

Abstract
Objectives
This study aimed to identify plasma proteomic signatures that differentiate active and inactive giant cell arteritis (GCA) from non-disease controls. By comprehensively profiling the plasma proteome of both patients with GCA and controls, we aimed to identify plasma proteins that (1) distinguish patients from controls and (2) associate with disease activity in GCA.
Methods
Plasma samples were obtained from 30 patients with GCA in a multi-institutional, prospective longitudinal study: one captured during active disease and another while in clinical remission. Samples from 30 age-matched/sex-matched/race-matched non-disease controls were also collected. A high-throughput, aptamer-based proteomics assay, which examines over 7000 protein features, was used to generate plasma proteome profiles from study participants.
Results
After adjusting for potential confounders, we identified 537 proteins differentially abundant between active GCA and controls, and 781 between inactive GCA and controls. These proteins suggest distinct immune responses, metabolic pathways and potentially novel physiological processes involved in each disease state. Additionally, we found 16 proteins associated with disease activity in patients with active GCA. Random forest models trained on the plasma proteome profiles accurately differentiated active and inactive GCA groups from controls (95.0% and 98.3% in 10-fold cross-validation, respectively). However, plasma proteins alone provided limited ability to distinguish between active and inactive disease states within the same patients.
Conclusions
This comprehensive analysis of the plasma proteome in GCA suggests that blood protein signatures integrated with machine learning hold promise for discovering multiplex biomarkers for GCA.
INTRODUCTION
Early in its disease course, giant cell arteritis (GCA) can cause irreversible vision loss and cerebrovascular events,1 highlighting the critical need for prompt diagnosis and treatment. Although diagnostic modalities such as temporal artery biopsy, ultrasonography, and other imaging techniques are available, there is still no specific laboratory marker for this disease.1 To mitigate the current challenge associated with the lack of a diagnostic biomarker for GCA, researchers have evaluated conventional acute phase markers, such as erythrocyte sedimentation rate (ESR) and C reactive protein (CRP), for their potential clinical applicability in GCA diagnosis. Notably, Kermani et al investigated the diagnostic performance of CRP and ESR for biopsy-confirmed cranial GCA.2 Their findings revealed that ESR and CRP, though sensitive at 84.2% and 86.4%, respectively, were limited by low specificity, at 29.5% and 30.5%, respectively. These results emphasise the need for more definitive and precise biomarkers in clinical practice.
Going beyond the scope of traditional acute phase markers, several recent studies have turned to blood proteins to identify indicators of disease. A study by van der Geest et al compared serum protein abundances between patients with GCA and controls, identifying seven proteins with significantly altered abundances.3 Similarly, Burja et al compared 65 immunological parameters between treatment-naïve patients with GCA and controls.4 The investigators found 27 proteins that were differentially abundant in the plasma of patients with GCA, identifying elevated levels of IFN-γ, IL-2, TNF-α and others. Another study by Wadström et al identified eight plasma proteins (CCL25, CD40, CXCL9, IL-2, IL10RB, IFN-γ, MCP3 and SCF) that were more abundant in individuals who eventually developed GCA.5
However, to date, studies have been limited to profiling a relatively small number of proteins, which might obscure the identification of other critical proteins or proteomic patterns that could serve as key markers for GCA. Therefore, comprehensive studies are essential to fully elucidate the diagnostic potential of plasma proteins. A recent study examined 1463 proteins in serum samples from 16 patients with new-onset GCA and 13 patients in remission.6 Through clustering analysis, the researchers identified distinct patient subgroups characterised by specific proteomic signatures and disease phenotypes.
While recent discoveries regarding plasma proteins advanced our understanding of the biomolecular hallmarks of GCA, the clinical transition from active disease to remission, and the potential recurrence of disease, remains complex and necessitates further investigation. Patients typically exhibit a favourable response to glucocorticoid therapy, with rapid symptom relief and a high rate of remission.7 8 Nonetheless, 20%–70% of patients experience disease relapse.9-12 Although acute phase markers have some utility in the assessment of disease activity, the lack of specificity is a significant limitation. Additionally, patients may exhibit smouldering inflammatory activity despite normal marker levels. Elevated cytokine levels have been reported in patients with GCA in clinical remission, and these increased levels were found to be associated with a higher risk of relapse.9 It remains uncertain whether other blood proteins persist at high levels in clinically quiescent disease. Consequently, compared with non-disease controls, a comprehensive analysis of the global blood proteome in patients with GCA (including those in later clinical remission) would represent a significant advancement in GCA research. Here, we present a detailed plasma proteome analysis in patients with GCA, aiming to identify proteins that differentiate active GCA, remission and control groups. This initial GCA versus control comparative analysis establishes a crucial starting point for future plasma proteomics studies to distinguish GCA from other inflammatory conditions.
METHODS
Patient enrolment, eligibility criteria and sample collection
The study population involved patients diagnosed with GCA from the multicentre, observational Vasculitis Clinical Research Consortium (VCRC) Longitudinal Study of GCA. Patients enrolled were over the age of 50 years and met the 1990 American College of Rheumatology classification criteria for GCA13 or had evidence of large-vessel vasculitis (LVV) by angiogram or biopsy. Data collected included demographics, medical history and measures of disease activity. Age-matched, sex-matched and race-matched non-disease individuals (controls) from the Mayo Clinic Biobank were also recruited for the study. None of the controls had autoimmune or inflammatory disease at the time of sample collection.
Plasma samples from 30 patients with GCA at two visits were included in this study. Samples were collected from patients between 2008 and 2018 (median (IQR): 2012 (2010, 2014)). The initial sample was drawn during the active stage of the disease; the second sample was taken when the patient was in clinical remission. For the purposes of this study, patients with GCA were ‘active’ if they had a Physician Global Assessment (PGA) score of 2 or higher at the study visit, as determined by the VCRC investigator. The PGA score was determined by clinical assessment while considering clinically available markers (ESR, CRP). The scale ranges from 0 (no disease activity) to 10 (maximum disease activity). There were no other disease characteristics or features for which the patients were selected. For the second sample, participants were required to have a PGA score of 0, an ESR of less than 30 mm/hour and a CRP level below 10 mg/L. (See online supplemental information for details on CRP reference ranges at each study site.) This sample was collected within 2.5–6.5 months after the initial sample. If multiple samples met these criteria, the sample collected closest in time to the initial sample was selected. If a participant had more than one qualifying paired sample, the pair with the highest PGA score at the initial sample was chosen. All plasma samples were stored in tubes containing EDTA at −80°C prior to use.
Plasma proteome profiling
The SomaScan Assay version 4 from SomaLogic was used to quantify over 7000 human proteins in 55 μL of plasma. This platform uses protein-capture SOMAmer (Slow Off-rate Modified Aptamer) reagents.14 15 Briefly, SOMAmers are single-stranded, chemically modified nucleic acids designed for high affinity, slow off-rate and high specificity to target proteins. This multiplexed, aptamer-based assay measures the relative binding of target proteins to aptamers. After protein concentrations were converted into corresponding DNA aptamer concentrations, protein abundance levels were quantified with a DNA microarray in relative fluorescence units (RFUs). Details regarding data standardisation, including normalisation and calibration, can be found in online supplemental information. A total of 90 plasma samples were profiled using the SomaScan assay, and protein abundance RFUs were quantile normalised prior to all statistical analyses.
Identification of differentially abundant proteins
Multiple linear regression models (MLRMs) were used to identify differentially abundant proteins between two study groups. Each MLRM aimed to characterise the association between protein abundance and the study group while concurrently adjusting for potential confounding factors. Preliminary steps in identifying potential confounders involved the construction of univariate (marginal) linear regression models for each protein, designating protein abundance as the response variable and one of six demographic or clinical features (ie, age, smoking status, sex, prednisone, methotrexate and aspirin use) as the predictor variable. Confounders were deemed significant if the p value of the predictor’s coefficient in the linear model was less than 0.05. After identifying significant confounders, MLRMs were formulated with protein abundance as the response variable, and the study group (eg, active GCA and controls) and any significant confounders as predictors. For example, if age and smoking status were both found to be significantly associated with the abundance of a certain protein, the corresponding MLRM would incorporate these variables alongside the study group as predictors. The significance of the association between protein abundance and the study group was determined by examining the p value of the study group’s coefficient in the MLRM, with a threshold set at p<0.01.
