

Abstract
Systems metabolic engineering, which recently emerged as metabolic engineering integrated with systems biology, synthetic biology, and evolutionary engineering, allows engineering of microorganisms on a systemic level for the production of valuable chemicals far beyond its native capabilities. Here, we review the strategies for systems metabolic engineering and particularly its applications in Escherichia coli. First, we cover the various tools developed for genetic manipulation in E. coli to increase the production titers of desired chemicals. Next, we detail the strategies for systems metabolic engineering in E. coli, covering the engineering of the native metabolism, the expansion of metabolism with synthetic pathways, and the process engineering aspects undertaken to achieve higher production titers of desired chemicals. Finally, we examine a couple of notable products as case studies produced in E. coli strains developed by systems metabolic engineering. The large portfolio of chemical products successfully produced by engineered E. coli listed here demonstrates the sheer capacity of what can be envisioned and achieved with respect to microbial production of chemicals. Systems metabolic engineering is no longer in its infancy; it is now widely employed and is also positioned to further embrace next-generation interdisciplinary principles and innovation for its upgrade. Systems metabolic engineering will play increasingly important roles in developing industrial strains including E. coli that are capable of efficiently producing natural and nonnatural chemicals and materials from renewable nonfood biomass.
INTRODUCTION
Escherichia coli has become not only one of the most characterized and understood organisms, but also the most sought organism for genetic manipulation since the success of the first ever genetically modified E. coli in 1973 that pioneered the field of genetic engineering (1, 2). As a Gram-negative facultative anaerobe, E. coli is one of the first colonizers in neonatal intestines and normally dwells in gastrointestinal microflora of many animals, including humans (3). Although these enteric bacteria might develop virulence through horizontal gene transfer (4), genetic manipulation in laboratory settings is simple and quick, rendering the strain attractive for research. Following the initial sequencing of the genome with 4.6 Mb size and about 4,300 genes annotated (5), the pangenome with a reservoir of almost 13,000 genes has been recently characterized (6). With the genome sequence readily available, the massive influx of omics information and the development of various engineering tools enabled our attempts to understand E. coli at the systems level.
Systems metabolic engineering emerged as the fields of metabolic engineering and biotechnology transitioned toward understanding the biology of organisms at the systems level. In 1991, metabolic engineering was first described and aimed to be a field concerning the application of recombinant DNA methods to develop high-performance strains for the production of useful chemicals and recombinant proteins (7). In the same year, the historical issuance of U.S. patent 5,000,000 appeared, granting rights to converting hexose or pentose to ethanol with a genetically modified E. coli expressing Zymomonas mobilis pyruvate decarboxylase and alcohol dehydrogenase (8). Together with the ever-growing omics data, rapidly advancing tools, and novel strategies, it is now possible to rationally design high-performance E. coli strains for producing not only ethanol but also higher alcohols (9, 10, 11, 12, 13, 14), hydrocarbons (15, 16, 17, 18), amino acids (19, 20, 21, 22, 23, 24, 25, 26), various polymer precursors (27, 28), recombinant proteins (29, 30), and plastics (31, 32, 33). Some of these production processes have achieved industrial-level production standards (greater than 100 g/liter production titer and 3 to 4 g/L/h productivity) and have been commercialized to meet the global market demands.
Systems metabolic engineering continues to expand at an unparalleled pace with the rapid development of state-of-the-art genetic manipulation strategies, high-throughput screening tools, computational analysis methods, cultivation strategies, and downstream processes (Fig. 1). In this contribution, we aim to outline both the traditional and the most recently developed genetic manipulation tools including zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN), multiplex automated genome engineering (MAGE), conjugative assembly genome engineering (CAGE), clustered regularly interspaced palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) systems, and global transcription machinery engineering (gTME), in light of systems metabolic engineering. We also cover strategies for systems metabolic engineering and approaches to arising issues including enhancement of biosynthetic pathway, improving carbon flux, creating novel pathways, transporter engineering, reducing acetic acid formation during high-cell-density cultivation (HCDC), and the use of plasmids without antibiotics. Additionally, the concerted application of these strategies for industrial-level production of chemical production is detailed. While we confine our discussion to focus on the systems metabolic engineering of E. coli, readers are encouraged to explore various publications on the successful applications of the strategies developed for other species as well (34, 35, 36, 37, 38, 39, 40, 41).
Figure 1.
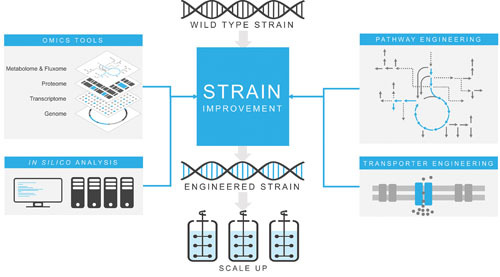
Overview of systems metabolic engineering. Systems metabolic engineering is the recursive process of improving a candidate strain via pathway engineering, transporter engineering, omics tools, and in silico analysis in an effort to increase the production of desired chemicals to industrial scales.
TOOLS FOR SYSTEMS METABOLIC ENGINEERING OF E. COLI
Here, we cover the various tools developed for genetic and genomic manipulations in E. coli. Some of these tools were not initially developed specifically for the purposes of systems metabolic engineering, but we listed them nonetheless, because these tools have become an integral part of strain development and, thus, a part of systems metabolic engineering. It must also be noted that some of these tools are very specific to E. coli, and applications of such tools on other organisms might not have been studied.
Recombineering: Recombination-Mediated Genetic Engineering
While the traditional cloning methodology, comprising digesting vectors with restriction enzymes and subsequent joining of the ends by ligation in vitro, is the starting point for almost all molecular biology experiments, it is not suitable for complex procedures (e.g., large-sized DNA manipulation). In recombination-mediated genetic engineering or “recombineering,” components derived from phages, λ and P1, have not only been the center of focus but also the most commonly used tools. Recombineering method has been used for engineering plasmids (42), genomic DNAs, and bacterial artificial chromosomes (BACs) (43, 44, 45), effectively using either single-stranded DNA (ssDNA)- (46, 47, 48, 49) or double-stranded DNA (dsDNA)-based techniques (50, 51, 52). In a metabolic engineering context, this process is used to alter expression levels of native proteins for flux optimization (53). In general, such methods rely on E. coli native recombination machinery originally used in DNA repair mechanisms.
The homologous recombination mechanism of E. coli that has most commonly been utilized in recombineering is the recABCD pathway. RecA is involved not only in homologous recombination but also in DNA repair and exhibits multiple functions, including DNA binding, ATP binding, DNA hydrolysis, protein degradation, and filament formation (54). RecBCD, also known as ExoV, exhibits helicase and exonuclease activities (55) and facilitates initial steps in DNA repair mechanism (for RecBCD reviews, see references 56, 57, 58). In E. coli, RecBCD degrades along linear DNA until it encounters a cis-element, chi (crossover hotspot instigator or χ) sequence, which facilitates the subsequent steps in recombination events (57, 59, 60). Linear dsDNA-based recombineering had been discouraged in the past, in particular because of E. coli RecBCD mechanisms, although linear dsDNA transformation in yeast had been common (61). While successful transformation with linear dsDNA in the recBC disrupted strain marks one of the first recombineering cases in historical perspective (62, 63), it required additional disruption of sbcB and sbcC because they often impede the use of linear dsDNA for homologous recombination template in the absence of functional RecBCD (62). While the strict requirement for recBC deficiency for linearized dsDNA transformation can be evaded by flanking linear dsDNA with the chi sequence, the chi-mediated recombineering suffers from low efficiency and positive-negative selection requirement (64). Use of λ recombination system for exonuclease deficiency requirement is one way to avoid this issue.
The λ recombination system designated “Red” consists of two proteins, the exonuclease (simply exo or sometimes α) and the bet protein (simply β) (65). Red is assisted by a third λ protein, gam (or γ), which increases exo and β activity on linear dsDNA by inhibiting E. coli RecBCD exonucleases (50, 55, 66). Since the presence of RecBCD no longer hinders stability of dsDNA, Red is universally applicable and greatly improves the linear dsDNA transformation efficiency as well as recombination (50). This system is often used in conjunction with site-specific recombination systems such as Cre/lox and Flp/FRT system.
Initially discovered in early 1980s, the Cre/lox system exploits the phage P1 infection mechanism in E. coli (67, 68). As a member of the λ recombinase family with a relatively small size (38 kDa), Cre (short for cause of recombination) protein from P1 recognizes and integrates the loxP (short for locus of crossover (x) in P1) sequence into a specific location of bacterial chromosome (loxB) in a Holliday junction-mediated event. Initially discovered from yeast plasmid “2μ circle” (69), the Flp/FRT (Flp recognition target) system is analogous to Cre/lox and uses two inverted repeats (70). While recognition sites in both systems are 34 base pairs (bp), Cre is more thermostable than Flp (71) and exhibits higher DNA binding affinity cooperativity (72). For engineering multiple genes, mutant lox sequences such as lox71, lox66, and lox2272 are also available for preventing further recombination events (73, 74). In conjunction with such site-specific recombination mechanisms in the above-mentioned homologous recombination, more efficient genetic engineering methods are being continuously developed.
The λ Red system combined with site-specific recombination was successful by the establishment of a one-step inactivation method for chromosomal deletion of E. coli genes (52). Widely accepted and used in systems metabolic engineering, this method employs the PCR-generated linear DNA including a drug selectable marker and homology extensions at both ends for recombination that are additionally flanked by lox (or FRT). Plasmid-based introduction of λ Red genes allows linear dsDNA integration into genome (52) followed by site-specific recombination for “pop out” of the selectable marker, successfully deleting the target gene (75, 76). Time taken for engineering of multiple genes has been improved recently using an integrated system for both recombination mechanisms (77). Because the two recombination systems were both integrated into one helper plasmid, there is no need for plasmid curing or repeated transformation for sequential genome-editing steps, thus greatly reducing the time taken for sequential recombinations (77). While this method boasts seamless efficiency and speed, the system leaves a scar in the edited genome. One way to overcome this challenge is to use a dual-selection system for the removal of the selection marker: a widely used dual-selection system involves the use of a gene named sacB.
Isolated from Bacillus subtilis, the sacB gene encoding levansucrase is used as a negative selection marker for an insertion sequence (78, 79). The sacB gene is usually used in conjunction with an antibiotic resistance gene for a two-step selection process to validate a homologous recombination of the host genomic DNA with the sequence of interest (80, 81). A plasmid harboring antibiotic resistance, sacB, and engineered sequence is introduced into a host cell, where the first level of screening is done on a medium plate containing the respective antibiotic to ensure proper genome integration of the plasmid. Once transformation of the plasmid is confirmed, the consequent host cells are then introduced into a sucrose-rich medium for a second level of screening; levansucrase converts sucrose into lethal levan. Only the host cells with successful second recombination will survive, thus validating the change in the genomic DNA of the host; some population with the second cross-over occurring at the same side as the first cross-over will revert to their original state and fail to survive. Survival of cells with spontaneous mutation in sacB coding region is the weakness of this system.
Meganucleases
Meganucleases, also known as homing endonucleases, are deoxyribonucleases that cleave recognition sites that are 12 to 40 bp long. Based on sequences and structural motifs, they are categorized into five classes: LAGLIDADG, GIY-YIG, HNH, His-Cys box, and PD-(D/E)XK; LAGLIDADG especially is the largest family that is found in all kingdoms of life (82). Meganucleases are easily distinguished from other restriction enzymes by special prefixes in their names: I- for those found in introns (83, 84, 85), PI- for those found in inteins (protein inserts) (86), F- for viral genes and those freestanding outside introns and inteins (87), and E- or H- for engineered or hybrid meganucleases (88). The first meganuclease identified is I-SceI, found in the mitochondria of baker’s yeast Saccharomyces cerevisiae (89). Subsequently, several other meganucleases, including I-CreI from the chloroplasts of a green alga Chlamydomonas reinhardtii (84) and I-DmoI from an archaeon Desulfurococcus mobilis (85), were identified and characterized.
The relatively long nature of meganuclease recognition sites explains their extremely low appearance frequency, which has inspired scientists to use them as a site-specific genome engineering or gene therapy tool. Mathematically, one recognition site 40 bp in length would only appear once in 1.2 × 1024 bp. In reality, one or more recognition sites occur in each genome of certain organisms. However, the scarcity of recognition sites in genomes also makes the use of meganucleases for direct engineering difficult because of the lack of variety and options for suitable use in a given genomic locus of interest. Two approaches have been taken to overcome this obstacle. In one approach, target DNA specificities were altered by either creating mutant meganucleases (90, 91) or chimeras using different parental nucleases (88, 92). In another approach, a meganuclease recognition sequence is inserted into the genomic locus of interest (89, 93). In either case, DNA cleavage followed by homologous recombinations at the target loci can be induced for desired chromosomal engineering (89, 92, 93). Alternatively, the scarcity of recognition sites in the genome that may pose a problem can be circumvented with the following novel strategy. When the organism of interest lacks the recognition sequence in the genome, the sequence can be introduced on a plasmid for in vivo cleavage, leading either to the loss of plasmids (94) or integrations of the linearized plasmids into the genome (94, 95).
Zinc Finger Nucleases (ZFNs)
As described in “Meganucleases” above, finding or synthesizing endonucleases that target and cleave desired sequences in a very specific manner has been a big concern in the field of genome engineering. Often described as “programmable nucleases,” such endonucleases have been designed and manufactured since the first report on the creation of a fusion endonuclease, named “zinc finger nucleases” (ZFNs) (Fig. 2). In a 1996 study, zinc finger domains were combined with fragments of a restriction endonuclease, producing the first ZFN (96). In this first article on ZFN, the authors employed zinc finger proteins CP-QDR and Sp1-QNR, which consist of three domains of Cys2His2 zinc fingers and thus bind to 9-bp target sequences (96). Moreover, removing the N terminus of FokI restriction endonuclease, which is responsible for the DNA binding activity (97), C-terminal 196 amino acids of FokI, which has the nonspecific DNA cleavage activity (98), was fused to the C terminus of each zinc finger protein (96). Maximally utilizing the requirement of FokI dimerization to work as a DNA cleaving unit (99), a genomic locus of interest can be more specifically targeted by using two ZFNs, which bind to both of the flanking regions of the target site separated 5 to 7 bp apart from each other. The homodimerization of the wild-type FokI domains, however, results in off-target cleavage issues, and this was solved by generating obligate heterodimeric FokI C-terminal domains by the modification of their dimerization interfaces (100, 101). Moreover, enhancement of the FokI domains by mutagenesis was reported (102, 103). In theory, each of the zinc fingers bind to 3-bp-long DNA in a modular fashion (104), and various zinc fingers generated by mutagenesis can be fused in tandem to generate zinc finger proteins targeting any of the desired sequences (105, 106). In reality, however, the zinc finger proteins modularly assembled from each of the zinc fingers often show off-target effects (107) or fail to target DNA (108). There are several methods to select out nonfunctional ZFNs among the new assemblies (109, 110, 111, 112); however, constructing a new functional ZFN is still a major challenge.
Figure 2.
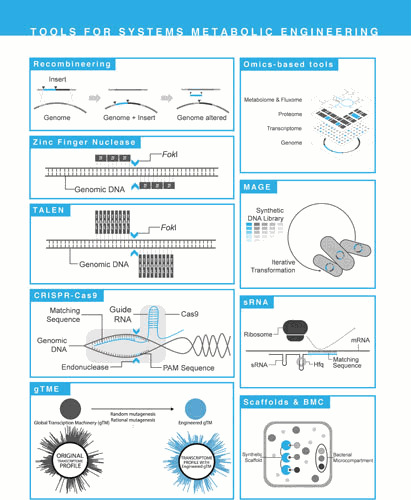
Tools for systems metabolic engineering. List of tools for genetic modification of candidate strains: recombineering, zinc finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system, global transcription machinery engineering (gTME), omics-based tools, multiplex automated genome engineering (MAGE), synthetic small regulatory RNA (sRNA), and scaffold proteins.
Transcription Activator-Like Effector Nucleases (TALENs)
Despite the great modularity of ZFNs, the biggest weakness of this group of nucleases is twofold. First, a single zinc finger domain recognizes 3 bp at once (104), requiring a library of at least 64 (=43) modules for a complete coverage on all possible triplets of base pairs. The massive size of the library renders difficulties for several research groups from easily accessing the library, especially for those consisting of zinc finger domains with high sequence specificity. More importantly, the assembly of zinc finger proteins from each of the zinc finger modules often results in nonfunctional or erroneous products (107, 108). An alternative engineered nuclease was developed to solve these problems with discovery of a group of proteins known as transcription activator-like effectors (TALEs). Initially discovered in plant pathogen Xanthomonas spp., TALEs are secreted from plant pathogens into the host cells and activate the transcription of plant genes, changing the physiology of the host plant (113). A TALE consists of an array of 33 to 35 amino acids repeats, and each repeat specifically binds to a single nucleotide in the major groove (114, 115). The 12th and 13th residues of each repeat, known as repeat variable diresidues (RVDs), determine the base pair specificities of each repeat (116, 117). These properties indicate the superiority of TALEs over zinc finger proteins: TALEs require only four types of repeats that bind to each of the four types of base pairs, while zinc finger proteins need a set of 64 zinc fingers binding to 64 types of 3-bp-long DNA. After unraveling of RVDs in 2009, the application to use TALEs as programmable nucleases was quickly achieved in the very next year (118). Transcription activator-like effector nucleases (TALENs)—programmable nucleases based on TALEs—follow exactly the same scheme as that of ZFNs; the C-terminal nuclease domain of FokI is fused to the C terminus of the TALE arrays (Fig. 2). The use of up to 20 repeats in each TALEN, however, makes the construction of TALENs challenging—the repetitive occurrence of the homologous sequences in TALEN DNA constructs leads to instability of the constructs and their intermediates (119). Nonetheless, attempts to improve the facile assembly of TALENs are continuing (120, 121, 122, 123, 124, 125, 126).
CRISPR/Cas System
In 2002, loci characterized by alternative occurrence of 21- to 37-bp-long repetitive sequences (repeats) and nonrepetitive sequences (spacers) were identified in prokaryotes by a bioinformatics analysis (127). Reflecting the noticeable structural features of theses loci, they are named as clustered regularly interspaced short palindromic repeats (CRISPRs), although the initial observation was made in 1987 (128). Moreover, protein-coding genes near CRISPRs were found together and named as CRISPR-associated (cas) genes (127). After the first discovery of CRISPR loci, many research groups suggested the role of CRISPR/Cas system as prokaryotic immunity (129, 130, 131). In 2007, the suggested CRISPR’s role as a prokaryotic acquired immune system was confirmed (132). The authors observed that Streptococcus thermophilus exposed to bacteriophages acquired new spacers derived from the phage DNA segments (later termed as protospacers) inside its CRISPR loci (132). Moreover, modification of particular spacers in CRISPR modulated the resistance profile of the host cells against various bacteriophages, indicating close correlation between bacterial resistance against virus and the CRISPR/Cas system (132). Subsequently, involvement of the CRISPR/Cas system in the resistance against plasmids and conjugation were also reported (133, 134). After years of brilliant research, now it is understood that CRISPR loci are transcribed and the resulting CRISPR RNAs (crRNAs) are processed by several components including Cas proteins and RNase III (135, 136, 137, 138). The processed crRNAs then work as effectors together with various other Cas proteins to cleave either DNA targets (135, 139) or RNA targets (140). When CRISPR/Cas systems are targeting their invader DNAs, they interrogate two points: the presence of protospacer adjacent motif (PAM)—the motif adjacent to protospacers that is required by effector Cas proteins to work as a nuclease—and complementarity of crRNA to the protospacer on target DNA (141, 142, 143).
After years of efforts to classify various CRISPR/Cas systems, the systems are now categorized into five types, classified into two classes: type I, type III, and type IV in class 1 CRISPR/Cas system and type II and type V in class 2 CRISPR/Cas system (144, 145, 146, 147). Among three of them, Cas9 protein from Streptococcus pyogenes belonging to type II CRISPR/Cas system (148), is especially the most widely used for genome engineering applications (Fig. 2). In type II CRISPR/Cas system, crRNA are associated with trans-activating crRNA (tracrRNA), and they together bind to the effector nuclease Cas9 (148). Elucidating the dual RNA systems in the CRISPR/Cas9 system, Doudna and her colleagues also created a chimeric guide RNA called single guide RNA (sgRNA) by joining essential motifs of crRNA and tracrRNA, making the system simpler (148). Nowadays, both crRNA/tracrRNA and sgRNA systems are widely used for various applications. While meganucleases, ZFNs, and TALENs require the manipulation of effector proteins to program them to target genomic loci of interest, simple exchange of crRNA or sgRNA for the programming of CRISPR/Cas9 system led to a boom in the field of genome engineering. Although originated from prokaryotes, CRISPR/Cas9 systems are the most actively used in engineering of eukaryotic genomes (149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159). There are relatively few examples of using CRISPR/Cas9 in prokaryotes (160, 161, 162, 163, 164, 165, 166). The first example of using the CRISPR/Cas9 system in genome engineering of E. coli was reported by Marraffini and colleagues (160). Their first trial utilized two plasmids: pCas9 containing tracrRNA, Cas9, and chloramphenicol resistance gene, and pCRISPR containing CRISPR spacer. Along with the application of recombineering strategy, these two plasmids and an oligo for point mutation were transformed, thus promoting successful recombination with a significant efficiency of 65 ± 14% (160).
The biggest concern in CRISPR/Cas9 systems is their off-target effects, as in other programmable nucleases. There have been many efforts to detect the off-target activity levels of the CRISPR/Cas9 systems (167, 168, 169, 170). Moreover, many trials to reduce off-target activities have been reported, including the use of Cas9 nickases and catalytically dead Cas9 (dCas9)-FokI nucleases (171, 172, 173, 174, 175). In addition, the relatively big size of the Cas9 gene (approximately 4.2 kb) makes gene work on Cas9 more difficult. There is a recent report on an identification of smaller (around 3 kb in gene length) Cas9 from a multidrug-resistant Staphylococcus aureus (MRSA) isolated at an Irish hospital (176).
Repurposed CRISPR/Cas Systems
The CRISPR system not only has been a crucial genome-editing tool, but it also has been modified to be used as a trans-acting gene knockdown system (177). The Cas9 protein with endonucleolytic activity was inactivated (dCas9) to repurpose this system into a gene expression regulating tool. This repurposed system knocks down gene expression by the following mechanism: when the target-oriented sgRNA and dCas9 are coexpressed, the scaffold of sgRNA binds with dCas9, leading the complex to the DNA sequence near the promoter site as in normal Cas9. After the complex binds the target DNA sequence, the CRISPRi R loop is formed, preventing the propagation of the incoming RNA polymerase and subsequently blocking the target gene transcription effectively. Fusing an activation domain or a repressor domain to dCas9 can be an alternative strategy for regulating gene expression using the CRISPR/Cas9 system. While this idea was successfully implemented in eukaryotic systems multiple times (178, 179, 180), only a single study reported to date has succeeded to activate a target gene with the dCas9-fusion protein system in E. coli (181). The authors fused the omega subunit of RNA polymerase with dCas9 and targeted this complex upstream of the target gene promoter by engineering the corresponding crRNA. The authors tested this system for overexpressing lacZ, which encodes beta-galactosidase, and obtained a 2.8-fold increase in expression when the omega subunit was fused to dCas9 at its C terminus.
Small Regulatory RNAs
While there have been significant advances in the regulation of gene expression by means of developing promoter, ribosome binding site, or intergenic regions between genes in a single operon for fine-tuning of gene expression, modification in chromosomal DNA is not only time consuming and laborious, but also unfit for engineering multiple targets simultaneously (182, 183, 184). As such, a new method for simultaneously studying multiple genes using trans-acting small regulatory RNAs (sRNA) for regulation and fine-tuning of inherent genes in E. coli was developed (185, 186). This strategy derives from native sRNAs in bacteria regulating gene expression by translational control (187). The structure of synthetic sRNA construct consists of three major constituents: a target-binding sequence, scaffold, and terminator (Fig. 2). sRNAs regulate gene expression by base-pairing its sequences to the target translation initiation region, consequently blocking the ribosome from taking action and thus significantly reducing translation efficiency (Fig. 2). The target-binding sequence is engineered into the antisense sequence (24 bp) of the translation initiation region of the target gene to be regulated. The scaffold within sRNA binds to an RNA chaperone Hfq protein, so as to enhance the binding of sRNA to the target mRNA sequence and to catalyze the degradation of mRNA by recruiting RNase E (187, 188). Among the 101 E. coli sRNAs discovered recently, the scaffold of MicC was selected for its strong repression capability. The advantages of using synthetic sRNAs as a tool is in its ability to test knockdown of essential genes and to fine-tune gene expressions, its ease in vector construction thus expediting the experiment process, and its ability to target multiple sites simultaneously. It has also been reported that not only repression, but also activation of certain genes by means of sRNAs exist in nature (189, 190).
Global Transcription Machinery Engineering (gTME)
After decades of extensive study on genotypes and phenotypes of various biological systems, it is now widely accepted that a majority of phenotypes are polygenic, rather than monogenic. Hence, an ability to simultaneously select and adjust multiple genes is very critical to engineer organisms for a desired trait, especially in the field of metabolic engineering. Intuition- and logic-based target gene selection to engineer a single phenotype is limited in many situations. Simultaneous manipulation of multiple target genes chosen is not affordable with traditional gene/genome engineering tools, either. In such situations, mimicking nature’s strategy to develop novel systems or components can be a wise choice—evolution, a series of random mutagenesis and selection. Stephanopoulos and colleagues introduced a concept named global transcription machinery engineering (gTME) that targets components in global transcription regulation for random mutagenesis to randomly manipulate gene expression profile in a transcriptome-wide manner followed by an application of proper selection powers (191, 192). They proved the system both in a eukaryotic microorganism yeast (191) and in a prokaryote E. coli (192). In gTME application in E. coli, one or multiple components involved in global transcription regulation, such as rpoD, are initially selected for construction of mutant libraries. The mutant libraries generated are then introduced into a host microorganism in the presence of a corresponding, nonmutagenized component. Subsequently, an individual organism exhibiting desired traits such as tolerance is selected by applying the proper screening method to the host containing the mutant libraries of the target global transcriptional constituents. The strength of gTME is that it enables engineering of multiple target genes, which are not often intuitive or logical, for desired phenotypes in a short period (191, 192).
