

Abstract
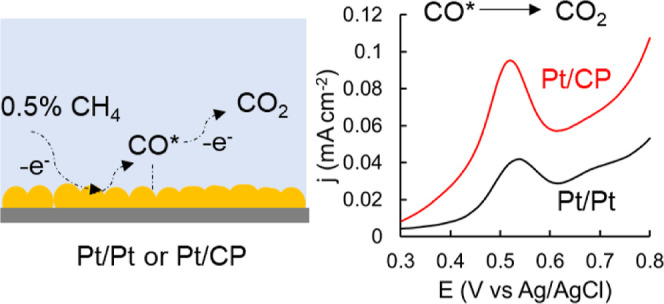
Methane is a potent greenhouse gas, and its rapid conversion at low concentrations under ambient conditions is a challenging process where combustion is not an option. Herein, we report an electrochemical method to address this problem. It was achieved by applying an oxidation potential to electrochemically activate methane followed by conducting an anodic cyclic voltammogram to fully oxidize activated methane to carbon dioxide on platinized Pt mesh (Pt/Pt) and carbon paper (Pt/CP). This “dynamic potential” oxidation approach enabled methane conversion with low energy consumption, thanks to the low activation potential. Effects of various experimental conditions (applied potential, reaction time, and methane concentration) were investigated. Pure methane and methane/nitrogen gas mixtures containing a series of low concentrations of methane were tested. It was found that methane conversion is independent of its concentration on both Pt/Pt and Pt/CP. Compared to Pt/Pt electrocatalysis, Pt/CP displayed approximately 10 times higher catalytic activity, which can be attributed to the stronger binding of intermediate CO* to Pt, leading to easier CO* activation in the presence of a carbon substrate. Carbon dioxide was the only compound detected during the electro-oxidation phase for Pt/Pt, while for Pt/CP, carbon dioxide and a small amount of formic acid (after 15 h reaction) were observed. Electrocatalytic conversion of methane to carbon dioxide on Pt/CP using 0.5% methane was measured, giving a methane conversion rate of 7.5 × 10–8 mol L–1 s–1 m–2, while the methane conversion rate on Pt/Pt with 1% methane was only 8.3 × 10–9 mol L–1 s–1 m–2.
1. Introduction
Methane (CH4) is a very significant greenhouse gas and is estimated to contribute 0.5 °C of the total net 1.2 °C warming above the preindustrial temperature.1,2 Despite the lower concentration of atmospheric CH4 compared to that of CO2, CH4 is approximately 80 times more powerful than CO2 at trapping heat (over a 20 year period).1 Moreover, rising temperatures have the potential to cause increasing CH4 emissions, worsening global warming.3 CH4 has a short average lifetime (approximately 12 years).2 Reducing CH4 emissions and enhancing CH4 oxidation under mild conditions using renewable energy can provide the opportunity to significantly reduce global warming and improve air quality, which will give more time to governments and businesses to deploy all the measures to ease climate change.4 However, research on advanced CH4 oxidation or, alternatively, CH4 extraction from the atmosphere is growing very slowly compared to CO2 removal, which is one of the hurdles to advance in the field.5 Our primary interest in CH4 is the conversion of low-concentration sources (below the combustion concentration, <∼5%), such as that produced from agriculture, contributing to ∼25% of total anthropogenic emissions.6 However, CH4 oxidation at low concentration under ambient conditions is very challenging and energy intensive due to its stable chemical structure.7 Converting low-concentration CH4 to value-added, easily managed, and less environmentally damaging products (e.g., CO2) under ambient conditions, ideally at the source before it enters the atmosphere, is an extremely valuable environmental target. Furthermore, there is potential to couple such CH4 oxidation with CO2 sequestration, minimizing negative effect on environment.8,9 Currently, direct CH4 conversion can be achieved by thermo-, photo-, or electrocatalysis.7,10
The earliest report of thermocatalysis involving methane (coal gas) in oxygen or air on hot fine Pt wire was published by Davy in 1801.11 CH4 oxidation/combustion in industry via thermocatalysis to form syngas requires high temperature (700–1100 °C) and pressure (typically above 10 bar) to break the C–H bond.12−14 Addition of expensive oxidants such as H2O215 and N2O16 can reduce the required temperature and pressure for thermocatalysis.
CH4 conversion via photocatalysis on semiconductor materials with or even without cocatalysts can be achieved at room temperature.17−19 Nevertheless, photocatalytic oxidation of CH4 normally results in low conversion rate and poor selectivity.17
Electrocatalysis is a popular alternative CH4 conversion method, thanks to low energy consumption, often high Faradaic efficiency, and a more controllable reaction.20 The most studied and commercially available electrochemical device is the solid oxide fuel cells (SOFCs), which provide fast kinetics for complete CH4 oxidation to CO2 and generate electricity.21,22 Nevertheless, SOFCs also require energy input due to high operating temperature and pressure and a relatively high concentration of CH4 (>30%).23 Therefore, SOFCs are not sufficient to meet the demand of achieving electrochemical oxidation of CH4 at low concentrations with minimum energy consumption.
The existing literature addressing the challenges of electrocatalysis is limited, with few reported studies available requiring relatively high-concentration CH4 or high temperature or complex electrocatalysts. For instance, Kim et al. employed 50 v/v % CH4 with 50 v/v % O2 to generate H2O2 electrochemically to partially oxidize CH4 to mixed oxygenates using a treated carbon electrode at 25 °C and 1 bar.24 Natinsky et al. reported CH4 electrochemical partial oxidation to methanol using an O2-sensitive metalloradical catalyst for a CH4/air mixture (PCH4/Pair = 35, P total = 1 bar) as the reactant at room temperature.25 Sarno et al. conducted electrochemical oxidation of 10 v/v % CH4 to methanol on a Rh single atom/NiO/V2O5 catalyst at 100 °C.26 To oxidize much lower concentration of CH4, a few studies used lean CH4 (approximately 0.2%–1% CH4) as the reactant based on thermal catalysis at relatively high temperature (>100 °C).13,27,28 Carlsson et al. managed to convert low-concentration CH4 (500 vol ppm) into oxygenates over iron molybdate via periodic operating conditions; however, high temperature (>673 K) was required.29 Huang et al. achieved CH4 wet reforming using CH4/H2O at 1:2 ratio catalyzed by ZrO2/Cu(111) at near room temperature.30 Despite this progress, significant effort is required to achieve low-concentration CH4 under ambient conditions.2
A less explored alternative to direct CH4 electrochemical conversion is combining CH4 activation and oxidation. It was reported that CH4 can be activated on the Pt surface under “dynamic potential” conditions by applying a suitable overpotential, followed by oxidization to CO2 using a more positive potential under ambient conditions without adding any oxidants or complicated catalysts.31−33 Pt(100) crystal facets exhibit the highest catalytic performance;31 and polycrystalline Pt is also active for CH4 oxidation to CO2, providing an easier approach for atmospheric CH4 mitigation. The reaction mechanism was briefly investigated on a pure Pt electrode by using pure CH4, but it is not fully understood yet. It is believed that CH4 can be activated by applying suitable adsorption potential to break C–H bond and form C–Hx fragments capable of further reaction with surface-bound oxygen species.32−34 The CH4 activation step, the breakage of the first C–H bond, has been experimentally demonstrated as the rate-determining step for CH4 reforming.35,36 Based on these prior art reports, this approach can potentially extend the electrocatalytic method to Pt particle-coated carbon electrodes capable of converting atmospheric/low-concentration CH4, which has not been reported yet to the best of our knowledge. The use of carbon support can reduce the amount of Pt and electrode cost. Carbon support with the Pt catalyst can work as a gas diffusion electrode to improve CH4 diffusion and enhance the catalytic activity. Although CO2 is the main product from this method instead of liquid products, it provides a few advantages. First, the gas product can be easily separated from the electrolyte solution. Second, unlike conventional combustion, no high temperature, pressure, or CH4 concentration is required, reducing energy consumption and operation cost. Finally, converting atmospheric CH4 to CO2 can contribute significantly to easing global warming.37,38 It has been estimated that atmospheric CH4 concentration (∼1.8 ppm) can be restored back to 750 ppb (preindustrial concentration) by removing approximately 3 Gt of CH4 from air, generating about 8 Gt additional atmospheric CO2, which is only equivalent to a few months of current industrial CO2 emission, but overall such CH4 conversion would eliminate approximately 17% of total radiative forcing.38 CO2 mitigation is relatively easier than CH4 due to the higher atmospheric CO2 concentration, easier separation of CO2, more active nature of CO2, and much more CO2-focused research being carried out.
