

Abstract
The frequency of errors upon decoding of messenger RNA by the bacterial ribosome is low, with one misreading event per 1 × 104 codons. In the universal genetic code, the AUN codon box specifies two amino acids, isoleucine and methionine. In bacteria and archaea, decoding specificity of the AUA and AUG codons relies on the wobble avoidance strategy that requires modification of C34 in the anticodon loop of isoleucine transfer RNAIleCAU (tRNAIleCAU). Bacterial tRNAIleCAU with 2-lysylcytidine (lysidine) at the wobble position deciphers AUA while avoiding AUG. Here we report cryo-electron microscopy structures of the Escherichia coli 70S ribosome complexed with elongation factor thermo unstable (EF-Tu) and isoleucine-tRNAIleLAU in the process of decoding AUA and AUG. Lysidine in tRNAIleLAU excludes AUG by promoting the formation of an unusual Hoogsteen purine–pyrimidine nucleobase geometry at the third position of the codon, weakening the interactions with the mRNA and destabilizing the EF-Tu ternary complex. Our findings elucidate the molecular mechanism by which tRNAIleLAU specifically decodes AUA over AUG.
The ribosome is the molecular machine that reads the messenger RNA, binds transfer RNA (tRNA) molecules carrying amino acids, and forms peptide bonds to generate polypeptides. Accurate decoding of mRNA is a crucial step in all cells. Binding of the aminoacyl-tRNA (aa-tRNA) to its cognate mRNA codon positioned in the decoding center of the small (30S) ribosomal subunit triggers rearrangements that stabilize the incoming ternary complex, composed of elongation factor thermo unstable (EF-Tu), GTP and aa-tRNA1,2,3. In the decoding center, nucleotides of the 16S ribosomal RNA (rRNA)—A1492, A1493 and G530—closely monitor the geometry of the first and second base pairs between aa-tRNA and the mRNA4,5,6. The stabilization and increased dwelling time of the EF-Tu•GTP•aa-tRNA ternary complex allows EF-Tu to associate with the GTP-activating center of the large (50S) subunit, including the sarcin–ricin loop, and to rapidly hydrolyze GTP1,2,3. The conformational change in EF-Tu that follows ensures delivery of the aa-tRNA to the aminoacyl (A) site of the ribosome7.
The structure of the decoding center restricts the geometry of the first and second base pairs between the aa-tRNA and the mRNA to the Watson–Crick (WC) conformation. Recent studies have shown that non-WC base pairs at these positions, such as G:U, are forced by the decoding center to adopt a WC-like geometry8,9,10. In contrast, the third position of the codon, which pairs with nucleotide 34 in tRNA, is not monitored as closely by the ribosome, allowing for the formation of noncanonical base pairs. The ‘wobble’ character of the third position is at the core of the degeneracy of the genetic code, allowing one tRNA to bind to multiple codons directing the incorporation of a single amino acid. It is, therefore, particularly challenging for tRNAs and the ribosome to discriminate A-site codons solely from the nucleotide identity at the third position of the mRNA.
To circumvent this fundamental issue, the cell uses post-transcriptional modifications of tRNAs. Ubiquitous across all kingdoms of life, modified nucleotides facilitate folding of tRNAs, recognition by aminoacyl-tRNA synthetases, maintenance of the translation reading frame and ensure protein synthesis accuracy11,12,13,14,15,16,17,18,19,20,21,22,23. Modifications of nucleotide 34 in the anticodon loop of tRNAs can either expand or restrict the decoding capacity of tRNAs. The presence of uridine 5-oxyacetic acid at the wobble position of tRNAValUAC, tRNAAlaUGC and tRNAProUGG allows pairing with adenosine, guanosine, uridine and cytosine at the third position of the mRNA24,25,26. Similarly, deamination of adenosine 34 to inosine allows it to pair with U, C and A (refs.27,28). Conversely, there are two examples in the genetic code where the wobble position of tRNA should discriminate between G and A in the mRNA. One occurs between the UGG (Trp) and UGA (stop) codons which is, however, accompanied by a high level of UGA readthrough29, illustrating the challenge of discrimination between purines at the wobble position of the mRNA. In the second case, the minor tRNAIleCAU decodes specifically its cognate AUA codon, while avoiding the AUG (Met) codon.
In bacteria, decoding of the rare AUA codon requires modification of tRNAIleCAU at position 34 of the anticodon loop. In Escherichia coli, and other eubacteria, the enzyme tRNAIle–lysidine synthetase (TilS)30,31 ligates lysine onto C34 converting it to 2-lysylcytidine, or lysidine (L34), allowing tRNAIleLAU to efficiently decode AUA and not AUG. Such conversion of C34 to L34 regulates not only codon specificity, but also ensures that isoleucyl-tRNA synthetase faithfully attaches isoleucine to tRNAIleLAU (refs.30,31,32). In archaea, C34 is instead modified to agmatidine (agm34), also conferring specificity toward AUA over AUG33,34,35,36. The structural basis for decoding of AUA by archaeal tRNAIle(agm34C)AU was elucidated earlier36, showing that the agmatine moiety at the C2 carbon of C34 enables it to pair with adenosine. In eubacteria, however, the molecular basis by which L34 pairs with adenosine and avoids guanosine at the wobble position remains speculative.
In this study, we use single-particle cryogenic electron microscopy (cryo-EM) to elucidate the mechanism of AUG codon avoidance by tRNAIleLAU. We report four cryo-EM reconstructions of the E. coli 70S ribosome bound to lysidine-containing tRNAIle representing various stages of AUA and AUG decoding (Fig. 1 and Table 1). The structures allow to visualize how tRNAIleLAU decodes AUA, confirming the ‘wobble avoidance’ hypothesis that enables efficient AUA translation and AUG exclusion.
Fig. 1: Cryo-EM structures of the ribosome bound to tRNAIleLAU.
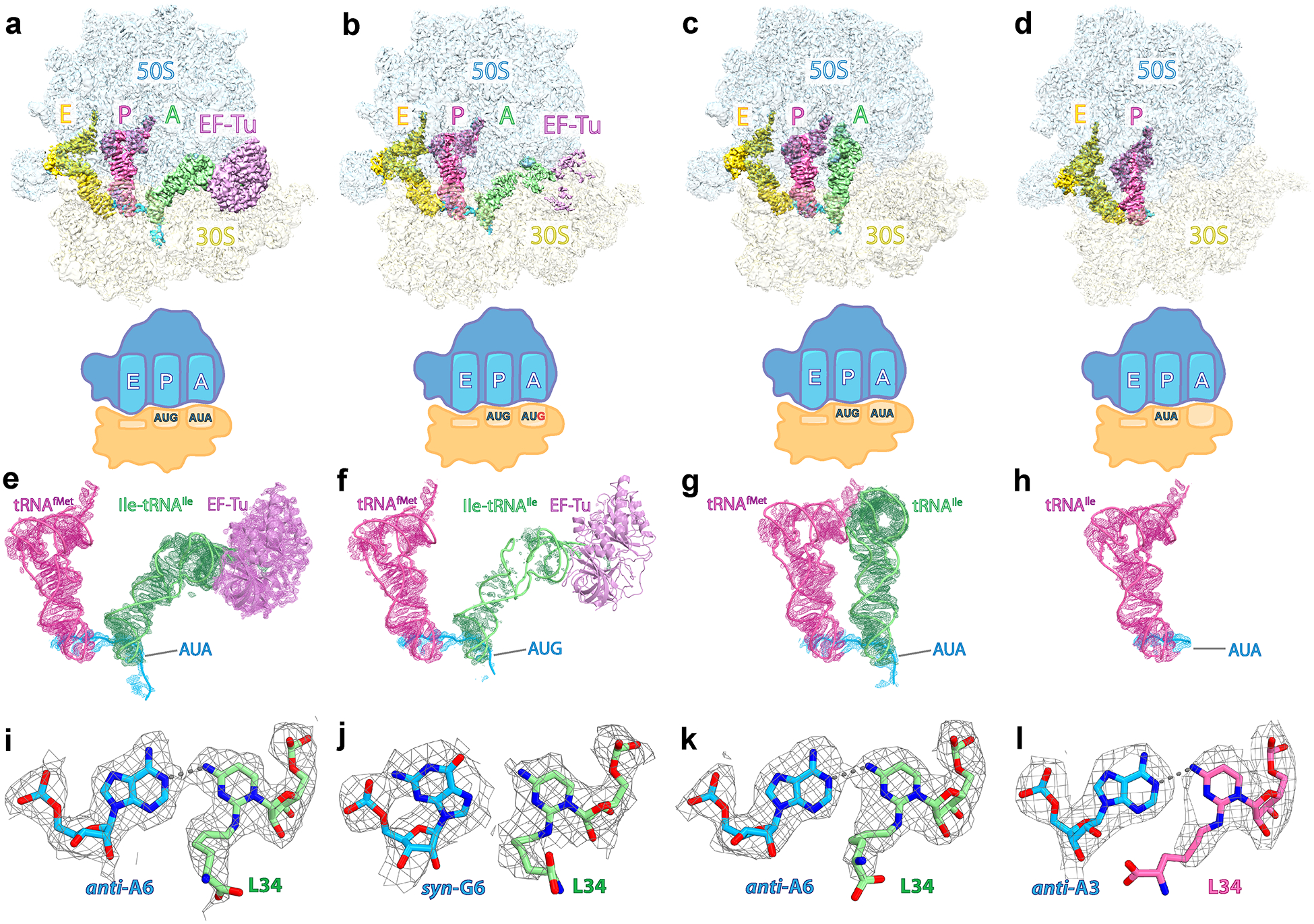
a, Density map of the E. coli 70S ribosome bound to EF-Tu•GDPCP•Ile-tRNAIleLAU in the presence of the cognate AUA codon (structure I) in the A site. The 50S and 30S subunits are shown in light blue and light yellow, respectively. The A-site Ile-tRNAIleLAU is green, the P-site initiator tRNAfMet is pink, the E-site Ile-tRNAIleLAU is yellow, the mRNA is cyan and EF-Tu is light magenta. b, Density map of the 70S ribosome bound to EF-Tu•GDPCP•Ile-tRNAIleLAU in the presence of paromomycin and the near-cognate AUG codon in the A site (structure II). c, Density map of the 70S ribosome bound to the A-site tRNAIleLAU in the presence of paromomycin and the cognate AUA codon in the A site (structure III). d, Density map of the 70S ribosome bound to the P-site tRNAIleLAU in the presence of the cognate AUA codon in the P site (structure IV). e–h, EM maps of the A and P sites in structures I–II–III–IV contoured at 4.0σ. i–l, Density maps of the wobble position for the AUA codon in structures I (i), III (k) and IV (l) or the AUG codon in structure III (j), paired with lysidine 34 (L34) in tRNAIleLAU. The gray dashed lines indicate putative hydrogen bonds. The EM density is contoured at 2.9σ (i and k) or 2.5σ (j and l).
Table 1.
Cryo-EM data collection, refinement and validation statistics
| Structure I (A/T Ile-tRNAIle, A-site AUA) (EMD-29819) (PDB 8G7P) | Structure II (A/T Ile-tRNAIle, A-site AUG) (EMD-29820) (PDB 8G7Q) | Structure III (A-site tRNAIle, A-site AUA) (EMD-29821) (PDB 8G7R) | Structure IV (P-site tRNAIle, P-site AUA) (EMD-29822) (PDB 8G7S) | |
|---|---|---|---|---|
| Data collection and processing | ||||
| Magnification | 96,000x | 96,000x | 96,000x | 96,000x |
| Voltage (kV) | 300 | 300 | 300 | 300 |
| Electron exposure (e−/Å2) | 40 | 40 | 40 | 40 |
| Defocus range (μm) | −1 to −2.3 | −1 to −2.3 | −1 to −2.3 | −1 to −2.3 |
| Detector | Falcon III | Falcon III | Falcon III | Falcon III |
| Pixel size (Å) | 0.86 | 0.86 | 0.86 | 0.85 |
| Symmetry imposed | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 682,429 | 764,480 | 923,703 | 586,922 |
| Final particle images (no.) | 135,882 | 47,694 | 466,262 | 131,911 |
| Map resolution (Å) | 2.9 | 3.1 | 2.8 | 3.1 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 |
| Refinement | ||||
| Initial model used (PDB code) | 7K00, 5UYM | 7K00, 5UYM | 7K00 | 7K00 |
| Model resolution (Å) | 3.0 | 3.2 | 2.9 | 3.3 |
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 |
| Map sharpening B factor (Å2) | −69 | −58 | −59 | −59 |
| Model composition | ||||
| Chains | 58 | 58 | 56 | 55 |
| Non-hydrogen atoms | 151,825 | 151,181 | 147,377 | 145,317 |
| Protein residues | 6,098 | 6,086 | 5,550 | 5,550 |
| RNA residues | 4,785 | 4,783 | 4,783 | 4,702 |
| Ligands: Mg2+/Zn2+/K+ | 876 / 2 / 2 | 950 / 2 / 0 | 734 / 2 / 2 | 564 / 2 / 0 |
| Waters | 716 | 505 | 331 | 219 |
| B factors (Å2) | ||||
| Protein residues | 60.2 | 55.2 | 82.0 | 32.5 |
| RNA residues | 58.2 | 58.2 | 78.2 | 37.3 |
| Ions | 44.8 | 41.4 | 52.9 | 20.7 |
| Waters | 39.5 | 35.9 | 46.0 | 15.6 |
| CCmask | 0.88 | 0.86 | 0.86 | 0.87 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.003 | 0.006 | 0.004 | 0.004 |
| Bond angles (°) | 0.631 | 0.689 | 0.598 | 0.572 |
| Validation | ||||
| MolProbity score | 1.5 | 1.6 | 1.5 | 1.6 |
| Clashscore | 4.1 | 4.7 | 3.9 | 5.3 |
| Rotamer outliers (%) | 0 | 0 | 0 | 0 |
| Cβ outliers (%) | 0.02 | 0 | 0 | 0 |
| Ramachandran plot | ||||
| Favored (%) | 95.54 | 94.07 | 95.94 | 95.83 |
| Allowed (%) | 4.38 | 5.88 | 4.03 | 4.14 |
| Disallowed (%) | 0.08 | 0.05 | 0.04 | 0.04 |
Results
70S ribosome structures bound to EF-Tu•GDPCP•Ile-tRNAIleLAU
To visualize the interactions between Ile-tRNAIleLAU and the AUA or AUG codon during decoding, we trapped the ternary complex EF-Tu•GDPCP•Ile-tRNAIleLAU bound to the E. coli 70S ribosome with the use of the nonhydrolyzable GTP analog, GDPCP. The mRNA-programmed ribosome complexes contain the AUA (sample I) or the AUG (sample II) codon in the A site. We collected cryo-EM data and proceeded with two-dimensional (2D) and 3D classification of the selected particles. This process yielded two main class averages in sample I with clear density for EF-Tu bound to the ribosome (Extended Data Fig. 1). However, the same approach with sample II did not yield any ribosome particle bound to EF-Tu, suggesting that the molecular recognition of the near-cognate AUG codon by Ile-tRNAIleLAU is transient and labile, confirming rejection of the AUG methionine codon. We therefore stabilized the ternary complex bound to AUG with the addition of the aminoglycoside antibiotic paromomycin, which strengthens the interactions between the 16S rRNA monitoring nucleotides A1492, A1493 and G530, and the minor groove of the codon–anticodon helix, thereby facilitating binding of near-cognate tRNAs6. Using this strategy, particle classification of dataset II with the AUG codon in the A site revealed one main class average with weak density for EF-Tu (Extended Data Fig. 2).
