

Abstract
Background
The emergence of Multidrug-Resistant Tuberculosis (MDR-TB) poses a significant threat to global tuberculosis control efforts. This study aimed to examine the influence of mutations in Toxin-Antitoxin system genes on the global transmission of MDR-TB caused by Mycobacterium tuberculosis (M. tuberculosis).
Methods
Whole-genome sequencing was conducted on 13,518 M. tuberculosis isolates. Genes of the Toxin-Antitoxin system were obtained from the National Center for Biotechnology Information (NCBI) Gene database. Techniques such as Random Forest, Gradient Boosting Decision Tree, and Generalized Linear Mixed Models were employed to identify mutation sites in Toxin-Antitoxin system-related genes that facilitated the transmission of MDR-TB.
Results
4,066 (30.08%) were identified as MDR-TB strains of all analyzed isolates. We found significant associations between specific gene mutations and MDR-TB transmission clusters including mutations in Rv0298 (G213A), Rv1959c (parE1, C88T), Rv1960c (parD1, C134T), Rv1991A (maze, G156A), Rv2547 (vapB, C54G), Rv2862A (vapB23, T2C), and Rv3385c (vapB46, G70A). Additionally, several gene mutations associated with MDR-TB transmission clades such as Rv1956 (higA, G445T), Rv1960c (parD1, C134T), and Rv1962A (vapB35, G99A) were noted. Certain gene mutations including vapB35 (G99A), higA (G445T), and parD1 (C134T) correlated with cross-regional transmission clades.
Conclusion
This study highlights the significant association between specific gene mutations in the Toxin-Antitoxin system and the global transmission of MDR-TB, providing valuable insights for developing targeted interventions to control MDR-TB.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12879-024-10142-4.
Keywords: Toxin-antitoxin system, Gene mutations, Multidrug-resistant tuberculosis, Transmission
Background
The emergence of drug-resistant tuberculosis (DR-TB) poses a significant challenge to global efforts in controlling tuberculosis (TB). It is estimated that 3.3% of newly diagnosed TB patients and 17% of those who have previously received treatment may be affected by multidrug-resistant tuberculosis (MDR-TB) [1]. MDR-TB refers to a type of TB caused by Mycobacterium tuberculosis (M. tuberculosis) strains that are resistant to at least two of the most potent anti-TB drugs, namely isoniazid and rifampicin. The ongoing transmission and development of MDR-TB present a severe threat to the global control of TB. Mono-resistant tuberculosis (MR-TB) refers to M. tuberculosis that shows resistance to a specific anti-tuberculosis drug, particularly isoniazid, while remaining sensitive to other anti-tuberculosis medications. This form of resistance poses significant challenges for treatment, as it can lead to prolonged illness and complicate management strategies. The toxin-antitoxin system is a genetic module composed of a toxin and its corresponding antitoxin, which has been found to play a role in various bacterial processes. M. tuberculosis stands out for having an exceptionally high number of toxin-antitoxin loci. Among the various toxin-antitoxin systems encoded by the M. tuberculosis H37Rv strain, most belong to the type II category, where both the toxin and antitoxin are proteins. This type of system is the most common and abundant among toxin-antitoxin systems. In type II toxin-antitoxin systems, an unstable antitoxin tightly binds to its specific toxin, forming a stable protein complex that deactivates it [2]. If the antitoxin is degraded, the toxin becomes active and inhibits essential processes, such as DNA replication or protein synthesis, until antitoxin production resumes [2]. Despite being widespread in bacteria, the precise physiological roles of toxin-antitoxin systems still need to be fully understood. Some have been associated with functions like plasmid maintenance, immunity against bacteriophages, and the formation of dormant persisters tolerant to antibiotics [3–10]. Toxin-antitoxin systems may enhance the ability of M. tuberculosis to resist host and antibiotic pressures by regulating growth under different stress conditions. These toxin-antitoxin systems in M. tuberculosis are considered promising targets for developing new anti-TB drugs. Disrupting the type II or III toxin-antitoxin complex could provide a new approach to antibacterial therapy. Overall, toxin-antitoxin systems in M. tuberculosis play a crucial role in the transmission and development of MDR-TB, making the study of genetic mutations in these systems highly significant [11, 12].
Whole genome sequencing (WGS) is increasingly being utilized in the field of epidemiology to provide comprehensive insights into the transmission dynamics of M. tuberculosis [13]. By allowing researchers to examine genetic variations at a high resolution, WGS enables the identification of specific mutations associated with virulence, resistance, and transmission patterns. In this study, we employed WGS not only to estimate the transmission dynamics but also to reevaluate the influence of gene mutations in the toxin-antitoxin system on the global transmission clusters of MDR strains. This approach provides a more detailed understanding of how genetic factors contribute to the spread of MDR-TB in different populations, thereby informing public health strategies for controlling outbreaks.
Methods
Clinical isolates
The present study analyzed M. tuberculosis strains from two sample sets. The first set consisted of a newly sequenced WGS dataset comprising 1,550 patients with culture-confirmed TB cases and drug-susceptibility testing results reported to the TB Surveillance System in Shandong Province between 2011 and 2018. Ethical approval for the study protocol (Approval No. 2017 − 337) was obtained from the Center for Ethics in Human Research at Shandong Provincial Hospital, affiliated with Shandong First Medical University. The second set aimed to encompass a comprehensive dataset of 12,411 M. tuberculosis strains from 52 countries and 18 regions [14–22]. Isolate metadata was retrieved from the Sequence Read Archive (SRA) repository, and additional filtering was performed using the outlined methods. The code used for this analysis has been uploaded to our GitHub repository, which can be accessed at https://github.com/shenmemingziheshi/Statistical-code.git.
