

Abstract
Ability of IL-17-producing CD8+ T cells (Tc17) to transform into cytotoxic anti-tumour effectors makes them a promising candidate for immune effector cell (IEC) therapy. However, key factors regulating Tc17 reprogramming remain poorly defined, hindering translation of Tc17-based IEC use from bench to bedside. We probed the effects of multiple cytokines and underlying signalling pathways on Tc17 cells and identified pivotal role for IL-4 and PI3K/AKT in promoting Tc17 transformation into cytotoxic IFN-γ-producing IECs, an effect dependent on Eomes expression. IL-4 not only triggered Tc17 cytotoxicity, but also induced cell expansion, which significantly improved the antitumour potential of Tc17 cells compared to that of IFN-γ-producing CD8+ T cells (Tc1) in a murine model. Furthermore, IL-4/AKT signalling drove the upregulation of the T-cell receptor-associated transmembrane adaptor 1 (Trat1) in Tc17 cells to promote IL-4-induced T-cell receptor stabilization and Tc17 cytotoxicity. Finally, we proposed a possible procedure to expand human Tc17 from peripheral blood of cancer patients, and confirmed the function of IL-4 in Tc17 reprogramming. Collectively, these results document a novel IL-4/AKT/Eomes/Trat1 axis that promotes expansion and transformation of Tc17 cells into cytotoxic effectors with a therapeutic potential. IL-4 priming of Tc17 cells should be further explored as a cell therapy engineering strategy to generate IECs to augment anti-tumour responses.
Keywords: adoptive cell therapy, Eomes, IL-4, IL-17, T cell reprogramming, Tc17, Trat1
INTRODUCTION
Advances in the immune effector cell (IEC) generation have dramatically changed the therapeutic landscape of cellular therapy of cancer [1–6]. However, success remains largely restricted to the field of chimeric antigen receptor engineered T cell therapy which has demonstrated efficacy in a narrow range of malignancies. Challenges in generation of polyclonal tumour antigen specific IECs remain, with results of recent clinical trials suggesting current strategies usually induce acute but transient responses [7–10]. Highly differentiated CD8+ T cells generated ex vivo using current standard protocols in adoptive cell therapy (ACT) usually exhibit limited in vivo T cell activation, proliferation, and survival, leading to decreased antitumour activity [11,12]. Therefore, an immediate need exists for generation of more effective IECs with a durable immune surveillance potential to maximize the long-term success of anti-cancer ACT.
IL-17-producing CD8+ T cells (Tc17) have recently been identified in mice [13] and humans [14]. They are characterized by reduced cytotoxicity and greater plasticity when compared to the typical cytotoxic T lymphocytes (CTLs). Recent studies have proposed that Tc17 cells can protect hosts against lethal influenza challenge and fungal pneumonia [15,16], worsen graft-versus-host disease [17,18], and modulate autoimmune diseases, including multiple sclerosis, type I diabetes, colitis, and psoriasis [19–21]. Although Tc17 cells expresses IFN-γ perforin, Fas ligand (FasL), and granzyme B at lower levels [13], evidence supports a comparable antitumour activity and greater persistence when compared to Tc1 cells in mouse models [22–26]. Furthermore, longer persistence of Tc17 may contribute to a more durable cytotoxicity under repeated tumour challenges [26]. Previous studies have indicated that certain cytokines, such as IL-23 or IL-12, can convert Tc17 cells from primarily IL-17A producers to IFN-γ producers [27,28], and this conversion promotes the antitumour activity of Tc17 cells. However, the functions of other cytokines in regulating Tc17 reprogramming and the mechanisms underlying cytotoxicity promotion remain unclear.
Herein, we sought to identify the critical factors and associated underlying molecular mechanisms promoting Tc17 cell expansion and reprogramming into anti-tumour effectors. We identified IL-4-induced PI3K/AKT pathway activity as critical for reprogramming of Tc17 cells into IFN-γ producers, in a process dependent upon Eomes upregulation. Notably, IL-4 treatment not only triggered Tc17 cytotoxicity but also induced cell expansion and promoted the expression of the T-cell receptor-associated transmembrane adaptor 1 (Trat1) which stabilizes the T-cell receptor (TCR). Observed effects may ultimately enhance cellular effector functions of Tc17 cells and augment their antitumour potential as that seen with Tc1 cells. At last, we found that the downregulation of Trat1 in cancer tissues is associated with poor survival among patients in several types of human cancer. We proposed a protocol to expand human Tc17 from peripheral blood of cancer patients, and confirmed the function of IL-4 in Tc17 reprogramming, suggesting its potential to development into novel anti-cancer ACT.
RESULTS
IL-4 promotes the conversion and expansion of Tc17 cells into IFN-γ-producing cells
IL-12 induces IFN-γ production in Tc17 cells [28]; however, the effects of other cytokines on the functional plasticity of Tc17 cells are not well-documented. To evaluate impact of distinct cytokines on Tc17 cells, polarized influenza haemagglutinin (HA)-specific Tc17 cells from TCR transgene mice (Clone 4 mice) were stimulated with common gamma chain cytokines (IL-2, IL-4, IL-7 and IL-15), the Th17 polarization cytokines IL-1β and IL-23 [29,30], or the Th1 polarization cytokine IL-12 in vitro (Figure 1a). IL-4, IL-7 and IL-12 increased the frequencies of IFN-γ-producing cells, whereas IL-2 decreased the frequencies of IL-17A- and IFN-γ-producing cells. IL-1β, IL-23 and IL-15 moderately increased the Tc17 percentage (Figure 1b). Notably, among these cytokines, IL-4 displayed the greatest efficiency in increasing the population of IFN-γ-producing cells (from 2.8 ± 1.3% to 45.6 ± 8.6%) and decreasing the frequencies of IL-17A cells (from 62.5 ± 2.9% to 10 ± 1.1%, Figure 1c). Furthermore, in a cytokine secretion assay, we confirmed that the IL-17A-secreting cells can transform into IFN-γ-producing cells, and the IFN-γ/IL-17A double positive cells could be a transient stage in the conversion (Figure 1d). Although IL-13 can transduce signals through IL-4Rα, which expressed on Tc17 cells (Figure S1a) [31], we found that IL-13 treatment only moderately increased the Tc17 population, whereas it failed to induce the IFN-γ-production, suggesting the distinct functions of IL-4 in Tc17 cell conversion. Titration of the IL-4 concentration for Tc17 conversion showed that the efficiency peaked when over 20 ng/ml of IL-4 was used (Figure S1b).
FIGURE 1.
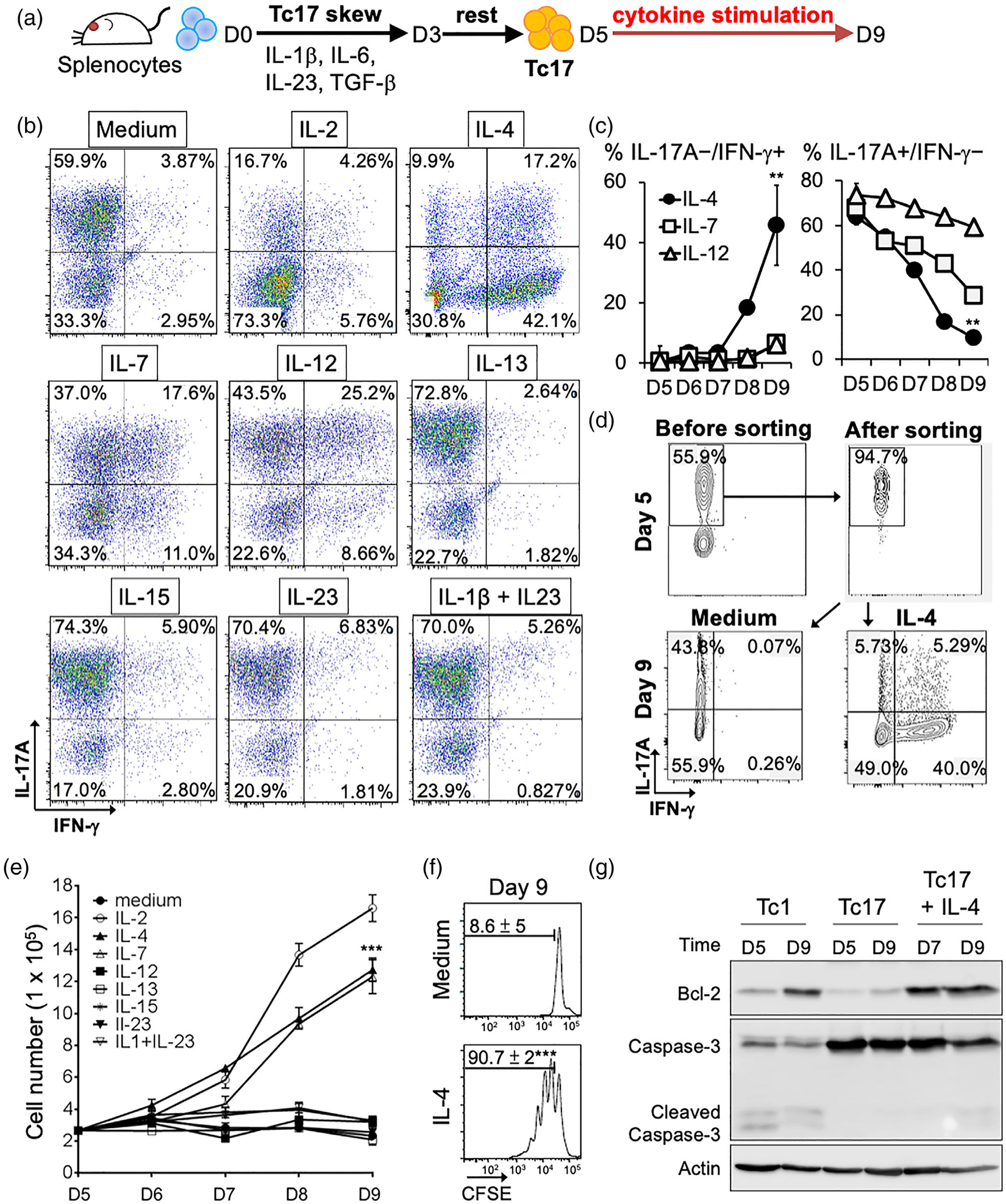
Effects of different cytokines on conversion and expansion of Tc17 cells. (a) Schema of Tc17 cells polarization and cytokine stimulation. Splenocytes were incubated in Tc17 skewing medium for 3 days (D3). After 2 days’ rest, different cytokines were added to Tc17 cells for further 4 days (D9). (b) Representative dot plots showing population changes in IL17A- and IFN-γ-producing cells following stimulation with different cytokines at day 9 (D9). (c) Average changes in IL-17A/IFN-γ population upon IL-4, IL-7, or IL-12 stimulation. Mean ± SD are shown form three independent experiments. **P < 0.01. (d) IL-4 treatment of IL-17A producing cells programs them into IFN-γ-producing cells. Cytokine secretion assay was used to label and sort IL-17A producing cells. Cells were treated with or without IL-4 for 4 days. Representative results were shown from one out of three independent experiments. (e) Effects of different cytokines on Tc17 expansion. Viable cells were counted using trypan blue exclusion assay. Mean ± SD of three independent experiments was shown. (f) IL-4 enhanced Tc17 proliferation. CFSE-labelled assay was used to measure proliferation of Tc17 cells. Mean ± SD of three independent experiments was shown at right. ***P < 0.001. (g) IL-4 induces anti-apoptosis signature in Tc17 cells. Expression of Bcl-2 and Caspase3 in Tc1 and Tc17 cells was analysed by Western blot. Actin was used as a loading control
The ex vivo expansion of antigen (Ag)-specific T cells and their in vivo persistence are crucial factors for the success of ACT in patients with cancer [1,32]. Among the cytokines effects of which we tested on mouse Tc17 cells, only IL-2, IL-4 and IL-7 significantly increased the number of viable cells (Figure 1e), with the carboxifluorescein diacetate succinimidyl ester (CFSE) dilution assay supporting the direct proliferative effect of IL-4 on the subset (Figure 1f). Furthermore, IL-4 treatment increased the expression of the anti-apoptotic protein Bcl-2 in Tc17 cells, whereas the levels of cleaved caspase-3 were lower compared to that in apoptosis-susceptible Tc1 population (Figure 1g). In summary, IL-4 promotes Tc17 polarization to IFN-γ-producing CD8+ T cells with enhanced anti-apoptotic potential and represents a viable strategy for their in vitro expansion.
