

Abstract
Platelets are critical mediators of hemostasis and thrombosis. Platelets circulate as discs in their resting form but change shape rapidly upon activation by vascular damage and/or soluble agonists such as thrombin. Platelet shape change is driven by a dynamic remodeling of the actin cytoskeleton. Actin filaments interact with the protein myosin, which is phosphorylated on the myosin light chain (MLC) upon platelet activation. Actin-myosin interactions trigger contraction of the actin cytoskeleton, which drives platelet spreading and contractile force generation. Filamin A (FLNA) is an actin cross-linking protein that stabilizes the attachment between subcortical actin filaments and the cell membrane. In addition, FLNA binds multiple proteins and serves as a critical intracellular signaling scaffold. Here, we used platelets from mice with a megakaryocyte/platelet-specific deletion of FLNA to investigate the role of FLNA in regulating platelet shape change. Relative to controls, FLNA-null platelets exhibited defects in stress fiber formation, contractile force generation, and MLC phosphorylation in response to thrombin stimulation. Blockade of Rho kinase (ROCK) and protein kinase C (PKC) with the inhibitors Y27632 and bisindolylmaleimide (BIM), respectively, also attenuated MLC phosphorylation; our data further indicate that ROCK and PKC promote MLC phosphorylation through independent pathways. Notably, the activity of both ROCK and PKC was diminished in the FLNA-deficient platelets. We conclude that FLNA regulates thrombin-induced MLC phosphorylation and platelet contraction, in a ROCK- and PKC-dependent manner.
Keywords: cytoskeleton, Filamin A, myosin, phosphorylation, platelets, protease-activated receptor signaling
Introduction
Platelets are anucleate cells produced by megakaryocytes in the bone marrow and are important regulators of hemostasis, wound healing, and inflammation [1–3]. Platelets circulate as discs and become activated in response to vascular damage and/or soluble agonists [4]. Platelet activation is tightly regulated and requires co-ordinated signaling pathways to promote efficient blood clot formation [5]. However, abnormal activation of platelets can lead to arterial thrombosis, a pathologic condition that underpins myocardial infarction and stroke [6]. In their quiescent state, platelets circulate in the blood as miniscule discoid cells. Once activated, platelets undergo a drastic shape change that is driven by the dynamic remodeling of the actin cytoskeleton in concert with actomyosin contraction [3,7]. This shape change is one of the earliest physiological responses upon platelet activation, which accompanies granule secretion, aggregation, and adhesion to the damaged blood vessels [7,8]. Despite the recognition that extensive actin reorganization and internal contraction occur during platelet activation [9,10], the precise biochemical signals that link membrane receptors and the actin cytoskeleton during platelet contraction remain undefined.
Nonmuscle myosin IIa (NM IIA) is the predominant heavy-chain myosin isoform expressed in platelets, and is the main driver for platelet contraction [11]. This contractility is essential for platelet function, since variants of the gene for NM IIA (MYH9) are associated with bleeding disorders in both humans and mice [12,13]. Activation of platelets by thrombin initiates signaling cascades that drive the phosphorylation of the myosin light chain (MLC); this step is required for NM IIA to adopt an active conformation with increased actin binding affinity [14]. The phosphorylation of MLC also increases the actin-stimulated ATPase activity in NM IIA; when sufficient ATP is present, NM IIA moves along the actin filaments providing cellular contractile forces [15]. Thrombin cleaves G-protein coupled receptors on the platelet surface, termed protease-activated receptors (PARs) [16]. Following PAR activation, there are two pathways to MLC phosphorylation. One is a calcium (Ca2+)-dependent pathway that activates MLC kinase (MLCK), which in turns phosphorylates MLC at serine 19 [16,17]. The second pathway is Ca2+-independent and involves the activation of the small GTPase RhoA and its effector Rho kinase (ROCK), which phosphorylates and thereby inactivates MLC phosphatase (MLCP). This phosphatase normally dephosphorylates MLC at serine 19 [18,19]. These co-ordinated signaling events ultimately lead to actomyosin contraction and platelet shape change.
Filamin A (FLNA) is a 280-kDa actin-binding protein that has a critical role in platelet function and morphology. Mice with a platelet-specific deletion of FLNA exhibit macrothrombocytopenia and increased tail bleeding time [20]. In humans, multiple variants of the FLNA gene are associated with aberrant platelet function [21]. FLNA is a homodimeric protein that cross-links and stabilizes actin filaments into orthogonal networks, thus conferring cellular structural integrity [22]. Moreover, FLNA reportedly interacts with over 50 protein partners [21]. These binding partners include cell surface receptors, other cytoskeletal proteins, and protein kinases, making FLNA a crucial signaling scaffold. Of note, FLNA interacts with multiple proteins in the MLC phosphorylation pathway, including RhoA and ROCK [23–26].
To define the role of FLNA in regulating actomyosin contraction in platelets, we studied platelets derived from mice with a megakaryocyte/platelet-specific deletion of FLNA. Here we show that thrombin-stimulated FLNA-null platelets have severe defects in stress fiber formation and contractile force generation, as measured by traction force microscopy. Biochemical studies of the FLNA-null platelets revealed that FLNA is a critical regulator of thrombin-induced MLC phosphorylation. Specifically, we determined that FLNA regulates the activity of ROCK and protein kinase C (PKC), both of which promote MLC phosphorylation. However, our data indicate that ROCK and PKC promote MLC phosphorylation independently of each other.
Results
FLNA-deficient platelets exhibit defects in stress fiber formation
To evaluate the role of FLNA in the platelet shape change reaction, we used platelets from conditional knockout (KO) mice. The deletion of FLNA from the platelets of the conditional KO mice was verified by immunoblotting (Figure 1A). Floxed (control) and FLNA-deficient (platelet-specific KO) platelets were activated with thrombin and seeded onto fibrinogen-coated coverslips; F-actin was labeled with Alexa-488-phalloidin (Figure 1B,C). Notably, the higher magnification images illustrated marked morphological differences between the control and FLNA-deficient platelets (Figure 1B,C). At 45 min, control platelets exhibited lamellipodia with thick stress fibers (Figure 1B) that are characteristic of advanced spreading [27]. Conversely, FLNA-deficient platelets exhibited spike-like filopodia without lamellipodia or stress fibers (Figure 1C), suggesting that these platelets were still in the early stages of spreading. Accordingly, the percentage of platelets with visible stress fibers was significantly reduced (P < 0.05) in FLNA-deficient platelets relative to floxed controls (Figure 1D).
Figure 1. FLNA-deficient platelets have diminished stress fiber formation compared to control platelets.

(A) Lysates from floxed (control) FLNA-deficient (platelet-specific KO) platelets were resolved by SDS–PAGE and probed with an anti-FLNA antibody. Beta-actin is shown as a loading control. (B and C) Floxed (control, B) and FLNA-deficient (platelet-specific KO, C) Platelets were allowed to spread on fibrinogen-coated coverslips for 45 min prior to labeling with Alexa-488-phalloidin. Insets: higher magnification images of the floxed (control) and FLNA-deficient (platelet-specific KO) allowed to spread for 45 min on immobilized fibrinogen. (D) Bar graph depicts the quantification of the presence of visible stress fibers in floxed (control, blue bar) and FLNA-deficient (platelet-specific KO, pink bar) platelets. Calculations were based on the mean percentage (%) of stress-fiber positive platelets per field of view. Data are mean ± SEM and represent three independent experiments. *P < 0.05, based on Student's t-test.