Whole genome sequencing and single nucleotide polymorphism identification
We analyzed 1,550 M. tuberculosis isolates, extracting their genomic DNA using the cetyltrimethylammonium bromide (CTAB) method [23], which is known for its effectiveness in isolating high-quality nucleic acids from resilient bacterial cell walls. CTAB (C₁₉H₄₂BrN, 95%) was purchased from Sigma-Aldrich. The quality of the extracted DNA was assessed with a Nanodrop spectrophotometer to measure the A260/A280 ratio, ensuring it fell within the range of 1.8 to 2.0, indicative of pure DNA. Additionally, we evaluated the integrity of the DNA by running a portion on an agarose gel, confirming that the majority of samples exhibited clear and distinct bands without significant degradation or shearing. All procedures were conducted according to standard protocols, and reagents were sourced from reputable manufacturers, including Sigma-Aldrich for CTAB. This stringent quality control ensured that the DNA met the necessary standards for subsequent sequencing. Nonetheless, 103 isolates were deemed unsuitable for further study due to extraction issues and subpar DNA integrity. The remaining 1,447 isolates were sequenced on the Illumina HiSeq 4000 platform (Illumina, San Diego, CA, USA) using an Illumina HiSeq 3000/4000 SBS kit with 300 cycles. Libraries were pooled for sequencing in a paired-end format with 250 bp reads, targeting a coverage of > 200×.The ensuing sequence data was archived in the NCBI database under the BioProject ID PRJNA1002108. Unfortunately, two samples were compromised during processing, leaving 1,445 isolates for in-depth analysis. The reads from the Shandong cohort and 12,073 worldwide strains were aligned against the H37Rv reference genome (NC_000962.3) using bwa mem (version 0.7.17-r1188). We utilized samclip (v 0.4.0) and samtools markup (v 1.15) to eliminate duplicate and truncated alignments. Samples with under 98% coverage or a depth of less than 20x were excluded [24, 25]. Additionally, any strain indicating mixed lineage or Mycobacterium bovis contamination was removed from the dataset. After applying these criteria, 13,518 isolates, including 12,073 from the global dataset and 1,445 from Shandong, qualified for the analysis. For a comprehensive inventory of the isolates, please consult Supplementary Data 1 and Supplementary Data 2.
Variant detection in our study was conducted using the combined efforts of Samclip (version 0.4.0) and SAMtools (version 1.15). Post variant detection, we further refined the quality of our findings by applying additional filters through Free Bayes (version 1.3.2) and Bcftools (version 1.15.1). The criterion set for this refinement process was stringent: only variants with a genotype of ‘1/1’, a quality score of at least 100, a depth of 10 or more, and an allele frequency greater than 0.5 were considered. We also excluded single nucleotide polymorphisms (SNPs) located within known repetitive regions, such as PPE, PE-PGRS genes, and those associated with mobile genetic elements and repeats, as identified by Tandem Repeat Finder (version 4.09) and Repeatmask (version 4.1.2-p1) [26, 27]. The variant call format (VCF) files that successfully passed through our filtration were then annotated with snpEff (version 4.3t), providing us with a curated set of SNPs for our analysis [28].
Genotypic drug resistance prediction and lineage assignments
To categorize the lineages and forecast drug resistance in M. tuberculosis WGS data, we employed TBProfiler (version 4.3.0), a web-based analytical tool [29, 30]. This tool leverages the Tuberculosis Database, a curated repository for drug-resistance identification validated with over 17,000 samples carrying genotypic and phenotypic data. Polymorphisms related to resistance (including SNPs and indels) unveiled by TBProfiler were reassessed according to the catalog endorsed by the World Health Organization (WHO). This catalog lists molecular targets for drug-susceptibility testing in the M. tuberculosis complex, providing standardized interpretations for resistance based on genotypic findings [31]. In our study, genotypic MDR was defined as detecting mutations resisting both isoniazid and rifampicin.
Phylogenomic analyses
Phylogenetic and phylogenomic evaluations were carried out utilizing the maximum-likelihood method facilitated by IQ-TREE software (version 1.6.12). We constructed the phylogenetic tree applying the general time reversible (GTR) model for nucleotide substitutions, adding the GAMMA distribution to account for rate heterogeneity across sites. To determine the robustness of the phylogenetic branches, we executed 1000 bootstrap replicates. The phylogenetic structure was anchored using the genome of Mycobacterium canettii strain CIPT 140,010,059 (NC_15848.1) as an outgroup, providing a comparative baseline for our analysis [32]. The resulting phylogenetic tree was created with IQ-TREE (v1.6.12).
Acquisition of toxin-antitoxin system genes
We sourced a comprehensive set of 78 toxin-antitoxin system genes from the NCBI database. Utilizing Python scripts, we detected mutations within these specific genes associated with the toxin-antitoxin systems (Supplementary Data 3).
Transmission dynamic
Transmission cluster and clade
In our novel research, we investigated the influence of toxin-antitoxin system gene mutations on the transmission of M. tuberculosis. We analyzed SNPs within branches leading to either leaf nodes or transmission clusters/clades. A transmission cluster, defined as strains with a genetic distance of 12 SNPs or less, indicates recent transmission events. On the other hand, a transmission clade, encompassing strains with up to 25 SNPs differences, captures both recent and more distant transmission occurrences. These findings, based on established precedents in the scientific literature [33–35], open new avenues for understanding the transmission of M. tuberculosis (Supplementary Data 1 and Supplementary Data 2).
Clade size
Transmission clades were classified into three categories based on the number of isolates within each clade: large (above the 75th percentile), medium (between the 25th and 75th percentile), and small (below the 25th percentile) [36].
Cross-regional clade
A cross-regional clade was defined as two or more M. tuberculosis strains within a clade isolated in at least two regions.
Statistical analysis and modeling
We presented categorical data in terms of frequencies and percentages. Our statistical analyses used generalized linear mixed models (GLMM) within the R statistical environment (version 4.2.3). In addition, we employed Python (version 3.7.4), specifically leveraging the Scikit-learn library to implement sophisticated machine learning algorithms such as random forest and gradient-boosting decision trees. We constructed predictive models using these machine learning techniques and validated their performance by partitioning the dataset into training and testing sets in a 7:3 ratio. The efficacy of the predictive models was meticulously evaluated using a suite of metrics, including Kappa, sensitivity, specificity, accuracy, as well as predictive values (positive and negative), likelihood ratios (positive and negative), and the area under the receiver operating characteristic curve (AUC). Post-modeling, we performed a feature importance analysis to discern the impact of each variable on the predictions, identifying the most significant predictors by intersecting the top-performing variables across both models. This methodological rigor allowed us to pinpoint the key features driving the accuracy of our risk factor predictions. We adhered to a stringent statistical significance threshold, recognizing P-values below 0.05 in two-tailed tests.
