

Abstract
Transcription factor 19 (TCF19) plays critical roles in type 1 diabetes and the maintenance of pancreatic β cells. Recent studies have also implicated TCF19 in cell proliferation of hepatic carcinoma and non-small cell lung carcinoma; however, the mechanism underlying this regulation remains elusive. At the molecular level, TCF19 contains two modules, the plant homeodomain (PHD) finger and the forkhead-associated (FHA) domain, of unclear function. Here, we show that TCF19 mediates hepatocellular carcinoma HepG2 cell proliferation through its PHD finger that recognizes trimethylated lysine 4 of histone 3 (H3K4me3). W316 of the PHD finger of TCF19 is one of the critical residues eliciting this function. Whole genome microarray analysis and orthogonal cell-based assays identified a large subset of genes involved in cell survival and proliferation that depend on TCF19. Our data suggest that TCF19 acts as a pro-oncogene in hepatocellular carcinoma cells and that its functional PHD finger is critical in cell proliferation.
Graphical Abstract
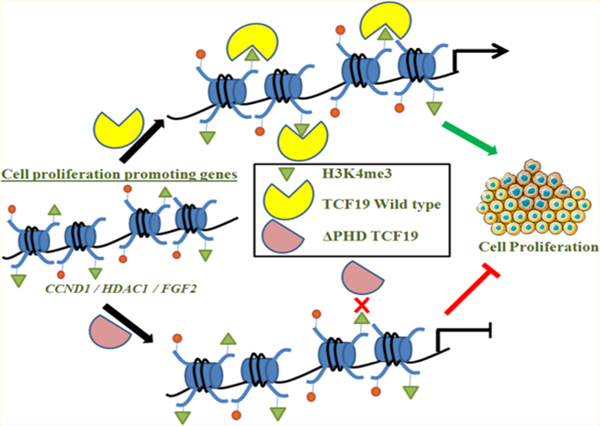
Cell growth, survival and proliferation require adequate environmental and external signals, including mutagens and nutrients such as glucose and amino acids.1 The level of cell proliferation in tumor tissues is increased due to an expedited rate of cell division. Hepatocellular carcinoma (HCC) represents the most abundant primary liver cancer that affects more than 500000 people worldwide.2 Chronic liver disease and cirrhosis are the predominant cause of HCC.3 HCC originates from the hepatic stem cells, although this hypothesis is still under investigation and needs to be confirmed. Tumors progress from the first stage with local expansion slowly through intrahepatic spread leading to advanced stages of distant metastasis.
HCC progresses through uncontrolled liver cell mass expansion and leads to chronic cirrhosis of liver. Over the years, several studies have been conducted to understand the involvement of different factors in HCC progression through cell proliferation. Various small molecules, including TGF-β4 and EGFL7,5 transcription factors and extracellular proteins such as TRIM52,6 mutated β-catenin,7 and survivin8,9 have been reported to regulate HCC cell proliferation. Our studies suggest that not only these molecules and proteins but also the epigenetic landscape in which they function is essential for HCC proliferation.
Transcription factor 19 (TCF19), also known as SC1, was originally identified as a growth-regulated cDNA and has emerged as a key epigenetic reader protein.10 A substantial increase in the TCF19 expression level could be achieved upon treating the cells with fetal bovine serum (FBS) used as a source of growth factor. A recent study of TCF19 has shown that a fine balance in the expression of cell proliferation and apoptotic genes is instrumental in the maintenance of pancreatic β cells.11 TCF19 was also identified as a type I diabetes-associated locus involved in the regulation of endoplasmic reticulum (ER) stress, and genomewide association studies revealed its role in chronic hepatitis B prognosis.12 Recently, two independent studies indicated the function of TCF19 as a cell proliferation promoter in hepatocellular carcinoma and non-small cell lung carcinoma.13,14 These studies reported that TCF19 targets the FOXO1 pathway in a differential manner. Furthermore, TCF19 is involved in regulating glucose homeostasis and repressing de novo glucose production in HepG2 cells by regulating key enzymes of gluconeogenesis.15 The study presented here highlights the role of the plant homeodomain (PHD) finger of TCF19 in cell proliferation. In addition, we show that the proliferation genes are under direct regulation of TCF19 through its H3K4me3 binding ability. Our data suggest that TCF19 functions as a pro-oncogenic factor in the progression of hepatocellular carcinoma.
MATERIALS AND METHODS
Cell Culture and Glucose Treatment.
HepG2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco), 1% Anti-Anti (Gibco), and 1% essential amino acid. Cells were kept in a 37 °C incubator with 5% CO2.
For high-glucose treatment,15 HepG2 cells were seeded in DMEM and replenished with fresh medium containing 40 mM glucose for the next 2 days.
Gene Silencing and Overexpression.
HepG2 cells were transfected with TCF19 siRNA (Santa Cruz, sc-63113) or negative control siRNA using transfection reagent Interferrin (Polyplus) following the manufacturer’s instructions. Cells were kept in 37 °C for 24 h with the supply of 5% CO2. After that, further experiments were carried out.15
To overexpress FLAG-conjugated constructs of WT-TCF19, ΔPHD-TCF19, or ΔFHA-TCF19, we transfected the DNA using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol and kept cells for 24 h at 37 °C.15
Cloning and Protein Purification.
The TCF19 PHD finger constructs (residues 296–342 with additional N-terminal DD or DDCAC added) were cloned into the pGEX-6P-1 (GE Healthcare) expression vector. The cDNA was purchased from Open Biosystems (Thermo Scientific). Unlabeled and 15N-labeled WT and mutant TCF19 PHD finger proteins were expressed in Escherichia coli Rosetta-2 (DE3) pLysS cells that were grown in Luria-Bertani broth or minimal medium supplemented with 15NH4Cl (Sigma) and 150 μM ZnCl2. Cells were induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside for 16–18 h at 18 °C. Following cell harvesting by centrifugation, cells were lysed by sonication. GST fusion proteins were purified with glutathione Sepharose 4B beads (Thermo Fisher Scientific), and the tag was cleaved with PreScission protease. Size exclusion chromatography was performed to further purify the protein until homogeneity. Millipore concentrators (Millipore) were used to concentrate the proteins.
Nuclear Magnetic Resonance (NMR) Titrations of Histone Peptides.
The 1H–15N HSQC spectra of 0.1 mM uniformly 15N-labeled WT and mutant TCF19 PHD finger proteins were recorded on a Varian INOVA 500 MHz spectrometer using pulse field gradients to suppress artifacts and eliminate the water signal. The binding was characterized by monitoring chemical shift changes in 1H–15N HSQC spectra of the PHD finger as chemically synthesized histone tail peptides were added in a stepwise manner. A nonlinear least-squares analysis in Kaleidagraph was performed to determine the dissociation constants using the equation
where is the concentration of the peptide, is the concentration of the protein, is the observed chemical shift change, and is the normalized chemical shift change at saturation. Normalized chemical shift changes were calculated using the equation , where is the change in chemical shift in parts per million.
Fluorescence Spectroscopy.