Reduced contractile force generation in FLNA-deficient platelets
Since contractile force generation is an integral part of the platelet shape change reaction, we used traction force microscopy to measure the force exerted by individual platelets. Specifically, we utilized a high-yield contraction force microscopy technique in which ‘black dots’ are microcontact-printed onto flexible polydimethylsiloxane (PDMS) substrates [28,29]. Control and FLNA-KO platelets were pre-stimulated with thrombin for 10 min, seeded onto the flexible black dot substrates (Figure 2A) and labeled with Alexa-488-phalloidin. The displacement of the black dots from the undeformed grid was used to calculate the magnitude and direction of forces using regularized Fourier transform traction cytometry [30,31]. The total force for each platelet was calculated by summing the force magnitudes within the boundary of each platelet. After 45 min, the mean force generated by the FLNA-KO platelets (mean = 10.4 nN) was significantly less (P < 0.05) than that generated by the control platelets (mean = 13.5 nN) (Figure 2B). Since force generation is affected by spread area [28,32,33], we used linear regression to analyze force generation as a function of area. After 45 min, the control platelets produced significantly more force per unit area than did FLNA-KO platelets (Figure 2C). These data indicate that the differences in force generation between the floxed (control) and FLNA-KO platelets are not merely due to differences in cell spreading.
Figure 2. Reduced contractile force generation by FLNA-deficient platelets.

(A) Floxed (control) and FLNA-deficient (platelet-specific KO) platelets were seeded onto the black dots and labeled with phalloidin (top panels; middle panels depict the cells on the black dots). The bottom panels depict the traction forces exerted by individual platelets (cyan arrows) that were calculated based on the displacement of the black dots. (B) Dot-plot depicts the forces generated by control platelets (blue, n = 251 at 20 min, n = 270 at 45 min) and FLNA-deficient platelets (platelet-specific KO, red, n = 242 at 20 min, n = 283 at 45 min). Gray dots represent all of the individual platelets measured. *P < 0.05; **P < 0.01; ****P < 0.0001, based on one-way ANOVA and Sidak's multiple comparisons tests. Data were obtained from six independent experiments. (C) Line graph depicts linear regression analysis of force generation by floxed (control, blue line) and FLNA-deficient (platelet-specific KO, red line) platelets. ****P < 0.001. (D) Photographs depict fibrin clot retraction by floxed (control) and FLNA-deficient (platelet-specific KO) platelets in fibrinogen (1 mg/ml), stimulated with thrombin (1 U/ml) after 45 min. (E) Bar graph depicts clot retraction by floxed platelets (control, blue bar) and FLNA-deficient platelets (platelet-specific KO, pink bar), based on the percentage (%) of the retracted clot area. Data are mean ± SEM and represent three independent experiments. ***P < 0.001, based on Student's t-test.
To validate the force measurements and further investigate platelet contraction on a larger scale, we performed a fibrin clot retraction assay to evaluate, in the context of a physiological assay, the contractile function of control and FLNA-KO platelets. We observed that clot retraction was virtually nonexistent with the FLNA-deficient platelets (Figure 2D,E). Collectively, these data suggest a key role of FLNA in regulating actomyosin contraction in platelets and fibrin clot maturation.
FLNA regulates RhoA activity and MLC phosphorylation via control of MLC phosphatase
Given the role of the small GTPase RhoA in actomyosin contraction [34], we wished to compare RhoA activity in control and FLNA-deficient platelets following stimulation by thrombin. A microplate-based colorimetric assay revealed significantly (P < 0.05) diminished levels of active, GTP-bound RhoA in FLNA-deficient platelets relative to controls (Figure 3A). To investigate the biochemical mechanism related to the weaker contractile force observed in FLN-KO platelets, we measured MLC phosphorylation by immunoblotting in both control and FLNA-KO platelets following thrombin stimulation. Since MLC phosphorylation is a relatively rapid event following platelet activation, we treated floxed (control) and FLNA-KO platelets with thrombin (0.1 U/ml) for 5 or 10 min. Phosphorylation of MLC at the Ser19 residue was visibly reduced in the FLNA-KO platelets relative to controls (Figure 3B); this finding was quantified by densitometry (Figure 3C). MLC phosphorylation was similarly reduced in FLNA-KO platelets following activation by collagen-related peptide (Supplementary Figure S1A,B). This suggests that FLNA is essential in regulating MLC phosphorylation downstream of PAR and GPVI signaling.
Figure 3. FLNA-null platelets exhibit diminished RhoA activation and MLC phosphorylation compared to control platelets.

(A) RhoA activation in response to thrombin stimulation was measured in floxed (control, blue bars) platelets and FLNA-deficient (platelet-specific KO, pink bars) platelets. The level of active, GTP-bound RhoA was measured using a G-LISA activation assay, and represented as relative fold change. Data are mean ± SEM and represent three independent experiments. ***P < 0.001, based on two-way ANOVA and Tukey post hoc multiple comparison tests. (B) Immunoblot depicts MLC phosphorylation at Ser19 in control platelets (control) and FLNA-null platelets (platelet-specific KO) after thrombin treatment for the indicated times. Total MLC is shown as a loading control. (C) Bar graph represents the densitometry quantification of phospho-MLC2 (Ser19) relative to total MLC protein in floxed platelets (control, blue bars) and FLNA-deficient platelets (platelet-specific KO, pink bars). Data are mean ± SEM, analyzed by two-way ANOVA and Tukey post hoc multiple comparison tests, and represent a minimum of three independent experiments. ****P < 0.0001, based on two-way ANOVA and Tukey post hoc multiple comparison tests. (D) Immunoblot depicts MYPT1 phosphorylated at Thr853 in floxed platelets (control) and FLNA-null platelets (platelet-specific KO) after thrombin treatment for the indicated times. Total MYPT1 is shown as a loading control. (E) Bar graph represents the densitometry quantification of phospho-MYPT1 (Thr853) relative to total MYPT1 in floxed platelets (control, blue bars) and FLNA-null platelets (platelet-specific KO, pink bars). Data are mean ± SEM, analyzed by two-way ANOVA and Tukey post hoc multiple comparison tests, and represent a minimum of three independent experiments. ***P < 0.001, ****P < 0.0001, based on two-way ANOVA and Tukey post hoc multiple comparison tests. NT, no treatment.
MLC is simultaneously phosphorylated by MLCK and de-phosphorylated by the corresponding MLC phosphatase (MLCP). Importantly, upon platelet activation by thrombin, Rho kinase (ROCK) phosphorylates the regulatory subunit of MLCP (MYPT1) at Thr696 and/or Thr853 the regulatory subunit of thereby inactivating the phosphatase activity of MLCP. Therefore, we evaluated the role of FLNA in modulating MLCP activity by immunoblotting platelet lysates for phospho-MYPT1 (Thr 853). Notably FLNA-KO platelets had significantly reduced (P < 0.05) levels of phospho-MYPT1 (Thr 853) relative to controls (Figure 3D,E). Similar results were observed in FLNA-KO platelets treated with collagen-related peptide (Supplementary Figure S2A,B). These data indicate that FLNA expression is required to maintain normal ROCK activity, and its subsequent ability to constrain the phosphatase activity of MLCP.
MLC phosphorylation is regulated by both PKC and ROCK
Protein kinase C (PKC) is another kinase responsible for MLC phosphorylation [35] and is also activated by thrombin stimulation [36,37]. Like ROCK, PKC binds FLNA [24,38]. We therefore sought to determine the individual and collective roles of PKC and ROCK in modulating MLC phosphorylation. We treated platelets with the ROCK inhibitor Y27632 and the PKC inhibitor bisindolylmaleimide I (BIM I) prior to thrombin stimulation. Inhibition of either ROCK or PKC resulted in a marked reduction in MLC phosphorylation (Figure 4A,B). However, the effects of blocking both ROCK and PKC (using both inhibitors simultaneously) were additive and essentially abrogated thrombin-induced MLC phosphorylation (Figure 4A,B). These data are consistent with the notion that PKC and ROCK control MLC phosphorylation via independent pathways despite both kinases binding FLNA. This notion is further exemplified by our finding that thrombin-induced phosphorylation (hence inactivation) of MLCP was inhibited by Y27632 but not by BIM (Figure 4C,D), suggesting that PKC regulation of MLC phosphorylation is independent of the ROCK-MLCP axis.
