

Abstract
The rare-earth metal dialkyl complexes (κ2-L1)RE(CH2SiMe3)2·(THF)2 [RE = Lu(1a), Yb(1b), Er(1c), Y(1d), Dy(1e)] (L1 = 1-(2-N-C5H10NCH2CH2)-3-(2,6-iPr2C6H3N CH)-C8H4N) and the rare-earth metal monoalkyl complexes (κ2-L1)2RE(CH2SiMe3)·(THF)n [n = 0, RE = Lu(2a), Yb(2b); n = 1, Er(2c), Y(2d), Dy(2e)], (κ2-L2)2RE(CH2SiMe3)·THF [RE = Yb(3a), Er(3b), Y(3c), Dy(3d), Gd(3e)] (L2 = 1-(2-N-C5H10NCH2CH2)-3-(AdN CH)-C8H4N) (Ad = adamantyl, C10H15) have been synthesized and fully characterized. These complexes feature chelate ligands having a conjugated system (–C C–C N) with an sp2 carbon, which enables both electrophilic and nucleophilic carbon centres to be directly connected to the highly electrophilic rare-earth metal ions. The reactions of these complexes with different pyridine derivatives have been systematically investigated with the discovery of reactivity patterns distinct from those of previously reported transition metal complexes. These unusual reactivity patterns include consecutive C–H activation/1,1-migratory insertion/C–N and C–H activation, C–H activation/1,1-migratory insertion/C–N bond activation, C–H activation/1,1-migratory insertion, and homolytic redox reactions. DFT calculation results together with experimental evidences support the key mechanistic proposal that the indol-2-yl carbon of the ligands exhibits electrophilic carbene character accounting for the subsequent 1,1-migratory insertion reaction after C–H activation.
The rare-earth metal complexes exhibited unique reactivity towards pyridine derivatives leading to the unveiling of consecutive C–H activation/1,1-migratory insertion/C–N bond activation via intramolecular redox, and homolytic redox reactions.
Introduction
Pyridine and its derivatives, as an important class of pharmaceutical platform, are widely used in drug design and also play crucial roles in organic synthesis, inorganic materials and organometallic chemistry. Transformation of pyridine derivatives is thus a great interest of current synthetic chemistry.1 Since the initial report on cyclometallation reactions of titanium alkyl complexes with pyridines in 1981,2 a range of transition metal and rare-earth metal alkyl or hydrido complexes have been studied in such cyclometallation reactions (Scheme 1a),3,5a,6d and 1,2- and 1,4-benzyl additions to the pyridine ring of 2-phenylpyridine were occasionally observed (Scheme 1a).4 After activation of the sp2 C–H bond on the pyridine or the phenyl ring of 2-phenylpyridine, the resulting three- or five-membered metallacycles can react with hydrogen, Lewis bases, alkenes, alkynes, selenium and carbon monoxide to produce various functionalized pyridine derivatives.5,6 However, to date, the migratory insertion of the three-membered metallacycles to the electrophilic carbon bonded to the rare-earth metal centre has not yet been reported, and the sp3 C–H activation of 2-N,N-dimethylaminopyridine followed by 1,1-migratory insertion and C–N bond cleavage also remains elusive.
Scheme 1. Reactivity of metal complexes with pyridine derivatives.

Meanwhile, the 2,2′-bipyridyl radical anion is commonly obtained by the reactions of low-valent rare-earth metal complexes with 2,2′-bipyridine (Scheme 2, method 1).7 Alternatively, 2,2′-bipyridyl anionic radical rare-earth or actinide metal complexes can also be obtained by treatment of the corresponding complexes with 2,2′-bipyridine in the presence of KC8 (Scheme 2, method 2).8 Following these two methods, a range of rare-earth metal and actinide complexes containing the 2,2′-bipyridyl anionic radical have been reported,8b,c,9–14 and they showed unique reactivity towards various organic or inorganic molecules.8b,d–g,9–14 Notably, in the reaction of 1,3-butadiene Sc(iii) complex with 2,2′-bipyridine,15 it was proposed to involve a Sc(iii) to Sc(i) reaction pattern to produce the scandium bipyridyl anionic radical (bipy˙−) complex. However, transformation of Sc(iii) to Sc(i) is unlikely under such conditions due to the high reducing potential of Sc(iii) to Sc(i) and the reactivity of the corresponding bipyridyl anionic radical complex has not been studied. Very recently, Zi and coworkers reported that the reaction of (Cp3tms)2ThIVI2 (Cp3tms = η5-1,2,4-(Me3Si)3C5H2) with a mixture of bipyridine and KC8 afforded the thorium 2,2′-bipyridyl metallocene (Cp3tms)2Th(bipy), in which the bipyridine is a dianionic ligand and the thorium is in the oxidation state of +4. This complex exhibited plenty of reactivities with various molecules to generate one electron or two electron reduction and C–C coupling products.16 In addition, it has been reported very recently that the reaction of 1,3-bisfunctionalized indol-2-yl rare-earth metal dialkyl complexes with 2,2′-bipyridine produced the bipyridyl anionic radical complexes (Scheme 2, method 3).3g However, the reactivity of the corresponding rare-earth metal 2,2′-bipyridyl radical anion complexes has yet been underexplored.
Scheme 2. Prior methods for the preparation of 2,2′-bipyridyl and 6,6′-dimethyl-2,2′-bipyridyl anionic radical complexes.

On the other hand, various types of rare-earth metal alkylidene complexes including bridged17 and mononuclear terminal ones18 were synthesized since the proposed formation of rare-earth alkylidene complexes by Schumann in 1979.19 These alkylidene complexes, however, displayed mainly nucleophilic properties. Meanwhile, rare-earth metal complexes with singlet N-heterocarbenes (NHCs)20 and cyclic (alkyl)(amino) carbenes (CAACs)21 as supporting ligands have also been reported. However, reactivities like 1,1-migratory insertion and nucleophilic substitution reactions, which are common in traditional late transition metal electrophilic carbenes, are rarely developed except for our recent reports by utilizing the 1,3-bisfunctionalized indol-2-yl ligands (Scheme 3).22 Also, the synthesis of high-oxidation state rare-earth metal complexes bearing both electrophilic carbene and nucleophilic alkyls faces a great challenge due to their extreme instability and reactivity, which results from the lack of electrons in the d-orbital to form π-backbonding with the rare-earth metal ions to stabilize the corresponding electrophilic carbene.
Scheme 3. Rare-earth metal electrophilic carbenes and their 1,1-migratory insertion reactions from our previous studies.

