

Abstract
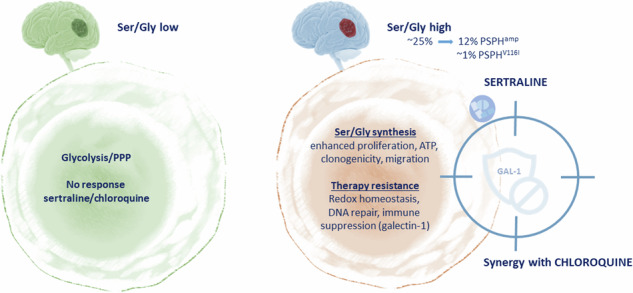
The serine/glycine (ser/gly) synthesis pathway branches from glycolysis and is hyperactivated in approximately 30% of cancers. In ~13% of glioblastoma cases, we observed frequent amplifications and rare mutations in the gene encoding the enzyme PSPH, which catalyzes the last step in the synthesis of serine. This urged us to unveil the relevance of PSPH genetic alterations and subsequent ser/gly metabolism deregulation in the pathogenesis of glioblastoma. Primary glioblastoma cells overexpressing PSPH and PSPHV116I showed an increased clonogenic capacity, cell proliferation, and migration, supported by elevated nucleotide synthesis and utilization of reductive NAD(P). We previously identified sertraline as an inhibitor of ser/gly synthesis and explored its efficacy at suboptimal dosages in combination with the clinically pretested chloroquine to target ser/glyhigh glioblastoma models. Interestingly, ser/glyhigh glioblastomas, including PSPHamp and PSPHV116I, displayed selective synergistic inhibition of proliferation in response to combination therapy. PSPH knockdown severely affected ser/glyhigh glioblastoma clonogenicity and proliferation, while simultaneously increasing its sensitivity to chloroquine treatment. Metabolite landscaping revealed that sertraline/chloroquine combination treatment blocks NADH and ATP generation and restricts nucleotide synthesis, thereby inhibiting glioblastoma proliferation. Our previous studies highlight ser/glyhigh cancer cell modulation of its microenvironment at the level of immune suppression. To this end, high PSPH expression predicts poor immune checkpoint therapy responses in glioblastoma patients. Interestingly, we show that PSPH amplifications in glioblastoma facilitate the expression of immune suppressor galectin-1, which can be inhibited by sertraline treatment. Collectively, we revealed that ser/glyhigh glioblastomas are characterized by enhanced clonogenicity, migration, and suppression of the immune system, which could be tackled using combined sertraline/chloroquine treatment, revealing novel therapeutic opportunities for this subgroup of GBM patients.
Subject terms: Cancer metabolism, Cell biology, Immunology
Introduction
Glioblastoma (GBM) is the most common and aggressive brain cancer in adults. The current standard of care for GBM includes tumor resection, followed by radiation and adjuvant temozolomide chemotherapy [1, 2]. Despite this multimodal treatment, the median overall survival of patients with GBM remains less than 15 months. As GBM has shown limited therapeutic improvement over the last decades, there is an urgent need to develop novel effective therapeutic strategies.
Alterations in tumor metabolism are an important feature of GBM, however, metabolism-targeting agents have not been included in clinical practice. GBM displays an enhanced glycolytic flux, with high glucose consumption and rapid lactic acid efflux to the tumor microenvironment [3]. GBM is characterized by a highly hypoxic tumor microenvironment, associated with a more aggressive tumor phenotype and therapy resistance [4]. Hypoxia-inducible transcription factors (HIFs) induce the expression of glycolytic enzymes, glucose transporters, lactate exporters, and pH regulators. This hypoxic and acidic microenvironment in metabolically altered GBMs affects radiotherapy efficacy and drug delivery, and plays a crucial role in biological mechanisms involved in cancer progression and treatment resistance, such as aberrant DNA repair, angiogenesis, and cancer stem cell maintenance [5–10]. Moreover, it negatively regulates the accessibility and infiltration of immune cells by affecting the metabolite availability for immune effector cells and by inducing an immunosuppressive phenotype via an upregulation of immune checkpoints on cancer cells, e.g., PD-L1 [11, 12]. Although efforts have been made to develop glycolysis-targeting agents, inhibitors of HIFs, and immune checkpoint inhibitors for treating GBM, all therapeutic approaches present elevated toxicity without improvement in the outcomes of patients with GBM [13–17]. Additionally, an important challenge in developing new therapeutic compounds is their poor blood-brain barrier (BBB) penetrance [18–20]. To solve this paradigm, using repurposed compounds with known BBB penetrance provides promising therapeutic opportunities for GBM, with a special focus on the design of combinational drugs that specifically target cancer cells while minimizing damage to healthy cells.
Currently, there is an increasing focus on therapeutic strategies targeting the side branches of glycolysis, especially the serine/glycine (ser/gly) synthesis pathway, since it is upregulated in approximately 30% of all cancers, including GBM [21]. This pathway is a direct regulator of glycolysis via pyruvate kinase M2 (PKM2), a key sensor controlling the glycolytic flux. With increasing serine levels, serine functions as an allosteric activator of PKM2, supporting aerobic glycolysis and regulating either pyruvate conversion to lactic acid or pyruvate catabolism into the tricarboxylic acid (TCA) cycle [22, 23]. The glycolytic intermediate, 3-phosphoglycerate (3PG) is the glycolytic intermediate fueling serine synthesis, in a series of three consecutive enzymatic steps catalyzed by phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), and phosphoserine phosphatase (PSPH). Further generation of glycine from serine is controlled by a bidirectional conversion catalyzed by serine hydroxymethyltransferases (SHMTs), SHMT1 in the cytosol and SHMT2 in mitochondria. Serine synthesis pathway hyperactivation is mainly restricted to cancer cells due to their high demands for serine and glycine and the downstream products, such as ATP and nucleotides, favoring their uncontrolled proliferation [24, 25]. Importantly, glycine is required for the synthesis of antioxidant glutathione (GSH), which enables cancer cells to cope with oxidative stress in a hypoxic microenvironment, and provides a defense mechanism against many therapeutic agents. Moreover, ser/gly synthesis and downstream one-carbon metabolism modulate the NAD + /NADH and NADp + /NADPH ratios, maintaining NADH and NADPH reductive equivalents in cancer cells [26, 27]. NADH is required for mitochondrial oxidative phosphorylation and ATP production, and NADPH provides reducing equivalents to generate reduced forms of antioxidant molecules, such as the reduction of glutathione disulfide (GSSC) to GSH [28].
In GBM, metabolic reprogramming via upregulation of PHGDH and SHMT2 has been found to provide a survival advantage under hypoxic and nutrient-deprived conditions. High levels of PHGDH protect GBM cells from hypoxia-induced cell death and starvation by regulating redox homeostasis and the NADPH/NADP+ ratio [29]. SHMT2 has been reported to be highly expressed in pseudopalisading cells surrounding ischemic necrotic foci in GBM, suppressing PKM2 activity and limiting pyruvate entry into the TCA cycle [30]. In addition, GBM cells have been shown to activate serine synthesis and one-carbon metabolism to survive glutamine deprivation. In this study, Tanaka K. et al. demonstrated that low glutamine conditions induce an upregulation of MTHFD2 and SHMT2, resulting in a high NAD + /NADH ratio for redox maintenance [31]. Enhanced NAD+ levels have been linked to improved mitochondrial function under stress, leading to protection against dietary limitations [32].
We recently repurposed sertraline, an antidepressant that efficiently crosses the BBB, as a specific SHMT1/2 inhibitor with robust anti-cancer properties, effectively targeting ser/gly-dependent cancers in vitro and in vivo [33–35]. Our group also recently demonstrated that cancer cells increase SHMT2-driven dependency on the ser/gly synthesis pathway upon radiotherapy treatment. This reshapes their intrinsic capacity for radiotherapy resistance while also establishing an immunosuppressive microenvironment, which are both efficiently targeted by (neo)adjuvant sertraline treatment. In addition, the tumor inhibitory effects of sertraline can be further enhanced when combined with compounds causing mitochondrial dysfunction, e.g., rotenone, antimycin A, or the anti-malarial artemether in ser/gly-dependent breast cancer mouse models [34]. Accumulating data also indicate the anti-tumor activity of another anti-malarial drug, i.e., chloroquine, by impairing mitochondrial function, suppressing autophagy-mediated elimination of damaged mitochondria, and inducing mitochondrial membrane potential loss [36–38]. Our institute has conducted a phase-IB clinical trial in GBM with promising results using chloroquine. This clinical trial investigated the addition of chloroquine to concurrent radiotherapy and temozolomide for newly diagnosed GBM cases, observing acceptable tolerability [39]. Therefore, the inhibition of serine/glycine synthesis using sertraline combined with mitochondria targeting by chloroquine may be a promising therapeutic strategy for GBM.
Among the 25% of GBMs exhibiting ser/gly pathway hyperactivation, ~13% of cases harbor genetic amplifications and mutations in PSPH, underscoring the significance of PSPH and de novo ser/gly synthesis pathway in the pathogenesis of GBM [21]. In this study, we demonstrate that genetic aberrations in PSPH induce higher clonogenic and migratory capacities with increased synthesis of nucleotides and antioxidants in GBM. We identified that the combination of sertraline and chloroquine effectively and synergistically restricts GBM cell proliferation and reduces the clonogenic capacity of GBM cell lines and patient-derived GBM cell models. Metabolite profiling revealed that sertraline, together with chloroquine, more efficiently blocked NADH and ATP generation, while restricting nucleotide synthesis, causing a drastic block in the proliferation capacity of GBM cells. Importantly, PSPH genetic alterations facilitated the expression of the immune checkpoint protein galectin-1. In this study, we highlight the contribution of ser/gly pathway alterations and hyperactivation in promoting an immunosuppressive and aggressive tumor phenotype, causing poor therapeutic responses and outcomes in GBM, which could be improved using combined sertraline/chloroquine treatment.
Materials & methods
Tumoral Glycine levels
Tumor tissue samples were collected at the Department of Neurosurgery, Umeå University Hospital, between 2004 and 2016. Intratumoral Glycine levels were quantified using cross-platform global metabolomic profiling. Glioma classification was performed in accordance with 2016 WHO classification of tumors of the CNS and subsequent cIMPACT-NOW additions. Further details describing tissue collection, tumor classification and metabolomic profiling analyses have been presented elsewhere [40]. Survival of included patients was followed up through electronic patient files linked to the Swedish national population register which covers virtually a 100% of deaths in Sweden on August 15, 2024 [41]. Ethical approval for this study was obtained from the Ethics Committee at Umeå University (218/2003; 2011/308-31 M). Patients were included in the study after signed informed consent according to good clinical practice and the Helsinki Declaration.
Array comparative genomic hybridization (CGH)
This study was approved by the medical ethics committee of our institute (KU Leuven #S59367). Genomic DNA (1 μg) was hybridized to 4 x 180 K CGH + SNP whole-genome microarrays (G4890A) (Agilent Technologies), according to the manufacturer’s protocol. Microarray slides were scanned using an Agilent G2565A DNA Microarray Scanner, and image analysis was performed using the Feature Extraction V10.10.1.1 software (Agilent Technologies). Array CGH data were analyzed using the Agilent Genomic Workbench 7.0.4.0 software (Agilent Technologies). The ADM-2 algorithm was applied to identify DNA copy number alterations (CNAs). Copy number loss was defined as a log2 ratio < –0.25, and copy number gain as a log2 ratio > 0.25, with at least 500 probes in a region. Amplification was defined as log2 ratio > 1.5. A female HapMap genotype (European Female, NA12878_V1) was used as a reference for the detection of LOH.