The particles with clear density for EF-Tu•GDPCP•Ile-tRNAIleLAU were sorted using focused 3D variability37 with either a segment mask around the A-site tRNA and EF-Tu (dataset I) or a spherical mask around the binding site of EF-Tu (dataset II). For each dataset, this approach37,38,39 yielded to one main 3D volume containing EF-Tu•GDPCP•A/T-Ile-tRNAIleLAU, P- and E-site tRNAs with the A-site AUA (structure I—135,882 particles) or AUG (structure II—47,694 particles) codon (Fig. 1a,b,e,f). Refinement of each reconstruction generated density maps with a nominal resolution of 2.9 Å (structure I) and 3.1 Å (structure II), with local resolution estimations ranging from 2 to 6 Å (Extended Data Fig. 3a,b). As expected, structure II with the near-cognate AUG codon in the A site has a lower nominal and local resolution attributed to the labile binding of the ternary complex. Correspondingly, while the EM density of the anticodon region paired with the AUG codon in structure II is clear, the density attributable to the remaining regions of the Ile-tRNAIleLAU and EF-Tu is rather poor (Fig. 1f and Extended Data Fig. 4b). Nevertheless, the map around the decoding center revealed the molecular basis of AUG codon rejection by Ile-tRNAIleLAU.
The base pairs between tRNAIleLAU and AUG are disrupted
In the presence of the cognate AUA codon in the A site, canonical WC base pairs form at both the first and second positions of the codon (Figs. 2a and 3a,b), leading to favorable interactions between G530, A1492 and A1493, and the codon–anticodon helix. The 30S subunit is in a ‘closed’ conformation as reported previously for the structure of the ribosome bound to archaeal tRNA2Ile modified with agmatidine 34 (agm34)36. There is clear density for 2-lysylcytidine (lysidine) in the anticodon loop of tRNAIle, which confirms that the tRNA was modified by TilS at position 34 (Figs. 1i–k and 2a–c). Lysidine 34 (L34) forms one hydrogen bond with the third nucleotide of the A-site codon, A6, adopting a base pair geometry similar to that seen between agm34 of archaeal tRNA2Ile and adenosine in the mRNA (Figs. 1i,k and 2a,c,f)36.
Fig. 2: Role of lysidine 34 in tRNAIleLAU during decoding.
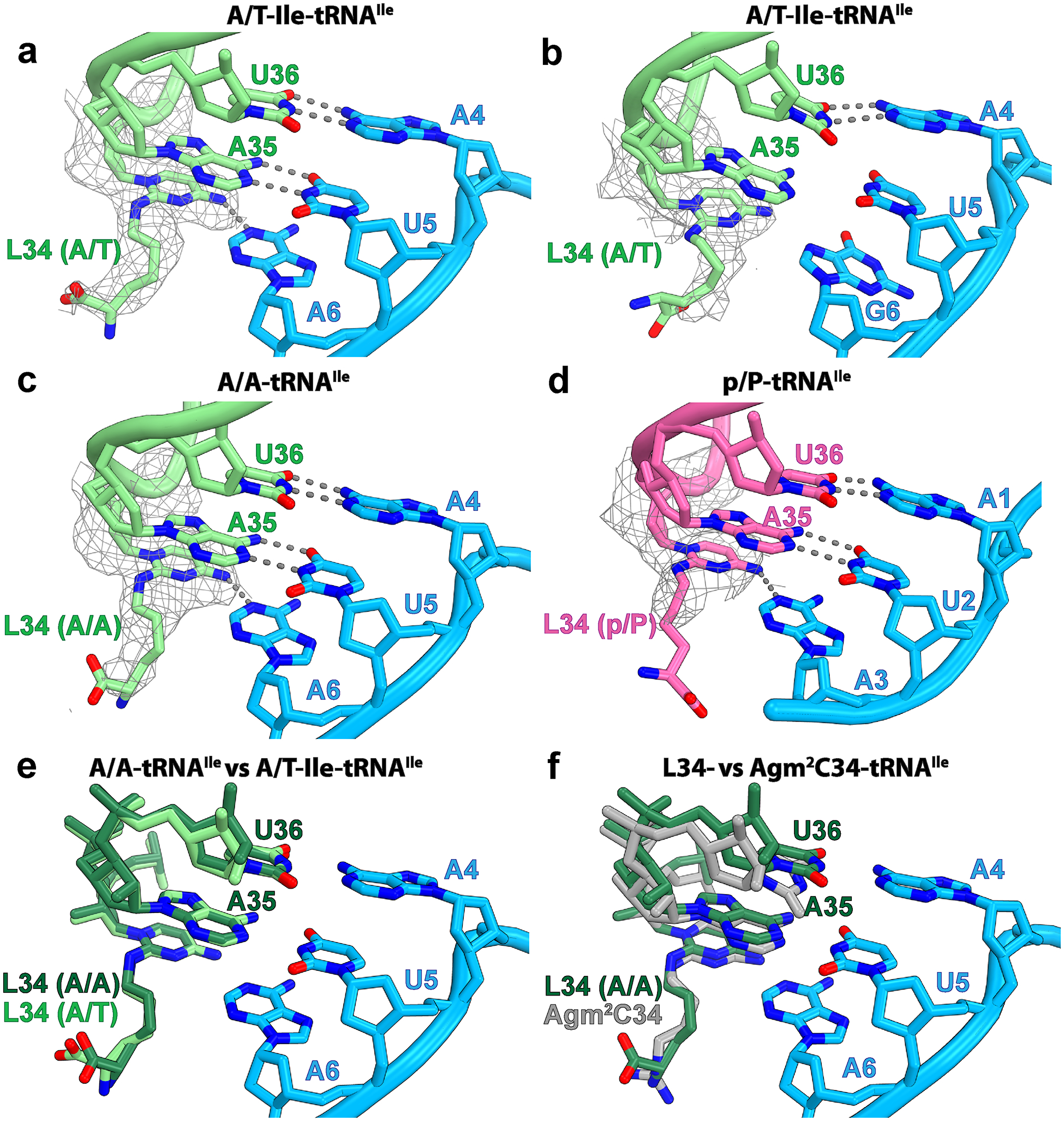
a, Base pairs between the A/T-Ile-tRNAIleLAU (light green) and the AUA codon (light blue) in structure I. b, Same as in a with the AUG codon in structure II. c, Base pairs between the accommodated A-site tRNAIleLAU and the AUA codon in structure III. d, Base pairs between the P-site tRNAIleLAU (magenta) and the AUA codon in structure IV. The Coulomb potential density of lysidine 34 (L34), contoured at 2.9σ, is shown as gray mesh. The gray dashed lines show putative hydrogen bonds. e, Relative conformation of the anticodon loop, including L34, of tRNAIleLAU in the A/T (light green, structure I) and A/A (dark green, structure III) bound states. f, Structure alignment of A-site tRNA2Ile containing agm34 from Haloarcula marismortui (gray, PDB 4V8N (ref.36)) with the A-site lysidine-containing tRNAIleLAU from E. coli (dark green, structure III).
Fig. 3: Conformation of codon–anticodon base pairs with the cognate AUA and near-cognate AUG codons.
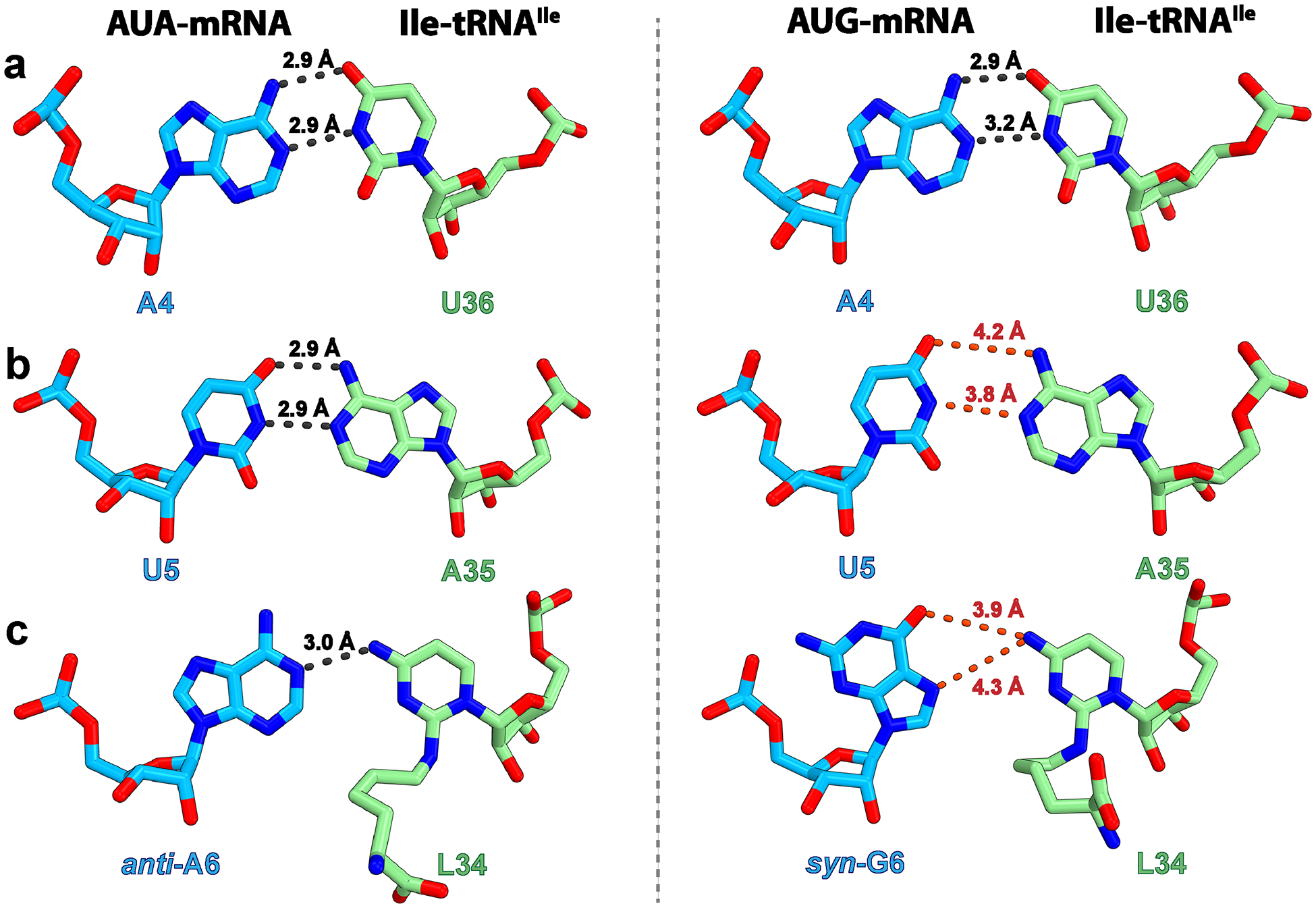
a–c, Base pair at the first (a), second (b) or third (c) position of the codon formed between the E. coli Ile-tRNAIleLAU and the cognate AUA (left, structure I) or near-cognate AUG (right, structure II) codon. The gray dashed lines indicate distances that are conducive to hydrogen bond formation (<3.5 Å threshold42), whereas those above this threshold, and therefore unlikely to form, are red.
Upon decoding of AUG by tRNAIleLAU, the lysine moiety of L34 would collide with the exocyclic amine of G6 in the mRNA (Extended Data Fig. 5d). Furthermore, the exocyclic amine of L34 would be facing the hydrogen bond donor N1 of G6 in the mRNA (Extended Data Fig. 5d), leading to a steric clash. Despite these potentially nonfavorable interactions at the wobble position of the A-site codon, the anticodon stem-loop (ASL) of tRNA and the mRNA occupy the same position in structures I (AUA) and II (AUG) (Extended Data Fig. 6a). Instead of perturbing the backbone geometry of the mRNA and tRNA, structure II shows that the nucleobase of G6 rotates around the glycosidic bond to the syn-conformation, deflecting the collision with L34 (Figs. 1j and 2b and Extended Data Figs. 5c and 7b). However, in spite of the seemingly favorable Hoogsteen geometry of G6–L34 at the wobble position, the distance between N7 or O6 of syn-G6 and the exocyclic amine of L34 (4.3 and 3.9 Å, respectively) is not conducive to form hydrogen bonds (Fig. 3c and Extended Data Fig. 5c). Interestingly, the nucleotide downstream of G6, A7, also flips to the syn-conformation, presumably maximizing π–π stacking interactions with G6 (Extended Data Fig. 7c).
In the syn-conformation, the sugar edge of G6 is facing the phosphate backbone of the mRNA, bringing the amine group and the nonbridging phosphate oxygen of G6 in close proximity (Extended Data Fig. 6b). To avoid a collision with the phosphate group, the nucleobase of G6 inclines by ~15° around the glycosidic bond in structure II relative to A6 in structure I. Because G6 forms π–π stacking interactions with A7 and U5, the inclined conformation of G6 propagates to the neighboring bases. The nucleobase of U5 at the second position of the AUG codon inclines by ~16° (Extended Data Fig. 6b), which increases the distance with A35 in tRNAIleLAU beyond the interaction threshold (Fig. 3b). The influence of the inclined base of G6 does not reach A4 at the first position of the codon, preserving a favorable distance for hydrogen bond formation with U36 in the ASL of tRNAIleLAU (Fig. 3a). These observations suggest that the presence of L34 in tRNAIleLAU excludes G at the wobble position by prohibiting formation of base pairs at the second and third positions of the AUG codon in the A-site.
Upon decoding of the cognate AUA codon, L34 in tRNAIleLAU packs against nucleotide G530 in the decoding center and forms water-mediated interactions with G530 and C518 in helix h18 of 16S rRNA (Fig. 4a,c and Extended Data Fig. 7a). On the contrary, the gap between the lysine moiety of L34 and G530 in structure II (Fig. 4b), presumably caused by the lack of hydrogen bond with G6, increases the flexibility of L34 and perturbs the molecular recognition of the AUG codon by tRNAIleLAU. In agreement with this premise, we observe fragmented density for EF-Tu and the acceptor domain of Ile-tRNAIleLAU in the A/T conformation (Fig. 1f and Extended Data Fig. 4b), suggesting unstable binding of the ternary complex to the ribosome programmed with AUG in the A site.