IL-4 enhanced Tc17 antitumour immunity in vitro and in vivo
Since IL-4 reprogrammed Tc17 cells to secret IFN-γ and enhanced their expansion, we next analysed whether it also promoted Tc17 cytotoxicity and antitumour activity. Using standard in vitro cytotoxicity assays, we found that IL-4 treatment significantly increased the tumour kill by the HA-specific Tc17 cells in a model of HA-expressing colon cancer (CT26-HA; Figure 2a,b). In a follow-up in vivo CTL assay, we observed that the IL-4-treated Tc17 cells exhibited enhanced cytotoxic potential when compared to that seen with untreated Tc17 cells (Figure 2c). Similar results were observed in vitro and in vivo using Tc17 cells responding to ovalbumin (generated from OT-I mice) and using a B16-OVA melanoma cell line as a target (Figure S2). IL-4-treatment of the pool of polarized Tc17 cells significantly enhanced expression of the core cytotoxic molecules to comparable levels of the pool of IL-12-polarized Tc1, including perforin, granzyme B, and FasL (Figure 2d)
FIGURE 2.
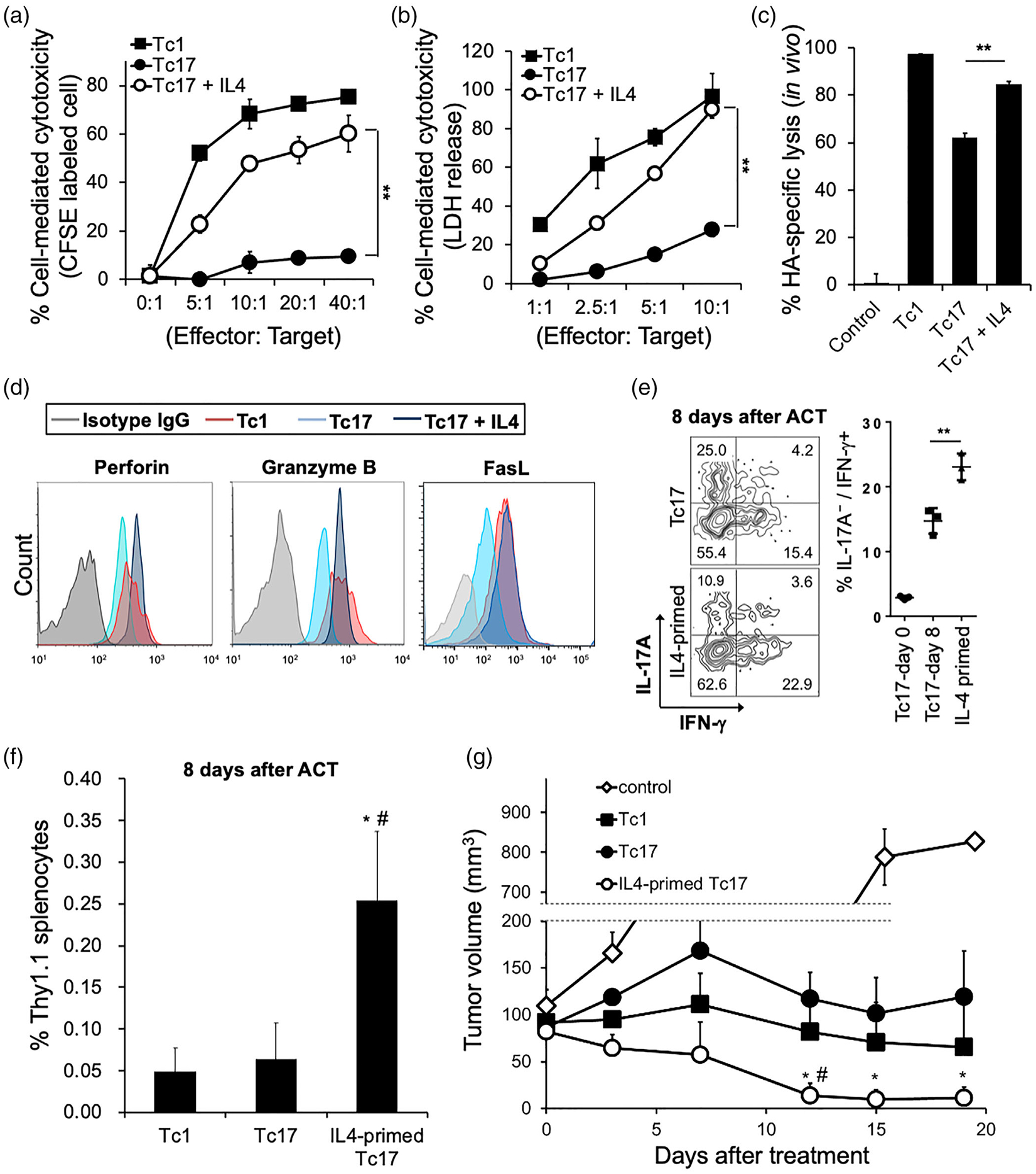
IL-4 enhances the anti-tumour cytotoxicity of Tc17 cells. (a and b) In vitro cytotoxicity of IL-4 programmed Tc17 cells. The HA-specific effectors were polarized under Tc1 and Tc17 skewing conditions. Target cells (CT26-HA) and non-target cells (CT26) were labelled and mixed in different effector: Target ratio for 16 h. Specific lysis of target cells was calculated as (1-target/nontarget) × 100%. Mean ± SD was shown from three independent experiments. **P < 0.01. (b) Target cells (CT26-HA) and control cells were mixed under different effector: Target ratio for 16 h and measured as a 492 nm absorbance. Percentage of specific lysis are average from three independent experiments; mean ± SD was shown. **P < 0.01. (c) In vivo cytotoxicity of IL-4 programmed Tc17 cells. Polarized HA-specific Tc1, Tc17, and IL-4-stimulated Tc17 were injected intravenously into recipient mice. HA-primed target cells and non-target cells were labelled by CFSE, and injected to the recipient mice 6 h later. Specific lysis of target cells was measured after 16 h, and calculated as (1-target/nontarget) × 100% (n = 4). Mean ± SD was shown. **P < 0.01. (d) Expression of Perforin, Granzyme B, and Fas-ligand (FasL) in polarized HA-specific Tc1, Tc17, and IL-4-stimulated Tc17 cells. One representative experiment of three with similar results is shown. (e) In vivo conversion of Tc17 cells into IFN-γ-producing cells. HA-specific Tc17 cells (Thy1.1) were stimulated with or without IL-4 for 24 h, then injected into the CT26HA tumour-bearing mice (Thy1.2) for 8 days (n = 3). IL-17A/IFN-γ producing populations of Thy1.1 positive lymphocytes were measured by flow cytometry. Experiments were independently repeated three times with similar results. Mean ± SD is shown. **P < 0.01. (f) Tc cell expansion after adoptive cell therapy. CT26-HA tumour-bearing mice received HA-specific Tc1, Tc17, or IL-4-primed Tc17 cells (1 106), and cell expansion analysed 8 days after cell transfer mean ± SD was shown. *P < 0.05 to Tc17. # P < 0.05 to Tc1. (g) Comparison of the antitumour efficacy of Tc1, Tc17, and IL-4-primed Tc17 in a CT26-HA tumour-bearing model. HA-specific Tc1, Tc17, and IL-4-stimulated Tc17 cells (1× 106) were injected intravenously into tumour-bearing mice (n = 7 per group). Tumour size was monitored serially after ACT. Mean ± SEM was shown. *P < 0.05 to Tc17. #P < 0.05 to Tc1
As Tc17 cells have been reported to spontaneously convert into IFN-γ-producing cells upon encountering tumour-specific target antigens in vivo [24,27,28], we next assessed whether IL-4 enhanced this phenotype in vivo. HA-specific Tc17 cells (Thy1.1+) were primed with or without IL-4 for 24 h and adoptively transferred to CT26-HA-tumour bearing mice (Thy1.2+). We found that IL-4 treatment augmented Tc17 cell conversion and enhanced the frequency of transferred cells in vivo (Thy1.1+ cells in spleen; Figure 2e,f). Notably, IL-4-treated Tc17 cells demonstrated superior antitumour activity when compared to that seen with adoptive transfer of untreated Tc17 and Tc1 cells (Figure 2g). Collectively, these findings suggest that IL-4 effectively induces Tc17 expansion and cytotoxicity, and promotes antitumour immune responses in vitro and in vivo.
