

Summary
Artificial intelligence (AI) and deep learning technologies hold promise for identifying effective drugs for human diseases, including pain. Here, we present an interpretable deep-learning-based ligand image- and receptor’s three-dimensional (3D)-structure-aware framework to predict compound-protein interactions (LISA-CPI). LISA-CPI integrates an unsupervised deep-learning-based molecular image representation (ImageMol) of ligands and an advanced AlphaFold2-based algorithm (Evoformer). We demonstrated that LISA-CPI achieved ∼20% improvement in the average mean absolute error (MAE) compared to state-of-the-art models on experimental CPIs connecting 104,969 ligands and 33 G-protein-coupled receptors (GPCRs). Using LISA-CPI, we prioritized potential repurposable drugs (e.g., methylergometrine) and identified candidate gut-microbiota-derived metabolites (e.g., citicoline) for potential treatment of pain via specifically targeting human GPCRs. In summary, we presented that the integration of molecular image and protein 3D structural representations using a deep learning framework offers a powerful computational drug discovery tool for treating pain and other complex diseases if broadly applied.
Keyword(s): artificial intelligence, compound-protein interaction, deep learning, drug repurposing, gut metabolite, G-protein coupled receptor, GPCR, ImageMol, pain, AlphaFold2, Evoformer
Graphical abstract
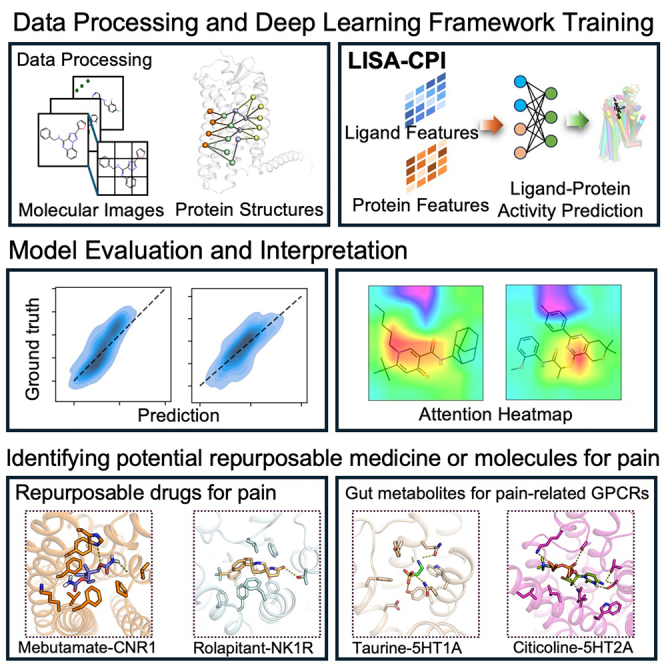
Highlights
-
•
LISA-CPI is a ligand- and receptor-structure-aware framework for target prediction
-
•
LISA-CPI is a deep learning model pretrained on ∼10 million unlabeled molecules
-
•
LISA-CPI shows high accuracy in prediction of compound-protein interactions
-
•
LISA-CPI identifies repurposable drugs and gut metabolites for pain-related GPCRs
Motivation
The rise of advanced artificial intelligence technologies motivated their application to drug discovery. One of the fundamental challenges is how to learn molecular representation from chemical structures. Traditional molecular representation methods rely on a large amount of domain knowledge, such as sequence-based and graph-based approaches, and their accuracy in extracting informative vectors is limited. As motivated by computer vision and image-based deep learning technologies, we presented a self-supervised image representation learning framework that combines molecular image and protein representations for the accurate prediction of compound-protein interactions.
Yang et al. develop a self-supervised deep learning framework (LISA-CPI) with chemical awareness to learn molecular images from ∼10 million unlabeled drug-like molecules and protein structural representations from AlphaFold2’s Evoformer. LISA-CPI offers a powerful drug discovery foundation model for computational drug discovery in pain and other diseases if broadly applied.
Introduction
Pain, especially chronic pain, afflicts 50 million adults in the United States1 and 20% of the population worldwide.2 Currently, available analgesics are mainly small molecules (such as opioids), relieving the pain but with deleterious side effects, in particular drug addiction.3 The opioid epidemic highlights an urgent need to develop non-opioid analgesics with less addiction for treating pain. G-protein-coupled receptors (GPCRs) are prevalent druggable targets for treating pain4 since they trigger intracellular signaling events in the sensory neurons and, thereby, participate in most pathophysiological processes in pain perception.5 Recent advances uncovered biased agonists of opioids or other GPCRs (such as the μ-opioid receptor) to avoid adverse effects, such as addiction and sedation.6,7,8 However, the identification of distinct chemotypes yielding analgesia without drug addiction side effects by targeting GPCRs is still a challenge.9 In addition to drugs, it is worth noting that gut microbiota and its metabolites have been reported to be involved in the morbidity of chronic pain.10 For example, decreased abundance of the short-chain fatty acid (such as butyrate) derived from Bacteroides is associated with long-term pain,11 which targeted several potential GPCRs (such as FFAR3 and GPR109A).12
Traditional bioactive ligands targeting disease-related proteins (including GPCRs) were determined by biological experiments, which are costly and time consuming.13 Recent advances suggested that artificial intelligence (AI)-based compound-protein interaction (CPI) predictions hold great promise in identifying potential drugs and drug repurposing.14 Traditional machine learning algorithms, such as support vector machine,15,16 random forest,17 and kernel regression,18 have been widely used by training handcrafted molecule fingerprint descriptors and protein sequence descriptors. Recent deep-learning-based end-to-end methods, such as DeepDTA19 and GraphDTA,20 were reported to improve predictive performance. However, these handcrafted chemical and protein sequence descriptors require significant domain expert knowledge and often fail to capture pharmacologically relevant features of CPIs due to low dimensionality. Recently, our team developed an unsupervised deep learning framework (ImageMol21) by capturing pharmacologically relevant features of ligands from molecular image representations. ImageMol showed improved accuracy in CPI predictions compared with sequence-based models and graph-based models.21 In addition, the recent AlphaFold2 model can systematically predict the structures of the whole human proteome based on amino acid sequences,22 suggesting it is possible to apply three-dimensional (3D) structural information for CPI prediction. Moreover, a few recently developed deep learning technologies that consider the 3D structure of the proteins were shown to offer promising improvement in CPI predictions.23
In this study, we present a deep learning framework to predict CPIs by integrating ligand image-based and protein 3D-structure-based representations (termed LISA-CPI). Our approach outperformed existing models on CPI prediction of GPCR and kinase benchmarks (ImageMol) through chemical awareness and 3D protein residue pair representations. In order to identify potential treatment approaches for pain, we utilized LISA-CPI to predict potential medicines from United States Food and Drug Administration (FDA)-approved drugs and gut-microbiota-derived metabolites. As a result, we prioritized potential repurposable drugs (such as methylergometrine) and gut metabolite-based (such as citicoline) candidate treatments for pain by specifically targeting pain-associated GPCRs. In summary, the LISA-CPI framework offers a useful computational drug discovery framework for pain and other human diseases if broadly applied.
