

Abstract

Cleavage of inert C–N bonds in unstrained azacycles such as pyrrolidine remains a formidable challenge in synthetic chemistry. To address this, we introduce an effective strategy for the reductive cleavage of the C–N bond in N-benzoyl pyrrolidine, leveraging a combination of Lewis acid and photoredox catalysis. This method involves single-electron transfer to the amide, followed by site-selective cleavage at the C2–N bond. Cyclic voltammetry and NMR studies demonstrated that the Lewis acid is crucial for promoting the single-electron transfer from the photoredox catalyst to the amide carbonyl group. This protocol is widely applicable to various pyrrolidine-containing molecules and enables inert C–N bond cleavage including C–C bond formation via intermolecular radical addition. Furthermore, the current protocol successfully converts pyrrolidines to aziridines, γ-lactones, and tetrahydrofurans, showcasing its potential of the inert C–N bond cleavage for expanding synthetic strategies.
Introduction
Cyclic amines, particularly pyrrolidines, stand as pivotal structures within both natural products and synthetic building blocks, serving as cornerstones in the synthesis of myriad N-containing molecules, profound biological and medicinal relevance (Figure 1A).1 Historically, the chemical transformation of these motifs has enriched the synthetic toolkit, offering a cascade of valuable derivatives ranging from therapeutics to biological agents. Recently, peripheral functionalization through late-stage C(sp3)–H functionalization has become a modern and popular method, offering versatile and efficient ways to embellish these amines.2−7 In contrast to such peripheral functionalization, “skeletal remodeling”, which involves deconstruction and re-editing the core ring structure, has recently garnered significant attention as a new approach in organic synthesis.8−14 Such a transformation can be divided into two phases: the cleavage of inert bonds and further transformations. This allows for the conversion of pyrrolidine frameworks into different-sized cyclic amines through insertion or contraction reactions or into carbocycles through replacement reactions. Therefore, this method of modifying ring systems can have a substantial impact by enabling access to diverse structurally edited amines and unexplored chemical spaces.15
Figure 1.
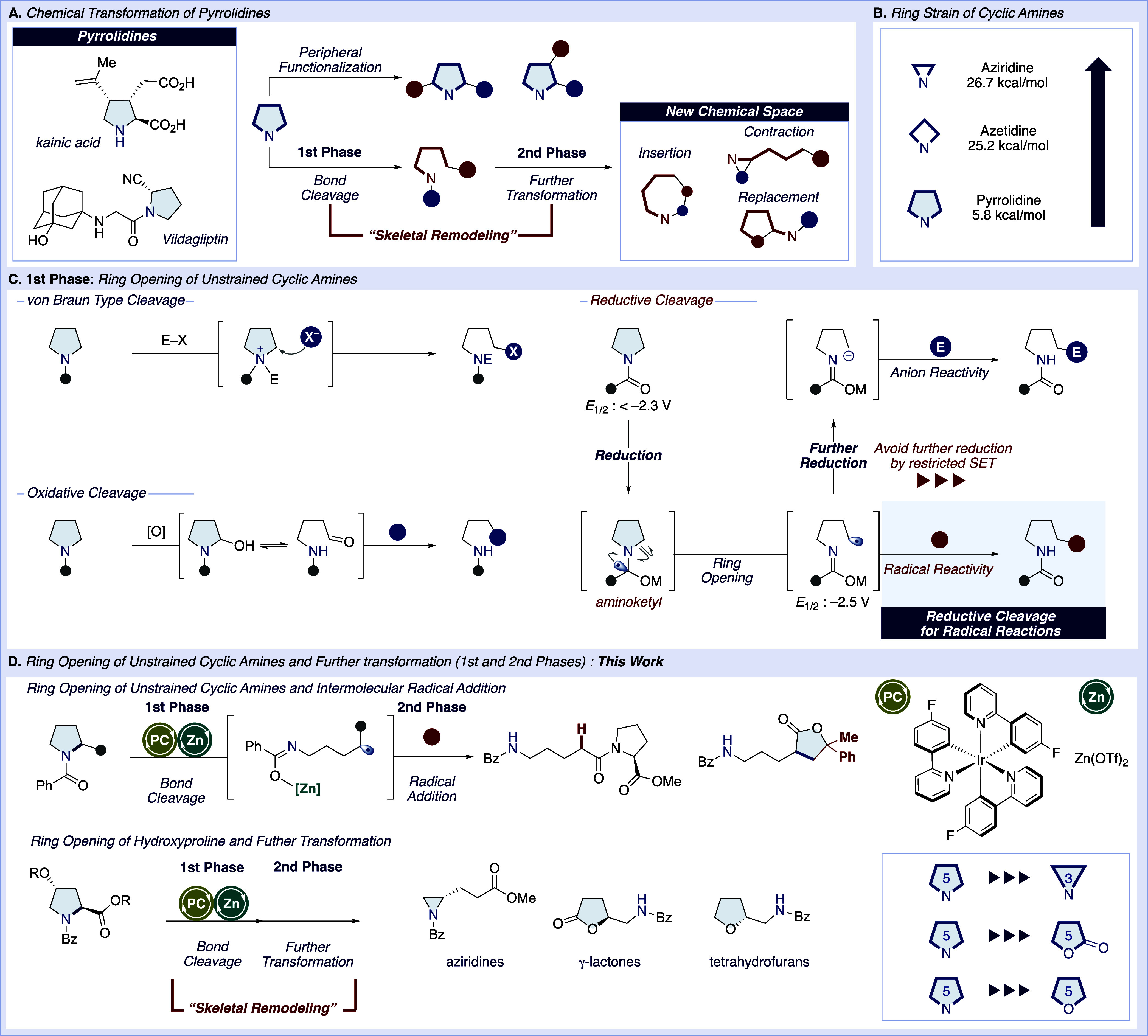
(A) Chemical transformation of pyrrolidines. (B) Ring strain of cyclic amines. (C) First phase: Ring opening of unstrained cyclic amines. (D) Ring opening of unstrained cyclic amines and further transformation (first and second phases).
However, the establishment of versatile methods for the transformation of pyrrolidines still faces significant challenges, particularly in the first phase involving C–N bond cleavage. For example, ring-opening reactions via homolytic cleavage using radicals are well-known for smaller rings such as aziridines and azetidines, driven by ring strain.16−26 These methods, however, are not applicable to pyrrolidines, making the process more challenging (Figure 1B).27 Although still limited to date, ingenious examples to tailor the unstrained pyrrolidine systems have been developed, which can be categorized into three mechanistically distinct approaches.
One approach is nucleophilic substitution of quaternary ammonium salts, von Braun type reactions (Figure 1C).28,29 This protocol was recently improved by using chloroformates,30 or difluorocarbene31,32 as more competent reagents. This transformation even facilitates the total synthesis of complex alkaloids.33,34 Additionally, BAr3-catalyzed ring opening has recently emerged as another approach exploiting ammonium intermediates.35,36 Another traditional example is the α-oxidation of cyclic amine followed by hemiaminal(ether) formation, where the resulting aldehyde further undergoes functionalization via oxidation and decarboxylative processes.8,11,37−44 These oxidative approaches have recently been highlighted by a series of elegant works from the Sarpong group.8,11−13,45
Contrasting with the above two strategies based on the electron-rich nature of amines, reductive C–N bond cleavage has been less employed. Early examples represented hydrogenolysis of cyclic amines using molecular hydrogen with transition metals.46 Thereafter, single-electron reduction of carbonyl handle affording aminoketyl radical has gained as a new alternative of reductive C–N bond cleavage. Pioneered by Szostak and Procter, the ring opening of N-acyl pyrrolidines using TmI2 (E°(TmIII/II) = −2.2 V vs SCE) more reducing than SmI2 was achieved.47 More recently, Yu and co-workers reported a protocol for the reductive ring opening of N-Boc pyrrolidines with an aryl or ester group at the C2-position employing consecutive photoinduced electron transfer (ConPET).48 These highly reductive approaches have faced the challenge of the choice of functionalization after reductive ring opening remaining limited to transformations involving carbanion intermediates. This limitation is likely due to the resulting radical being more susceptible to further reduction than the parent compound. The requirement for strong reduction conditions and a stoichiometric reductant could further reduce the accompanying carbon radical into a carbanion. We assumed that successfully avoiding multiple reductions could engage the reductive opening of cyclic amines in radical-mediated functionalization.
