

Abstract
In this study, we report the synthesis and characterization of 12 novel rhenium(I) complexes with the general formula fac-[Re(CO)3(NN)X]n+ where (NN) is a 2,2′-bipyridine analogue ligand, X = Cl–, Br–, or H2O, and n = 0 or 1, focusing on their speciation in an aqueous solution. The prepared organorhenium complexes are stable in a wide pH range in an aqueous solution, and no release of the bidentate ligands or the carbonyl ligands was observed. The stability of the complexes in various biologically relevant matrices (cell culture medium and real blood serum) was also demonstrated. However, the simultaneous substitution of the halido ligand by water and slow hydrolysis of the ester bonds in the ligands were observed, affecting both the solubility and the lipophilicity of the compounds. The aqua complexes became more lipophilic in the presence of chloride ions, while the hydrophilicity increased significantly with time due to the hydrolysis of the ester bonds, which probably contributed to their weak pharmacological activity. The results also showed kinetically hindered aqueous solvation of the halido complexes and low chloride ion affinity of the aqua complexes. The deprotonation of the coordinated aqua ligand in the complexes occurs in the pH = 7–10 range, leading to significant formation (18–30%) of hydroxido species at pH = 7.4. The halido complexes showed somewhat higher cytotoxicity (IC50 = 60–99 μM) on human colon adenocarcinoma cancer cells (Colo205 and Colo320) than the corresponding aqua complexes (IC50 > 100 μM). In all cases, no antibacterial effect was observed (MIC > 100 μM), but some of the complexes showed moderate antiviral activity (IC50 ∼ 50 μM) on Herpes simplex virus 2.
Introduction
Organorhenium(I) tricarbonyl complexes have gained considerable attention in the last decades due to their wide range of biological activity, including antitumor,1−7 antiviral,8,9 and antibacterial10,11 properties. These complexes have been reported to exert their pharmacological action via enzyme inhibition, phototoxicity, DNA damage, mitochondrial effects, or regulation of oxidative stress. Particular fac-Re(CO)3 complexes were reported to exhibit strong activity against SARS-CoV-2, and inhibition of the main protease of the virus appears to be a key factor in their mechanism of action.8,9,12 A series of Re(CO)3 complexes with the general formula fac-[Re(CO)3(NN)(azole)]+, where (NN) is a bidentate oligopyridine ligand and azole is a monodentate bioactive ligand (clotrimazole or ketoconazole), showed excellent activity against epimastigotes and trypomastigotes of Trypanosoma cruzi, the parasitic agent responsible for Chagas disease.13 In recent years, we developed metal complexes of different pyridine-4,5-dicarboxylate ester ligands with (NN) bidentate coordination mode and screened their biological properties. The various biological tests (antimicrobial activity, enzyme inhibition, interactions with various biological molecules) performed for organoruthenium(II), silver(I), copper(II), and zinc(II) complexes of these ligands revealed some promising results.14−18
Herein, we report the synthesis, structural characterization, and solution speciation studies of 12 new organorhenium(I) tricarbonyl complexes with 2,2′-bipyridine analogue pyridine-4,5-dicarboxylate ligands (Chart 1). Although numerous fac-[Re(CO)3] complexes have been developed in recent years, their behavior in aqueous solutions has been relatively unexplored. The novel compounds were tested on the Colo205 human colon adenocarcinoma cell line and its drug-resistant counterpart (Colo320). As multidrug resistance (MDR) is a serious drawback not only in chemotherapy but also in bacterial infections, the antibacterial activity of the compounds was monitored against various bacterial strains, including methicillin-resistant Staphylococcus aureus (MRSA), known to be responsible for difficult-to-treat hospital-acquired infections.19
Chart 1. (a) Chemical Structures of the Pyridine-4,5-dicarboxylate Ligands L1–L4 Used for the Synthesis of the (b) fac-[Re(CO)3] Complexes.

In addition to drug resistance, the lack of selectivity toward cancer cells over healthy cells is a serious problem, leading to a significant weakening of the immune system during chemotherapy. Viral infections are also recognized as a potential threat to patients. Examples of compounds with dual or multiple therapeutic effects were also reported, demonstrating synergy between anticancer, antibacterial, and antiviral properties.20,21 Consequently, developing compounds with simultaneous pharmacological properties would obviously be beneficial.22 Considering these factors, the title compounds were evaluated not only for their anticancer and antibacterial activities but also for their antiviral effects.
Results and Discussion
Synthesis and Characterization of Compounds
Ligands L1–4 (Chart 1a and S1) were synthesized according to the reported procedure.14,23 Organorhenium(I) fac-tricarbonyl chlorido, bromido, and aqua complexes 1–4Cl, 1–4Br, and 1–4Aq (Chart 1b and S1) were newly prepared through one-step synthesis for chlorido and bromido or two-step procedure for aqua complexes (Scheme 1). Neutral halido complexes 1–4Cl and 1–4Br were prepared according to a previously reported procedure24,25 with some modifications. The reaction mixture was stirred in toluene for 2 h at 120 °C in a high-pressure tube. The precipitate formed was collected, and the red-to-orange solids were found to be light, air, and moisture stable.
Scheme 1. Preparation of The Halido and Aqua Complexes.

The positively charged aqua complexes 1–4Aq were prepared according to the literature procedure starting from 1–4Cl and silver triflate in CH2Cl2.26 The reaction was carried out in the dark overnight at room temperature. Formed AgCl was removed by vacuum filtration over Celite. The solvent was evaporated, and the residue was dissolved in a mixture of methanol and water in a ratio of 99:1 and left to slowly evaporate. The product was obtained after 5 days when all the solvent evaporated.
All the prepared compounds were analyzed using 1H NMR spectroscopy (Figures S1–S12), mass spectrometry (MS), infrared (IR) and UV–visible (UV–vis) spectroscopy, and elemental analysis (CHN), and where we were able to obtain suitable crystals, X-ray structure analysis was also performed. The formation of the complexes such as fac-[Re(CO)3(L)Cl] (1–4Cl), fac-[Re(CO)3(L)Br] (1–4Br), and fac-[Re(CO)3(L)H2O]CF3SO3 (1–4Aq) (Chart 1b) is fully supported by ESI-MS and the spectroscopic experiments.
Crystallization and X-Ray Structure Analysis
Single crystals of 2Cl, 3Br, 4Br, and 4Aq (Figure 1) were prepared with different methods of crystallization. For the structures of 3Br and 4Br crystals were obtained from the mother liquor of toluene by slow evaporation. Similarly, single crystals of 4Aq and 2Cl were grown by slowly evaporating solvents from a solution of methanol and water and from a solution of acetone and water, respectively. Compounds 3Cl and 4Cl were successfully crystallized from the solution of chloroform with vapor diffusion of n-heptane utilizing the CrystalBreeder multireactor crystallization platform.27 Compounds 1Aq and 3Aq have been crystallized as methanesulfonate salts from a solution of methanol and water. Methanesulfonate complexes were prepared with the same procedure as triflate aqua complexes substituting silver triflate with silver methanesulfonate as a reagent and were used as crude products only for the crystallization experiments. A different silver salt was used to stabilize the structure with hydrogen bonding. The images of the analyzed single crystals are tabulated in Table S1, and the crystal structures of the eight organometallic compounds were determined by X-ray crystallography (Figure 1, Table S2–S4).
Figure 1.

Crystal structures of rhenium(I) tricarbonyl complexes in the top row: 2Cl, 3Cl, and 4Cl, in the middle row: 3Br and 4Br, and in the bottom row: 1Aq, 3Aq (both as methanesulfonate salts), and 4Aq (as the triflate salt). Thermal ellipsoids are drawn at the 50% probability level. All hydrogens except H2O hydrogen atoms in aqua complexes are omitted.
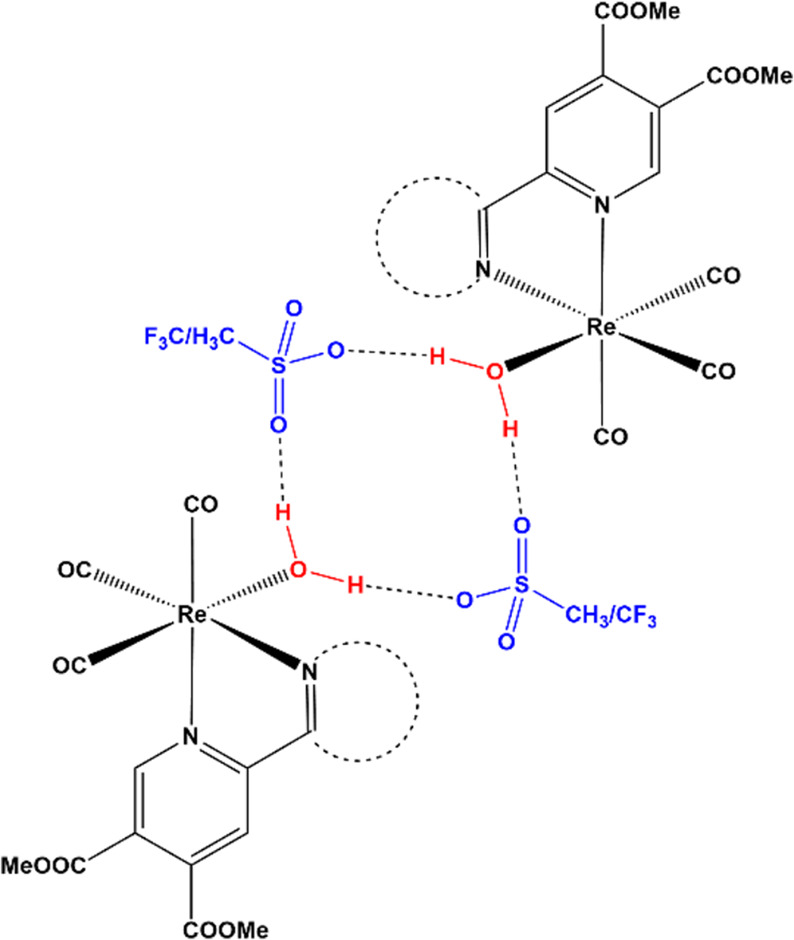
All compounds present a slightly distorted octahedral geometry mainly attributed to the constrained geometry and relatively low bite angle (ca. 75°) of the chelating ligand coordinated via the (NN) donor set. The carbonyl ligands are expectedly bound in facial geometry. Bond lengths and angles are all within the expected range. In all cases (except 3Aq where bond lengths are equal), the Re–N bond lengths show a slightly longer bond (0.01–0.03 Å) between the pyridine bearing two-electron acceptor ester groups in comparison to the second (unsubstituted) heterocycle (Table S5). The only other observable trend in bond length changes where in the reported structures we can observe a gradual increase in the Re–C bond length and a decrease in the respective C–O carbonyl bond length of the carbonyl ligand opposite the halido or aqua ligand X (Table S5). This can be attributed to the increasing trans effect of the ligand X (Br– > Cl– > OH2). In the case of all aqua complexes, we can observe the formation of a hydrogen bond network between the aqua ligand and the oxygen atoms of the respective counterion (methanesulfonate or triflate) with the O–O distance being ca. 0.2 Å shorter for the more basic methanesulfonate ion. Each aqua ligand acts as a hydrogen bond donor to two separate sulfonate groups, which in turn act as bridging hydrogen bond acceptors to the neighboring rhenium species. The tetrameric supramolecular arrangement is illustrated in Scheme 2.
Scheme 2. Supramolecular Tetrameric Hydrogen Bond Network Present in Rhenium Aqua Complexes 1Aq, 3Aq, and 4Aq.