The samples containing the PHD fingers and progressively increasing concentrations of the histone peptide were excited at 280 nm on a FluoroLog-3 spectrofluorometer (HORIBA) at 25 °C. Emission spectra were recorded over a range of wavelengths between 310 and 405 nm with a 0.5 nm step size and a 1 s integration time and averaged over three scans. A nonlinear least-squares analysis was performed to determine the values using the equation
where is the concentration of the histone peptide, is the concentration of the PHD finger, is the observed change in signal intensity, and is the difference in signal intensity of the free and bound states of the PHD finger. Three separate experiments were performed to obtain the average value with the error calculated as the standard deviation between the runs.
Homology Modeling.
The PHD finger of Jumonji/ARID domain-containing protein 1A was used as a template to perform a homology model of TCF19-PHD employing SWISS-MODEL,16 and its structural quality was verified using PROCHECK and WHATIF.17 Pymol was employed to visualize the structure, and the critical residues obtained from NMR were highlighted.
Microarray Reanalysis and Validation.
RNA isolation and microarray analysis were performed in the previous study from our laboratory.15,18 Here the differentially regulated gene list was reanalyzed and categorized into different biological processes. A p value of <0.05 and a fold change cutoff of ≥2 were set for this analysis. The genes that came under cell proliferation were taken into account for further analysis and validation.
Biological Pathways and Gene Ontology Analysis.
The KEGG pathway analyzer tool was used to identify the genes from the TCF19 knocked down differentially expressed gene set, involved in KEGG Ontology. Genes in cell cycle pathway regulated by TCF19 were color coded using the data mapper within the software.19 Differentially expressed genes under a cell proliferation subset were ascertained into proper functional classification using DAVID20,21 and PANTHER22,23 Gene Ontology analysis tools. A statistically significant differentially expressed cell proliferation gene set was imported into Cytoscape version 2.8 to characterize the connections and modeling the network. Gene ontology processes with specific genes were subjected to color codes relating to their fold change, and the major significant biological processes were highlighted to understand the importance of TCF19 in cell proliferation. A statistically significant set of genes were overrepresented to generate a biological network using BiNGO, a Java-based plugin for Cytoscape to determine the Gene Ontology categories.24
Chromatin Immunoprecipitation (ChIP) Assay.
Chromatin immunoprecipitation was performed using a previously mentioned protocol.25 In brief, cells were cross-linked using 1% formaldehyde and the reaction was stopped using 0.125 M glycine. Then cells were lysed using cell lysis buffer [5 mM PIPES (pH 8.0), 85 mM KCl, and 0.5% NP40 added with protease inhibitor cocktail (PIC) (Roche)]. After that, nuclear lysis buffer [50 mM Tris-HCl (pH 8.0), 10 mM EDTA, and 1% sodium dodecyl sulfate (SDS) supplemented with PIC] was added to complete the nuclear lysis. Thereafter, sonication and preclearing were performed. The anti-TCF19 antibody (sc-390923) and anti-histone 3 lysine trimethylation antibody (ab8580) were used for immunoprecipitation. To pull down the complex, blocked DYNA beads were added and, after binding for 2 h, washed with RIPA buffer, high-salt buffer, LiCl wash buffer, and TE buffer, successively. RNase A- and Proteinase K-treated beads were subjected to de-cross-linking at 65 °C for at least 6 h. Next, DNA was prepared by the phenol/chloroform method and subjected to quantitative real-time polymerase chain reaction (qRT-PCR) or stored at 4 °C. Primers for the promoters of CCND1, HDAC1, and FGF2 genes were used for qRT-PCR. Experiments were repeated thrice, and the error was calculated as ± standard deviation (SD) within the biological triplicates.
Quantitative Real-Time PCR.
Total RNA was isolated from HepG2 cells using TRIzol reagent (Hi-Media) and then reverse transcribed using the Revertaid First Strand cDNA synthesis kit (Thermo Fischer Scientific) following the manufacturer’s protocol.15 Then qRT-PCR was performed using ABI-SYBR GREEN mix in a StepONE plus FAST real-time PCR machine. All of the samples were analyzed three times independently.
Glucose Uptake Assay.
Cellular glucose uptake was measured by a previously described procedure.26 It was done in HepG2 cells under three different conditions: stably expressing either non-targeting shRNA, TCF19 shRNA, or TCF19 shRNA complemented with WT-TCF19. HepG2 cells were plated into 12-well dishes, incubated for 24 h, and then kept to starve for 6 h in glucose free medium. After that, 10 μM 2-NBDG was introduced for 2 h. Then cell lysis was performed using lysis buffer [10 mM Tris-HCl (pH 8.0) and 0.2% SDS], and the uptake was quantified using a Synergy HTX Multimode reader.
Cell Proliferation Assay.
Control and transfected cells were plated in triplicate using 24-well plates at a density of 0.2 × 106 per well. MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was added to the wells at the time points indicated in this study, and the assay was performed as previously described.27
BrdU Incorporation Assay.
For the BrdU incorporation assay, a previously described protocol was used.28 HepG2 cells were seeded and trasfected for 24 h on coverslips placed in 12-well dishes. Then BrdU was added at a concentration of 18 μg/mL for 30 min at 37 °C. After that, the cells were taken out of the CO2 incubator and washed twice with ice-cold phosphate-buffered saline (PBS). Then they were fixed with ice-cold 100% ethanol for 30 min followed by acid treatment with 2 N HCl. Cells were fixed with 3% BSA followed by immunostaining with the anti-BrdU antibody. Following PBST washes, the cells were incubated with the Alexa Fluor-488-conjugated secondary antibody for 1 h at room temperature. Coverslips were then washed with PBST, stained with DAPI, and mounted on glass slides. The imaging was performed using a Zeiss LSM 710 confocal microscope.
Wound Healing Assay.
The wound healing assay was performed with HepG2 cells following the procedure described previously.27 Cells were seeded in a six-well dish in triplicate sets, grown to a confluency of 80–90%, transfected, and after 24 h the cell surface was scratched with a 20–200 μL sterile tip. Images were captured at time points after scratching the surface at 0, 8, and 16 h to measure the extent of wound healing by the cells with a Nikon T1 E100 microscope. The 5-fluorouracil treatment of the cells at a final concentration of 1 mM was performed on another triplicate set 8 h prior to making the scratch. Using ImageJ developed by the National Institutes of Health, the images were quantified to calculate the wound recovery by the cells with their proliferating ability.
RESULTS
TCF19 Is a Regulator of Hepatic Cell Proliferation.
To ascertain the role of TCF19 in cell proliferation, we analyzed the differential gene expression network that was obtained by a microarray in the absence of TCF19 under high-glucose stimulation (40 mM glucose treatment for 48 h). In comparison to low-glucose treatment (cells maintained in 5.5 mM glucose), in high-glucose treatment we identified a large subset of genes involved in cell proliferation. We performed KEGG cell cycle pathway analysis and using pathway mapper identified a subset of 26 genes that were differentially regulated upon knocking down TCF19 (Figure S1A,B). We performed gene ontology analysis employing DAVID and BiNGO software (Figure 1A–C) and confirmed the transcription levels of the candidates by qRT-PCR (Figure 1D). Chromatin immunoprecipitation (ChIP) assays revealed that TCF19 occupies promoters, but not the gene body of proliferation genes, such as CCND1, HDAC1, and FGF2 in HepG2 (Figure 1E) and Huh7 (Figure S1C) cell lines.