Additionally, mixed-effects linear regression models (MeLRMs) were used to identify differentially abundant proteins between active and inactive visits within patients with GCA while adjusting for significant confounders and intrasubject longitudinal variation. Significant confounders were identified in the same manner as described above. MeLRMs were then constructed with protein abundance as the response variable, the study group and all identified significant confounders as fixed-effect predictors and patient ID as the random-effect variable.
For these analyses, a protein was considered to be significantly associated with the study group, indicating differential abundance between the two phenotypes, if its p value met the significance threshold (p<0.01).
Identification of plasma proteins correlated with PGA and CRP
Spearman’s rank correlation coefficient (ρ) was calculated to quantify the correlation between protein abundance and PGA or CRP (mg/dL) in patients with active GCA patients. Additionally, MLRMs were used to identify proteins associated with PGA or CRP Following the protocol for identifying significant confounders, linear models were constructed with protein abundance as the response variable, and either PGA or CRP along with any of the identified significant confounders, as the predictor variables. The linear models provided the p value of the coefficient for PGA or CRP Proteins were classified as correlated with PGA or CRP if they met two criteria: (1) an absolute Spearman’s ρ (or |ρ|) exceeding 0.4 and (2) a p<0.05 from the coefficient in the linear regression model.
Leave-one-patient-out cross-validation
The active versus inactive GCA classification scenario involves the same individuals at different disease stages. This scenario requires a cross-validation technique that controls for patient identity and ensures the model does not identify patient-specific variations rather than disease-related features. Therefore, classification accuracy between active and inactive GCA was assessed through ‘leave-one-patient-out’ cross-validation. In this method, patient-matched active and inactive GCA samples served as the test set (2 samples per iteration) while the remaining samples formed the training set (58 samples per iteration). This process was iteratively applied across all unique patients. This approach effectively controls for intrasubject similarities by leveraging repeated measurements from the same patient, ensuring that the model’s performance is assessed on truly independent data points.
Feature selection using linear regression modelling
Feature selection was performed within each training fold of the cross-validation process prior to training a random forest classifier. This process involved constructing univariate linear regression models to identify the significance of the association between protein abundance and study group. The linear regression model treated protein abundance as the response variable and the study group as the predictor. Proteins were rank-ordered according to the product of their –log10(p value) and log2(fold-change) in descending order. The p value corresponds to the coefficient associated with the study group in the linear regression model; and the fold-change was defined as the ratio of mean abundance in GCA patients to that in controls, thus prioritising proteins with higher abundance in GCA for the top set of features. To examine how the number of input proteins affects the classifier’s performance, varying numbers of features (ranging from the top 10–250 significant proteins) were selected to be included in the model.
RESULTS
Participant characteristics
The study cohort included 30 patients with GCA and 30 age- matched/sex-matched/race-matched non-disease controls. Of the 60 study participants, 76.7% (46 of 60) were female and the majority were white (table 1). For patients with GCA, the two visits were spaced an average of 123.2 days apart (figure 1A). Patients with active GCA had mean CRP levels and PGA scores of 3.2 mg/dL (SD=2.6) and 4.4 (SD=2.1), respectively (table 1). Patients in clinical remission had mean CRP levels and PGA scores of 0.5 mg/dL (SD=0.3) and 0.0 (SD=0.0), respectively. Among 30 patients with active GCA, 24 (or 80%) were in the early disease course (ie, within 1 year of initial diagnosis) with a mean disease duration of 7.6 months. Notably, 9 (or 30%) were confirmed to have LVV by angiogram or biopsy at baseline while the remaining 21 had cranial GCA. Table 1 and online supplemental table 1 describe the study participants’ demographic and clinical characteristics, including race, smoking status (ie, smokers or non-smokers), body mass index and medication use.
Table 1.
Demographic and clinical characteristics of study participants
| Characteristic | Active GCA (n=30) |
Inactive GCA (n=30) |
Non-disease controls (n=30) |
|---|---|---|---|
| Age (years) | |||
| Mean±SD | 70.5±9.3 | 70.9±9.3 | 70.0±7.8 |
| IQR† (Q1, Q3) | (63.7, 77.2) | (64.1, 70.9) | (65.3, 72.8) |
| Sex | |||
| Female, n (%) | 23 (76.7) | 23 (76.7) | 23 (76.7) |
| Male, n (%) | 7 (23.3) | 7 (23.3) | 7 (23.3) |
| Race | |||
| White, n (%) | 29 (96.7) | 29 (96.7) | 30 (100.0) |
| Asian, n (%) | 1 (3.3) | 1 (3.3) | 0 (0.0) |
| Smokers, n (%) | 17 (56.7) | 17 (56.7) | 10 (33.3) |
| Unknown, n | 0 | 0 | 1 |
| Body mass index | |||
| Mean±SD | 27.2±5.6 | 27.6±6.0 | 24.2±5.2 |
| IQR (Q1, Q3) | (23.1, 31.0) | (22.7, 31.1) | (20.6, 27.5) |
| Unknown, n | 4 | 5 | 1 |
| C reactive protein (mg/dL) | |||
| Mean±SD | 3.2±2.6 | 0.5±0.3 | N/A |
| IQR (Q1, Q3) | (1.4, 3.7) | (0.3, 0.8) | |
| Disease stage in active GCA group | |||
| Early disease course§, n (%) | 24 (80) | ||
| Disease duration (months) | N/A | N/A | |
| Mean±SD | 7.6±11.9 | ||
| IQR (Q1, Q3) | (0.4, 10.8) | ||
| Flare history, n (%) | 9 (27.3) | ||
| Physician global assessment | |||
| Mean±SD | 4.4±2.1 | 0.0±0.0 | N/A |
| Treatment | |||
| Prednisone, n (%) | 22 (73.3) | 21 (70.0) | N/A |
| Prednisone dose (mg/day) | |||
| Median | 10 | 10 | |
| IQR | (2.75–26) | (8.75–10.0) | |
| Unknown, n | 11 | 9 | |
| Aspirin/clopidogrel, n (%) | 19 (63.3) | 17 (56.7) | |
| Aspirin low dose (mg/day) | 325.0 | 325.0 | |
| Abatacept/lefluomide/ methotrexate, n (%) | 5 (16.7) | 9 (30.0) | |
| Statin, n (%) | 4 (13.3) | 5 (16.7) | |
| Oestrogen, n (%) | 1 (3.3) | 0 (0.0) |
IQR, interquartile range.
Within 1 year of initial GCA diagnosis.
GCA, giant cell arteritis; N/A, not applicable.
Figure 1.