Results
A deep learning framework of ligand image- and 3D-structure-based representation
To predict interactions between compounds (e.g., drugs or gut metabolites) and pain-associated GPCRs (Figure 1A), we developed a deep learning framework that incorporated an unsupervised deep learning algorithm (ImageMol)21 and a neural network-based algorithm (Evoformer) derived from AlphaFold222 (cf. STAR Methods). ImageMol was utilized to extract key molecular structure features from ∼10 million molecular images with high accuracy, while Evoformer outputs protein sequence alignment and pair representations. These structure representations contain key information about the residue location and the relation between the residue pairs. The LISA-CPI framework is illustrated in Figure 1B. Overall, LISA-CPI consists of four steps: (1) extracting high-dimensional latent features with chemical awareness from encoded molecular images by ImageMol,21 (2) encoding structural representations from the protein amino acid sequence by Evoformer and then projecting them into low-dimension space, (3) integrating the features from steps 1 and 2 and constructing a neural network, and (4) utilizing a multi-layer perceptron (MLP) to predict CPIs (activity is regarded as the label) from the combined features of compounds and proteins.
Figure 1.

Schematic illustration of the LISA-CPI framework
(A) A diagram depicting the roles of GPCRs in pain. Our work aims to predict the interactions between approved drugs/gut metabolites (left) and pain-associated GPCRs (right).
(B) Model architecture. Arrows indicate the flow of the information from the input through both the ligand image learning part and the receptor structure learning part to the final prediction part.
Performance evaluation of LISA-CPI on benchmark ligand-GPCR interactions
To validate the performance of the LISA-CPI framework, we first evaluated the top 20 GPCRs (regression task) that have the most binding activity data retrieved from the ChEMBL and GLASS databases24,25 (see STAR Methods). The training dataset contains 71,757 ligand-GPCR pairs, ranging from 1,761 pairs for OX2R to 6,897 pairs for DRD2 (Table S1). We only kept potent bioactive compounds (inhibition constant/potency, Ki < 10 μM) with an average pKi of 7.18 (Figure S1A). We utilized 70% of the dataset of each GPCR as the training set and the rest of the dataset as the test set. 10-fold cross-validation was carried out on the training set. The mean absolute error (MAE) and Pearson correlation coefficient (R) of the predicted and ground-truth activity values were calculated to evaluate the predictive performance. Here, we took the state-of-the-art ImageMol,21 CHEM-BERT,26 and MolCLR27 models as the comparison. For each GPCR dataset, we observed that the predicted MAE of binding activity via LISA-CPI is smaller than that of the other three models (Figure S1C), suggesting the high accuracy of LISA-CPI. Specifically, combining ligand image- and protein structure-based representations improves the MAE by ∼20% (0.248 vs. 0.199; Figure S1C) compared to ligand image-based representation alone (ImageMol),21 the second-best performing model.
We next used t-distributed stochastic neighbor embedding (t-SNE) to visualize the distribution of the embedding space of the compounds and their corresponding MAEs on all GPCR test datasets. We found that the MAEs of 90% of datasets are lower than 0.414 (Figure S1D). Furthermore, we revealed a strong correlation between the experimental activity values and the predicted activity values across 17 GPCR datasets via LISA-CPI (R > 0.5; Figures 2A and S2A). In particular, LISA-CPI exhibited stronger correlations across all 5 pain-associated GPCR datasets (R > 0.7), including HRH3, MC4R, OPRK, OPRM, and OX2R. In comparison to ImageMol, LISA-CPI also achieved lower MAEs (higher accuracy) for HRH3 (0.150 [LISA-CPI] vs. 0.234 [ImageMol]), MC4R (0.174 vs. 0.269), OPRK (0.212 vs. 0.287), OPRM (0.208 vs. 0.278), and OX2R (0.163 vs. 0.238). These results suggested that LISA-CPI outperformed ImageMol after we integrated protein 3D-structure-based representation in predicting experimentally determined ligand-GPCR interactions.
Figure 2.

Predictive performance on selected GPCR target receptors
(A) Predictive performance of the proposed LISA-CPI on 6 selected top-20 GPCR targets. Predicted pKi and ground-truth pKi of each compound for each GPCR target are contour plotted with point density. Pearson’s correlation coefficient R and p values are labeled.
(B) Predictive performance of the proposed LISA-CPI on 8 selected pain-associated GPCR targets. Predicted pKi and ground-truth pKi of each compound for each GPCR target are contour plotted with point density. Pearson’s correlation coefficient R and p values are labeled.
(C) Receiver operating characteristic (ROC) curves showcasing the predictive performance of the proposed LISA-CPI and three other models (ImageMol, CHEM-BERT, and MolCLR) on 2 selected kinase targets and the entire kinase dataset. Solid lines and shades represent the mean and one standard deviation of ROC curves obtained from 10-fold cross-validation, respectively.
We next turned to interpret LISA-CPI models and generated the heatmaps of molecular images using Grad-CAM (gradient-weighted class activation mapping)28 to visualize the attention pattern of LISA-CPI on compounds with different activity values. We selected 3 example compounds with high affinity (pKi > 8) or low affinity (pKi < 6), individually shown in Figure S2B. We found that for compounds exhibiting high affinities, higher attention areas (depicted by warmer color areas) cover the majority of the compound structures. These high-attention areas are particularly focused on the important functional substructure of active ligands, such as hydroxy groups, phenyl groups, carbonyl groups, and ether groups (Figure S2B). For GPCR ligands with low affinities, most areas of the compounds are covered by lower-attention areas (depicted by cooler color areas), and only very few functional groups are covered by higher-attention areas (Figure S2C). These findings confirm that LISA-CPI captured meaningful features that can be used to help interpret predictive results. In addition, we also visualized GPCR structural representations along the amino acid sequence. For example, we observed that most peaks (marked in blue vertical lines) of structural representations from CCR2 and NK1R resided in transmembrane (TM) helical domains (marked in light blue areas, Figure S2D), which contain the main ligand-binding sites.29,30 Taking these results together, LISA-CPI offers an accurate tool to predict ligand-GPCR interactions.
Performance evaluation of LISA-CPI on benchmark compound-kinase interactions
To further validate our proposed LISA-CPI model, we also tested 10 kinase targets with a classification task. The training dataset contains 1,046 compound-kinase pairs, ranging from 80 pairs for CDK4 to 110 pairs for FLT3 (Table S1). We trained and assessed LISA-CPI following the procedure outlined in the STAR Methods. We continue to use the state-of-the-art ImageMol model,21 CHEM-BERT model,26 and MolCLR model27 as the comparison. LISA-CPI achieved high area under receiver operating characteristic (AUROC) scores for 8 kinase targets (AUROC > 0.75, best AUROC of 0.90 on EGFR). In particular, LISA-CPI improves the AUROC by 11.6% (0.77 vs. 0.69) across all kinases compared to ImageMol, the second-best performing model (Figures 2C and S3A). Altogether, these results show that LISA-CPI offers an accurate tool to predict ligand-kinase interactions as below.
