

Abstract
Ulcerative colitis (UC) is a refractory, chronic inflammatory bowel disease of unknown etiology. Although platelets are activated in UC, their relevance in pathophysiology remains unclear. We analyzed the correlation of platelet activation and platelet–monocyte complexes (PMCs) with severity of mucosal inflammation using the Mayo endoscopic subscore (MES). Platelet activation marker, CD62P was upregulated in patients with UC compared with that in healthy controls (P < 0.05). CD62P expression was significantly higher in patients with MES3 (severe inflammation) than in those with MES ≤ 2 (endoscopic remission to moderate inflammation) (P < 0.001). The concentration of sCD62P in patients with MES0 (endoscopic remission) was significantly higher than in those with MES ≥ 1 (P < 0.01). The expression of CD40L, CD63, PAC-1, annexin V, and CD36, and the concentrations of sCD40L, PF4, and RANTES did not correlate with MES. The proportion of PMCs in patients with MES3 was higher than in those with MES ≤ 2 (P < 0.05). CD16 expression on monocytes with platelets was significantly higher than in monocytes without platelets (P < 0.001). Patients with complete remission after treatment showed significant reduction in PMCs 3 months after treatment (P < 0.05) but had no change in CD62P and sCD62P. Our data suggest that platelet activation via the CD62P–PMC axis is involved in UC pathophysiology.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-78462-8.
Keywords: Ulcerative colitis, CD62P, Platelet–monocyte complexes, Mayo endoscopic subscore, CD16
Subject terms: Autoimmunity, Inflammatory bowel disease
Introduction
Ulcerative colitis (UC) is an intractable chronic inflammatory bowel disease (IBD), characterized by relapse and remission, requiring lifelong treatment1. Although the pathogenesis of UC has not been completely elucidated, accumulating evidence suggests that changes in the genetics, gut microbiota, and environmental factors underlying a dysregulated immune response in the colonic mucosa play important roles in its development. Besides steroids and immunosuppressants, anti-TNF-α, -IL-12, -IL-23, and -α4β7 integrin have been used for UC treatment2–5. Despite recent advances in treatment, UC remains a disease with unmet medical needs that compromise the quality of life, not only in the short term but also in the long term, owing to the existence of unidentified factors that drive gut inflammation.
Platelets are one of the main blood cells involved in hemostasis and thrombosis. They also act as immune cells and promote inflammation6. Moreover, chronic inflammatory milieu activates platelets, leading to secondary thrombocytosis and hypercoagulability7,8. Patients with naïve or undercontrolled UC often exhibit high platelet counts9. Thromboembolism, such as in ischemic heart disease, stroke, and mesenteric ischemia, is a burdensome complication of UC10. Moreover, activated platelets conjugate with monocytes, leading to augmentation of innate and acquired immunity11. Platelet–monocyte complexes (PMCs) are sensitive in vivo markers of platelet activation12. These findings suggest that platelets and PMCs could serve as novel therapeutic targets and disease biomarkers for UC.
CD62P, also known as P-selectin or GMP140, is a member of the selectin family of adhesion molecules that is highly expressed on activated platelets13. In addition to CD62P, other surface markers, including CD40L, CD63, PAC-1, annexin V, and CD36, are associated with activated platelets14–18. Although activated platelets are characterized by several surface markers, platelets are easily activated by weak stimuli. Surface markers of activated platelets should therefore be rapidly measured after peripheral blood samples are obtained from patients. Hence, only a small number of cases are included in studies on activated platelets and PMCs in patients with IBD, and few studies have analyzed multiple surface markers simultaneously. Few studies have investigated the significance of activated platelets and PMCs in IBD. This study was aimed at comprehensively exploring useful surface marker and cytokine candidates in activated platelets to evaluate IBD severity.
Methods
Study subjects and blood sample collection
Patients with UC (n = 54), diagnosed according to clinical, radiological, endoscopic, and pathological basis, and healthy controls (HCs) (n = 11) were enrolled in this study. Patient demographic and disease characteristics data, including age, sex, and blood biomarkers (platelet count, C-reactive protein level, Lichtiger index, Mayo endoscopic subscore, and ulcerative colitis endoscopic index of severity and treatment), were collected. Patients with UC who had cardiac disease or an overt infection, or were taking antiplatelet or anticoagulant medications were not included in this study. Healthy volunteers with no history of current illness or medication served as healthy controls. The clinical backgrounds of the patients with UC and HC are presented in Table 1. All participants provided written informed consent. This study was approved by the Kansai Medical University Hospital Committee before its initiation (approval number: 2021011) and was performed according to the Declaration of Helsinki. After blood was collected in tubes with a separation gel, whole blood was collected in Acid Citrate Dextrose blood collection tubes (BD Pharmingen, San Jose, CA, USA) using a 21G needle without a tourniquet to avoid platelet activation.
