

SUMMARY
Viral encephalitis is a growing public health threat with limited diagnostic and treatment options. Simian immunodeficiency virus (SIV)-infected macaques are an established model for human immunodeficiency virus (HIV), and approximately 60% of untreated pigtail macaques rapidly progress to characteristic SIV encephalitis (SIVE). The immune responses of SIV-infected macaques are investigated in plasma, cerebrospinal fluid (CSF), and brain tissue to determine correlates with SIVE pathology. Macaques with SIVE show myeloid-dominant brain lesions with inflammasome activation in infected and bystander cells, as assessed by interleukin (IL)-1β, IL-18, and apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC), and elevations in monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, and tumor necrosis factor alpha (TNF-α). SIV-specific immunoglobulin (Ig)G in plasma and CSF is predictive of SIVE as early as 21 days post-inoculation; animals with SIVE continue to show negligible seroconversion 3 months after infection. This dichotomy in immune responses, wherein some macaques fail to initiate robust IgG responses and subsequently develop SIVE, provides insight into the pathogenesis and heterogeneous outcomes in viral encephalitis.
Graphical abstract

In brief
Outcomes in the SIV-infected macaque model of HIV, including SIV encephalitis (SIVE), are highly variable between animals. Castell et al. find that SIV-specific antibody responses predict protection from SIVE. Plasma and cerebrospinal fluid SIV-specific IgG correlates with reduced brain viral RNA and inflammasome activation and lower compartmentalized CNS cytokine release.
INTRODUCTION
Viral infections of the central nervous system (CNS) can cause clinical manifestations of encephalitis, with an incidence of approximately 19 cases per 100,000 person years globally.1 Although full recovery is possible, permanent neurological impairment is common, and the fatality rate has been estimated to be 10% in the US.2,3 Despite the severity of clinical disease, few evidence-based treatments are available to address viral encephalitis beyond supportive care.4,5
Viruses are the most common infectious cause of encephalitis, but due to diagnostic limitations, a specific virus is identified in less than 50% of patients.6,7 Possible etiologies include arboviruses, herpesviruses, paramyxoviruses, rhabdoviruses, and retroviruses. The epidemiology of these viral infections varies, but the frequency with which encephalitis occurs once an individual is infected is often low and highly unpredictable. The environment, viral virulence, and immune responses all play roles in disease pathogenesis.3 This complicates both the diagnosis and management of viral CNS infections.6
Human immunodeficiency virus (HIV) is known to invade the CNS early in infection, and roughly half of people experience neurological symptoms during acute infection.8 If untreated, infection can progress to HIV encephalitis (HIVE), which manifests clinically as HIV-associated dementia (HAD). Histopathologically, HIVE is characterized by myeloid-dominant encephalitis with multinucleated giant cells, which may be accompanied by microglial nodules.9 Infected macrophages and microglia represent a major source of virus within the brain, wherein viral replication and inflammasome activation lead to pyroptosis and tissue damage.10–12 Development of robust adaptive immune responses to infection has been demonstrated to be critical for protection against poor CNS outcomes; in particular, failure to seroconvert has been described as an early predictor of rapid progression in macaque models.13,14 However, the interactions of innate and adaptive immunity in determining the outcomes of HIV infection are complex and still poorly understood.
Despite the development of highly effective anti-retroviral therapies (ART) that prevent the development of HAD, people with HIV on ART continue to experience cognitive consequences of infection, most commonly limited to mild cognitive impairment but which also includes CNS viral escape and immune reconstitution inflammatory syndrome (IRIS).15–17 Uncovering the mechanisms of ongoing CNS dysfunction is needed to develop appropriate prevention and therapies to improve quality of life for people with HIV. A complete understanding of HIV-induced CNS inflammation is also important for improving outcomes in patients with limited access to the necessary diagnostic and treatment resources.18–20
Simian immunodeficiency virus (SIV) is a retrovirus closely related to HIV in structure, cellular tropism, and pathology.21 Both SIV-infected rhesus and pigtail macaques are well-established and valuable models for understanding early infection, the viral reservoir, and the complex CNS pathology of HIV.22,23 Neuropathogenic models, such as inoculation with the immunosuppressive swarm SIVdeltaB670 and neurovirulent clone SIV/17E-Fr, are especially useful for investigating the pathogenesis of viral infection in the CNS compartment, as approximately 60% of untreated infected macaques progress to prototypical SIV encephalitis (SIVE).24,25
Non-human primate models recapitulate important aspects of the human population, such as an immune system highly homologous to our own and a wide genetic diversity within the population. This allows for the investigation of how host factors affect outcomes of infection with a characterized virus in a controlled environment.21 Therefore, we sought to utilize the established SIV-infected macaque model as a tool to elucidate which aspects of the innate and adaptive immune response drive the variable CNS clinical outcomes seen in retroviral infection.
RESULTS
Cohort characteristics
Forty pigtail (Macaca nemestrina, n = 23, experimental cohort) and rhesus (Macaca mulatta, n = 17, validation cohort) macaques of mixed sex and age were assessed in this study (Figure 1). All macaques within this cohort were infected with the same viral stocks and received no treatment to control infection. The characteristics of individual animals are listed in Table S1. Macaques were classified as having encephalitis (SIVE) or no encephalitis (no SIVE) based on brain pathology at the study endpoint.
Figure 1. Cohort characteristics.
Demographics and available immunologic outcomes of historical cohorts (n = 40) of SIV-inoculated (SIVdeltaB670 and SIV/17E-Fr), untreated rhesus and pigtail macaques grouped by clinical outcome (no SIVE or SIVE). Measures are grouped by peripheral vs. CNS compartment, with the sample size for each indicated. Gray boxes indicate the figure number(s) where data are presented. Created with BioRender.com.
All pigtail macaques were young adult males that were inoculated with both SIVdeltaB670 and SIV/17E-Fr. The end of study for all pigtails was approximately 84 days post-inoculation (DPI); any individuals that were removed from study before 50 DPI were excluded from this cohort (range: 50–101 DPI). A subset of male, young adult pigtail macaques (n = 16) with similar terminal time points and readily available samples were selected for further characterization of CNS immune activation and pathology, as well as to study the mechanism using in vitro assays.
Macaques with SIVE have higher CSF viral loads compared to those without SIVE
Known clinical, virological, and immunological features were compared between the CNS outcome groups (SIVE and no SIVE) within the experimental cohort to identify any predisposing characteristics. Animal weights and plasma viral load were similar between groups over the course of infection, with the exception a single time point of higher plasma virus in macaques with SIVE 70 DPI (Figures 2A and 2B). Cerebrospinal fluid (CSF) viral loads were also similar until after the acute infection period, when viral load decreased to approximately 100-fold lower in macaques without SIVE, while macaques with SIVE maintained elevated CSF virus throughout infection (Figure 2C). These differences were significant starting 42 days post-infection through to the end of the study. A high CSF viral load is a well-established correlate of severe SIVE.26 The major histocompatibility complex (MHC) allele Mane-A1*084:01:01, which has previously been associated with lower CSF viral load in the pigtail model,27 was present in individuals in both groups (Figure 2D). All macaques had similar levels of circulating CD4 T cells, B cells, and CD8 T cells (Figures 2E, 2F, and S1).
Figure 2. Macaques with SIVE have higher CSF viral loads compared to those without SIVE (n = 23).
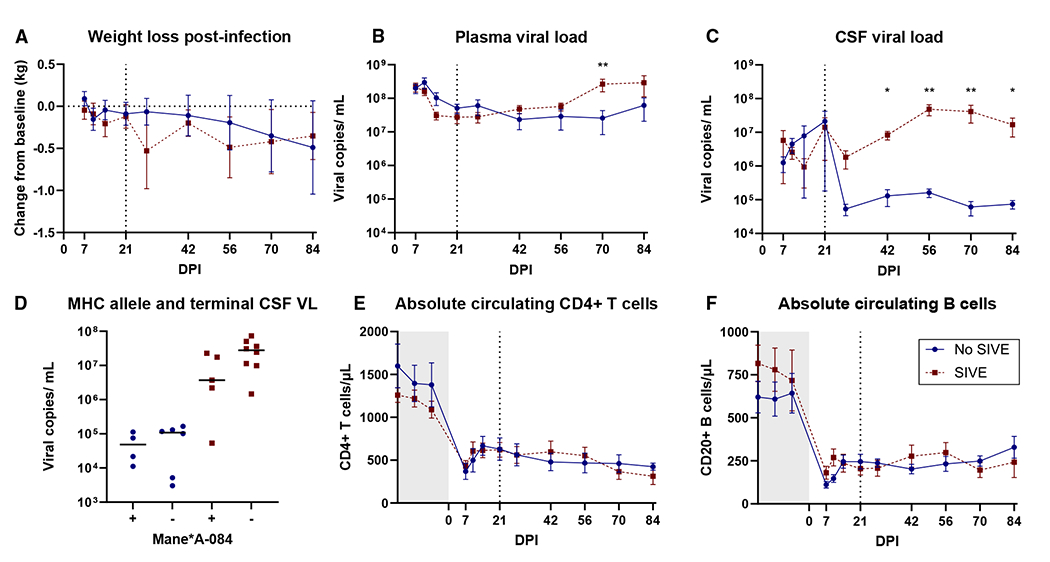
(A) No differences between experimental cohort groups were found in post-infection weight loss (n = 16).
(B) Plasma viral loads were different only at 70 days post-infection.
(C) CSF viral load consistently distinguished groups starting 42 days post-infection.
(D) CSF viral loads separated by Mane-A1*084:01:01 allele.
(E and F) Circulating CD4+ T cell (E) and B cell (F) counts did not distinguish between groups over the course of infection. Gray box indicates pre-infection measures, and the line at 21 DPI indicates when CNS outcome groups became distinguishable.
All comparisons between outcome groups were assessed by Mann-Whitney using Holm-šídák correction for multiple comparisons; points depict mean values with the standard error of the mean (SEM). *p < 0.05 and **p < 0.01. See also Figure S1.