Figure 4. PKC and ROCK regulate MLC phosphorylation.

(A) Immunoblot depicts MLC phosphorylation at Ser19 in wild-type mouse platelets pre-incubated with vehicle, the PKC inhibitor BIM (15 µM), and/or the ROCK inhibitor Y27632 (50 µM) for 30 min prior to stimulation with thrombin (0.1 U/ml) for 10 min. Total MLC is shown as a loading control. (B) Bar graph represents the densitometric quantification of p-MLC2 (Ser19) relative to total MLC in thrombin-activated platelets pre-treated with vehicle (TR, orange), BIM (BIM, green bar), Y27632 (Y27632, blue bar), or BIM plus Y27632 (BIM + Y27632, purple bar). Resting platelets serve as the negative control (NT, red bar). Data are mean ± SEM and represent a minimum of four independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, based on one-way ANOVA and Tukey post hoc multiple comparison tests. (C) Immunoblot depicts MYPT1 phosphorylation at Thr853 in wild-type mouse platelets pre-incubated with vehicle, the PKC inhibitor BIM (15 µM), and/or the ROCK inhibitor Y27632 (50 µM) for 30 min prior to stimulation with thrombin (0.1 U/ml) for 10 min. Total MYPT1 is shown as a loading control. (D) Bar graph represents the densitometric quantification of p-MYPT1 (Thr853) relative to total MYPT1 in thrombin-activated platelets pre-treated with vehicle (TR, orange), BIM (BIM, green bar), Y27632 (Y27632, blue bar), or BIM plus Y27632 (BIM + Y27632, purple bar). Resting platelets serve as the negative control (NT, red bar). Data are mean ± SEM and represent a minimum of four independent experiments. *P < 0.05, ****P < 0.001, based on one-way ANOVA and Tukey post hoc multiple comparison tests.
FLNA is required for thrombin-induced PKC activity in platelets
Since FLNA is a ligand for PKC [38], we wished to compare the activity of PKC in control platelets and FLNA-deficient platelets. We evaluated the phosphorylation status of downstream effectors of PKC using a primary antibody that targets proteins with phospho-serine residues surrounded by an arginine (Arg) or lysine (Lys) at the −2 and +2 positions, and a hydrophobic residue at the +1 position [39]. We observed that the loss of FLNA expression in platelets conferred a significant (P < 0.05) reduction in PKC substrate phosphorylation following thrombin activation (Figure 5A,B), indicating that FLNA is essential for PKC activity in response to stimulation by thrombin.
Figure 5. FLNA regulates the function of PKC in platelets.
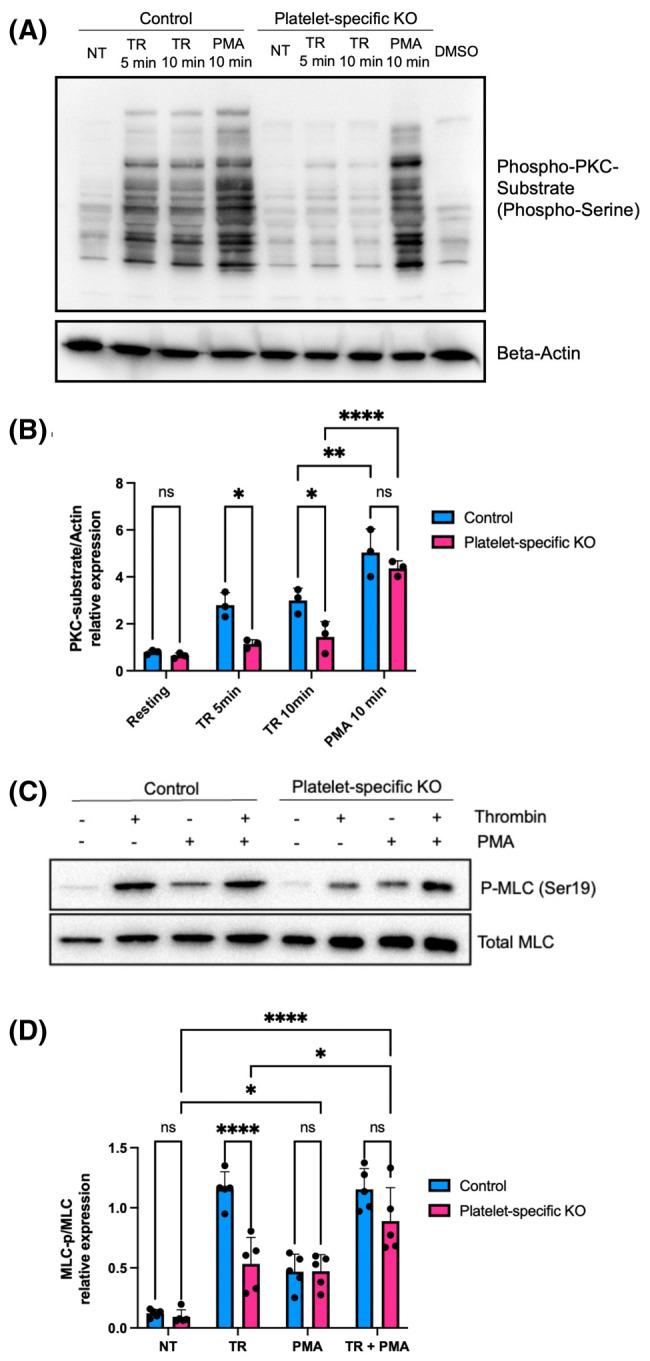
(A) Immunoblot depicts phospho-serine PKC substrates in floxed platelets (control) and FLNA-null platelets (platelet-specific KO) after treatment with thrombin or the phorbol ester PMA for the times indicated. Beta-actin is shown as a loading control. (B) Bar graphs depict the densitometric quantification of phospho-serine PKC substrates relative to beta-actin in floxed (control, blue bars) and FLNA-null platelets (platelet-specific KO, pink bars). Data are mean ± SEM, and represent a minimum of three independent experiments. *P < 0.05, **P < 0.01, ****P < 0.0001, based on two-way ANOVA and Tukey post hoc multiple comparison tests. (C) Immunoblot depicts MLC phosphorylation at Ser19 in floxed platelets (control) and FLNA-null platelets (platelet-specific KO) after treatment with thrombin and/or PMA for 10 min. Total MLC is shown as the loading control. (D) Bar graph depicts the densitometric quantification of phospho-MLC2 (Ser19) relative to total MLC in floxed (control, blue bars) and FLNA-null platelets (platelet-specific KO, pink bars). Platelets were untreated (NT), treated with thrombin (TR), treated with PMA (PMA), or treated with thrombin and PMA (TR + PMA). Data are mean ± SEM and represent a minimum of five independent experiments. *P < 0.05, ****P < 0.0001, based on two-way ANOVA and Tukey post hoc multiple comparison tests.