Performance in identifying ligands for pain-associated GPCRs
Next, we sought to examine the performance of LISA-CPI on pain-associated GPCRs. We trained new LISA-CPI models using the collected experimental CPI dataset specifically for the 13 pain-associated GPCRs. In total, 13 reported acute-pain- or chronic-pain-associated GPCRs were identified based on previous reports31,32 (see STAR Methods and Table S2), including opioid receptors (OPRM, OPRD, and OPRK),33 serotonin receptors (5HT1A, 5HT1B, 5HT1D, 5HT2A, and 5HT7R),34 cannabinoid receptors (CNR1 and CNR2),35 a metabotropic glutamate receptor (mGluR5),36 a chemokine receptor (CCR2),37 and a tachykinin receptor NK-1 (NK1R).38 As shown by Figure S3B, 5HT1A and 5HT2A and the opioid receptors are also listed in the top 20 most well-studied GPCRs. As is shown in Figures 2B and S3D, LISA-CPI achieved high performance (higher R) for most of the pain-associated GPCRs (R > 0.65, best R of 0.81 on 5HT1D) in the binding affinity predictions. Compared to ImageMol, the MAE values of LISA-CPI models have been improved by 20.8% on average and 32.2% at best (5HT1D) over the ImageMol (Figure S3B). We used t-SNE to visualize the distribution of the embedding space of the compounds and their corresponding MAEs on pain-associated GPCR test datasets. Similar to Figure S1D, the MAEs of 90% of datasets are lower than 0.406 (Figure S3C). For the potential treatment of pain, either an agonist or antagonist for pain-associated GPCRs was predicted (Table S2). Thus, we next trained a LISA-CPI classification model on a dataset featuring 12 pain-associated GPCRs (excluding CCR2 because of the antagonist-only dataset for CCR2). The dataset contains 10,816 compound-GPCR pairs. LISA-CPI also showed a better AUROC on 12 pain-associated GPCRs than that of ImageMol (Figure S4). Specifically, LISA-CPI improves the AUROC by 19.2% on average compared to ImageMol. Overall, LISA-CPI proves to be generalizable on GPCRs, in particular for pain-associated GPCRs.
We next turned to check the top 10 example compounds interacting with 5 pain-associated GPCRs, including CNR1, CNR2, 5HT1B, NK1R, and 5HT7R, because the predicted correlation of these GPCRs ranges from 0.57 to 0.75. For each GPCR, we randomly selected one compound with high activity (pKi > 8) and one with low activity (pKi < 6). Consistent with Figures S2B and S2C, we observed that the 5 high bioactive compounds (pKi >8; Figures 3 and S5A) capture more structural information on molecular images compared to the 5 low bioactive compounds (pKi < 6; Figures 3 and S5B). Furthermore, we inspected the binding modes to structurally visualize the CPI using structure-based molecular docking simulations. We modeled GPCR structures by AlphaFold2 and performed molecular docking for each druggable pocket in each GPCR structural model (see STAR Methods). We found that high bioactive compound-protein pairs exhibit superior binding modes and docking scores (Figures 3 and S5A). For example, CHEMBL1909850 was reported to inhibit the CNR1 receptor with higher affinity (pKi: 8.5239) compared to CHEMBL497392 (pKi: 5.00). We found that CHEMBL1909850 showed a stronger chemical structure awareness in the image representation than a low bioactive molecule of CHEMBL497392 (Figure S5B). CHEMBL1909850 has a stronger molecular docking score (−7.49) with the CNR1 receptor than CHEMBL497392 (docking score: −5.67), further supporting our predictions. To validate our predicted binding modes, we first compared the structure similarity between AlphaFold2 and literature-reported crystal structures. AlphaFold2 models of pain-associated GPCRs showed high structural confidence (predicted local distance difference test [pLDDT] score > 70) and high quality in TM regions (TM root mean standard deviation [TM-RMSD] < 1 Å; Figures 3 and S6A). The predicted position of the high-affinity molecule aligned well with the reported ligand in the crystal structure compared to the low-affinity molecule (Figure 3). Beyond the binding affinity, we also checked the predicted agonists of CNR2 (Figure 3A) and antagonists of NK1R (Figure 3B). Consistently, high-affinity functional molecules showed a strong chemical awareness in image representation, a high docking score, and good alignment with the crystal structures (Figure 3). Taken together, combined with ligand-GPCR binding mode analysis, we demonstrated that our LISA-CPI model achieved high performance in identifying both agonists and antagonists for pain-associated GPCRs.
Figure 3.

Representative heatmap of molecules and putative binding modes for pain
(A) Heatmaps of attention levels on ligand images with high activity value (pKi > 8), low activity value (pKi < 6), and agonist (first column). Putative binding structures of these molecules with the CNR2 receptor are shown in the second to fourth columns. Structural comparison between AlphaFold2 and crystal structure for CNR2 at binding positions is shown in the third and fourth columns.
(B) Heatmaps of attention levels on ligand images with high activity value (pKi > 8), low activity value (pKi < 6), and antagonist (first column). Putative binding structures of these molecules with NK1R receptor are shown in the second to fourth columns. Structural comparison between AlphaFold2 and crystal structure for NK1R at binding positions is shown in the third and fourth columns.
Discovery of repurposable drugs via targeting pain-associated GPCRs
We next sought to uncover potential FDA-reported drugs that may act on pain-associated GPCRs as candidate treatments for pain. We used all the compounds in the pain dataset and the 13 pain-associated GPCRs to train LISA-CPI. Subsequently, we employed this trained model to predict ligand-GPCR interactions between 2,308 FDA-approved drugs and 13 pain-associated GPCRs, as we presented earlier. The top 20 drugs with the highest predicted binding affinity for each GPCR were considered the candidate repurposable drugs. As a result, of a total of 42 prioritized drugs, brexpiprazole, ergometrine, fondaparinux, mebutamate, meprobamate, methylergometrine, rolapitant, and sucralfate were predicted to interact with all 13 GPCRs (Figure 4A; Table S2). Here, we prioritized several top-predicted drug-GPCR pairs that may hold potential for treating pain (Figures 4B and 4C). In particular, 4 drugs exhibited superior chemical awareness in molecular image representation (Figure 4B).
Figure 4.

Drug repurposing predictions targeting pain-associated GPCRs
(A) A network illustrating the interaction between the 13 pain-associated GPCR targets and the 20 FDA-approved drugs with the highest predicted activity values (Table S3). Orange lines represent agonists to the GPCR targets, and green lines indicate antagonists to the GPCR target.
(B) Four drugs were selected from the 20 FDA-approved drugs with the highest predicted activity values, and the heatmaps of attention levels on these 4 selected drugs are illustrated. A warmer color indicates a higher attention level, and a cooler color indicates a lower attention level.
(C) Putative binding structures of the molecules in (B) and their corresponding GPCR targets.
Mebutamate is an anxiolytic and sedative drug with anti-hypertensive effects.40 We predicted that mebutamate is a strong agonist with CNR1 (predicted activity score: 10.03), including two hydrogen bonds with residues Asp149 and Tyr328 (Figure 4C). Buprenorphine has been reported to treat acute pain, chronic pain, and opioid use disorder.41 It was reported as a μ-opioid receptor partial agonist,42 consistent with our findings (interacting with OPRM, predicted activity score: 8.80). Apart from the opioid receptors, we also found the drugs that potentially interact with non-opioid receptors. For example, methylergometrine was reported to benefit both the prevention and acute treatment of migraine.43 We predicted that methylergometrine is an antagonist of the 5HT2A receptor (predicted activity score: 9.47; Figure 4C). In addition, we also found that ergometrine (used for postpartum hemorrhage44) has a high antagonistic affinity (predicted activity score: 8.61) with 5HT2A, aligning with the previous report45 (Figure S5C). Rolapitant is used to prevent delayed chemotherapy-induced nausea and vomiting.46 We predicted that it is an antagonist of NK1R (predicted activity score: 8.90), which is consistent with rolapitant being an antagonist of NK1R.47 Vilazodone, an anti-depression drug,48 was predicted to be an agonist of the 5HT1A receptor by forming a hydrogen bond with Asn386 and strong hydrophobic interactions (predicted activity score: 9.31; Figure S5D). Collectively, these FDA-approved drugs prioritized by LISA-CPI may potentially interact with pain-associated receptors, especially non-opioid receptors.