Global cytokine elevations are present in CSF in SIVE
Circulating levels of ten proinflammatory cytokines and chemokines were measured in CSF from the experimental cohort through the first month of infection as well as the terminal time point using a Meso Scale Discovery (MSD) multiplex assay. There were no differences in the CSF levels of these cytokines between groups during the first month of infection; however, all measured cytokines (interleukin [IL]-15, IL-18, IL-1β, IL-6, interferon-gamma inducible protein (IP)-10, MCP-1, MIP-1α, MIP-1β, and tumor necrosis factor alpha [TNF-α]), with the exception of interferon (IFN)-γ, were elevated in the CSF of macaques with SIVE by the end of the study, while viremic macaques without SIVE overall had levels more similar to their pre-infection baselines (Figures 3A, 3C–3G, and S2).
Figure 3. Global cytokine elevations are present in CSF in encephalitis (n = 16).

(A and B) Experimental cohort terminal CSF cytokines (A) and terminal CSF:plasma ratios (B) were measured for ten cytokines. Bars indicate group median, and black symbols represent values below the lower limit of detection (LLOD).
(C–G) Longitudinal plasma and CSF levels are shown for IL-18 (C), IL-1β (D), MCP-1 (E), MIP-1α (F), and TNF-α (G). Points depict geometric mean with 95% confidence interval, and gray line indicates the LLOD.
Differences between CNS outcome groups were assessed by Mann-Whitney using Holm-šídák correction for multiple comparisons. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figures S2 and S3.
The same cytokine panel was used to assess plasma through the first month of infection and from the terminal time point (Figure S2). There were no differences in the plasma levels of these cytokines between groups during acute infection or at the study endpoint, with the exception of higher IL-15 in macaques with SIVE.
To assess whether cytokine elevations in the CSF were likely to have been driven by local production, CSF:plasma cytokine ratios were calculated and compared between groups. Most of the measured cytokines in macaques showed a baseline CSF:plasma ratio near 1, suggesting that the blood-CSF barrier is permissive to the passage of these cytokines even in an immune quiescent state. In contrast to the other analytes, the CSF:plasma ratio of IL-18 had a median value of 0.05 in uninfected macaques, indicating that IL-18 may be specifically excluded from the CNS compartment.
When CSF cytokines were normalized to plasma cytokine levels for each analyte, only a subset distinguished the two CNS outcome groups (Figure 3B). These included inflammasome-associated cytokines IL-18 and IL-1β, macrophage trafficking cytokines MCP-1 and MIP-1α, and the proinflammatory cytokine TNF-α. IL-18, 1L-1β, and MCP-1 all showed selective elevations (median ratios >2) in the CNS compartment of macaques with SIVE, indicating that these cytokines may be locally produced specifically in the context of SIVE. In particular, IL-18 showed a 115-fold higher CSF:plasma ratio in macaques with SIVE compared to those without SIVE. Previous work from our lab demonstrated that MCP-1 and IL-6 were specifically elevated in the CSF of macaques with SIVE.28 Our results recapitulate these findings, although they suggest that CSF IL-6 may be derived partially from elevated plasma IL-6, while MCP-1 may be released more locally within the CNS.
To confirm that the cytokine responses observed after SIV infection were not a species-specific phenomenon, the same cytokine panel was also assessed in the terminal plasma and CSF of SIV-infected rhesus macaques as a validation cohort (Figure S3). The inflammatory profile within the CNS was less pronounced in rhesus macaques with SIVE but similarly showed elevations in IL-18, MIP-1α, and MIP-1β. When CSF:plasma ratios were compared, IL-18 remained significantly elevated in these animals. Although IL-6 and MCP-1 were not different between outcome groups, high inter-animal variability and a small sample size may have limited our ability to identify a difference in the trend toward higher levels of these cytokines in the CSF of rhesus macaques with SIVE.
Inflammasome proteins are associated with myeloid cells in the brain parenchyma of macaques with SIVE
Immunohistochemistry (IHC) for inflammasome proteins and a myeloid cell marker was performed to assess the extent of inflammatory change within the parenchyma of the brain. Basal ganglia collected at necropsy from macaques with and without SIVE were stained for the myeloid marker ionized calcium binding adaptor (Iba)-1, as well as for inflammatory cytokines IL-1β and IL-18 and the inflammasome adaptor protein ASC. Iba-1 is constitutively expressed by myeloid lineage cells, including monocytes, macrophages, and microglia.29 Macaques with SIVE showed subjectively elevated numbers of Iba-1-positive cells commonly concentrated in perivascular areas; these cells showed a more rounded and less ramified phenotype than in non-encephalitic brains (Figure 4A). The signal for IL-1β was minimal in all sections of basal ganglia for both outcome groups (Figure 4B). Given that absolute levels of IL-1β are lower than many other inflammatory cytokines in the CSF, this finding may be consistent with expected levels of protein within the tissue. Although IL-18 is constitutively expressed in myeloid cells,30 the signal for IL-18 protein appeared to be increased in cells within the basal ganglia of macaques with SIVE (Figure 4C). Similarly, as an adaptor protein for inflammasome activation, ASC is constitutively expressed within the cytoplasm of myeloid cells.31 However, there was a dramatic difference in the level of ASC expression in the basal ganglia between macaques with and without SIVE (Figure 4D). The quantified whole-tissue area of both IL-18- and ASC-positive staining was greater in macaques with SIVE, and ASC levels were closely correlated with the CSF levels of IL-18, suggesting a general upregulation of the inflammasome pathway within the CNS compartment (Figures 4E–4G). In addition to increased positive staining for both IL-18 and ASC within individual cells, the overall increase in brain myeloid cells, as indicated by Iba-1, further increased the levels of these inflammasome components within the basal ganglia of macaques with SIVE.
Figure 4. Inflammasome proteins are associated with myeloid cells in the brain parenchyma of macaques with SIVE (n = 16).

(A–D) Images show white matter at 4× (left) and 10× (right) from experimental cohort basal ganglia without (top) and with (bottom) SIVE stained against Iba-1 (A), IL-1β (B), IL-18 (C), and ASC (D) with DAB. Inset images and arrows indicate multinucleate giant cells.
(E and F) Quantified IL-18 (E) and ASC (F) signals over total slide area were different between outcome groups by Mann-Whitney U (IL-18 p = 0.0047, ASC p < 0.001).
(G) ASC correlated with CSF IL-18 using linear regression (R2 = 0.9631, slope ≠ 0 p < 0.0001).
SIV RNA co-localizes with inflammasome proteins but is largely excluded from the brain parenchyma of macaques without SIVE
Dual in situ hybridization (ISH) for SIV RNA was performed in conjunction with IHC for either the IL-18 or ASC protein to investigate the localization of activated innate cells with infected cells. ISH revealed large differences in the distribution of viral RNA within the brain parenchyma between outcome groups. In macaques with SIVE, SIV RNA was present throughout the basal ganglia and showed a similar pattern of distribution to both IL-18 (Figures 5A and 5B) and ASC (Figures 5C and 5D) proteins. This primarily reflected the fact that for all three markers, positive cells were most frequently located in perivascular areas. While cells double positive for either SIV and IL-18 or SIV and ASC were present, they were not the predominant cell population, indicating that direct infection may not be a requirement for increased expression of inflammasome components. In contrast, macaques without SIVE had negligible SIV RNA detected within the parenchyma of the basal ganglia. Consistent with IHC alone, constitutive levels of ASC and IL-18 were present but at much lower levels than in macaques with SIVE. Due to the limited sensitivity of ISH for the detection of a virus, qPCR of basal ganglia was used to quantify the brain viral load (Figure 5E). The level of SIV RNA detected within tissue correlated with the same measure in CSF; CSF levels of SIV RNA and IL-18 both corresponded with the ISH results observed in each outcome group (Figures 3A and 5F).
Figure 5. SIV RNA co-localizes with inflammasome proteins but is largely excluded from the brain parenchyma of macaques without encephalitis.

(A–D) Images show 4× (left) and 10× (right) ISH for SIV RNA (red) from experimental cohort basal ganglia without (top) and with (bottom) SIVE stained against IL-18 (A and B) and ASC (C and D) with DAB. Arrows indicate multinucleate giant cells.
(E) Basal ganglia SIV RNA was different between outcomes by Mann-Whitney; ***p < 0.001.
(F) SIV RNA in CSF and basal ganglia was correlated using Spearman’s rank (r = 0.7811, p = 0.0009).
SIV-specific IgG responses define CNS outcomes of SIV-infected macaques
Because the onset of innate immune signaling appeared similar between outcome groups during the acute period and diverged later in the course of infection, we sought to assess if the adaptive immune system played a role in determining if our experimental cohort would develop SIVE. Striking differences in plasma SIV-specific immunoglobulin (Ig)G titers were observed between macaques with and without SIVE. By the study endpoint, the majority of macaques without SIVE had developed robust responses against SIV. However, macaques with SIVE showed negligible anti-SIV IgG antibody responses, which were often at comparable levels to their pre-inoculation reactivity (Figure 6A). The same pattern was observed in our validation cohort of SIV-infected rhesus macaques (Figure S4). In contrast, while overall lower in magnitude, plasma anti-SIV IgM responses persisted in infected macaques at the study endpoint regardless of CNS outcome, albeit at lower titers in macaques with SIVE (Figure 6B). The absence of SIV-specific IgG in the plasma of macaques with SIVE did not appear to be the consequence of a global deficiency in B cells or their antibody-secreting function, as these animals showed similar levels of circulating B cells and total plasma IgG to those that were able to mount robust antiviral antibody responses (Figures 2F, 6C, and S4).
Figure 6. SIV-specific IgG responses define CNS outcomes of SIV-infected macaques, likely derived from the periphery, and differentiate groups early in infection.

(A and B) Experimental cohort plasma SIV-specific IgG (A, n = 23) and IgM (B, n = 14) titration curves were graphed using nonlinear regression to generate sigmoidal (4PL) curves; the extra sum-of-squares F test rejected the null that one curve would fit both CNS outcomes (p < 0.0001).
