

Abstract
The 3D organization of mammalian chromatin was described more than 30 years ago by visualizing sites of DNA synthesis at different times during the S phase of the cell cycle. These early cytogenetic studies revealed structurally stable chromosome domains organized into subnuclear compartments. Active-gene-rich domains in the nuclear interior replicate early, whereas more condensed chromatin domains that are largely at the nuclear and nucleolar periphery replicate later. During the past decade, this spatiotemporal DNA replication programme has been mapped along the genome and found to correlate with epigenetic marks, transcriptional activity and features of 3D genome architecture such as chromosome compartments and topologically associated domains. But the causal relationship between these features and DNA replication timing and the regulatory mechanisms involved have remained an enigma. The recent identification of cis-acting elements regulating the replication time and 3D architecture of individual replication domains and of long non-coding RNAs that coordinate whole chromosome replication provide insights into such mechanisms.
Before each cell division, eukaryotic chromosomes must be entirely duplicated. Duplication involves the replication not only of DNA but also of all chromatin components, appropriate epigenetic information and 3D organization. Almost 60 years ago, the first insights into this process revealed that large segments of mammalian chromosomes replicate at distinct times during the S phase of the cell cycle1. Modern genomics approaches have revealed that multi-megabase segments of chromosomes, referred to as constant timing regions (CTRs), replicate via the coordinate activation of adjacent replicons at characteristic times during the S phase. These CTRs are delimited by large timing transition regions (TTRs) along which DNA synthesis progressively advances2–8 (FIG. 1a).
Fig. 1 |. Replication timing relationship to 3D chromatin structure.
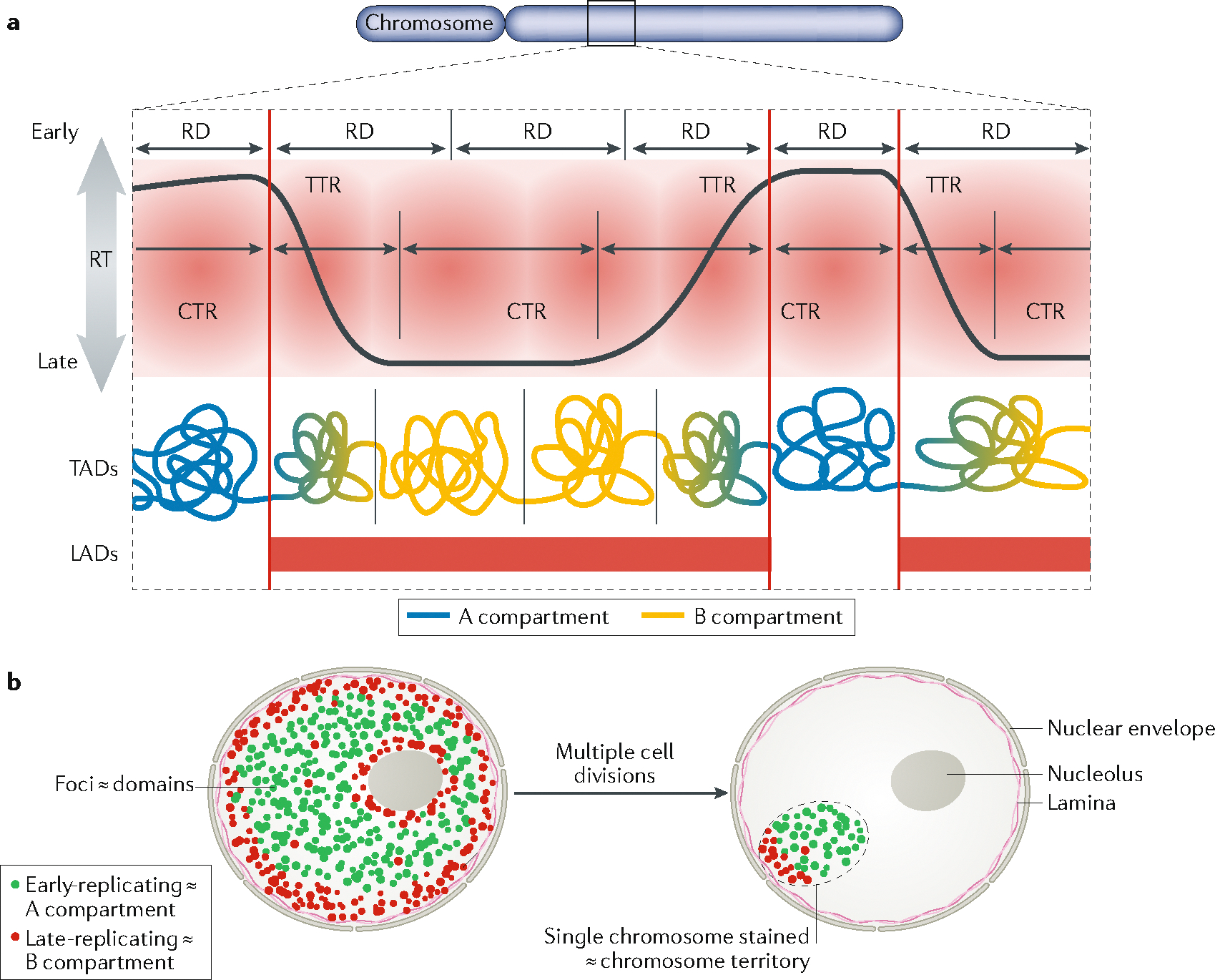
a | Current model of the relationship between replication timing (RT) and chromatin structure. Early and late constant timing regions (CTRs) are 1–5 Mb regions separated by timing transition regions (TTRs) as demarcated within the red shaded area. These CTRs consist of one to several replication domains (RDs), defined as chromatin segments that coordinately switch RT during cell fate changes (that is, between different cell types; see FIG. 2a). RDs share the properties and approximate boundaries of a subset of topologically associated domains (TADs), aligning most closely with TADs that are at compartment boundaries. Early CTRs correspond to the A compartment, but late CTRs correspond to the B compartment and TTRs correspond to the transitions between compartments. Both TTRs and late CTRs correspond to lamina-associated domains (LADs). b | RT illuminates genome architecture: nuclei after an early S pulse label (green) followed by several hours of a chase period and then a late S pulse label (red). In this model, observable foci of DNA synthesis correspond to the replication domains in panel a and early/late-replicating chromatin corresponds to A/B compartments. After multiple passages, only one chromosome per cell remains labelled, marking the chromosome territory47,202. However, the foci retain their label intensity46–49 and genetic continuity203, demonstrating that the DNA that is synthesized during one cell cycle remains clustered together as a structural unit of chromosomes for many generations.
All known eukaryotes have such a ‘DNA replication timing programme’, but the size of CTRs and TTRs varies, being scaled to the size of their genome9. This programme is extremely robust — almost every attempt to disrupt it, using chemical inhibitors, gene knockdown or gene knockout, has been unsuccessful (TABLE 1). The programme is also highly conserved between closely related eukaryotic species9–16. Altogether, these observations suggest that the timing of replication of discrete regions of chromosomes has an important biological function. Replicating different segments of the genome at different times ensures that the number of replication forks does not exceed the availability of limiting factors such as nucleotides and proteins required for DNA replication17, consolidates forks for rapid response to replication stress18 and ensures that the genome is fully replicated18. However, these constraints that lead to segmental replication of the genome do not explain why the genome should be replicated in a defined and evolutionarily conserved temporal order. Another proposed function of controlling DNA replication timing is to regulate the gene dosage19. However, most genes are subject to mechanisms that reduce the expression of post-replicated genes two-fold20,21 so it is not clear how extensively such a mechanism could drive the positive evolutionary selection of a specific genome-wide timing programme. Of note, mutation frequencies vary strongly during the S phase22. The frequency of point mutations is much higher for late-replicating DNA, possibly owing to the downregulation of mismatch repair (MMR) proteins during mid S phase23, and different types of structural mutation are correlated with early or late replication22–27. Thus, one hypothesis for the biological function of a defined replication timing programme is that it concentrates genomic variation to particular parts of the genome.
Table 1 |.
Gene deletion effects on global replication timing and chromatin architecture in mammals
| Gene deletion | Effect on replication timing | Effect on 3D structure |
|---|---|---|
| Rif1 (KO and KD) | Genome-wide changes (MEF, mESC, HeLa)164–166 | Localized changes (mESC)166 |
| Cohesin (KD and KO of factors from the complex) | No change (HCT116)77 | Loss of a part of TAD boundaries, no compartment change (HCT116, mouse hepatocytes)73,80 |
| CTCF (KD and KO) | No change (KO: D.M.G., unpublished data, mESC)75 | Loss of CTCF loops and local interactions, no compartment change (mESC, mNPC, HEK293T)151,205 |
| Suz12 (KO) | No change (mESC)66 | N/A |
| MeCP2 (KO) | No change (D.M.G., unpublished data) | N/A |
| G9a (KO) | Few local changes (mESC, mNPC)112 | No change (mESC)71 |
| H1a-H1d-H1e (triple KO) | No change (D.M.G., unpublished data) | Local changes (mESC)206 |
| cMyc-nMyc (double KO) | No change (D.M.G., unpublished data) | N/A |
| BAF53a (KO) | No change (mESC)207 | N/A |
| BAF250a (KO) | Local changes (mESC)207 | N/A |
| Brg1 (KO) | Local changes (mESC)207 | N/A |
CTCF, CCCTC-binding factor; KD, knockdown; KO, knockout; MEF, mouse embryonic fibroblast; mESC, mouse embryonic stem cell; mNPC, mouse neural precursor cell (derived from ESC); N/A, data not available; TAD, topologically associated domain.
In mammalian cells, considerable attention has been given to the relationship of replication timing to chromatin architecture and cell differentiation. The replication timing programme remains highly stable within a cell type and even between individual cells of the same type28,29, unlike transcription and many epigenetic marks30, but the timing of replication of at least 50% of the genome changes during cell fate transitions. These developmental switches occur in smaller units of 400–800 kb that are referred to as ‘replication domains’4,13,31–33. Replication domains consist of 1–4 replicons that synchronously initiate DNA replication, forming ‘replicon clusters’ that can be observed on isolated DNA fibres34,35. The larger CTRs are believed to consist of multiple adjacent replication domains that replicate at similar times4 (FIG. 1a). The replication timing of specific replication domains becomes stably altered in several diseases. Certain diseases, as well as individual patients, can therefore be identified by ‘replication timing signatures’36–38. As not just DNA but the entire structure of the chromosome must be replicated, a second logical hypothesis for the biological function for robust cell-type-specific spatial and temporal control of DNA replication is that where and when chromatin replicates is important for the maintenance of cell-type-specific epigenetic states and provides a mechanism to alter them during cell fate transitions. Testing this hypothesis will require an understanding of the mechanisms regulating the coordinated firing of replicons at defined times and their organization into replication domains. Thus, decrypting the relationship between replication timing, chromatin architecture, transcription and cell differentiation is an important challenge for the future.
The regulation of DNA replication timing in mammalian cells also occurs at the level of entire chromosomes39. Asynchronous replication of chromosomes was also first observed nearly 60 years ago, but it was originally thought to be unique to X chromosomes in female mammals. However, it was later discovered that most cancer cells have at least one chromosome for which replication is severely delayed and often continues when cells enter the G2 phase or even mitosis40–45. The mechanisms that control entire chromosome replication, and their relationship to the mechanisms that regulate the temporal order of replication of individual chromosomal domains, are poorly understood.
In this Review, we discuss our current understanding of the relationships between DNA replication timing and the 3D architecture of the genome in mammalian cells. First, we describe how replication timing is related to the spatial organization of the genome. Second, we discuss the dynamics of replication timing and 3D chromatin interactions during cell differentiation and during progression through the cell cycle. We then present a general overview of the regulation of the replication timing, followed by a discussion of recent discoveries of cis elements and trans-acting factors that regulate replication timing. Finally, we discuss the interplay between replication timing and transcription.
Links to spatial genome organization
Until recently, studying large-scale chromatin architecture was restricted to cytogenetic methods, with genome organization often highlighted by labelling sites of DNA replication. Pulse labelling of cells with nucleotide analogues for a short period of time enabled the visualization of ‘replication foci’ (FIG. 1b), which were sites of coordinated replicon activation. After chasing for multiple cell divisions, these foci retained their size, shape and intensity, suggesting that they are stable chromosomal units of structural organization46–49. When chased for longer periods of time, these pulse–chase experiments resulted in cells in which only one or a few chromosomes retained the label, revealing the nuclear organization of chromosomes into discrete territories (FIG. 1b), which could also be visualized by fluorescence in situ hybridization (FISH) with chromosome-specific probes50. Finally, the spatial distribution of the labelled foci was seen to vary during the S phase, most dramatically when comparing early with late S phase, suggesting the existence of two organizational compartments of chromatin in the nucleus that replicate in the first or second half of the S phase51–53 (FIG. 1b). This spatial organization of late-replicating chromatin at the nuclear periphery and around the nucleolus, and of early-replicating chromatin in more central regions of the nucleus, is remarkably conserved from single-cell ciliates to humans54. Once these patterns are established during early G1 phase49, they are maintained throughout interphase owing to the immobile nature of mammalian interphase chromatin55. Over the past decade, several genomics technologies to study the molecular organization of chromatin in the nucleus were developed, which enabled these cytogenetic observations to be related to 1D molecular maps of chromosomal DNA and its epigenetic marks, as well as to 3D genome architecture revealed by Hi-C (high-throughput chromosome conformation capture) methods (BOX 1). Here we discuss how these different scales of genome organization are related to different aspects of DNA replication.
Box 1 |. compartments and topologically associated domains in cell population and single cells.