To this end, we envisioned that restricted single-electron transfer (SET), which is difficult with a stoichiometric reductant or conPET strategy, would provide access to radical-mediated transformations. To avoid the problematic further reduction of the susceptible carbon radical (−0.8 ∼ −2.5 V vs SCE),49,50 we focused on catalytic approach enabled by photoredox catalysis. Generally, the reduction of amide requires a high reduction power far beyond the range of standard photocatalysts. However, aromatic amide possesses a relatively less negative reduction potential, making them a feasible option (E1/2 = −2.3 V vs SCE).51 Thus, we envisioned that employing a highly reducing photoredox catalyst for the reduction of aromatic amides would be a successful combination to achieve radical-based C–N bond cleavage of pyrrolidines.
In this study, we report the successful generation of carbon radicals using a combination of zinc triflate and a photoredox catalyst. This approach not only facilitated carbon–carbon bond formation with alkenes and alkynes but also enabled the “skeletal remodeling” of pyrrolidines into aziridines, γ-lactones, and tetrahydrofurans (Figure 1D).
Results and Discussion
We commenced our investigation by screening reaction conditions in the ring-opening reaction of N-benzoyl-2-methylpyrrolidine 1a (Table 1). Irradiation with blue LEDs (λmax = 456 nm) in the presence of Ir(ppy)3 (E1/2red(IrIII*/IrIV) = −1.73 V vs SCE)52 and γ-terpinene yielded no product (Table 1, entry 1). We attributed this result to the difficulty of single-electron amide reduction and tested several Lewis acids to activate the amide carbonyl group. The desired acyclic product 2a was obtained, albeit in a considerably low yield accompanied by unreacted 1a (Table 1, entries 2–4). The yield of 2a was markedly improved when Zn(OTf)2 was used (Table 1, entry 5). Relevant additives, Zn(OAc)2 and TfOH, were less effective compared to Zn(OTf)2 (Table 1, entries 6 and 7). To our delight, we found that the combination of Zn(OTf)2 and Ir(4-Fppy)3 (E1/2red(IrIII*/IrIV) = −1.91 V vs SCE) dramatically improved the conversion, providing 2a in 92% yield (Table 1, entry 8). Switching to Ir(dFppy)3 (E1/2red(IrIII*/IrIV)= = −1.28 V vs SCE) failed to produce the desired product (Table 1, entry 9). THF and DMF were not suitable, presumably because the interaction between Zn(OTf)2 and these solvents hampered the desired transformation (Table 1, entries 10 and 11).53 Replacing γ-terpinene with 1,4-cyclohexadiene (1,4-CHD) slightly reduced the yield (84%) of 2a (Table 1, entry 12). This result may be attributed to the faster HAT rate of γ-terpinene than 1,4-CHD.54 Control experiments revealed the requirement for both visible light and Lewis acid (Table 1, entries 13 and 14).
Table 1. Optimization of the Reaction Conditionsa.

| Entry | Photocatalyst | Lewis acid | Solvent | 2a/% |
|---|---|---|---|---|
| 1 | Ir(ppy)3 | none | CH2Cl2 | 0 |
| 2 | Ir(ppy)3 | BF3·OEt2 | CH2Cl2 | 5 |
| 3 | Ir(ppy)3 | TMSOTf | CH2Cl2 | 6 |
| 4 | Ir(ppy)3 | Sc(OTf)3 | CH2Cl2 | 1 |
| 5 | Ir(ppy)3 | Zn(OTf)2 | CH2Cl2 | 30 |
| 6 | Ir(ppy)3 | Zn(OAc)2 | CH2Cl2 | trace |
| 7 | Ir(ppy)3 | TfOH | CH2Cl2 | 13 |
| 8 | Ir(4-Fppy)3 | Zn(OTf)2 | CH2Cl2 | 92 |
| 9 | Ir(dFppy)3 | Zn(OTf)2 | CH2Cl2 | 0 |
| 10 | Ir(4-Fppy)3 | Zn(OTf)2 | THF | 2 |
| 11 | Ir(4-Fppy)3 | Zn(OTf)2 | DMF | 0 |
| 12b | Ir(4-Fppy)3 | Zn(OTf)2 | CH2Cl2 | 84 |
| 13c | Ir(4-Fppy)3 | Zn(OTf)2 | CH2Cl2 | 0 |
| 14 | Ir(4-Fppy)3 | none | CH2Cl2 | 0 |
Conditions: 1a (0.10 mmol), 1.0 mol % Photocatalyst, 5.0 mol % Lewis acid, γ-terpinene (3.0 equiv) in solvent (0.10 M), blue LEDs (456 nm), 12 h, and under a N2 atmosphere. Yields were determined by 1H NMR analysis.
1,4-Cyclohexadiene was used instead of γ-terpinene.
Without irradiation.
(E1/2 (IrIV/IrIII*) and E1/2 (IrIV/IrIII) V vs SCE).52
With the optimal conditions in hand, we evaluated the substrate scope of the reductive ring opening of pyrrolidines (Scheme 1). 2-Alkyl pyrrolidines, including 2-methyl pyrrolidine (1a), prolinol derivatives (1b–1d) and 2-phenyl pyrrolidine (1e), underwent ring opening in moderate to good yields. Additionally, sterically demanding spirocyclic pyrrolidine 1f reacted smoothly to furnish 2f without the need for an extended reaction time. Proline derivatives such as esters (1g–1i) and amides (1j and 1k) were tolerated under these conditions, and the ring-opened products 2 were obtained in excellent yields, except for 2j, which was insoluble to CH2Cl2 and obtained in a moderate yield by using a CH2Cl2/DMF mixed solvent. Pyrrolidine 1l possessing quaternary carbon at the C2-position was less reactive compared to 1h, but prolonging the reaction time to 24 h led to an increase in the yield of 2l (90%). Taken together with the result of 1b–d, the reduction of the pendant C2 ester is not essential, unlike other reductive approaches in the C–N bond cleavage of proline derivatives.55−57 In the reaction of a fused bicycle 1m, regioselective C–N bond cleavage occurred at the ester-substituted C2-carbon and afforded the sole product 2m in a good yield. This regioselectivity could be attributed to the stability of the resulting radical intermediate.58 Proline containing dipeptides (1n–1r) participated in this protocol, and the corresponding products 2n–2r were obtained in moderate to excellent yields (53–95%). Oxidizable methionine and nucleophilic serine and tyrosine residues were all accommodated, demonstrating the high level of chemoselectivity of this catalytic system. Notably, one pyrrolidine of 1r remained intact under the reaction conditions, probably due to different susceptibilities for the reduction between aromatic and aliphatic amides. Interestingly, the pyrrolidine containing trifluoro acetyl group 1s gave the product 2s in a moderate yield without affecting the trifluoro acetyl group.59 While indoline 1t was less reactive than pyrrolidines, raising the reaction temperature and altering the benzoyl group to the 4-methoxybenzoyl group improves the yield of the product 2t. On the other hand, other azaheterocycles, such as aziridine 1u, azetidine 1v, and piperidine 1w, were also reacted, furnishing the product in moderate to good yields. In the reaction of 1u, phenyl oxazoline was obtained via intramolecular nucleophilic ring opening reaction, which can deactivate the photocatalyst by substitution of its ligand, leading to the modest conversion.60 The ring opening of azepane 1x proceeded, albeit in a low yield. Notably, unsubstituted azetidine 1y and pyrrolidine 1z were applicable, highlighting that this catalytic system can generate a diverse range of radicals, including even unstable primary alkyl radicals.