(C) Total IgG was similar between groups using Mann-Whitney (gray area indicates estimated reference range for normal macaque plasma IgG).
(D and E) CSF SIV-specific IgG (D, n = 16) and IgM (E) titration curves were graphed with the same method; the null that one curve would fit both datasets was rejected (p < 0.0001) for IgG only. Points depict mean values with SEM.
(F) Total IgGs in CSF were similar between outcome groups using Mann-Whitney.
(G) SIV-specific IgG titers were correlated between the plasma and CSF using linear regression (R2 = 0.8944, slope ≠ 0 p < 0.001).
(H) The ratios of SIV-specific IgG to total IgG were similar between compartments within outcome groups.
(I) Plasma SIV-specific IgG titer was measured over the course of acute infection in untreated pigtail macaques.
Differences between CNS outcome groups were assessed by Mann-Whitney using Holm-šídák correction for multiple comparisons. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S4.
SIV-specific IgG in the CNS may cross passively from the periphery and differentiates groups early in infection
Since the majority of inflammatory changes in macaques with SIVE appeared to be localized to the CNS compartment, antibody responses within this space were also assessed. Although antibody titers were consistently much lower in the CSF when compared with plasma, the same pattern in anti-SIV IgG and IgM was observed using CSF from the study endpoint. Macaques without SIVE consistently expressed anti-SIV IgG responses in the CSF, while animals with SIVE had negligible reactivity (Figure 6D). CSF anti-SIV IgM antibodies were only present at very low levels and showed no differences between CNS outcome groups, consistent with the size of the IgM molecule and poor diffusion across the blood-brain barrier (BBB) (Figure 6E).32 To assess the likelihood that CSF anti-SIV IgG may be primarily derived from the periphery, total IgG was measured in both compartments and compared to anti-SIV titers (Figure 6F). When anti-SIV IgG titers between plasma and CSF were directly correlated using the data from macaques without SIVE, macaques with SIVE were omitted because they did not have significant titers; a close correlation (R2 = 0.89) between antibody responses in these compartments was observed (Figure 6G). When SIV-specific IgG:total IgG ratios were compared between plasma and CSF, macaques showed similar ratios between compartments, consistent with the passage of anti-SIV IgG occurring non-selectively across the blood-CSF barrier. Total IgG levels within the CSF were equivalent between CNS outcome groups, with the exception of two macaques with SIVE who showed 3.0- to 5.5-fold elevation in CSF IgG relative to the cohort mean (Figure 6H). Whether this difference is consistent with BBB dysfunction or local production of IgG requires further investigation.
After characterizing the SIV-specific antibody responses at the study endpoint, IgG responses in macaque plasma samples from the acute infection period were measured to determine how early in infection CNS outcome groups could be distinguished (Figure 6I). Based on the longitudinal anti-SIV IgG plasma titers, macaques with SIVE showed minimal seroconversion against SIV and no additional development of antibody responses 14 days post-infection. In contrast, macaques that did not develop SIVE showed a steady increase in anti-SIV IgG plasma titers over the course of infection. CNS outcome groups were distinguishable as early as 21 days post-infection using this assay. Additionally, the plateau of SIV-specific antibody responses occurred prior to the increase in IL-18 production in the CNS, suggesting that SIV-specific antibodies may have a protective role in this space.
SIV-specific antibodies may protect the CNS by preventing CNS macrophage infection
To determine if direct neutralization of SIV infection played a role in protecting the CNS, two in vitro assays were used. The efficacy of macaque plasma from the study endpoint in blocking SIV integration was directly assessed using a TZM-bl reporter cell line; these cells luminesce through the expression of luciferase when HIV or SIV viral integration occurs. When grouped by CNS outcome, plasma neutralization curves from this assay show the same pattern as SIV-specific IgG titers, with macaques without SIVE showing a robust neutralization capacity that prevents viral integration and macaques with SIVE showing minimal inhibition of SIV (Figure 7A). At 1:105 and 1:106 dilutions, plasma from macaques with SIVE showed a lower neutralization capacity than the IgG control, suggesting a potential for antibody-dependent enhancement in SIV infection in these individuals.
Figure 7. SIV-specific antibodies may protect the CNS by preventing CNS macrophage infection.

(A) The in vitro neutralization capacity (n = 12) of plasma showed CNS outcome accounted for the largest portion of variation using a two-way ANOVA (p < 0.0001). Multiple comparisons were performed using Dunnett’s correction; differences from IgG control are designated with red asterisks for the encephalitis group and blue asterisks for no encephalitis.
(B) Plasma immunoglobulin was used to inhibit infection in pigtail MDMs (n = 8). Analysis was performed as above; DPI × CNS outcome and CNS outcome collectively accounted for the largest portion of variation (p = 0.0009 and 0.0192, respectively). Points depict mean values with SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S5.
To confirm that IgG was playing a direct role in neutralization and that SIV infection could be inhibited in CNS target cells, we purified plasma Ig from both outcome groups and assessed its neutralization capacity in macaque monocyte-derived macrophages (MDMs). Ig from macaques without SIVE showed a robust capacity to prevent SIV infection and proliferation in MDMs, while Ig from macaques with SIVE showed similar viral kinetics to both nonspecific Ig treatment and SIV infection alone (Figure 7B). Interestingly, this protective effect appears to be limited to SIVdeltaB670 and did not affect SIV/17E-Fr infection in vitro (Figure S5). The results from both cell culture experiments support a role for SIV-specific IgG in directly inhibiting SIV infection in macrophages and protecting the CNS.
DISCUSSION
Although the occurrence of viral encephalitis is sporadic, the consequences are often devastating for patients.2,33 In 2019, an estimated 1.4 million cases of encephalitis resulted in about 90,000 patient deaths globally.1 The World Health Organization emphasizes that the true incidence of encephalitis is difficult to estimate and that this condition represents a growing global threat due to factors such as emerging viruses and climate change.34 A better understanding of the determinants and pathogenesis of infectious encephalitis is critical to addressing the unpredictability of clinical disease and the limited treatment options available.
This study demonstrates the importance of antibody responses in protecting the CNS from viral infection, as shown in two species of macaques wherein antibodies help to exclude virus from invading the CNS. In untreated SIV-infected macaques, SIVE was characterized by highly upregulated innate immune responses in the CNS compartment, including elevated CSF cytokines, increased macrophage trafficking, and upregulated inflammasome proteins within brain tissue. Despite these changes, the immune response of macaques with SIVE was ineffective at curtailing viral spread, as these animals also showed viral RNA distributed throughout the brain parenchyma. In contrast, in infected macaques that did not develop SIVE, immune activation within the CNS compartment remained relatively quiescent, with indicators of innate immune inflammation remaining more similar to those of uninfected macaques. The antibody responses of these clinical outcome groups differed markedly; macaques with SIVE failed to effectively seroconvert against SIV, while macaques without SIVE showed robust anti-SIV IgG responses. Anti-SIV IgG titers could be used to predict CNS clinical outcome as early as 21 days post-infection. Importantly, the anti-SIV antibodies were capable of neutralizing viral infection in vitro in macrophages, while the cells in the brain were shown to be infected.35
In our animal model of SIVE, uncontrolled viral replication within the CNS was accompanied by diffuse upregulation of inflammasome proteins within microglia and invading macrophages. Inflammatory lesions were concentrated around blood vessels, where both infected and bystander perivascular macrophages showed activation. Concurrently, locally elevated levels of the inflammasome cytokines IL-18 and IL-1β were found in the CSF. These findings are consistent with the prototypical pathology of SIVE, which is characterized by microglial nodules, myeloid-dominant perivascular infiltrates, and multinucleate giant cells.36 This inflammatory pattern is similar to HIVE, which classically presents with white matter lesions of microglial nodules and multinucleate giant cells sometimes accompanied by astrocytosis and myelin loss.9 Similarly, this investigation recapitulated previous findings that correlated the cytokines MCP-1 and IL-6, associated with macrophage trafficking and pyrogenic signaling, respectively, within the CNS with SIVE.28,37 These cytokines are also found in encephalitis of other viral etiologies.38–40
There is a diverse array of viruses that infect the CNS and lead to encephalitis, and some similarities have been observed in the pathogenesis and immune responses to these infections, including inflammasome activation within the brain.6,41,42 Although these innate immune pathways can play an important role in reducing viral replication, the nonspecific nature of inflammasome signaling can contribute to disease pathogenesis through tissue damage and pyroptosis of bystander cells in a variety of conditions, including HIV.10,42–44 Due to the unique structure of the brain and its limited capacity for repair, extensive activation of inflammasome pathways can be especially detrimental to CNS outcomes in inflammatory disease. In particular, activation of the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome has been associated with the pathogenesis of a wide range of brain insults, including stroke, traumatic brain injury, drugs of abuse, neurodegenerative diseases, and encephalitic viruses.45 Because of the role of inflammasome activation in tissue damage, understanding and interrupting the mechanisms that initiate these pathways represents a promising therapeutic target for many inflammatory conditions.