Table 1.
Clinical features.
| UC (n = 54) | HC (n = 11) | |
|---|---|---|
| Age (median, IQR) | 47 (29–57) | 44 (30–45) |
| Gender (female/male) | 25/29 | 5/6 |
| Platelet counts ×104/µL (median, IQR) | 29.0 (24.4–34.3) | |
| C-Reactive Protein mg/ml (median, IQR) | 0.32 (0.08–2.83) | |
| Lichtiger index (median, IQR) | 7.5 (3–12) | |
| Mayo endoscopic subscore : MES | ||
| MES0 | 11 | |
| MES1 | 6 | |
| MES2 | 9 | |
| MES3 | 26 | |
| Not examined | 2 | |
| Current medication | ||
| No medication | 4 | |
| 5ASA only | 19 | |
| Currently using any corticosteroids, thiopurine, biologics, or Janus kinase inhibitor | 31 | |
CRP, C-reactive protein; 5-ASA, 5-Aminosalicylic acid.
Antibodies and reagents
Fluorochrome-conjugated antibodies specific to CD61 (VI-PL2), CD62P (AK-4), CD40L (89 − 76), CD63 (MEM-259), PAC-1 (SP-2), annexin V, CD36 (CB38), CD14 (M5E2), CD16 (3G8), IgG1 κ Isotype Control (MOPC-21), IgG1, κ Isotype Control (X40), and IgM, κ Isotype Control (G155-228) were purchased from BD Pharmingen and Thermo Fisher Scientific (Waltham, MA, USA).
Cell and sera preparation
For flow cytometry, platelets were prepared as follows: 5 µl of whole blood was mixed with 5 µl of fluorochrome-conjugated antibody cocktail, incubated at 24 °C for 15 min in the dark, fixed with 250 µl of 4% paraformaldehyde at 4 °C for 30 min in the dark, and analyzed using flow cytometry on BD FACSCalibur (BD Biosciences). PMCs were prepared according to previous reports, with slight modifications19. Briefly, 900 µl of whole blood was fixed with 1000 µl of 4% paraformaldehyde in the dark at room temperature for 15 min, followed by washing twice with PBS. Fixed samples were added to 3000 µl of ACK lysis buffer (Abcam Plc, Cambridge, UK) at 24 °C for 5 min in the dark and washed twice with 3000 µl of PBS. Sera were retrieved by centrifuging the sample tubes containing separation gel at 1700 × g for 10 min at 4 °C and stored at − 80 °C until use.
Cytokine quantification
The soluble forms of CD62P (sCD62P), sCD40L, PF4, and RANTES in the sera were quantified using a cytokine-specific ELISA kit, according to the manufacturer’s instructions (BD Biosciences and R&D Systems). All cytokines were quantified using standard curves provided with the corresponding ELISA kit.
Immunohistochemistry
Total colectomy specimens from 12 cases, seven females and five males, the median age (IQR) 51(44–57) of ulcerative colitis from 2009 to 2022 were used for immunohistochemistry. Specimens were fixed with 10% formalin, followed by paraffin embedding and section. The sectioned specimens were deparaffinized in xylene, rehydrated in graded ethanol. Primary antibodies, anti-human CD62P (AB6632, abcam) (1:200), and CD14 (ab133335, abcam) (1:2000), were diluted in tris-buffered saline with 0.1% Tween-20 and incubated at 4 °C in a humidified chamber for 16 h. The appropriate species-specific AlexaFluor (488 or 555)-conjugated antibodies (Thermo Fisher Scientific) (1:250) were used as the secondary antibodies and incubated at 24 °C in a humidified chamber for 1 h. Colon images were captured using an Olympus BX53 fluorescence microscope (Olympus).
Collection of clinical data
Patient demographic and disease characteristic data, including sex, age, clinical severities (Lichtiger index: range 0–21), blood biomarkers (hemoglobin, platelet count, serum albumin, and CRP level), and medical treatments were collected from medical charts. The severity of mucosal inflammation was classified using the Mayo endoscopic subscore (MES)20. MES is an item of Mayo scores, which is a composite of four items, namely the stool frequency subscore, rectal blooding subscore, MES, and Physician Global Assessment subscore, which ranges from 0 to 12, with each of the four subscores ranging from 0 to 3. The MES was classified as MES0 (endoscopic remission), MES 1 (mild mucosal inflammation), MES2 (moderate mucosal inflammation), and MES3 (severe mucosal inflammation). The most severely affected portion of the mucosa was scored. The typical endoscopic findings in patients with MES scores of 0, 1, 2, and 3 are shown in Supplementary Fig. S1.