In view of these points, we designed the chelate ligands bearing a conjugated system (–C C–C N), which can produce an sp2 carbon showing electrophilic character to bond with the rare-earth metal centre. Thus, a series of 1,3-bisfunctionalized indol-2-yl rare-earth metal complexes bearing electrophilic and nucleophilic carbon centres were synthesized, and their reactivities towards pyridine derivatives were extensively studied with findings of unusual reactivity patterns. Herein, we report these results.
Results and discussion
Synthesis and characterization of the rare-earth metal complexes
The stoichiometric (1 : 1) reactions of the proligand22c HL1 (L1 = 1-(2-N-C5H10NCH2CH2)-3-(2,6-iPr2C6H3N CH)-C8H5N) (see the ESI†) with RE(CH2SiMe3)3·(THF)2 (RE = Lu, Yb, Er, Y, Dy) at room temperature afforded the corresponding rare-earth metal dialkyl complexes (κ2-L1)RE(CH2SiMe3)2·(THF)2 [RE = Lu(1a), Yb(1b), Er(1c), Y(1d), Dy(1e)] (Scheme 4 and Fig. 1).22c In addition, the stoichiometric (2 : 1) reactions of the proligand HL1 (L1 = 1-(2-N-C5H10NCH2CH2)-3-(2,6-iPr2C6H3N CH)-C8H5N) and HL2 (L2 = 1-(2-N-C5H10NCH2CH2)-3-(AdN CH)-C8H5N) (Ad = adamantyl, C10H15) (see the ESI†) with RE(CH2SiMe3)3·(THF)2 (RE = Lu, Yb, Er, Y, Dy, Gd) at room temperature afforded the corresponding rare-earth metal monoalkyl complexes (κ2-L1)2RE(CH2SiMe3)·(THF)n [n = 0, RE = Lu(2a), Yb(2b); n = 1, Er(2c), Y(2d), Dy(2e)] and (κ2-L2)2RE(CH2SiMe3)·THF [RE = Yb(3a), Er(3b), Y(3c), Dy(3d), Gd(3e)] (Scheme 4 and Fig. 1). These complexes are very sensitive to moisture and air. They are soluble in THF and toluene, but slightly soluble in hexane. All complexes were fully characterized by spectroscopic methods, elemental analyses, and single crystal X-ray diffraction studies. The diamagnetic complexes 1a, 1d, 2a, 2d and 3c were further characterized by the NMR spectroscopic method (see the ESI†). The methylene protons of the –CH2SiMe3 moiety in 3c exhibited a signal at −0.11 ppm. The methylene protons of the Y–CH2SiMe3 (1d) gave doublet resonances at −0.20 ppm due to coupling with the yttrium ion (2JY–H = 5.0 Hz), which is comparable with the results reported in the literature (−0.43 ppm for the methylene protons of the Y–CH2SiMe3, 2JY–H = 3.0 Hz in {[η1–μ–η1-3-(CyNCH(CH2SiMe3))Ind]Y(THF)2(CH2SiMe3)}2).23 The signals centred at 40.8, 50.7 and 55.1 ppm in the 13C{1H} spectra for 1d, 2d and 3c were assigned to the methylene carbons of the Y–CH2SiMe3 coupled to the yttrium nucleus with 1JY–C = 36.3 Hz, 37.5 Hz, and 37.5 Hz, respectively. In addition, the signals centred at 204.1, 205.8, and 205.2 ppm for the corresponding 1d, 2d and 3c in the 13C{1H} spectrum were assigned to the indol-2-yl carbon (C2-ind) coupled to the yttrium nucleus with 1JY–C = 43.8 Hz, 50.0 Hz, and 50.0 Hz, respectively, which correlate with previous results (202.0 ppm for the indol-2-yl carbon (C2-ind)) with 1JY–C = 50.0 Hz,22b but are shifted downfield in comparison with the resonance at 197.1 ppm as observed in the NHC carbene carbon 1JY–C = 25.0 Hz in LMesYCH2TMS(THF) (LMes = 1-(3-(2,6-iPr2C6H3N CH)C8H4N)-CH2CH2-3-(2-CH2-4,6-Me2C6H2)-(N(CH)2NC)).22b The 89Y NMR spectra of 1d and 3c have resonances at δ = 1101.5 and 773.0 ppm, respectively, which are within the wide range of reported organometallic yttrium complexes30 (Fig. S14 and S22 in the ESI†). The 1,3-bisfunctionalized indol-2-yl ligands are all bonded to the rare-earth metal ion in the κ2 form, which is different from the κ3 bonding mode found in the dimeric chloride complexes,24 probably due to the coordination of THF and steric effects.
Scheme 4. Synthesis of the rare-earth metal complexes 1–3.

Fig. 1. Representative molecular structures of 1d (left), 2d (middle) and 3c (right) with the thermal ellipsoid at 30% probability level. All hydrogen atoms are omitted and the diisopropylphenyl (Dipp) group, the adamantyl (Ad) group and the carbon atoms of THF are drawn in wireframe style for clarity.

Reactivity of the rare-earth metal complexes with pyridine derivatives
Given the importance of pyridine derivatives as molecular platforms in drug design and the motive to acquire meaningful insights in understanding catalytic reaction mechanisms, we performed stoichiometric reactions of the above-synthesized complexes with pyridine derivatives.
Reactions of the rare-earth metal complexes with 2-N,N-dimethylaminopyridine. Unique consecutive sp3 C–H activation/1,1-migratory insertion/C–N activation through intramolecular redox
The stoichiometric (1 : 1) reactions of the dialkyl complexes 1 with 2-N,N-dimethylaminopyridine in toluene at room temperature afforded the complexes L1-aRE(η2-N,N-2-MeNPy)(THF) [RE = Lu(4a), Yb(4b), Er(4c), Y(4d)] (L1-a = 1-(2-N-C5H10NCH2CH2)-3-(2,6-iPr2C6H3NCH)-2-(methinyl)-C8H4N) in good yields (Scheme 5 and Fig. 2). The 89Y NMR spectrum of 4d shows a resonance at δ = 880.7 ppm, which is in the range of reported yttrium complexes30 (see Fig. S29 in the ESI†). Single crystal X-ray structural analyses reveal that these complexes are composed of an unusual 1,3-bisfunctionalized indol-2-methinyl ligand, and 2-N-methylamidopyridine. The latter possibly originated from 2-N,N-dimethylaminopyridine through consecutive sp3 C–H activation and C–N bond cleavage. The unexpected 1,3-bisfunctionalized indol-2-methinyl ligand is proposed to result from a unique sequence of sp3 C–H activation of 2-N,N-dimethylaminopyridine/1,1-migratory insertion/C–N bond cleavage through intramolecular redox, C–H activation and dimerization (see Scheme S5 in the ESI†). To the best of our knowledge, this reactivity pattern is unprecedented in the reactions of transition metal complexes with pyridine derivatives.
Scheme 5. Reactions of 1 with 2-N,N-dimethylaminopyridine.

Fig. 2. Representative molecular structure of 4d. All hydrogen atoms are omitted and the diisopropylphenyl (Dipp) group and the carbon atoms of THF are drawn in wireframe style for clarity.

The reaction process of 1d with 2-N,N-dimethylaminopyridine is then investigated by DFT calculations, which is in agreement with experimental results. The calculations reveal that the C1 (indol-2-yl carbon) of the indolyl shows electrophilic character with a natural charge of −0.19823 owing to the lack of electrons in the p-orbital, while the Y–C65 and Y–C80 bonds of the Y–CH2SiMe3 are strongly polarized with the natural charges of −1.66362 and −1.64763 for the C65 and C80 (see the ESI†), respectively, exhibiting strong nucleophilic properties. First, the dissociation of two THF molecules provides the vacant site for the coordination of the pyridine amine in B, in which the C1 (indol-2-yl carbon) of the indolyl displays even higher electrophilicity (natural charge −0.15301). This indicates that the coordination of the strongly electron-donating ligand with the central metal facilitates the formation of the rare-earth metal electrophilic carbene. The formation of intermediate B is computed to be endothermic by 6.7 kcal mol−1. Second, the C–H activation of the methyl group of the pyridine amine in TS(B–C) results in the formation of tetramethyl silane. Intrinsic coordinate calculation for this transition state yields methylene (pyridine amine) connection to the yttrium (C) and dissociation of silane, which is favoured by 21.5 kcal mol−1. The release of silane and re-coordination of the THF molecule to the metal centre in the next step leads to the formation of a stable intermediate (D), in which the C1 (indol-2-yl carbon) is more electrophilic than the one in 1d (natural charge: −0.15925 in Dvs. −0.19823 in 1d), and is exothermic by 12.2 kcal mol−1. In the next step, the barrier for the 1,1-migratory insertion of the methylene group of the pyridine amine to the electrophilic carbon C1 [TS(D–E)] is favoured over the 1,1-migratory insertion of the CH2SiMe3 group to the C1 [TS(D–E′)] by 5.9 kcal mol−1. This suggests that the coordination of a donor THF not only promotes the formation of the electrophilic carbene, but also stabilizes the intermediate thereby promoting the selective 1,1-migratory insertion. The consecutive transition state [TS(E–F)] via the Y–C1 (indol-2-yl carbon) bond cleavage leads to the C–N bond breakage (intramolecular redox) giving a stable intermediate (F), exothermic by 44.2 kcal mol−1. Electron rearrangement from F to G followed by C–H activation of the methylene group [TS(G–H), barrier = 24.3 kcal mol−1] leads to the formation of tetramethyl silane and H. The latter isomerizes and dimerizes to form the final product, 4d (Scheme 6 and see more details in the ESI†).
Scheme 6. Computed enthalpy profile for the formation of 4d from 1d at room temperature. The enthalpy is given in kcal mol−1.