The phorbol ester, phorbol-12-myristate-13-acetate (PMA), is well-established as a potent activator of PKC [40]. PMA mimics the effect of diacylglycerol, thereby bypassing the requirement for the upstream signals that activate PKC [41]. Importantly, treatment with PMA effectively rescued the defect in PKC activation that was observed in the FLNA-deficient platelets (Figure 5A,B). We then wished to determine whether the rescue of PKC activity would also rescue the defect in MLC phosphorylation observed in FLNA-deficient platelets. In the FLNA-null platelets, PMA largely restored the defect in thrombin-induced MLC phosphorylation (Figure 5C,D), which is consistent with the notion that FLNA regulates MLC phosphorylation at least in part via PKC. Collectively, our data support a proposed model in which FLNA modulates MLC phosphorylation via two parallel pathways: by controlling PKC activity and by inhibiting MLCP (Figure 6).
Figure 6. Proposed model of FLNA as a regulator of MLC phosphorylation.

Schematic diagram illustrating a proposed model in which FLNA regulates MLC phosphorylation and cell contraction in platelets. (A) FLNA acts as a major scaffolding protein for both RhoA and ROCK to facilitate the phosphorylation (and therefore inhibition) of MLC phosphatase after thrombin activation. (B) FLNA modulates the activity of PKC which also contributes to MLC phosphorylation.
Discussion
Actomyosin contraction plays an essential role in multiple aspects of platelet function including shape change [7], clot retraction [9], and granule secretion [36,42–44]. However, the precise nature of the signals that underpin actomyosin contraction remain unclear although we hypothesized that this process would be regulated in part by actin-binding proteins. Here we found that the actin cross-linking protein FLNA is required for both the efficient phosphorylation of MLC and actomyosin contraction downstream of activation by thrombin. Moreover, our data provide further insights into the role of FLNA in regulating the RhoA/ROCK and PKC signaling pathways that lead to MLC phosphorylation.
FLNA is required for contractile force generation
To the best of our knowledge, our study is the first to report a role for FLNA in regulating MLC phosphorylation and actomyosin contraction in platelets. Myosin IIA is the predominant myosin heavy chain expressed in platelets and the actomyosin contraction mediated by this protein plays significant roles in platelet physiology and hemostasis [11,45]. As platelets contract, the associated fibrin clot becomes smaller, thus increasing thrombus density and stability. This compaction allows for maintenance of blood vessel patency and tissue reperfusion following vascular injuries [46]. Interestingly, FLNA-deficient mice exhibit a similar phenotype to those mice lacking functional myosin IIA, which exhibit macrothrombocytopenia, defective platelet clot retraction, and prolonged bleeding time [12]. Furthermore, platelets devoid of functional myosin IIA reportedly exhibit defective stress fiber formation while spreading on fibrinogen [12], which is similar to our observations in FLNA-deficient platelets. Our previous finding of diminished granule secretion by FLNA-deficient platelets [47] is also consistent with a reported reduction in granule secretion resulting from the inhibition of myosin IIA [48]. The striking similarity in the phenotype of platelets between these two mouse strains supports the existence of a functional relationship between FLNA and myosin IIA.
The notion that FLNA is responsible for cellular contractility is supported by a report that mice with a deletion of FLNA in smooth muscle exhibit a significant reduction of blood pressure and vascular tension [49]. Conversely, mice with impaired FLNA mRNA editing demonstrated increased vascular contraction and hypertension, notably accompanied by an increase in MLC phosphorylation [50]. Together, these findings point to a role for FLNA in the regulation of actomyosin activation in diverse cell types. However, ours is the first report of FLNA regulation of MLC phosphorylation and force generation in platelets; this central finding is supplemented by mechanistic data on the MLC-associated kinases and phosphatases.
FLNA regulates the ROCK-MLCP pathway
ROCK is a critical mediator of megakaryocyte and platelet function [19,51,52]. For example, published data indicate that ROCK activity and MLC phosphorylation in megakaryocytes is essential for the formation of proplatelets [52], which give rise to new platelets. Moreover, in platelets, defects in ROCK activity are associated with impaired MLC phosphorylation, clot retraction, and platelet aggregation [19,53,54]. Furthermore, ROCK is regulated upstream by the small GTPase RhoA [55]. Both RhoA and ROCK have been identified as FLNA ligands [25,56]; the Ig repeat 24 interacts with the carboxy-terminal pleckstrin homology domain of ROCK to control actin remodeling [21,23–25]. Our finding of increased MLCP activity in FLNA-deficient platelets suggests that FLNA influences ROCK activity since phosphorylation of MYPT1 (Thr853) is an accepted marker for ROCK activity [57]. Since both RhoA and ROCK bind FLNA near its C-terminus, it is plausible that FLNA brings RhoA and ROCK into proximity, sustaining their activation and culminating in the phosphorylation (and inactivation) of MLCP thus promoting MLC phosphorylation. Interestingly, FLNA reportedly interacts with Trio [58], a guanine nucleotide exchange factor with documented RhoA-activating properties [59] thus suggesting another mechanism by which FLNA may promote RhoA activity.
FLNA regulates PKC activity
PKC is another critical effector of platelet function [60] that also binds FLNA [38]. The notion that PKC activity is contingent on FLNA expression is supported by our finding of diminished PKC substrate phosphorylation in FLNA-deficient platelets. Importantly, the rescue of PKC function in FLNA-deficient platelets by PMA suggests that FLNA has a synergistic role with PKC. Traditionally, the activation of PKC involves a transition from the inactive latent form to an active membrane-bound form [61]. Since FLNA is ideally positioned between the membrane and F-actin [22], it is conceivable that FLNA also serves as a scaffold to approximate PKC with its downstream targets. Specifically, PKC reportedly phosphorylates the smooth muscle protein CPI-17; this protein binds and inhibits MLCP at its catalytic subunit (PP1) thereby increasing cellular contraction [62]. Our data in platelets partially corroborate this concept since PKC inhibition reduced MLC phosphorylation. However, blockade of PKC did not affect MLCP phosphorylation, which is consistent with published data indicating that CPI-17 inhibits MLCP by direct binding, i.e. in a phosphorylation-independent manner [62,63].
An important and novel finding from our study is that PKC and ROCK both regulate MLC phosphorylation but appear to do so independently. Moreover, PKC does not appear to promote MLC phosphorylation by inhibiting MLCP. In one study, the activity of MLCK was enhanced by PKC, although PKC-specific phosphorylation sites that directly activate myosin have not been identified [64]. In light of the functional diversity of PKC isoforms and the complexity of signaling cross-talk [60], it is conceivable that PKC could drive MLC phosphorylation independently of MLCP.
A limitation of our current study is that the exact binding site of ROCK on FLNA was not identified. Due to the many proteins with which FLNA interacts [21], a complete inhibition of FLNA would likely perturb other FLNA-containing signaling pathways, and would therefore be of limited practical use. Therefore, identification of the exact binding interface(s) between ROCK and FLNA would provide valuable insight into facilitating novel drug designs to specifically disrupt (or enhance) the FLNA-ROCK interaction. Furthermore, we did not identify the specific PKC isoform responsible for FLNA-mediated MLC phosphorylation. However, since FLNA-deficient platelets exhibit a normal calcium rise in response to thrombin stimulation [47], one can surmise that a calcium-independent isoform — such as from the novel PKC subfamily [65] — could be the operative isoform. Additional research is required to elucidate the mechanisms of action that govern FLNA and PKC in the context of platelet shape change.
In summary, this study identifies FLNA as a critical mediator for the generation of contractile force and shape change in thrombin-stimulated platelets. We propose that FLNA is a major scaffold for the subcellular localization of ROCK and PKC to facilitate the appropriate downstream signaling required to phosphorylate MLC and to generate actomyosin contraction. It should be noted that therapies aimed at blocking FLNA function also confer the potential for off-target effects since FLNA is expressed in multiple tissues. Nonetheless, given the critical scaffolding role played by FLNA, a better understanding of this complex signaling system (and related protein-protein interactions) — combined with ongoing advances in targeted tissue-specific drug delivery [66] — will facilitate the identification of therapeutic targets for improved management of bleeding disorders and/or thrombotic diseases.