Sustained Akt activation is essential for Tc17 reprogramming
We focused our studies on understanding of signalling changes induced by IL-4 during the process of Tc17 polarization into cytotoxic effectors. Stat, Akt and ERK are possible pathways triggered by IL-4 [33]. A comparison of IL-2-, IL-4-, or IL-7-triggered cell signalling in Tc17 cells revealed that IL-4 activated multiple Stats (Figure 3a and Figure S3a), with Stat2 and Stat6 exclusively activated by IL-4. In addition, IL-4 also induced Akt activation, but did not significantly trigger MAP kinases (ERK, p38 and JNK; Figure 3b and Figure S3a).
FIGURE 3.
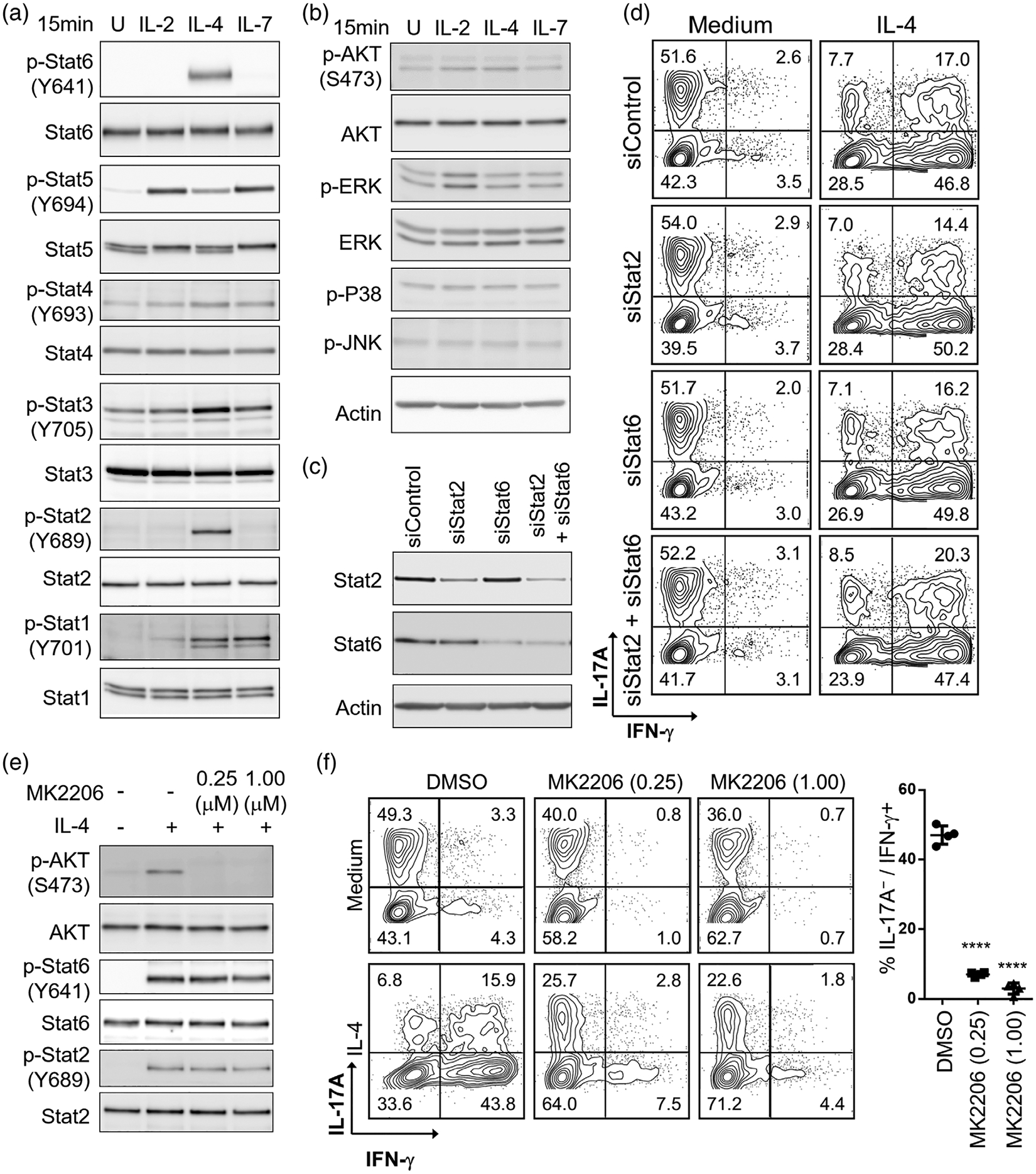
Analysis of signalling pathways induced by IL-4 during the Tc17 reprogramming. (a) Activation of Stat signalling. (b) Activation of Akt, ERK, P38, and JNK. IL-2-, IL-4-, or IL-7-triggered Stat, Akt, and MAPK signalling in polarized Tc17 cells. Tc17 cells were treated with designated cytokines for 15 min, then analysed by Western blotting. Actin was used as loading control. (c) Western blot analysis of Tc17 cells following siRNA knockdown of Stat2 and Stat6. (d) Representative contour plots show IL-4-induced Tc17 conversion to IFN-γ-producing cells in Stat2- and Stat6-cells following siRNA knockdown. (e) Specific inhibition of IL-4-induced AKT signalling in Tc17 cells using MK2206. (f) MK2206 suppresses IL-4-induced Tc17 reprogramming. DMSO was used as solvent control. Percentage of IL-17A−/IFN-γ+ cells after IL-4 treatment was shown at right. ****P < 0.0001. All experiments were independently repeated three times with similar results
To evaluate the impact of Stat2 and Stat6 in mediating IL-4 effects on Tc17 reprogramming, we pursued their silencing using siRNA approach (Figure 3c). Neither single nor double Stat2 and Stat6 knockdowns suppressed IL-4-induced Tc17 conversion (Figure 3d). We next focused our studies on the Akt pathway and using MK2206, we documented a selective Akt blockade without an impact on Stat2 or Stat6 phosphorylation (Figure 3e). Notably, Akt pathway blockade significantly suppressed IL-4-induced Tc17 reprogramming (Figure 3f) and cell expansion (data not shown). Moreover, treatment with PI3K or mTOR inhibitors also suppressed IL-4-induced Tc17 reprogramming, whereas no significant effects were observed upon treatment with the TCF/β-catenin inhibitor ICG-001 (Figure S3b). Additionally, we observed that IL-4 treatment sustained long-term Akt activation during Tc17 reprogramming (Figure S3c). Akt pathway blockade at earlier time points suppressed Tc17 reprogramming more significantly (Figure S3d). Collectively, these data indicate that the Tc17 reprogramming effects of IL-4 are critically regulated by active Akt signalling.
Eomes is an important regulator for IL-4-induced Tc17 reprogramming
We next studied the impact of IL-4 on the regulation of key transcriptional factors during the Tc17 cell differentiation. Following IL-4 stimulation, we observed downregulation of the key Tc17 transcription factor RORγt, with parallel upregulation of Eomes. Tc1 transcription factor T-bet and the Tc2 transcription factor GATA3 were not affected significantly (Figure 4a). On a protein level, flow cytometric analyses showed that IL-4 treatment also increased the frequency of Eomes-positive cells (Figure 4b). Concordant with our observation of IL-4 mediating its Tc17 conversion through Akt signalling, we found that PI3K-Akt–mTOR pathway blockade suppressed Eomes expression (Figure 4b). We saw no impact of inhibition of the β-catenin/TCF-mediated signal with ICG-001 on Eomes expression. To confirm the functions of Eomes in IL-4-induced Tc17 conversion, we relied on the CD8+ T cells where conditional deletion of Eomes in mature T cells is driven by CD4-Cre transgene. Despite CD8+ T cell stimulated with anti-CD3/anti-CD28 monoclonal antibodies yielded lower Tc17 polarizing efficiency than TCR transgene mice, we saw that Eomes ablation did not significantly affect Tc17 skewing; but it significantly suppressed the generation of IFN-γ-producing cells (Figure 4c). Thus, IL-4-Akt axis effects on Tc17 polarization to cytotoxic effectors are dependent upon regulation of transcriptional factor Eomes.
FIGURE 4.