Statistical analysis
The Shapiro–Wilk test, Student’s t-test, Mann–Whitney U test, Pearson correlation test, Spearman correlation test, Wilcoxon signed-rank test, Dunn test, and Tukey’s multiple comparison test were performed for data analysis using SPSS version 26 (IBM, Armonk, NY, USA). P-values < 0.05 were considered to indicate statistically significant differences.
Results
Comprehensive measurement of platelet activation markers
Platelets were identified in whole blood based on their forward scatter/side scatter and CD61 positivity characteristics (Fig. 1a). First, we compared the expression of platelet activation markers, including CD62P, PAC1, CD40L, annexin V, CD63, and CD36, between HC and patients with UC. CD62P was significantly upregulated in the UC group (30.3, IQR 24.3–39.6%) compared with that in the HC group (24.8, IQR 22.5–26.6%) (P = 0.01) (Fig. 1b, c), while the expression of other surface molecules, namely CD40L, CD63, PAC1, annexin V, and CD36 was not significantly different between the two groups.
Fig. 1.

CD62P on platelets is upregulated in patients with UC. Expression of platelet activation markers in patients with UC was compared with that in healthy controls using flow cytometry. (a) Dot plots represent the platelet gating strategy. Platelets were identified in whole blood according to their FSC/SSC and CD61 positivity characteristics. (b) Representative patterns of expression of CD62P, CD40L, CD63, PAC1, annexin V, and CD36 on platelets in patients with UC and healthy controls. (c) Expression of the markers shown in (b) in patients with UC (n = 54) was quantified and compared with that in healthy controls (n = 11). The bar graphs represent the median expression with the IQR. P-values were calculated using the Mann–Whitney U test; *P < 0.05. UC, ulcerative colitis; FSC, forward scatter; SSC, side scatter; IQR, interquartile range.
Correlation between mucosal inflammation and platelet surface molecules
To clarify the correlation between platelet activation markers and endoscopic severities in UC patients, we compared the expression of the platelet activation markers in UC patients, classified by MES. The expression of CD62P increased as the endoscopic activity became more severe, whereas there were no tendencies in the expression of CD40L, CD63, PAC1, annexin V, and CD36 between each MES (Supplementary Fig. S2). To focus on severe inflammation, we divided patients in the UC group into those with MES ≤ 2 and MES3, and compared the expression of activation markers on platelets collected on the day of colonoscopy. The expression of CD62P was significantly higher in MES3 than in MES ≤ 2 (P < 0.001) (Fig. 2). However, the expression of CD40L, CD63, PAC1, annexin V, and CD36 was not significantly different between patients with MES ≤ 2 and MES3 (Fig. 2).
Fig. 2.

Potential contribution of platelet activation markers to endoscopic severity. The endoscopic findings represented by MES ≤ 2 (n = 26) and MES3 (n = 26) were compared with the expression of CD62P, CD40L, CD63, PAC1, annexin V, and CD36 in platelets collected on the day of colonoscopy. P-values were calculated using the Mann–Whitney U test; ***P < 0.001. MES, Mayo endoscopic subscore.
Correlation between mucosal inflammation and platelet-derived soluble factors
We also quantified platelet-derived soluble factors, namely sCD62P, sCD40L, PF4, and RANTES, in the sera of patients with UC and compared them with the MES. Contrary to the results for the activation markers, concentration of sCD62P tended to be negatively correlated to MES, while no tendency was found in CD40L, PF4, and RANTES (Supplementary Fig. S3). Moreover, concentration of sCD62P in patients with MES0 was significantly higher than that in patients with MES ≥ 1 (P = 0.002) (Fig. 3). However, concentrations of secreted CD40L, PF4, and RANTES were not different between patients with MES0 and MES ≥ 1 (Fig. 3).
Fig. 3.
