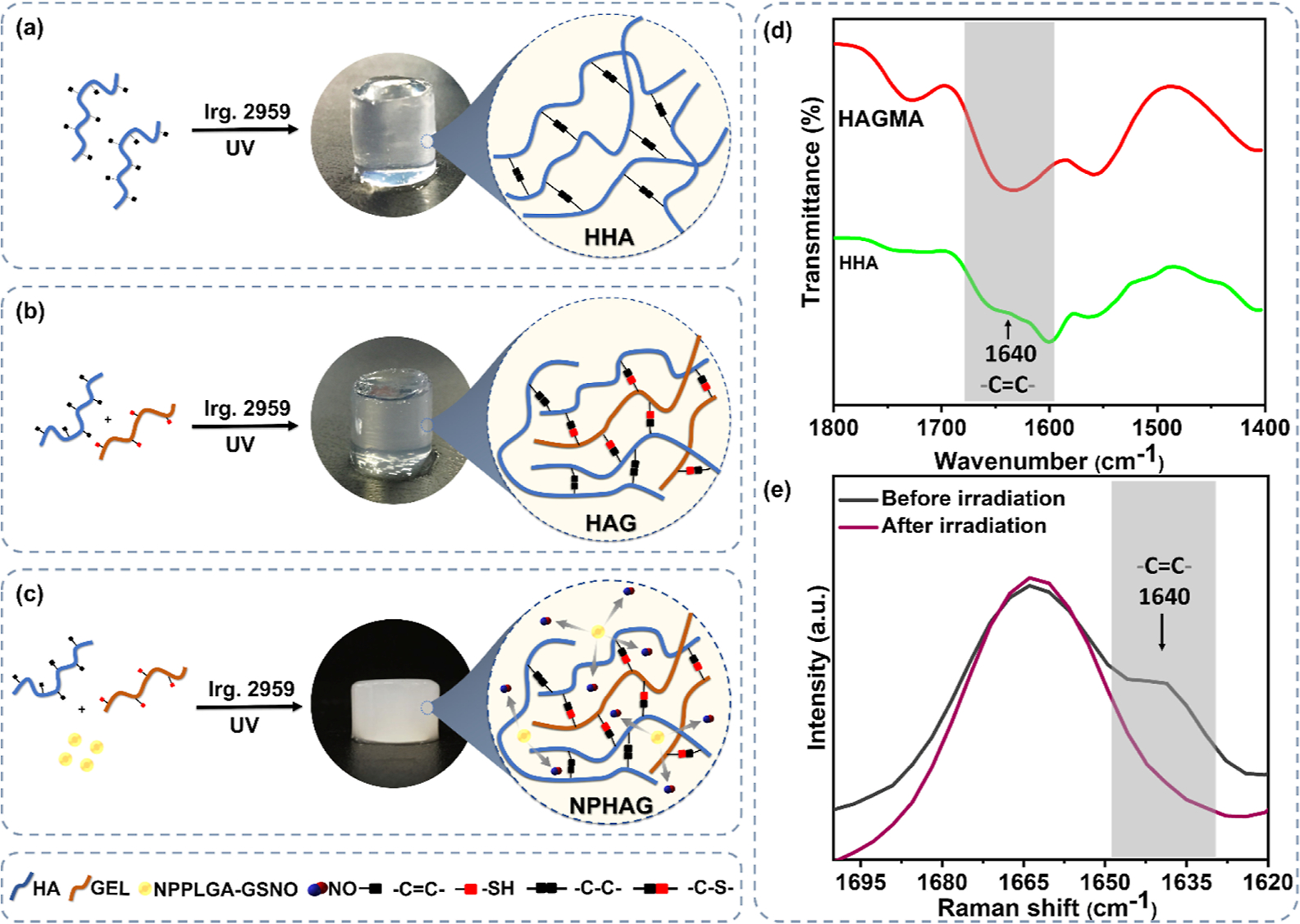
Schematic representation of the photo-cross-linking reactions between the vinyl groups in the (a) formation of HHA, and between the vinyl and SH groups in the formation of HAG in the (b) absence and (c) presence of NPPLGA-GSNO. (d) FTIR spectra of HAGMA and HHA and (e) Raman spectra of HAG1, highlighting the reduction or full disappearance of the band assigned to the vinyl group at 1640 cm−1 after photo-cross-linking.
It should be noted that while the HHA and HAG are completely transparent, the NPHAG is turbid. This turbidity can be attributed to the light scattering caused by NPPLGA-GSNO. The formation of HHA and HAG1 was confirmed by the disappearance of the band assigned to the vinyl group from HAGMA at 1640 cm−1 in the FTIR (Figure 3d) and Raman (Figure 3e) spectra, respectively.37 Based on the special biological properties of HA and its relevance in biomedical applications,8 we decided to keep the HA concentration at 1.5 wt % in HAG formulations while keeping the GELSH concentrations below 1.5 wt % so that HA is always the major component.
3.5. Rheological Measurements.
Rheological measurements were performed to characterize the gelation process of HA promoted by the photo-cross-linking reactions in the absence and presence of GELSH and NPPLGA-GSNO. Figure 4a shows representative photographs of the sol−gel transition of the HHA hydrogel when irradiated with ultraviolet light and removed from the tube. Figure 4b–d shows the curves of the elastic or storage (G′) and viscous or loss (G″) modules as a function of the irradiation time during the sol−gel transition of HHA and NPHHA (Figure 4b), HAG0.5 and NPHAG0.5 (Figure 4c), and HAG1 and NPHAG1 (Figure 4d), promoted by photo-cross-linking. In all cases, the materials are initially fluid solutions with liquid-like behavior and have G″ higher than G′. With the progression of the photo-cross-linking process, G′ increases sharply, becoming higher than G″ and the fluid solutions acquire a solid-like behavior. As the photo-cross-linking process is completed the curves tend toward a plateau. The gelation times, obtained from the point at which the G′ and G″ curves intersect, are 334 ± 19 s for HHA, 433 ± 70 s for NPHHA, 72 ± 11 s for HAG0.5, 76 ± 13 s for NPHAG0.5, 73 ± 3 s for HAG1, and 75 ± 6 s for NPHAG1 (Figure 4e).
Figure 4.