Results
MDR population structure
A total of 13,518 M. tuberculosis isolates were analyzed, out of which 7,732 isolates (57.20%) were susceptible to all tested drugs, indicating pan-susceptibility. Among the isolates, 4,066 (30.08%) were identified as MDR-TB strains. Eight hundred fifty-eight isolates (6.35%) showed resistance to isoniazid only, which are classified as MR-TB. Additionally, 862 isolates (6.38%) were categorized as other DR strains, as shown in Table 1.
Table 1.
The characteristics of TB isolates
| Characteristic | N (%) | ||
|---|---|---|---|
|
MDR-TB 4066(30.08%) |
Lineage | Lineage1 | 64(1.57%) |
| Lineage2 | 2509(61.71%) | ||
| Lineage3 | 84(2.07%) | ||
| Lineage4 | 1390(37.19%) | ||
| Lineage5 | 19(0.47%) | ||
| Cluster | Yes | 1803(44.34%) | |
| No | 2263(55.66%) | ||
| Clade | Yes | 2328(57.26%) | |
| No | 1738(42.74%) | ||
| Clade size | Small | 582(25%) | |
| Medium | 956(41.07%) | ||
| Large | 790(33.93%) | ||
| Cross-regional | Yes | 227(9.75%) | |
| No | 2101(9.02%) | ||
| MR-TB | 858(6.35%) | ||
| Sensitive | 7732(57.20%) | ||
| Other | 862(6.38%) | ||
MDR, multidrug resistance; MDR-TB, multidrug-resistant tuberculosis; MR, Mono-resistant tuberculosis
The incidence of MDR-TB, a global health concern, varied significantly among different lineages. In lineage 1, the rate of MDR-TB was observed to be 7.52% (n = 64), while in lineage 2, it was significantly higher at 48.88% (n = 2509). Lineage 3 had an MDR rate of 8.67% (n = 84), whereas lineage 4 had a rate of 21.42% (n = 1390). Lineage 5 exhibited a rate of 47% (n = 19), while both lineage 6 and lineage 7 had a 0% rate of MDR-TB. In the 18 regions designated by the United Nations (UN) Geoscheme, the highest incidence of MDR-TB was found in Central Asia (95.27%), South America (88.49%), Melanesia (79.63%), and Middle Africa (79.61%). On the other hand, Northern Europe (4.12%), Eastern Africa (3.87%), Northern America (1.82%), and Western Europe (1.65%) exhibited the lowest rates of MDR-TB, as shown in Fig. 1. The construction of the phylogenetic tree for MDR-TB isolates was described in Supplementary Fig. 1.
Fig. 1.

The distribution of M. tuberculosis strains in a specific region and the proportion of MDR strains
Relationship between toxin-antitoxin system gene mutations and MDR-TB transmission clusters
Our study refined our analysis by filtering out loci with fewer than 10 mutations, ultimately focusing on 72 SNPs that showed potential relevance to the transmission clusters of MDR-TB. Using GLMM, we identified seven SNPs significantly associated with MDR-TB clusters (P < 0.05), see Table 2. These seven SNPs comprised two nonsynonymous SNPs, one start loss mutation, one-stop gain mutation, and three synonymous SNPs. Specifically, these mutations included Rv0298 (G213A) with a frequency of 26.76% (n = 1088), Rv1959c (parE1, C88T) at 4.53% (n = 184), Rv1960c (parD1, C134T) at 1.08% (n = 44), Rv1991A (maze, G156A) at 0.3% (n = 13), Rv2547 (vapB, C54G) at 4.67% (n = 190), Rv2862A (vapB23, T2C) at 0.2% (n = 8), and Rv3385c (vapB46, G70A) also at 0.3% (n = 12). Moreover, we developed prediction models using the random forest and gradient boosting decision tree methods, as detailed in the supplementary materials (Supplementary Table 4, Supplementary Tables 9, and Supplementary Fig. 2). The modeling outcomes highlighted that the SNPs Rv0298 G213A, parE1 C88T, parD1 C134T, maze G156A, vapB C54G, vapB23 T2C, and vapB46 G70A were significant contributors to the predictive accuracy of both models. In summary, our findings showed a positive correlation between the SNPs Rv0298 G213A, parE1 C88T, parD1 C134T, maze G156A, vapB C54G, vapB23 T2C, and vapB46 G70A, and the transmission clusters within MDR-TB.
Table 2.
Generalized linear mixed model analysis was conducted on clustered and non-clustered MDR-TB isolates
| Rv number | Gene | Position | SNP | Amino acid changes | P value | OR (95%CI) |
|---|---|---|---|---|---|---|
| Rv0239 | vapB24 | 289,179 | A76C | Thr26Pro | 0.994 | - |
| Rv0239 | vapB24 | 289,300 | C197T | Ser66Phe | 9.999E-01 | - |
| Rv0298 | - | 363,375 | C124A | Leu42Met | 0.776 | 1.192(0.355–4.004) |
| Rv0298 | - | 363,464 | G213A | Arg71Arg | 0.022 | 6.104(1.295–28.782) |
| Rv0300 | vapB2 | 363,996 | T171C | Gly57Gly | 0.177 | 2.174(0.704–6.716) |
| Rv0657c | vapB6 | 753,562 | T56C | Ile19Thr | 0.202 | 0.547(0.216–1.383) |
| Rv0657c | vapB6 | 753,589 | A29C | Asp10Ala | 0.420 | 0.604(0.177–2.057) |
| Rv0662c | vapB7 | 755,873 | C151T | Arg51Cys | 0.730 | 1.137(0.547–2.366) |
| Rv0662c | vapB7 | 755,947 | G77A | Arg26His | 0.530 | 1.473(0.439–4.946) |
| Rv0664 | vapB8 | 758,548 | G17T | Cys6Phe | 0.580 | 1.375(0.445–4.247) |
| Rv1241 | vapB33 | 1,384,315 | G38A | Arg13Gln | 0.169 | 3.575(0.582–21.976) |
| Rv1246c | relE | 1,388,827 | A152T | Asn51Ile | 0.998 | - |
| Rv1494 | mazE4 | 1,686,299 | T29G | Ile10Ser | 0.061 | 0.434(0.181–1.039) |