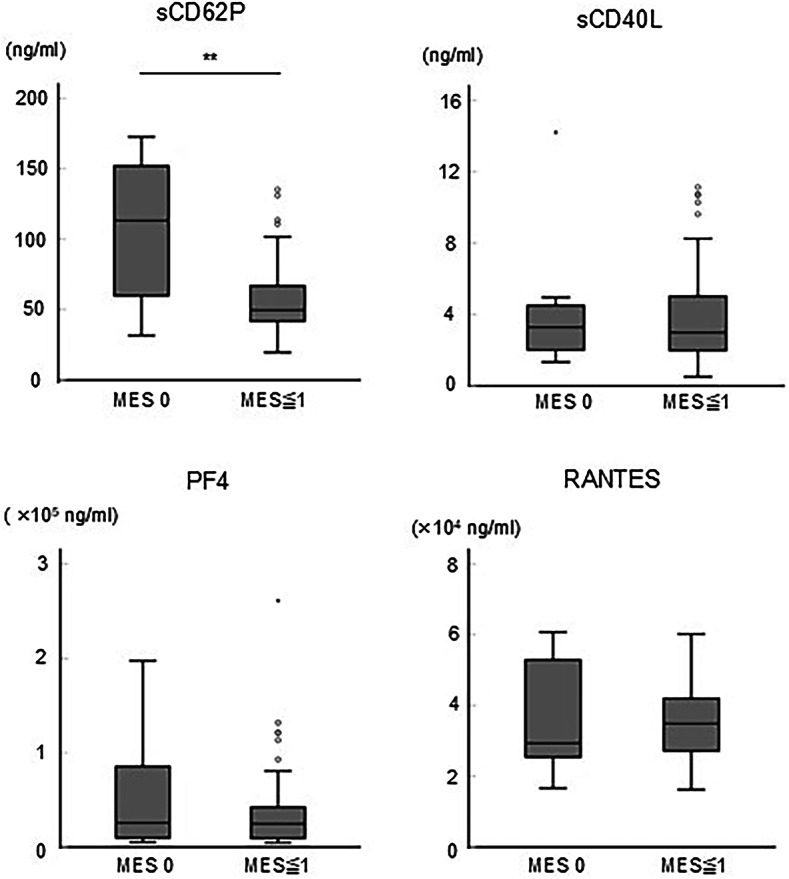
Potential contribution of platelet-derived soluble factors to endoscopic severity. The endoscopic findings represented by MES0 (n = 11) and MES ≥ 1 (n = 41) were compared with the concentrations of sCD62P, sCD40L, PF4, and RANTES in the sera collected on the day of colonoscopy. P-values were calculated using the Mann–Whitney U test; **P < 0.01.
Correlation of mucosal inflammation and platelet–monocyte complex
We identified PMCs as CD14+ and CD61+ using flow cytometry (Fig. 4a). Patients with MES3 showed significantly higher proportion of PMCs than those with MES ≤ 2 (P = 0.02) (Fig. 4b). The expression of CD16 on PMCs, which reflects monocyte maturation, was significantly increased than that on unconjugated platelets in the patients (P < 0.001) (Fig. 4c). We investigated whether the presence of PMCs in the intestinal mucosa also depends on the severity of inflammation. It was revealed that PMCs were mainly present in areas of mucosal defects, and the number of PMCs was significantly increased in a depth-dependent fashion of mucosal damage (Fig. 4d).
Fig. 4.

Correlation of platelet–monocyte complex with endoscopic severity. (a) Dot plots represent the gating strategy of the PMCs. PMCs were identified in whole blood based on their FSC, CD14, and CD61 positivity. (b) The proportion of CD14+ platelets in peripheral blood from patients UC having MES ≤ 2 (n = 26) and MES3 (n = 26) was determined using flow cytometry. (c) Dot plots show representative patterns of monocytes with (upper) and without (lower) platelets (left), and their CD16 expression (middle). The bar graph (right) shows significant upregulation of CD16 in PMCs compared with that in unconjugated platelets in patients with UC (n = 52). P-values were calculated using the Mann–Whitney U test: *P < 0.05, ***P < 0.001. PMC, platelet–monocyte complex. (d) Immunofluorescence staining was performed to compare the presence of PMCs as CD62P + platelets aggregation (green) with CD14 + monocytes (red) among no erosion (left), erosion within mucosal layer (middle), and ulceration reaching submucosal layer (right). The dot plot illustrates the number of PMC in colon mucosa. Data were derived from randomized views (×200) (no erosion; n = 10, erosion; n = 19, ulcer; n = 11) from 9 independent surgical specimens. P-values were calculated using the Tukey’s multiple comparison test: **P < 0.01, ***P < 0.001. Arrowheads indicate PMCs. Bars indicate 50 μm. PCI, phase contrast image. DAPI, 4’,6-diamidino-2-phenylindole.