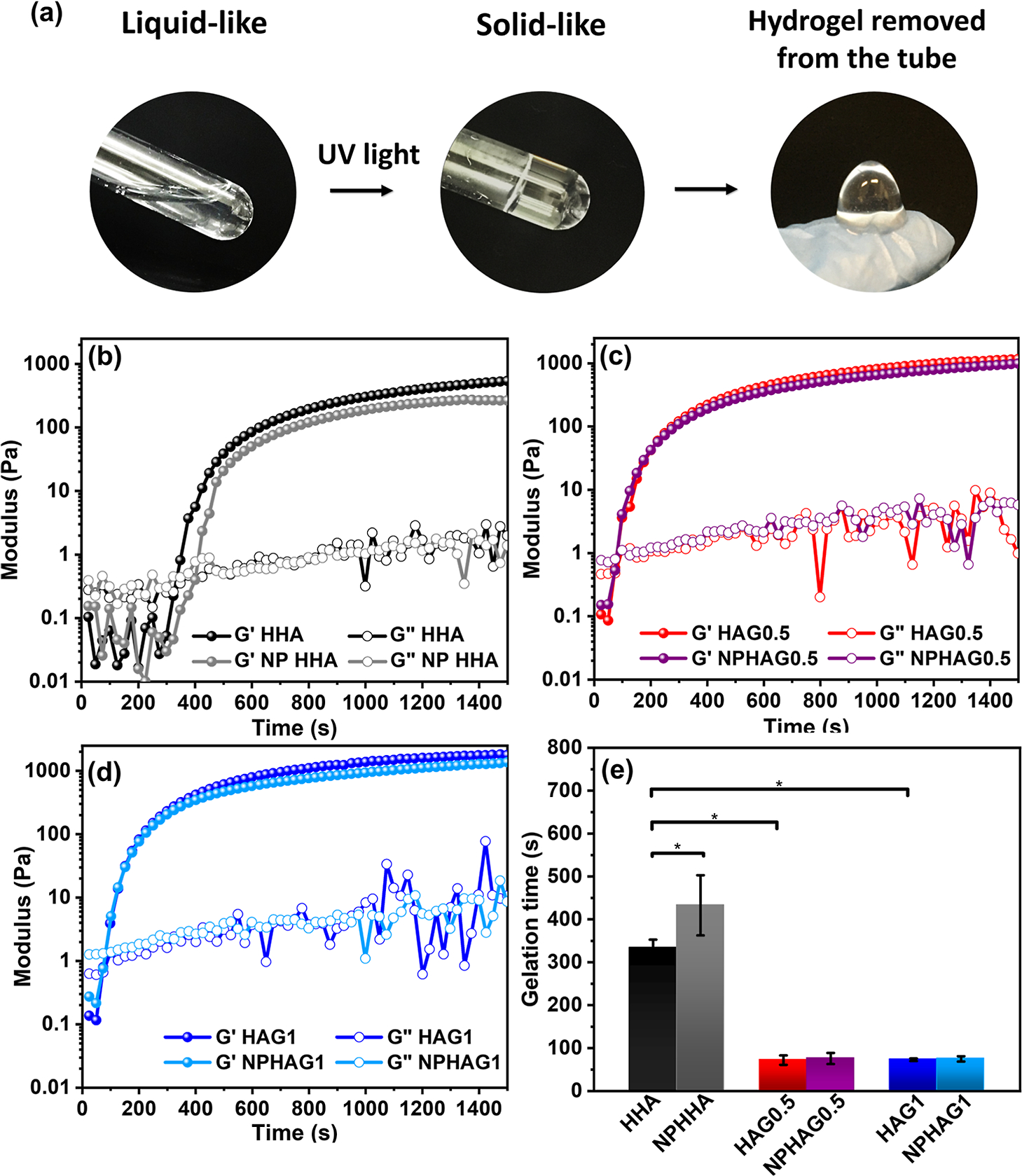
(a) Representative photographs of the sol−gel transition of HAGMA inside a test tube and of formed HHA after its removal from the tube. Elastic (G′) and viscous (G″) modulus as a function of time of UV light irradiation of the (b) HHA and NPHHA, (c) HAG0.5 and NPHAG0.5, and (d) HAG1 and NPHAG1. (e) Gelation time of the hydrogels obtained at the crossing point of modules G′ and G″.
First, the presence of GELSH accelerates the gelation process, making the gelation time 4.6 times shorter than the gelation time of HHA (there was no statistically significant difference between the gelation times of HAG0.5 and HAG1). This reduction in gelation time can be attributed to the lower activation energy for hydrogen abstraction from the thiol group of GELSH. While the −C═C− bond energy is 728 kJ mol−1,38 the S−H bond energy is 368 kJ mol−1.36 Therefore, the presence of GELSH along with HAGMA increases the reactivity of the system as a whole and accelerates the gelation process, as already reported elsewhere.36 This is a desirable result for the proposed application of the present NO-releasing HA/GEL for tissue regeneration since the in situ irradiation time should be as short as possible.
Second, the increase in the gelation time of HHA in the presence of NPPLGA-GSNO can be attributed to the photochemical release of NO from GSNO exposed on the surface of NPPLGA-GSNO and consequent partial suppression of the photo-cross-linking process. To check this assumption, we performed rheological monitoring of HHA gelation in the presence of free GSNO. The modulus vs irradiation time curves shown in Figure S6a confirmed that the presence of unprotected GSNO leads to a 4-fold increase (from 334 to 1240 s) in the gelation time of HHA, as expected for the well-known effect of RSNOs in the inactivation of radical species.23 The absence of this effect in HAG0.5 and HAG1 can be attributed to the formation of thiol–ene bonds in these hydrogels, whose acceleration of the photo-crosslinking process overcomes the effect of the suppression of photo-cross-linking by the NO released from GSNO. Moreover, the presence of NPPLGA-GSNO led to a statistically significant reduction of 26 and 28% in the elastic modulus G′ of the HAG0.5 and HAG1, respectively (Figure S6b). This trend for the hydrogels containing NPPLGA-GSNO suggests that this effect is due to the light scattering caused by NPPLGA-GSNO.
It can also be seen in Figure S6b that HAG0.5, NPHAG0.5, HAG1, and NPHAG1 have G′ values greater than the modules of HHA and NPHHA. This result agrees with the expected mechanical strengthening in the structure of the hydrogels as a result of the formation of thiol–ene bonds in the presence of GELSH. This hypothesis was confirmed by comparing the rheological properties of HAG1 and a physical mixture of HAGMA and nonthiolated gelatin, named HHA/GEL, at the same mass ratio as used for HAG1 (Figure S6c). As can be seen, the gelation time of HHA/GEL is about 4 times longer than that of HAG1, which clearly demonstrates the contribution of the thiol–ene bond formation. This conclusion is reinforced by similarity between the rheological properties of HHA and HHA/GEL, where in both cases only cross-linking between the vinyl groups is involved (Figure S6d).
3.6. Mechanical Properties.
Figure 5a,b shows the stress–strain curves and corresponding elastic moduli of HHA, NPHHA, HAG0.5, NPHAG0.5, HAG1, and NPHAG1. First, the addition of GELSH led to a 1.7- and 4.0-fold increase in the elastic modulus of HAG0.5 and HAG1, respectively, compared to HHA, in accordance with the increase in the elastic modulus described above due to the presence of GELSH and assigned to the formation of thiol–ene bonds in the photo-cross-linking process. Second, in all cases, the presence of the NPPLGA-GSNO led to a 3.8-, 6.8-, and 8.6-fold increase in the elastic moduli of NPHHA (8.3 kPa), NPHAG0.5 (15 kP), and NPHAG1 (19 kPa), respectively, compared to HHA (2.2 kPa).
Figure 5.