Multiplex Automated Genome Engineering (MAGE)
Instead of creating mutant global transcription factors for genome engineering, direct multiplex editing of genomic loci can also be achieved. Church and his colleagues provided the way to simultaneously manipulate multiple genomic loci either randomly or rationally mutagenized (53). Unlike gTME, where transcriptome profiles are altered by mutant global transcription machineries, MAGE can generate a library of hosts with multiple genomic loci edited directly in the genomic sequence where the editing includes residue substitution, deletion, and insertion. With bacteriophage λ Red ssDNA binding protein β expressed, mutation-containing ssDNA oligonucleotides for targeting the lagging strand of replication fork during DNA replication at designated genomic loci are introduced into a host by electrophoresis, generating a daughter population of hosts containing targeted sequences edited. A pool of various oligonucleotides can be delivered into the host at once, and repeated delivery of the oligo library results in a set of hosts with genomic sequences differentially modified in each generation. It is noticeable that the whole procedure of repetitive oligo delivery is automatically operated with the help of machinery and an operating program developed in the research (53). Once experimental conditions are optimized, more than 30% of the host population incorporates mutation for a single locus, which only takes 2 to 2.5 h (53). Modified versions of MAGE for improved modification efficiency have also been reported (193, 194).
Conjugative Assembly Genome Engineering (CAGE)
While recombinant plasmids can be transferred among strains after they are constructed in one host strain, modifications made directly on genomic DNA, however, are hard coded and difficult to be transferred in parallel. In order to reintroduce the same chromosomal modifications in a different strain, the same modification procedures must be performed repeatedly. This makes parallel genomic modification-based engineering inefficient and time consuming compared with plasmid-based engineering. In 2011, Church group reported genome-wide substitution of every TGA stop codon to TAA by accumulating pieces of partial genomic modifications made on each host cell line into a single, final host strain (195). They named the system employed for this achievement as conjugative assembly genome engineering (CAGE), which literally describes the core mechanism involved in this system.
CAGE maximally utilizes the virtue of DNA transfer from one cell to another during a process called conjugation. Isaacs et al. split the F+ plasmid, which enables the host cell working as a donor cell during conjugation, into two parts: oriT and the other constituents. oriT, the starting point of DNA replication and subsequent transfer of F+ plasmid to a recipient cell during conjugation, is integrated into the host genome to make the host competent for a donor role (195). This causes the whole genomic DNA to be replicated and transferred to the recipient cell during conjugation. The remaining components in F+ plasmid, in contrast, are retained as a closed-circular plasmid and introduced to the donor cell during CAGE process. While they help the process of conjugation, they are not transferred to the recipient since they are physically separated from oriT. The prepared donor cells are then cocultured with recipient cells, promoting conjugation between two different cell populations to transfer the modified genomic DNA from donors to the recipients. Subsequently, the transferred donor genomic DNA substitutes the corresponding region in recipient genome with the help of recipient cell’s endogenous recombination machinery (195).
The recombination, however, is a stochastic process and the points where recombination occurs are randomly chosen. Consequently, the probability for a locus on donor genomic DNA to replace the corresponding recipient DNA segment is higher as the locus is located further from both ends of the linearized donor DNA fragment, following simple probability theories. To make sure the engineered regions on donor genomic DNA successfully substitutes the recipient DNA, the engineered region on donor cells is bounded by oriT and a positive selection marker, including antibiotic resistance genes (195). Moreover, the starting point of a region to be replaced on recipient genome is defined by a positive-negative selection marker, such as tolC (196) and galK (45). Together with these markers, resulting recombinant cells with the first recombination point occurring too far away from oriT are selected out by negative selection effect of the positive-negative marker. On the other hand, ones with the second recombination occurring too close to the first recombination site are selected out since they cannot survive without a positive marker. In this manner, the only final recombinant strains containing the whole engineered segment of the donor genome are obtainable. These processes can be repeated until all of the pieces of partially engineered regions of each cell line are accumulated in a single cell line.
Trackable Multiplex Recombineering (TRMR)
The random engineering of multiple genomic loci allows isolation of new strains with unexpected target genes modified in an unexpected way. Obtaining such a brand new strain itself is a great work, but it will be more amazing to know how the new strain is changed. A method called trackable multiplex recombineering (TRMR), suggested by Warner et al., provides a way to manipulate multiple genomic loci, detect the loci affected, and elucidate the manner in which they are modified (197). In TRMR, 20-bp-long “barcode sequences” are inserted upstream or downstream of the target genomic loci while the target loci are simultaneously modified with the help of the recombineering techniques. The barcodes are used for detecting the changes that occurred at the target loci. Combined with microarray techniques, they can give information on allelic frequencies of the modified genes in a bacterial population subjected to TRMR (197, 198).
Synthetic Scaffolds and Bacterial Microcompartments
The idea of spatial organization of enzymes was first conceived from mammalian signal transduction pathways where grouping of enzymes by spatial organization is needed to handle signal transductions from one billion proteins (199); similarly, metabolism within a cell is a highly complex network of chemical reactions. With this complexity, problems may occur in regard to unwanted side reactions (200), limiting metabolic flux due to low substrate specificity (201), low productivity and yield (9), and hindrance of cell growth by toxic materials (202). Handling these problems would require channeling of substrates or directing target chemicals or enzymes to a confined area. To cope with these problems, examples from nature were readily adopted (203), which include tryptophan synthase inherent in Salmonella enterica serovar Typhimurium (204), carbamoyl phosphate synthetase, and glutamine phosphoribosylpyrophosphate aminotransferase in E. coli, and dihydrofolate reductase-thymidylate synthase inherent in Leishmania major (205). Additionally, carboxysomes were discovered and characterized in cyanobacteria and chemolithotrophic bacterial species, in which rate-limiting steps involved in the Calvin cycle take place (206). Inspired by these examples, synthetic scaffolds and bacterial microcompartments (BMCs) were developed. Synthetic scaffolds aim at localizing enzymes in order to increase the local intermediate concentration of a desired biochemical reaction or to prevent toxic materials from damaging the cell. Starting from the development of synthetic protein scaffold (201), synthetic scaffolds using RNA (207) and DNA (208) as building blocks were sequentially developed, all of which were demonstrated in E. coli.
BMCs, which resemble organelles within eukaryotic cells, tend to encapsulate large numbers of certain enzymes to form distinct reaction environments. This tendency increases the concentration of intermediates such as in carboxysome, in which CO2 is concentrated around the otherwise inefficient RuBisCO (209, 210). Additionally, toxic intermediates are effectively blocked from incorporating into the host metabolism as in examples of Eut and Pdu microcompartments (211, 212, 213). It is interesting to note that BMCs are thought to regulate the amount and types of chemicals that can go through the compartment, while intermediates and toxic materials seemed to be kept inside of the bacterial microcompartments (BMCs), nontoxic native substrates, and products diffuse across the compartment relatively easily (210). The precise mechanism of how this happens is yet to be determined, although several hypotheses exist (212, 214, 215). Encapsulins and lumazine synthase complexes are two other simpler microcompartment systems reported (216, 217).
Omics Tools
In systems metabolic engineering, omics-based tools are system-wide strategies that utilize data from x-omes to analyze hosts on the systems level and unravel unknown mechanisms to enhance the production of target chemicals. Since a tremendous amount of data is associated with multiomics-based approaches, computational simulations often aid in the omics analysis. Readers are encouraged to refer to several excellent reviews on multiomics studies and incorporation of in silico simulations (218, 219, 220, 221).
The first bacterial genome sequencing of Haemophilus influenzae opened the field of genomics, starting a new era in omics-based approaches (222). Numerous sequencing technologies have emerged since this first onset, facilitating the development of genomics (223). With the increasing amount of genome data, comparative genomics comparing the genome sequences of two organisms has become feasible. From the genomic data open and available for all, genome-scale modeling has been developed actively, which will be explained in later sections (224, 225, 226).
However, the expression levels of each gene cannot be identified through genomics. Transcriptomics analyzes the mRNA levels of various genes at a time, particularly the change of gene expression levels when given perturbation (218, 227). The development of transcriptomics has had a deep relationship with the development of high-throughput microarray development (228). By measuring the fluorescence in the microarray, the expression levels of each gene can be examined. For transcriptome modeling, a reverse-engineering strategy named network identification by regression (NIR) was used (229). Transcriptomics has several applications, such as the elucidation of useful metabolic genes (230) or the identification of target genes or pathways to be engineered (231).
Unlike transcriptome, proteome reflects the posttranscriptional regulations by small RNAs, mRNA degradation, and represents the real phenotype of the cell much more clearly than the transcriptome does. Readers are invited to read an excellent review on the proteome of E. coli covering aspects not explained in this section (232). Two-dimensional (2D) electrophoresis is a widely used method for analyzing an organism’s proteome (233). For highly expressed proteins, however, biased results were often shown (234), leading to the development of non-gel-based approaches such as mass spectrometry (MS) (235), matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) (236), and liquid chromatography-tandem mass spectrometry (LC-MS/MS) (237). The main interest in proteomics analysis is to examine the change in cellular and metabolic states during the production of metabolites of interest.
Since the proteome of an organism still does not precisely represent the physiology of the cell and enzyme activities, additional omics was needed, leading to the emergence of metabolomics (238). Several analytical methods were adopted for metabolomic analyses including nuclear magnetic resonance (NMR) (239), high-performance liquid chromatography (HPLC) (240), and MS (241). Metabolomics, however, contains a serious problem arising from the dynamic turnover events of cellular metabolites. Therefore, an instant quenching method is required (242). Other problems such as cultivation techniques and metabolite extraction methods are some of the ongoing challenges.
Fluxomics is widely modeled and applied in metabolic engineering, because it analyzes the reaction rates of different metabolic pathways (243, 244). Because it is almost impossible to obtain the dynamic intracellular fluxes of a cellular system, indirect approaches are used for metabolic flux analysis (MFA): 13C-based flux analysis and constraints-based flux analysis. The basic assumption in these analyses is that the cellular fluxes are at steady state. In 13C-based analysis, an isotope-labeled carbon source is fed to the medium, letting 13C-incorporated metabolites to be detected and patterns to be analyzed by NMR and gas chromatography mass spectrometry (GC/MS) (245). However, expensive isotope samples, difficulty in conducting experiments, and large-scale calculations are the bottlenecks in this approach. Constraints-based flux analysis is a set of algorithms for optimization-based simulation (246). Metabolic flux analysis can also be used for elucidating the metabolic characteristics of a newly identified organism such as in the case of Mannheimia succiniciproducens (247).
Other kinds of omics including glycomics, lipidomics, and interactomics have also been actively researched to date. Because each omics study has its own drawbacks, the integrated analysis and use of omics are required for a comprehensive analysis of an organism. The first attempt to integrate multiple omics was by Hood and colleagues to characterize the phenotype of S. cerevisiae (248). Several other significant multiomics studies regarding E. coli are as listed (249, 250). A prominent tool that deals with the multiomics data of E. coli is EcoCyc (251, 252, 253). Through the latest version of EcoCyc, a tremendous amount of data including genes, metabolites, reactions, operons, metabolic pathways, and metabolic flux models of E. coli can be accessed (253).
In Silico Genome-Scale Metabolic Network Models and Tools
A massive amount of genome data has been accumulating since the beginning of the genomic era. The availability of such information allowed researchers to understand and predict the physiology of organisms. One of the initial studies used such information to predict the growth of bacteria (254, 255). Currently, these data are routinely applied to predict and analyze detailed physiological and metabolic status of bacteria (22, 25, 225, 256, 257, 258). There are several comprehensive reviews covering the diverse applications of genome data for various purposes (259, 260, 261, 262, 263).
A single genome-scale metabolic network model consists of an immense amount of data from various literature, experimental sets, and databases. The data integrated into a single genome-scale model includes information on genome-wide metabolic genes, hundreds to thousands of metabolites, and thousands of stoichiometric metabolic reactions. Constructing such models is beyond the scope of this review, and descriptions of the methods to construct such models can be found from other reviews and book chapters (259, 260, 264, 265). During past three decades of E. coli genome-scale reconstruction history, several versions of thorough E. coli genome-scale metabolic network models have been published (225, 256, 257, 266), such as EcoMBEL979 (266), iAF1260 (225), iJO1366 (256), and EcoCyc-18.0-GEM (267) containing thousands of metabolites, metabolic enzymes, and stoichiometric reaction rulesets.
An E. coli genome-scale model alone cannot exhibit the metabolic state of this bacterium in a given condition. To determine the metabolic state, flux information of each metabolic reaction should be determined. However, there are many more metabolic reactions than metabolites in the models, as in real E. coli, rendering the complete determination of the exact flux solutions impossible; for example, the most thorough E. coli K-12 MG1655 genome-scale model, iJO1366, contains 1,136 metabolites and 1,473 metabolic reactions (256). In bacteria, the complex sets of regulatory components fine-tune the metabolic fluxes. In current E. coli genome-scale models, unfortunately, such complicated regulations have not been implemented yet. Therefore, during in silico simulations, an algorithm mimicking or representing such regulations should be employed to select the best prediction on the real metabolic state among the vast solution space.
Flux balance analysis (FBA) is the most widely used algorithm. Based on genome-scale models, FBA applies stoichiometric information as major constraints and builds a solution space for metabolic flux distribution (255, 268, 269). To specify a solution that best represents the actual state among the possible solutions, FBA generally employs an objective function of maximum biomass formation. This is based on the assumption that E. coli and other bacteria have evolved to maximize their growth and replication, reflecting the generally acknowledged expression “The goal of every bacterium is to become bacteria” as said by microbiologist Stanley Falkow. Simulation of how a genetic perturbation modulates the flux distribution is also an important aspect in metabolic engineering, especially for the selection of engineering targets. In addition to FBA, another useful algorithm for this purpose is the minimization of metabolic adjustment (MOMA) algorithm (270). This algorithm is based on the idea that when perturbation is introduced in a bacterial strain by knocking out one or more genes, the bacteria redistribute their metabolic fluxes to minimize flux changes (270). Based on the reference flux distribution data obtained from FBA, MOMA can be employed to predict modifications in the flux distribution. The third noticeable algorithm is the regulatory on/off minimization (ROOM) algorithm (271). This algorithm is similar to MOMA, but differs in that ROOM focuses on minimizing the number of fluxes significantly changed, i.e., turned on or off, while MOMA focuses on minimizing the total magnitude of changes throughout the whole metabolic fluxes (271). All these algorithms predict the physiology of bacteria fairly well, and each has its strengths and weaknesses in different conditions, such as initial stages or steady-state phases (271).
These genome-scale metabolic models and metabolic state prediction algorithms can be used together to select engineering targets for improving strain performances. For these purposes, several algorithms have been developed and reported. These include OptForce (272), OptKnock (273), OptGene (274), OptSwap (275), FSEOF (flux scanning based on enforced objective flux) (258), FVSEOF (flux variability scanning based on enforced objective flux) (276), and CosMos (277). Most algorithms contain one of the three metabolic flux prediction algorithms FBA, MOMA, and ROOM within. Moreover, there are numerous environments for in silico simulation, such as MatLab-based COBRA Toolbox (278, 279) and a stand-alone program suite MetaFluxNet (280, 281). With the help of these models, algorithms, and programs, targets of gene knockout, amplification, and flux level adjustment to improve strains can be predicted.
Design and construction of novel biosynthetic pathways can also be assisted by in silico pathway prediction tools. Such algorithms consist of two components: reaction rule sets and heuristics for narrowing down most probable pathway candidates. Currently, numerous pathway prediction tools are available for designing novel biosynthetic pathways for chemicals of interest, including PathoLogic (282), PathMiner (283), BNICE (284), Pathway Hunter Tool (285), DESHARKY (286), UM-PPS (287), MetaRoute (288), Rahnuma (289), ReTrace (290), Atom Metanet (291), Cho et al. 2010 (292), PathPred (293), RetroPath (294), SymPheny Biopathway Predictor (27), XTMS (295), GEM-Path (296), and RouteSearch (297). There are a number of indepth reviews describing and analyzing such useful tools (259, 260, 261, 262, 263, 298, 300).
Plasmid Addiction System
Selection for plasmid-transformed bacteria has been the foundation in development of biotechnology, and the use of plasmids for industrial application is ever crucial (301, 302). The plasmid-harboring bacteria are often selected in the presence of antibiotics. However, the use of antibiotics for industrial-scale systems is often undesired due in part to the cost of antibiotics, contamination of products with residual antibiotics, and possible degradation of antibiotics leading to instability of plasmids during cultivation (303). In light of problems with antibiotics in industrial systems, the plasmid addiction system delivers an alternative mode of selection while maintaining plasmids in an antibiotics-free environment.
The plasmid addiction system can be largely categorized into three types: toxin/antitoxin-based, metabolism-based, or operator repressor titration systems (301). An example for the toxin/antitoxin-based system adapted from phage P1 protein called Doc (death on curing) and Phd (prevent host death) where the former is toxin and latter is antitoxin (304, 305). The toxin is stable and causes cell death in the absence of plasmid, whereas the unstable antitoxin prevents cell death in the presence of plasmid. Cell propagation thus only depends on the presence of plasmid, which harbors both toxin and antitoxin, where the latter inhibits the activity of the former. In the operator repressor titration system, a gene essential for metabolism, such as dapD, is controlled under the lac operator/promoter (306). Repression of dapD expression prevents L-lysine biosynthesis and peptidoglycan chain cross-linking (307). The host lacI repressor thus inhibits growth unless it is sequestered or “titrated” away from the lac operator/promoter controlling dapD expression. This titration is achieved by introducing another lac operator/promoter on a multicopy plasmid (303). Similarly, an essential gene for cell growth and metabolism is deleted in the host chromosome in the metabolism-based system. (308). A notable study using this system involves the deletion of dapE in the host chromosome and complementing it with dapL from Synechocystis sp. PCC 6308 (301). Using this strategy, the transformed E. coli was able to maintain the plasmid during cultivation at a 400-liter fermentation in a 650-liter tank for cyanophycin production. The plasmid stability reported here is at 98%, and up to 18% (w/w) of dry cell weight (DCW) for the polymer was demonstrated. It must be noted that this study serves as an important example in the field of systems metabolic engineering for producing value-added chemicals at a semi-industrial scale without any antibiotics (301).
STRATEGIES FOR SYSTEMS METABOLIC ENGINEERING OF E. COLI
The hallmark of systems metabolic engineering is to produce desired chemicals with engineered microbes not only to industrial-scale standards, but also to bring changes imperative to the chemical industry. To achieve such goals, various strategies described here have been developed to engineer both native and synthetic pathways of E. coli for the production of valuable chemicals (Fig. 3). The strategies described here utilize various tools including those listed in the previous section. Table 1 summarizes notable achievements in the production of valuable chemicals in E. coli strains with metabolic engineering strategies described in genotypic notation.
Figure 3.
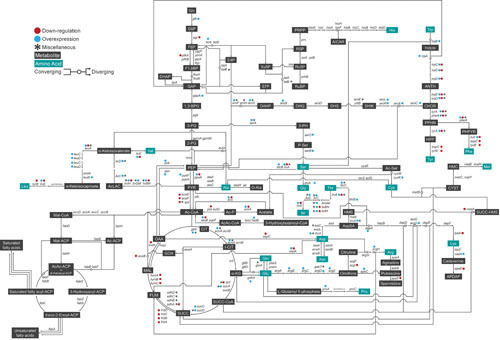
The endogenous metabolism of E. coli. The endogenous metabolic pathway of E. coli mapping products (metabolites in black boxes and amino acids in turquoise boxes) and the genes (italicized) of the enzymes responsible for the reactions based on EcoCyc E. coli database. Every overexpression (blue circle), downregulation (red circle), and all other miscellaneous modifications including feedback-release (asterisk) attempted in E. coli for systems metabolic engineering purposes are noted. Convergence and divergence of metabolites are denoted by circular nodes, where some reactions are reversible. As an example for reversible reaction, F6P and GAP converge to form E4P and Xu5P; conversely, E4P and Xu5P converge to form F6P and GAP. Glc, glucose; G6P, glucose 6-phosphate; F6P, fructose 6-phosphate; F1,6BP, fructose 1,6-bisphosphate; GAP, glyceraldehyde 3-phosphate; DHAP, dihydroxyacetone phosphate; 1,3-BPG, 1,3-bisphosphoglycerate; 3-PG, 3-phosphoglycerate; 2-PG, 2-phosphoglycerate; PEP, phosphoenolpyruvate; PYR, pyruvate; Ac-CoA, acetyl-CoA; CIT, citrate; I-CIT, isocitrate; α-KG, α-ketoglutarate; SUCC-CoA, succinyl-CoA; SUCC, succinate; FUM, fumarate; MAL, malate; OAA, oxaloacetate; GOX, glyoxylate; E4P, erythrose 4-phosphate; Xu5P, xylulose 5-phosphate; S7P, sedoheptulose 7-phosphate; R5P, ribose 5-phosphate; Ru5P, ribulose 5-phosphate; RuBP, ribulose 1,5-bisphosphate; PRPP, 5-phosphoribose 1-pyrophosphate; AICAR, 5-amino-1-(5-phosphoribosyl)imidazole-4-carboxamide; His, L-histidine; DAHP, 3-deoxy-arabino-heptulosonate 7-phosphate; DHQ, 3-dehydroquinate; DHS, dehydroshikimate; SHIK, shikimate; CHOR, chorismate; PPHN, prephenate; HPP, 4-hydroxyphenylpyruvate; Tyr, L-tyrosine; PHPYR, phenylpyruvate; Phe, L-phenylalanine; ANTH, anthranilate; Trp, L-tryptophan; 3-PH, 3-phosphohydroxypyruvate; P-Ser, 3-phosphoserine; Ser, L-serine; Ac-Ser, acetyl-serine; AcLAC, acetolactate; Val, L-valine; Leu, L-leucine; Ala, L-alanine; D-Ala, D-alanine; Ac-P, acetyl phosphate; AcAc-CoA, acetoacetyl-CoA; Glu, L-glutamate; Gln, L-glutamine; Arg, L-arginine; Pro, L-proline; Asp, L-aspartate; Asn, L-asparagine; AspSA, aspartate-semialdehyde; Lys, L-lysine; HMS, homoserine; Thr, L-threonine; Ile, L-isoleucine; SUCC-HMS, succinylhomoserine; CYST, cystathionine; HMC, homocysteine; Met, L-methionine; Ac-ACP, acetyl-acyl carrier protein (ACP); Mal-CoA, malonyl-CoA; Mal-ACP, malonyl-ACP; AcAc-ACP, acetoacetyl-ACP.
Table 1.