Solution Chemical Properties of the Halido Complexes
We first chose the halido derivatives to study their solution chemical properties in detail, since their behavior in aqueous solutions might be more complex, due to the presence of coordinated chloride coligand, compared to aqua species. When the halido complexes are dissolved in water, several transformation processes can take place. The coordinated halido coligand may be replaced by water, then the aqua ligand may deprotonate, the (NN) bidentate ligand may dissociate, and the ester bonds in the ligand scaffold may be hydrolyzed. These processes are obviously dependent on the conditions (e.g., solvent, pH, temperature) and do not necessarily occur instantaneously. The stability of a series of antiparasitic Re(I) tricarbonyl complexes of chelators with (NN) donor set such as bpy, 1,10-phenantroline, and their derivatives was investigated in different media (e.g., DMSO, various DMSO/blood serum mixtures).13 These compounds showed high stability in the biologically relevant matrices tested without releasing the bidentate ligand. However, there is much less known about the possible coligand exchange processes in such types of complexes studied in this work, in particular the exchange of the chlorido/bromido (or other monodentate) coligand with water and its subsequent deprotonation.
As a first step, the solubility of the halido complexes (1–4Cl, 3Br) was studied in an aqueous solution. Based on previous solubility tests, the aqua analogues were completely soluble in 20% (v/v) DMSO/H2O medium at a concentration of 5 mM, whereas the halido complexes were practically insoluble and were soluble only in DMSO at the same concentration. This finding suggests that replacing the chlorido coligand with water increases the solubility of the complexes, since halido complexes are neutral compounds, while aqua derivatives are positively charged with a chloride counterion. However, for aqua complexes, the use of triflate as a counteranion is also beneficial for the water solubility. It was observed that the dissolution/solvation of the halido complexes in water at pH 7.4 and at 0.1 M KCl ionic strength is rather slow (>24 h needed to reach equilibrium); thus, the solubility was determined after a waiting time of 24 h (Figure S13), revealing the low aqueous solubility of halido complexes (<160 μM). The solubility was significantly decreased due to the additional methyl group on the ligand scaffold (3Cl vs 4Cl). Complexes 3Cl and 3Br showed the highest solubility and were therefore selected for further investigation, while 3Aq and 1Cl were also included for comparison in certain cases.
Initially, the changes in the UV–vis spectrum of complexes 3Cl and 3Br dissolved in water (at pH = 6.1) at low concentration (55 μM) were followed over time, and no significant spectral changes were observed over ca. 58.5 h (Figure S14). Interestingly, the normalized spectra of both complexes are identical to that of 3Aq (Figure S15), suggesting that the compounds have the same composition under the conditions used; they are most likely in the aqua complex form. This suggestion was further confirmed by 1H NMR spectroscopy, and the same peaks were found for all three complexes (Figure S16). However, minor peaks appeared after a waiting time of 24 h, most probably due to the partial hydrolysis of the ester bond in the bidentate ligand (vide infra), which did not affect the charge transfer bands in the UV–vis spectra. In order to follow the time dependence of the exchange of the coordinated chlorido coligand to H2O, complexes 1Cl and 1Aq were dissolved in DMSO. In this solvent, their spectra significantly differ due to the coordination of the different coligands and remained intact up to 16 h (Figure S17). Nevertheless, partial coordination of DMSO to the metal ion is possible, since 1H NMR spectra of 3Cl recorded in DMSO-d6 and acetonitrile-d3 significantly differ (shifts in peaks, not shown), and in this case, the solvent effect must be taken into consideration, which can be significant. The DMSO stock solutions were then diluted with water up to 5% (v/v) DMSO/H2O. The changes in the charge transfer bands were followed over time, and for comparison, fac-[Re(CO)3(bpy)(Cl)] was also studied (Figure 2). The spectrum of the chlorido complexes was changed over 2 h in 5% (v/v) DMSO/H2O, while 1Aq remained intact over the monitored time (16 h). Moreover, the spectrum of 1Cl after 2 h is very similar to that of 1Aq, and both findings confirm our suggestion on the exchange process of the chlorido coligand to H2O (Figures 2 and S17). Based on the 1H NMR spectra recorded for 1Aq and 1Cl (Figure S18), the two sets of peaks appeared for 1Cl after 5 min, indicating the copresence of aqua and chlorido complexes, and as it was expected the peaks belonging to the chlorido complex disappeared with time. This exchange process is not instantaneous. Therefore, the solvation of the halido complexes is kinetically hindered. To obtain information on the behavior of the chlorido complexes in the cell culture medium, the stock solution of 3Cl (in DMSO-d6) was diluted with Eagle’s Minimum Essential Medium (EMEM) to 10% (v/v) DMSO-d6 and the 1H NMR spectra were recorded over time (Figure S19). For comparison, the spectra for 3Aq were also recorded in the same way. Although the hydrolysis of ester bonds (vide infra) and the coordination of EMEM component(s) are clearly seen in parallel in both cases (and the replacement of Cl– with H2O in the case of 3Cl after 25 min), the 1H NMR spectra of 3Cl and 3Aq are almost identical in EMEM after 72 h.
Figure 2.
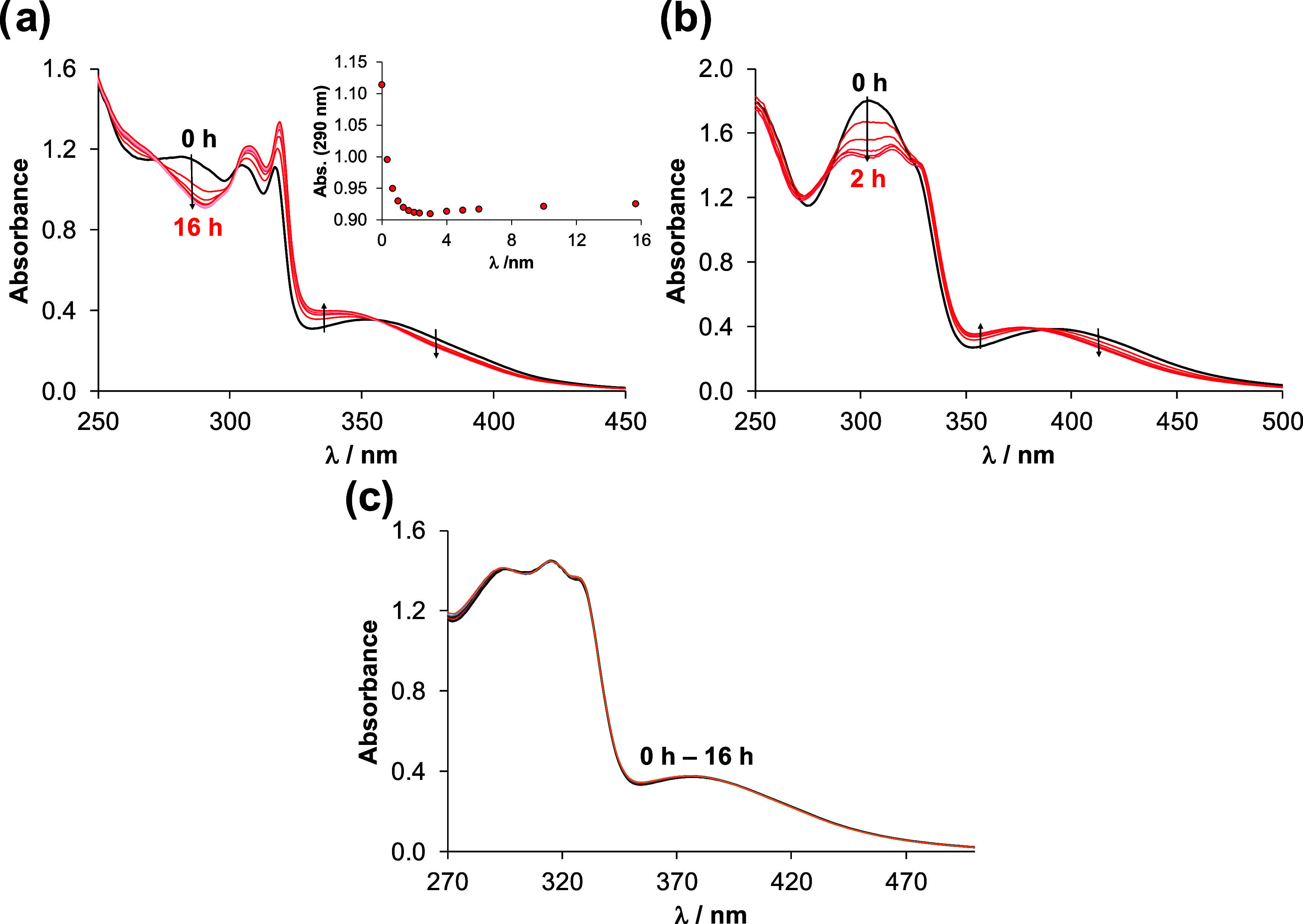
UV–vis spectra of (a) fac-[Re(CO)3(bpy)(Cl)] chlorido complex, (b) chlorido complex 1Cl and (c) aqua complex 1Aq in 5% (v/v) DMSO/H2O followed over time. Inserted figure in (a) shows absorbance values
at 290 nm (●) plotted against time. {ccomplex = 50 μM; pH ∼ 6.2; 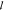 = 1 cm; T = 25.0 °C}.
= 1 cm; T = 25.0 °C}.
Solution Chemical Properties of The Aqua Complexes
As the aqua complexes are readily soluble in water, further stability measurements were carried out on them. The UV–vis spectrum of aqua complexes 1–4Aq in water (pH ∼ 6.2) was followed over time. No significant spectral changes were observed during 24 h in the case of 1Aq and 2Aq, and after 36 h the spectra slightly changed (Figure S20), while for 3Aq and 4Aq, the spectra remain intact for 48 h (Figure S21). Additionally, 1H NMR spectra were recorded for 1Aq (Figure S22), revealing the appearance of a new set of peaks after 24 h. The ratio of this new product is about 4% after 48 h, calculated from the integration of the corresponding peaks of the substituted pyridine ring protons. These changes indicate the hydrolysis of the ester bond of the coordinated ligand under the conditions used rather than the dissociation of the complex. The UV–vis spectra of 1–4Aq were also recorded at pH 7.4 (in 20 mM phosphate buffer) showing significant changes over time. The reaction was completed within ∼36 h (Figure S23). The 1H NMR spectra also showed the decreasing signals belonging to the aqua complexes and the increasing signals of new peaks (Figure 3), which is suggested to be due to the coordination of the phosphate ions as coligand. Parallel to the coordination of the phosphate ion, slow hydrolysis of the ester bonds also occurred. Notably, for the ligand alone negligible (∼5%) hydrolysis could be observed after 48 h (Figure 3).
Figure 3.

1H NMR spectrum of 1Aq in the low-field region in phosphate buffer (20 mM, pH = 7.4) followed over time and that of the corresponding ligand is also shown. Rectangles with colored background show the appearance of a new set of peaks. {ccomplex = 500 μM; 10% (v/v) D2O/H2O; T = 25.0 °C}.
The effect of the HEPES buffer was also studied at pH 7.4. Both the UV–vis and 1H NMR spectra indicated changes during the monitored 48 h (Figure S24) although less new peaks appeared in the 1H NMR spectra compared to the presence of the phosphate ions. Analysis of the direct comparison of the effect of the two kinds of mediums (Figure S25) led to the conclusion that the two ester bonds in the coordinated ligand are hydrolyzed differently. The behavior of the aqua complexes 1–4Aq was also studied in EMEM. The UV–vis spectra show a rather fast reaction (within 1 h), most probably, one of the components of the cell culture medium coordinates to the metal ion (Figure S26). It was also confirmed by 1H NMR spectroscopy, namely, a new set of peaks appeared in the spectrum (Figure S27). Interestingly, signals of hydrolyzed products were not identified most probably due to the slightly acidic conditions (pH ∼ 6.5). It should also be noted that precipitation occurred in the case of complexes 1Aq and 4Aq after 48 h, while complexes 2Aq and 3Aq remained dissolved (Figure S26). The spectral changes of the aqua complexes in blood serum were also monitored. The changes were found to be almost negligible for 1Aq and 2Aq, while they were more significant for 3Aq and 4Aq, but the charge transfer bands (∼360–500 nm) did not change much over time (Figure S28). 1H NMR spectrum of 2Aq (Figure S29) in serum shows the appearance of new sets(s) of peaks; however, the Δδ between the original and the new chemical shift is small (∼0.06 ppm), which suggests the coordination of a serum component to the metal ion. In the case of complex 3Aq (Figure S29b), a new peak set appeared further from the original peaks (Δδ = 0.24–0.36 ppm), which is related to the hydrolysis of the ester bond.