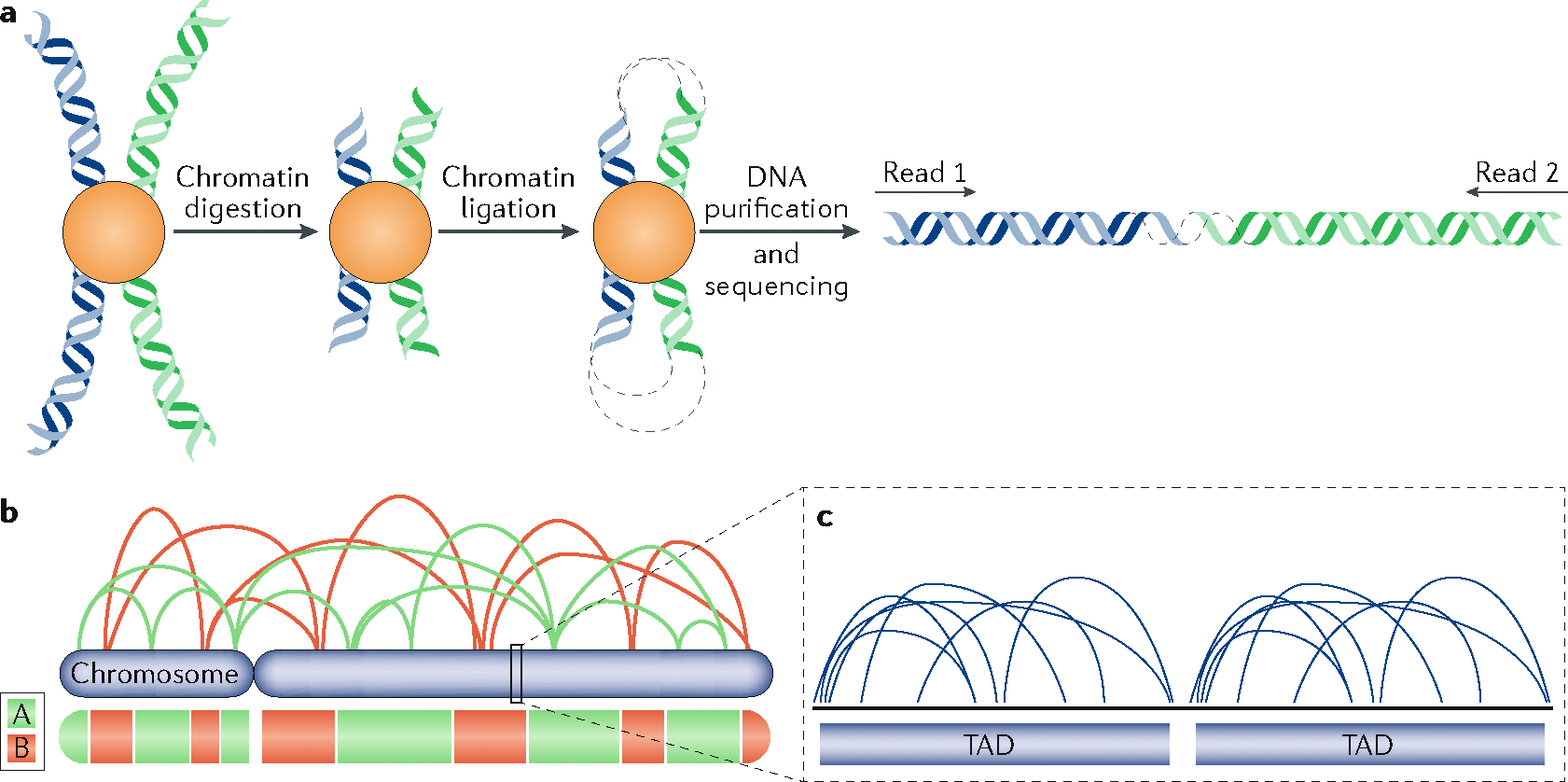
In the past decade, the development of chromatin conformation capture methods, particularly Hi-C (high-throughput chromosome conformation capture) and its derivatives, has enabled exquisite quantification of the frequency with which pairs of chromosomal sites interact genome-wide187, allowing the molecular identification of 3D chromatin architecture. These experiments consist of cleaving DNA within immobilized chromatin and re-ligating it to create chimeric DNA fragments composed of two or more interacting regions. These chimeric DNA fragments are then analysed by quantitative PCR or sequencing (see the figure, part a). The first component (PC1) of the principal component analysis (PCA), also named the eigenvector, of the pairwise interactions identifies the two major folding compartments termed A and B56, enriched for interactions within each group (see the figure, part b)56. This division of the chromatin correlates well with different chromatin features such as histone marks, gene transcription and DNase hypersensitivity, and particularly well with replication timing, likely representing the euchromatin and heterochromatin compartments illuminated by observing DNA synthesis cytologically13,56,57. These compartments identified through PCA can be further sub-stratified via a clustering analysis into several sub-compartments called A1, A2, B1, B2 and B3 (REF.58). Similar compartments can be observed when Hi-C is done in single cells where only a snapshot of interactions can be observed per cell28,188. On a finer scale, chromosomes can be seen to consist of a series of sub-megabase-sized self-interacting units70,71 termed topologically associated domains (TADs), which appear to be punctuated by boundaries that are stable in different cell types (see the figure, part c). These units may correspond to the stable units of chromosome structure also illuminated by labelling DNA synthesis cytologically, termed replication foci35. Within TADs, even finer clusters of interactions, or sub-TADs, can be identified58,189 that are less conserved among cell types83,190,191 and may represent cell-type-specific chromatin hubs. TADs can also be observed at the single-cell level97, but this overlap is not perfect and both single-cell Hi-C188 and Oligo-STORM (oligo-fluorescence in situ hybridization-mediated tracing of large chromosome segments through individual cell nuclei assisted by super-resolution microscopy) show that the positions of TAD boundaries vary from cell to cell78. CCCTC-binding factor (CTCF) and the cohesin complex are central to TAD structure. Depletion of CTCF or a subunit of the cohesin complex leads to an important perturbation of TADs without affecting chromatin compartments73,151 or replication timing75,77, highlighting the fact that compartments and TADs are being maintained by different mechanisms.
Early- and late-replicating chromatin compartments.
From its initial inception, Hi-C identified the first principal component of large-scale chromatin folding in the nucleus that separates chromatin into two nuclear compartments, defined as A and B56 (BOX 1). These A and B compartments roughly correlate with actively transcribed open chromatin and more silent compact chromatin, respectively, and very closely correlate with early- and late-replicating DNA13,57, as predicted by the cytogenetic studies summarized above (FIG. 1). At higher resolution, Hi-C can resolve these compartments into several sub-compartments (BOX 1), which continue to correlate with DNA regions that replicate at different times58. Interestingly, when compartments are defined at very high resolution, they overlap with active and inactive transcription units and no longer correlate with DNA replication timing59, indicating that the resolution of analysis of genomics data must be scale-appropriate for the biological process being investigated.
Chromosome territories.
In the nucleus, chromosomes occupy limited areas defined as chromosome territories. These territories can be observed by FISH60, but they can also be visualized by labelling replicating DNA during one cell cycle and then letting cells divide multiple times47. Indeed, after each division, labelled chromosomes will be randomly distributed among the daughter cells, eventually resulting in nuclei containing only one labelled chromosome, for which the signal will remain undiluted and will highlight the entire chromosome. Thus, the observed labelled region of the nucleus corresponds to the territory of that chromosome (FIG. 1b).
It is possible that the temporal window within which each whole chromosome replicates is determined by long non-coding RNAs (lncRNAs) coating the chromosome from which they are expressed. Three lncRNAs have been shown to be essential for the regulation of the replication time window of the chromosome from which they are transcribed. First, Xist is necessary for late replication of the whole inactive X chromosome in mice61, but also its deletion in differentiated human cells results in the inactive X being replicated even later in S phase (that is, the entire window of time during which the chromosome replicates is shifted)61,62. Secondly, it was recently discovered that deletion of two lncRNAs (ASAR6 and ASARA15) known as asynchronous replication and autosomal RNAs (ASARs) drastically delay the replication of the chromosomes they are coating40–42.
Lamina-associated domains.
A large subnuclear compartment is the chromatin near the nuclear periphery63. This compartment, consisting of lamina-associated domains (LADs), can be identified either cytogenetically64,65 or using genomics methods66,67 as chromatin that is bound by proteins of the inner nuclear envelope. As much of the chromatin that is near the nuclear lamina can also be in proximity to the nucleolar periphery or at other internal heterochromatic sites in alternating cell cycles65,68, LADs comprise most of the late-replicating compartment observed by cytogenetic labelling (FIG. 1b), also defined as the B compartment by Hi-C69. However, LADs do not contain only late-replicating chromatin and do not correlate as well with replication timing as do the A/B compartments. This puzzle was resolved by the finding that the borders of the earliest replication domains overlap strongly with the borders of LADs. Therefore, replication forks originating from early-replicating domains move rapidly into the adjacent LADs, causing them to replicate early even though they lack active origins of replication66. Moreover, the transitions from LADs (lamina-associated) to inter-LADs (not lamina-associated) are sharp, whereas transitions from the A to B compartments are gradual and correspond to the gradual TTRs between early- and late-replicating CTRs. Thus LADs, which comprise peripheral, nucleolar and other heterochromatic foci, consist of TTRs and late CTRs (FIG. 1a).
Topologically associated domains.
Along the length of chromosomes, 3D chromatin interactions are clustered within chromatin domains known as topologically associated domains (TADs)70,71 (BOX 1). The boundaries of TADs, which can be identified as the sites where chromatin interactions change in directionality, align with the boundaries of replication domains, suggesting that TADs correspond to the stable units of chromosome structure identified cytogenetically as replication foci66,72 (FIG. 1a). Indeed, replication foci labelled in living cells and tracked by correlative live and super-resolution microscopy displayed biophysical parameters consistent with TADs35. However, the relationship between TAD structure and replication domains remains obscure and, similar to compartments, TADs also lose their alignment with replication domains at high resolution as they become further divided into sub-TADs or individual chromatin loops. An important challenge is to understand the biological significance of TADs defined at different scales, and their relationship to chromosome functions, including DNA replication.
TADs are delimited by boundaries and are defined as regions within which chromatin interactions occur with higher frequency (that is, within the TAD) than in other regions (outside the TAD) (BOX 1). The CCCTC-binding factor (CTCF) zinc finger protein and the cohesin complex are the best-studied factors that play a role in determining TAD structure. These factors bind to some TAD boundaries and form chromatin loops through the extrusion of chromatin through the doughnut-shaped cohesin molecule until a second CTCF site of the opposite polarity is encountered73. Although their binding at these boundaries can restrict enhancer–promoter interactions to within TADs74, there is no evidence that the TAD boundaries provide a mechanism for confining coordinated replicon activation to within the domain. Indeed, deletion of chromatin sites localized at the TAD boundaries did not affect replication timing of several domains75. Furthermore, although depleting CTCF or cohesin can severely diminish insulation of chromatin interactions between adjacent TADs measured by Hi-C, CTCF75,76 and cohesin77 depletion has no effect on replication timing. Moreover, high-resolution oligopaint tracing of individual chromosome segments revealed that there is a high degree of cell-to-cell heterogeneity in the positioning of TAD boundaries. These usually coincide with CTCF and cohesin binding sites but, in the absence of cohesin, TAD boundaries persist but no longer form at specific sites78.
In principle, TAD boundaries do not need to be fixed, but could instead simply be the site of transition between two chromosomal domains enriched for self-interaction79. In terms of replication timing, cis elements required for early replication, called early-replication control elements (ERCEs), were identified within TADs, located far from the boundaries. ERCEs interact with each other independently of CTCF/cohesin, and their interactions persist in the absence of CTCF75. Moreover, ERCEs are sites of master transcription regulatory factor binding and resemble transcriptional super-enhancers that have been shown to maintain their interactions in the absence of cohesin73,80. In the case of the analysed TAD, deletion of its boundaries had no effect on its replication timing, but deletion of ERCEs affected the local TAD architecture. Thus, ERCEs promote early replication within the confines of a TAD, and interact to form loops that contribute to TAD structure, independently of any specific boundary elements. Together, these observations suggest that replication timing may be an additional mechanism to link genome architecture to function, and interfering with ERCEs should provide a means to test this hypothesis and identify the underlying mechanisms.
Replication timing in cell fate transitions
Whereas TAD boundaries have been claimed to be largely conserved between cell types58,70,81,82, the A/B compartments and the regions associating with the nuclear lamina frequently differ between cell types67,83. Similarly, replication domains retain their boundaries in different cell types, but differ in their replication timing, correlating with A/B compartments and lamina association66. These observations support a ‘replication domain model’84, in which replication domains are invariant structural units, but the time at which they replicate, and their higher order organization and positioning within the nucleus, is developmentally regulated.
Spatiotemporal domain consolidation and compartment switching during differentiation.
Stem cells provide a system to track changes in properties (for example, replication timing and subnuclear localization of replication domains) relative to each other. Early observations demonstrated that when replication timing changes during a switch in cell fate, it does so in units of approximately 0.5 Mb (replication domains, FIG. 2a), and as stem cells become progressively more restricted in their differentiation potential, an increasing number of adjacent replication domains replicate at similar times. This results in fewer and larger early and late CTRs through a process termed ‘domain consolidation’4. Replication domains also consolidate spatially, which was initially visualized by FISH — as the physical compaction and movement of domains closer to their neighbours — and by targeted chromatin conformation capture (4C) — as increased interactions with neighbouring domains85. Compartment consolidation is coincident with changes in the subnuclear position of replication domains (interior to periphery or vice versa), also originally visualized by FISH85,86. Later, it was confirmed genome-wide by Hi-C that adjacent TADs consolidate spatially into A and B compartments, correlated with changes in replication timing that consolidate temporally83. These findings, together with the cytogenetic studies from the 1990s, have suggested an intimate link between subnuclear compartmentalization and replication timing.
Fig. 2 |. 3D chromatin structure and replication timing are dynamic during cell differentiation and during the cell cycle.