| Product | Strain designation (vector if any) | Note | Titer (g liter−1) | Reference |
|---|---|---|---|---|
| Metabolites from central carbon metabolism and derivatives | ||||
| Malic acid | XZ658 | KJ060 (ATCC 8739 ΔldhA ΔackA ΔadhE ΔpflB) ΔmgsA ΔpoxB ΔfrdBC ΔsfcA ΔmaeB ΔfumB ΔfumAC | 34 | 476 |
| Fumaric acid | CWF812 (pTac15kppc) | W3110 ΔiclR ΔfumC ΔfumA ΔfumB ΔarcA ΔptsG ΔaspA ΔlacI PgalP::Ptrc; vector-based overexpression of ppc | 28.2 | 309 |
| Succinic acid | JCL1208 (pPC201) | JCL1208 (E. coli K-12 F– λ– rpoS396(Am) rph-1 lacIq lacZ::Tn5 ΔrecA zfi::Tn10); vector-based overexpression of ppc | 10.7 | 310 |
| AFP111 (pTrc99A-pyc) | AFP111 (E. coli F– λ– rpoS396(Am) rph-1ΔpflAB::CmR ldhA::KmR ptsG); vector-based overexpression of pyc (Rhizobium etli) | 99.2 | 477 | |
| KJ122 | KJ073 (E. coli C ΔldhA::FRT ΔadhE::FRT Δ(focA-pflB)::FRT ΔackA ΔmsgA ΔpoxB) ΔackA ΔadhE ΔldhA ΔfocA-pflB ΔtdcDE ΔcitF ΔaspC ΔsfcA | 82.7 | 337 | |
| D-Lactic acid | ALS974 | YYC202 (DSM 14335, Hfr zbi::Tn10 poxB1 Δ(aceEF) rpsL pps-4 pfl-1) frdABCD | 138 | 478 |
| L-Lactic acid | LA20ΔlldD (pZSglpKglpD) | LA02 (MG1655 Δpta::FRT ΔadhE::FRT ΔfrdA::FRT-KmR-FRT) ΔmgsA::FRT ΔldhA::ldh (Streptococcus bovis) ΔlldD::FRT; vector-based overexpression of glpK and glpD | 50.1 | 479 |
| Butyric acid | BuT-8L-ato | BL21 ΔptsG ΔpoxB HK022::PRPL-glf (Zymomonas mobilis) ΔldhA ΔfrdA φ80attB::PL-ter (Terponema denticola DSM14222) λattB::PL-crt (Clostridium acetobutylicum DSM792) ΔadhE::φ80attB::PL-phaA (Cupriavidus necator)-hbd (C. acetobutylicum DSM792) PL-atoDABD Δpta | >10 | 480 |
| Itaconic acid | BW25113(DE3) Δpta ΔldhA (pKV-CGA) | BW25113 (E. coli K-12 F– λ– rph-1Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) Δ(rhaD-rhaB)568 hsdR514)(DE3) Δpta ΔldhA; vector-based overexpression of cadA (opt, Aspergillus terreus), acnA (opt, Corynebacterium glutamicum), and gltA (opt, C. glutamicum) | 0.69 | 481 |
| SO11 (pLysS, pETHis-cad, pRSF-acnB) | SO02 (BW25113(DE3) icd); vector-based overexpression of cad (A. terreus) and acnB | 4.34 | 482 | |
| Adipic acid | AA7 (pTrc-ter-paaJ, pZS*27ptb-buk1-paaH1-ech) | QZ1111 (MG1655 ΔptsG ΔpoxB Δpta ΔsdhA ΔiclR); vector-based overexpression of paaJ (E. coli), paaH1 (Ralstonia eutropha H16), ech (R. eutropha H16), ter (Euglena gracilis), ptb (C. acetobutylicum), and buk1 (C. acetobutylicum) | 0.000639 | 324 |
| Ethanol | KO12 | E. coli W pfl+ pfl::(pdc (Z. mobilis)-adhB (Z. mobilis)-CmR) (Selected for high CmR (600 ng/liter) and hyperexpressive for pdc and adh) frd recA; | 54 | 483, 484 |
| Isopropanol | TA11 (pTA39, pTA36) | ATCC 11303 (E. coli B) lacIq TcR; vector-based overexpression of thl (C. acetobutylicum), adc (C. acetobutylicum), atoA (E. coli), atoD (E. coli), and adh (Clostridium beijerinckii) | 4.9 | 315 |
| Same as above. Highest titer achieved by gas stripping recovery method | 143 | 316 | ||
| 1,2-Propanediol | AG1 (pNEA30) | AG1 (F- λ. endA1 hsdR17 [rK−, mK+] supE44 thi-1 recA1 gyrA96 relA1); vector-based overexpression of mgs and gldA | 0.7 | 485 |
| ‘Evolved E. coli tpiArc’ Ptrc16-gapA (pJB137-PgapA-ppsA) | ‘Evolved E. coli tpiArc’ (MG1655 lpdfbr (low sensitivity to NADH) tpiArc (reconstructed tpiA) ΔpflAB ΔadhE ΔldhA::CmR ΔgloA Δald ΔaldB Δedd ΔarcA Δndh evolved under microaerobic conditions) Ptrc16-gapA; vector-based overexpression of ppsA | 3.7 | 486 | |
| 1,3-Propanediol | FMP′ 1.5gapA ΔmgsA (pSYCO106) | FM5 glpK gldA ndh pstHIcrr galP-Ptrc glk-Ptrc* arcA edd gapA-P1.5 mgsA; vector-based overexpression of DAR1 (Saccharomyces cerevisiae), GPP2 (S. cerevisiae), dhaB1 (Klebsiella pneumoniae), dhaB2 (K. pneumoniae), dhaB3 (K. pneumoniae), dhaX (K. pneumoniae) | 135.3 | 487 |
| 1-Butanol | JCL187 (pJCL17, pJCL60) | BW25113 F′ [traD36 proAB+ lacIq ZΔM15 TcR] ΔadhE, ΔldhA, ΔfrdBC, Δfnr, Δpta; vector-based overexpression of atoB (E. coli), adhE2 (C. acetobutylicum), crt (C. acetobutylicum), bcd (C. acetobutylicum), etfA (C. acetobutylicum), etfB (C. acetobutylicum), and hbd (C. acetobutylicum) | 0.552 | 334 |
| JCL299 (pEL11, pIM8, pCS138) | JCL299 (BW25113 F′ [traD36 proAB+ lacIq ZΔM15 TcR] ΔadhE ΔldhA ΔfrdBC Δpta); vector-based overexpression of atoB (E. coli), adhE2 (C. acetobutylicum), crt (C. acetobutylicum), hbd (C. acetobutylicum), ter (Treponema denticola), and fdh (Candida boidinii) | 30 | 488 | |
| 1,4-Butanediol | ECKh-422 (pZS*13-sucCD-sucD-4hbd/sucA, pZE23S-025B-34) | MG1655 lacIq ΔadhE ΔldhA ΔpflB ΔlpdA::lpdD354K (K. pneumonia) Δmdh ΔarcA gltAR163L SmR; vector-based overexpression of sucC (E. coli), sucD (E. coli), sucD (Porphyromonas gingivalis W83),4hbd (P. gingivalis W83), sucA (Mycobacterium bovis), cat2 (P. gingivalis W83), and 025B (aldehyde dehydrogenase, C. beijerinckii) | 18 | 27 |
| 1-Hexanol | JCL299 (pEL11, pEL102, pCS138) | JCL299; vector-based overexpression of atoB (E. coli), adhE2 (C. acetobutylicum), crt (C. acetobutylicum), hbd (C. acetobutylicum), bktB (R. eutropha), and fdh (C. boidinii) | 0.047 | 336 |
| Ethylene | DH5α (pEFE30) | DH5α; ethylene-forming enzyme (EFE) gene(Pseudomonas syringae pv. phaseolicola PK2) | Detected | 363 |
| Ectoine | DH5α (pAKECT1) | DH5α; vector-based overexpression of lysC (C. glutamicum MH20-22B), ectA (Marinococcus halophilus), ectB (M. halophilus), and ectC (M. halophilus) | 57 mg/g DCW | 489 |
| 2-Pentanone | JCL299 (pDK69 and pEL142) | JCL299; vector-based overexpression of ter (T. denticola), crt (C. acetobutylicum), hbd (C. acetobutylicum), bktB (R. eutropha), pcaI (Pseudomonas putida), pcaJ (P. putida), and adc (C. acetobutylicum) | 0.24 | 333 |
| Amino acids and derivatives | ||||
| l-Serine | YF-7 (pYF-1) | DH5α ΔsdaA ΔiclR ΔarcA ΔaceB; vector-based overexpression of serAfbr, serB, and serC | 8.3 | 490 |
| l-Alanine | ALS887 (pTrc99A-alaD) | CGSC 6162 (DC80 aceF10 fadR200 tyrT5(AS) adhE80 mel-1) aceF ldhA; vector-based overexpression of alaD (Bacillus sphaericus) | 32 | 491 |
| l-Valine | VAMF (pKBRilvBNCED, pTrc184ygaZHlrp) | W3110 ΔlacI ilvGatt ::Ptac ilvBatt::Ptac ilvHG41A,C50T ΔilvA ΔpanB ΔleuA ΔaceF Δmdh ΔpfkA::KmR; vector-based overexpression of ilvB, ilvN, ilvC, ilvE, ilvD, ygaZ, ygaH, and lrp | 7.55 | 22 |
| WLA (pKBRilvBNmutCED, pTrc184ygaZHlrp) | E. coli W ΔlacI ΔilvA; vector-based overexpression of ilvB, ilvNfbr, ilvC, ilvE, ilvD, ygaZ, ygaH, and lrp | 60.7 | 24 | |
| l-Threonine | TH28C (pBRThrABCR3) | WL3110 (W3110 ΔlacI) thrAC1034T lysCC1055T Pthr::Ptac ΔlysA ΔmetA ilvAC290T Δtdh ΔiclR Pppc::Ptrc ΔtdcC Pacs::CmR-Ptrc; vector-based overexpression of thrAC1034T, thrB, thrC, rhtA, rhtB, and thtC | 82.4 | 25 |
| l-Isoleucine | ILE03 (pBRthrABCygaZH, pTacilvAIH) | TH20 (W3110 ΔlacI, thrAC1034T, lysCC1-55T, Pthr::Ptac, ΔlysA, ΔmetA, ilvAC290T, Δtdh, ΔiclR, Pppc::Ptrc) ilvAC1339T, G1341T, C1351G, T1352C (fbr), PilvEDA::Ptrc, PilvC::Ptrc, Plrp::Ptrc; vector-based overexpression of thrAC1034T, thrB, thrC, ygaZ, ygaH, ilvAfbr , ilvI, and ilvH | 9.46 | 21 |
| l-Homoalanine | ATCC 98082 ΔrhtA (pZElac_ilvABS_GDH) | Threonine overproducer ATCC 98082 (VNIIgenetika 472T23; ilvA442 supE spot thrC1010 Sac+ thrR) ΔrhtA; vector-based overexpression of ilvA (Bacillus subtilis) and gdhA (K92V, T195S) | 5.4 | 19 |
| l-Lysine | NT1003 (pSC101-ppc-pntB-aspA) | NT1003 (l-Lysine strain, ΔMet ΔThr, CCTCC No. M 2013239); vector-based overexpression of ppc, pntB, and aspA | 134.9 | 492 |
| l-Phenylalanine | AR-G91 | BW25113(DE3) tyrR::PT7-aroFfbr-pheAfbr | 1.255 | 313 |
| YP1617 (pACYA177 harboring aroF and pheA) | YP1617 (l-tyrosine auxotroph, CCTCC No. M 2013320); vector-based overexpression of aroF and pheA | 56.2 | 493 | |
| l-Tyrosine | AR-G2 | BW25113(DE3) tyrR::PT7-aroFfbr-tyrAfbr | 0.852 | 313 |
| S17-1 (pTyr-a, pTyr-b) | S17-1; vector-based overexpression of tyrA, tyrC, aroG, aroK, aroF, ppsA, tktA, and anti-csrA and anti-tyrR sRNAs | 21.9 | 185 | |
| l-Tryptophan | NST100 | E. coli K-12 | ∼1.4 | 494 |
| Dpta/mtr-Y (pSV-709, pMEL03-Y) | TRTH0709 (E. coli K-12 ΔtrpR Δtna; vector-based overexpression of aroGfbr, trpEfbr, trpD, trpC, trpB, trpA, and serA) Δpta Δmtr; vector-based overexpression of tktA, ppsA, and yddG | 46.68 | 20 | |
| 3-Aminopropionic acid | CWF4NA2 (pTac15kPTA, pSynPPC7) | W3110 ΔiclR ΔfumC ΔfumA ΔfumB ΔptsG ΔlacI PaspA::Ptrc Pacs::Ptrc; vector-based overexpression of panD (C. glutamicum ATCC 13032), aspA, and ppc | 32.3 | 495 |
| Cadaverine | XQ56 (p15CadA) | WL3110 - ΔspeE ΔspeG ΔygjG ΔpuuPA PdapA::Ptrc; vector-based overexpression of cadA | 9.61 | 312 |
| Putrescine | XQ52 (p15SpeC) | W3110 ΔspeE ΔspeG ΔargI ΔpuuPA ΔrpoS PargECBH::Ptrc PspeF-potE::Ptrc PargD::Ptrc PspeC::Ptrc; vector-based overexpression of speC | 24.2 | 311 |
| 1-Propanol | CRS-BuOH 23 (pCS49, pSA62, pSA55I) | JCL16 (BW25113 F′ [traD36 proAB+ lacIq ZΔM15 TcR]) ΔmetA Δtdh ΔilvB ΔilvI ΔadhE: vector-based overexpression of thrAfbr (E. coli ATCC 21277) thrB (ATCC 21277), thrC (ATCC 21277), ilvA, leuA, leuB, leuC, leuD, kivd (Lactococcus lactis), and ADH2 (S. cerevisiae); The strain also produces equimolar ratio of 1-butanol | ∼1 | 496 |
| PRO2 (pBRthrABC-tac-cimA-tac-ackA, pTacDA-tac-adhEmut) | W3110 strain ΔlacI thrAC1034T lysCC1055T Pthr::Ptac ΔlysA ΔmetA ilvAC1139T, G1341T, C1351G, T1352C)T ΔtdhA ΔiclR Pppc::Ptrc ΔilvIH ΔilvBN ΔrpoS; vector-based overexpression of thrA, thrB, thrC, cimA (Methanocaldococcus jannaschii), ackA, atoD, atoA, and adhEmut | 10.8 | 314 | |
| Acetoin | XL1-Blue (pAL306) | XL1-Blue; vector-based overexpression of alsS (B. subtilis) and alsD (Aeromonas hydrophila) | 21 | 497 |
| 2,3-Butanediol | BL21(DE3) (p18COR) | BL21(DE3); vector-based overexpression of budA (co, K. pneumonia) and budC (co, K. pneumoniae) | 1.04 | 498 |
| 2-Butanone | AL1458 (pAL544, pAL597) | MG1655 lacIq ; vector-based overexpression of alsS (B. subtilis), alsD (Enterobacter aerogenes), adh (Leuconostoc pseudomesenteroides), dhaB1 (Klebsiella pneumonia MGH 78578), dhaB2 (K. pneumonia MGH 78578), dhaB3 (K. pneumonia MGH 78578), orfz (K. pneumonia MGH 78578), and orf2B (K. pneumonia MGH 78578) | 0.151 | 368 |
| Isobutyl acetate | JCL260 (pAL603, pAL676) | JCL260 (BW25113 F′ [traD36 proAB+ lacIq ZΔM15 TcR] ΔadhE ΔldhA ΔfrdBC Δfnr Δpta ΔpflB); vector-based overexpression of alsS (B. subtilis), ilvC, ilvD, kivd (L. lactis), adhA (L. lactis), and ATF1 (S. cerevisiae) | 17.2 | 332 |
| Isobutanol | JCL260 (pSA55, pSA69) | JCL16 ΔadhE ΔldhA ΔfrdBC Δfnr Δpta ΔpflB; vector-based overexpression of PDC6 (S. cerevisiae), ADH2 (S. cerevisiae), kivd (L. lactis), ilvC, ilvD, and alsS (B. subtilis) | 22 | 9 |
| JCL260 (pSA65, pSA69) | JCL260; vector-based overexpression of kivd (L. lactis), adhA (L. lactis), ilvC, ilvD, and alsS (B. subtilis) | 50.8 | 335 | |
| 1-Heptanol | ATCC 98082 ΔrhtA (pZS_thrO, pZE_LeuA*BCDKA6, a medium copy plasmid harboring ilvA and leuAfbr) | ATCC 98082 ΔrhtA; vector-based overexpression of thrAG433R (fbr), thrB, thrC, leuAG462D (fbr), H97A, S139G, N167G, P169A, leuB, leuC, leuD, kivd (L. lactis), ADH6 (S. cerevisiae), leuAfbr, and ilvA (B. subtilis) | 0.0802 | 14 |
| 1-Octanol | 0.002 | |||
| 3-Phenylpropanol | ATCC31884 (pZE_LeuA*BCDKA6) | ATCC 31884 (phenylalanine overproducer); vector-based overexpression of leuAG462D (fbr), H97A, S139G, N167G, P169A, leuB, leuC, leuD, kivd (L. lactis), ADH6 (S. cerevisiae) | 0.0041 | |
| 2-Methyl-1-butanol | CRS22 (pAFC3, pAFC46, pCS49) | BW25113 F′ [proAB+ lacIq ZΔM15 Tn10 (TcR)] ΔmetA Δtdh; vector-based overexpression of ilvG (Salmonella enterica serovar Typhimurium), ilvM (S. enterica serovar Typhimurium), ilvC (S. enterica serovar Typhimurium), ilvD (S. typhimurium), ilvA (C. glutamicum), thrABC | 1.25 | 11 |
| 3-Methyl-1-butanol | AL2 (pIAA11, pIAA13, pIAA16) | Second round NTG-created mutant of JCL16; vector-based overexpression of alsS (Bacillus subtilis), ilvC, ilvD, kivd (L. lactis), ADH2 (S. cerevisiae), leuAfbr, leuB, leuC, and leu D | 9.5 | 351 |
| 3-Methyl-1-pentanol | ATCC 98082 (pZS_thrO, pZE_LeuABCDKA6, pZAlac_tdcBilvGMCD) | ATCC 98082 ΔilvE ΔtyrB; vector-based overexpression of thrAG433R (fbr), thrB, thrC, leuAG462D (fbr),S139G, leuB, leuC, leuD, kivdV461A, F381L (L. lactis), ADH6 (S. cerevisiae), tdcB, ilvG, ilvM, ilvC, and ilvD | 0.7935 | 10 |
| 4-Methyl-1-pentanol | Same as 3-methyl-1-pentanol | Same as 3-methyl-1-pentanol except LeuAG462D, S139G, H97L and kivdV461A, F381L | 0.2024 | |
| 4-Methyl-1-hexanol | Same as 3-methyl-1-pentanol | Same as 3-methyl-1-pentanol except LeuAG462D, S139G, H97A, N167A and kivdV461A, F381L | 0.0573 | |
| 5-Methyl-1-heptanol | Same as 3-methyl-1-pentanol | 0.022 | ||
| 1-Pentanol | Same as 3-methyl-1-pentanol | Same as 3-methyl-1-pentanol except LeuAG462D and kivdV461A, M538A | 0.7505 | |
| Catechol | AB2834 (pKD136, pKD9.069A) | AB2834 ( F–tsx-352 glnV42(AS) λ– aroE353 malT352(λR); vector-based overexpression of tkt, aroF, aroB, aroZ (K. pneumoniae), and aroY (K. pneumoniae), | 2.0 | 317 |
| W3110 9923 (pJLBaroGfbr tktA, pTrc-ant3 | W3110 trpD9923; vector-based overexpression of tktA, aroGfbr, antA (Peudomonas aeruginosa), antB (P. aeruginosa), and antC (P. aeruginosa) | 4.47 | 326 | |
| cis,cis-Muconic acid | WN1 (pWN2.248) | KL7 (AB2834 serA::aroB-aroZ) lacZ::tktA-aroZ; vector-based overexpression of catA, aroY, serA, and aroFfbr | 36.8 | 369 |
| p-Hydroxybenzoic acid | JB161 (pJB2.274) | D2704 ((ΔtrpC-E)trpR tyrA Δ(pheA)) serA:: Ptac-aroA-aroL-aroC-aroB-KmR; vector-based overexpression of tktA, ubiC, serA, aroFfbr | 12 | 318 |
| p-Aminobenzoic acid (PABA) | PABA-25 | acs::PT7lac-pabAB (opt, Corynebacterium efficiens) ascF::PT7lac-pabAB (opt, C. efficiens) mtlA::PT7lac-pabAB (opt, C. efficiens) pflBA::PT7lac-pabC tyrR::PT7lac-aroFfbr | 4.8 | 328 |
| 4-Hydroxycoumarin | Strain D (pZE-EP-APTA and pCS-PS) | BW25113 F′ [proAB+ lacIq ZΔM15 Tn10 (TcR)]; vector-based overexpression of luc, entC, pfpchB, aroL, ppsA, tktA, aroGfbr, pqsD, and sdgA | 0.4831 | 327 |
| 2-Phenylethanol (PE) | JCL16 (pSA55) | JCL16; vector-based overexpression of kivd (L. lactis) | 0.149 | 9 |
| DCU4 (pCL1920-pheAfbr-aroFwt, pUC19-adh1-kdc) | DH5α; vector-based overexpression of pheAfbr , aroF, adh1 (S. cerevisiae S288c), and kdc (Pichia pastoris GS115) | 0.285 | 331 | |
| PAR-105 | BW25113(DE3) tyrR::PT7-aroFfbr-pheAfbr mtlA::PT7-ipdC (Azospirillum brasilense NBRC102289) acs::PT7-yahK ΔfeaB ΔtyrA | 0.842 | 26 | |
| 2-(4-Hydroxyphenyl)ethanol (4HPE) | PAR-104 | BW25113(DE3) tyrR::PT7-aroFfbr-tyrAfbr mtlA::PT7-ipdC (A. brasilense NBRC102289) acs::PT7-yahK ΔfeaB ΔpheA | 1.14 | |
| Phenyllactic acid (PLA) | PAR-58 | BW25113(DE3) tyrR::PT7-aroFfbr-pheAfbr acs::PT7-ldhA (C. necator JCM20644) mtlA:: PT7-ldhA (C. necator JCM20644) | 1.00 | |
| 4-Hydroxyphenyllactic acid (4HPLA) | PAR-3 | BW25113(DE3) tyrR::PT7-aroFfbr-tyrAfbr; acs::PT7-ldhA (C. necator JCM20644) | 1.48 | |
| Phenylacetic acid (PAA) | PAR-100 | BW25113(DE3) tyrR::PT7-aroFfbr-pheAfbr mtlA::PT7-ipdC (A. brasilense NBRC102289) acs::PT7-feaB ΔtyrA | 1.20 | |
| 4-Hydroxyphenylacetic acid (4HPAA) | PAR-102 | BW25113(DE3) tyrR::PT7-aroFfbr-tyrAfb mtlA::PT7-ipdC (A. brasilense NBRC102289) acs::PT7-feaB ΔyahK ΔpheA | 1.12 | |
| Styrene | NST74 (pSpal2At, pTfdc1Sc) | NST74 (aroH367 tyrR366 tna-2 lacY5 aroF394 (fbr) malT384 pheA101 (fbr) pheO352 aroG397 (fbr)); vector-based overexpression of PAL2 (Arabidopsis thaliana) and FDC1 (S. cerevisiae) | 0.26 | 329 |
| Styrene oxide | N74dASO (pTpal-fdc, pTKstyAB) | NST74 ΔtyrA; vector-based overexpression of FDC1 (S. cerevisiae), PAL2 (A. thaliana), styA (P. putida S12), and styB (P. putida S12) | 1.32 | 330 |
| Phenylethanethiol | N74dAPED (pTal-fdc, pTKnah) | NST74 ΔtyrA; vector-based overexpression of FDC1 (S. cerevisiae), PAL2 (A. thaliana), and nahAaAbAcAd (P. putida NCBI 9816) | 1.23 | |
| Phenol | PheBL21 (pTyr-a-TPL, pTyr-b) | BL21(DE3); vector-based overexpression of aroK, tyrC, aroGA146N, and tyrAA354V, M53I, tpl (Pasteurella multocida 36950), ppsA, tktA, and aroF; vector-based regulated expression of anti-csrA synthetic sRNA, anti-tyrR synthetic sRNA | 3.79 | 319 |
| Fatty acids and derivatives | ||||
| Free fatty acids (FFA) | RB03 (pTHfadBA.fadM-) | MG1655 fadR atoCc ΔarcA Δcrp::crp* ΔadhE Δpta ΔfrdA ΔfucO ΔyqhD ΔfadD; vector-based overexpression of fadB, fadA, and fadM | ∼7 | 12 |
| Fatty acid ethyl esters (FAEEs) | TOP10 (pMicrodiesel) | TOP10; vector-based overexpression of pdc (Z. mobilis), adhB (Z. mobilis), and atfA (Acinetobacter baylyi) | 1.28 | 322 |
| Heptadecene (and other C13–C17 alkanes and alkenes) | MG1665 (pCL1920 derivative harboring PCC7942_orf1594, pACYC derivative harboring PCC7942_orf1593) | MG1665; vector-based overexpression of PCC7942_orf1593 (Synechocystis elongates) and PCC7942_orf1594 (S. elongates) | 0.042 | 321 |
| Short-chain alkanes (C10-C12) | GAS3 (pTacCer1FadD, pTrcAcR′TesA(L109P)) | W3110 ΔfadE ΔfadR PfadD::Ptrc; vector-based overexpression of CER1 (A. thaliana), fadD, acr (C. acetobutylicum), and tesA (mutant leaderless version) | 0.580 | 15 |
| Propane | ProFΔA (pET-TPC4, pCDF-ADO, pACYC-petF-fpr) | BL21(DE3) ΔyqhD Δahr; vector-based overexpression of tes4 (Bacteroides fragilis), sfp (B. subtilis), car (Mycobacterium marinum), ADO (Prochlorococcus marinus), petF (Synechocystis sp. PCC 6803), and fpr (E. coli) | 0.032 | 18 |
| Secondary metabolites | ||||
| Isoprene | Rosetta (pJF-kIspS, pET28 derivative harboring hmgS, hmgR, atoB, fni, mk, pmk, and pmd) | Rosetta; Vector-based overexpression of kIspS (opt, Peuraria montana), hmgS (Enterococcus faecalis), hmgR (E. faecalis), atoB (E. coli), fni (Streptococcus pneumoniae), mk (S. pneumoniae), pmk (S. pneumoniae), and pmd (S. pneumoniae) | 0.32 | 373 |
| Geraniol | GEOL(0.5K) (pSNAK, pT-GPES(0.5K)) | MGΔyjgB (MG1655 ΔyjgB); vector-based fine-controlled overexpression of mvaK1 (Staphylococcus pneumoniae), mvaK2 (S. pneumoniae), mvaD (S. pneumoniae), idi (E. coli), mvaE (E. faecalis), mvaS (E. faecalis), ispA (mutated, E. coli), and tGES (truncated, Ocimum basilicum) | 1.119 | 499 |
| Limonene | 1pC (pJBEI-6409) | DH1; vector-based overexpression of atoB (E. coli), HMGS (opt, Staphylococcus aureus) and HMGR (opt, S. aureus), MK (Saccharomyces cerevisiae), PMK (S. cerevisiae), PMD (S. cerevisiae), idi (E. coli), trGPPS (opt, truncated, Abies grandis), and LS (opt, Mentha spicata) | 0.435 | 367 |
| Perillyl alcohol | 1pA+P (pJBEI-6410, pBbB8k-P450) | DH1; vector-based overexpression of atoB (E. coli), HMGS (opt, Staphylococcus aureus) and HMGR (opt, S. aureus), MK (Saccharomyces cerevisiae), PMK (S. cerevisiae), PMD (S. cerevisiae), idi (E. coli), trGPPS (opt, truncated, Abies grandis), LS (opt, Mentha spicata), ahpG (opt, Mycobacterium HXN 1500), ahpH (opt, M. HXN 1500), and ahpI (opt, M. HXN 1500) | 0.1 | |
| α-Farnesene | ispALaFS-NA (pTispALaFS and pSNA) | DH5α; vector-based overexpression of FS (opt, Malus x domestica)-(GGGGS)2-ispA (E. coli), mvaE (E. faecalis), mvaS (E. faecalis), mvaK1 (S. pneumoniae), mvaK2 (S. pneumoniae), mvaD (S. pneumoniae), and idi (E. coli) | 0.380 | 500 |
| Amorphadiene (Artemisinic acid precursor) | DH10B (pMevT, pMBIS, pADS) | DH10B; vector-based overexpression of atoB (E. coli), ERG13 (S. cerevisiae), tHMGR (truncated, S. cerevisiae) ERG12 (S. cerevisiae), ERG8 (S. cerevisiae), MVD1 (S. cerevisiae), idi (E. coli), ispA (E. coli), and ADS (Artemisia annua L) | 0.024 | 372 |
| B86 (pAM52, pMBIS, pADS) | DH1; vector-based overexpression of atoB (E. coli), mvaS (S. aureus), mvaA (S. aureus), ERG12 (S. cerevisiae), ERG8 (S. cerevisiae), MVD1 (S. cerevisiae), idi (E. coli), ispA (E. coli), and ADS (Artemisia annua L) | 27.4 | 473 | |
| Artemisinic acid | DH1 (pAM92, pCWori-A13AMO-aaCPRct) | DH1; vector-based overexpression of A13AMO (opt, N-terminal transmembrane domain engineering on CYP71AV1, A. annua), aaCPRct (opt, A. annua), atoB (E. coli), ERG13 (S. cerevisiae), tHMGR (truncated, S. cerevisiae) ERG12 (S. cerevisiae), ERG8 (S. cerevisiae), MVD1 (S. cerevisiae), idi (E. coli), ispA (E. coli), and ADS (Artemisia annua L) | 0.1 | 474 |
| Taxadiene | Strain #26 (EDE3Ch1TrcMEPp5T7TG) (pSC101 derivative harboring TS and GGPPS) | K-12 MG1655 ΔrecA ΔendA (DE3) Ptrc-dxs-idi-ispDF; vector-based overexpression of TS (Taxus brevifolia) and GGPPS (Taxus canadensis) | 1 | 346 |
| Taxadien-5α-ol | Strain #26 (EDE3Ch1TrcMEPp5T7TG)-At24T5αOH-tTCPR (pSC101 derivative harboring TS and GGPPS, p10T7At24T5 αOH-tTCPR) | K-12 MG1655 ΔrecA ΔendA (DE3) Ptrc-dxs-idi-ispDF; vector-based overexpression of TS (Taxus brevifolia), GGPPS (Taxus canadensis), At24T5αOH (truncation at 24th amino acid residue, T5αOH, Taxus cuspidate), and tTCPR (74 amino acids truncated TCPR, T. cuspidate) | 0.6 | |
| Levopimaradiene | MG1655 ΔendA ΔrecA (p10TrcMEP, pTrcGGPPS*-CRT) | MG1655 ΔendA ΔrecA; vector-based overexpression of dxs, idi, ispD, ispF, GGPPSS239C, G295D (Sequences for N-terminal 98 amino acids removed, T. canadensis) and LPSM593I, Y700F (Sequences for N-terminal 40 amino acids removed, Ginkgo biloba) | 0.7 | 423 |
| Phytoene | JM101 (pACCRT-EB, pHP11) | JM101; vector-based overexpression of crtE (Erwinia uredovora), crtB (E. uredovora), and ipp (Haematococcus pluvialis) | < 0.005 | 501 |
| Lycopene | JM101 (pACCRT-EIB, pHP11) | JM101; vector-based overexpression of crtE (Erwinia uredovora), crtB (E. uredovora), crtI (E. uredovora), and ipp (Haematococcus pluvialis) | < 0.01 | |
| BW18302 (p2IDI, pPSG184) | BW18302 (F– λ– lacX74 glnL2001); vector-based, controlled expression of glnAp2-idi, glnAp2-pps | 0.16 | 343 | |
| LYC010 | CAR001 (ATCC 8739 ldhA::M1-93::crtEXYIB (Pantoea agglomerans)::ldhA M1-37::dxs M1-37::idi) ΔcrtXY M1-46::sucAB M1-46::talB M1-46::sdhABCD RBSL9::crtE RBSL12::dxs RBSL7::idi | 3.53 | 323 | |