The above proposed and computed mechanism involved the formation of the intermediate G, which bears a methylene group being connected to the indol-2-yl carbon. Therefore, we have carried out a VT-NMR test to track the reaction of complex 1a with 2-N,N-dimethylaminopyridine from 263 K to 293 K (Fig. S25 in the ESI†). The formation and disappearance of intermediate G can be clearly observed from the spectra as indicated by the appearance and disappearance of Hb of the CH2SiMe3 (see Fig. S25 in the ESI†), which provides support for the results of DFT calculations and the proposed mechanism. Unfortunately, our attempts to isolate the intermediate G from the reaction of 1 with 2-N,N-dimethylaminopyridine were unsuccessful. However, the stoichiometric (1 : 1) reactions of the complexes 2 with 2-N,N-dimethylaminopyridine in toluene at room temperature afforded a mixture of (L1-b)2RE(η2-N,N-2-MeNPy) [RE = Lu(5a), Yb(5b), Er(5c), Y(5d)] (L1-b = 1-(2-N-C5H10NCH2CH2)-3-(2,6-iPr2C6H3N CH)indol-2-methylenyl) (Fig. 3) and (L1)2RE(η2-N,N-2-MeNPy) [RE = Lu(6a), Yb(6b), Er(6c), Y(6d)]. Complexes 6 can also be directly obtained through the reaction of complexes 2 with 2-methylaminopyridine with improved yields (Scheme 7), which is similar to the results reported recently.3g X-ray analyses reveal that complexes 5 contain a novel functionalized indol-2-methylenyl ligand, which provides indirect support for the proposed intermediate G in the formation of 4. The formation of the complexes 5 may involve a sequence as follows: the interaction of the nucleophilic alkyl CH2SiMe3 with the sp3 C–H bond of the 2-N,N-dimethylaminopyridine through σ-bond metathesis gives the intermediate A2, which then undergoes 1,1-migratory insertion to produce the intermediate A4 possibly driven by the electrophilic character of the indol-2-yl carbon (see the ESI†). Then the breakage of the Y–C bond leads to the cleavage of the C–N bond (intramolecular redox), followed by electron rearrangement to afford the intermediate A5, which then interacts with another molecule of A5 to deliver the final ligand redistribution complexes 5 and 6 (Scheme 8).
Fig. 3. Representative molecular structure of 5d. All hydrogen atoms are omitted and the diisopropylphenyl (Dipp) group is drawn in wireframe style for clarity.

Scheme 7. Reactions of 2–3 with 2-N,N-dimethylaminopyridine and 2-N-methylaminopyridine.

Scheme 8. Possible mechanism for the reaction between 2d and 2-N,N-dimethylaminopyridine.

The formation of complexes 5d and 6d from 2d has also been investigated computationally (see Scheme S6 in the ESI†). The reaction mechanism is quite similar to that presented in Scheme 6, except that in the last step the lack of a second silyl ligand stops the following C–H activation and dimerization and favours the formation of complexes 5d and 6d (−57.8 kcal mol−1) (see the ESI†). Notably, the indol-2-yl carbon of 2d still exhibits some electrophilic character (natural charge of −0.25) as compared to the highly nucleophilic alkyl carbon (natural charge of −1.69). This charge difference might explain why the first step is the proton transfer from 2-N,N-dimethylaminopyridine to the alkyl with a kinetically accessible barrier of 23.4 kcal mol−1. After releasing the silane from the coordination sphere, a C–C bond is formed via 1,1-migratory insertion between the nucleophilic carbon of ortho-metallated 2-N,N-dimethylaminopyridine and the electrophilic carbon of the indol-2-yl ligand in A3 (natural charge of −0.21) with an accessible barrier of 29.6 kcal mol−1.
When complexes 3, which bear an adamantyl group on the imino nitrogen, were reacted with 2-N,N-dimethylaminopyridine in a 1 : 1 ratio in toluene at room temperature, different complexes L2L2-aRE(THF) [RE = Er(7a), Y(7b)] (L2-a = 1-(2-N-C5H10NCH2CH2)-3-(AdNCH)-2-(3-Me2NC5H3N)-C8H4N) (Ad = adamantyl, C10H15) were isolated in moderate yields (Scheme 7 and Fig. 4). Comparison of the structures between 5 and 7 highlights the influence of the electronic and steric effects of the ligands on the reactions. The formation of 7 involves the activation of the sp2 C–H bond of the ortho pyridine ring by the nucleophilic alkyl CH2SiMe3via σ-bond metathesis, forming a three-membered ring intermediate B1, which then undergoes a 1,1-migratory insertion reaction to the electrophilic carbon of the indol-2-yl carbon to form the intermediate B2. Finally, coordination of the 2-dimethylamino group to the metal centre cleaves the Y–C (indol-2-yl carbon) bond accompanied by an intramolecular redox reaction resulting in the reduction of the pyridine ring, and the final complexes 7 bearing the dearomatic pyridine and indolyl rings are obtained (Scheme 9). From the structural parameters of 7b (Fig. 4), it can be seen that the imino group of one of the ligands transforms into an amido group with a bond length of Y–N(3) 2.250(3) Å, which can be compared with the Y–N single bond distance (2.254(4) Å) in the complex [κ3-(N,N,N)-2-(Me2NCH2CH2N CH)C8H5N]Y[N(SiMe3)2]25 with the same coordination number. In addition, the indol-2-yl carbon of the ligand bonds with the ortho-carbon of pyridine ring with a bond length of C(1)–C(53) 1.362(5) Å, suggesting a double bond character. Thus, dearomatization of the pyridine ring forms a negative nitrogen bonding to the metal with a bond length of Y–N(7) 2.296(3) Å, which is quite shorter than that of 2.376(5) Å found in 5, in which the bond between the rare-earth metal centre and pyridine nitrogen is a coordination bond (Scheme 7 and Fig. 4).
Fig. 4. Representative molecular structure of 7b. All hydrogen atoms are omitted and the adamantyl (Ad) group is drawn in wireframe style for clarity.

Scheme 9. Possible mechanism for the reaction between 3c and 2-N,N-dimethylaminopyridine.