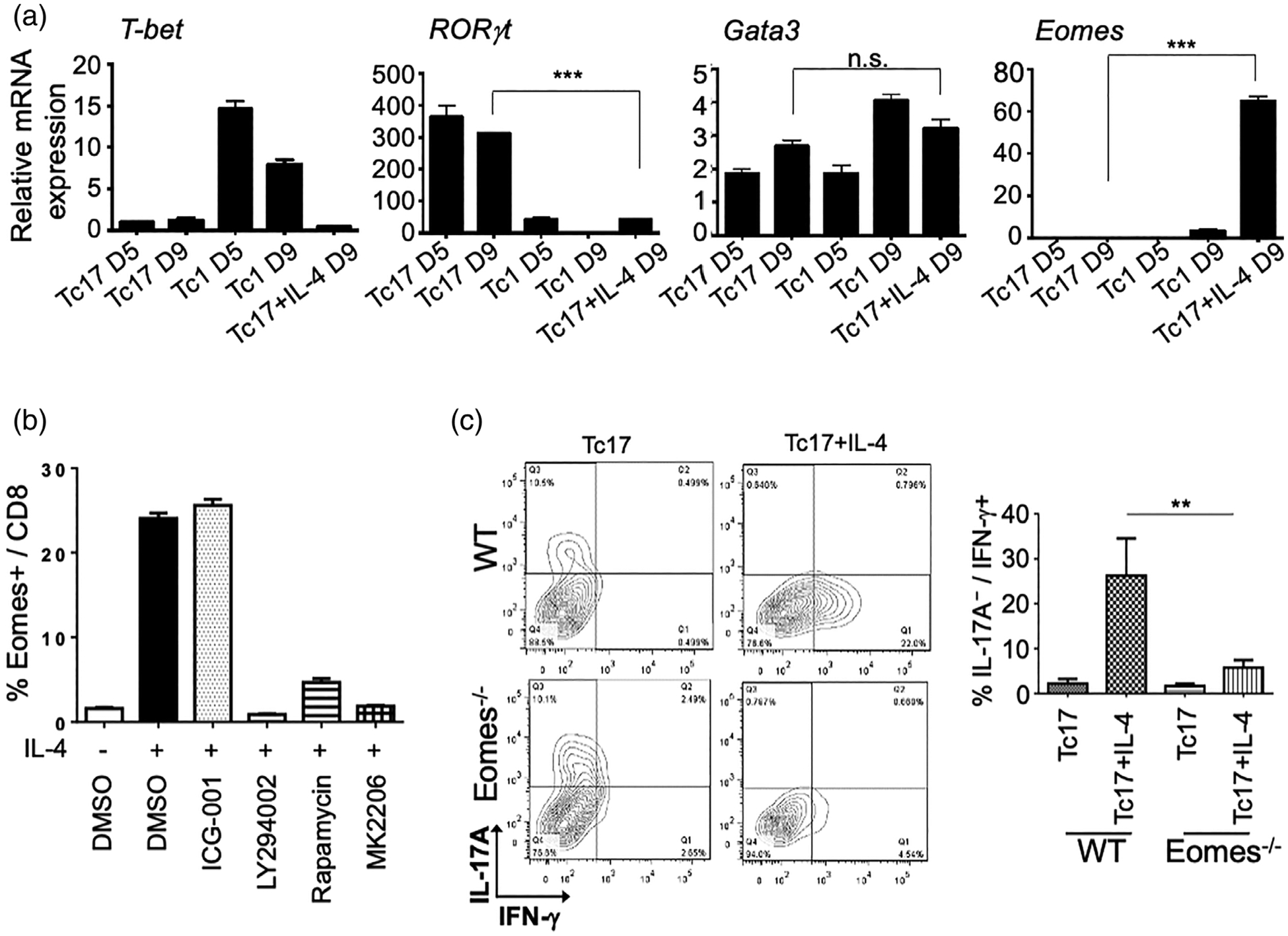
Eomes regulates IL-4-induced Tc17 reprogramming. (a) qPCR of mouse Tbx21 (T-bet), Rorc (RORγt), Gata3, and Eomes in naïve CD8 cells and polarized Tc1, Tc17, and IL-4-treated Tc17 cells. Relative fold changes to naïve CD8 cells are shown. ***P < 0.001; n.s.: not significance. (b) Inhibition of PI3K/AKT/mTOR pathway suppressed IL-4-induced Eomes expression in Tc17 cells. Polarized Tc17 were treated with or without IL-4 and with indicated compounds (1.0 μM each). Cumulative flow cytometry data showing percentage of Eomes positive cells in CD8+ T cells is shown. (c) Conditional knockout of Eomes in Tc cells hampers IL-4 triggered Tc17 reprogramming. Contour plots and cumulative flow cytometry data show loss of Tc17 reprogramming upon Eomes ablation. **P < 0.01. These experiments were independently repeated three times with similar results
Trat1 contributes to IL-4-induced Tc17 cytotoxicity and associates with high survival rates of cancer patients
In the above analysis, we noticed that IL-4 treatment of Tc17 cells led to a significant increase in surface expression of CD3 (1.7-fold; Figure 5a). TCR signalling in mature T cells promotes multiple cellular events, including cytokine production, proliferation, and trafficking [34]. We next focused on impacts of Tc17 reprogramming on TCR events. We found that the CD3e mRNA levels were reduced marginally (Figure 5b), suggesting that regulation is likely post-transcriptional in origin. c-Cbl and Cbl-b ubiquitin ligases have been proposed to modulate TCR degradation [35,36]. However, their expression were only slightly altered after IL-4 treatment (Figure S4). We next measured the gene expression levels of the transmembrane TCR adaptor proteins, Trat1 and Lat, both of which exert TCR-stabilizing functions [37–39]. We found that the expression of Trat1 significantly upregulated in Tc17 cells under IL-4 stimulation, while Lat, was downregulated (Figure 5c,d and Figure S4). Trat1 is also a chaperone of the costimulatory molecule CTLA-4, which can participate in Tc17 development [40,41], however, our studies showed that IL-4-induced Trat1 expression in Tc17 cells did not significantly influence CTLA-4 expression (Figure S5a). Trat1 upregulation by IL-4-was dependent upon the intact functioning of IL-4-Akt axis crucial for Tc17 reprogramming, as Akt pathway blockade induced within 24 h of IL-4 stimulation suppressed Trat1 upregulation (Figure S5b,c). To evaluate the functions of Trat1, we silenced its expression in Tc17 cells using the corresponding siRNA (Figure 5e). Trat1 silencing reversed the IL-4-induced expression of CD3 on the cell surface (Figure 5f), supporting its role in TCR stabilization during Tc17 reprogramming. Interestingly, Trat1 silencing attenuated Akt activation at later time points after IL-4 stimulation (Figure S5d), whereas it only marginally suppressed IL-4-induced Tc17 reprogramming (Figure S5e), suggesting existence of temporally restricted feed-forward loop between Akt and Trat1 that is dispensable for Tc17 polarization. We next evaluated whether regulation of Trat1 influenced the antitumour properties of the IL-4 polarized Tc17 cells. CTL assay revealed that Trat1 silencing significantly suppressed IL-4-induced Tc17 cytotoxicity (Figure 5g). This effect was associated with suppression of the signalling molecules downstream of TCR, including ERK, p38 and JNK activation, but not Zap70/Syk (Figure 5h), suggesting Trat1 modulates TCR signalling and results in promoting antitumour activity.
FIGURE 5.
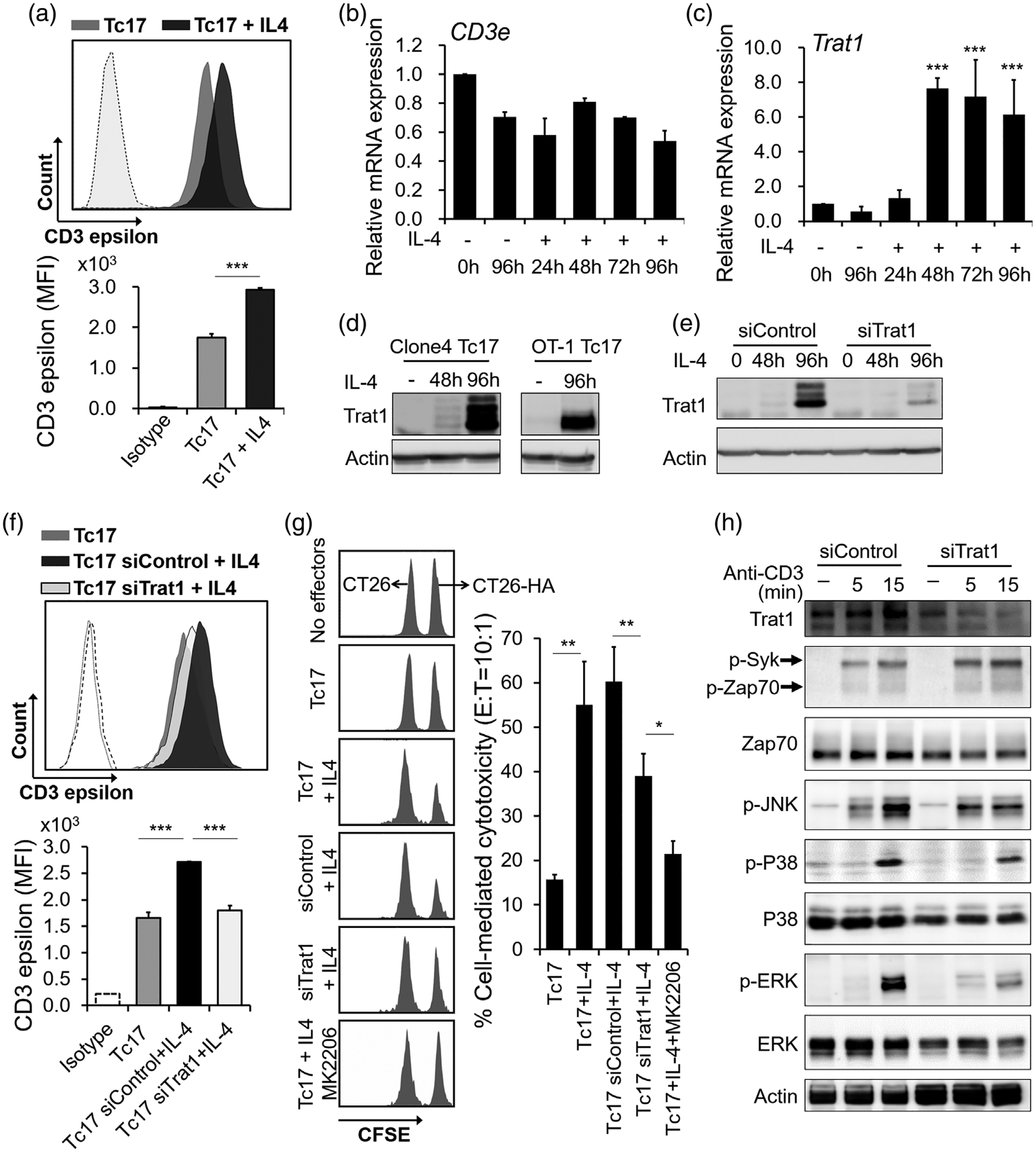
Upregulation of Trat1 during IL-4-induced Tc17 polarization in cytotoxic anti-tumour effectors. (a) Histogram flow cytometry plot and cumulative data show expression of CD3ε in Tc17 cells and IL-4-treated Tc17 cells. Mean fluorescence intensities were shown at the lower panel. Isotype IgG was used as staining control (dash line). ***P < 0.001. (b and c) relative mRNA expression of Cd3ε and Trat1 in Tc17 cells after IL-4 stimulation. ***P < 0.001, comparing to un-treated control cells. (d) Protein levels of Trat1 in polarized Tc17 cells from Clone 4 mice (left) and OT-1 mice (right). Cells were treated with or without IL-4 for indicated period. (e) Knockdown of Trat1 by siRNA (siTrat1). Non-specific siRNA was used as transfection control (siControl). (f) Knockdown of Trat1 reversed IL-4-induced surface expression of CD3ε on Tc17 cells. ***P < 0.001. (g) Knockdown of Trat1 and AKT pathway blockage suppress IL-4-induced cytotoxicity in Tc17. HA-specific effectors were mixed with target cells (CT26-HA) or nontarget cells (CT26) which were labelled by CFSE in 10:1 ratio. Cells were analysed by FACS after 16 h. Percent specific lysis was calculated as (1-target/nontarget) × 100%. A representative data from one out of three experiments is shown. Mean ± SD was shown. *P < 0.05, **P < 0.01. (h) Knockdown of Trat1 suppresses TCR signalling. Trat1 was knocked down by siRNA in IL-4-reprogrammed Tc17 cells which were then stimulated with anti-CD3 antibody for indicated time points. The activation of JNK, P38 and ERK were suppressed, comparing to the control cells (siControl). These experiments were independently repeated three times with similar results, and representative results are shown
Establish human Tc17 expansion and reprogramming protocol from peripheral blood
In human peripheral blood mononuclear cells (PBMC), Tc17 cell is a very rare population (<0.2% of CD8+ T cells) [14]. To increase number of Tc17 and examine the effects of IL-4 in human Tc17 reprogramming, we established an ex vivo culture protocol to expand and transform Tc17 from PBMC into IFN-γ-producing CD8+ T cells (Figure 6a). In this condition, the frequency of Tc17 cells from healthy donors was increased over 20 times (0.3% to 6%, n = 5, Figure 6b). In addition, after 7 days IL-4 stimulation, the IFN-γ-producing CD8+ T cells increased along with decrease of IL-17A-producing cells. To further evaluate this protocol for human IEC therapy, we applied this protocol on PBMC from colon cancer patients (n = 10). The frequencies of patients’ Tc17 cells were increased from 0.1% to 2.2%. Importantly, IL-4 treatment for 7 days increased population of IFN-γ-producing cells and decrease frequencies of IL-17A cells (Figure 6c). These results suggest IL-4 may have similar function to reprogram Tc17 cells in human and murine.