In this work, we present an experimental study of electro-oxidation of various concentrations of CH4 on platinized Pt (Pt/Pt) and carbon paper electrodes (Pt/CP) under ambient conditions (room temperature and atmospheric pressure). The electrochemical experiments were conducted to determine the catalytic activity of Pt particles for CH4 oxidation on Pt/Pt and Pt/CP electrodes as functions of potential and time, as well as CH4 concentrations. The acidic medium was chosen for the electrolyte solution. This is because Pt catalyst degradation in alkaline medium is severe,39 CH4 electrochemical oxidation activity on Pt is likely to be pH-dependent and, hence, acidic conditions would provide increased activity over time.33
Using the “dynamic potential” method, the C–H bond was broken to form a C–Hx fragment during activation and oxidized with surface oxygen species to CO*, which was further fully oxidized to CO2. Optimal reaction potential and time were obtained. At low CH4 concentrations (0.5 and 1 v/v %), the conversion rate of CH4 to CO2 was estimated. It was observed that Pt/CP exhibited catalytic activity that was approximately 10 times higher than that of Pt/Pt. CH4 electrochemical oxidation was found to be likely independent of its concentration on Pt in the presence of inert balance gas N2(g). This study presents an enhanced understanding of electrochemical methane oxidation at low concentrations under ambient conditions using simple electrocatalysts. It provides a promising and cost-effective approach for the full utilization of methane in the future.
2. Experiment and Materials
2.1. Materials and Equipment
Unless stated otherwise, Ag/AgCl (3 M KCl) (BASi Inc., USA) was used as a reference electrode, platinum mesh was used as the counter electrode (CE), Pt/Pt or Pt/CP was used as the working electrode (WE), and 0.5 M HClO4 was used as an electrolyte. Carbon paper (Sigracet 28AA, Fuel Cell Store, USA) was used as the substrate for the Pt/CP electrodes. H2PtCl6 (>99.9% trace metals basis, Sigma-Aldrich, USA) and HClO4 (70% in water, Thermo Fisher Scientific, USA) were used without further purification. Ultrapure water (>18.2 MΩ cm at 25 °C) was used for all aqueous solutions. CH4 (99.995%, BOC New Zealand) and carbon monoxide (CO) (99.97%, BOC New Zealand) were used as the reactant gases.
A Gamry Interface 5000E potentiostat was used for all electrochemical measurements in this work. Scanning electron microscopy (SEM) was used to characterize the electrode surface after electrodeposition. X-ray diffraction (XRD) was used to characterize the chemical structure of Pt on the electrodes.
Gas chromatography (GC) (SRI Instruments, GC 8610C, USA) was used to measure the gas composition. SRI GC is equipped with a methanizer flame ionization detector and thermal conductivity detector, HayeSep-D, and molecular sieve columns, while H2 is the carrier gas. Standard gases with known concentrations of CH4 (80 ppm and 2.5% mol balanced with air, APC Techsafe, New Zealand) and CO2 (1% vol balanced with air, APC Techsafe, New Zealand) were used to calibrate the GC so that accurate estimates of CH4 and CO2 concentrations in the headspace above the electrolytes could be obtained.
Proton nuclear magnetic resonance (1H NMR) and high-performance liquid chromatography (HPLC) were used to detect any liquid oxygenates in the electrolyte generated during the CH4 electrochemical oxidation process.
2.2. Electrode Preparation
Prior to use, Pt mesh was flame annealed using a butane torch. Carbon paper was sonicated in acetone, methanol, and isopropanol for 1 min each prior to use. The WE and CE were platinized following a modified procedure reported by Boyd et al. to generate Pt electrodes with high surface area.32 It was achieved via Pt electrodeposition using chronoamperometry (CA) by applying −0.3 V for 60 s and −0.25 V for 60 s and repeated for 10 cycles in 0.5 M HClO4 (purged with N2) containing 10 mM H2PtCl6. After deposition, the platinized electrodes were rinsed in ultrapure water and cleaned by cyclic voltammetry (CV) between −0.3 and 1.2 V at 50 mV s–1 until stable CVs were obtained in 0.5 M HClO4 purged with N2(g). The electrochemically active surface area (ECSA) of Pt was estimated from H-adsorption peaks on the CVs. A similar process was used to electrodeposit Pt on the carbon paper electrode.
2.3. CH4 Electroadsorption and Oxidation Experiments
The experimental setup is shown in Figure 1. The electrolyte 0.5 M HClO4 was recirculated between the electrochemical cell and reservoir via peristaltic pumps to ensure that the electrolyte was saturated with reactant gas purged via a gas bubbler. CH4 electrochemical oxidation was conducted by applying an activation potential for a fixed time followed by anodic CV scans under ambient conditions. During the experiment, the headspace gas in the reservoir was continuously transferred to the GC. The headspace gas was sampled 2 min after each CA and CV cycle. Figure 2 shows the electrochemical cell configuration when Pt/CP was used as the WE. The reactant gas was purged to Pt/CP via a graphite gas channel assembled in a stack cell.
Figure 1.
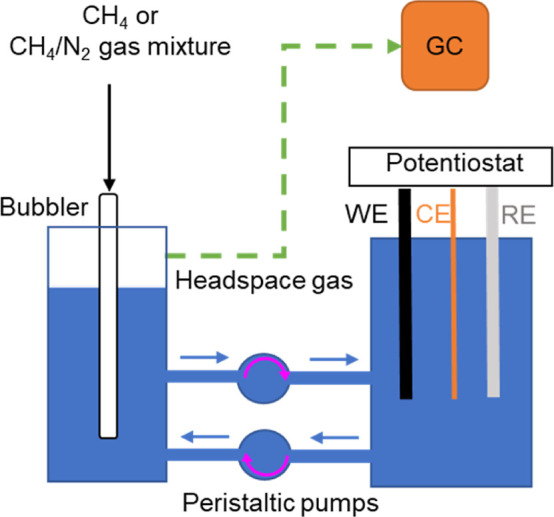
Schematic illustration of the experiment setup for conducting CH4 electrochemical oxidation and measuring the gas composition during the experiment.
Figure 2.
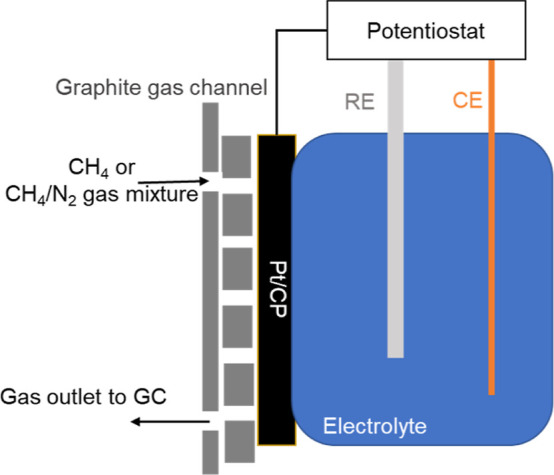
Schematic illustration of the electrochemical cell configuration using Pt/CP as the WE.