In summary, in the biological media used, there was no evidence for the release of the original bidentate and carbonyl ligands, while the labile aqua coligand underwent ligand exchange processes. On the other hand, the ester bonds of the ligand were hydrolyzed in a time-dependent manner.
The effect of the pH on the stability and ligand exchange processes of the complexes was studied using UV–vis spectrophotometry. No spectral changes were observed between pH 2 and 6 indicating that the complexes remained intact. However, the spectra showed a batochromic shift from pH ∼ 6 up to ∼9.5, and isosbestic points were also observed. (For a representative spectrum series for 1Aq see Figure S30). Most probably this pH-dependent process is the deprotonation of the aqua ligand, and proton dissociation constants (pKa) could be calculated based on the spectral changes (Table 1).
Table 1. Proton Dissociation Constants of The Coordinated Water Co-Ligand (pKa (H2O)) and Water/Chloride Ion Exchange Constants (log K′ (H2O/Cl–)) for the Aqua Complexes Determined by UV–vis and 1H NMR Spectroscopya.
| pKa (H2O) |
||||
|---|---|---|---|---|
| method | complex | 0.1 M KNO3 | 0.1 M KClb | log K′ (H2O/Cl–)a |
| UV–vis | 1Aq | 8.07 ± 0.04 | 8.02 ± 0.08 | 0.80 ± 0.04 |
| 2Aq | 7.78 ± 0.04 | 7.78 ± 0.09 | 1.16 ± 0.11 | |
| 3Aq | 7.99 ± 0.03 | 8.04 ± 0.04 | 0.74 ± 0.11 | |
| 4Aq | 8.03 ± 0.02 | n.d. | 0.74 ± 0.05 | |
| 1H NMR | 3Aq | 8.08 ± 0.02 | n.d. | n.d. |
{I = 0.1 M KNO3 or KCl; T = 25.0 °C}.
Equilibrium time: 1 h.
The pKa values of the complexes are similar, and only for 2Aq a somewhat lower value was obtained due to the negative inductive effect caused by the additional nitrogen in the heterocyclic ring. Notably, pKa = 8.3 was reported for the analogous aqua complex of a 1,10-phenanthroline derivative.281H NMR titration was also carried out for the complex 3Aq (Figure 4), and the calculated pKa (Table 1) is in good agreement with that obtained by the UV–vis titrations. The hydrolysis products (only two kinds) also appeared with increasing pH, and based on the integration of the corresponding peaks at pH 9.9, ∼80% of the 3Aq complex is hydrolyzed and the ratio of the two hydrolysis products is 1:3. Based on the spectra recorded, the fully hydrolyzed form (when both functional groups are in their carboxylate form) could not be detected.
Figure 4.
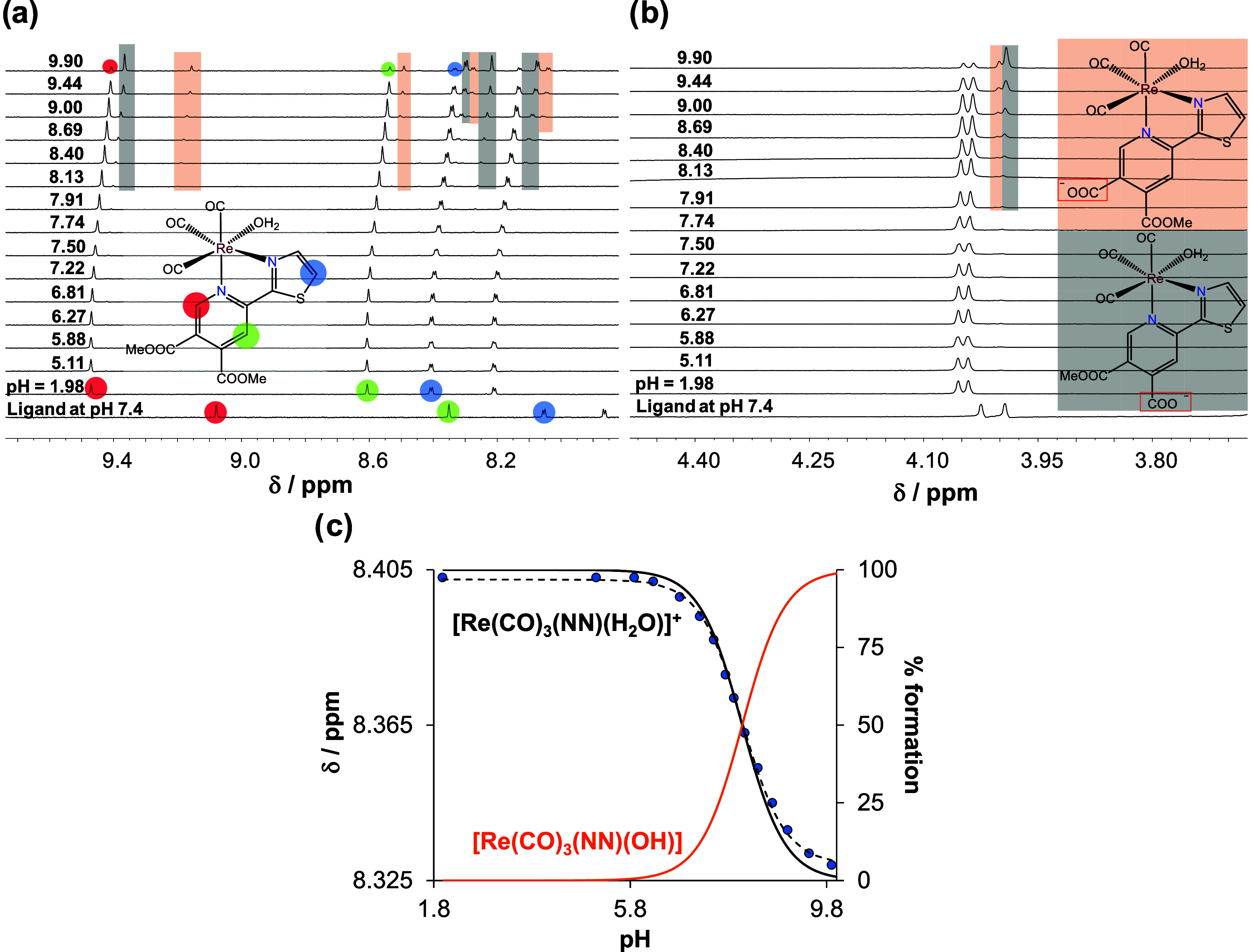
1H NMR spectra in the (a) low-field and (b) high-field region of 3Aq complex at different pH values (1.98 → 9.90). The spectrum of the free ligand is also shown at pH = 7.4. Rectangles with colored backgrounds indicate the appearance of new sets of peaks related to hydrolyzed products. (c) Chemical shift values of C(5′)H proton (●) along with the fitted line and concentration distribution curves (black and orange solid lines) as a function of pH. It is noteworthy that traces of methanol were present in the solvent, but its peak intensity was increased in the basic pH range (not shown). {ccomplex = 660 μM, I = 0.1 M KNO3, 10% (v/v) D2O/H2O, T = 25.0 °C}.
The pKa values of the complexes (1–3Aq) were also determined at 0.1 M KCl ionic strength (Table 1) and similar values were obtained as in the absence of chloride ions, indicating a weak chloride ion affinity of the complexes. Moreover, UV–vis titrations were also performed for 3Cl and 3Br, and the determined pKa values (7.98 ± 0.03 and 8.06 ± 0.03 for 3Cl and 3Br, respectively, in 0.1 M KNO3) are very similar to that of 3Aq. These findings also confirm the formation of aqua complexes from halido complexes in water.
Based on the determined pKa values, concentration distribution curves were calculated (for 1Aq see Figure 4c) and the fractions of the aqua and mixed-hydroxido complexes are shown in Table S6 at pH 7.4. According to the calculations, the fraction of the mixed hydroxido complex is significant in all cases (18–30%).
It is noteworthy that deprotonation of the aqua ligand modifies the charge of the complex. However, ester hydrolysis also affects the overall charge. For 1Aq, at pH 7.4 after 48 h (Figure S24b), only ∼12% of the complex remained in a nonhydrolyzed form (with COOMe substituents, +1 charge) and the rest (∼88%) are hydrolyzed products (with COOMe and COO– substituents). Based on further calculations involving the pKa (H2O) value, the amount of neutral complex (with COOMe and COO– substituents, and H2O as coligand) is ∼72% (82% of the 88%), and the rest amount (16%; 18% of the 88%) of the complex is negatively charged (with COOMe and COO– substituents, and OH– as coligand).
The exchange of the coordinated H2O with chlorido coligand was investigated by UV–vis and 1H NMR spectroscopy at pH 6.0. (This condition was chosen since, at this pH, based on the pKa values (Table 1), the complexes are in their aqua form). Minor but tendentious changes in the UV–vis spectra were followed over time, and the equilibrium reached within 1 h (Figure S31). The 1H NMR spectra showed the appearance of a new set of peaks related to the formation of the chlorido complex (Figure S32a). Based on the integration of the corresponding peaks belonging to the aqua and chlorido forms, their molar ratio was plotted against time (Figure S32b), revealing a similar reaction time as seen in the case of the UV–vis studies (∼1 h). It is noteworthy that after 2 h precipitation occurred since the applied concentration (500 μM) is much higher compared to the UV–vis experiment (25 μM). Therefore, in order to characterize the chloride ion affinity of the aqua complexes, UV–vis spectra of individual samples containing the aqua complex at different chloride ion concentrations were recorded after a 1 h waiting time. A representative UV–vis spectrum series for complex 3Aq is shown in Figure 5, and from the spectral changes relatively low exchange constants (log K′ (H2O/Cl–) ∼ 1) were computed (Table 1), indicating the weak chloride ion affinity of the aqua complexes.
Figure 5.

UV–vis
spectra of aqua complex 3Aq in the HEPES
buffer (pH = 6.0) with increasing chloride ion concentration. Inserted
figure shows absorbance values at 385 nm (●) plotted against cCl–/ccomplex along with the fitted (dashed) line. {ccomplex = 40 μM cCl– = 0–800 mM;  = 1 cm; T = 25.0 °C}.
= 1 cm; T = 25.0 °C}.
Lipophilicity of The Organorhenium Complexes
The lipophilicity of the complexes was characterized at pH 7.4. The traditional shake-flask method was applied in HEPES buffer (pH 7.4) to determine the distribution coefficient (D), via n-octanol/buffered aqueous solution partitioning. It is noteworthy, when the chloride ion is coordinated to Re(I), the actual charge changes from +1 to neutral, which probably has an impact on the lipophilicity; therefore, various chloride ion concentrations were applied, namely, cCl– = 4, 24, and 100 mM, which are relevant to different biofluids, such as nucleus, cytosol, and serum, respectively (Figure S33). The measurements were also conducted in the absence of chloride ions.
On the one hand, these complexes possess a lipophilic character (log D7.4 = +0.28 – +1.92); moreover, the additional methyl group present in complex 4Aq increased the lipophilicity compared to the analogous 3Aq. On the other hand, it is clearly seen that the higher the chloride ion concentration, the higher the lipophilicity of the complexes; however, the halido complexes were found to be more lipophilic compared to the corresponding aqua complexes even at cCl– = 100 mM, which is in line with the relatively low chloride ion affinity of the aqua complexes (Table 1). Notably, due to the low aqueous solubility of halido complexes, they were preliminarily dissolved in the n-octanol phase (Figure S33).