| Rv1740 | vapB34 | 1,967,783 | A79G | Thr27Ala | 0.997 | - |
| Rv1740 | vapB34 | 1,967,844 | C140A | Ala47Glu | 0.466 | 1.235(0.7-2.181) |
| Rv1740 | vapB34 | 1,967,897 | G193C | Glu65Gln | 0.674 | 1.349(0.335–5.438) |
| Rv1943c | mazE5 | 2,195,025 | A323G | Glu108Gly | 0.839 | 0.92(0.411–2.058) |
| Rv1943c | mazE5 | 2,195,321 | G27A | Thr9Thr | 0.863 | 0.891(0.241–3.289) |
| Rv1955 | higB | 2,201,720 | T2C | Val1? | 0.408 | 1.332(0.675–2.63) |
| Rv1955 | higB | 2,201,808 | C90G | Asp30Glu | 0.853 | 0.936(0.465–1.883) |
| Rv1956 | higA | 2,202,491 | A354G | Ala118Ala | 0.995 | - |
| Rv1956 | higA | 2,202,500 | C363T | His121His | 0.265 | 3.36(0.399–28.29) |
| Rv1956 | higA | 2,202,582 | G445T | Ala149Ser | 0.990 | - |
| Rv1959c | parE1 | 2,203,869 | G109A | Glu37Lys | 0.053 | 8.02(0.975–65.957) |
| Rv1959c | parE1 | 2,203,890 | C88T | Gln30* | 5.510E-09 | 33.215(10.233–107.81) |
| Rv1960c | parD1 | 2,204,092 | C134T | Thr45Ile | 0.002 | 4.464(1.693–11.768) |
| Rv1962A | vapB35 | 2,205,283 | G267A | Arg89Arg | 0.154 | 3.743(0.609–23.024) |
| Rv1962A | vapB35 | 2,205,371 | T179G | Val60Gly | 0.999 | - |
| Rv1962A | vapB35 | 2,205,451 | G99A | Lys33Lys | 0.185 | 0.471(0.155–1.434) |
| Rv1962A | vapB35 | 2,205,511 | G39A | Thr13Thr | 0.359 | 0.716(0.35–1.463) |
| Rv1991A | mazE6 | 2,234,736 | G156A | Arg52Arg | 4.610E-04 | 38.321(4.981-294.819) |
| Rv2009 | vapB15 | 2,258,096 | C67T | Arg23Trp | 0.116 | 2.379(0.808–7.005) |
| Rv2142c | parE2 | 2,402,439 | C72T | Asp24Asp | 0.175 | 0.479(0.165–1.387) |
| Rv2274c | mazF8 | 2,546,601 | A205G | Ile69Val | 0.576 | 0.62(0.116–3.313) |
| Rv2274c | mazF8 | 2,546,684 | G122T | Gly41Val | 0.986 | - |
| Rv2274c | mazF8 | 2,546,803 | G3A | Met1? | 0.204 | 0.479(0.154–1.493) |
| Rv2526 | vapB17 | 2,851,303 | G213C | Glu71Asp | 0.060 | 3.33(0.95-11.677) |
| Rv2545 | vapB18 | 2,867,913 | T131G | Leu44Arg | 0.075 | 2.357(0.918–6.055) |
| Rv2547 | vapB19 | 2,868,659 | C54G | Ala18Ala | 0.003 | 2.066(1.278–3.339) |
| Rv2547 | vapB19 | 2,868,787 | A182G | Asp61Gly | 0.083 | 3.022(0.865–10.554) |
| Rv2595 | vapB40 | 2,925,594 | G103A | Asp35Asn | 0.997 | - |
| Rv2601A | vapB41 | 2,930,254 | G185A | Gly62Asp | 0.973 | 1.018(0.357–2.903) |
| Rv2653c | - | 2,976,616 | C294G | His98Gln | 1.000E + 00 | - |
| Rv2653c | - | 2,976,654 | C256T | Leu86Phe | 0.983 | - |
| Rv2653c | - | 2,976,830 | A80C | Gln27Pro | 0.201 | 0.337(0.064–1.783) |
| Rv2654c | - | 2,977,157 | T78G | Ala26Ala | 0.021 | 0.041(0.003–0.611) |
| Rv2830c | vapB22 | 3,137,058 | C167T | Ala56Val | 0.581 | 0.78(0.323–1.884) |
| Rv2862A | vapB23 | 3,174,748 | T2C | Ile1? | 0.047 | 2.735(1.012–7.388) |
| Rv2865 | relF | 3,177,660 | G124A | Ala42Thr | 0.172 | 0.541(0.224–1.306) |
| Rv2866 | relG | 3,177,884 | C63A | Arg21Arg | 0.986 | - |
| Rv2871 | vapB43 | 3,183,210 | A73G | Thr25Ala | 0.991 | - |
| Rv3181c | - | 3,550,092 | G52T | Val18Phe | 0.915 | 1.069(0.313–3.656) |
| Rv3385c | vapB46 | 3,799,821 | C123T | Asp41Asp | 9.998E-01 | - |
| Rv3385c | vapB46 | 3,799,874 | G70A | Ala24Thr | 0.004 | 7.973(1.911–33.263) |
| Rv3407 | vapB47 | 3,826,316 | G65T | Arg22Leu | 0.994 | - |
| Rv3407 | vapB47 | 3,826,501 | C250T | Arg84Cys | 0.203 | 0.809(0.584–1.121) |
| Rv3697A | vapB48 | 4,140,319 | C145T | Arg49Cys | 0.990 | - |
| Rv3697A | vapB48 | 4,140,384 | G80A | Gly27Glu | 0.998 | - |
SNP, single nucleotide polymorphism; OR, odds ratio; CI, confidence interval
Relationship between toxin-antitoxin system gene mutations and MDR-TB transmission clades
After excluding sites with fewer than 10 mutations, a total of 72 SNPs within genes related to the toxin-antitoxin system, the main objective was to investigate the association between these SNPs and transmission clades in MDR-TB. Our GLMM analysis found that five SNPs exhibited statistically significant associations with MDR-TB transmission clades(P < 0.05), see Supplementary Table 1. These significant SNPs included Rv0298(G213A, n = 1422, 34.97%), Rv1959c (parE1, C88T, n = 191, 4.70%), Rv1960c (parD1, C134T, n = 47, 1.16%), Rv1991A (maze6, G156A, n = 13, 0.32%), and Rv2547(vapB19, C54G, n = 229, 5.63%). Specifically, Rv0298 G213A, maze6 G156A, and vapB19 C54G were synonymous SNPs, while parE1 C88T represented a stop gain mutation and parD1 C134T was a nonsynonymous SNP. Additionally, prediction models were developed using random forest and gradient-boosting decision tree methods. Detailed information about these models can be found in the supplementary materials (Supplementary Table 5, Supplementary Table 10, Supplementary Fig. 3). The modeling results highlighted that the SNPs Rv0298 G213A, maze6 G156A, vapB19 C54G, parE1 C88T, and parD1 C134T significantly contributed to the accuracy of both models in predicting transmission clades. To summarize, our findings suggested a positive correlation between the SNPs Rv0298 G213A, maze6 G156A, vapB19 C54G, parE1 C88T, parD1 C134T, and the transmission clades within MDR-TB.