The reactant gases include pure CH4, N2, and CH4/N2 gas mixture containing various selected concentrations (0.1, 0.2, 0.5, and 1%) of CH4. Gases were continuously purged at a rate of 50 mL min–1 and controlled by Alicat mass flow controllers. Control experiments were carried out either in the absence of CH4 (with electrochemistry) or with CH4 present but without applying a potential. The charge transferred during CA and from the oxidation peak during the first CV scan after CA was calculated using Gamry Echem Analyst software. The gas composition of samples analyzed by GC was estimated by using PeakSimple software.
Electrochemical oxidation of CO on Pt/Pt and Pt/CP was carried out in a CO-saturated 0.5 M HClO4 solution. CVs were recorded at 50 mV s–1.
3. Results and Discussion
Figure 3 shows the SEM images of a platinized Pt mesh and carbon paper prepared via electrochemical Pt deposition. The stable surface was produced by applying electrochemical CV cycling for consecutive experiments. The SEM images of unmodified Pt mesh and carbon paper are shown in Figure S1. Clearly, the images delineate the Pt particles from the underlying substrates. The surface roughness (and thus the ECSA) of Pt for electrocatalysis has been increased. With similar Pt deposition conditions on both substrates, the Pt/Pt surface is relatively smooth, and the distribution appears uniform with micrometer-sized Pt particles on the surface. Spherical Pt particles with high density were observed on carbon paper, including carbon fibers and graphite flakes. These features can potentially provide more edge and defect structures as active sites on carbon paper, potentially making Pt/CP more active for catalysis than Pt/Pt. Yet, there are areas where carbon support (black in color) is clearly visible underneath bright white Pt particles. XRD patterns of Pt/Pt and Pt/CP were obtained and are shown in Figure S2. It exhibits typical XRD patterns of a polycrystalline Pt face-centered cubic phase and Pt on carbon paper support.40,41
Figure 3.

SEM images, platinized Pt wire mesh [scale bars (a) 10 and (b) 1 μm], and platinized carbon paper [scale bars (c) 10 and (d) 1 μm].
The presence of Pt is also confirmed by the CVs shown in Figure 4. The CVs were obtained after electrochemical CV cycling cleaning. Consistent with the literature, typical polycrystalline Pt electrochemical features were observed, including H adsorption and desorption, double-layer charging, oxide formation, and oxide reduction.42 Similar Pt electrochemistry features can be observed from both Pt/Pt and Pt/CP, but the double-layer current on Pt/CP (∼0.05 mA cm–2) is greater than that on Pt/Pt (∼0.01 mA cm–2), which is attributed to the higher resistance from the carbon substrate still in contact with electrolyte, resulting in a slightly negative oxide reduction and H adsorption potentials of approximately 50 mV. It was reported that surface physicochemical properties and the structure of carbon materials play an important role in the activity of Pt/carbon catalysts. This is because the interaction between carbon and Pt and perimeter of the Pt/C contact interface exposed to electrolyte can modify the physicochemical and electronic structure of Pt and its catalytic activity as a result.43 The carbon support can alter the galvanic potential and raise the electron density in the Pt catalyst particles, which favor the electron transfer at the electrode–electrolyte interface and thus accelerate the electrochemical processes.43
Figure 4.
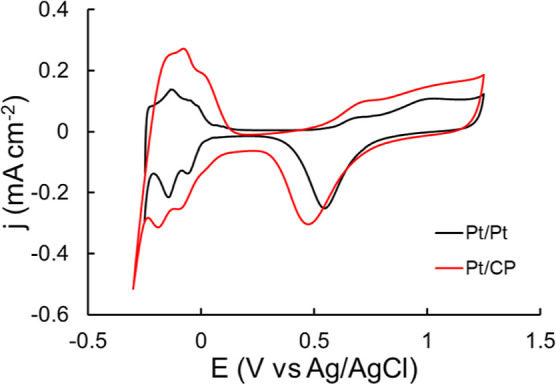
CVs of Pt/Pt and Pt/CP in 0.5 M HClO4 degassed with N2. Scan rate = 50 mV s–1.
3.1. Electrochemical Oxidation of CH4
The fabricated electrodes Pt/Pt and Pt/CP were used as the WE for CH4 electrochemical activation and oxidation.
It has been previously reported that CH4 oxidation can be achieved under ambient conditions on Pt through CH4 activation by applying a suitable potential and followed by CV scans to a more positive potential to remove CH4 oxidation intermediates on the electrode surface. This “dynamic potential” approach combines a longer (ca. 30 min), relatively low potential activation phase at an applied potential of Eapp = 0.3 or 0.4 V [versus reversible hydrogen electrode (RHE)] and a shorter (approximately 1 min) and more positive oxidation potential with an oxidation peak at Ep = 0.6–0.8 V vs RHE.32,33
According to Figures 4 and 5, a series of activation potentials in the non-Faradaic region (∼0.1–0.3 V vs Ag/AgCl) where no redox processes occur can be chosen to find the optimal activation potential. Additionally, the non-Faradaic region of Pt may be dependent on the structure, morphologies, and its electrochemical response, which can lead to slightly different optimal activation potential for different Pt-based electrodes. For instance, the non-Faradaic region of the Pt/CP CVs shifted to a slightly more positive potential compared to Pt/Pt CVs, resulting in a corresponding shift in the CH4 activation potential, which is discussed in detail in Section 3.2.
Figure 5.

(a) CVs of Pt/Pt under N2 and CH4 after 30 min adsorption at 0.15 V in 0.5 M HClO4; (b) CVs of Pt/CP under N2 and CH4 after 30 min adsorption at 0.3 V in 0.5 M HClO4. Scan rate = 50 mV s–1.
A sequence of CA experiments was conducted by applying an activation potential of 0.15 V for Pt/Pt and 0.3 V for Pt/CP at different activation times. It was found that the oxidation peak charge density increases (∼5.5 μC cm–2 at 2 min) with activation time, and it reaches a plateau (∼16 μC cm–2) between 30 and 45 min (Figure S3). Considering that the oxidation peak densities obtained at 30 and 45 min are similar, 30 min duration was chosen in this study for the electrochemical activation to reduce energy consumption.
Figure S4 shows the current density versus time recorded in CH4-saturated 0.5 M HClO4 at Eapp = 0.15 and 0.3 V, respectively, for 30 min for the Pt/Pt (Figure S4a) and Pt/CP (Figure S4b) electrodes. The current densities were obtained after subtracting the current density from the control experiment by using N2 in place of CH4. The positive current corresponds to CH4 activation and oxidation to intermediate species. From the chronoamperometry, the current density did not decrease after reaching steady state, suggesting that CO poisoning is not an issue during the 30 min electrochemical activation process.
Figure 5 shows the first two CVs obtained directly after CA. For both electrodes, an oxidation peak at Ep of approximately 0.5 V appeared on the first anodic CV scan and disappeared on the second CV scan. This phenomenon was not observed in the control experiment under N2 in the absence of CH4. The oxidation peak disappeared on the second CV scan for the Pt/Pt electrode and decreased for the Pt/CP electrode. Repeated CV scans were required to eliminate the oxidation peak completely for the Pt/CP electrode.
