

Abstract
The US3 protein is a viral kinase that is conserved among the Alphaherpesvirinae. Here, we show that US3 of the swine alphaherpesvirus pseudorabies virus causes dramatic alterations in the cytoskeleton, resulting in the formation of long actin- and microtubule-containing cell projections in infected and transfected cells. Analysis with a GFP-labeled virus showed that multiple virus particles move inside the projections toward the tip. GFP-labeled virus could also be found in the cytoplasm of neighboring cells that were in contact with the projections. In addition, projection formation could be inhibited by using the actin-stabilizing drug jasplakinolide and could be induced by using the Rho kinase inhibitor Y27632. Analyzing the effect of these drugs on intercellular virus spread indicated that the observed US3-induced alterations in the host cytoskeleton are associated with enhanced intercellular virus spread, thereby suggesting a previously undescribed aspect of alphaherpesvirus spread.
Keywords: cytoskeleton, herpes, projections
Some viruses have been reported to exploit the host cytoskeleton in often very fascinating ways to facilitate important aspects of their life cycle like entry, egress, and intercellular spread. The polyomavirus simian virus 40 interacts with actin and microtubules to facilitate virus transport to the nucleus (1, 2), and vaccinia virus interacts with microtubules and induces the formation of actin tails to allow efficient egress and intercellular spread in the presence of virus-neutralizing antibodies (3–9). In addition, members of the herpesviruses, adenoviruses, and retroviruses have been shown to use dynein-dependent microtubule-associated transport for efficient delivery of the viral genome at the nucleus (10–13). The pseudorabies virus (PRV), a member of the alphaherpesvirus subfamily of the herpesviruses, has been shown to use bidirectional microtubule-mediated transport during entry and egress stages of infection in neurons (13, 14).
The alphaherpesviruses are a subfamily of the herpesviruses, containing closely related and important viruses of man and different animal species, including the human herpes simplex virus (HSV) and varicella-zoster virus (VZV) as well as several important animal viruses such as the porcine pseudorabies virus, bovine herpesvirus 1, and equine herpesvirus 1, which can cause mild, but sometimes devastating diseases, such as encephalitis. Alphaherpesviruses cause latent infections, and specific stress stimuli may result in reactivation of virus from latency (15). These reactivation events are responsible for the recurrent symptoms in many alphaherpesvirus-infected individuals (e.g., cold sores for HSV and shingles for VZV). These recurrent symptoms indicate that alphaherpesviruses have developed pathogenetically important strategies that enable them to spread in the presence of an active antiviral immunity, including virus-neutralizing antibodies (16).
Here, we report that the US3 protein kinase of the pseudorabies virus, which is conserved among alphaherpesviruses, causes the formation of long actin- and microtubule-containing cell projections that can contact neighboring cells. Newly formed virus particles move inside these projections, generally toward the tip, and projection formation was found to be associated with an increase in intercellular virus transmission in the presence of virus-neutralizing antibodies, especially in cells seeded at low density, thereby suggesting a previously uncharacterized aspect of alphaherpesvirus intercellular spread.
Materials and Methods
Virus Strains, Cells, and Inoculation. Wild-type PRV NIA3 strain, isogenic NIA3 US3-null and NIA3 US3-rescue strains, and a recombinant PRV Becker virus expressing the VP26 capsid protein fused to GFP were used (14, 17–19). The latter strain was a kind gift of Greg Smith (Northwestern University, Chicago) and Lynn Enquist (Princeton University, Princeton). The NIA3 strains were kindly provided by ID-DLO (Lelystad, The Netherlands). Swine testicle (ST) and rabbit kidney (RK13) cells were seeded at low (35,000 cells per ml) or high (250,000 cells per ml) density, as indicated, and cultivated in supplemented Eagle's MEM (medium A) as described (20). Cells were inoculated at a multiplicity of infection (MOI) of 10 unless stated otherwise.
Antibodies, Chemicals, and Buffers. FITC-labeled porcine polyclonal anti-PRV antibodies and PRV-specific hyperimmune serum 1955 were produced at our laboratory as described (21). Mouse monoclonal anti-US3 IgG1 antibodies were kindly provided by LeighAnne Olsen and Lynn Enquist (Princeton University), whereas rabbit polyclonal antibodies directed against the US3 protein were a kind gift from Bruce Banfield (University of Colorado, Denver). Phalloidin–Texas red, FITC-labeled goat anti-mouse IgG, Texas red-labeled goat anti-rabbit IgG, propidium iodide, and jasplakinolide were purchased from Molecular Probes. Monoclonal anti-alpha-tubulin IgG1, cytochalasin D, and nocodazole were from Sigma. FITC-labeled rat anti-mouse IgG1 was obtained from Becton Dickinson. Rho kinase inhibitor Y27632 [(R)-(+)-transN-(4-pyridyl)-4-(1-aminoethyl)-cyclohexane-carboxamide dihydrochloride] was purchased from Calbiochem. Mouse anti-V5 antibody, pcDNA3.1D:V5-His:lacZ and Lipofectamine reagent were from Invitrogen.