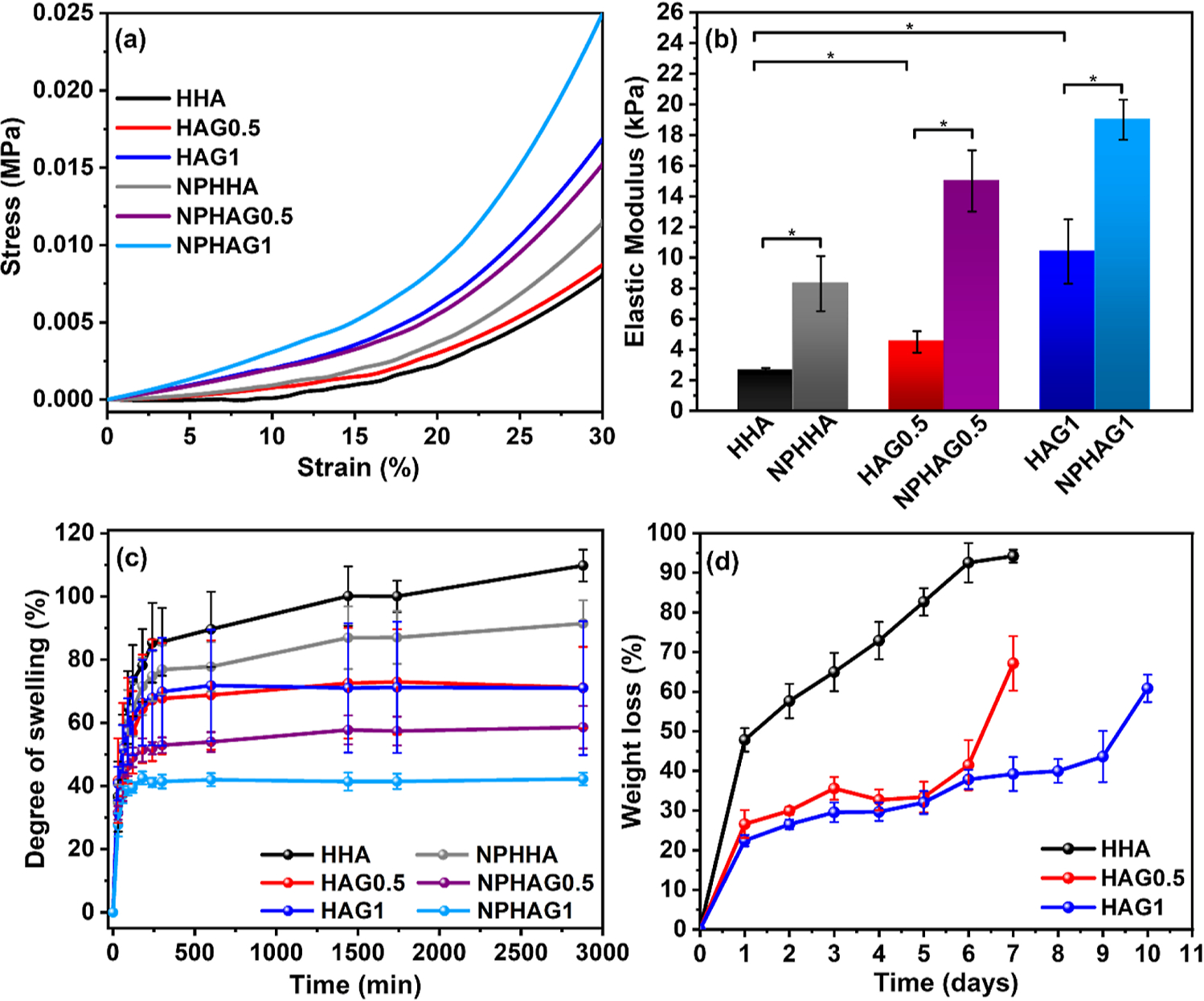
(a) Stress–strain curves of HHA, NPHHA, HAG0.5, NPHAG0.5, HAG1, and NPHAG1 (b) elastic modulus extracted from the curves of (a). (c) Swelling curves of HHA, NPHHA, HAG0.5, NPHAG0.5, HAG1, and NPHAG1 in PBS solution (pH 7.4). (d) ML of HHA, HAG0.5, and HAG1 during the in vitro degradation assay in the presence of hyaluronidase.
Although this increase in the compression modulus of the hydrogels with the introduction of NPPLGA-GSNO does not reflect the reduction in elastic modulus measured by rheology for NPHHA, NPHAG0.5, and NPHAG1 compared to HHA (Figure S6b), it should be noted that the two techniques used measure different mechanical responses.
The elastic modulus of hydrogels used in tissue regeneration strongly influences cell behavior through the mechanotransduction process that operates when cells come into contact with the material. Thus, based on the similarity of elastic moduli, it can be considered that the present hydrogels have the greatest potential for guiding tissue regeneration in the kidney (5–10 kPa), brain (1–4 kPa), heart (10–15 kPa), intestine (20–40 kPa), and nucleus pulposus of intervertebral disks.19,39
3.7. Swelling and Degradation Behavior.
The water absorption capacity of the present hydrogels is essential for their proposed biomedical applications since the penetration of biological fluids in the freshly formed hydrogels favors the infiltration and seeding of cells.40 The swelling behavior of HHA, NPHHA, HAG0.5, NPHAG0.5, HAG1, and NPHAG1 in PBS solution (pH 7.4), over 10 h, after their formation by photo-cross-linking is shown in Figure 5c. The presence of GELSH in the formulations led to a decrease in the swelling degree of HHA, which is directly proportional to the GELSH amount. When comparing the hydrogels with and without NPPLGA-GSNO, it can be noted that the incorporation of NPs led to an additional reduction in swelling degree. Although this result may have a contribution to the known hydrophobicity of PLGA in the NPPLGA-GSNO,41 it is worth considering that it is more likely a result of the grafting of the NPPLGA-GSNO to the photo-cross-linked HA/GEL network. This grafting is in fact expected to occur through the establishment of thiol–ene bonds between carbon radicals HAGMA and thiyl radicals formed in the photoejection of NO from GSNO, leading to an increase in the cross-linking degree and a consequent decrease in the swelling degree.
Ideally, the rate of degradation of prosthetic hydrogels used for tissue repair should match the rate of the formation of new tissue. To evaluate the degradability of the present hydrogels under enzymatic action, HHA, HAG0.5, and HAG1 were subjected to in vitro degradation assays which were carried out in phosphate buffer (pH 7.4) in the presence of hyaluronidase over 10 days. Hyaluronidase degrades HA by cleaving the β 1,4 glycosidic bond.42 Figure 5d shows that HHA lost 48 ± 3% of its initial mass on first day of testing in the presence of the enzyme and was completely degraded after the eighth day. The degradation rate, on the other hand, decreased with the addition and increasing concentration of GELSH. HAG0.5 retained approximately 58% of its initial mass until the sixth day of incubation. On the tenth day, HAG1 had the lowest observed degradation rate, losing 61 ± 3.5% of its mass. This difference in degradation rate can be attributed to hyaluronidase’s decreased accessibility to HA cleavage sites with the addition of GELSH. It must be noted that the hyaluronidase concentration used in the in vitro assay was 10 U mL−1, which is much higher than the concentration of hyaluronidase present in the human body, which can range from 2.8 × 10−6 to 3.8 × 10−3 U mL−1 in plasma, for example.20 Therefore, HHA, HAG0.5, and HAG1 are expected to have a longer residence time in the human body, preserving their three-dimensional structure.
3.8. Morphological Analysis.
SEM was used to examine the morphology of hydrogels prepared with and without NPPLGA-GSNO. The dry hydrogels displayed a honeycomb morphology with interconnected pores, as shown in the micrographs of Figures 6a,b and S7a–f. These micrographs show no evidence of phase separation between HAGMA and GELSH. The pore size distribution ranged from 32 to 56 μm. This porosity can help the transport of nutrients and gases, as well as provide a large surface area for cell adhesion and migration.18
Figure 6.