Fig. 4: The decoding center stabilizes L34 at the wobble position.
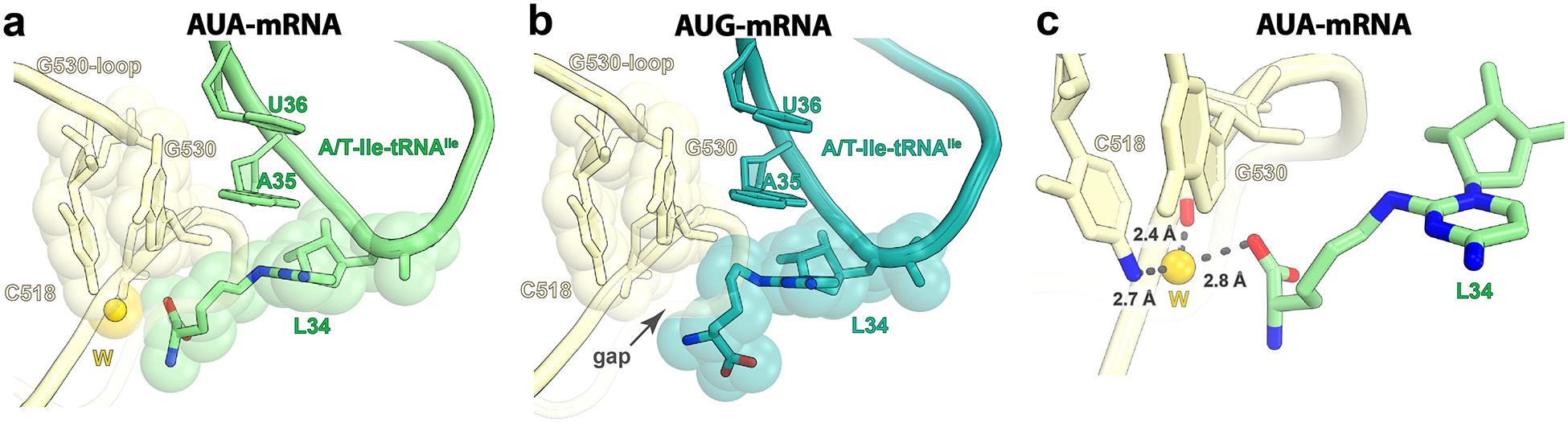
a, Upon decoding of the cognate AUA codon, the universal nucleotide G530 in helix h18 stacks with L34 of Ile-tRNAIleLAU bound in the A/T conformation. b, The near-cognate AUG codon weakens the interactions with the decoding center, evidenced by the gap between G530 and L34 in structure II. c, Water-mediated interactions (gray dashed lines) between L34 of Ile-tRNAIleLAU in structure I and the G530 loop in the decoding center. The putative water ‘W’ is shown as a yellow sphere.
Cryo-EM of the ribosome with accommodated tRNAIleLAU
Following the recognition of the cognate AUA codon, EF-Tu hydrolyzes GTP and inorganic phosphate (Pi) is released, leading to a conformational change in EF-Tu allowing Ile-tRNAIleLAU to accommodate into the A site. We set to determine the structure of a post-accommodated state ribosome programmed with AUA and bound to tRNAIleLAU in the A site (structure III). Similarly, complex IV was programmed with the AUA codon and tRNAIleLAU in the P site, mimicking a post-translocated ribosome state.
Our pre-accommodated structure I represents a state similar to that previously reported with Phe-tRNAPhe bound to EF-Tu•GDPCP (Extended Data Fig. 8a)1. In structure I, the ternary complex of EF-Tu•GDPCP•Ile-tRNAIleLAU is in the GTP-activated state with an ordered switch I in the G-domain of EF-Tu and the catalytic His84 within interaction distance from the nonbridging oxygen of A2662 in the sarcin–ricin loop (Extended Data Fig. 8b,c). The EM map shows clear density for GDPCP bound to EF-Tu and for the isoleucine residue attached to A76 of the Ile-tRNAIleLAU in the A/T conformation (Extended Data Figs. 4a and 8d,e).
The A-site tRNAIleLAU in structure III is fully accommodated into the A site (Fig. 1c,g), and the lysine chain of L34 does not change its position during tRNA accommodation (Figs. 1i,k and 2c,e). This is consistent with the nature of the conformational change upon aa-tRNA transitioning from the A/T to the A/A conformation during the accommodation into the A site, tRNA movement that is mediated through the kink at the junction of the ASL and the D-stem (Extended Data Fig. 6c)3. The base pairs between the ASL and the AUA codon in the A site are preserved, thereby maintaining the translation reading frame. In both structures I and III with the A-site AUA, clear density for L34 is seen with the carboxyl group of the lysine moiety forming a water-mediated interaction with the G530 loop (Fig. 4a,c and Extended Data Fig. 7a,d). The structures suggest that one additional interaction may form between the amine of the lysine side chain and the 2′OH of A7 in the mRNA (Extended Data Fig. 7e,f). In both structures, the polar side chain of L34 does not alter the recognition of the third nucleotide of the A-site codon (A6), with clear density for the potassium ion coordinating O6 of G530, O2 of C518 and the carbonyl oxygen of Pro45 in ribosomal protein uS12. This K+ ion is observed in elongation ribosome complexes and stabilizes nucleotide A6 of mRNA (Supplementary Fig. 1)40. The packing between L34 and nucleotide G530 observed in both structures I and III probably fastens the codon–anticodon helix into the decoding center, leading to rapid GTP hydrolysis by EF-Tu and tRNA accommodation upon AUA decoding.
Structure IV contains tRNAIleLAU bound to the P site, mimicking a post-translocated state (Fig. 1d,h). Despite the fact that the EM density attributable to L34 is weaker in the P site than in the A site, it is clear that L34 forms one hydrogen bond with the wobble position of the AUA codon, identical to that seen in the A site (Fig. 1l and Supplementary Fig. 2). Lysidine in the P-site tRNAIleLAU may also interact with the backbone of mRNA as previously proposed with tRNAIle(agm34C)AU (ref.36); however, the carboxyl and the amine groups of lysidine are not resolved in this map, indicating that these interactions, if any, are not responsible for the stabilization of tRNAIleLAU in the P site of the ribosome. Thus, structure IV suggests that once tRNAIleLAU is transferred to the P site, the primary role of L34 in the anticodon loop is to assure that the integrity of the base pairs with the mRNA is preserved.
Discussion
Structures of the 70S ribosome captured during decoding of the cognate AUA (structure I) and near-cognate AUG (structure II) codons reveal the molecular mechanism by which lysidine-containing tRNAIleLAU specifically decodes AUA over AUG. Ribosome pelleting assays of the complexes show that EF-Tu•GDPCP•Ile-tRNAIleLAU associates with ribosomes programmed with the AUG codon in the A site independently whether paromomycin is present or not (Supplementary Fig. 3). Based on this observation, we first collected cryo-EM data of EF-Tu•GDPCP•Ile-tRNAIleLAU bound to the ribosome in the process of decoding the near-cognate AUG codon in the absence of paromomycin. However, despite extensive particle sorting using focused 3D variability analysis and 3D classification, no class average displayed the EF-Tu-ternary complex bound to the ribosome. It is likely that the co-pelleting of EF-Tu•GDPCP•Ile-tRNAIleLAU with ribosomes programmed with the near-cognate AUG codon in the A site represents transient binding events, failing to form a stable complex for structure determination. We then assembled the complex in the presence of paromomycin, which allowed to capture EF-Tu•GDPCP•Ile-tRNAIleLAU bound to the near-cognate AUG codon on the ribosome. Despite the poor overall density of EF-Tu in this structure (Fig. 1f and Extended Data Fig. 4b), the density for the ASL and the decoding center region is well defined (Extended Data Fig. 7b,c), allowing us to elucidate the mechanism of AUG avoidance. In structures I and II, the 30S subunit has the same conformation, in which the shoulder and the head domains tighten the decoding center as previously observed upon decoding of cognate or near-cognate codons in the presence of paromomycin1,5,7,10.
In structure I with the cognate AUA codon in the A site, the first and second positions of the codon–anticodon helix form canonical WC base pairs. At the wobble position, L34 in tRNAIleLAU mispairs with adenosine forming one hydrogen bond between the exocyclic amine of L34 and the N1 of A6 in mRNA (Fig. 3c). The same base pair geometry has been observed in the decoding center at the wobble position upon UGA decoding by the Hirsh suppressor tRNATrp (ref.41) and decoding of AUA by the archaeal tRNA2Ile containing agmatidine36. Seemingly, the reduced stability of the wobble A:C base pair forming one hydrogen bond is compensated by the stacking interaction mediated by the lysine moiety of L34 and nucleotide G530 (Fig. 4a). The lysine side chain is further stabilized in the decoding center by water-mediated interactions with the nonbridging phosphate oxygen of nucleotide G530 and the N4 of C518 (Fig. 4c).
Upon decoding of the near-cognate AUG codon, the presence of L34 makes it incompatible to interact with G at the wobble position of the codon. As previously proposed36, if L34 takes the conformation seen in structure I, the lysine side chain of L34 would collide with the exocyclic amine of G and the exocyclic amine of L34 would be facing the hydrogen bond donor N3 of G (Extended Data Fig. 5d). It is believed that these repulsive interactions at the wobble position would lead to tRNA rejection. While plausible, such chemical incompatibility would probably be ‘absorbed’ by the wobble character of the third position of the mRNA, which is not as closely monitored by the decoding center allowing nonstandard base pairs to form. Structure II visualizes the structural defects in the decoding center caused by the presence of L34 during decoding of the near-cognate AUG codon and provides the basis for its rejection.
Whereas canonical WC interactions are seen at the first position between the AUG codon and the anticodon of tRNAIleLAU, the second and third positions are deprived of hydrogen bonds between the tRNA and mRNA. The third nucleotide of the mRNA, G6, flips to the syn-conformation relieving the steric hindrance with L34. In this conformation, the distance between the exocyclic amine of L34 and the acceptors of hydrogen bonds N7 or O6 of G is ~4.3 Å and 3.9 Å, respectively, too long to establish hydrogen bonds (Fig. 3c). In the syn-conformation, the nucleobase of G6 inclines to avoid a collision between its exocyclic amine and the sugar-phosphate backbone of the mRNA. Through π–π stacking interactions, U5 also inclines bringing its WC edge away from A35 of tRNA, with distances between hydrogen bond donors and acceptors exceeding 3.5 Å, the threshold for hydrogen-bond formation (Fig. 3b and Extended Data Fig. 6a,b)42. Similarly, through stacking interactions, syn-G6 affects A7 which also flips to the syn-conformation, maximizing the contact with G6 (Extended Data Figs. 6b and 7c). The lack of hydrogen bond between L34 and syn-G6 in structure II introduces flexibility in L34, which is characterized by the altered position of the lysine moiety relative to that in structures I and III with AUA. Correspondingly, the gap between L34 and G530 suggests a lack of stabilization of tRNA by the decoding center upon AUG decoding (Fig. 4b), evidenced by the weak EM density of EF-Tu and the acceptor stem of Ile-tRNAIleLAU (Extended Data Fig. 4b).
The aminoglycoside antibiotic paromomycin stabilizes the binding of near-cognate aminoacyl-tRNAs to the ribosome, promoting decoding errors6. One legitimate concern is that paromomycin may affect the conformation of the mRNA-tRNAIleLAU duplex in structure II with the near-cognate AUG codon. Two observations suggest that the drug does not alter decoding of mRNA by the ribosome: (1) the conformation of the mRNA and the A/T-Ile-tRNAIleLAU in structures I (no paromomycin) and II (with paromomycin) is identical (Extended Data Fig. 6a), and (2) the conformation of the mRNA and the A-site ASL in structure I is identical to that in structure III with accommodated A-site tRNAIleLAU and paromomycin (Extended Data Fig. 6c).
In this study, we elucidated the mechanism of accurate AUA decoding and how the AUG codon is avoided in E. coli by the minor tRNAIleLAU. The geometry of the mispair at the wobble position mediated by L34 in eubacterial tRNAIleLAU, and agm34 in archaeal tRNA2Ile (ref.36), exemplifies the role of post-transcriptional modifications of tRNAs for accurate reading of the genetic code and maintenance of the reading frame.
Methods
Preparation of 70S ribosomes, EF-Tu, TilS, IleRS, Ile-tRNAIleLAU and mRNA
The full-length EF-Tu, TilS and IleRS sequences were polymerase chain reaction amplified from Escherichia coli MRE600 genomic DNA and cloned into pET21a plasmid (Novagen) carrying C-terminal 6x His tag. E. coli BL21 (DE3) Star (C601003, Invitrogen) cells transformed with these constructs were grown in the lysogeny broth medium supplemented with 50 μg ml−1 carbenicillin (C-103–25, Gold Biotechnology) and 1 mM ZnCl2 (for the expression of IleRS to ensure formation of the zinc finger domain43) to an absorbance of 0.6 at 600 nm before inducing expression of C-His–E. coli (Ec)EF-Tu/TilS/IleRS with 0.25 mM (EF-Tu and TilS) or 1 mM (for IleRS) isopropylthiol-β-D-galactoside (12481C50, Gold Biotechnology) for 4 h at 37 °C. Purification of C-terminal 6x His-tagged E. coli EF-Tu was done as previously described1, with some modifications. Briefly, cells were lysed in buffer A (50 mM HEPES–KOH, pH 7.5, 60 mM NH4Cl, 7 mM MgCl2, 15 mM imidazole, 500 mM KCl and 5% glycerol), supplemented with one tablet of complete EDTA-free protease inhibitor cocktail (Thermo Scientific) and lysozyme. The resuspended cells were lysed by passing several times through an LM20 high-pressure homogenizer (Microfluidics) operated at 15,000 psi. The cell debris was removed by centrifugation at 30,625g at 4 °C for 30 min. The filtered lysate (through a 0.22 μm filter, Millipore) was applied to an equilibrated 5 ml HisTrap HP (GE Healthcare) column. EcEF-Tu (C-His) was eluted with a linear gradient of imidazole to 250 mM. Fractions containing EcEF-Tu (C-His) were pooled, concentrated and further purified on a Source 15Q (HR 16/10) (Cytiva) anion exchange column (20 ml), pre-equilibrated in buffer B (50 mM HEPES–KOH, pH 7.5, 60 mM NH4Cl, 7 mM MgCl2, 7 mM β-mercaptoethanol and 5% glycerol) and eluted with a linear gradient of NaCl to 1 M. Peak fractions were concentrated and purified on a Superdex 200 HiLoad 16/60 (GE Healthcare) size-exclusion chromatography column equilibrated in buffer 5 mM HEPES–KOH, pH 7.5, 60 mM NH4Cl, 7 mM MgCl2, 7 mM β-mercaptoethanol and 50 mM NaCl, and finally flash-frozen and stored at −80 °C.