Transfection Assays. ST cells were transiently transfected with a V5-encoding control plasmid (pcDNA3.1D:V5-His:lacZ) (Invitrogen) or with a US3-encoding expression vector (pcDNA3.1D:V5-His:US3; pKG1) (22) by use of Lipofectamine reagent according to the manufacturer's instructions. At 24 h after transfection, cells were paraformaldehyde-fixed, Triton X-100-permeabilized, and processed for immunofluorescence staining.
Immunofluorescence Stainings. Dilution of antibodies or probes were always made in PBS. Staining filamentous actin (F-actin) of cells inoculated with the PRV VP26-GFP virus was done by incubating them with phalloidin–Texas red (1:100; 1 h at 37°C). Double staining of F-actin and PRV antigens or F-actin and the US3 protein was principally done as described (20). For double staining of F-actin and microtubules, the cells were processed as follows. At the indicated time points, cells were washed in cytoskeleton-stabilizing buffer (CSB) (10 mM Pipes buffer/150 mM NaCl/5 mM EGTA/5 mM MgCl2/5 mM glucose monohydrate), fixed with paraformaldehyde diluted in CSB (3%, 10 min), Triton X-100-permeabilized, and washed. Then, cells were incubated (1 h at 37°C) with a mixture of phalloidin–Texas red (1:100) and anti-tubulin IgG1 antibodies (1:100) and subsequently washed twice. Finally, cells were incubated (1 h at 37°C) with FITC-labeled rat anti-mouse IgG1 (1:50) and washed twice. For double staining of the US3 protein and microtubules, cells were processed in a similar way, with the exception that cells were incubated with a mixture of rabbit anti-US3 antibodies (1:75) and mouse anti-tubulin IgG1 (1:100), and subsequently with a mixture of Texas red-labeled goat anti-rabbit IgG (1:50) and FITC-labeled rat anti-mouse IgG1 (1:50).
Confocal and Time-Lapse Microscopy. Images of immunofluorescence stainings were acquired on a Leica TCS SP2 confocal microscope (Leica). An Argon 488-nm laser line and Gre/Ne 543-nm laser line were used to excite FITC and Texas red fluorochromes. Merged images were made by use of Leica confocal software. For time-lapse recording of living cells an ultraview Live Cell Imager (PerkinElmer) connected to an inverted fluorescence microscope (Zeiss Axiovert 200) equipped with a temperature and CO2 incubation chamber (Zeiss) was used. Excitation of GFP was at 488 nm with an Argon–Krypton laser.
Induction and Inhibiton of Projection Formation Using Y27632 or Jasplakinolide. Y27632, a specific inhibitor of the Rho-controlled actin signaling cascade (23, 24), was used to artificially induce US3-like cell projections in US3-null PRV-infected ST cells. Briefly, ST cells (1 day after seeding at low density) were inoculated with US3-null PRV, incubated for 1 h, washed once, and further incubated at 37°C with medium A. At 5 h postinoculation (hpi), cells were incubated for 1 h with 30 μM Y27632 (diluted in medium A). Jasplakinolide, a cyclic peptide isolated from the marine sponge Jaspis johnstoni that has the capacity to stabilize F-actin (25), was used to counteract formation of US3-induced cell projections in cells infected with wild-type or US3-rescue PRV. From 2 hpi onwards until fixation, cells were cultivated in the presence of a final concentration of 50 nM jasplakinolide (diluted in medium A). After fixation (6 hpi), Y27632- and jasplakinolide-treated cells were permeabilized and stained for both F-actin and PRV antigens. Two hundred cell clusters (consisting of 5–10 cells) were scored for the presence of cell projections, and data shown represent means ± SD of three independent observations.