FIGURE 6.
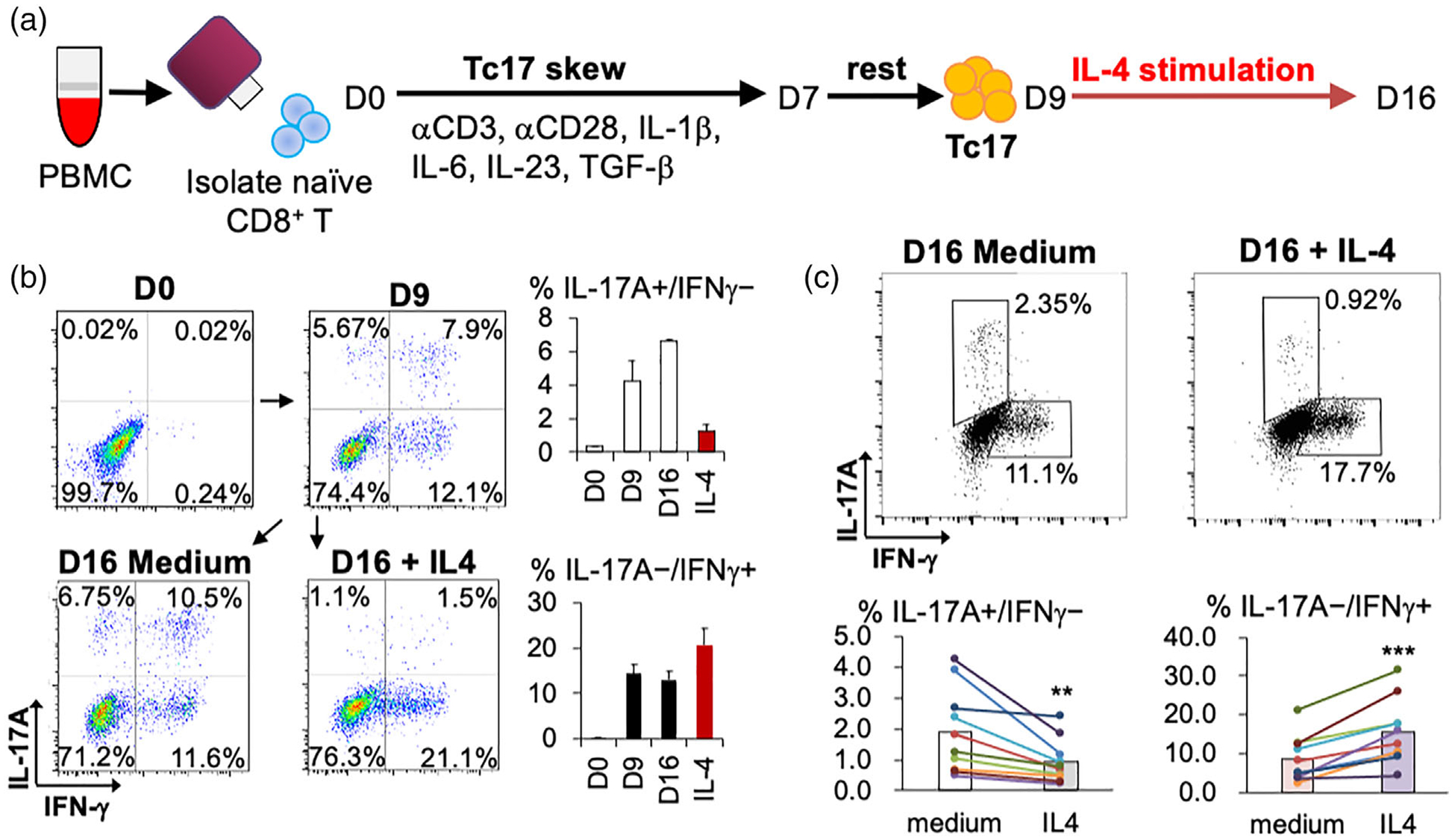
Ex vivo culture protocol for transforming Tc17 from human PBMC. (a) Procedure of polarization human PBMC into Tc17 and secondary IL-4 stimulation. (b) Frequency of human IL-17A and IFN-γ producing Tc cells with or without IL-4 stimulation from healthy donors. Representative graphs are shown at left. Mean ± SD was shown at right (n = 5). (c) Frequency of human IL-17A and IFN-γ producing Tc cells with or without IL-4 stimulation from PBMC of colorectal cancer patients. Representative graphs were shown at upper panel. Cell population changes of each patient are shown at lower panel (n = 10). **P < 0.01, ***P < 0.001
TRAT1 associates with high survival rates of cancer patients
Referring to the Human Protein Atlas database (https://www.proteinatlas.org) and Monaco scaled dataset [42], TRAT1 is exclusively expressed in human T cells, especially in memory CD8+ and memory CD4+ T cells, whereas it is downregulated in terminal effector CD8+ T cells. To further elucidate the clinical significance of TRAT1, we analysed the ONCOMINE database and the TCGA RNA samples. Data indicated that TRAT1 expression was downregulated in colorectal cancer (CRC) tissues compared to that in normal tissues in several independent datasets (Figure S6a). Notably, lower TRAT1 expression in patients with rectal cancer showed a significantly low survival rate (Figure S6b), while not in colon cancer patients. Whether if this is due to the poor prognosis of rectal cancer than colon cancer [43] deserves future investigations. Analysis of the other cancer types using the TCGA RNA samples also revealed that TRAT1 downregulation was associated with poor survival in patients with breast cancer (p = 0.00015), head and neck cancer (p = 0.0022), and liver cancer (p = 0.0019, Figure S7). Immunohistochemistry (IHC) was used to evaluate the TRAT1 protein expression in a CRC tissue array containing 69 primary tumour tissues and eight non-tumour colon tissues. The staining patterns revealed that TRAT1 was expressed exclusively in diffused stromal cells, but not in parenchymal cells and cancer cells (Figure S6c). Results indicated that 86% (7/8) of the non-tumour colon tissue samples contained TRAT1-expressing stromal cells, whereas only 20% (14/69) of the CRC tissues contained TRAT1-positive cells (Table S1). Collectively, these results suggest that TRAT1 is frequently downregulated in cancer tissue, which may associate with immunosuppressive microenvironment.
DISCUSSION
While the anti-tumour effects of Tc17 cells have been well documented, mechanisms that can enhance anti-tumour benefits of Tc17 cells and strategies to facilitate their use as IECs in ACT remain poorly defined [22–24,44]. Here, we document the benefits of IL-4 on promoting the differentiation and expansion of Tc17 cells into IFN-γ-producing CTL effectors with augmented cytotoxicity in vitro and in vivo, thus highlighting its role as a vehicle for improved Tc17 engineering for use in cellular immunotherapy of cancer. In our work, we demonstrate that PI3K/Akt pathway plays a central role in IL-4-induced Tc17 plasticity, inducing Eomes expression essential to direct Tc17 reprogramming and upregulating Trat1 to stabilize TCR and enhance its signalling, critical for improvement in antitumour immune responses. We further revealed that TRAT1 is frequently downregulated in cancerous tissues, which implied an immunosuppressive microenvironment. Notably, we proposed a novel ex vivo culture protocol to transform Tc17 from human PBMC, indicating the possible applicability of Tc17 in ACT for cancer patients.
IL-4 is a well-defined type-II T cell cytokine that inhibits IFN-γ production and Th1 responses [45,46]. Recent studies have proposed a requirement for IL-4 in the development of innate memory CD8+ cells in the thymus [47,48]. IL-4 also regulates the development of the memory phenotypes of peripheral CD8+ T cells during infection [49], a process accompanied striking upregulation of the transcription factor Eomes, but not T-bet, whereas Tc17 development relies on the induction of RORγt while the T-bet and Eomes silencing [50,51]. Concordant with these findings, we documented that Eomes expression is dispensable for Tc17 polarization, but surprisingly necessary to mediate IL-4 driven Tc17 plasticity and reprogramming into cytotoxic effectors. Based on the prior reports [52,53] and the results of our study, we hypothesize Eomes upregulation may facilitate development of the memory phenotypes of Tc17 cells. Indeed, we observed that the Tc17 cells converted into IFN-γ producing effectors exhibited reduced pro-apoptotic hallmarks and greater persistence in vivo, a desired trait in IEC therapy. Furthermore, these features provide a clear distinction from the low Eomes- and high T-bet-expressing Tc1 cell populations often exploited in adoptive immunotherapy.