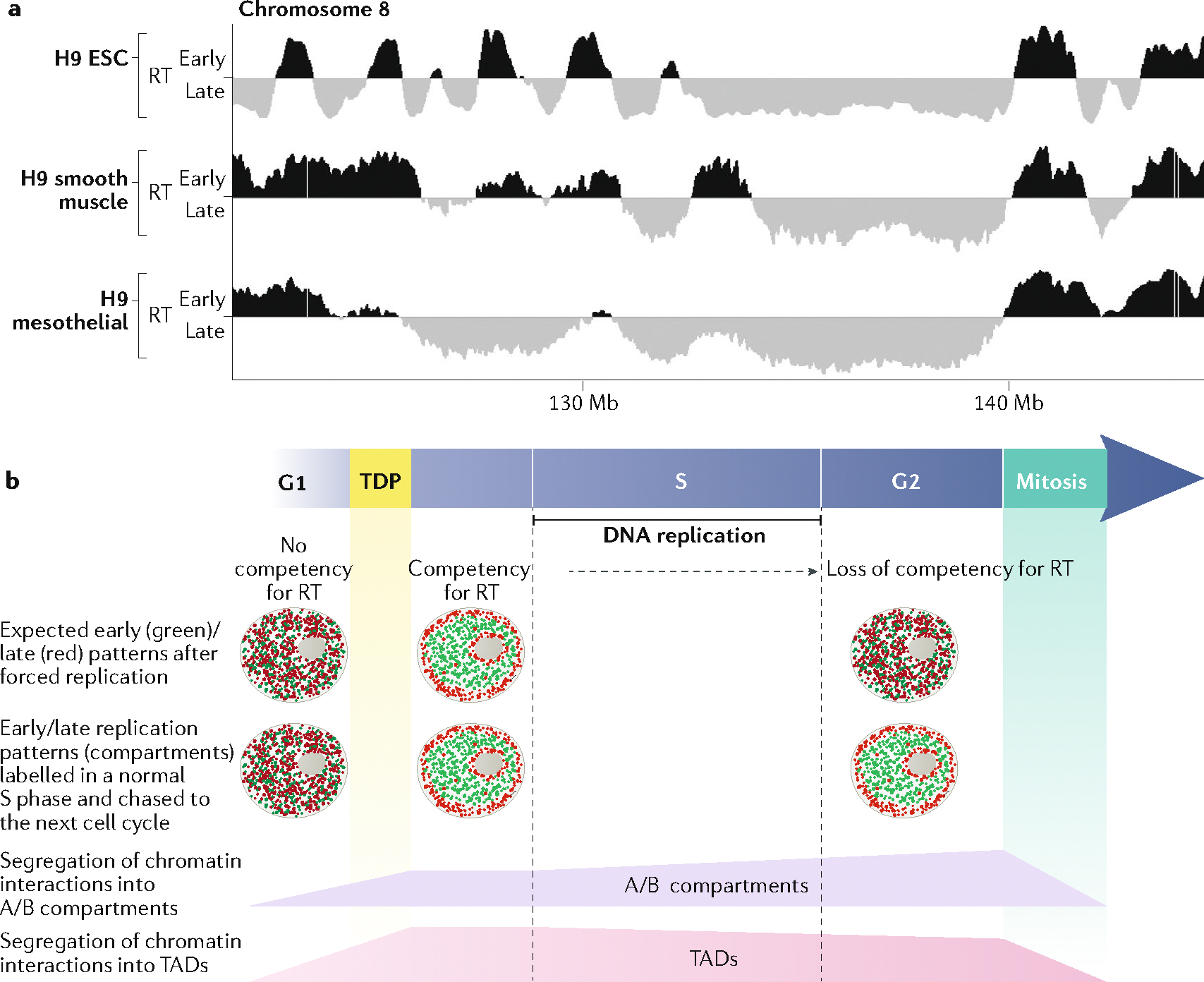
a | Replication timing (RT) is regulated during differentiation. Here, RT is shown for a region of chromosome 8, in human embryonic stem cells H9 (H9 ESC) and two differentiated H9 cells. Some regions switch RT during differentiation (black to grey, or grey to black), whereas others remain constant. RT data are available at www.replicationdomain.com. b | Both a defined RT programme and interphase chromatin architecture are set up coincidently at the timing decision point (TDP) during the G1 phase49,100. The information that defines RT is lost during the G2 phase99. In nuclei that are artificially forced to replicate their DNA before the TDP or after the S phase, DNA replication does not follow any specific RT49,99. The early- and late-replicating 3D compartments illuminated by replication labelling in the prior S phase are re-established at the TDP and persist through the remainder of interphase into the G2 phase, demonstrating that this spatial organization is not sufficient to dictate an RT programme. 3D chromatin interactions — both the separation between large-scale spatial compartments and the distinction between topologically associated domains (TADs) — are dismantled during mitosis and re-formed at the TDP, coincident with the establishment of RT100. Whereas compartments and TADs become slightly more or less distinct, respectively, during the course of the S phase101, the major architectural changes in genome architecture occur during entry into and exit from mitosis.
Interestingly, it has been found that the correlation between replication timing and chromatin A/B compartments is weaker in human embryonic stem cells (ESCs) than in differentiated cells and that during lineage commitment the first cell cycles are accompanied by uncoordinated changes in compartments and replication timing that become resolved in later cell cycles87. Thus, either the compartments or the replication timing can switch prior to the other during the early period of germ layer commitment, demonstrating that the two can be uncoupled, at least during periods of high plasticity. To understand the mechanisms underlying the close but indirect connection between compartments and replication timing, it will be necessary to be able to independently manipulate the compartment in which a domain resides and the compartment’s replication timing. The identification of cis elements that control replication timing and compartments provides the first molecular tool with the potential to enable such manipulation75.
Uncoupling between replication timing and 3D chromatin interactions during early embryogenesis.
Replication timing can be uncoupled from TAD structures. A recent study revealed that TADs form after the four-cell stage of mouse embryonic development, in a DNA replication-dependent manner88. However, spatiotemporal patterns of replication can already be observed in one-cell mouse embryos89, suggesting that A/B compartments and a replication timing programme are present already in the mammalian zygote, in the absence of TAD structures or zygotic transcription. Similarly, in early embryos undergoing rapid cleavage divisions, distinct patterns of replication timing and chromatin architecture are observed before (in zebrafish) or concomitant with (in Drosophila melanogaster) zygotic genome activation (ZGA), but it is not yet known when they are established during these early cleavage stages90–95. The study of genome architecture and DNA replication timing in cleavage-stage embryos will be facilitated by the recent development of single-cell Hi-C, single-cell RNA sequencing and single-cell replication timing28,96,97.
Replication timing during the cell cycle
The replication programme is established early during the G1 phase of the cell cycle, at the timing decision point (TDP)49,98, and is dismantled during the S phase99. If DNA replication is forced to initiate very early during G1 (before the TDP) or later during G2, this results in replication taking place in a random temporal order49,99.
The TDP: replication timing and chromatin architecture are established during early G1 phase.
The TDP is a short window of time during which chromatin domains move to the positions where they will reside for the remainder of interphase, which can be identified cytogenetically49. Moreover, targeted chromatin conformation capture (4C) showed that the re-establishment of 3D chromatin interactions to form both TADs and A/B compartments following cell division occurs within the same time window as the TDP100 (FIG. 2b), and single-cell Hi-C technology confirmed that chromatin interactions are re-established during the early G1 phase, which is the time window of the TDP101. Interestingly, this time window precedes the time when replication origin sites are selected, known as the origin decision point102. Forcing replication to initiate between the TDP and the origin decision point results in a correct programme of DNA replication — that is, early- and late-replication regions replicate at the expected time — but replication initiation (BOX 2) occurs at random sites103. This demonstrates that replication timing and genome architecture are established prior to and independently of mechanisms that control the sites where replication initiates. Consistent with independent mechanisms, replication timing is highly deterministic from cell to cell28,29, while initiation sites are chosen in a highly stochastic fashion with many sites used in less than 2% of cell cycles and no two chromosomes replicated from same cohort of initiation sites104 (BOX 2).
Box 2 |. Initiation of DNA replication; from origin licensing to origin firing.
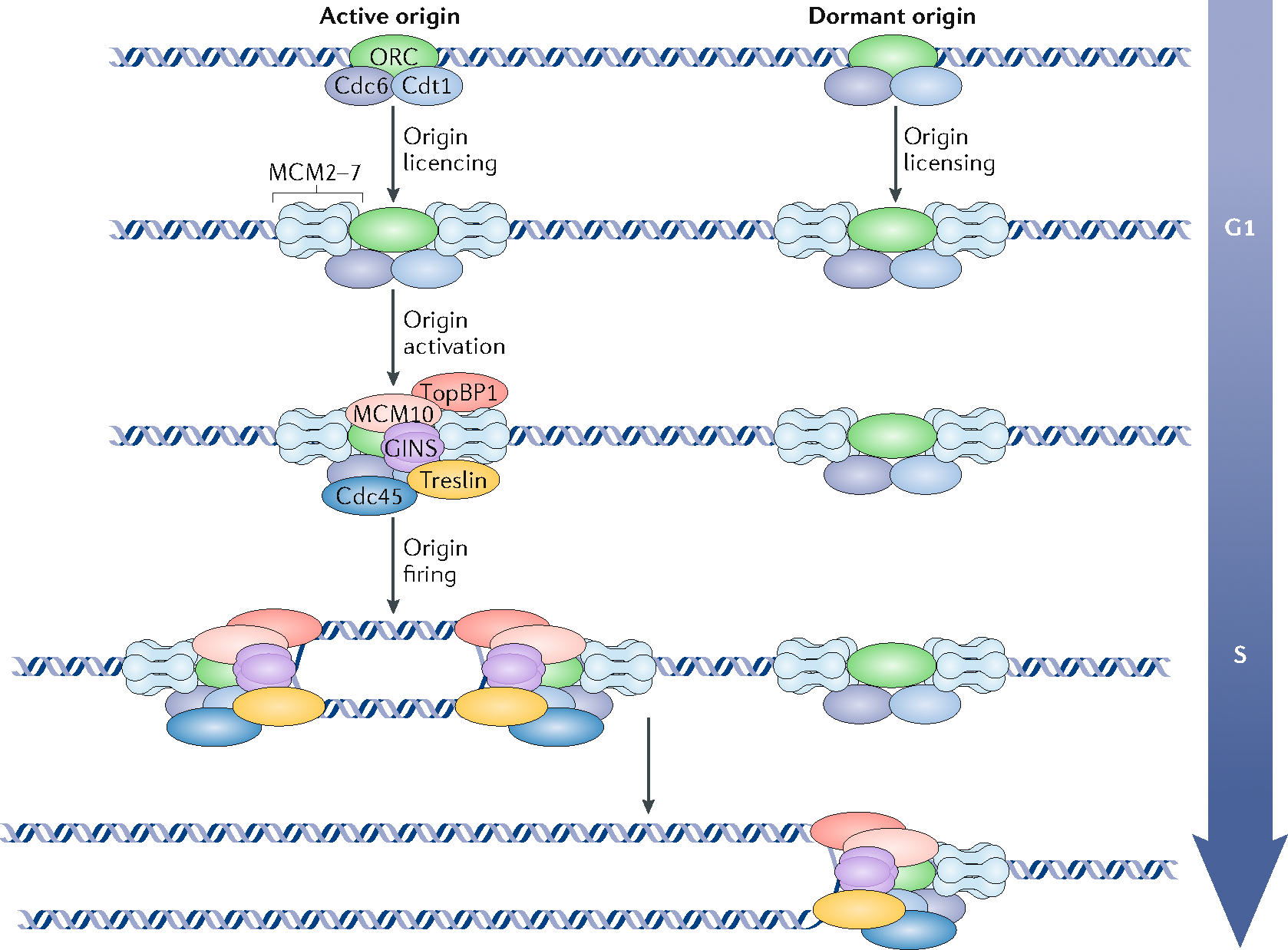
In eukaryotes, origin recognition complex (ORC), Cdc6 and Cdt1 cooperate to load two copies of the ring-shaped heterohexameric MCM replicative helicase around DNA in a process called origin licensing, and the fully loaded double hexamer is then referred to as a pre-replication complex (pre-RC). Pre-RCs are assembled during telophase but can continue to assemble throughout most of the G1 phase. A subset of origins is then activated (origin activation) during the G1/S transition whereas others remain dormant and are removed during passage of the replication fork (see the figure). Origin firing occurs at different times during S phase in a defined temporal order. Pre-RCs assembled as early as telophase49,192 are fully functional to initiate replication193,194 but remain inactive until they are phosphorylated by the Dbf4-dependent kinase (DDK)155, and are activated when cyclin-dependent kinase (CDK), MCM10, Treslin and TopBP1 participate in the assembly of Cdc45 and the GINS complex with the MCM hexamer to form the active CMG helicase157,195,196. Restricting initiation to only once per cell cycle is elegantly accomplished with a two-cycle engine; pre-RCs assemble under conditions that do not permit initiation, and initiation does not occur until conditions are no longer permissive for pre-RC assembly197. In mammals, the features of DNA or chromatin that dictate where pre-RCs assemble is unknown, but assembly sites appear to be highly flexible as many more pre-RCs are assembled than are activated and sites of initiation are chosen in a highly stochastic fashion; indeed, the same chromosomes in different cells do not use the same cohort of replication origins104,198. It is also not known how certain pre-RCs are chosen for initiation199. The excess pre-RCs are called ‘dormant origins’200,201 and they are used to ensure that all DNA is replicated in a timely manner, by serving as a backup when encountering conditions that slow replication forks or to complete fortuitous large regions that have not initiated as cells approach the G2 phase. Although replication origins do not have an intrinsic firing time, elements that regulate replication timing promote or inhibit the activation of pre-RCs in their chromatin domain.
Replication timing but not chromatin architecture is lost after S phase.
The position of subnuclear domains observed cytogenetically and the chromatin interactions observed by Hi-C are preserved during the G2 phase, whereas replication timing is lost99,100, indicating that the basic scaffold of interphase genome architecture is not sufficient to dictate replication timing (FIG. 2b). Thus, during the cell cycle, replication timing becomes established at the moment of compartmentalization and TAD formation, but this temporal programme is lost prior to the dismantling of 3D structure that occurs in mitosis99,100. It is worth noting that single-cell Hi-C studies found that 3D chromatin interactions are modified during the S phase, as TAD boundaries are weaker whereas compartments are more strongly separated in the G2 phase compared with G1 phase101. The finding that the partitioning of compartments becomes stronger when the replication timing programme becomes erased further emphasizes the indirect relationship between nuclear compartments and replication timing.
These results suggest a model in which 3D chromatin interactions create a scaffold during early G1 phase, with which additional cell cycle-regulated proteins interact to establish the DNA replication timing programme and are then cleared from chromatin during DNA replication. Possible candidates for these factors, the nature of which remains to be determined, are suggested below.
Regulation of replication timing
Identifying the regulators of DNA replication timing is challenging. First, almost all gene knockouts and knockdowns assessed in the literature do not cause any major alterations in the timing of replication, which suggests that a robust maintenance system is in place (TABLE 1). Second, studying origins of replication is technically difficult in mammals as the pool of origins that are used for replication differs from cell to cell (BOX 2), obscuring genome-wide studies on cell populations.
Epigenetic mechanisms as replication timing regulators.