The filtered crude lysate, containing C-terminal 6x His-tagged E. coli TilS, was applied onto a HisTrap HP column, pre-equilibrated in buffer A (20 mM Tris–HCl pH 8.0, 500 mM NaCl, 5 mM imidazole and 1 mM PMSF) supplemented with 1 tablet of EDTA-free protease inhibitor cocktail, washed with 20 mM imidazole, and eluted with a linear gradient of imidazole to 500 mM. Fractions containing EcTilS (C-His) were pooled, concentrated and purified on a Source 15Q column, pre-equilibrated in buffer B (20 mM Tris–HCl pH 8.0 and 10 mM β-mercaptoethanol) and eluted with a linear gradient of NaCl to 1 M. Finally, EcTilS (C-His) was purified on a Superdex 200 HiLoad 16/60 column in buffer 20 mM Tris–HCl pH 8.0, 10 mM β-mercaptoethanol, 150 mM NaCl, and flash-frozen and stored at −80 °C.
E. coli IleRS was purified as described previously44, with some modifications. Briefly, the lysate containing EcIleRS (C-His) was applied onto a HisTrap HP column, pre-equilibrated in buffer A (20 mM Tris–HCl pH 7.0, 500 mM NaCl, 10 mM imidazole, 0.1 mM PMSF and 5 mM β-mercaptoethanol) supplemented with one tablet of EDTA-free protease inhibitor cocktail and eluted with a linear gradient of imidazole to 250 mM. The fractions containing EcIleRS were pooled and purified on a Source 15Q column in buffer B (20 mM Tris–HCl pH 7.0 and 10 mM β-mercaptoethanol) and eluted with a linear gradient of NaCl to 1 M. Finally, EcIleRS (C-His) was purified on a Superdex 200 HiLoad 16/60 column in buffer 20 mM Tris–HCl pH 7.0, 10 mM β-mercaptoethanol and 200 mM NaCl, and flash-frozen and stored at −80 °C.
E. coli initiator tRNAfMet was purified using previously established procedures45. E. coli tRNAIleCAU was cloned into the pBSTNAV plasmid and expressed under the constitutive lpp promoter overnight in E. coli JM109 cells in 2× yeast extract tryptone medium. Total tRNA was extracted using phenol-chloroform, followed by 96% ethanol precipitation. tRNAIleCAU was purified by anion exchange on Source 15Q (HR 16/10) (Cytiva), and reversed-phase chromatography was performed on PROTO 300 C4 high-performance liquid chromatography (10 mm × 250 mm) (Higgins Analytical) columns. The deacylated-tRNAIleCAU was lysidinylated with TilS in buffer containing 100 mM HEPES pH 8.0, 10 mM KCl, 10 mM MgCl2, 10 mM DTT, 2 mM ATP and 10.9 mM L-lysine for 60 min at 37 °C, then aminoacylated with E. coli isoleucyl-tRNA synthetase (50 μM tRNAIleLAU, 2.5 μM IleRS in the aminoacylation buffer supplemented with 2.5 mM ATP and 2.5 mM L-isoleucine for 30 min at 37 °C) and finally purified by reversed-phase high-performance liquid chromatography on the C4 column. The purity of deacylated and lysidinylated tRNAIleLAU was assessed in the aminoacylation reaction with the E. coli methionyl-tRNA synthetase (50 μM tRNAIleLAU, 7.7 μM MetRS in aminoacylation buffer supplemented with 10 mM ATP and 10 mM L-methionine for 60 min at 37 °C). The resulting profile on the C4 column did not show any peak for aminoacylated tRNA, confirming that our tRNAIleLAU was lysidinylated and thus recognized only by IleRS, and not by MetRS (Supplementary Fig. 4).
E. coli 70S ribosomes, isolated from strain MRE600, were prepared as previously described46. The 27-mer mRNA (27-M-AUA), with the sequence 5′-GGC AAG GAG GUA AAA AUG AUA AAA GAA-3′, and the AUG codon in the P site and the AUA codon in the A site (underlined), was used to assemble ribosome complexes resulting in structures I and III. The 27-mer mRNA (27-M-AUG), with the sequence 5′-GGC AAG GAG GUA AAA AUG AUG AAA GAA-3′, and the AUG codon in the A and P sites (underlined), was used to generate structure II. The 21-mer mRNA (21-AUA-F), with the sequence 5′-C AAG GAG GUA AAA AUA UUC UA-3′ and the AUA codon in the P site (underlined), was used in structure IV. The mRNAs were chemically synthesized by Integrated DNA Technologies.
Ribosome binding assays
E. coli 70S ribosomes (final concentration 0.5 μM) were programmed with Met-AUA or Met-AUG mRNA (1.25 μM) and fMet-tRNAfMet (1 μM) by incubation at 37 °C for 10 min in 5 mM Tris–HCl pH 7.4, 60 mM NH4Cl, 10 mM MgCl2 and 6 mM β-mercaptoethanol. Then, paromomycin (P9297, Sigma-Aldrich) was added (to Met-AUG-programmed ribosomes) to the final concentration of 50 μM and incubated for 10 min at room temperature. In parallel, 0.5 μM EF-Tu was pre-incubated with 250 μM GDPCP for 5 min at 37 °C, and then incubated for 2 min with Ile-tRNAIleLAU at 37 °C. The elongation ternary complex was added to the initiation complex mixture and incubated for 10 min at room temperature. The mixture was centrifuged through a cold sucrose cushion buffer (5 mM Tris–HCl pH 7.4, 60 mM NH4Cl, 10 mM MgCl2, 6 mM β-mercaptoethanol and 30% sucrose) in the TLS55 rotor (Beckman Coulter) for 1 h at 214,000g at 4 °C. The top 40 μl sample solution and the bottom 20 μl were precipitated with 100% trichloroacetic acid (02215618, Thermo Scientific Chemicals), pelleted by centrifugation, washed with ice-cold acetone and dissolved in 1× sodium dodecyl sulfate protein-loading buffer. Results were analyzed via 4–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (M00654, SurePage, GenScript) and stained with Coomassie. The data are summarized in Supplementary Fig. 3.
Sample preparation, cryo-EM and data acquisition
Ribosome complexes (20 μl) with A-site tRNAIleLAU (70S-A-site Ile-tRNAIleLAU) and P-site tRNAfMet were prepared by incubating 2 μM E. coli MRE600 70S ribosomes with 8 μM mRNA (27-M-AUA for complexes I and III or 27-M-AUG for complex II) in 1× ribosome buffer (5 mM Tris–HCl pH 7.4, 60 mM NH4Cl, 10 mM MgCl2 and 6 mM β-mercaptoethanol) at 37 °C for 5 min. fMet-tRNAfMet was added to the final concentration of 8 μM and incubated for 5 min. Paromomycin (P9297, Sigma-Aldrich) (complexes II and III) was added to the final concentration of 100 μM and incubated at room temperature for 10 min. Finally, 15 μM Ile-tRNAIleLAU was added and incubated for 10 min (complex III). For complexes I and II (with EF-Tu), 10 μl of the elongation mix obtained by incubating 60 μM (final—30 μM) with 2 mM GDPCP (final—1 mM) for 5 min at 37 °C, and then with 40 μM Ile-tRNAIleLAU (final—20 μM) for 1 min at 37 °C, was mixed with initiation complexes (10 μl) and incubated for 10 min at room temperature.
For the ribosome complex IV with P-site tRNAIleLAU (70S-P-site tRNAIleLAU), 2 μM E. coli MRE600 70S ribosomes were programmed with 21-AUA-Phe mRNA by incubation in 1× ribosome buffer at 37 °C for 5 min. Then, Ile-tRNAIleLAU was added to the final concentration of 20 μM and incubated for 10 min at room temperature. All complexes were kept on ice for a few minutes before being applied onto cryo-EM grids.
Quantifoil R2/1 gold 200 mesh grids (Q2100AR1, Electron Microscopy Sciences) were pre-cleaned for 30 s in an H2O2 atmosphere using the Solarus 950 plasma cleaner (Gatan). The mixture (4 μl), containing 2 μM E. coli 70S•A-site tRNAIleLAU/70S•EF-Tu•GDPCP•A-site Ile-tRNAIleLAU/70S•P-site tRNAIleLAU, was applied onto grids, blotted in 85% humidity at 22 °C for 24 s and plunged-frozen in liquid nitrogen-cooled ethane using a Leica EM GP2 cryo-plunger. Grids were transferred into a Titan Krios G3i electron microscope (ThermoFisher Scientific) operating at 300 keV and equipped with a Falcon III direct electron detector camera (ThermoFisher Scientific). The image stacks (movies) were acquired with a pixel size of 0.86 Å px−1 (structures I, II and III) or 0.85 Å px−1 (structure IV). Data collection was done in the EPU software (ThermoFisher Scientific) setup to record movies with 39 (for structures I, II, IV) or 20 fractions (for structure II) with a total accumulated dose of 40 e− Å−2 per movie. A total of 10,381 (I)/10,734 (II)/13,536 (III)/9,711 (IV) image stacks were collected with a defocus ranging between −1 and −2.3 μm. The statistics of data acquisition are summarized in Table 1.
Cryo-EM data processing
Data processing was done in cryoSPARC 3.3.2 (ref.47). The image stacks were imported into cryoSPARC and gain corrected. Image frames (fractions) were motion corrected with dose weighting using the patch motion correction, and patch contrast transfer function (CTF) estimation was performed on the motion-corrected micrographs. Based on the relative ice thickness, CTF fit, length and curvature of motion trajectories, 10,207 (dataset I—EF-Tu•GDPCP•Ile-tRNAIleLAU with the AUA codon in the A site)/10,417 (dataset II—EF-Tu•GDPCP•Ile-tRNAIleLAU with the AUG codon in the A site)/13,462 (dataset III—A-site tRNAIleLAU with the AUA codon in the A site)/9,486 (dataset IV—P-site tRNAIleLAU with the AUA codon in the P site) micrographs were selected for further processing (Extended Data Figs. 1, 2, 9 and 10).
For dataset I (70S•EF-Tu•GDPCP•Ile-tRNAIleLAU•AUA), 1,094,211 particles were picked using the circular ‘blob’ picker in cryoSPARC and filtered on the basis of defocus-adjusted power and pick scores to 1,070,785 particles. Then, particles were subjected to two rounds of reference-free 2D classification. After discarding bad particles, 682,429 particles were selected from 2D classification and used to generate the ab initio volume. Using ‘heterogeneous refinement’ in cryoSPARC with five groups, these particles were further classified into four ribosome class averages. One class average did not yield anything meaningful and was considered not specimen-related (6,992 particles), and the other class average contained 50S subunits (2,713 particles), which were both discarded from further processing. Class average I contained solid density for EF-Tu, A-, P- and E-site tRNAs (611,980 particles); the density of EF-Tu in class average II (60,744 particles) was weak and not further processed. To further classify particles in class average I, we performed focused 3D variability analysis with a mask around EF-Tu bound to A-site Ile-tRNAIleLAU. This process allowed the removal of 33,690 ‘bad’ noisy particles and 160,188 particles with weak density for EF-Tu, leaving one main class with 418,102 particles displaying solid density for EF-Tu, A-, P- and E-site tRNAs. We further polished the class containing solid EF-Tu density with a second round of 3D variability using a new mask around the A-site Ile-tRNAIleLAU and EF-Tu. This approach allowed us to remove 54,465 ‘bad’ or noisy particles, 200,010 particles with noisy/no bound factor and 30,745 particles with weak density for EF-Tu, leaving 135,882 particles with solid density for EF-Tu and the A-site tRNA. These particles were re-extracted to full size (512 × 512 pixel box), and nonuniform with CTF refinement in cryoSPARC yielded reconstructions with a nominal resolution of 2.9 Å for structure I (70S•EF-Tu•GDPCP•Ile-tRNAIleLAU•AUA) (Extended Data Figs. 1 and 3).
Dataset II (70S•EF-Tu•GDPCP•Ile-tRNAIleLAU•AUG) was similarly processed; 1,080,870 particles were picked using the circular ‘blob’ picker in cryoSPARC, which were filtered on the basis of defocus-adjusted power and pick scores to 1,073,026 particles. Two rounds of 2D classification selected 764,480 particles, which were used to generate ab initio volumes with four groups. ‘Heterogeneous refinement’ was used to sort particles into one class average (749,653 particles), discarding 14,827 ‘bad’ particles. The class average, composed of two groups from the ‘heterogeneous refinement’ job, represents nonrotated 70S ribosomes that appear to be weakly bound to EF-Tu and A-site tRNA, and with solid density for P- and E-site tRNAs. The particles were classified using focused 3D variability with a spherical mask (45 Å) around the A-site tRNA and EF-Tu binding regions. Within this class, 420,466 particles had P- and E-site tRNAs and no EF-Tu bound; 281,493 particles had no EF-Tu bound, with weak density for the A-site tRNA and solid density for P- and E-site tRNAs; and 47,694 particles had bound A-, P- and E-site tRNAs, with weak density for EF-Tu. Re-extraction of these particles to full size followed with nonuniform and CTF refinement in cryoSPARC yielded a reconstruction with a nominal resolution of 3.1 Å for structure II (70S•EF-Tu•GDPCP•Ile-tRNAIleLAU•AUG) (Extended Data Figs. 2 and 3).
Processing of dataset III (70S•A-site tRNAIleLAU•AUA) followed a similar workflow. The 1,402,100 particles picked were filtered on the basis of defocus-adjusted power and pick scores to 1,371,433 particles. Two rounds of 2D classification selected 923,703 particles, which were used to generate ab initio volumes with five groups. ‘Heterogeneous refinement’ was used to sort particles into two ribosome class averages. Class I (853,477 particles) contained solid density for A-, P- and E-site tRNAs; class II (62,160 particles) had weak density for A-, P-, and E-site tRNAs, and 8,066 ‘bad’ noisy particles were discarded. The particles from class average I were classified using focused 3D variability with a mask around A-, P- and E-site tRNA regions. Within this class, 313,235 particles had P- and E-site tRNAs bound, 316,622 particles had only E-site tRNA bound, and 466,262 particles had solid density for A-, P- and E-site tRNAs. Re-extraction of these particles to full size followed with nonuniform and CTF refinement in cryoSPARC yielded a reconstruction with a nominal resolution of 2.8 Å for structure III (70S•A-site tRNAIleLAU•AUA) (Extended Data Figs. 3 and 9).