Early kinetics of platelet activation upon therapeutic intervention
We compared the expression of CD62P and sCD62P and the proportion of PMCs before and 3 months after therapeutic intervention in patients with endoscopic remission (MES = 0). These patients received corticosteroids, biologics, or Janus kinase inhibitors. Although the expression of CD62P and sCD62P was not altered (Fig. 5a), the proportion of PMCs was significantly reduced in 3 months, with mucosal remission (P = 0.03) (Fig. 5a-b). The expression of other platelet activation markers was not altered after treatment (data not shown).
Fig. 5.

Early change in PMCs but not in CD62P and sCD62P in patients with remission. (a) The graphs show the changes in CD62P, sCD62P, and PMCs before and 3 months after treatment in patients with UC (n = 6) who achieved endoscopic remission. P-values were calculated using the Wilcoxon signed rank test, *P < 0.05. (b) Representative endoscopic findings (upper) and dot plots (lower) before (left) and after 3 months (right) of treatment in patients with UC are shown.
Discussion
In addition to their role in hemostasis, platelets are also immune modulators. They can promote either pro- or anti-inflammatory responses through diverse mechanisms based on the individual immunological milieu21–24. Platelets are involved in the pathogenesis of various autoimmune diseases25–27. Our findings in this study are important because they emphasize the fact that platelet activation correlates to disease aggravation, which involves abnormal immunity similar to that observed in other autoimmune diseases. The expression of CD62P in platelets was upregulated in patients with UC, which is consistent with the findings in previous studies28,29. Herein, we demonstrate that this upregulation is correlated with the severity of mucosal inflammation. CD62P, a member of the selectin family, is a 140 kDa membrane glycoprotein located in the α-granules and dense granules of platelets and endothelial cells. Upon platelet activation, the α-granule membrane fuses with the platelet plasma membrane, leading to upregulation of CD62P on the platelet surface30. In contrast, the expression levels of CD40L, CD63, PAC1, annexin V, and CD36 did not differ between the HC and UC groups. These data suggest that the kinetics of CD62P is different from those of other platelet activation markers in patients with UC, which might be relevant to the understanding of disease pathophysiology and aid in identifying new therapeutic targets. Originally, platelet activation mediates wound healing by secretion of cytokines, chemokines, and growth factors that control recruitment and activation of cell populations involved in tissue repairs31. CD62P-intact rather than CD62-deficient platelets ameliorated experimental gastrointestinal injury model32, suggesting that platelet activation and CD62P play a critical role in mucosal healing of UC. On the other hand, wound healing is inseparably linked to inflammation and platelets also play a role in potential immune activator6. One possibility is that chronic inflammation and disease progression may increase the number of factors that result in the exposure of CD62P on platelets leading to empower immune activation rather than mucosal healing. For instance, platelets can be activated by shear stress on the wall of blood vessels, which is induced by endothelial dysfunction during chronic inflammation33. Notably, biochemicals, such as adenosine diphosphate (ADP), arachidonic acid, thrombin receptor activating peptide 6, and thrombin, rather than shear stress contribute to the exposure of CD62P on platelets34. ADP is released from various cell populations, such as apoptotic cells, activated immune cells, necrotic cells, and activated platelets, in the inflammatory milieu. Although ADP plays a role in tissue repair and hemostasis, it is also involved in inflammatory responses and thrombosis35. Arachidonic acid is released from cell membranes and mediates wound healing and inflammatory responses36,37. Thrombin, a serine protease synthesized in the liver, plays a role in hemostasis and promotes platelet aggregation in inflamed colon mucosa38. Taken together, we speculate that mucosal injuries trigger further activation of the immune system and mucosal healing, leading to the upregulation of CD62P on platelets by biochemicals rather than CD62P being constitutively active in UC. In contrast to CD62P expression, the concentration of the soluble form of CD62P decreased as mucosal damage progressed. A tendency for downregulation of sCD62P with disease progression has also been observed in gastric cancer; the expression of sCD62P was lower in stage III and IV patients than in stage I and II patients39. Because sCD62P is shed from platelet membranes, elevated sCD62P levels in mucosal remission might reflect reduced CD62P expression on the platelet membranes in patients with UC.