Replication timing is extremely robust, remaining largely unaltered in a given cell type following genetic knockouts and drug perturbations, but it is extensively modified during cell fate transitions. It is also erased during the S phase, but restored during the following G1 phase of the cell cycle99. Altogether, this suggests the involvement of epigenetic mechanisms. Several observations support this hypothesis. First, replication timing has been cytologically observed to differ between the two alleles of imprinted genes in a cell-type-dependent manner105,106. Allele-specific replication timing can also be observed on non-imprinted loci107,108. In some cases, the asynchrony observed between alleles could be explained either by epigenetic mechanisms or by single-nucleotide polymorphisms or structural variation that influence the replication timing (see below), but in cases of random allelic differences such as X-chromosome inactivation39 or allele switching109 it presumably must occur via specific epigenetic marks. Second, some copies of the ribosomal RNA (rRNA) genes replicate at different times than other copies, and this difference is dependent on their DNA methylation state (FIG. 3a). rRNA genes are present in multiple copies in eukaryotes. In mammals they are regulated by the chromatin remodeller complex NoRC, which establishes silenced chromatin to repress transcription. Each rRNA gene can be present in either of two states: either not associated with NoRC, presenting a low level of methylation at the promoter and replicating early; or bound by NoRC, highly methylated and replicating late. Overexpression of NoRC leads to an increase in the proportion of rRNA gene copies per cell that are highly methylated and replicating late, indicating that DNA methylation and chromatin silencing can delay DNA replication110. It has been shown that for each rRNA gene, the activity is different on each allele: one allele is repressed and replicates late, whereas the other allele is active and replicates early111. Third, differential replication timing of the inactive (Xi) and active X chromosomes is a clear example where epigenetic mechanisms regulate replication timing. Delayed replication of the inactive X chromosome seems to be dependent on the expression of the lncRNA Xist, which coats the entire chromosome. However, Xist is also required to silence the Xi, raising the possibility that the effect on replication timing might be indirect. Indeed, direct effects of Xist on replication timing remain to be confirmed. Moreover, deletion of Xist in mouse fibroblasts causes the Xi to replicate even later in the S phase61, highlighting the complex role of Xist in controlling DNA replication. Finally, it has recently been shown that random allelic asynchrony of replication is common and regulated by lncRNAs43.
Fig. 3 |. Epigenetic versus sequence-specific regulation of replication timing.
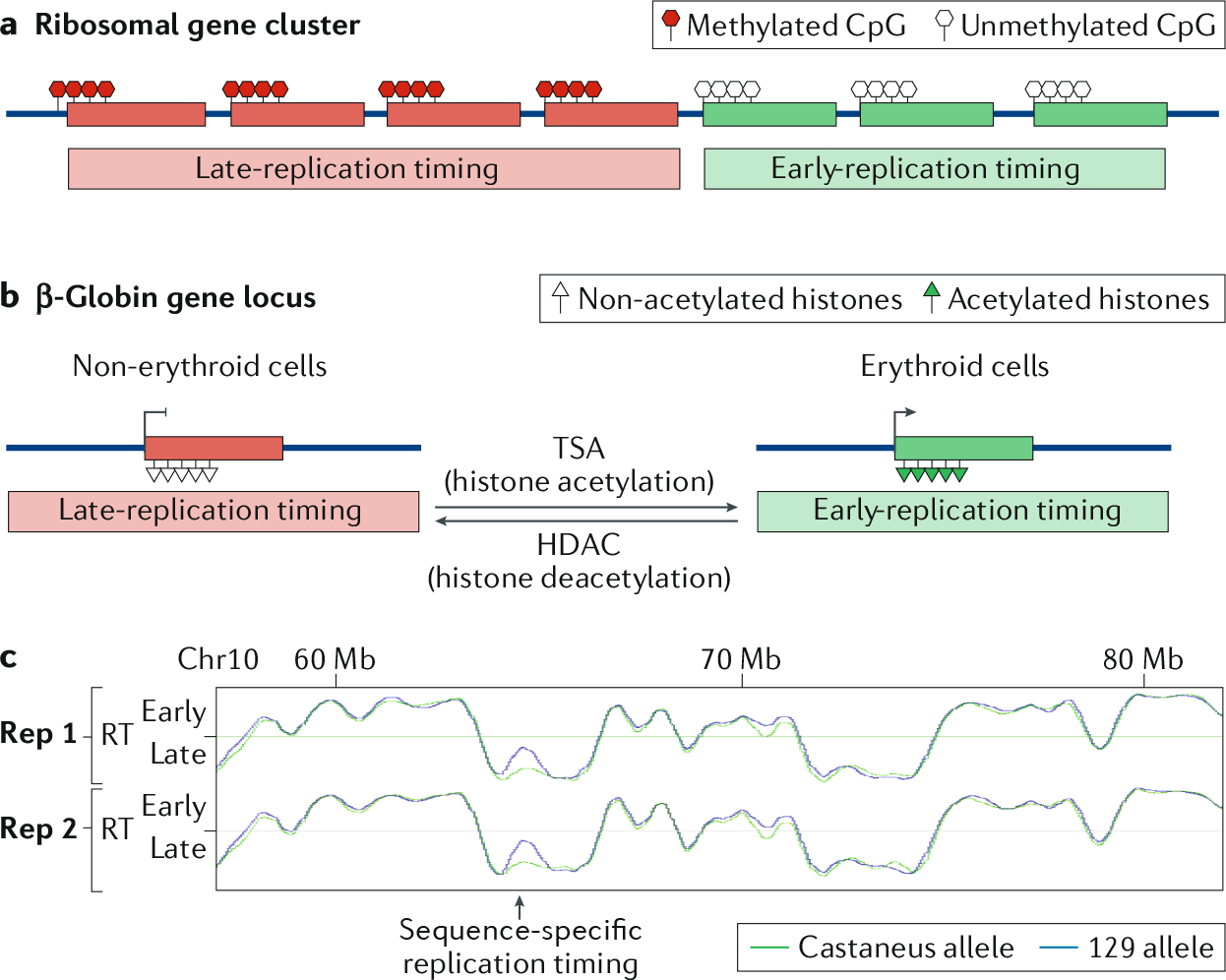
a | The cluster of ribosomal DNA consists of repeated genes coding for ribosomal RNAs. Correlated with the methylation status of each copy of these genes, they will replicate late (highly methylated) or early (low methylation level)110,111. b | The β-globin locus is an example of developmental regulation of replication timing (RT): in erythroid cells, the locus is activated, rich in histone acetylation and early replicating. In non-erythroid cells, the locus is silenced, without histone acetylation and late replicating. Inducing histone acetylation in non-erythroid cells with trichostatin A (TSA) treatment promotes early replication of the locus, whereas depleting the locus for histone acetylation by targeting histone deacetylases (HDACs) to a β-globin transgene in erythroid cells induces late replication of the locus117–120. c | Sequence-specific RT can be observed using subspecies F1 hybrid mouse embryonic stem cells (ESCs). Here, some non-imprinted regions have different RT, depending on the allele. Data from GEO series GSE95091 (REF.204).
The genomics era has enabled correlation of many features of chromatin and DNA sequence to replication timing. These studies have suggested many intriguing hypotheses, but also highlight that even very strong correlations do not imply causal relationships (for example112). Several histone post-translational modifications are associated with replication timing. Marks of active transcription such as histone H3 lysine 4 monomethylation (H3K4me1), dimethylation (H3K4me2) and trimethylation (H3K4me3), H3K20me1 and H3K36me3, and H3K9 and H3K27 acetylation, correlate with early replication113. Interestingly, the only mark strongly associated with late replication is H3K9me2 (REF.13), but removing detectable H3K9me2 by knocking out the methyltransferase responsible for the deposition of this mark has no effect on replication timing112, despite the fact that it may dissociate LADs from the nuclear periphery68 and can cause spreading of phosphorylated H3S10, which is normally restricted to early-replicating regions, into TTRs and late-replicating regions114. However, histone methylation on H4K20 is required for the proper replication of some late-replicating domains: impairing H4K20 methylation leads to a further delay in the replication of several late-replicating regions115.
A simple explanation for some marks being correlated with early DNA replication is that if these marks are rapidly established during chromatin maturation, the density of such marks detected by chromatin immunoprecipitation (ChIP) would be directly proportional to the copy number of each locus, and thus their profile along the genome would exactly match replication timing. For example, monomethylation is deposited rapidly, whereas dimethylation and trimethylation occur much later, sometimes early in the following cell cycle116. This could be the reason why H3K9me1 is predicted to correlate very closely with early DNA replication, but H3K9me2 and H3K9me3 do not. This hypothesis does not explain the H3K9me2 correlation, but does provide a clear example of the logical fallacy of concluding causality from correlation.
Early studies on the β-globin domain in human cells pointed to a role for histone acetylation in the control of replication timing117. This chromosomal domain is replicated early in erythroid cells that express the β-globin gene, but replicates late in cell types that do not express β-globin118 (FIG. 3b). In HeLa cells, inhibition of histone deacetylation by trichostatin A (TSA) advanced the time of origin firing in this locus119, whereas forcing deacetylation of this locus on a transgene integrated into the genomes of erythroid cells by targeting a histone deacetylase to the transgene delayed its replication120.
In mouse fibroblasts, TSA treatment of mouse cells slightly advanced the timing of replication of the normally late-replicating pericentromeric heterochromatin measured by immunofluorescence121. Also, in yeast, two histone deacetylases, Sir2 and Rpd3, regulate the replication timing of ribosomal DNA (rDNA)122. These studies indicate that DNA acetylation levels have an impact on the timing of its replication. Furthermore, the reverse may also be true; replication timing can influence the acetylation levels of chromatin123,124. Thus, histone acetylation seems to be a bona fide player in the regulation of early replication, which is now also supported by the discovery of ERCEs, which are strongly enriched in H3K27Ac marks75. Importantly, however, histone acetylation, especially H3K27Ac, is commonly found at all enhancer regions, whether or not they are ERCEs75, so histone acetylation alone is not sufficient to advance replication timing.
Interestingly, using mathematical modelling it is possible to predict cell-type-specific replication timing on the basis of DNase hypersensitivity profiles125. Although there is a general correlation between early replication and chromatin nuclease accessibility, domains whose replication timing is developmentally regulated are nuclease inaccessible, similar to late-replicating domains, and have many epigenetic features of late-replicating domains, even in cell types where they are replicated early (and thus predicted to have features of early-replicating domains)85,100,126.
Sequence-dependent mechanisms regulating replication timing.
At the genome-wide level, different techniques have tried to establish maps of initiation sites, or active origins of replication in mammalian cells, by sequencing different elements — purified small nascent DNA strands (SNS-seq; RNA-primed single-stranded DNA fragments of 800–1200 bp)127, replication bubbles (Bubble-seq; DNA fragments containing replication bubble structures)128, Okazaki fragments (OK-seq; nascent, EdU-labelled, single-stranded DNA fragments smaller than 250 bp)129 or initiation sites (ini-seq; first nucleotides to replicate at the onset of S phase)130 — or by chromatin immunoprecipitation of pre-replication complex proteins (ChIP-seq)131,132. The main observations are that the mapped sites are often associated with G-quadruplexes (secondary structures formed by guanines)127,130,133, high GC density134,135 or a nucleotide distribution asymmetry, with a bias in G/C or in A/T content, with different features often found at different origins136. Despite these differences in nucleotide content, no origin-specific sequence has been identified in eukaryotes other than in budding yeast137. There is great flexibility in the sites at which replication can initiate (BOX 2) and any DNA sequence can function as an origin in the right context138.
The relationship of origins to replication timing in mammalian cells remains elusive. The density of replication origins is generally higher in open early-replicating chromatin128–132. However, replication domains that switch replication timing during development (approximately 50% of the genome) have the same origin density as constitutively late-replicating regions, even when they replicate very early during the S phase100,127. Moreover, there is ample evidence to indicate that regulation of DNA replication timing acts upstream of the machinery that regulates where replication initiates18. This is also true in budding yeast, where factors binding to sites nearby but separable from origins influence replication timing139,140. However, in mammalian cells, the regulation is likely at the level of entire domains rather than individual origins and, even before the discovery of ERCEs, there was evidence that specific DNA sequences do play a role in replication timing. For example, ectopic insertion of the β-globin locus control region (LCR) advanced local replication timing141,142, demonstrating that exogenous DNA sequences can modify replication timing. In a mouse strain carrying a copy of human chromosome 21, the exogenous human chromosome conserved its species-specific replication timing in various mouse tissues, suggesting that the DNA sequence of the human chromosome dictates its characteristic pattern of replication timing143. To identify DNA sequences that potentially control replication timing, asynchronously replicating regions were identified by constructing separate replication timing profiles for each chromosome homologue in human erythroblasts, and these regions were then linked to large structural variations107. Consistent with the independent regulation of origin sites and timing of origin firing, these regions of asynchrony, although rare, could be linked to changes in origin efficiency, but not to the use of different origins136. A similar study identified rare replication timing quantitative trait loci as clusters of single-nucleotide polymorphisms and short indels linked to replication timing differences in human individuals144. These observations suggest a role for specific short DNA sequence alterations in the regulation of replication timing, in addition to epigenetic mechanisms. One prediction from these results is that crosses between more distantly related individuals should enhance asynchronous replication in the F1 hybrid offspring. However, an extensive study of cells derived from crosses between Mus musculus castaneus and Mus musculus musculus in different parental configurations, although clearly identifying replication timing asynchronies linked to subspecies genome (FIG. 3c), did not detect an enhanced degree of replication asynchrony, which was cell-type specific and varied from 1% to 12% in different cell types108. These results support a role for DNA sequences that is modulated by epigenetic events occurring during development.
Altogether, there is substantial evidence in support for both genetic and epigenetic having roles in regulating DNA replication timing. Interestingly, as replication timing can affect histone acetylation and genetic mutation frequency22,23, there is also evidence for replication timing affecting the epigenetic status and DNA sequence composition.
Cis and trans regulation of replication timing
Several elements are known to contribute to the regulation of DNA replication timing, acting either in cis, such as ASARs and ERCEs, or in trans, such as factors that promote or inhibit replication initiation (BOX 2). First, at the chromosome level, replication timing could require the presence of single long interspersed nuclear element 1 (LINE1), in the antisense orientation, within long non-coding ASARs, which are present in multiple copies on each chromosome, as observed on human chromosome 6 (REFS41–43). Although expressed from only one transcription unit on one of the two homologous chromosomes, ASARs identified on human chromosomes 6 and 15 associate in cis with the entire chromosome, forming an ‘RNA cloud’ that coats the entire chromosome (FIG. 4a), but if the LINE1 element within the ASAR is deleted or inverted, replication of the entire chromosome is severely delayed. Second, early replication of individual domains requires the presence of multiple ERCEs that interact in 3D space and, when all ERCEs within a domain are deleted, the entire domain replicates very late in the S phase75. Finally, trans regulatory elements comprise the factors directly involved in the regulation of origin firing, allowing or inhibiting the activation of the replicative helicase (BOX 2).