| β-Carotene | JM101 (pACCAR16ΔcrtX, pHP11) | JM101; vector-based overexpression of crtE (E. uredovora), crtB (E. uredovora), crtI (E. uredovora), crtY (E. uredovora), and ipp (Haematococcus pluvialis) | ∼0.01 | 501 |
| Oxytetracycline | BAP1 (pDCS11, pMRH08) | BAP1 (BL21(DE3) ΔprpRBCD::PT7-sfp (B. subtilis) PT7-prpE); vector-based overexpression of rpoN and oxytetracycline biosynthetic operons (oxyTA1ABCDE, oxyIHGF, oxyJKLMLO, oxtRQP, and oxyST; Streptomyces rimosus) | 0.002 | 404 |
| Resveratrol | BW27784 (pUCo-Vvsts-At4cl1) | BW27784 (F– Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ– Δ(araH-araF)570(::FRT) ΔaraEp-532::FRT φPcp18-araE553 Δ(rhaD-rhaB)568 hsdR514; vector-based overexpression of sts (V. venifera) and 4cl-1 (A. thaliana) | 2.3 | 407 |
| Violacein | Vio-4 (pBvioABCE-Km) | MG1655 ΔtrpR::FRT ΔtnaA::FRT ΔsdaA::FRT Δlac::Ptac-aroFBL-FRT ΔtrpL trpEfbr Δgal::Ptac-tktA-FRT Δxyl::Ptac-serAfbr-FRT Δfuc::Ptac-vioD-FRT; vector-based overexpression of vioA (Chromobacterium violaceum), vioB (C. violaceum), vioC (C. violaceum), and vioE (C. violaceum) | 0.71 | 325 |
| Deoxyviolacein | dVio-6 (pBvioABCE-Km) | MG1655 ΔtrpR::FRT ΔtnaA::FRT ΔsdaA::FRT Δlac::Ptac-aroFBL-FRT ΔtrpL trpEfbr Δgal::Ptac-tktA-FRT Δxyl::Ptac-serAfbr-FRT; vector-based overexpression of vioA (C. violaceum), vioB (C. violaceum), vioC (C. violaceum), and vioE (C. violaceum) | 0.32 | |
| dVio-8 (pBvioABCE-Km) | MG1655 ΔtrpR::FRT ΔtnaA::FRT ΔsdaA::FRT ΔΔlac::Ptac-aroFBL-FRT ΔtrpL trpEfbr Δgal::Ptac-tktA-FRT Δxyl::Ptac-serAfbr-FRT; vector-based overexpression of vioA (C. violaceum), vioB (C. violaceum), vioC (C. violaceum), and vioE (C. violaceum) | 1.6 | 502 | |
| Pinocembrin | DH5α (pQlinkN_hPAL_h4CL_hCHS_lCHIopt) | DH5α; vector-based overexpression of hPAL (A. thaliana), h4CL (S. coelicolor), hCHS (A. thaliana), and lCHI (opt, A. thaliana) | 0.024 | 436 |
| Strain 4 (pRSF-aroF-pheA, pCDF-PAL-4CL, pET-CHS-CHI, pACYC-matB-matC) | BL21 Star (DE3); vector-based overexpression of aroF, pheAfbr, PAL (Rhodotorula glutinis), 4CL (Petroselinum crispum), CHS (Petunia x hybrida), CHI (Medicago sativa), matB (Rizobium trifolii), and matC (R. trifolii). | 0.040 | 503 | |
| Polymers | ||||
| Polylactic acid (PLA) | JLX10 (pPs619C1400-CpPCT532 | XL1-Blue ΔackA PldhA::Ptrc Δppc ΔadhE Pacs::Ptrc; vector-based expression of phaC1E130D, S325T, S477R, Q481M (Pseudomonas sp. MBEL 6-19), and pctA243T (one silent mutation: A1200G; Clostridium propionicum) | 11 wt% | 338 |
| P(3HB-co-lactate) | JLX10 (pMCS103CnAB, pPs619C1310-CpPCT540) | XL1-Blue ΔackA PldhA::Ptrc Δppc ΔadhE Pacs::Ptrc; vector-based expression of phaAB (C. neactor), phaC1E130D, S477F, Q481K (Pseudomonas sp. MBEL 6-19), and pctV193A (four silent mutations: T78C, T669C, A1125G, and T1158C; C. propionicum) | 46 wt% | |
| Proteins | ||||
| Spider silk protein (dragline, 96mer) | BL21(DE3) (pSH96, pTetgly2-glyAn) | BL21(DE3); vector-based overexpression of glyV, glyX, glyY, glyA, and 96 repeats of a spider dragline silk protein monomer; glycine-rich (44.9%) and ultra-high molecular weight protein (284.9 kDa) production, | 0.5 | 29 |
| Human leptin | BL21(DE3) (pAC104CysK, pEDOb5) | BL21(DE3); vector-based overexpression of cysK (E. coli) and human obese gene | 51 | 30 |
| Interleukin-12 β chain | BL21(DE3) (pAC104CysK, PEDIL-12p40) | BL21(DE3); vector-based overexpression of cysK (E. coli) and human IL-12p40 gene | 24 | |
| Miscellaneous | ||||
| D-1,2,4-Butanetriol | DH5α (pWN6.186A) | DH5α; vector-based overexpression of mdlC (P. putida); from d-xylonic acid | 1.6 | 365 |
| BL21(DE3) (30a-mtkABM. petroleiphilum-sucD-4hbD, 184-abfT-2-adhE2 | BL21(DE3); vector-based overexpression of mtkA (Methylibium petroleiphilum PM1), mtkB (M. petroleiphilum PM1), sucD (P. gingivalis W38), 4hbD (P. gingivalis W38), abfT-2 (P. gingivalis W38), adhE2 (C. acetobutylicum DSM1731); from glucose | 0.00000012 | 366 | |
| Monoethylene glycol (MEG) | EWE3 (pXdh-YqhD) | W3110 ΔxylA (DE3); vector-based overexpression of xdh and yqhD; from D-xylose | 11.69 | 364 |
| d-Glucaric acid | JT9 (pJD727, pJD765) | BL21 Star (DE3) (F– ompT hsdSB (rB–mB–) gal dcm rne131 (DE3)); vector-based overexpression of ino1 (S. cerevisiae), MIOX (mouse), udh (P. syringae), and a scaffold composed of one GBD domain, four SH3 domains, and four PDZ domains | 2.5 | 371 |
| Riboflavin | RF05S-M40 (p20C-EC10) | MG1655 Δpgi Δedd Δeda Ptrc-acs; vector-based overexpression of ribA (B. subtilis), ribB (B. subtilis), ribD (B. subtilis), ribE (B. subtilis), and ribC (B. subtilis) | 2.7 | 504 |
All overexpressed genes mentioned are derived from E. coli unless specified otherwise in parentheses.
Parental strains previously reported in other studies are indicated as strain names reported, with genotypes described in parentheses. For strains repetitively mentioned in this table, descriptions on genotypes are omitted after the first notation.
opt, codon-optimized; att, attenuation relieved; fbr, feedback-resistant; KmR, kanamycin resistance; TcR, tetracycline resistance; SmR, streptomycin resistance; -(GGGGS)2-, protein linker with amino acid sequence GGGGSGGGGS.
Engineering Native E. coli Metabolism
Before engineering E. coli as a host organism to produce various chemicals and materials of interest, it is important to engineer and upgrade its native metabolism. Here we present the strategies to engineer the native E. coli metabolism that have been designed with tools developed over the evolving field of systems metabolic engineering (Fig. 3 and Table 1). These strategies are indeed fundamental for the production of any candidate chemicals and should serve and be treated as the baseline approaches to strain development.
Engineering endogenous biosynthetic pathways for increasing precursor pools
In systems metabolic engineering, the exchange of native promoter with a stronger promoter for increased expression of native gene is a common strategy to increase metabolic flux toward a desired compound. For further improved expression, the genes can also be cloned in a multicopy plasmid. Here, the gene targets are often metabolic genes in central carbon metabolism (27, 309, 310), biosynthesis of amino acids (19, 20, 21, 22, 23, 24, 25, 311, 312, 313, 314, 315, 316, 317, 318, 319), and biosynthesis of fatty acids (12, 15, 16, 17, 18, 320, 321, 322) (Fig. 3 and Table 1).
Metabolic genes targeted for overexpression depend on metabolic pathways that the products of interest are derived from. The most common target genes for overexpression, however, are the ones from the central carbon metabolism, involving glycolysis, pentose phosphate pathway (PPP), tricarboxylic acid (TCA) cycle, and glyoxylate pathway (Fig. 4). Since all of the chemicals produced in systems metabolic engineering are derived from glucose and other renewable carbon sources, the enhancement of the central carbon metabolism, which provides essential key intermediates for all chemicals, is one of the most basic strategies in this field. Still, overexpression strategies of genes in the central carbon metabolism depend on the target product and key intermediates of the product chemicals.
Figure 4.
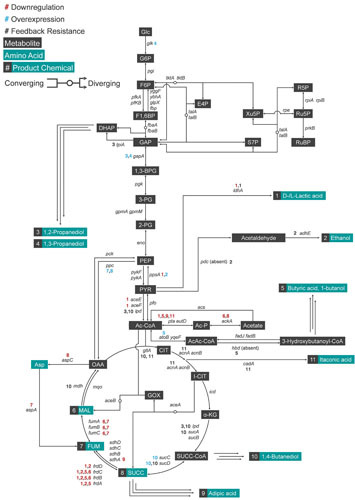
Examples of metabolic engineering strategies to produce chemicals of interest from central carbon metabolism. Target products included are (1) D- and L-lactic acid, (2) ethanol, (3) 1,2-propanediol, (4) 1,3-propanediol, (5) butyric acid and 1-butanol, (6) malic acid, (7) fumaric acid, (8) succinic acid, (9) adipic acid, (10) 1,4-butanediol, and (11) itaconic acid. Numbers labeled beside gene names indicate the products that targeted each gene for engineering during engineering. The colors of the numbers indicate modes of engineering: red, downregulation including knockout; blue, upregulation; black, all other miscellaneous modifications including feedback-release, heterologous gene introduction, and mutagenesis for modified enzyme activities. Convergence and divergence of metabolites are denoted by circular nodes, where some reactions are reversible. As an example for reversible reaction, F6P and GAP converge to form E4P and Xu5P; conversely, E4P and Xu5P converge to form F6P and GAP. For abbreviations, see Fig. 3.
In the TCA cycle, the increased influx of carbons through acetyl coenzyme A (acetyl-CoA) alone does not expand the pool of constituent chemicals such as oxaloacetate, malate, and fumarate (Fig. 4). Rather, the pool itself is fueled through a couple of direct routes from glycolytic intermediates phosphoenolpyruvate and pyruvate via the reductive branches. The production of chemicals that deplete TCA cycle metabolites thus often requires the refilling of the TCA intermediates. However, increasing TCA cycle flux should be taken with precaution since it can lead to unwanted acetate accumulation due to overflow, offsets the redox balance, and decreases product formation due to increased flux toward biomass. Increase of TCA flux can be achieved by overexpression of ppc (21, 25, 309, 310) and pck (310) to convert phosphoenolpyruvate to oxaloacetate (Figs. 3 and 4). In addition to elevating the TCA cycle metabolite levels, overexpression of sucA and sucB for converting α-ketoglutarate to succinyl-CoA (27, 323), sucC and sucD (27), and sdhA, sdhB, sdhC, and sdhD for increased reducing power supply (323) have been reported. Glyoxylate pathway genes aceA and aceB can be indirectly upregulated by deleting a global regulator iclR (25, 309, 312, 324).
The pentose phosphate pathway provides valuable precursors for essential chemicals in cells, including aromatic amino acids and nucleotides, in addition to providing reducing powers and energy (Fig. 4). Among the various metabolic genes involved in pentose phosphate pathway, the most frequent overexpression targets are tktA and tktB (20, 318, 319, 325, 326, 327) encoding transketolases, which produces erythrose 4-phosphate, an important precursor of aromatic metabolites, together with xylose 5-phosphate, from fructose 6-phosphate and glyceraldehyde 3-phosphate. In addition, talA and talB (323) encoding transaldolases were reported to be overexpressed.
During the production of amino acids and derivatives, desired products can be selectively produced either by enriching the precursor metabolites in central carbon metabolism or increasing the flux after the branch point metabolites (Figs. 3 and 5). Increasing fluxes toward precursors from central carbon metabolism such as phosphoenolpyruvate and pentose phosphate pathway (20, 318, 319, 325, 326, 327), and pyruvate (20, 319, 327) can lead to the enhanced production of amino acids and derivatives (Figs. 3–7).
Figure 5.
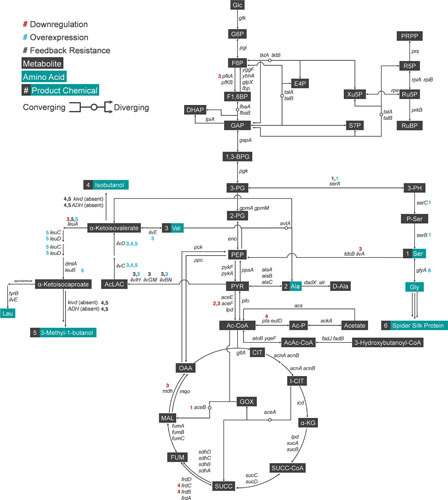
Examples of metabolic engineering strategies to produce chemicals of interest derived from glycolytic intermediates. Target products included are (1) L-serine, (2) L-alanine, (3) L-valine, (4) isobutanol, (5) 3-methyl-1-butanol, and (6) glycine-rich spider silk protein. The colors of the numbers indicate modes of engineering: red, expression downregulation including knockout; blue, expression upregulation; black, all other miscellaneous modifications including feedback-release, heterologous gene introduction, and mutagenesis for modified enzyme activities. Convergence and divergence of metabolites are denoted by circular nodes, where some reactions are reversible. As an example for reversible reaction, F6P and GAP converge to form E4P and Xu5P; conversely, E4P and Xu5P converge to form F6P and GAP. For abbreviations, see Fig. 3.
Figure 7.
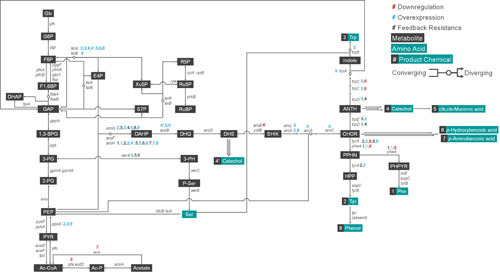
Examples of metabolic engineering strategies to produce chemicals of interest derived from PPP intermediates. Target products included are (1) L-phenylalanine, (2) L-tyrosine, (3) L-tryptophan, (4) catechol, (5) cis,cis-muconic acid, (6) p-hydroxybenzoic acid, (7) p-aminobenzoic acid, and (8) phenol. The colors of the numbers indicate modes of engineering: red, expression downregulation including knockout; blue, expression upregulation; black, all other miscellaneous modifications including feedback-release, heterologous gene introduction, and mutagenesis for modified enzyme activities. Convergence and divergence of metabolites are denoted by circular nodes, where some reactions are reversible. As an example for reversible reaction, F6P and GAP converge to form E4P and Xu5P; conversely, E4P and Xu5P converge to form F6P and GAP. For abbreviations, see Fig. 3.
Once the carbons from renewable carbon sources are converted to the precursor chemicals in the central carbon metabolism, the fluxes are further diverged into each specific amino acid and its derivatives. Since many of the amino acids share common upstream metabolic pathways, overproduction of a specific amino acid often requires the overexpression of metabolic genes downstream from the branching points (Figs. 3 and 5). For example, a branched amino acid L-valine is derived from pyruvate in the central carbon metabolism. Park et al. overexpressed ilvBN, which converts 2 molecules of pyruvate into a single acetolactate, with ilvC and ilvD, enhancing the carbon flux from pyruvate to α-ketoisovalerate, the branching point of three important pathways producing L-isoleucine, L-valine, and pantothenate (24). In addition, only a single route toward L-valine was reinforced by overexpressing ilvE, which directly converts α-ketoisovalerate into L-valine, to specifically produce L-valine in the engineered E. coli (Fig. 5) (24). In another example, another branched amino acid L-isoleucine is derived from a hydroxyl amino acid L-threonine, sharing a biosynthetic pathway from oxaloacetate to L-aspartate to L-threonine. The authors first increased the flux from L-aspartate to L-threonine by overexpression of thrA, thrB, and thrC genes in a plasmid (Fig. 6) (21). The carbon flux from L-threonine to the target product L-isoleucine was then increased together by overexpression of ilvA and ilvH in another plasmid, and ilvC and ilvEDA with its promoter exchanged (Fig. 6) (21).
Figure 6.
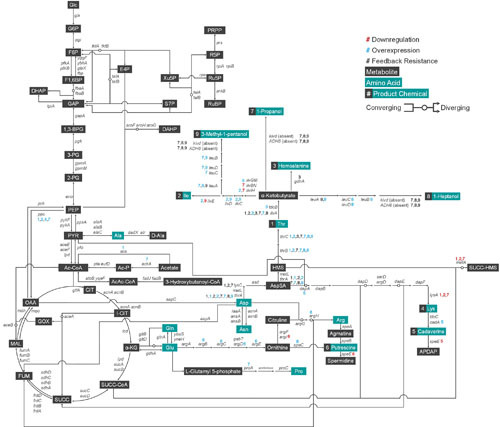
Examples of metabolic engineering strategies to produce chemicals of interest derived from TCA cycle intermediates. Target products included are (1) L-threonine, (2) L-isoleucine, (3) L-homoalanine, (4) L-lysine, (5) cadaverine, (6) putrescine, (7) 1-propanol, (8) 1-heptanol, and (9) 3-methyl-1-propanol. The colors of the numbers indicate modes of engineering: red, expression downregulation including knockout; blue, expression upregulation; black, all other miscellaneous modifications including feedback-release, heterologous gene introduction, and mutagenesis for modified enzyme activities. Convergence and divergence of metabolites are denoted by circular nodes, where some reactions are reversible. As an example for reversible reaction, F6P and GAP converge to form E4P and Xu5P; conversely, E4P and Xu5P converge to form F6P and GAP. For abbreviations, see Fig. 3.
Instead of overexpressing individual genes specifically, a set of related biosynthetic genes can be overexpressed in a combined manner by manipulating their global regulator genes. In aromatic amino acids and derivatives biosynthesis, deletion of tyrR can globally upregulate tyrA, tyrB, aroF, aroL, and aroG, and is a common strategy employed (26, 313, 328, 329, 330). Similarly, deletion of a global regulator trpR can upregulate aroL, trpE, trpD, trpC, trpB, trpA, and aroH, the genes involved in the production of aromatic amino acids, and more specifically, L-tryptophan (20, 318, 325). In contrast, the overexpression, instead of knockout, of a global regulator leucine responsive protein (Lrp) can lead to upregulation of ilvI, serC, aroA, gltB, gltD, gltF, kbl, tdh, serA, and cadA, globally increasing the fluxes in various amino acids and derivatives (21, 22, 24).
Despite the overexpression strategies on various target genes, the enhancement of metabolic fluxes toward a desired product is not always successful. A significant fraction of metabolic enzymes are regulated by cellular metabolic states in the transcription, translation, and catalytic activity levels. The use of inducible promoters, including Plac, Ptac, Ptrc, or artificial promoters, including J23100, allows successful expression of the target genes. However, the feedback inhibition of the fully expressed enzymes cannot be circumvented in this way. For example, enzymes in L-isoleucine biosynthesis LysC, ThrA, ThrB, IlvA, and IlvIH are feedback-inhibited by the cellular concentration of L-lysine, L-threonine, L-threonine, L-isoleucine, and L-isoleucine, respectively. To overcome the feedback inhibition of the metabolic genes and increase the desired metabolic fluxes toward L-isoleucine, the authors overexpressed genes encoding feedback-resistant enzymes lysCC1055T, thrAC1034T, ilvAC1339T, G1341T, C1351G, T1352C, and ilvHG41A, C50T (Figs. 3 and 6) (21). So far, the identification and introduction of various feedback-resistant genes for thrA (10, 21, 25), ilvA (21), ilvH (21, 22, 23), lysC (21, 25), thrL (25), ilvN (23, 24), aroF (26, 313, 318, 328, 330), pheA (26, 313, 330, 331), aroG (20, 319, 326, 327), trpE (20), and tyrA (319) have been reported.
Elimination of competing, degradation, and by-product formation pathways
Enhancement of pathway fluxes toward the formation of target product biosynthesis alone is often insufficient to develop a high-performance strain. There exist metabolic branch points draining important precursors or intermediates through by-product formation pathways or degradation pathways. The final product once produced can also be subjected to these draining pathways. Proper blockage of such competitive by-product formation pathways and degradation pathways should be conducted along with metabolic gene overexpression to obtain the synergistic enhancement of the target biosynthetic fluxes.
In central carbon metabolism, the most common target for pathway deletion is the conversion of acetyl-CoA into acetyl phosphate, which is catalyzed by pta (9, 12, 16, 20, 324, 332, 333, 334, 335, 336, 337). The following action of ackA converts acetyl phosphate into acetate (Fig. 4). The formation of acetate not only sequesters valuable carbon fluxes from acetyl-CoA, but also leads to cell toxicity by accumulating organic acid acetate and retards successful growth of the cells. In the same context as the deletion of pta, deletion of ackA (337, 338) and acs (26, 328) has been reported (Figs. 4 and 7). In the TCA cycle, the cyclic pathway is sometimes linearized by knocking out some of the constituent pathways. For this purpose, deletion of frdA (12) and fumA (309), fumB (309), and fumC (309) has been conducted (Fig. 4). In addition, deletion of mdh that is responsible to the reversible conversion of malate to oxaloacetate was reported (Figs. 3 and 4) (22, 27).
Gene deletion strategies for flux enhancement in amino acids and derivatives biosynthesis, as in gene overexpression strategies, can be divided twofold: the deletion of genes to enrich the precursors from central carbon metabolism and the deletion of genes after branching points toward various amino acids sharing the same upper pathways. To increase the flux toward erythrose 4-phosphate and thus PPP to extract enough reducing powers, pfkA that competes for the use of fructose 6-phosphate with the initial steps in PPP involving tktA and tktB was knocked out (Fig. 5) (22). On the other hand, to reduce the drainage of pyruvate, an important precursor for L-valine, L-isoleucine, and L-alanine, toward acetyl-CoA and subsequently toward TCA cycle, fatty acid biosynthesis, and acetic acid, aceF has been knocked out (Fig. 5) (22).
A combinatorial strategy to enrich precursors in central carbon metabolism and an overexpression of metabolic genes after branching points do increase the flux toward the amino acids of interest. However, it is not enough to specifically overproduce a single amino acid. The fluxes toward other amino acids sharing the same upper pathway increase coincidently, consuming the carbon fluxes for the synthesis of unwanted related amino acids. To prevent such waste, competing pathways from the key branching points are deleted.
The downstream pathways toward L-valine and L-leucine are divided at α-ketoisovalerate (Fig. 5). Deletion of ilvE (10) converting α-ketoisovalerate to L-valine and leuA (11) directing the carbon flux toward L-leucine removed the fluxes toward L-valine and L-leucine, respectively. Chorismate is another branching hot spot for aromatic amino acids biosynthesis. Knockout of trpE, trpC, and trpD in the long chorismate-to-tryptophan conversion process successfully blocked the waste of carbon fluxes toward L-tryptophan and reinforced the fluxes toward L-phenylalanine and L-tyrosine (Fig. 7) (318). The branching point for L-phenylalanine and L-tyrosine is prephenate, and the initial steps toward each amino acid are catalyzed by pheA and tyrA. Knockout of pheA (26) and tyrA (26, 330) have been tried to overproduce L-tyrosine and L-phenylalanine, respectively (Fig. 7). Biosynthesis of L-methionine shares the same biosynthetic pathway with l-threonine and L-isoleucine down to homoserine. Deletion of metA, which guides the carbon flux from homoserine to L-methionine, successfully redirects the whole carbon flux from homoserine toward L-threonine, L-isoleucine, and derivatives (11, 25).