Cell culture
GBM cell lines U118-MG, GA-MG, LN-18, and GL261 were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and maintained at 37 °C and 5% CO2.These cell lines all presented active glucose fractional contribution to the serine/glycine synthesis pathway. 1919 and 2012 patient derived GBM cells were kindly provided by Prof. C.G. Hubert, Department of Biochemistry, School of Medicine, Case Western Reserve University, Cleveland, Ohio, USA. 1919 and 2012.2 GBM cell models with comparable proliferation rates, were obtained from an intermediate proneural (PN) GBM tumor and a PN GBM tumor, respectively, as previously described [42]. The 1919 GBM presented TERT and EGFR mutations, low expression of HES5 and unmethylated MGMT, while the 2012 GBM is characterized by CDK4 and MDM2 hallmark mutations, high expression of HES5 and unmethylated MGMT. GBM cells were cultured at 37 °C and 5% CO2 in Neurobasal medium (ThermoFisher) supplemented with 1x B27 without vitamin A (Invitrogen), 10 ng/ml recombinant human EGF (R&D systems), 10 ng/ml basic FGF (Bio-Connect), 1x L-glutamine (VWR), indicated further as complete NB medium [43, 44]. For low glucose experiments cells were plated in Neurobasal™-A Medium, no D-glucose, no sodium pyruvate, supplemented with 0,5 mg/ml D-Glucose, 1 mM sodium pyruvate (ThermoFisher), and the components previously described for complete NB media, and incubated at 37° and 5% CO2 for 72 h. For hypoxic experiments, cells were exposed to normoxia (21% O2) and moderate hypoxia (0.2% O2) for 48 h using a modular atmosphere-controlled system Whitley H85 hypoxystation- Don Whitley Scientific. All cell lines were regularly tested for Mycoplasma contamination.
Plasmids and generation of stable cell lines
pMSCV-IRES-GFP plasmids containing PSPH WT or V116I encoding cDNA were ordered from GenScript. For transfection, 8 μg pMSCV- PSPH WT-IRES-GFP, pMSCV-PSPH V116I-IRES GFP and pMSCV-IRES-GFP and 8 μg of packaging plasmid pCLAmpho were transfected into 293 T cells using lipofectamine (ThermoFisher). Packaging 293 T cells were cultured in DMEM (Sigma-Aldrich) with 10% FBS and after 24 h the medium was replaced with complete NB medium supplemented with 5% FBS. Medium was collected 24 h after media renewal and filtered through a 0.45μm filter. 1919 cells were incubated with retroviral supernatants overnight, supplemented with 2 μg/mL polybrene. Transduction efficiency was measured by FACS analysis, which demonstrated an efficiency of ~30% for all inserts, representative of one integration site per cell. Cells were GFP-sorted using S3e™ Cell Sorter (Bio-Rad) to generate stable cell lines with > 90% of cells expressing GFP. pLKO.1 mCherry plasmids containing either scrambled control or PSPH shRNAs were generated as previously described and now stably integrated into 2012.2 GBM cells [33]. Efficiencies 72 h post transduction varied between 60–75% mCherry expressing cells. PSPH western blot analysis confirmed functional knockdown in samples that were taken 72 h and 7 days after transduction.
13C6-Glucose tracing metabolomics
Cells were cultured in complete NB medium (1 × 106 cells/T-25 flask), without D-glucose supplemented with 4.5 g/L 13C6-glucose (Sigma) and incubated at 37 °C for 24 h. For the preparation of medium samples for metabolomics, 10 µl of media was collected and 990 µl of medium extraction buffer (80% methanol, containing 2 µM d27 myristic acid) was added. The samples were centrifuged for 15 min at 4 °C at 20.000 x g and 250 µl were transferred into a new tube. For the preparation of cell extracts for metabolomics, cells were washed with an ice-cold (4 °C) 0.9% NaCl solution. Subsequently, 300 µl of cellular extraction buffer (80% methanol, containing 2 µM d27 myristic acid) were added to the cells and incubated for 2–3 min on ice before scraping the cells. The samples were centrifuged at 20,000 × g for 15 min at 4 °C. The protein pellet was analyzed using Pierce BCA Protein Assay kit (Thermofisher) to determine protein concentrations, and the supernatant was transferred into a new tube for the identification of metabolite levels by mass spectrometry analysis at the Metabolomics Expertise Center (MEC - KU Leuven/VIB). Mass spectrometry was performed using a Q Exactive™ Focus hybrid quadrupole-Orbitrap. Peak identification was performed by the core facility. Data were normalized for the protein input.
Flow cytometry
To measure mitochondrial ROS levels, cells were seeded (500,000 cells/well) in 6-well plates and allowed to attach overnight. After 24 h, mitochondrial ROS levels were analyzed by incubating GBM cells with 250 nM of Mitotracker Red CM-H2XRos in serum-free medium at 37 °C for 30 min. To measure ROS levels, cells were incubated with 5uM cellROX Deep Red (ThermoFisher, #C10422) for 30 min in serum-free medium at 37 °C for 30 min in normoxia or hypoxia. For γH2AX and cleaved-PARP staining, cells were seeded and allowed to attach overnight. Next, cells were collected and stained with the Alexa Fluor® 647conjugated Phospho-Histone H2A.X (Ser139), or Cleaved PARP (Asp214) (D64E10) XP rabbit mAb and Anti-Rabbit IgG (H + L) F(ab’) fragment antibodies (Cell Signaling, #5625 and #4412, respectively) after fixation and permeabilization with 100% methanol. The analysis was performed on a FACS Canto II cytometer and BD FACS Diva 6.1.1 software was used. FlowJo V10.8 was used to exclude doublets and cellular debris and to analyze the mean fluorescence intensity (MFI).
Proliferation assay
1919 isogenic cells, MDA-MB-468 BRCA PHGDH wild type cells and MDA-MB-231 BRCA cells characterized by PHGDH amplification were seeded (7500 cells/well) in 96-well plates (Sigma-Aldrich) and incubated at 37 °C for 24 h to obtain optimal adherence to the surface. Next, BRCA cells were treated with 3 µM sertraline and 4 µM chloroquine, using DMSO as control. During the following days, cell proliferation was assessed by real-time imaging of confluence using an IncuCyte Zoom system (Essen BioScience) with 6 technical replicates per condition. The area under the curve (AUC) was calculated using GraphPad Prism (v.9.2.0), and the fold-change difference was calculated between the AUC of DMSO and the different treatments.
Alvetex 3D clonogenic
The long-term effects of sertraline and chloroquine on GBM cells were investigated using AlvetexTM (Reprocell) 3D colony formation assay [45]. GBM cells (300.000 cells/plate) were seeded in 6 cm culture dishes. 1919 and 2012.2 were cultured in complete NB medium, and 1919 GBM isogenic models, GFP Ctrl, PSPHamp and PSPHV116I were cultured in low glucose complete NB medium. After 24 h, 2012.2 and 1919 cells were treated with sertraline (5 µM), Chloroquine (5 µM) and DMSO as control. 1919 GBM isogenic models were treated with sertraline (5 µM), Chloroquine (4 µM) and DMSO as control. The reduction in chloroquine concentration is due to the higher sensitivity to chloroquine as monotherapy in low glucose conditions. After 48 h, cells were counted and seeded as single cells, 300 and 600 cells/well for 1919 and 2012.2 cells in normoxia and hypoxia, respectively, and 200 cells/well for 1919 isogenic models in low glucose, followed by re-addition of the treatments. Cells were plated in a 24 well plate on a polystyrene scaffold previously permeabilized with 70% ethanol followed by 2× washed with PBS and coated with Matrigel (1:40 with NB medium). For the hypoxic experiments, medium was incubated in the hypoxic chamber for 48 h before use for the treatment of 1919 and 2012.2 cells. 1919 and 2012.2 cells were treated with sertraline (5 µM), Chloroquine (5 µM) and DMSO as control in the hypoxic chamber and incubated for 48 h before placing them back in the incubator under normal oxygen conditions until the end of the clonogenic experiment. After 14 days of incubation, cells were stained with MTT (Sigma-Aldrich) and fixated with 2% paraformaldehyde (PFA). Visible cell clusters were counted as colonies using ImageJ software (Fiji). Cells were seeded in triplicates and n ≥ 3 independent assays were carried out.
Migration scratch assay
Cells were seeded (60,000 cells/well) in a 96-well plate (Greiner) coated with 0.3 mg/ml matrigel, allowed to attach overnight, and scratched using the IncuCyte Wound maker (Essen Bioscience). The migration of cancer cells was assessed in low glucose medium. These cells were pre-incubated for 48 h in low glucose medium before seeding for the migration scratch assay. Cell migration was monitored using Incucyte FLR (Essen Bioscience). The width, representing the distance covered by migrating cells, was calculated by measuring the area at the initial time point of the scratch and the subsequent time points, along with the length of the scratch.
Immunoblotting
Cells were lysed in protein lysis buffer (Cell Signaling) supplemented with Roche complete™ Protease Inhibitor Cocktail tablets (Sigma-aldrich). Proteins were boiled with Laemmli sample buffer (Biorad) plus 2-mercaptoethanol as loading buffer. Equal amounts of proteins were loaded in 4–15% Criterion TGX Precast Midi Protein gels (Biorad). Proteins were transferred onto PVDF membranes (Thermofisher) using a Power Blotter–Semi-dry Transfer System (Thermofisher) and incubated overnight with PSPH (Bioconnect, #14513-1-AP, 1:1000) rabbit polyclonal antibody,galectin-1/LGALS1 (D608T) Rabbit mAb (Cell Signaling, #12936S, 1:1000), SOD2 (D3X8F) XP rabbit mAb (Cell Signaling, #13141S), vinculin mouse mAb (Sigma-aldrich, #V9131, 1:4000), actin (clone C4) Mouse mAb (MP biomedicals, #691001, 1:4000) and rabbit anti-lamin A (C-term) (Sigma, cat. #L1293, 1:1000) antibodies diluted in 5% skimmed milk in Tris-Buffered NaCl Solution with Tween 20 (TBST). After washing with TBST, membranes were incubated for 1 h at room temperature with the secondary antibodies conjugated to horseradish peroxidase (HRP) anti-rabbit IgG (Cell Signaling) or anti-mouse IgG (Cell Signaling). ECL reagents (Sigma-Aldrich) were used to visualize proteins in the Azure C600 immunoblot imager. Image Studio Lite was used for quantitative analysis of protein expression.
Quantitative RT-PCR
RNA was isolated using a NucleoSpin kit (Machery-Nagel, 740955.50) and converted in cDNA using an iScript™ cDNA Synthesis Kit (Biorad). PSPH and SHMT2 mRNA expression together with RPL13A as reference gene were analyzed using the SensiMix™ SYBR® Low-ROX Kit (Meridian Bioscience) in the CFX connect Real-Time System (Biorad). Relative mRNA expression from four technical repeats was determined using the ΔΔCt method (primer sequences for PSPH, F: TGTCAGAAATGACACGGCGA and R: GGGGGTTGCTCTGCTATGAG, RPL13A, F: CGAGGTTGGCTGGAAGTACC and R: CTTCTCGGCCTGTTTCCGTAG).