Scheme 1. Scope of the Ring Opening of Pyrrolidines.
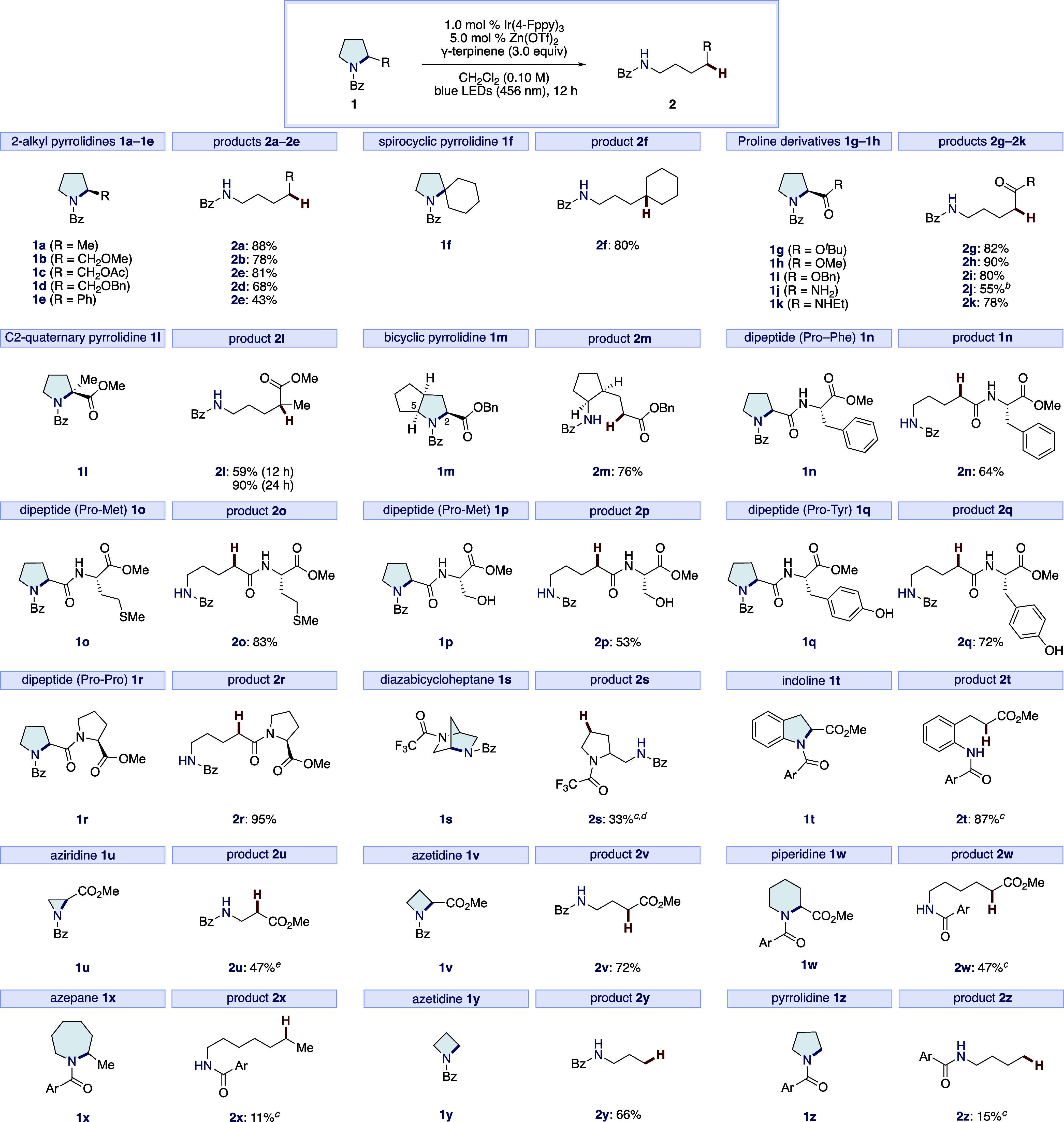
Conditions: 1 (0.20 mmol), 1.0 mol % Ir(4-Fppy)3, 5.0 mol % Zn(OTf)2, γ-terpinene (3.0 equiv) in CH2Cl2 (0.10 M) under a N2 atmosphere and blue LEDs (456 nm) irradiation for 12 h. Isolated yields.
CH2Cl2/DMF (0.10 M, 9:1).
Fan off (40 °C).
24 h.
3.0 mol % Ir(4-Fppy)3. Ar = 4-MeOC6H4.
To provide insight into the mechanistic details of this reductive C–N bond cleavage, we performed a radical clock experiment (Figure 2A). Treatment of pyrrolidine 1aa with a cyclopropyl moiety afforded olefin 2aa in a good yield, suggesting the intermediacy of the cyclopropylcarbinyl radical in the ring opening of pyrrolidine. We next examined the effect of N-acyl groups in this reaction (Figure 2B). 2-Methyl substituted pyrrolidines bearing three different N-acyl substituents, 1a, 1ab, and 1ac were subjected to the established conditions. 1a was converted into the corresponding product 2a in 88% isolated yield. In contrast, no reaction was observed when acetyl pyrrolidine 1ab and trifluoro acetyl pyrrolidine 1ac were used as the starting materials. We presumed that in the cases of 1ab and 1ac, single-electron transfer from the excited photocatalyst to amide carbonyl did not occur. To gain insights into the interaction between Zn(OTf)2 and pyrrolidines 1a, 1ab, and 1ac, we examined the sensitivity of 13C NMR to the addition of Zn(OTf)2 (Figure 2C). The result indicated that the amide carbonyl carbon of 1a and 1ab undergo a downfield chemical shift with increasing the amount of Zn(OTf)2. In contrast, no change was observed in the experiment for 1ac. These results are consistent with the successful reduction of benzoyl pyrrolidine 1a facilitated by the coordination of Zn(OTf)2 to the amide carbonyl. On the other hand, no reaction progress was observed with 1ab despite the successful coordination of Zn(OTf)2. To better understand the different reactivities of 1a, 1ab, and 1ac, we measured cyclic voltammetry (CV) (Figure 2D). The reduction peak of 1a was observed at −2.68 V. While no apparent peak was detected with 1ab, the beginning of the reduction was observed with 1ac. Considering that the acetyl group is more difficult to reduce compared to the benzoyl group with a similar tertiary amide, the reduction peak of 1ab seems to be far from the measurable range under the present conditions. Although trifluoroacetyl is more reducible than acetyl, 1ac has no ability to coordinate with Zn(OTf)2, according to the NMR experiment. Taken together, coordination Zn(OTf)2 to the amide carbonyl of 1a would facilitate single-electron transfer and enable the present reductive C–N bond cleavage.59,61
Figure 2.