However, the blocking of such branch pathways is powerful enough that essential products for cell proliferation from those deleted pathways cannot be produced in such engineered strains. One of the most traditional solutions to circumvent these auxotrophy problems is supplementing the minimal amount for each of the auxotrophic metabolites or expensive rich components such as yeast extract (10, 11, 25, 26, 311, 314, 318, 330). Using the more recent tactic to avoid such problems is knocking down the essential metabolic genes. Na et al. recently applied sRNA for L-tyrosine and cadaverine overproduction, and successfully knocked down two essential genes murE and ackA (185). Another recent invention, CRISPRi, can also be employed for this purpose. For more information on CRISPRi, refer to the subsection “Repurposed CRISPR/Cas systems” under “Tools for Systems Metabolic Engineering of E. coli.”
In addition to the precursors and intermediates in amino acids and derivatives biosynthesis, the final products themselves are either subjected to degradation or recruited for other product formations. L-Threonine itself is a final amino acid product, but can be further metabolized to produce L-isoleucine. A gene catalyzing the first step from L-threonine toward L-isoleucine, ilvA, was mutated to prevent the loss of L-threonine overproduced (25). To prevent the degradation of L-threonine, tdh, which is responsible for converting L-threonine to an unstable intermediate 2-amino-3-ketobutanoate was removed (25). Moreover, in the synthesis of L-arginine derivative putrescine and L-lysine derivative cadaverine, Qian et al. eliminated metabolic genes that degrade or utilize these chemicals: speE, speG, puuP, puuA, and ygjG (311, 312).
In the biosynthesis of fatty acids and derivatives, securing the pool of acetyl-CoA, the monomer of fatty acids, is one of the most important factors. In this context, pta encoding phosphate acetyltransferase, an enzyme converting acetyl-CoA into acetyl phosphate, is frequently eliminated for fatty acid and derivative synthesis (12, 16). To prevent the degradation and utilization of fatty acids produced, fadD coding for fatty acid acyl-CoA synthetase is commonly deleted (12, 15, 16, 17, 320). In addition, fadE (15, 320), arcA (12), adhE (12), frdA (12), yqhD (12, 18), fucO (12), and ahr (18) have been deleted for free fatty acid and alkane biosynthesis in E. coli.
Transporter engineering
Engineering of transporters including exporters and importers is one of the common strategies employed in systems metabolic engineering especially for production of amino acids and related compounds (Table 1). This strategy has been applied in the production of L-valine (22, 23, 24), L-isoleucine (21), L-threonine (25), putrescine (311), and cadaverine (312). Specifically, target exporters for overexpression include amino acid transporters ygaZ, ygaH (22, 23, 24), rhtA, rhtB, rhtC (25), and putrescine/ornithine antiporter potE (311). Transporters that are involved in uptake of amino acids, on the other hand, must be eliminated for preventing reintroduction of secreted compounds back into the cells. Deletion of tdcC for L-threonine production (25) and puuP for diamine production fall into this category (311, 312). In particular, in the production of L-threonine, deletion of the tdcC and overexpression of rhtC alone (one of the three exporters) resulted in 15.6% and 50.2% increase in production titer, respectively (25). Engineering transporters, however, do not always guarantee an increase in production titer because overexpression of certain transporters has been shown to be detrimental to cell growth. In the production of cadaverine, overexpression of cadB retarded cell growth, possibly because of compromise in the membrane integrity (312).
Metabolic state-responsive flux balancing
All biological systems continuously interact with environmental changes and adapt accordingly. In the same context, the metabolic state of host microorganisms is dynamic, not only because of its own cellular functions, but also in response to the constant changes in the medium composition, both macroscopic and microscopic. However, the engineered metabolic circuits introduced during metabolic engineering are often driven by strong promoters that are not responsive to the intracellular perturbations. The expression levels of each metabolic gene can be controlled with various tools, including induction systems and libraries of promoters and ribosome binding sites, but it cannot be regulated in response to the dynamically changing metabolic states of the host cells in an automatic or synchronized manner. Consequently, introduction of heterologous genes or frequent manipulation of endogenous genes could yield metabolic imbalances, thus leading to the retardation of cell growth (320, 339) or loss of functional genes introduced in the population (339, 340, 341), decreasing the final product titer. Utilization of the deregulated expression system is intuitive and straightforward for pathway design and construction, but is suboptimal for the production of target products with the best titer.
To integrate the host metabolic state into the metabolic gene expression and flux control, natural expression-regulatory systems, including two-component regulatory systems (342), can be maximally utilized. In 2001, Liao borrowed regulatory components from Ntr regulon in E. coli (343), the function of which is to adjust the host to nitrogen-deficient conditions (344). To increase the titer of lycopene in E. coli, Farmer et al. utilized NRI (nitrogen regulator I) coded by glnG to detect free acetyl phosphate level. By deleting another Ntr regulon component NRII, encoded by glnL, the authors maximized the capacity of NRI for detecting free acetyl phosphate level and expressed the genes under the control of glnAp2 promoter. The introduction of glnAp2 promoter to the upstream of pps and idi with NRII eliminated allowed the 50% enhanced production of lycopene and threefold increase in its productivity, compared with a strain possessing the same genetic construct, with the exception that pps and idi are under the control of Ptac (343).
Keasling reported another system that regulates expression of metabolic genes in response to the cellular physiology (320). Zhang et al. engineered a natural fatty acid production-related gene expression regulator FadR, of which repressor activity is antagonized by the binding of acyl-CoA (345), to stably produce fatty acid ethyl esters (FAEEs). In this report, chimeric promoters PLR and PAR were constructed by adding regulatory component or fadBA promoter (PfadBA) into phage lambda promoter (PL) and phage T7 promoter (PA1), respectively. Moreover, PFL1, PFL2, and PFL3 were constructed by further combining PLR and PAR with PlacUV5. These newly produced three chimeric promoters PFL1, PFL2, and PFL3 enabled different expression of genes in response to the isopropyl-β-D-thiogalactopyranoside and fatty acid concentrations (320). By combinatorial adaptation of these promoters to pdc, adhB, atfA, and fadD, a stable, threefold increase in production titer was observed (320). With a large number of biosensors and transcription factors available, strategies for regulating fluxes in response to physiological status are a promising regulatory system to be implemented in many other biosynthetic pathways.
High-throughput engineering
Biological systems are complex, and a huge number of components interact together, forming an immense, intertwined network. Even in a biosynthetic pathway of a single product, at least several to tens of serial catalytic steps are involved, along with competing reactions and regulatory points in the same pathway. Moreover, multiple biosynthetic routes exist to produce a single chemical from glucose or other renewable carbon sources in nature. Therefore, numerous possibilities and strategies have to be tested for designing and constructing an optimal strain, leading to the emergence of systematic and high-throughput approaches.
There have been many trials to systematically examine huge number of possible combinations and choose the single best strain among them. In 2010, Stephanopoulos reported production of taxadiene, a precursor of an important anticancer drug Taxol, in E. coli (346). Ajikumar et al. designed and constructed a nine-step taxadiene biosynthetic pathway from pyruvate and glyceraldehyde 3-phosphate, consisting of an upstream 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway and a downstream module. To maximize the flux, and thus the final titer of taxadiene, the authors selected four enzymatic bottleneck steps consisting of dxs, ispD, ispF, and idi gene products from the upstream module down to isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). Different expression level combinations of these four upstream genes and two downstream genes were tested by using a multivariate-modular approach. The final strain selected showed 15,000-fold increase in the taxadiene titer, with the final concentration of 1.02 g/liter (346). Further conversion of taxadiene to taxadien-5α-ol yielded 58 mg/liter, which is 2,400-fold higher than the previously reported taxadien-5α-ol production in yeast, proving the value of a systematic approach (346).
In addition to modifying promoter strength, ribosome binding sites can be targeted for systematic adjustment of gene expression level. In 2014, Sun et al. utilized libraries of ribosome binding sequences (RBSs) and promoters to systematically optimize the expression level of lycopene production genes (323). From a previously engineered strain CAR0001 with dxs, idi, and heterologous crtEXYIB under control of M1-37, M1-46, and M1-93 promoters from a promoter library (347), authors further modulated the expression level of central carbon metabolism genes sucAB, sdhABCD, and talB by introducing M1-46 promoter, to adjust cellular ATP and NADPH levels (323). The resulting strain showed 76% improvement in lycopene production. The following exchange of RBSs upstream of crtE, dxs, and idi with RBSL9, RBSL12, and RBSL7 from an RBS library, respectively, further increased the titer by 32%, with the final titer of 3.52 g/liter (323). This report demonstrates that further systematic adjustment of RBS strength improves the titer of the target chemicals.
Systematic repression or knockout of potential targets can also be a powerful approach to improve final titers. However, the traditional knockout strategies are often time consuming when multiple targets are examined in a combinatorial manner. Recently, new technologies to downregulate or knock out multiple targets in a more facile and rapid manner have been developed. One of them is a synthetic regulatory sRNA system. Recently, synthetic regulatory sRNA system was applied for overproducing L-tyrosine and cadaverine by screening the knockdown gene targets (185). By reducing the expression of certain genes, the metabolic flux is rebalanced, so the research was aimed at screening the gene target that concentrates the metabolic flux toward the product of interest. First, four intuitive knockdown targets tyrR, csrA, pgi, and ppc genes and additional 84 target genes were screened. The best strain that produces L-tyrosine up to 21.9 g/liter was made by sRNA-mediated fine-tuned knockdown of tyrR and csrA. The fine tuning was achieved by altering the binding energy between the sRNA and the mRNA. Second, for the production of cadaverine, 122 synthetic sRNAs were tested which target individual genes related to cadaverine production or regulatory pathways. Through this procedure, three best strains with ackA, pdhR, and murE individually knocked down, and subsequent fine tuning of murE knockdown level resulted in the best strain with cadaverine titer 12.6 g/liter. It is notable that the gene targets ackA and pdhR were not the genes involved in the major cadaverine-producing pathway, and that murE and ackA were essential genes (185, 186).
In addition to the sRNA system, CRISPR-Cas system and CRISPRi together allows more convenient knockout and knockdown target screening. This year, Lv et al. applied CRISPRi system to screen possible knockdown target genes for improving P(3HB-co-4HB) biosynthesis (348). They introduced various sgRNAs targeting multiple genetic loci with dCas9, and successfully controlled 4HB content by regulating the knockdown strengths of TCA cycle genes sucC, sucD, sdhA, and sdhB (348). In another study, multiplex deletion and insertion of E. coli genomic loci using CRISPR/Cas9 system have been demonstrated, and up to three targets were successfully modified (349). Contrary to the routine application of CRISPR/Cas-based systems for downregulation studies, CRISPR/Cas9 system has also been employed for upregulation as well (181). By the fusion of RNA polymerase ω subunit to dCas9, controlled activation of specific genes can be achieved by simply exchanging sgRNAs or crRNA-tracrRNAs, targeting the upstream of the target genes (181). With these systems allowing more convenient modification of multiple genomic loci, systematic approaches to maximize the capacity of a metabolically engineered strain will become more feasible in the near future.
Multiplex-level genome engineering
One of the most powerful ways to improve strains has been random mutagenesis, treating strains with mutagens such as ultraviolet (UV) light (350) and N-methyl-N′-nitro-N-nitroguanidine (NTG) (351) in the presence of proper selection powers. These genome-wide evolutionary approaches allowed tremendous improvement of strains and still propel the strain developments in many industrial fields. In most cases, however, it is difficult to track genetic modifications introduced into the strain, prohibiting proper understanding of the newly developed strains. The seemingly advantageous characteristics introduced during the mutagenesis may impede further enhancements of the strains. In addition, random mutagenesis often leads to retarded growth of the strains and screening for the desired improvement can be laborious. Accordingly, more rational approaches attracted huge attention in academic fields as molecular biology techniques accumulated. Because of the strength of evolutionary strain development, more rational or at least trackable genome-wide evolutionary approaches have been developed and used, including random mutagenesis in preselected, restricted genomic loci and mutagenesis of which consequences are trackable (53, 191, 192, 193, 197, 352).
One of the earliest achievements in multiplex genome engineering was made by using gTME, which elegantly changes the global gene expression pattern by simply mutating a single global transcription machinery (191, 192). By targeting a global cellular transcription machinery σ70 for mutagenesis in E. coli, Alper et al. successfully selected and reported various novel strains showing ethanol tolerance, improved lycopene production, and enhanced tolerance for both ethanol and sodium dodecyl sulfate (SDS) (192). In the case of lycopene, its production was also improved with the application of MAGE (53). During MAGE, simultaneous overexpression and knockout of multiple genes lead to strains with both improved growth rate and lycopene content (53). Coselection MAGE (CoS-MAGE), a modified version of MAGE, improved indigo production by targeting 12 operons for promoter exchange with T7 promoters (193). TRMR focuses more specifically on the facile tracking of genetic changes in a host population after a multiplex genome engineering procedure (197). By detecting barcode sequences inserted together with modification regions using microarray, gene expression level adjustments responsible for tolerance against salicin, D-fucose, methylglyoxal, and L-valine were identified (197). In addition, four novel tolerance genes against furfural, a growth inhibitor often found in acid-treated lignocellulosic biomass, were identified with the help of TRMR (352). As the above examples indicate, the use of multiplex genome engineering tools not only improves strain performances with the help of evolutionary approaches, but also provides new insights to understand genotypes corresponding to the desired phenotypes.
Omics-assisted approaches
Traditional methodologies for strain improvement include both random and rational approaches (353). However, random mutations often accompany undesired alterations of the genome, and rational manipulation of the genome is limited only to a small region of the genome. Thus, the emergence of omics-based approaches was viewed as one of the solutions for these limitations, and a reflection of the increasing need for a holistic and high-throughput approach (37). Since the first complete genome sequencing of E. coli K-12 strain, a tremendous amount of data on E. coli x-omes began to accumulate (5). There are many reports on producing valuable chemicals or proteins through omics study, and the notable examples are described in this section.
A representative application of transcriptome into systems metabolic engineering in E. coli is the production of L-valine (22). Through the transcriptome profile, ilvCDE genes, which are part of L-valine biosynthetic pathway, were shown to be overexpressed in the base Val strain. The ilvCDE genes were thus overexpressed, resulting in improved L-valine titer from 1.31 g/liter to 3.43 g/liter. Lrp, which positively regulates the level of ilvIH, was also examined. The expression levels of lrp were shown to have decreased, which was thought to be due to L-leucine fed to the medium for complementing the L-leucine auxotrophy. Thus, lrp was overexpressed in order to offset the negative effect of L-leucine on ilvIH expression, showing an increased L-valine titer. Previously unknown valine transporter was investigated by homology searching for the need of transporter engineering. It turned out that the most probable candidate was ygaZH. Through transcriptome profile, ygaZH was shown to be downregulated, which was thought to be blocking the L-valine excretion. Hence, ygaZH was overexpressed, resulting in an even higher L-valine titer. By transcriptome analysis, L-valine titer was increased to a final titer of 7.61 g/liter, about a 5.8-fold increase compared with that of the Val strain (22). Followed by in silico simulation, this strain was developed to produce even more L-valine. Production of L-threonine from E. coli followed a similar approach as described above: following rational engineering, transcriptome analysis, and in silico flux response analysis were conducted (25).
Proteomics has also been employed in E. coli for producing various valuable products. Examples include antibody fragment production in E. coli (354), determination of a fusion partner for overproducing recombinant proteins in extracellular medium (355), and successful production of serine-rich human proteins leptin and interleukin-12 β subunit in E. coli (30). In the particular example of serine-rich protein production, the protein levels of protein elongation factors (including EF-Tu), serine biosynthetic enzymes (GlyA and CysK), and many others were decreased, which was alleviated by overexpressing cysK. Another notable example is the production of spider silk with the help of transcriptomics (29). The stress-response proteins were upregulated when spider silk proteins, with high contents of L-glycine, were produced. Along with the fact that L-glycine biosynthetic enzymes GlyA and GlyS were overexpressed, it was hypothesized that the shortage of L-glycine and glycyl-tRNA pool was the main bottleneck in spider silk production. Therefore, glyVXY and glyA were overexpressed, successfully increasing the level of glycyl-tRNA and L-glycine, respectively. This led to the successful production of spider silk in a large amount in E. coli (29).
Utilizing fluxomics for metabolic engineering is important since concentrating flux to the final product would be an ultimate goal in systems metabolic engineering. Analyzing the whole cell fluxome is mainly done by 13C-based flux analysis. Several studies, including fatty acid production (356) and L-phenylalanine production (357), employed the 13C-based flux analysis to examine the flux profile of engineered E. coli and suggested further engineering targets. In another study about L-isoleucine production, 13C-based flux analysis was conducted and found that intermediates accumulated intracellularly in E. coli (358). Therefore, dihydroxy acid dehydrogenase (DH) and transaminase-B (TA-B) were introduced to redirect the flux toward the product L-isoleucine increasing the titer significantly.
In silico genome-scale metabolic simulation and algorithm-based rational approaches
The metabolic network within E. coli is enormous and complex. There have been numerous research and reports on the metabolic pathways and engineering of such pathways in E. coli. Nevertheless, it is still almost impossible to intuitively predict the comprehensive or close to near-holistic metabolic profiles of a recombinant E. coli and changes in metabolic fluxes once modifications are introduced. With computers and appropriate algorithms representing the metabolic networks and their properties, fair approximations on certain recombinant strains can nonetheless be obtained. Currently, there are plenty of in silico tools and resources for analyzing and predicting the metabolic fluxes in E. coli (25, 225, 255, 256, 257, 258, 266, 268, 270, 217, 272, 273, 274, 275, 276, 277, 278, 279, 280, 281).
During metabolic engineering procedures, the identification of the appropriate gene knockout targets is an important step. Some of the targets are relatively straightforward once a metabolic network and a target overproduction product are given. However, selection of valuable targets based on intuition is limited for a few initial targets, and unexpected results may arise after a period of time-consuming gene work. Prediction of reliable knockout targets can be delivered with various objective functions and algorithms, including MOMA (22, 258, 338, 359), OptKnock (27, 360), and OptSwap (275). Among various amino acids, L-valine was an exemplary study that employed in silico gene perturbation analysis to select appropriate knockout targets (22). To construct L-valine overproducing E. coli strain, Park et al. chose engineering targets with intuitive reasoning, followed by a second round of engineering with targets selected based on transcriptomic changes. The resulting strain produced 7.61 g/liter of L-valine (22). To further increase the L-valine titer, in silico gene perturbation analysis adopting MOMA was conducted, using an in silico genome-scale metabolic network EcoMBEL979 (266). Among the knockout targets proposed by the algorithm were aceE/F and lpdA, pfkA/B, and mdh. Interestingly, pfkA/B genes code phosphofructokinase, an enzyme in the initial steps of glycolysis. The genes pfkA/B are unlikely selected as knockout targets based on intuitive reasoning, since knockout of them might prune off the glycolytic pathway, impairing the central carbon metabolic pathways that play a central role in energy supply to the host cell. The in silico simulation, however, suggested pfkA/B since the carbon flux from glucose can circumvent through PPP, eventually reaching the downstream central carbon metabolism. As a result, additional knockout of aceF, pfkA (corresponding to 90% of the phosphofructokinase activity in native E. coli), and mdh led to L-valine titer improvement up to 20 g/liter in batch culture (Fig. 5) (22). Similarly, Yim et al. successfully constructed a 1,4-butanediol (BDO)-producing strain with the help of in silico simulations (27).
Appropriate amplification targets can also be suggested by in silico simulations based on genome-scale models. For this purpose, flux response analysis (309), FSEOF (258), and FVSEOF (276) are exemplary algorithms to be employed. Lycopene, an antioxidant, was reported to be produced by a recombinant E. coli of which amplification targets were identified by in silico simulations. Choi et al. developed FSEOF, an algorithm for gene amplification target selection, and applied it to enhance lycopene production in E. coli (258). They introduced crtEBI into an E. coli strain for lycopene production, and performed FSEOF on the genome-scale metabolic model EcoMBEL979 (266) with information on four additional heterologous reactions using the MetaFluxNet platform (280, 281). The FSEOF simulation suggested 35 target genes involved in central carbon metabolism and 1-deoxy-D-xylulose 5-phosphate (DXP) pathway (or MEP pathway) among 983 metabolic genes of possible target candidates (258). Further verification of the 35 targets by flux variability test narrowed down the candidates into a set of genes responsible for 21 metabolic reactions. Among those 21 genes, Choi et al. examined the overexpression of seven genes, pfkA, pgi, fbaA, tpiA, icdA, mdh, and idi, and a combinatorial overexpression of idi and mdh, which showed the best improvement in single-gene overexpression, exhibited 2.7- and 3.2-fold improvements in lycopene concentration and content, respectively (258).
Identification of gene deletion and gene amplification targets are meaningful, but biological systems are not as simple—they are not like binary systems composed of on’s and off’s. Some metabolic steps have linear relationships with the target product production capacity: the stronger the flux, the higher the production capacity; or, the weaker the flux, the higher the production capacity. Many metabolic reactions, however, have a nonlinear relationship to the production capacity: there exists an optimal flux level for the best production capacity. Therefore, to construct the best strain for a target chemical production, the expression level of the major pathways involved should be fine-tuned. Algorithms such as flux response analysis (23, 25, 338), OptReg (361), CosMos (277), and k-OptForce (362) can help the target gene selection and optimal flux level determination. Production of an amino acid L-threonine was optimized by applying flux response analysis. To produce L-threonine at a high concentration, Lee et al. developed an E. coli strain TH07 (pBRThrABC) based on rational metabolic pathway engineering, resulting in a strain producing 10.1 g/liter of L-threonine in flask culture (25). To further increase the performance of the strain, Lee et al. performed transcriptome analysis, and obtained ppc as an engineering target (Fig. 6). Both knockout and plasmid-based amplification of ppc, however, resulted in the decrease of L-threonine titer, indicating the presence of optimal ppc level for L-threonine production (25). In silico flux response analysis on PPC flux level indicated an optimal flux of 12.2 mmol/g DCW/h with a flux of 4 mmol/g DCW/h in unengineered strain. Exchange of the ppc promoter with Ptrc allowed higher expression of ppc but lower than plasmid-based amplification in this lacI-mutant strain, Th19C (pBRThrABC), and the L-threonine titer was improved by 27.7% (25). Similarly, based on transcriptome analysis and flux response analysis, ICL flux was adjusted by iclR deletion, and the resulting strain TH20C (pBRThrABC) showed 51.4% improvement in L-threonine production, compared with the original TH07 (pBRThcABC) strain (25). These examples prove the capabilities of in silico genome-scale model-based simulations for narrowing down appropriate engineering targets and guiding the path for further engineering. Combined use of these strategies will allow more comprehensive and thorough approaches to develop the best strains producing desired chemicals at high titers.
Expansion of E. coli Metabolism with Synthetic Pathways
Many valuable chemicals nonnative to E. coli have been attempted for production in E. coli (Table 1) and the innovative strategies developed and employed to produce these chemicals at high titers (Table 1) are described in this section. Strategies that have been undertaken to expand E. coli metabolism with nonnative synthetic pathways should serve as a guide to aspiring endeavors to produce valuable chemicals noninherent in E. coli metabolism.
Production of small chemicals noninherent to E. coli
Since the initial suggestion of concept (7), expansion of E. coli metabolism by heterologous expression of proteins for producing desired chemicals enabled production of various noninherent compounds (Table 1). Compounds including ethylene (363), monoethylene glycol (364), 1,4-butanediol (1,4-BDO) (27), 1,2,4-butanetriol (365, 366), ethanol (8), higher alcohols (9, 10, 11, 12, 13, 14), homoalanine (19), adipic acid (324), styrene (329), p-aminobenzoic acid (328), limonene (367), 2-butanone (368), phenol (319), catechol (317, 326), styrene oxide (330), cis,cis-muconic acid (369), D-glucaric acid (370, 371), p-hydroxybenzoic acid (318), phenyllactic acid (26), 4-hydroxyphenyllactic acid (26), 2-(4–hydroxyphenyl)ethanol (26), phenylacetic acid (26), 4-hydroxyphenylacetic acid (26), amorphadiene (372), alkanes (15, 16, 17, 18), FAEEs (320), isoprene (373), lycopene (258, 323, 343), polyhydroxyalkanoates (31, 32, 33), cyanophycin (374, 375), and various recombinant proteins (29, 30) have been produced, and many of them are not only noninherent in E. coli, but they are also noninherent in any naturally occurring organisms. Production of noninherent molecules generally requires (i) identification of noninherent biochemical reaction(s), (ii) construction of synthetic pathway(s) leading toward the compound(s), and (iii) optimization of the pathway. The noninherent reactions can either be brought in from a different host organism for a known reaction or developed by engineering an enzyme to have nonnatural functions.
Expression of heterologous gene in E. coli allows the extension of metabolic capacity by introducing nonnative reactions and further extension is possible with modification of substrate specificity of the gene product. A notable example is production of wide range of higher alcohols that do not naturally occur in E. coli metabolism by introducing heterologous biochemical reaction for expanding the metabolic space. Noninherent alcohols produced using this strategy include 1-pentanol (10, 14), (S)-2-methyl-1-butanol (10), 3-methyl-1-butanol (10), 1-hexanol (10, 14), (S)-3-methyl-1-pentanol (10), 4-methyl-1-pentanol (10), 1-heptanol (14), (S)-4-methyl-1-hexanol (10), 1-octanol (14), (S)-5-methyl-1-heptanol (10), 2-phenylethanol (9, 14, 331), and 3-phenylpropanol (14). The main strategy is to use 2-keto acid decarboxylase (KDC) and alcohol dehydrogenase reactions for converting 2-keto acid substrates into corresponding alcohols (9). Since certain KDC such as Kivd from Lactococcus lactis exhibit broad substrate specificity, it is able to convert various 2-keto acids into corresponding aldehydes by decarboxylation reaction. The aldehydes are subsequently converted to corresponding alcohols since the alcohol dehydrogenase also exhibits broad substrate specificity. The broad substrate specificity can be altered by reaction pocket modification for accepting a broader range of substrates. Specifically, the F381L/V461A mutant of Kivd is able to accommodate larger-size substrates for subsequent conversion (10). Furthermore, modification of binding pocket residues in LeuA along with feedback resistance (G462D) allows in vivo generation of diverse 2-keto acid variants to be converted by Kivd (14). With the feedback-resistant mutant, additional H97A/S139G/N167A LeuA binding pocket mutant allowed microbial production of 4-methyl-1-hexanol and 5-methyl-1-heptanol (10), and additional H97A/S139G/N167G/P169A lead to 1-heptanol, 1-octanol, 2-phenylethanol, and 3-phenylpropanol production (14). While the enzymes with modified binding pockets are introduced to L-threonine- or L-phenylalanine-producing strains for enhanced precursor availability (10, 14), using strains capable of industrial level production coupled with fed-batch fermentation (25) would further improve the noninherent alcohol production.