Similar to innate immunity, common trends in adaptive immune response occur across agents that cause viral encephalitis.46–48 These include the importance of B cells and antibody responses in mediating protection or recovery from encephalitis.49,50 For example, in tick-borne encephalitis virus (TBEV)-infected patients with encephalitis, serum anti-TBEV IgG responses have been found to be predictive of neurologic outcomes. A subset of these patients failed to seroconvert against TBEV, and this group showed more severe disease and a higher rate of mortality.51 Mouse models of alphavirus encephalomyelitis using Sindbis virus have been used to extensively characterize the role of antibodies in resolving CNS infection. While IgM and IgG knockout mouse strains were both able to clear brain virus, mice lacking both Ig subtypes showed persistent brain infection.52,53 Similarly, knockout mouse strains were used to demonstrate the critical role of the antibody response in protection from encephalitis caused by Rift Valley fever virus.47 In addition, earlier work in SIV-infected pigtail macaques supports the role of the antibody response in the specific context of retroviral encephalitis; seroconversion against SIV was closely correlated with lower brain viral loads in this model.13
Antibodies can mediate immune responses through multiple mechanisms in addition to their role in direct neutralization of pathogens. These signaling functions enable antibodies to play a role in the elimination of infected cells by providing targets to innate and adaptive effectors. Antibody binding can promote antigen presentation to T cells and complement activation. Complement not only leads to target cell lysis but has also been shown to play an important role in both B cell activation and myeloid cell signaling.54–56 Non-neutralizing antibodies that bind to virus or viral proteins can also tag them for uptake and degradation by myeloid cells or antibody-mediated cytotoxicity.57 A growing body of research on antiviral responses is supporting the importance of non-neutralizing antibody responses in control and recovery from infection.58 In vitro investigation of plasma IgG from macaques demonstrated the direct efficacy of anti-SIV antibodies in neutralizing cell-free virus. While this represents a mechanism by which antibodies mediate protection of the CNS, it is possible that antibodies or other B cell functions also contribute to indirect mechanisms of protection from encephalitis, such as through immune crosstalk. Previous work in the dual-inoculated pigtail model demonstrated that both viral strains, SIVdeltaB670 and SIV/17E-Fr, are present in macaques with SIVE; CNS virus in macaques without SIVE is not as well characterized.59,60
The traditional role of B cells is in antigen presentation and antibody production; however, activated B cells can migrate to sites of inflammation and engage in the local immune response though cellular interactions that are an area of ongoing research.50,61 The role of B cells in local immune crosstalk and cytokine production can be challenging to define due to the broad diversity of phenotypes, which is becoming appreciated.62 Investigations focused on the role of B cells in neurodegenerative conditions have identified both proinflammatory subsets, which can drive pathology, and immunomodulatory subsets, which may be neuroprotective. For example, B cells have become particularly salient in the context of multiple sclerosis (MS), an autoimmune condition largely characterized by T cell pathology which, for some patients, is highly responsive to B cell-depleting therapies such as rituximab.63 Conversely, B cells have demonstrated neuroprotective effects in the context of traumatic brain injury, where their dynamic cytokine expression profile modulated the inflammatory phenotype of infiltrating myeloid cells.64 In HIV infection, B cell responses and antibody production, particularly within the intrathecal compartment, are known to be dysregulated, and the role they play in neuropathology is still unclear.65 As in other infectious disease contexts, current evidence suggests that robust early antibody responses are protective, while ongoing immune activation during chronic disease may contribute to damage and dysfunction within the CNS.66,67 Our work indicates that differential B cell and antibody responses are an important predictor of CNS outcome from viral infection; future investigations could elucidate the specific cell phenotypes and tissue niches that contribute to protection.
With the exception of herpes simplex encephalitis, for which acyclovir is a critical treatment, there are few evidence-based options for the treatment of viral encephalitis.5 Intravenous Ig administration (IVIG) is one therapeutic option sometimes pursued for patients with encephalitis of multiple etiologies. Reports on the efficacy of this treatment in viral encephalitis are inconsistent, which may be a factor of the diverse protocols, comorbidities, and viral agents for which IVIG has been applied.68 One study in pediatric enteroviral encephalitis reported that IVIG administration resulted in lower levels of a subset of plasma cytokines, including IL-6 and IFN-γ, 12–24 h after administration, suggesting a role in immune crosstalk.69 In support of the immunomodulatory role of antibody administration, IVIG is has been demonstrated to be an effective option for the treatment of autoimmune encephalitis.70 Although a number of hypotheses for this effect have been proposed, the mechanism of action has yet to be established.
Here, we have demonstrated a dichotomy in CNS outcomes in SIV-inoculated macaques that provides insights into the pathogenesis of viral encephalitis. Adequate initiation of the adaptive immune response is necessary for the production of antigen-specific IgG antibodies, but this fails to occur in a subset of individuals. Control of cell-free virus within the CNS by antibodies reduces nonspecific innate inflammatory responses, including inflammasome activation, which play a primary role in driving inflammation and tissue disruption within the brain parenchyma. Further work is needed to elucidate what aspects of the early immune response interrupt the development of effective antibody responses.
Limitations of the study
With the development of highly effective ART for HIV, the neuropathology of chronic HIV infection has become less severe without a concurrent decrease in the prevalence of cognitive impairment.16 Although the inflammatory and histopathologic mechanisms likely driving neurocognitive changes are more subtle in this setting, SIVE also represents a valuable tool to gain a broader understanding of the immunology and pathogenesis of viruses that cause encephalitis.24 In addition, although late-stage HAD and the accompanying HIVE have become rare clinical occurrences in the US, significant barriers to both diagnosis of and accessible treatment for HIV persist globally.20 Therefore, a robust understanding of the predisposing factors and lasting consequences of HIVE remains important.
Similarly, this investigation does not explore how the development of antibody responses is affected by ART. In particular, initiation of treatment very early in infection could potentially reduce the amount of viral antigen present and abrogate the protective effect of the early adaptive immune response. This could be a contraindication for early ART if therapy does not provide a commensurate benefit in reduction of the viral reservoir.
In people with HIV, time to diagnosis, anti-retroviral treatment history, comorbid medical conditions, sociodemographics, and other factors all come together to mediate patient clinical outcomes.71 In addition, understanding the neurocognitive consequences of HIV is further restricted by limitations in accessing the CNS. Non-human primate models of HIV closely recapitulate pathology while controlling many of these extrinsic factors and allow direct study of the brain in different phases of infection. For these reasons, these models represent valuable tools for understanding the complex interplay of virus, host, and environment that determines outcomes of infection.
Although the aim of this study was to utilize an existing animal model to understand generalizable aspects of viral CNS pathology, SIV infection involves distinct mechanisms of infection that may distinguish it from other etiologies of viral encephalitis. Because of this, it is possible that the findings of this work may not be generalizable beyond SIV and similar retroviruses. The importance of specific B cell subsets or antibody specificities in the context of CNS outcomes remains unexplored and represents an important area for future studies.
In addition, the macaque samples in this investigation were all banked tissues from completed studies utilized in a retrospective manner. This limited the ability of this work to explore a larger cohort of animals or to ask prospective questions regarding pathologic outcomes. The inclusion of macaques from different years and study cohorts introduces the possibility of bias from uncontrolled extrinsic factors, such as animal origin or changes in husbandry practices. Similarly, the individuals available have limited our ability to investigate potential sex differences in this work.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rebecca T. Veenhuis (rterill1@jhmi.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
STAR★METHODS
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
A retrospective analysis of banked plasma, CSF, and brain tissue samples was performed using 40 rhesus (Macaca mulatta, n = 17) and pigtail (Macaca nemestrina, n = 23) macaques from multiple independent study cohorts. Both male and female macaques of varied ages were included in this analysis; summary demographics are given in Figure S1 and individual demographics are listed in Table S1. All macaques were specific pathogen free for SIV, simian T cell leukemia virus, and simian type D retrovirus before inoculation. For all study cohorts, macaques were intravenously inoculated with a single stock of the neurovirulent clone SIV/17E-Fr (10,000 50% animal infectious dose [AID50]) and immunosuppressive swarm SIVdeltaB670 (50 AID50), as previously described,72,73 and did not receive any ART. For this investigation, macaques were selected and assigned to a binary clinical outcome group based on the development of SIVE as assessed by a veterinary pathologist using histopathological results post necropsy. Hematoxylin and eosin stained frontal and parietal cortex, basal ganglia, thalamus, midbrain, and cerebellum were reviewed by two pathologists blinded to experimental intervention.
All macaques were housed in the same animal facility and received ad libitum food (Purina 5038) and water, as well as daily enrichment from behavior staff. Macaques were able to visualize and interact with conspecifics in the room but were otherwise individually housed.
Sample collection was performed using a standardized timeline for all cohorts; this included 3 baseline samples, each at least 2 weeks apart, before inoculation followed by post-inoculation samples at days 7, 10, 14, 21, and 28. Macaques were sedated with 10 mg/kg intramuscular ketamine at each timepoint for CSF collection from the cisterna magna and blood collection from femoral venipuncture. Blood was collected into syringes with acid citrate dextrose solution at a 5:1 ratio of blood to anticoagulant. At study endpoint, macaques were deeply anesthetized with ketamine and pentobarbital. CSF and blood were collected as above before euthanasia with pentobarbital and whole-body perfusion using phosphate-buffered saline (PBS). Brain tissue was either flash frozen in liquid nitrogen or fixed using Streck tissue fixative (STF) (2005–2012) or Histochoice (2012–2013) for 1 week before tissues were trimmed and embedded in paraffin.
All procedures were approved by the Johns Hopkins University Institutional Animal Care and Use Committee and were conducted in accordance with the guidelines set forth in the Animal Welfare Regulations and the Guide for the Care and Use of Laboratory Animals. These studies were conducted within a fully AAALAC accredited facility.
Cell lines
The use of the TZM-bl cell line is described further in the method details. TZM-bl cells were maintained using complete DMEM media (Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, and 100 U/mL penicillin-streptomycin). Cells were incubated with 0.05% trypsin in PBS for 5 min to resuspend cells for passage. CEM cells were used for SIV growth and purification; cells were maintained suspended in flasks using R10 media (Roswell Park Memorial Institute (RPMI) 1640 Medium supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, and 100 U/mL penicillin-streptomycin). All cells were incubated at 37°C in a humidified, 5% CO2 atmosphere.
Primary cell cultures
Pigtail monocyte-derived macrophages were derived from healthy pigtail macaque whole blood. The differentiation and use of these cells are described further in the method details; cells were maintained in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 0.2 mM sodium pyruvate, 50 ng/mL M-CSF, and 4 μg/mL gentamicin.
