

Abstract
Charge transport characteristics for metal–molecule–metal junctions containing two structurally related π-conjugated systems were studied to probe π–π interactions in molecular junctions. The first molecule contains a typical π-conjugated framework derived from phenylene vinylene units, whereas the second has the phenylene vinylene structure interrupted by a [2.2]paracyclophane (pCp) core. Electrochemical investigations were used to characterize the defects and packing density of self-assembled monolayers of the two molecules on gold surfaces and to enable quantitative comparison of their transport characteristics. Current–voltage measurements across molecular junctions containing the two species demonstrate that the pCp moiety yields a highly conductive break in through-bond π-conjugation. The observed high conductivity is consistent with density functional theory calculations, which demonstrate strong through-space π–π coupling across the pCp moiety.
Keywords: charge transport, molecular electronics, surface science
Although the importance of intermolecular π–π overlap in thin-film organic electronics is well understood (1, 2), the role that such intermolecular interactions play in molecular electronics is less clear. Experiments have demonstrated that molecules addressed in parallel act as independent conductance channels (3–5); namely, the conductance of an ensemble of molecules is simply the sum of the individual molecular conductances. Calculations predict, however, that more facile charge transport could be achieved by a monolayer of strongly interacting conjugated molecules due to the formation of a pseudo two-dimensional band structure in the molecular layer (6). In particular, strong π-orbital coupling is calculated for structures in which the intermolecular distance is reduced to <4 Å (7). It is believed that the herringbone packing structure (8) exhibited in monolayers of rigid-rod π-conjugated molecular wires precludes strong π–π coupling between neighboring molecules, and thus a different motif is needed to experimentally address such interactions.
In this work, we use molecular design to explicitly address the issue of electronic coupling by π–π interactions in a molecular tunnel junction. The structures of the two molecules under study are 1,4-bis[4′-(acetylthio)styryl]benzene (1) and 4,12-bis[4′-(acetylthio)styryl][2.2]paracyclophane (2) (Fig. 1). The acetate groups are removed during the self-assembly protocol to provide thiol groups that bind to the Au electrodes. Our molecular design incorporates conjugated structures with similar length (sulfur–sulfur distance is 19.3 and 19.9 Å for 1 and 2, respectively) and identical surface binding groups but with different electronic structures. Compound 1 contains a typical π-conjugated framework, whereas 2 may be viewed as a pair of π-conjugated stilbene units attached through an ethylene bridge, a [2.2]paracyclophane (pCp) core. The 3.09-Å cofacial ring–ring spacing imposed by pCp provides a controlled experimental system to investigate strongly coupled π-systems. Unlike previous measurements that demonstrated the charge transport properties of two adjacent self-assembled monolayer-modified electrodes (9, 10), in 2, the orientation of π–π interaction along the molecular backbone (path of conduction) is defined and can be viewed as two conjugated films between the Au contacts (Fig. 1). It is known that the optical properties of well-defined, linked conjugated oligomers such as 2 are often perturbed by transannular extension of conjugation (11–14). Although these previous optical measurements have been used to determine the extent of through-space delocalization, nothing is known about how this arrangement influences the conductance of such molecules. The charge transport efficiency of 2 relative to 1, therefore provides insight into π–π (through-space) coupling in molecular tunnel junctions.
Fig. 1.

The structures of compounds 1 and 2 used in this study and the structure of the metal–molecule–metal junctions formed in the crossed-wire tunnel junction.
Materials and Methods
General Considerations. Electrochemical measurements were performed in a standard three-electrode electrochemical cell by using a commercially available Au working electrode (1.6-mm diameter), a Ag/AgCl reference electrode, and a Pt auxiliary electrode. Solutions were degassed with Ar for at least 20 min. Blocking experiments were performed in 1 mM 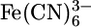 in 1 M KCl at a sweep rate of 100 mV/s. Desorption experiments were performed in 0.1 M H2SO4 at a sweep rate of 100 mV/s. Electrodes were cleaned by polishing to a mirror surface with 0.05-μm alumina and sonicating in deionized water, followed by repeated electrochemical cycling in 0.1 M H2SO4 until there was no change in the area of the Au oxide removal peak. The electrode area was calculated for each electrode immediately before they were functionalized with the monolayer. Commercially available 10-μm Au wires were used in crossed-wire measurements. Au wires were cleaned in a solution of 30% (vol/vol) hydrogen peroxide, followed by rinsing with deionized water and anhydrous ethanol.