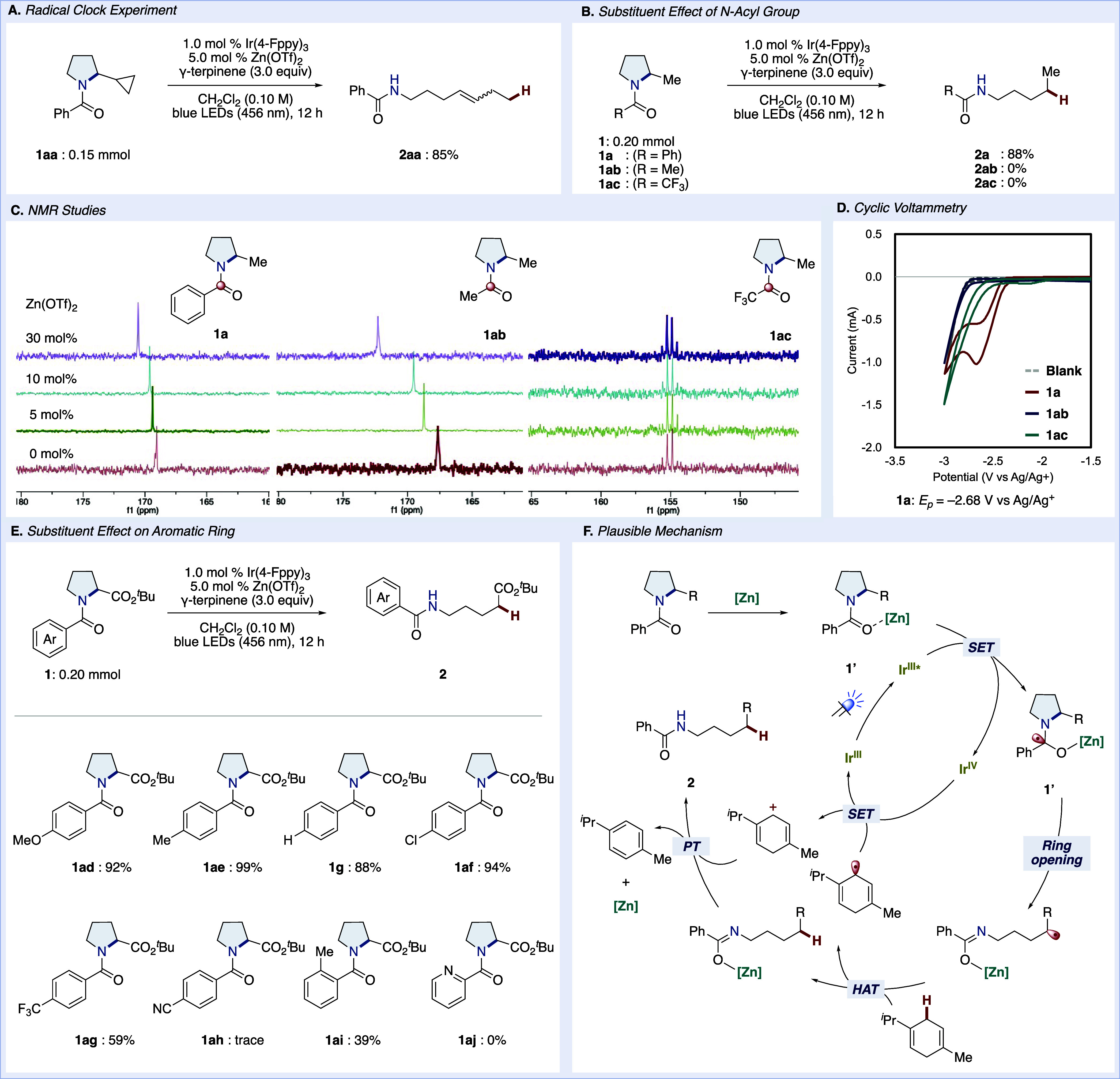
Mechanistic investigations. (A) Radical clock experiment. (B) Substituent effect of the N-acyl group. (C) NMR studies. (D) Cyclic voltammetry experiments. See the Supporting Information for the details of the experiments. (E) Substituent effect on the aromatic ring. (F) Plausible mechanism.
We next explored the effect of the aroyl group (Figure 2E). Pyrrolidines with electron rich or neutral aroyl substituents (1ad, 1af, and 1g) were suitable for the reaction to provide the corresponding products in good to excellent yields. The Cl-substituted pyrrolidine 1af also reacted without a decrease in yield, while CF3-substituted benzoyl pyrrolidine 1ag afforded the product in a moderate yield, and the reaction with highly electron deficient CN-substituted 1ah did not proceed. Additionally, the reaction was inhibited using ortho-substituted benzoyl pyrrolidine 1ai, likely because the methyl group caused the aryl ring of the benzoyl group to rotate, resulting in an electronic state different from other benzoyl groups.62 Consequently, we believe that the 4-methoxybenzoyl group as an N-substituent in the substrate scope facilitates Zn(OTf)2 coordination, thereby improving reaction efficiency. On the other hand, picoline amide 1aj did not provide any products, likely because although Zn(OTf)2 coordinates might be easy, the electron-deficient nature of the pyridine ring hindered the reaction from proceeding.
A plausible mechanism for the ring opening reaction is outlined in Figure 2F. First, the excited state photoredox catalyst *IrIII is generated under the irradiation of blue LEDs (IrIII → *IrIII). Single-electron transfer (SET) from *IrIII to 1′, a complex of 1 and Zn(OTf)2, occurs, followed by the ring opening of pyrrolidine to furnish a radical intermediate (*IrIII → IrIV). We believe that β-scission of the generated aminoketyl radical necessitates the ring strain of pyrrolidine based on the observation of a significantly lower reactivity of piperidine under these ring opening conditions (see the Supporting Information). The alkyl radical generated from the ring opening would then undergo hydrogen-atom transfer (HAT) from γ-terpinene, leading to zinc imidate along with a γ-terpinene-derived radical, which would subsequently be oxidized to a cation by IrIV (IrIV → IrIII). Finally, proton transfer from the γ-terpinene-derived cation to the zinc imidate would provide desired ring-opened product 2 along with regeneration of Zn(OTf)2.