Alleviation of metabolic burdens and toxicity
Adaptation of heterologous pathways often elicits stress response to the cell because of toxic intermediates or products, or because of the metabolic burden imposed on the cell. Cell growth hindrance is a serious problem, because biomass and chemical production are deeply related with each other. Thus, several breakthrough strategies have been devised and applied for solving these problems. Excessive metabolic burden comes from several factors such as biased codon usage and overexpression of genes and proteins via plasmids (376, 377). In recombinant spider silk production in E. coli, repetitive incorporation of L-glycine imposed a huge metabolic burden on the cell (378). The authors thus increased the L-glycine and glycyl-tRNA pool by overexpressing glyVXY and glyA (29). Also, production of serine-rich leptin and interleukin-12 β chain was enhanced by overexpressing cysK, alleviating the metabolic burden imposed on the cell (30). By overexpressing cysK, protein biosynthetic capacity was notably enhanced by activation of EF-Tu, and serine biosynthetic pathway was also activated. In this research, proteomic analysis revealed that simply transforming a plasmid can induce stress response to the cell (30). Thus, the plasmid design for gene overexpression is an important factor to be considered. An example in this case was demonstrated in the total synthesis of erythromycin A from E. coli, where reducing the number of plasmids and redesigning them increased the erythromycin A production by 5-fold (379). Toxic intermediates or products can also be detrimental to cell growth and chemical production. In the early days while metabolic engineering was at its infancy, adaptive evolution techniques were used to increase tolerance toward desired chemicals. After the historical construction of the first ethanol-producing E. coli (8), Ingram and colleagues constructed another strain with much higher ethanol tolerance by subculturing the parent strain in media with high ethanol concentration (380). Mimicking natural selection can be facilitated in a much more organized way by using systematic approaches. Following the pioneering work using gTME in yeast to enhance ethanol tolerance (191), a similar strategy was used in E. coli to increase butanol tolerance (381).
Although these random approaches were quite effective, limitations in directed evolution and laborious characterization of each mutation have led to development of more systematic and rational approaches. One notable example is the engineering efflux pumps targeting toxic products, especially fuels and pharmaceuticals. Mukhopadhyay and colleagues manifested this idea with biofuel production, where 43 pumps were tested to increase the tolerance along with the production of five biofuels in E. coli (382). The bactericidal effect of plant-derived terpenoids can be overcome by modulating various cellular machineries. Toxicity of geraniol in E. coli was successfully alleviated by overexpressing marA, which subsequently upregulates the expression of AcrAB-TolC efflux pump (383). In contrast, knockout of recA resulted in the extreme sensitivity to geraniol in E. coli, inferring the role of RecA in geraniol tolerance (384). Similarly, acute toxicity of limonene was alleviated by point mutating (L177Q) an alkyl hydroperoxidase AhpC, reducing the generation of toxic limonene-hydroperoxide (385). Toxic intermediate leakage is another serious problem. In taxol precursor production, the whole biosynthetic pathway was divided into two modules with varying expression levels in order to minimize the cellular indole level, which inhibits taxol biosynthesis and retards cell growth (346). Also, metabolic burden was decreased by reducing the plasmid copy number while maintaining a similar expression level. Synthetic scaffolds can also modulate the metabolic flux to minimize the leakage of toxic intermediate to the cytoplasm. Keasling and colleagues reduced the level of toxic intermediate hydroxymethylglutaryl-CoA (HMG-CoA) by bringing the biosynthetic enzymes in close proximity, thus successfully enhancing the mevalonate production (201). Bacterial microcompartment can be an alternative strategy since this synthetic compartment can successfully sequester toxic materials within (211, 212, 386). Alleviation of toxicity can be further enhanced by two-phase cultivation, where an example can be found in phenol production from E. coli (319).
Apart from heterologous pathways and noninherent materials, native chemicals also impose toxicity to the cell when produced in excessive amount. For putrescine and propanol production in E. coli, enhancement of cell growth was successfully achieved by deleting the stress-responsive rpoS encoding RNA polymerase sigma factor (311, 314).
Production of secondary metabolites in E. coli
While the term secondary metabolite cannot be concisely defined, the common characteristics are that they are generally not involved in central metabolism and not required in cell survival. Many secondary metabolites are also in charge of the defense mechanism or the reproduction of the host, and many of them are important pharmaceuticals, cosmetics, and health-promoting nutraceuticals, among others (387, 388). Using E. coli as a chassis organism is advantageous in that it has a clean genetic background regarding secondary metabolite biosynthesis in which complex feedback regulations do not exist. However, production of secondary metabolites in E. coli has been hampered because of high GC contents of native genes, insoluble proteins, noninherent precursors, and lack of modification machinery. The most notable cases—polyketides, terpenoids and alkaloids—are described in this section.
Polyketides, compounds containing β-keto groups with various degrees of reduction, are classified into three types (389). Among these categories, bacterial modular type I polyketide synthases (PKS) are characterized by their multiple modular assembly lines of PKS. Several cases of type I polyketide production in E. coli were reported (390, 391) among which the production of erythromycin is the most renowned (392, 393). Production of erythromycin in E. coli had been hindered because of the large size of the enzymes (all larger than 300 kDa), posttranslational modification, and the availability of precursors (propionyl-CoA and (S)-methylmalonyl-CoA) (394, 395). The first success in producing erythromycin aglycone, 6-deoxyerythronolide B (6-dEB) in E. coli was achieved with the productivity of 0.01 mmol g DCW−1 day−1. The protein solubility problem was solved by lowering the temperature and expressing the enzymes under PT7. Also, posttranslational modification system and heterologous precursor biosynthetic pathways were introduced (396). Following this initial success, many studies focused on overproducing the noninherent precursor, (2S)-methylmalonyl-CoA (395, 397. 398, 399). The highest titer of 6-dEB obtained was 1.1 g/liter, which was comparable to the commercialized host, showing a great potential toward commercialization (400). In type II PKS, three crucial enzymes—KSα, KSβ, and ACP—known as minimal PKS, are iteratively used to synthesize the carbon backbone of their products (401). In contrast to type I polyketides, production of type II polyketides in E. coli was rather a disappointment because of the aggregation of the minimal PKS (401, 402). One of the only two minor successes was achieved by expressing fungal PKS instead of insoluble type II minimal PKS for producing SEK26, an anthraquinone (403). Another success was reported by overexpressing an alternative sigma factor, σ54, for the production of oxytetracycline (404). This was possible by increasing the mRNA level of oxyB, encoding KSβ. However, expressing minimal PKS in soluble form was still unsuccessful. The third family of PKS, also called the chalcone synthase (CHS)-like PKS, has also been problematic in terms of expression in E. coli because of factors such as low glycosylation efficiency and pH conditions (405). Examples of products from PKSs in this category includes stilbenes (406, 407), flavonoids (408, 409, 410, 411, 412, 413, 414), anthocyanins (405, 415), curcuminoids (416), and warfarin precursors (327). Studies focusing on overproducing precursors, which are much more complex than in type I and II, have also been active (411, 412, 416).
The second category of secondary metabolites is alkaloids. Not as much progress has been made in producing these compounds in E. coli because plant enzymes are not easily amenable to expression in bacterial systems. Thus, E. coli was used to produce (S)-reticuline, an important intermediate in producing benzylisoquinoline, from L-dopamine and yeast was cocultivated for conversion of (S)-reticuline to benzylisoquinoline alkaloids. The subsequent study showed production of (S)-reticuline directly from glucose using E. coli (417, 418).
Isoprenoids, composed of terpenoids and carotenoids, are an important group of chemicals used for fragrances, fuels, or pharmaceuticals (387, 419). The conversion from the native 1-deoxy-D-xylulose 5-phosphate (DXP) pathway (or MEP pathway) in E. coli (420) into the heterologous mevalonate pathway from S. cerevisiae (372) was a major breakthrough in this category. Primary studies of terpenoid production started from monoterpenes (421, 422) which has moved on to diterpenes including the acclaimed examples, artemisinic acid (419), taxol precursor (346), and levopimaradiene (423). Of these, taxol and artemisinin precursor production are explained in later sections of the chapter.
Producing complex secondary metabolites is often assisted with using microbial consortium such as in the production of taxol precursor (424) and benzylisoquinoline alkaloid precursor (S)-reticuline (417). In this strategy, highly intertwined metabolic pathways are distributed to E. coli and S. cerevisiae, each taking in charge of the parts that suit the host. Thus, by using this coculture strategy, production of challenging chemicals can be enhanced significantly.
Enhancement of desired biochemical reactions by spatial localization of enzymes
Spatial localization of enzymes in a modular way can be a brilliant solution for heterologous biochemical reactions hindered by side reactions, inefficient distribution of metabolic flux, or unfavorable reaction conditions. One of the notable earlier trials for spatial localization of enzymes is fusing two or more related enzymes together. Several examples demonstrated include the production of galactonolactone (425) and glycerol (426). Another representative example is engineering type I polyketide synthase (PKS) in which various enzymes are expressed in a fusion polypeptide. This modular system was adopted for producing a single-form alkane and alkenes, pentadecaheptane and pentadecaheptane (427), and for producing several long-chain methyl-branched esters in E. coli (428). However, fusing proteins still arouses controversy since the fusion may significantly reduce the original activity of the enzyme. Also, the larger the size of the polypeptide, the harder it gets to be expressed inside the cell. Another feasible engineering strategy for substrate channeling is to couple modular reactions with N- and C-terminal peptide linkers to enhance the interaction of enzymes (38). Engineering type I polyketide synthase can give another example in this category (429). By engineering N- and C-terminal linkers, epothilone (391) and ansamycin precursor (390) could be produced successfully. Large-size rifA (14.4 kb) and epoD (21.8 kb) were divided into two parts, and intermodular linker sequences from 6-deoxyerythronolide B synthase (DEBS) were linked to the C and N termini of the two split enzymes to facilitate the interaction between the two fragments.
A more efficient system for substrate channeling, synthetic protein scaffold, was devised by Keasling and colleagues (201). Synthetic protein scaffolds comprised three types of interaction domains—GBD, SH3, and PDZ—which originated from metazoan cells. This system was applied for overproduction of mevalonate by targeting acetoacetyl-CoA (AtoB), hydroxylmethylglutaryl-CoA synthase (HMGS), and hydroxylmethylglutaryl-CoA reductase (HMGR) to be used with the scaffold. They were expressed with peptide ligand attached to their C-terminal specific for the corresponding protein-protein interaction domain within the protein scaffold assembly (Fig. 2). Additional effort of altering the architecture of the scaffold increased the mevalonate titer by 77-fold (around 5 mM), compared with the control strain without scaffold. Glucaric production was also greatly enhanced by using synthetic protein scaffold (371). To cope with the rate-limiting myo-inositol oxygenase (MIOX), protein scaffolds were used to enhance the substrate channeling which was successful at increasing the titer about 50% more than previously reported (371). Then, a novel strategy, yet using the similar mechanism to protein scaffold, was developed: DNA scaffold system (208). Onto the custom designed DNA scaffold, target enzymes are bound that are fused with distinct zinc finger domains designed to specifically bind to the scaffold sequence. This system was demonstrated by increasing the titer of resveratrol, 1,2-propanediol, and mevalonate (208). Because profound development is being achieved in technologies to design and construct a 2D or 3D structure of DNA molecules (430), DNA scaffold is gaining a potential to be developed into 2D or 3D scaffold system, as was demonstrated in the RNA scaffold system (207). RNA scaffold is an alternative methodology for organizing intracellular sequential reactions (207). The RNA scaffold module has two RNA aptamer domains, PP7 and MS2, which bind to PP7 and MS2 proteins with respective cargo enzymes fused (D0). Extended RNA scaffolds could also be created into one-dimensional (D1) and two-dimensional (D2) assemblies. When applied to hydrogen production using two enzymes, [Fe-Fe]-hydrogenase and ferredoxin (431), a significant increase in hydrogen production (11- and 48-fold, respectively) was achieved by adopting D1 and D2 architectures. The RNA scaffold system can be characterized by its organelle-like structured D2 assembly, which had been previously mimicked by BMCs only.
Whereas active research has been done for engineering these synthetic proteins by various means, the development in engineering BMCs for spatial localization of enzymes was rather slow. Although there are several reports about engineering the characteristics of BMCs (432) and heterologous expression of BMCs in E. coli (433, 434), there is only one report of using this system for biosynthesis of chemicals. After successfully characterizing the signal peptide and fusing this peptide with Pdc (pyruvate decarboxylase) and Adh (alcohol dehydrogenase), ethanol production was enhanced (435). Increase in titer was possible because of enzyme localization and regional anaerobic condition made by the compartment that is favorable to ethanol production.
In silico algorithm-assisted synthetic pathway design
Many of the chemicals noninherent to E. coli are naturally produced and observed in other organisms, including other bacterial, archaeal, or eukaryotic species. In such cases, the natural, heterogeneous biosynthetic pathways for the chemicals of interest in other organisms can be artificially introduced into an E. coli host strain. In some cases, however, the heterologous biosynthetic pathways may be unnecessarily circumventing, or the chemicals are indeed found in nature but their biosynthesis may have yet to be elucidated. In more difficult cases, the desired chemicals may not be found in nature. In such situations, novel biosynthetic pathways must be designed. Sometimes the design process is simple: the desired products share most of their structures with inherent metabolites in E. coli and a couple of steps of conversions from such intermediates produces the desired products. In many cases, unfortunately, chemicals of interest are far from inherent metabolites in E. coli such that long series of catalytic steps are required, rendering the design of novel pathways difficult. Thus, in silico pathway prediction tools are used to present potential novel biosynthetic pathways to researchers.
One good example of employing an in silico pathway prediction tool to find a novel biosynthetic pathway is the production of flavonoid pinocembrin (436). Faulon employed RetroPath (294) to screen feasible novel pathways producing pinocembrin (436). The application of RetroPath suggested 11 novel sets of metabolic reactions, with the best pathway identical to the natural flavonoid biosynthetic pathway. While a combination of various isozymes applicable in the best pathway suggests nine million possibilities, application of RetroPath allowed selecting the 12 best enzyme combinations with the highest scores (436). Following construction and examination of the novel biosynthetic pathways in E. coli led to a strain producing 1.39 mg/liter pinocembrin (436). Additional pathway optimization using ReproPath allowed a 17-fold increase in titer, with a specific value of 24.14 mg/liter (436). Similarly, SymPheny Biopathway Predictor allowed the design and construction of a novel 1,4-BDO biosynthetic pathway in E. coli (27).
Process Engineering Aspects
Process engineering is an integral part of systems metabolic engineering. While strain development by systems metabolic engineering is important for increasing production of chemicals in a laboratory setting, it is certainly not enough to bring these microbial systems to industrial standards without optimal process engineering. As such, two strategies—high cell density culture and utilization of various carbon sources—are described in this section to allow these microbial systems to be viable in the production of chemicals in the world market.
High cell density cultivation
High cell density cultivation (HCDC) is an essential part for the microbial production of relevant chemicals on an industrial scale. Fed-batch culture technique is most often used to achieve HCDC in contemporary systems for metabolically engineered microbes. Critical studies for understanding E. coli fermentation technology and subsequently increasing cell mass were carried out in 1970s and 1980s in an effort to improve productivity (437, 438, 439). Growing E. coli to achieve density as high as 100 g/liter cell mass in liquid medium usually refers to this method of cultivation, and certain cases for producing various compounds and proteins requires this cultivation technique (438, 440). Production of proteins including growth hormones (441, 442), interleukins (30, 443, 444), and compounds including poly(3-hydroxybutyric acid) (445, 446) are some of the notable examples that employed HCDC for high production titers. The feeding mechanisms include exponential feeding, constant feeding, increased feeding, pH stat, dissolved oxygen (DO) stat, and substrate concentration control (438). The widely accepted biggest problem in HCDC process performance is the accumulation of acetate that inhibits growth and prevents recombinant protein product formation (438, 440). While glycerol prevents acetate formation, possibly because of the relatively slower intake compared with that of glucose (447), glycerol is generally more expensive than glucose (448) and a slower growth rate is observed (447). Although crude glycerol is abundantly produced in the biodiesel industry as a by-product, impurities make it less attractive as a universal carbon source (449). However, acetate formation can be greatly reduced when growth rate is slowed down (438, 440). In a study where exponential feeding fed-batch cultivation was combined with pH stat, the problem with acetate accumulation was solved by reducing the specific growth rate (μ = 0.093 h−1) and 101 g/liter cell biomass was also achieved (450). Although the drawback was carbon flux going mostly toward biomass formation instead of product formation, this study presents a potential strategy for reducing acetate formation for future applications.
Utilization of various carbon sources
One of the ultimate goals envisioned in industrial bioprocesses is to use renewable nonfood biomass for the production of desired chemicals. It is therefore of utmost importance for microbes to be able to utilize not only glucose, but also a large portfolio of carbon sources. There have been several successful efforts to engineer E. coli such that it can utilize carbon sources including glycerol (451), xylose (365, 452, 453), sucrose (454, 455, 456), cellobiose (453), xylan (457), and cellulose (457). One notable example is the development of sucrose-utilizing E. coli K-12 strain to produce L-threonine while maintaining its production integrity (455). In this example, sucrose-utilizing operons were introduced to the host, where sucrose was observed to be hydrolyzed into glucose and fructose in the extracellular space and subsequently transported into the cell by the respective uptake systems (455). This study is a huge leap in strain development because a limited number of E. coli strains such as E. coli W (458) and E. coli EC3132 (459) have been characterized to utilize sucrose despite the depth of accumulated knowledge for many of the well-studied model strains including K-12, B, and C (455, 456). Another notable example describes the consolidation of cellulolytic enzyme expression, pretreated biomass hydrolysis, and production of biofuels from cellulosic biomass (457). Implementing cellulolytic components including cellulase and β-glucosidase allowed the utilization of cellulose, while xylobiosidase and endoxylanase allowed the utilization of xylan. With the use of this system, E. coli was able to grow on ionic liquid-treated switchgrass, Eucalyptus globulus and yard waste to produce value-added fuels. While the product titers (28 mg/liter butanol, 1.7 mg/liter pinene, and 71 mg/liter FAEEs) are low for industrial applications, this study demonstrates important strides toward utilizing pretreated biomass for renewable chemical production.
INDUSTRIAL LEVEL PRODUCTION OF CHEMICALS BY SYSTEMS METABOLIC ENGINEERING OF E. COLI : CASE STUDIES
With the use of the strategies of systems metabolic engineering, an increasing number of engineered E. coli strains are being developed for the production of diverse chemicals and materials. In this section, we present the two examples acclaimed in this field of metabolic engineering, where the microbial production of these chemicals demonstrates the sheer capacity of what can be envisioned and achieved with systems metabolic engineering. The success of 1,4-butanediol and artemisinin certainly showcases what systems metabolic engineering can deliver to the sustainable chemical industry.
1,4-BDO
The most notable example for developing a microorganism to produce chemicals and successful transition to industrial scale is 1,4-BDO production in E. coli (27, 460, 461). 1,4-BDO is an industrially important compound for butadiene, tetrahydrofuran, polytetrahydrofuran, polybutylene terephthalate, polyurethane, and other polyester applications. The petrochemical world market production for 1,4-BDO has reached 1.3 million tons per year (462). A metabolic pathway prediction tool SymPheny was used to design a novel pathway leading to 1,4-BDO from E. coli central metabolism. The novel pathway consists of E. coli sucCD, adh, and sucA, Porphyromonas gingivalis sucD and 4hdb, and codon-optimized version of Clostridium beijerinckii ald. To improve flux toward TCA cycle, R163L mutation was also introduced to citrate synthase encoded by gltA. The strain was further optimized by OptKnock simulation identifying deletion candidates (463). Accordingly, endogenous adhE, pflB, ldh, mdh, and arcA deletions were carried out. Endogenous lpdA was also replaced with Klebsiella pneumonia lpdA for improved anaerobic activity. While this demonstrated the highest reported 1,4-BDO with 18 g/liter in 5 days of fermentation in 2 liter-scale (27), the strain was continuously optimized and an accumulation of more than 50 genetic changes was made to the final production strain (461). From the study, most of the carbon flux has been determined to be directed toward 1,4-BDO by radiolabeled 13C-based flux analysis, suggesting that there are little by-products to be eliminated for downstream processes. Moreover, the oxygen uptake rate was also optimized to reduce cell biomass formation since additional cost is required to remove biomass at the downstream processes. It is important to optimize oxygen uptake rate as it determines carbon propagation toward cell biomass formation—a reduced respiration signifies a fewer number of cells producing the target chemical, which in turn requires more cultivation time.
Successful optimization of strain performance and process engineering has led to the production titer of 120 g/liter with 3 g/liter/h productivity which are feasible values for industrial applications (461). This technology has been recently licensed to BASF for industrial scale-up process and further development. BASF additionally possesses the capability to purify 1,4-BDO using a three-stage fractional distillation (464), and the production of butadiene and polytetrahydrofuran for spandex applications with this renewable 1,4-BDO produced is also under development.
Artemisinic Acid
Artemisinin is a potent antimalarial drug extracted from plant Artemisia annua (465). While artemisinic acid production in S. cerevisiae was a brilliant success in order to adjust the production cost to be sold in the developing world (466, 467, 468), it would not have been possible without the initial development of artemisinic acid biosynthetic pathways and precursors in E. coli (372). Biosynthesis of artemisinic acid was put forward by the first success of amorpha-4,11-diene production achieved by introducing the mevalonate pathway from S. cerevisiae in E. coli to produce IPP and DMAPP, the building blocks for isoprenoid production (372). In this pioneering study, the heterologous mevalonate pathway was distributed into two operons: upstream (atoB, HMGS, tHMGR) and downstream (ERG12, ERG8, MVD1, idi, and ispA). Codon optimization of ADS (amorphadiene synthase) and addition of glycerol in the medium enabled the production of amorphadiene up to 24 μg caryophyllene equivalents/ml. Following this initial success, many trials were done to increase the production of terpenes by optimizing the mevalonate pathway (469, 470, 471).
In addition to these metabolic engineering strategies, endeavors undertaken to maximize the mevalonate production includes the utilization of synthetic protein scaffold to modulate the metabolic flux (201), and engineering the tunable intergenic regions (TIGRs) to regulate the expression of genes inside an operon (182). However, at this point, Keasling and colleagues speculated that the production of artemisinin in E. coli had been underestimated because of the volatility of amorphadiene in water. Therefore, they set up a two-phase partitioning bioreactor using a dodecane organic phase, successfully preventing the evaporation of amorphadiene (472). In 2009, by using alternative mevalonate biosynthetic enzymes and optimization of fermentation process in regard to glucose and nitrogen restriction, amorphadiene titer had significantly escalated to as high as 29.7 g/liter (473).
Obtaining such high titer was a remarkable success, and commercialization was expected to be at hand; however, chemical conversion from amorphadiene to artemisinic acid was tough, and only a minor success was achieved for this conversion in vivo. Keasling and colleagues successfully expressed plant P450s in E. coli heterologously and directly produced artemisinic acid at titers of >100 mg/liter with the E. coli DH1 strain, engineering amorphadiene oxidase (AMO) and using appropriate P450 reductase (CPR) from A. annua as a redox partner (474). Despite much higher production of amorphadiene in E. coli than in S. cerevisiae, the inability of E. coli to express P450 enzymes efficiently and its low conversion rate from amorphadiene to artemisinic acid has led to change of the chassis organism to yeast (475). Following the shift in host organism, a revolutionary step-up toward commercialization was achieved in yeast where 25 g/liter of artemisinic acid was produced followed by chemical conversion to artemisinin with 40 to 45% of overall yield (475).
CONCLUSIONS
With the advent of systems metabolic engineering, E. coli has been thoroughly exploited for the production of useful chemicals and materials far beyond its native capabilities. Systems metabolic engineering not only adopts tools extensively from various disciplines, but also uses emerging tools tailored to meet the specific needs for strain improvement. Complementing the exhaustive depository of tools readily available, innovative strategies for systems metabolic engineering of E. coli have since also been demonstrated to produce chemicals in pioneering approaches. Taken together, several of the chemicals produced by these engineered strains have met industrial production standards, already contributing a substantial supply to the world market. It must be noted that many of the chemical products attempted for production in E. coli admittedly serve only as proofs-of-concept, and further engineering efforts are therefore required to manifest significance viable in industrial production standards.
Engineered microbes such as E. coli have the potential to dominate the production of chemicals—from petroleum derivatives to pharmaceuticals. Driven by the influx of new tools and strategies, the capacity with which systems metabolic engineering of microbes holds for the future is certainly limitless. While systems metabolic engineering is no longer in its infancy, it is now positioned to embrace the next generation of interdisciplinary principles and innovation. This new era of biotechnology, in its consummate maturity, promises the sustainable production of useful chemicals and materials from renewable resources.
ACKNOWLEDGMENTS
S.Y. Lee conceived the project. K.R. Choi, J.H. Shin, J.S. Cho, D. Yang and S.Y. Lee wrote the manuscript. All authors read and approved the final manuscript. K.R. Choi and J.H. Shin contributed equally to this manuscript.
Authors thank Dr. J. Lee and J.Y. Ryu for critical discussion. This work was supported by the Technology Development Program to Solve Climate Changes on Systems Metabolic Engineering for Biorefineries (NRF-2012M1A2A2026556 and NRF-2012M1A2A2026557) and also by the Intelligent Synthetic Biology Center through the Global Frontier Project (2011-0031963) from the Ministry of Science, ICT and Future Planning (MSIP) through the National Research Foundation (NRF) of Korea.
The authors declare no competing interests.