Dataset IV (70S•P-site tRNAIleLAU•AUA) was processed as above. A total of 941,432 particles were picked and filtered on the basis of defocus-adjusted power and pick scores to 931,907 particles. Two rounds of 2D classification selected 586,922 particles, which were used to generate ab initio volumes with five groups. ‘Heterogeneous refinement’ was used to sort particles into two main ribosome class averages. Class I (338,813 particles) had solid density for P- and E-site tRNAs; class II (207,069 particles) had density for only E-site tRNA. The 50S subunits (34,690 particles) and 6,350 ‘bad’ noisy particles were discarded. The particles from class average I were further classified using focused 3D variability with a mask encompassing the P- and E-site tRNA binding sites. Within this class, 167,286 particles had P- and E-site tRNAs bound; 102,964 particles contained only the E-site tRNA. A second round of focused 3D variability was applied with a mask around the P-site tRNA. A total of 131,911 particles had solid density for P- and E-site tRNAs, 29,004 particles contained only the E-site tRNA and 5,552 particles were discarded. Re-extraction of these particles to full size followed with nonuniform and CTF refinement in cryoSPARC yielded a reconstruction with a nominal resolution of 3.1 Å for structure IV (70S•P-site tRNAIleLAU•AUA) (Extended Data Fig. 3 and 10).
Model building and refinement of the E. coli 70S ribosome complexes
The models were assembled from individual parts. As a starting model, the 30S and 50S subunits were taken from the high-resolution structure of the E. coli 70S ribosome (PDB 7K00 (ref.48)) and rigid-body fitted into the 2.8 Å resolution density map of structure I using UCSF Chimera 1.14 (ref.49). The tRNAifMet and EF-Tu were taken from PDB 5UYM1, and the model of the Ile-tRNAIleLAU was generated in Coot50 by mutating the corresponding nucleotide sequence of Phe-tRNAPhe from 5UYM1. Then, the tRNAs were rigid-body fitted into the EM density using UCSF Chimera49 and adjusted in Coot50. We observe clear density for the isoleucine residue attached to A76 of the E-site tRNA in all four complexes (I–IV). This is probably caused by the excess of Ile-tRNAIleLAU relative to ribosomes used during complex formation. Similarly, a potentially aminoacylated tRNA bound in the E site has recently been reported in the structure of a translating E. coli ribosome stalled with the polyketide tetracenomycin X (ref.51). The resulting model of the E. coli 70S ribosome was then rigid-body fitted into the maps of complexes II, III and IV. The complete models of the E. coli 70S ribosome complexes with ordered solvent and bound EF-Tu•GDPCP, A-site Ile-tRNAIleLAU, P-site tRNAifMet, and E-site Ile-tRNAIleLAU (structures I and II), A-site tRNAIleLAU, P-site tRNAifMet, and E-site Ile-tRNAIleLAU (structure III), P-site tRNAIleLAU and E-site Ile-tRNAIleLAU (structure IV) were real-space refined into the EM maps for five cycles using PHENIX 1.19.2 (ref.52), including global energy minimization and group ADP refinement strategies along with base pair restraints for rRNA and tRNA together with Ramachandran and secondary structure restraints. The resulting models were validated using the comprehensive validation tool for cryo-EM in PHENIX53 (Table 1).
Extended Data
Extended Data Fig. 1.
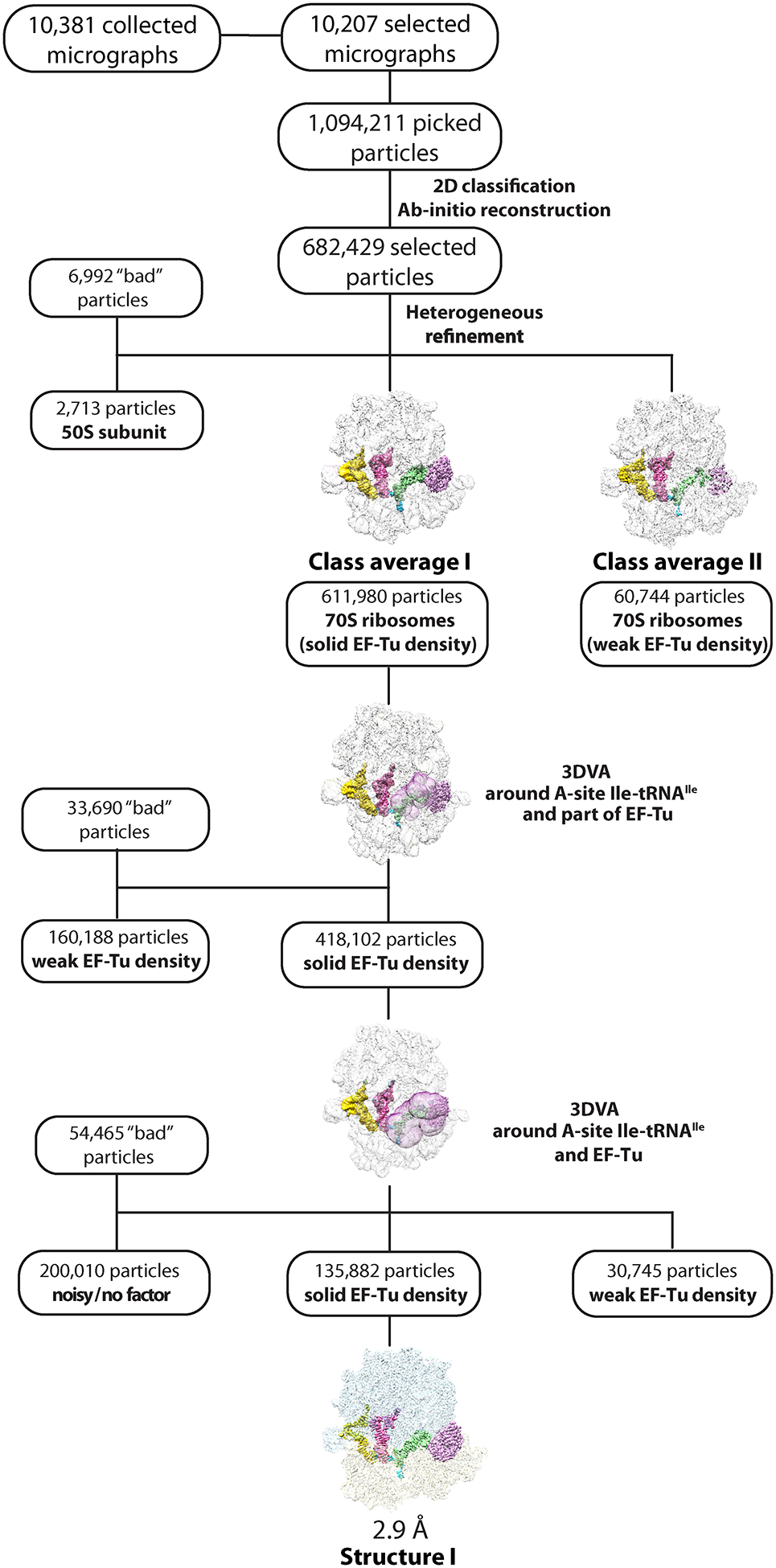
Cryo-EM data processing and particle classification workflow for structure I.
In this 70S ribosome complex, EF-Tu•GDPCP•Ile-tRNAIleLAU is bound to the cognate AUA codon in the A site. See Methods for details.
Extended Data Fig. 2.
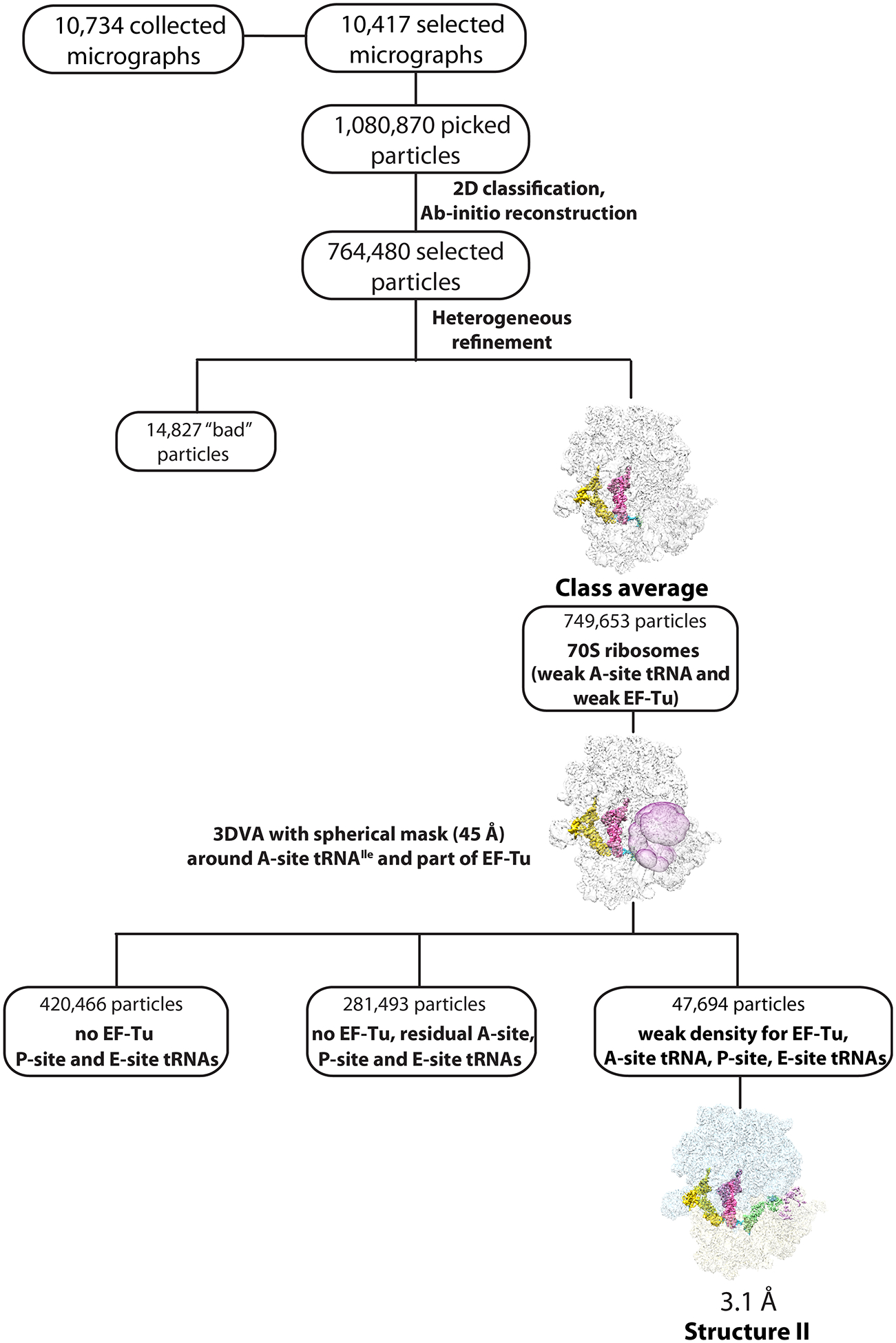
Cryo-EM data processing and particle classification workflow for structure II.
In this 70S ribosome complex, EF-Tu•GDPCP•Ile-tRNAIleLAU is bound to the near-cognate AUG codon in the A site in the presence of paromomycin. See Methods for details.
Extended Data Fig. 3.
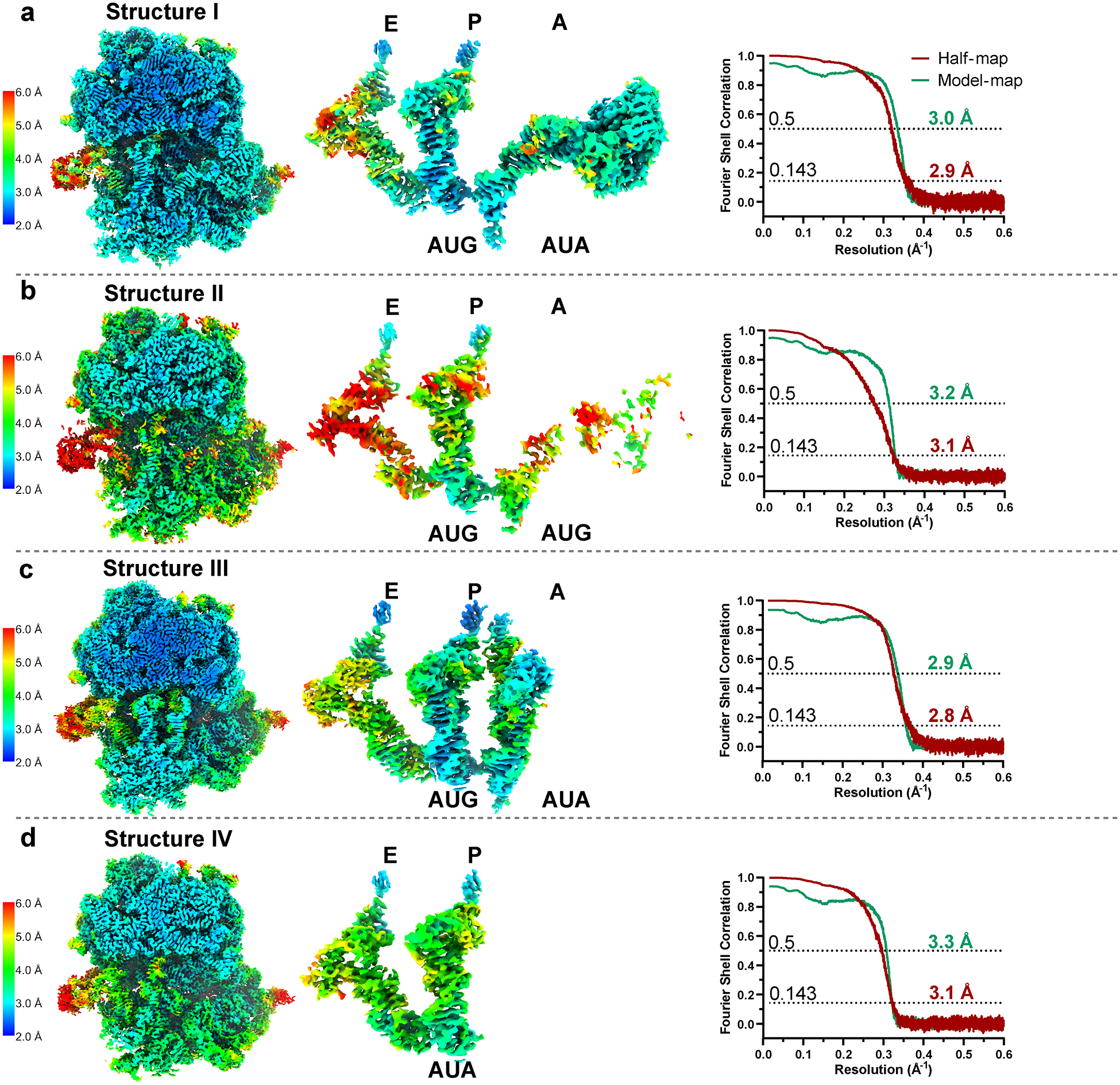
Local resolution estimation and Fourier Shell Correlation (FSC) validation.