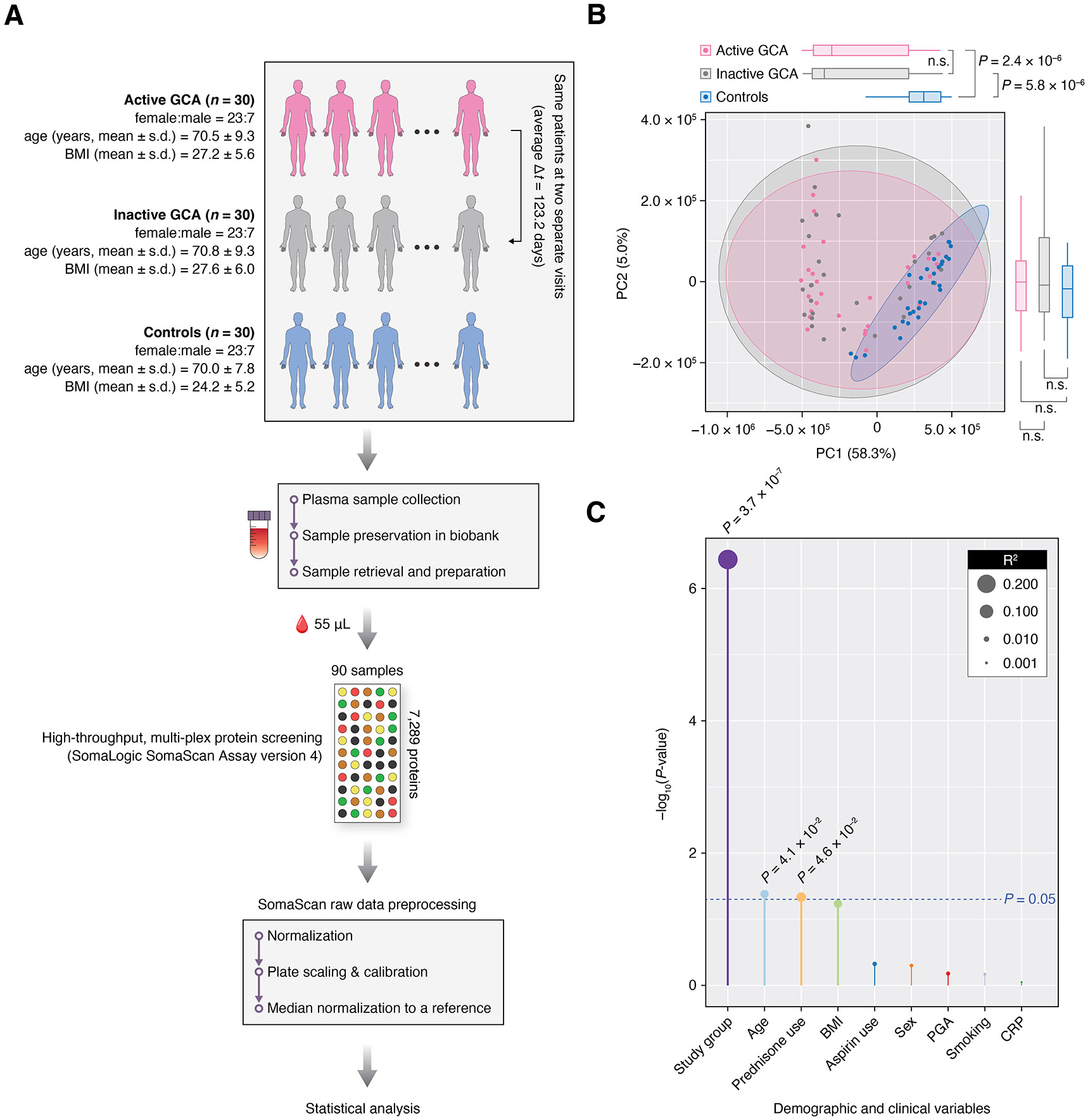
Analysis of plasma proteome profiles in patients with giant cell arteritis (GCA) and controls. (A) Study design overview: Plasma samples were collected from patients with GCA in two instances: during an active disease state (active GCA group, n=30) and while in clinical remission (inactive GCA group, n=30). Concurrently, plasma samples from controls (n=30) were also collected. Proteome profiling of 7289 proteins in all 90 samples was performed using the SomaScan Assay version 4 (SomaLogic), and these profiles were subsequently used in downstream statistical analyses. (B) Principal component analysis (PCA) on the plasma proteome profiles, where each point on the scatter plot represents an individual sample. Box-and-whisker plots displayed on the top and to the right of the plot represent the distribution of principal component 1 (PC1) and principal component 2 (PC2) values, respectively. (C) Clinical and demographic factors associated with PC1 values. Bar height represents the –log10 transformation of the p value of the association, with the significance threshold indicated in blue (p=0.05). Additionally, the size of the data points on the plot corresponds to the R2 value from each linear regression model. BMI, body mass index; PGA, physician global assessment; CRP, C reactive protein.
Plasma proteome profiles differ between GCA groups and controls
Figure 1B displays a principal component analysis (PCA) plot of all 90 plasma proteome profiles (online supplemental table 2) projected onto the first two principal components (PC1 and PC2). By reducing the high dimensionality of the dataset, each PC, representing a linear combination of a set of proteins, captures a significant amount of variance in protein abundances. Specifically, PC1 and PC2 together account for 63.3% of the total variance in the dataset. Notably, the PCA plot shows a clear separation between the GCA groups (active and inactive) and controls along PC1 (p<0.05, Mann-Whitney U test). However, no significant difference was observed between the active and inactive GCA groups along either PC1 or PC2 (p≥0.05, Wilcoxon signed-rank test), suggesting that differences between these disease states may be subtle and involve a relatively small set of proteins. As shown in figure 1C, the first PC was mainly driven by the study group (ie, active GCA, inactive GCA, controls) while the age of participants and prednisone use (by patients with GCA only) were also significantly associated with PC1 (p<0.05, univariate linear regression models).
Plasma protein differences in active and inactive GCA compared with controls
We identified a total of 537 plasma proteins that were significantly different between the active GCA group and controls (p<0.01, MLRM; figure 2A-C and online supplemental table 3). Among these, 202 proteins, including CCL14, CD34, CD59, CD8A, IL17C, IL1RL1, IL5, IL31, MMP19 and TNFRSF1A (designations for all protein symbols are provided in online supplemental information), were more abundant in patients with active GCA than in controls. Notably, 7 of the 202 proteins (CD206, endostatin, IL-31, PRTN3, PTX3, TNF R1 and VCAM-1) had previously been reported to be more abundant in the blood of active GCA patients than controls (online supplemental table 4) while the rest are reported here for the first time using a much higher throughput assay. Conversely, 335 proteins, such as CDK2, IFNG, IFNL1, STAT3, S100A4 and S100A6, were less abundant in patients with active GCA. Between the nine LVV and 21 cranial GCA (CGCA) patients, only 32 proteins were found to be differentially abundant during the active disease state: 25 proteins were more abundant in LVV and 7 proteins were more abundant in CGCA (online supplemental table 5).
Figure 2.