Figure 1.
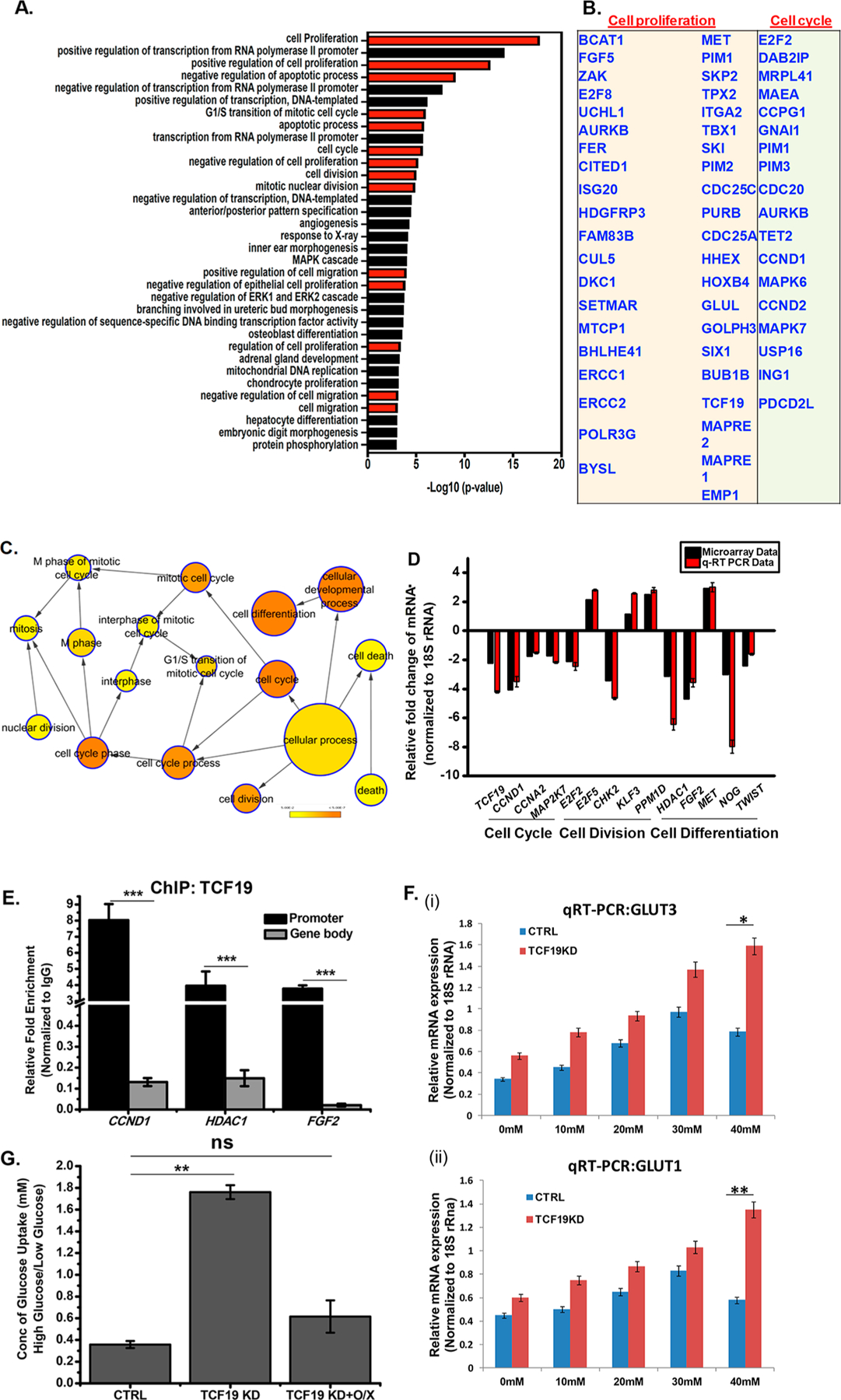
TCF19 is an important player in cellular proliferation. (A) Gene ontology (biological process) categories showing the most significant enrichment in a list of proliferation-associated genes obtained after microarray analysis upon TCF19 knockdown. GO terms are indicated on the Y-axis. The −log 10 P value on the X-axis indicates the level of significance of each process as obtained from the DAVID tool. (B) List of key cell proliferation- and cell cycle-related genes. (C) Genes regulating cellular proliferation that were differentially expressed at HG after knocking down TCF19 were analyzed by BiNGO. Fold change 2.0; p ≤ 0.05. (D) Validation of microarray genes for proliferation was performed by knocking down TCF19 under high-glucose conditions. (E) TCF19 is recruited to the promoter of the proliferation genes, but not on the gene body as observed by the ChIP assay. (F) qRT-PCR analysis of GLUT3 and GLUT1 in the presence and absence of TCF19 with an increasing glucose concentration. The presence of TCF19 represses the overexpression of GLUT receptors. (G) Glucose uptake assay to measure the cellular glucose uptake ability in the presence and absence of TCF19 under low- and high-glucose conditions. TCF19 KD stable cells were complemented with the FL FLAG-TCF19 construct that compensated for the cellular glucose uptake. Cells were glucose starved for 6 h followed by treatment with 10 μM 2-NBDG for 2 h.
To gain insight into the differentially regulated pathways upon high-glucose induction, we monitored the expression of the GLUT receptors (GLUT1 and GLUT3) that are involved in the transport of glucose in cells and were differentially expressed in the microarray analysis. The result indicated that in the presence of TCF19 the level of expression of GLUT1 and GLUT3 increased till 30 mM glucose; however, GLUT1 and GLUT3 were downregulated at higher glucose concentrations. In the absence of TCF19, GLUT receptor expression was upregulated parallel to the increasing glucose concentration (Figure 1F). Concomitant with the altered expression of GLUT receptors under high-glucose conditions in a TCF19 null background, we observed that the cellular glucose uptake was significantly enhanced (Figure 1G). TCF19 knocked down (KD) stable cells (Figure S1D,E) were complemented with the FL FLAG-TCF19 construct (O/X) that compensated for the cellular glucose uptake (Figure 1G). These data indicated that upon high-glucose induction TCF19 is overexpressed and partially represses the excess glucose uptake by the cells, although the mechanism by which TCF19 regulates glucose-mediated cell proliferation remains unclear.
The PHD Finger of TCF19 Is Crucial for TCF19-Mediated Hepatic Cell Proliferation.