Local resolution heat maps on slices of density from structures I (a, EF-Tu•GDPCP•Ile-tRNAIleLAU with A-site AUA), II (b, EF-Tu•GDPCP•Ile-tRNAIleLAU with A-site AUG), III (c, tRNAIleLAU with A-site AUA), and IV (d, tRNAIleLAU with P-site AUA) shown in the range of 2–6 Å resolution, calculated with cryoSPARC 3.3.2 implementation of BlocRes56. The gold-standard FSC curves of each half-map (red), using a ‘soft mask’ excluding solvent and model-map (green), are plotted across resolution. Map and model validation were performed in PHENIX 1.19.2 (ref.53).
Extended Data Fig. 4.
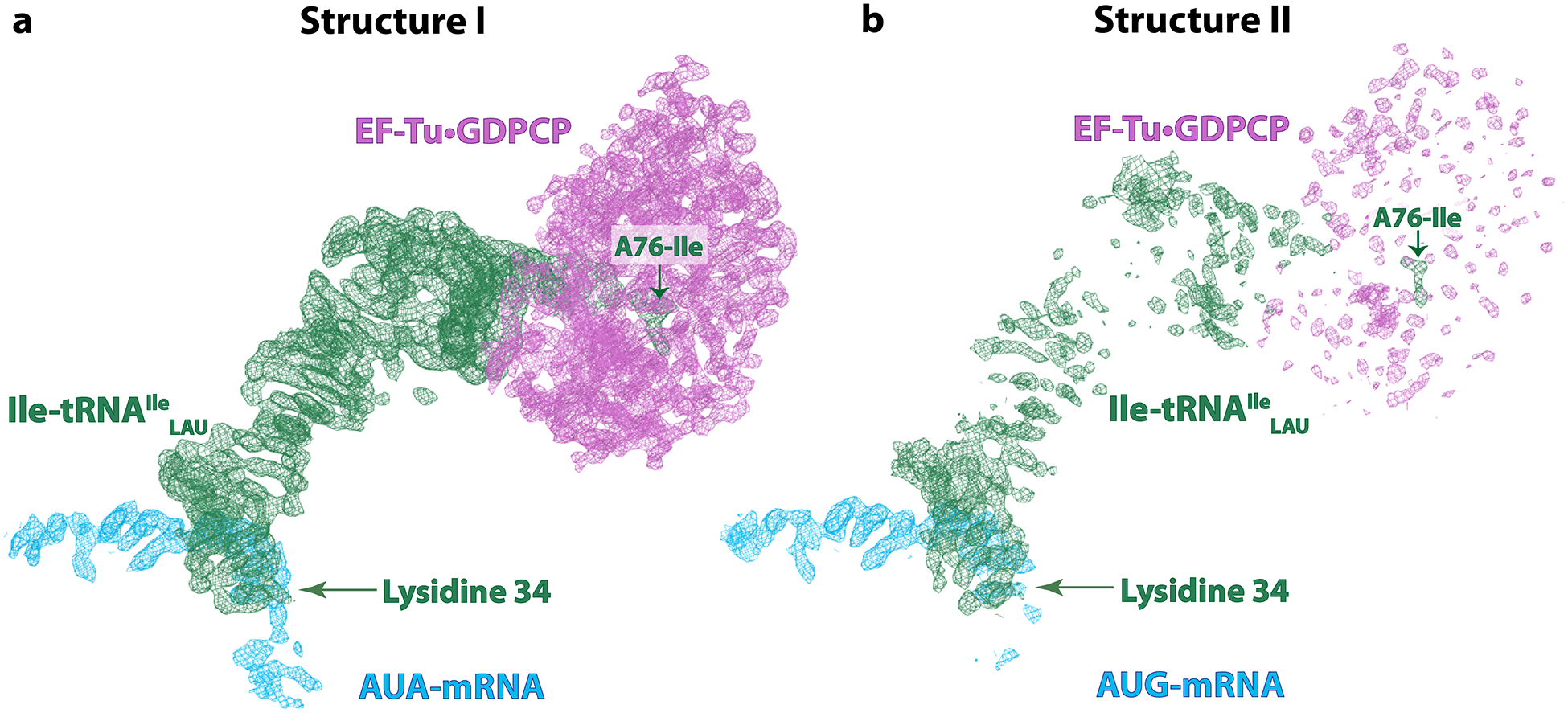
Cryo-EM density of the EF-Tu ternary complex in structures I (A-site AUA) and II (A-site AUG).
a, Well-defined density of EF-Tu, Ile-tRNAIleLAU and GDPCP in structure I upon decoding of the cognate AUA codon in the A site. b, In structure II with the near-cognate AUG codon, the presence of paromomycin stabilizes the anticodon loop in the decoding center, while the density for the other parts of Ile-tRNAIleLAU and EF-Tu is poorly resolved, suggesting unstable binding to the ribosome. The Coulomb potential density (shown as mesh and contoured at 4.0σ) of EF-Tu·GDPCP is magenta, that of the A/T-Ile-tRNAIleLAU is green, and that of the AUA- or AUG-mRNA is blue. The isoleucine attached to residue A76 of tRNAIleLAU and the lysidine 34 modification in the anticodon loop are indicated.
Extended Data Fig. 5.
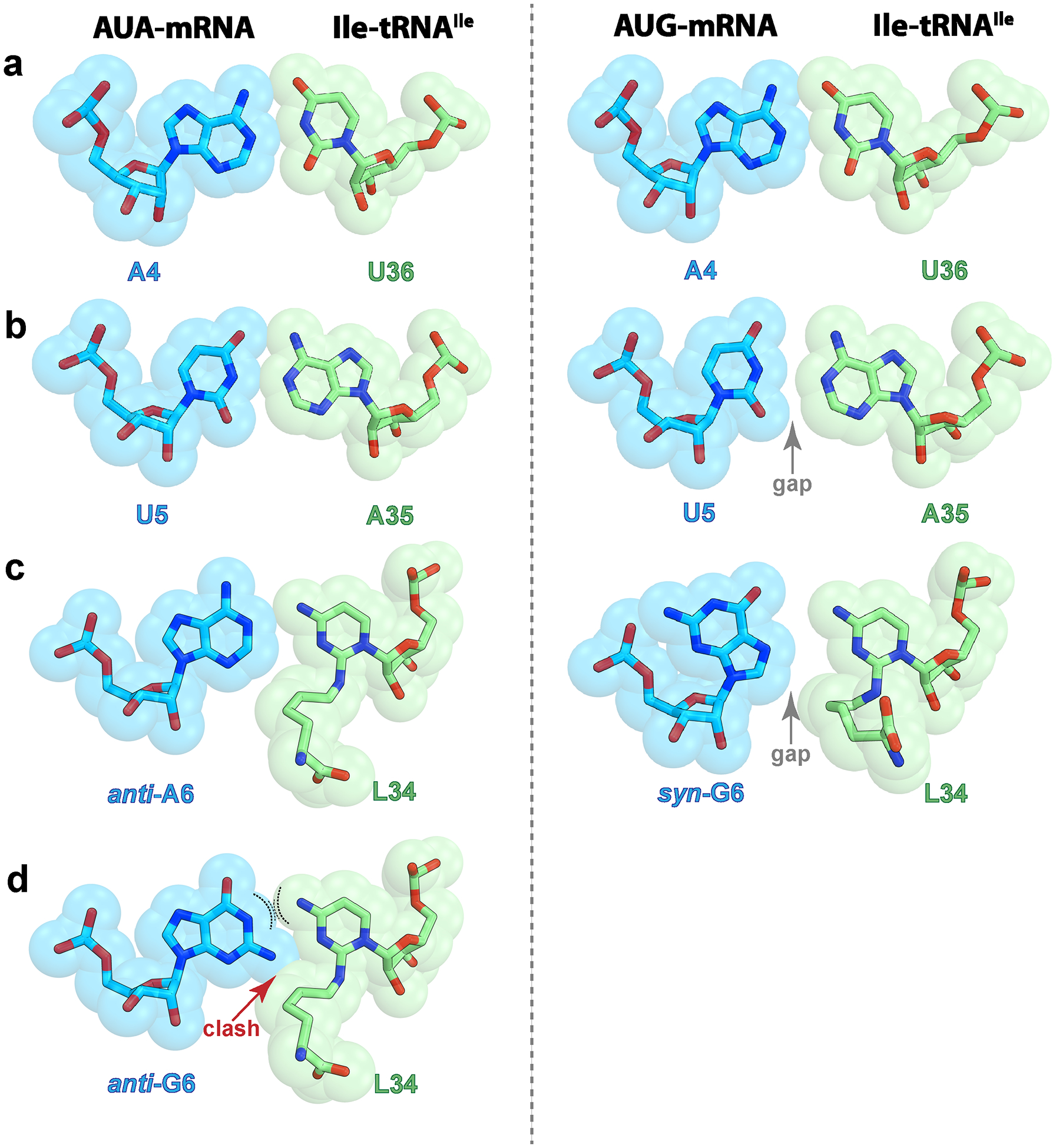
Base pair geometries in the codon-anticodon helix.
First (a), second (b), or third (c) position of the codon upon decoding of the cognate AUA (left, structure I) or the near-cognate AUG (right, structure II) codon. The gap that forms at the second and third positions in the complex with the AUG codon is indicated with a gray arrow. The hydrogen bonds between nucleotides are shown in Fig. 4. d, Potential non-favorable interactions between G6 in the usual syn-conformation and lysidine 34 during decoding of the near-cognate AUG codon. The curved dashed lines indicate repulsive forces between two donors of hydrogen bond.
Extended Data Fig. 6.
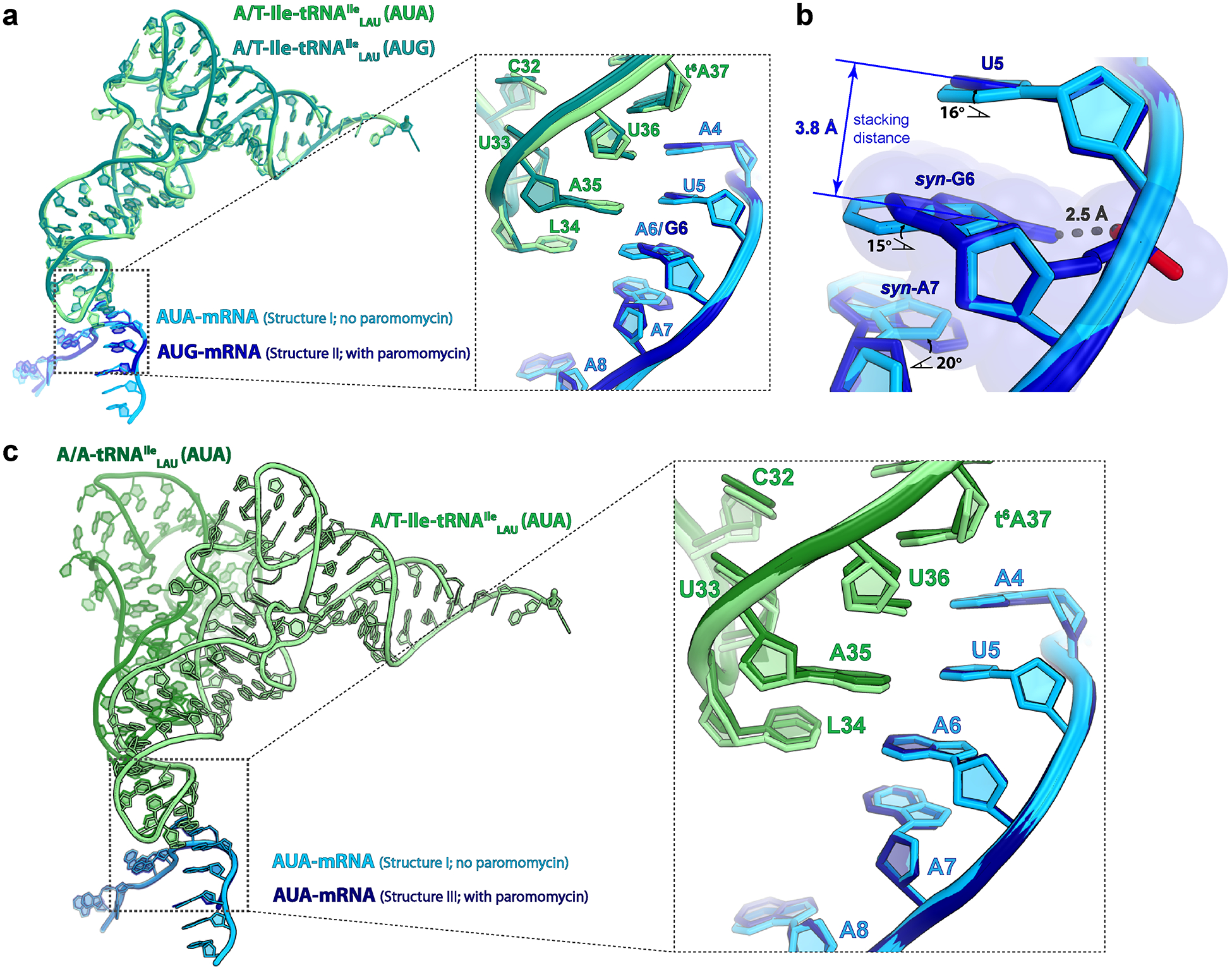
Comparison of the Ile-tRNAIleLAU in the A/T state and bound to EF-Tu in structures I (cognate A-site AUA) and II (near-cognate A-site AUG), and tRNAIleLAU in the A/A state in structure III (cognate A-site AUA).
a, Structure alignment based on the 23S rRNA shows that the Ile-tRNAIleLAU has the exact same conformation in structures I and II. The conformation of the mRNA bases in the A site diverges between structures I and II at the second and third position of the codon (inset). b, Close-up view of the second and third positions of the A-site codon. Relative to A6 in the AUA codon (structure I), syn-G6 inclines by ~15° to avoid a collision between the exocyclic amine and the phosphate oxygen of the mRNA backbone in the AUG codon (structure II). Through π-π stacking interactions, the effects of the inclined conformation of syn-G6 propagate to the neighboring bases, weakening the codon-anticodon base pairs with Ile-tRNAIleLAU. c, Structure alignment based on 23S rRNA showing that AUA-mRNA has the same conformation in structures I and III, regardless of the presence (structure III) or the absence (structure I) of paromomycin in the decoding center, confirming that paromomycin has no obvious effect on the conformation of the decoding center.
Extended Data Fig. 7.