The observation for the Pt/Pt electrode is consistent with previous studies of CH4 electrochemical oxidation, indicating that all intermediate species from activated CH4 are oxidized in the first CV scan.32 The CH4 activation and oxidation mechanisms have been briefly investigated by several groups. It was reported that CH4 decomposes at the Pt surface and forms *CO with surface oxygen species at a suitable oxidation potential, which is then oxidized with adsorbed H2O and/or O to form CO2 in the following anodic CV scan.31−33
Although most of the studies reported that the electrochemical oxidation of CO adsorbed on a smooth electrode is usually achieved in one CV cycle,44,45 this is not the case for the Pt/CP electrode. We found that repeated CV cycles were required to eliminate the oxidation peak (at ∼0.5 V) at the Pt/CP electrode, or in other words, to completely oxidize absorbed CO. There are two possible causes behind this observation. First, this is attributed to oxidation of CO adsorbed in different pores of the porous materials in repeated CVs.46 Second, the CO2 generated from the previous anodic CV scan is reduced to CO, and/or more CH4 can be adsorbed to the surface and oxidized by the cathodic CV scan and reoxidized CO2. Carbon paper is a porous material; therefore, it is not surprising to observe such diffusion-related phenomena similar to the one reported for Pt-modified zeolite by Mojović et al.46 In addition, compared to the oxidation peak obtained from the Pt/Pt electrode, the oxidation peak from the Pt/CP electrode is broader, which is attributed to the existence of two types of CO* adsorbed species (weakly and strongly adsorbed CO*) on polycrystalline Pt supported by a carbon electrode, which was also reported in the literature.47 CO electrochemical oxidation was conducted on Pt/Pt and Pt/CP, as shown in Figure 6. Two oxidation peaks at Ep = 0.57 and 0.83 V were seen on the first CV scan on Pt/Pt, which are assigned to the oxidation of adsorbed CO on different facets of the polycrystalline Pt surface.48 These oxidation peaks almost completely disappeared in successive CV scans, indicating that adsorbed CO was almost completely removed in the first CV cycle. However, only one broad and asymmetric oxidation peak at Ep = 0.89 V was observed on the first CV scan using Pt/CP as the WE, which disappeared on the second CV scan. A new oxidation peak at Ep = 0.57 V on the second CV scan for Pt/CP was observed, which gradually decreased in the later CV scans, yet it did not disappear completely. This oxidation peak is attributed to the oxidation of the absorbed CO*. This phenomenon is similar to that observed for CH4 electrochemical oxidation on Pt/CP. It suggests that adsorbed CO* can be continuously oxidized on porous Pt/CP surface. Additionally, it indicates that the platinized electrode can avoid CO* poisoning by applying an anodic CV scan to oxidize adsorbed CO*. The “dynamic potential” method used in this study can regenerate the active surface during every cycle to avoid CO poison.
Figure 6.

CVs (1st CV scan: black line, 2nd: red, 3rd: green, and 4th: purple) of CO electrochemical oxidation on (a) Pt/Pt and (b) Pt/CP in CO-saturated 0.5 M HClO4. Scan rate = 50 mV s–1.
The adsorbed species on the Pt/Pt surface were quantified by integrating charge transferred to them based on the oxidation peak on the first CV scan. The ratio of charge transferred during activation (Qa) and oxidation peak from the CV (Qo) is approximately 3, which is consistent with 6 electrons transferred during CA (eq 1) and two electrons transferred during oxidation (eq 2).32 Hence, we concluded that all of the activated CH4 is converted to CO2, which was also confirmed by GC.
| 1 |
| 2 |
On the other hand, for Pt/CP, the ratio of charge transferred during the activation and oxidation peak from the CV (Qa/Qo) is approximately 50, which is an order of magnitude higher than that on the Pt/Pt electrode. This could be due to a few factors that are discussed below.
For example, side reactions on both Pt and the carbon surface, such as trace amount of O2 adsorption (O2 + 2H2O + 4e– = 4OH–, E = 0.401 V vs standard hydrogen electrode),49 which is close to the CH4 adsorption potential, can compete with CH4 adsorption. Activated CH4 on the surface can react with reactive oxygen species generated from oxygen reduction reaction to form oxygenates instead of CO2.18,50,51 To assess production of any liquid oxygenate generation during the activation, a large Pt/CP (4 cm × 4 cm) with an ECSA of ∼200 cm2 was used as the WE, the electrochemical activation potential was applied for 15 h, and the electrolyte was used for H NMR and HPLC measurement. There were no clear peaks observed in 1H NMR spectra, but there was a small peak assigned to formic acid (0.5 mM HCOOH from the calibration curve) identified in HPLC chromatogram (Figure S5), suggesting that oxygenate was generated from CH4 electrochemical oxidation on Pt/CP. However, there were no products detected in the liquid phase after 1 cycle of reaction using Pt/CP, which is likely due to the extreme low concentration.
A significant difference was observed in charge density transfer between the Pt/Pt and Pt/CP electrodes of approximately 15 and 170 μC cm–2, respectively, during the oxidation phase, after applying the optimal potentials of 0.15 and 0.3 V, respectively, for the same activation time. The much higher catalytic activity evidenced by such higher current in the case of Pt/CP could be partially due to the potential difference. Although at the same potential of 0.15 V, the Pt/CP charge transfer density of approximately 120 μC cm–2 is still much higher than that for Pt/Pt. This improved catalytic activity could be attributed to the carbon support providing higher pore volume, altering the electronic structure of Pt to enhance the electron transfer, providing unique perimeter interface between Pt particles and carbon support and potentially enhancing the diffusion of CH4 to Pt via spillover from the hydrophobic carbon surface acting as a collector of the CH4 for the Pt active sites.52,53
3.2. Electrocatalyst Performance as a Function of Activation Potential
According to previous studies, the electrocatalytic activation of CH4 depends on applied potentials. It is also affected by the catalyst morphology and the nature of electrolyte used. Therefore, a series of feasible potentials were chosen to activate CH4 in this work. The electrocatalytic efficiency is compared according to the oxidation peak charge density from the first anodic CV scan after electrochemical activation. Obtained data show that 0.15 and 0.3 V are the optimal potentials for CH4 adsorption on Pt/Pt and Pt/CP, respectively, for holding the potential for 30 min (Figure 7). Consistent with previous studies that the applied activation potential and time affect the charge transferred during the oxidation process (the generation of oxidation products), it suggests that the CH4 activation to break the first C–H bond in the chronoamperometry process is likely the rate-determining step.32,34
Figure 7.

Oxidation peak charge density calculated from the first CV scan (50 mV s–1) at selected activation potentials (a) Pt/Pt and (b) Pt/CP for 30 min in CH4-saturated 0.5 M HClO4.
Overall, Pt/CP exhibited a much higher catalytic activity than Pt/Pt. The oxidative performance of these materials could be correlated with the electronic properties of Pt particles interacting with different supports.54 As reported by Mehdi et al., according to studies of vibration frequencies, Mulliken populations, charge transfer, charge density differences, and density of states, carbon support enhanced the back-donation of electrons to CO* via donating electrons to Pt, resulting in stronger CO* binding to Pt and enhanced CO* activation as discussed above.55 To further understand the role of carbon support for Pt catalysis, we performed the electrochemical oxidation of CO catalyzed by Pt/Pt and Pt/CP in a CO-saturated 0.5 M HClO4 solution as mentioned above. Figure 8 compares the first two CV scans obtained on two of these electrodes. As discussed in Section 3.1, different oxidation peaks were observed on the Pt/Pt and Pt/CP electrodes. The oxidation peak on Pt/CP is approximately 60 mV more positive than that on Pt/Pt, suggesting that the adsorbed CO* on the Pt/CP surface is more stable than that on the Pt/Pt surface.56 In addition, the charge density calculated from the CO oxidation peak on Pt/CP is 2 times higher than that on Pt/Pt, suggesting that Pt/CP is more efficient for CO oxidation. These results support the hypothesis that Pt/CP can enhance CO conversion and improve CH4 activation and overall oxidation as a result.