Besides, our efforts to effect the reactions of 2-N,N-dimethylaminopyridine with the rare-earth metal monoalkyl complexes bearing 1-methyl (or benzyl)-3-(2,6-iPr2C6H3N CH) indolyl ligands resulted only in unidentified mixtures, indicating that the piperidyl moiety attached to the 1-indolyl motif might render the indol-2-yl carbon more electrophilic.
Reactions of the complexes with 2-phenylpyridine. Tandem sp2 C–H activation, 1,1-migratory insertion and proton abstraction reactions
Encouraged by the above findings, we then studied the stoichiometric (1 : 1) reactions of the complexes 2 with 2-phenylpyridine in toluene at room temperature. To our surprise, two different kinds of complexes, namely, (L1)2RE(η2-N,C-2-phenylpyridyl) [RE = Er(8a), Y(8b)], and L1L1-cDy(THF) (9) (L1-c = 1-(2-N-C5H10NCH2CH2)-3-(2,6-iPr2C6H3NCH)-2-(2-phenyl-C5H3N)-C8H5N) were isolated in moderate yields (Scheme 10). The formation of complexes 8 involves only the activation of the sp2 C–H bond, and this reactivity pattern is similar to that in previous reports.3g,5c–e,g In addition, the signal centred at 192.0 ppm for the complex 8b in the 13C{1H} spectrum was assigned to the phenyl carbon coupled to the yttrium nucleus with 1JY–C = 37.5 Hz. This is obviously located upfield as compared to those (203.2 to 205.8 ppm) of the indol-2-yl carbon observed in 1d, 2d, and 3c, which may be attributed to the electrophilic character of the indol-2-yl carbon. But the resonance at 192.0 ppm for the carbanion in 8b is comparable with the result reported in the literature (190.8 ppm for the phenyl carbon and 1JY–C = 41.6 Hz in {N2O}Y(η2-N,C-2′-phenylpyridyl)2(THF), ({N2O}−= bis(imino)phenoxy)).5e Compared to 1d and 3c, the resonance of the 89Y NMR of 8b (501.0 ppm, Fig. S43 in the ESI†) shifted high field possibly due to the coordination of 2-phenylpyridine.30 However, when the rare-earth metal is dysprosium(iii), which has a larger ionic radius than Er3+ and Y3+, the corresponding monoalkyl complex reacted with 2-phenylpyridine in toluene at room temperature to produce complex 9. This differs from not only the formation of 8 but also the result reported recently,3g and might be attributed to the electronic and steric effects of the supporting ligand. Specifically, we propose that the interaction of the ortho sp2 C–H bond of the 2-phenylpyridine ring with the nucleophilic alkyl CH2SiMe3 through σ-bond metathesis forms a three-membered ring C1, which is similar to that in the formation of 7 and previously reported reaction process of rare-earth metal alkyls or hydrides with pyridine derivatives.3a–d,5b Then, a 1,1-migratory insertion to the electrophilic carbon of the indol-2-yl gives the tertiary carbon bonded to the metal centre C2, which then activates the sp2 C–H bond of the phenyl ring to afford the final complex 9 (Scheme 10). To our knowledge, this reactivity pattern is also unprecedented in the reactions of transition metal complexes with 2-phenylpyridine. From the structural parameters of 9 (Fig. 5), it is found that the indolyl system was dearomatized and the imino nitrogen was transformed to amido with a Dy–N(3) bond length of 2.333(4) Å, which can be compared with the Dy–N distance of 2.250(3) Å found in the literature.22b Such a process is also different from that leading to 7, which involves an intramolecular redox reaction leading to dearomatization of the pyridine ring.
Scheme 10. Reaction of 2 with 2-phenylpyridine.

Fig. 5. The molecular structure of 9. All hydrogen atoms are omitted and the diisopropylphenyl (Dipp) group is drawn in wireframe style for clarity.

Reactions of the complexes with 4-N,N-dimethylaminopyridine (DMAP). Tandem sp2 C–H activation and 1,1-migratory insertion reactions
In the mechanism proposed above, there are some important intermediates such as E, A4, B2, and C2 (Scheme 12), all of which bear a tertiary indol-2-yl carbon bonding (RE–Ctertiary bond) with the central metal ion. Cleavage of the RE–Ctertiary bond plays a crucial role in the formation of 4, 5 and 7via intramolecular redox, and in the formation of 9via σ-bond metathesis. In order to isolate these key intermediates bearing the RE–Ctertiary bond to get more insights into the above reaction processes, we next studied the reactivity of rare-earth metal complexes bearing both electrophilic and nucleophilic carbon centres with other pyridine derivatives like 4-N,N-dimethylaminopyridine (DMAP), which is less sterically demanding than 2-N,N-dimethylaminopyridine and has only one coordination site to the central metal ions. When complexes 2 and 3 were treated with 2 equiv. of DMAP in toluene at room temperature, the complexes L1L1-dRE(DMAP) [RE = Er(10a), Y(10b), Dy(10c)] (L1-d = 1-(2-N-C5H10NCH2CH2)-3-(2,6-iPr2C6H3NCH)-2-(4-N,N-Me2NC5H3N)-C8H4N) and L2L2-bDy(DMAP) (11, L2-b = 1-(2-N-C5H10NCH2CH2)-3-(AdNCH)-2-(4-N,N-Me2NC5H3N)-C8H4N) (Ad = adamantyl, C10H15) were isolated in moderate yields (Scheme 11). All of these complexes were fully characterized by spectroscopic methods, elemental analyses and single crystal X-ray analyses. From the crystal structures of these complexes (Fig. 6), it can be seen that the complexes 10 and 11 feature a tertiary indol-2-yl carbon connected to the metal centre with a Y–C bond length of 2.565(6) Å. C(30)–C(44)–N(6) [C(30)–C(44) (1.386(8) Å), C(44)–N(6) (1.392(7) Å)] forms an aza-allyl (or amido-ene) conjugated system bonded with the central metal with the Y(1)–C(30) distance of 2.656(6) Å, Y(1)–C(44) distance of 2.597(6) Å, and Y(1)–N(6) distance of 2.229(5) Å, which can be compared with the Y–N bond length (2.242(5) Å) in the literature.22b In addition, the C(29)–C(30) and C(29)–C(57) distances are 1.491(8) Å and 1.468(8) Å, respectively, which can be compared with the C–C single bond distance. The formation of the ligand systems (L1-d or L2-b) in 10 and 11 is proposed to involve the interaction of the nucleophilic alkyl CH2SiMe3 with the ortho sp2 C–H of the pyridine ring through σ-bond metathesis to form a three-membered ring metallacycle D1. This metallacycle would then undergo 1,1-migratory insertion to the indol-2-yl carbon to produce the final L1-d or L2-b (Scheme 11) after ligand exchange of DMAP with THF. The driving force for the migration might be partly due to electrophilic character of the indol-2-yl carbon as revealed by DFT calculations of complex 2d. This reactivity pattern is different from the cross-carbanion coupling reaction of the ortho substituted pyridine derivatives at a rare-earth metal centre.26 These results also contrast those in previously reported reactions of rare-earth metal alkyls or hydrides with pyridine derivatives, where only the three-membered ring metallacycles3a–d,5b or the pyridine coupled products6 were isolated. While the 1,1-(2-pyridyl) migratory insertion is parallel to the 1,1-alkyl migratory insertion reported previously,22a this reaction involves an aryl sp2 carbon migratory insertion. These results thus provide strong evidence for the existence of E, A4, B2, and C2 (Scheme 12) as key intermediates in the formation of complexes 4, 5, 7, and 9, as well as the ensuing proposed mechanistic ways leading to these complexes.
Scheme 12. Structure comparison between important intermediates E, A4, B2, C2 and complexes 10–11.

Scheme 11. Reaction of 2 and 3 with DMAP.

Fig. 6. Representative molecular structure of 10b. All hydrogen atoms are omitted and the diisopropylphenyl (Dipp) group is drawn in wireframe style for clarity.