Fig. 4 |. Cis- and trans-acting elements regulating replication timing.
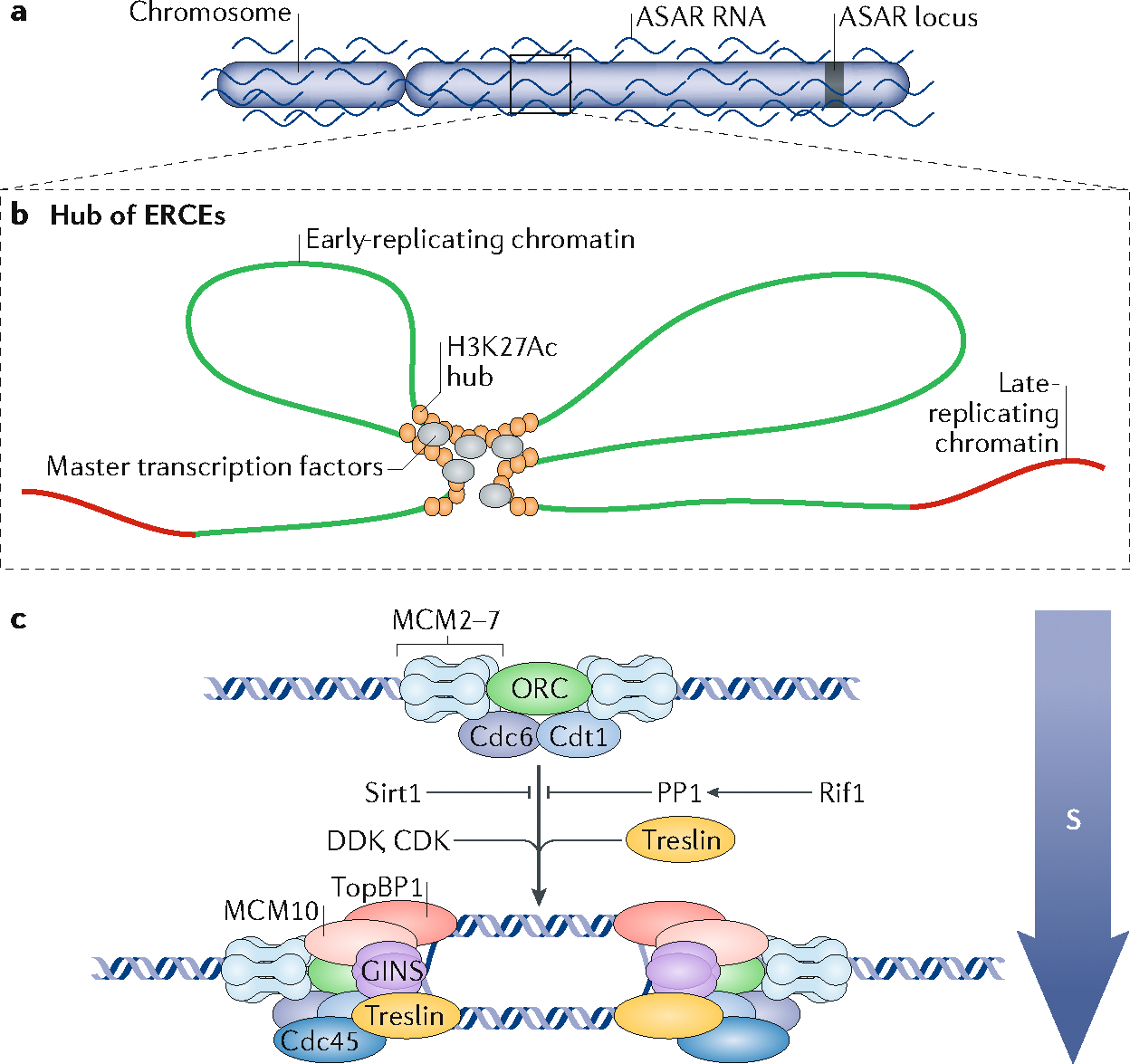
a | Long non-coding RNA asynchronous replication and autosomal RNAs (ASARs) are expressed from one locus specific to each homologous chromosome, and coat the whole chromosome from which they are expressed. This coating is necessary to define the temporal window during which DNA replication can occur on each chromosome43. b | Early-replication control elements (ERCEs) are sequences that are found to interact with each other within a megabase-sized domain, forming a cluster or ‘hub’ of ERCEs. They are large domains of acetylated histones and are bound by master transcription factors. ERCEs are necessary for the early replication of the chromosome domain in which they reside75. c | Replication origins are activated by recruitment of S-phase promoting factors (SPFs) such as the kinases Dbf4-dependent kinase (DDK) and cyclin-dependent kinase (CDK), and the protein Treslin, and inhibited by factors such as Rif1 and the deacetylase Sirt1 (see also BOX 2) that inhibit SPFs. All known trans-acting regulators of replication timing act on DDK, CDK or Treslin but potentially other such regulators may exist that act upon other SPFs such as TopBP1, Cdc45, Mcm10 and GINS.
lncRNAs that coordinate autosomal DNA synthesis.
The inactive Xi chromosome in female mammals is replicated with considerable delay compared with the active X chromosome. This delay is due to the expression of the Xist gene from the Xi chromosome, which then coats the Xi in cis. However, Xist gene deletion in a differentiated cell line leads to an increase in the delay of the replication of the Xi chromosome61. This observation has been largely ignored by the research community until recently, when lncRNAs with similar deletion phenotypes to Xist — named ASARs — were found to be expressed from human autosomes 6 and 15 and coat the chromosome from which they are expressed in cis41–43. Like Xist RNA145, ASARs also coat the chromosome from which they are expressed (FIG. 4a) when they are ectopically expressed and can delay the replication timing of the chromosome into which they are inserted. Also similar to Xist, loss of ASAR expression from the endogenous chromosome leads to a drastic delay in replication of the entire chromosome42,43. In some cases, the chromosome continues to be replicated into mitosis and fails to condense properly, probably because condensation requires completion of replication41,42. On human chromosome 6, maintenance of a normal replication timing programme was found to be dependent on a single LINE1 element within the ASAR6 gene. Normal timely chromosome 6 replication required antisense expression of this LINE1; deletion or inversion of the LINE1 resulted in delayed replication of the entire chromosome. When inserted ectopically into a different chromosome, ASAR6 suppressed the expression of endogenous LINE1 across the entire chromosome, suggesting that ASAR6 suppresses expression of the endogenous ASARs43. Interestingly, ASARs reside in regions of chromosomes that are subject to random asynchronous replication and are expressed mono-allelically from the allele that replicates later41,42. The molecular mechanisms by which ASARs function remain to be elucidated, but a working hypothesis is that homologous chromosomes express different ASARs, which coat the chromosome they are expressed from to ensure the synchronous replication of homologous chromosomes43. Thus, in this model, ectopic expression suppresses the resident ASAR, explaining why both deletion of the resident ASAR and ectopic expression of a second ASAR give rise to the same phenotype.
The search for cis elements that regulate origin firing.
The discovery of cis-acting lncRNAs that coordinate the synthesis of entire autosomes during the cell cycle was entirely unexpected; however, cis elements that regulate the firing of replication origins have been sought for decades. Early yeast studies indicated that origins replicated at times dictated by their chromosomal context146,147. However, the mechanisms of action of the yeast elements remained elusive and attempts to identify similar elements in higher eukaryotes have failed for a long period of time. In 1990, much excitement followed the discovery that cells from patients with Hispanic thalassaemia harboured a naturally occurring 35-kb deletion removing most of the human β-globin LCR. This chromosomal deletion was associated with the loss of β-globin gene transcription, the gain of closed chromatin structure and a shift to late replication across the β-globin locus in erythroid cells148,149. Ten years later, targeted deletion of the human LCR was achieved. This deletion led to the loss of β-globin gene transcription, but the domain still switched to early replication and open chromatin during erythroid cell differentiation150. The lack of effect of this deletion on replication timing at the native locus was never resolved and this setback, coupled with the failure to identify other cis elements regulating replication timing, led many to speculate that replication timing may be regulated exclusively by epigenetic mechanisms, as suggested by the observed random and imprinted allelic differences (discussed above), or by complex sequence features that are very difficult to identify by chromosome engineering methods (for example, AT content and G4 quadruplex density).
This longstanding open question was resolved by recent studies using CRISPR–Cas9 to generate a large series of deletions and inversions in a replication domain in mouse ESCs, which revealed that replication timing, transcription and the 3D architecture of the domain are under the control of defined cis-acting elements termed ERCEs (discussed above)75 (FIG. 4b). A single ERCE has partial activity to advance replication timing, but at least two ERCEs are required for replication early in the S phase.
ERCEs resemble super-enhancers as they contain broad domains of acetylated histones (mainly H3K27ac) and multiple sites that are co-occupied by master transcription factors Oct4, Sox2 and Nanog (OSN) as well as by the histone acetyltransferase p300. Interestingly, ERCEs interact with each other in 3D but are not bound by CTCF and cohesin, which are the major known players in mediating chromatin interactions73,151. On the basis of these properties, ERCE loci were predicted genome-wide, and deletions of several predicted ERCEs further validated their existence75. Moreover, deletion of ERCEs results in a domain-wide switch from A to B Hi-C compartments, disruption of local TAD architecture and loss of all transcription throughout the domain75. These results show that replication timing, spatial chromatin organization and transcription are intimately linked and co-regulated. They also provide an explanation for the enigmatic results of the past: the β-globin LCR may be one of several redundant erythroid-activated ERCEs in the region of the β-globin domain that are missing in the Hispanic thalassaemia deletion.
Trans-acting regulators of replication timing recruit or antagonize essential initiation factors.
Replication is initiated when the cyclin-dependent kinases152,153, the Dbf4-dependent kinase Cdc7-Dbf4 (DDK)154,155, the protein complex GINS156 and the initiation factors Treslin157,158, Mcm10 and Cdc45 (REF.156) cooperate to activate a subset of pre-loaded MCM helicases, known as pre-replication complexes (pre-RCs) (BOX 2, FIG. 4c). The remainder of the pre-RCs are either activated later in S phase or remain dormant and are removed when a replication fork passes through them (in vertebrates, the vast majority remain dormant).
Most of our knowledge of trans-acting factors that can control replication timing comes from studies in yeasts, where the common function is to promote or inhibit pre-RC activation. For example, the fission yeast telomere binding factor TAZ1 impairs DDK activation during early S phase at a set of late origins by blocking the recruitment of Sld3 (the homologue of mammalian Treslin) to the pre-RC, thus preventing origin firing until late S phase159. In budding yeast and fission yeast, pericentromeric heterochromatin is replicated early, following the recruitment of Dbf4 by Swi6 (the homologue of mammalian HP1) in fission yeast160 or Ctf19 (the homologue of CENP-P) in budding yeast161. In budding yeast, the forkhead 1 (Fkh1) and Fkh2 transcription factors recruit Dbf4 to a set of early-firing origins139. In mammalian cells, phosphorylated SIRT1 prevents dormant origin firing162, possibly through its ability to deacetylate and destabilize MCM10 (REF.163). Finally, the chromatin-binding factor RIF1 has a conserved role in replication timing control from yeast to humans164–166. In yeast, D. melanogaster, mouse and human, RIF1 has been shown to interact with the PP1 phosphatase167–170 (FIG. 4c), which has the ability to dephosphorylate the MCM complex at its Cdc7-dependent phosphorylation sites168, suggesting that RIF1 may delay DNA replication initiation by antagonizing phosphorylation by Cdc7 (REF.168). However, in budding yeast, RIF1 binds to DNA in the proximity of early- and late-replicating origins and may affect the activity of both171. Moreover, in mammalian cells, PP1 recruitment by RIF1 is also required for pre-RC formation167. In mouse ESCs, Rif1 is broadly enriched in late-replicating chromatin, but Rif1 depletion has effects on both early and late replication166. Overall, it seems that, in many cases, regulation of replication timing involves either promoting or antagonizing the function of proteins that activate pre-RCs.
In yeast, the ability of trans-acting factors to advance or delay replication is often linked to the 3D coalescence of commonly regulated origins. TAZ1-regulated origins localize near telomeres during G1 and early S phase172, and various genetic manipulations in this study indicated that the delayed initiation of DNA replication from these origins is dependent on their spatial proximity in the 3D nuclear space172. In addition, Fkh1 and Fkh2 transcription factors mediate the 3D clustering of a set of early origins173, and the ability of Fkh1 and Fkh2 to dimerize is essential for origin clustering and early replication independent of its ability to activate transcription140. The fact that mammalian ERCEs were identified by virtue of their 3D interactions could suggest that they function similarly. It is possible that spatial clustering promotes the formation of subnuclear domains that concentrate replication initiation factors. Human Treslin was recently shown to interact with Brd4 (REF.174), a bromodomain protein with high affinity for acetylated histones. As ERCEs are characterized by the presence of large stretches of H3K27Ac, this suggests a clear link between ERCE clustering and the formation of Treslin-rich domains that promote pre-RC activation.
Replication timing and transcription
The positive correlation between early replication and transcriptional activity in higher eukaryotes is well known4–8,31,175–179. However, several studies indicate that the two processes can be uncoupled in many contexts and are almost certainly indirectly related. Indeed, most genes that switch replication timing in mammals can be expressed when late replicating32. ASAR lncRNA genes are asynchronously replicated and expressed from the late-replicating allele41. In yeast, no correlation between the level of transcription and the timing of replication has been observed180. Moreover, although budding yeast Fkh1 and Fkh2 transcription factors are required for early replication of a large number of origins, mutations that impair their ability to dimerize without affecting their transcription regulatory function delay early origin firing without affecting transcription140,173.
In mammals, several studies have attempted to establish a causality between these two processes, but the results have been difficult to reconcile. A study in ESCs showed that targeting a strong transactivator to a late-replicating site led to transcription activation and re-localization of that chromosomal region towards the interior of the nucleus, whereas targeting a related peptide that could cause re-localization but not transcription did not anticipate DNA replication178. This led the authors to conclude that transcription activation is sufficient to advance replication timing178. However, studies deleting the β-globin LCR have shown that disrupting transcription at this locus does not prevent the switch to early replication of the β-globin locus during erythropoiesis117,150,181. Moreover, targeting a histone acetyltransferase to the β-globin locus in non-erythroid cells advanced replication timing whereas transcription of the β-globin gene remained low120. There is some evidence in support of the idea that transcriptional activity and the transcript length might have to reach a minimal threshold for it to have an effect on replication timing182. At the DppA2/4 locus in mouse ESCs, deletion of all ERCEs within a domain had an impact on both replication timing and transcription, but other deletions inactivated transcription with no changes in replication timing, or substantially delayed replication timing with no changes in transcription75. Overall, these studies show that transcription activation can be associated with earlier replication, but not systematically, reinforcing the hypothesis of an indirect relationship between these two processes.