Figure 8.
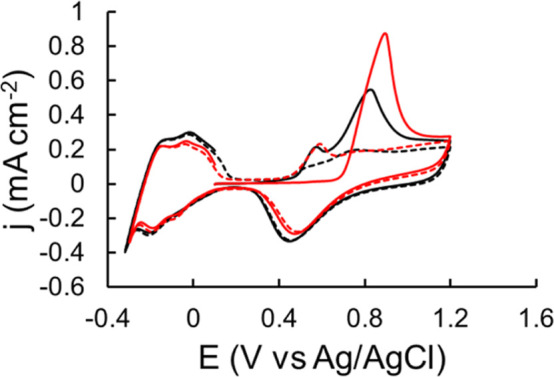
CVs of CO electrochemical oxidation on Pt/Pt (black line: 1st CV scan; black dashed line: 2nd CV scan) and Pt/CP (red line: 1st CV scan; red dashed line: 2nd CV scan) in CO-saturated 0.5 M HClO4. Scan rate = 50 mV s–1.
This is consistent with previous studies that employing favorable support materials can provide a synergistic effect of enhanced electron transfer and stronger metal anchoring, leading to higher electrocatalytic activities.57
For both electrocatalysts, the catalytic activity gradually increases by shifting the potential to more positive values but decreases for both electrocatalysts when the potential is about 100 mV more positive than the optimal potential. This is attributed to the competing adsorption of H2O/O2 molecules at higher potential and/or the decreased coverage of CO* as it can directly be oxidized to CO2 at higher potential.
3.3. Electro-oxidation of Low-Concentration CH4 Balanced with N2
The experiments discussed in Sections 3.1 and 3.2 deal with pure CH4. However, it is essential to be able to oxidize CH4 at low concentrations under ambient conditions for practical applications, for instance, lean CH4 from mining, CH4 generated from farms, and ultimately, atmospheric CH4. Hence, we used CH4/N2 gas mixtures with different yet low concentrations of CH4 for electro-oxidation on Pt/Pt and Pt/CP as a model study for further study on CH4/air gas mixture.
The oxidation charge density obtained from the first CV scan after applying 0.15 V for 30 min obtained using a CH4/N2 gas mixture with different concentrations of CH4 on the Pt/Pt electrode is shown in Figure 9a. Evidently, there is no significant difference in oxidation peak charge density as the CH4 concentration changes compared to that of pure CH4. The same phenomenon is recorded on the Pt/CP electrode with different concentrations of CH4 in N2, as shown in Figure 9b. Pt/CP displayed much higher catalytic performance than the Pt/Pt surface for a low concentration of CH4 in N2. A similar trend is observed from CA for selected concentration of CH4 during the activation phase. Despite the fact that we cannot fully eliminate the possibility that the Pt active site is saturated with CH4 at low concentration, it can be seen from Figure S3 that the oxidation peak charge density increases with electrochemical activation time and reaches the plateau at approximately 2700 s in solution saturated with pure CH4, so the surface is unlikely saturated with low-concentration CH4 at 1800 s. This indicates that the surface coverage with adsorbed species is independent of CH4 concentrations in the CH4/N2 gas mixture at the same potential for the same period.
Figure 9.

(a) Plot of charge density from the oxidation peak on the first CV scan (scan rate = 50 mV s–1) after applying 0.15 V for 30 min on the Pt/Pt surface; (b) plot of charge density from the oxidation peak on the first CV scan after applying 0.3 V for 30 min on the Pt/CP surface for CH4/N2 gas mixtures with various CH4 concentrations.
Abbasi et al. described a similar phenomenon that the ignition, extinction, and fractional conversion of CH4 via catalytic oxidation on Pt are independent of CH4 concentration, which was because Pt is not sensitive to water produced during thermal oxidation.58 Thus, the reaction rate may be mainly dependent on the binding affinity between the catalyst and the intermediates. As shown in Figure 8, Pt/CP exhibits higher capability of CO adsorption and oxidation, resulting in higher CH4 activation/oxidation performance than Pt/Pt. Our finding is consistent with the calculation reported by Boyd et al. that CH4 activation can be influenced by the binding of CO*.32
Furthermore, N2 is effectively an inert gas in our system, and it will not affect CH4 adsorption on Pt active sites. This has been shown by several studies in which N2 has to be activated to be adsorbed on Pt at room temperature or only at low temperature.43,59 It was also reported that N2 does not chemisorb on the nondefective Pt(111) surface, which is the main phase of Pt observed in the XRD pattern (Figure S1). Both experimental and theoretical results reported by Tripa et al. showed that chemisorption of N2 on step sites at low temperature (88 K) and the predicted binding energy of N2 on these sites is very small, and other adsorbates with higher binding energy displace N2.60 Therefore, N2 adsorption is not expected to compete with CH4 adsorption on Pt at room temperature in this work.
To monitor the products generated from CH4 electrochemical oxidation, GC was used to measure the composition of the headspace gas before and after the reaction. As expected, only CO2 was detected by GC in the headspace gas during the CH4 oxidation. Over the course of the experiment, we observed the expected decrease in CH4 concentration and increase in the CO2 concentration.
For the Pt/Pt electrode, 1 v/v % CH4 diluted with N2 was used as the reactant in order to reduce the experimental error. As shown in Figure S6a, a CH4 peak was eluted at 0.85 and 4.73 min through two columns, while CO2 was detected at 5.03 min. There was approximately 10 ppm of CO2 on average measured from the peak area after 1 cycle of electrochemical oxidation. This CO2 peak area increased by repeating the electrochemical oxidation for three cycles, as shown in Figure S6b, and the CO2 peak is clearly more pronounced and estimated to be approximately 30 ppm. The consumption of CH4 can be estimated from the decrease of the CH4 peak after the reaction. For each electrochemical cycle, approximately 10 ppm mol CH4 in average was consumed, corresponding to 8.3 × 10–9 mol L–1 s–1 m–2 CH4, with 7.8 × 10–9 mol L–1 s–1 m–2 CO2 generated, giving a conversion rate of approximately 94% for the Pt/Pt electrode. Two control experiments were carried out. In the absence of CH4, no detectable change of CH4 concentration and CO2 was recorded, as shown in Figure S7.
CO2 generation was also detected from CH4 oxidation on Pt/CP by GC. As shown in Figure S8, 0.5% CH4 balanced with N2 was used as the reactant for CH4 electrochemical oxidation, and the CO2 peak increased after one cycle. It should be noted that there was some CO2 residue in the sealed reactor before the reaction, and only the difference of peak area for CO2 and CH4 before and after the reaction was used for calculation. As mentioned in Section 2.3, a 0.5% CH4/N2 gas mixture was directly purged to the Pt/CP electrode instead of purging the electrolyte, with the Pt/CP electrode operating as a gas diffusion electrode. In this case, the post-reaction CO2 concentration was approximately 5.3 × 10–8 mol L–1 s–1 m–2 per cycle, and CH4 concentration decreased by 7.5 × 10–8 mol L–1 s–1 m–2, giving a conversion rate of approximately 70% to CO2. This is lower conversion to CO2 compared to 1% CH4 in N2 conversion on the Pt/Pt surface, which is consistent with the high ratio of Qa/Qo for Pt/CP discussed in Section 3.1. Although CO2 is the only product detected from the headspace during reaction using GC, more CH4 consumed than CO2 generated might be attributed to CH4 partial oxidation to oxygenates in the presence of reactive oxygen species, as formic acid was detected in the electrolyte after a long reaction time.