Discovery of gut microbial metabolite via targeting pain-associated GPCRs
To uncover microbial metabolites10,49,50,51 for the potential prevention and treatment of pain, we used LISA-CPI to predict the CPIs between 13 pain-associated GPCRs and 379 human gut-derived metabolites retrieved from a previous study.52 For each GPCR, we prioritized the top 20 gut metabolites that may interact with the GPCR via the LISA-CPI models. The gut bacteria that have the largest level of the investigated metabolites were inspected. Figure 5A shows the network between the gut metabolites and their potential binding GPCRs with the bacteria information (Table S3). We grouped the metabolites by the microbiota genera (see STAR Methods). In total, 18 genera were achieved. Of those, Clostridium and Bacteroides have the most metabolites (11 and 7, respectively). Previous studies have reported that Clostridium and Bacteroides were highly associated with chronic pain by producing butyrate and propionate.11,53 Citicoline (cytidine 5′-diphosphocholine) and NAD (nicotinamide adenine dinucleotide) are the two most abundant metabolites in the bacterium Bacteroides (log2 fold changes are 13.4 and 14.5, respectively, bacteria vs. germ-free control). They also have high attention levels (warmer color) on the metabolites and even higher attention levels on important functional groups, especially hydroxyl groups, amines, carboxyl groups, and carbonyl groups, as suggested by the LISA-CPI framework (Figure 5B). We predicted that these gut microbial metabolites may interact with all 13 pain-associated GPCRs, and the best predicted candidate GPCRs were 5HT2A (predicted activity score of 8.35 as an antagonist) and NK1R (predicted activity score of 8.19 as an antagonist), respectively (Figure 5C). In addition, citicoline and NAD metabolism have been reported to prevent peripheral neuropathic pain in animal models.54,55 For instance, 10 gut metabolites, such as tryptamine and indoleacrylic acid, were prioritized from the bacterium Clostridium. Of those, tryptamine has the highest level in Clostridium (log2 fold change: 12.2). Tryptamine may be involved in alleviating chronic pain by mediating the kynurenine signaling pathway.56 Another tryptophan metabolite, indoleacrylic acid, was reported to mitigate the inflammation response.57 We predicted indoleacrylic acid as an agonist of OPRK (predicted activity score: 7.07) by forming three hydrogen bonds with residues His108, Asn109, and Tyr287 (Figure S5E). Clostridium metabolite 5-aminoimidazole-4-carboxamide-1-beta-ribofuranosyl 5′-monophosphate (AICAR) is an AMPK activator that attenuates inflammatory pain.58 We found that it may inhibit the NK1R with a predicted activity score of 7.31 (Figure S5F). Prevotella, a well-studied genus of bacteria, is found to be significantly associated with increased abdominal pain.59 Furthermore, we found a high level of taurine in Prevotella and interaction with 5HT1A (Figure 5C). Taurine was discovered to regulate inflammatory diseases with joint pain.60 Another fecal bacterium, Clostridiales, was reported to be significantly related to irritable bowel syndrome, which is characterized by abdominal pain. We discovered that the N-acetyltryptophan derived from Clostridiales showed a strong agonistic activity with the 5HT1B receptor (Figure 5C). Together, these results show that gut metabolites identified by LISA-CPI may offer potential molecular therapy for pain treatment.
Figure 5.

Gut-microbiota-derived metabolite predictions targeting pain-associated GPCRs
(A) A network illustrating the interaction between the 13 pain-associated GPCR targets and selected gut-microbiota-derived metabolites (Table S4). Orange lines represent agonists to the GPCR targets, and green lines indicate antagonists to the GPCR target.
(B) Heatmaps of attention levels on 4 selected gut-microbiota-derived metabolites. A warmer color indicates a higher attention level, and a cooler color indicates a lower attention level.
(C) Binding structures of the metabolites in (B) and their corresponding GPCR targets.
Discussion
In this study, we developed the prototype of a deep-learning-based drug discovery framework that integrates both molecular image representation for ligands and protein 3D structure representation in predicting the binding activity using ligand-GPCR interactions. The proposed LISA-CPI framework leverages the pretrained molecular encoder of ImageMol21 and the pretrained Evoformer from AlphaFold2,22 which can take advantage of pretrained models to achieve low computational cost and high accuracy. We demonstrated that the new LISA-CPI framework has superior performance compared to state-of-the-art models in predicting the binding activities for both benchmark and pain-associated ligand-GPCR interaction datasets. Via LISA-CPI models, we computationally prioritized new potential repurposable drugs or gut microbial metabolites as candidate non-addictive treatments for pain by specifically targeting pain-associated GPCRs.
The advantage of the LISA-CPI framework over the ImageMol framework is that it handled not only molecular images but also protein structure representations for each compound-protein (ligand-GPCR) pair. The structural representation encoded by Evoformer captures latent structural and functional information, while the latent features of molecular images provide insights into the global and local structural information of molecules, along with important chemical properties. This integration enables LISA-CPI to capture structural information from both receptors and ligands, which is the key mechanism underlying its good performance. Additionally, the integration of receptors’ structure representations and molecular images allows the LISA-CPI framework to be applicable to multiple protein targets simultaneously, while ImageMol is limited to one protein target at a time. Besides, with receptor structure and function information, the LISA-CPI framework can predict not only accurate CPI activity (binding affinity) but also functionality (agonist/antagonist) without knowledge of structural binding site information. Furthermore, the LISA-CPI framework displayed a superior performance to the state-of-the-art ImageMol. The LISA-CPI framework achieved a 20% improvement in the MAE compared to the ImageMol framework on average, with only one exception: NK1R. For NK-1R, the LISA-CPI framework achieved a comparable performance to ImageMol. For functional prediction, we predicted 12 pain-associated GPCRs, except for CCR2 because of the antagonist-only dataset. The LISA-CPI framework also outperformed state-of-the-art molecular representation models: sequence-based26 and graph-based models.27
Limitations of the study
We acknowledge several potential limitations in the current LISA-CPI framework. First, the model only encodes 2D images of molecules, lacking 3D information on the spatial atomic positions of molecules. Furthermore, single protein representations derived from the Evoformer of AlphaFold2 were employed rather than 3D protein structures of GPCR targets or the ligand-receptor binding complexes. One possible approach to overcome these limitations is to incorporate the 3D structural information of both ligands and receptors by using graph or 3D mesh data of molecules and proteins. The recent advancement of AlphaFold361 in biomolecular interaction prediction holds promise for improving GPCR model accuracy. We believe that by integrating AlphaFold3, we could potentially elevate the performance of LISA-CPI. A previous study showcased a graph neural network model by considering the spatial interactions between ligands, paving a way for effectively leveraging 3D information.62 Another way to improve the performance and generalization of the LISA-CPI framework is to expand our model to a multi-modal deep learning framework. This framework would not only consider information in ligand images or receptor structures but also other representations, such as the physical or chemical properties of both ligands and receptors, Simplified Molecular Input Line Entry System (SMILES) strings of the ligands, and amino acid sequences of the receptors. Using vision transformers,63 which consider more “global” information of the molecular images, to replace the currently used convolutional-neural-network-based molecular encoder may also provide benefits to the LISA-CPI framework. Additionally, while some studies suggest AlphaFold2-generated protein structures may not be universally applicable for structure-based drug design due to relatively low accuracy in side chains,64,65 a recent paper highlights AlphaFold2’s potential in structure-based drug discovery, especially for the GPCR protein family.66 Additional investigation is necessary to determine the case-by-case effectiveness of AlphaFold2 structures for drug discovery.