Genome-wide analyses reporting widespread correlation between transcription and replication timing in the context of cell differentiation have also provided abundant examples of uncoupling between transcription and replication timing. In human and mouse cells, it is the constitutively early-replicating genes — which are early replicating in every cell type and constitute two-thirds of all genes — that drive the genome-wide correlation of early replication with transcription because they are, by definition, always early replicating when expressed. Very few genes are constitutively late replicating. By contrast, the majority of genes for which the timing of replication changes during differentiation (approximately one-third of genes) can be found to be transcribed in one or more cell types in which they are late replicating31,32. Moreover, the global correlation between replication timing and gene expression tends to decrease during the early stages of human stem cell fate commitment and differentiation32. Recently, taking close time points during the first few cell divisions of human ESCs undergoing a cell fate transition, many replication timing changes were found to be independent of changes in gene expression87, albeit often being only a temporary uncoupling. Finally, in early animal embryos undergoing rapid cleavage divisions, a DNA replication timing programme is established prior to the onset of transcription at the mid-blastula transition90,91,183,184.
A recent study assembling novel ‘replication timing networks’ provides a potential resolution to some of these complex observations. This study linked transcriptional changes that are coordinated with replication timing changes across many human ESC lines differentiating towards different lineages. Sets of genes were identified whose expression predicted early replication of a separate, unlinked, set of replication domains185. These genes almost exclusively encoded transcription factors. When ChIP data for these transcription factors were analysed, remarkably, all of them were found to co-occupy specific sites in the affected domains. This is reminiscent of the observation that transcription factors of the major pluripotency transcription-related network all bind together to ERCEs75. From these observations, we suggest that the imperfect correlation between replication timing and transcription could result from early replication being regulated by combinations of transcription factors, independently of their roles in transcription. Perhaps replication is influenced by transcription-related network transcription factors that, like Fkh1 and Fkh2 in budding yeast140, have separable roles in transcription and replication timing. Altogether, the most plausible conclusion from these studies is that transcription and replication timing are regulated by closely related but separable mechanisms.
Conclusion and perspectives
Studies using multiple model systems in the past few years have provided numerous new insights into the regulation of DNA replication timing and its relationship to 3D genome architecture that promise to fuel rapid future progress in this research field. We have discussed here how the timing of replication reflects chromosome architecture: early- and late-replicating chromatin coincides with A and B compartments, respectively, whereas replication domains are stable units of replication corresponding to a certain category of TADs. Coordinated firing of DNA replication origins within defined replication domains occurs at characteristic times that are conserved between homologous chromosomes and between individual cells. Replication timing is regulated at the level of these replication domains, independently of known 3D genome architectural factors or boundaries. Instead, association of cis-acting elements with transcriptional regulatory factors and their interaction in 3D space may establish subnuclear environments that recruit replication initiation factors. The independence of their roles in transcription versus 3D clustering (FIG. 5) could explain, in part, the imperfect correlation between transcription and replication timing. For example, the super-enhancer nature of ERCEs could create platforms for their self-association and recruitment of replication initiation proteins such as Treslin and DDK. A single ERCE may create a weak platform that can advance replication timing slightly, whereas additional ERCEs synergize by interacting to create a strong platform for recruitment, potentially through phase separation186. Likewise, factors that create transcriptionally repressed subnuclear environments may recruit factors that antagonize initiation proteins and delay replication. At the chromosome level, ASARs coat the entire chromosome to set the overall temporal window of replication during the S phase. Their mechanism of action and their relationship with ERCEs remain to be elucidated.
Fig. 5 |. Proposed model of organization within the nucleus.
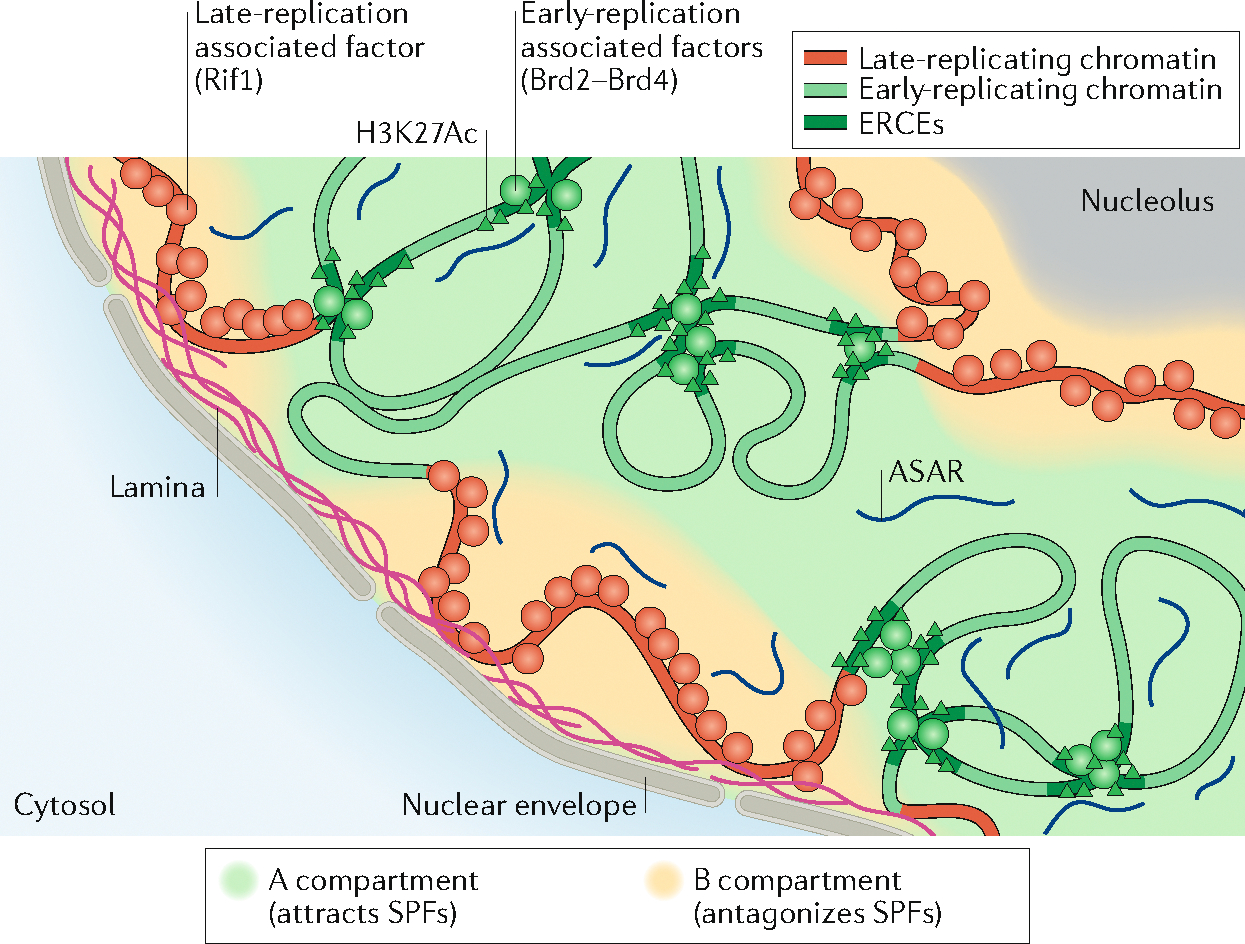
Model for the compartmentalization of the nucleus into and early- and late-replicating chromatin regions. In this model, early-replicating regions, within the A compartment, contain elements rich in H3K27Ac, bound by factors such as Brd2 and Brd4 that recruit S-phase-promoting factors (SPFs; see FIG. 4) such as Treslin. These elements, called early-replication control elements (ERCEs), form a platform for recruiting SPFs within the domain in which they belong. In this model, the replication of late-replicating regions, within the B compartment and associated with the nuclear lamina and the nucleolus, would be delayed by factors inhibiting origin firing (that is, antagonizing SPFs), such as Rif1 (which antagonizes DDK). At the chromosome level, coating of each chromosome by long non-coding RNAs known as asynchronous replication and autosomal RNAs (ASARs) could ensure the presence of replication machinery factors within each chromosome territory.
The recent discovery of ERCEs and ASARs and the recently available tools to dissect their structure and underpinning molecular mechanisms will fuel many future discoveries. First, both ERCEs and ASARs are expected to be widespread elements, and the systematic identification of such elements, at the genome-wide level, is an important and achievable goal. Second, ERCEs will be dissected molecularly to identify critical sequences and, ultimately, trans-acting binding partners, while proteins and RNAs that interact with ASARs will be identified. The question of whether ASARs delay entire chromosomes without disrupting the temporal order of replication will be addressed, and ultimately whether ASARs and ERCEs cooperate or whether ASARs are a global chromosome-wide threshold to ERCE activity. Third, our model predicts that there are different ERCEs in different tissues and that deletion of ERCEs specific to one cell type will have no effect on early replication in other cell types. Ultimately, artificially creating the individual components of ERCEs by targeting proteins that can induce chromatin loops, histone acetylation as well as manipulations that separate the functions of transcription versus chromatin replication of transcription factors, such as those recently initiated in budding yeast, will dissect the mechanisms regulating replication timing and how it is related to transcription and 3D organization.
Acknowledgements
This work was supported by National Institutes of Health (NIH) grants GM083337 and DK107965 to D.M.G. The authors thank M. Thayer for helpful discussions and critique of the manuscript.
Glossary
- Constant timing regions
(CTRs). Regions of the chromatin containing one or more adjacent replication domains that are replicating at nearly the same time.
- Replicons
Units of replication comprising a replication origin and the DNA being replicated.
- Timing transition regions
(TTRs). Regions of the chromatin between two replications domains replicating at different times.
- Replication forks
Complexes comprising template DNA, oligonucleotide primers and proteins necessary for DNA replication. Two ‘sister’ replication forks are assembled at each replication origin and may be associated in 3D space.
- Replication stress
Stalling of replication forks, which can be caused by chemical interference or radiation, or can be the result of a lack of nucleotides or proteins necessary for DNA replication.
- Mismatch repair
(MMR). DNA repair mechanism occurring at the replication fork involved in the repair of base mismatch and small insertions/deletions occurring during DNA replication or recombination.
- Hi-C
A genome-wide chromatin conformation capture technology that uses chromatin digestion, re-ligation of sequences in close proximity and sequencing to identify chromatin pairwise 3D chromatin interactions in the nucleus.
- Principal component analysis
(PCA). Mathematical transformation applied on complex (with multiple variables) datasets, to extract values describing the data called PC1, PC2 and so forth, with PC1 the value best describing the data.
- Xist
Long non-coding RNA that is expressed in female cells from only one x chromosome. xist inactivates the x chromosome from which it is expressed, enabling dosage compensation (similar x-linked gene expression levels in male and female cells).
- Asynchronous replication and autosomal RNAs
(ASARs). Long non-coding RNAs that are essential for the timely replication and condensation of the entire chromosome from which they are expressed.
- Lamina-associated domains
(LADs). Domains of chromatin that come in close proximity to the nuclear lamina.
- Inter-LADs
Domains between two adjacent lamina-associated domains.
- Topologically associated domains
(TADs). Domains of chromatin enriched for 3D interactions within the domain as compared with between domains.
- Correlative live and super-resolution microscopy
Combines the temporal resolution of time-lapse fluorescence microscopy with the spatial resolution of super-resolution microscopy.
- CCCTC-binding factor
(CTCF). Protein highly conserved in eukaryotes that associates with chromatin to mediate transcription and chromatin insulation.
- Cohesin
Protein complex composed of SMC1 and SMC3 that forms a large ring structure that accommodates two strands of chromatin. Cohesin has a key architectural role, forming chromatin loops and maintaining sister chromatids tied together after DNA replication.
- Early replication control elements
(ERCEs). DNA sequences necessary for the early replication of entire replication domains.
- Super-enhancers
Chromatin region rich in enhancers, with a high level of transcription associated factors and acetylated histones.
- Chromatin conformation capture
Technologies that assess 3D interactions within chromatin. These technologies are based on the cutting and re-ligation of chromatin immobilized in intact nuclei and identification of pairwise interactions by quantitative PCR or sequencing.
- Zygotic genome activation
(ZGA). Stage of embryonic development at which the transcription of the zygotic genome becomes activated.
- Imprinted genes
Genes that are expressed only from one chromosome homologue, the choice of which is dependent on its parental origin. The mechanism behind genomic imprinting relies on DNA methylation.
- β-Globin domain
A megabase-sized locus containing the β-globin gene, which is expressed only in erythroid cells. DNA replication properties of this domain have been extensively studied as it replicates early in erythroid cells and late in other cell types.
- Ribosomal DNA
(rDNA). Part of the genome coding for ribosomal RNA. rDNA is constituted of several copies of the same genes, the expression of each varying depending on cell type.
- Indels
Insertions or deletions in the genome of a cell or an individual.