METHOD DETAILS
Cytokine measurement
A panel of ten inflammatory and chemoattractant cytokines were measured in plasma and CSF samples using a custom Meso Scale Discovery (MSD) U-PLEX non-human primate multiplex assay. This platform uses electrochemiluminescent detection to perform a sandwich immunoassay for multiple analytes in a single well. IFN-γ, IL-15, IL-18, IL-1β, IL-6, IP-10, MCP-1, MIP-1α, MIP-1β, and TNF-α were assessed using a 96-well assay per the manufacturer’s recommended protocol. Values which fell below the lower limit of detection (LLOD) for each analyte were assigned a value of ½ the LLOD.
Immunohistochemistry and in situ hybridization
All staining was performed using the Leica Bond RXm. Recommended Leica protocols were followed for IHC; Advanced Cell Diagnostics (ACD)’s recommended protocol was followed for use of RNAscope for ISH. In brief, basal ganglia was sectioned onto positively charged slides before placement in the Leica Bond RXm for baking and dewaxing. Primary antibodies against Iba-1, IL-18, IL-1β, and ASC were diluted (Table S2) in antibody diluent (Leica AR9359) and staining was performed with the Bond Polymer Refine Detection Kit (Leica DS9800) using IHC Protocol F or F rabbit for mouse and rabbit antibodies, respectively. Per manufacturer product sheets, antibodies against IL-18 and IL-1β were nonspecific for precursor versus cleaved cytokine. For dual staining using ISH and IHC, staining was performed sequentially for SIV RNA (ACD 312818) followed by IHC for IL-18 or ASC. ISH was performed using the Bond Polymer Refine Red Detection Kit (Leica DS9390) and the RNAscope 2.5 LS reagent kit RED (ACD 322150) using a 20 min epitope retrieval (ER2 at 95°C) and 1 min hybridization. After staining, slides were rinsed in deionized water before dehydration in alcohol (IHC only) or baking (ISH and IHC). Slides were submerged in xylene before coverslips were applied using the Leica Cv5030.
Slides were scanned using a Zeiss Axioscan 7 and uploaded to a central repository (Proscia Concentriq). Image analysis of whole-slide images was performed using QuPath 0.4.3 using a single threshold to calculate the percentage of tissue with positive signal for all slides.74
SIV-specific antibody and total IgG measurement
An enzyme-linked immunoassay (ELISA) was performed to detect SIV-specific antibodies within the plasma and CSF of SIV-infected macaques. Adsorbent 96-well plates (Invitrogen) were coated overnight with 1.5 μg/mL SIVmac251 lysate (ZeptoMetrix) followed by a 3-h blocking incubation (BD OptEIA Assay Diluent). A single lot of viral lysate was used for all assays and demonstrated robust cross-reactivity using plasma from pigtail macaques inoculated with SIVdeltaB670 and SIV/17E-Fr. Plates were washed between steps using BD OptEIA Wash solution. A serial dilution of each sample was performed in triplicate and incubated for 2 h, before the addition of a 1:5000 dilution of IgG (Goat anti-monkey IgG-HRP, Abcam #ab112767) or IgM (Goat anti-monkey IgM-HRP, Sigma #SAB3700780) secondary antibody for 1 h (at 37°C and 5% CO2). A 1M phosphoric acid stop solution was added after 15 min of development with TMB substrate (BD OptEIA Substrate Reagents A and B), and plate absorbance was read at 450nm. Sigmoidal 4PL antibody titration curves were generated for each macaque sample and used to interpolate a calculated titer for that individual using Prism 10.1.2 (GraphPad Software, Boston, Massachusetts). The threshold for determining antibody titer was assigned a value 3-times the mean absorbance of the negative control.
Total IgG was measured using a commercial ELISA (abcam IgG Monkey ELISA kit, ab190549). Manufacturer’s protocol was followed to measure IgG from plasma (diluted 1:80000) and CSF (diluted 1:800).
Immunoglobulin purification from plasma
Plasma immunoglobulin was purified using a Nab Protein A/G Spin Kit (ThermoFisher 89950) and concentrated using Amicon Ultra Centrifugal Filters (Millipore UFC8100). Purified immunoglobulin was quantitated by ELISA (abcam IgG Monkey ELISA kit, ab190549) per the manufacturer’s protocol using a 1:200000 dilution.
TZM-bl SIV neutralization assay
TZM-bl cells were obtained from the Center for AIDS Reagents (CFAR ID 5011). This cell HeLa-derived cell line was developed from JC.53 cells (which stably express CD4 and CCR5) to express luciferase and galactosidase under the control of the HIV-1 promotor. As this cell line is highly sensitive to infection with both HIV and SIV, it was utilized to assess virus neutralization following previously validated methods.75 Briefly, cells were plated 1 x 104 cells/well in 100 μL of growth medium (Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, and 100 U/mL penicillin-streptomycin) in 96-well plates and allowed to incubate for at least 6 h before beginning the assay. A SIVdeltaB670 and SIV/17E-Fr combined viral stock, of the same virus ratio used in the animal studies, was used to inoculate TZM-bl cells at a concentration of 8 x 106 viral RNA copies/mL after 1 h incubation with serial dilutions of each heat-inactivated plasma sample. Luminescence was developed using a luciferase kit (Promega Bright-glo E2620) 48 h after inoculation per manufacturer’s directions; signal was read using a SpectraMax iD5 (ROM v1.2 b64).
Primary MDM SIV neutralization assay
Phlebotomy was performed as described above to collect healthy pigtail macaque whole blood. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using a Percoll density gradient. PBMCs were plated at 2 x 106 cells/well in 3 mL of medium (DMEM supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 0.2 mM sodium pyruvate, 50 ng/mL M-CSF, and 4 μg/mL gentamicin) and allowed to differentiate for 7–10 days. Plates were then washed with PBS to remove non-adherent cells. A SIVdeltaB670 and SIV/17E-Fr combined viral stock, of the same virus ratio used in the animal studies, was incubated for 1 h with 1 μg/mL purified immunoglobulin from each plasma sample before inoculating cells at a concentration of 8 x 106 viral RNA copies/mL. Cell supernatants were collected after 3, 7, 10, and 14 days of infection. Viral RNA was isolated from supernatant using the QIAamp MinElute Virus Spin Kit (Qiagen 57704 per manufacturer’s recommendations) and quantified by RT-qPCR using a BIORAD CFX96. The following primers were used for SIV gag (forward, SIV21F 5′-GTCTGCGTCATCTGGTGCATTC-3′ reverse, SIV22R, 5′-CACTAGGTGTCTCTGCACTATCTGTTTTG-3′ probe, SIV23, FAM-5′-CTTCCTCAGTGTGTTTCACTTTCTCTTCTG-3′-BH1) with cycle conditions of 50°C for 30 min, 94°C for 15 min to reverse transcribe RNA, followed by 45 cycles of PCR at 94°C for 15 s, 55°C for 15 s, and 60°C for 30 s.
Flow cytometry, viral load measurement, and MHC genotyping
Flow cytometry to phenotype circulating leukocytes, viral load measurement in plasma, CSF, and basal ganglia using RT-qPCR for SIV gag, and pigtail MHC genotyping for the Mane-A1*084:01:01 allele were performed as part of the original study design for these macaque cohorts. These data were utilized in the current work as previous studies did not assess macaques on the basis of clinical outcomes (i.e., SIVE). More information on the specific techniques used to obtain these results is available in previous publications.76–78
In brief, flow cytometry was performed using whole blood stained with antibodies for CD3, CD4, CD8, and CD20 (Table S2) for 20 min at room temperature, followed by a 10-min fixation with BD FACS Lysing Solution (BD Biosciences). The samples were analyzed by a single technician using a FACSCalibur (BD Biosciences) or a BD LSRFortessa (BD Biosciences) flow cytometer with each animal run on a consistent platform.
Viral RNA was isolated from 140 μL of plasma or CSF using the QIAamp Viral RNA Minikit (Qiagen) according to the manufacturer’s protocol. For basal ganglia, 50 mg of tissue was homogenized for 30 s in lysing matrix D (MP Bio) and RNA STAT −60 using the Fast Prep-24 homogenizer (MP Bio). After a 5 min incubation, 200 μL chloroform was added for 15 s of shaking and 3 min of incubation. Samples were then centrifuged at 14,000g for 15 min at 4°C. The aqueous portion was incubated overnight at −20°C with 500uL cold 2-isopropanol. Samples were centrifuged as before to remove the 2-isopropanol, washed in cold 70% ethanol, and centrifuged at 10,000g for 5 min at 4°C to remove the ethanol. Pellets air dried for 15 min, then total RNA was isolated using the RNeasy kit (Qiagen) according to the manufacturer’s protocol. Isolated RNA was assessed using Nanodrop. SIV gag RNA was quantified by RT-qPCR using an Applied Biosystems Prism 5700. The following primers were used (forward, SGAG03 5′-CAGGGAAIIAAGCAGATGAATTAG-3′ reverse, SGAG04 5′-GTTTCACTTTCTCTTCTGCGTG-3′ probe, pSUS05 5′-(6-carboxyfluorescein[FAM])ATTTGGATTAGCAGAAAGCCTGTTGGAG (6-carboxytetramethylrhodamine [TAMRA])-3′) with cycle conditions of 50°C for 30 min, 94°C for 15 min to reverse transcribe RNA, followed by 45 cycles of PCR at 94°C for 15 s, 55°C for 15 s, and 60°C for 30 s.