SEM micrographs of (a) HAG0.5 and (b) NPHAG0.5. The magnifications show details of the morphology of the pore structure and reveal the presence of embedded PLGA-GSNO nanoparticles protruding from the surface of the pore walls (b).
Although the incorporation of NPPLGA-GSNO did not affect the pore morphology, the NPPLGA-GSNO changed the texture of the surface of the pores, where they can be clearly seen underneath the pore walls (Figure 6a,b).
3.9. NO Release.
Figure 7a shows a representative real-time NO release curve, obtained after adding NPPLGA-GSNO in PBS solution (pH 7.40) at 37 °C. The NO signal that appears immediately after the introduction of the samples into the reaction flask of the NO analyzer can be assigned to the thermal dimerization of the terminal GSNO molecules at the water-NPPLGA-GSNO interface as the nanoparticles swell, and water starts penetrating the NPPLGA-GSNO, providing mobility to the terminal GSNO molecules. A proof of concept that GSNO is the NO source is shown in Figure 7b, based on a demonstration of its photochemical NO release. As the hydration of the NPPLGA-GSNO becomes progressively slower, depending on the hydration rate, as well as on the hydrolysis of the NPPLGA-GSNO, the NO signal decays slowly, reaching the baseline after ca. 35 h, as can be seen in the inset of Figure 7a. In this analysis, it should be considered that this release profile may be governed by the hydrolysis rate of PLGA, which is relatively high.43 The corresponding cumulative NO release curve from NPPLGA-GSNO is shown in Figure 7c. The slope of the curve of Figure 7c allows us to estimate a rate of NO release of 1.23 pmol mg−1 min−1.
Figure 7.
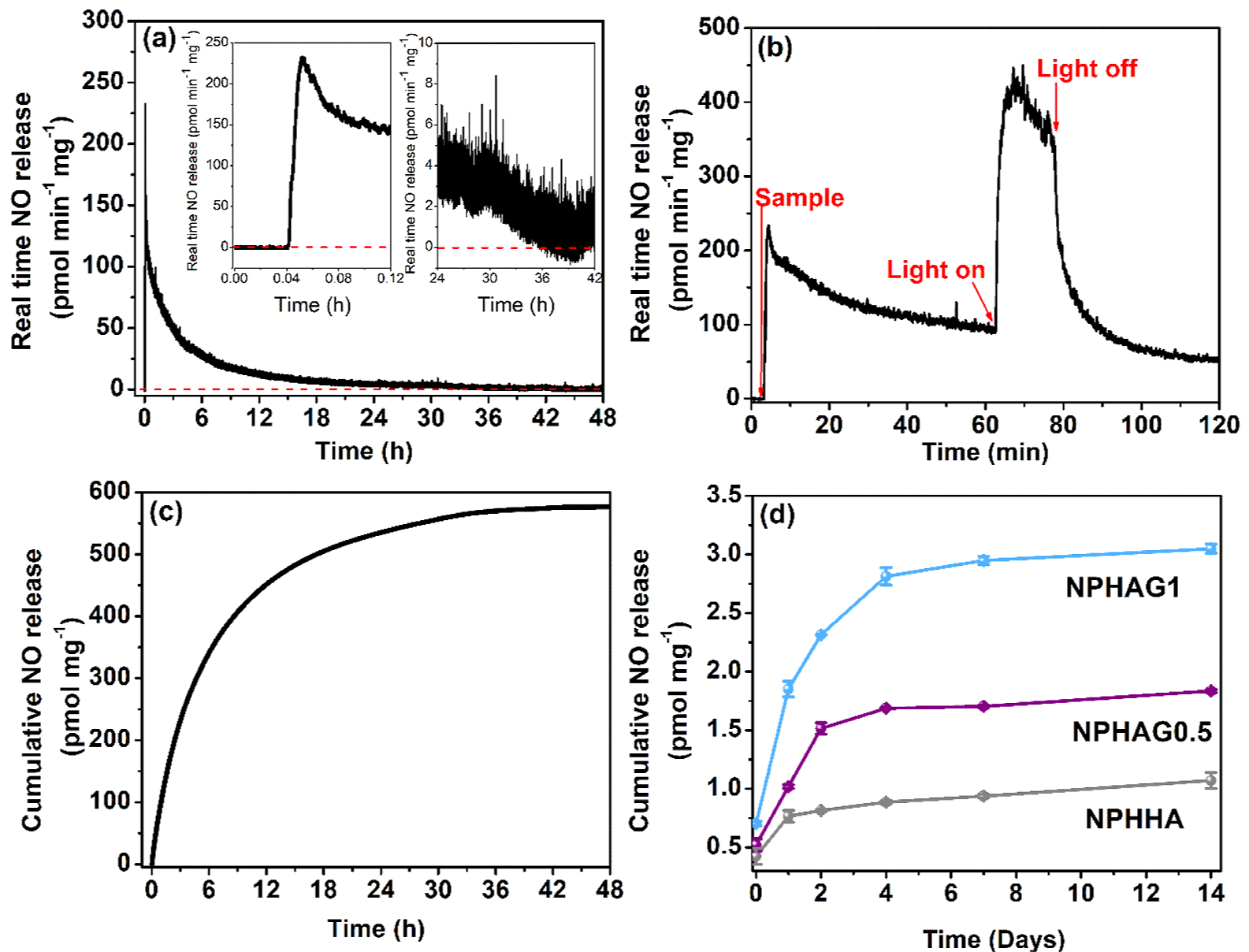
(a) Real-time NO release from NPPLGA-GSNO analyzed for 48 h in PBS at 37 °C. (b) Proof of concept for the NO release from NPPLGA-GSNO suspended in a PBS solution (pH 7.40) at 37 °C. (c) Cumulative release of NO from NPPLGA-GSNO analyzed over 48 h. (d) Cumulative release of NO from NPHHA, NPHAG0.5, and NPHAG1.
Figure 7d shows the long-term NO release profiles of NPHHA, NPHAG0.5, and NPHAG1 over a 14 day monitoring period under incubation in PBS solution (pH 7.4) at 37 °C. In these experiments, the cumulative NO released from the hydrogels was quantified after 1, 2, 4, 7, and 14 days of incubation, by reducing the NO byproducts, such as nitrite (NO2−) and nitrate (NO3−) back to NO with vanadium chloride.27,44 Figure 7d unveils two noteworthy findings. First, the release of NO from NPPLGA-GSNO incorporated into cross-linked HAG matrices exhibits a significantly prolonged profile (extending up to 14 days) when compared to the NO release from NPPLGA-GSNO directly added to a PBS solution under identical conditions (with a release duration of up to 35 h). Second, it is evident that hydrogels subjected to photo-cross-linking in the presence of GELSH (specifically, NPHAG0.5 and NPHAG1) released significantly higher quantities of NO. Furthermore, the NO release amounts are directly proportional to the mass proportion of GELSH used in the hydrogel formulation. This outcome can be comprehended by considering a well-established phenomenon: the hydrolysis of PLGA (and polyesters in general) is catalyzed by the localized acidification generated during hydrolysis.45 This catalytic effect is particularly pronounced when the free acids produced during hydrolysis are not promptly removed from the polymer–solution interface through diffusion or washing.