The contact between platelets and monocytes is mediated by adhesion via CD62P on platelets and PSGL1 on monocytes, which leads to the formation of PMCs and activation of monocytes11,12,40. Because platelet activation is a primary factor in the aggregation of platelets and monocytes, PMCs are a sensitive indicator of platelet activation. There are conflicting reports that PMC kinetics is positively or inversely correlated with disease severity in UC41,42. In the present study, the highest proportion of PMCs was observed in patients with MES3. The expression of CD16 was higher in monocytes that were conjugated with platelets than in the unconjugated ones. The proportion of PMCs in the blood was significantly higher in the MES3 cases, and PMCs were more prevalent in areas with severe mucosal damage, suggesting that the proportion of PMCs in the blood and the expression of PMCs in the intestinal tract might be correlated. Moreover, the proportion of PMCs in patients with mucosal remission after treatment was significantly reduced, but CD62P and sCD62P levels were not altered. Taken together, our findings indicate that PMCs, promoted by platelet activation via CD62P upregulation in the inflammatory milieu, sensitively reflect mucosal healing; however, platelets are latently activated within a short period after mucosal remission. Therefore, maintenance therapy after mucosal remission is reasonable based on CD62P kinetics.
This study had some limitations. We did not assess the role of PMCs in the pathogenesis of UC. Whether PMCs accelerate or inhibit inflammatory responses remains controversial43,44. Further studies are needed to clarify this issue. Interestingly, patients with severe COVID-19 show platelet activation and PMC formation, which correlate with poor prognosis45.
Modulation of CD62P expression in platelets may be a potential target for therapeutic interventions aimed at preventing inappropriate platelet activation and controlling excessive clotting in UC. Crizanlizumab is a monoclonal antibody against human CD62P that has been approved for the treatment of vaso-occlusive crisis in Sickle cell disease46. A phase 2 study on the efficacy of crizanlizumab in sickle cell disease revealed no serious adverse effects, including bleeding events47. However, considering their pharmacological action, it is not confirmed whether anti-CD62P antibodies are usable for patients with an underlying intestinal mucosal injury and risk of intestinal bleeding, and this must be rigorously evaluated.
In summary, CD62P is upregulated in patients with UC. We show that CD62P expression is positively and sCD62P levels are negatively correlated with mucosal inflammation. Notably, PMCs, which express CD16, also correlate with mucosal injury. The CD62P–PMCs axis may be explored for understanding the UC pathogenesis and for the development of new therapeutics against this intractable disease. The accumulation of a larger number of cases is needed, and phenotype studies of PMCs will be required.
CRP, C-reactive protein; 5-ASA, 5-Aminosalicylic acid.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thank Editage for English language editing. All authors have read the journal’s authorship agreement and policy. This study was funded by a Grant-in-Aid for Scientific Research (C) of JSPS KAKENHI Grant Numbers JP22K07995. All participants provided written informed consent, and the study was approved by the Kansai Medical University Hospital Committee before its initiation (approval number: 2021011).
Author contributions
M.N. conceived the study. Y.S. and T.T. designed the study and prepared initial drafts of the manuscript. Y.S., T.T., N.Y., and Y.I. performed the experiments. Y.H., T.T., T.I., T.F., and S.S. analyzed the data and assisted in the preparation of the manuscript. M.N. revised the initial draft of the manuscript. All other authors collected and interpreted the data and critically reviewed the manuscript.
Data availability
The data for this study are available from the corresponding author upon reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Le Berre, C., Honap, S. & Peyrin-Biroulet, L. Ulcerative colitis. Lancet. 402, 571–584. 10.1016/S0140-6736(23)00966-2 (2023). [DOI] [PubMed] [Google Scholar]
- 2.Rutgeerts, P. et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl. J. Med.353, 2462–2476. 10.1056/NEJMoa050516 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Sands, B. E. et al. Ustekinumab as induction and maintenance therapy for Ulcerative Colitis. N Engl. J. Med.381, 1201–1214. 10.1056/NEJMoa1900750 (2019). [DOI] [PubMed] [Google Scholar]
- 4.D’Haens, G. et al. Mirikizumab as induction and maintenance therapy for Ulcerative Colitis. N Engl. J. Med.388, 2444–2455. 10.1056/NEJMoa2207940 (2023). [DOI] [PubMed] [Google Scholar]