Seahorse oxymetry
Cells were plated on XF96 culture plates (20,000 cells/well) in complete NB medium. After 24 h, the culture medium was replaced with DMEM containing 10 mM glucose, 2 mM glutamine, 1.85 g/L NaCl, and 3 mg/L phenol red, pH 7.4. Cells were incubated for 1 h in a CO2-free incubator prior analysis. Basal, maximal, and ATP-linked oxygen consumption rates (OCRs) were quantified using the XF Cell Mito Stress Test Kit (Agilent) on a Seahorse XF96 bioenergetics analyzer (Agilent). Sequentially, basal OCR was acquired without treatment; ATP-linked OCR after the addition of 1 µM of ATP synthase inhibitor oligomycin; maximal OCR after mitochondrial potential disruption using 1 µM of ionophore carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP); and non-mitochondrial OCR after the addition of 0.5 µM of Complex I inhibitor rotenone together with 0.5 µM of Complex III inhibitor antimycin A. All data were normalized by total protein content (Bio-Rad Protein Assay).
Tetramethylrhodamine Methyl Ester (TMRM) fluorescence live imaging
Cells were plated (50,000 cells/well) in 8-wells chamber ibiTreat (Ibidi). Cells were pre-treated with DMSO, sertraline (5 µM) and chloroquine (4 µM) in low glucose complete NB medium. After 48 h, cells were incubated with 100 nM of TMRM T668 (ThermoFisher) and Hoechst 34589 (Sigma-Aldrich) in low glucose complete NB medium for 10 min at 37 °C and 5% CO2. After 10 min, the medium was replaced before imaging. For live cell imaging, we used a FEI Corsight microscope with wide field and an Andromeda spinning disk. The FEI MAPS software and Live Acquisition software (LA, FEI) were used to control the microscope and capture images. The microscope was further equipped with an incubation system able to control temperature at 37 °C (Ibidi) and CO2 at 5% (Digital Pixel, UK) at the specimen level. TMRM and Hoechst staining was imaged using laser line 561 nm and 461 nm, respectively, and the 446/523/600/677 nm BrightLine® quad-band bandpass filter. Mitochondrial volume and TMRM intensity quantification were determined using a similar MATLAB script as previously reported [46]. To quantify mitochondrial volume, cells were imaged with z-stacks consisting of 250 planes and an interval of 0.147μm. Representative images were edited using Fiji software.
Public data analysis
Data of mutations, copy number variations, and mRNA overexpression of PSPH were collected from the cBioPortal (http://www.cbioportal.org/) data repository using the publicly available TCGA (The Cancer Genome Atlas) Glioblastoma Firehose Legacy dataset. GBM samples were included for ranking of samples according to mRNA expression levels of PSPH together with the PTEN mutational status of these samples. PSPH gene expression analysis was generated in the GEPIA platform comparing GBM tumor samples with TCGA normal and GTEx data. Kaplan-Meier curves were generated with SPSS using the GBM dataset (PIMD: 30996326) publicly available in TIGER database. Median expression was used as cut-off for high versus low PSPH and galectin-1 analyses.
Statistics
Survival analysis was conducted using the Kaplan-Meier method to estimate overall survival, defined as the time from operation until the occurrence of death, loss to follow-up, or the end of the study period (August 15, 2024), whichever occurred first. To illustrate the association between Glycine levels and survival, patients were categorized into three groups based on quantiles of the metabolites. The log-rank test was used to compare overall survival among these groups. Additionally, Cox proportional hazards regression was performed to estimate hazard ratios (HR) and 95% confidence intervals (CIs). The model included age at operation and glioma subtype as covariates, and interactions between these covariates and metabolite levels were assessed.
The number of biological replicates per experiment, the number of experiments performed for each dataset, and the statistical analyses performed are stated in the figure legends. Results are presented as means ± standard deviation (SD) unless otherwise stated. Statistical analyses were performed using GraphPad Prism (v.10.1.2) or IBM SPSS software based upon F-test for Equality of Variances unless otherwise stated. Box plots present five sample statistics: minimum, lower quartile, median, upper quartile, and maximum. Whiskers/inner fences are defined based on the distribution of the data points, up to a maximum of 1.5 times the height of the box. Outliers are shown in each plot when exceeding the whiskers. Rounded outliers lie in the range of 1.5–3 times the height of the box and asterisks or stars are extreme outliers that have values more than three times the height of the boxes. Comparative analyses between different experimental groups were performed using Student’s t-test and one-way ANOVA with Tukey’s and Dunnett’s post hoc tests for intergroup comparisons unless otherwise stated. Fisher’s exact test was performed for PTEN enrichment in GBM samples ranked according to PSPH expression levels. The statistical analysis of survival curves was performed using Log-rank (Mantel-Cox) test. Pearson’s correlation analysis was used to determine the correlation of PSPH and galectin-1 mRNA expression levels. Results were considered significant if the p-value was <0.05 (*), < 0.01 (**), < 0.001 (***), or <0.0001 (****).
Results
GBM is characterized by genetic ser/gly pathway alterations in PSPH
Ser/gly pathway involvement in GBM is not well understood. This study aims to provide in-depth insights into the contribution of ser/gly pathway in the pathogenesis of GBM. As glycine is the last metabolite in the ser/gly pathway, we explored intra-tumoral glycine levels in glioma patients using a previously published dataset [40], included 216 patients diagnosed between 2004 and 2016, with a median age of 60 years at the time of operation (IQR 49–67). The median survival time is 1.23 years (95%CI = 0.99–1.41). The majority of patients had glioblastoma (144 with IDH-wildtype and 9 IDH-mutated). Baseline data is summarized in Table S1. We found that higher intra-tumoral glycine levels were associated with worse survival in glioma patients across all subtypes (Fig. 1A, p < 0.001). After adjusting for glioma subtype and age at operation, we observed that higher levels of glycine were associated with an increased risk of death (HR = 1.20, 95%CI = 1.02–1.41) (Table S2).
Fig. 1. Intratumoral glycine levels and ser/gly pathway PSPH genetic alterations in the pathogenesis of GBM.

A Kaplan-Meier curves illustrating differences in survival in 216 glioma patients divided into three groups based on quantiles of intratumoral glycine levels irrespective of glioma subtype. B The main PSPH mutation observed in GBM patients is the recurrent PSPH V116I mutation (cBioPortal, TCGA, Firehose Legacy). C The PSPH V116I mutation is located at the inner core of the PSPH hydrolase domain. D PSPH gene expression analysis from GEPIA data platform comparing GBM tumor samples with normal tissue controls. Statistical significance p < 0.05 (*).
We recently described that 25% of GBM patients present genetic alterations in the genes encoding ser/gly synthesis enzymes [21], observing common PSPH amplifications (cBioPortal, TCGA PanCancer Atlas dataset) (Figure S1) [25]. However, PSPH ser/gly pathway enzyme genetic aberrations are not acknowledged as important drivers of GBM because of the close proximity of EGFR (chr7:55019017–55211628) to PSPH (chr7:56011051–56051604), which is a common driver oncogene in GBM [47–49]. Yet, PSPH is recurrently amplified in a genomic region of focal copy number gain in 13% of the GBM brain tumors (TCGA copy number portal, 2013–08–16 TCGA Pan-Cancer data set, Glioblastoma multiforme, n = 580, q value = 5.77 × 10−61). This finding was consistent with the possible role of PSPH in cancer initiation, growth, and/or survival. In our cohort of 48 array-CGH GBM cases, we observed one patient with a distinct amplification peak of PSPH (chr7: 55846273–56155279), in which EGFR was not included. The minimal amplification region included 8 genes, of which PSPH was located in the middle. The other protein-coding genes were SEPT14, MRPS17, GBAS, CCT6A, SUMF2, PHKG1, and CHCHD2 (Table S3). Interestingly, one patient in the TCGA dataset presented a similar distinct PSPH amplification peak, containing only 6 protein-coding genes: GBAS, PSPH, CCT6A, SUMF2, PHKG1, and CHCHD2 (Figure S1), again not including EGFR. In addition, we observed a recurrent mutation in PSPH that leads to an amino acid change from valine to isoleucine (V116I). This mutation was mainly observed in GBM, present in 3 GBM cases out of 8 patient samples with this mutation (cBioportal, COSMIC study - 329 COSU329 Glioblastoma Multiforme, TCGA), and was also observed in lung squamous cell carcinoma, clear cell renal cell carcinoma, colon adenocarcinoma, liver neoplasm and prostate carcinoma (Table S4). This mutation has not been described as potentially pathogenic, as the amino acid change is predicted to not affect protein function (PolyPhen-2, HumDiv method, score of 0.002, sensitivity: 0.99; specificity: 0.30) (Fig. 1B, C). However, the PSPH protein structure highlights the position of the V116I mutation at the inner core of the hydrolase domain (Fig. 1C). Furthermore, consulting the publicly available GEPIA data source platform, it can be appreciated that PSPH is highly expressed in GBM tumors compared to normal tissue (Fig. 1D, p < 0.05). Taken together, these data emphasize the pathogenic nature of high tumoral glycine levels and genetic PSPH alterations in GBM, warranting further investigation to unravel its contribution to the GBM pathogenesis.
Ser/glyhigh GBM cells present increased synthesis of ser/gly-derived nucleotides and antioxidants
Next, we aimed to investigate the significance of the detected PSPH amplifications (PSPHamp) or mutations (PSPHV116I) in patients with GBM in the pathogenesis of GBM. Therefore, we required a GBM model more reliant on ser/gly uptake rather than de novo ser/gly synthesis. We characterized the dependency on ser/gly synthesis of two patient-derived primary GBM models by performing 13C-glucose tracing. We found a clear difference between the two GBM models in glucose shuttling towards de novo serine and glycine synthesis, reflected as m + 3 and m + 2, respectively. GBM 2012.2 displayed 4- and 2-fold higher glucose-derived synthesis of serine and glycine, respectively, compared to GBM 1919, which showed low levels of baseline ser/gly synthesis, suggesting dependency on serine and glycine uptake (Fig. 2A, p < 0.0001, p = 0.0002). This was supported by the decreased levels of serine in the medium of 1919 cells compared to input media and 2012.2 cells (p = 0.0129, p = 0.0027, respectively), as well as reduced synthesized serine and glycine into the media (Fig. 2B, Figure S2).
Fig. 2. The ser/gly synthesis-dependent patient-derived GBM model presents higher serine and glycine derived synthesis of nucleotides and antioxidants, as well as ser/gly synthesis metabolic adaptation under hypoxia.