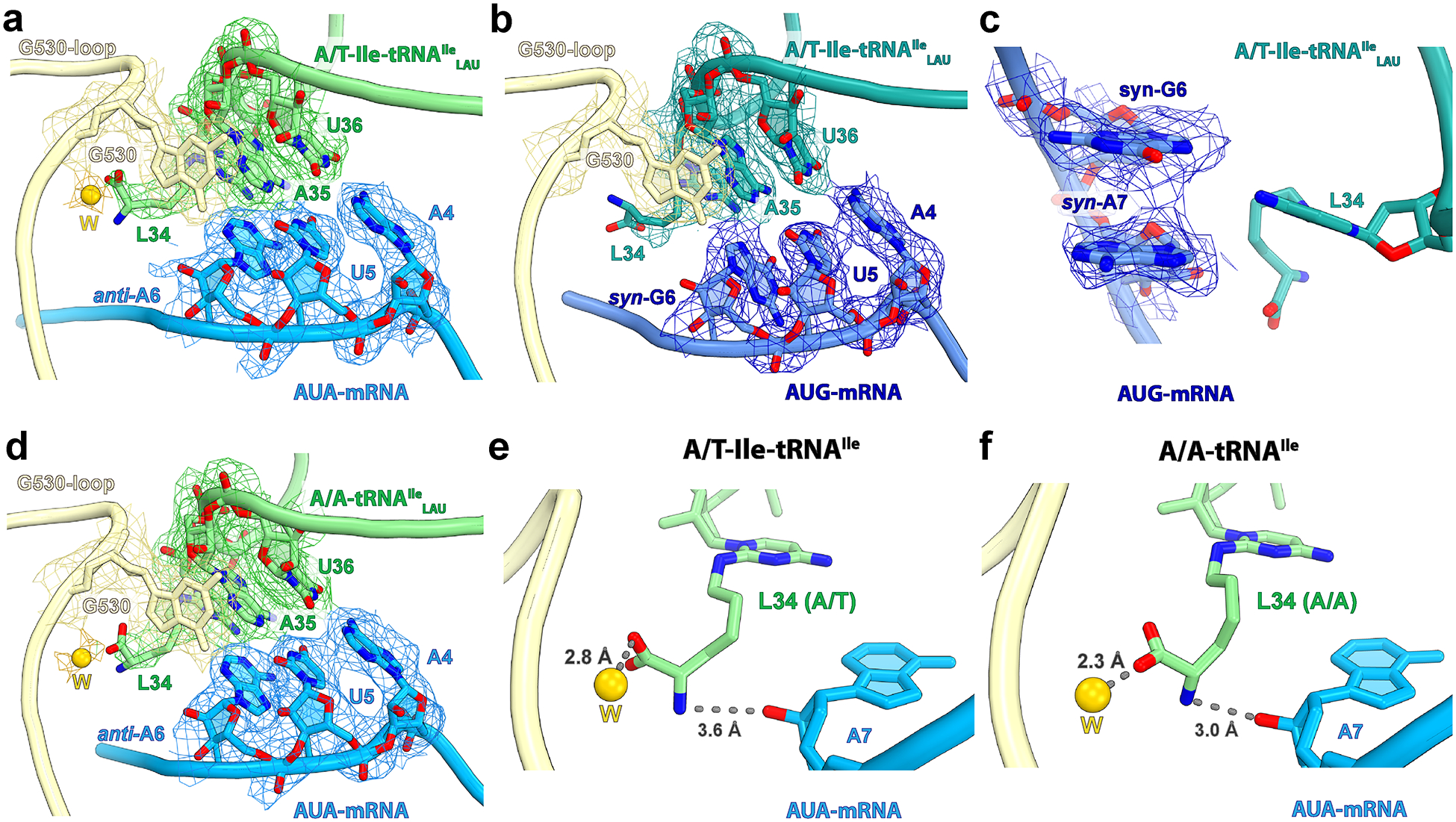
Cryo-EM density of the codon-anticodon region.
a, Structure I with the cognate AUA codon in the A site. Note that nucleotide A6 at the third position of the A-site codon is in the usual anti-conformation. b, Structure II with the near-cognate AUG codon in the A site. In this structure, nucleotide G6 at the wobble position of the A-site codon adopts the unusual syn-conformation. c, Nucleotide A7 immediately downstream of the A-site codon also adopts the syn-conformation in structure II, which maximizes stacking with syn-G6. d, EM map of the decoding center in structure III with accommodated A-site tRNAIleLAU bound to the cognate AUA codon. The Coulomb potential density is contoured at 2.9σ. e-f, The amine of the lysidine side chain in structures I (with the A/T-Ile-tRNAIleLAU) and III (with the A/A-tRNAIleLAU) may interact with the 2’OH group of A7 immediately downstream of the A-site codon. The putative water ‘W’ is shown as a yellow sphere and the gray dashed lines depict hydrogen bonds.
Extended Data Fig. 8.
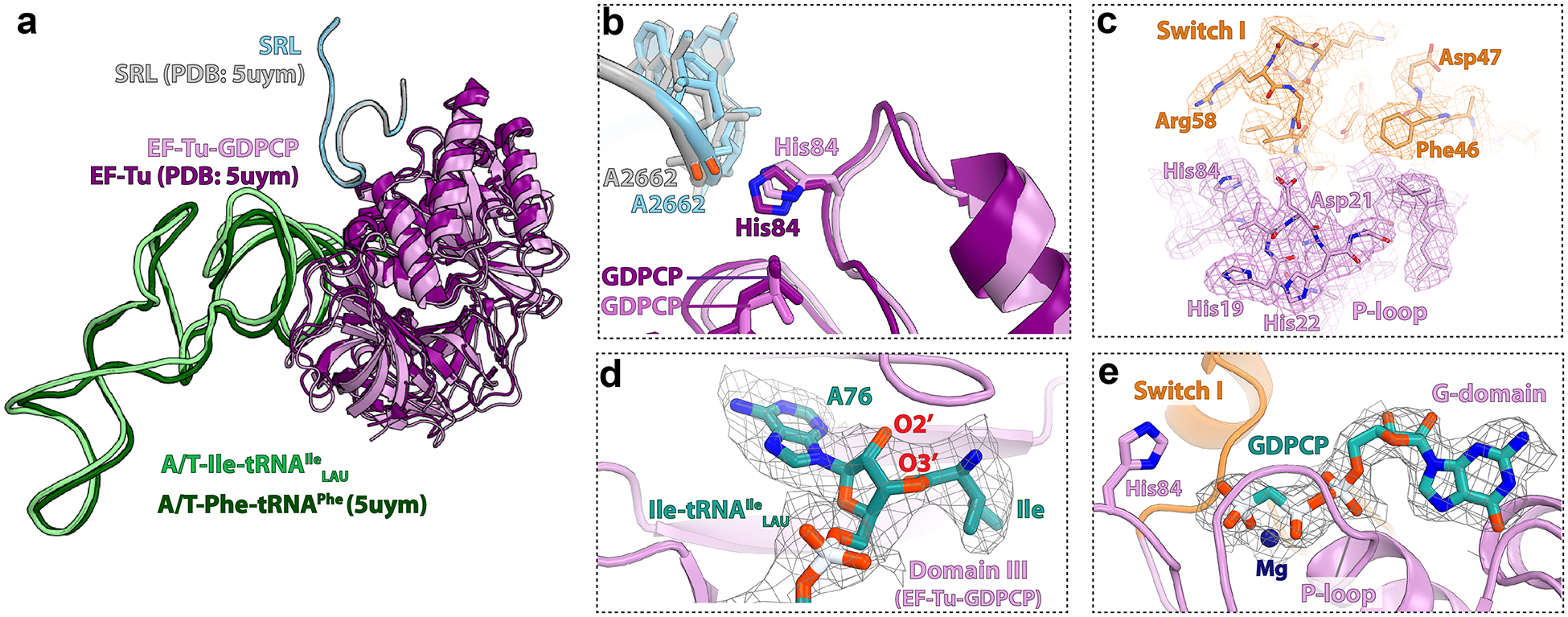
Comparison of the EF-Tu•GDPCP•Ile-tRNAIleLAU ternary complex in structure I with that from a previous study (5UYM1).
The conformation of EF-Tu and tRNA bound in the A/T state is the same as in the 70S ribosome•EF-Tu•GDPCP•Phe-tRNAPhe complex upon decoding of the cognate phenylalanine codon (PDB 5UYM)1. The structures are aligned based on the sarcin-ricin loop (SRL) region in the 50S subunit. a, The conformation of the EF-Tu•GDPCP•Ile-tRNAIleLAU ternary complex in structure I is in the GTP-activated state as reported previously1. b, The catalytic His84 in the G-domain of EF-Tu is within interaction distance from A2662 in the SRL. c, The quality of the EM map for EF-Tu in structure I allows to unambiguously model amino acid side chains. The density of the P-loop region is shown with magenta mesh, and that of the ordered switch I region with orange mesh. d, The EM density (gray mesh) of the isoleucine residue attached to nucleotide A76 of Ile-tRNAIleLAU bound to EF-Tu in structure I. e, Nucleotide-binding pocket in the G-domain of EF-Tu. The EM density of the GDPCP nucleotide is shown with gray mesh. In panels c-e, the Coulomb potential density is contoured at 2.8σ.
Extended Data Fig. 9.
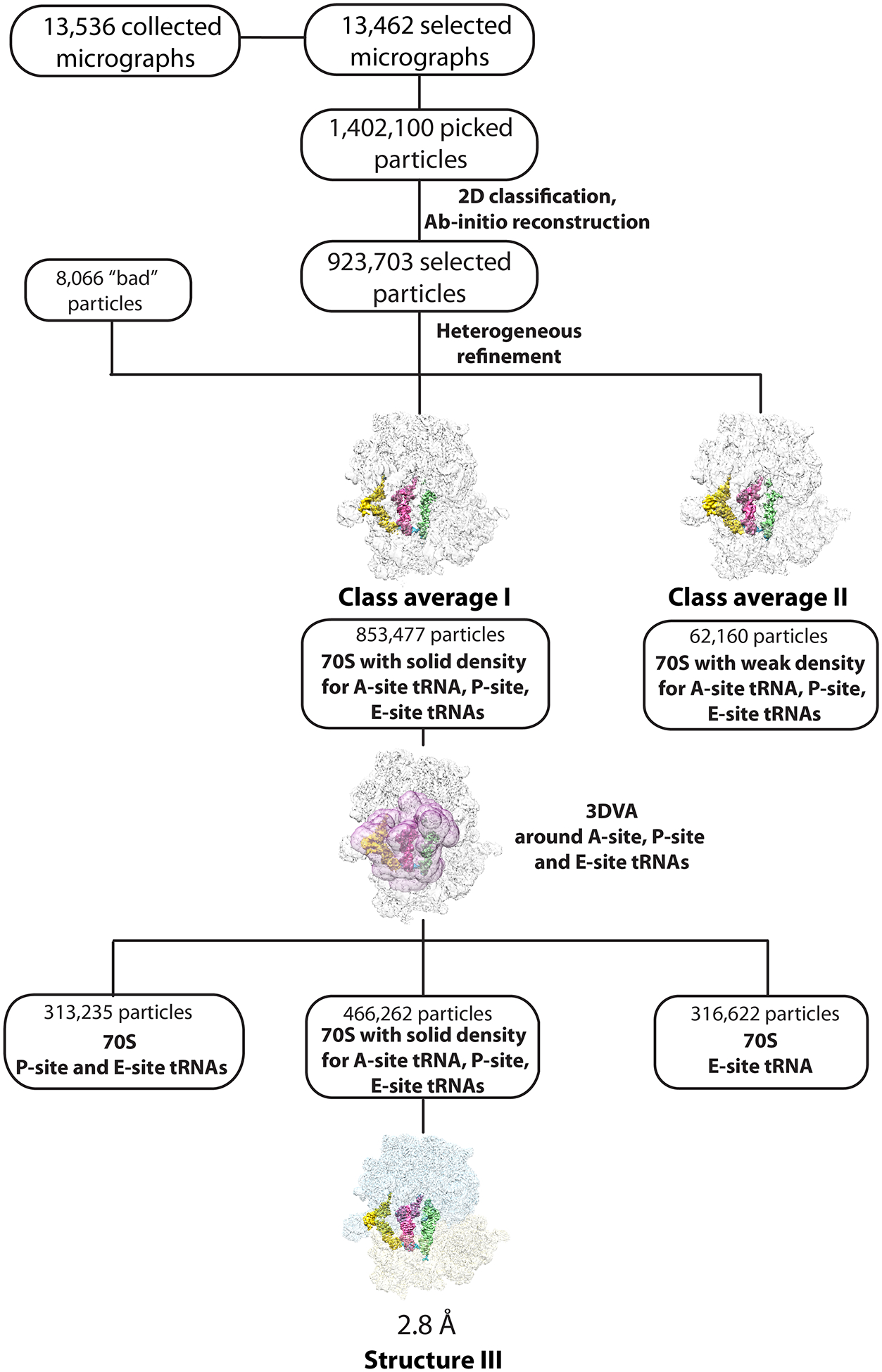
Cryo-EM data processing and particle classification workflow for structure III.
In this 70S ribosome complex, tRNAIleLAU is bound to the cognate AUA codon in the A site in the presence of paromomycin. See Methods for details.
Extended Data Fig. 10.