Methods
Reagents
Antibodies against p-MLC2 Ser19 (cat. #3671), MLC2 (cat. #8505), p-MYPT1 Thr853 (cat. #4563), MYPT1 (cat. #2634), and p-Ser PKC substrate (cat. #2261) were purchased from Cell Signaling Technologies (Danvers, MA, U.S.A.). HRP-conjugated anti-beta-actin was purchased from Santa Cruz Biotechnology (Dallas, TX, U.S.A.). Anti-rabbit IgG, HRP-linked antibody (cat. #7074) was purchased from Cell Signaling Technologies (Danvers, MA, U.S.A.). Fibrinogen (F3879), BIM I, phorbol 1,2-myristate 1,3-acetate (PMA), and formaldehyde solution were purchased from MilliporeSigma (Oakville, ON, Canada). Thrombin was purchased from Chrono-Log (Havertown, PA, U.S.A.). The GPVI-specific collagen-related peptide was obtained from the Versiti Blood Research Institute Protein Chemistry Core Laboratory (Milwaukee, WI, U.S.A.). Bovine serum albumin (BSA) was purchased from Cytiva HyClone Laboratories (Logan, UT, U.S.A.). 100X Halt protease and phosphatase inhibitor cocktail, Alexa Fluor® 488, ProLong™ Gold Antifade Mountant, and Fluoromount-G™ Mounting Medium were obtained from Thermo Fisher Scientific (Mississauga, ON, Canada). The ROCK inhibitor Y27632 (ab120129) was purchased from Abcam (Cambridge, MA, U.S.A.).
Mice
Conditional KO mice were generated where FLNA expression was deleted in the megakaryocyte/platelet cell lineage [39,47]. All parental strains were obtained from The Jackson Laboratories: STOCK Flnatm1.1Caw/J (Strain #010907), with the allele simply called Flnafl hereafter (note Flna is on the X chromosome); C57BL/6-Tg(Pf4-icre)Q3Rsko/J (Strain #008535), with the transgene simply called Pf4-cre hereafter. Both parental strains were backcrossed onto 129S1/SvImJ (Strain #002448) at least six times for genetic background standardization. Then, female Flnafl/fl mice that also carried Pf4-cre/+ were bred with male Flnafl/Y mice. All experiments used FLNA-null platelets from Flnafl/fl, Pf4-cre/+ females and Flnafl/Y, Pf4-cre/+ males; littermate Flnafl/fl females and Flnafl/Y males were employed as controls. Approval for animal work was obtained from the University of British Columbia's Animal Care Committee.
Mouse platelet preparation
Mouse blood was drawn by retro-orbital plexus bleeding using heparinized capillary tubes and collected into acid citrate dextrose buffer. Whole blood was diluted with Tyrode's buffer (0.32 mM NaH2PO4, 150 mM NaCl, 2.65 mM KCl, 10.5 mM HEPES, 12 mM NaHCO3, 2.1 mM MgCl2, and 5 mM glucose) and centrifuged at 200 g for 7 min. The platelet-rich plasma was harvested and centrifuged at 800 g for 10 min to isolate the platelet pellet which was then gently resuspended in Tyrode's buffer. The platelet suspension was adjusted to a concentration of 2 × 108 - 5 × 108 platelets/ml and allowed to rest at room temperature for 20 min. Washed platelets were supplemented with 2 mM CaCl2 prior to stimulation with agonists.
Fluorescence microscopy and quantification of stress fibers
Coverslips were coated with fibrinogen (0.1 mg/ml), followed by a gentle wash with PBS. Platelets were stimulated with 0.05 U/ml thrombin for 1 min, plated on to fibrinogen-coated coverslips, and allowed to spread at 37°C for 45 min. After incubation, platelets were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and labeled with Alexa Fluor-488 phalloidin. Samples were mounted with ProLong™ Gold Antifade (ThermoFisher Scientific, Grand Island, NY, U.S.A.) mounting medium. Platelets were imaged using a Leica SP5 laser scanning confocal, with a 100× oil immersion objective. To quantify stress fiber formation, multiple fields of view (FOV) were analyzed to determine the percentage (%) of platelets (control and FLNA-deficient) with visible stress fibers per FOV.
Clot retraction assay
Washed platelets in Tyrode's buffer (3 × 108/ml) were incubated with 1% BSA, 1 mg/ml fibrinogen, and 2 mM calcium chloride in an aggregometer tube for 5 min. Platelets were then stimulated with 1 U/ml thrombin and incubated at 37°C for 45 min. Photographic images of the retracting clots were obtained, and the areas of the retracted clots were analyzed with ImageJ software.
Black dot fabrication
Black dots were fabricated by microcontact printing of fluorescent BSA onto flexible PDMS substrates as previously described [28]. Briefly, Sylgard 184 PDMS (Dow Corning) was prepared at a base to curing agent ratio of 10 to 1 by weight, cast against a silicon master structure containing an array of 850 nm diameter posts with 2 µm center-to-center spacing, and baked at 110°C for 20 min. Cured PDMS was peeled from the master structure, resulting in a PDMS stamp with an array of holes. Alexa-Fluor 594-conjugated BSA diluted in PBS to 2.5 µg/ml was pipetted onto the PDMS stamp and incubated for 45 min. The stamp was then dipped into dishes of PBS 3X and gently dried with nitrogen. To transfer the pattern, a polyvinyl alcohol film was plasma-etched for 1 min, pressed onto the BSA-adsorbed stamp, and incubated for 20 min under a 50 g weight. The polyvinyl alcohol film was peeled away from the stamp and pressed against a flexible (6.54 kPa) PDMS substrate. The flexible PDMS substrate was fabricated by mixing 98% Sylgard 527 (prepared at a part A to part B ratio of 1 to 1 by weight) with 2% Sylgard 184 (prepared at a base to curing agent ratio of 10 to 1 by weight), pipetting onto glass coverslips, and curing overnight at 65°C. After allowing 20 min for the fluorescent pattern to transfer onto the flexible PDMS substrate, the substrate was submerged in PBS. Upon rehydration, the polyvinyl film detached from the flexible PDMS and was discarded. The black dot substrates were stored in PBS at 4°C and were used within 3 weeks.
Mouse platelet seeding onto black dots
Black dot substrates were incubated with 1 mg/ml of human fibrinogen at room temperature for 1.5 h in the dark and submerged in PBS after incubation. Washed platelets (3.2 × 106/ml per substrate) were pre-stimulated with 0.05 U thrombin in the presence of 1 mM Ca2+ for 1 min and seeded directly onto the black dot substrates for 10 min at room temperature. After 10 min, unbound platelets were removed by dipping the substrate in PBS and immediately transferring to Tyrode's buffer for further incubation at 37°C. Substrates were then fixed with 4% paraformaldehyde at room temperature and gently rinsed with PBS twice. Fixed platelet substrates were permeabilized with 0.1% Triton X-100 at room temperature and washed 3x with PBS. Permeabilized platelets were stained with Alexa Fluor-488 phalloidin prior to mounting with Fluoromount-G mounting medium (ThermoFisher Scientific, Grand Island, NY, U.S.A.).
Black dot imaging and single-platelet force analysis
Fixed and stained platelets were imaged on a Leica SP5 confocal microscope with a 63× oil immersion objective (NA = 1.4). The images of platelets were taken with a large enough field of view to ensure the clarity of the platelet spreading morphology. All image analysis was conducted in MATLAB. Cell boundaries were determined from the fluorescent F-actin image with a user-adjusted threshold and shape fill. Black dot boundaries and centroids were determined via automated detection. Black dot displacement from the undeformed grid was used to calculate the magnitude and direction of forces using regularized Fourier transform traction cytometry. Total force for each platelet was calculated by summing the force magnitudes within each platelet boundary.