A Metabolic tracing using 13C-glucose in 1919 and 2012.2 GBM cells. Serine is synthesized from the glycolytic intermediate 3-PG, producing labeled serine (m + 3) and serine can generate labeled glycine (m + 2). Two-way ANOVA with Šídák’s multiple comparisons test has been performed. B Serine levels in media of 1919 and 2012.2 cells, corrected by levels of serine in NB media. One-way ANOVA with Tukey’s multiple comparison test has been performed. C Percentage (%) of 13C-derived triphosphate nucleotides and GSH mass distribution in 1919 and 2012.2. cells. Two-way ANOVA with Šídák’s multiple comparisons test has been performed. D Relative PSPH gene expression measured by RT-qPCR in n = 3 biological replicates of 2012.2 and 1919 cells after 48 h exposure to hypoxia. Gene expression under hypoxia is normalized to normal oxygen conditions. Unpaired 2-tailed t-test has been performed. E PSPH protein expression measured by immunoblot in n = 3 biological replicates and n = 3 technical repeats of 2012.2 and n ≥ 3 biological replicates and n = 3 technical repeats for 1919 cells after 48 h exposure to hypoxia. Protein levels were corrected to the loading control vinculin with the comparison to PSPH expression levels under normal oxygen conditions using an unpaired 2-tailed t-test. Representative images of PSPH protein expression in 1919 and 2012.2 cell models under normal oxygen and hypoxic conditions. F The oxygen consumption rate (OCR) of 2012.2 and 1919 cells after pre-treatment with sertraline for 48 h was measured using XFe96 Seahorse technology. Mito Stress Test was performed, and the graph represents the OCR measurements over time with the sequential injection of oligomycin, FCCP, and rotenone/antimycin A. G The fold change of basal, maximal, and ATP-linked respiration was calculated in n = 4 biological replicates with n = 4 technical replicates in each experiment. Unpaired 2-tailed t-test has been performed. Statistical significance p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
The ser/gly synthesis pathway provides one-carbon precursors for the synthesis of nucleotides (Figure S3A). Consistent with their high de novo ser/gly synthesis, 2012.2 cells showed the preference for incorporating glucose-derived serine and glycine into the synthesis of purine and pyrimidine nucleotides, i.e., ATP, GTP, CTP, UTP, TTP, reflected as m + 6 to m + 9 labeled nucleotides (Fig. 2C, S3B). In contrast, 1919 cells appear to preferentially channel glucose carbons into the pentose phosphate pathway rather than serine and glycine synthesis for nucleotide synthesis, as indicated by higher levels of m + 5 isotopologues (Fig. 2C). Antioxidant GSH can be labeled from glucose-derived glycine (m + 2). We observed an increased proportion of m + 2 and m + 4 labeled GSH in 2012.2 cells compared to 1919 cells, reflecting the increase in ser/gly synthesis activity and production of labeled glycine (Fig. 2C). In contrast, compared to 2012.2 cells, 1919 GBM cells appeared to be metabolically dependent on the urea cycle, as suggested by the higher intracellular levels of citrulline, ornithine and arginosuccinate (Figure S4).
As previously reported, ser/gly synthesis is important for GBM cells as an adaptation and survival mechanism to hypoxia [29]. Interestingly, we observed a 2-3 fold elevated PSPH gene and protein expression in 2012.2 cells under hypoxic conditions (p = 0.0098 and p = 0.0082), whereas 1919 cells did not exhibit upregulation of PSPH expression (Fig. 2D, E). In addition, we measured the protein levels of manganese superoxide dismutase 2 (SOD2, or MnSOD) in 1919 and 2012.2 cells following exposure to hypoxia. SOD2 is a mitochondrial antioxidant enzyme, which converts O2•− to hydrogen peroxide (H2O2) and molecular oxygen, reducing oxidative stress [50]. Our results indicated that SOD2 protein levels decreased in 2012.2 cells under hypoxia compared to normoxia, whereas SOD2 levels remained unchanged in 1919 cells (Figure S5). The decreased SOD2 levels might be indicative for reduced oxidative stress in 2012.2 cells, which may relate to 2012.2’s enhanced ser/gly-synthesis capacity to respond to the cellular oxidative stress, especially under hypoxia.
Next, we determined the sensitivity of our two patient-derived GBM models to ser/gly pathway inhibition by targeting SHMT using sertraline. GBM 2012.2 cells showed high sensitivity to sertraline, as shown by a significant impairment of its mitochondrial function: we observed a 2-fold reduction in the basal oxygen consumption rate (OCR), mitochondrial-linked ATP production, and maximal respiration in 2012.2 cells treated with sertraline compared to non-treated cells (p = 0.0022, p < 0.0001, p < 0.0001, respectively), whereas 1919 GBM cells did not show significant differences (Fig. 2F, G). Collectively, these data demonstrated that 2012.2 are ser/gly synthesis-dependent GBM cells, highlighted by increased serine- and glycine-derived synthesis of nucleotides and GSH, as well as adaptive upregulation of PSPH under hypoxic conditions, which could be efficiently targeted by sertraline treatment.
PSPH facilitates proliferation, clonogenicity, migration and redox homeostasis in GBM
To determine the contribution of PSPHamp and PSPHV116I PSPH genetic defects to GBM pathogenesis, we used the 1919 GBM model with low ser/gly synthesis. The establishment of 1919 GBM cells with overexpression of wildtype PSPH and PSPHV116I was confirmed by enhanced expression of PSPH by immunoblot and consecutive upregulation of ser/gly pathway enzyme SHMT2, most pronounced in PSPHV116I cells (Fig. 3A, B, Figure S6). 13C6-glucose tracing was used to investigate the metabolic consequences of these PSPH genetic alterations in GBM cells. We observed metabolic rewiring towards the pentose phosphate pathway in 1919 PSPHamp and PSPHV116I GBM cells, as evidenced by the enrichment of m + 5 purine and pyrimidine nucleotides. PSPHamp GBM 1919 cells showed higher synthesis of AMP, GTP, CDP, CTP, and UMP, whereas PSPHV116I GBM 1919 cells exhibited enhanced synthesis of CDP and CTP (Fig. 3C). Additionally, these cells showed upregulation of the synthesis of ser/gly synthesis-derived nucleotides, as indicated by the enrichment of m + 6 isotopologues, i.e., AMP in 1919 PSPHamp cells, CTP in 1919 PSPHV116I cells and GTP in both cell models, compared to 1919 control cells (Fig. 3C). Purines and pyrimidines play an important role in DNA and RNA synthesis, which is required for high-rate cancer cell proliferation. Given this, we investigated the long-term clonogenic ability of these cells using an alvetex clonogenic assay that resembles the physiological three-dimensional (3D) tumor structure. We observed that PSPHamp and PSPHV116I GBM 1919 cells possessed a 2- and 3-fold increased clonogenic capacity, respectively (Fig. 3D, p < 0.05).
Fig. 3. PSPH overexpression and mutation favor GBM stem cell clonogenicity and growth.

A The GBM 1919 was stably transduced with GFP containing vectors to test PSPH overexpression effects (PSPHamp) and the V116I mutant form of PSPH (PSPHV116I), using GFP control cells (GFP Ctrl). B Immunoblot analysis of PSPH protein expression in the different 1919 isogenic models, i.e. Ctrl, PSPHamp and PSPHV116I in n ≥ 3 biological replicates and n = 4 technical repeats. One-way ANOVA with Dunnett’s multiple comparison test has been performed. C Heatmap showing the percentage (%) of 13C-glucose derived mass distribution of m + 5 and m + 6 purines and pyrimidines nucleotides in PSPHamp and PSPHV116I GBM 1919 cells compared to 1919 GFP control cells under basal high glucose conditions in three biological replicates for each cell model. Two-way ANOVA test has been performed comparing each nucleotide to the 1919 GFP Ctrl. D Alvetex 3D clonogenic assays of PSPHamp and PSPHV116I GBM 1919 cells. Each individual dot represents and independent experiment. Unpaired 2-tailed t-test has been performed. E Representative images and graph of the migration rate of 1919 GFP control cells and 1919 PSPHamp and PSPHV116I cells in low glucose media. The dotted line limits the area of scratch at time 0. Area under the curve (AUC) of n = 3 independent experiments with n ≥ 10 wells analyzed/experiment has been calculated and normalized to 1919 GFP control cells. One-way ANOVA with Dunnet’s multiple comparisons test has been performed. F 13C-glucose derived mass distribution of NAD and NADP synthesis in three biological replicates for each cell model. Two-way ANOVA with Dunnett’s multiple comparisons test has been performed. G Intracellular levels of NAD and NADP in the isogenic models. One-way ANOVA with Dunnett’s multiple comparisons test has been performed. H Mitochondrial ROS measured using Mitotracker Red CM-H2XRos in GBM isogenic models. One-way ANOVA with Tukey’s multiple comparisons test has been performed. I Mito Stress Test was performed in 1919 isogenic models, measuring the oxygen consumption rate (OCR) using XFe96 Seahorse technology. Proton leak OCR was determined in n = 4 biological replicates and n = 4 technical replicates in each experiment. One-way ANOVA with Dunnett’s multiple comparisons test has been performed. Statistical significance p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****).
Furthermore, we triggered ser/gly metabolic rewiring in GBM 1919 cells by reducing glucose levels, aligning with the physiological conditions of the tumor microenvironment characterized by low oxygen and glucose availability [51, 52]. Several studies have reported that glucose-starvation activates gluconeogenesis for serine and glycine synthesis [53, 54]. The enhanced dependency on the ser/gly metabolic pathway under low glucose conditions in 1919 PSPHamp and PSPHV116I GBM cells was confirmed by an increased sensitivity to the inhibition of this pathway using sertraline, measured as a reduction in the proliferative capacity of GBM cells (Figure S7A). We found that especially PSPHV116I mutant cells accelerated their 2D growth under physiological (low) glucose conditions (Figure S7B, p = 0.0013 for PSPHV116I), and both GBM isogenic models showed a 2-fold increase in clonogenic capacity in 3D alvetex assays under low glucose conditions (Fig. 3D, p < 0.05). Interestingly, we observed an increased migratory capacity of 1919 cells with PSPH and PSPHV116I overexpression, as measured by the extent of wound closure in a time-lapse scratch assay (Fig. 3E).
Next, we focused on the reductive capacity of these GBM cell models. NAD and NADP function as electron carriers in a multitude of redox reactions. We observed increased NAD synthesis in both PSPHamp and PSPHV116I 1919 GBM cell models, as indicated by the enrichment in m + 10 NAD isotopologues. Notably, the intracellular levels of NAD were decreased in 1919 PSPHamp cells, indicating a high turnover of NAD (Fig. 3F, G). Furthermore, 1919 PSPHV116I GBM cells exhibited enhanced synthesis of NADP compared to 1919 GFP control cells, reflected by the higher levels of m + 10 NADP isotopologues, while no changes in cellular NADP levels were observed. Conversely, 1919 PSPHamp cells displayed increased utilization of NADP, as evidenced by decreased levels compared to 1919 GFP control cells (Fig. 3F, G). NADH is required for mitochondrial oxidative phosphorylation and ATP production. Cancer cells require increased levels of NADPH for increased nucleotide synthesis and as protection against ROS [28]. Therefore, these results suggested that 1919 GBM cells overexpressing PSPH and PSPHV116I presented a more aggressive phenotype with increased synthesis of nucleotides and utilization of NAD and NADP, which leads to superior mitochondrial function in support of fueling the pool of ATP, nucleotides, and antioxidants. The increase in antioxidant defense was supported by the reduced ROS levels observed in 1919 GBM PSPHamp and 1919 GBM PSPHV116I cells compared to parental cells (Fig. 3H). In addition, investigation of the mitochondrial function of 1919 PSPHamp and PSPHV116I GBM cells revealed an elevated proton leak in 1919 GBM PSPHV116I cells (Fig. 3I). A high proton leakage has been associated with cancer stem cells (CSCs), uncoupling oxidative phosphorylation to control the production of ROS. This, in turn, can sustain CSC self-renewal while limiting oxidative stress and preserving mitochondrial function [55–57].