Thus, three different reactivity patterns of the ortho-pyridine C–H bond with the synthesized rare-earth metal alkyls were found in this work. (1) The ortho pyridine C–H bond activation and 1,1-migratory insertion followed by the intramolecular redox provided complexes 7 when complexes 3 bearing an adamantyl functionalized ligand were treated with 2-N,N-dimethylaminopyridine. (2) The ortho pyridine C–H bond activation and 1,1-migratory insertion followed by abstraction of H produced complex 9 when complex 2e was reacted with 2-phenylpyridine. (3) The ortho pyridine C–H bond activation accompanied by 1,1-migratory insertion delivered complexes 10 and 11 when complexes 2 and 3 were reacted with 4-N,N-dimethylaminopyridine, indicating that steric, electronic and coordination effects of substrates, substituents of the ligands, and ionic radii of the central metal have significant influence on the reactivity patterns. The isolation and characterization of some complexes structurally similar to the proposed key intermediates provide important evidence in understanding the reaction processes.
Reactions of the rare-earth metal monoalkyl complexes with 2,2′-bipyridine. The bipyridyl anionic radical complexes produced by a homolytic redox reaction and their redox properties
As pointed out above, the reported methods for the generation of the bipyridyl anionic radical complexes utilized the reactions of low-valent metal complexes with 2,2′-bipyridine7 or treatment of a mixture of metal complexes and 2,2′-bipyridine in the presence of an alkaline metal or KC8.8 We have reported the coordination-promoted homolytic reactions of the RE–X (RE = rare-earth metal, X = N, C) bonds to produce the divalent rare-earth metal complexes27 and homolytic cerium(iv) alkyl and aryl complexes can also provide cerium(iii) products.29 Therefore, we proposed a conceptual idea that the reaction of an external, unsaturated and strong donor ligand such as 2,2′-bipyridine with rare-earth metal alkyls, where the RE3+/RE2+ has high reductive potential, would promote the homolytic cleavage of the RE–alkyl bonds while reducing the unsaturated 2,2′-bipyridine (Scheme 13). In line with this hypothesis, our recent studies on the reactions of rare-earth metal dialkyl complexes with 2,2′-bipyridine indeed delivered the bipyridyl anionic radical complexes.3g However, the reactivity of monoalkyl rare-earth metal complexes with 2,2′-bipyridine and reactivity of the resulting bipyridyl radical rare-earth metal complexes have not been investigated. Then, we tried the stoichiometric (1 : 1) reactions of the complexes 2 and 3 with 2,2′-bipyridine in toluene at room temperature, the corresponding reactions indeed afforded complexes (L1)2RE(bipy˙−) [RE = Yb(12a), Er(12b), Y(12c), Dy(12d)] and (L2)2Er(bipy˙−) (13) with bipyridyl anionic radical ligation in moderate yields (Scheme 14 and Fig. 7). The GC-MS analysis of the reaction mixtures after hydrolysis shows an m/z peak at 174.25, which might be attributed to the alkyl Me3SiCH2 radical coupling product of [(Me3SiCH2)2]+ (see the ESI†). Therefore, the reaction provides a synthetic method for the generation of the bipyridyl radical anions. Compared with the sterically induced reduction (SIR) method proposed by Evans's group,31 both methods release free radicals and reduce unsaturated substrates. In the present case, complexes 2 are inert to the addition of a weak coordination substrate like chlorobenzene to the toluene solution (see Fig. S50 in ESI†), while the homolytic redox reaction simultaneously happens upon the addition of 2,2′-bipyridine. This implies that the coordination capacity of the substrate may play a key role in the reaction while steric effects cannot be ruled out. From the crystal structural parameters of complexes 12–13, it can be seen that the bond lengths of the 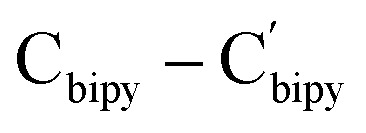 are 1.414(5) Å, 1.413(8) Å, 1.412(6) Å, 1.407(9) Å and 1.41(2) Å, respectively, which are consistent with the reported bond length of bipy˙− (1.43 Å) in the literature,3g,13,28 but deviate from those of bipy0 and bipy2− (1.49 Å and 1.38 Å).8f,11,13,28 The generation of the bipyridyl anionic radical was further proved by EPR analysis (see Fig. S98 in the ESI†) with a resonance at g = 2.004, which is similar to that of (NNfc)Y(bipyPh) (NNfc = 1,1′-fc(NSitBuMe2)2) (g = 2.0048) produced from the reaction of a corresponding yttrium alkyl complex with 2 equiv. of 2-phenylpyridine.6d The UV/vis spectra of complexes 12–13 in THF solution have been studied (see Fig. S99 in the ESI†). The strong absorption band at 200–400 nm can be attributed to the transition of the aromatic ring's π–π* or the charge transfer of the ligand to the metal center. The absorption band at 500 nm and several weak broad absorption bands in the range of 700–1000 nm are due to the characteristic absorption band of the bipyridyl radical anion, which are consistent with the reported results.3g,7a,8a,14
are 1.414(5) Å, 1.413(8) Å, 1.412(6) Å, 1.407(9) Å and 1.41(2) Å, respectively, which are consistent with the reported bond length of bipy˙− (1.43 Å) in the literature,3g,13,28 but deviate from those of bipy0 and bipy2− (1.49 Å and 1.38 Å).8f,11,13,28 The generation of the bipyridyl anionic radical was further proved by EPR analysis (see Fig. S98 in the ESI†) with a resonance at g = 2.004, which is similar to that of (NNfc)Y(bipyPh) (NNfc = 1,1′-fc(NSitBuMe2)2) (g = 2.0048) produced from the reaction of a corresponding yttrium alkyl complex with 2 equiv. of 2-phenylpyridine.6d The UV/vis spectra of complexes 12–13 in THF solution have been studied (see Fig. S99 in the ESI†). The strong absorption band at 200–400 nm can be attributed to the transition of the aromatic ring's π–π* or the charge transfer of the ligand to the metal center. The absorption band at 500 nm and several weak broad absorption bands in the range of 700–1000 nm are due to the characteristic absorption band of the bipyridyl radical anion, which are consistent with the reported results.3g,7a,8a,14
Scheme 13. Proposed homolytic redox reaction (HRR) for the generation of the bipyridyl radical anion.

Scheme 14. Reactions of 2–3 with 2,2′-bipyridine and reactions of the bipyridyl complexes with diphenyldiazomethane.

Fig. 7. Representative molecular structure of 12c. All hydrogen atoms are omitted and the diisopropylphenyl (Dipp) group is drawn in wireframe style for clarity.

Although the bipyridyl anionic radical rare-earth metal complexes can be obtained with different methods, the reactivity of the corresponding bipyridyl anionic radical rare-earth metal complexes is rarely developed. Then, the reactivity of the bipyridyl anionic radical ligated complexes were explored. The stoichiometric (1 : 3) reactions of the rare-earth metal bipyridyl anionic radical ligated complexes 12 with diphenyldiazomethane afforded the redox complexes bearing an iminato group (L1-e)2RE(N CPh2) [RE = Yb(14a), Er(14b), Y(14c)] (L1-e = 1-(2-N-C5H10NCH2CH2)-3-(2,6-iPr2C6H3N CH)-2-(Ph2C N2)-C8H4N) (Scheme 14 and Fig. 8). Similar to the formation of 7, the reactions of diphenyldiazomethane with the indol-2-yl carbon may follow a sequence of coordination, 1,1-migratory insertion and RE–C2-ind bond cleavage with an intramolecular redox process. This indicates that the bipyridyl ligated complexes show redox properties to reduce the N N double bond of the diphenyldiazomethane to produce the iminato ligated complexes 14, which is parallel to the reported reactions of the bipyridyl actinide complexes with unsaturated small molecules.8b,d–g However, the reactivity of the bipyridyl anionic radical rare-earth metal complexes with unsaturated substrates is rare.
Fig. 8. Representative molecular structure of 14c. All hydrogen atoms are omitted and the diisopropylphenyl (Dipp) group is drawn in wireframe style for clarity.