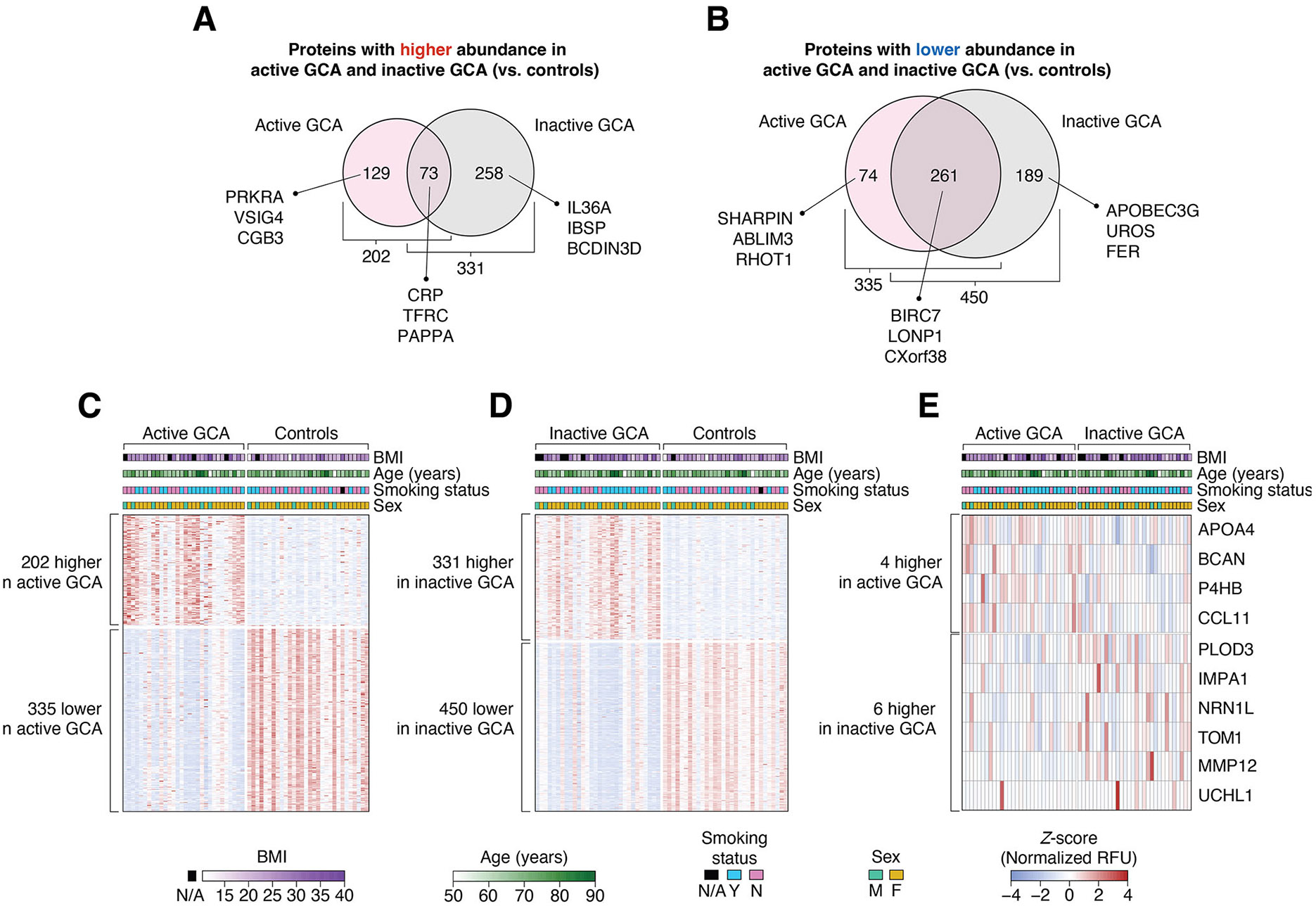
Differentially abundant plasma proteins between study groups. (A, B) Of the 7289 measured plasma proteins, 984 showed differential abundance between GCA groups (ie, active or inactive GCA) and controls. In active GCA, the abundance of 202 and 335 proteins were higher and lower compared with controls, respectively. Conversely, in inactive GCA, the abundance of 331 and 450 proteins were higher and lower than in controls, respectively. Heatmaps showing the Z-score-normalised relative fluorescence unit (RFU) abundances of differentially abundant proteins between (C) active GCA and controls (537 proteins); (D) inactive GCA and controls (781 proteins) and (E) active and inactive GCA (10 proteins). In each heatmap, the columns represent study participants while the rows correspond to proteins. Demographic variables (BMI, age, smoking status, and sex) of each study subject are shown at the top of each heatmap. BMI, body mass index; GCA, giant cell arteritis.
Our analysis also revealed 781 plasma proteins differentially abundant between patients of the inactive GCA group and controls (p<0.01, MLRM; figure 2A,B and D and online supplemental table 6). Of these, 331 proteins, including CCL1, CCL18, CCL23, CD14, CD8A, CXCL16, CXCL9, IL12RB1, IL18R1, IL18R1, IL36A, IL5, LILRB5, TLR1 and TNFRSF1B, were significantly higher in the inactive GCA group, while 450 proteins, including BCL2, CD2AP, CD38, CDK2, IFNG, IFNL1, IL2RG, STAT3, S100A4 and S100A6, were of lower abundance in inactive GCA. Notably, 73 of the 202 proteins that were more abundant in active GCA compared with controls, and 261 of the 335 proteins that were less abundant in active GCA compared with controls, remained so even in the inactive disease state (figure 2A,B). This suggests that certain blood proteins maintain altered levels even during remission of GCA.
To further address potential confounding effects of immunosuppressive medications, we performed an additional analysis restricted to the seven inactive subjects with GCA who were not taking any immunosuppressants (abatacept, leflunomide, methotrexate or prednisone) at the time of sample collection. We found 551 and 414 proteins that were significantly higher and lower, respectively, in patients in remission and off treatment (online supplemental table 7). Notably, 165 and 312 of these proteins displayed directional changes consistent with those observed in the analysis that included all inactive subjects with GCA (n=30). These overlapping proteins, despite arising from a smaller sample size comparison, may provide greater confidence in identifying truly differentially abundant proteins in patients in remission, as they are less likely to be confounded by treatment effects.
In addition to the differences observed between the GCA groups and controls, we identified 10 plasma proteins that showed significant differences in abundance between the active and inactive GCA groups (p<0.01, MeLRM; figure 2E and online supplemental table 8). Specifically, four proteins (APOA4, BCAN, P4HB and CCL11) significantly decreased on remission. In contrast, six proteins (PLOD3, IMPA1, NRN1L, TOM1, MMP12 and UCHL1) significantly increased in abundance when patients transitioned from active GCA to remission.
We performed an additional analysis focusing on patients who were taking immunosuppressive medications (abatacept, leflunomide, methotrexate or prednisone) at both their active disease and remission visits (n=18). Using the same statistical approach as previously described for all patients (n=30), we found nine proteins (activated protein C, CA056, CLC4G, CYTF, HMGN1, IGFBP-6, Met, protein C and TFPI) that were significantly higher and one protein (SOM2) that was significantly lower in patients with active GCA compared with those in remission (online supplemental table 9). Notably, none of these proteins overlapped with those identified in the analysis that included all subjects with GCA, despite adjusting for medication use in the statistical models. This finding underscores the complexity of disentangling disease activity from treatment effects and highlights the importance of considering medication status in blood proteomic studies in GCA.
Functional enrichment analysis of differentially abundant proteins
Having identified differentially abundant plasma proteins, we next investigated the biological processes associated with these proteins through gene ontology (GO) enrichment analysis. Using the DAVID online tool,16 we identified 193 significantly enriched (ie, over-represented) GO terms associated with proteins higher in the active GCA group compared with controls (p<0.01, modified one-tailed Fisher’s exact test; online supplemental table 10); and 357 GO terms enriched in proteins higher in the inactive GCA group (online supplemental table 11). Figure 3A,B illustrates the top 20 enriched biological processes for proteins more abundant in active and inactive GCA groups, respectively, compared with controls. Most of these enriched GO terms were found to be related to immune processes and inflammation, including ‘humoral immune response’, ‘response to bacterium’ and ‘inflammatory response’. Altogether, plasma proteins enriched in immune functions were elevated in patients with active GCA and remained elevated even after patients achieved remission.
Figure 3.