Ser/glyhigh GBMs are selectively sensitive to combined sertraline/chloroquine treatment
High ser/gly synthesis pathway levels and enzyme expression supports GBM progression, causing lower overall patient survival [58, 59]. Therefore, we aimed to define a novel and selective targeted combination therapy for these cases. We previously showed the high efficacy of sertraline in combination with the anti-malarial drug artemether in breast cancer models, which affected mitochondrial function [34]. In this study, we aimed to combine sertraline with the anti-malarial drug chloroquine, which was previously investigated in a clinical trial for GBM by our department. The efficacy of this therapeutic combination was first validated in our breast cancer models, demonstrating that PHGDH wild type breast cancer cells do not respond to the therapeutic combination, whereas PHGDH-amplified breast cancer cells exhibited a synergistic sensitivity to suboptimal dosages of sertraline plus chloroquine (Figure S8A). Compared to the combination of sertraline with artemether, the combination of sertraline and chloroquine showed a stronger synergy with less toxicity to PHGDH wild type cells. Interestingly, we observed a high synergistic efficacy of sertraline and chloroquine combination therapy in all of the ser/gly-producing GBM cell lines. The suboptimal dosage of sertraline showed effective pathway targeting by increasing secretion of excess serine and lowering its synthesis (Figure S8B). The combination treatment, using dosages of both compounds that as monotherapies hardly affected GBM proliferation, resulted in complete inhibition of GBM cancer cell proliferation (Fig. 4A). Furthermore, monitoring cell confluence using an incucyte revealed that GBM cells treated with the combination therapy did not recover over time.
Fig. 4. Combined sertraline and chloroquine treatment efficiently targets ser/glyhigh GBMs.

A Incucyte confluency monitoring of human and mouse GBM cell models as readout for cell proliferation in response to sertraline (red), chloroquine (blue) or combined treatment (green) as compared to a DMSO control (gray). B Alvetex 3D clonogenic assays in ser/glyhigh 2012.2 GBM cells and ser/glylow 1919 GBM cells upon combined sertraline and chloroquine treatment, under both normal oxygen and hypoxic conditions. Each individual dot represents an independent experiment. One-way ANOVA with Tukey’s multiple comparison test has been performed. C Alvetex 3D clonogenic assay in GFP Ctrl, PSPHamp, PSPHV116I GBM 1919 cells treated with DMSO, sertraline, chloroquine, or the combination treatment in low glucose conditions. Each individual dot represents an independent experiment. One-way ANOVA with Tukey’s multiple comparison test has been performed, comparing each treatment to their respective DMSO controls in each model. D Alvetex 3D clonogenic assay and the quantification of ser/glyhigh 2012.2 GBM cells comparing scrambled control cells to PSPH knockdown cells for their clonogenic capacity using two independent shRNAs, shPSPH #1 and #2 respectively (representative of 3 biological repeats). One-way ANOVA with Tukey’s multiple comparison test has been performed compared to the scrambled control. E Incucyte confluency monitoring of GBM cells as readout for cell proliferation 72 h after transduction plating 2000 cells per well in 6 replicates to profile the effect of PSPH knockdown (shPSPH1 blue, shPSPH2 light blue) in ser/glyhigh 2012.2 GBM cells as compared to scrambled control cells and in response to chloroquine (striped line) treatment. One-way ANOVA with Tukey’s multiple comparison test has been performed to determine the effects of PSPH targeting compared to the scrambled control. For CQ effectiveness over time multiple unpaired t tests with False Discovery Rate (FDR) was performed with q-values < 0.05 determining the actual point of CQ impact on growth curve deviation. Statistical significance p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and q < 0.05 (*), q < 0.001 (***).
Next, we assessed the therapeutic responses to sertraline/chloroquine combination therapy in the patient-derived GBM models, i.e., 1919 and 2012.2, using 3D alvetex assays. Notably, only the ser/gly-producing 2012.2 GBM cells exhibited high sensitivity to sertraline/chloroquine treatment, with a 60% reduction in survival, whereas ser/glylow synthesis GBM 1919 cells were not significantly affected by the combination therapy (Fig. 4B). Moreover, the efficacy of sertraline plus chloroquine in 2012.2 cells was highly significant under hypoxic conditions (Fig. 4B). 3D clonogenic assays demonstrated that the isogenic PSPHamp and PSPHV116I GBM 1919 cells display high sensitivity to the combination therapy under physiologically relevant low concentrations of glucose, a response that was not significantly observed in wildtype 1919 GBM cells (Fig. 4C). The assessment of cleaved-PARP protein levels as apoptosis marker by flow cytometry suggested that the combination treatment primarily affects proliferation rather than inducing apoptosis (Figure S9). Collectively, these data highlight combined sertraline/chloroquine treatment as a novel targeted therapy for ser/gly-producing GBMs under hypoxia and low glucose levels, i.e., conditions that reflect the physiological aspects of the GBM microenvironment.
As proof-of-principle, we assessed the effect of PSPH knockdown in 2012.2 ser/glyhigh GBM cells. To enable this, we transduced 2012.2 GBM cells with a scrambled control and two independent PSPH targeting shRNAs, shPSPH1 and shPSPH2 respectively (Figure S10) [33]. Knockdown of PSPH inhibited the clonogenic capacity of ser/glyhigh 2012.2 GBM cells in 3D clonogenic assays. The ability of these ser/glyhigh GBM cells to form colonies was reduced, however, mainly affected the colony size (Fig. 4D). We further determined the effect of PSPH knockdown on GBM proliferation. PSPH knockdown restricted GBM cell proliferation and sensitized GBM cells to chloroquine treatment, while scrambled controls remained unaffected by chloroquine treatment. When PSPH knockdown was too strong the additive effect of CQ was only mildly present, as in shPSPH1 GBM cells (Fig. 4E). These data show that PSPH knockdown reduces the proliferation and clonogenicity in ser/glyhigh GBM. In addition, shPSPH cells are more sensitive to CQ treatment as in the case of sertraline, where sertraline treatment shows beneficial properties with respect to therapeutic window of opportunity at low dosages for synergistic treatment efficacy with chloroquine.
Sertraline/chloroquine efficacy can be attributed to restriction of nucleotides and impairment of mitochondrial function in ser/glyhigh GBMs
To elucidate the mode-of-action for the effectiveness of this therapeutic combination, we performed metabolomic profiling of GBM cell lines in response to sertraline and chloroquine as monotherapies, as well as the combination therapy. The significant inhibitory effects observed in the group treated with sertraline plus chloroquine included a reduction in purine and pyrimidine nucleotides, i.e., ATP and GTP, CTP and UTP (Fig. 5A). Additionally, sertraline and chloroquine as monotherapies, as well as their combination, strongly reduced the cellular levels of the nucleotide TTP by 50–70%. Adequate cellular TTP pools have been shown to be essential to preserve nuclear and mitochondrial genome stability [60]. Ser/gly synthesis dependence led to an enhanced synthesis of nucleotides in 2012.2 GBM cells, as well as in 1919 GBM isogenic models with overexpression of PSPH and the PSPHV116I mutation, which explains their high sensitivity to the inhibitory effects of sertraline plus chloroquine treatment.
Fig. 5. Combined sertraline and chloroquine treatment efficacy relies on the blocking of nucleotide synthesis and the impairment of mitochondrial function.

A Mass spectrometry analysis of the cellular metabolome in response to treatment modalities in U118-MG, GA-MG, LN-18 and GL261 cells. Sertraline is indicated in red, chloroquine treatment in blue and the combination therapy in green, indicated as relative metabolite fold changes (FC) compared to DMSO treated GBM cells. A one-way ANOVA test with Fisher’s Least Significant Difference (LSD) test has been performed. B TMRM mitochondrial staining of 1919 isogenic models. Graphs represent the intensity of TMRM staining corrected by the area in µm3. One-way ANOVA with Dunnett’s multiple comparisons test has been performed. C TMRM intensity/µm3 in 1919 GBM isogenic models treated with DMSO, sertraline, chloroquine, and combination treatment in n = 3 independent experiments and n ≥ 8 pictures per condition in each experiment. One-way ANOVA with Tukey’s multiple comparison test has been performed comparing GFP Ctrl, PSPHamp and PSPHV116I for each treatment group. D Representative images of TMRM staining in 1919 GBM isogenic models upon different treatments using Fire lookup table in Image J. Statistical significance p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****).
Moreover, sertraline and chloroquine combination resulted in diminished levels of TCA metabolites fumarate, malate, and citrate, as well as aspartate (Fig. 5A). Interestingly, impairment of mitochondrial respiration limits the synthesis of aspartate, which is required for protein and nucleotide synthesis [61, 62]. Of note, GBM 2012.2 cells showed higher basal levels of aspartate compared to 1919 GBM cells, which could explain their sensitivity to the blockage of aspartate by combining sertraline and chloroquine treatment (Figure S4). Elevated fumarate levels have also been linked to mitochondrial metabolic rewiring in aggressive high-grade prostate cancer [63]. The treatment of GBM cells with sertraline plus chloroquine strongly depleted the cellular levels of fumarate. These results, along with the reduced levels of NADH solely in the combination treatment, further support the impairment of the mitochondrial function.
Therefore, we used the fluorescent dye Tetramethylrhodamine Methyl Ester (TMRM) to study mitochondrial function in living cells using fluorescence imaging. First, we observed a reduction in TMRM intensity in 1919 GBM PSPHamp and PSPHV116I cells under low glucose conditions compared to 1919 GFP control cells, reflecting a lower mitochondrial membrane potential (ΔΨm) (Fig. 5B). ΔΨm plays a role in supporting ATP synthesis since the conversion of ADP to ATP by the ATP synthase is coupled to ΔΨm dissipation. The lower ΔΨm in 1919 GBM PSPHamp and PSPHV116I cells could explain the use of ATP by these cells for proliferation rather than to sustain the ΔΨm via ATP hydrolysis by ATP synthase. Interestingly, a low ΔΨm has been associated with enhanced self-renewal capabilities and radioresistance [64, 65]. The treatment of 1919 GBM isogenic models with sertraline and chloroquine led to increased mitochondrial size and TMRM intensity (Fig. 5D). This observation was especially significant with chloroquine treatment as monotherapy and the combination of sertraline and chloroquine in 1919 PSPHamp and PSPHV116I cell models, leading to a disrupted mitochondrial morphology, which appeared swollen and fragmented compared to healthy mitochondria (Fig. 5D). Stable ΔΨm is essential for the maintenance of normal mitochondria structures and physiological function. As such, a deficiency in ATP synthase activity leads to increased ΔΨm and restriction of mitochondrial respiration, thereby limiting the generation of ATP and NAD+ via mitochondrial respiration [66]. These results indicate that PSPH-amplified or mutant ser/glyhigh synthesis GBMs are strongly dependent on their superior physiological mitochondrial function, which is effectively targeted by sertraline plus chloroquine.
PSPH as biomarker for immunotherapy responses in patients with GBM
Besides providing intrinsic benefits to cancer cells, we previously observed that ser/gly producing cancer cells actively modulate their microenvironment. While the cancer cells provide their microenvironment with secreted metabolites, sertraline treatment could target the galectin-1 mediated immunosuppressive marks of ser/gly metabolically dependent lung cancer [35, 67]. This observation prompted us to investigate the immune-phenotyping contribution of PSPH in GBM. To this end, we analyzed the impact of PSPH gene expression levels in a cohort of GBM patients who received anti-PD1 immunotherapy. We found that GBM patients with high tumor PSPH expression showed poor immunotherapy responses and therefore a worse overall survival compared to those with low tumor PSPH expression (TIGER immune database, Fig. 6A, p = 0.010). GBM is characterized by a strong local immunosuppressive tumor microenvironment. Several reports highlight galectin-1 as a key protein in the regulation of GBM tumor microenvironment, promoting immune escape, as well as migration, invasion, and angiogenesis [68–71]. Hence, galectin-1 is correlated with GBM aggressiveness and poor patient survival [72]. In the anti-PD1-treated cohort of GBM patients, we confirmed the worse overall survival of patients with a high expression of LGALS1, the gene encoding the protein galectin-1 (Fig. 6B, p = 0.018). Supporting these findings, we observed a positive correlation between PSPH and LGALS1 mRNA expression in patients with GBM (p = 0.005, R = 0.472) (Fig. 6C). The performance comparison of PSPH expression against known immune response signatures revealed that PSPH expression in GBM patients functions as a potential biomarker to predict immunotherapy responses (PSPH, HR = 4.475, 95% CI = 1.334–15.02, p = 0.0104).