Conclusions
In summary, a series of rare-earth metal complexes bearing both electrophilic and nucleophilic moieties were synthesized by applying the conjugated system (–C C–C N) with an sp2 carbon being bonded to the metal centres. DFT calculations and experimental results reveal that the indol-2-yl carbon displays electrophilic character that can undergo a 1,1-migratory insertion reaction as the late transition metal electrophilic carbenes did, while the carbon of alkyls bonded to the metal centre exhibits nucleophilic properties to activate sp3 and sp2 C–H bonds of the pyridine derivatives via σ-bond metathesis. Studies on the reactions of the synthesized complexes with pyridine derivatives lead to disclosure of different reactivity patterns including: (1) unprecedented consecutive C–H activation/1,1-migratory insertion/C–N and C–H activation delivering the 1,3-bisfunctionalized indol-2-methinyl rare-earth metal complexes when the dialkyl complexes were treated with 2-N,N-dimethylaminopyridine; (2) the consecutive C–H activation/1,1-migratory insertion/C–N bond activation producing the indol-2-methylenyl rare-earth metal complexes when the monoalkyl complexes were reacted with 2-N,N-dimethylaminopyridine; (3) the tandem C–H activation/1,1-migratory insertion affording the 2-(2-[4-N,N-dimethylamino]pyridyl)-indol-2-yl rare-earth metal complexes when the monoalkyl complexes were reacted with 4-N,N-dimethylaminopyridine, and (4) homolytic redox reaction (HRR) generating the 2,2′-bipyridyl anionic radical complexes when the monoalkyl complexes were treated with 2,2′-bipyridine. These reactions provide helpful insights in understanding and designing relevant catalytic reactions. It is also found that the coordination of the strongly electron-donating ligand with the central metal facilitates the formation of the rare-earth metal electrophilic carbene. Isolation and characterization of some intermediate equivalents provide important evidence for understanding these multiple-step reaction processes. The findings in this work provide useful methods to access different dearomatic indolyl or functionalized indolyl complexes. Furthermore, the reactions of the rare-earth metal monoalkyl complexes with 2,2′-bipyridine provided a conceptually new route to bipyridyl anionic radical ligated complexes, which show redox properties towards diphenyldiazomethane delivering the iminato group by reduction of the N N double bond of the diphenyldiazomethane. To the best of our knowledge, the reactivity patterns found in this paper are completely distinct from those of transition metal alkyls or hydrides with pyridine derivatives previously reported, highlighting the uniqueness of the rare-earth metal complexes bearing both electrophilic and nucleophilic carbon centres.
Data availability
Experimental procedures, X-ray crystallographic details, NMR and infrared spectroscopy data, and computational details are available in the ESI.† CCDC 2278697–2278701, 2348805–2348814 and 2348816–2348844 contain the supplementary crystallographic data for this paper.
Author contributions
W. Wu led and performed the synthesis, reactivity study of the complexes, characterization experiments of complexes, and manuscript draft writing. T. Rajeshkumar performed the DFT calculations under the supervision of L. Maron. D. Hong and Z. Huang assisted in the X-ray data collection and refinement. F. Chai, S. Zhu and Q. Yuan assisted in the analysis of experimental data. L. Maron participated in the discussion on DFT calculation results. S. Wang was responsible for the project design, discussion, manuscript writing, and editing.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful for the financial support from the National Natural Science Foundation of China (No. 22031001 and U23A2079) and the grants from the Anhui Province (202305a12020018, GXXT-2021-052).
Electronic supplementary information (ESI) available. CCDC 2278697–2278701, 2348805–2348814 and 2348816–2348844. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04197f
References
- (a) Bull J. A. Mousseau J. J. Pelletier G. Charette A. B. Chem. Rev. 2012;112:2462–2713. doi: 10.1021/cr200251d. [DOI] [PubMed] [Google Scholar]; (b) Allais C. Grassot J. Rodriguez J. Constantieux T. Chem. Rev. 2014;114:10829–10868. doi: 10.1021/cr500099b. [DOI] [PubMed] [Google Scholar]; (c) Escolano M. Gaviña D. Alzuet-Piña G. Díaz-Oltra S. Sánchez-Roselló M. Del Pozo C. Chem. Rev. 2024;124:1122–1246. doi: 10.1021/acs.chemrev.3c00625. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nakao Y. Kanyiva K. S. Hiyama T. J. Am. Chem. Soc. 2008;130:2448–2449. doi: 10.1021/ja710766j. [DOI] [PubMed] [Google Scholar]; (e) Nakao Y. Yamada Y. Kashihara N. Hiyama T. J. Am. Chem. Soc. 2010;132:13666–13668. doi: 10.1021/ja106514b. [DOI] [PubMed] [Google Scholar]; (f) Chen Q. Jourdin X. M. D. Knochel P. J. Am. Chem. Soc. 2013;135:4958–4961. doi: 10.1021/ja401146v. [DOI] [PubMed] [Google Scholar]; (g) Wang G. Cao J. Gao L. Chen W. Huang W. Cheng X. Li S. J. Am. Chem. Soc. 2017;139:3904–3910. doi: 10.1021/jacs.7b00823. [DOI] [PubMed] [Google Scholar]; (h) Sun Q. Chen P. Wang Y. Luo Y. Yuan D. Yao Y. Inorg. Chem. 2018;57:11788–11800. doi: 10.1021/acs.inorgchem.8b01959. [DOI] [PubMed] [Google Scholar]; (i) Kundu A. Inoue M. Nagae H. Tsurugi H. Mashima K. J. Am. Chem. Soc. 2018;140:7332–7342. doi: 10.1021/jacs.8b03998. [DOI] [PubMed] [Google Scholar]; (j) Han C. Liu Y. Tian X. Rominger F. Hashmi A. S. K. Org. Lett. 2021;23:9480–9484. doi: 10.1021/acs.orglett.1c03667. [DOI] [PubMed] [Google Scholar]; (k) Liu J. Li Y. Jiang J. Liu Y. Ke Z. ACS Catal. 2021;11:6186–6192. [Google Scholar]; (l) Ma J.-B. Zhao X. Zhang D. Shi S.-L. J. Am. Chem. Soc. 2022;144:13643–13651. doi: 10.1021/jacs.2c04043. [DOI] [PubMed] [Google Scholar]; (m) Cao H. Bhattacharya D. Cheng Q. Studer A. J. Am. Chem. Soc. 2023;145:15581–15588. doi: 10.1021/jacs.3c05242. [DOI] [PubMed] [Google Scholar]
- Klei E. Teuben J. H. J. Organomet. Chem. 1981;214:53–64. [Google Scholar]