3.4. Stability Test of the Platinized Electrodes for CH4 Electrochemical Oxidation
Pt/CP was reused 30 times to oxidize 0.5% CH4 balanced with N2 to test its stability. As shown in Figure 10, the oxidation peak charge density remained relatively stable over 30 cycles (CA and CV, each 30 min), suggesting that Pt/CP has reasonably good stability and, thus, is a promising candidate material as a gas diffusion electrode for electro-oxidation of low-concentration CH4. The headspace gas composition change during repeated cycles was recorded by GC (Figure S9). Decrease of CH4 concentration and increase of CO2 concentration were observed.
Figure 10.

Plot of q vs number of cycles for Pt/CP using 0.5% CH4 in N2 as the reactant and applying 0.3 V for 30 min.
To further understand the stability of Pt/CP, SEM, XRD, and CV results were collected and compared. Figure 11a shows CVs of Pt/CP after the 1st and 30th cycles of CH4 electrochemical oxidation using 0.5% CH4 in N2 as the reactant. There are no clear changes in terms of the Pt features and activated CH4 oxidation peak. Meanwhile, X-ray diffractograms (Figure 11b) and SEM images (Figure 11c,d) were collected for Pt/CP as prepared and after 30 cycles of CH4 electrochemical oxidation.
Figure 11.

(a) CVs of Pt/CP after the 1st cycle (black) and 30 cycles (red); (b) XRD patterns and inlet is the zoom-in from 30 to 100°; (c,d) SEM images [scale bar 1 μm] of Pt/CP at a scan rate of 50 mV s–1, as prepared and after 30 cycles of electrochemical activation and oxidation in 0.5 M HClO4 with 0.5% CH4 balanced with N2.
There is no clear observed difference in the morphology and crystal structure for Pt/CP before and after the stability test. However, for Pt/Pt, the SEM images (Figure S10) show a smoother surface with some pinholes compared to Pt/Pt as prepared, suggesting that the surface has experienced mild restructure and lost some Pt particles during the electrochemical reaction. In the case of Pt/CP, there was no clear change from CVs and XRD. These results suggest that Pt/CP electrodes are stable for 30 cycles of CH4 electrochemical reactions. Additionally, according to the results shown in Figures S4, 7, and 8, the current density during 30 min activation did not decrease, CO can be oxidized via anodic CV scan, and the surface can be regenerated; CO poisoning is not an issue of CH4 electrochemical oxidation in this study.
4. Conclusions
Through a combination of various experiments, the electrochemical oxidation of pure CH4 and low-concentration CH4 in N2 balancing gas mixtures on both Pt/Pt and Pt/CP under ambient conditions has been investigated. The results indicated that electro-oxidation of CH4 on Pt/Pt and Pt/CP is dependent on the applied potential and time but independent of the CH4 concentrations within the range studied using N2 as the balance gas. Pt/CP tends to exhibit higher catalytic performance than Pt/Pt, which could be attributed to the altered electronic structure due to the presence of a carbon support and hydrophobic carbon support surface, which enhances gas diffusion to the active sites. Both Pt/Pt and Pt/CP can catalyze conversion of low-concentration CH4. The stability test suggests that Pt/CP is a cost-effective electrocatalyst with high activity and stability for CH4 activation and oxidation. These observations indicated that it is possible to reduce the usage of Pt without scarifying the catalytic activity. It was found that while >90% CH4 was converted to CO2 on Pt/Pt, approximately 70% of CH4 was converted to CO2 on Pt/CP, and formic acid was detected after long reaction time. Therefore, future work will be essential to understand the impact of other reactions during CH4 activation on Pt/CP with a low-concentration CH4/air gas mixture used as the reactant.
Acknowledgments
We acknowledge the funding support from the New Zealand Agricultural Greenhouse Gas Research Centre. We also would like to express appreciation to Dr. Kim Eccleston for his constructive comments and suggestions to refine the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c06665.
Additional SEM images and XRD results of Pt/Pt and Pt/CP, chronoamperometry results of methane activation on Pt/Pt and Pt/CP, GC results of CH4 consumption and CO2 generation, and CV results for Pt/Pt stability testing (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Control methane to slow global warming-fast. Nature 2021, 596, 461. 10.1038/d41586-021-02287-y [DOI] [PubMed] [Google Scholar]
- Abernethy S.; Kessler M. I.; Jackson R. B. Assessing the potential benefits of methane oxidation technologies using a concentration-based framework. Environ. Res. Lett. 2023, 18 (9), 094064. 10.1088/1748-9326/acf603. [DOI] [Google Scholar]
- Dean J. F.; Middelburg J. J.; Röckmann T.; Aerts R.; Blauw L. G.; Egger M.; Jetten M. S. M.; de Jong A. E. E.; Meisel O. H.; Rasigraf O.; Slomp C. P.; in’t Zandt M. H.; Dolman A. J. Methane feedbacks to the global climate system in a warmer world. Rev. Geophys. 2018, 56 (1), 207–250. 10.1002/2017RG000559. [DOI] [Google Scholar]
- IEA . Global Methane Tracker; Licence: CC BY 4.0, Paris, 2022. https://www.iea.org/reports/global-methane-tracker-2022.
- Vass A.; Mul G.; Katsoukis G.; Altomare M. Challenges in the selective electrochemical oxidation of methane: too early to surrender. Curr. Opin. Electrochem. 2024, 47, 101558. 10.1016/j.coelec.2024.101558. [DOI] [Google Scholar]
- IEA . Methane Tracker; Licence: CC BY 4.0, Paris, 2021. https://www.iea.org/reports/methane-tracker-2021.