Although we only explore the interactions between drug/gut metabolites and pain-associated GPCR targets in this study, we believe that the LISA-CPI framework has broader applications beyond modulation of pain. To exhibit our predictions at 3D scale, we also showed the putative binding modes of the selected cases, while the accuracy of molecular docking is still limited.67 These predicted drug/gut metabolite-GPCR interactions may shed insight into further functional validations. Gut metabolites have been implicated in various diseases, such as diabetes,68 depression,69 and Alzheimer’s disease (AD).70 A previous study revealed the molecular relationships between gut metabolite and GPCR targets in AD.71 Thus, it is essential to predict the targets of gut metabolites to shed light on the roles of gut metabolites in disease pathology and aid in the identification of novel therapeutic strategies. Beyond GPCRs, the LISA-CPI framework is able to predict other targets by utilizing Evoformer. For example, many targets for AD that have been derived from genetic analysis, such as PLCG272 and SORL1,73 have no reported bioactive ligands. Importantly, our predictions on repurposable drugs and gut metabolites targeting pain-associated GPCRs require further experimental validations in the future.
Resource availability
Lead contact
Further information and requests for resources and software should be directed to and will be fulfilled by the lead contact, Feixiong Cheng (chengf@ccf.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All predicted GPCR-drug and GPCR-metabolite pairs are freely available in Tables S3 and S4. Accession numbers or web links for the public available datasets are listed in the key resources table.
-
•
All code and datasets used in training and testing are available at https://github.com/ChengF-Lab/LISA-CPI and https://github.com/yuxin212/GPCR-public. Archival DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
This work was primarily supported by the National Institute on Aging (NIA) under award numbers R01AG084250, R56AG074001, U01AG073323, R01AG066707, R01AG076448, R01AG082118, RF1AG082211, and R21AG083003 and the National Institute of Neurological Disorders and Stroke (NINDS) under award number RF1NS133812 to F.C. This work was also supported by the National Science Foundation (NSF) under grant numbers 2217104 and 2212465 to Q.G.
Author contributions
F.C. conceived the study. Y.Y. and Y.Q. implemented the pipeline, constructed the databases, developed the codes, and performed all experiments. Y.Y., Q.G., and F.C. performed data analyses and discussed and interpreted all results. J.H., M.R.-Z., Y.Y., Y.Q., Q.G., and F.C. wrote and critically revised the manuscript.
Declaration of interests
J.H. and M.R.-Z. are full-time employees of IBM Research.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Gut microbial metabolites dataset | Han et al.52 | https://sonnenburglab.github.io/Metabolomics_Data_Explorer |
| GLASS database | Chan et al.24 | https://zhanggroup.org/GLASS/ |
| ChEMBL database | Gaulton et al.25 | https://www.ebi.ac.uk/chembl/ |
| BindingDB database | Gilson et al.74 | https://www.bindingdb.org/ |
| DrugBank | Wishart et al.75 | https://go.drugbank.com/ |
| AlphaFold2 Protein Structure Database | Jumper et al.22 | https://alphafold.ebi.ac.uk/ |
| Software and algorithms | ||
| Open Babel | https://github.com/openbabel/openbabel | Version 3.1.1 |
| Protein Preparation Wizard | Schrödinger Inc. | Version 2020.1 |
| Fpocket suite | https://github.com/Discngine/fpocket | Version 2.0 |
| AutoDock Vina | https://github.com/ccsb-scripps/AutoDock-Vina | Version 1.1.2 |
| ImageMol | Zeng et al.21 | https://doi.org/10.1038/s42256-022-00557-6 |
| AlphaFold2 | Jumper et al.22 | https://doi.org/10.1038/s41586-021-03819-2 |
| Python | https://www.python.org/ | Version 3.8.15 |
| LISA-CPI | This paper | https://github.com/ChengF-Lab/LISA-CPI and https://doi.org/10.5281/zenodo.13551268 |
Method details
Description of dataset
Total 71,757 ligand-GPCR pairs for 20 top-ranked GPCRs and 33,212 pairs for 13 pain-associated GPCRs were retrieved from the ChEMBL and BindingDB databases.25,74 To be accurate, only pairs with Ki value were retained. Duplicate ligand-GPCR pairs were removed based on InChIKey and UniProt ID. The mean of activities was adopted if several values were for one pair. For the top-20 GPCR dataset, 76.6% of compounds (55,001 compounds in total) have an activity value between 6 and 9, and the mean activity value of all compounds is 7.18 (Figure S1A). For the 13 pain-associated GPCR dataset, 74.9% of compounds (25,826 compounds in total) have an activity value between 6 and 9, and the mean activity value of all compounds is 7.28 (Figure S1B). 10,816 ligand-GPCR pairs featuring agonist/antagonist for 13 pain-associated GPCRs were obtained. As only antagonist is available for CCR2, we excluded CCR2 from the training dataset of agonist/antagonist prediction to keep the fairness of the LISA-CPI classification model. 2,308 FDA-approved drugs (only small molecules) were assembled from Drugbank (version 2021.1).75 379 microbial metabolites from human gut strains in vitro were collected from the previous study.52 We further collected compound-kinase interactions for 10 human kinases (Table S1) from ChEMBL database.
Description of the LISA-CPI framework
As shown in Figure 1B, the LISA-CPI framework consists of 4 parts, ligand molecular image feature extraction part based on ImageMol,21 receptor protein structure representation extraction part based on Evoformer of AlphaFold2,22 feature combination and processing part, and CPI prediction part. The ligand molecular image feature extraction part is based on the pretrained molecular encoder from ImageMol
| (Equation 1) |
where stands for the trainable parameters of the molecular encoder , stands for the input molecular image with the shape of and 3 channels, stands for the latent feature, and stands for the number of latent feature channels. The receptor protein structure representation extraction part uses the first part of AlphaFold2, which consists of first searching for MSA representation and pair representation using the amino acid sequence and then using 48 Evoformer blocks to produce the intermediate representations. The intermediate representations include a single representation and a pair representation , where stands for the number of residues of the protein, and and stand for the number of single representation channels and the number of pair representation channels, respectively. The pair representations can become extremely large for proteins with long amino acid sequences. To keep a low computational cost, we only use the single representation in the rest of the model. Next, we calculate the mean value over the residue dimension of to obtain , the 1D structure representation. We then perform min-max normalization to scale the range of to . We observe that is highly noisy with a lot of spikes. Figure S6B (left) shows a highly noisy 1D of 5HT1A. We apply Gaussian smoothing to to reduce spike noises
| (Equation 2) |
| (Equation 3) |
where stands for the smoothed structure representation, is the Gaussian filter with standard deviation , stands for the position at the single representation, and stands for the free variable during the integral. Figure S6B (middle) shows the smoothed structure representation of 5HT1A. To make sure all the structure representations have the same dimension for convenient training, we perform zero padding to both sides of to a dimension of 1,024. Figure S6B (right) shows the zero-padded of 5HT1A with a dimension of 1,024.
We then concatenate the latent feature and the smoothed single representation to get the combined features
| (Equation 4) |
which includes both ligand molecular information and receptor protein structure information. We use the combined features to feed into the activity value prediction model
| (Equation 5) |
where is the trainable parameters of the activity value prediction model, and is the predicted activity value.
For regression tasks, we used Mean Squared Error (MSE) to calculate the loss between the predicted activity value and the ground truth activity value to measure the performance of our model and update trainable parameters in our model through backpropagation
| (Equation 6) |
where is the number of samples in the training dataset. The reason we choose MSE instead of Mean Absolute Error (MAE) is that MAE is minimized by conditional median which may lead to bias during optimization while MSE is minimized by conditional mean which avoids such issue.
For classification tasks, we used Binary Cross Entropy (BCE) to calculate the loss between the predicted class and the ground truth class to update trainable parameters in our model through backpropagation
| (Equation 7) |
where is the number of samples in the training dataset.