Genotyping for the MHC Mane-A1*084:01:01 allele was performed using cDNA generated from PBMC RNA. Initial PCR was performed for Sequence Specific Primer (SSP) 2 (forward 5′-CGG GTC TCA CAC CTT CCA GAG GAT GTA T-3′ reverse 5′-CGG TCC AGG AAC GCA GGT CCC-3′) and GAPDH (forward 5′-TGC CAT CAA TGA CCC CTT CAT TGA CCT C-3′ reverse 5′-CCC AGC CTT CTC CAT GGT GGT GAA GAC-3′) under the following cycling conditions: 95°C 20 min followed by 35 cycles 94°C 20 s, 72°C 40 s and final extension 72°C 10 min. Any samples with a 134 bp amplified product were subsequently run for SSP1 (forward 5′-GGC CAA CAC ACA GAC CTA CCG AGA GAG-3′ reverse 5′-CCC TGC CGT CGT AGG CGT ACT GGC TAT AT-3′ 161 bp) and SSP3 (forward 5′-GGC GCC TCC TCC GCG GAT ATA G-3′ reverse 5′-GGC ACT CGC CCT CCA CGT AGG T-3′ 174 bp) in addition to repeat SSP2 and GAPDH. Samples which were negative for SSP2 were repeated to verify their Mane-A1*084:01:01 status while samples positive for SSP2, SSP1, and SSP3 were considered Mane-A1*084:01:01 positive.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis and graphs were generated using GraphPad Prism 10.1.2. Specific analyses and sample sizes for each experiment are described in the figure legends. A p-value <0.05 was considered significant for all comparisons. Inclusion of subjects is described in in the “animals” section; any animals receiving ART or that were euthanized before 50 days post-infection were excluded from this investigation.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Iba-1 | Wako | Cat#019–19741; RRID: AB_839504 |
| Rabbit polyclonal anti-IL-18 | Abcam | Cat# ab191152; RRID: AB_2737346 |
| Mouse monoclonal anti-IL-1β (clone E7-2-hIL1β) | Santa Cruz Biotechnology | Cat# sc-32294; RRID: AB_627790 |
| Rabbit monoclonal anti-ASC (clone E1E3I) | Cell Signaling Technology | Cat#13833; RRID: AB_2798325 |
| Goat polyclonal anti-monkey IgG, HRP conjugated | Abcam | Cat# ab112767; RRID: AB_10866625 |
| Goat polyclonal anti-monkey IgM, HRP conjugated | Sigma | Cat# SAB3700780; RRID: AB_3101871 |
| Mouse monoclonal anti-CD3 (SP34-2), PerCP-Cy5.5 conjugated | BD Biosciences | Cat#552852; RRID: AB_394493 |
| Mouse monoclonal anti-CD4 (L200), FITC conjugated | BD Biosciences | Cat#550628; RRID: AB_393789 |
| Mouse monoclonal anti-CD8a (RPA-T8), FITC conjugated | BD Biosciences | Cat#561948; RRID: AB_11154582 |
| Mouse monoclonal anti-CD20 (2H7), FITC conjugated | BD Biosciences | Cat#556632; RRID: AB_396501 |
| Mouse monoclonal anti-CD3 (SP34-2), V500 conjugated | BD Biosciences | Cat#560770; RRID: AB_1937322 |
| Mouse monoclonal anti-CD3 (S3.5), Qdot 655 conjugated | Molecular Probes/Life Technologies | Cat#Q10007; RRID: AB_11180600 |
| Mouse monoclonal anti-CD4 (OKT4), Brilliant Violet 650 conjugated | Biolegend | Cat#317436; RRID: AB_2563050 |
| Mouse monoclonal anti-CD8a (RPA-T8), Qdot 650 conjugated | Molecular Probes/Life Technologies | Cat# Q10152; RRID: AB_1500499 |
| Mouse monoclonal anti-CD8a (RPA-T8), Brilliant Violet 570 conjugated | Biolegend | Cat#301038; RRID: AB_2563213 |
| Mouse monoclonal anti-CD20 (2H7), eFluor 450 conjugated | Invitrogen | Cat#48-0209-42; RRID: AB_1633384 |
| Virus strains | ||
| SIVdeltaB670 | Grown in CEM cell line | N/A |
| SIV/17E-Fr | Grown in CEM cell line | N/A |
| SIVmac251 lysate | ZeptoMetrix | Cat#0810011 |
| Biological samples | ||
| SIV-infected macaque plasma | In-house sample archive | See Table S1 for IDs |
| SIV-infected macaque cerebrospinal fluid | In-house sample archive | See Table S1for IDs |
| SIV-infected pigtail macaque basal ganglia (paraffin-embedded tissue blocks) | In-house sample archive | See Table S1 for IDs |
| Uninfected pigtail macaque whole blood | Healthy blood donors | - |
| Critical commercial assays | ||
| MSD U-PLEX Biomarker Group 1 (NHP) Assays, custom 10-cytokine panel | Meso Scale Discovery | Cat#K15068M |
| Bond Polymer Refine Detection Kit | Leica | Cat#DS9800 |
| Bond Polymer Refine Red Detection Kit | Leica | Cat#DS9390 |
| RNAscope 2.5 LS reagent kit RED | ACD | Cat#322150 |
| RNAscope 2.5 LS Probe- SIVmac239 | ACD | Cat#312818 |
| BD OptEIA Reagent Set B | BD Biosciences | Cat#550534 |
| Monkey IgG ELISA kit | Abcam | Cat#ab190549 |
| Nab Protein A/G Spin kit, 0.2 mL | ThermoFisher Scientific | Cat#89950 |
| Bright-Glo Luciferase Assay System | Promega | Cat#E2620 |
| QIAamp MinElute Virus Spin kit | Qiagen | Cat#57704 |
| Experimental models: Cell lines | ||
| Human: T1 (174 x CEM.T1) cells | ATCC | ID#CRL-1991, RRID: CVCL_7904 |
| Human: TZM-bl cells | NIBSC | ID#5011 |
| Experimental models: Organisms/strains | ||
| Pigtail macaques (Macaca nemestrina) | Bred for research | See Table S1 for IDs |
| Rhesus macaques (Macaca mulatta) | Bred for research | See Table S1 for IDs |
| Software and algorithms | ||
| GraphPad Prism (10.1.2) | GraphPad Software | https://www.graphpad.com/ |
| QuPath (0.4.3) | Open source | https://qupath.github.io |
| Concentriq for Research | Proscia | https://proscia.com/concentriq-platform/ |
Highlights.
SIVE is associated with compartmentalized viral replication and cytokine production
Myeloid-dominant lesions show inflammasome activation in infected and bystander cells
SIV-specific IgG responses in the plasma and CSF predict protection from SIVE
ACKNOWLEDGMENTS
The authors would like to thank the Johns Hopkins Research Animal Resources staff for their ongoing assistance in conducting these studies. In particular, Rock Scarborough, Bess Carlson, Isabella Baumann, and Bruce Baldwin have provided invaluable assistance in animal care and sample collection. We would also like to thank Drs. Jacqueline Brockhurst and Katie Mulka for generously sharing their experimental methods for use in this work. This study was supported by funding from NIH grants R01MH127981, R01DA050529, R01NS055651, R01HL078479-01, P01AI131306, T32OD011 089, and U42OD13117.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114833.
REFERENCES
- 1.Wang H, Zhao S, Wang S, Zheng Y,Wang S, Chen H, Pang J, Ma J, Yang X, and Chen Y (2022). Global magnitude of encephalitis burden and its evolving pattern over the past 30 years. J. Infect 84, 777–787. 10.1016/j.jinf.2022.04.026. [DOI] [PubMed] [Google Scholar]
- 2.Hansen MA, Samannodi MS, and Hasbun R (2020). Predicting inpatient mortality among encephalitis patients: A novel admission risk score. Open Forum Infect. Dis 7, ofaa471. 10.1093/ofid/ofaa471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Granerod J, Huang Y, Davies NWS, Sequeira PC, Mwapasa V, Rupali P, Michael BD, Solomon T, and Easton A (2023). Global landscape of encephalitis: key priorities to reduce future disease burden. Clin. Infect. Dis 77, 1552–1560. 10.1093/cid/ciad417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tunkel AR, Glaser CA, Bloch KC, Sejvar JJ, Marra CM, Roos KL, Hartman BJ, Kaplan SL, Scheld WM, and Whitley RJ; Infectious Diseases Society of America (2008). The management of encephalitis: clinical practice guidelines by the Infectious Diseases Society of America. Clin. Infect. Dis 47, 303–327. 10.1086/589747. [DOI] [PubMed] [Google Scholar]
- 5.Aksamit AJ (2021). Treatment of viral encephalitis. Neurol. Clin 39, 197–207. 10.1016/j.ncl.2020.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Yong HYF, Pastula DM, and Kapadia RK (2023). Diagnosing viral encephalitis and emerging concepts. Curr. Opin. Neurol 36, 175–184. 10.1097/WCO.0000000000001155. [DOI] [PubMed] [Google Scholar]
- 7.George BP, Schneider EB, and Venkatesan A (2014). Encephalitis hospitalization rates and inpatient mortality in the United States, 2000-2010. PLoS One 9, e104169. 10.1371/journal.pone.0104169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hellmuth J, Fletcher JLK, Valcour V, Kroon E, Ananworanich J, Intasan J, Lerdlum S, Narvid J, Pothisri M, Allen I, et al. (2016). Neurologic signs and symptoms frequently manifest in acute HIV infection. Neurology 87, 148–154. 10.1212/WNL.0000000000002837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucas S (2023). Historical and current issues in HIV encephalitis, and the role of neuropathology in HIV disease: a pathological perspective. J. Neurol 270, 1337–1345. 10.1007/s00415-022-11503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG, Broadhurst DI, and Power C (2014). Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology 11, 35. 10.1186/1742-4690-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mamik MK, Hui E, Branton WG, McKenzie BA, Chisholm J, Cohen EA, and Power C (2017). HIV-1 Viral Protein R Activates NLRP3 Inflammasome in Microglia: implications for HIV-1 Associated Neuroinflammation. J. Neuroimmune Pharmacol 12, 233–248. 10.1007/s11481-016-9708-3. [DOI] [PubMed] [Google Scholar]
- 12.Fernandes JP, Branton WG, Cohen EA, Koopman G, Kondova I, Gelman BB, and Power C (2024). Caspase cleavage of gasdermin E causes neuronal pyroptosis in HIV-associated neurocognitive disorder. Brain 147,717–734. 10.1093/brain/awad375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Neil SP, Suwyn C, Anderson DC, Niedziela G, Bradley J, Novembre FJ, Herndon JG, and McClure HM (2004). Correlation of acute humoral response with brain virus burden and survival time in pig-tailed macaques infected with the neurovirulent simian immunodeficiency virus SIVsmmFGb. Am. J. Pathol 164, 1157–1172. 10.1016/S0002-9440(10)63204-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong JJ, Amancha PK, Rogers KA, Courtney CL, Havenar-Daughton C, Crotty S, Ansari AA, and Villinger F (2014). Early lymphoid responses and germinal center formation correlate with lower viral load set points and better prognosis of simian immunodeficiency virus infection. J. Immunol 193, 797–806. 10.4049/jimmunol.1400749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowen L, Nath A, and Smith B (2018). CNS immune reconstitution inflammatory syndrome. Handb. Clin. Neurol 152,167–176. 10.1016/B978-0-444-63849-6.00013-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irollo E, Luchetta J, Ho C, Nash B, and Meucci O (2021). Mechanisms of neuronal dysfunction in HIV-associated neurocognitive disorders. Cell. Mol. Life Sci 78, 4283–4303. 10.1007/s00018-021-03785-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pérez-Valero I, Ellis R, Heaton R, Deutsch R, Franklin D, Clifford DB, Collier A, Gelman B, Marra C, McCutchan JA, et al. (2019). Cerebrospinal fluid viral escape in aviremic HIV-infected patients receiving antiretroviral therapy: prevalence, risk factors and neurocognitive effects. AIDS 33, 475–481. 10.1097/QAD.0000000000002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winston A, and Spudich S (2020). Cognitive disorders in people living with HIV. Lancet. HIV 7, e504–e513. 10.1016/S2352-3018(20)30107-7. [DOI] [PubMed] [Google Scholar]
- 19.Nightingale S, Ances B, Cinque P, Dravid A, Dreyer AJ, Gisslén M, Joska JA, Kwasa J, Meyer A-C, Mpongo N, et al. (2023). Cognitive impairment in people living with HIV: consensus recommendations for a new approach. Nat. Rev. Neurol 19, 424–433. 10.1038/s41582-023-00813-2. [DOI] [PubMed] [Google Scholar]
- 20.World Health Organization (2023). Global HIV Programme- HIV Data and Statistics. https://www.who.int/teams/global-hiv-hepatitis-and-stis-programmes/hiv/strategic-information/hiv-data-and-statistics.