Since 3Br and 3Cl have the highest solubility in the water among the halido complexes (Figure S13), the measurements for these compounds were also performed when they were dissolved in water. As a result, the determined log D7.4 values are very similar to those obtained in the case of the 3Aq aqua complex. The measurements were conducted after 4 h waiting time, and within this time frame, the hydrolysis of the ester bonds in the complexes at pH = 7.4 is negligible. However, using a longer waiting time, it becomes more significant; therefore, the lipophilicity of 3Aq, 3Br, and 3Cl was investigated also at various time points, namely, after 9, 25, 50, and 77 h in the absence of chloride ions, and the results are shown in Figure 6.
Figure 6.
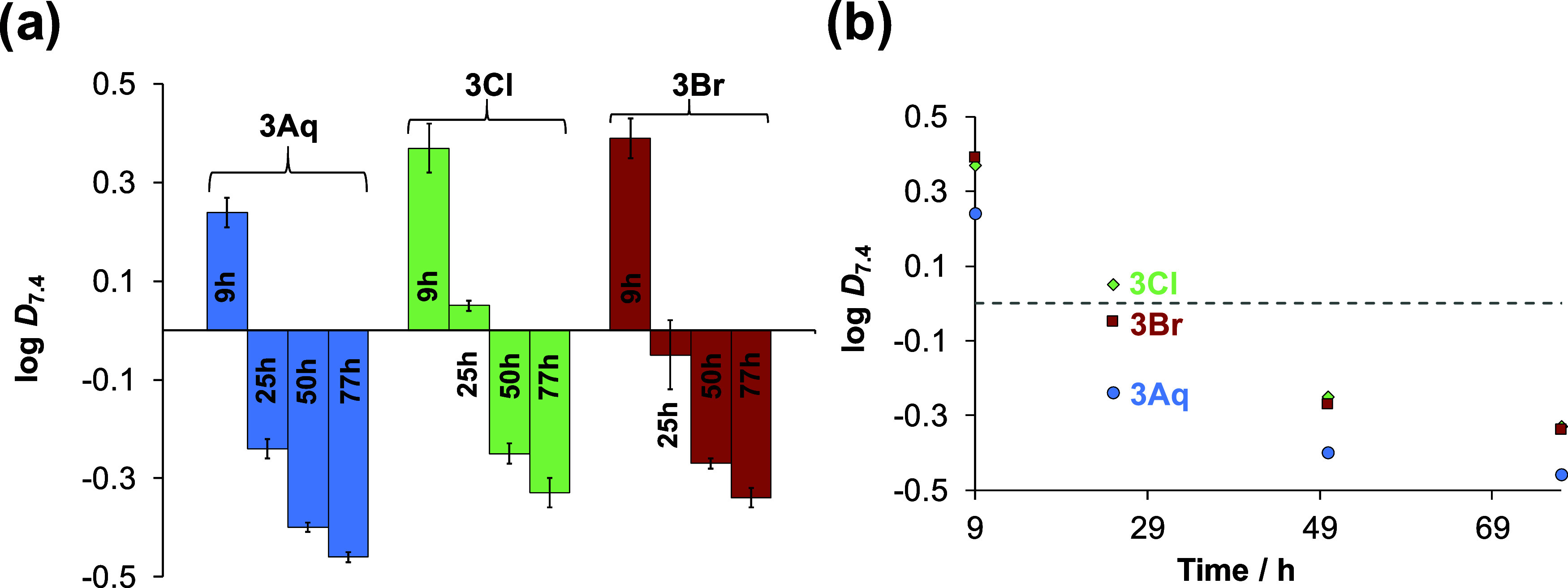
Lipophilicity (a) of selected complexes (3Aq, 3Cl, and 3Br) expressed as log D7.4 measured via n-octanol/buffered aqueous solution partitioning at pH = 7.4 (HEPES buffer) in the absence of chloride ions, investigated over time (9, 25, 50, and 77 h). For better visualization, all the log D7.4 values are plotted against time in (b). The complexes were previously dissolved in the water phase (buffered). {ccomplex = 50 μM; T = 25.0 °C}.
Over the 77 h time period, the lipophilicity of the three complexes significantly changes and becomes more hydrophilic (at 77 h, log D7.4 = −0.34 – −0.46). It is noteworthy that 3Aq is the most hydrophilic compound at each time point compared to 3Cl and 3Br.
Evaluation of The Pharmacological Activity of The Organorhenium Complexes
In Vitro Anticancer Activity of The Complexes on Human Colon Adenocarcinoma Cancer Cells
The cytotoxic activity of selected halido (1–4Cl, and 3Br with chlorido and bromido coligand, respectively) and aqua (1–4Aq) complexes was evaluated in the drug-sensitive Colo205 and its resistant counterpart Colo320 human colonic adenocarcinoma cell lines using the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay. For comparison, fac-[Re(CO)3(bpy)(Cl)] (in which bpy denotes 2,2′-bipyridine) was also tested, and doxorubicin was used as a positive control. IC50 values for the halido complexes using 72 h incubation time along with their resistance factors (RF = IC50 (Colo320)/IC50 (Colo205)) are shown in Table 2. Based on these data, it can be concluded that the halido complexes (1–4Cl and 3Br) showed weak cytotoxic activity (IC50 = 60–99 μM) against both drug-sensitive and -resistant cancer cells. The resistance factors of these complexes fall in the range of 1.03–1.25, which are significantly lower than that of doxorubicin. The aqua complexes (1–4Aq) were characterized by even higher IC50 values (>100 μM). The somewhat higher cytotoxic activity of the halido complexes might be the result of their kinetic inertness compared to aqua derivatives, based on the previous solution chemical studies.
Table 2. In Vitro Cytotoxicity of The Halido Complexes (1–4Cl and 3Br), and fac-[Re(CO)3(bpy)(Cl)] as a Reference Compound, Tested in The Drug-Sensitive Colo205 and Its Resistant Counterpart Colo320 Human Colon Adenocarcinoma Cell Lines, In Addition to The Resistance Factors (RF = IC50(Colo320)/ IC50(Colo205))a.
| IC50 (μM) | Colo205 | Colo320 | RF |
|---|---|---|---|
| 1Cl | 87.24 ± 0.92 | 97.1 ± 1.6 | 1.11 |
| 2Cl | 96.48 ± 0.10 | 99.16 ± 0.25 | 1.03 |
| 3Cl | 66.0 ± 1.7 | 83.03 ± 0.50 | 1.26 |
| 3Br | 62.6 ± 3.7 | 72.66 ± 0.40 | 1.16 |
| 4Cl | 60.2 ± 1.3 | 76.3 ± 2.2 | 1.27 |
| fac-[Re(CO)3(bpy)(Cl)] | 25.2 ± 1.1 | 49.1 ± 1.7 | 1.95 |
| doxorubicin | 0.58 ± 0.06 | 3.13 ± 0.29 | 5.17 |
IC50 values of 1–4Aq > 100 μM in both cell lines. {incubation time: 72 h}.
On the one hand, the exchange of the pyridine (1Cl) heterocyclic ring to pyrazine (2Cl) has no considerable effect on the anticancer activity, while the presence of thiazole (3Cl, 4Cl) increased the cytotoxicity, as IC50 values were decreased by 24 ± 3% in the case of the Colo205 cell line. On the other hand, the exchange of the chlorido for the bromido coligand (3Cl vs 3Br) as well as the presence of an additional methyl group on the thiazole ligand scaffold (3Cl vs 4Cl) had only a minor effect on the cytotoxicity. The IC50 values of the reference complex fac-[Re(CO)3(bpy)(Cl)] and its 1Cl analogue show some differences, and despite the somewhat higher IC50 values of 1Cl, it shows a lower resistance factor as well as the other 2,2′-bipyridine analogues.
Antibacterial Activity of The Complexes
The antibacterial activity of the selected complexes was tested in gram-negative Escherichia coli and gram-positive S. aureus as well as its methicillin-resistant counterpart (MRSA). The compounds showed no activity against the tested bacterial strains, as the minimum inhibitory concentration (MIC) was above 100 μM in each case. It is worth mentioning that silver complexes with the ligands used in this study showed remarkable activity against the standard panel of microorganisms (bacteria, fungi) and a selection of clinical isolates from the milk of the cow diagnosed with mastitis.16 Antifungal and especially antibacterial activities of corresponding copper(II) and zinc(II) complexes were much lower.17,18 Obviously, the type of metal ion bonded to these ligands is very important for biological activity.
Antiviral Activity of The Complexes
Selected compounds (1–4Aq, 1–4Cl, and 3Br) and the reference complex fac-[Re(CO)3(bpy)(Cl)] were tested for their antiviral activity against Herpes simplex virus 2 (HSV-2), which is a double-stranded DNA virus that infects mammals, including humans.29 The antiviral activity of complexes against HSV-2 was investigated by using Vero cells (originally isolated from kidney epithelial cells of an African green monkey) to host the growing viruses. First, the cytotoxicity of the complexes against Vero cells was assessed by an MTT assay (Figure S34), and it was found that the complexes display no significant activity at the applied concentrations. The maximum nontoxic concentration was found to be 50 μM for complex 1Aq, 100 μM for the others, and 25 μM for fac-[Re(CO)3(bpy)(Cl)].
In the next step, Vero cells were subjected to HSV-2 infection (at a multiplicity of infection (MOI) of 0.1), followed by treatment with the complexes at varying concentrations for 24 h. Then the cells were lysed, and the antiviral efficacy of the compounds was evaluated by comparing the viral yield reduction against untreated Vero cells. The HSV-2 growth was monitored by direct quantitative polymerase chain reaction (qPCR) analysis. The higher cycle threshold (Ct) values mean that more PCR cycles were required to detect the target nucleic acid, indicating stronger antiviral activity. The complexes capable of inhibiting HSV-2 growth were 1–4Cl and 3Br, while the aqua complexes were much less effective (see the average Ct values obtained for 1–4Cl and 3Br at the different complex concentrations in Figure 7 and for 1–4Aq in Figure S35).
Figure 7.

Antiviral effect of complexes 1–4Cl and 3Br against HSV-2 at different concentrations. As a comparison, the reference complex fac-[Re(CO)3(bpy)(Cl)] (ref) was also involved. The HSV-2 DNA concentration was measured by direct qPCR. Data represent the average −Ct values ± standard deviations. {Treatment of the Vero cells: 24 h; n = 3}.
In this case, the IC50 value means the complex concentration at which the DNA content decreased by 50%, interpreted as the concentration at which the qPCR Ct value increased by ca. one cycle. The complex concentration that inhibited the HSV-2 growth by 90% (IC90) increased the Ct value by ca. 3.32 cycles. The maximum HSV-2 growth corresponded to a DNA concentration of Ct ca. 15.5 as detected by direct qPCR. Inhibition curves showed that the most potent complexes were 4Cl and 3Br (Figure 7), exceeding the activity of the reference complex fac-[Re(CO)3(bpy)(Cl)]. In the case of both complexes, the IC50 were c.a. 50 μM and IC90 was between 50 and 100 μM.
The direct impact of complexes on DNA polymerase of qPCR was also examined to ensure the reliability of our antiviral studies (Figure S36), since metal ions and metal complexes can affect the qPCR reaction by interacting with the polymerase enzyme.30 Based on the similar Ct (and relatively low dCt) values obtained for the treated and untreated samples, there is no stimulation or inhibition during the qPCR.
Conclusions
Twelve new tricarbonylrhenium(I) complexes with the general formula fac-[Re(CO)3(NN)X]n+ (X = Cl–, Br– or H2O and n = 0 or 1) were synthesized and characterized. X-ray crystallographic analysis of the complexes revealed the expected chelating binding mode of the bidentate nitrogen ligands, and the coordination sphere was completed by the binding of a chlorido, bromido, or aqua ligand.