In this study, we compared the cellular signals induced by common gamma chain cytokines IL-2, IL-4, and IL-7 and observed that IL-4 activated multiple Stats, consistent with the findings of a previous study [54]. Although Stat2 and Stat6 were exclusively activated by IL-4, their transient siRNA-mediated silencing had no impact on Tc17 reprogramming, and we revealed Akt pathway is essential for IL-4-induced Tc17 reprogramming (Figure 3e,f). Notably, in a conventional T cell ACT mouse model, Akt inhibition in IL-2-expanded Tc cells promoted the development of the gene expression signature and metabolic profile characteristics of long-lived memory T cells, which are associated with enhanced persistence and antitumour immunity [55]. In contrast to the development of ex vivo transformed Tc17 cells, IL-4-induced Akt pathway activation promoted the antitumour immunity of Tc17 cells. Furthermore, we postulate that prolonged Akt activation, sustained for several days after the IL-4 treatment of Tc17 cells (Figure S3c), is essential for repression of the transcription factor RORγt and induction of Eomes. Accordingly, this mechanism could be unique to Tc17 cells, but not to conventional CTLs. In addition, different intrinsic resident Tc17 cell population may exhibit various plasticity. For example, antifungal Tc17 cells seems more durable and stable with the assistance of Toll-like receptor signalling [16,56]. Commensal-specific Tc17 cells displayed type-II T cell gene signature [57]. While in our IL-4 transformed Tc17 cells, these type-II T cell genes, such as Gata3, IL-5, and IL-13, have not been induced (Figure 4a and data not shown). The exquisite variety of Tc17 cells and underlying mechanisms are worth to further exploration.
We propose that temporal effects of IL-4 early during the Tc17 reprogramming sustain the Akt activation, where the initial 24 h are crucial for upregulation of Trat1 (Figure S5c). This leads to a strengthened TCR signalling and may contribute to resulting Tc17 functionality, including enhanced antigenic response and anti-tumour efficacy. Interestingly, transient siRNA-mediated silencing of Trat1 attenuated Akt activity at later time points and partially suppressed IL-4-induced Tc17 reprogramming, suggesting Trat1 also contributes to the maintenance of Akt activation during reprogramming. The observation of Trat1 upregulation could, in addition to an enhanced anti-apoptotic signature, also help explain greater persistence of reprogrammed Tc17 cells. This phenotype may carry a greater potential for sustenance of long-term anti-tumour efficacy, owing to an enhanced immune surveillance. Whether similar phenotypic implications of Trat1 could be applied to other IECs, remains to be seen.
TRAT1 is a TCR-associated protein that stabilizes TCR expression; however, its pathological function has never been explored [37–39]. Data from the microarray and RNAseq databases indicated that TRAT1 is frequently downregulated in cancer tissues, and its downregulation tends to correlate with poor survival among patients in several types of cancer. As TRAT1 was only expressed on CD4+ and CD8+ T cells, the findings from the IHC experiments indicated that TRAT1-positive cells are observed in only 20% of CRC tissues, which implies that the number of tumour infiltrated TRAT1-positive T cells are frequently decreased or the TRAT1 is downregulated in the infiltrated T cells. Although further examination is needed, we reasonably suggest that TRAT1 expression in cancer tissues is associated with antitumour immunity in solid tumours. Interestingly, using cBioPortal (https://www.cbioportal.org/) to analyse co-expressed genes of TRAT1 in TCGA CRC samples [58], we found that TRAT1 expression was positively correlated with EOMES expression but not RORC expression (data not shown), which is in agreement with our finding that EOMES and TRAT1 are possibly regulated by the same signal axis.
Our study describes a novel IL-4/Akt/Eomes/Trat1 signalling loop that is critical for reprogramming of Tc17 cells to augment antitumour immunity (Figure 7). While Tc17 cells are rarely detected (<0.2% of CD8+ T cells) in human peripheral blood [14], our discovery highlights new approaches to enhance the Tc17 reprogramming efficacy, maximize their expansion and persistence, and allow for novel ACT strategies to enhance cellular immunotherapy benefits.
FIGURE 7.
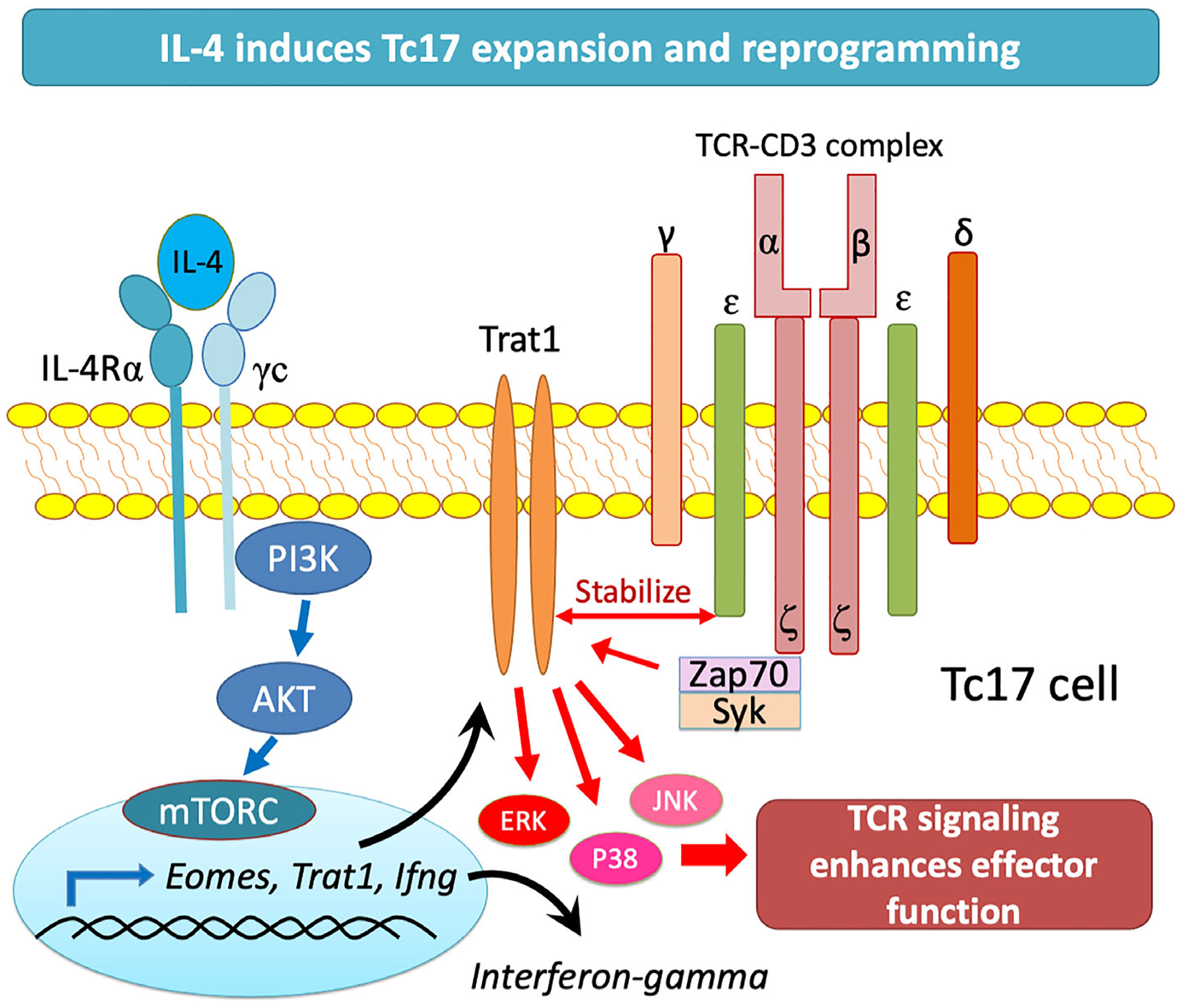
IL4 mediated Trat1 expression enhances Tc17 anti-tumour phenotype. The IL-4/AKT/Eomes/Trat1 axis is a novel signalling loop that promotes expansion and reprogramming of Tc17 cells into cytotoxic effectors with a therapeutic potential for adoptive cell transfer immunotherapy
MATERIALS AND METHODS
Animals and cell lines
BALB/c and C57BL/6 mice were purchased from National Laboratory Animal Centre, Taiwan. CD4-Cre C57BL/6 mice were kindly provided from Dr. Charles Drake (Columbia University Irving Medical Centre). CD8+ TCR transgenic mice (Clone 4) with Thy1.1 as a congenic marker which express a TCR recognizing Kd-restricted HA epitope (518IYSTVASSL526) [59], C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I) mice, and Eomesfl/fl mice were obtained from the Jackson Laboratory (Bar Harbor, ME). CD4-Cre mice were bred to Eomesfl/fl mice to generate CD4-Cre x Eomesfl/fl mice. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of China Medical University, Taiwan. Carcinoma cell lines CT26HA and its paternal CT26 were gifts form Dr. Mempel Thorsten (Harvard Medical School) [60]; B16-OVA and its paternal B16 cell lines were gifts from Dr. Jonathan Powell (The Johns Hopkins University School of Medicine).
Reagents and antibodies
All cytokines were purchased from Peprotech except for IL-23 (R&D Systems). Small molecule inhibitors MK2206, LY294002, ICG-001 and rapamycin were purchased from Selleck Chemicals. Anti-IFN-γ and anti-IL-4 antibodies were purchased from eBioscience. The following fluorescently labelled antibodies against murine antigens were purchased from Biolegend: anti-CD3ϵ (145-2C11), anti-CD8α (53–6.7) (BD Biosciences, USA). Anti-IL-17 (Tc11–18H10), anti-IFN-γ (XMG1.2), anti-Fas-L (MFL3), and anti-CD90.1 (Thy1.1) (OX-7). Anti-granzyme B (NG2B) and anti-perforin (eBio0MAK-D) were from eBioscience. All antibodies used in western blots were purchased form Cell Signalling Technology except for anti-Trat1 (Santa Cruz Biotechnology) and anti-STAT2 pY689 (Millipore). Western blots were repeated at least three times, and representative figures were shown.