We only optimize the ligand molecular image learning part and the activity value prediction part with all parameters of the protein structure learning part frozen, because Evoformer has a relatively large size which can take a long time to train.
Molecular docking
3D structure models of GPCRs were retrieved from AlphaFold2 Website (https://alphafold.ebi.ac.uk/). 2D structures of small molecules were processed by Open Babel. All protein structures were prepared by using the Protein Preparation Wizard module (Schrödinger Inc, version 2020.1). Fpocket suite (version 2.0) was utilized to characterize potential druggable binding sites.76 Molecular docking was processed by AutoDock Vina (version 1.1.2).77
Model tuning and hyperparameter selection
To train our models, a scheduled learning rate was set. The initial learning rate was set to , and the weight decay was set to . The first 10 epochs were scheduled to warm up the learning rate, with three learning rate milestones at 10 epochs, 20 epochs, and 30 epochs. The AdamW78 optimizer was used to find the optimal trainable parameters of the models. Each model was trained for 80 epochs in total, with early stopping implemented.
For baseline comparison methods, the models were trained based on the pre-trained models provided by the original studies. The default hyperparameters of these models, as provided in the original code, were used to train baseline comparison methods.
Quantification and statistical analysis
Performance evaluations of the methods for binding affinity prediction tasks (regression tasks) were measured using mean absolute error (MAE) and Pearson’s correlation coefficient (R).
The MAE is calculated as follows:
| (Equation 8) |
where is the number of compounds in the test dataset, which is 30% of the total dataset, is the actual binding affinity value, and is the predicted binding affinity value. Specifically, 10-fold cross validation was performed on the training dataset to compute the mean and standard deviations of the MAEs.
Pearson’s correlation coefficient (R) is calculated as follows:
| (Equation 9) |
where is the mean of all actual binding affinity values in the test dataset, and is the mean of all predicted binding affinity values in the test dataset. The model with the best performance during 10-fold cross validation was used to predict the binding affinity values and plot contour plots with Pearson’s correlation coefficient (R) in Figure 2.
For the classification tasks, including agonist-antagonist classification and compound-Kinase interaction classification, the performance evaluations were measured with the area under receiver operating characteristic (AROC) curve. The ROC curves were plotted by calculating the true positive rate against false positive rate at all possible intervals.
Published: September 27, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2024.100865.
Contributor Information
Qiang Guan, Email: qguan@kent.edu.
Feixiong Cheng, Email: chengf@ccf.org.
Supplemental information
References
- 1.Duca L.M., Helmick C.G., Barbour K.E., Nahin R.L., Von Korff M., Murphy L.B., Theis K., Guglielmo D., Dahlhamer J., Porter L., et al. A Review of Potential National Chronic Pain Surveillance Systems in the United States. J. Pain. 2022;23:1492–1509. doi: 10.1016/j.jpain.2022.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GBD 2015 Disease and Injury Incidence and Prevalence Collaborators national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1545–1602. doi: 10.1016/s0140-6736(16)31678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Volkow N.D., McLellan A.T. Opioid Abuse in Chronic Pain--Misconceptions and Mitigation Strategies. N. Engl. J. Med. 2016;374:1253–1263. doi: 10.1056/NEJMra1507771. [DOI] [PubMed] [Google Scholar]
- 4.Jeon M., Jagodnik K.M., Kropiwnicki E., Stein D.J., Ma'ayan A. Prioritizing Pain-Associated Targets with Machine Learning. Biochemistry. 2021;60:1430–1446. doi: 10.1021/acs.biochem.0c00930. [DOI] [PubMed] [Google Scholar]
- 5.Geppetti P., Veldhuis N.A., Lieu T., Bunnett N.W. G Protein-Coupled Receptors: Dynamic Machines for Signaling Pain and Itch. Neuron. 2015;88:635–649. doi: 10.1016/j.neuron.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Brust T.F., Morgenweck J., Kim S.A., Rose J.H., Locke J.L., Schmid C.L., Zhou L., Stahl E.L., Cameron M.D., Scarry S.M., et al. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci. Signal. 2016;9:ra117. doi: 10.1126/scisignal.aai8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manglik A., Lin H., Aryal D.K., McCorvy J.D., Dengler D., Corder G., Levit A., Kling R.C., Bernat V., Hübner H., et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Draper-Joyce C.J., Bhola R., Wang J., Bhattarai A., Nguyen A.T.N., Cowie-Kent I., O'Sullivan K., Chia L.Y., Venugopal H., Valant C., et al. Positive allosteric mechanisms of adenosine A(1) receptor-mediated analgesia. Nature. 2021;597:571–576. doi: 10.1038/s41586-021-03897-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen D.D., Lieu T., Halls M.L., Veldhuis N.A., Imlach W.L., Mai Q.N., Poole D.P., Quach T., Aurelio L., Conner J., et al. Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci. Transl. Med. 2017;9:eaal3447. doi: 10.1126/scitranslmed.aal3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li S., Hua D., Wang Q., Yang L., Wang X., Luo A., Yang C. The Role of Bacteria and Its Derived Metabolites in Chronic Pain and Depression: Recent Findings and Research Progress. Int. J. Neuropsychopharmacol. 2020;23:26–41. doi: 10.1093/ijnp/pyz061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garvey M. The Association between Dysbiosis and Neurological Conditions Often Manifesting with Chronic Pain. Biomedicines. 2023;11:748. doi: 10.3390/biomedicines11030748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodgkinson K., El Abbar F., Dobranowski P., Manoogian J., Butcher J., Figeys D., Mack D., Stintzi A. Butyrate's role in human health and the current progress towards its clinical application to treat gastrointestinal disease. Clin. Nutr. 2023;42:61–75. doi: 10.1016/j.clnu.2022.10.024. [DOI] [PubMed] [Google Scholar]
- 13.Paul S.M., Mytelka D.S., Dunwiddie C.T., Persinger C.C., Munos B.H., Lindborg S.R., Schacht A.L. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 14.Ye Q., Hsieh C.Y., Yang Z., Kang Y., Chen J., Cao D., He S., Hou T. A unified drug-target interaction prediction framework based on knowledge graph and recommendation system. Nat. Commun. 2021;12:6775. doi: 10.1038/s41467-021-27137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacob L., Vert J.P. Protein-ligand interaction prediction: an improved chemogenomics approach. Bioinformatics. 2008;24:2149–2156. doi: 10.1093/bioinformatics/btn409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bock J.R., Gough D.A. Virtual screen for ligands of orphan G protein-coupled receptors. J. Chem. Inf. Model. 2005;45:1402–1414. doi: 10.1021/ci050006d. [DOI] [PubMed] [Google Scholar]
- 17.Pahikkala T., Airola A., Pietilä S., Shakyawar S., Szwajda A., Tang J., Aittokallio T. Toward more realistic drug-target interaction predictions. Brief. Bioinform. 2015;16:325–337. doi: 10.1093/bib/bbu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamanishi Y., Araki M., Gutteridge A., Honda W., Kanehisa M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24:i232–i240. doi: 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Öztürk H., Özgür A., Ozkirimli E. DeepDTA: deep drug-target binding affinity prediction. Bioinformatics. 2018;34:i821–i829. doi: 10.1093/bioinformatics/bty593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen T., Le H., Quinn T.P., Nguyen T., Le T.D., Venkatesh S. GraphDTA: predicting drug-target binding affinity with graph neural networks. Bioinformatics. 2021;37:1140–1147. doi: 10.1093/bioinformatics/btaa921. [DOI] [PubMed] [Google Scholar]