- Long interspersed nuclear element 1
(LINE1). Class of transposable elements estimated to be present at around 500,000 copies in the human and mouse genomes.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Taylor JH Asynchronous duplication of chromosomes in cultured cells of Chinese hamster. J. Biophys. Biochem. Cytol. 7, 455–464 (1960). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desprat R et al. Predictable dynamic program of timing of DNA replication in human cells. Genome Res. 19, 2288–2299 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farkash-Amar S et al. Global organization of replication time zones of the mouse genome. Genome Res. 18, 1562–1570 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hiratani I et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLOS Biol. 6, e245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacAlpine DM, Rodríguez HK & Bell SP Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 18, 3094–3105 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schübeler D et al. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat. Genet. 32, 438–442 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Woodfine K et al. Replication timing of the human genome. Hum. Mol. Genet. 13, 191–202 (2004). [DOI] [PubMed] [Google Scholar]
- 8.White EJ et al. DNA replication-timing analysis of human chromosome 22 at high resolution and different developmental states. Proc. Natl Acad. Sci. USA 101, 17771–17776 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhind N & Gilbert DM DNA replication timing. Cold Spring Harb. Perspect. Biol. 5, a010132 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller CA & Nieduszynski CA Conservation of replication timing reveals global and local regulation of replication origin activity. Genome Res. 22, 1953–1962 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y et al. Continuous-trait probabilistic model for comparing multi-species functional genomic data. Cell Syst. 7, 208–218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryba T et al. Replication timing: a fingerprint for cell identity and pluripotency. PLOS Comput. Biol. 7, e1002225 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryba T et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 20, 761–770 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farkash-Amar S & Simon I Genome-wide analysis of the replication program in mammals. Chromosome Res. 18, 115–125 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Xu J et al. Genome-wide identification and characterization of replication origins by deep sequencing. Genome Biol. 13, R27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agier N et al. The evolution of the temporal program of genome replication. Nat. Commun. 9, 2199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantiero D, Mackenzie A, Donaldson A & Zegerman P Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 30, 4805–4814 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rivera-Mulia JC & Gilbert DM Replicating large genomes: divide and conquer. Mol. Cell 62, 756–765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Müller CA & Nieduszynski CA DNA replication timing influences gene expression level. J. Cell Biol. 216, 1907–1914 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voichek Y, Bar-Ziv R & Barkai N Expression homeostasis during DNA replication. Science 351, 1087–1090 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Padovan-Merhar O et al. Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Mol. Cell 58, 339–352 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sima J & Gilbert DM Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr. Opin. Genet. Dev. 25, 93–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Supek F & Lehner B Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521, 81–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kazanov MD et al. APOBEC-induced cancer mutations are uniquely enriched in early-replicating, gene-dense, and active chromatin regions. Cell Rep. 13, 1103–1109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blumenfeld B, Ben-Zimra M & Simon I Perturbations in the replication program contribute to genomic instability in cancer. Int. J. Mol. Sci. 18, E1138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J et al. The distribution of genomic variations in human iPSCs is related to replication-timing reorganization during reprogramming. Cell Rep. 7, 70–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu J et al. Influence of ATM-mediated DNA damage response on genomic variation in human induced pluripotent stem cells. Stem Cells Dev. 25, 740–747 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dileep V & Gilbert DM Single-cell replication profiling to measure stochastic variation in mammalian replication timing. Nat. Commun. 9, 427 (2018). This article develops methods to quantify the degree of cell-to-cell and homologue-to-homologue variation in the replication timing programme in single-haploid and inter-species F1 hybrid mouse cells, demonstrating high conservation of replication timing and chromatin compartments, conclusions that were later confirmed in Takahashi et al. (2019).
- 29.Takahashi S et al. Genome-wide stability of the DNA replication program in single mammalian cells. Nat. Genet. 51, 529–540 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Ozgyin L, Horvath A, Hevessy Z & Balint BL Extensive epigenetic and transcriptomic variability between genetically identical human B-lymphoblastoid cells with implications in pharmacogenomics research. Sci. Rep. 9, 4889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hiratani I et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 20, 155–169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivera-Mulia JC et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25, 1091–1103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson KA, Elefanty AG, Stanley EG & Gilbert DM Spatio-temporal re-organization of replication foci accompanies replication domain consolidation during human pluripotent stem cell lineage specification. Cell Cycle 15, 2464–2475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berezney R, Dubey DD & Huberman JA Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 108, 471–484 (2000). [DOI] [PubMed] [Google Scholar]
- 35. Xiang W et al. Correlative live and super-resolution imaging reveals the dynamic structure of replication domains. J. Cell Biol. 217, 1973–1984 (2018). This article labels replication foci and tracks their dynamic movements through the cell cycle at super-resolution in living cells, demonstrating that replication foci, consisting of several simultaneously labelled replication forks clustered in space, have properties resembling the behaviour of self-associating chromatin units or TADs, consistent with the genomic studies in Pope et al. (2014).
- 36.Ryba T et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22, 1833–1844 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rivera-Mulia JC et al. DNA replication timing alterations identify common markers between distinct progeroid diseases. Proc. Natl Acad. Sci. USA 114, E10972–E10980 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sasaki T et al. Stability of patient-specific features of altered DNA replication timing in xenografts of primary human acute lymphoblastic leukemia. Exp. Hematol. 51, 71–82 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morishima A, Grumbach MM & Taylor JH Asynchronous duplication of human chromosomes and the origin of sex chromatin. Proc. Natl Acad. Sci. USA 48, 756–763 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoffregen EP, Donley N, Stauffer D, Smith L & Thayer MJ An autosomal locus that controls chromosome-wide replication timing and mono-allelic expression. Hum. Mol. Genet. 20, 2366–2378 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donley N, Stoffregen EP, Smith L, Montagna C & Thayer MJ Asynchronous replication, mono-allelic expression, and long range cis-effects of ASAR6. PLOS Genet. 9, e1003423 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donley N, Smith L & Thayer MJ ASAR15, A cis-acting locus that controls chromosome-wide replication timing and stability of human chromosome 15. PLOS Genet. 11, e1004923 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Platt EJ, Smith L & Thayer MJ L1 retrotransposon antisense RNA within ASAR lncRNAs controls chromosome-wide replication timing. J. Cell Biol. 217, 541–553 (2018). This article demonstrates that the activity of ASARs, mono-allelically expressed lncRNAs, coating the chromosome from which they are expressed and required for coordination of replication timing between chromosome homologues, can be traced to much smaller L1 retrotransposons expressed in the antisense direction within the ASAR lncRNAs.
- 44.zur Hausen H Chromosomal changes of similar nature in seven established cell lines derived from the peripheral blood of patients with leukemia. J. Natl Cancer Inst. 38, 683–696 (1967). [PubMed] [Google Scholar]
- 45.Smith L, Plug A & Thayer M Delayed replication timing leads to delayed mitotic chromosome condensation and chromosomal instability of chromosome translocations. Proc. Natl Acad. Sci. USA 98, 13300–13305 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sparvoli E, Levi M & Rossi E Replicon clusters may form structurally stable complexes of chromatin and chromosomes. J. Cell Sci. 107, 3097–3103 (1994). [DOI] [PubMed] [Google Scholar]
- 47.Ferreira J, Paolella G, Ramos C & Lamond AI Spatial organization of large-scale chromatin domains in the nucleus: a magnified view of single chromosome territories. J. Cell Biol. 139, 1597–1610 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jackson DA & Pombo A Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 140, 1285–1295 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dimitrova DS & Gilbert DM The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol. Cell 4, 983–993 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Cremer T & Cremer C Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2, 292–301 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Nakamura H, Morita T & Sato C Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp. Cell Res. 165, 291–297 (1986). [DOI] [PubMed] [Google Scholar]
- 52.O’Keefe RT, Henderson SC & Spector DL Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific α-satellite DNA sequences. J. Cell Biol. 116, 1095–1110 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hiratani I, Takebayashi S, Lu J & Gilbert DM Replication timing and transcriptional control: beyond cause and effect—part II. Curr. Opin. Genet. Dev. 19, 142–149 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solovei I et al. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137, 356–368 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Abney JR, Cutler B, Fillbach ML, Axelrod D & Scalettar BA Chromatin dynamics in interphase nuclei and its implications for nuclear structure. J. Cell Biol. 137, 1459–1468 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yaffe E et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLOS Genet. 6, e1001011 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rao SSP et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rowley MJ et al. Evolutionarily conserved principles predict 3D chromatin organization. Mol. Cell 67, 837–852 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cremer T et al. Role of chromosome territories in the functional compartmentalization of the cell nucleus. Cold Spring Harb. Symp. Quant. Biol. 58, 777–792 (1993). [DOI] [PubMed] [Google Scholar]
- 61.Diaz-Perez SV et al. A deletion at the mouse Xist gene exposes trans-effects that alter the heterochromatin of the inactive X chromosome and the replication time and DNA stability of both X chromosomes. Genetics 174, 1115–1133 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Diaz-Perez S et al. The element(s) at the nontranscribed Xist locus of the active X chromosome controls chromosomal replication timing in the mouse. Genetics 171, 663–672 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vogel MJ, Peric-Hupkes D & van Steensel B Detection of in vivo protein–DNA interactions using DamID in mammalian cells. Nat. Protoc. 2, 1467–1478 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Farkash-Amar S et al. Systematic determination of replication activity type highlights interconnections between replication, chromatin structure and nuclear localization. PLOS ONE 7, e48986 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ragoczy T, Telling A, Scalzo D, Kooperberg C & Groudine M Functional redundancy in the nuclear compartmentalization of the late-replicating genome. Nucleus 5, 626–635 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pope BD et al. Topologically associating domains are stable units of replication-timing regulation. Nature 515, 402–405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peric-Hupkes D et al. Molecular maps of the reorganization of genome–nuclear lamina interactions during differentiation. Mol. Cell 38, 603–613 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kind J et al. Single-cell dynamics of genome–nuclear lamina interactions. Cell 153, 178–192 (2013). [DOI] [PubMed] [Google Scholar]
- 69.Rivera-Mulia JC & Gilbert DM Replication timing and transcriptional control: beyond cause and effect—part III. Curr. Opin. Cell Biol. 40, 168–178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nora EP et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moindrot B et al. 3D chromatin conformation correlates with replication timing and is conserved in resting cells. Nucleic Acids Res. 40, 9470–9481 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rao SSP et al. Cohesin loss eliminates all loop domains. Cell 171, 305–320.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rodríguez-Carballo E et al. The HoxD cluster is a dynamic and resilient TAD boundary controlling the segregation of antagonistic regulatory landscapes. Genes Dev. 31, 2264–2281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sima J et al. Identifying cis elements for spatiotemporal control of mammalian DNA replication. Cell 176, 816–830 (2019). This article describes the discovery and characterization of cis-acting elements controlling early replication, chromatin compartment, TAD architecture and transcription of entire replication domains in mouse ESCs.
- 76.Ma J & Duan Z Replication timing becomes intertwined with 3D genome organization. Cell 176, 681–684 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Oldach P & Nieduszynski CA Cohesin-mediated genome architecture does not define DNA replication timing domains. Genes 10, 196 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bintu B et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018). This article demonstrates that TAD boundaries are variable from cell to cell, and that when cohesin is depleted, which has no effect on replication timing, TAD boundaries are still detectable in single cells but their positions are too stochastic to be detectable in ensemble/population genomics studies.