in 1 M KCl at a sweep rate of 100 mV/s. Desorption experiments were performed in 0.1 M H2SO4 at a sweep rate of 100 mV/s. Electrodes were cleaned by polishing to a mirror surface with 0.05-μm alumina and sonicating in deionized water, followed by repeated electrochemical cycling in 0.1 M H2SO4 until there was no change in the area of the Au oxide removal peak. The electrode area was calculated for each electrode immediately before they were functionalized with the monolayer. Commercially available 10-μm Au wires were used in crossed-wire measurements. Au wires were cleaned in a solution of 30% (vol/vol) hydrogen peroxide, followed by rinsing with deionized water and anhydrous ethanol.
Preparation of Monolayer Films. Working under a nitrogen atmosphere, 10 ml of a tetrahydrofuran solution containing 2–4 mg of 1 or 2 was treated with ≈50 μl of concentrated NH4OH (aq). The solution was passed through a 0.2-μm membrane into a vial containing a clean Au electrode or a clean Au wire. The substrate was submerged in the solution overnight (≈14 h), rinsed with tetrahydrofuran and absolute ethanol, and stored under ethanol.
Current–Voltage (I–V) Characterization. A photograph of the crossed-wire apparatus used in these experiments is shown in Fig. 2. The crossed-wire apparatus is housed in a nitrogen-purged bell jar inside a Faraday cage. Two triaxial and two bnc bulkhead electrical connectors are mounted onto an aluminum plate in a cross pattern. The 10-μm-diameter Au wires, one modified with a self-assembled monolayer of the molecule of interest, are mounted to the electrical connectors with indium solder. A rare earth magnet mounted onto the upper triaxial connector supplies a constant magnetic field (0.04 T at the junction) parallel to the fixed wire. The deflection wire is mounted so that it has a curved section perpendicular to the applied magnetic field and is ≈1 mm from the fixed wire. The junction separation is controlled by bending the deflection wire with the Lorentz force generated from a small dc deflection current. The deflection current is slowly increased, while monitoring the tunneling current at a fixed voltage of 0.5 V, to bring the wires gently together to form a junction at the contact point. After initial contact, the junction “relaxes” over the time scale of seconds to a minute to a stable conductance state. The current–voltage characteristics of the molecular junctions are recorded by an Agilent 4155B semiconductor parameter analyzer (Agilent Technologies, Palo Alto, CA) by means of the two triaxial connectors. All measurements were made at room temperature.
Fig. 2.

Photograph of the crossed-wire apparatus. Note that the 10-μm-diameter Au fixed wire (mounted vertically between the small red leads) and deflection wire (mounted horizontally between the green leads) are not visible.
Density Functional Theory Calculations. The electronic structure of the two molecules coupled to one Au atom at either end was calculated within the density functional theory approximation (15). The density functional theory calculations were performed by using the B3LYP functional (16) coupled with the LANL2DZ basis set (17) for all atoms. The entire “extended molecule” (molecule plus Au atoms) was relaxed to find the final structure before performing the electronic structure calculations.
Results and Discussion
Monolayer Preparation and Characterization. The synthesis of compounds 1 and 2 is described in detail in ref. 18. Briefly, the synthetic strategy involves protecting the reactive thiol group as its S-methyl derivative, which is more tolerant of synthetic manipulations that require strong or nucleophilic bases. As shown in Fig. 3, the conjugated fragments were formed by a twofold coupling under Wittig (19) or Heck-type (20) conditions for 1 and 2, respectively. The S-methyl termini were converted to S-acetyl termini following a dealkylation protocol (21, 22). The thermal conditions of this step were found to fully isomerize the olefinic linkages providing only all-E α,ω-bis(thioacetyl)phenylenevinylenes. To avoid working with the more reactive free thiols, the S-acetyl groups were deprotected to free thiols in situ under a nitrogen atmosphere, and the resulting solutions were immediately exposed to the Au surfaces (23).
Fig. 3.

Synthetic route to compounds 1 and 2. The reagents and conditions are as follows: (i) methyltriphenylphosphonium bromide, NaH, tetrahydrofuran; (ii) 1,4-bis[(methyl)triphenylphosphonium chloride]benzene, NaH, tetrahydrofuran; (iii) pseudo-p-dibromo[2.2]paracyclophane, Pd(OAc)2, tri-o-tolylphosphine, Et3N, dimethylformamide; and (iv) (1) NaSCH3, dimethylformamide, and (2) AcCl.