- 21.Zeng X., Xiang H., Yu L., Wang J., Li K., Nussinov R., Cheng F. Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework. Nat. Mach. Intell. 2022;4:1004–1016. doi: 10.1038/s42256-022-00557-6. [DOI] [Google Scholar]
- 22.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim S., Lu Y., Cho C.Y., Sung I., Kim J., Kim Y., Park S., Kim S. A review on compound-protein interaction prediction methods: Data, format, representation and model. Comput. Struct. Biotechnol. J. 2021;19:1541–1556. doi: 10.1016/j.csbj.2021.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan W.K.B., Zhang H., Yang J., Brender J.R., Hur J., Özgür A., Zhang Y. GLASS: a comprehensive database for experimentally validated GPCR-ligand associations. Bioinformatics. 2015;31:3035–3042. doi: 10.1093/bioinformatics/btv302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaulton A., Bellis L.J., Bento A.P., Chambers J., Davies M., Hersey A., Light Y., McGlinchey S., Michalovich D., Al-Lazikani B., Overington J.P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim H., Lee J., Ahn S., Lee J.R. A merged molecular representation learning for molecular properties prediction with a web-based service. Sci. Rep. 2021;11:11028. doi: 10.1038/s41598-021-90259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y., Wang J., Cao Z., Barati Farimani A. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 2022;4:279–287. doi: 10.1038/s42256-022-00447-x. [DOI] [Google Scholar]
- 28.Selvaraju R.R., Cogswell M., Das A., Vedantam R., Parikh D., Batra D. 2017 IEEE International Conference on Computer Vision (ICCV) 2017. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization; pp. 618–626. [DOI] [Google Scholar]
- 29.Schöppe J., Ehrenmann J., Klenk C., Rucktooa P., Schütz M., Doré A.S., Plückthun A. Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists. Nat. Commun. 2019;10:17. doi: 10.1038/s41467-018-07939-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng Y., Qin L., Zacarías N.V.O., de Vries H., Han G.W., Gustavsson M., Dabros M., Zhao C., Cherney R.J., Carter P., et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature. 2016;540:458–461. doi: 10.1038/nature20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Che T. Advances in the Treatment of Chronic Pain by Targeting GPCRs. Biochemistry. 2021;60:1401–1412. doi: 10.1021/acs.biochem.0c00644. [DOI] [PubMed] [Google Scholar]
- 32.Gottesman-Katz L., Latorre R., Vanner S., Schmidt B.L., Bunnett N.W. Targeting G protein-coupled receptors for the treatment of chronic pain in the digestive system. Gut. 2021;70:970–981. doi: 10.1136/gutjnl-2020-321193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.James A., Williams J. Basic Opioid Pharmacology - An Update. Br. J. Pain. 2020;14:115–121. doi: 10.1177/2049463720911986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sommer C. Serotonin in pain and analgesia: actions in the periphery. Mol. Neurobiol. 2004;30:117–125. doi: 10.1385/mn:30:2:117. [DOI] [PubMed] [Google Scholar]
- 35.Pertwee R.G. Cannabinoid receptors and pain. Prog. Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 36.Sevostianova N., Danysz W. Analgesic effects of mGlu1 and mGlu5 receptor antagonists in the rat formalin test. Neuropharmacology. 2006;51:623–630. doi: 10.1016/j.neuropharm.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Abbadie C., Lindia J.A., Cumiskey A.M., Peterson L.B., Mudgett J.S., Bayne E.K., DeMartino J.A., MacIntyre D.E., Forrest M.J. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc. Natl. Acad. Sci. USA. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dionne R.A., Max M.B., Gordon S.M., Parada S., Sang C., Gracely R.H., Sethna N.F., MacLean D.B. The substance P receptor antagonist CP-99,994 reduces acute postoperative pain. Clin. Pharmacol. Ther. 1998;64:562–568. doi: 10.1016/s0009-9236(98)90140-0. [DOI] [PubMed] [Google Scholar]
- 39.Piscitelli F., Ligresti A., La Regina G., Gatti V., Brizzi A., Pasquini S., Allarà M., Carai M.A.M., Novellino E., Colombo G., et al. 1-Aryl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide: an effective scaffold for the design of either CB1 or CB2 receptor ligands. Eur. J. Med. Chem. 2011;46:5641–5653. doi: 10.1016/j.ejmech.2011.09.037. [DOI] [PubMed] [Google Scholar]
- 40.Tetreault L., Richer P., Bordeleau J.M. Hypnotic properties of mebutamate: a comparative study of mebutamate, secobarbital and placebo in psychiatric patients. Can. Med. Assoc. J. 1967;97:395–398. [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson R.E., Fudala P.J., Payne R. Buprenorphine: considerations for pain management. J. Pain Symptom Manage. 2005;29:297–326. doi: 10.1016/j.jpainsymman.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Cowan A., Lewis J.W., Macfarlane I.R. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br. J. Pharmacol. 1977;60:537–545. doi: 10.1111/j.1476-5381.1977.tb07532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niño-Maldonado A.I., Caballero-García G., Mercado-Bochero W., Rico-Villademoros F., Calandre E.P. Efficacy and tolerability of intravenous methylergonovine in migraine female patients attending the emergency department: a pilot open-label study. Head Face Med. 2009;5:21. doi: 10.1186/1746-160x-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spencer S.P.E., Lowe S.A. Ergometrine for postpartum hemorrhage and associated myocardial ischemia: Two case reports and a review of the literature. Clin. Case Rep. 2019;7:2433–2442. doi: 10.1002/ccr3.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson M.P., Loncharich R.J., Baez M., Nelson D.L. Species variations in transmembrane region V of the 5-hydroxytryptamine type 2A receptor alter the structure-activity relationship of certain ergolines and tryptamines. Mol. Pharmacol. 1994;45:277–286. [PubMed] [Google Scholar]
- 46.Goldberg T., Fidler B., Cardinale S. Rolapitant (Varubi): A Substance P/Neurokinin-1 Receptor Antagonist for the Prevention of Chemotherapy-Induced Nausea and Vomiting. P T. 2017;42:168–172. [PMC free article] [PubMed] [Google Scholar]
- 47.Duffy R.A., Morgan C., Naylor R., Higgins G.A., Varty G.B., Lachowicz J.E., Parker E.M. Rolapitant (SCH 619734): a potent, selective and orally active neurokinin NK1 receptor antagonist with centrally-mediated antiemetic effects in ferrets. Pharmacol. Biochem. Behav. 2012;102:95–100. doi: 10.1016/j.pbb.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 48.Chauhan M., Parry R., Bobo W.V. Vilazodone for Major Depression in Adults: Pharmacological Profile and an Updated Review for Clinical Practice. Neuropsychiatr. Dis. Treat. 2022;18:1175–1193. doi: 10.2147/ndt.S279342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin B., Wang Y., Zhang P., Yuan Y., Zhang Y., Chen G. Gut microbiota regulates neuropathic pain: potential mechanisms and therapeutic strategy. J. Headache Pain. 2020;21:103. doi: 10.1186/s10194-020-01170-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen P., Wang C., Ren Y.-n., Ye Z.-j., Jiang C., Wu Z.-b. Alterations in the gut microbiota and metabolite profiles in the context of neuropathic pain. Mol. Brain. 2021;14:50. doi: 10.1186/s13041-021-00765-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo R., Chen L.-H., Xing C., Liu T. Pain regulation by gut microbiota: molecular mechanisms and therapeutic potential. Brit. Br. J. Anaesth. 2019;123:637–654. doi: 10.1016/j.bja.2019.07.026. [DOI] [PubMed] [Google Scholar]