Relationship between toxin-antitoxin system gene mutations and MDR-TB cross-regional transmission clades
Upon eliminating sites with fewer than 10 mutations, our analysis included 49 SNPs within genes linked to the toxin-antitoxin system. The study was designed to investigate the correlation between these SNPs and cross-regional transmission clades in MDR-TB. Applying the GLMM analysis, revealed that three SNPs were significantly associated with MDR-TB cross-regional transmission clades (P < 0.05), see Table 3. These significant SNPs were Rv1956(higA, G445T, n = 12, 0.52%), Rv1960c (parD1, C134T, n = 12, 0.52%), and Rv1962A (vapB35, G99A, n = 5, 0.21%). VapB35 G99A was identified as a synonymous SNP, while higA G445T and parD1 C134T were classified as nonsynonymous SNPs. Additionally, prediction models were developed using random forest and gradient- boosting decision tree techniques. Further details of these models can be found in the supplementary materials (Supplementary Table 6, Supplementary Tables 11, and Supplementary Fig. 4). The results drawn from the models emphasized the important contribution of the SNPs vapB35 G99A, higA G445T, and parD1C134T towards the predictive accuracy of both models. In conclusion, our study suggested a positive association between the SNPs vapB35 G99A, higA G445T, parD1C134T, and the cross-regional transmission clades within MDR-TB.
Table 3.
Generalized linear mixed model analysis was conducted on cross-regional and non-cross-regional MDR-TB isolates
| Rv number | Gene | Position | SNP | Amino acid changes | P value | OR (95%CI) |
|---|---|---|---|---|---|---|
| Rv0239 | vapB24 | 289,179 | A76C | Thr26Pro | 1.000E + 00 | |
| Rv0298 | - | 363,464 | G213A | Arg71Arg | 0.999 | |
| Rv0300 | vapB2 | 363,996 | T171C | Gly57Gly | 0.999 | |
| Rv0657c | vapB6 | 753,562 | T56C | Ile19Thr | 1.000E + 00 | |
| Rv0662c | vapB7 | 755,873 | C151T | Arg51Cys | 0.835 | 0.847(0.485–11.204) |
| Rv0664 | vapB8 | 758,548 | G17T | Cys6Phe | 9.996E-01 | |
| Rv1246c | relE | 1,388,827 | A152T | Asn51Ile | 1.000E + 00 | |
| Rv1494 | mazE4 | 1,686,299 | T29G | Ile10Ser | 0.999 | |
| Rv1740 | vapB34 | 1,967,844 | C140A | Ala47Glu | 0.900 | 1.081(0.882–9.854) |
| Rv1943c | mazE5 | 2,195,025 | A323G | Glu108Gly | 0.999 | |
| Rv1955 | higB | 2,201,720 | T2C | Val1? | 0.080 | 2.274(3.878–24.336) |
| Rv1955 | higB | 2,201,808 | C90G | Asp30Glu | 9.997E-01 | |
| Rv1956 | higA | 2,202,491 | A354G | Ala118Ala | 9.998E-01 | |
| Rv1956 | higA | 2,202,500 | C363T | His121His | 0.999 | |
| Rv1956 | higA | 2,202,582 | G445T | Ala149Ser | 1.280E-04 | 6.347(221.676-1469.987) |
| Rv1959c | parE1 | 2,203,869 | G109A | Glu37Lys | 9.997E-01 | |
| Rv1959c | parE1 | 2,203,890 | C88T | Gln30* | 9.999E-01 | |
| Rv1960c | parD1 | 2,204,092 | C134T | Thr45Ile | 0.003 | 3.777(17.989-106.139) |
| Rv1962A | vapB35 | 2,205,451 | G99A | Lys33Lys | 0.003 | 7.057(326.409-4128.076) |
| Rv1962A | vapB35 | 2,205,511 | G39A | Thr13Thr | 9.996E-01 | |
| Rv1991A | mazE6 | 2,234,736 | G156A | Arg52Arg | 0.999 | |
| Rv2142c | parE2 | 2,402,439 | C72T | Asp24Asp | 0.999 | |
| Rv2274c | mazF8 | 2,546,684 | G122T | Gly41Val | 0.999 | |
| Rv2274c | mazF8 | 2,546,803 | G3A | Met1? | 0.999 | |
| Rv2526 | vapB17 | 2,851,303 | G213C | Glu71Asp | 0.999 | |
| Rv2547 | vapB19 | 2,868,659 | C54G | Ala18Ala | 0.747 | 1.155(1.324–7.603) |
| Rv2601A | vapB41 | 2,930,254 | G185A | Gly62Asp | 0.999 | |
| Rv2653c | - | 2,976,616 | C294G | His98Gln | 1.000E + 00 | |
| Rv2830c | vapB22 | 3,137,058 | C167T | Ala56Val | 0.998 | |
| Rv2862A | vapB23 | 3,174,748 | T2C | Ile1? | 0.978 | 0.97(0.298–23.36) |
| Rv2865 | relF | 3,177,660 | G124A | Ala42Thr | 0.439 | 0.539(0.359–8.194) |
| Rv2871 | vapB43 | 3,183,210 | A73G | Thr25Ala | 1.000E + 00 | |
| Rv3385c | vapB46 | 3,799,874 | G70A | Ala24Thr | 9.996E-01 | |
| Rv3407 | vapB47 | 3,826,316 | G65T | Arg22Leu | 0.999 | |
| Rv3407 | vapB47 | 3,826,501 | C250T | Arg84Cys | 4.660E-06 | 0.34(0.886–2.229) |
SNP, single nucleotide polymorphism; OR, odds ratio; CI, confidence interval
Relationship between toxin-antitoxin system gene mutations and clade size
Upon exclusion of sites with fewer than 10 mutations, our analysis encompassed 49 SNPs within the toxin-antitoxin system. The data revealed that 31 SNPs were significantly associated with clade size (P < 0.05). Among these influential SNPs were 21 nonsynonymous SNPs, one stop gained SNP, one start gained SNP, an initiator codon variant, and seven synonymous SNPs. These all demonstrated a positive correlation with clade size. Key SNPs among these include vapB35 G99A, higA G445T, parD1C134T, Rv0298 G213A, maze6 G156A, vapB19 C54G, and parE1 C88T. For a more comprehensive understanding, please refer to Fig. 2.