RhoA activation assay
RhoA activity was determined in control and FLNA-deficient platelets using the absorbance-based G-LISA® RhoA Activation Assay Kit, cat. #BK124 (Cytoskeleton Inc., Denver, CO, U.S.A.). Briefly, 100 µl of control and FLNA-deficient mouse platelets (3 × 108 cells/ml) were activated with 0.1 U thrombin for 5 min at 37°C. The platelets were solubilized with ice-cold lysis buffer supplied by the manufacturer, and the levels of active, GTP-bound RhoA were determined in the lysates in accordance with the manufacturer's specifications. Absorbance was read at 490 nm using a SpectraMax i3 microplate reader.
Immunoblotting
Washed platelets (3 × 108 cells/ml) were solubilized and resuspended in 5X sample buffer. Samples were heated at 95°C for 5 min. Proteins were resolved using SDS–PAGE and transferred to a PVDF membrane. The membranes were incubated with various primary antibodies overnight at 4°C, followed by incubation with HRP-conjugated secondary antibodies for 1 h at room temperature. Bands were detected by enhanced chemiluminescence (Cell Signaling Technologies, Danvers, MA, U.S.A.) and visualized with a Bio-Rad ChemiDoc imaging system (Mississauga, ON, Canada).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9 software. Differences between two groups were analyzed using the Student's t-test. Differences between three or more groups were analyzed using an analysis of variance (ANOVA) and Tukey or Sidak's post hoc multiple comparison tests. Statistical significance was set at P < 0.05.
Acknowledgements
The authors thank Tess Lengyell and Beth Simpson (UBC Centre for Molecular Medicine and Therapeutics) as well as Takehide Murakami (UBC Biomedical Research Centre) for assistance with mouse breeding and genotyping, and Ingrid Ellis (UBC Faculty of Dentistry) for editorial assistance. The authors also thank Hervé Falet (Versiti Blood Research Institute) for the provision of the collagen-related peptide.
Abbreviations
- ANOVA
analysis of variance
- BIM I
bisindolylmaleimide I
- BSA
bovine serum albumin
- FLNA
Filamin A
- FOV
fields of view
- KO
knockout
- MLC
myosin light chain
- MLCK
MLC kinase
- MLCP
MLC phosphatase
- MYPT1
protein phosphatase 1 regulatory subunit 12A
- PAR
protease-activated receptor
- PDMS
polydimethylsiloxane
- PKC
protein kinase C
- PMA
phorbol-12-myristate-13-acetate
Data Availability
The relevant data that support the conclusions of this study are contained within the article. The original, full-sized unprocessed images for all of the immunoblots presented in the Figures are provided as Supplementary Figures.
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Funding
This study was supported by a Canadian Institutes of Health Research (CIHR) Project Grant (PJT-156341) to H.K. F.H. acknowledges support from the UBC Centre for Blood Research Graduate Award Program, and a Research Studentship from the Canadian Venous Thromboembolism Research Network (CanVECTOR). M.Y.M. received support from the National Institutes of Health (HL007093). E.D.S. received support from a Training Graduate PhD Award (19-0491) from The Arthritis Society. NS acknowledges support from the National Institutes for Health (HL145262, HL149734, AR074990) and the National Science Foundation (CMMI1824792). H.K. acknowledges salary support from a Scholar Award (17650) from Michael Smith Health Research BC.
Open Access
Open access for this article was enabled by the participation of University of British Columbia in an all-inclusive Read & Publish agreement with Portland Press and the Biochemical Society under a transformative agreement with Individual.
CRediT Author Contribution
Hugh Kim: Conceptualization, Supervision, Funding acquisition, Writing - original draft, Writing - review & editing. Felix Hong: Conceptualization, Formal analysis, Investigation, Methodology, Writing - original draft, Writing - review & editing. Molly Y. Mollica: Conceptualization, Formal analysis, Methodology. Kalyan Golla: Conceptualization, Methodology. Enoli De Silva: Conceptualization, Methodology. Nathan J. Sniadecki: Supervision, Methodology, Writing - review & editing. José A. López: Conceptualization, Supervision, Writing - review & editing.
Ethical Approval
Ethical approval was obtained from the University of British Columbia's Animal Care Committee (#A21-0177). Animal work took place on UBC's Point Grey Campus. Mice were anesthetised with 250 mg/kg Avertin in a single intraperitoneal injection as per standard operating procedures. After blood collection, the animals were immediately euthanized by cervical dislocation.
Supplementary Material
References
- 1.Machlus, K.R. and Italiano, Jr, J.E. (2013) The incredible journey: from megakaryocyte development to platelet formation. J. Cell Biol. 201, 785–796 10.1083/jcb.201304054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brewer, D.B. (2006) Max Schultze (1865), G. Bizzozero (1882) and the discovery of the platelet. Br. J. Haematol. 133, 251–258 10.1111/j.1365-2141.2006.06036.x [DOI] [PubMed] [Google Scholar]
- 3.Gremmel, T., Frelinger, III, A. L., and Michelson, A. D. (2016) Platelet physiology. Semin. Thromb. Hemost. 42, 191–204 10.1055/s-0035-1564835 [DOI] [PubMed] [Google Scholar]
- 4.Tomaiuolo, M., Brass, L.F. and Stalker, T.J. (2017) Regulation of platelet activation and coagulation and its role in vascular injury and arterial thrombosis. Interv. Cardiol. Clin. 6, 1–12 10.1016/j.iccl.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bye, A.P., Unsworth, A.J. and Gibbins, J.M. (2016) Platelet signaling: a complex interplay between inhibitory and activatory networks. J. Thromb. Haemost. 14, 918–930 10.1111/jth.13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amalia, L. and Dalimonthe, N.Z. (2020) Clinical significance of Platelet-to-White Blood Cell Ratio (PWR) and National Institute of Health Stroke Scale (NIHSS) in acute ischemic stroke. Heliyon 6, e05033 10.1016/j.heliyon.2020.e05033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shin, E.K., Park, H., Noh, J.Y., Lim, K.M. and Chung, J.H. (2017) Platelet shape changes and cytoskeleton dynamics as novel therapeutic targets for anti-thrombotic drugs. Biomol. Ther. 25, 223–230 10.4062/biomolther.2016.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beaumont, D.O., Whitten, K.W., Mock, R.G. and Slattery, C.W. (1989) Measuring platelet function with platelet shape change, an early event in aggregation. Thromb. Res. 53, 109–127 10.1016/0049-3848(89)90373-3 [DOI] [PubMed] [Google Scholar]
- 9.Johnson, G.J., Leis, L.A., Krumwiede, M.D. and White, J.G. (2007) The critical role of myosin IIA in platelet internal contraction. J. Thromb. Haemost. 5, 1516–1529 10.1111/j.1538-7836.2007.02611.x [DOI] [PubMed] [Google Scholar]