Fig. 6. PSPH expression for the prediction of immunotherapy responses in GBM patients.

A The graphs show the Kaplan-Meier curve comparing the overall survival of GBM patients, equally divided in high versus low 50% PSPH expression using the GBM dataset n = 34 [107] of patients that were treated with anti-PD1. P value was calculated using a Log-rank Mantel–Cox test. B The graph shows the Kaplan-Meier curve comparing the overall survival equally divided in high versus low (50%) LGALS1 (Galectin-1) expression. P value was calculated using a Log-rank Mantel–Cox test. C Pearson correlation of LGALS1 and PSPH expression in patients with GBM (PIMD: 30996326). D Immunoblot analysis of the immunosuppressive protein Galectin-1 expression in 1919 GBM isogenic models with overexpression of PSPH and PSPHV116I in n ≥ 3 biological replicates and n = 3 technical repeats. One-way ANOVA with Dunnett’s multiple comparisons test has been performed. E Immunoblot analysis of the immunosuppressive protein Galectin-1 expression in 1919 GBM isogenic models with overexpression of PSPH and PSPHV116I. Cells were treated with DMSO as a control and 7.5 µM sertraline for 48 h. The graph shows the protein quantification corrected by the loading control lamin A and relative to the 1919 GFP Ctrl cells treated with DMSO in n = 3 biological replicates and n = 2 technical replicates. A one-way ANOVA with Šídák’s multiple comparisons test, comparing the GFP Ctrl cells treated with DMSO to the PSPHamp and PSPHV116I DMSO-treated cells, as well as comparing each cell model with their respective sertraline-treated groups. Statistical significance p < 0.05 (*), p < 0.01 (**), p < 0.001 (***).
Finally, in our isogenic GBM models, we confirmed that overexpression of PSPH and PSPHV116I enhanced galectin-1 expression, measured by immunoblot (Figs. 2A, 6D). Inhibition of ser/gly synthesis using sertraline led to a reduction of galectin-1 expression in PSPH and PSPHV116I overexpressing cells, with a more pronounced and significant effect observed in the PSPHV116I cell model (Fig. 6E). This suggests that PSPH, at least indirectly, is involved in regulating galectin-1 expression, which can be targeted using sertraline. This data underscores the significance of PSPH genetic aberrations leading to ser/gly synthesis metabolic rewiring to support and sustain the aggressive and immunosuppressive phenotype of GBM tumors. They emphasize the potential of targeting ser/gly synthesis pathway using (neo)adjuvant sertraline plus chloroquine treatment to improve the future outcomes of patients with GBM.
Discussion
In this study, we demonstrated that PSPH and PSPHV116I overexpression in patient-derived GBM cells enhanced purine and pyrimidine nucleotide synthesis, as well as synthesis and utilization of the reductive equivalents NAD(P). These metabolic changes resulted in increased proliferation, clonogenicity, and migration, enhanced mitochondrial function, and immune evasive status of PSPHamp and PSPHV116I cells compared to 1919 GBM control cells. Interestingly, for the first time, we report that PSPH amplifications in GBM dictate the expression of immune checkpoint galectin-1, supported by poor anti-PD1 immunotherapy responses and outcome in patients with GBM. These data indicate the implication of PSPH in the regulation of the aggressive and immunosuppressive tumor microenvironment in GBM, which could be targeted by sertraline/chloroquine combination treatment.
Although predicted not to be pathogenic using the bioinformatics tool PolyPhen-2, the PSPH V116I mutation is located at the inner core of the PSPH hydrolase domain. Valine and isoleucine are both aliphatic, hydrophobic amino acid that can play a role in substrate recognition. However, some studies have described an enhanced or dysregulated activity of proteins presenting valine to isoleucine substitutions. For instance, the pathogenic V122I variant of the protein transthyretin can induce cardiomyopathy due to destabilization of the transthyretin quaternary structure, i.e., the tetramer-folded monomer equilibrium, and an increased rate of tetramer dissociation [73]. Moreover, the V717I substitution in amyloid precursor protein is a common mutation associated with familial Alzheimer’s disease worldwide [74–76]. V284I mutation in the enzyme gentisate 1,2-dioxygenase I in lower eukaryotes leads to superior enzyme function, up to 260% [77]. We observed that GBM cells with amplification of both PSPH and PSPHV116I shared metabolic and phenotypic characteristics, such as augmented nucleotides and NAD synthesis and, subsequently, enhanced proliferative and clonogenic capabilities. Upon increased synthesis in these cells, NAD might be used as a co-factor to synthesize nucleotides and amino acids [78–80]. Both PSPH isogenic models showed a higher synthesis of ribose-5-phophate-derived CTP and ser/gly synthesis-derived GTP. Elevated CTP and GTP pools enable adequate DNA and RNA synthesis necessary for cancer cell proliferation and DNA repair in response to genotoxic stresses in multiple cancer types [81]. It has been reported that increased CTP pools in pancreatic ductal adenocarcinoma cells lead to acquired resistance to chemotherapeutic gemcitabine [82]. In addition, triple-negative breast cancer cells increase their CTP and TTP pools in response to doxorubicin or cisplatin in vitro, thereby optimizing DNA repair [83]. In GBM, the metabolic profiling of GBM cell lines showed that the abundance of GTP nucleotides correlates with resistance to radiation therapy, while inhibition of GTP synthesis sensitized GBM cells to irradiation by impairing DNA repair [84]. Therefore, the increased levels of CTP and GTP nucleotides in GBM tumors with genetic alterations of PSPH might induce resistance to chemotherapeutics and genotoxic stresses. As such, we observed a reduction in DNA damage, measured by γH2AX staining using flow cytometry, in 1919 cells with amplification of PSPH and PSPHV116I, both at baseline and in response to a 4 Gy irradiation (Figure S11). Apart from DNA and RNA synthesis, augmented GTP nucleotide pools have been associated with promoting metastasis across several cancer types via the activation of RHO-family GTPases, which are involved in extracellular matrix degradation, basement membrane invasion, and cell migration [85, 86]. In line with this, we observed a superior migratory capacity of 1919 PSPHamp and 1919 PSPHV116I GBM cells. Moreover, overexpression of PSPH V116I led to differences in their requirement for elevated NADP synthesis. Cancer cells typically maintain high levels of NADP, primarily providing the reducing power for anabolic reactions and redox balance, crucial for sustaining their rapid growth and providing protection against oxidative stress. The reliance on protection against oxidative stress in 1919 PSPHV116I cells was further evidenced by an increased proton leak compared to 1919 control cells and 1919 PSPHamp since mitochondrial uncoupling and proton leakage have been proposed to protect against oxidative damage by limiting the ROS formation [87]. This is supported by the lower levels of mitochondrial ROS of 1919 PSPHV116I compared to 1919 PSPHamp GBM cells.
The data obtained in this study is not restricted to GBM, as we obtained similar data in the A549 lung cancer model. PSPH amplifications and mutations also occur in lung cancer cases, therefore, we generated overexpression models of PSPH and PSPHV116I in A549 ser/glylow lung cancer cells [35]. The generated models showed a clear protein overexpression of PSPH (Figure S12A). In addition, we observed an induction of ser/gly synthesis by metabolic tracing of 13C6-glucose, detecting enhanced secretion of synthesized serine and glycine into the medium (Figure S12B). Downstream of ser/gly synthesis, A549 PSPHamp and PSPHV116I cells exhibited increased labeled GSH synthesis and secretion into the media and intracellular levels of NADH (Figure S12C, D). This observation highlights the need for stratification methods in cancer to define ser/glyhigh cancers that can benefit from (neo)adjuvant sertraline treatment.
Here, we explored the use of sertraline in combination with chloroquine, including investigating this therapeutic strategy under hypoxic and low glucose conditions. Most therapeutic strategies are studied in settings with high glucose and oxygen conditions. However, the GBM tumor microenvironment is known to be depleted of amino acids, especially in poorly vascularized areas [31]. These nutrient-deprived areas are characterized by hypoxia, which all-together, represent key factors in treatment resistance [88]. Therefore, this research approach hinders progression in the development of effective treatments for patients with GBM and, more importantly, the clinical translation of therapeutic findings. Hypoxia and low glucose have been shown to induce metabolic reprogramming towards ser/gly synthesis and mitochondrial metabolism to gain a survival advantage [29, 53]. We found that the combination therapy successfully targeted metabolic rewiring towards ser/gly synthesis and mitochondrial metabolism, rendering GBM cancer cells sensitive to this therapeutic strategy. The efficacy is attributed to the reduction in the synthesis of purine and pyrimidines, ATP, GTP, CTP, and TTP, along with the targeting of the TCA cycle metabolites, i.e., fumarate, malate, aspartate, and citrate, as well as NADH, essential for proper mitochondrial function. Consequently, alterations in ΔΨm and mitochondrial morphology were observed, supporting mitochondrial dysfunction.
Sertraline treatment did not affect glycolysis in these GBM cells, while SHMT2 knockdown has previously been shown to increase PK activity in GBM [30]. Although sertraline targets SHMT2, we cannot exclude the possibility that off-target effects might have contributed to the mitochondrial blockade observed in these GBM cells. Previous studies identify mTOR signaling as target of sertraline; however, since mTOR is also described as a nutrient stress sensor, it is difficult to determine whether this effect is due to direct targeting or the result of metabolic rewiring [23, 89–92]. In addition, SHMT2 depletion has been shown to induce mitochondrial respiratory chain deficiency by reducing basal respiration and respiratory capacity, decreasing NAD + /NADH ratios, and lowering the expression levels of respiratory complex I, IV, and V proteins [93]. The NAD + /NADH balance is also affected by both, the ser/gly synthesis pathway inhibition and chloroquine, based on the availability of aspartate. Aspartate is an important amino acid for tumor growth [94]. Chloroquine treatment has been shown to cause aspartate depletion, which limits nucleotide synthesis [95, 96]. The synthesis of aspartate requires electron acceptors that are maintained by electron transport chain reactions. Complex I regenerates NAD+ from NADH, and a pharmacological inhibition of that reaction disturbs the NAD + /NADH balance and the subsequent reduction in aspartate synthesis [62]. Aspartate depletion has also been reported to disrupt the malate-aspartate shuttle and to induce mitochondrial dysfunction [97]. Interestingly, serine synthesis has been found to be inhibited upon genetic inhibition of malate-aspartate shuttle components due to a low NAD + /NADH ratio [98]. In line with these studies, we observed that the combination of sertraline plus chloroquine diminished the levels of NADH, and both sertraline and chloroquine treatments induced depletion of aspartate and malate, with an even more pronounced effect in the combination therapy. Moreover, lowering GSH levels by targeting the ser/gly synthesis pathway using sertraline may also increase the sensitivity of GBM cells to the redox-dependent effects of chloroquine. Chloroquine-induced alterations in redox potential and chloroquine resistance have been associated with higher levels of GSH. High GSH levels might 1) reduce the accumulation of chloroquine, for instance, by increasing the formation of chloroquine-GSH adducts; and 2) counteract the oxidative damage caused by chloroquine [99, 100]. In addition, chloroquine has been described to compete with NADH for binding to glycolytic enzyme lactate dehydrogenase, occupying the NADH-binding pocket of this enzyme, which was illustrated in the malarial parasite Plasmodium falciparum [101]. In our GBM models, depletion of the cellular NADH pool resulting from the combined treatment of sertraline and chloroquine might enhance the sensitivity of resistant GBM cells to chloroquine by increasing the availability of the NADH-binding pocket to exert its function. In summary, the combination of sertraline and chloroquine targets different aspects of metabolic rewiring in GBM cells, i.e., nucleotide synthesis, redox homeostasis, and mitochondrial function, causing severe impairment of their proliferation and survival capabilities.