REFERENCES
- 1.Cohen SN, Chang AC. 1973. Recircularization and autonomous replication of a sheared R-factor DNA segment in Escherichia coli transformants. Proc Natl Acad Sci USA 70:1293–1297. [PubMed] 10.1073/pnas.70.5.1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen SN, Chang AC, Boyer HW, Helling RB. 1973. Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad Sci USA 70:3240–3244. [PubMed] 10.1073/pnas.70.11.3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guaraldi F, Salvatori G. 2012. Effect of breast and formula feeding on gut microbiota shaping in newborns. Front Cell Infect Microbiol 2:94. 10.3389/fcimb.2012.00094 [PubMed] 10.3389/fcimb.2012.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed SA, Awosika J, Baldwin C, Bishop-Lilly KA, Biswas B, Broomall S, Chain PS, Chertkov O, Chokoshvili O, Coyne S, Davenport K, Detter JC, Dorman W, Erkkila TH, Folster JP, Frey KG, George M, Gleasner C, Henry M, Hill KK, Hubbard K, Insalaco J, Johnson S, Kitzmiller A, Krepps M, Lo CC, Luu T, McNew LA, Minogue T, Munk CA, Osborne B, Patel M, Reitenga KG, Rosenzweig CN, Shea A, Shen X, Strockbine N, Tarr C, Teshima H, van Gieson E, Verratti K, Wolcott M, Xie G, Sozhamannan S, Gibbons HS, Threat Characterization Consortium. 2012. Genomic comparison of Escherichia coli O104:H4 isolates from 2009 and 2011 reveals plasmid, and prophage heterogeneity, including Shiga toxin encoding phage Stx2. PLoS One 7:e48228. 10.1371/journal.pone.0048228. 10.1371/journal.pone.0048228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. [PubMed] 10.1126/science.277.5331.1453 [DOI] [PubMed] [Google Scholar]
- 6.Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, Crabtree J, Sebaihia M, Thomson NR, Chaudhuri R, Henderson IR, Sperandio V, Ravel J. 2008. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 190:6881–6893. [PubMed] 10.1128/JB.00619-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey JE. 1991. Toward a science of metabolic engineering. Science 252:1668–1675. [PubMed] 10.1126/science.2047876 [DOI] [PubMed] [Google Scholar]
- 8.Ingram LO, Conway T, Alterthum F. 1991. Ethanol production by Escherichia coli strains co-expressing Zymomonas PDC and ADH genes. US patent 5,000,000.
- 9.Atsumi S, Hanai T, Liao JC. 2008. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451:86–89. [PubMed] 10.1038/nature06450 [DOI] [PubMed] [Google Scholar]
- 10.Zhang KC, Sawaya MR, Eisenberg DS, Liao JC. 2008. Expanding metabolism for biosynthesis of nonnatural alcohols. Proc Natl Acad Sci USA 105:20653–20658. [PubMed] 10.1073/pnas.0807157106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cann AF, Liao JC. 2008. Production of 2-methyl-1-butanol in engineered Escherichia coli. Appl Microbiol Biotechnol 81:89–98. [PubMed] 10.1007/s00253-008-1631-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dellomonaco C, Clomburg JM, Miller EN, Gonzalez R. 2011. Engineered reversal of the beta-oxidation cycle for the synthesis of fuels and chemicals. Nature 476:355–359. [PubMed] 10.1038/nature10333 [DOI] [PubMed] [Google Scholar]
- 13.Machado HB, Dekishima Y, Luo H, Lan EI, Liao JC. 2012. A selection platform for carbon chain elongation using the CoA-dependent pathway to produce linear higher alcohols. Metab Eng 14:504–511. [PubMed] 10.1016/j.ymben.2012.07.002 [DOI] [PubMed] [Google Scholar]
- 14.Marcheschi RJ, Li H, Zhang KC, Noey EL, Kim S, Chaubey A, Houk KN, Liao JC. 2012. A synthetic recursive “+1” pathway for carbon chain elongation. ACS Chem Biol 7:689–697. [PubMed] 10.1021/cb200313e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi YJ, Lee SY. 2013. Microbial production of short-chain alkanes. Nature 502:571–574. [PubMed] 10.1038/nature12536 [DOI] [PubMed] [Google Scholar]
- 16.Torella JP, Ford TJ, Kim SN, Chen AM, Way JC, Silver PA. 2013. Tailored fatty acid synthesis via dynamic control of fatty acid elongation. Proc Natl Acad Sci USA 110:11290–11295. [PubMed] 10.1073/pnas.1307129110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howard TP, Middelhaufe S, Moore K, Edner C, Kolak DM, Taylor GN, Parker DA, Lee R, Smirnoff N, Aves SJ, Love J. 2013. Synthesis of customized petroleum-replica fuel molecules by targeted modification of free fatty acid pools in Escherichia coli. Proc Natl Acad Sci USA 110:7636–7641. [PubMed] 10.1073/pnas.1215966110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kallio P, Pasztor A, Thiel K, Akhtar MK, Jones PR. 2014. An engineered pathway for the biosynthesis of renewable propane. Nat Commun 5:4731. 10.1038/ncomms5731. [PubMed] 10.1038/ncomms5731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang KC, Li H, Cho KM, Liao JC. 2010. Expanding metabolism for total biosynthesis of the nonnatural amino acid L-homoalanine. Proc Natl Acad Sci USA 107:6234–6239. [PubMed] 10.1073/pnas.0912903107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Cheng LK, Wang J, Liu Q, Shen T, Chen N. 2013. Genetic engineering of Escherichia coli to enhance production of L-tryptophan. Appl Microbiol Biotechnol 97:7587–7596. [PubMed] 10.1007/s00253-013-5026-3 [DOI] [PubMed] [Google Scholar]
- 21.Park JH, Oh JE, Lee KH, Kim JY, Lee SY. 2012. Rational design of Escherichia coli for L-isoleucine production. ACS Synth Biol 1:532–540. [PubMed] 10.1021/sb300071a [DOI] [PubMed] [Google Scholar]
- 22.Park JH, Lee KH, Kim TY, Lee SY. 2007. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc Natl Acad Sci USA 104:7797–7802. [PubMed] 10.1073/pnas.0702609104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JH, Kim TY, Lee KH, Lee SY. 2011. Fed-batch culture of Escherichia coli for L-valine production based on in silico flux response analysis. Biotechnol Bioeng 108:934–946. [PubMed] 10.1002/bit.22995 [DOI] [PubMed] [Google Scholar]
- 24.Park JH, Jang YS, Lee JW, Lee SY. 2011. Escherichia coli W as a new platform strain for the enhanced production of L-valine by systems metabolic engineering. Biotechnol Bioeng 108:1140–1147. [PubMed] 10.1002/bit.23044 [DOI] [PubMed] [Google Scholar]
- 25.Lee KH, Park JH, Kim TY, Kim HU, Lee SY. 2007. Systems metabolic engineering of Escherichia coli for L-threonine production. Mol Syst Biol 3:149. 10.1038/msb4100196. [PubMed] 10.1038/msb4100196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koma D, Yamanaka H, Moriyoshi K, Ohmoto T, Sakai K. 2012. Production of aromatic compounds by metabolically engineered Escherichia coli with an expanded shikimate pathway. Appl Environ Microbiol 78:6203–6216. [PubMed] 10.1128/AEM.01148-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yim H, Haselbeck R, Niu W, Pujol-Baxley C, Burgard A, Boldt J, Khandurina J, Trawick JD, Osterhout RE, Stephen R, Estadilla J, Teisan S, Schreyer HB, Andrae S, Yang TH, Lee SY, Burk MJ, Van Dien S. 2011. Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol 7:445–452. [PubMed] 10.1038/nchembio.580 [DOI] [PubMed] [Google Scholar]
- 28.Biebl H, Menzel K, Zeng AP, Deckwer WD. 1999. Microbial production of 1,3-propanediol. Appl Microbiol Biotechnol 52:289–297. [PubMed] 10.1007/s002530051523 [DOI] [PubMed] [Google Scholar]
- 29.Xia XX, Qian ZG, Ki CS, Park YH, Kaplan DL, Lee SY. 2010. Native-sized recombinant spider silk protein produced in metabolically engineered Escherichia coli results in a strong fiber. Proc Natl Acad Sci USA 107:14059–14063. [PubMed] 10.1073/pnas.1003366107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han MJ, Jeong KJ, Yoo JS, Lee SY. 2003. Engineering Escherichia coli for increased productivity of serine-rich proteins based on proteome profiling. Appl Environ Microbiol 69:5772–5781. [PubMed] 10.1128/AEM.69.10.5772-5781.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SY. 1996. Bacterial polyhydroxyalkanoates. Biotechnol Bioeng 49:1–14. [PubMed] 10.1002/(SICI)1097-0290(19960105)49:1<1::AID-BIT1>3.3.CO;2-1 [DOI] [PubMed] [Google Scholar]
- 32.Yang TH, Jung YK, Kang HO, Kim TW, Park SJ, Lee SY. 2011. Tailor-made type II Pseudomonas PHA synthases and their use for the biosynthesis of polylactic acid and its copolymer in recombinant Escherichia coli. Appl Microbiol Biotechnol 90:603–614. [PubMed] 10.1007/s00253-010-3077-2 [DOI] [PubMed] [Google Scholar]
- 33.Yang JE, Choi YJ, Lee SJ, Kang KH, Lee H, Oh YH, Lee SH, Park SJ, Lee SY. 2014. Metabolic engineering of Escherichia coli for biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) from glucose. Appl Microbiol Biotechnol 98:95–104. [PubMed] 10.1007/s00253-013-5285-z [DOI] [PubMed] [Google Scholar]
- 34.Lee JW, Kim TY, Jang YS, Choi S, Lee SY. 2011. Systems metabolic engineering for chemicals and materials. Trends Biotechnol 29:370–378. [PubMed] 10.1016/j.tibtech.2011.04.001 [DOI] [PubMed] [Google Scholar]
- 35.Lee JW, Na D, Park JM, Lee J, Choi S, Lee SY. 2012. Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol 8:536–546. [PubMed] 10.1038/nchembio.970 [DOI] [PubMed] [Google Scholar]
- 36.Lee JW, Kim HU, Choi S, Yi J, Lee SY. 2011. Microbial production of building block chemicals and polymers. Curr Opin Biotechnol 22:758–767. [PubMed] 10.1016/j.copbio.2011.02.011 [DOI] [PubMed] [Google Scholar]
- 37.Park JH, Lee SY. 2008. Towards systems metabolic engineering of microorganisms for amino acid production. Curr Opin Biotechnol 19:454–460. [PubMed] 10.1016/j.copbio.2008.08.007 [DOI] [PubMed] [Google Scholar]
- 38.Felnagle LA, Chaubey A, Noey EL, Houk KN, Liao JC. 2012. Engineering synthetic recursive pathways to generate non-natural small molecules. Nat Chem Biol 8:518–526. [PubMed] 10.1038/nchembio.959 [DOI] [PubMed] [Google Scholar]
- 39.Becker J, Wittmann C. 2015. Advanced biotechnology: metabolically engineered cells for the bio-based production of chemicals and fuels, materials, and health-care products. Angew Chem Int Ed Engl 54:3328–3350. [PubMed] 10.1002/anie.201409033 [DOI] [PubMed] [Google Scholar]
- 40.Becker J, Wittmann C. 2012. Bio-based production of chemicals, materials and fuels – Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol 23:631–640. [PubMed] 10.1016/j.copbio.2011.11.012 [DOI] [PubMed] [Google Scholar]
- 41.Wendisch VF. 2014. Microbial production of amino acids and derived chemicals: synthetic biology approaches to strain development. Curr Opin Biotechnol 30:51–58. [PubMed] 10.1016/j.copbio.2014.05.004 [DOI] [PubMed] [Google Scholar]
- 42.Thomason LC, Costantino N, Shaw DV, Court DL. 2007. Multicopy plasmid modification with phage lambda Red recombineering. Plasmid 58:148–158. [PubMed] 10.1016/j.plasmid.2007.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swaminathan S, Ellis HM, Waters LS, Yu D, Lee EC, Court DL, Sharan SK. 2001. Rapid engineering of bacterial artificial chromosomes using oligonucleotides. Genesis 29:14–21. [PubMed] 10.1002/1526-968X(200101)29:1<14::AID-GENE1001>3.0.CO;2-X [DOI] [PubMed] [Google Scholar]
- 44.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56–65. [PubMed] 10.1006/geno.2000.6451 [DOI] [PubMed] [Google Scholar]
- 45.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using gaIK selection. Nucleic Acids Res 33:e36. 10.1093/nar/gni035. [PubMed] 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ellis HM, Yu DG, DiTizio T, Court DL. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci USA 98:6742–6746. [PubMed] 10.1073/pnas.121164898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costantino N, Court DL. 2003. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc Natl Acad Sci USA 100:15748–15753. [PubMed] 10.1073/pnas.2434959100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Buchholz F, Muyrers JP, Stewart AF. 1998. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet 20:123–128. [PubMed] 10.1038/2417 [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Pfeifer BA. 2008. 6-Deoxyerythronolide B production through chromosomal localization of the deoxyerythronolide B synthase genes in E. coli. Metab Eng 10:33–38. [PubMed] 10.1016/j.ymben.2007.09.002 [DOI] [PubMed] [Google Scholar]
- 50.Murphy KC. 1998. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol 180:2063–2071. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy KC, Campellone KG, Poteete AR. 2000. PCR-mediated gene replacement in Escherichia coli. Gene 246:321–330. [PubMed] 10.1016/S0378-1119(00)00071-8 [DOI] [PubMed] [Google Scholar]
- 52.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645. [PubMed] 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. 2009. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460:894–898. [PubMed] 10.1038/nature08187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. 1994. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev 58:401–465. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murphy KC. 1991. Lambda Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme. J Bacteriol 173:5808–5821. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amundsen SK, Smith GR. 2003. Interchangeable parts of the Escherichia coli recombination machinery. Cell 112:741–744. [PubMed] 10.1016/S0092-8674(03)00197-1 [DOI] [PubMed] [Google Scholar]
- 57.Dillingham MS, Kowalczykowski SC. 2008. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev 72:642–671. [PubMed] 10.1128/MMBR.00020-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Myers RS, Stahl FW. 1994. Chi and the RecBCD enzyme of Escherichia coli. Annu Rev Genet 28:49–70. [PubMed] 10.1146/annurev.ge.28.120194.000405 [DOI] [PubMed] [Google Scholar]
- 59.Lao PJ, Forsdyke DR. 2000. Crossover hot-spot instigator (Chi) sequences in Escherichia coli occupy distinct recombination/transcription islands. Gene 243:47–57. [PubMed] 10.1016/S0378-1119(99)00564-8 [DOI] [PubMed] [Google Scholar]
- 60.Ponticelli AS, Schultz DW, Taylor AF, Smith GR. 1985. Chi-dependent DNA strand cleavage by RecBC enzyme. Cell 41:145–151. [PubMed] 10.1016/0092-8674(85)90069-8 [DOI] [PubMed] [Google Scholar]
- 61.Moerschell RP, Tsunasawa S, Sherman F. 1988. Transformation of yeast with synthetic oligonucleotides. Proc Natl Acad Sci USA 85:524–528. [PubMed] 10.1073/pnas.85.2.524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jasin M, Schimmel P. 1984. Deletion of an essential gene in Escherichia coli by site-specific recombination with linear DNA fragments. J Bacteriol 159:783–786. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Winans SC, Elledge SJ, Krueger JH, Walker GC. 1985. Site-directed insertion and deletion mutagenesis with cloned fragments in Escherichia coli. J Bacteriol 161:1219–1221. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dabert P, Smith GR. 1997. Gene replacement with linear DNA fragments in wild-type Escherichia coli: enhancement by Chi sites. Genetics 145:877–889. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carter DM, Radding CM. 1971. The role of exonuclease and beta protein of phage Lambda in genetic recombination. II. Substrate specificity and mode of action of Lambda exonuclease. J Biol Chem 246:2502–2512. [PubMed] [PubMed] [Google Scholar]
- 66.Karu AE, Sakaki Y, Echols H, Linn S. 1975. The gamma protein specified by bacteriophage gamma. Structure and inhibitory activity for the recBC enzyme of Escherichia coli. J Biol Chem 250:7377–7387. [PubMed] [PubMed] [Google Scholar]
- 67.Sternberg N, Hamilton D. 1981. Bacteriophage P1 site-specific recombination. I. recombination between loxP sites. J Mol Biol 150:467–486. 10.1016/0022-2836(81)90375-2 [DOI] [PubMed] [Google Scholar]
- 68.Hoess RH, Ziese M, Sternberg N. 1982. P1 site-specific recombination: nucleotide sequence of the recombining sites. Proc Natl Acad Sci USA 79:3398–3402. [PubMed] 10.1073/pnas.79.11.3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hartley JL, Donelson JE. 1980. Nucleotide sequence of the yeast plasmid. Nature 286:860–865. [PubMed] 10.1038/286860a0 [DOI] [PubMed] [Google Scholar]
- 70.Broach JR, Guarascio VR, Jayaram M. 1982. Recombination within the yeast plasmid 2μ circle is site-specific. Cell 29:227–234. [PubMed] 10.1016/0092-8674(82)90107-6 [DOI] [PubMed] [Google Scholar]
- 71.Buchholz F, Ringrose L, Angrand PO, Rossi F, Stewart AF. 1996. Different thermostabilities of FLP and Cre recombinases: implications for applied site-specific recombination. Nucleic Acids Res 24:4256–4262. [PubMed] 10.1093/nar/24.21.4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ringrose L, Lounnas V, Ehrlich L, Buchholz F, Wade R, Stewart AF. 1998. Comparative kinetic analysis of FLP and cre recombinases: mathematical models for DNA binding and recombination. J Mol Biol 284:363–384. [PubMed] 10.1006/jmbi.1998.2149 [DOI] [PubMed] [Google Scholar]
- 73.Araki K, Araki M, Yamamura K. 2002. Site-directed integration of the cre gene mediated by Cre recombinase using a combination of mutant lox sites. Nucleic Acids Res 30:e103. 10.1093/nar/gnf102. [PubMed] 10.1093/nar/gnf102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Albert H, Dale EC, Lee E, Ow DW. 1995. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J 7:649–659. [PubMed] 10.1046/j.1365-313X.1995.7040649.x [DOI] [PubMed] [Google Scholar]
- 75.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. [PubMed] 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 76.Palmeros B, Wild J, Szybalski W, Le Borgne S, Hernandez-Chavez G, Gosset G, Valle F, Bolivar F. 2000. A family of removable cassettes designed to obtain antibiotic-resistance-free genomic modifications of Escherichia coli and other bacteria. Gene 247:255–264. [PubMed] 10.1016/S0378-1119(00)00075-5 [DOI] [PubMed] [Google Scholar]
- 77.Song CW, Lee SY. 2013. Rapid one-step inactivation of single or multiple genes in Escherichia coli. Biotechnol J 8:776–784. [PubMed] 10.1002/biot.201300153 [DOI] [PubMed] [Google Scholar]
- 78.Gay P, Lecoq D, Steinmetz M, Ferrari E, Hoch JA. 1983. Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis - expression of the gene in Escherichia coli. J Bacteriol 153:1424–1431. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gay P, Lecoq D, Steinmetz M, Berkelman T, Kado CI. 1985. Positive selection procedure for entrapment of insertion-sequence elements in Gram-negative bacteria. J Bacteriol 164:918–921. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blomfield IC, Vaughn V, Rest RF, Eisenstein BI. 1991. Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol Microbiol 5:1447–1457. [PubMed] 10.1111/j.1365-2958.1991.tb00791.x [DOI] [PubMed] [Google Scholar]
- 81.Link AJ, Phillips D, Church GM. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol 179:6228–6237. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Paques F. 2011. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 11:11–27. [PubMed] 10.2174/156652311794520111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Colleaux L, Dauriol L, Betermier M, Cottarel G, Jacquier A, Galibert F, Dujon B. 1986. Universal code equivalent of a yeast mitochondrial intron reading frame is expressed into Escherichia coli as a specific double strand endonuclease. Cell 44:521–533. [PubMed] 10.1016/0092-8674(86)90262-X [DOI] [PubMed] [Google Scholar]
- 84.Thompson AJ, Yuan XQ, Kudlicki W, Herrin DL. 1992. Cleavage and recognition pattern of a double-strand-specific endonuclease (I-CreI) encoded by the chloroplast 23s-ribosomal-RNS intron of Chlamydomonas reinhardtii. Gene 119:247–251. 10.1016/0378-1119(92)90278-W [DOI] [PubMed] [Google Scholar]
- 85.Dalgaard JZ, Garrett RA, Belfort M. 1993. A site-specific endonuclease encoded by a typical archaeal intron. Proc Natl Acad Sci USA 90:5414–5417. [PubMed] 10.1073/pnas.90.12.5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gimble FS, Stephens BW. 1995. Substitutions in conserved dodecapeptide motifs that uncouple the DNA-binding and DNA cleavage activities of PI-SceI endonuclease. J Biol Chem 270:5849–5856. [PubMed] 10.1074/jbc.270.11.5849 [DOI] [PubMed] [Google Scholar]
- 87.Belfort M, Roberts RJ. 1997. Homing endonucleases: keeping the house in order. Nucleic Acids Res 25:3379–3388. [PubMed] 10.1093/nar/25.17.3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chevalier BS, Kortemme T, Chadsey MS, Baker D, Monnat RJ, Stoddard BL. 2002. Design, activity, and structure of a highly specific artificial endonuclease. Mol Cell 10:895–905. [PubMed] 10.1016/S1097-2765(02)00690-1 [DOI] [PubMed] [Google Scholar]
- 89.Posfai G, Kolisnychenko V, Bereczki Z, Blattner FR. 1999. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res 27:4409–4415. [PubMed] 10.1093/nar/27.22.4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Seligman LM, Chisholm KM, Chevalier BS, Chadsey MS, Edwards ST, Savage JH, Veillet AL. 2002. Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res 30:3870–3879. [PubMed] 10.1093/nar/gkf495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Doyon JB, Pattanayak V, Meyer CB, Liu DR. 2006. Directed evolution and substrate specificity profile of homing endonuclease I-SceI. J Am Chem Soc 128:2477–2484. [PubMed] 10.1021/ja057519l [DOI] [PubMed] [Google Scholar]
- 92.Epinat JC, Arnould S, Chames P, Rochaix P, Desfontaines D, Puzin C, Patin A, Zanghellini A, Paques F, Lacroix E. 2003. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res 31:2952–2962. [PubMed] 10.1093/nar/gkg375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Richard GF, Dujon B, Haber JE. 1999. Double-strand break repair can lead to high frequencies of deletions within short CAG CTG trinucleotide repeats. Mol Gen Genet 261:871–882. [PubMed] 10.1007/s004380050031 [DOI] [PubMed] [Google Scholar]
- 94.Lee DJ, Bingle LE, Heurlier K, Pallen MJ, Penn CW, Busby SJ, Hobman JL. 2009. Gene doctoring: a method for recombineering in laboratory and pathogenic Escherichia coli strains. BMC Microbiol 9:252. 10.1186/1471-2180-9-252. [PubMed] 10.1186/1471-2180-9-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Herring CD, Glasner JD, Blattner FR. 2003. Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene 311:153–163. 10.1016/S0378-1119(03)00585-7 [DOI] [PubMed] [Google Scholar]
- 96.Kim YG, Cha J, Chandrasegaran S. 1996. Hybrid restriction enzymes: zinc finger fusions to FokI cleavage domain. Proc Natl Acad Sci USA 93:1156–1160. [PubMed] 10.1073/pnas.93.3.1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li L, Chandrasegaran S. 1993. Alteration of the cleavage distance of Fok I restriction endonuclease by insertion mutagenesis. Proc Natl Acad Sci USA 90:2764–2768. [PubMed] 10.1073/pnas.90.7.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li L, Wu LP, Chandrasegaran S. 1992. Functional domains in Fok I restriction endonuclease. Proc Natl Acad Sci USA 89:4275–4279. [PubMed] 10.1073/pnas.89.10.4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. 1998. FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci USA 95:10570–10575. [PubMed] 10.1073/pnas.95.18.10570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miller JC, Holmes MC, Wang J, Guschin DY, Lee Y-L, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, Gregory PD, Pabo CO, Rebar EJ. 2007. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotech 25:778–785. [PubMed] 10.1038/nbt1319 [DOI] [PubMed] [Google Scholar]
- 101.Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. 2007. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotech 25:786–793. [PubMed] 10.1038/nbt1317 [DOI] [PubMed] [Google Scholar]
- 102.Guo J, Gaj T, Barbas CF. 2010. Directed evolution of an enhanced and highly efficient FokI cleavage domain for zinc finger nucleases. J Mol Biol 400:96–107. [PubMed] 10.1016/j.jmb.2010.04.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, Miller JC, Urnov FD, Gregory PD, Holmes MC. 2011. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods 8:74–79. [PubMed] 10.1038/nmeth.1539 [DOI] [PubMed] [Google Scholar]
- 104.Pavletich NP, Pabo CO. 1991. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252:809–817. [PubMed] 10.1126/science.2028256 [DOI] [PubMed] [Google Scholar]
- 105.Segal DJ, Stege JT, Barbas CF, 3rd. 2003. Zinc fingers and a green thumb: manipulating gene expression in plants. Curr Opin Plant Biol 6:163–168. [PubMed] 10.1016/S1369-5266(03)00007-4 [DOI] [PubMed] [Google Scholar]
- 106.Bae KH, Kwon YD, Shin HC, Hwang MS, Ryu EH, Park KS, Yang HY, Lee DK, Lee Y, Park J, Kwon HS, Kim HW, Yeh BI, Lee HW, Sohn SH, Yoon J, Seol W, Kim JS. 2003. Human zinc fingers as building blocks in the construction of artificial transcription factors. Nat Biotechnol 21:275–280. [PubMed] 10.1038/nbt796 [DOI] [PubMed] [Google Scholar]
- 107.Cornu TI, Thibodeau-Beganny S, Guhl E, Alwin S, Eichtinger M, Joung JK, Cathomen T. 2008. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol Ther 16:352–358. [PubMed] 10.1038/sj.mt.6300357 [DOI] [PubMed] [Google Scholar]
- 108.Ramirez CL, Foley JE, Wright DA, Muller-Lerch F, Rahman SH, Cornu TI, Winfrey RJ, Sander JD, Fu F, Townsend JA, Cathomen T, Voytas DF, Joung JK. 2008. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods 5:374–375. [PubMed] 10.1038/nmeth0508-374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, Unger-Wallace E, Sander JD, Muller-Lerch F, Fu F, Pearlberg J, Gobel C, Dassie JP, Pruett-Miller SM, Porteus MH, Sgroi DC, Iafrate AJ, Dobbs D, McCray PB, Jr., Cathomen T, Voytas DF, Joung JK. 2008. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31:294–301. [PubMed] 10.1016/j.molcel.2008.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, Curtin SJ, Blackburn JS, Thibodeau-Beganny S, Qi Y, Pierick CJ, Hoffman E, Maeder ML, Khayter C, Reyon D, Dobbs D, Langenau DM, Stupar RM, Giraldez AJ, Voytas DF, Peterson RT, Yeh JR, Joung JK. 2011. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods 8:67–69. [PubMed] 10.1038/nmeth.1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gupta A, Christensen RG, Rayla AL, Lakshmanan A, Stormo GD, Wolfe SA. 2012. An optimized two-finger archive for ZFN-mediated gene targeting. Nat Methods 9:588–590. [PubMed] 10.1038/nmeth.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bhakta MS, Henry IM, Ousterout DG, Das KT, Lockwood SH, Meckler JF, Wallen MC, Zykovich A, Yu Y, Leo H, Xu L, Gersbach CA, Segal DJ. 2013. Highly active zinc-finger nucleases by extended modular assembly. Genome Res 23:530–538. [PubMed] 10.1101/gr.143693.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kay S, Hahn S, Marois E, Hause G, Bonas U. 2007. A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science 318:648–651. [PubMed] 10.1126/science.1144956 [DOI] [PubMed] [Google Scholar]