Mouse Tc17 cell polarization
Leukocytes from spleens and peripheral lymph nodes were harvested from 6 to 9-week old mice. For Ag-specific activation, Clone-4 splenocytes were pulsed with 2 μg/ml HA class I Kd peptide 518–526 (HA-1) and OT-1 splenocytes were pulsed with OVA peptide 323–339. For wild-type and Eomes-deficient mice, CD8+ lymphocytes were isolated by negative selection kit (STEMCELL Technologies), and activated on an anti-CD3/anti-CD28 monoclonal antibodies-coated plate. Skewing conditions for Tc1 and Tc17 were as previously described [13]. Cells were cultured for 3 days in skewing medium and then rested for 2 days. The efficiency of Tc1 and Tc17 polarization were measured by flow cytometry. Tc17 cells were adjusted to 1 × 106/ml and stimulated with different cytokines, including IL-2, IL-4, IL-7, IL-12, IL-13, IL-15, IL-23 and IL-1 plus IL23 (20 ng/ml of each) for another 4 days.
Human Tc17 cell polarization
Human CD8 T cells were isolated from peripheral blood of healthy donors and newly diagnosed CRC patients before standard treatment by EasySepTM Human Naïve CD8 T Cell Isolation Kit (STEMCELL Technologies Inc). Cells were cultured in IMDM medium (Invitrogen) with supplementation of sodium pyruvate (1.0 mM), nonessential amino acid (0.1 mM), penicillin and streptomycin (100 IU/ml) (Gibco), 2-ME (50 μM; Sigma-Aldrich), heat-inactivated FBS (5%; HyClone), and recombinant human IL-2 (5 ng/ml). The polarization condition for human Tc17 cells were as follows: plate-coated anti-CD3 (OKT3, 1 μg/ml), soluble anti-CD28 antibody (2 μg/ml), anti-human IFNγ antibody (20 μg/ml), anti-human IL-4 antibody (20 μg/ml) (BioLegend Inc), and recombinant human cytokines including TGF-β1 (1.25 ng/ml), IL-1β (20 ng/ml), IL-6 (20 ng/ml) and IL-23 (20 ng/ml) (PeproTech Inc) for 5 days. Then cells were moved to new plates in medium with above cytokines, anti-human IFNγ, and anti-human IL-4 antibodies for further 2 days. The cells were resting in culture medium for 2 days, and cultured for a further 7 days in medium or medium with human IL-4 (20 ng/ml, PeproTech).
The study protocol was reviewed and approved by the Research Ethics Committee of China Medical University and Hospital (CMUH106-REC3-107) with ClinicalTrials.gov Identifier (NCT03291639), and the informed consent of all participating patients was obtained.
Flow cytometry and intracellular staining
Before ICS, cells were restimulated for 5 h in the presence of PMA (50 ng/ml), ionomycin (500 ng/ml), and GolgiStop (BD Biosciences). The cell surface markers were labelled first, intracellular proteins were stained by fluorochrome-conjugated antibodies after fixation and permeabilization. Flow cytometry was performed using a FACSVerse (BD Biosciences) and analysed using FlowJo software (Tree Star). Statistical analyses were performed using Prism 5.0 (GraphPad Software).
Cell sorting of viable IL-17 producing cells
Mouse IL-17 and IFN-γ secretion assay was carried out according to the manufacturer’s protocol (Miltenyi Biotec). Cells were sorted by using BD FACSAria instrument. Propidium iodide staining was used to exclude dead/dying cells, and IL-17+IFN-γ− CD8 T cells were sorted and incubated overnight for further experiments.
In vitro CTL assay
The HA-specific and OVA-specific effector cells were prepared from Clone 4 and OT-I mice as above description. CFSE (Invitrogen, Life Technologies) labelled target cells (CT26HA or B16-OVA, labelled by 5 μM CFSE) and control cells (CT26 and B16-F10 cells, labelled by 0.5 μM CFSE) were mixed under different effector: target (ET) ratio for 16 h and analysed by FACS. Percent specific lysis was calculated as (1-targets/control) × 100%. LDH Cytotoxicity Detection Kit (Takara) was used for LDH release CTL assay according to manufacturer’s protocol. Cells were mixed under different effector: target (ET) ratio for 16 h and measured as a 492 nm absorbance reading on a microplate reader.
In vivo CTL assay
Target cells were prepared from wild type mice splenocytes pulsed with HA-1 peptide or OVA peptide for 2 h, and labelled with 5 μM or 0.5 μM CFSE. 1 × 107 Effector cells were i.v. injected into wild type mice. Six hours later, 1 × 107 CFSE labelled target cells and 1 107 non-target cells were i.v. into recipients. Mice were sacrificed and analysed by FACS after 16 h. Percent specific lysis was calculated as (1-target/nontarget) × 100%.
Tumour growth and adoptive transfer treatment
Eight-week-old BALB/c mice were injected subcutaneously with 1 × 106 CT26HA tumour cells, and allowed tumour size reach around 100 mm3 (7–10 days), at which point 106 in vitro activated Clone 4 T cells programmed as Tc1, Tc17, or IL-4-stimulated Tc17 cells (7 mice per group) were administered intravenously. Serial tumour measurements were obtained, and tumour volume was calculated by (L × W2) × 1/2.
Small interfering RNA
Accell mouse siRNA against Stat2, Stat6, Trat1, and control siRNA were purchased from Thermo Scientific. According to the manufacturer’s manual, 1 × 106/ml skewed mouse Tc17 cells were mixed with Accell siRNA (1 μM) for 3 to 4 days without transfection reagent. The knockdown efficiency was examined by Western blotting.
Quantitative real-time PCR
Total RNA was extracted using the RNeasy Micro Kit (Qiagen), and cDNA was synthesized with the Super-Script III enzyme (Invitrogen). All primers were purchased from Applied Biosystems; reactions were performed in triplicate using an Applied Biosystems 7900 instrument.
Immunohistochemistry
A paraffin-embedded colorectal cancer tissues array including clinicopathological information was purchased from Shanghai Outdo Biotech and Pantomics, Inc (HCol-Ade90Sur). Array was incubated with anti-TRAT1 antibody (Santa Cruz Biotechnology, sc-393 175, 1:100) in 5% bovine serum albumin/PBS and 0.1% Triton X-100 (Sigma) for 16 h at 4°C. UltraVision Quanto Detection System (Thermo Fisher Scientific Inc.) was used to amplify the signal. The specific immunostaining was visualized with 3,3-diaminobenzidine and counterstained with haematoxylin (Sigma). The distribution of TRAT1 positive cells were measured by two scorers blinded to the clinical parameters.
Statistical analysis
Data analysis was performed using GraphPad Prism 8 (GraphPad Software, Inc). One way ANOVA multiple comparisons were used for analysing multiple experimental groups. Student t test was used for statistical analyses. Two-sided Fisher exact test was used to analyse clinical features and TRAT1 expression in CRC tissue array. Kaplan–Meier analysis and the log-rank test were used to estimate survival rate. P < 0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Professor Mien-Chie Hung and Professor Lu-Hai Wang for giving excellent scientific advice. The corresponding author thank the tumor immunology group led by Professor Drew Pardoll for enlightening his research in tumor immunology. This work was financially supported by a Career Developing Grant (NHRI-EX101-10124SC, NHRI-EX102-10124SC, NHRI-EX103-10124SC, and NHRI-EX104-10124SC) from the National Health Research Institutes, Taiwan to Hung-Rong Yen. This study was also partially supported by China Medical University (CMU107-TU-04), China Medical University Hospital (DMR-108-011), ‘Chinese Medicine Research Center, China Medical University’ from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (CMRC-CHM-1), Ministry of Science and Technology (MOST 108-2320-B-039-024-MY3), and health and welfare surcharge of tobacco products, China Medical University Hospital Cancer Research Center of Excellence, Ministry of Health and Welfare (MOHW109-TDU-B-212-134024), Taiwan to Hung-Rong Yen; National Heart, Lung, and Blood Institute (NHLBI, 1K08HL145116) and Huntsman Cancer Foundation to Vedran Radojcic; Ministry of Science and Technology post-doctoral fellowship grants MOST 109-2811-B-039-500 and MOST 109-2811-B-039-509 to CHL, and MOST 106-2811-B-039-023 to CTL. Experiments and data analysis were performed in part through the use of the Medical Research Core Facilities, Office of Research & Development at China medical University, Taichung, Taiwan. None of the funders and institutions listed had a role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Funding information
China Medical University Hospital, Grant/Award Number: DMR-108-011; China Medical University, Taiwan, Grant/Award Number: CMU107-TU-04; Higher Education Sprout Project by the Ministry of Education, Grant/Award Number: CMRC-CHM-1; Huntsman Cancer Foundation; NHLBI, Grant/Award Number: 1K08HL145116; Ministry of Health and Welfare, Grant/Award Number: MOHW109-TDU-B-212-134024; Ministry of Science and Technology, Taiwan, Grant/Award Numbers: MOST 106-2811-B-039-023, MOST 108-2320-B-039-024-MY3, MOST 109-2811-B-039-500, MOST109-2811-B-039-509; National Health Research Institutes, Grant/Award Numbers: NHRI-EX101-10124SC, NHRI-EX102-10124SC, NHRI-EX103-10124SC, NHRI-EX104-10124SC
Footnotes
CONFLICT OF INTEREST
The authors have declared that no competing interests exist.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
REFERENCES
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castrationresistant prostate cancer. N Engl J Med. 2010;363(5):411–22. [DOI] [PubMed] [Google Scholar]
- 4.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandran SS, Paria BC, Srivastava AK, Rothermel LD, Stephens DJ, Dudley ME, et al. Persistence of CTL clones targeting melanocyte differentiation antigens was insufficient to mediate significant melanoma regression in humans. Clin Cancer Res. 2015;21(3):534–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer-what clinicians need to know. 2011;8(10):577–Nat Rev Clin Oncol, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, et al. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. 2010;16(24):6122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015;33:445–74. [DOI] [PubMed] [Google Scholar]