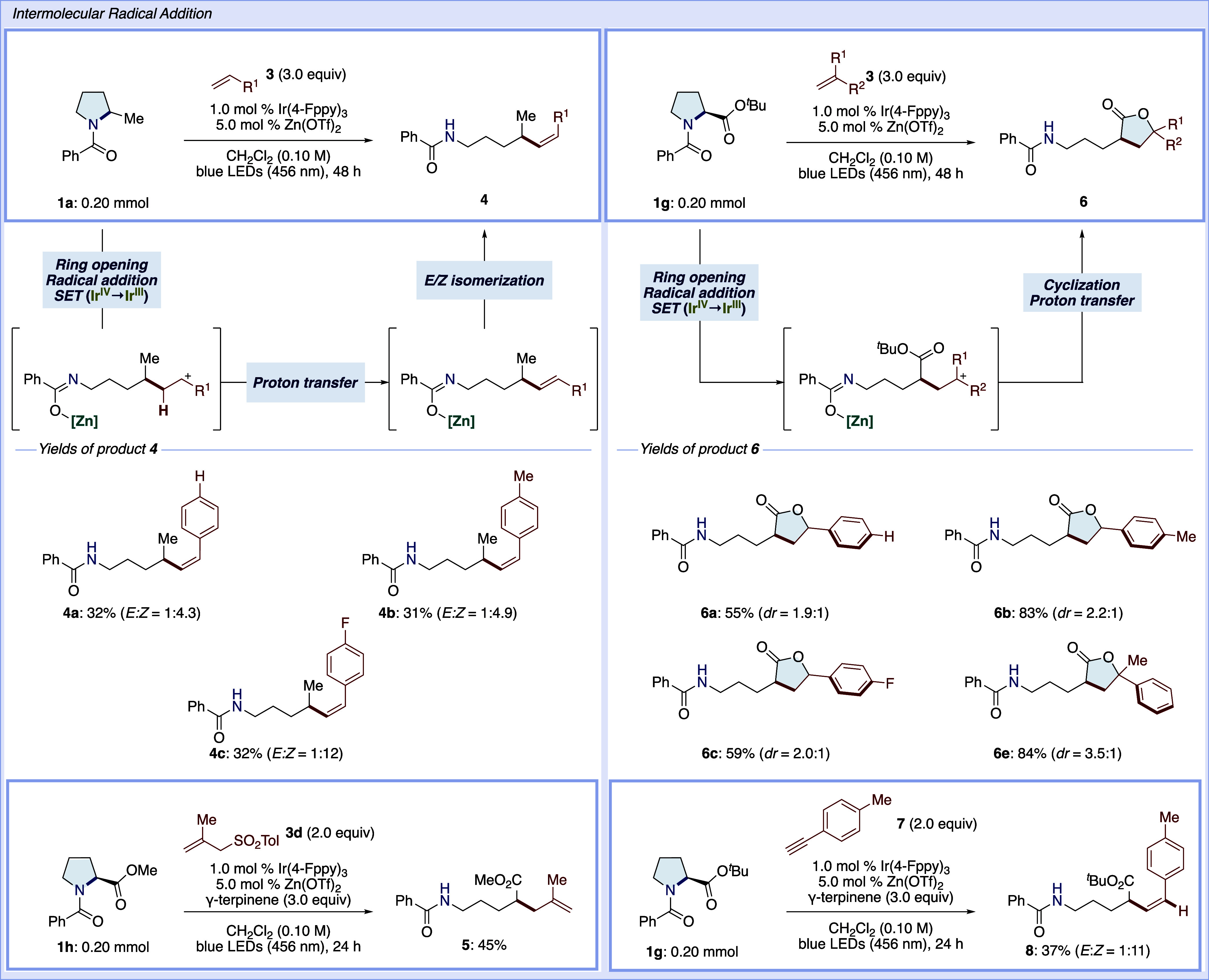
To further explore the radical reactivity of this catalytic system, we investigated C–C bond formation through intermolecular radical addition (Scheme 2). First, we conducted alkenylation using styrene derivatives. The reaction of pyrrolidine 1a with styrene (3a) in the absence of γ-terpinene afforded the alkenylated product 4a with good (Z)-isomer selectivity, albeit in low yield. This alkenylation presumably occurs via sequential steps involving ring opening/radical addition to 3a, oxidation of the generated benzyl radical, proton transfer, and followed by photoinduced E–Z isomerization. The E–Z isomerization is supported by experiments of a similar π-system under the current reaction conditions (see the Supporting Information).63 In addition to 3a, 4-methylstylene (3b) and 1-fluoro-4-vinylbenzene (3c) are also tolerated to yield the corresponding products 4b and 4c. Additionally, treatment of pyrrolidine 1h with allyl sulfone 3d afforded alkene 5 via the extrusion of an aryl sulfonyl radical.64 Moreover, the reaction of pyrrolidine 1g with 3a furnished lactone 6a via the generation of benzyl cation, followed by cyclization and the release of isobutene.65 This transformation highlights the utility of photoredox catalysis, which enables a restricted single-electron transfer. Other substituted styrenes, such as 3b, 3c, and 3e, were tolerated in this lactone formation reaction. Furthermore, 1-ethynyl-4-methylbenzene (7) was also accommodated in the radical addition reaction, predominantly yielding the (Z)-isomer of styrene 8 from pyrrolidine 1g. This sequence of exploration underscores the versatility and efficiency of our catalytic system in facilitating a variety of radical-mediated transformations, expanding the scope of potential synthetic applications.
Scheme 2. Intermolecular Radical Addition.
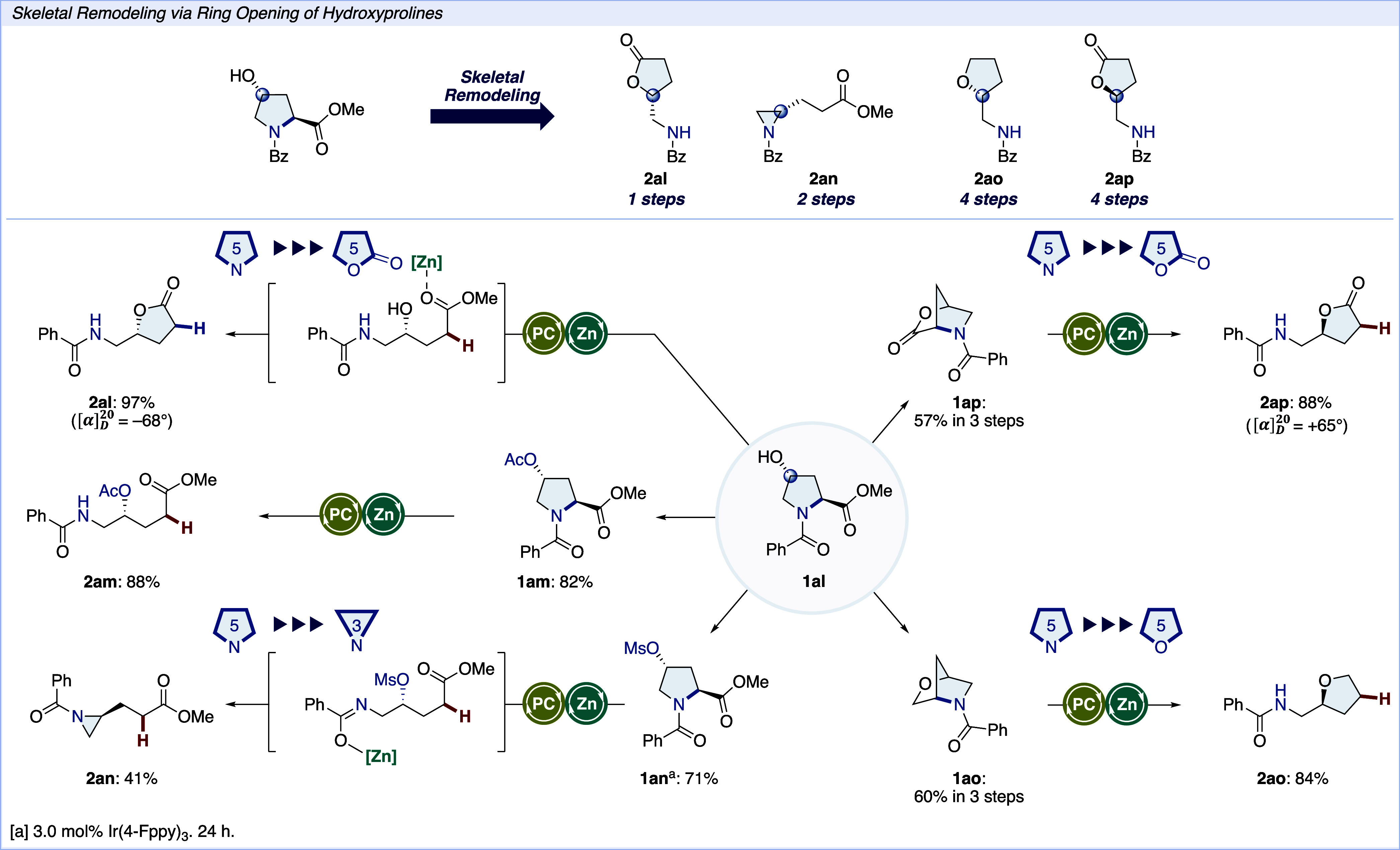
Next, to demonstrate the synthetic utility of this protocol, we subjected l-hydroxyproline derivatives to optimal conditions to convert into the skeletal edited compounds (Scheme 3). The reaction of alcohol 1al afforded lactone 2al in 97% yield, presumably forged via Lewis acid assisted lactonization. The O-acetyl variant 1am was efficiently converted into the ring-opened product 2am. When the acetyl group was replaced with a mesyl group (1an), aziridine 2an was obtained via intramolecular SN2 fashion. Notably, this aziridination occurred during the purification process by column chromatography. Bridged bicyclic compounds 1ao and 1ap reacted smoothly and gave enantiomerically pure tetrahydrofuran 2ao and γ-lactone 2ap in good yields, respectively. Notably, 2ap is the enantiomer of 2al, as confirmed by optical rotation measurements. This protocol successfully produces optically active compounds, leveraging the stereochemistry derived from l-hydroxyproline. This ability to manipulate the stereochemistry and achieve high yields underscores the robustness and versatility of our method in generating diverse and enantiomerically pure heterocycles.