- (a) Waston L. J. Chem. Soc., Chem. Commun. 1983;6:276–277. [Google Scholar]; (b) Thompson M. E. Baxter S. M. Bulls A. R. Burger B. J. Nolan M. C. Santarsiero B. D. Schaefer W. P. Bercaw J. E. J. Am. Chem. Soc. 1987;109:203–219. [Google Scholar]; (c) Haan K. H. D. Wielstra Y. Teuben J. H. Organometallics. 1987;6:2053–2060. [Google Scholar]; (d) Tsurugi H. Yamamoto K. Mashima K. J. Am. Chem. Soc. 2011;133:732–735. doi: 10.1021/ja1100118. [DOI] [PubMed] [Google Scholar]; (e) Kaneko H. Nagae H. Tsurugi H. Mashima K. J. Am. Chem. Soc. 2011;133:19626–19629. doi: 10.1021/ja208293h. [DOI] [PubMed] [Google Scholar]; (f) Qu L. Roisnel T. Cordier M. Yuan D. Yao Y. Zhao B. Kirillov E. Inorg. Chem. 2020;59:16976–16987. doi: 10.1021/acs.inorgchem.0c02112. [DOI] [PubMed] [Google Scholar]; (g) Zhu S. Wu W. Hong D. Chai F. Huang Z. Zhu X. Zhou S. Wang S. Inorg. Chem. 2024;63:14860–14875. doi: 10.1021/acs.inorgchem.4c00981. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Zhang J. Hong J. Zhang F. Weng L. Zhou X. Organometallics. 2014;33:7052–7058. [Google Scholar]
- (a) Deelman B. J. Stevels W. M. Teuben J. H. Lakin M. T. Spek A. L. Organometallics. 1994;13:3881–3891. [Google Scholar]; (b) Lu E. Chu J. Chen Y. Borzov M. V. Li G. Chem. Commun. 2011;47:743–745. doi: 10.1039/c0cc03212c. [DOI] [PubMed] [Google Scholar]; (c) Guan B.-T. Hou Z. J. Am. Chem. Soc. 2011;133:18086–18089. doi: 10.1021/ja208129t. [DOI] [PubMed] [Google Scholar]; (d) Luo G. Luo Y. Qu J. Hou Z. Organometallics. 2012;31:3930–3937. [Google Scholar]; (e) Song G. Wylie W. N. O. Hou Z. J. Am. Chem. Soc. 2014;136:12209–12212. doi: 10.1021/ja504995f. [DOI] [PubMed] [Google Scholar]; (f) Song G. Wang B. Nishiura M. Hou Z. Chem.–Eur. J. 2015;21:8394–8398. doi: 10.1002/chem.201501121. [DOI] [PubMed] [Google Scholar]; (g) Azpíroz R. Di Giuseppe A. Urriolabeitia A. Passarelli V. Polo V. Perez-Torrente J. J. Oro L. A. Castarlenas R. ACS Catal. 2019;9:9372–9386. [Google Scholar]
- (a) Carver C. T. Diaconescu P. L. J. Am. Chem. Soc. 2008;130:7558–7559. doi: 10.1021/ja802728k. [DOI] [PubMed] [Google Scholar]; (b) Carver C. T. Benitez D. Miller K. L. Williams B. N. Tkatchouk E. Goddard III W. A. Diaconescu P. L. J. Am. Chem. Soc. 2009;131:10269–10278. doi: 10.1021/ja902794w. [DOI] [PubMed] [Google Scholar]; (c) Carver C. T. Williams B. N. Ogilby K. R. Diaconescu P. L. Organometallics. 2010;29:835–846. [Google Scholar]; (d) Williams B. N. Huang W. Miller K. L. Diaconescu P. L. Inorg. Chem. 2010;49:11493–11498. doi: 10.1021/ic101493k. [DOI] [PubMed] [Google Scholar]; (e) Shibata Y. Nagae H. Sumiya S. Rochat R. Tsurugi H. Mashima K. Chem. Sci. 2015;6:5394–5399. doi: 10.1039/c5sc01599e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Schultz M. Boncella J. M. Berg D. J. Don Tilley T. Andersen R. A. Organometallics. 2002;21:460–472. [Google Scholar]; (b) Booth C. H. Walter M. D. Kazhdan D. Hu Y.-J. Lukens W. W. Bauer E. D. Maron L. Eisenstein O. Andersen R. A. J. Am. Chem. Soc. 2009;131:6480–6491. doi: 10.1021/ja809624w. [DOI] [PubMed] [Google Scholar]; (c) Evans W. J. Drummond D. K. J. Am. Chem. Soc. 1989;111:3329–3335. [Google Scholar]
- (a) Xiao Y. Sun R. Liang J. Fang Y. Liu Z. Jiang S. Wang B. Gao S. Huang W. Inorg. Chem. Front. 2021;8:2591–2602. [Google Scholar]; (b) Heng Y. Li T. Wang D. Hou G. Zi G. Walter M. D. Organometallics. 2023;42:91–113. [Google Scholar]; (c) Kraft S. J. Fanwick P. E. Bart S. C. Inorg. Chem. 2010;49:1103–1110. doi: 10.1021/ic902008w. [DOI] [PubMed] [Google Scholar]; (d) Zhang L. Zhang C. Hou G. Zi G. Walter M. D. Organometallics. 2017;36:1179–1187. [Google Scholar]; (e) Li T. Wang D. Heng Y. Hou G. Zi G. Walter M. D. Organometallics. 2023;42:392–406. [Google Scholar]; (f) Ren W. Song H. Zi G. Walter M. D. Dalton Trans. 2012;41:5965–5973. doi: 10.1039/c2dt00051b. [DOI] [PubMed] [Google Scholar]; (g) Ren W. Zi G. Walter M. D. Organometallics. 2012;31:672–679. [Google Scholar]
- Mehdoui T. Berthet J.-C. Thuery P. Salmon L. Riviere E. Ephritikhine M. Chem.–Eur. J. 2015;11:6994–7006. doi: 10.1002/chem.200500479. [DOI] [PubMed] [Google Scholar]
- Jacquot L. Xemard M. Clavaguera C. Nocton G. Organometallics. 2014;33:4100–4106. [Google Scholar]
- Stennett C. R. Nguyen J. Q. Ziller J. W. Evans W. J. Organometallics. 2023;42:696–707. [Google Scholar]
- Lv Y. Kefalidis C. E. Zhou J. Maron L. Leng X. Chen Y. J. Am. Chem. Soc. 2013;135:14784–14796. doi: 10.1021/ja406413d. [DOI] [PubMed] [Google Scholar]
- Rosenzweig M. W. Heinemann F. W. Maron L. Meyer K. Inorg. Chem. 2017;56:2792–2800. doi: 10.1021/acs.inorgchem.6b02954. [DOI] [PubMed] [Google Scholar]
- Diaconescu P. L. Cummins C. C. Dalton Trans. 2015;44:2676–2683. doi: 10.1039/c4dt02422b. [DOI] [PubMed] [Google Scholar]
- Beetstra D. J. Meetsma A. Hessen B. Teuben J. H. Organometallics. 2003;22:4372–4374. [Google Scholar]
- Wang S. Wang D. Heng Y. Li T. Ding W. Zi G. Walter M. D. Inorg. Chem. 2024;63:7473–7492. doi: 10.1021/acs.inorgchem.4c00635. [DOI] [PubMed] [Google Scholar]
- (a) Dietrich H. M. Törnroos K. W. Anwander R. J. Am. Chem. Soc. 2006;128:9298–9299. doi: 10.1021/ja062523y. [DOI] [PubMed] [Google Scholar]; (b) Ma W. Yu C. Chi Y. Chen T. Wang L. Yin J. Wei B. Xu L. Zhang W.-X. Xi Z. Chem. Sci. 2017;8:6852–6856. doi: 10.1039/c7sc02018j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhou J. Li T. Maron L. Leng X. Chen Y. Organometallics. 2015;34:470–476. [Google Scholar]; (d) Levine D. S. Tilley T. D. Andersen R. A. Organometallics. 2017;36:80–88. [Google Scholar]; (e) Bonath M. Maichle-Mössmer C. Sirsch P. Anwander R. Angew. Chem., Int. Ed. 2019;58:8206–8210. doi: 10.1002/anie.201902063. [DOI] [PubMed] [Google Scholar]; (f) Hong J. Zhang L. Yu X. Li M. Zhang Z. Zheng P. Nishiura M. Hou Z. Zhou X. Chem.–Eur. J. 2011;17:2130–2137. doi: 10.1002/chem.201002670. [DOI] [PubMed] [Google Scholar]; (g) Zhang W.-X. Wang Z. Nishiura M. Xi Z. Hou Z. J. Am. Chem. Soc. 2011;133:5712–5715. doi: 10.1021/ja200540b. [DOI] [PubMed] [Google Scholar]; (h) Li T. Nishiura M. Cheng J. Li Y. Hou Z. Chem.–Eur. J. 2012;18:15079–15085. doi: 10.1002/chem.201202796. [DOI] [PubMed] [Google Scholar]; (i) Birkelbach V. M. Kracht F. Dietrich H. M. Stuhl C. Maichle-Mössmer C. Anwander R. Organometallics. 2020;39:3490–3504. [Google Scholar]; (j) Hollfelder C. O. Jende L. N. Dietrich H.-M. Eichele K. Maichle-Mössmer C. Anwander R. Chem.–Eur. J. 2019;25:7298–7302. doi: 10.1002/chem.201901269. [DOI] [PubMed] [Google Scholar]; (k) Buschmann D. A. Schumacher L. Anwander R. Chem. Commun. 2022;58:9132–9135. doi: 10.1039/d2cc03532d. [DOI] [PubMed] [Google Scholar]; (l) Scott J. Fan H. Wicker B. F. Fout A. R. Baik M.-H. Mindiola D. J. J. Am. Chem. Soc. 2008;130:14438–14439. doi: 10.1021/ja806635x. [DOI] [PubMed] [Google Scholar]; (m) Litlabø R. Zimmermann M. Saliu K. Takats J. Törnroos K. W. Anwander R. Angew. Chem., Int. Ed. 2008;47:9560–9564. doi: 10.1002/anie.200803856. [DOI] [PubMed] [Google Scholar]; (n) Rieser T. E. Thim-Spöring R. Schädle D. Sirsch P. Litlabø R. Törnroos K. W. Maichle-Mössmer C. Anwander R. J. Am. Chem. Soc. 2022;144:4102–4113. doi: 10.1021/jacs.1c13142. [DOI] [PubMed] [Google Scholar]; (o) Schadle D. Litlabø R. Meermann-Zimmermann M. Thim-Spöring R. Schädle C. Maichle-Mössmer C. Törnroos K. W. Anwander R. Inorg. Chem. 2024;63:9624–9637. doi: 10.1021/acs.inorgchem.3c04422. [DOI] [PubMed] [Google Scholar]