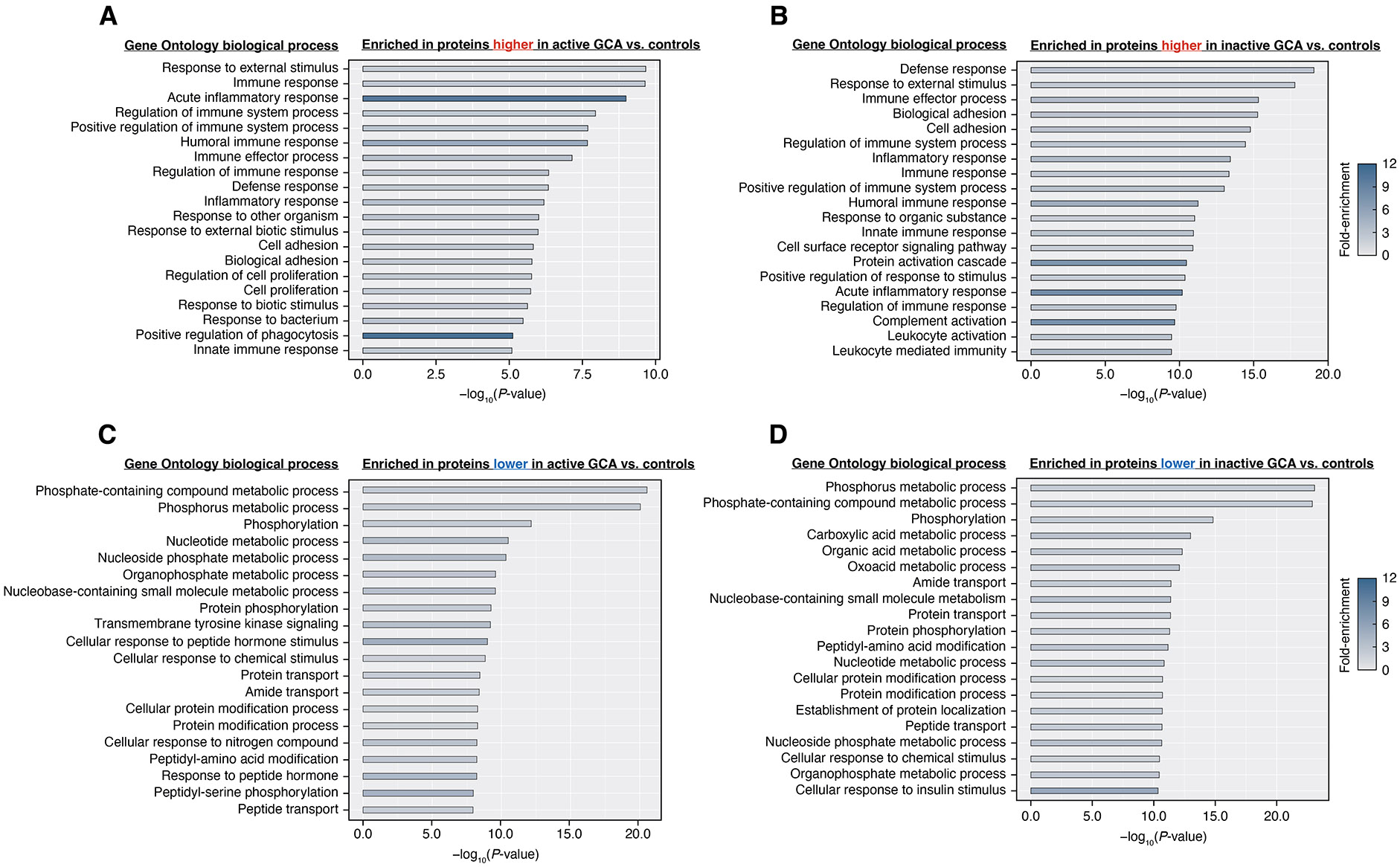
Gene ontology (GO) enrichment analysis of differentially abundant plasma proteins in giant cell arteritis. (A, B) Top 20 enriched GO biological processes of proteins with significantly higher abundance in active (or inactive) GCA compared with controls. Proteins with higher abundance in either GCA group are mostly linked to immune processes. (C, D) Top 20 enriched biological processes of proteins with significantly lower abundance in active (or inactive) GCA than in controls. Most of these proteins were enriched in functions related to metabolism, cell signalling and transport. GO biological processes (located to the left of each bar graph) are arranged in descending order based on significance, represented by the –log10 of the p values obtained from a modified one-tailed Fisher’s exact test. Longer bars indicate greater significance while shading illustrates fold-enrichment. GCA, giant cell arteritis.
Additionally, we found a total of 432 (online supplemental table 12) and 578 (online supplemental table 13) enriched GO terms in proteins significantly lower in patients with active and inactive GCA compared with controls, respectively. Figure 3C,D displays the top 20 enriched biological processes for proteins less abundant in the active and inactive GCA groups compared with controls. Many of these GO terms were linked to metabolism, including ‘phosphorous metabolic process’, ‘carboxylic acid metabolic process’ and ‘nucleotide metabolic process’ and also to biological processes involved in cell signalling and biomolecular transport.
Correlation of plasma proteins with PGA and CRP in patients with active GCA
To determine whether plasma proteins reflect disease activity in active GCA, we examined the correlation of their abundances with PGA. We found 16 proteins with significant correlations (|Spearman’s ρ|>0.4 and p<0.05 in MLRMs; figure 4A and online supplemental table 14): 5 (including IHGA1 and NUMBL) showed positive correlations with PGA while 11 (including FAM171B, HS6ST2 and COLEC12) displayed negative correlations. Notably, only one protein (LAMA3) overlapped between the sets of proteins differentially abundant in active GCA versus controls and those correlating with PGA. These findings suggest that LAMA3 may serve as a potential biomarker for disease activity in GCA.
Figure 4.
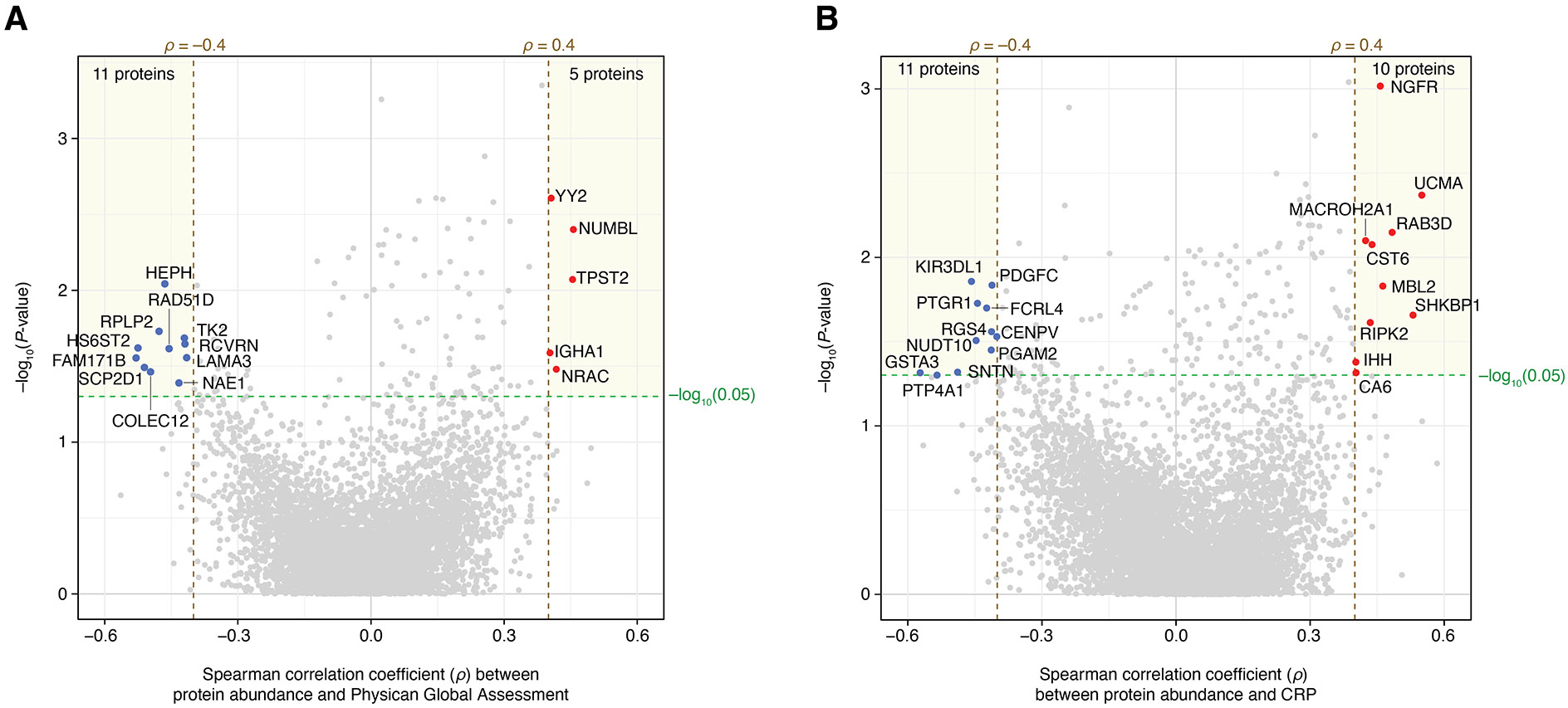
Plasma protein correlations with physician global assessment (PGA) and C reactive protein (CRP) in patients with active giant cell arteritis. (A) Five proteins displayed positive correlations with PGA scores (Spearman’s ρ>0.4 and p<0.05 in MLRMs), whereas 11 proteins exhibited negative correlations (Spearman’s ρ<−0.4 and p<0.05 in MLRMs). (B) 10 proteins were positively correlated with blood CRP levels (measured in mg/dL) while 11 proteins showed negative correlations. MLRMs, multiple linear regression models.