To define the role of the two domains of TCF19, the PHD finger and FHA domain, in the regulation of hepatic cell proliferation, we deleted these domains individually, ectopically expressed either the PHD finger, or the FHA domain deleted constructs of TCF19 in cells, and carried out cell growth and wound healing assays. Overexpression of WT-TCF19, ΔPHD-TCF19, and ΔFHA-TCF19 was confirmed by qRT PCR (Figure 2SB) and Western blotting (Figure 2SC). As shown in Figure 2A, overexpression of FLAG-TCF19 significantly upregulated hepatic cell growth as compared to overexpression of FLAG-ΔPHD-TCF19, whereas overexpression of FLAG-ΔFHA-TCF19 had no significant effect (Figure 2A). In support, BrdU incorporation assays showed that the deletion of the PHD finger resulted in the compromised uptake of BrdU in the cells expressing comparable levels of WT, ΔPHD-TCF19, and ΔFHA-TCF19 (Figure S2A), indicating that this domain affects cell growth (Figure 2B,C). Subsequently, the critical role of the TCF19 PHD finger was corroborated by wound healing assays. Overexpression of FLAG-TCF19 substantially accelerated wound healing (Figure 2D,E). In contrast, FLAG-ΔPHD-TCF19 overexpressed cells showed a notable decrease in wound healing ability, whereas FLAG-ΔFHA-TCF19 had no effect on the extent of healing (Figure 2D). Furthermore, treatment of hepatic cells with 5-fluorouracil, a potent proliferation blocker, led to a decrease in the extent of wound healing upon overexpression of WT FLAG-TCF19 (Figure 2F,G).
Figure 2.
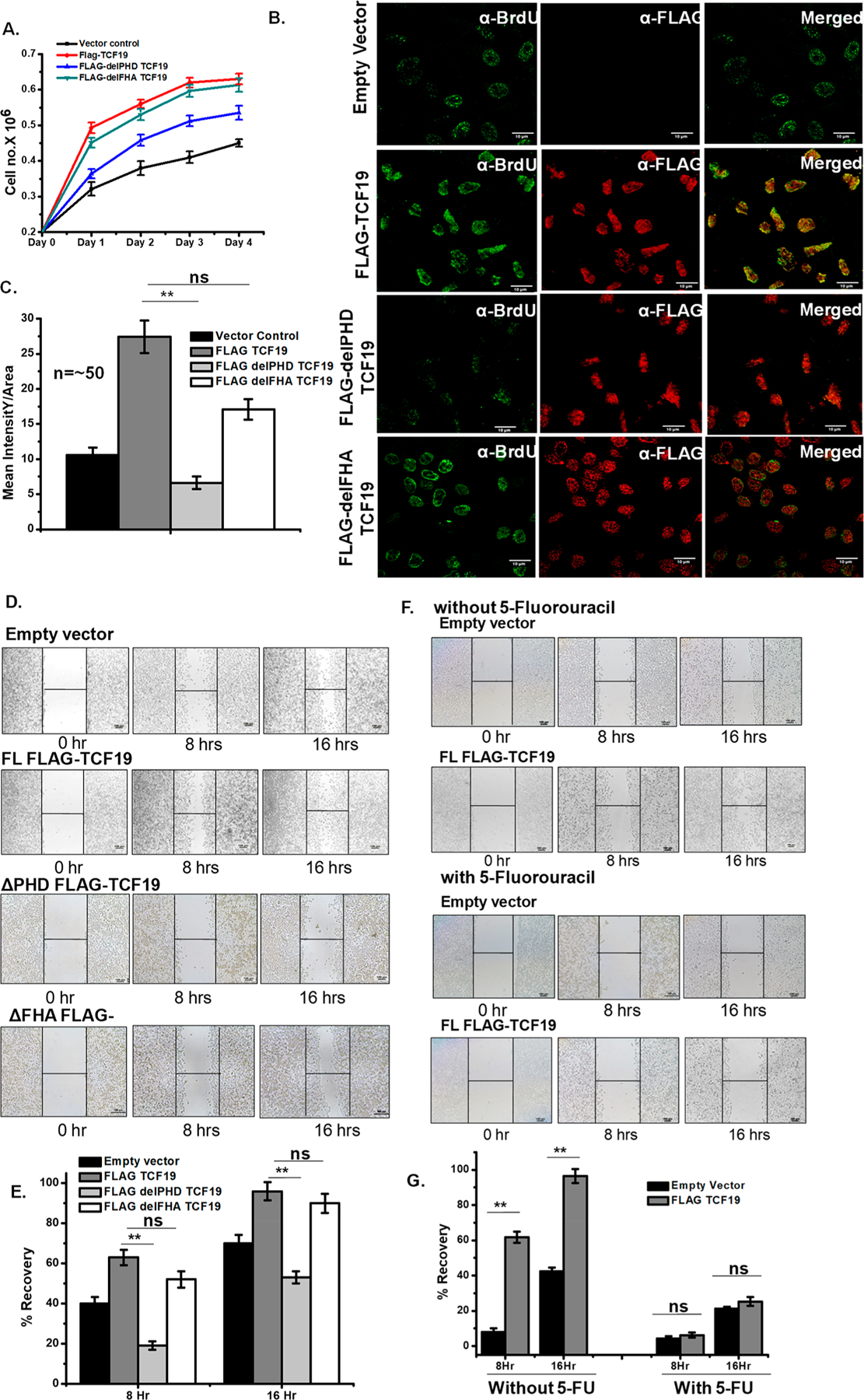
TCF19 promotes cellular proliferation. (A) Cell proliferation assay by the cell number counting technique. The WT-TCF19 overexpressed cell (red) proliferates at a rate higher than those of delPHD-TCF19 (blue) overexpressed and control (black) cells. (B) BrdU incorporation assay by confocal microscopy measurement also showed higher proliferation in WT-TCF19 overexpressed cells. (C) Quantification of the microscopy data (n ∼ 50). (D) The wound healing assay also showed a positive result on proliferation in the case of wild type overexpressed cells. (E) Quantification of recovery by wound healing. (F) Wound healing assay in the presence and absence of potential proliferation blocker 5-fluorouracil in empty vector transfected cells as well as WT-TCF19 transfected cells. (G) Quantification of wound healing in the presence and absence of 5-fluorouracil.
The rate of cell proliferation was not dependent on the presence or absence of glucose in TCF19 knocked down cells as siRNA-mediated silencing of TCF19 (Figure S3A) showed a significant reduction in the rate of cell proliferation (Figure 3A), as well as the rate of BrdU incorporation (Figure 3B,C) under low- and high-glucose conditions. A similar result of glucose-independent function of TCF19 in cellular growth was confirmed by the wound healing assay (Figure 3D–G). Collectively, these results point to the role of TCF19, and its PHD finger in particular, in mediating cell proliferation in a glucose concentration-independent manner.
Figure 3.