- Meng X.; Cui X.; Rajan N. P.; Yu L.; Deng D.; Bao X. Direct methane conversion under mild condition by thermo-electro-or photocatalysis. Chem 2019, 5 (9), 2296–2325. 10.1016/j.chempr.2019.05.008. [DOI] [Google Scholar]
- Verma G.; Chetri J. K.; Reddy K. R. Spatial variation of methane oxidation and carbon dioxide sequestration in landfill biogeochemical cover. Environ. Technol. 2024, 1–17. 10.1080/09593330.2024.2372052. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Wang H.; Liu Y.; Liu P.; Zhu B.; Zheng Y.; Li J.; Chistoserdova L.; Ren Z. J.; Zhao F. Electrochemically coupled CH4 and CO2 consumption driven by microbial processes. Nat. Commun. 2024, 15 (1), 3097. 10.1038/s41467-024-47445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher Shah M. S. A.; Oh C.; Park H.; Hwang Y. J.; Ma M.; Park J. H. Catalytic oxidation of methane to oxygenated products: recent advancements and prospects for electrocatalytic and photocatalytic conversion at low temperatures. Advanced Science 2020, 7 (23), 2001946. 10.1002/advs.202001946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy H. VIII. Some new experiments and observations on the combustion of gaseous mixtures, with an account of a method of preserving a continued light in mixtures of inflammable gases and air without flame. Philos. Trans. R. Soc. London 1817, 107, 77–85. 10.1098/rstl.1817.0009. [DOI] [Google Scholar]
- Tang P.; Zhu Q.; Wu Z.; Ma D. Methane activation: the past and future. Energy Environ. Sci. 2014, 7 (8), 2580–2591. 10.1039/C4EE00604F. [DOI] [Google Scholar]
- Yuan S.; Li Y.; Peng J.; Questell-Santiago Y. M.; Akkiraju K.; Giordano L.; Zheng D. J.; Bagi S.; Román-Leshkov Y.; Shao-Horn Y. Conversion of methane into liquid fuels—bridging thermal catalysis with electrocatalysis. Adv. Energy Mater. 2020, 10 (40), 2002154. 10.1002/aenm.202002154. [DOI] [Google Scholar]
- Tarasov A.; Root N.; Lebedeva O.; Kultin D.; Kiwi-Minsker L.; Kustov L. Platinum nanoparticles on sintered metal fibers are efficient structured catalysts in partial methane oxidation into synthesis Gas. ACS Omega 2020, 5 (10), 5078–5084. 10.1021/acsomega.9b04020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Wu B.; Huang M.; Lu Z.; Li J.; Zhong L.; Sun Y. IrFe/ZSM-5 synergistic catalyst for selective oxidation of methane to formic acid. Energy Fuels 2021, 35 (5), 4418–4427. 10.1021/acs.energyfuels.0c04198. [DOI] [Google Scholar]
- Zhao G.; Adesina A.; Kennedy E.; Stockenhuber M. Formation of surface oxygen species and the conversion of methane to value-added products with N2O as oxidant over Fe-ferrierite catalysts. ACS Catal. 2020, 10 (2), 1406–1416. 10.1021/acscatal.9b03466. [DOI] [Google Scholar]
- Hu D.; Ordomsky V. V.; Khodakov A. Y. Major routes in the photocatalytic methane conversion into chemicals and fuels under mild conditions. Appl. Catal., B 2021, 286, 119913. 10.1016/j.apcatb.2021.119913. [DOI] [Google Scholar]
- Song H.; Meng X.; Wang S.; Zhou W.; Wang X.; Kako T.; Ye J. Direct and selective photocatalytic oxidation of CH4 to oxygenates with O2 on cocatalysts/ZnO at room temperature in water. J. Am. Chem. Soc. 2019, 141 (51), 20507–20515. 10.1021/jacs.9b11440. [DOI] [PubMed] [Google Scholar]
- Xie J.; Jin R.; Li A.; Bi Y.; Ruan Q.; Deng Y.; Zhang Y.; Yao S.; Sankar G.; Ma D.; et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 2018, 1 (11), 889–896. 10.1038/s41929-018-0170-x. [DOI] [Google Scholar]
- Prajapati A.; Collins B. A.; Goodpaster J. D.; Singh M. R. Fundamental insight into electrochemical oxidation of methane towards methanol on transition metal oxides. Proc. Natl. Acad. Sci. U.S.A. 2021, 118 (8), e2023233118 10.1073/pnas.2023233118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Su C.; Wu Y.; Ran R.; Shao Z. Progress in solid oxide fuel cells with nickel-based anodes operating on methane and related fuels. Chem. Rev. 2013, 113 (10), 8104–8151. 10.1021/cr300491e. [DOI] [PubMed] [Google Scholar]
- Park S.; Vohs J. M.; Gorte R. J. Direct oxidation of hydrocarbons in a solid-oxide fuel cell. Nature 2000, 404 (6775), 265–267. 10.1038/35005040. [DOI] [PubMed] [Google Scholar]
- Jiao Y.; Wang L.; Zhang L.; An W.; Wang W.; Zhou W.; Tadé M. O.; Shao Z.; Bai J.; Li S.-D. Direct operation of solid oxide fuel cells on low-concentration oxygen-bearing coal-bed methane with high stability. Energy Fuels 2018, 32 (4), 4547–4558. 10.1021/acs.energyfuels.7b02968. [DOI] [Google Scholar]
- Kim J.; Kim J. H.; Oh C.; Yun H.; Lee E.; Oh H.-S.; Park J. H.; Hwang Y. J. Electro-assisted methane oxidation to formic acid via in-situ cathodically generated H2O2 under ambient conditions. Nat. Commun. 2023, 14 (1), 4704. 10.1038/s41467-023-40415-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natinsky B. S.; Lu S.; Copeland E. D.; Quintana J. C.; Liu C. Solution catalytic cycle of incompatible steps for ambient air oxidation of methane to methanol. ACS Cent. Sci. 2019, 5 (9), 1584–1590. 10.1021/acscentsci.9b00625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarno M.; Ponticorvo E.; Funicello N.; De Pasquale S. Methane electrochemical oxidation at low temperature on Rh single atom/NiO/V2O5 nanocomposite. Appl. Catal., A 2020, 603, 117746. 10.1016/j.apcata.2020.117746. [DOI] [Google Scholar]
- Li K.; Liu K.; Xu D.; Ni H.; Shen F.; Chen T.; Guan B.; Zhan R.; Huang Z.; Lin H. Lean methane oxidation over Co3O4/Ce0.75Zr0.25 catalysts at low-temperature: synergetic effect of catalysis and electric field. Chem. Eng. J. 2019, 369, 660–671. 10.1016/j.cej.2019.03.059. [DOI] [Google Scholar]
- Li S.; Zhang Y.; Wang Z.; Du W.; Zhu G. Morphological effect of CeO2 catalysts on their catalytic performance in lean methane combustion. Chem. Lett. 2020, 49 (5), 461–464. 10.1246/cl.200049. [DOI] [Google Scholar]
- Carlsson P. A.; Jing D.; Skoglundh M. Controlling selectivity in direct conversion of methane into formaldehyde/methanol over iron molybdate via periodic operation conditions. Energy Fuels 2012, 26 (3), 1984–1987. 10.1021/ef300007n. [DOI] [Google Scholar]
- Huang E.; Rui N.; Rosales R.; Liu P.; Rodriguez J. A. Activation and conversion of methane to syngas over ZrO2/Cu(111) catalysts near room temperature. J. Am. Chem. Soc. 2023, 145 (15), 8326–8331. 10.1021/jacs.3c01980. [DOI] [PubMed] [Google Scholar]
- Ma H. B.; Sheng T.; Yu W. S.; Ye J. Y.; Wan L. Y.; Tian N.; Sun S. G.; Zhou Z. Y. High catalytic activity of Pt (100) for CH4 electrochemical conversion. ACS Catal. 2019, 9 (11), 10159–10165. 10.1021/acscatal.9b02738. [DOI] [Google Scholar]
- Boyd M. J.; Latimer A. A.; Dickens C. F.; Nielander A. C.; Hahn C.; Nørskov J. K.; Higgins D. C.; Jaramillo T. F. Electro-oxidation of methane on platinum under ambient conditions. ACS Catal. 2019, 9 (8), 7578–7587. 10.1021/acscatal.9b01207. [DOI] [Google Scholar]