- 52.Han S., Van Treuren W., Fischer C.R., Merrill B.D., DeFelice B.C., Sanchez J.M., Higginbottom S.K., Guthrie L., Fall L.A., Dodd D., et al. A metabolomics pipeline for the mechanistic interrogation of the gut microbiome. Nature. 2021;595:415–420. doi: 10.1038/s41586-021-03707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minerbi A., Gonzalez E., Brereton N.J.B., Anjarkouchian A., Dewar K., Fitzcharles M.A., Chevalier S., Shir Y. Altered microbiome composition in individuals with fibromyalgia. Pain. 2019;160:2589–2602. doi: 10.1097/j.pain.0000000000001640. [DOI] [PubMed] [Google Scholar]
- 54.Emril D.R., Wibowo S., Meliala L., Susilowati R. Cytidine 5'-diphosphocholine administration prevents peripheral neuropathic pain after sciatic nerve crush injury in rats. J. Pain Res. 2016;9:287–291. doi: 10.2147/jpr.S70481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai Y., Lin J., Ren J., Zhu B., Wu C., Yu L. NAD(+) metabolism in peripheral neuropathic pain. Neurochem. Int. 2022;161:105435. doi: 10.1016/j.neuint.2022.105435. [DOI] [PubMed] [Google Scholar]
- 56.Jovanovic F., Candido K.D., Knezevic N.N. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. Int. J. Mol. Sci. 2020;21:6045. doi: 10.3390/ijms21176045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wlodarska M., Luo C., Kolde R., d'Hennezel E., Annand J.W., Heim C.E., Krastel P., Schmitt E.K., Omar A.S., Creasey E.A., et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe. 2017;22:25–37.e6. doi: 10.1016/j.chom.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang H.-C., Lin L.-X., Hu X.-F., Zhu H., Li H.-P., Zhang R.-Y., Hu L., Liu W.-T., Zhao Y.-L., Shu Y., et al. AMPK activation attenuates inflammatory pain through inhibiting NF-κB activation and IL-1β expression. J. Neuroinflammation. 2019;16:34. doi: 10.1186/s12974-019-1411-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choo C., Mahurkar-Joshi S., Dong T.S., Lenhart A., Lagishetty V., Jacobs J.P., Labus J.S., Jaffe N., Mayer E.A., Chang L. Colonic mucosal microbiota is associated with bowel habit subtype and abdominal pain in patients with irritable bowel syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 2022;323:G134–G143. doi: 10.1152/ajpgi.00352.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schaffer S., Kim H.W. Effects and Mechanisms of Taurine as a Therapeutic Agent. Biomol. Ther. (Seoul) 2018;26:225–241. doi: 10.4062/biomolther.2017.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abramson J., Adler J., Dunger J., Evans R., Green T., Pritzel A., Ronneberger O., Willmore L., Ballard A.J., Bambrick J., et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630:493–500. doi: 10.1038/s41586-024-07487-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li S., Zhou J., Xu T., Huang L., Wang F., Xiong H., Huang W., Dou D., Xiong H. Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining. 2021. Structure-aware Interactive Graph Neural Networks for the Prediction of Protein-Ligand Binding Affinity; pp. 975–985. [DOI] [Google Scholar]
- 63.Dosovitskiy A., Beyer L., Kolesnikov A., Weissenborn D., Zhai X., Unterthiner T., Dehghani M., Minderer M., Heigold G., Gelly S. An image is worth 16x16 words: Transformers for image recognition at scale. arXiv. 2020 doi: 10.48550/arXiv.2010.11929. Preprint at. 2010.11929. [DOI] [Google Scholar]
- 64.He X.-h., You C.-z., Jiang H.-l., Jiang Y., Xu H.E., Cheng X. AlphaFold2 versus experimental structures: evaluation on G protein-coupled receptors. Acta Pharmacol. Sin. 2023;44:1–7. doi: 10.1038/s41401-022-00938-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H., Sun X., Cui W., Xu M., Dong J., Ekundayo B.E., Ni D., Rao Z., Guo L., Stahlberg H., et al. Computational drug development for membrane protein targets. Nat. Biotechnol. 2024;42:229–242. doi: 10.1038/s41587-023-01987-2. [DOI] [PubMed] [Google Scholar]
- 66.Lyu J., Kapolka N., Gumpper R., Alon A., Wang L., Jain M.K., Barros-Álvarez X., Sakamoto K., Kim Y., DiBerto J., et al. AlphaFold2 structures guide prospective ligand discovery. Science. 2024;384:eadn6354. doi: 10.1126/science.adn6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Su M., Yang Q., Du Y., Feng G., Liu Z., Li Y., Wang R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019;59:895–913. doi: 10.1021/acs.jcim.8b00545. [DOI] [PubMed] [Google Scholar]
- 68.Hu J., Ding J., Li X., Li J., Zheng T., Xie L., Li C., Tang Y., Guo K., Huang J., et al. Distinct signatures of gut microbiota and metabolites in different types of diabetes: a population-based cross-sectional study. eClinicalMedicine. 2023;62:102132. doi: 10.1016/j.eclinm.2023.102132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu L., Wang H., Chen X., Zhang Y., Zhang H., Xie P. Gut microbiota and its metabolites in depression: from pathogenesis to treatment. EBioMedicine. 2023;90:104527. doi: 10.1016/j.ebiom.2023.104527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferreiro A.L., Choi J., Ryou J., Newcomer E.P., Thompson R., Bollinger R.M., Hall-Moore C., Ndao I.M., Sax L., Benzinger T.L.S., et al. Gut microbiome composition may be an indicator of preclinical Alzheimer’s disease. Sci. Transl. Med. 2023;15:eabo2984. doi: 10.1126/scitranslmed.abo2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qiu Y., Hou Y., Gohel D., Zhou Y., Xu J., Bykova M., Yang Y., Leverenz J.B., Pieper A.A., Nussinov R., et al. Systematic characterization of multi-omics landscape between gut microbial metabolites and GPCRome in Alzheimer's disease. Cell Rep. 2024;43:114128. doi: 10.1016/j.celrep.2024.114128. [DOI] [PubMed] [Google Scholar]
- 72.Andreone B.J., Przybyla L., Llapashtica C., Rana A., Davis S.S., van Lengerich B., Lin K., Shi J., Mei Y., Astarita G., et al. Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat. Neurosci. 2020;23:927–938. doi: 10.1038/s41593-020-0650-6. [DOI] [PubMed] [Google Scholar]
- 73.Pottier C., Hannequin D., Coutant S., Rovelet-Lecrux A., Wallon D., Rousseau S., Legallic S., Paquet C., Bombois S., Pariente J., et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol. Psychiatry. 2012;17:875–879. doi: 10.1038/mp.2012.15. [DOI] [PubMed] [Google Scholar]
- 74.Gilson M.K., Liu T., Baitaluk M., Nicola G., Hwang L., Chong J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016;44:D1045–D1053. doi: 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Le Guilloux V., Schmidtke P., Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformat. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loshchilov I., Hutter F. Decoupled weight decay regularization. arXiv. 2017 doi: 10.48550/arXiv.1711.05101. Preprint at. 1711.05101. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All predicted GPCR-drug and GPCR-metabolite pairs are freely available in Tables S3 and S4. Accession numbers or web links for the public available datasets are listed in the key resources table.
-
•
All code and datasets used in training and testing are available at https://github.com/ChengF-Lab/LISA-CPI and https://github.com/yuxin212/GPCR-public. Archival DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.