- 21.Garcia-Tellez T, Huot N, Ploquin MJ, Rascle P, Jacquelin B, and Müller-Trutwin M (2016). Non-human primates in HIV research: Achievements, limits and alternatives. Infect. Genet. Evol 46,324–332. 10.1016/j.meegid.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Klatt NR, Canary LA, Vanderford TH, Vinton CL, Engram JC, Dunham RM, Cronise HE, Swerczek JM, Lafont BAP, Picker LJ , et al. (2012). Dynamics of simian immunodeficiency virus SIVmac239 infection in pigtail macaques. J. Virol 86, 1203–1213. 10.1128/JVI.06033-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beck SE, Queen SE, Metcalf Pate KA, Mangus LM, Abreu CM, Gama L, Witwer KW, Adams RJ, Zink MC, Clements JE, and Mankowski JL. (2018). An SIV/macaque model targeted to study HIV-associated neurocognitive disorders. J. Neurovirol. 24, 204–212. 10.1007/s13365-017-0582-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beck SE, Queen SE, Witwer KW, Metcalf Pate KA, Mangus LM, Gama L, Adams RJ, Clements JE, Christine Zink M, and Mankow- ski JL (2015). Paving the path to HIV neurotherapy: Predicting SIV CNS disease. Eur. J. Pharmacol. 759, 303–312. 10.1016/j.ejphar.2015.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clements JE, Gama L, Graham DR, Mankowski JL, and Zink MC (2011). A simian immunodeficiency virus macaque model of highly active antiretroviral treatment: viral latency in the periphery and the central nervous system. Curr. Opin. HIV AIDS 6, 37–42. 10.1097/COH.0b013e3283412413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zink MC, Suryanarayana K, Mankowski JL, Shen A, Piatak M, Spelman JP, Carter DL, Adams RJ, Lifson JD, and Clements JE (1999). High viral load in the cerebrospinal fluid and brain correlates with severity of simian immunodeficiency virus encephalitis. J. Virol 73, 10480–10488. 10.1128/JVI.73.12.10480-10488.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith MZ, Fernandez CS, Chung A, Dale CJ, De Rose R, Lin J, Brooks AG, Krebs KC, Watkins DI, O’Connor DH, et al. (2005). The pigtail macaque MHC class I allele Mane-A*10 presents an immundominant SIV Gag epitope: identification, tetramer development and implications of immune escape and reversion. J. Med. Primatol 34, 282–293. 10.1111/j.1600-0684.2005.00126.x. [DOI] [PubMed] [Google Scholar]
- 28.Mankowski JL, Queen SE, Clements JE, and Zink MC (2004). Cerebrospinal fluid markers that predict SIV CNS disease. J. Neuroimmunol 157, 66–70. 10.1016/j.jneuroim.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 29.Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, and Dickson DW (2007). Actin-binding proteins coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J. Histochem. Cytochem 55, 687–700. 10.1369/jhc.6A7156.2007. [DOI] [PubMed] [Google Scholar]
- 30.Pirhonen J (2001). Regulation of IL-18 expression in virus infection. Scand. J. Immunol 53, 533–539. 10.1046/j.1365-3083.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- 31.Bryan NB, Dorfleutner A, Rojanasakul Y, and Stehlik C (2009). Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J. Immunol 182, 3173–3182. 10.4049/jimmu-nol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reiber H (1994). Flow rate of cerebrospinal fluid (CSF)-a concept common to normal blood-CSF barrier function and to dysfunction in neurological diseases. J. Neurol. Sci 122, 189–203. 10.1016/0022-510x(94)90298-4. [DOI] [PubMed] [Google Scholar]
- 33.Pandya D, and Johnson TP (2023). Chronic and delayed neurological manifestations of persistent infections. Curr. Opin. Neurol 36, 198–206. 10.1097/WCO.0000000000001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.World Health Organization (2023). Why Encephalitis Matters? Report of the Virtual Meeting, 28–29 June 2022 (World Health Organization). License: CC BY-NC-SA 3.0 IGO. https://iris.who.int/handle/10665/366223. [Google Scholar]
- 35.Abreu CM, Veenhuis RT, Avalos CR, Graham S, Parrilla DR, Ferreira EA, Queen SE, Shirk EN, Bullock BT, Li M, et al. (2019). Myeloid and CD4 T Cells Comprise the Latent Reservoir in Antiretroviral Therapy-Suppressed SIVmac251-Infected Macaques. mBio 10, e01659–19. 10.1128/mBio.01659-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mankowski JL, Clements JE, and Zink MC (2002). Searching for clues: tracking the pathogenesis of human immunodeficiency virus central nervous system disease by use of an accelerated, consistent simian immunodeficiency virus macaque model. J. Infect. Dis 186, S199–S208. 10.1086/344938. [DOI] [PubMed] [Google Scholar]
- 37.Hammoud DA, Sinharay S, Shah S, Schreiber-Stainthorp W, Maric D, Muthusamy S, Lee DE, Lee CA, Basuli F, Reid WC, et al. (2019). Neuroinflammatory changes in relation to cerebrospinal fluid viral load in simian immunodeficiency virus encephalitis. mBio 10, e00970–19. 10.1128/mBio.00970-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma H, Albe JR, Gilliland T, McMillen CM, Gardner CL, Boyles DA, Cottle EL, Dunn MD, Lundy JD, Salama N, et al. (2022). Long-term persistence of viral RNA and inflammation in the CNS of macaques exposed to aerosolized Venezuelan equine encephalitis virus. PLoS Pathog. 18, e1009946. 10.1371/journal.ppat.1009946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao Y, Gu J, Li M, Li G, Zhao L, and Ai J (2022). MRI changes and expressions of neuron-specific enolase and monocyte chemoattractant protein-1 in cerebrospinal fluid in patients with severe herpes simplex virus encephalitis. Cell. Mol. Biol 68, 78–82. 10.14715/cmb/2022.68.11.13. [DOI] [PubMed] [Google Scholar]
- 40.Albe JR, Ma H, Gilliland TH, McMillen CM, Gardner CL, Boyles DA, Cottle EL, Dunn MD, Lundy JD, O’Malley KJ, et al. (2021). Physiological and immunological changes in the brain associated with lethal eastern equine encephalitis virus in macaques. PLoS Pathog. 17, e1009308. 10.1371/journal.ppat.1009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suwanmanee S, Ghimire S, Edwards J, and Griffin DE (2023). Infection of Pro- and Anti-Inflammatory Macrophages by Wild Type and Vaccine Strains of Measles Virus: NLRP3 Inflammasome Activation Independent of Virus Production. Viruses 15, 260. 10.3390/v15020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang W, Li G, De W, Luo Z, Pan P, Tian M, Wang Y, Xiao F, Li A, Wu K, et al. (2018). Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1 β secretion. Nat. Commun 9, 106. 10.1038/s41467-017-02645-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayes CK, Wilcox DR, Yang Y, Coleman GK, Brown MA, and Longnecker R (2021). ASC-dependent inflammasomes contribute to immunopathology and mortality in herpes simplex encephalitis. PLoS Pathog. 17, e1009285. 10.1371/journal.ppat.1009285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar M, Roe K, Orillo B, Muruve DA, Nerurkar VR, Gale M, and Verma S (2013). Inflammasome adaptor protein Apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in west Nile virus encephalitis. J. Virol 87, 3655–3667. 10.1128/JVI.02667-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiarini A, Gui L, Viviani C, Armato U, and Dal Prà I (2023). NLRP3 Inflammasome’s Activation in Acute and Chronic Brain Diseases-An Update on Pathogenetic Mechanisms and Therapeutic Perspectives with Respect to Other Inflammasomes. Biomedicines 11, 999. 10.3390/biomedicines11040999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balint E, Feng E, Giles EC, Ritchie TM, Qian AS, Vahedi F, Montemarano A, Portillo AL, Monteiro JK, Trigatti BL, and Ashkar AA (2024). Bystander activated CD8+ T cells mediate neuropathology during viral infection via antigen-independent cytotoxicity. Nat. Commun 15, 896. 10.1038/s41467-023-44667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbeau DJ, Cartwright HN, Harmon JR, Spengler JR, Spiropoulou CF, Sidney J, Sette A, and McElroy AK (2021). Identification and Characterization of Rift Valley Fever Virus-Specific T Cells Reveals a Dependence on CD40/CD40L Interactions for Prevention of Encephalitis. J. Virol 95, e0150621. 10.1128/JVI.01506-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garber C, Soung A, Vollmer LL, Kanmogne M, Last A, Brown J, and Klein RS (2019). T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat. Neurosci 22, 1276–1288. 10.1038/s41593-019-0427-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tyor WR, Wesselingh S, Levine B, and Griffin DE (1992). Long term intraparenchymal Ig secretion after acute viral encephalitis in mice. J.Immunol 149, 4016–4020. [PubMed] [Google Scholar]