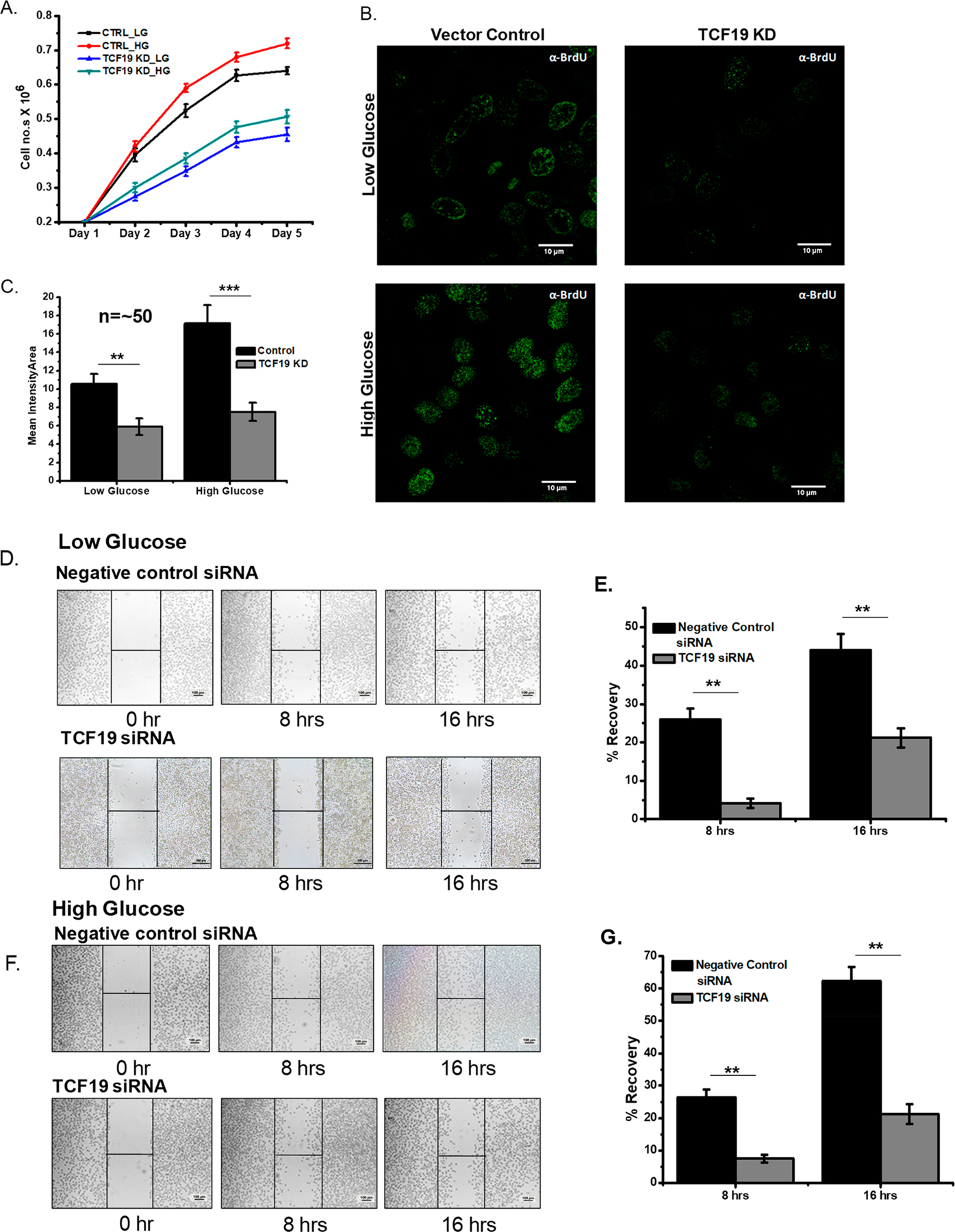
TCF19 enhances cellular proliferation irrespective of glucose. (A) Cell proliferation assay by the cell number counting technique showing the reduced rate of proliferation in the absence of TCF19 under low-glucose (blue) and high-glucose (cyan green) conditions. (B) BrdU incorporation assay by confocal microscopy measurement also showed a lower rate of proliferation in TCF19 KD cells under low- and high-glucose conditions. (C) Quantification of the microscopy data (n ∼ 50). (D–G) Wound healing assay and their corresponding quantification under low- and high-glucose conditions.
Molecular Analysis of the Histone Binding Site of the TCF19 PHD Finger.
The sequence of the PHD finger of TCF19 is similar to that of the PHD fingers that recognize histone H3K4me3 and was previously found to have chromatin binding ability.15 Interestingly, our ChIP data show the enrichment of H3K4me3 on promoters of TCF19 target genes, including CCND1, HDAC1, and FGF2, suggesting that the histone binding function of its PHD finger is essential for their transcription regulation (Figure 4A). To characterize this function in detail, we assayed the TCF19 PHD finger by NMR. Large chemical shift changes in 1H–15N heteronuclear single-quantum coherence (HSQC) spectra of the uniformly 15N-labeled wild type PHD finger were observed when the H3K4me3 peptide (residues 1–12) was progressively added to the NMR sample (Figure 4B). Molecular modeling of the TCF19 PHD finger revealed an aromatic cage composed of W307 and W316 residues that are likely involved in the interaction with trimethylated Lys4. As shown in Figure 4B (iii), mutation of W316 to a tyrosine essentially abolished the interaction between the TCF19 PHD finger and H3K4me3, and the H3K4me3 binding activity of the W307Y mutant was substantially decreased (Kd = 2 mM) (Table 1). We note that a number of residues in and around the H3K4me3 binding site appear to be necessary for the structural stability of the PHD finger, as A306Y, Q309R, D311R, D314R, W307A, and W316A mutants were unstable or unfolded on the basis of their 1H–15N HSQC spectra (data not shown). Additionally, the three folded V305Y, C324A, and R330Y mutants were impaired in terms of their binding to H3K4me3, suggesting that the TCF19 PHD finger structure and its histone binding function are very sensitive to mutations and could be easily disrupted. The critical residues identified by NMR have been mapped on the homology model (Figure 4C). Together, our findings underscore the critical role of the H3K4me3-selective PHD finger of TCF19 in regulating hepatic cell proliferation.
Figure 4.