The behavior of the Re(CO)3 complexes was studied in water and in different biological matrices such as cell culture medium and blood serum to get deeper insights into their stability and the different chemical forms present in solution. The compounds were found to be stable in different biological matrices and no dissociation of the bidentate ligands and/or carbonyl ligands was observed. This finding suggests rather high stability, but the coordinated ligands were not intact, as hydrolysis of the ester bonds was detected over time at pH = 7.4, which decreased the lipophilicity and thus possibly the cytotoxic activity when 1Cl was compared to the fac-[Re(CO)3(bpy)(Cl)]. The complexes were stable over a wide pH range, and when the pH was lowered to 2 there was no evidence of release of the bidentate ligand, even at the relatively low concentrations, which hindered the determination of their formation constants. However, the coordinated water coligand deprotonates at pH > 6 (pKa = 7.8–8.1), and based on the determined pKa values (7.8–8.1), the formation of mixed hydroxido complexes is significant (18–30%) at pH 7.4. The halido complexes transform into the corresponding aqua complexes in an aqueous solution, which is consistent with the relatively low chloride ion affinity of the aqua complexes.
The organorhenium complexes with the halido ligands (1–4Cl, 3Br) showed weak cytotoxic activity on the Colo205 (IC50 = 60–96 μM) and the resistant Colo320 (IC50 = 73–99 μM) cancer cell lines and exhibited moderate antiviral activity against Herpes simplex virus type 2. It was found that the presence of a thiazole moiety in the complexes can increase the cytotoxicity. However, the complexes with the aqua coligand (1–4Aq) displayed even weaker cytotoxicity (IC50 > 100 μM) and antiviral activity. However, on the basis of the solution studies, the dissolution of the halido complexes also results in aqua complexes, but this process is not spontaneous. Therefore, the somewhat stronger bioactivity of the halido complexes might be the result of their kinetic inertness compared to that of the aqua species, which may hinder their immediate interaction with bioligands in the cell culture medium.
This study represents the first systematic investigation of the solution speciation of tricarbonyl organorhenium(I) complexes bearing bipyridine analogues. This type of information is crucial for the interpretation of the pharmacological effects of these compounds, and it helps elucidate the relationships between the solution speciation and biological properties and also helps the design and development of a next-generation of compounds.
Materials and Methods
Chemical and General Information
All starting materials for the synthesis were purchased from commercial sources (Merck, Fluorochem, and Strem Chemicals) and were used as received. Solvents for the reactions were dried over sodium sulfate and molecular sieves, while solvents for isolation of the compounds were used without further purification or drying. All other solvents were of analytical grade and used without further purification. KOH, HEPES, D2O, EMEM media, doxorubicin, and human serum (from human male AB plasma) were Merck (St. Louis, MO, USA) products and were used without further purification. DMSO, KCl, KNO3, HCl, HNO3, NaH2PO4, Na2HPO4, KH2PO4, and n-octanol were Molar Chemicals (Halásztelek, Hungary) products. DMSO-d6 was purchased from VWR Chemicals (Radnor, Pennsylvania, USA).
NMR spectroscopy was performed on a Bruker Avance III 500 spectrometer or Bruker Avance Neo 600 spectrometer at room temperature. 1H NMR spectra were recorded at 500 and 600 MHz. Chemical shifts are referenced to residual peaks of the deuterated solvent CDCl3, CD3CN at 7.26 and 1.94 ppm, 1.94 ppm respectively. Chemical shifts (δ) and coupling constants (J) are given in ppm and Hz, respectively. All NMR data were processed using MestRe-Nova version 11.0.4 or 14.3.0. Infrared spectra were recorded on a PerkinElmer Spectrum Two with ATR. High-resolution mass spectra (HRMS) were recorded on an Agilent 6224 Accurate Mass TOF LC/MS instrument. Elemental analyses (CHN) were carried out on a PerkinElmer 2400 II instrument. UV/vis spectra were collected on a PerkinElmer Lambda 750 UV/vis/near-IR spectrophotometer. IR and UV–vis data were processed using Spectragryph.31
Single crystal X-ray diffraction data was collected at 150 K on a SuperNova diffractometer with Atlas detector using CrysAlis software with monochromated Mo Kα (0.71073 Å).32 The initial structural models were solved with direct methods implemented in SHELXT using the Olex2 graphical user interface.33 A full-matrix least-squares refinement on F2 magnitudes with anisotropic displacement parameters for all non-hydrogen atoms using Olex2 or SHELXL-2018/3 was performed.33,34 All non-hydrogen atoms were refined anisotropically, while hydrogen atoms were placed at calculated positions and treated as riding on their parent atoms. Details on the crystal data, data acquisition, and refinement are presented in Table S2–S4. Mercury35 was used for the preparation of the figures. CCDC 2366664–2366671 contains the supplementary crystallographic data for this paper.
General Procedure for The Synthesis of Ligands
Chelating pyridine dicarboxylic acid methyl ester ligands (pdce) were synthesized according to the literature.14,23 The starting 2-acetylheterocycle was mixed with N,N-dimethylformamide dimethyl acetal (DMF-DMA, 1.2 mol equiv) in 20 mL of dry toluene, and the reaction mixture was refluxed for 24 h. The respective enaminone was isolated by removing the toluene under reduced pressure and filtering the solid product with vacuum filtration. The product was washed with n-hexane and dried for 1 h at 45 °C. The enaminone was then refluxed with dimethyl acetylenedicarboxylate (DMAD, 2 molar equiv) in 20 mL of acetonitrile for 3 h. After the reaction was finished the reaction mixture was concentrated under reduced pressure and purified with column chromatography on silica gel. The product was eluted with the mobile phase ethyl acetate/petrol ether in the ratio of 2:1. After combining of the appropriate fractions, the mobile phase was evaporated under reduced pressure. To the product 15 mL of methanol was added together with ammonium acetate (10 mol equiv) and the reaction was stirred at room temperature overnight. The formed precipitate was filtered with vacuum filtration and washed with distilled water and n-hexane. The obtained product was dried overnight at 45 °C.
General Procedure for the Synthesis of the fac-Tricarbonylrhenium(I) Halido Complexes with General Formula fac-[Re(CO)3(NN)X] X = Cl or Br
Rhenium complexes were synthesized according to the literature procedure.24,25 A reaction mixture containing the appropriate ligand L1–L4 (0.2 mmol), a metal precursor [Re(CO)5X] (X = Cl or Br; 0.2 mmol), and 10 mL of dry toluene was heated to 120 °C in 20 mL high-pressure tubes for 2 h. The reaction mixtures were then cooled in an ice bath, and the precipitates formed were collected by vacuum filtration and washed with n-hexane. The products were left to dry overnight at 45 °C.
fac-[Re(CO)3(L1)Cl]
1Cl Yield 73.9% (282 mg), red-orange solid. 1H NMR (500 MHz, acetonitrile-d3) δ = 9.32 (d, J = 0.7 Hz, 1H), 9.06 (ddd, J = 5.4, 1.6, 0.8 Hz, 1H), 8.62 (d, J = 0.7 Hz, 1H), 8.51 (dt, J = 8.2, 1.1 Hz, 1H), 8.25 (td, J = 7.9, 1.5 Hz, 1H), 7.71 (ddd, J = 7.7, 5.4, 1.2 Hz, 1H), 3.98 (s, 3H), 3.97 (s, 3H) ppm. IR selected bands (ATR): ú = 2018, 1930, 1892, 1871, 1739, 1437, 1392, 1270, 1145, 1096, 959, 833, 784, 642, 628, 482 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, CH2Cl2): 307.35 (25 400), 429.27 (4000). ESI-HRMS (acetonitrile) for [M–Cl + CH3CN]+ C19H15N3O7Re+ found: 584.0454 (calculated: 584.0468). Elemental analysis for C17H12ClN2O7Re calculated (%): C: 35.33 H: 2.09 N; 4.85; found (%): C: 35.23; H, 1.93; N: 4.71.
fac-[Re(CO)3(L2)Cl]
2Cl Yield 77.6% (89.9 mg), red solid. 1H NMR (500 MHz, acetonitrile-d3) δ = 9.72 (d, J = 1.3 Hz, 1H), 9.35 (d, J = 0.8 Hz, 1H), 9.03 (dd, J = 3.0, 1.3 Hz, 1H), 8.89 (d, J = 3.0 Hz, 1H), 8.77 (d, J = 0.7 Hz, 1H), 4.00 (s, 3H), 3.98 (s, 3H) ppm. IR selected bands (ATR): ú = 2021, 1939, 1926, 1887, 1727, 1438, 1281, 1242, 1144, 788, 480 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 2.5 × 10–5 M, CH2Cl2): 313.62 (30 000), 455.87 (4800). ESI-HRMS (acetonitrile) for [M–Cl + CH3CN]+ C18H14N4O7Re+ found: 585.0410 (calculated: 585.0420). Elemental analysis for C16H11ClN3O7Re calculated (%): C: 33.19; H, 1.92; N, 7.29; found (%): C: 33.31; H: 1.79; N: 7.21. Single crystals were obtained by slow evaporation from a mixture of acetone and water.
fac-[Re(CO)3(L3)Cl]
3Cl Yield 89.3% (104.3 mg), red solid. 1H NMR (500 MHz, acetonitrile-d3) δ = 9.30 (d, J = 0.7 Hz, 1H), 8.41 (d, J = 0.7 Hz, 1H), 8.24 (d, J = 3.3 Hz, 1H), 8.05 (d, J = 3.3 Hz, 1H), 3.97 (s, 3H), 3.97 (s, 3H) ppm. IR selected bands (ATR): ú = 2024, 1889, 1734, 1615, 1545, 1438, 1300, 1142, 1092, 781, 737, 524, 481 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 4 × 10–5 M, CH2Cl2): 326.80 (24500), 444.66 (4750). ESI-HRMS (acetonitrile) for [M-Cl + CH3CN]+ C17H13N3O7ReS+ found: 590.0019 (calculated: 590.0032). Elemental analysis for C15H10ClN2O7ReS calculated (%): C: 30.85; H, 1.73; N, 4.80; found (%): C: 30.80; H: 1.69; N: 4.94. Single crystals were obtained from the solution of chloroform with vapor diffusion of n-heptane utilizing the CrystalBreeder multireactor crystallization platform.27
fac-[Re(CO)3(L4)Cl]
4Cl Yield 59.0% (70.6 mg), red solid. 1H NMR (500 MHz, CDCl3) δ = 9.39 (d, J = 0.7 Hz, 1H), 8.09 (s, 1H), 7.43 (d, J = 1.1 Hz, 1H), 4.04 (s, 3H), 4.03 (s, 3H), 2.82 (d, J = 0.9 Hz, 3H) ppm. IR selected bands (ATR): ú = 2023, 1898, 1867, 1730, 1434, 1280, 1261, 1091, 777, 645, 527, 474 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, CH2Cl2): 341.14 (18 600), 438.08 (3800). ESI-HRMS (acetonitrile) for [M-Cl + CH3CN]+ C18H15N3O7ReS+ found: 604.016 (calculated: 604.0188). Elemental analysis for C16H12ClN2O7ReS calculated (%): C: 32.14; H, 2.02; N, 4.68; found (%): C: 32.55; H: 1.85; N: 4.63. Single crystals were obtained from the solution of chloroform with vapor diffusion of n-heptane utilizing the CrystalBreeder multireactor crystallization platform.27
fac-[Re(CO)3(L1)Br]
1Br Yield 36.8% (45.8 mg), orange solid. 1H NMR (500 MHz, CDCl3) δ = 9.43 (d, J = 0.6 Hz, 1H), 9.13 (ddd, J = 5.5, 1.6, 0.8 Hz, 1H), 8.35 (d, J = 0.7 Hz, 1H), 8.30 (dt, J = 8.3, 1.0 Hz, 1H), 8.13 (td, J = 7.9, 1.6 Hz, 1H), 7.64 (ddd, J = 7.7, 5.5, 1.3 Hz, 1H), 4.04 (d, J = 6.3 Hz, 6H) ppm. IR selected bands (ATR): ú = 2019, 1874, 1739, 1601, 1550, 1436, 1271, 1145, 1096, 783, 640, 635, 533, 481 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, CH2Cl2): 308 (21 600), 437 (3200). ESI-HRMS (acetonitrile) for [M-Br + CH3CN]+ C19H15N3O7Re+ found: 584.0460 (calculated: 584.0468). Elemental analysis for C17H12BrN2O7Re calculated (%): C: 32.81 H: 1.49 N; 4.50; found (%): C: 32.88; H: 1.58; N: 4.55.