In the case of the NPHAG0.5 and NPHAG1, it is logical to expect that as the equilibrium swelling degree of the hydrogels decreases, there is an increase in diffusion constraints. This leads to higher concentrations of free acids accumulating in close proximity to the NPPLGA-GSNO. Consequently, the autocatalytic acceleration of NPPLGA-GSNO hydrolysis increases the rate of NO release. This phenomenon arises from the mobilization of PLGA-GSNO oligomers formed at the interface of NPPLGA-GSNO, facilitating bimolecular reactions between terminal GSNO moieties. These reactions result in the formation of S−S bonds and the consequent release of free NO. This interpretation is further supported by the observation that the equilibrium swelling degrees of the three NO-releasing hydrogels follow the sequence: NPHHA > NPHAG0.5 > NPHAG1 (as shown in Figure 5c), which directly corresponds to the increasing amounts of NO released at each time point in Figure 7d. Ultimately, the results depicted in Figure 7d illustrate that the quantities of NO released from these hydrogels can be precisely controlled by varying the presence and amount of GELSH in the formulations.
In this study, we maintained a constant NPPLGA-GSNO load (20% by mass relative to the dry mass of HA) across all three hydrogel compositions examined. However, it is worth noting that the release of NO from each of these hydrogels might also be effectively controlled by incorporating varying amounts of NPPLGA-GSNO, either lower or higher than the fixed concentration used.
Controlling the rate of NO release from biomaterials is fundamental for allowing their biomedical applications since the actions of NO depend strongly on its local concentration in the tissues.46 When a tissue is damaged, the inducible form of NO synthase (iNOS) produces a high level of NO (local NO concentration >800 nM) in response to inflammatory mediators, which may lead to nitrosative stress. On the other hand, if released at low rates, achieving local concentrations <1 nmol L−1, NO may mediate protective and proliferative effects.47 Therefore, the possibility of modulating the NO release rates of the present hydrogels, as shown in Figure 7d, reinforces the prospects of using these formulations to obtain in situ anti-inflammatory and proliferative actions in tissue regeneration.48
3.10. In Vitro Cytocompatibility and Cell Adhesion.
To quantify the cytocompatibility of the hydrogels, leachates of each sample were collected, diluted, and exposed to NIH 3T3 mouse fibroblast cells. The results showed that cells exposed to the leachate dilutions from HHA, HAG0.5, HAG1, NPHHA, NPHAG0.5, and NPHAG1 all had a relative cell viability greater than 70% (Figure 8a), exceeding the ISO threshold for cytocompatibility.28 The results also confirm that the photoinitiator concentration used for producing these hydrogels is safe for the present application, showing no toxic effect due to the radicals generated in the photo-cross-linking process, in accordance with other works.49 Therefore, all of these materials are suitable for biological use.
Figure 8.
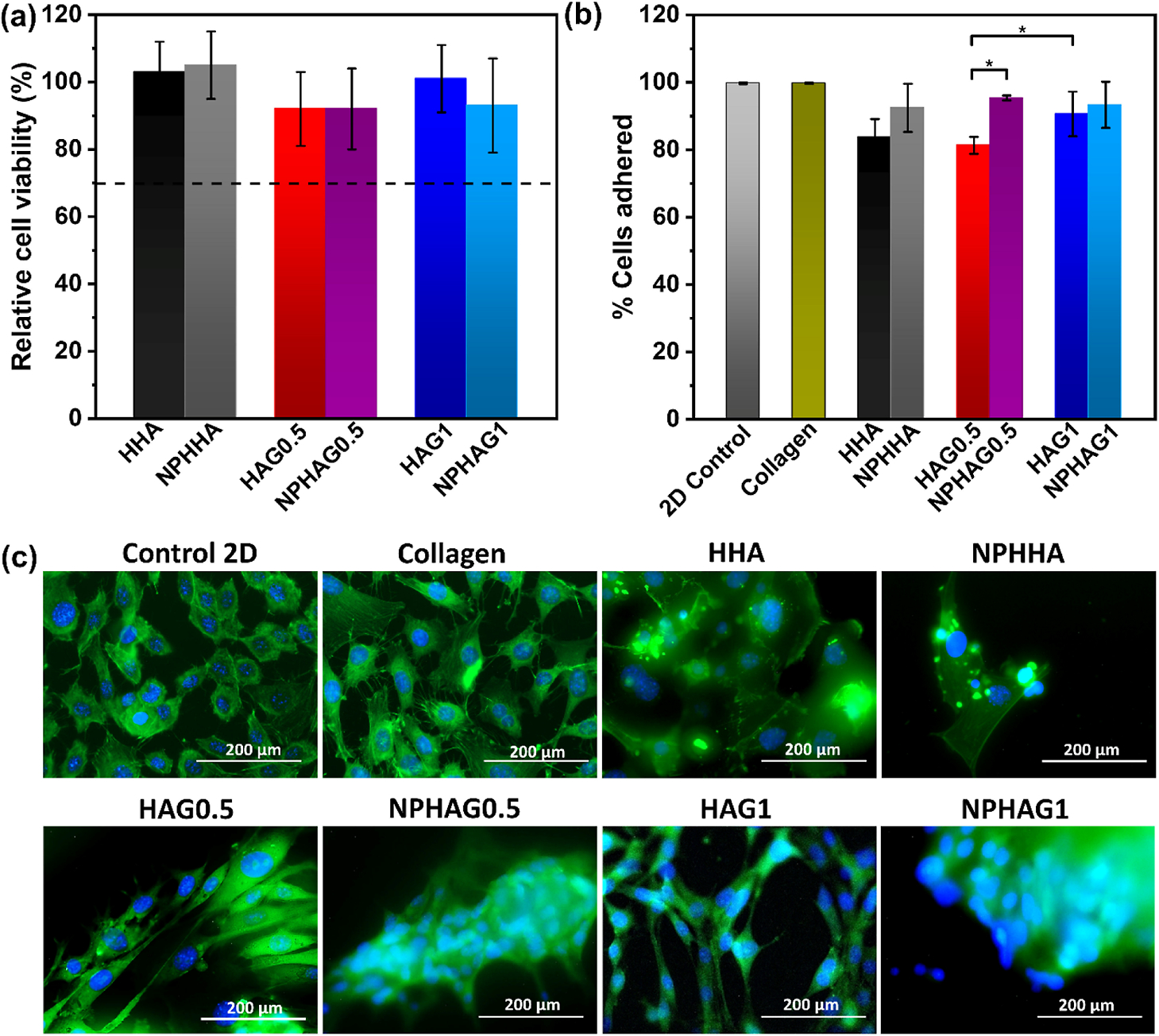
(a) Relative biocompatibility of fibroblastic cells from cell contact with hydrogel leachates. (b) Percentage of fibroblasts adhered to the control and hydrogel groups after 24 h of incubation. (c) Fluorescence image of controls (control 2D and collagen) and hydrogels after 72 h of incubation. Cells were stained with DAPI (blue) and Alexa Fluor 488 phalloidin (green) dyes
The adhesion of fibroblasts to the surface of the hydrogels was quantified after 24 h of seeding the cells on the formed hydrogel surface. Figure 8b shows that all hydrogels had greater than 80% cell adhesion. Two controls were used for this assay: the culture plate (2D control) and collagen gel (3D control). The release of NO, from the inclusion of NPPLGA-GSNO, increased the cell adhesion. The NPHHA and NPHAG1 hydrogels showed cell adhesion statistically similar to the controls, and the percentage of adhered cells in NPHAG0.5 was higher than in HAG0.5. The increase in GEL concentration also promoted greater adhesion when comparing the data for HAG0.5 and HAG1, due to the interaction of GEL with integrins, cell adhesion proteins.50
The morphology of the adhered cells was analyzed with an optical microscope at 0, 24, and 72 h of incubation. Figure S8 shows that after 24 h, only the cells that adhered to the 2D control showed spread-out spindle morphology, with typical fibroblast morphology, while the cells on the other materials remained rounded, indicative of primitive actin filament maturation.