- (a) Mao W. Xiang L. Maron L. Leng X. Chen Y. J. Am. Chem. Soc. 2017;139:17759–17762. doi: 10.1021/jacs.7b11097. [DOI] [PubMed] [Google Scholar]; (b) Aparna K. Ferguson M. Cavell R. G. J. Am. Chem. Soc. 2000;122:726–727. [Google Scholar]; (c) Mills D. P. Soutar L. Lewis W. Blake A. J. Liddle S. T. J. Am. Chem. Soc. 2010;132:14379–14381. doi: 10.1021/ja107958u. [DOI] [PubMed] [Google Scholar]; (d) Fustier M. Le Goff X. F. Le Floch P. Mézailles N. J. Am. Chem. Soc. 2010;132:13108–13110. doi: 10.1021/ja103220s. [DOI] [PubMed] [Google Scholar]; (e) Mao W. Xiang L. Lamsfus C. A. Maron L. Leng X. Chen Y. J. Am. Chem. Soc. 2017;139:1081–1084. doi: 10.1021/jacs.6b13081. [DOI] [PubMed] [Google Scholar]; (f) Wang C. Zhou J. Zhao X. Maron L. Leng X. Chen Y. Chem.–Eur. J. 2016;22:1258–1261. doi: 10.1002/chem.201504725. [DOI] [PubMed] [Google Scholar]
- Schumann H. Muller J. J. Organomet. Chem. 1979;169:C1–C4. [Google Scholar]
- (a) Xie W. Hu H. Cui C. Angew. Chem., Int. Ed. 2012;51:11141–11144. doi: 10.1002/anie.201205317. [DOI] [PubMed] [Google Scholar]; (b) Zhang M. Zhang J. Ni X. Shen Z. RSC Adv. 2015;5:83295–83303. [Google Scholar]; (c) Lv K. Cui D. Organometallics. 2008;27:5438–5440. [Google Scholar]; (d) Yao H. Zhang J. Zhang Y. Sun H. Shen Q. Organometallics. 2010;29:5841–5846. [Google Scholar]; (e) Fegler W. Spaniol T. P. Okuda J. Dalton Trans. 2010;39:6774–6779. doi: 10.1039/c001699c. [DOI] [PubMed] [Google Scholar]; (f) Lapshin I. V. Cherkasov A. V. Lyssenko K. A. Fukin G. K. Trifonov A. A. Inorg. Chem. 2022;61:9147–9161. doi: 10.1021/acs.inorgchem.2c00698. [DOI] [PubMed] [Google Scholar]; (g) Pan Y. Zhao A. Li Y. Li W. So Y.-M. Yan X. He G. Dalton Trans. 2018;47:13815–13823. doi: 10.1039/c8dt02130a. [DOI] [PubMed] [Google Scholar]; (h) Lapshin I. V. Cherkasov A. V. Asachenkob A. F. Trifonov A. A. Chem. Commun. 2020;56:12913–12916. doi: 10.1039/d0cc05424k. [DOI] [PubMed] [Google Scholar]; (i) Huang Z. Wang S. Zhu X. Yuan Q. Wei Y. Zhou S. Mu X. Inorg. Chem. 2018;57:15069–15078. doi: 10.1021/acs.inorgchem.8b02067. [DOI] [PubMed] [Google Scholar]; (j) Yao C. Lin F. Wang M. Liu D. Liu B. Liu N. Wang Z. Long S. Wu C. Cui D. Macromolecules. 2015;48:1999–2005. [Google Scholar]; (k) Yuan J. Hu H. Cui C. Chem.–Eur. J. 2016;22:5778–5785. doi: 10.1002/chem.201600512. [DOI] [PubMed] [Google Scholar]; (l) Pan Z. Zhang J. Guo L. Yang H. Li J. Cui C. Inorg. Chem. 2021;60:12696–12702. doi: 10.1021/acs.inorgchem.1c01780. [DOI] [PubMed] [Google Scholar]
- (a) Xiao Y. Liu Z. Liang J. Yang K. Huang W. Dalton Trans. 2022;51:15873–15882. doi: 10.1039/d2dt02759c. [DOI] [PubMed] [Google Scholar]; (b) Pan Y. Jiang X. So Y.-M. To C. T. He G. Catalysts. 2020;10:71–85. [Google Scholar]
- (a) Huang Z. Wang R. Sheng T. Zhong X. Wang S. Zhu X. Yuan Q. Wei Y. Zhou S. Inorg. Chem. 2021;60:18843–18853. doi: 10.1021/acs.inorgchem.1c02589. [DOI] [PubMed] [Google Scholar]; (b) Hong D. Rajeshkumar T. Zhu S. Huang Z. Zhou S. Zhu X. Maron L. Wang S. Sci. China: Chem. 2023;66:117–126. [Google Scholar]; (c) Wu W. Rajeshkumar T. Hong D. Zhu S. Huang Z. Chai F. Wang W. Yuan Q. Wei Y. Xie Z. Maron L. Wang S. Inorg. Chem. 2024;63:18365–18378. doi: 10.1021/acs.inorgchem.4c02316. [DOI] [PubMed] [Google Scholar]
- Zhang G. Deng B. Wang S. Wei Y. Zhou S. Zhu X. Huang Z. Mu X. Dalton Trans. 2016;45:15445–15456. doi: 10.1039/c6dt02922a. [DOI] [PubMed] [Google Scholar]
- Huang Z. Wang S. Zhu X. Wei Y. Yuan Q. Zhou S. Mu X. Wang H. Chin. J. Chem. 2021;39:3360–3368. [Google Scholar]
- Wei Y. Gao J. Jiang L. Huang Z. Bao Q. Yuan Q. Zhang L. Zhou S. Wang S. Organometallics. 2022;41:2985–2996. [Google Scholar]
- Liu W. Zhao Y. Ma W. Xu L. Wei J. Zhang W.-X. Cell Rep. Phys. Sci. 2023;4:101479–101492. [Google Scholar]
- (a) Sheng E. Wang S. Yang G. Zhou S. Cheng L. Zhang K. Huang Z. Organometallics. 2003;22:684–692. [Google Scholar]; (b) Zhang K. Zhang W. Wang S. Sheng E. Yang G. Xie M. Zhou S. Feng Y. Mao L. Huang Z. Dalton Trans. 2004;7:1029–1037. doi: 10.1039/b314115b. [DOI] [PubMed] [Google Scholar]; (c) Wang S. Tang X. Vega A. Saillard J.-Y. Zhou S. Yang G. Yao W. Wei Y. Organometallics. 2007;26:1512–1522. [Google Scholar]; (d) Zhou S. Wu Z. Zhou L. Wang S. Zhang L. Zhu X. Wei Y. Zhai J. Wu J. Inorg. Chem. 2013;52:6417–6426. doi: 10.1021/ic4003109. [DOI] [PubMed] [Google Scholar]
- Fortier S. Veleta J. Pialat A. Le Roy J. Ghiassi K. B. Olmstead M. M. Metta-Magana A. Murugesu M. Villagran D. Chem.–Eur. J. 2016;22:1931–1936. doi: 10.1002/chem.201504982. [DOI] [PubMed] [Google Scholar]
- Feng B. Ye L.-W. Wang N. Hu H.-S. Li J. Tamm M. Chen Y. CCS Chem. 2024 doi: 10.31635/ccschem.024.202404131. [DOI] [Google Scholar]
- (a) Schaverien C. J. Organmetallics. 1994;13:69–82. [Google Scholar]; (b) White R. E. Hanusa T. P. Organometallics. 2006;25:5621–5630. [Google Scholar]; (c) Latsch L. Lam E. Coperet C. Chem. Sci. 2020;11:6724–6735. doi: 10.1039/d0sc02321c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Evans W. J. Nyce G. W. Ziller J. W. Angew. Chem., Int. Ed. 2000;39:240–242. [PubMed] [Google Scholar]; (b) Evans W. J. Kozimor S. A. Ziller J. W. Kaltsoyannis N. J. Am. Chem. Soc. 2004;126:14533–14547. doi: 10.1021/ja0463886. [DOI] [PubMed] [Google Scholar]; (c) Evans W. J. Inorg. Chem. 2007;46:3435–3449. doi: 10.1021/ic062011k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental procedures, X-ray crystallographic details, NMR and infrared spectroscopy data, and computational details are available in the ESI.† CCDC 2278697–2278701, 2348805–2348814 and 2348816–2348844 contain the supplementary crystallographic data for this paper.