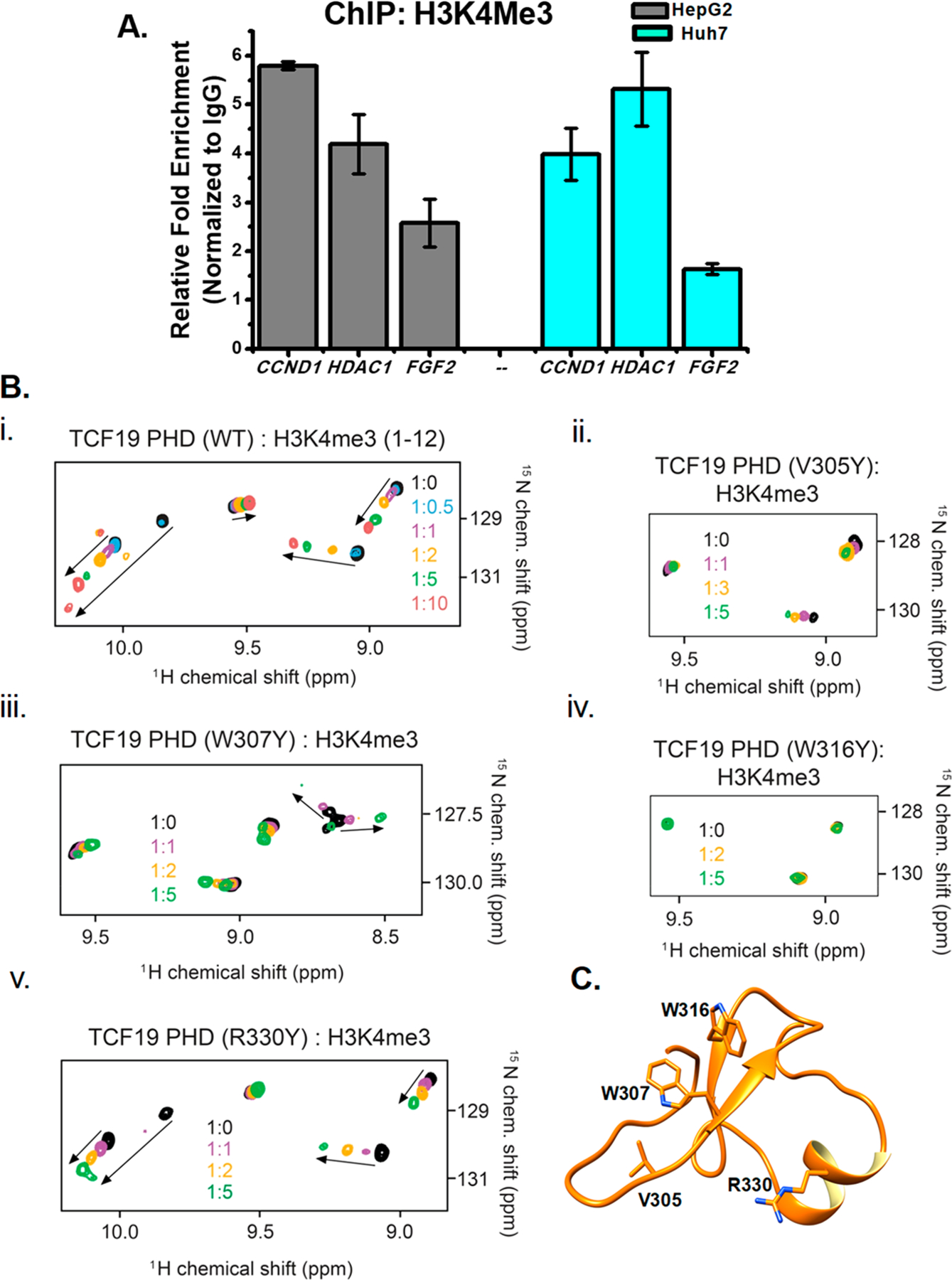
TCF19 binds to H3K4me3 through its PHD finger and regulates expression of proliferation genes. (A) The proliferation genes are also enriched with H3K4me3 at the cognate sites where TCF19 is recruited. (B) Superimposed 1H–15N HSQC spectra of uniformly 15N-labeled WT or mutated TCF19 PHD finger, collected as H3K4me3 peptide (residues 1–12) was added stepwise. Spectra are color-coded according to the protein:peptide molar ratio. (C) Structure of TCF19 as visualized by molecular modeling with the critical residues observed through NMR highlighted.
Table 1.
Measurement of the Binding Affinities of TCF19-PHD WT and Its Mutants with H3K4me3 Peptides As Obtained from NMR
| protein | peptide | |
|---|---|---|
| TCF19 (DDCAC) PHD | H3K4me3 | 4.7 ± 0.4 μMa |
| TCF19 PHD V305Y | H3K4me3 | weak binding |
| TCF19 PHD A306Y | unstable | |
| TCF19 PHD Q309R | unstable | |
| TCF19 PHD D311R | unstable | |
| TCF19 PHD D314R | unstable | |
| TCF19 PHD C324A | H3K4me3 | 440 ± 170 μM |
| TCF19 PHD R330Y | H3K4me3 | 400 ± 60 μM |
| TCF19 PHD W307A | unfolded | |
| TCF19 PHD W307Y | H3K4me3 | 2.0 ± 0.6 mM |
| TCF19 PHD W316A | unfolded | |
| TCF19 PHD W316Y | H3K4me3 | no binding |
Obtained from tryptophan fluorescence spectroscopy.
A Structurally Elucidated Critical Residue of TCF19 in Vitro Can Compromise Hepatic Cell Proliferation.
We subsequently tested the contribution of the critical residue W316 of the TCF19 PHD finger, which had shown a complete abrogation of binding to H3K4Me3 by NMR, toward hepatic cell proliferation. To negate the contribution of the endogenous TCF19, the cell proliferation assays were performed in a system in which the endogenous TCF19 expression was at first knocked down from the hepatic cells and then complemented back with either FLAG-TCF19 wild type or FLAG-TCF19 W316Y mutant constructs. We ensured comparable levels of expression of the wild type and W316Y mutant (Figure S4B) before we proceeded with cell proliferation assays. As shown in Figure 5A, overexpression of FLAG-TCF19 W316Y showed significantly reduced hepatic cell growth as compared to that of the FLAG-TCF19 wild type. Subsequently, BrdU incorporation assays showed that this site specific mutation could dramatically reduce the rate of uptake of BrdU in the cells as compared to wild type TCF19 (Figure 5B,C and Figure S4A). The critical role played by the W316 residue was further tested by wound healing assays. As compared to the FLAG-TCF19 wild type, the FLAG-TCF19 W316Y mutant showed a notable decrease in wound healing capability (Figure 5D,E). We finally tested whether the FLAG-TCF19 W316Y mutant construct could be recruited to the TCF19 target genes, including CCND1, HDAC1, and FGF2, by ChIP assays. As represented in Figure 5F, the level of recruitment of the FLAG-TCF19 W316Y mutant to the H3K4Me3-enriched candidate genes was significantly reduced compared to that of the FLAG-TCF19 wild type. Collectively, our results clearly indicate that the critical residue W316 of TCF19 that mediates the interaction with H3K4Me3 is also a key regulator of the hepatic cell proliferation function as exhibited by TCF19.
Figure 5.