Next, to evaluate whether plasma proteins in active GCA reflect inflammatory conditions, we analysed the correlation of their abundances with CRP. We identified 21 proteins significantly correlated with CRP (|Spearman’s ρ|>0.4 and p<0.05 in MLRMs), with 10 (including NGFR, RAB3D and RIPK2) and 11 (including FCRL4, KIR3DL1, PDGFC, PTP4A1 and PGAM2) exhibiting positive and negative correlations, respectively (figure 4B and online supplemental table 15). There was no overlap between the proteins significantly correlated with CRP and those differentially abundant in the active GCA versus control comparison. Moreover, no proteins overlapped between those correlated with either PGA or CRP and the proteins differentially abundant between active and inactive GCA states.
Plasma proteome profiles with machine learning distinguish patients with GCA from controls
For our final analysis, we developed two random forest classification models: one using plasma proteome profiles from patients with active GCA and controls; and the other using profiles from patients with inactive GCA and controls. These models were tasked with classifying participants as either active GCA versus controls; or inactive GCA versus controls. In 10-fold cross-validation, these approaches achieved a classification accuracy of 95.0% and 98.3% in distinguishing active and inactive GCA groups from controls, respectively (table 2). Moreover, the models achieved very high (≥90.0%) specificity, sensitivity, positive predictive value and negative predictive value, as detailed in table 2.
Table 2.
Random forests classification metrics in cross-validation
| Classification task* | Validation method | Accuracy | Specificity | Sensitivity | PPV | NPV |
|---|---|---|---|---|---|---|
| Active GCA versus controls | 10-fold CV | 95.0% | 96.7% | 93.3% | 96.7% | 93.5% |
| Inactive GCA versus controls | 10-fold CV | 98.3% | 100% | 96.7% | 100% | 96.8% |
| Active GCA versus inactive GCA | Leave-one-patient-out | 51.7% | 46.7% | 56.7% | 51.5% | 51.9% |
Classification tasks were performed using the default configurations of the random forest classifier from the sci-kit learn library (V.1.3.2) in Python. Classification metrics were calculated as the ratio of total correct predictions to total predictions, as defined for each metric, across each fold of cross-validation method.
GCA, giant cell arteritis; NPV, negative predictive value; PPV, positive predictive value.
We next addressed the question of the minimal set of plasma proteins necessary to effectively differentiate between the GCA patient groups from controls. For this, we used linear regression models for feature selection within each fold of cross-validation (see the ‘Methods’ section). We conducted multiple instances of 10-fold cross-validation to thoroughly evaluate the impact of varying the size of the feature set (of the most significant proteins) on model performance. We found that classification accuracy remained relatively stable across a wide range of feature set sizes (figure 5). Interestingly, as few as 10 plasma proteins were sufficient to distinguish active and inactive GCA groups from controls with accuracies of approximately 95.0% and 98.3%, respectively.
Figure 5.
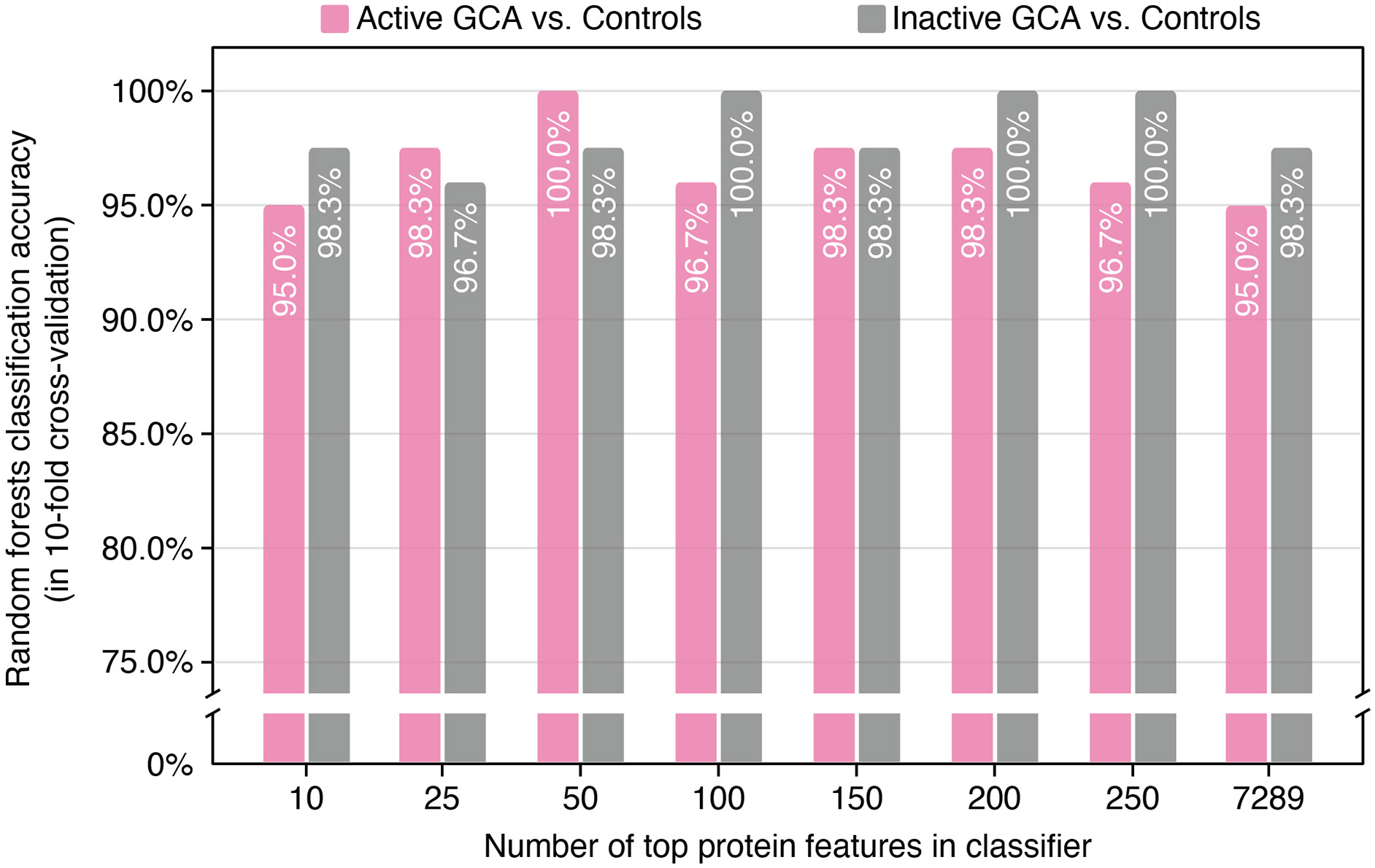
Classification accuracy of random forest classifiers with varying sizes of input feature sets. Pink bars indicate the accuracy in classifying active giant cell arteritis (GCA) versus controls, and grey represent accuracy in classifying inactive GCA versus controls. This process involved preselecting a specified number of input plasma protein features. Different numbers of top-selected features were chosen in each training set fold, ranging from 10 to 250. These selected features were then applied in a random forest classifier, with accuracy assessed on the test set across all ten folds of cross-validation.