Plaque Assays in the Absence or Presence of Different Drugs. ST cells (at 1 day after seeding when seeded at low density or 5 days after seeding when seeded at high density) were inoculated with wild-type PRV, US3-null PRV, or US3-rescue PRV at a MOI of 0.001. After a 1-h adsorption period at 37°C, the inoculum was removed and extracellular virus was inactivated by treatment of the monolayer with citrate buffer (40 mM Na citrate/10 mM KCl/135 mM NaCl, pH 3) for 1 min. Thereafter, cells were incubated in prewarmed medium A supplemented with 20% PRV-specific hyperimmune serum 1955 (SN titer 512; medium B) to neutralize extracellular virus. To test the effect of Y27632 on plaque size, wild-type PRV, US3-null PRV, and US3-rescue PRV-infected ST cells were incubated with Y27632 (diluted to a final concentration of 30 μM in prewarmed medium B) between 5 and 6 hpi. Afterward, cells were washed twice and maintained in medium B. To study the effect of jasplakinolide, cytochalasin D, or nocodazole on plaque size, infected ST cells were incubated with a final concentration of 50 nM jasplakinolide, 1 μM cytochalasin D, or 30 μM nocodazole (diluted in medium B) from 3 hpi onwards until the time of fixation. At 24 hpi, plaques were methanol-fixed (100%, 20 min, –20°C), stained for PRV antigens by use of FITC-labeled polyclonal anti-PRV antibodies, counterstained for nuclei with propidium iodide, and visualized by fluorescence microscopy. In cells seeded at low density, the number of nuclei per plaque was used as a measure for plaque size (a minimum of 100 plaques was scored per sample). In confluent monolayers of cells, plaques were measured as described (26). Data shown always represent means ± SD of triplicate assays.
Results
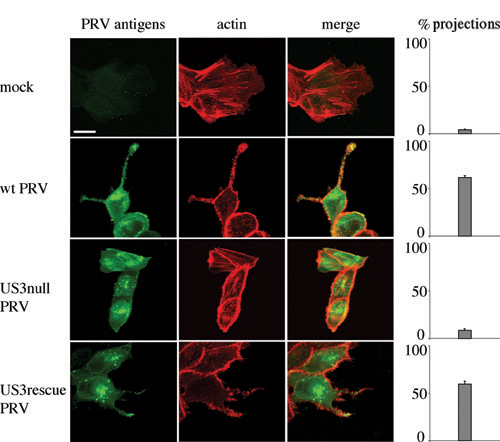
The PRV US3 Protein Kinase Causes the Formation of Actin-Containing Cell Projections. PRV infection of ST cells that were seeded at low cell density resulted in the formation of long, actin-containing cell projections that were often branched and were between 5 and 40 μm in length (Fig. 1A). Projections typically appeared from 4 to 6 hpi onwards. Projections were not observed in cells infected with a US3-null virus or mock-infected cells, whereas a US3-rescue virus showed projection formation like observed using wild-type virus (Fig. 1 A). Similar US3-mediated cell projections were observed in rabbit kidney (RK-13) cells, indicating that the formation of projections is not restricted to ST cells (Fig. 6, which is published as supporting information on the PNAS web site). In addition, in both cell types, US3-mediated formation of cell projections was accompanied with the disassembly of actin stress fibers, in agreement with what we described earlier (20).
Fig. 1.

US3 mediates the formation of long, actin-containing cell projections in PRV-infected and US3-transfected cells. (A) ST cells that were seeded at low density (cell clusters of 5–10 cells) were either mock-infected or infected with wild-type, US3-null, or US3-rescue PRV at a MOI of 10. Cells were paraformaldehyde-fixed at 7 hpi, permeabilized, and stained for viral proteins (green) and actin (red). Arrowheads indicate cell projections. (Bar, 10 μm.) (Right) Graphs showing percentages of cell clusters that display projections (means ± SD of triplicate assays). (B) Live cell imaging of the formation of cell projections in PRV-infected cells. ST cells were inoculated with PRV at a MOI of 10 and analyzed at different time intervals by using phase contrast microscopy. Arrows indicate cell projections. (Bar, 10 μm.) (C) ST cells were transfected with a eukaryotic expression vector encoding the PRV US3 protein (Upper) or a control protein (V5, Lower). At 24 h after transfection, cells were paraformaldehyde-fixed, permeabilized, and stained for US3 or V5 (green) and actin (red). Arrowheads indicate cell projections. (Bar, 10 μm.) (Right) Graphs showing percentages of transfected cells that display projections (means ± SD of triplicate assays).
Time-lapse microscopy showed that the projections in PRV-infected cells did not result from passive retraction of the cell but that they were actively produced during infection (Fig. 1B).
In addition, the US3 protein was found to be able to induce the formation of cell projections in the absence of other viral proteins. Indeed, ST cells that were transfected with a mammalian expression vector encoding the US3 protein showed actin-containing cell projections that were similar as observed in PRV-infected cells (Fig. 1C). Cell projections were not observed in cells transfected with a control plasmid (Fig. 1C). Transfection of the US3-encoding expression vector showed that cell projections are also formed in cells that make part of a monolayer (Fig. 1C).