- 11.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115(6):1616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yen HR, Harris TJ, Wada S, Grosso JF, Getnet D, Goldberg MV, et al. Tc17 CD8 T cells: functional plasticity and subset diversity. J Immunol. 2009;183(11):7161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kondo T, Takata H, Matsuki F, Takiguchi M. Cutting edge: phenotypic characterization and differentiation of human CD8+ T cells producing IL-17. J Immunol. 2009;182(4):1794–8. [DOI] [PubMed] [Google Scholar]
- 15.Hamada H, Garcia-Hernandez Mde L, Reome JB, Misra SK, Strutt TM, McKinstry KK, et al. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol. 2009;182(6):3469–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nanjappa SG, Hernandez-Santos N, Galles K, Wuthrich M, Suresh M, Klein BS. Intrinsic MyD88-Akt1-mTOR signaling coordinates disparate Tc17 and Tc1 responses during vaccine immunity against fungal pneumonia. PLoS Pathog. 2015;11(9):e1005161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gartlan KH, Markey KA, Varelias A, Bunting MD, Koyama M, Kuns RD, et al. Tc17 cells are a proinflammatory, plastic lineage of pathogenic CD8+ T cells that induce GVHD without antileukemic effects. Blood. 2015;126(13):1609–20. [DOI] [PubMed] [Google Scholar]
- 18.Chasset F, Le Buanec H, Sicre de Fontbrune F, de Masson A, Rivet J, Bergeron A, et al. Evidence of Th1, Th17 and Tc17 cells in psoriasiform chronic graft-versus-host disease. Exp Dermatol. 2016;25(1):64–5. [DOI] [PubMed] [Google Scholar]
- 19.Yaochite JN, Caliari-Oliveira C, Davanso MR, Carlos D, Malmegrim KC, Cardoso CR, et al. Dynamic changes of the Th17/Tc17 and regulatory T cell populations interfere in the experimental autoimmune diabetes pathogenesis. Immunobiology. 2013;218(3):338–52. [DOI] [PubMed] [Google Scholar]
- 20.Cheuk S, Wiken M, Blomqvist L, Nylen S, Talme T, Stahle M, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol. 2014;192(7):3111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang Y, Pan HF, Ye DQ. Tc17 cells in immunity and systemic autoimmunity. Int Rev Immunol. 2015;34(4):318–31. [DOI] [PubMed] [Google Scholar]
- 22.Yu Y, Cho HI, Wang D, Kaosaard K, Anasetti C, Celis E, et al. Adoptive transfer of Tc1 or Tc17 cells elicits antitumor immunity against established melanoma through distinct mechanisms. J Immunol. 2013;190(4):1873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Hernandez Mde L, Hamada H, Reome JB, Misra SK, Tighe MP, Dutton RW. Adoptive transfer of tumor-specific Tc17 effector T cells controls the growth of B16 melanoma in mice. J Immunol. 2010;184(8):4215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinrichs CS, Kaiser A, Paulos CM, Cassard L, Sanchez-Perez L, Heemskerk B, et al. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood. 2009;114(3):596–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flores-Santibanez F, Cuadra B, Fernandez D, Rosemblatt MV, Nunez S, Cruz P, et al. In vitro-generated Tc17 cells present a memory phenotype and serve as a reservoir of Tc1 cells in vivo. Front Immunol. 2018;9:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X, Zawidzka EM, Li H, Lesch CA, Dunbar J, Bousley D, et al. RORgamma agonists enhance the sustained antitumor activity through intrinsic Tc17 cytotoxicity and Tc1 recruitment. Cancer Immunol Res. 2019;7(7):1054–63. [DOI] [PubMed] [Google Scholar]
- 27.Nelson MH, Kundimi S, Bowers JS, Rogers CE, Huff LW, Schwartz KM, et al. The inducible costimulator augments Tc17 cell responses to self and tumor tissue. J Immunol. 2015;194(4):1737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowers JS, Nelson MH, Kundimi S, Bailey SR, Huff LW, Schwartz KM, et al. Dendritic cells in irradiated mice trigger the functional plasticity and antitumor activity of adoptively transferred Tc17 cells via IL-12 signaling. Clin Cancer Res. 2015;21(11):2546–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203(7):1685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31(2):331–41. [DOI] [PubMed] [Google Scholar]
- 31.Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immun. 2000;105(6):1063–70. [DOI] [PubMed] [Google Scholar]
- 32.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3(9):666–U2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCormick SM, Heller NM. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine. 2015;75(1):38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krummel MF, Bartumeus F, Gerard A. T cell migration, search strategies and mechanisms. Nat Rev Immunol. 2016;16(3):193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myers MD, Sosinowski T, Dragone LL, White C, Band H, Gu H, et al. Src-like adaptor protein regulates TCR expression on thymocytes by linking the ubiquitin ligase c-Cbl to the TCR complex. Nat Immunol. 2006;7(1):57–66. [DOI] [PubMed] [Google Scholar]
- 36.Naramura M, Jang IK, Kole H, Huang F, Haines D, Gu H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. 2002;3(12):1192–9. [DOI] [PubMed] [Google Scholar]
- 37.Bruyns E, Marie-Cardine A, Kirchgessner H, Sagolla K, Shevchenko A, Mann M, et al. T cell receptor (TCR) interacting molecule (TRIM), a novel disulfide-linked dimer associated with the TCR-CD3-zeta complex, recruits intracellular signaling proteins to the plasma membrane. J Exp Med. 1998;188(3):561–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirchgessner H, Dietrich J, Scherer J, Isomaki P, Korinek V, Hilgert I, et al. The transmembrane adaptor protein TRIM regulates T cell receptor (TCR) expression and TCR-mediated signaling via an association with the TCR zeta chain. J Exp Med. 2001;193(11):1269–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jahan AS, Lestra M, Swee LK, Fan Y, Lamers MM, Tafesse FG, et al. Usp12 stabilizes the T-cell receptor complex at the cell surface during signaling. Proc Natl Acad Sci USA. 2016;113(6):E705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valk E, Leung R, Kang H, Kaneko K, Rudd CE, Schneider H. T cell receptor-interacting molecule acts as a chaperone to modulate surface expression of the CTLA-4 coreceptor. Immunity. 2006;25(5):807–21. [DOI] [PubMed] [Google Scholar]
- 41.Pick J, Arra A, Lingel H, Hegel JK, Huber M, Nishanth G, et al. CTLA-4 (CD152) enhances the Tc17 differentiation program. Eur J Immunol. 2014;44(7):2139–52. [DOI] [PubMed] [Google Scholar]
- 42.Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carre C, et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019;26(6):1627–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paschke S, Jafarov S, Staib L, Kreuser ED, Maulbecker-Armstrong C, Roitman M, et al. Are colon and Rectal cancer two different tumor entities? A proposal to abandon the term colorectal cancer. Int J Mol Sci. 2018;19(9):2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu Y, Hong BX, Li HY, Zheng YH, Zhang MJ, Wang SQ, et al. Tumor-specific IL-9-producing CD8(+) Tc9 cells are superior effector than type-I cytotoxic Tc1 cells for adoptive immunotherapy of cancers. Proc Natl Acad Sci USA. 2014;111(6):2265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carvalho LH, Sano G, Hafalla JC, Morrot A, Curotto de Lafaille MA, Zavala F. IL-4-secreting CD4+ T cells are crucial to the development of CD8+ T-cell responses against malaria liver stages. Nat Med. 2002;8(2):166–70. [DOI] [PubMed] [Google Scholar]
- 46.Stager S, Alexander J, Carter KC, Brombacher F, Kaye PM. Both interleukin-4 (IL-4) and IL-4 receptor alpha signaling contribute to the development of hepatic granulomas with optimal antileishmanial activity. Infect Immun. 2003;71(8):4804–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lai D, Zhu J, Wang T, Hu-Li J, Terabe M, Berzofsky JA, et al. KLF13 sustains thymic memory-like CD8(+) T cells in BALB/c mice by regulating IL-4-generating invariant natural killer T cells. J Exp Med. 2011;208(5):1093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee YJ, Jameson SC, Hogquist KA. Alternative memory in the CD8 T cell lineage. Trends Immunol. 2011;32(2):50–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Renkema KR, Lee JY, Lee YJ, Hamilton SE, Hogquist KA, Jameson SC. IL-4 sensitivity shapes the peripheral CD8+ T cell pool and response to infection. J Exp Med. 2016;213(7):1319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Intlekofer AM, Banerjee A, Takemoto N, Gordon SM, Dejong CS, Shin H, et al. Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science. 2008;321(5887):408–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mielke LA, Liao Y, Clemens EB, Firth MA, Duckworth B, Huang Q, et al. TCF-1 limits the formation of Tc17 cells via repression of the MAF-RORgammat axis. J Exp Med. 2019;216(7):1682–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller CH, Klawon DEJ, Zeng S, Lee V, Socci ND, Savage PA. Eomes identifies thymic precursors of self-specific memory-phenotype CD8(+) T cells. Nat Immunol. 2020;21(5):567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kavazovic I, Han H, Balzaretti G, Slinger E, Lemmermann NAW, Ten Brinke A, et al. Eomes broadens the scope of CD8 T-cell memory by inhibiting apoptosis in cells of low affinity. PLoS Biol. 2020;18(3):e3000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Sa A, Pinheiro A, Morrot A, Chakravarty S, Overstreet M, Bream JH, et al. IL-4 induces a wide-spectrum intracellular signaling cascade in CD8+ T cells. J Leukoc Biol. 2007;81(4):1102–10. [DOI] [PubMed] [Google Scholar]
- 55.Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015;75(2):296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nanjappa SG, McDermott AJ, Fites JS, Galles K, Wuthrich M, Deepe GS Jr, et al. Antifungal Tc17 cells are durable and stable, persisting as long-lasting vaccine memory without plasticity towards IFNgamma cells. PLoS Pathog. 2017;13(5):e1006356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrison OJ, Linehan JL, Shih HY, Bouladoux N, Han SJ, Smelkinson M, et al. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science. 2019;363(6422):eaat6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morgan DJ, Liblau R, Scott B, Fleck S, McDevitt HO, Sarvetnick N, et al. CD8(+) T cell-mediated spontaneous diabetes in neonatal mice. J Immunol. 1996;157(3):978–83. [PubMed] [Google Scholar]
- 60.Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest. 2014;124(6):2425–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.