fac-[Re(CO)3(L2)Br]
2Br Yield 75.9% (94.6 mg), orange solid. 1H NMR (500 MHz, CDCl3) δ = 9.60 (d, J = 1.3 Hz, 1H), 9.44 (d, J = 0.6 Hz, 1H), 9.07 (dd, J = 3.0, 1.3 Hz, 1H), 8.87 (d, J = 3.0 Hz, 1H), 8.50 (s, 1H), 4.06 (d, J = 1.6 Hz, 6H) ppm. IR selected bands (ATR): ú = 2022, 1925, 1888, 1727, 1438, 1278, 1107, 854, 638, 459 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, CH2Cl2): 314 (22 200), 462 (3400). ESI-HRMS (acetonitrile) for [M-Br + CH3CN]+ C18H14N4O7Re+ found: 584.0407 (calculated: 585.0420). Elemental analysis for C16H11BrN3O7Re calculated (%): C: 30.83 H: 1.78 N; 6.74; found (%): C: 30.97; H: 1.65; N: 6.65.
fac-[Re(CO)3(L3)Br]
3Br Yield 83.6% (105.5 mg), orange solid. 1H NMR (500 MHz, CDCl3) δ = 9.41 (d, J = 0.7 Hz, 1H), 8.26 (d, J = 3.3 Hz, 1H), 8.13 (d, J = 0.8 Hz, 1H), 7.85 (d, J = 3.3 Hz, 1H), 4.04 (d, J = 5.1 Hz, 6H) ppm. IR selected bands (ATR): ú = 2019, 1906, 1737, 1615, 1435, 1302, 1264, 1142, 1093, 768, 645, 531, 488 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, dichloromethane): 327 (19 000), 452 (3600). ESI-HRMS (acetonitrile) for [M-Br + CH3CN]+ C17H13N3O7ReS+ found: 590.0018 (calculated: 590.0032). Elemental analysis for C15H10BrN2O7ReS calculated (%): C: 28.67 H: 1.60 N; 4.46; found (%): C: 28.76; H: 1.29; N: 4.46. Single crystals were obtained by the slow evaporation from toluene.
fac-[Re(CO)3(L4)Br]
4Br Yield: 69.1% (90.5 mg); orange-brown solid. 1H NMR (500 MHz, CDCl3) δ = 9.41 (d, J = 0.7 Hz, 1H), 8.10 (d, J = 0.8 Hz, 1H), 7.44 (d, J = 0.9 Hz, 1H), 4.03 (d, J = 6.4 Hz, 7H), 2.82 (d, J = 0.9 Hz, 3H) ppm. IR selected bands (ATR): ú = 2022, 1801, 1930, 1733, 1614, 1435, 1291, 1141, 1093, 951, 794, 644, 486 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, CH2Cl2): 342 (17200), 444 (3200). ESI-HRMS (acetonitrile) for [M-Br + CH3CN]+ C18H15N3O7ReS+ found: 604.0178 (calculated: 604.0188). Elemental analysis for C16H12BrN2O7ReS calculated (%): C: 29.91 H: 1.88 N; 4.36; found (%): C: 29.65; H: 1.66; N: 4.32. Single crystals were obtained by slow evaporation from toluene.
General Procedure for The Synthesis of fac-Tricarbonylrhenium(I) Aqua Complexes with General Formula fac-[Re(CO)3(NN)(H2O)]CF3SO3
Complexes were synthesized according to the literature procedure.26 A reaction mixture containing appropriate chlorido complex 1Cl–4Cl (0.1 mmol), silver triflate (0.12 mmol), and 10 mL of dichloromethane was stirred at room temperature overnight. The precipitate of AgCl was removed by filtration over Celite, and dichloromethane was removed under reduced pressure. The product was dissolved in a mixture of methanol and water in a ratio of 99:1 and left to slowly evaporate.
fac-[Re(CO)3(L1)(H2O)]CF3SO3
1Aq Yield 28.9% (16.0 mg), red-orange solid. 1H NMR (500 MHz, acetonitrile-d3) δ = 9.34 (d, J = 0.7 Hz, 1H), 9.12–9.01 (m, 1H), 8.67 (d, J = 0.7 Hz, 1H), 8.56 (dt, J = 8.1, 1.0 Hz, 1H), 8.34 (td, J = 7.9, 1.5 Hz, 1H), 7.80 (ddd, J = 7.8, 5.5, 1.2 Hz, 1H), 4.01 (s, 3H), 4.00 (s, 3H) ppm. IR selected bands (ATR): ú = 3121, 2033, 1906, 1732, 1436, 1275, 1232, 1144, 1096, 1025, 781, 634 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, methanol): 312.19 (15 200), 379.02 (3200). ESI-HRMS (acetonitrile) for [M-Cl + CH3CN]+ C19H15N3O7Re+ found: 584.0450 (calculated: 584.0468). Elemental analysis for C18H14F3N2O11ReS calculated (%): C: 30.47; H: 1.99; N: 3.95; found (%): C: 31.31; H: 1.83; N: 4.10.
fac-[Re(CO)3(L2)(H2O)]CF3SO3
2Aq Yield 26.9% (11.3 mg), red-orange solid. 1H NMR (500 MHz, acetonitrile-d3) δ = 9.77 (d, J = 1.4 Hz, 1H), 9.36 (s, 1H), 9.03 (dd, J = 3.1, 1.3 Hz, 1H), 9.00 (d, J = 3.1 Hz, 1H), 8.82 (s, 1H), 4.02 (s, 3H), 4.00 (s, 3H) ppm. IR selected bands (ATR): ú = 2925, 2036, 1910, 1732, 1437, 1276, 1235, 1144, 1027, 636, 477 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, methanol): 316.08 (15 000), 397.02 (5200). ESI-HRMS (acetonitrile) for [M-Cl + CH3CN]+ C18H14N4O7Re+ found: 584.0405 (calculated: 585.0420). Elemental analysis for C17H13F3N3O11ReS·0.03 C7H8 calculated (%): C: 30.62; H, 2.04; N, 5.74; found (%): C: 30.13; H: 1.85; N: 5.78.
fac-[Re(CO)3(L3)(H2O)]CF3SO3
3Aq Yield 23.1% (14.7 mg), red-orange solid. 1H NMR (500 MHz, acetonitrile-d3) δ = 9.32 (d, J = 0.7 Hz, 1H), 8.48 (d, J = 0.7 Hz, 1H), 8.25 (d, J = 3.3 Hz, 1H), 8.16 (d, J = 3.3 Hz, 1H), 3.99 (s, 6H) ppm. IR selected bands (ATR): ú = 3120, 2034, 1906, 1732, 1439, 1285, 1224, 1141, 1024, 634, 526, 514 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, methanol): 332.74 (16 800), 340.75 (17 000). ESI-HRMS (acetonitrile) for [M-Cl + CH3CN]+ C17H13N3O7ReS+ found: 590.0015 (calculated: 590.0032). Elemental analysis for C16H12F3N2O11ReS2 calculated (%): C: 26.86; H: 1.69; N: 3.92; found (%): C: 27.43; H: 1.30; N: 3.91.
fac-[Re(CO)3(L4)(H2O)]CF3SO3
4Aq Yield 45.9% (35.2 mg), red-orange solid. 1H NMR (500 MHz, acetonitrile-d3) δ = 9.31 (d, J = 0.7 Hz, 1H), 8.43 (d, J = 0.7 Hz, 1H), 7.80 (d, J = 1.0 Hz, 1H), 3.99 (s, 3H), 3.99 (s, 3H), 2.75 (d, J = 0.9 Hz, 3H) ppm. IR selected bands (ATR): ú = 3133, 2035, 1945, 1914, 1733, 1449, 1434, 1285, 1224, 1140, 1093, 1021, 632, 521 cm–1. UV–vis (λ (nm) (ε (M–1cm–1)), c = 5 × 10–5 M, methanol): 354.38 (18200). ESI-HRMS (acetonitrile) for [M-Cl + CH3CN]+ C18H15N3O7ReS+ found: 604.0170 (calculated: 604.0188). Elemental analysis for C17H14F3N2O11ReS2 calculated (%): C: 27.99; H, 1.93; N, 3.84; found (%): C: 28.44; H: 1.43; N: 4.01. Single crystals were obtained by slow evaporation from a mixture of methanol and water.
Stock Solutions and Sample Preparation
Milli-Q water or DMSO was used for the preparation of stock and sample solutions. Aqueous stock solutions of aqua complexes were obtained by dissolving an exact amount in water, and their concentrations were calculated based on a weight-in-volume basis. A 4-fold dilution of human serum was carried out with PBS’ buffer for the stability measurements.
UV–visible Spectrophotometry
An Agilent Cary
3500 spectrophotometer was utilized to obtain UV–vis spectra
in the wavelength range of 200–1100 nm. The path length ( ) was 1 cm
in most cases (the actual
) was 1 cm
in most cases (the actual  is always
indicated in the legends of the
figures). The concentrations of the complexes were between 50 and
100 μM. Spectra were always background- and baseline corrected.
The computer program HypSpec36 was used
to obtain equilibrium constants.
is always
indicated in the legends of the
figures). The concentrations of the complexes were between 50 and
100 μM. Spectra were always background- and baseline corrected.
The computer program HypSpec36 was used
to obtain equilibrium constants.
Determination of Distribution Coefficients
The traditional shake-flask method was used to obtain distribution coefficients of the complexes in n-octanol/buffered aqueous solution (20 mM HEPES, pH = 7.4) using UV–vis spectrophotometry (Agilent Cary 3500 spectrophotometer, Santa Clara, CA, USA) for the analysis. Aqua complexes were dissolved in buffered aqueous solutions previously saturated with n-octanol. In the case of halido complexes, compounds were dissolved in the presaturated n-octanol phase due to their high lipophilicity and low aqueous solubility. Then, the aqueous and n-octanol phases were gently mixed in different volume ratios (using 1:1, 1:10, 1:20, 1:40 for aqua and 1:40 n-octanol-to-buffered aqueous solution volume ratio for halido complexes) for 4 h, followed by phase separation. Three different concentrations of chloride ions (4, 24, or 100 mM) were applied in a similar manner as described in our former reports, taking into consideration the different chloride ion concentrations found in the biofluids,37−39 and measurements were also done without chloride ions. Then, the UV–vis spectra of the aqueous or n-octanol phase were recorded and compared to a reference spectrum. Distribution coefficients (DpH) were calculated by the following equations.
When the stock solution was made in an aqueous buffer
When the stock solution was made in the n-octanol
Determination of Thermodynamic Solubility
Thermodynamic solubility (S7.4) of the halido complexes was assessed by measuring the saturation levels in water at pH = 7.4 (10 mM HEPES buffer and 0.1 M KCl) at 25.0 ± 0.1 °C. The concentration of the compounds was determined by UV–vis spectrophotometry. For the calibration, stock solutions of the compounds were used with known concentrations dissolved in 100% DMSO, 75 and 50% (v/v) DMSO/buffered aqueous solutions.
Solution Studies Using 1H NMR Spectroscopy
A Bruker Avance III HD Ascend 500 Plus instrument (Billerica, MA, USA) was used for the NMR studies. Spectra were recorded with a WATERGATE water suppression pulse scheme in the presence of 10% (v/v) D2O in most cases. In some cases, 5–10% (v/v) DMSO-d6/H2O was also used. DSS internal standard was added to samples to obtain reference peaks. 1H NMR titration was carried out in the presence of 0.1 M KNO3. The computer program HypSpec36 was used to obtain equilibrium constants.