Thus, the PRV US3 protein kinase causes the formation of long, actin-containing cell projections, both in infected and transfected cells.
US3-Induced Cell Projections Contain Microtubules. Alphaherpesviruses are known to be able to travel long intracellular distances along microtubules, e.g., in neuronal axons (13, 14). If the cell projections that we observe during infection contain microtubules, this may open the possibility that PRV can travel in these projections. Double labeling of the cell projections for actin and tubulin showed that the projections are filled with microtubule bundles up to the tip of the projection. Only the small side branches appeared to be devoid of tubulin (Fig. 2A). Cell projections that were formed in US3-transfected cells were also found to contain microtubule bundles (Fig. 2 A).
Fig. 2.

US3-induced cell projections contain microtubules in PRV-infected and US3-transfected cells and virus particles in PRV-infected cells. (A) ST cells, at 7 hpi with wild-type PRV (Upper) or 24 h after transfection with a US3-encoding plasmid (Lower), were paraformaldehyde-fixed, permeabilized, and stained for microtubules (green) and actin (red, Upper) or US3 (red, Lower). Arrowheads indicate cell projections. (Bar, 10 μm.) (B) ST cells, at 7 hpi with a PRV strain that encodes a GFP-tagged VP26 capsid protein (green), were paraformaldehyde-fixed, permeabilized, and stained for actin (red). Small green dots represent virus particles. Regions with virus particles are indicated with arrowheads. (Bar, 10 μm.) (Inset) An enlargement of the boxed area.
Thus, cell projections observed in PRV-infected and PRV US3-transfected cells contain microtubules, possibly allowing virus travel in the projections.
Cell Projections Contain Virus Capsids. Infecting cells with a PRV strain in which the VP26 capsid protein has been tagged with GFP (14) showed that the projections contain numerous VP26-GFP punctae (Fig. 2B). The observed VP26-GFP punctae have been shown earlier to represent single PRV capsids (13, 14).
Thus, PRV capsids are located in the microtubule-containing cell projections induced by PRV.
Virus Capsids in the Cell Projections Move Inside the Projections, Generally Toward the Tip. We performed fluorescence live cell imaging on ST cells infected at a high MOI with the VP26-GFP PRV virus. At 6 hpi, cells that had formed projections were selected and followed by fluorescence live cell imaging over a 15-min period. VP26-GFP particles were frequently found to move at high speed through the projection in the direction of the tip (particles 2 and 3 in Fig. 3 and Movie 1, which is published as supporting information on the PNAS web site). Average speed of the particles (analyzed by following different individual particles in different projections) was 0.29 ± 0.08 μm/s. Although general movement of the particles was clearly toward the tip (Movie 1), rare short movements of capsids in the opposite direction, toward the nucleus, could be observed (particle 1 in Fig. 3 B–D), sometimes followed by a subsequent change in direction, moving toward the tip of the projection (particle 1 in Fig. 3 H–J), corresponding well with data on bidirectional movement of alphaherpesviruses in neuronal axons during egress (14). In line with the observed general movement of virus particles toward the tip of projections is the distribution of the virus particles in the projections, which we analyzed at 6 and 8 hpi. At 6 hpi, only 17% of projections (5 of 30) displayed predominant localization of virus particles (>50%) in the tip of the projection (top third of the projection). This percentage of projections with the majority of virus particles located in the tip increased to 50% (15 of 30) at 8 hpi.
Fig. 3.

PRV capsids travel in the US3-induced cell projections. ST cells were infected at a MOI of 10 with a PRV strain that expresses a GFP-tagged VP26 capsid protein. At 6 hpi, a live cell imaging video was taken from a cell that displays a cellular projection (A, phase contrast image of the cell; B–J, time lapse recording images, total duration, 910 s, taken with 10-s intervals). Different capsid movements could be observed (see Movie 1). Individual movement of three capsids are summarized (tracking number is located underneath particle). Capsids 2 (indicated with an arrow in Movie 1) and 3 show fast, saltatory migration toward the tip of the projection. Capsid number 1 shows short migration toward the nucleus (B–D), which was only rarely observed, later followed by movement toward the tip of the projection (H–J). (Bar, 5 μm.)
Together, these data show that virus particles migrate inside the projections, with a clear overall migration in the direction of the tip.