Fig. 2.
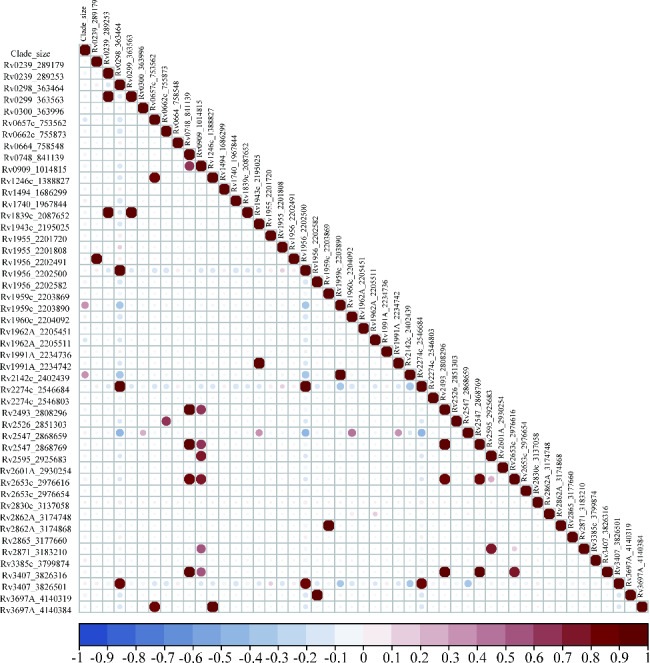
Correlation analysis of toxin-antitoxin system gene mutations and clade size of MDR-TB
MR isolates, MDR isolates and toxin-antitoxin system gene mutations
After filtering out SNPs with a mutation frequency below 10, we identified and kept 82 SNPs for subsequent analysis. We then analyzed the relationship between these 82 SNPs and the development of MDR isolates, comparing them to MR isolates. The GLMM showed that three SNPs were statistically significant for the development of MDR isolates(P < 0.05), among which two nonsynonymous SNPs and one stop gained SNP were positively correlated with the development of MDR isolates, including Rv1959c (parE1, C88T, n = 229, 5.63%), Rv1962A (vapB35, G39A, n = 88, 2.16%) and Rv3385c (vapB46, C123T, n = 17, 0.42%), see Supplementary Table 2. Two prediction models were established using random forest and gradient boosting decision tree, see Supplementary Table 7, Supplementary Tables 12 and Supplementary Fig. 5 for details. Based on the importance scores of the models, we found that parE1 C88T, vapB35 G39A and vapB46 C123T contributed most to the random forest and gradient-boosting decision tree. The results indicated that compared with MR isolates, parE1 C88T, vapB35 G39A and vapB46 C123T increased the risk of development of MDR isolates.
Sensitive isolates, MDR isolates and toxin-antitoxin system gene mutations
Following excluding SNPs with a mutation frequency below 10, 163 SNPs were identified and retained for further analysis. We subsequently investigated the correlation between these SNPs and the emergence of MDR isolates compared to sensitive isolates. The GLMM analysis revealed that 16 out of these 163 SNPs held statistical significance for the evolution of MDR isolates(P < 0.05), see Supplementary Table 3. Among these, nine were nonsynonymous SNPs and seven were synonymous SNPs that exhibited a positive correlation with the development of MDR isolates, including: Rv0657c (vapB6, T56C), Rv0662c (vapB7, C151T), Rv0748 (vapB31, C234T), Rv1740 (vapB34, C140A, G193C), Rv1960c (parD1, C134T), Rv1962A (vapB35, G267A), Rv1991A (maze6, G156A), Rv2104c (vapB37, G205C), Rv2274c (mazF8, A108T), Rv2545(vapB18, T131G), Rv2547 (vapB19, C54G), Rv2550c (vapB20, A54C), Rv3385c (vapB46, C123T, G70A). We established two prediction models using the random forest and gradient-boosting decision tree algorithms (Supplementary Table 8, Supplementary Tables 13, and Supplementary Table 5). Supplementary Fig. 6. Our findings indicated that the SNPs vapB6 T56C, vapB7 C151T, vapB34 C140A, vapB34 G193C, parD1 C134T, vapB35 G267A, maze6 G156A, mazF8 A108T, vapB18 T131G, vapB19 C54G, vapB20 A54C and vapB46(C123T, G70A) significantly contributed to both models. However, the contributions of vapB31 C234T and vapB37 G205C were not observed in the gradient-boosting decision tree model. Our results suggested an increased risk of developing MDR isolates when associated with these specific SNPs, compared to sensitive isolates.
Discussion
The toxin-antitoxin system plays a crucial role in the proliferation of MDR isolates [11]. Our aimed to investigate the association between MDR development, MDR transmission, and mutations within the toxin-antitoxin system genes. We analyzed 13,518 globally sourced M. tuberculosis isolates to achieve this, and examined 78 distinct toxin-antitoxin system genes. Our research outcomes reveal significant insights into the relationship between these genetic variations and MDR-related factors. For specific details regarding the observed mutations, please refer to Fig. 3.
Fig. 3.