In Vitro Cytotoxicity Assay
Cell Lines and Culture Conditions
Doxorubicin-sensitive Colo205 (ATCC-CCL-222) and ABCB1 (MDR1)-LRP-expressing resistant Colo320/MDR-LRP (ATCC-CCL-220.1) human colonic adenocarcinoma cell lines were purchased from LGC Promochem (Teddington, UK). The cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 1 mM Na-pyruvate, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Sigma, Steinheim, Germany). Cell lines were incubated at 37 °C in a 5% CO2, 95% air atmosphere. The semiadherent human colon cancer cells were detached with Trypsin-Versene (EDTA, Sigma, Steinheim, Germany) solution for 5 min at 37 °C.
MTT Assay on Colo205 and Colo320 Cells
The tested compounds were dissolved in DMSO or 20% (v/v) DMSO/H2O mixture to prepare 5 mM stock solutions, which were diluted in complete culture medium, to study the effect of compounds on the cell growth of human colonic adenocarcinoma cell lines (doxorubicin-sensitive Colo205 and resistant Colo320 colonic adenocarcinoma cells). Doxorubicin (Merck, Darmstadt, Germany) was used as a positive control. The cells were treated with Trypsin-Versene (EDTA) solution. They were adjusted to a density of 1 × 104 cells in 100 μL of the appropriate culture medium and were added to each well, with the exception of the medium control wells. Then stock solutions were diluted in the appropriate culture medium, and 2-fold serial dilutions of compounds were prepared in 100 μL of the medium, horizontally. The final volume of the wells containing compounds and cells was 200 μL. The plates containing the cancer cells were incubated at 37 °C for 72 h; at the end of the incubation period, 20 μL of MTT solution (from a stock solution of 5 mg/mL) were added to each well. After incubation at 37 °C for 4 h, 100 μL of SDS solution (10% in 0.01 M HCI) were added to each well, and the plates were further incubated at 37 °C overnight. Cell growth was determined by measuring the optical density (OD) at 540/630 nm with a Multiscan EX ELISA reader (Thermo Labsystems, Cheshire, WA, USA). Inhibition of the cell growth (expressed as IC50: inhibitory concentration that reduces by 50% the growth of the cells exposed to the tested compounds) was determined from the sigmoid curve where 100 – ((ODsample – ODmedium control)/(ODcell control – ODmedium control)) × 100 values were plotted against the logarithm of compound concentrations. Curves were fitted by GraphPad Prism software (2021, GraphPad Prism Software, San Diego, CA, USA)40 using the sigmoidal dose–response model (comparing variable and fixed slopes). The IC50 values were obtained from at least 3 independent experiments.
Antibacterial Activity Assay
The following bacterial strains were used in our experiments: gram-positive S. aureus American Type Culture Collection (ATCC) 25923 as the methicillin-susceptible reference bacterial strain; the methicillin-resistant S. aureus ATCC 43300 (MRSA) strain; E. coli ATCC 25922 as a gram-negative bacterial strain.
MIC values of the complexes were determined in 96-well plates based on the Clinical and Laboratory Standard Institute guidelines (CLSI guidelines).41 The stock solutions of the compounds (dissolved in DMSO or a 20% (v/v) DMSO/H2O mixture in 5 mM concentration) were diluted in 100 μL of Mueller Hinton Broth. Then 10–4 dilutions of an overnight bacterial culture in 100 μL of the medium was added to each well with the exception of the medium control wells. The plates were further incubated at 37 °C for 18 h; at the end of the incubation period, the MIC values of tested compounds were determined by visual inspection.
Antiviral Activity Assay
Cultivation and Quantification of Herpes Simplex Virus Type 2
HSV-2 strain was (gift from Dr. Ilona Mucsi, University of Szeged, Szeged, Hungary) grown in Vero (ATCC) cells and the infectivity was determined by using the plaque titration method.42 The virus titer was expressed as plaque-forming units (PFU).43
Culture of Vero Cells and MTT Assay
Vero cells (ATCC) were placed into the 96-well plate (Sarstedt, Nümbrecht, Germany) at a density of 4 × 106 cells/plate. The cells at a density of 4 × 104 cells per well were in 100 μL of minimal essential medium (MEM) (Sigma; USA) with Earle salts supplemented with 25 μg/mL gentamicin, 10% heat-inactivated fetal bovine serum (FBS) (Gibco; Germany), 8 mM HEPES, 2 mM l-glutamine, 1× nonessential amino acids, and 1 μg/mL fungisone. The cells were incubated for 60 min at room temperature (RT) to avoid the edge effect and then for 24 h at 37 °C, 5% CO2 that secure a 90% confluent cell layer.44
MTT assay was used to determine the maximum non-toxic concentration of the complexes on Vero cells. The cells were grown in 96-well plate at density of 4 × 104 cells per well were in 100 μL of MEM with Earle salts supplemented with 25 μg/mL gentamicin, 10% heat-inactivated FBS, 8 mM HEPES, 2 mM l-glutamine, 1× nonessential amino acids, and 1 μg/mL fungisone. The cells were incubated for 1 h at RT and then overnight at 37 °C, 5% CO2. When the cell layer reached around 90% confluency the medium was complemented with the serial 2-fold dilutions of complexes. Three parallels were applied for each concentration in the range of 100–0.048 μM for each complex. 10 μL of the MTT labeling reagent was added into wells at 0.5 mg/mL final concentration. The plate was incubated for 240 min at 37 °C, 5% CO2 and then 100 μL of the solubilization solution (10% SDS in 1 M HCl) was added to each well. Next day the absorbance of the wells was determined by a microtiter plate reader (Labsystems Multiskan Ex 355, Thermo Fisher Scientific, Waltham, MA USA). The absorbance of the formazan product was measured at 540 nm.45
Investigation of The Impact of Complexes on HSV-2 Growth in Vero Cells
The Vero cells were transferred into the wells of the 96-well plate at a density of 4 × 104 cells/well in 100 μL of Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma; USA) containing 100 U/mL penicillin, 100 mg/mL streptomycin sulfate, 5% FBS, 0.25 g/mL amphotericin B and 0.14% NaHCO3. Prior to infection, the cells were washed with PBS and then were incubated with HSV-2 (MOI 0.1) for 60 min at 37 °C under a 5% CO2 atmosphere. Then the cells were washed with PBS again, and the culture medium was complemented with the serial 2-fold dilutions of the complexes was added to triplicate wells in the concentration range of 100–0.078 μM. The plates were incubated at 37 °C, 5% CO2 for 24 h. The plates were evaluated with RT-qPCR.
Cell Lysis and Direct Quantitative PCR
The supernatans of infected cells were removed and washed with PBS twice, and then 100 μL of high-quality ultrapure water was added to wells at the end of 24 h infection. The cells were subjected to two freeze–thaw cycles. Templates for qPCR reactions originated from mixed cell lysates. Process of PCR was described previously.46 Briefly, we used 5× HOT FIREPol EvaGreen qPCR Supermix (Solis Solis BioDyne, Tartu, Estonia) and HSV-2 gD2 gene specific primer in a Bio-Rad CFX96 real time PCR system for the qPCR reaction. Sequences of the gD2 gene specific primer pair were the following: gD2-F: 5′-TCAGCGAGGATAACCTGGGA-3′, and gD2-R 5′-GGGAGAGCGTACTTGCAGGA-3′. Annealing-extension temperature was 69 °C. Cycle where the amplification curve stepped over the baseline can correspond to the Ct value of a given sample.
Acknowledgments
Project no TKP2021-EGA-32 has been implemented with the support provided by the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme. ÚNKP-23-3-SZTE-496 New National Excellence program of the Ministry for Innovation and Technology, and the “Lendület” Programme (HUN-REN Hungarian Research Network, LP2019-6/2019) are also acknowledged. The authors also acknowledge the support of the Centre for Research Infrastructure at the University of Ljubljana, Faculty of Chemistry and Chemical Technology, which is part of the Network of Research and Infrastructural Centres UL (MRIC UL) and is financially supported by the Slovenian Research and Innovation Agency (ARIS; Infrastructure programme no. I0-0022) for the use of the Supernova diffractometer. This research was also funded by program grant P1-0175, of the ARIS. This article is also based upon work from COST Action EURESTOP, CA21145, supported by COST (European Cooperation in Science and Technology). We would like to acknowledge support from Erasmus+ program for providing funding for student exchange (to SCR and DCA). We would like to thank Žiga Poklečki (University of Ljubljana) for his help in the laboratory. University of Szeged Open Access Fund (grant number: 7186) is also acknowledged (to É.A.E.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c07117.