Transfer of Virus Capsids to Uninfected Cells in the Presence of Virus-Neutralizing Antibodies. To obtain indications on whether the US3-induced cell projections allow passage of virus from infected to uninfected cells, ST cells were infected at a low MOI (0.001) with VP26-GFP virus and, from 2 hpi onwards, incubated in the presence of virus-neutralizing antibodies to inhibit infection by virus that is released or present in the medium. Projections originating from infected cells that contacted uninfected cells could be observed. Some capsids could be observed in the cytoplasm of the originally uninfected cells, indicative for virus transfer from projections to uninfected cells in the presence of virus-neutralizing antibodies (Fig. 4).
Fig. 4.

Presence of virus particles in uninfected cells that are contacted by projections originating from a PRV-infected cell. ST cells were inoculated at a MOI of 0.001 with a PRV strain that expresses a GFP-tagged VP26 capsid protein (green). At 2 hpi, virus-neutralizing antibodies were added to the medium. At 7 hpi, cells were paraformaldehyde-fixed, permeabilized, and stained for actin (red). Infected cells could be identified by a strong VP26-GFP signal in the nucleus. Pictures show an infected cell with projections contacting an originally uninfected cell, with Upper images showing an overlay of different confocal sections through the cells, and Lower images showing a single confocal section through the center of the originally uninfected cells. (Inset) An enlargement of the boxed area. Arrowheads indicate virus particles in the cytoplasm of the originally uninfected cell. (Bar, 10 μm.)
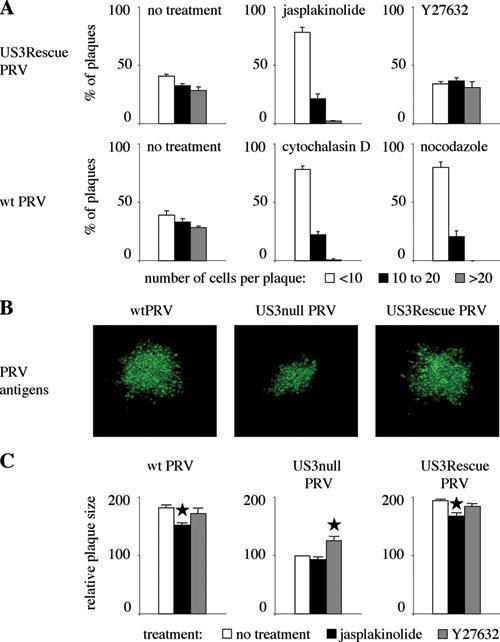
Inhibition and Induction of Projection Formation and Associated Effect on Intercellular Virus Spread. A US3-null virus has been reported before to display a defect in direct intercellular spread (27). In agreement with this finding, we found that wild-type virus spreads much more efficient in sparsely seeded ST cells (cultures consisting mainly of cell clusters of 5–10 cells) compared to US3-null virus (Fig. 5 A and C Left). To examine whether US3-induced projections may be associated with enhanced virus spread, we evaluated whether a specific drug was able to suppress the formation of cell projections, and, if so, whether addition of this drug had an effect on the efficiency of antibody-resistant virus spread. The actin stabilizing drug jasplakinolide was found to suppress the formation of US3-induced cell projections in wild-type PRV-infected cells (Fig. 5B). In addition, this drug was found to reduce the efficiency of intercellular spread of wild-type virus in sparsely seeded ST cells to the level of US3-null virus (Fig. 5C Center). Jasplakinolide also reduced the efficiency of US3-rescue virus to the level of US3-null virus in sparsely seeded ST cells (Fig. 7A Center, which is published as supporting information on the PNAS web site).
Fig. 5.

The US3-induced cell projections are associated with enhanced virus intercellular spread. (A) Plaques of wild-type (Upper) and US3-null (Lower) PRV in sparsely plated ST cells in the presence of virus-neutralizing antibodies. Cells were methanol-fixed at 24 hpi and stained for viral antigens (green), and nuclei were counterstained by using propidium iodide (red). Arrows indicate projections in cells infected with wild-type virus, arrowheads indicate nuclei that make part of the virus plaques. (Bar, 10 μm.) (B) Addition of 50 nM of the actin-stabilizing drug jasplakinolide to ST cells infected with wild-type PRV (Upper) inhibits formation of projections, whereas addition of 30 μM of the Rho kinase inhibitor Y27632 to cells infected with US3-null PRV induces the formation of cell projections. (Bar, 10 μm.) Graphs on the right indicate the percentage of cell clusters that display projections (compare with Fig. 1 A). Data represent means ± SD of triplicate assays. (C) Effect of addition of 50 nM jasplakinolide or 30 μM Y27632 on plaque size of wild type (Upper) and US3-null (Lower) PRV in sparsely plated ST cells in the presence of virus-neutralizing antibodies. Plaque sizes were measured by determining the number of cells per plaque (white bars, <10; black bars, 10 to 20; gray bars, >20).