- 5.Feagan, B. G. et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl. J. Med.369, 699–710. 10.1056/NEJMoa1215734 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Morrell, C. N., Aggrey, A. A., Chapman, L. M. & Modjeski, K. L. Emerging roles for platelets as immune and inflammatory cells. Blood. 123, 2759–2767. 10.1182/blood-2013-11-462432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrison, C. N. et al. Guideline for investigation and management of adults and children presenting with a thrombocytosis. Br. J. Haematol.149, 352–375. 10.1111/j.1365-2141.2010.08122.x (2010). [DOI] [PubMed] [Google Scholar]
- 8.Aksu, K., Donmez, A. & Keser, G. Inflammation-induced thrombosis: mechanisms, disease associations and management. Curr. Pharm. Des.18, 1478–1493. 10.2174/138161212799504731 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Talstad, I., Rootwelt, K. & Gjone, E. Thrombocytosis in ulcerative colitis and Crohn’s disease. Scand. J. Gastroenterol.8, 135–138 (1973). [PubMed] [Google Scholar]
- 10.Singh, S., Kullo, I. J., Pardi, D. S. & Loftus, E. V. Epidemiology, risk factors and management of cardiovascular diseases in IBD. Nat. Rev. Gastroenterol. Hepatol.12, 26–35. 10.1038/nrgastro.2014.202 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Passacquale, G. et al. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS One. 6, e25595. 10.1371/journal.pone.0025595 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michelson, A. D., Barnard, M. R., Krueger, L. A., Valeri, C. R. & Furman, M. I. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation. 104, 1533–1537. 10.1161/hc3801.095588 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Théorêt, J. F., Yacoub, D., Hachem, A., Gillis, M. A. & Merhi, Y. P-selectin ligation induces platelet activation and enhances microaggregate and thrombus formation. Thromb. Res.128, 243–250. 10.1016/j.thromres.2011.04.018 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Freedman, J. E. CD40-CD40L and platelet function: beyond hemostasis. Circ. Res.92, 944–946. 10.1161/01.RES.0000074030.98009.FF (2003). [DOI] [PubMed] [Google Scholar]
- 15.Azorsa, D. O., Hyman, J. A. & Hildreth, J. E. CD63/Pltgp40: a platelet activation antigen identical to the stage-specific, melanoma-associated antigen ME491. Blood 78, 280–284 (1991). [PubMed]
- 16.Anderson, G. P., van de Winkel, J. G. & Anderson, C. L. Anti-GPIIb/IIIa (CD41) monoclonal antibody-induced platelet activation requires fc receptor-dependent cell-cell interaction. Br. J. Haematol.79, 75–83. 10.1111/j.1365-2141.1991.tb08010.x (1991). [DOI] [PubMed] [Google Scholar]
- 17.Thiagarajan, P. & Tait, J. F. Binding of annexin V/placental anticoagulant protein I to platelets. Evidence for phosphatidylserine exposure in the procoagulant response of activated platelets. J. Biol. Chem.265, 17420–17423 (1990). [PubMed] [Google Scholar]
- 18.Kehrel, B. et al. Glycoprotein VI is a major collagen receptor for platelet activation: it recognizes the platelet-activating quaternary structure of collagen, whereas CD36, glycoprotein IIb/IIIa, and Von Willebrand factor do not. Blood. 91, 491–499 (1998). [PubMed] [Google Scholar]
- 19.Singh, M. V., Davidson, D. C., Kiebala, M. & Maggirwar, S. B. Detection of circulating platelet-monocyte complexes in persons infected with human immunodeficiency virus type-1. J. Virol. Methods. 181, 170–176. 10.1016/j.jviromet.2012.02.005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schroeder, K. W., Tremaine, W. J. & Ilstrup, D. M. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N Engl. J. Med.317, 1625–1629. 10.1056/NEJM198712243172603 (1987). [DOI] [PubMed] [Google Scholar]
- 21.Bakogiannis, C., Sachse, M., Stamatelopoulos, K. & Stellos, K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine. 122, 154157. 10.1016/j.cyto.2017.09.013 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Wang, X., Luo, Y., Masci, P. P., Crawford, R. & Xiao, Y. Influence of Interleukin-1 Beta on platelet-poor plasma clot formation: a potential impact on Early Bone Healing. PLoS One. 11, e0149775. 10.1371/journal.pone.0149775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science. 303, 1532–1535. 10.1126/science.1092385 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Sadallah, S., Amicarella, F., Eken, C., Iezzi, G. & Schifferli, J. A. Ectosomes released by platelets induce differentiation of CD4 + T cells into T regulatory cells. Thromb. Haemost. 112, 1219–1229. 10.1160/TH14-03-0281 (2014). [DOI] [PubMed] [Google Scholar]
- 25.El Bannoudi, H. et al. Platelet LGALS3BP as a mediator of myeloid inflammation in systemic Lupus Erythematosus. Arthritis Rheumatol.75, 711–722. 10.1002/art.42382 (2023). [DOI] [PubMed] [Google Scholar]