Scheme 3. Skeletal Remodeling via Ring Opening of Hydroxy Pyrrolidine Derivatives.
Conclusions
In conclusion, we have developed a reductive C–N bond cleavage of N-benzyl pyrrolidines using photoredox catalysis with Lewis acid.66 This reaction enabled unique transformations via a radical mechanism, which was previously unattainable through traditional reductive pyrrolidine C–N bond cleavage, using widely available starting materials. In the context of amide bond activation, the present protocol represents a rare example of σC–N bond cleavage.28,48,67−70 The critical role of the Lewis acid was elucidated by NMR studies and cyclic voltammetry. Additionally, we successfully synthesized γ-lactones, aziridines, and tetrahydrofurans through “skeletal remodeling” reactions, starting from hydroxyproline derivatives. Ongoing efforts in our laboratory are focused on exploring new transformations of nitrogen-containing compounds using photoredox catalysis, further expanding the synthetic utility of this approach.
Acknowledgments
This work was supported by JSPS KAKENHI Grant Numbers JP21H05213 (Digi-TOS) (to J.Y.), JP23K13752 (to E.O.), The Uehara Memorial Foundation (to E.O.), and Satomi Foundation (to E.O.). This work was partly supported by JST ERATO Grant Number JPMJER1901 (to J.Y.). Materials Characterization Central Laboratory in Waseda University is acknowledged for the support of HRMS measurement. We acknowledge Kessil Lighting for lights used in this study.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c13210.
Experimental procedures and spectroscopic data for compounds including 1H, 13C, and 19F NMR spectra (PDF)
Author Contributions
# K.A. and M.H. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Campos K. R. Direct sp3 C–H Bond Activation Adjacent to Nitrogen in Heterocycles. Chem. Soc. Rev. 2007, 36, 1069–1084. 10.1039/B607547A. [DOI] [PubMed] [Google Scholar]
- Mitchell E. A.; Peschiulli A.; Lefevre N.; Meerpoel L.; Maes B. U. W. Direct α-Functionalization of Saturated Cyclic Amines. Chem.—Eur. J. 2012, 18, 10092–10142. 10.1002/chem.201201539. [DOI] [PubMed] [Google Scholar]
- Cordier C. J.; Lundgren R. J.; Fu G. C. Enantioconvergent Cross-Couplings of Racemic Alkylmetal Reagents with Unactivated Secondary Alkyl Electrophiles: Catalytic Asymmetric Negishi α-Alkylations of N-Boc-Pyrrolidine. J. Am. Chem. Soc. 2013, 135, 10946–10949. 10.1021/ja4054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Zheng Z.; Yang J.; Zhang X.; Fan X. Recent Advances in the Functionalization of Saturated Cyclic Amines. Org. Chem. Front. 2021, 8, 4582–4606. 10.1039/D1QO00171J. [DOI] [Google Scholar]
- Chen W.; Paul A.; Abboud K. A.; Seidel D. Rapid Functionalization of Multiple C–H Bonds in Unprotected Alicyclic Amines. Nat. Chem. 2020, 12, 545–550. 10.1038/s41557-020-0438-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M. H.; Shurtleff V. W.; Terrett J. A.; Cuthbertson J. D.; MacMillan D. W. C. Native Functionality in Triple Catalytic Cross-Coupling: sp3 C–H Bonds as Latent Nucleophiles. Science 2016, 352, 1304–1308. 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque J. B.; Kuroda Y.; Göttemann L. T.; Sarpong R. Deconstructive Diversification of Cyclic Amines. Nature 2018, 564, 244–248. 10.1038/s41586-018-0700-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy S. H.; Dherange B. D.; Berger K. J.; Levin M. D. Skeletal Editing through Direct Nitrogen Deletion of Secondary Amines. Nature 2021, 593, 223–227. 10.1038/s41586-021-03448-9. [DOI] [PubMed] [Google Scholar]
- Jurczyk J.; Lux M. C.; Adpressa D.; Kim S. F.; Lam Y. H.; Yeung C. S.; Sarpong R. Photomediated Ring Contraction of Saturated Heterocycles. Science 2021, 373, 1004–1012. 10.1126/science.abi7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque J. B.; Kuroda Y.; Göttemann L. T.; Sarpong R. Deconstructive Fluorination of Cyclic Amines by Carbon-Carbon Cleavage. Science. 2018, 361, 171–174. 10.1126/science.aat6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque J. B.; Sarpong R.; Musaev D. G. Key Mechanistic Features of the Silver(I)-Mediated Deconstructive Fluorination of Cyclic Amines: Multistate Reactivity versus Single-Electron Transfer. J. Am. Chem. Soc. 2021, 143, 3889–3900. 10.1021/jacs.0c13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaledin A. L.; Roque J. B.; Sarpong R.; Musaev D. G. Computational Study of Key Mechanistic Details for a Proposed Copper (I)-Mediated Deconstructive Fluorination of N-Protected Cyclic Amines. Top. Catal. 2022, 65, 418–432. 10.1007/s11244-021-01443-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurczyk J.; Woo J.; Kim S. F.; Dherange B. D.; Sarpong R.; Levin M. D. Single-Atom Logic for Heterocycle Editing. Nat. Synth. 2022, 1, 352–364. 10.1038/s44160-022-00052-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos K. R.; Coleman P. J.; Alvarez J. C.; Dreher S. D.; Garbaccio R. M.; Terrett N. K.; Tillyer R. D.; Truppo M. D.; Parmee E. R. The Importance of Synthetic Chemistry in the Pharmaceutical Industry. Science 2019, 363, eaat0805 10.1126/science.aat0805. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Q.; Vogelsang E.; Qu Z. W.; Grimme S.; Gansäuer A. Titanocene-Catalyzed Radical Opening of N-Acylated Aziridines. Angew. Chem., Int. Ed. 2017, 56, 12654–12657. 10.1002/anie.201707673. [DOI] [PubMed] [Google Scholar]
- Hao W.; Wu X.; Sun J. Z.; Siu J. C.; Macmillan S. N.; Lin S. Radical Redox-Relay Catalysis: Formal [3 + 2] Cycloaddition of N-Acylaziridines and Alkenes. J. Am. Chem. Soc. 2017, 139, 12141–12144. 10.1021/jacs.7b06723. [DOI] [PubMed] [Google Scholar]