Inversely, we tested whether a specific drug could induce US3-like projections in US3-null PRV-infected cells, and, if so, whether this drug had an effect on the efficiency of antibody-resistant intercellular virus spread. The Rho kinase inhibitor Y27632 was found to induce formation of actin- and microtubule-containing cell projections in US3-null PRV-infected cells that were very reminiscent to those observed in wild-type PRV-infected cells (Fig. 5B). In addition, this drug was able to increase the efficiency of antibody-resistant spread of US3-null virus in sparsely seeded ST cells (Fig. 5C Right). Although Y27632 also induced projections in noninfected cells (data not shown), which is in line with other reports (28), the effect of Y27632 on noninfected cells is not likely to have a major influence on virus intercellular spread, because this drug did not obviously increase the efficiency of spread of wild-type or US3-rescue virus in sparsely seeded ST cells (Figs. 5C Right and 7A Right). The effects of jasplakinolide and Y27632 on efficiency of intercellular spread were not caused by effects on virus growth, as assessed by one-step growth curves (data not shown).
Although much less pronounced, similar effects of jasplakinolide and Y27632 on the efficiency of spread of wild-type, US3-null, or US3-rescue virus could be observed in confluent monolayers of cells (Fig. 7 B and C).
In further support of a possible involvement of actin and microtubule rearrangements in efficient intercellular spread was the observation that cytochalasin D (inhibitor of actin polymerisation) and nocodazole (inhibitor of microtubule polymerisation) reduced the efficiency of spread of wild-type virus to the level of US3-null virus in sparsely seeded ST cells (Fig. 7A).
Together, these data indicate that the US3-induced projections are associated with enhanced virus intercellular spread in the presence of virus-neutralizing antibodies.
Discussion
In the current study, we show that the US3 protein kinase of the alphaherpesvirus PRV induces profound alterations in the cellular cytoskeleton, resulting in the formation of actin- and microtubule-containing cell projections. In PRV-infected cells, we show that virus capsids move in these projections in a bidirectional fashion with preferential movement toward the tip of the projection. In addition, US3-induced projections are associated with enhanced virus intercellular spread in the presence of virus-neutralizing antibodies, thereby suggesting a previously uncharacterized aspect of alphaherpesvirus intercellular spread.
One of the important questions regarding our current findings is whether cytoskeleton- and virus-filled cell projections may occur in cells infected with other alphaherpesviruses. Interestingly, it has been demonstrated recently that cells infected with the human alphaherpesvirus HSV-1 also display the formation of long cell projections (29). Although it remains to be studied whether these projections contain actin, microtubules, and/or virus, or what proteins are involved, the projections have been speculated to facilitate intercellular spread of HSV (29), which is in line with our current results. It will be interesting to determine whether the role of US3 in this process may be conserved. The US3 protein kinase is conserved among alphaherpesviruses. It is a multifunctional protein, because, apart from our observations that it causes formation of actin- and microtubule-containing cell projections and actin stress fiber disassembly, it has also been reported to be implicated in egress of virus particles from the nucleus and in protecting cells from apoptosis (22, 30–35). Because the functions of the US3 protein in viral egress from the nucleus and in protecting cells from apoptosis are conserved among different alphaherpesviruses, including HSV and PRV, it is plausible that its function that we describe here is also conserved. In this context, it is interesting that transfection of the HSV-2 US3 ORF has been shown to result in actin stress fiber disassembly and that the US3 protein of Marek's disease virus has very recently been shown to be involved in actin stress fiber disassembly, which is similar to what we observed for PRV US3 (refs. 20, 36, and 37, and the current study).