Due to the soft nature of hydrogels (elastic modulus in the range of 2–19 kPa), cells take longer to spread and migrate across the material’s surface. In the present case, this process was achieved after 72 h of incubation. The dependence of morphology on substrate stiffness has been reported elsewhere.51,52 Incubating the hydrogels with adhered cells for 72 h allowed the cells to proliferate and spread. At this point, the cells were stained to analyze the nucleus (DAPI) and the cytoskeleton, specifically the F-actin (Alexa Fluor 488 Phalloidin).
The fluorescent image shows cells grown on the 3D hydrogel substrates with morphologies similar to those of the 2D and 3D controls, Figure 8c. The similarities between the morphologies support the ability of hydrogels not only to allow cell adhesion but also to promote cell growth. Interestingly, cells in NPHHA, NPHAG1, and NPHAG0.5 tend to aggregate in close proximity. This may be due to increased cell–cell interactions and signaling in the presence of NO, which has been shown to promote cell proliferation at low concentrations.53 The biological results confirmed that these hydrogels are safe for the biological environment and can be applied in situ to stimulate adhesion and proliferation of the cells.
4. CONCLUSIONS
In summary, we have successfully developed photo-cross-linkable NO-releasing hydrogels by combining HAGMA and GELSH. The incorporation of GELSH not only reduced the photogelation time of the hydrogels through thiol–ene bond formation but also imparted them with improved mechanical properties, enhanced resistance to enzymatic degradation, and reduced water absorption rates. The utilization of PLGA nanoparticles functionalized with GSNO has emerged as a highly efficient and innovative strategy. This approach effectively shielded GSNO from the free radicals generated during the photo-cross-linking process, resulting in controlled and sustained NO release profiles spanning up to 14 days. Additionally, the presence of PLGA-GSNO nanoparticles provided an extra layer of mechanical reinforcement to the hydrogels. The photo-cross-linking process of HAGMA/GELSH solutions gave rise to hydrogels characterized by porous structures that distinctly indicated the presence of PLGA-GSNO nanoparticles embedded within the pore walls. Swelling of these hydrogels led to NO release, driven by the dimerization of GSNO moieties chemically bound at the chain ends of the PLGA. This process facilitated a gradual NO release, dictated by the kinetics of hydration and hydrolysis of the PLGA-GSNO nanoparticles. Moreover, our HAGMA/GELSH hydrogels exhibited good cytocompatibility, actively promoting fibroblast adhesion. Consequently, these hydrogels hold substantial promise as a novel approach to tissue regeneration therapies. Their unique capability of localized NO release offers the potential for beneficial actions that can significantly contribute to tissue repair and regeneration.
Supplementary Material
ACKNOWLEDGMENTS
D.M.C. received fellowships from the Brazilian Coordination for the Improvement of Higher Education Personnel (CAPES) and the National Council for Scientific and Technological Development (CNPq) (grant no. 200187/2022-8). L.C.E.d.S. and M.F.d.O. received fellowships from the São Paulo Research Foundation (FAPESP), Brazil (grant nos. 2018/14142-5 and 2018/025205, respectively). M.G.d.O. acknowledges FAPESP, Brazil, for the financial support (grant no. 2016/02414-5). E.J.B. and H.H. acknowledge the National Institutes of Health, USA, for the financial support (grant nos. R01HL134899 and R01HL151473).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.3c10030.
1H NMR spectrum of HAGMA; schemes of the reaction between HA and GMA, and GEL and HCT; FTIR-ATR spectra of HA, GMA, HAGMA, GEL, and GELSH; mean hydrodynamic diameters of NPPLGA-GSNO obtained by dynamic light scattering; surface charge measured by zeta potential of NPPLGA-GSNO as a function of pH; calibration curve of GSNO in DMSO in the concentration range of 0.1–0.7 mmol L−1 measured at 336 nm; UV–vis absorption spectrum of NPPLGA-GSNO dispersed in DMSO; schemes of the photo-crosslinking reactions between vinyl groups of HAGMA, leading to the formation of HHA, and the reaction between the vinyl group of HGGMA and the thiol group of GELSH, leading to the formation of thiol–ene bond in HAG; variation of the G′ and G″ moduli as a function of the irradiation time for HHA in the absence and in the presence of free GSNO at the concentration of 0.15 mmol L−1; bar graph of the elastic modulus of HHA, NPHHA, HAG0.5, NPHAG0.5, HAG1, and NPHAG1; SEM images of the HHA, NPHHA, HAG0.5, NPHAG0.5, HAG1, and NPHAG1 hydrogels; and optical microscopy images of fibroblast adhesion on the controls (2D control and collagen) and HHA, NPHHA, HAG0.5, NPHAG0.5, HAG1, NPHAG1 hydrogels after 0 h, 24 and 72 h of incubation (PDF)
Contributor Information
Daniele M. Catori, Institute of Chemistry, University of Campinas, UNICAMP, Campinas 13083-970 São Paulo, Brazil
Laura C. E. da Silva, Institute of Chemistry, University of Campinas, UNICAMP, Campinas 13083-970 São Paulo, Brazil
Matheus F. de Oliveira, Institute of Chemistry, University of Campinas, UNICAMP, Campinas 13083-970 São Paulo, Brazil
Grace H. Nguyen, School of Chemical, Materials and Biomedical Engineering, College of Engineering, University of Georgia, Athens 30602 Georgia, United States
Joseph C. Moses, School of Chemical, Materials and Biomedical Engineering, College of Engineering, University of Georgia, Athens 30602 Georgia, United States
Elizabeth J. Brisbois, School of Chemical, Materials and Biomedical Engineering, College of Engineering, University of Georgia, Athens 30602 Georgia, United States
Hitesh Handa, School of Chemical, Materials and Biomedical Engineering, College of Engineering, University of Georgia, Athens 30602 Georgia, United States; Pharmaceutical and Biomedical Sciences Department, College of Pharmacy, University of Georgia, Athens 30602 Georgia, United States.