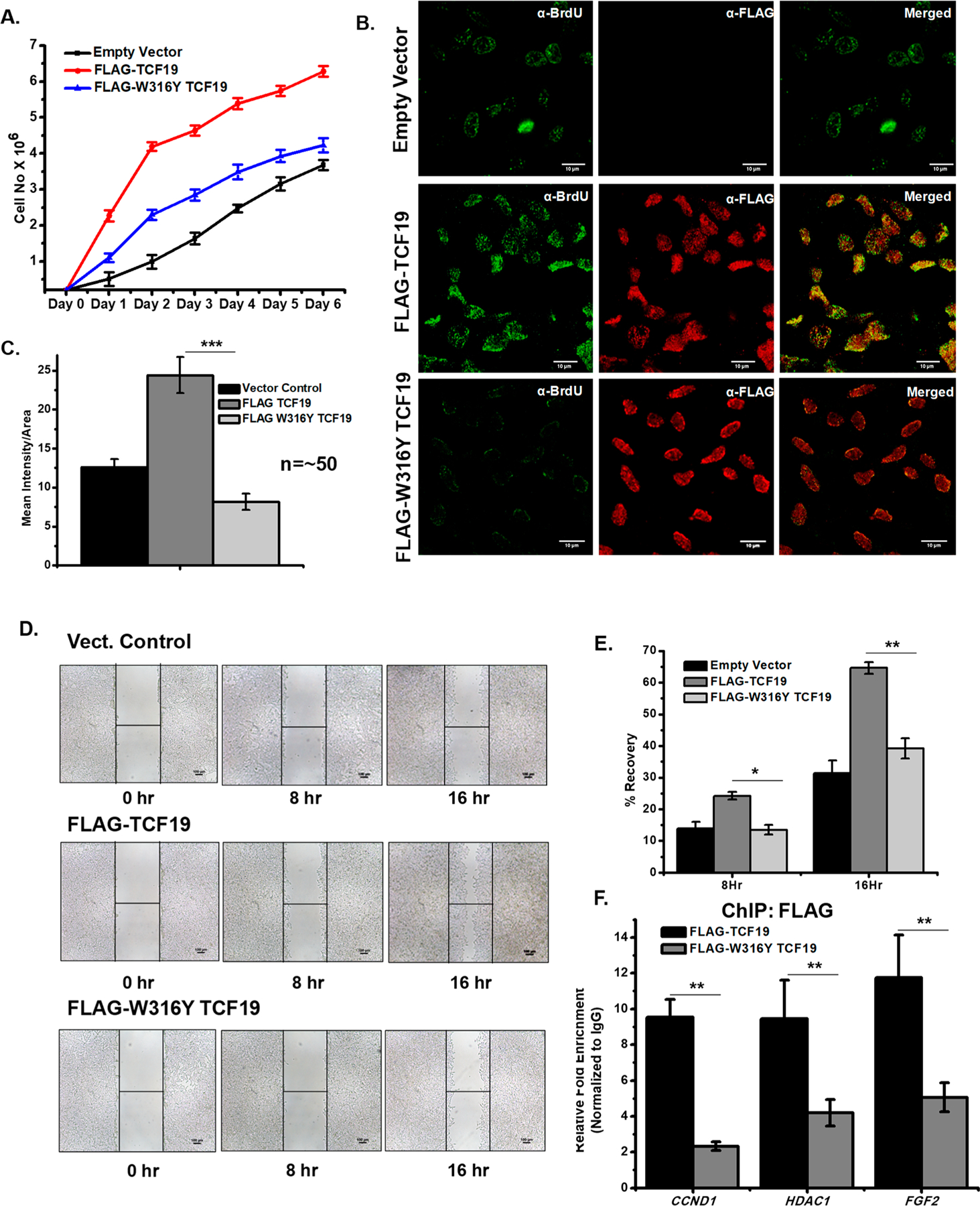
(A) Cell proliferation assay by the cell number counting technique. FLAG-TCF19 overexpressed cells (red) proliferate at a rate higher than those of FLAG-W316Y TCF19 (blue) overexpressed and control (black) cells. (B) BrdU incorporation assay by confocal microscopy measurement also showed a lower rate of proliferation in FLAG-W316Y TCF19 overexpressed cells compared to WT FLAG-TCF19 overexpressed cells. (C) Quantification of the microscopy data (n ∼ 50). (D) Wound healing assay showing decreases in the rate of proliferation in the case of FLAG-W316Y TCF19 overexpressed cells compared to FL FLAG-TCF19 overexpressed cells. (E) Quantification of recovery by wound healing. (F) ChIP qRT-PCR analysis shows a high occupancy of TCF19 on cell proliferation genes, while the W316Y mutation in TCF19 reduces its occupancy on the same targets.
DISCUSSION
TCF19 was found to be involved in the proliferation of INS-β,11 non-small cell lung carcinoma,14 and hepatocellular carcinoma13 cells; however, the link between its ability to regulate cell growth and the chromatin binding potential of TCF19 was not investigated. Our microarray results in HepG2 cells in the presence and absence of TCF19 have identified a large subset of genes that are implicated in cell proliferation. We validated several candidates from this subset and confirmed that many of the downregulated genes are well-known pro-proliferative genes and the upregulated ones are antiproliferative genes (Figure 1). There are a number of studies showing the involvement of glucose in the context of cell proliferation,29–31 and we observed that TCF19 is overexpressed upon glucose stimulation.15 Our data suggest that TCF19 regulates the optimum uptake levels of glucose to maintain a sustainable concentration of glucose in the hepatic cell. We observed that the rate of cellular glucose uptake was substantially increased when TCF19 was abrogated from the cells, and this finding establishes the role of TCF19 in maintaining glucose homeostasis. Through wound healing assays, we also found that the rate of cell proliferation was almost comparably affected in the absence of TCF19 in cells under low- and high-glucose conditions. These data indicate that TCF19 itself is an important regulator of hepatic cell proliferation.
TCF19 has recently been shown to recognize H3K4me3,15 and our data demonstrate that TCF19 exerts its downstream functions in maintaining glucose homeostasis in the hepatic cell through the interaction of the PHD finger with the H3K4me3 mark enriched at promoters of proliferation genes. We propose that through H3K4me3 recognition, the TCF19 PHD finger reprograms the expression of glucose-responsive genes. In agreement, wound healing and BrdU incorporation assays confirmed that deletion of the PHD finger results in a lower rate of hepatic cell proliferation (Figure 2). Using mutagenesis, NMR spectroscopy, and modeling, we identified residues in the PHD finger that are involved in this interaction. We further show that the TCF19 PHD finger structure and its histone H3K4me3 binding function are sensitive to mutations and could be easily disrupted (Figure 4). Through a cell-based assay, we further show that mutating the critical residue W316 in the PHD finger of the TCF19 protein significantly reduced its occupancy on cell proliferation-promoting genes and concomitantly weakened its cell proliferation ability (Figure 5). Collectively, in this study, we report that TCF19 is a pro-oncogenic epigenetic regulator. Through the PHD finger-mediated recognition of H3K4me3, TCF19 regulates expression of the proliferation genes. Using a small molecule-based approach to perturb this interaction could restore an antiproliferative state in cells by reprogramming these genes, and thus, TCF19 could be an attractive therapeutic target.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the laboratory members, Isha Sengupta, Sulagna Sanyal, Santanu Adhikary, Amrita Sengupta, and Vipin Singh, for their help with experiments. The authors extend their thanks to Dr. Sangram Bagh and his laboratory members at SINP, Kolkata, for their assistance with the use of the multimode reader.
Funding
This study was funded by Biomolecular Assembly, Recognition and Dynamics (BARD) (Grant 12-R&D-SIN-5.04–0103) by the Department of Atomic Energy (DAE), Government of India, and Science and Engineering Research Board File CRG/2018/000985 of the Department of Science and Technology (DST), Government of India (to C.D.), and by grants from Cancer Prevention and Research Institute of Texas (CPRIT; RR170020) (to S.S.G.) and the National Institutes of Health (to T.G.K.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.9b00771.
Figures S1–S4 (PDF)
Accession Codes
UniProt Accession ID for TCF19, Q9Y242.
The authors declare no competing financial interest.