- Steiman T. J.; Liu J.; Mengiste A.; Doyle A. G. Synthesis of β-Phenethylamines via Ni/Photoredox Cross-Electrophile Coupling of Aliphatic Aziridines and Aryl Iodides. J. Am. Chem. Soc. 2020, 142, 7598–7605. 10.1021/jacs.0c01724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada J. G.; Williams W. L.; Ting S. I.; Doyle A. G. Role of Electron-Deficient Olefin Ligands in a Ni-Catalyzed Aziridine Cross-Coupling to Generate Quaternary Carbons. J. Am. Chem. Soc. 2020, 142, 8928–8937. 10.1021/jacs.0c02237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood D. P.; Guan W.; Lin S. Titanium and Cobalt Bimetallic Radical Redox Relay for the Isomerization of N-Bz Aziridines to Allylic Amides. Synthesis 2021, 53, 4213–4220. 10.1055/s-0037-1610779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongbang S.; Doyle A. G. Ni/Photoredox-Catalyzed C(sp3)-C(sp3) Coupling between Aziridines and Acetals as Alcohol-Derived Alkyl Radical Precursors. J. Am. Chem. Soc. 2022, 144, 20067–20077. 10.1021/jacs.2c09294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams W. L.; Gutiérrez-Valencia N. E.; Doyle A. G. Branched-Selective Cross-Electrophile Coupling of 2-Alkyl Aziridines and (Hetero)Aryl Iodides Using Ti/Ni Catalysis. J. Am. Chem. Soc. 2023, 145, 24175–24183. 10.1021/jacs.3c08301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y.; Hayashi M.; Sato R.; Tai K.; Nagase T. Development of Photoredox Cross-Electrophile Coupling of Strained Heterocycles with Aryl Bromides Using High-Throughput Experimentation for Library Construction. Org. Lett. 2023, 25, 5569–5573. 10.1021/acs.orglett.3c01821. [DOI] [PubMed] [Google Scholar]
- Hao W.; Harenberg J. H.; Wu X.; MacMillan S. N.; Lin S. Diastereo- and Enantioselective Formal [3 + 2] Cycloaddition of Cyclopropyl Ketones and Alkenes via Ti-Catalyzed Radical Redox Relay. J. Am. Chem. Soc. 2018, 140, 3514–3517. 10.1021/jacs.7b13710. [DOI] [PubMed] [Google Scholar]
- McCallum T.; Wu X.; Lin S. Recent Advances in Titanium Radical Redox Catalysis. J. Org. Chem. 2019, 84, 14369–14380. 10.1021/acs.joc.9b02465. [DOI] [PubMed] [Google Scholar]
- Wu X.; Chang Y.; Lin S. Titanium Radical Redox Catalysis: Recent Innovations in Catalysts, Reactions, and Modes of Activation. Chem. 2022, 8, 1805–1821. 10.1016/j.chempr.2022.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Lai Z.; Adijiang A.; Zhao H.; An J. Selective C–N σ Bond Cleavage in Azetidinyl Amides under Transition Metal-Free Conditions. Molecules 2019, 24, 459. 10.3390/molecules24030459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elderfield R. C.; Hageman H. A. The Von Braun Cyanogen Bromide Reaction I. Application to Pyrrolidines and Ethyleneimines (1). J. Org. Chem. 1949, 14, 605–637. 10.1021/jo01156a015. [DOI] [Google Scholar]
- Elderfield R. C.; Green M. The Von Braun Cyanogen Bromide Reaction. II. Application to N-Arylpyrrolidines. J. Org. Chem. 1952, 17, 431–441. 10.1021/jo01137a016. [DOI] [Google Scholar]
- Yu C.; Shoaib M. A.; Iqbal N.; Kim J. S.; Ha H. J.; Cho E. J. Selective Ring-Opening of N-Alkyl Pyrrolidines with Chloroformates to 4-Chlorobutyl Carbamates. J. Org. Chem. 2017, 82, 6615–6620. 10.1021/acs.joc.7b00681. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Heo J.; Kim D.; Chang S.; Seo S. Ring-Opening Functionalizations of Unstrained Cyclic Amines Enabled by Difluorocarbene Transfer. Nat. Commun. 2020, 11, 4761. 10.1038/s41467-020-18557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J.; Ma X.; Ou Z.; Song Q. Deconstructive Functionalizations of Unstrained Carbon–Nitrogen Cleavage Enabled by Difluorocarbene. ACS Cent. Sci. 2020, 6, 1819–1826. 10.1021/acscentsci.0c00779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong S.; Lim H.; Han S. Biosynthetically Inspired Transformation of Iboga to Monomeric Post-Iboga Alkaloids. Chem. 2019, 5, 353–363. 10.1016/j.chempr.2018.10.009. [DOI] [Google Scholar]
- Lim H.; Seong S.; Kim Y.; Seo S.; Han S. Biopatterned Reorganization of Alkaloids Enabled by Ring-Opening Functionalization of Tertiary Amines. J. Am. Chem. Soc. 2021, 143, 19966–19974. 10.1021/jacs.1c10205. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chang S. cine-Silylative Ring-Opening of α-Methyl Azacycles Enabled by the Silylium-Induced C–N Bond Cleavage. J. Am. Chem. Soc. 2020, 142, 12585–12590. 10.1021/jacs.0c05241. [DOI] [PubMed] [Google Scholar]
- Peng Y.; Oestreich M. B(C6F5)3-Catalyzed Regioselective Ring Opening of Cyclic Amines with Hydrosilanes. Chem.—Eur. J. 2023, 29, e202203721 10.1002/chem.202203721. [DOI] [PubMed] [Google Scholar]
- Han G.; McIntosh M. C.; Weinreb S. M. A Convenient Synthetic Method for Amide Oxidation. Tetrahedron Lett. 1994, 35, 5813–5816. 10.1016/S0040-4039(00)78191-3. [DOI] [Google Scholar]
- Boto A.; Hernández R.; Suárez E. Tandem Radical Decarboxylation-Oxidation of Amino Acids: A Mild and Efficient Method for the Generation of N-Acyliminium Ions and Their Nucleophilic Trapping. J. Org. Chem. 2000, 65, 4930–4937. 10.1021/jo000356t. [DOI] [PubMed] [Google Scholar]
- Cocquet G.; Ferroud C.; Guy A. A Mild and Efficient Procedure for Ring-Opening Reactions of Piperidine and Pyrrolidine Derivatives by Single Electron Transfer Photooxidation. Tetrahedron 2000, 56, 2975–2984. 10.1016/S0040-4020(00)00048-X. [DOI] [Google Scholar]
- Ito R.; Umezawa N.; Higuchi T. Unique Oxidation Reaction of Amides with Pyridine-N-Oxide Catalyzed by Ruthenium Porphyrin: Direct Oxidative Conversion of N-Acyl-L-Proline to N-Acyl-L-Glutamate. J. Am. Chem. Soc. 2005, 127, 834–835. 10.1021/ja045603f. [DOI] [PubMed] [Google Scholar]
- Kaname M.; Yoshifuji S.; Sashida H. Ruthenium Tetroxide Oxidation of Cyclic N-Acylamines by a Single Layer Method: Formation of ω-Amino Acids. Tetrahedron Lett. 2008, 49, 2786–2788. 10.1016/j.tetlet.2008.02.127. [DOI] [Google Scholar]
- Osberger T. J.; Rogness D. C.; Kohrt J. T.; Stepan A. F.; White M. C. Oxidative Diversification of Amino Acids and Peptides by Small-Molecule Iron Catalysis. Nature 2016, 537, 214–219. 10.1038/nature18941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Man Y.; Wang K.; Wan X.; Tong L.; Li N.; Tang B. Hydrogen Bond Directed Aerobic Oxidation of Amines via Photoredox Catalysis. Chem. Commun. 2018, 54, 10989–10992. 10.1039/C8CC06603E. [DOI] [PubMed] [Google Scholar]
- Liu R. H.; He Y. H.; Yu W.; Zhou B.; Han B. Silver-Catalyzed Site-Selective Ring-Opening and C–C Bond Functionalization of Cyclic Amines: Access to Distal Aminoalkyl-Substituted Quinones. Org. Lett. 2019, 21, 4590–4594. 10.1021/acs.orglett.9b01496. [DOI] [PubMed] [Google Scholar]