Finally, we investigated the potential of machine learning to classify patients as either having active or inactive GCA based on their plasma proteome profiles. This classification was performed using a ‘leave-one-patient-out’ cross-validation strategy, which accounts for the presence of multiple plasma samples from different visits for the same patient (see the ‘Methods’ section). Using this approach, the accuracy of distinguishing between active and inactive GCA based on plasma proteome profiles was 51.7% (table 2).
DISCUSSION
While several studies have identified key inflammatory molecules as potential biomarkers for GCA,3-5 the exploration of the blood proteome could provide additional options for disease assessment. Furthermore, the distinction between plasma protein profiles of patients with GCA while active and in remission, compared with control groups, remains largely unknown. In the current study, we performed proteomic profiling of plasma from patients with GCA enrolled in a prospective longitudinal cohort study, and non-disease controls, to identify signature proteins and their associated functions. Our study design uses a longitudinal approach, following the same patients over two time points as their disease activity transitions from active to inactive. This within-patient design offers advantages in controlling for certain confounding factors, especially individual genetic variations. We also identified proteins linked to PGA and CRP in active GCA. Additionally, we present a proof-of-concept machine-learning strategy that could potentially enhance diagnostic accuracy for GCA. Promisingly, our random forest-based machine-learning models were able to distinguish active GCA from controls, and inactive GCA from controls, with a classification accuracy exceeding 90%, even when limited to a small number of proteins. We share the plasma proteome profiles from our study (online supplemental table 2) with the scientific community to encourage further research into GCA.
Our study identified immune response proteins that are common to both active and inactive GCA, as well as those that differ between these states. We observed that 23 immune response proteins, including B2M, C9, CD8A, LBP IL5 and TRIL, were more abundantly present in both active and inactive GCA compared with controls. (Designations for all protein symbols are provided in online supplemental information). However, we identified 28 immune response proteins (eg, C8G, CCL14, CFD, CD59, IL1RL1 and PDCD1) that were exclusively more abundant in active GCA (but not in inactive GCA) relative to controls. Interestingly, our analysis revealed that certain coinhibitory receptors that regulate T cell activation, specifically PDCD1,17 were more abundant only in active GCA. The PDCD1 gene was reported to be highly expressed in the arteries of patients with untreated GCA, whereas its expression was nearly absent in healthy vessels.18 This could potentially align with our observation that plasma PDCD1 exhibits higher abundance in active GCA (but not in inactive GCA) compared with controls, potentially reflecting abnormal immune cell activity within the arteries affected by GCA.
Our analysis revealed a higher number of proteins differentiating inactive GCA from controls compared with active GCA. This unexpected finding warrants further investigation. One potential explanation lies in our study design. We used a much larger feature space, encompassing a wider range of proteins than previous studies that often focus on inflammatory panels. This broader approach might be more sensitive to subtle proteomic changes associated with inactive GCA. For example, we identified 51 immune response proteins (eg, CCL3, CFB, CXCL3, CXCL9, CCL23, HLA-G and IL18R1) that were exclusively elevated in inactive GCA (compared with both active GCA and controls). Intriguingly, these proteins differentiating inactive GCA could point towards previously unexplored areas of human physiology, which were potentially influenced by medications, the time elapsed since the active state or other factors. Notably, complement proteins unique to inactive GCA are components of pathways that are upstream and downstream of complement activation, including classic and lectin pathways (C1RL, C1S, C2 and C4BPA), regulatory proteins (CR1, CFI and CLU), the alternative pathway (CFB, CFHR1 and CFHR5) and the terminal complement cascade (C5 and C6). Conversely, complement proteins unique to active GCA were related to lectin pathway (MASP1), alternative pathway (CFD), regulatory protein (CD59) and terminal complement cascade (C8G). Of note, both C3 and C9 were higher in active GCA and inactive GCA compared with controls. Although we found a higher number of complement proteins involved in inactive GCA, further investigation is necessary to address whether the different landscape of complement proteins between active and inactive GCA is mainly a consequence of treatment19 or a unique characteristic of the inactive state of GCA.
We identified a substantial number of plasma proteins associated with GCA in both active (537 proteins) and inactive states (781 proteins) compared with controls. However, a direct comparison between active and inactive GCA revealed only 10 proteins showing significant differences. Within this small set, a notable finding was matrix metallopeptidase 12 (MMP12), which may hold potential in distinguishing inactive GCA from active GCA. We observed a higher abundance of MMP12 in inactive GCA compared with both controls (p=0.010) and active GCA (p=0.003), whereas its levels in active GCA did not significantly differ from controls. This specific association of MMP12 with inactive GCA is a novel discovery, contrasting with previous research which did not identify MMP12 gene expression in ascending aortic tissues as a distinguishing factor between inflammatory aneurysms (including GCA and clinically isolated aortitis) and noninflammatory aneurysms.20 Similarly, monocytes/macrophages in blood did not exhibit gene expression differences in MMP12 between patients with GCA and healthy individuals.21 Our results, therefore, suggest that the plasma protein MMP12 may warrant experimental validation as an indicator of the remission state of GCA.
We acknowledge several limitations of our study that could influence the scope and applicability of our results. First and foremost, the patients who participated in this study were not treatment-naïve. As the rapid systemic effects of glucocorticoids can impact the plasma proteome profile, disease state comparisons should ideally be conducted on patients who have not undergone therapy. Furthermore, the remaining effects of prior treatments and the different nature of GCA relapses compared with GCA at first presentation need further investigation. However, enrolling untreated patients in our longitudinal database is logistically challenging because suspected GCA patients require immediate treatment to prevent complications such as blindness, even before a confirmed diagnosis. Additionally, only patients with a confirmed diagnosis were enrolled on the longitudinal VCRC study to ensure accurate disease characterisation. Second, while we used cross-validation to assess the performance of our random forest classifiers, the ideal scenario would involve testing the classifiers on an external set to validate the generalisability and robustness of our classification model. Third, the modest sample size curtails our ability to fully encapsulate the diversity of patients with GCA. A larger and more heterogeneous cohort size would allow for a broader characterisation of the GCA patient population. Fourth, potential confounding factors may have influenced our results, such as geographical or cultural biases specific to our cohorts. The inclusion of predominantly white participants from the Midwest region of the USA narrows the generalisability of our findings. Fifth, we chose not to use multiple hypothesis correction methods on our nominal p values because we observed a large loss of significant results after applying the Benjamini-Hochberg correction procedure. The resulting absence may reflect the lack of stark differences in blood proteins between study groups; the modest sample size of our exploratory pilot study; the high dimensionality of the number of proteins being tested (many of which will not be relevant to autoimmune disease); and the lack of independence among individual tests (a key assumption in the Bonferroni and Benjamini-Hochberg procedures). Although we acknowledge that multiple hypothesis correction is crucial for reducing false positives, we opted to avoid possibly overlooking true (yet subtle) differences. Instead, we took steps to minimise potential confounders by carefully selecting our cohorts and adjusting for demographic and medication effects in our statistical models. Sixth, while validating the elevated plasma proteins in GCA would significantly strengthen our work, the experimental confirmation of our key findings falls outside the scope of this investigation. Our primary focus was to establish ‘molecular signatures’,22 or algorithm-based, multiplex biomarkers, in GCA using comprehensive plasma proteome profiling and machine-learning. Validation using common bench techniques (eg, ELISA) would be necessary to make specific claims about individual proteins (eg, identifying novel disease mechanisms or a single definitive clinical marker); however, this is not aligned with our study’s broad, high-throughput approach to identify disease-specific, proteomic patterns which could later be developed into a blood proteomic panel for GCA. Additionally, while both SomaLogic’s platform and ELISA measure protein abundance, they use fundamentally different methodologies, leading to challenges in directly comparing results. Seventh, our study lacks a disease control group, hindering the differentiation of GCA from non-GCA inflammatory conditions. Future studies will aim to identify blood proteins for distinguishing GCA from other inflammatory mimics. Eighth, the low classification accuracy in distinguishing between active and inactive GCA suggests that the complexity and subtlety of proteomic changes in GCA may require more sophisticated machine-learning methods or the integration of other omics technologies to improve classification performance. Last, our analysis was conducted without comprehensive data on comorbid conditions within our patient cohort, including prevalent conditions in older adults such as type 2 diabetes and cardiovascular disease. This omission limits our ability to fully understand how these comorbidities may have influenced the plasma proteomic landscape in GCA.