We previously characterized the monolayer formation of 1 and 2 on Au surfaces (18). In the present work, electrochemical studies were performed to determine the molecular packing and defect density in the monolayers. Such analysis enables the conductivity of the molecular species to be evaluated on a per-molecule basis from charge transport measurements on monolayer junctions. Porter et al. (24) demonstrated that the ability of a monolayer to block a redox process at an electrode surface is related to the thickness and defect density of the film. Because 1 and 2 have nominally the same length and thus monolayers of the two also have nominally the same thickness, the blocking characteristics of the two films can be used to probe their relative defect density. Cyclic voltammetry measurements of bare Au and electrodes treated with 1 and 2 are shown in Fig. 4. Electrodes functionalized with 1 show no redox wave in the presence of 1 mM 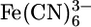 in 1 M KCl aqueous solution. A steadily increasing current (versus potential) is observed, consistent with electron tunneling across the monolayer to the redox couple in solution (24). Under the same conditions, cyclic voltammetry reveals that electrodes treated with 2 have a less perfect monolayer structure. However, the absence of a current peak and the sigmoidal shape of the wave indicate only minor “pinhole” defects in the monolayer film (25–29). The blocking characteristics of 2 are in good agreement with results recently reported for monolayers of oligo(phenylene ethynylene) molecules such as 4,4′-di(phenylene ethynylene)benzenethiol (30), which are known to form highly ordered densely packed monolayers (8). The fractional surface coverage (θ) and fractional pinhole area (1–θ) were calculated relative to the anodic peak of the bare electrode (24). The Au/1 electrode has a fractional surface coverage of 0.9996 and a fractional pinhole area of 4.1 × 10–4, whereas the Au/2 electrode has a fractional surface coverage of 0.98 and a fractional pinhole area of 2.0 × 10–2.
in 1 M KCl aqueous solution. A steadily increasing current (versus potential) is observed, consistent with electron tunneling across the monolayer to the redox couple in solution (24). Under the same conditions, cyclic voltammetry reveals that electrodes treated with 2 have a less perfect monolayer structure. However, the absence of a current peak and the sigmoidal shape of the wave indicate only minor “pinhole” defects in the monolayer film (25–29). The blocking characteristics of 2 are in good agreement with results recently reported for monolayers of oligo(phenylene ethynylene) molecules such as 4,4′-di(phenylene ethynylene)benzenethiol (30), which are known to form highly ordered densely packed monolayers (8). The fractional surface coverage (θ) and fractional pinhole area (1–θ) were calculated relative to the anodic peak of the bare electrode (24). The Au/1 electrode has a fractional surface coverage of 0.9996 and a fractional pinhole area of 4.1 × 10–4, whereas the Au/2 electrode has a fractional surface coverage of 0.98 and a fractional pinhole area of 2.0 × 10–2.
Fig. 4.

Cyclic voltammetry for Au/1 (red), Au/2 (blue), and untreated Au electrodes (green) in 1 mM 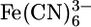 M KCl (aq) solution.
M KCl (aq) solution.
Although these blocking experiments reveal that 1 and 2 form nearly complete monolayers with low defect density, they do not directly address the question of how many molecules per unit area are on the surface. To better understand the packing density of the two monolayer films, we modeled the monolayer packing according to the van der Waals radii of the molecules (31). The calculated molecular density was 5.53 × 10–10 and 2.68 × 10–10 mol/cm2 for films containing 1 and 2, respectively, assuming a nearly perpendicular orientation of the molecules (see Fig. 7, which is published as supporting information on the PNAS web site). These calculations imply that perfect monolayers of 1 would have approximately twice as many molecules (2.06) per unit area as monolayers of 2. These calculated packing densities are supported by electrochemical desorption measurements that determine the molecular surface coverage from the current needed to irreversibly oxidize the molecules from the electrode (32, 33). The desorption current associated with the Au/1 electrode is consistent with that reported for alkane thiols, whereas the desorption current for Au/2 is roughly half the expected value. Electrochemical desorption thus confirms that monolayers of 1 have roughly twice the number of molecules (1.86) per unit area than monolayers of 2 (see Fig. 8, which is published as supporting information on the PNAS web site).