- 26.Boilard, E. et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 327, 580–583. 10.1126/science.1181928 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Postlethwaite, A. E. & Chiang, T. M. Platelet contributions to the pathogenesis of systemic sclerosis. Curr. Opin. Rheumatol.19, 574–579. 10.1097/BOR.0b013e3282eeb3a4 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Schürmann, G. M. et al. Increased expression of cell adhesion molecule P-selectin in active inflammatory bowel disease. Gut. 36, 411–418. 10.1136/gut.36.3.411 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tekelioglu, Y., Uzun, H. & Sisman, G. Activated platelets in patients suffering from inflammatory bowel disease. Bratisl Lek Listy. 115, 83–85. 10.4149/bll_2014_018 (2014). [DOI] [PubMed] [Google Scholar]
- 30.de Bruijne-Admiraal, L. G., Modderman, P. W., Von dem Borne, A. E. & Sonnenberg, A. P-selectin mediates ca(2+)-dependent adhesion of activated platelets to many different types of leukocytes: detection by flow cytometry. Blood. 80, 134–142 (1992). [PubMed] [Google Scholar]
- 31.Gawaz, M. & Vogel, S. Platelets in tissue repair: control of apoptosis and interactions with regenerative cells. Blood. 122, 2550–2554. 10.1182/blood-2013-05-468694 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Pethaperumal, S., Hung, S. C., Lien, T. S., Sun, D. S. & Chang, H. H. P-Selectin is a critical factor for platelet-mediated Protection on Restraint stress-Induced Gastrointestinal Injury in mice. Int. J. Mol. Sci.2310.3390/ijms231911909 (2022). [DOI] [PMC free article] [PubMed]
- 33.Zanoli, L. et al. Vascular consequences of inflammation: a position statement from the ESH Working Group on Vascular structure and function and the ARTERY Society. J. Hypertens.38, 1682–1698. 10.1097/HJH.0000000000002508 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roka-Moiia, Y. et al. Platelet activation via shear stress exposure induces a differing pattern of biomarkers of activation versus biochemical agonists. Thromb. Haemost. 120, 776–792. 10.1055/s-0040-1709524 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le, T. T. et al. Purinergic Signaling in Pulmonary inflammation. Front. Immunol.10, 1633. 10.3389/fimmu.2019.01633 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh, S. Y., Lee, S. J., Jung, Y. H., Lee, H. J. & Han, H. J. Arachidonic acid promotes skin wound healing through induction of human MSC migration by MT3-MMP-mediated fibronectin degradation. Cell. Death Dis.6, e1750. 10.1038/cddis.2015.114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, B. et al. Metabolism pathways of arachidonic acids: mechanisms and potential therapeutic targets. Signal. Transduct. Target. Ther.6, 94. 10.1038/s41392-020-00443-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Motta, J. P. et al. Active thrombin produced by the intestinal epithelium controls mucosal biofilms. Nat. Commun.10, 3224. 10.1038/s41467-019-11140-w (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Da Silva, J. P. A. et al. Evaluation of platelet activation marker expression and its correlation with tumorigenesis and tumor progression in patients with gastric cancer. J. Surg. Oncol.126, 125–131. 10.1002/jso.26908 (2022). [DOI] [PubMed] [Google Scholar]
- 40.Rolling, C. C., Barrett, T. J. & Berger, J. S. Platelet-monocyte aggregates: molecular mediators of thromboinflammation. Front. Cardiovasc. Med.10, 960398. 10.3389/fcvm.2023.960398 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pamuk, G. E. et al. Increased circulating platelet-neutrophil, platelet-monocyte complexes, and platelet activation in patients with ulcerative colitis: a comparative study. Am. J. Hematol.81, 753–759. 10.1002/ajh.20655 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Zamora, C. et al. Inverse Association between Circulating Monocyte-Platelet Complexes and inflammation in Ulcerative Colitis patients. Inflamm. Bowel Dis.24, 818–828. 10.1093/ibd/izx106 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Carestia, A. et al. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell. Rep.28, 896–908e895. 10.1016/j.celrep.2019.06.062 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Lee, S. J. et al. Activated platelets convert CD14 + CD16- into CD14 + CD16 + monocytes with enhanced FcγR-Mediated phagocytosis and skewed M2 polarization. Front. Immunol.11, 611133. 10.3389/fimmu.2020.611133 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hottz, E. D. et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 136, 1330–1341. 10.1182/blood.2020007252 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ataga, K. I. et al. Crizanlizumab for the Prevention of Pain crises in Sickle Cell Disease. N Engl. J. Med.376, 429–439. 10.1056/NEJMoa1611770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanter, J. et al. Pharmacokinetics, pharmacodynamics, safety, and efficacy of crizanlizumab in patients with sickle cell disease. Blood Adv.7, 943–952. 10.1182/bloodadvances.2022008209 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data for this study are available from the corresponding author upon reasonable request.