We previously described how targeting the ser/gly synthesis pathway using sertraline not only affected tumor cells but also modulated the anti-tumor immune response when combined with radiotherapy. This mechanism involves galectin-1 depletion, leading to induction of NK cell signatures in the tumor microenvironment and improved tumor control. This is especially interesting for GBM patients, since galectin-1 has been identified as a poor prognostic factor, fulfilling a key role in the aggressiveness and immune escape of GBM cells [68, 72, 102–104]. One of the main reasons for the limited regression of GBM induced by immunotherapy is the ability of GBM cells to evade immune surveillance. The immunosuppressive effects of galectin-1 are mediated by several mechanisms, including polarization of macrophages towards an M2 anti-inflammatory phenotype, inhibition of the capacity of NK cells to kill cancer cells, and induction of regulatory T-cell infiltration [102, 105, 106]. Consequently, galectin-1 has become an emerging therapeutic target in GBM to induce a more robust anti-tumor immune response. Beyond our expectations, we found a positive correlation between PSPH and galectin-1 expression in patients with GBM undergoing anti-PD1 treatment. PSPH expression in these GBM cases may function as a biomarker for anti-PD1 treatment responses. Interestingly, both PSPH and PSPH V116I amplifications were capable of inducing galectin-1 expression in isogenic primary GBMs, providing evidence for PSPH as (in)direct regulator of immune evasive galectin-1 expression. Recent genomic and transcriptomic profiling of tumors of patients with GBM following treatment with PD-1 inhibitors nivolumab or pembrolizumab revealed a significant enrichment of PTEN mutations associated with immunosuppressive expression signatures in non-responders [107]. Interestingly, using the TCGA GBM patient-dataset, we observed that GBM tumors with high PSPH expression are strongly enriched for PTEN mutations (p = 0.002) (Figure S13). This finding suggests that PTEN mutations are an additional subgroup of ser/glyhigh GBMs, along with PSPH amplification and PSPH V116I mutation, which could benefit from (neo)adjuvant sertraline treatment. Collectively, these findings underscore the regulatory role of PSPH, facilitating the increased expression of galectin-1, offering therapeutic opportunities to overcome phenotypically immunosuppressive GBMs, and augment the efficacy of immunotherapies.
In summary, we highlight the importance of PSPH genetic alterations in GBM in promoting tumor aggressiveness and immunosuppression, which could be effectively targeted by a novel combinatorial therapeutic strategy of sertraline plus chloroquine.
Supplementary information
Acknowledgements
We thank all researchers and clinicians for their contributions to the field and apologize to those whose work we did not describe or cite. At UCLouvain, authors thank Marie Bedin and Thibaut Vazeille for technical assistance.
Author contributions
ASC designed and performed research, analyzed data, and wrote the manuscript. KGS, AB, and JH performed experiments. BD provided GBM sample collection and the array comparative genomic hybridization (CGH) data. BB, BM, WYW, RLS, re-analyzed their previously published glioma dataset, analyzed data, and wrote manuscript. PS, PV, KDK, KMAR, MPGB and MV wrote manuscript. KRK designed and performed research, analyzed data, supervised the study, and wrote the manuscript. All authors reviewed, edited and agreed with the content of the manuscript.
Funding
ASC received funding from the European Union’s Horizon Theradnet 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No # 860245 THERADNET (to MV and PS). Metabolomics mass spectrometry analysis was funded by FWO 2019-2021 krediet aan navorsers to KRK at KU Leuven, KNAW early career award 2021 and FEBS excellence award 2021 to KRK at Maastricht University. PS is a Research Director of the Belgian Fonds National de la Recherche Scientifique (F.R.S.-FNRS).
Data availability
Western blots are available in the original data file. Publicly available data is mentioned in the materials and methods section and/or indicated in the figure legend. Data is available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41389-024-00540-3.
References
- 1.Wen PY, Reardon DA. Progress in glioma diagnosis, classification and treatment. Nat Rev Neurol. 2016;12:69–70. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, Van Den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. [DOI] [PubMed] [Google Scholar]
- 3.Hölzer T, Herholz K, Jeske J, Heiss W-D. FDG-PET as a prognostic indicator in radiochemotherapy of glioblastoma. J computer Assist Tomogr. 1993;17:681–7. [DOI] [PubMed] [Google Scholar]
- 4.Jensen RL. Brain tumor hypoxia: tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a therapeutic target. J neuro-Oncol. 2009;92:317–35. [DOI] [PubMed] [Google Scholar]
- 5.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncol. 2005;7:134–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer cell. 2009;15:501–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay R, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α. Oncogene. 2009;28:3949–59. [DOI] [PubMed] [Google Scholar]
- 8.Boyd NH, Tran AN, Bernstock JD, Etminan T, Jones AB, Gillespie GY, et al. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics. 2021;11:665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marampon F, Gravina GL, Zani BM, Popov VM, Fratticci A, Cerasani M, et al. Hypoxia sustains glioblastoma radioresistance through ERKs/DNA-PKcs/HIF-1α functional interplay. Int J Oncol. 2014;44:2121–31. [DOI] [PubMed] [Google Scholar]
- 10.Hjelmeland AB, Wu Q, Heddleston J, Choudhary G, MacSwords J, Lathia J, et al. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011;18:829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samanta D, Semenza GL. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochimica et Biophysica Acta (BBA)-Rev Cancer. 2018;1870:15–22. [DOI] [PubMed] [Google Scholar]
- 12.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunbar E, Coats B, Shroads A, Langaee T, Lew A, Forder J, et al. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investigational N. drugs. 2014;32:452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro-Oncol. 2022;24:1935–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020;6:1003–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michelakis E, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra4. [DOI] [PubMed] [Google Scholar]
- 17.Strowd R, Ellingson B, Raymond C, Yao J, Wen PY, Ahluwalia M, et al. Activity of a first-in-class oral HIF2-alpha inhibitor, PT2385, in patients with first recurrence of glioblastoma. J neuro-Oncol. 2023;165:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harder BG, Blomquist MR, Wang J, Kim AJ, Woodworth GF, Winkles JA, et al. Developments in blood-brain barrier penetrance and drug repurposing for improved treatment of glioblastoma. Front Oncol. 2018;8:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Upton DH, Ung C, George SM, Tsoli M, Kavallaris M, Ziegler DS. Challenges and opportunities to penetrate the blood-brain barrier for brain cancer therapy. Theranostics. 2022;12:4734–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, Qiu W, Sun T, Wang L, Du C, Hu Y, et al. Therapeutic strategies of glioblastoma (GBM): The current advances in the molecular targets and bioactive small molecule compounds. Acta Pharmaceutica Sin B. 2022;12:1781–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sánchez-Castillo A, Vooijs M, Kampen KR. Linking serine/glycine metabolism to radiotherapy resistance. Cancers. 2021;13:1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012;491:458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, et al. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci USA. 2012;109:6904–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi S, et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell death Dis. 2013;4:e877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geeraerts SL, Heylen E, De Keersmaecker K, Kampen KR. The ins and outs of serine and glycine metabolism in cancer. Nat Metab. 2021;3:131–41. [DOI] [PubMed] [Google Scholar]
- 26.Meiser J, Tumanov S, Maddocks O, Labuschagne CF, Athineos D, Van Den Broek N, et al. Serine one-carbon catabolism with formate overflow. Sci Adv. 2016;2:e1601273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L, Canaveras JCG, Chen Z, Wang L, Liang L, Jang C, et al. Serine catabolism feeds NADH when respiration is impaired. Cell Metab. 2020;31:809–21.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ju H-Q, Lin J-F, Tian T, Xie D, Xu R-H. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduct Target Ther. 2020;5:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engel AL, Lorenz NI, Klann K, Münch C, Depner C, Steinbach JP, et al. Serine-dependent redox homeostasis regulates glioblastoma cell survival. Br J Cancer. 2020;122:1391–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D, Fiske BP, Birsoy K, Freinkman E, Kami K, Possemato RL, et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015;520:363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka K, Sasayama T, Nagashima H, Irino Y, Takahashi M, Izumi Y, et al. Glioma cells require one-carbon metabolism to survive glutamine starvation. Acta Neuropathologica Commun. 2021;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cantó C, Menzies KJ, Auwerx J. NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kampen KR, Fancello L, Girardi T, Rinaldi G, Planque M, Sulima SO, et al. Translatome analysis reveals altered serine and glycine metabolism in T-cell acute lymphoblastic leukemia cells. Nat Commun. 2019;10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geeraerts SL, Kampen KR, Rinaldi G, Gupta P, Planque M, Louros N, et al. Repurposing the antidepressant sertraline as SHMT inhibitor to suppress serine/glycine synthesis–addicted breast tumor growth. Mol cancer therapeutics. 2021;20:50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heylen E, Verstraete P, Van Aerschot L, Geeraerts SL, Venken T, Timcheva K, et al. Transcription factor NKX2–1 drives serine and glycine synthesis addiction in cancer. British Journal of Cancer. 2023:1-17. [DOI] [PMC free article] [PubMed]
- 36.Hori YS, Hosoda R, Akiyama Y, Sebori R, Wanibuchi M, Mikami T, et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J neuro-Oncol. 2015;122:11–20. [DOI] [PubMed] [Google Scholar]
- 37.Vessoni AT, Quinet A, de Andrade-Lima LC, Martins DJ, Garcia CC, Rocha CR, et al. Chloroquine-induced glioma cells death is associated with mitochondrial membrane potential loss, but not oxidative stress. Free Radic Biol Med. 2016;90:91–100. [DOI] [PubMed] [Google Scholar]
- 38.Kim EL, Wüstenberg R, Rübsam A, Schmitz-Salue C, Warnecke G, Bücker E-M, et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro-Oncol. 2010;12:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Compter I, Eekers DB, Hoeben A, Rouschop KM, Reymen B, Ackermans L, et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: a phase IB trial. Autophagy. 2021;17:2604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Björkblom B, Wibom C, Eriksson M, Bergenheim AT, Sjöberg RL, Jonsson P, et al. Distinct metabolic hallmarks of WHO classified adult glioma subtypes. Neuro-Oncol. 2022;24:1454–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ludvigsson JF, Almqvist C, Bonamy A-KE, Ljung R, Michaëlsson K, Neovius M, et al. Registers of the Swedish total population and their use in medical research. Eur J Epidemiol. 2016;31:125–36. [DOI] [PubMed] [Google Scholar]