Molecular orientation is also of importance, particularly with respect to enabling electrical contact to both ends of the molecule in the crossed-wire tunnel junction. Recent angle-resolved near-edge x-ray adsorption fine structure spectroscopy measurements confirm that both molecules have their long axis oriented ≈37° from the surface normal in the monolayer films (A. Hexemer, D.S.S., E. J. Kramer, and G.C.B., unpublished data). This finding compares favorably with the molecular tilt determined from infrared spectroscopy for the more extensively studied oligo(phenylene ethynylene) systems (30).
Molecular Junction Measurements. Previously, we have shown that crossed-wire tunneling junctions have the ability to examine charge transport (34) and inelastic electron tunneling spectroscopy (35) for a variety of molecular species in a well controlled, reproducible manner. Specifically, we have demonstrated the utility of this technique to compare the conductivity of aliphatic and aromatic molecules (36) as well as aromatic and organometallic species (37). A crossed-wire tunneling junction is formed when one 10-μm diameter Au wire, modified with a monolayer of the molecule of interest, is brought into gentle contact with a second unmodified Au wire controlled by a small dc deflection current in the presence of an external magnetic field. Experimentally, this approach offers several advantages to other conductivity measurement techniques. Primarily, because the top metal–molecule contact is made mechanically, we avoid exposing the molecules to a metal evaporation that can disturb or chemically modify the thin molecular layer (38). Furthermore, because formation of molecular junctions in this manner is relatively easy, we are able to repeatedly measure the sample of interest generating multiple data sets where statistics can be applied. The number of molecules contained within the ≈300-nm2 junction depends on the packing density of the monolayer film. Although the measurements are made on an ensemble of molecules in the monolayer, previous studies have shown excellent correlation between crossed-wire measurements and single-molecule STM measurements (39).
I–V characteristics for molecular junctions formed from monolayers of 1 and 2 are shown in Fig. 5. The reported I–V characteristics are the average from multiple traces on three independently formed junctions. The error bars shown in Fig. 5 Inset are one standard deviation in the measurement. Multiple traces on the same junction show little variation. The main contribution to the uncertainty in the measured value results from variations in the number of molecules contacted in the independently formed junctions. Both molecules yielded molecular junctions stable to repeated measurements for bias voltages in the ±1 V range. As expected from the symmetric nature of the molecular junctions, the measured I–V characteristics are symmetric with respect to bias voltage polarity (34). The symmetry of the I–V characteristics demonstrates that the Au–S contacts at either end of the molecules are chemically similar (40).
Fig. 5.

I–V characteristics (linear and log scale, Upper Inset) for Au/1/Au (red) and Au/2/Au (blue) averaged from three independently formed junctions. The error bars show one standard deviation of the data. Graphic illustration of transport in Au/1/Au and Au/2/Au junctions (Lower Inset).
The measured, efficient charge transport across the delocalized structure of 1 is consistent with transport measurements on similar molecules (36, 41, 42). The most striking aspect of the I–V characteristics is the high conductivity measured for 2 relative to 1. Although it is not surprising that 1 is a good molecular wire, the excellent conductance of 2 is noteworthy because of the break in π-conjugation imposed by the internal pCp core. The conductivity of Au/1/Au, calculated from the slope of the linear low bias region, is only 2.3 times greater than Au/2/Au. Additionally, when considering that the packing density of the monolayer of 2 is approximately half the packing density of monolayer of 1, the conductivity per molecule for the two structures is the same given the uncertainties of the measurements. For comparison, orders of magnitude differences in conductivity are observed between saturated alkanes and π-conjugated oligomers of similar length (36, 43). The similar measured I–V characteristics of 1 and 2, specifically the magnitude of the conductance, indicate that the pCp arrangement provides an efficient mechanism for charge transport.
Electronic Structure Calculations. A number of researchers have demonstrated the utility of ab initio calculations for examining the charge transport efficiency of a molecular system (44–47). These previous studies have shown that delocalized orbitals that span the entire molecule and are positioned close to the Fermi level of the metal electrodes facilitate charge transport across molecular junctions. To evaluate the potential conductance channels for the molecules investigated in this study, we calculated the electronic structure of molecules 1 and 2 at the B3LYP/LANL2DZ level of density functional theory (15–17) with single Au atoms attached to the terminal S on either side of the molecule. The Au atoms are used to represent the interaction of the molecules with the Au electrodes. Although single Au atoms are clearly not equivalent to an extended Au surface, previous studies have demonstrated the utility of such calculations (45, 47). Charge density plots for the frontier orbitals of molecules 1 and 2 bound to two Au atoms as well as their energy are shown in Fig. 6. For reference, the Fermi level of Au is –5.31 eV (48).