Chemical structure of the ligands and the fac-[Re(CO)3] complexes. 1H NMR spectra of the prepared complexes; picture and crystallographic data for the complexes obtained as single crystal with selected bond lengths; aqueous solubility data, time-dependent UV–vis, and 1H NMR spectra of the halido and aqua complexes in different biologically relevant matrices; UV–vis spectra of 1Aq in water at different pH values; ratio of the aqua and hydroxido complexes at pH = 7.4; UV–vis and 1H NMR spectra of the aqua complex 1Aq in the presence of chloride ions; lipophilicity data for the studied complexes; cytotoxicity of the complexes against Vero cells and antiviral effect of complexes 1–4Aq against HSV-2 and Ct and dCt values of the halido complexes (PDF)
X-ray data (ZIP)
NMR data (ZIP)
Author Contributions
T.P.: Investigation, data curation, writing. J.K.: Investigation, data curation, writing, supervision. S.C.R.: Investigation. D.C.A.: Investigation. U.R.: Investigation. M.N.: Investigation. E.F.V.: Investigation. J.N.: Investigation. A.B.: Investigation, data curation. G.S.: Investigation, supervision. I.T.: Conceptualization, review, supervision. É.A.E.: Conceptualization, review, supervision. All authors have read and approved the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Leonidova A.; Gasser G. Underestimated Potential of Organometallic Rhenium Complexes as Anticancer Agents. ACS Chem. Biol. 2014, 9 (10), 2180–2193. 10.1021/cb500528c. [DOI] [PubMed] [Google Scholar]
- Gasser G.; Ott I.; Metzler-Nolte N. Organometallic Anticancer Compounds. J. Med. Chem. 2011, 54 (1), 3–25. 10.1021/jm100020w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew H. S.; Mai C.-W.; Zulkefeli M.; Madheswaran T.; Kiew L. V.; Delsuc N.; Low M. L. Recent Emergence of Rhenium(I) Tricarbonyl Complexes as Photosensitisers for Cancer Therapy. Molecules 2020, 25 (18), 4176. 10.3390/molecules25184176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkankit C. C.; King A. P.; Knopf K. M.; Southard T. L.; Wilson J. J. In Vivo Anticancer Activity of a Rhenium(I) Tricarbonyl Complex. ACS Med. Chem. Lett. 2019, 10 (5), 822–827. 10.1021/acsmedchemlett.9b00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopf K. M.; Murphy B. L.; MacMillan S. M.; Baskin J. M.; Barr M. P.; Boros E.; Wilson J. J. In Vitro Anticancer Activity and in Vivo Biodistribution of Rhenium(I) Tricarbonyl Aqua Complexes. J. Am. Chem. Soc. 2017, 139 (40), 14302–14314. 10.1021/jacs.7b08640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinman P. S.; Welsh A.; Omondi R. O.; Khan S.; Prince S.; Nordlander E.; Smith G. S. Aminoquinoline-based Re(I) tricarbonyl complexes: Insights into their antiproliferative activity and mechanisms of action. Eur. J. Med. Chem. 2024, 266, 116094 10.1016/j.ejmech.2023.116094. [DOI] [PubMed] [Google Scholar]
- Collery P.; Desmaele D.; Vijaykumar V. Design of Rhenium Compounds in Targeted Anticancer Therapeutics. Curr. Pharm. Des. 2019, 25 (31), 3306–3322. 10.2174/1381612825666190902161400. [DOI] [PubMed] [Google Scholar]
- Karges J.; Cohen S. M. Metal Complexes as Antiviral Agents for SARS-CoV-2. ChemBioChem 2021, 22 (16), 2600–2607. 10.1002/cbic.202100186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karges J.; Kalaj M.; Gembicky M.; Cohen S. M. ReI Tricarbonyl Complexes as Coordinate Covalent Inhibitors for the SARS-CoV-2 Main Cysteine Protease. Angew. Chem., Int. Ed. 2021, 60 (19), 10716–10723. 10.1002/anie.202016768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler K.; Zobi F. Anticancer and Antibiotic Rhenium Tri- and Dicarbonyl Complexes: Current Research and Future Perspectives. Molecules 2022, 27 (2), 539. 10.3390/molecules27020539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łyczko K.; Pogorzelska A.; Częścik U.; Koronkiewicz M.; Rode J. E.; Bednarek E.; Kawęcki R.; Węgrzyńska K.; Baraniak A.; Milczarek M.; Dobrowolski J. C. Tricarbonyl rhenium(i) complexes with 8-hydroxyquinolines: structural, chemical, antibacterial, and anticancer characteristics. RSC Adv. 2024, 14 (25), 18080–18092. 10.1039/D4RA03141E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karges J.; Giardini M. A.; Blacque O.; Woodworth B.; Siqueira-Neto J. L.; Cohen S. M. Tricarbonyl Enantioselective inhibition of the SARS-CoV-2 main protease with rhenium(i) picolinic acid complexes. Chem. Sci. 2023, 14 (3), 711–720. 10.1039/D2SC05473F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soba M.; Scalese G.; Casuriaga F.; Pérez N.; Veiga N.; Echeverría G. A.; Piro O. E.; Faccio R.; Pérez-Díaz L.; Gasser G.; Machado I.; Gambino D. Multifunctional organometallic compounds for the treatment of Chagas disease: Re(I) tricarbonyl compounds with two different bioactive ligands. Dalton Trans. 2023, 52, 1623–1641. 10.1039/D2DT03869B. [DOI] [PubMed] [Google Scholar]
- Traven K.; Sinreih M.; Stojan J.; Seršen S.; Kljun J.; Bezenšek J.; Stanovnik B.; Turel I.; Rižner T. L. Ruthenium complexes as inhibitors of the aldo-keto reductases AKR1C1–1C3. Chem.-Biol. Interact. 2015, 234, 349–359. 10.1016/j.cbi.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Traven K.; Eleftheriadis N.; Seršen S.; Kljun J.; Bezenšek J.; Stanovnik B.; Turel I.; Dekker F. J. Ruthenium complexes as inhibitors of 15-lipoxygenase-1. Polyhedron 2015, 101, 306–313. 10.1016/j.poly.2015.09.019. [DOI] [Google Scholar]
- Andrejević T. P.; Milivojevic D.; Glišić BĐ.; Kljun J.; Stevanović N. L.; Vojnovic S.; Medic S.; Nikodinovic-Runic J.; Turel I.; Djuran M. I. Silver(i) complexes with different pyridine-4,5-dicarboxylate ligands as efficient agents for the control of cow mastitis associated pathogens. Dalton Trans. 2020, 49 (18), 6084–6096. 10.1039/D0DT00518E. [DOI] [PubMed] [Google Scholar]
- Andrejević T. P.; Aleksic I.; Počkaj M.; Kljun J.; Milivojevic D.; Stevanović N. L.; Nikodinovic-Runic J.; Turel I.; Djuran M. I.; Glišić BĐ. Tailoring copper(ii) complexes with pyridine-4,5-dicarboxylate esters for anti-Candida activity. Dalton Trans. 2021, 50 (7), 2627–2638. 10.1039/D0DT04061D. [DOI] [PubMed] [Google Scholar]
- Andrejević T. P.; Aleksic I.; Kljun J.; Pantović B. V.; Milivojevic D.; Vojnovic S.; Turel I.; Djuran M. I.; Glišić BĐ. Zinc(II) Complexes with Dimethyl 2,2′-Bipyridine-4,5-dicarboxylate: Structure, Antimicrobial Activity and DNA/BSA Binding Study. Inorganics 2022, 10 (6), 71. 10.3390/inorganics10060071. [DOI] [Google Scholar]
- Bal A. M.; David M. Z.; Garau J.; Gottlieb T.; Mazzei T.; Scaglione F.; Tattevin P.; Gould I. M. Future trends in the treatment of methicillin-resistant Staphylococcus aureus (MRSA) infection: An in-depth review of newer antibiotics active against an enduring pathogen. J. Global Antimicrob. Resist. 2017, 10, 295–303. 10.1016/j.jgar.2017.05.019. [DOI] [PubMed] [Google Scholar]
- Mirabelli P.; Coppola L.; Salvatore M. Cancer Cell Lines Are Useful Model Systems for Medical Research. Cancers 2019, 11 (8), 1098. 10.3390/cancers11081098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alibek K.; Bekmurzayeva A.; Mussabekova A.; Sultankulov B. Using antimicrobial adjuvant therapy in cancer treatment: a review. Infect. Agents Cancer 2012, 7 (1), 33. 10.1186/1750-9378-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser K. E.; Eichten A.; Coussens L. M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6 (1), 24–37. 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- Bezenšek J.; Prek B.; Grošelj U.; Kasunič M.; Svete J.; Stanovnik B. A simple metal-free synthesis of 2-substituted pyridine-4,5-dicarboxylates and their N-oxides. Tetrahedron 2012, 68 (24), 4719–4731. 10.1016/j.tet.2012.04.013. [DOI] [Google Scholar]
- Wrighton M.; Morse D. L. Nature of the lowest excited state in tricarbonylchloro-1,10-phenanthrolinerhenium(I) and related complexes. J. Am. Chem. Soc. 1974, 96 (4), 998–1003. 10.1021/ja00811a008. [DOI] [Google Scholar]
- Smieja J. M.; Kubiak C. P. Re(bipy-tBu)(CO)3Cl–improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies. Inorg. Chem. 2010, 49 (20), 9283–9289. 10.1021/ic1008363. [DOI] [PubMed] [Google Scholar]
- Carrington S. J.; Chakraborty I.; Bernard J. M.; Mascharak P. K. A Theranostic Two-Tone Luminescent PhotoCORM Derived from Re(I) and (2-Pyridyl)-benzothiazole: Trackable CO Delivery to Malignant Cells. Inorg. Chem. 2016, 55 (16), 7852–7858. 10.1021/acs.inorgchem.6b00511. [DOI] [PubMed] [Google Scholar]
- Technobis, CrystalBreeder. https://www.crystallizationsystems.com. (accessed Aug 1, 2024).
- Sharma S A.; N V.; Kar B.; Das U.; Paira P. Target-specific mononuclear and binuclear rhenium(i) tricarbonyl complexes as upcoming anticancer drugs. RSC Adv. 2022, 12 (31), 20264–20295. 10.1039/D2RA03434D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter T. C.; Ehlers B.; Müller T.; Yoon K.-J.; Teifke J. P.. Herpesviruses. In Diseases of Swine, 11th ed.; Zimmermann J. J.; Karriker L. A.; Ramirez A.; Schwartz K. J.; Stevenson G. W.; Zhang J., Eds.; Wiley, 2019. [Google Scholar]
- Zhang X.; Guo J.; Song B.; Zhang F. Spatiotemporal Regulation of Metal Ions in the Polymerase Chain Reaction. ACS Omega 2022, 7 (37), 33530–33536. 10.1021/acsomega.2c04507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menges F.Spectragryph—Optical Spectroscopy Software″, Version 1.2.16, 2022http://www.effemm2.de/spectragryph. (accessed Aug 1, 2024).
- Oxford Diffraction Ltd, CrysAlis PRO . Yarnton, Oxfordshire, England 2011.
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Sheldrick G. M.Shelxl 2018/3, Program for Crystal Structure Refinement; University of Göttingen: Germany, 2018.
- Macrae C. F.; Edgington P. R.; McCabe P.; Pidcock E.; Shields G. P.; Taylor R.; Towler M.; Van De Streek J. Mercury: visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. 10.1107/S002188980600731X. [DOI] [Google Scholar]
- Gans P.; Sabatini A.; Vacca A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43 (10), 1739–1753. 10.1016/0039-9140(96)01958-3. [DOI] [PubMed] [Google Scholar]
- Pivarcsik T.; Pósa V.; Kovács H.; May N. V.; Spengler G.; Pósa S. P.; Tóth S.; Yazdi Z. N.; Özvegy-Laczka C.; Ugrai I.; Szatmári I.; Szakács G.; Enyedy É.A. Metal Complexes of a 5-Nitro-8-Hydroxyquinoline-Proline Hybrid with Enhanced Water Solubility Targeting Multidrug Resistant Cancer Cells. Int. J. Mol. Sci. 2023, 24 (1), 539. 10.3390/ijms24010593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivarcsik T.; Dömötör O.; Mészáros J. P.; May N. V.; Spengler G.; Csuvik O.; Szatmári I.; Enyedy É.A. 8-Hydroxyquinoline-Amino Acid Hybrids and Their Half-Sandwich Rh and Ru Complexes: Synthesis, Anticancer Activities, Solution Chemistry and Interaction with Biomolecules. Int. J. Mol. Sci. 2021, 22 (20), 11281. 10.3390/ijms222011281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros J. P.; Poljarević J. M.; Szatmári I.; Csuvik O.; Fülöp F.; Szoboszlai N.; Spengler G.; Enyedy É.A. An 8-hydroxyquinoline-proline hybrid with multidrug resistance reversal activity and the solution chemistry of its half-sandwich organometallic Ru and Rh complexes. Dalton Trans. 2020, 49 (23), 7977–7992. 10.1039/D0DT01256D. [DOI] [PubMed] [Google Scholar]
- GraphPad Prism Version 7.00 for Windows, Graph Pad Software; Graph Pad Software: La Jolla, California USA, 2018https://www.graphpad.com. (accessed Aug 1, 2024).
- CLSI . Susceptibility Testing Process. In Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 10th ed.; Christopher P. J.; Polgar E. P., Eds.; Clinical and Laboratory Standards Institute: Wayne, MI, USA, 2015; Vol. 32, pp 15–19. [Google Scholar]
- Abdelmageed A. A.; Ferran M. C. The Propagation, Quantification, and Storage of Vesicular Stomatitis Virus. Curr. Protoc. Microbiol. 2020, 58 (1), e110 10.1002/cpmc.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucsi I.; Molnár J.; Motohashi N. Combination of benzo[a]phenothiazines with acyclovir against herpes simplex virus. Int. J. Antimicrob. Agents 2001, 18 (1), 67–72. 10.1016/S0924-8579(01)00323-5. [DOI] [PubMed] [Google Scholar]
- Mansoury M.; Hamed M.; Karmustaji R.; Al Hannan F.; Safrany S. T. The edge effect: A global problem. The trouble with culturing cells in 96-well plates. Biochem. Biophys. Rep. 2021, 26, 100987 10.1016/j.bbrep.2021.100987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayarathna S.; Sasidharan S. Cytotoxicity of methanol extracts of Elaeis guineensis on MCF-7 and Vero cell lines. Asian Pac. J. Trop. Biomed. 2012, 2 (10), 826–829. 10.1016/S2221-1691(12)60237-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virók D. P.; Eszik I.; Mosolygó T.; Önder K.; Endrész V.; Burián K. A direct quantitative PCR-based measurement of herpes simplex virus susceptibility to antiviral drugs and neutralizing antibodies. J. Virol. Methods 2017, 242, 46–52. 10.1016/j.jviromet.2017.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.