REFERENCES
- (1).Broach JR (2012) Nutritional control of growth and development in yeast. Genetics 192, 73–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, de Oliveira AC, Santoro A, Raoul J-L, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz J-F, Borbath I, Haüssinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, and Bruix J (2008) Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med 359, 378–390. [DOI] [PubMed] [Google Scholar]
- (3).Wang K, and Sun D (2018) Cancer stem cells of hepatocellular carcinoma. Oncotarget 9, 23306–23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Arrese M, Hernandez A, Astete L, Estrada L, Cabello-Verrugio C, and Cabrera D (2018) TGF-β and Hepatocellular Carcinoma: When A Friend Becomes An Enemy. Curr. Protein Pept. Sci 19, 1172–1179. [DOI] [PubMed] [Google Scholar]
- (5).Li Z, Xue T-Q, Yang C, Wang Y-L, Zhu X-L, and Ni C-F (2018) EGFL7 promotes hepatocellular carcinoma cell proliferation and inhibits cell apoptosis through increasing CKS2 expression by activating Wnt/β-catenin signaling. J. Cell. Biochem 119, 10327–10337. [DOI] [PubMed] [Google Scholar]
- (6).Zhang Y, Tao R, Wu S-S, Xu C-C, Wang J-L, Chen J, Yu Y-S, Tang Z-H, Chen X-H, and Zang G-Q (2018) TRIM52 upregulation in hepatocellular carcinoma cells promotes proliferation, migration and invasion through the ubiquitination of PPM1A. J. Exp. Clin. Cancer Res 37, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang Z, Sheng Y-Y, Gao X-M, Wang C-Q, Wang X-Y, Lu XU, Wei J-W, Zhang K-L, Dong Q-Z, and Qin L-X (2015) β-catenin mutation is correlated with a favorable prognosis in patients with hepatocellular carcinoma. Mol. Clin. Oncol 3, 936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Su C (2016) Survivin in survival of hepatocellular carcinoma. Cancer Lett 379, 184–190. [DOI] [PubMed] [Google Scholar]
- (9).Yenkejeh R, Sam M, and Esmaeillou M (2017) Targeting survivin with prodigiosin isolated from cell wall of Serratia marcescens induces apoptosis in hepatocellular carcinoma cells. Hum. Exp. Toxicol 36, 402–411. [DOI] [PubMed] [Google Scholar]
- (10).Ku DH, Chang CD, Koniecki J, Cannizzaro LA, Boghosian-Sell L, Alder H, and Baserga R (1991) A new growth-regulated complementary DNA with the sequence of a putative trans-activating factor. Cell Growth Differ 2, 179–186. [PubMed] [Google Scholar]
- (11).Krautkramer KA, Linnemann AK, Fontaine DA, Whillock AL, Harris TW, Schleis GJ, Truchan NA, Marty-Santos L, Lavine JA, Cleaver O, Kimple ME, and Davis DB (2013) Tcf19 is a novel islet factor necessary for proliferation and survival in the INS-1 β-cell line. Am. J. Physiol. Endocrinol. Metab 305, E600–E610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kim YJ, Kim HY, Lee J-H, Yu SJ, Yoon J-H, Lee H-S, Kim CY, Cheong JY, Cho SW, Park NH, Park BL, Namgoong S, Kim LH, Cheong HS, and Shin HD (2013) A genome-wide association study identified new variants associated with the risk of chronic hepatitis B. Hum. Mol. Genet 22, 4233–4238. [DOI] [PubMed] [Google Scholar]
- (13).Zeng CX, Fu SB, Feng WS, Zhao JY, Li FX, and Gao P (2019) TCF19 enhances cell proliferation in hepatocellular carcinoma by activating the ATK/FOXO1 signaling pathway. Neoplasma 66, 46–53. [DOI] [PubMed] [Google Scholar]
- (14).Zhou Z, Chen G, Deng C, Tang J, Xie L, Zhou H, Ye X, Zhang D, Shi R, Tian D, Qiao G, and Ben X (2019) TCF19 contributes to cell proliferation of non-small cell lung cancer by inhibiting FOXO1. Cell Biol. Int 43, 1416–1424. [DOI] [PubMed] [Google Scholar]
- (15).Sen S, Sanyal S, Srivastava DK, Dasgupta D, Roy S, and Das C (2017) Transcription factor 19 interacts with histone 3 lysine 4 trimethylation and controls gluconeogenesis via the nucleosome-remodeling-deacetylase complex. J. Biol. Chem 292, 20362–20378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, and Schwede T (2014) SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42, W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Davis IW, Murray LW, Richardson JS, and Richardson DC (2004) MolProbity: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 32, W615–W619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sengupta I, Das D, Singh SP, Chakravarty R, and Das C (2017) Host transcription factor speckled 110 kDa (Sp110), a nuclear body protein, is hijacked by Hepatitis B virus protein X for viral persistence. J. Biol. Chem 292, 20379–20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kanehisa M, Furumichi M, Tanabe M, Sato Y, and Morishima K (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Huang DW, Sherman BT, and Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- (21).Huang DW, Sherman BT, and Lempicki RA (2009) Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, and Narechania A (2003) PANTHER: A library of protein families and subfamilies indexed by function. Genome Res 13, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Thomas PD, Kejariwal A, Guo N, Mi H, Campbell MJ, Muruganujan A, and Lazareva-Ulitsky B (2006) Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res 34, W645–W650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Maere S, Heymans K, and Kuiper M (2005) BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 21 (16), 3448–3449. [DOI] [PubMed] [Google Scholar]
- (25).Adhikary S, Sanyal S, Basu M, Sengupta I, Sen S, Srivastava DK, Roy S, and Das C (2016) Selective recognition of H3.1K36 dimethylation/H4K16 acetylation facilitates the regulation of all-trans-retinoic acid (ATRA)-responsive genes by putative chromatin reader ZMYND8. J. Biol. Chem 291, 2664–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zou C, Wang Y, and Shen Z (2005) 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J. Biochem. Biophys. Methods 64, 207–215. [DOI] [PubMed] [Google Scholar]
- (27).Adhikary S, Chakravarti D, Terranova C, Sengupta I, Maitituoheti M, Dasgupta A, Srivastava DK, Ma J, Raman AT, Tarco E, Sahin AA, Bassett R, Yang F, Tapia C, Roy S, Rai K, and Das C (2019) Atypical plant homeodomain of UBR7 functions as an H2BK120Ub ligase and breast tumor suppressor. Nat. Commun 10 (1), 1398–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Basu M, Khan MW, Chakrabarti P, and Das C (2017) Chromatin reader ZMYND8 is a key target of all trans retinoic acid-mediated inhibition of cancer cell proliferation. Biochim. Biophys. Acta, Gene Regul. Mech 1860, 450–459. [DOI] [PubMed] [Google Scholar]
- (29).Han J, Zhang L, Guo H, Wysham WZ, Roque DR, Willson AK, Sheng X, Zhou C, and Bae-Jump VL (2015) Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol. Oncol 138, 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhou W, Ramachandran D, Mansouri A, and Dailey MJ (2018) Glucose stimulates intestinal epithelial crypt proliferation by modulating cellular energy metabolism. J. Cell. Physiol 233, 3465–3475. [DOI] [PubMed] [Google Scholar]
- (31).Zhang B, Shi Y, Zou J, Chen X, Tang W, Ye F, and Liu Z (2017) High glucose stimulates cell proliferation and Collagen IV production in rat mesangial cells through inhibiting AMPK-KATP signaling. Int. Urol. Nephrol 49, 2079–2086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.