Marcelo G. de Oliveira, Institute of Chemistry, University of Campinas, UNICAMP, Campinas 13083-970 São Paulo, Brazil
REFERENCES
- (1).Napoli C; Paolisso G; Casamassimi A; Al-Omran M; Barbieri M; Sommese L; Infante T; Ignarro L Effects of Nitric Oxide on Cell Proliferation: Novel Insights. J. Am. Coll. Cardiol. 2013, 62 (2), 89–95. [DOI] [PubMed] [Google Scholar]
- (2).Vercelino R; Cunha TM; Ferreira ES; Cunha FQ; Ferreira SH; de Oliveira MG Skin Vasodilation and Analgesic Effect of a Topical Nitric Oxide-Releasing Hydrogel. J. Mater. Sci. Mater. Med. 2013, 24 (9), 2157–2169. [DOI] [PubMed] [Google Scholar]
- (3).Champeau M; Póvoa V; Militão L.; Cabrini FM.; Picheth GF.; Meneau F.; Jara CP.; de Araujo EP.; de Oliveira MG. Supramolecular Poly(acrylic acid)/F127 Hydrogel with Hydration-Controlled Nitric Oxide Release for Enhancing Wound Healing. Acta Biomater. 2018, 74, 312–325. [DOI] [PubMed] [Google Scholar]
- (4).Póvoa VCO; Santos GJ; Picheth GF; Jara CP; Silva LC; Araújo EP; Oliveira MG Wound Healing Action of Nitric Oxide-Releasing Self-Expandable Collagen Sponge. J. Tissue Eng. Regen. Med. 2020, 14 (6), 807–818. [DOI] [PubMed] [Google Scholar]
- (5).Giglio LP; Picheth GF; Løvschall KB; Zelikin AN; de Oliveira MG S-Nitrosothiol-Terminated Poly(Vinyl Alcohol): Nitric Oxide Release and Skin Blood Flow Response. Nitric Oxide 2020, 98, 41–49. [DOI] [PubMed] [Google Scholar]
- (6).Estes Bright LM; Griffin L; Mondal A; Hopkins S; Ozkan E; Handa H Biomimetic Gasotransmitter-Releasing Alginate Beads for Biocompatible Antimicrobial Therapy. J. Colloid Interface Sci. 2022, 628, 911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ghalei S; Douglass M; Handa H Nitric Oxide-Releasing Gelatin Methacryloyl/Silk Fibroin Interpenetrating Polymer Network Hydrogels for Tissue Engineering Applications. ACS Biomater. Sci. Eng. 2022, 8 (1), 273–283. [DOI] [PubMed] [Google Scholar]
- (8).Dovedytis M; Liu ZJ; Bartlett S Hyaluronic Acid and Its Biomedical Applications: A Review. Eng. Regen. 2020, 1, 102–113. [Google Scholar]
- (9).Vasvani S; Kulkarni P; Rawtani D Hyaluronic Acid: A Review on Its Biology, Aspects of Drug Delivery, Route of Administrations and a Special Emphasis on Its Approved Marketed Products and Recent Clinical Studies. Int. J. Biol. Macromol. 2020, 151, 1012–1029. [DOI] [PubMed] [Google Scholar]
- (10).Zhang Z; Wei X; Gao J; Zhao Y; Zhao Y; Guo L; Chen C; Duan Z; Li P; Wei L Intra-Articular Injection of Cross-Linked Hyaluronic Acid-Dexamethasone Hydrogel Attenuates Osteoarthritis: An Experimental Study in a Rat Model of Osteoarthritis. Int. J. Mol. Sci. 2016, 17 (4), 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Necas J; Bartosikova L; Brauner P; Kolar J Hyaluronic Acid (Hyaluronan): A Review. Vet. Med. 2008, 53 (8), 397–411. [Google Scholar]
- (12).Levett PA; Melchels FP; Schrobback K; Hutmacher DW; Malda J; Klein TJ A Biomimetic Extracellular Matrix for Cartilage Tissue Engineering Centered on Photocurable Gelatin, Hyaluronic Acid, and Chondroitin Sulfate. Acta Biomater. 2014, 10 (1), 214–223. [DOI] [PubMed] [Google Scholar]
- (13).Sanmartín-Masiá E; Poveda-Reyes S; Gallego Ferrer G Extracellular Matrix-Inspired Gelatin/Hyaluronic Acid Injectable Hydrogels. Int. J. Polym. Mater. Polym. Biomater. 2017, 66 (6), 280–288. [Google Scholar]
- (14).Segura T; Anderson BC; Chung PH; Webber RE; Shull KR; Shea LD Crosslinked Hyaluronic Acid Hydrogels: A Strategy to Functionalize and Pattern. Biomaterials 2005, 26 (4), 359–371. [DOI] [PubMed] [Google Scholar]
- (15).Muir VG; Burdick JA Chemically Modified Biopolymers for the Formation of Biomedical Hydrogels. Chem. Rev. 2021, 121 (18), 10908–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dimatteo R; Darling NJ; Segura T In Situ Forming Injectable Hydrogels for Drug Delivery and Wound Repair. Adv. Drug Delivery Rev. 2018, 127, 167–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Phan VHG; Thambi T; Duong HTT; Lee DS Poly (Amino Carbonate Urethane)-Based Biodegradable, Temperature, and pH-Sensitive Injectable Hydrogels for Sustained Human Growth Hormone Delivery. Sci. Rep. 2016, 6 (1), 29978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mei Q; Rao J; Bei HP; Liu Y; Zhao X 3D Bioprinting Photo-Crosslinkable Hydrogels for Bone and Cartilage Repair. Int. J. Bioprinting 2021, 7 (3), 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chen P; Ning L; Qiu P; Mo J; Mei S; Xia C; Zhang J; Lin X; Fan S Photo-Crosslinked Gelatin-Hyaluronic Acid Methacrylate Hydrogel-Committed Nucleus Pulposus-Like Differentiation of Adipose Stromal Cells for Intervertebral Disc Repair. J. Tissue Eng. Regen. Med. 2019, 13 (4), 682–693. [DOI] [PubMed] [Google Scholar]
- (20).Zhang C; Dong Q; Liang K; Zhou D; Yang H; Liu X; Xu W; Zhou Y; Xiao P Photopolymerizable Thiol-Acrylate Maleiated Hyaluronic Acid/Thiol-Terminated Poly (Ethylene Glycol) Hydrogels as Potential In Situ Formable Scaffolds. Int. J. Biol. Macromol. 2018, 119, 270–277. [DOI] [PubMed] [Google Scholar]
- (21).Ganzarolli de Oliveira M S-Nitrosothiols as Platforms for Topical Nitric Oxide Delivery. Basic Clin. Pharmacol. Toxicol. 2016, 119, 49–56. [DOI] [PubMed] [Google Scholar]
- (22).de Souza GFP; Denadai JP; Picheth GF; de Oliveira MG Long-term Decomposition of Aqueous S-Nitrosoglutathione and S-Nitroso-N-Acetylcysteine: Influence of Concentration, Temperature, pH, and Light. Nitric Oxide 2019, 84, 30–37. [DOI] [PubMed] [Google Scholar]
- (23).Simplicio FI; Seabra AB; de Souza GFP; Oliveira MG In Vitro Inhibition of Linoleic Acid Peroxidation by Primary S-Nitrosothiols. J. Braz. Chem. Soc. 2010, 21, 1885–1895. [Google Scholar]
- (24).Reis AV; Fajardo AR; Schuquel IT; Guilherme MR; Vidotti GJ; Rubira AF; Muniz EC Reaction of Glycidyl Methacrylate at the Hydroxyl and Carboxylic Groups of Poly(Vinyl Alcohol) and Poly(Acrylic Acid): Is This Reaction Mechanism Still Unclear? J. Org. Chem. 2009, 74 (10), 3750–3757. [DOI] [PubMed] [Google Scholar]
- (25).Vlierberghe SV; Schacht E; Dubruel P Reversible Gelatin-Based Hydrogels: Fine-Tuning of Material Properties. Eur. Polym. J. 2011, 47 (5), 1039–1047. [Google Scholar]