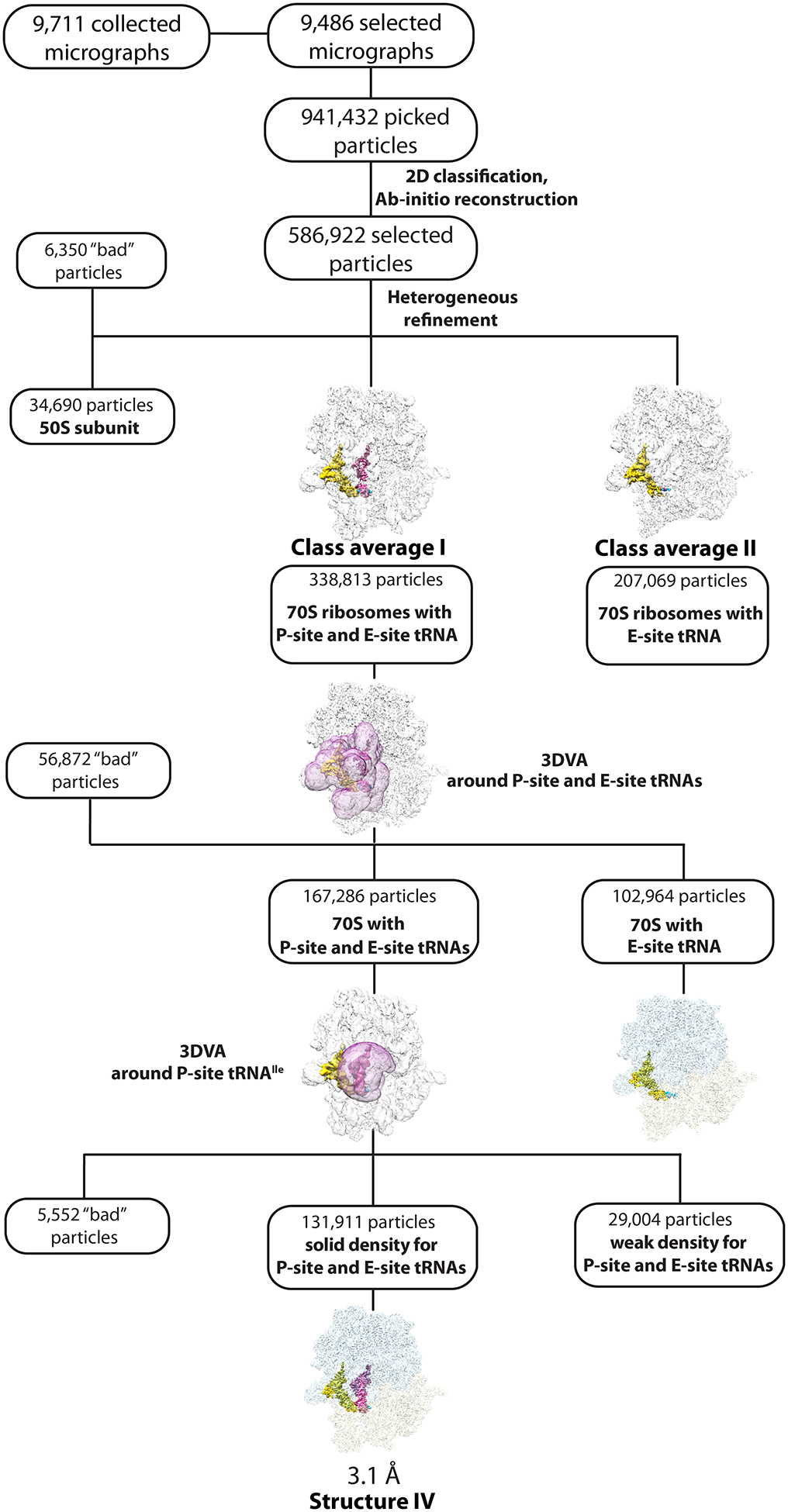
Cryo-EM data processing and particle classification workflow for structure IV.
In this 70S ribosome complex, tRNAIleLAU is bound to the cognate AUA codon in the P site. See Methods for details.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank R. Basu for critical reading of the paper and useful suggestions. We thank the staff of the Sealy Center for Structural Biology and Molecular Biophysics cryo-electron microscopy facility at the University of Texas Medical Branch for advice and support. Special thanks to K.-Y. (Clem) Wong and J. Perkyns (University of Texas Medical Branch) for computational support and to the Sealy and Smith Foundation for supporting the Sealy Center for Structural Biology at the University of Texas Medical Branch. We are grateful to T. Suzuki and N. Akiyama for kindly sharing their unpublished cryo-EM data of the ribosome complexed with the lysidine-modified tRNAIleLAU. This work was supported by NIH grant R01GM136936 (to M.G.G.), the Welch Foundation grant H-2032-20230405 (to M.G.G.), startup funds from the University of Texas Medical Branch (to M.G.G.), Rising Science and Technology Acquisition and Retention Program award from the University of Texas system (to M.G.G.), NIH grant P41GM103311 (to the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco) for developing UCSF Chimera, and NIH grant R01GM129325 (to the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco) and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases for developing UCSF ChimeraX.
Footnotes
Competing interests
The authors declare no competing interests.
Figure generation
All figures showing atomic models were rendered using the PyMol software (www.pymol.org), UCSF Chimera 1.14 (ref.49) or UCSF ChimeraX 1.4 (ref.54) and assembled with Adobe Illustrator (Adobe). The EM density is shown within a radius of 2 Å around atoms.
Supplementary information
Supplementary Figs. 1–4 and references.
Micrograph_Structure_I.
Micrograph_Structure_II.
Micrograph_Structure_III.
Micrograph_Structure_IV.
Data availability
The atomic coordinates were deposited in the RCSB Protein Data Bank (PDB) under accession codes 8G7P (structure I; 70S ribosome with EF-Tu•GDPCP•A/T-Ile-tRNAIleLAU bound to the cognate AUA codon), 8G7Q (structure II; 70S ribosome with EF-Tu•GDPCP•A/T-Ile-tRNAIleLAU bound to the near-cognate AUG codon), 8G7R (structure III; 70S ribosome with A-site tRNAIleLAU bound to the cognate AUA codon) and 8G7S (structure IV; 70S ribosome with the P-site tRNAIleLAU bound to the cognate AUA codon). The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-29819 (structure I; 70S ribosome with EF-Tu•GDPCP•A/T-Ile-tRNAIleLAU bound to the cognate AUA codon), EMD-29820 (structure II; 70S ribosome with EF-Tu•GDPCP•A/T-Ile-tRNAIleLAU bound to the near-cognate AUG codon), EMD-29821 (structure III; 70S ribosome with A-site tRNAIleLAU bound to the cognate AUA codon) and EMD-29822 (structure IV; 70S ribosome with the P-site tRNAIleLAU bound to the cognate AUA codon). The unaligned multi-frame cryo-EM micrographs have been deposited in the Electron Microscopy Public Image Archive (EMPIAR)55 with the accession codes EMPIAR-11582 (structure I), EMPIAR-11583 (structure II), EMPIAR-11584 (structure III) and EMPIAR-11585 (structure IV).
References
- 1.Loveland AB, Demo G, Grigorieff N & Korostelev AA Ensemble cryo-EM elucidates the mechanism of translation fidelity. Nature 546, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Voorhees RM, Schmeing TM, Kelley AC & Ramakrishnan V The mechanism for activation of GTP hydrolysis on the ribosome. Science 330, 835–838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmeing TM et al. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science 326, 688–694 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ogle JM & Ramakrishnan V Structural insights into translational fidelity. Annu. Rev. Biochem 74, 129–77 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Ogle JM, Murphy IV FV, Tarry MJ & Ramakrishnan V Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111, 721–732 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Ogle JM et al. Recognition of cognate transfer RNA by the 30 S ribosomal subunit. Science 292, 897–902 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Loveland AB, Demo G & Korostelev AA Cryo-EM of elongating ribosome with EF-Tu• GTP elucidates tRNA proofreading. Nature 584, 640–645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rozov A, Westhof E, Yusupov M & Yusupova G The ribosome prohibits the G*U wobble geometry at the first position of the codon-anticodon helix. Nucl. Acids Res 44, 6434–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rozov A, Demeshkina N, Westhof E, Yusupov M & Yusupova G Structural insights into the translational infidelity mechanism. Nat. Commun 6, 7251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demeshkina N, Jenner L, Westhof E, Yusupov M & Yusupova G A new understanding of the decoding principle on the ribosome. Nature 484, 256–9 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Nilsson EM & Alexander RW Bacterial wobble modifications of NNA-decoding tRNAs. IUBMB Life 71, 1158–1166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klassen R, Bruch A & Schaffrath R Independent suppression of ribosomal +1 frameshifts by different tRNA anticodon loop modifications. RNA Biol. 14, 1252–1259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Powell CA et al. TRMT5 Mutations Cause a Defect in Post-transcriptional Modification of Mitochondrial tRNA Associated with Multiple Respiratory-Chain Deficiencies. Am. J. Hum. Genet 97, 319–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Motorin Y & Helm M tRNA stabilization by modified nucleotides. Biochemistry 49, 4934–44 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Voigts-Hoffmann F et al. A methyl group controls conformational equilibrium in human mitochondrial tRNA(Lys). J. Am. Chem. Soc 129, 13382–3 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Alexandrov A et al. Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21, 87–96 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Kadaba S et al. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 18, 1227–40 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madore E et al. Effect of modified nucleotides on Escherichia coli tRNAGlu structure and on its aminoacylation by glutamyl-tRNA synthetase. Predominant and distinct roles of the mnm5 and s2 modifications of U34. Eur. J. Biochem 266, 1128–35 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Helm M, Giege R & Florentz C A Watson-Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry 38, 13338–46 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Kruger MK & Sorensen MA Aminoacylation of hypomodified tRNAGlu in vivo. J. Mol. Biol 284, 609–20 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Sylvers LA, Rogers KC, Shimizu M, Ohtsuka E & Soll DA 2-thiouridine derivative in tRNAGlu is a positive determinant for aminoacylation by Escherichia coli glutamyl-tRNA synthetase. Biochemistry 32, 3836–41 (1993). [DOI] [PubMed] [Google Scholar]
- 22.Rogers MJ et al. Selectivity and specificity in the recognition of tRNA by E coli glutaminyl-tRNA synthetase. Biochimie 75, 1083–90 (1993). [DOI] [PubMed] [Google Scholar]
- 23.Bjork GR, Wikstrom PM & Bystrom AS Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244, 986–9 (1989). [DOI] [PubMed] [Google Scholar]
- 24.Weixlbaumer A et al. Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat. Struct. Mol. Biol 14, 498–502 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nasvall SJ, Chen P & Bjork GR The wobble hypothesis revisited: uridine-5-oxyacetic acid is critical for reading of G-ending codons. RNA 13, 2151–64 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nasvall SJ, Chen P & Bjork GR The modified wobble nucleoside uridine-5-oxyacetic acid in tRNAPro(cmo5UGG) promotes reading of all four proline codons in vivo. RNA 10, 1662–73 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy F.V.t. & Ramakrishnan V Structure of a purine-purine wobble base pair in the decoding center of the ribosome. Nat. Struct. Mol. Biol 11, 1251–2 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Crick FH Codon--anticodon pairing: the wobble hypothesis. J. Mol. Biol 19, 548–55 (1966). [DOI] [PubMed] [Google Scholar]
- 29.Parker J Errors and alternatives in reading the universal genetic code. Microbiol. Rev 53, 273–98 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soma A et al. An RNA-Modifying Enzyme that Governs Both the Codon and Amino Acid Specificities of Isoleucine tRNA. Mol. Cell 12, 689–698 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Nakanishi K et al. Structural basis for translational fidelity ensured by transfer RNA lysidine synthetase. Nature 461, 1144–1148 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Muramatsu T et al. A novel lysine-substituted nucleoside in the first position of the anticodon of minor isoleucine tRNA from Escherichia coli. J. Biol. Chem 263, 9261–9267 (1988). [DOI] [PubMed] [Google Scholar]
- 33.Köhrer C et al. Identification and characterization of a tRNA decoding the rare AUA codon in Haloarcula marismortui. RNA 14, 117–126 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeuchi Y et al. Agmatine-conjugated cytidine in a tRNA anticodon is essential for AUA decoding in archaea. Nat. Chem. Biol 6, 277–282 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Mandal D et al. Agmatidine, a modified cytidine in the anticodon of archaeal tRNAIle, base pairs with adenosine but not with guanosine. Proc. Natl. Acad. Sci. USA 107, 2872–2877 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voorhees RM et al. The structural basis for specific decoding of AUA by isoleucine tRNA on the ribosome. Nat. Struc. Mol. Biol 20, 641–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Punjani A & Fleet DJ 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol 213, 107702 (2021). [DOI] [PubMed] [Google Scholar]
- 38.von Loeffelholz O et al. Focused classification and refinement in high-resolution cryo-EM structural analysis of ribosome complexes. Curr. Opin. Struct. Biol 46, 140–148 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Serna M Hands on methods for high resolution cryo-electron microscopy structures of heterogeneous macromolecular complexes. Front. Mol. Biosci 6, 33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rozov A et al. Importance of potassium ions for ribosome structure and function revealed by long-wavelength X-ray diffraction. Nat. Commun 10, 2519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmeing TM, Voorhees RM, Kelley AC & Ramakrishnan V How mutations in tRNA distant from the anticodon affect the fidelity of decoding. Nat. Struct. Mol. Biol 18, 432–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kajander T et al. Buried charged surface in proteins. Structure 8, 1203–14 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Dulic M, Cvetesic N, Perona JJ & Gruic-Sovulj I Partitioning of tRNA-dependent Editing between Pre- and Post-transfer Pathways in Class I Aminoacyl-tRNA Synthetases J. Biol. Chem 285, 23799–23809 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang C, Yashiro Y, Sakaguchi Y, Suzuki T & Tomita K Substrate specificities of Escherichia coli ItaT that acetylates aminoacyl-tRNAs. Nucl. Acids Res 48, 7532–7544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jünemann R et al. In vivo deuteration of transfer RNAs: overexpression and large-scale purification of deuterated specific tRNAs. Nucl. Acids Res 24, 907–913 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basu RS, Sherman MB & Gagnon MG Compact IF2 allows initiator tRNA accommodation into the P site and gates the ribosome to elongation. Nat. Commun 13, 3388 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Watson ZL et al. Structure of the bacterial ribosome at 2 Å resolution. Elife 9(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pettersen EF et al. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Struct. Biol 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leroy EC, Perry TN, Renault TT & Innis CA Tetracenomycin X sequesters peptidyl-tRNA during translation of QK motifs. Nat Chem Biol (2023). [DOI] [PubMed] [Google Scholar]
- 52.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Afonine PV et al. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. D Struct. Biol 74, 814–840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pettersen EF et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iudin A et al. EMPIAR: the Electron Microscopy Public Image Archive. Nucleic Acids Res. 51, D1503–D1511 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cardone G, Heymann JB & Steven AC One number does not fit all: mapping local variations in resolution in cryo-EM reconstructions. J. Struct. Biol 184, 226–36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates were deposited in the RCSB Protein Data Bank (PDB) under accession codes 8G7P (structure I; 70S ribosome with EF-Tu•GDPCP•A/T-Ile-tRNAIleLAU bound to the cognate AUA codon), 8G7Q (structure II; 70S ribosome with EF-Tu•GDPCP•A/T-Ile-tRNAIleLAU bound to the near-cognate AUG codon), 8G7R (structure III; 70S ribosome with A-site tRNAIleLAU bound to the cognate AUA codon) and 8G7S (structure IV; 70S ribosome with the P-site tRNAIleLAU bound to the cognate AUA codon). The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-29819 (structure I; 70S ribosome with EF-Tu•GDPCP•A/T-Ile-tRNAIleLAU bound to the cognate AUA codon), EMD-29820 (structure II; 70S ribosome with EF-Tu•GDPCP•A/T-Ile-tRNAIleLAU bound to the near-cognate AUG codon), EMD-29821 (structure III; 70S ribosome with A-site tRNAIleLAU bound to the cognate AUA codon) and EMD-29822 (structure IV; 70S ribosome with the P-site tRNAIleLAU bound to the cognate AUA codon). The unaligned multi-frame cryo-EM micrographs have been deposited in the Electron Microscopy Public Image Archive (EMPIAR)55 with the accession codes EMPIAR-11582 (structure I), EMPIAR-11583 (structure II), EMPIAR-11584 (structure III) and EMPIAR-11585 (structure IV).