- Soro D. M.; Roque J. B.; Rackl J. W.; Park B.; Payer S.; Shi Y.; Ruble J. C.; Kaledin A. L.; Baik M. H.; Musaev D. G.; Sarpong R. Photo- and Metal-Mediated Deconstructive Approaches to Cyclic Aliphatic Amine Diversification. J. Am. Chem. Soc. 2023, 145, 11245–11257. 10.1021/jacs.3c01318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Betsbrugge J.; Van Den Nest W.; Verheyden P.; Tourwé D. New Amino Acids Derived from L-Pyroglutamic Acid: Synthesis of Trans-4-Benzyl-cis-5-Phenyl-L-Proline, L-α-(2-Benzyl-3-Phenylpropyl)-Glycine and L-α-(3-Phenylpropyl)-Glycine. Tetrahedron 1998, 54, 1753–1762. 10.1016/S0040-4020(97)10399-4. [DOI] [Google Scholar]
- Szostak M.; Spain M.; Procter D. J. Uncovering the Importance of Proton Donors in TmI2-Promoted Electron Transfer: Facile C–N Bond Cleavage in Unactivated Amides. Angew. Chem., Int. Ed. 2013, 52, 7237–7241. 10.1002/anie.201303178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Qu Q.; Ran C. K.; Wang W.; Zhang W.; He Y.; Liao L. L.; Ye J. H.; Yu D. G. Photocatalytic Carboxylation of C–N Bonds in Cyclic Amines with CO2 by Consecutive Visible-Light-Induced Electron Transfer. Angew. Chem., Int. Ed. 2023, 62, e202217918 10.1002/anie.202217918. [DOI] [PubMed] [Google Scholar]
- Fu Y.; Liu L.; Yu H. Z.; Wang Y. M.; Guo Q. X. Quantum-Chemical Predictions of Absolute Standard Redox Potentials of Diverse Organic Molecules and Free Radicals in Acetonitrile. J. Am. Chem. Soc. 2005, 127, 7227–7234. 10.1021/ja0421856. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Lu L.; Zhang W.; Wang Y.; Ware S. D.; Mondragon J.; Rein J.; Strotman N.; Lehnherr D.; See K. A.; Lin S. Electrochemically Driven Cross-Electrophile Coupling of Alkyl Halides. Nature 2022, 604, 292–297. 10.1038/s41586-022-04540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.; Szostak R.; Szostak M. Proton-Coupled Electron Transfer in the Reduction of Carbonyls Using SmI2-H2O: Implications for the Reductive Coupling of Acyl-Type Ketyl Radicals with SmI2-H2O. Org. Biomol. Chem. 2016, 14, 9151–9157. 10.1039/C6OB01621A. [DOI] [PubMed] [Google Scholar]
- Teegardin K.; Day J. I.; Chan J.; Weaver J. Advances in Photocatalysis: A Microreview of Visible Light Mediated Ruthenium and Iridium Catalyzed Organic Transformations. Org. Process Res. Dev. 2016, 20, 1156–1163. 10.1021/acs.oprd.6b00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laturski A. E.; Gaffen J. R.; Demay-Drouhard P.; Caputo C. B.; Baumgartner T. Probing the Impact of Solvent on the Strength of Lewis Acids via Fluorescent Lewis Adducts. Precis. Chem. 2023, 1, 49–56. 10.1021/prechem.2c00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Q.; Lee D. D.; Dinh A. N.; Seguin R. P.; Zhang R.; Xu L. Development and Application of a Peroxyl Radical Clock Approach for Measuring Both Hydrogen-Atom Transfer and Peroxyl Radical Addition Rate Constants. J. Org. Chem. 2021, 86, 153–168. 10.1021/acs.joc.0c01920. [DOI] [PubMed] [Google Scholar]
- Honda T.; Ishikawa F. Reductive Deamination of α-Amino Carbonyl Compounds by Means of Samarium Iodide. Chem. Commun. 1999, 2, 1065–1066. 10.1039/a903073e. [DOI] [Google Scholar]
- Honda T.; Takahashi R.; Namiki H. Syntheses of (+)-Cytisine, (−)-Kuraramine, (−)-Isokuraramine, and (−)-Jussiaeiine A. J. Org. Chem. 2005, 70, 499–504. 10.1021/jo048365f. [DOI] [PubMed] [Google Scholar]
- Traoré M.; Mietton F.; Maubon D.; Peuchmaur M.; Francisco Hilário F.; Pereira de Freitas R.; Bougdour A.; Curt A.; Maynadier M.; Vial H.; Pelloux H.; Hakimi M.-A.; Wong Y.-S. Flexible Synthesis and Evaluation of Diverse Anti-Apicomplexa Cyclic Peptides. J. Org. Chem. 2013, 78, 3655–3675. 10.1021/jo4001492. [DOI] [PubMed] [Google Scholar]
- Hioe J.; Zipse H. Radical Stability and Its Role in Synthesis and Catalysis. Org. Biomol. Chem. 2010, 8, 3609–3617. 10.1039/c004166a. [DOI] [PubMed] [Google Scholar]
- Campbell M. W.; Polites V. C.; Patel S.; Lipson J. E.; Majhi J.; Molander G. A. Photochemical C–F Activation Enables Defluorinative Alkylation of Trifluoroacetates and -Acetamides. J. Am. Chem. Soc. 2021, 143, 19648–19654. 10.1021/jacs.1c11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dik-Kung M.; Chung-Hang L.; Hai-Jing Z.; Bingyong H.. Method of Detecting Helicase Activity. US2017145472A1, 2017.
- Williams O. P.; Chmiel A. F.; Mikhael M.; Bates D. M.; Yeung C. S.; Wickens Z. K. Practical and General Alcohol Deoxygenation Protocol. Angew. Chem., Int. Ed. 2023, 62, e202300178 10.1002/anie.202300178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agasti S.; Beattie N. A.; McDouall J. W.; Procter D. J. SmI2-Catalyzed Intermolecular Coupling of Cyclopropyl Ketones and Alkynes: A Link between Ketone Conformation and Reactivity. J. Am. Chem. Soc. 2021, 143, 3655–366. 10.1021/jacs.1c01356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K.; Staig S. J.; Weaver J. D. Facile Synthesis of Z-Alkenes via Uphill Catalysis. J. Am. Chem. Soc. 2014, 136, 5275–5278. 10.1021/ja5019749. [DOI] [PubMed] [Google Scholar]
- Huang X.; Luo S.; Burghaus O.; Webster R. D.; Harms K.; Meggers E. Combining the Catalytic Enantioselective Reaction of Visible-Light-Generated Radicals with a by-Product Utilization System. Chem. Sci. 2017, 8, 7126–7131. 10.1039/C7SC02621H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X.-J.; Yang D.-T.; Wang L.; Song T.; Wu L.-Z.; Liu Q. A Novel Intermolecular Synthesis of γ-Lactones via Visible-Light Photoredox Catalysis. Org. Lett. 2013, 15, 6054–6057. 10.1021/ol402954t. [DOI] [PubMed] [Google Scholar]
- An initial version of this work was deposited in ChemRxiv on July 27, 2024. 10.26434/chemrxiv-2024-q5f3h. [DOI]
- Lei Y.; Wrobleski A. D.; Golden J. E.; Powell D. R.; Aubé J. Facile C–N Cleavage in a Series of Bridged Lactams. J. Am. Chem. Soc. 2005, 127, 4552–4553. 10.1021/ja050214m. [DOI] [PubMed] [Google Scholar]
- Hu F.; Lalancette R.; Szostak M. Structural Characterization of N-Alkylated Twisted Amides: Consequences for Amide Bond Resonance and N–C Cleavage. Angew. Chem., Int. Ed. 2016, 55, 5062–5066. 10.1002/anie.201600919. [DOI] [PubMed] [Google Scholar]
- Li G.; Ma S.; Szostak M. Amide Bond Activation: The Power of Resonance. Trends Chem. 2020, 2, 914–928. 10.1016/j.trechm.2020.08.001. [DOI] [Google Scholar]
- Amide Bond Activation; Szostak M., Ed.; Wiley: 2022. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.