- 10.Bearer, E.L., Prakash, J.M. and Li, Z. (2002) Actin dynamics in platelets. Int. Rev. Cytol. 217, 137–182 10.1016/s0074-7696(02)17014-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berg, J.S., Powell, B.C. and Cheney, R.E. (2001) A millennial myosin census. Mol. Biol. Cell 12, 780–794 10.1091/mbc.12.4.780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leon, C., Eckly, A., Hechler, B., Aleil, B., Freund, M., Ravanat, C.et al. (2007) Megakaryocyte-restricted MYH9 inactivation dramatically affects hemostasis while preserving platelet aggregation and secretion. Blood 110, 3183–3191 10.1182/blood-2007-03-080184 [DOI] [PubMed] [Google Scholar]
- 13.Pal, K., Nowak, R., Billington, N., Liu, R., Ghosh, A., Sellers, J.R.et al. (2020) Megakaryocyte migration defects due to nonmuscle myosin IIA mutations underlie thrombocytopenia in MYH9-related disease. Blood 135, 1887–1898 10.1182/blood.2019003064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vicente-Manzanares, M., Ma, X., Adelstein, R.S. and Horwitz, A.R. (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790 10.1038/nrm2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scholey, J.M., Taylor, K.A. and Kendrick-Jones, J. (1980) Regulation of non-muscle myosin assembly by calmodulin-dependent light chain kinase. Nature 287, 233–235 10.1038/287233a0 [DOI] [PubMed] [Google Scholar]
- 16.Feghhi, S., Tooley, W.W. and Sniadecki, N.J. (2016) Nonmuscle myosin IIA regulates platelet contractile forces through Rho kinase and myosin light-chain kinase. J. Biomech. Eng. 138, 1045061 10.1115/1.4034489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizuno, Y., Isotani, E., Huang, J., Ding, H., Stull, J.T. and Kamm, K.E. (2008) Myosin light chain kinase activation and calcium sensitization in smooth muscle in vivo. Am. J. Physiol. Cell Physiol. 295, C358–C364 10.1152/ajpcell.90645.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alvarez-Santos, M.D., Alvarez-Gonzalez, M., Estrada-Soto, S. and Bazan-Perkins, B. (2020) Regulation of myosin light-chain phosphatase activity to generate airway smooth muscle hypercontractility. Front. Physiol. 11, 701 10.3389/fphys.2020.00701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aburima, A., Wraith, K.S., Raslan, Z., Law, R., Magwenzi, S. and Naseem, K.M. (2013) cAMP signaling regulates platelet myosin light chain (MLC) phosphorylation and shape change through targeting the RhoA-Rho kinase-MLC phosphatase signaling pathway. Blood 122, 3533–3545 10.1182/blood-2013-03-487850 [DOI] [PubMed] [Google Scholar]
- 20.Falet, H., Pollitt, A.Y., Begonja, A.J., Weber, S.E., Duerschmied, D., Wagner, D.D.et al. (2010) A novel interaction between FlnA and Syk regulates platelet ITAM-mediated receptor signaling and function. J. Exp. Med. 207, 1967–1979 10.1084/jem.20100222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Silva, E., Hong, F., Falet, H. and Kim, H. (2022) Filamin A in platelets: bridging the (signaling) gap between the plasma membrane and the actin cytoskeleton. Front. Mol. Biosci. 9, 1060361 10.3389/fmolb.2022.1060361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura, F., Stossel, T.P. and Hartwig, J.H. (2011) The filamins: organizers of cell structure and function. Cell Adh. Migr. 5, 160–169 10.4161/cam.5.2.14401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim, H. and McCulloch, C.A. (2011) Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Lett. 585, 18–22 10.1016/j.febslet.2010.11.033 [DOI] [PubMed] [Google Scholar]
- 24.Ueda, K., Ohta, Y. and Hosoya, H. (2003) The carboxy-terminal pleckstrin homology domain of ROCK interacts with filamin-A. Biochem. Biophys. Res. Commun. 301, 886–890 10.1016/s0006-291x(03)00048-2 [DOI] [PubMed] [Google Scholar]
- 25.Ohta, Y., Hartwig, J.H. and Stossel, T.P. (2006) FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat. Cell Biol. 8, 803–814 10.1038/ncb1437 [DOI] [PubMed] [Google Scholar]
- 26.Sun, C., Forster, C., Nakamura, F. and Glogauer, M. (2013) Filamin-A regulates neutrophil uropod retraction through RhoA during chemotaxis. PLoS One 8, e79009 10.1371/journal.pone.0079009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bender, M. and Palankar, R. (2021) Platelet shape changes during thrombus formation: role of actin-based protrusions. Hamostaseologie 41, 14–21 10.1055/a-1325-0993 [DOI] [PubMed] [Google Scholar]
- 28.Beussman, K.M., Mollica, M.Y., Leonard, A., Miles, J., Hocter, J., Song, Z.et al. (2023) Black dots: high-yield traction force microscopy reveals structural factors contributing to platelet forces. Acta Biomater. 163, 302–311 10.1016/j.actbio.2021.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mollica, M.Y., Beussman, K.M., Kandasamy, A., Rodriguez, L.M., Morales, F.R., Chen, J.et al. (2023) Distinct platelet F-actin patterns and traction forces on von Willebrand factor versus fibrinogen. Biophys. J. 122, 3738–3748 10.1016/j.bpj.2023.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabass, B., Gardel, M.L., Waterman, C.M. and Schwarz, U.S. (2008) High resolution traction force microscopy based on experimental and computational advances. Biophys. J. 94, 207–220 10.1529/biophysj.107.113670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han, S.J., Oak, Y., Groisman, A. and Danuser, G. (2015) Traction microscopy to identify force modulation in subresolution adhesions. Nat. Methods 12, 653–656 10.1038/nmeth.3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brill-Karniely, Y., Nisenholz, N., Rajendran, K., Dang, Q., Krishnan, R. and Zemel, A. (2014) Dynamics of cell area and force during spreading. Biophys. J. 107, L37–L40 10.1016/j.bpj.2014.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanke, J., Probst, D., Zemel, A., Schwarz, U.S. and Koster, S. (2018) Dynamics of force generation by spreading platelets. Soft Matter 14, 6571–6581 10.1039/C8SM00895G [DOI] [PubMed] [Google Scholar]
- 34.Derksen, P.W.B. and van de Ven, R.A.H. (2020) Shared mechanisms regulate spatiotemporal RhoA-dependent actomyosin contractility during adhesion and cell division. Small GTPases 11, 113–121 10.1080/21541248.2017.1366966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ringvold, H.C. and Khalil, R.A. (2017) Protein kinase C as regulator of vascular smooth muscle function and potential target in vascular disorders. Adv. Pharmacol. 78, 203–301 10.1016/bs.apha.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Offermanns, S. (2006) Activation of platelet function through G protein-coupled receptors. Circ. Res. 99, 1293–1304 10.1161/01.RES.0000251742.71301.16 [DOI] [PubMed] [Google Scholar]
- 37.Smyth, S.S., Woulfe, D.S., Weitz, J.I., Gachet, C., Conley, P.B., Goodman, S.G.et al. (2009) G-protein-coupled receptors as signaling targets for antiplatelet therapy. Arterioscler. Thromb. Vasc. Biol. 29, 449–457 10.1161/atvbaha.108.176388 [DOI] [PubMed] [Google Scholar]
- 38.Tigges, U., Koch, B., Wissing, J., Jockusch, B.M. and Ziegler, W.H. (2003) The F-actin cross-linking and focal adhesion protein filamin A is a ligand and in vivo substrate for protein kinase C alpha. J. Biol. Chem. 278, 23561–23569 10.1074/jbc.m302302200 [DOI] [PubMed] [Google Scholar]
- 39.De Silva, E., Devine, D.V., Jan, E., Roskelley, C.D. and Kim, H. (2022) Filamin A regulates caspase-3 cleavage in platelets in a protein kinase C (PKC)-dependent manner. Biochem. J. 479, 2351–2364 10.1042/BCJ20220177 [DOI] [PubMed] [Google Scholar]