SNPs in toxin-antitoxin system genes that confer to MDR isolates. Synonymous mutations were marked as dark blue stars, stop codons were represented by light blue stars, and missense mutations were indicated by red stars
Our study has identified a non-synonymous SNP, C134T, occurring at position 2,204,092 in parD1, which significantly escalates the risk of MDR strain transmission. This mutation has a substantial impact on various aspects related to MDR transmission, including the formation of transmission clusters, transmission clades, clade size, and cross-regional transmission. The occurrence of the Thr45Ile mutation induced by this SNP is a key mechanism that leads to structural changes in parD1, disrupting the delicate balance between toxins and antitoxins [37]. This disruption ultimately increases the risk of MDR strain transmission.
Furthermore, we identified a stop-gained SNP, C88T, at position 2,203,890 of parE1, and a synonymous SNP, C54G, at position 2,868,659 of vapB19, both of which correlate positively with an increased risk of MDR strain transmission, particularly in terms of transmission cluster, transmission clade, and clade size. The stop-gained SNP could result in a truncated ParE1 protein, altering its ability to neutralize the corresponding toxin effectively and perturbing the balance within the toxin-antitoxin system [38]. Similarly, the synonymous SNP may impact the secondary structure or stability of the VapB19 protein, influencing its interaction with other molecules or proteins involved in MDR strain transmission. These findings suggest the potential functional significance of synonymous mutations in drug resistance dissemination.
Our study has identified two synonymous SNPs, G39A (Thr13Thr) at position 2,205,511 in vapB35 and C123T (Asp41Asp) at position 3,799,821 in vapB46, as pertinent to MDR development. Although these mutations do not alter the amino acid sequence, they can still influence protein expression levels, folding, and function. We propose that these SNPs may affect the DNA-binding properties of the respective antitoxins, potentially disrupting the delicate balance within the toxin-antitoxin system, a disruption that promotes bacterial persistence and contributes to the emergence of DR-TB.
Despite the encouraging findings from our study, certain limitations merit consideration. Firstly, our understanding of the toxin-antitoxin system genes in M. tuberculosis is still emerging. Therefore, our insights should be interpreted cautiously, and more research is needed to confirm these preliminary observations. Additionally, we focused specifically on SNPs; hence, our analysis did not include other genetic variations potentially impacting drug resistance. Furthermore, our study used globally sourced M. tuberculosis isolates, which may introduce geographic and demographic biases affecting the observed associations. Finally, while our study reveals associations between specific SNPs and MDR factors, it does not elucidate the precise molecular mechanisms underlying these relationships. Future studies should thus focus on validating our results and exploring the exact molecular pathways involved, thereby providing deeper insight into the development and transmission of multi-drug resistance in M. tuberculosis.
Conclusion
Our study significantly contributes to understanding the factors underlying drug resistance in M. tuberculosis. Identifying SNPs within the toxin-antitoxin system genes highlights their substantial role in MDR development and transmission. Further validation and detailed investigations into the precise molecular mechanisms underlying the impact of these SNPs will advance our knowledge of genetic variation, bacterial persistence, and drug resistance. Ultimately, this research can pave the way for developing more effective strategies to combat TB.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Shandong Public Health Clinical Research Center and Weifang Respiratory Disease Hospital for providing us with the clinical sample data. Additionally, we extend our thanks to all the authors who have shared their sequence datasets on NCBI.
Abbreviations
- M. tuberculosis
Mycobacterium tuberculosis
- MDR-TB
Multidrug-Resistant Tuberculosis
- WGS
Whole genome sequencing
- SNP
Single nucleotide polymorphism
- SNPs
Single nucleotide polymorphisms
- NCBI
National Center for Biotechnology Information
- PPV
Positive Predictive Value
- NPV
Negative Predictive Value
- PLR
Positive Likelihood Ratio
- NLR
Negative Likelihood Ratio
- AUC
Area Under Curve
Author contributions
HCL, FL, YML, YS, and YFL participated in the study design. FL, YL, HCL, YML, XLK, NNT, and YFL performed data collection and statistical analyses. YL, YYL, YZZ, and YWH helped draft the manuscript. YWH, QLH, and YZZ overviewed and supervised the project. All authors read and approved the final manuscript.
Funding
This study was supported by grants from the Shandong Provincial Natural Science Foundation (No. ZR2020KH013; No. ZR2021MH006), the Department of Science & Technology of Shandong Province (CN) (No. 2007GG30002033; No. 2017GSF218052), and the Jinan Science and Technology Bureau (CN) (No. 201704100). It is important to note that the funding bodies provided financial support for the study but were not involved in the study design, data collection, analysis, interpretation, or manuscript writing process.
Data availability
The whole genome sequences have been submitted to the NCBI under the accession number PRJNA1002108.
Declarations
Ethics approval and consent to participate
This study complied with the Declaration of Helsinki, and was approved by the Ethics Committee of Shandong Provincial Hospital, affiliated with Shandong University (SPH), the Ethics Weifang Respiratory Disease Hospital (WRDH), and the Ethics Committee of Shandong Provincial Chest Hospital (SPCH), which waived informed patient consent because all patient records and information were anonymized and deidentified before the analysis.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yameng Li and Yang Shao contributed equally to this work.
Contributor Information
Yao Liu, Email: doctorliu@126.com.
Huaichen Li, Email: lihuaichen@163.com.