- Gurses S. M.; Kronawitter C. X. Electrochemistry of the interaction of methane with platinum at room temperature investigated through operando FTIR spectroscopy and voltammetry. J. Phys. Chem. C 2021, 125 (5), 2944–2955. 10.1021/acs.jpcc.0c09433. [DOI] [Google Scholar]
- Cant N. W.; Lukey C. A.; Nelson P. F.; Tyler R. J. The rate controlling step in the oxidative coupling of methane over a lithium-promoted magnesium oxide catalyst. J. Chem. Soc. Chem. Commun. 1988, (12), 766–768. 10.1039/c39880000766. [DOI] [Google Scholar]
- Bartholomew C. H.; Farrauto R. J.. Hydrogen Production and Synthesis Gas Reactions: Fundamentals of Industrial Catalytic Processes, 2nd ed.; John Wiley & Sons, Inc., 2005; Chapter 6, pp 339–486. [Google Scholar]
- Wei J.; Iglesia E. Isotopic and kinetic assessment of the mechanism of reactions of CH4 with CO2 or H2O to form synthesis gas and carbon on nickel catalysts. J. Catal. 2004, 224 (2), 370–383. 10.1016/j.jcat.2004.02.032. [DOI] [Google Scholar]
- Sirigina D. S. S. S.; Goel A.; Nazir S. M. Process concepts and analysis for co-removing methane and carbon dioxide from the atmosphere. Sci. Rep. 2023, 13 (1), 17290. 10.1038/s41598-023-44582-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R. B.; Solomon E.; Canadell J.; Cargnello M.; Field C. Methane removal and atmospheric restoration. Nat Sustainability 2019, 2 (6), 436–438. 10.1038/s41893-019-0299-x. [DOI] [Google Scholar]
- Zadick A.; Dubau L.; Sergent N.; Berthomé G.; Chatenet M. Huge instability of Pt/C catalysts in alkaline medium. ACS Catal. 2015, 5 (8), 4819–4824. 10.1021/acscatal.5b01037. [DOI] [Google Scholar]
- Ji W.; Qi W.; Tang S.; Peng H.; Li S. Hydrothermal synthesis of ultrasmall Pt nanoparticles as highly active electrocatalysts for methanol oxidation. Nanomaterials 2015, 5 (4), 2203–2211. 10.3390/nano5042203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Wang X.; Tan M.; Liu H.; Ma Q.; Xu Q.; Pollet B. G.; Su H. Electrodeposited platinum with various morphologies on carbon paper as efficient and durable self-supporting electrode for methanol and ammonia oxidation reactions. Int. J. Hydrogen Energy 2023, 48 (7), 2617–2627. 10.1016/j.ijhydene.2022.10.157. [DOI] [Google Scholar]
- Climent V.; Feliu J. M. Thirty years of platinum single crystal electrochemistry. J. Solid State Electrochem. 2011, 15 (7–8), 1297–1315. 10.1007/s10008-011-1372-1. [DOI] [Google Scholar]
- Wang Y. J.; Zhao N.; Fang B.; Li H.; Bi X. T.; Wang H. Carbon-supported Pt-based alloy electrocatalysts for the oxygen reduction reaction in polymer electrolyte membrane fuel cells: particle size, shape, and composition manipulation and their impact to activity. Chem. Rev. 2015, 115 (9), 3433–3467. 10.1021/cr500519c. [DOI] [PubMed] [Google Scholar]
- García G.; Koper M. T. Stripping voltammetry of carbon monoxide oxidation on stepped platinum single-crystal electrodes in alkaline solution. Phys. Chem. Chem. Phys. 2008, 10 (25), 3802–3811. 10.1039/b803503m. [DOI] [PubMed] [Google Scholar]
- Ciapina E. G.; Santos S. F.; Gonzalez E. R. The electrooxidation of carbon monoxide on unsupported Pt agglomerates. J. Electroanal. Chem. 2010, 644 (2), 132–143. 10.1016/j.jelechem.2009.09.022. [DOI] [Google Scholar]
- Mojović Z.; Banković P.; Jović-Jovičić N.; Rabi-Stanković A. A.; Milutinović-Nikolić A.; Jovanović D. Carbon monoxide electrooxidation on Pt and PtRu modified zeolite X. J. Porous Mater. 2012, 19, 695–703. 10.1007/s10934-011-9521-6. [DOI] [Google Scholar]
- Sethuraman V. A.; Lakshmanan B.; Weidner J. W. Quantifying desorption and rearrangement rates of carbon monoxide on a PEM fuel cell electrode. Electrochim. Acta 2009, 54 (23), 5492–5499. 10.1016/j.electacta.2009.04.049. [DOI] [Google Scholar]
- Couto A.; Rincón A.; Pérez M. C.; Gutiérrez C. Adsorption and electrooxidation of carbon monoxide on polycrystalline platinum at pH 0.3–13. Electrochim. Acta 2001, 46 (9), 1285–1296. 10.1016/S0013-4686(00)00714-3. [DOI] [Google Scholar]
- Bard A. J.; Faulkner L. R.; White H. S.. Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons, 2022. [Google Scholar]
- Zhu S.; Li X.; Pan Z.; Jiao X.; Zheng K.; Li L.; Shao W.; Zu X.; Hu J.; Zhu J.; et al. Efficient photooxidation of methane to liquid oxygenates over ZnO nanosheets at atmospheric pressure and near room temperature. Nano Lett. 2021, 21 (9), 4122–4128. 10.1021/acs.nanolett.1c01204. [DOI] [PubMed] [Google Scholar]
- Sun X.; Chen X.; Fu C.; Yu Q.; Zheng X.-S.; Fang F.; Liu Y.; Zhu J.; Zhang W.; Huang W. Molecular oxygen enhances H2O2 utilization for the photocatalytic conversion of methane to liquid-phase oxygenates. Nat. Commun. 2022, 13 (1), 6677. 10.1038/s41467-022-34563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Li S. Performance enhancement of proton exchange membrane fuel cell through carbon nanofibers grown in situ on carbon paper. Molecules 2023, 28 (6), 2810. 10.3390/molecules28062810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samad S.; Loh K. S.; Wong W. Y.; Lee T. K.; Sunarso J.; Chong S. T.; Wan Daud W. R. Carbon and non-carbon support materials for platinum-based catalysts in fuel cells. Int. J. Hydrogen Energy 2018, 43 (16), 7823–7854. 10.1016/j.ijhydene.2018.02.154. [DOI] [Google Scholar]
- Chen W.; Cao J.; Yang J.; Cao Y.; Zhang H.; Jiang Z.; Zhang J.; Qian G.; Zhou X.; Chen D.; et al. Molecular-level insights into the electronic effects in platinum-catalyzed carbon monoxide oxidation. Nat. Commun. 2021, 12 (1), 6888. 10.1038/s41467-021-27238-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoodinia M.; Åstrand P. O.; Chen D. Influence of carbon support on electronic structure and catalytic activity of pt catalysts: binding to the CO molecule. J. Phys. Chem. C 2016, 120 (23), 12452–12462. 10.1021/acs.jpcc.6b02001. [DOI] [Google Scholar]
- Kien N. T.; Yara H.; Chiku M.; Higuchi E.; Inoue H. Effect of surface composition on electrochemical oxidation reaction of carbon monoxide and ethanol of PtxRh1–x solid solution electrodes. Int. J. Electrochem. 2023, 2023 (1), 2386013. 10.1155/2023/2386013. [DOI] [Google Scholar]
- Elangovan A.; Xu J.; Sekar A.; Liu B.; Li J. Enhancing methanol oxidation reaction with Platinum-based catalysts using a N-doped three-dimensional graphitic carbon support. ChemCatChem 2020, 12 (23), 6000–6012. 10.1002/cctc.202001162. [DOI] [Google Scholar]
- Abbasi R.; Huang G.; Istratescu G. M.; Wu L.; Hayes R. E. Methane oxidation over Pt, Pt: Pd, and Pd based catalysts:effects of pre-treatment. Can. J. Chem. Eng. 2015, 93 (8), 1474–1482. 10.1002/cjce.22229. [DOI] [Google Scholar]
- Schwaha K.; Bechtold E. The adsorption of activated nitrogen on platinum single crystal faces. Surf. Sci. 1977, 66 (2), 383–393. 10.1016/0039-6028(77)90026-7. [DOI] [Google Scholar]
- Tripa C. E.; Zubkov T. S.; Yates J. T. Jr; Mavrikakis M.; Nørskov J. K. Molecular N2 chemisorption—specific adsorption on step defect sites on Pt surfaces. J. Chem. Phys. 1999, 111 (18), 8651–8658. 10.1063/1.480204. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.