- (26).Lee J; Kwak D; Kim H; Kim J; Hlaing SP; Hasan N; Cao J; Yoo J-W Nitric Oxide-Releasing S-Nitrosoglutathione-Conjugated Poly(Lactic-Co-Glycolic Acid) Nanoparticles for the Treatment of MRSA-Infected Cutaneous Wounds. Pharmaceutics 2020, 12 (7), 618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Basu S; Ricart K; Gladwin MT; Patel RP; Kim-Shapiro DB Tri-Iodide and Vanadium Chloride Based Chemiluminescent Methods for Quantification of Nitrogen Oxides. Nitric Oxide 2022, 121, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).International Organization for Standardization. Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity; ISO 10993–5:2009, 2009;. [Google Scholar]
- (29).Niloy KK; Gulfam M; Compton KB; Li D; Huang GT-J; Lowe TL Methacrylated Hyaluronic Acid-Based Hydrogels Maintain Stemness in Human Dental Pulp Stem Cells. Regen. Eng. Transl. Med. 2020, 6 (3), 262–272. [Google Scholar]
- (30).da Silva EP; Guilherme MR; Garcia FP; Nakamura CV; Cardozo-Filho L; Alonso CG; Rubira AF; Kunita MH Drug Release Profile and Reduction in the In Vitro Burst Release from Pectin/HEMA Hydrogel Nanocomposites Crosslinked with Titania. RSC Adv. 2016, 6 (23), 19060–19068. [Google Scholar]
- (31).Sochilina A; Savelyev A; Akasov R; Zubov V; Khaydukov E; Generalova A Preparing Modified Hyaluronic Acid with Tunable Content of Vinyl Groups for Use in Fabrication of Scaffolds by Photoinduced Crosslinking. Russian J. Bioorganic Chem. 2021, 47 (4), 828–836. [Google Scholar]
- (32).Silverstein RM; Webster FX; Kiemle DJ; Bryce DL Spectrometric Identification of Organic Compounds, 7th; The State University of New York, College of Environmental Science and Forestry, 2005. [Google Scholar]
- (33).Wang PG; Xian M; Tang X; Wu X; Wen Z; Cai T; Janczuk AJ Nitric Oxide Donors: Chemical Activities and Biological Applications. Chem. Rev. 2002, 102 (4), 1091–1134. [DOI] [PubMed] [Google Scholar]
- (34).Van Hoorick J; Tytgat L; Dobos A; Ottevaere H; Van Erps J; Thienpont H; Ovsianikov A; Dubruel P; Van Vlierberghe S (Photo-) Crosslinkable Gelatin Derivatives for Biofabrication Applications. Acta Biomater. 2019, 97, 46–73. [DOI] [PubMed] [Google Scholar]
- (35).Lee HJ; Fernandes-Cunha GM; Myung D In Situ-Forming Hyaluronic Acid Hydrogel through Visible Light-Induced Thiol-Ene Reaction. React. Funct. Polym. 2018, 131, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Machado TO; Sayer C; Araujo PH Thiol-ene Polymerisation: A Promising Technique to Obtain Novel Biomaterials. Eur. Polym. J. 2017, 86, 200–215. [Google Scholar]
- (37).Okamura H; Yamagaki M; Nakata K Analysis of Network Structures in Thiol-Ene UV Curing System Using Reworkable Resins. Polymers 2018, 11 (1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bruice PY Organic Chemistry; Pearson/Prentice Hall, 2004; Vol. 1. [Google Scholar]
- (39).Carotenuto F; Politi S; Ul Haq A; De Matteis F; Tamburri E; Terranova ML; Teodori L; Pasquo A; Di Nardo P From Soft to Hard Biomimetic Materials: Tuning Micro/Nano-Architecture of Scaffolds for Tissue Regeneration. Micromachines 2022, 13 (5), 780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Asadi N; Alizadeh E; Salehi R; Khalandi B; Davaran S; Akbarzadeh A Nanocomposite Hydrogels for Cartilage Tissue Engineering: A Review. Artif. Cells Nanomed. Biotechnol. 2018, 46 (3), 465–471. [DOI] [PubMed] [Google Scholar]
- (41).Makadia HK; Siegel SJ Poly Lactic-Co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3 (3), 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Stern R; Jedrzejas MJ Hyaluronidases: Their Genomics, Structures, and Mechanisms of Action. Chem. Rev. 2006, 106 (3), 818–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Liu G; McEnnis K Glass Transition Temperature of PLGA Particles and the Influence on Drug Delivery Applications. Polymers 2022, 14 (5), 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Fukuto JM; Cho JY; Switzer CH Nitric Oxide; Elsevier, 2000, pp 23–40.The Chemical Properties of Nitric Oxide and Related Nitrogen Oxides [Google Scholar]
- (45).Rescignano N; Tarpani L; Romani A; Bicchi I; Mattioli S; Emiliani C; Torre L; Kenny J; Martino S; Latterini L; et al. In-Vitro Degradation of PLGA Nanoparticles in Aqueous Medium and in Stem Cell Cultures by Monitoring the Cargo Fluorescence Spectrum. Polym. Degrad. Stab. 2016, 134, 296–304. [Google Scholar]
- (46).Spiller F; Oliveira Formiga R; Fernandes da Silva Coimbra J.; Alves-Filho JC.; Cunha TM.; Cunha FQ. Targeting Nitric Oxide as a Key Modulator of Sepsis, Arthritis, and Pain. Nitric Oxide 2019, 89, 32–40. [DOI] [PubMed] [Google Scholar]
- (47).Thomas DD; Ridnour LA; Isenberg JS; Flores-Santana W; Switzer CH; Donzelli S; Hussain P; Vecoli C; Paolocci N; Ambs S; et al. The Chemical Biology of Nitric Oxide: Implications in Cellular Signaling. Free Radic. Biol. Med. 2008, 45 (1), 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Turesin F; Del Soldato P; Wallace JL Enhanced Anti-Inflammatory Potency of a Nitric Oxide-Releasing Prednisolone Derivative in the Rat. Br. J. Pharmacol. 2003, 139 (5), 966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Xu H; Casillas J; Krishnamoorthy S; Xu C Effects of Irgacure 2959 and Lithium Phenyl-2, 4, 6-Trimethylbenzoylphosphinate on Cell Viability, Physical Properties, and Microstructure in 3D Bioprinting of Vascular-Like Constructs. Biomed. Mater. 2020, 15 (5), 055021. [DOI] [PubMed] [Google Scholar]
- (50).Spicer CD Hydrogel Scaffolds for Tissue Engineering: The Importance of Polymer Choice. Polym. Chem. 2020, 11 (2), 184–219. [Google Scholar]
- (51).Yeung T; Georges PC; Flanagan LA; Marg B; Ortiz M; Funaki M; Zahir N; Ming W; Weaver V; Janmey PA Effects of Substrate Stiffness on Cell Morphology, Cytoskeletal Structure, and Adhesion. Cell Motil. Cytoskeleton 2005, 60 (1), 24–34. [DOI] [PubMed] [Google Scholar]
- (52).Skardal A; Mack D; Atala A; Soker S Substrate Elasticity Controls Cell Proliferation, Surface Marker Expression, and Motile Phenotype in Amniotic Fluid-Derived Stem Cells. J. Mech. Behav. Biomed. Mater. 2013, 17, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Thomas DD; Ridnour LA; Isenberg JS; Flores-Santana W; Switzer CH; Donzelli S; Hussain P; Vecoli C; Paolocci N; Ambs S; et al. The Chemical Biology of Nitric Oxide: Implications in Cellular Signaling. Free Radic. Biol. Med. 2008, 45 (1), 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.