Fig. 6.

Charge density plots of the frontier orbitals of 1 and 2 attached to two Au atoms. The through-space conjugation across the pCp core is clearly evident in the HOMO-1 orbital (–5.8 eV) of Au/2/Au.
Analysis of orbital topology shows that the highest occupied molecular orbital (HOMO) of 1 bound to two Au atoms (Fig. 6) is the most likely conductance channel for this system, because this delocalized orbital (i) spans the entire molecule, (ii) has appreciable charge density on the terminal Au atoms, and (iii) is close in energy to the Fermi level of Au. This analysis is consistent with previous studies, which determined that hole transport through filled molecular orbitals such as the HOMO dominate charge transport in π-conjugated molecular wire systems (44–47). A similar argument can be made for the HOMO and HOMO-1 orbitals of 2 bound to two Au atoms (Fig. 6). These two orbitals are nearly degenerate in energy, are within 0.5 eV of the Au Fermi level, and span the entire molecule. These calculated charge density plots of 2 demonstrate that although there is a break in through-bond conjugation at the pCp core, the π-system of the two stilbene units spans the entire molecule because of through-space conjugation. The HOMO has a node in the π-system at the pCp core, whereas the HOMO-1 clearly demonstrates delocalization across the pCp core. Although it can be argued that the HOMO-1 orbital is probably more important to charge transport because it has a larger electron density on the terminal Au atoms, and hence better coupling to the metal electrodes, both orbitals undoubtedly contribute to the measured charge transport. The calculated electronic structure for 2 bound to two Au atoms, demonstrates that the low-lying through-space delocalized state (11–14) can effectively facilitate charge transport in a molecular junction.
Conclusions
The measured charge transport across metal–molecule–metal junctions formed from a pair of chromophores held together by a pCp core is surprisingly conductive and comparable with that of an intact π-delocalized structure. Although previous spectroscopic measurements on pCp containing chromophores have demonstrated strong through-space coupling (11–14), the results reported here enable the electronic conduction across the pCp linkage to be compared to its fully conjugated analog. The junction I–V characteristics demonstrate that the Au/1/Au junction is approximately a factor of two more conductive than the Au/2/Au junction. Considering that the number of molecules contacted in the Au/2/Au junction is approximately half the number contacted in the Au/1/Au junction suggests that the conductivity on a permolecule basis is nearly the same. The similar conductivity is likely due to charge-transport through energetically similar filled molecular orbitals. The molecular structure of 2 demonstrates that suitably arranged π-systems can exhibit strong through-space π–π coupling that is efficient at promoting charge transport across the system. Thus, the cofacial alignment and ring-to-ring spacing of 3.09 Å in the pCp core provide one example of an orientation and packing that would provide intermolecular π-coupling. Although it is currently difficult to imagine how one could drive a monolayer system to adapt such molecular packing, this result should provide motivation for such research as well as for designing future molecular species that incorporate more than one conjugated subunit.
Supplementary Material
Acknowledgments
This work was supported by the Office of Naval Research, National Science Foundation Grant DMR 0097611 (to D.S.S. and G.C.B.), and the Defense Advanced Research Projects Agency (S.A.T. and J.G.K.).
Author contributions: G.C.B. and J.G.K. designed research; D.S.S., S.A.T., and J.G.K. performed research; D.S.S. and G.C.B. contributed new reagents/analytic tools; D.S.S., S.A.T., G.C.B., and J.G.K. analyzed data; and D.S.S., G.C.B., and J.G.K. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HOMO, highest occupied molecular orbital; I–V, current–voltage; pCp, [2.2]paracyclophane.