Another important issue is how the US3 protein exerts its effect on the cytoskeleton that results in the formation of actin- and microtubule-containing branched cell projections. We found that addition of Y27632, an inhibitor of the Rho-associated kinase ROCK, was able to restore the formation of these cell projections in cells infected with US3-null PRV, and was associated with enhanced intercellular spread of this mutant virus. This finding suggests that US3 may act by interfering with the Rho GTPase-regulated signaling pathways (e.g., Rho, Rac, Cdc42) that control actin dynamics. Interestingly, the HSV-2 orthologue of US3 has been described to display homology to p21-activated kinase (PAK), a downstream effector of Rac/Cdc42, and is thought to act as a viral analog of activated PAK, thereby explaining the observed actin stress fiber disassembly in cells transfected with HSV-2 US3 (36). PAKs, especially PAK1, are known to be associated with actin stress fiber disassembly and concomitant formation of cellular extensions (reviewed in ref. 38). In this context, it may be of interest that PAK is a crucial factor in the hepatocyte growth factor (HGF)-induced formation of actin- and microtubule-containing branched cell projections in epithelial cells, which very closely resemble the US3-induced branched cell projections that we describe here (28). In line with this, Y27632, the ROCK inhibitor that induces US3-like projections in US3-null PRV-infected cells, has been reported to induce HGF-like branching in epithelial cells (28). In general, PAKs and their substrates are located at the plasma membrane. PRV US3 has been reported to contain different localization signals, including membrane, mitochondrial, and nuclear localization domains (20, 39). In a recent report on the different localization signals in PRV US3 there was mention of filamentous processes that were observed in cells transfected with PRV US3, especially with membrane-associated US3 (39). These processes most likely represent the US3-induced cell projections that we describe here, suggesting that membrane-associated US3 may have a crucial role in the processes we observe. Future experiments are aimed at dissecting the exact role of Rho signaling and PAKs in the US3-induced cell projections.
In conclusion, our current results show that PRV US3, during infection and transfection, induces profound cytoskeletal rearrangements that result in the formation of actin- and microtubule-containing cell projections. In infected cells, these projections contain virus particles that migrate in a directed fashion toward the tip of the projections and projection formation is associated with enhanced intercellular spread of the virus in the presence of virus-neutralizing antibodies. This virus–cell interaction of alphaherpesviruses with potential implications for efficiency of antibody-resistant alphaherpesvirus intercellular spread may represent an important target for the development of antiviral drugs that interfere with recurrent alphaherpesvirus symptoms.
Supplementary Material
Acknowledgments
We thank Greg Smith and Lynn Enquist for VP26-GFP PRV; the ID-DLO Institute in The Netherlands for NIA3 strains; and LeighAnne Olsen, Lynn Enquist, and Bruce Banfield for US3-specific antibodies. We also thank Sarah Glorieux for help with performing some of the plaque assays and Carine Boonen, Lieve Sys, and Chantal Vanmaercke for excellent technical assistance. This study was supported by a concerted research action fund of the research council of Ghent University.
Author contributions: H.W.F. designed research; H.W.F. and G.V.M. performed research; D.A. contributed new reagents/analytic tools; H.W.F., G.V.M., and H.J.N. analyzed data; and H.W.F. and G.V.M. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PRV, pseudorabies virus; HSV, herpes simplex virus; ST, swine testicle; MOI, multiplicity of infection; hpi, hours postinnoculation.
References
- 1.Pelkmans, L., Kartenbeck, J. & Helenius, A. (2001) Nat. Cell Biol. 3, 473–483. [DOI] [PubMed] [Google Scholar]
- 2.Pelkmans, L., Puntener, D. & Helenius, A. (2002) Science 296, 535–539. [DOI] [PubMed] [Google Scholar]
- 3.Cudmore, S., Cossart, P., Griffiths, G. & Way, M. (1995) Nature 378, 636–638. [DOI] [PubMed] [Google Scholar]
- 4.Wolffe, E. J., Katz, E., Weisberg, A. & Moss, B. (1997) J. Virol. 71, 3904–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolffe, E. J., Weisberg, A. S. & Moss, B. (1998) Virology 244, 20–26. [DOI] [PubMed] [Google Scholar]