References
- 1.World Health Organization. Global tuberculosis report 2023. Geneva: World Health Organization; 2023. [Google Scholar]
- 2.Fraikin N, Goormaghtigh F, Van Melderen L, Type, II Toxin-Antitoxin Systems. : Evolution and Revolutions. Margolin W, editor. J Bacteriol [Internet]. 2020 [cited 2024 Mar 25];202. 10.1128/JB.00763-19 [DOI] [PMC free article] [PubMed]
- 3.T T, K G. Mechanism of post-segregational killing by the hok/sok system of plasmid R1. Sok antisense RNA regulates hok gene expression indirectly through the overlapping mok gene. Journal of molecular biology [Internet]. 1992 [cited 2024 Mar 25];223. https://pubmed.ncbi.nlm.nih.gov/1370544/ [DOI] [PubMed]
- 4.Germain E, Castro-Roa D, Zenkin N, Gerdes K. Molecular mechanism of bacterial persistence by HipA. Mol Cell. 2013;52:248–54. [DOI] [PubMed] [Google Scholar]
- 5.Harms A, Maisonneuve E, Gerdes K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science. 2016;354:aaf4268. [DOI] [PubMed] [Google Scholar]
- 6.Page R, Peti W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat Chem Biol. 2016;12:208–14. [DOI] [PubMed] [Google Scholar]
- 7.Hazan R, Engelberg-Kulka H. Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol Genet Genomics. 2004;272:227–34. [DOI] [PubMed] [Google Scholar]
- 8.Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GPC. The phage abortive infection system, ToxIN, functions as a protein–RNA toxin–antitoxin pair. Proc Natl Acad Sci USA. 2009;106:894–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koga M, Otsuka Y, Lemire S, Yonesaki T. Escherichia coli rnlA and rnlB Compose a Novel Toxin–Antitoxin System. [DOI] [PMC free article] [PubMed]
- 10.Guegler CK, Laub MT. Shutoff of host transcription triggers a toxin-antitoxin system to cleave phage RNA and abort infection. Mol Cell. 2021;81:2361–e23739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sala A, Bordes P, Genevaux P. Multiple toxin-antitoxin systems in Mycobacterium tuberculosis. Toxins (Basel). 2014;6:1002–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundaram K, Vajravelu LK, Paul AJ. Functional characterization of toxin-antitoxin system in Mycobacterium tuberculosis. Indian J Tuberc. 2023;70:149–57. [DOI] [PubMed] [Google Scholar]
- 13.Cheng B, Behr MA, Howden BP, Cohen T, Lee RS. Reporting practices for genomic epidemiology of tuberculosis: a systematic review of the literature using STROME-ID guidelines as a benchmark. Lancet Microbe. 2021;2:e115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, He G, Wang S, Lin S, Chen J, Zhang W. Evaluation of whole-genome sequence method to Diagnose Resistance of 13 anti-tuberculosis drugs and characterize resistance genes in clinical Multi-drug Resistance Mycobacterium tuberculosis isolates from China. Front Microbiol. 2019;10:1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang C, Luo T, Shen X, Wu J, Gan M, Xu P, et al. Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect Dis. 2017;17:275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koster KJ, Largen A, Foster JT, Drees KP, Qian L, Desmond E, et al. Genomic sequencing is required for identification of tuberculosis transmission in Hawaii. BMC Infect Dis. 2018;18:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hicks ND, Yang J, Zhang X, Zhao B, Grad YH, Liu L, et al. Clinically prevalent mutations in Mycobacterium tuberculosis alter propionate metabolism and mediate multidrug tolerance. Nat Microbiol. 2018;3:1032–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q, Ma A, Wei L, Pang Y, Wu B, Luo T, et al. China’s tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis. Nat Ecol Evol. 2018;2:1982–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang H, Ding N, Yang T, Li C, Jia X, Wang G, et al. Cross-sectional whole-genome sequencing and epidemiological study of Multidrug-resistant Mycobacterium tuberculosis in China. Clin Infect Dis. 2019;69:405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo T, Comas I, Luo D, Lu B, Wu J, Wei L, et al. Southern East Asian origin and coexpansion of Mycobacterium tuberculosis Beijing family with Han Chinese. Proc Natl Acad Sci USA. 2015;112:8136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang Q, Liu Q, Ji L, Li J, Zeng Y, Meng L, et al. Citywide transmission of Multidrug-resistant tuberculosis under China’s Rapid Urbanization: a Retrospective Population-based genomic spatial epidemiological study. Clin Infect Dis. 2020;71:142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coll F, Phelan J, Hill-Cawthorne GA, Nair MB, Mallard K, Ali S, et al. Genome-wide analysis of multi- and extensively drug-resistant Mycobacterium tuberculosis. Nat Genet. 2018;50:307–16. [DOI] [PubMed] [Google Scholar]
- 23.Paterson AH, Brubaker CL, Wendel JF. A Rapid Method for extraction of cotton (Gossypium spp.) genomic D N A Suitable for RFLP or PCR Analysis.
- 24.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung Y, Han D. BWA-MEME: BWA-MEM emulated with a machine learning approach. Bioinformatics. 2022;btac137. [DOI] [PubMed]
- 26.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu F, Zhang Y, Zhang L, Li Z, Fang Q, Gao R, et al. Systematic comparative analysis of single-nucleotide variant detection methods from single-cell RNA sequencing data. Genome Biol. 2019;20:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tayyab N, Zaman G, Satti L, Ikram A, Gardezi AH, Khadim MT. Direct susceptibility testing on MGIT 960 TB system: a Rapid Method for detection of drug resistant tuberculosis. J Coll Physicians Surg Pak. 2018;28:590–3. [DOI] [PubMed] [Google Scholar]
- 30.Phelan JE, O’Sullivan DM, Machado D, Ramos J, Oppong YEA, Campino S, et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 2019;11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker TM, Miotto P, Köser CU, Fowler PW, Knaggs J, Iqbal Z, et al. The 2021 WHO catalogue of Mycobacterium tuberculosis complex mutations associated with drug resistance: a genotypic analysis. Lancet Microbe. 2022;3:e265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guerra-Assunção J, Crampin A, Houben R, Mzembe T, Mallard K, Coll F, et al. Large-scale whole genome sequencing of M. Tuberculosis provides insights into transmission in a high prevalence area. eLife. 2015;4:e05166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jajou R, de Neeling A, van Hunen R, de Vries G, Schimmel H, Mulder A, et al. Epidemiological links between tuberculosis cases identified twice as efficiently by whole genome sequencing than conventional molecular typing: a population-based study. PLoS ONE. 2018;13:e0195413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiner-Oms Á, Sánchez-Busó L, Corander J, Gagneux S, Harris SR, Young D et al. Genomic determinants of speciation and spread of the Mycobacterium tuberculosis complex. Sci Adv. 2019. [DOI] [PMC free article] [PubMed]
- 37.Mahatha AC, Banerjee SK, Ghosh A, Lata S, Saha S, Basu J et al. A systems approach to decipher a role of transcription factor RegX3 in the adaptation of Mycobacterium tuberculosis to hypoxic stress. Microbiol (Reading). 2022;168. [DOI] [PubMed]
- 38.da Silva CAPT, Lourenço RF, Mazzon RR, Ribeiro RA, Marques MV. Transcriptomic analysis of the stationary phase response regulator SpdR in Caulobacter crescentus. BMC Microbiol. 2016;16:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole genome sequences have been submitted to the NCBI under the accession number PRJNA1002108.