- 42.Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016;76:2465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verduin M, Hoosemans L, Vanmechelen M, van Heumen M, Piepers JAF, Astuti G, et al. Patient-derived glioblastoma organoids reflect tumor heterogeneity and treatment sensitivity. Neuro-Oncol Adv. 2023;5:vdad152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez-Roman N, Stevenson K, Gilmour L, Hamilton G, Chalmers AJ. A novel 3D human glioblastoma cell culture system for modeling drug and radiation responses. Neuro-Oncol. 2016;19:229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iannetti EF, Smeitink JA, Beyrath J, Willems PH, Koopman WJ. Multiplexed high-content analysis of mitochondrial morphofunction using live-cell microscopy. Nat Protoc. 2016;11:1693–710. [DOI] [PubMed] [Google Scholar]
- 47.Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eskilsson E, Røsland GV, Solecki G, Wang Q, Harter PN, Graziani G, et al. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncol. 2018;20:743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim YS, Gupta Vallur P, Phaëton R, Mythreye K, Hempel N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants. 2017;6:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16:1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bikfalvi A, da Costa CA, Avril T, Barnier J-V, Bauchet L, Brisson L, et al. Challenges in glioblastoma research: focus on the tumor microenvironment. Trends cancer. 2023;9:9–27. [DOI] [PubMed] [Google Scholar]
- 53.Vincent EE, Sergushichev A, Griss T, Gingras M-C, Samborska B, Ntimbane T, et al. Mitochondrial phosphoenolpyruvate carboxykinase regulates metabolic adaptation and enables glucose-independent tumor growth. Mol cell. 2015;60:195–207. [DOI] [PubMed] [Google Scholar]
- 54.Keshet R, Lee JS, Adler L, Iraqi M, Ariav Y, Lim LQJ, et al. Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nat Cancer. 2020;1:894–908. [DOI] [PubMed] [Google Scholar]
- 55.Brookes PS. Mitochondrial H+ leak and ROS generation: an odd couple. Free Radic Biol Med. 2005;38:12–23. [DOI] [PubMed] [Google Scholar]
- 56.Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, et al. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–9. [DOI] [PubMed] [Google Scholar]
- 57.Echtay KS. Mitochondrial uncoupling proteins—what is their physiological role? Free Radic Biol Med. 2007;43:1351–71. [DOI] [PubMed] [Google Scholar]
- 58.Liu J, Guo S, Li Q, Yang L, Xia Z, Zhang L, et al. Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J neuro-Oncol. 2013;111:245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamizo A, Miyamatsu Y, Hirose H, Amano T, Matsuo S, Fujiwara M, et al. Metabolic remodeling of pyrimidine synthesis pathway and serine synthesis pathway in human glioblastoma. Sci Rep. 2022;12:16277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fiddler JL, Xiu Y, Blum JE, Lamarre SG, Phinney WN, Stabler SP, et al. Reduced Shmt2 expression impairs mitochondrial folate accumulation and respiration, and leads to uracil accumulation in mouse mitochondrial DNA. J Nutr. 2021;151:2882–93. [DOI] [PubMed] [Google Scholar]
- 61.Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162:540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. 2015;162:552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schöpf B, Weissensteiner H, Schäfer G, Fazzini F, Charoentong P, Naschberger A, et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat Commun. 2020;11:1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016;23:63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuwahara Y, Tomita K, Roudkenar MH, Roushandeh AM, Urushihara Y, Igarashi K, et al. Decreased mitochondrial membrane potential is an indicator of radioresistant cancer cells. Life Sci. 2021;286:120051. [DOI] [PubMed] [Google Scholar]
- 66.Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol cell. 2021;81:691–707.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sánchez-Castillo A, Heylen E, Hounjet J, Savelkouls KG, Lieuwes NG, Biemans R, et al. Targeting serine/glycine metabolism improves radiotherapy response in non-small cell lung cancer. British Journal of Cancer. 2023:1–17. [DOI] [PMC free article] [PubMed]
- 68.Verschuere T, Toelen J, Maes W, Poirier F, Boon L, Tousseyn T, et al. Glioma‐derived galectin‐1 regulates innate and adaptive antitumor immunity. Int J cancer. 2014;134:873–84. [DOI] [PubMed] [Google Scholar]
- 69.Camby I, Belot N, Lefranc F, Sadeghi N, De Launoit Y, Kaltner H, et al. Galectin-1 modulates human glioblastoma cell migration into the brain through modifications to the actin cytoskeleton and levels of expression of small GTPases. J Neuropathol Exp Neurol. 2002;61:585–96. [DOI] [PubMed] [Google Scholar]
- 70.Toussaint LG, Nilson AE, Goble JM, Ballman KV, James CD, Lefranc F, et al. Galectin-1, a gene preferentially expressed at the tumor margin, promotes glioblastoma cell invasion. Mol cancer. 2012;11:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mercier ML, Mathieu V, Haibe-Kains B, Bontempi G, Mijatovic T, Decaestecker C, et al. Knocking down galectin 1 in human hs683 glioblastoma cells impairs both angiogenesis and endoplasmic reticulum stress responses. J Neuropathol Exp Neurol. 2008;67:456–69. [DOI] [PubMed] [Google Scholar]
- 72.Chou S-Y, Yen S-L, Huang C-C, Huang E-Y. Galectin-1 is a poor prognostic factor in patients with glioblastoma multiforme after radiotherapy. BMC cancer. 2018;18:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang X, Buxbaum JN, Kelly JW. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc Natl Acad Sci USA. 2001;98:14943–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoshioka K, Miki T, Katsuya T, Ogihara T, Sakaki Y. The 717Val→ Ile substitution in amyloid precursor protein is associated with familial Alzheimer’s disease regardless of ethnic groups. Biochemical biophysical Res Commun. 1991;178:1141–6. [DOI] [PubMed] [Google Scholar]
- 75.Dai M-H, Zheng H, Zeng L-D, Zhang Y. The genes associated with early-onset Alzheimer’s disease. Oncotarget. 2018;9:15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qin Q, Yin Y, Wang Y, Lu Y, Tang Y, Jia J. Gene mutations associated with early onset familial Alzheimer’s disease in China: An overview and current status. Mol Genet Genom Med. 2020;8:e1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan CL, Yeo CC, Khoo HE, Poh CL. Replacement of tyrosine 181 by phenylalanine in gentisate 1, 2-dioxygenase I from Pseudomonas alcaligenes NCIMB 9867 enhances catalytic activities. J Bacteriol. 2005;187:7543–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hosios AM, Vander Heiden MG. The redox requirements of proliferating mammalian cells. J Biol Chem. 2018;293:7490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bao XR, Ong S-E, Goldberger O, Peng J, Sharma R, Thompson DA, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. elife. 2016;5:e10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Diehl FF, Lewis CA, Fiske BP, Vander Heiden MG. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat Metab. 2019;1:861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mullen NJ, Singh PK. Nucleotide metabolism: a pan-cancer metabolic dependency. Nat Rev Cancer. 2023;23:275–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer cell. 2017;32:71–87.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brown KK, Spinelli JB, Asara JM, Toker A. Adaptive reprogramming of de novo pyrimidine synthesis is a metabolic vulnerability in triple-negative breast cancer. Cancer Discov. 2017;7:391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou W, Yao Y, Scott AJ, Wilder-Romans K, Dresser JJ, Werner CK, et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat Commun. 2020;11:3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun. 2015;6:7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–86. [DOI] [PubMed] [Google Scholar]
- 87.Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling. Int J Mol Med. 2019;44:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Junttila MR, De Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–54. [DOI] [PubMed] [Google Scholar]
- 89.Lin C-J, Robert F, Sukarieh R, Michnick S, Pelletier J. The antidepressant sertraline inhibits translation initiation by curtailing mammalian target of rapamycin signaling. Cancer Res. 2010;70:3199–208. [DOI] [PubMed] [Google Scholar]
- 90.Jiang X, Lu W, Shen X, Wang Q, Lv J, Liu M, et al. Repurposing sertraline sensitizes non–small cell lung cancer cells to erlotinib by inducing autophagy. JCI insight. 2018;3. [DOI] [PMC free article] [PubMed]
- 91.Hwang H-Y, Shim JS, Kim D, Kwon HJ. Antidepressant drug sertraline modulates AMPK-MTOR signaling-mediated autophagy via targeting mitochondrial VDAC1 protein. autophagy. 2021;17:2783–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Magaway C, Kim E, Jacinto E. Targeting mTOR and metabolism in cancer: lessons and innovations. Cells. 2019;8:1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morscher RJ, Ducker GS, Li SH-J, Mayer JA, Gitai Z, Sperl W, et al. Mitochondrial translation requires folate-dependent tRNA methylation. Nature. 2018;554:128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol. 2018;20:775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Elliott IA, Dann AM, Xu S, Kim SS, Abt ER, Kim W, et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc Natl Acad Sci USA. 2019;116:6842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eloranta K, Cairo S, Liljeström E, Soini T, Kyrönlahti A, Judde J-G, et al. Chloroquine triggers cell death and inhibits PARPs in cell models of aggressive hepatoblastoma. Front Oncol. 2020;10:1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cheng C-T, Qi Y, Wang Y-C, Chi KK, Chung Y, Ouyang C, et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun Biol. 2018;1:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Broeks MH, Meijer NW, Westland D, Bosma M, Gerrits J, German HM, et al. The malate-aspartate shuttle is important for de novo serine biosynthesis. Cell Rep. 2023;42:113043. [DOI] [PubMed] [Google Scholar]
- 99.Lehane AM, McDevitt CA, Kirk K, Fidock DA. Degrees of chloroquine resistance in Plasmodium–Is the redox system involved? Int J Parasitology: Drugs Drug Resistance. 2012;2:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kasozi D, Mohring F, Rahlfs S, Meyer AJ, Becker K. Real-time imaging of the intracellular glutathione redox potential in the malaria parasite Plasmodium falciparum. PLoS Pathog. 2013;9:e1003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Read JA, Wilkinson KW, Tranter R, Sessions RB, Brady RL. Chloroquine binds in the cofactor binding site of Plasmodium falciparum lactate dehydrogenase. J Biol Chem. 1999;274:10213–8. [PubMed] [Google Scholar]
- 102.Baker GJ, Chockley P, Yadav VN, Doherty R, Ritt M, Sivaramakrishnan S, et al. Natural killer cells eradicate galectin-1–deficient glioma in the absence of adaptive immunity. Cancer Res. 2014;74:5079–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Verschuere T, De Vleeschouwer S, Lefranc F, Kiss R, Van Gool SW. Galectin-1 and immunotherapy for brain cancer. Expert Rev neurotherapeutics. 2011;11:533–43. [DOI] [PubMed] [Google Scholar]
- 104.Yu X, Qian J, Ding L, Yin S, Zhou L, Zheng S. Galectin-1: A Traditionally Immunosuppressive Protein Displays Context-Dependent Capacities. Int J Mol Sci. 2023;24:6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Van Woensel M, Mathivet T, Wauthoz N, Rosière R, Garg AD, Agostinis P, et al. Sensitization of glioblastoma tumor micro-environment to chemo-and immunotherapy by Galectin-1 intranasal knock-down strategy. Sci Rep. 2017;7:1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grauer OM, Nierkens S, Bennink E, Toonen LW, Boon L, Wesseling P, et al. CD4+ FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J cancer. 2007;121:95–105. [DOI] [PubMed] [Google Scholar]
- 107.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25:462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Western blots are available in the original data file. Publicly available data is mentioned in the materials and methods section and/or indicated in the figure legend. Data is available from the corresponding author on reasonable request.