Despite its limitations, this investigation reflects ongoing efforts to comprehensively characterise the blood proteome of patients with GCA. We anticipate that the exploration of GCA proteomic signatures and their validation in larger patient cohorts holds significant promise for deepening our understanding of the disease, developing new diagnostic approaches and identifying new therapeutic targets.
Supplementary Material
WHAT IS ALREADY KNOWN ON THIS TOPIC
Acute phase inflammatory response markers such as C reactive protein (CRP) and erythrocyte sedimentation rate have been used to evaluate patients with giant cell arteritis (GCA) for both diagnosis and disease monitoring, although with limited specificity. Blood proteins have emerged as a potential source of novel biomarkers for GCA.
WHAT THIS STUDY ADDS
This study provides an extensive examination of plasma proteins in both active and inactive GCA compared with non-disease individuals.We identified plasma proteins newly associated with GCA, as well as those correlated with disease activity and blood CRP levels. Additionally, we identified a machine-learning strategy that uses plasma proteome profiles to differentiate patients with GCA from controls with high sensitivity and specificity.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study used plasma proteome profiling as a promising approach for multiplex biomarker discovery in GCA. The findings support the need for further validation in larger patient cohorts and with disease controls to solidify its clinical applicability.
Acknowledgements
We thank all study participants who volunteered for this study, especially our dear patients. We also thank the Mayo Clinic Division of Rheumatology study coordinators and the Mayo Clinic Center for Individualized Medicine Biobank staff for facilitating plasma sample identification, packaging, and shipment. This work was previously presented at the American College of Rheumatology Convergence 2023 (see reference 23 for the published conference abstract).
Funding
This study was supported in part by the Mayo Clinic Division of Rheumatology and the Mayo Clinic Center for Individualized Medicine. The VCRC has received funding from the National Center for Advancing Translational Science, the National Institute of Arthritis and Musculoskeletal and Skin Diseases (U54 AR057319), the National Center for Research Resources (U54 RR019497). KJW is supported by the John F. Finn MN Arthritis Foundation Professorship.
Footnotes
► Additional supplemental material is published online only. To view, please visit the journal online (https://doi.org/10.1136/ard-2024-225868).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Consent obtained directly from patient(s).
Ethics approval This study involves human participants and was approved by Mayo Clinic Institutional Review Board (no. 05-004050 and no. 08-007049). Participants gave informed consent to participate in the study before taking part.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Source code and datasets used to generate the statistical analyses and machine learning results in this study are available at: https://github.com/cunni319/Plasma_GCA_2024.
REFERENCES
- 1.Pugh D, Karabayas M, Basu N, et al. Large-vessel vasculitis. Nat Rev Dis Primers 2022;7:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kermani TA, Schmidt J, Crowson CS, et al. Utility of erythrocyte sedimentation rate and C-reactive protein for the diagnosis of giant cell arteritis. Semin Arthritis Rheum 2012;41:866–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Geest KSM, Abdulahad WH, Rutgers A, et al. Serum markers associated with disease activity in giant cell arteritis and polymyalgia rheumatica. Rheumatol (Oxford) 2015;54:1397–402. [DOI] [PubMed] [Google Scholar]
- 4.Burja B, Feichtinger J, Lakota K, et al. Utility of serological biomarkers for giant cell arteritis in a large cohort of treatment-naïve patients. Clin Rheumatol 2019;38:317–29. [DOI] [PubMed] [Google Scholar]
- 5.Wadström K, Jacobsson LTH, Mohammad AJ, et al. Analyses of plasma inflammatory proteins reveal biomarkers predictive of subsequent development of giant cell arteritis: a prospective study. Rheumatology (Oxford) 2023;62:2304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zingg F, Ryser FS, Gloor AD, et al. Serum protein profiling reveals distinct patient clusters in giant cell arteritis. Rheumatol (Oxford) 2024;7. [DOI] [PubMed] [Google Scholar]
- 7.Borchers AT, Gershwin ME. Giant cell arteritis: a review of classification, pathophysiology, geoepidemiology and treatment. Autoimmun Rev 2012;11:A544–54. [DOI] [PubMed] [Google Scholar]
- 8.Garvey TD, Koster MJ, Warrington KJ. My treatment approach to giant cell arteritis. Mayo Clin Proc 2021;96:1530–45. [DOI] [PubMed] [Google Scholar]
- 9.García-Martínez A, Hernández-Rodríguez J, Espígol-Frigolé G, et al. Clinical relevance of persistently elevated circulating cytokines (tumor necrosis factor α and interleukin-6) in the long-term followup of patients with giant cell arteritis. Arthritis Care Res (Hoboken) 2010;62:835–41. [DOI] [PubMed] [Google Scholar]
- 10.Hoffman GS, Cid MC, Rendt-Zagar KE, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007;146:621–30. [DOI] [PubMed] [Google Scholar]
- 11.Sugihara T, Hasegawa H, Uchida HA, et al. Associated factors of poor treatment outcomes in patients with giant cell arteritis: clinical implication of large vessel lesions. Arthritis Res Ther 2020;22:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreel L, Betrains A, Molenberghs G, et al. Epidemiology and predictors of relapse in giant cell arteritis: a systematic review and meta-analysis. Joint Bone Spine 2023;90:105494. [DOI] [PubMed] [Google Scholar]
- 13.Hunder GG, Bloch DA, Michel BA, et al. The American college of rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990;33:1122–8. [DOI] [PubMed] [Google Scholar]
- 14.Gold L, Ayers D, Bertino J, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010;5:e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Candia J, Daya GN, Tanaka T, et al. Assessment of variability in the plasma 7k somascan proteomics assay. Sci Rep 2022;12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sherman BT, Hao M, Qiu J, et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021update). Nucleic Acids Res 2022;50:W216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuertes Marraco SA, Neubert NJ, Verdeil G, et al. Inhibitory receptors beyond T cell exhaustion. Front Immunol 2015;6:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Watanabe R, Berry GJ, et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci USA 2017;114:E970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Triggianese P, Conigliaro P, De Martino E, et al. Overview on the link between the complement system and auto-immune articular and pulmonary disease. Open Access Rheumatol 2023;15:65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hur B, Koster MJ, Jang JS, et al. Global transcriptomic profiling identifies differential gene expression signatures between inflammatory and noninflammatory aortic aneurysms. Arthritis Rheumatol 2022;74:1376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe R, Maeda T, Zhang H, et al. MMP (matrix metalloprotease)-9-producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ Res 2018;123:700–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sung J, Wang Y, Chandrasekaran S, et al. Molecular signatures from omics data: from chaos to consensus. Biotechnol J 2012;7:946–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Identification of giant cell arteritis using plasma proteome profiles integrated with machine learning. ACR Meeting Abstracts; 2023. Available: https://acrabstracts.org/abstract/identification-of-giant-cell-arteritis-using-plasma-proteome-profiles-integrated-with-machine-learning/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source code and datasets used to generate the statistical analyses and machine learning results in this study are available at: https://github.com/cunni319/Plasma_GCA_2024.