- 50.Barnett BE, Staupe RP, Odorizzi PM, Palko O, Tomov VT, Mahan AE, Gunn B, Chen D, Paley MA, Alter G, et al. (2016). Cutting Edge: B Cell-Intrinsic T-bet Expression Is Required To Control Chronic Viral Infection. J. Immunol. 197, 1017–1022. 10.4049/jimmunol.1500368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bogovič P, Lotrič-Furlan S, Avšič-Županc T, Korva M, Lusa L, Strle K, and Strle F (2021). Low Virus-Specific IgG Antibodies in Adverse Clinical Course and Outcome of Tick-Borne Encephalitis. Microorganisms 9, 332. 10.3390/microorganisms9020332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kulcsar KA, Baxter VK, Abraham R, Nelson A, and Griffin DE (2015). Distinct Immune Responses in Resistant and Susceptible Strains of Mice during Neurovirulent Alphavirus Encephalomyelitis. J. Virol 89, 8280–8291. 10.1128/JVI.00173-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nilaratanakul V, Chen J, Tran O, Baxter VK, Troisi EM, Yeh JX, and Griffin DE (2018). Germ Line IgM Is Sufficient, but Not Required, for Antibody-Mediated Alphavirus Clearance from the Central Nervous System. J. Virol 92, e02081–17. 10.1128/JVI.02081-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Erdei A, Kovács KG, Nagy-Baló Z, Lukácsi S, Mácsik-Valent B, Kurucz I, and Bajtay Z (2021). New aspects in the regulation of human B cell functions by complement receptors CR1, CR2, CR3 and CR4. Immunol. Lett 237, 42–57. 10.1016/j.imlet.2021.06.006. [DOI] [PubMed] [Google Scholar]
- 55.Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, Morgan BP, Sivasankar B, and Mortellaro A (2013). Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1 β release. J. Immunol 191, 1006–1010. 10.4049/jimmunol.1300489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arbore G, and Kemper C (2016). A novel “complement-metabolism-in-flammasome axis” as a key regulator of immune cell effector function. Eur. J. Immunol 46, 1563–1573. 10.1002/eji.201546131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayr LM, Su B, and Moog C (2017). Non-Neutralizing Antibodies Directed against HIV and Their Functions. Front. Immunol 8, 1590. 10.3389/fimmu.2017.01590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warter L, Appanna R, and Fink K (2012). Human poly- and cross-reactive anti-viral antibodies and their impact on protection and pathology. Immunol. Res 53, 148–161. 10.1007/s12026-012-8268-8. [DOI] [PubMed] [Google Scholar]
- 59.Babas T, Muñoz D, Mankowski JL, Tarwater PM, Clements JE, and Zink MC (2003). Role of microglial cells in selective replication of simian immunodeficiency virus genotypes in the brain. J. Virol 77, 208–216. 10.1128/jvi.77.1.208-216.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Babas T, Dewitt JB, Mankowski JL, Tarwater PM, Clements JE, and Zink MC (2006). Progressive selection for neurovirulent genotypes in the brain of SIV-infected macaques. AIDS 20, 197–205. 10.1097/01.aids.0000198078.24584.21. [DOI] [PubMed] [Google Scholar]
- 61.Gao X, Shen Q, Roco JA, Dalton B, Frith K, Munier CML, Ballard FD, Wang K, Kelly HG, Nekrasov M, et al. (2024). Zeb2 drives the formation of CD11c+ atypical B cells to sustain germinal centers that control persistent infection. Sci. Immunol 9, eadj4748. 10.1126/sciimmunol.adj4748. [DOI] [PubMed] [Google Scholar]
- 62.Haas KM (2023). Noncanonical B cells: characteristics of uncharacteristic B cells. J. Immunol 211,1257–1265. 10.4049/jimmunol.2200944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aspden JW, Murphy MA, Kashlan RD, Xiong Y, Poznansky MC, and Sîrbulescu RF (2023). Intruders or protectors - the multifaceted role of B cells in CNS disorders. Front. Cell. Neurosci 17, 1329823. 10.3389/fncel.2023.1329823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dwyer LJ, Maheshwari S, Levy E, Poznansky MC, Whalen MJ, and Sîrbulescu RF (2023). B cell treatment promotes a neuroprotective microenvironment after traumatic brain injury through reciprocal immuno-modulation with infiltrating peripheral myeloid cells. J. Neuroinflammation 20, 133. 10.1186/s12974-023-02812-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anthony IC, Crawford DH, and Bell JE (2003). B lymphocytes in the normal brain: contrasts with HIV-associated lymphoid infiltrates and lymphomas. Brain 126, 1058–1067. 10.1093/brain/awg118. [DOI] [PubMed] [Google Scholar]
- 66.Lackner P, Kuenz B, Reindl M, Morandell M, Berger T, Schmutzhard E, and Eggers C (2010). Antibodies to myelin oligodendrocyte glycoprotein in HIV-1 associated neurocognitive disorder: a crosssectional cohort study. J. Neuroinflammation 7, 79. 10.1186/1742-2094-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bonnan M, Barroso B, Demasles S, Krim E, Marasescu R, and Miquel M (2015). Compartmentalized intrathecal immunoglobulin synthesis during HIV infection - a model of chronic CNS inflammation? J. Neuroimmunol 285, 41–52. 10.1016/j.jneuroim.2015.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wagner JN, Leibetseder A, Troescher A, Panholzer J, and von Oertzen TJ (2022). Efficacy and safety of intravenous immunoglobulins for the treatment of viral encephalitis: a systematic literature review. J. Neurol 269, 712–724. 10.1007/s00415-021-10494-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang S-M, Lei H-Y, Huang M-C, Su L-Y, Lin H-C, Yu C-K, Wang J-L, and Liu C-C (2006). Modulation of cytokine production by intravenous immunoglobulin in patients with enterovirus 71-associated brainstem encephalitis. J. Clin. Virol 37, 47–52. 10.1016/j.jcv.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 70.Morales-Ruiz V, Juárez-Vaquera VH, Rosetti-Sciutto M, Sanchez-Murñoz F, and Adalid-Peralta L (2022). Efficacy of intravenous immunoglobulin in autoimmune neurological diseases. Literature systematic review and meta-analysis. Autoimmun. Rev 21, 103019. 10.1016/j.autrev.2021.103019. [DOI] [PubMed] [Google Scholar]
- 71.Watson CW-M, Sundermann EE, Hussain MA, Umlauf A, Thames AD, Moore RC, Letendre SL, Jeste DV, Morgan EE, and Moore DJ (2019). Effects of trauma, economic hardship, and stress on neurocognition and everyday function in HIV. Health Psychol. 38, 33–42. 10.1037/hea0000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zink MC, Amedee AM, Mankowski JL, Craig L, Didier P, Carter DL, Muñoz A, Murphey-Corb M, and Clements JE (1997). Pathogenesis of SIV encephalitis. Selection and replication of neurovirulent SIV. Am. J. Pathol 151,793–803. [PMC free article] [PubMed] [Google Scholar]
- 73.Kelly KM, Beck SE, Metcalf Pate KA, Queen SE, Dorsey JL, Adams RJ, Avery LB, Hubbard W, Tarwater PM, and Mankowski JL (2013). Neuroprotective maraviroc monotherapy in simian immunodeficiency virus-infected macaques: reduced replicating and latent SIV in the brain. AIDS 27, F21–F28. 10.1097/QAD.0000000000000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG,et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci. Rep 7, 16878. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sarzotti-Kelsoe M, Bailer RT, Turk E, Lin C.l., Bilska M, Greene KM, Gao H, Todd CA, Ozaki DA, Seaman MS, et al. (2014). Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods 409, 131–146. 10.1016/j.jim.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mankowski JL, Queen SE, Fernandez CS, Tarwater PM, Karper JM, Adams RJ, and Kent SJ (2008). Natural host genetic resistance to lentiviral CNS disease: a neuroprotective MHC class I allele in SIV-infected macaques. PLoS One 3,e3603. 10.1371/jour-nal.pone.0003603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Queen SE, Mears BM, Kelly KM, Dorsey JL, Liao Z, Dinoso JB, Gama L, Adams RJ, Zink MC, Clements JE, et al. (2011). Replication-competent simian immunodeficiency virus (SIV) Gag escape mutations archived in latent reservoirs during antiretroviral treatment of SIV-infected macaques. J. Virol 85, 9167–9175. 10.1128/JVI.00366-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Castell N, Guerrero-Martin SM, Rubin LH, Shirk EN, Brockhurst JK, Lyons CE, Najarro KM, Queen SE, Carlson BW, Adams RJ, et al. (2022). Effect of Single Housing on Innate Immune Activation in Immunodeficiency Virus-Infected Pigtail Macaques ( Macaca nemestrina ) as a Model of Psychosocial Stress in Acute HIV Infection. Psychosom. Med. 84, 966–975. 10.1097/PSY.0000000000001132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.