- 6.Roper, R. L., Wolffe, E. J., Weisberg, A. & Moss, B. (1998) J. Virol. 72, 4192–4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moss, B. & Ward, B. M. (2001) Nat. Cell Biol. 3, E245–E246. [DOI] [PubMed] [Google Scholar]
- 8.Rietdorf, J., Ploubidou, A., Reckmann, I., Holmstrom, A., Frischknecht, F., Zetll, M., Zimmermann, T. & Way, M. (2001) Nat. Cell Biol. 3, 992–1000. [DOI] [PubMed] [Google Scholar]
- 9.Newsome, T. P., Scaphlehorn, N. & Way, M. (2004) Science 306, 124–129. [DOI] [PubMed] [Google Scholar]
- 10.Sodeik, B., Ebersold, M. W. & Helenius, A. (1997) J. Cell. Biol. 136, 1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suomalainen, M., Nakano, M. Y., Keller, S., Boucke, K., Stidwill, R. P. & Greber, U. F. (1999) J. Cell. Biol. 144, 657–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonald, D., Vodicka, M. A., Lucero, G., Svitkina, T. M., Borisy, G. G., Emerman, M. & Hope, T. J. (2002) J. Cell. Biol. 159, 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith, G. A., Pomeranz, L., Gross, S. P. & Enquist, L. W. (2004) Proc. Natl. Acad. Sci. USA 101, 16034–16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith, G. A., Gross, S. P. & Enquist, L. W. (2001) Proc. Natl. Acad. Sci. USA 98, 3466–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones, C. (2003) Clin. Microbiol. Rev. 16, 79–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Favoreel, H. W., Nauwynck, H. J. & Pensaert, M. B. (2000) Arch. Virol. 145, 1269–1290. [DOI] [PubMed] [Google Scholar]
- 17.Baskerville, A., McFerran, J. B. & Dow, C. (1973) Vet. Bull. 43, 465–480. [Google Scholar]
- 18.van Zijl, M., van der Gulden, H., de Wind, N., Gielkens, A. & Berns, A. (1990) J. Gen. Virol. 71, 1747–1755. [DOI] [PubMed] [Google Scholar]
- 19.Kimman, T. G., de Wind, N., Oei-Lie, N., Pol, J. M. A., Berns, J. M. & Gielkens, A. L. J. (1992) J. Gen. Virol. 73, 243–251. [DOI] [PubMed] [Google Scholar]
- 20.Van Minnebruggen, G., Favoreel, H. W., Jacobs, L. & Nauwynck, H. J. (2003) J. Virol. 77, 9074–9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nauwynck, H. J. & Pensaert, M. B. (1995) Arch. Virol. 140, 1137–1146. [DOI] [PubMed] [Google Scholar]
- 22.Geenen, K., Favoreel, H. W., Olsen, L., Enquist, L. W. & Nauwynck, H. J. (2005) Virology 331, 144–150. [DOI] [PubMed] [Google Scholar]
- 23.Ishizaki, T., Uehata, M., Tamechika, I., Keel, J., Nonomura, K., Maekawa, M. & Narumiya, S. (2001) Mol. Pharmacol. 57, 976–983. [PubMed] [Google Scholar]
- 24.Narumiya, S., Ishizaki, T. & Uehata, M. (2000) Methods Enzymol. 325, 273–284. [DOI] [PubMed] [Google Scholar]
- 25.Bubb, M. R., Spector, I., Beyer, B. B. & Fosen, K. M. (1994) J. Biol. Chem. 275, 5163–5170. [DOI] [PubMed] [Google Scholar]
- 26.Favoreel, H. W., Van Minnebruggen, G., Nauwynck, H. J., Enquist, L. W. & Pensaert, M. B. (2002) J. Virol. 76, 6845–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demmin, G. L., Clase, A. C., Randall, J. A., Enquist, L. W. & Banfield, B. W. (2001) J. Virol. 75, 10856–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miao, H., Nickel, C. H., Cantley, L. G., Bruggeman, L. A., Bennardo, L. N. & Wang, B. (2003) J. Cell. Biol. 162, 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.La Boissière, S., Izeta, A., Malcomber, S. & O'Hare, P. (2004) J. Virol. 78, 8002–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klupp, B. G., Granzow, H. & Mettenleiter, T. C. (2001) J. Gen. Virol. 82, 2363–2371. [DOI] [PubMed] [Google Scholar]
- 31.Reynolds, A. E., Wills, E. G., Roller, R. J., Ryckman, B. J. & Baines, J. D. (2002) J. Virol. 76, 8939–8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Granzow, H., Klupp, B. G. & Mettenleiter, T. C. (2004) J. Virol. 78, 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leopardi, R., Van Sant, C. & Roizman, B. (1997) Proc. Natl. Acad. Sci. USA 94, 7891–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hata, S., Koyama, A. H., Shiota, H., Adachi, A., Goshima, F. & Nishiyama, Y. (1999) Microbes Infect. 1, 601–607. [DOI] [PubMed] [Google Scholar]
- 35.Cartier, A., Broberg, E., Komai, T., Henriksson, M. & Masucci, M. G. (2003) Cell Death Differ. 10, 1320–1328. [DOI] [PubMed] [Google Scholar]
- 36.Murata, T., Goshima, F., Daikoku, T., Takakuwa, H. & Nishiyama, Y. (2000) Genes Cells 5, 1017–1027. [DOI] [PubMed] [Google Scholar]
- 37.Schumacher, D., Tischer, B. K., Trapp, S. & Osterrieder, N. (2005) J. Virol. 79, 3987–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daniels, R. H. & Bokoch, G. M. (1999) Trends Biochem. Sci. 24, 350–355. [DOI] [PubMed] [Google Scholar]
- 39.Calton, C. M., Randall, J. A., Adkins, M. W. & Banfield, B. W. (2004) Virus Genes 29, 131–145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}