References
- 1.Bredas, J. L., Calbert, J. P., da Silva Filho, D. A. & Cornil, J. (2002) Proc. Natl. Acad. Sci. USA 99, 5804–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moon, H., Zeis, R., Bokent, E.-J., Besnard, C., Lovinger, A. J., Siegrt, T., Kloc, C. & Bao, Z. (2004) J. Am. Chem. Soc. 126, 15322–15323. [DOI] [PubMed] [Google Scholar]
- 3.Cui, X. D., Primak, A., Zarate, X., Tomfohr, J., Sankey, O. F., Moore, A. L., Moore, T. A., Gust, D., Harris, G. & Lindsay, S. M. (2001) Science 294, 571–574. [DOI] [PubMed] [Google Scholar]
- 4.Kushmerick, J. G., Naciri, J., Yang, J. C. & Shashidhar, R. (2003) Nano Lett. 3, 897–900. [Google Scholar]
- 5.Xu, B. & Tao, N. J. (2003) Science 301, 1221–1223. [DOI] [PubMed] [Google Scholar]
- 6.Yaliraki, S. N. & Ratner, M. A. (1998) J. Chem. Phys. 109, 5036–5043. [Google Scholar]
- 7.Rochefort, A., Martel, R. & Avouris, P. (2002) Nano Lett. 2, 877–880. [Google Scholar]
- 8.Yang, G., Qian, Y., Engtrakul, C., Sita, L. R. & Liu, G.-Y. (2000) J. Phys. Chem. B 104, 9059–9062. [Google Scholar]
- 9.Wold, D. J. & Frisbie, C. D. (2001) J. Am. Chem. Soc. 123, 5549–5556. [DOI] [PubMed] [Google Scholar]
- 10.Holmlin, R. E., Haag, R., Chabinyc, M. L., Ismagilov, R. F., Cohen, A. E., Terfort, A., Rampi, M. A. & Whitesides, G. M. (2001) J. Am. Chem. Soc. 123, 5075–5085. [DOI] [PubMed] [Google Scholar]
- 11.Bazan, G. C., Oldham, W. J., Lachicotte, R. J., Tretiak, S., Chernyak, V. & Mukamel, S. (1998) J. Am. Chem. Soc. 120, 9188–9204. [DOI] [PubMed] [Google Scholar]
- 12.Zyss, J., Ledoux, I., Volkov, S., Chernyak, V., Mukamel, S., Bartholomew, G. P. & Bazan, G. C. (2000) J. Am. Chem. Soc. 122, 11956–11962. [Google Scholar]
- 13.Wang, S., Bazan, G. C., Tretiak, S. & Mukamel, S. (2000) J. Am. Chem. Soc. 122, 1289–1297. [Google Scholar]
- 14.Bartholomew, G. P. & Bazan, G. C. (2001) Acc. Chem. Res. 34, 30–39. [DOI] [PubMed] [Google Scholar]
- 15.Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Zakrzewski, V. G., Montgomery, J. A., Jr., Stratmann, R. E., Burant, J. C., et al. (2002) gaussian 98 rev a.11.3 (Gaussian, Pittsburgh).
- 16.Becke, A. D. (1993) J. Chem. Phys. 98, 5648–5652. [Google Scholar]
- 17.Hay, P. J. & Wadt, W. R. (1985) J. Chem. Phys. 82, 299–310. [Google Scholar]
- 18.Seferos, D. S., Banach, D. A., Alcantar, N. A., Israelachvili, J. N. & Bazan, G. C. (2004) J. Org. Chem. 69, 1110–1119. [DOI] [PubMed] [Google Scholar]
- 19.Maercker, A. (1965) Org. React. 14, 270–490. [Google Scholar]
- 20.Heck, R. F. (1982) Org. React. 27, 345–390. [Google Scholar]
- 21.Testaferri, L., Tiecco, M., Tingoli, M., Chianelli, D. & Montanucci, M. (1983) Synthesis, 751–755.
- 22.Tiecco, M., Tingoli, M., Testaferri, D. C. & Maiolo, F. (1982) Synthesis, 478–480.