- 79.Kimura A & Horikoshi M Partition of distinct chromosomal regions: negotiable border and fixed border. Genes Cells 9, 499–508 (2004). [DOI] [PubMed] [Google Scholar]
- 80.Schwarzer W et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bonev B et al. Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stadhouders R et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet. 50, 238–249 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dixon JR et al. Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pope BD & Gilbert DM The replication domain model: regulating replicon firing in the context of large-scale chromosome architecture. J. Mol. Biol. 425, 4690–4695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takebayashi S, Dileep V, Ryba T, Dennis JH & Gilbert DM Chromatin–interaction compartment switch at developmentally regulated chromosomal domains reveals an unusual principle of chromatin folding. Proc. Natl Acad. Sci. USA 109, 12574–12579 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fisher CL & Fisher AG Chromatin states in pluripotent, differentiated, and reprogrammed cells. Curr. Opin. Genet. Dev. 21, 140–146 (2011). [DOI] [PubMed] [Google Scholar]
- 87.Dileep V et al. Rapid irreversible transcriptional reprogramming in human stem cells accompanied by discordance between replication timing and chromatin compartment. Stem Cell Rep. 13, 193–206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ke Y et al. 3D Chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell 170, 367–381.e20 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Ferreira J & Carmo-Fonseca M Genome replication in early mouse embryos follows a defined temporal and spatial order. J. Cell Sci. 110, 889–897 (1997). [DOI] [PubMed] [Google Scholar]
- 90.Siefert JC, Georgescu C, Wren JD, Koren A & Sansam CL DNA replication timing during development anticipates transcriptional programs and parallels enhancer activation. Genome Res. 27, 1406–1416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaaij LJT, van der Weide RH, Ketting RF & de Wit E Systemic loss and gain of chromatin architecture throughout zebrafish development. Cell Rep. 24, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hug CB, Grimaldi AG, Kruse K & Vaquerizas JM Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 169, 216–228 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Sasaki T, Sawado T, Yamaguchi M & Shinomiya T Specification of regions of DNA replication initiation during embryogenesis in the 65-kilobase DNApolα-dE2F locus of Drosophila melanogaster. Mol. Cell. Biol. 19, 547–555 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hyrien O, Maric C & Méchali M Transition in specification of embryonic metazoan DNA replication origins. Science 270, 994–997 (1995). [DOI] [PubMed] [Google Scholar]
- 95.Seller CA, Cho C-Y & O’Farrell PH Rapid embryonic cell cycles defer the establishment of heterochromatin by Eggless/SetDB1 in Drosophila. Genes Dev. 33, 403–417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hiratani I & Takahashi S DNA replication timing enters the single-cell era. Genes 10, 221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagano T et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Raghuraman MK, Brewer BJ & Fangman WL Cell cycle-dependent establishment of a late replication program. Science 276, 806–809 (1997). [DOI] [PubMed] [Google Scholar]
- 99.Lu J, Li F, Murphy CS, Davidson MW & Gilbert DM G2 phase chromatin lacks determinants of replication timing. J. Cell Biol. 189, 967–980 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dileep V et al. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res. 25, 1104–1113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagano T et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu JR & Gilbert DM Lovastatin arrests CHO cells between the origin decision point and the restriction point. FEBS Lett. 484, 108–112 (2000). [DOI] [PubMed] [Google Scholar]
- 103.Li F, Chen J, Solessio E & Gilbert DM Spatial distribution and specification of mammalian replication origins during G1 phase. J. Cell Biol. 161, 257–266 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Demczuk A et al. Regulation of DNA replication within the immunoglobulin heavy-chain locus during B cell commitment. PLOS Biol. 10, e1001360 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kitsberg D et al. Allele-specific replication timing of imprinted gene regions. Nature 364, 459–463 (1993). [DOI] [PubMed] [Google Scholar]
- 106.Kawame H, Gartler SM & Hansen RS Allele-specific replication timing in imprinted domains: absence of asynchrony at several loci. Hum. Mol. Genet. 4, 2287–2293 (1995). [DOI] [PubMed] [Google Scholar]
- 107.Mukhopadhyay R et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLOS Genet. 10, e1004319 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rivera-Mulia JC et al. Allele-specific control of replication timing and genome organization during development. Genome Res. 28, 800–811 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Masika H et al. Programming asynchronous replication in stem cells. Nat. Struct. Mol. Biol. 24, 1132–1138 (2017). [DOI] [PubMed] [Google Scholar]
- 110.Li J, Santoro R, Koberna K & Grummt I The chromatin remodeling complex NoRC controls replication timing of rRNA genes. EMBO J. 24, 120–127 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schlesinger S, Selig S, Bergman Y & Cedar H Allelic inactivation of rDNA loci. Genes Dev. 23, 2437–2447 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yokochi T et al. G9a selectively represses a class of late-replicating genes at the nuclear periphery. Proc. Natl Acad. Sci. USA 106, 19363–19368 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yue F et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen CCL et al. H3S10ph broadly marks early-replicating domains in interphase ESCs and shows reciprocal antagonism with H3K9me2. Genome Res. 28, 37–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brustel J et al. Histone H4K20 tri-methylation at late-firing origins ensures timely heterochromatin replication. EMBO J. 36, 2726–2741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Alabert C et al. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 29, 585–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schübeler D et al. Nuclear localization and histone acetylation: a pathway for chromatin opening and transcriptional activation of the human β-globin locus. Genes Dev. 14, 940–950 (2000). [PMC free article] [PubMed] [Google Scholar]
- 118.Epner E, Forrester WC & Groudine M Asynchronous DNA replication within the human beta-globin gene locus. Proc. Natl Acad. Sci. USA 85, 8081–8085 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kemp MG, Ghosh M, Liu G & Leffak M The histone deacetylase inhibitor trichostatin A alters the pattern of DNA replication origin activity in human cells. Nucleic Acids Res. 33, 325–336 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Goren A, Tabib A, Hecht M & Cedar H DNA replication timing of the human β-globin domain is controlled by histone modification at the origin. Genes Dev. 22, 1319–1324 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Casas-Delucchi CS et al. Histone hypoacetylation is required to maintain late replication timing of constitutive heterochromatin. Nucleic Acids Res. 40, 159–169 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yoshida K et al. The histone deacetylases sir2 and rpd3 act on ribosomal DNA to control the replication program in budding yeast. Mol. Cell 54, 691–697 (2014). [DOI] [PubMed] [Google Scholar]
- 123.Lande-Diner L, Zhang J & Cedar H Shifts in replication timing actively affect histone acetylation during nucleosome reassembly. Mol. Cell 34, 767–774 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang J, Xu F, Hashimshony T, Keshet I & Cedar H Establishment of transcriptional competence in early and late S phase. Nature 420, 198–202 (2002). [DOI] [PubMed] [Google Scholar]
- 125.Gindin Y, Valenzuela MS, Aladjem MI, Meltzer PS & Bilke S A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol. Syst. Biol. 10, 722 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Takebayashi S, Ryba T & Gilbert DM Developmental control of replication timing defines a new breed of chromosomal domains with a novel mechanism of chromatin unfolding. Nucleus 3, 500–507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Besnard E et al. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 19, 837–844 (2012). [DOI] [PubMed] [Google Scholar]
- 128.Mesner LD et al. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res. 23, 1774–1788 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Petryk N et al. Replication landscape of the human genome. Nat. Commun. 7, 10208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Langley AR, Gräf S, Smith JC & Krude T Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq). Nucleic Acids Res. 44, 10230–10247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dellino GI et al. Genome-wide mapping of human DNA-replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res. 23, 1–11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Miotto B, Ji Z & Struhl K Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc. Natl Acad. Sci. USA 113, E4810–E4819 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cayrou C et al. The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res. 25, 1873–1885 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cadoret J-C et al. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl Acad. Sci. USA 105, 15837–15842 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Picard F et al. The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLOS Genet. 10, e1004282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bartholdy B, Mukhopadhyay R, Lajugie J, Aladjem MI & Bouhassira EE Allele-specific analysis of DNA replication origins in mammalian cells. Nat. Commun. 6, 7051 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stinchcomb DT, Struhl K & Davis RW Isolation and characterisation of a yeast chromosomal replicator. Nature 282, 39–43 (1979). [DOI] [PubMed] [Google Scholar]
- 138.Gilbert DM Making sense of eukaryotic DNA replication origins. Science 294, 96–100 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fang D et al. Dbf4 recruitment by forkhead transcription factors defines an upstream rate-limiting step in determining origin firing timing. Genes Dev. 31, 2405–2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ostrow AZ et al. Conserved forkhead dimerization motif controls DNA replication timing and spatial organization of chromosomes in S. cerevisiae. Proc. Natl Acad. Sci. USA 114, E2411–E2419 (2017). This article demonstrates the separation of transcription and replication timing functions of forkhead proteins; mutations that disrupt the dimerization of forkhead proteins disrupt the 3D clustering of early replication origins and their early replication but do not affect transcriptional control by these proteins.
- 141.Simon I et al. Developmental regulation of DNA replication timing at the human beta globin locus. EMBO J. 20, 6150–6157 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hassan-Zadeh V et al. USF binding sequences from the HS4 insulator element impose early replication timing on a vertebrate replicator. PLOS Biol. 10, e1001277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pope BD et al. Replication-timing boundaries facilitate cell-type and species-specific regulation of a rearranged human chromosome in mouse. Hum. Mol. Genet. 21, 4162–4170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Koren A et al. Genetic variation in human DNA replication timing. Cell 159, 1015–1026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wutz A & Jaenisch R A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol. Cell 5, 695–705 (2000). [DOI] [PubMed] [Google Scholar]
- 146.Friedman KL et al. Multiple determinants controlling activation of yeast replication origins late in S phase. Genes Dev. 10, 1595–1607 (1996). [DOI] [PubMed] [Google Scholar]
- 147.Yompakdee C & Huberman JA Enforcement of late replication origin firing by clusters of short G-rich DNA sequences. J. Biol. Chem. 279, 42337–42344 (2004). [DOI] [PubMed] [Google Scholar]
- 148.Forrester WC et al. A deletion of the human β-globin locus activation region causes a major alteration in chromatin structure and replication across the entire β-globin locus. Genes Dev. 4, 1637–1649 (1990). [DOI] [PubMed] [Google Scholar]
- 149.Aladjem MI et al. Participation of the human β-globin locus control region in initiation of DNA replication. Science 270, 815–819 (1995). [DOI] [PubMed] [Google Scholar]
- 150.Cimbora DM et al. Long-distance control of origin choice and replication timing in the human β-globin locus are independent of the locus control region. Mol. Cell. Biol. 20, 5581–5591 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nora EP et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tanaka S et al. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 445, 328–332 (2007). [DOI] [PubMed] [Google Scholar]
- 153.Mailand N & Diffley JFX CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 122, 915–926 (2005). [DOI] [PubMed] [Google Scholar]
- 154.Owens JC, Detweiler CS & Li JJ CDC45 is required in conjunction with CDC7/DBF4 to trigger the initiation of DNA replication. Proc. Natl Acad. Sci. USA 94, 12521–12526 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jiang W, McDonald D, Hope TJ & Hunter T Mammalian Cdc7–Dbf4 protein kinase complex is essential for initiation of DNA replication. EMBO J. 18, 5703–5713 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Moyer SE, Lewis PW & Botchan MR Isolation of the Cdc45/Mcm2–7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl Acad. Sci. USA 103, 10236–10241 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kumagai A, Shevchenko A, Shevchenko A & Dunphy WG Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 140, 349–359 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Guo C et al. Interaction of Chk1 with Treslin negatively regulates the initiation of chromosomal DNA replication. Mol. Cell 57, 492–505 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tazumi A et al. Telomere-binding protein Taz1 controls global replication timing through its localization near late replication origins in fission yeast. Genes Dev. 26, 2050–2062 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hayashi MT, Takahashi TS, Nakagawa T, Nakayama J & Masukata H The heterochromatin protein Swi6/HP1 activates replication origins at the pericentromeric region and silent mating-type locus. Nat. Cell Biol. 11, 357–362 (2009). [DOI] [PubMed] [Google Scholar]
- 161.Natsume T et al. Kinetochores coordinate pericentromeric cohesion and early DNA replication by Cdc7–Dbf4 kinase recruitment. Mol. Cell 50, 661–674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Utani K et al. Phosphorylated SIRT1 associates with replication origins to prevent excess replication initiation and preserve genomic stability. Nucleic Acids Res. 45, 7807–7824 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fatoba ST et al. Human SIRT1 regulates DNA binding and stability of the Mcm10 DNA replication factor via deacetylation. Nucleic Acids Res. 41, 4065–4079 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cornacchia D et al. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 31, 3678–3690 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yamazaki S et al. Rif1 regulates the replication timing domains on the human genome. EMBO J. 31, 3667–3677 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Foti R et al. Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol. Cell 61, 260–273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hiraga S-I et al. Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep. 18, 403–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hiraga S-I et al. Rif1 controls DNA replication by directing protein phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev. 28, 372–383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sreesankar E, Bharathi V, Mishra RK & Mishra K Drosophila Rif1 is an essential gene and controls late developmental events by direct interaction with PP1–87B. Sci. Rep. 5, 10679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sukackaite R et al. Mouse Rif1 is a regulatory subunit of protein phosphatase 1 (PP1). Sci. Rep. 7, 2119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hiraga S-I et al. Budding yeast Rif1 binds to replication origins and protects DNA at blocked replication forks. EMBO Rep. 19, e46222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ogawa S et al. Shelterin promotes tethering of late replication origins to telomeres for replication-timing control. EMBO J. 37, e98997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Knott SRV et al. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell 148, 99–111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Sansam CG et al. A mechanism for epigenetic control of DNA replication. Genes Dev. 32, 224–229 (2018). This article shows that human Treslin directly interacts with the histone acetylation readers BRD2/BRD4 and this interaction is necessary for the normal spatiotemporal distribution of replication foci, providing a clear link between histone acetylation, pre-RC activation and replication timing.
- 175.Goldman MA, Holmquist GP, Gray MC, Caston LA & Nag A Replication timing of genes and middle repetitive sequences. Science 224, 686–692 (1984). [DOI] [PubMed] [Google Scholar]
- 176.Hansen RS, Canfield TK, Fjeld AD & Gartler SM Role of late replication timing in the silencing of X-linked genes. Hum. Mol. Genet. 5, 1345–1353 (1996). [DOI] [PubMed] [Google Scholar]
- 177.Woodfine K et al. Replication timing of human chromosome 6. Cell Cycle 4, 172–176 (2005). [DOI] [PubMed] [Google Scholar]
- 178.Therizols P et al. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science 346, 1238–1242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hatton KS et al. Replication program of active and inactive multigene families in mammalian cells. Mol. Cell. Biol. 8, 2149–2158 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Raghuraman MK et al. Replication dynamics of the yeast genome. Science 294, 115–121 (2001). [DOI] [PubMed] [Google Scholar]
- 181.Reik A et al. The locus control region is necessary for gene expression in the human β-globin locus but not the maintenance of an open chromatin structure in erythroid cells. Mol. Cell. Biol. 18, 5992–6000 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Blin M et al. Transcription-dependent regulation of replication dynamics modulates genome stability. Nat. Struct. Mol. Biol. 26, 58–66 (2019). [DOI] [PubMed] [Google Scholar]
- 183.Pourkarimi E, Bellush JM & Whitehouse I Spatiotemporal coupling and decoupling of gene transcription with DNA replication origins during embryogenesis in C. elegans. eLife 5, e21728 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Seller CA & O’Farrell PH Rif1 prolongs the embryonic S phase at the Drosophila mid-blastula transition. PLOS Biol. 16, e2005687 (2018). This article presents an elegant study of how the onset of a replication timing programme during Drosophila mid-blastula transition is regulated through the interplay of Rif1 and S-phase kinases.
- 185.Rivera-Mulia JC, Kim S, Gabr H, Kahveci T & Gilbert DM Replication timing networks: a novel class of gene regulatory networks. Genome Res. in the press (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sabari BR et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Denker A & de Laat W The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev. 30, 1357–1382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Stevens TJ et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Phillips-Cremins JE et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Battulin N et al. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 16, 77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Berlivet S et al. Clustering of tissue-specific sub-TADs accompanies the regulation of HoxA genes in developing limbs. PLOS Genet. 9, e1004018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Méndez J & Stillman B Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol. 20, 8602–8612 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Okuno Y, McNairn AJ, den Elzen N, Pines J & Gilbert DM Stability, chromatin association and functional activity of mammalian pre-replication complex proteins during the cell cycle. EMBO J. 20, 4263–4277 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dimitrova DS, Prokhorova TA, Blow JJ, Todorov IT & Gilbert DM Mammalian nuclei become licensed for DNA replication during late telophase. J. Cell Sci. 115, 51–59 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Im J-S et al. Assembly of the Cdc45–Mcm2–7–GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl Acad. Sci. USA 106, 15628–15632 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Douglas ME, Ali FA, Costa A & Diffley JFX The mechanism of eukaryotic CMG helicase activation. Nature 555, 265–268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Diffley JF Once and only once upon a time: specifying and regulating origins of DNA replication in eukaryotic cells. Genes Dev. 10, 2819–2830 (1996). [DOI] [PubMed] [Google Scholar]
- 198.Anglana M, Apiou F, Bensimon A & Debatisse M Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell 114, 385–394 (2003). [DOI] [PubMed] [Google Scholar]
- 199.Sasaki T et al. The Chinese hamster dihydrofolate reductase replication origin decision point follows activation of transcription and suppresses initiation of replication within transcription units. Mol. Cell. Biol. 26, 1051–1062 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Woodward AM et al. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 173, 673–683 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ge XQ, Jackson DA & Blow JJ Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 21, 3331–3341 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Maya-Mendoza A & Jackson DA Labeling DNA. replication foci to visualize chromosome territories in vivo. Curr. Protoc. Cell Biol. 75, 22.21.1–22.21.16 (2017). [DOI] [PubMed] [Google Scholar]
- 203.Maya-Mendoza A, Olivares-Chauvet P, Shaw A & Jackson DA S phase progression in human cells is dictated by the genetic continuity of DNA foci. PLOS Genet. 6, e1000900 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Marchal C et al. Genome-wide analysis of replication timing by next-generation sequencing with E/L Repli-seq. Nat. Protoc. 13, 819–839 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zuin J et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl Acad. Sci. USA 111, 996–1001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Geeven G et al. Local compartment changes and regulatory landscape alterations in histone H1-depleted cells. Genome Biol. 16, 289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Takebayashi S-I et al. Murine esBAF chromatin remodeling complex subunits BAF250a and Brg1 are necessary to maintain and reprogram pluripotency-specific replication timing of select replication domains. Epigenetics Chromatin 6, 42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]