- 40.Way, K.J., Chou, E. and King, G.L. (2000) Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol. Sci. 21, 181–187 10.1016/S0165-6147(00)01468-1 [DOI] [PubMed] [Google Scholar]
- 41.Gray, R.D., Lucas, C.D., MacKellar, A., Li, F., Hiersemenzel, K., Haslett, C., et al. (2013) Activation of conventional protein kinase C (PKC) is critical in the generation of human neutrophil extracellular traps. J. Inflamm. 10, 12 10.1186/1476-9255-10-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moskalensky, A.E. and Litvinenko, A.L. (2019) The platelet shape change: biophysical basis and physiological consequences. Platelets 30, 543–548 10.1080/09537104.2018.1514109 [DOI] [PubMed] [Google Scholar]
- 43.Zucker, M.B. and Nachmias, V.T. (1985) Platelet activation. Arteriosclerosis 5, 2–18 10.1161/01.atv.5.1.2 [DOI] [PubMed] [Google Scholar]
- 44.Sanborn, K.B., Rak, G.D., Maru, S.Y., Demers, K., Difeo, A., Martignetti, J.A.et al. (2009) Myosin IIA associates with NK cell lytic granules to enable their interaction with F-actin and function at the immunological synapse. J. Immunol. 182, 6969–6984 10.4049/jimmunol.0804337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nurden, A.T. (2022) Molecular basis of clot retraction and its role in wound healing. Thromb. Res. 231, 159–169 10.1016/j.thromres.2022.08.010 [DOI] [PubMed] [Google Scholar]
- 46.Samson, A.L., Alwis, I., Maclean, J.A.A., Priyananda, P., Hawkett, B., Schoenwaelder, S.M.et al. (2017) Endogenous fibrinolysis facilitates clot retraction in vivo. Blood 130, 2453–2462 10.1182/blood-2017-06-789032 [DOI] [PubMed] [Google Scholar]
- 47.Golla, K., Paul, M., Lengyell, T.C., Simpson, E.M., Falet, H. and Kim, H. (2023) A novel association between platelet filamin A and soluble N-ethylmaleimide sensitive factor attachment proteins regulates granule secretion. Res. Pract. Thromb. Haemost. 7, 100019 10.1016/j.rpth.2022.100019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kenny, M., Pollitt, A.Y., Patil, S., Hiebner, D.W., Smolenski, A., Lakic, N.et al. (2024) Contractility defects hinder glycoprotein VI-mediated platelet activation and affect platelet functions beyond clot contraction. Res. Pract. Thromb. Haemost. 8, 102322 10.1016/j.rpth.2024.102322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Retailleau, K., Arhatte, M., Demolombe, S., Peyronnet, R., Baudrie, V., Jodar, M.et al. (2016) Arterial myogenic activation through smooth muscle filamin A. Cell Rep. 14, 2050–2058 10.1016/j.celrep.2016.02.019 [DOI] [PubMed] [Google Scholar]
- 50.Jain, M., Mann, T.D., Stulic, M., Rao, S.P., Kirsch, A., Pullirsch, D.et al. (2018) RNA editing of Filamin A pre-mRNA regulates vascular contraction and diastolic blood pressure. EMBO J. 37, e94813 10.15252/embj.201694813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aslan, J.E. (2019) Platelet Rho GTPase regulation in physiology and disease. Platelets 30, 17–22 10.1080/09537104.2018.1475632 [DOI] [PubMed] [Google Scholar]
- 52.Chang, Y., Aurade, F., Larbret, F., Zhang, Y., Le Couedic, J.P., Momeux, L.et al. (2007) Proplatelet formation is regulated by the Rho/ROCK pathway. Blood 109, 4229–4236 10.1182/blood-2006-04-020024 [DOI] [PubMed] [Google Scholar]
- 53.Suzuki-Inoue, K., Hughes, C.E., Inoue, O., Kaneko, M., Cuyun-Lira, O., Takafuta, T.et al. (2007) Involvement of Src kinases and PLCgamma2 in clot retraction. Thromb. Res. 120, 251–258 10.1016/j.thromres.2006.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sladojevic, N., Oh, G.T., Kim, H.H., Beaulieu, L.M., Falet, H., Kaminski, K.et al. (2017) Decreased thromboembolic stroke but not atherosclerosis or vascular remodelling in mice with ROCK2-deficient platelets. Cardiovasc. Res. 113, 1307–1317 10.1093/cvr/cvx071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aslan, J.E. and McCarty, O.J. (2013) Rho GTPases in platelet function. J. Thromb. Haemost. 11, 35–46 10.1111/jth.12051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohta, Y., Suzuki, N., Nakamura, S., Hartwig, J.H. and Stossel, T.P. (1999) The small GTPase RalA targets filamin to induce filopodia. Proc. Natl Acad. Sci. U.S.A. 96, 2122–2128 10.1073/pnas.96.5.2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu, P.Y. and Liao, J.K. (2008) A method for measuring Rho kinase activity in tissues and cells. Methods Enzymol. 439, 181–189 10.1016/s0076-6879(07)00414-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bellanger, J.M., Astier, C., Sardet, C., Ohta, Y., Stossel, T.P. and Debant, A. (2000) The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat. Cell Biol. 2, 888–892 10.1038/35046533 [DOI] [PubMed] [Google Scholar]
- 59.Medley, Q.G., Serra-Pages, C., Iannotti, E., Seipel, K., Tang, M., O'Brien, S.P.et al. (2000) The trio guanine nucleotide exchange factor is a RhoA target. Binding of RhoA to the trio immunoglobulin-like domain. J. Biol. Chem. 275, 36116–36123 10.1074/jbc.m003775200 [DOI] [PubMed] [Google Scholar]
- 60.Harper, M.T. and Poole, A.W. (2010) Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J. Thromb. Haemost. 8, 454–462 10.1111/j.1538-7836.2009.03722.x [DOI] [PubMed] [Google Scholar]
- 61.Rosse, C., Linch, M., Kermorgant, S., Cameron, A.J., Boeckeler, K. and Parker, P.J. (2010) PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11, 103–112 10.1038/nrm2847 [DOI] [PubMed] [Google Scholar]
- 62.Watanabe, Y., Ito, M., Kataoka, Y., Wada, H., Koyama, M., Feng, J.et al. (2001) Protein kinase C-catalyzed phosphorylation of an inhibitory phosphoprotein of myosin phosphatase is involved in human platelet secretion. Blood 97, 3798–3805 10.1182/blood.v97.12.3798 [DOI] [PubMed] [Google Scholar]
- 63.Eto, M. and Kitazawa, T. (2017) Diversity and plasticity in signaling pathways that regulate smooth muscle responsiveness: paradigms and paradoxes for the myosin phosphatase, the master regulator of smooth muscle contraction. J. Smooth Muscle Res. 53, 1–19 10.1540/jsmr.53.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clement, O., Puceat, M., Walsh, M.P. and Vassort, G. (1992) Protein kinase C enhances myosin light-chain kinase effects on force development and ATPase activity in rat single skinned cardiac cells. Biochem. J. 285, 311–317 10.1042/bj2850311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vedula, P., Kurosaka, S., Leu, N.A., Wolf, Y.I., Shabalina, S.A., Wang, J.et al. (2017) Diverse functions of homologous actin isoforms are defined by their nucleotide, rather than their amino acid sequence. Elife 6, e31661 10.7554/eLife.31661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao, Z., Ukidve, A., Kim, J. and Mitragotri, S. (2020) Targeting strategies for tissue-specific drug delivery. Cell 181, 151–167 10.1016/j.cell.2020.02.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The relevant data that support the conclusions of this study are contained within the article. The original, full-sized unprocessed images for all of the immunoblots presented in the Figures are provided as Supplementary Figures.