- 23.Tour, J. M., Jones, L., II, Pearson, D. L., Lamba, J. J. S., Burgin, T. P., Whitesides, G. M., Allara, D. L., Parikh, A. N. & Atre, S. (1995) J. Am. Chem. Soc. 117, 9529–9534. [Google Scholar]
- 24.Porter, M. D., Bright, T. B., Allara, D. L. & Chidsey, C. E. D. (1987) J. Am. Chem. Soc. 109, 3559–3568. [Google Scholar]
- 25.Sabatani, E., Rubinstein, I., Maoz, R. & Sagiv, J. (1987) J. Electroanal. Chem. 219, 365–371. [Google Scholar]
- 26.Finklea, H. O., Snider, D. A., Fedyk, J., Sabatani, E., Gafni, Y. & Rubinstein, I. (1993) Langmuir 9, 3660–3667. [Google Scholar]
- 27.Che, G., Li, Z., Zhang, H. & Cabrera, C. R. (1998) J. Electroanal. Chem. 453, 9–17. [Google Scholar]
- 28.Finklea, H. O., Avery, S., Lynch, M. & Furtsch, T. (1987) Langmuir 3, 409–413. [Google Scholar]
- 29.Sabatani, E. & Rubinstein, I. (1987) J. Phys. Chem. 91, 6663–6669. [Google Scholar]
- 30.Stapleton, J. J., Harder, P., Daniel, T. A., Reinard, M. D., Yao, Y., Price, D. W., Tour, J. M. & Allara, D. L. (2003) Langmuir 19, 8245–8255. [Google Scholar]
- 31.Bondi, A. (1964) J. Phys. Chem. 68, 441–451. [Google Scholar]
- 32.Widrig, C. A., Chung, C. & Porter, M. D. (1991) J. Electroanal. Chem. 310, 335–359. [Google Scholar]
- 33.Sabatani, E., Cohen-Boulakia, J., Bruening, M. & Rubinstein, I. (1993) Langmuir 9, 2974–2981. [Google Scholar]
- 34.Kushmerick, J. G., Holt, D. B., Yang, J. C., Naciri, J., Moore, M. H. & Shashidhar, R. (2002) Phys. Rev. Lett. 89, 086802. [DOI] [PubMed] [Google Scholar]
- 35.Kushmerick, J. G., Lazorcik, J., Patterson, C. H., Shashidhar, R., Seferos, D. S. & Bazan, G. C. (2004) Nano Lett. 4, 639–642. [Google Scholar]
- 36.Kushmerick, J. G., Holt, D. B., Pollack, S. K., Ratner, M. A., Yang, J. C., Schull, T. L., Naciri, J., Moore, M. H. & Shashidhar, R. (2002) J. Am. Chem. Soc. 124, 10654–10655. [DOI] [PubMed] [Google Scholar]
- 37.Schull, T. L., Kushmerick, J. G., Patterson, C. H., George, C., Moore, M. H., Pollack, S. K. & Shashidhar, R. (2003) J. Am. Chem. Soc. 125, 3202–3203. [DOI] [PubMed] [Google Scholar]
- 38.Walker, A. V., Tighe, T. B., Stapleton, J. J., Haynie, B. C., Upilli, S., Allara, D. L. & Winograd, N. (2004) Appl. Phys. Lett. 84, 4008–4010. [Google Scholar]
- 39.Szuchmacher Blum, A., Kushmerick, J. G., Pollack, S. K., Yang, J. C., Moore, M. H., Naciri, J., Shashidhar, R. & Ratna, B. (2004) J. Phys. Chem. B 108, 18124–18128. [Google Scholar]
- 40.Kushmerick, J. G., Whitaker, C. M., Pollack, S. K., Schull, T. L. & Shashidhar, R. (2004) Nanotechnology 15, S489–S493. [Google Scholar]
- 41.Bumm, L. A., Arnold, J. J., Cygan, M. T., Dunbar, T. D., Burgin, T. P., Jones, L., II, Allara, D. L., Tour, J. M. & Weiss, P. S. (1996) Science 271, 1705–1707. [Google Scholar]
- 42.Reichert, J., Ochs, R., Beckman, D., Weber, H. B., Mayor, M. & Löhneyesen, H. V. (2002) Phys. Rev. Lett. 88, 176804. [DOI] [PubMed] [Google Scholar]
- 43.Kushmerick, J. G., Pollack, S. K., Yang, J. C., Naciri, J., Holt, D. B., Ratner, M. A. & Shashidhar, R. (2003) Ann. N.Y. Acad. Sci. 1006, 277–290. [DOI] [PubMed] [Google Scholar]
- 44.Xue, Y., Datta, S. & Ratner, M. A. (2001) J. Chem. Phys. 115, 4292–4299. [Google Scholar]
- 45.Derosa, P. A. & Seminario, J. M. (2001) J. Phys. Chem. B 105, 471–481. [Google Scholar]
- 46.Taylor, J., Brandbyge, M. & Stokbro, K. (2002) Phys. Rev. Lett. 89, 138301. [DOI] [PubMed] [Google Scholar]
- 47.Heurich, J., Cuevas, J. C., Wenzel, W. & Schon, G. (2002) Phys. Rev. Lett. 88, 256803. [DOI] [PubMed] [Google Scholar]
- 48.Lide, D. R. (1998) CRC Handbook of Chemistry and Physics (CRC, Boca Raton, FL).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.