- 114.Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. 2012. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335:716–719. [PubMed] 10.1126/science.1216211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deng D, Yan C, Pan X, Mahfouz M, Wang J, Zhu JK, Shi Y, Yan N. 2012. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335:720–723. [PubMed] 10.1126/science.1215670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. 2009. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512. [PubMed] 10.1126/science.1178811 [DOI] [PubMed] [Google Scholar]
- 117.Moscou MJ, Bogdanove AJ. 2009. A simple cipher governs DNA recognition by TAL effectors. Science 326:1501. 10.1126/science.1178817. [PubMed] 10.1126/science.1178817 [DOI] [PubMed] [Google Scholar]
- 118.Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. 2010. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186:757–761. [PubMed] 10.1534/genetics.110.120717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C, Recchia A, Cathomen T, Goncalves MA. 2013. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res 41:e63. 10.1093/nar/gks1446. [PubMed] 10.1093/nar/gks1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. 2011. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39:e82. 10.1093/nar/gkr218. [PubMed] 10.1093/nar/gkr218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. 2012. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30:460–465. [PubMed] 10.1038/nbt.2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schmid-Burgk JL, Schmidt T, Kaiser V, Honing K, Hornung V. 2013. A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes. Nat Biotechnol 31:76–81. [PubMed] 10.1038/nbt.2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Briggs AW, Rios X, Chari R, Yang L, Zhang F, Mali P, Church GM. 2012. Iterative capped assembly: rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res 40:e117. 10.1093/nar/gks624. 10.1093/nar/gks624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ding Q, Lee Y-K, Schaefer EAK, Peters DT, Veres A, Kim K, Kuperwasser N, Motola DL, Meissner TB, Hendriks WT, Trevisan M, Gupta RM, Moisan A, Banks E, Friesen M, Schinzel RT, Xia F, Tang A, Xia Y, Figueroa E, Wann A, Ahfeldt T, Daheron L, Zhang F, Rubin LL, Peng LF, Chung RT, Musunuru K, Cowan CA. 2013. A TALEN genome editing system to generate human stem cell-based disease models. Cell Stem Cell 12:238–251. [PubMed] 10.1016/j.stem.2012.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E, Kim D, Lee MS, Go EM, Song HJ, Kim H, Cho N, Bang D, Kim S, Kim JS. 2013. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 31:251–258. [PubMed] 10.1038/nbt.2517 [DOI] [PubMed] [Google Scholar]
- 126.Wang Z, Li J, Huang H, Wang G, Jiang M, Yin S, Sun C, Zhang H, Zhuang F, Xi JJ. 2012. An integrated chip for the high-throughput synthesis of transcription activator-like effectors. Angew Chem Int Ed Engl 51:8505–8508. [PubMed] 10.1002/anie.201203597 [DOI] [PubMed] [Google Scholar]
- 127.Jansen R, Embden JD, Gaastra W, Schouls LM. 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575. [PubMed] 10.1046/j.1365-2958.2002.02839.x [DOI] [PubMed] [Google Scholar]
- 128.Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. 1987. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. 2005. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151:2551–2561. [PubMed] 10.1099/mic.0.28048-0 [DOI] [PubMed] [Google Scholar]
- 130.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60:174–182. [PubMed] 10.1007/s00239-004-0046-3 [DOI] [PubMed] [Google Scholar]
- 131.Pourcel C, Salvignol G, Vergnaud G. 2005. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151:653–663. [PubMed] 10.1099/mic.0.27437-0 [DOI] [PubMed] [Google Scholar]
- 132.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. [PubMed] 10.1126/science.1138140 [DOI] [PubMed] [Google Scholar]
- 133.Marraffini LA, Sontheimer EJ. 2008. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322:1843–1845. [PubMed] 10.1126/science.1165771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, Moineau S. 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71. [PubMed] 10.1038/nature09523 [DOI] [PubMed] [Google Scholar]
- 135.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. [PubMed] 10.1126/science.1159689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Carte J, Wang RY, Li H, Terns RM, Terns MP. 2008. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev 22:3489–3496. [PubMed] 10.1101/gad.1742908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. 2010. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329:1355–1358. [PubMed] 10.1126/science.1192272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. [PubMed] 10.1038/nature09886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Marraffini LA, Sontheimer EJ. 2010. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463:568–571. [PubMed] 10.1038/nature08703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. 2009. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139:945–956. [PubMed] 10.1016/j.cell.2009.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–1400. [PubMed] 10.1128/JB.01412-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–740. [PubMed] 10.1099/mic.0.023960-0 [DOI] [PubMed] [Google Scholar]
- 143.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. 2014. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67. [PubMed] 10.1038/nature13011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Haft DH, Selengut J, Mongodin EF, Nelson KE. 2005. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol 1:e60. 10.1371/journal.pcbi.0010060. [PubMed] 10.1371/journal.pcbi.0010060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. 2006. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1:7. 10.1186/1745-6150-1-7. 10.1186/1745-6150-1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Makarova KS, Aravind L, Wolf YI, Koonin EV. 2011. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct 6:38. 10.1186/1745-6150-6-38 [PubMed] 10.1186/1745-6150-6-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13:722–736. [PubMed] 10.1038/nrmicro3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. [PubMed] 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. [PubMed] 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. [PubMed] 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. 2013. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods 10:1028–1034. [PubMed] 10.1038/nmeth.2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ren X, Sun J, Housden BE, Hu Y, Roesel C, Lin S, Liu LP, Yang Z, Mao D, Sun L, Wu Q, Ji JY, Xi J, Mohr SE, Xu J, Perrimon N, Ni JQ. 2013. Optimized gene editing technology for Drosophila melanogaster using germ line-specific Cas9. Proc Natl Acad Sci USA 110:19012–19017. [PubMed] 10.1073/pnas.1318481110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nakayama T, Fish MB, Fisher M, Oomen-Hajagos J, Thomsen GH, Grainger RM. 2013. Simple and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus tropicalis. Genesis 51:835–843. [PubMed] 10.1002/dvg.22720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu J-L, Gao C. 2013. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotech 31:686–688. [PubMed] 10.1038/nbt.2650 [DOI] [PubMed] [Google Scholar]
- 155.Li W, Teng F, Li T, Zhou Q. 2013. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotech 31:684–686. [PubMed] 10.1038/nbt.2652 [DOI] [PubMed] [Google Scholar]
- 156.Flowers GP, Timberlake AT, McLean KC, Monaghan JR, Crews CM. 2014. Highly efficient targeted mutagenesis in axolotl using Cas9 RNA-guided nuclease. Development 141:2165–2171. [PubMed] 10.1242/dev.105072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, Luo B, Alvarez-Carbonell D, Garcia-Mesa Y, Karn J, Mo X, Khalili K. 2014. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci USA 111:11461–11466. [PubMed] 10.1073/pnas.1405186111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ryan OW, Skerker JM, Maurer MJ, Li X, Tsai JC, Poddar S, Lee ME, DeLoache W, Dueber JE, Arkin AP, Cate JH. 2014. Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife 3. 10.7554/eLife.03703. [PubMed] 10.7554/eLife.03703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, Haass C, Schmid B. 2013. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140:4982–4987. [PubMed] 10.1242/dev.099085 [DOI] [PubMed] [Google Scholar]
- 160.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. [PubMed] 10.1038/nbt.2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Vercoe RB, Chang JT, Dy RL, Taylor C, Gristwood T, Clulow JS, Richter C, Przybilski R, Pitman AR, Fineran PC. 2013. Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet 9:e1003454. 10.1371/journal.pgen.1003454. 10.1371/journal.pgen.1003454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Oh JH, van Pijkeren JP. 2014. CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res 42:e131. 10.1093/nar/gku623. [PubMed] 10.1093/nar/gku623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tong Y, Charusanti P, Zhang L, Weber T, Lee SY. 2015. CRISPR-Cas9 based engineering of Actinomycetal genomes. ACS Synth Biol 4:1020–1029. [PubMed] 10.1021/acssynbio.5b00038 [DOI] [PubMed] [Google Scholar]
- 164.Cobb RE, Wang Y, Zhao H. 2015. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol 4:723–728. [PubMed] 10.1021/sb500351f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Huang H, Zheng G, Jiang W, Hu H, Lu Y. 2015. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim Biophys Sin (Shanghai) 47:231–243. [PubMed] 10.1093/abbs/gmv007 [DOI] [PubMed] [Google Scholar]
- 166.Xu T, Li Y, Shi Z, Hemme CL, Li Y, Zhu Y, Van Nostrand JD, He Z, Zhou J. 2015. Efficient genome editing in Clostridium cellulolyticum via CRISPR-Cas9 nickase. Appl Environ Microbiol 81:4423–4431. [PubMed] 10.1128/AEM.00873-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31:822–826. [PubMed] 10.1038/nbt.2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, Jaenisch R, Zhang F, Sharp PA. 2014. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32:670–676. [PubMed] 10.1038/nbt.2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. 2014. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol 32:677–683. [PubMed] 10.1038/nbt.2916 [DOI] [PubMed] [Google Scholar]
- 170.Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, Hwang J, Kim JI, Kim JS. 2015. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12:237–243, 231 p following 243. [DOI] [PubMed] [Google Scholar]
- 171.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. 2014. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32:279–284. [PubMed] 10.1038/nbt.2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. 2013. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389. [PubMed] 10.1016/j.cell.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, Skarnes WC. 2014. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11:399–402. [PubMed] 10.1038/nmeth.2857 [DOI] [PubMed] [Google Scholar]
- 174.Guilinger JP, Thompson DB, Liu DR. 2014. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32:577–582. [PubMed] 10.1038/nbt.2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK. 2014. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32:569–576. [PubMed] 10.1038/nbt.2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. 2015. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520:186–191. [PubMed] 10.1038/nature14299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. [PubMed] 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gilbert LA, Larson MH, Morsut L, Liu ZR, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154:442–451. [PubMed] 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M, Tsai JC, Weissman JS, Dueber JE, Qi LS, Lim WA. 2015. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 160:339–350. [PubMed] 10.1016/j.cell.2014.11.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517:583–588. [PubMed] 10.1038/nature14136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bikard D, Jiang WY, Samai P, Hochschild A, Zhang F, Marraffini LA. 2013. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41:7429–7437. [PubMed] 10.1093/nar/gkt520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pfleger BF, Pitera DJ, D Smolke C, Keasling JD. 2006. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol 24:1027–1032. [PubMed] 10.1038/nbt1226 [DOI] [PubMed] [Google Scholar]
- 183.Sharon E, Kalma Y, Sharp A, Raveh-Sadka T, Levo M, Zeevi D, Keren L, Yakhini Z, Weinberger A, Segal E. 2012. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat Biotechnol 30:521–530. [PubMed] 10.1038/nbt.2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sandoval NR, Kim JYH, Glebes TY, Reeder PJ, Aucoin HR, Warner JR, Gill RT. 2012. Strategy for directing combinatorial genome engineering in Escherichia coli. Proc Natl Acad Sci USA 109:10540–10545. [PubMed] 10.1073/pnas.1206299109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. 2013. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol 31:170–174. [PubMed] 10.1038/nbt.2461 [DOI] [PubMed] [Google Scholar]
- 186.Yoo SM, Na D, Lee SY. 2013. Design and use of synthetic regulatory small RNAs to control gene expression in Escherichia coli. Nat Protoc 8:1694–1707. [PubMed] 10.1038/nprot.2013.105 [DOI] [PubMed] [Google Scholar]
- 187.Gottesman S. 2004. The small RNA regulators of Escherichia coli: roles and mechanisms. Annu Rev Microbiol 58:303–328. [PubMed] 10.1146/annurev.micro.58.030603.123841 [DOI] [PubMed] [Google Scholar]
- 188.Brennan RG, Link TM. 2007. Hfq structure, function and ligand binding. Curr Opin Microbiol 10:125–133. [PubMed] 10.1016/j.mib.2007.03.015 [DOI] [PubMed] [Google Scholar]
- 189.Frohlich KS, Vogel J. 2009. Activation of gene expression by small RNA. Curr Opin Microbiol 12:674–682. [PubMed] 10.1016/j.mib.2009.09.009 [DOI] [PubMed] [Google Scholar]
- 190.Urban JH, Vogel J. 2008. Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS Biol 6:e64. 10.1371/journal.pbio.0060064. 10.1371/journal.pbio.0060064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Alper H, Moxley J, Nevoigt E, Fink GR, Stephanopoulos G. 2006. Engineering yeast transcription machinery for improved ethanol tolerance and production. Science 314:1565–1568. [PubMed] 10.1126/science.1131969 [DOI] [PubMed] [Google Scholar]
- 192.Alper H, Stephanopoulos G. 2007. Global transcription machinery engineering: a new approach for improving cellular phenotype. Metab Eng 9:258–267. [PubMed] 10.1016/j.ymben.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 193.Wang HH, Kim H, Cong L, Jeong J, Bang D, Church GM. 2012. Genome-scale promoter engineering by coselection MAGE. Nat Methods 9:591–593. [PubMed] 10.1038/nmeth.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bonde MT, Kosuri S, Genee HJ, Sarup-Lytzen K, Church GM, Sommer MO, Wang HH. 2015. Direct mutagenesis of thousands of genomic targets using microarray-derived oligonucleotides. ACS Synth Biol 4:17–22. [PubMed] 10.1021/sb5001565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Isaacs FJ, Carr PA, Wang HH, Lajoie MJ, Sterling B, Kraal L, Tolonen AC, Gianoulis TA, Goodman DB, Reppas NB, Emig CJ, Bang D, Hwang SJ, Jewett MC, Jacobson JM, Church GM. 2011. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 333:348–353. [PubMed] 10.1126/science.1205822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.DeVito JA. 2008. Recombineering with tolC as a selectable/counter-selectable marker: remodeling the rRNA operons of Escherichia coli. Nucleic Acids Res 36:e4. 10.1093/nar/gkm1084. [PubMed] 10.1093/nar/gkm1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Warner JR, Reeder PJ, Karimpour-Fard A, Woodruff LBA, Gill RT. 2010. Rapid profiling of a microbial genome using mixtures of barcoded oligonucleotides. Nat Biotechnol 28:856–862. [PubMed] 10.1038/nbt.1653 [DOI] [PubMed] [Google Scholar]
- 198.Mansell TJ, Warner JR, Gill RT. 2013. Trackable multiplex recombineering for gene-trait mapping in E. coli. Methods Mol Biol 985:223–246. [PubMed] 10.1007/978-1-62703-299-5_12 [DOI] [PubMed] [Google Scholar]
- 199.Good MC, Zalatan JG, Lim WA. 2011. Scaffold proteins: hubs for controlling the flow of cellular information. Science 332:680–686. [PubMed] 10.1126/science.1198701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jiang HX, Wood KV, Morgan JA. 2005. Metabolic engineering of the phenylpropanoid pathway in Saccharomyces cerevisiae. Appl Environ Microbiol 71:2962–2969. [PubMed] 10.1128/AEM.71.6.2962-2969.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KLJ, Keasling JD. 2009. Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol 27:753–759. [PubMed] 10.1038/nbt.1557 [DOI] [PubMed] [Google Scholar]
- 202.Zhu MM, Skraly FA, Cameron DC. 2001. Accumulation of methylglyoxal in anaerobically grown Escherichia coli and its detoxification by expression of the Pseudomonas putida glyoxalase I gene. Metab Eng 3:218–225. [PubMed] 10.1006/mben.2001.0186 [DOI] [PubMed] [Google Scholar]
- 203.Conrado RJ, Varner JD, DeLisa MP. 2008. Engineering the spatial organization of metabolic enzymes: mimicking nature’s synergy. Curr Opin Biotechnol 19:492–499. [PubMed] 10.1016/j.copbio.2008.07.006 [DOI] [PubMed] [Google Scholar]
- 204.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. 1988. 3-Dimensional structure of the tryptophan synthase alpha-2-beta-2 multienzyme complex from Salmonella typhimurium. J Biol Chem 263:17857–17871. [PubMed] [PubMed] [Google Scholar]
- 205.Miles EW, Rhee S, Davies DR. 1999. The molecular basis of substrate channeling. J Biol Chem 274:12193–12196. [PubMed] 10.1074/jbc.274.18.12193 [DOI] [PubMed] [Google Scholar]
- 206.Kerfeld CA, Heinhorst S, Cannon GC. 2010. Bacterial microcompartments. Annu Rev Microbiol 64:391–408. [PubMed] 10.1146/annurev.micro.112408.134211 [DOI] [PubMed] [Google Scholar]
- 207.Delebecque CJ, Lindner AB, Silver PA, Aldaye FA. 2011. Organization of intracellular reactions with rationally designed RNA assemblies. Science 333:470–474. [PubMed] 10.1126/science.1206938 [DOI] [PubMed] [Google Scholar]
- 208.Conrado RJ, Wu GC, Boock JT, Xu HS, Chen SY, Lebar T, Turnsek J, Tomsic N, Avbelj M, Gaber R, Koprivnjak T, Mori J, Glavnik V, Vovk I, Bencina M, Hodnik V, Anderluh G, Dueber JE, Jerala R, DeLisa MP. 2012. DNA-guided assembly of biosynthetic pathways promotes improved catalytic efficiency. Nucleic Acids Res 40:1879–1889. [PubMed] 10.1093/nar/gkr888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dou ZC, Heinhorst S, Williams EB, Murin CD, Shively JM, Cannon GC. 2008. CO2 fixation kinetics of Halothiobacillus neapolitanus mutant carboxysomes lacking carbonic anhydrase suggest the shell acts as a diffusional barrier for CO2. J Biol Chem 283:10377–10384. [PubMed] 10.1074/jbc.M709285200 [DOI] [PubMed] [Google Scholar]
- 210.Lee H, DeLoache WC, Dueber JE. 2012. Spatial organization of enzymes for metabolic engineering. Metab Eng 14:242–251. [PubMed] 10.1016/j.ymben.2011.09.003 [DOI] [PubMed] [Google Scholar]
- 211.Penrod JT, Roth JR. 2006. Conserving a volatile metabolite: a role for carboxysome-like organelles in Salmonella enterica. J Bacteriol 188:2865–2874. [PubMed] 10.1128/JB.188.8.2865-2874.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sampson EM, Bobik TA. 2008. Microcompartments for B-12-dependent 1,2-propanediol degradation provide protection from DNA and cellular damage by a reactive metabolic intermediate. J Bacteriol 190:2966–2971. [PubMed] 10.1128/JB.01925-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chen AH, Silver PA. 2012. Designing biological compartmentalization. Trends Cell Biol 22:662–670. [PubMed] 10.1016/j.tcb.2012.07.002 [DOI] [PubMed] [Google Scholar]
- 214.Kerfeld CA, Sawaya MR, Tanaka S, Nguyen CV, Phillips M, Beeby M, Yeates TO. 2005. Protein structures forming the shell of primitive bacterial organelles. Science 309:936–938. [PubMed] 10.1126/science.1113397 [DOI] [PubMed] [Google Scholar]
- 215.Tanaka S, Kerfeld CA, Sawaya MR, Cai F, Heinhorst S, Cannon GC, Yeates TO. 2008. Atomic-level models of the bacterial carboxysome shell. Science 319:1083–1086. [PubMed] 10.1126/science.1151458 [DOI] [PubMed] [Google Scholar]
- 216.Bacher A, Baur R, Eggers U, Harders HD, Otto MK, Schnepple H. 1980. Riboflavin synthases of Bacillus subtilis – purification and properties. J Biol Chem 255:632–637. [PubMed] [PubMed] [Google Scholar]
- 217.Sutter M, Boehringer D, Gutmann S, Guenther S, Prangishvili D, Loessner MJ, Stetter KO, Weber-Ban E, Ban N. 2008. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat Struct Mol Biol 15:939–947. [PubMed] 10.1038/nsmb.1473 [DOI] [PubMed] [Google Scholar]
- 218.Kim TY, Kim HU, Lee SY. 2010. Data integration and analysis of biological networks. Curr Opin Biotechnol 21:78–84. [PubMed] 10.1016/j.copbio.2010.01.003 [DOI] [PubMed] [Google Scholar]
- 219.Lee SY, Lee DY, Kim TY. 2005. Systems biotechnology for strain improvement. Trends Biotechnol 23:349–358. [PubMed] 10.1016/j.tibtech.2005.05.003 [DOI] [PubMed] [Google Scholar]
- 220.Vemuri GN, Aristidou AA. 2005. Metabolic engineering in the -omics era: elucidating and modulating regulatory networks. Microbiol Mol Biol Rev 69:197–216. [PubMed] 10.1128/MMBR.69.2.197-216.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhang WW, Li F, Nie L. 2010. Integrating multiple ‘omics’ analysis for microbial biology: application and methodologies. Microbiology 156:287–301. [PubMed] 10.1099/mic.0.034793-0 [DOI] [PubMed] [Google Scholar]
- 222.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, Mckenney K, Sutton G, Fitzhugh W, Fields C, Gocayne JD, Scott J, Shirley R, Liu LI, Glodek A, Kelley JM, Weidman JF, Phillips CA, Spriggs T, Hedblom E, Cotton MD, Utterback TR, Hanna MC, Nguyen DT, Saudek DM, Brandon RC, Fine LD, Fritchman JL, Fuhrmann JL, Geoghagen NSM, Gnehm CL, Mcdonald LA, Small KV, Fraser CM, Smith HO, Venter JC. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496–512. [PubMed] 10.1126/science.7542800 [DOI] [PubMed] [Google Scholar]
- 223.Metzker ML. 2010. Sequencing technologies — the next generation. Nat Rev Genet 11:31–46. [PubMed] 10.1038/nrg2626 [DOI] [PubMed] [Google Scholar]
- 224.Patil KR, Akesson M, Nielsen J. 2004. Use of genome-scale microbial models for metabolic engineering. Curr Opin Biotechnol 15:64–69. [PubMed] 10.1016/j.copbio.2003.11.003 [DOI] [PubMed] [Google Scholar]
- 225.Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BØ. 2007. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol 3:121. 10.1038/msb4100155. 10.1038/msb4100155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BØ. 2004. Integrating high-throughput and computational data elucidates bacterial networks. Nature 429:92–96. [PubMed] 10.1038/nature02456 [DOI] [PubMed] [Google Scholar]
- 227.Khodursky AB, Peter BJ, Cozzarelli NR, Botstein D, Brown PO, Yanofsky C. 2000. DNA microarray analysis of gene expression in response to physiological and genetic changes that affect tryptophan metabolism in Escherichia coli. Proc Natl Acad Sci USA 97:12170–12175. [PubMed] 10.1073/pnas.220414297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Tjaden B, Saxena RM, Stolyar S, Haynor DR, Kolker E, Rosenow C. 2002. Transcriptome analysis of Escherichia coli using high-density oligonucleotide probe arrays. Nucleic Acids Res 30:3732–3738. [PubMed] 10.1093/nar/gkf505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gardner TS, di Bernardo D, Lorenz D, Collins JJ. 2003. Inferring genetic networks and identifying compound mode of action via expression profiling. Science 301:102–105. [PubMed] 10.1126/science.1081900 [DOI] [PubMed] [Google Scholar]
- 230.Serrania J, Vorhölter FJ, Niehaus K, Pühler A, Becker A. 2008. Identification of Xanthomonas campestris pv. campestris galactose utilization genes from transcriptome data. J Biotechnol 135:309–317. [PubMed] 10.1016/j.jbiotec.2008.04.011 [DOI] [PubMed] [Google Scholar]
- 231.Park JH, Lee SY, Kim TY, Kim HU. 2008. Application of systems biology for bioprocess development. Trends Biotechnol 26:404–412. [PubMed] 10.1016/j.tibtech.2008.05.001 [DOI] [PubMed] [Google Scholar]
- 232.Han MJ, Lee SY. 2006. The Escherichia coli proteome: past, present, and future prospects. Microbiol Mol Biol Rev 70:362–439. [PubMed] 10.1128/MMBR.00036-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Choe LH, Lee KH. 2000. A comparison of three commercially available isoelectric focusing units for proteome analysis: the multiphor, the IPGphor and the protean IEF cell. Electrophoresis 21:993–1000. 10.1002/(SICI)1522-2683(20000301)21:5<993::AID-ELPS993>3.0.CO;2-9 [DOI] [PubMed] [Google Scholar]
- 234.Gygi SP, Corthals GL, Zhang Y, Rochon Y, Aebersold R. 2000. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc Natl Acad Sci USA 97:9390–9395. [PubMed] 10.1073/pnas.160270797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Aebersold R, Mann M. 2003. Mass spectrometry-based proteomics. Nature 422:198–207. [PubMed] 10.1038/nature01511 [DOI] [PubMed] [Google Scholar]
- 236.Birch RM, O’Bryne C, Booth IR, Cash P. 2003. Enrichment of Escherichia coli proteins by column chromatography on reactive dye columns. Proteomics 3:764–776. [PubMed] 10.1002/pmic.200300397 [DOI] [PubMed] [Google Scholar]
- 237.Gevaert K, Van Damme J, Goethals M, Thomas GR, Hoorelbeke B, Demol H, Martens L, Puype M, Staes A, Vandekerckhove J. 2002. Chromatographic isolation of methionine-containing peptides for gel-free proteome analysis - identification of more than 800 Escherichia coli proteins. Mol Cell Proteomics 1:896–903. 10.1074/mcp.M200061-MCP200 [DOI] [PubMed] [Google Scholar]
- 238.Oldiges M, Lutz S, Pflug S, Schroer K, Stein N, Wiendahl C. 2007. Metabolomics: current state and evolving methodologies and tools. Appl Microbiol Biotechnol 76:495–511. [PubMed] 10.1007/s00253-007-1029-2 [DOI] [PubMed] [Google Scholar]
- 239.De Graaf AA, Striegel K, Wittig RM, Laufer B, Schmitz G, Wiechert W, Sprenger GA, Sahm H. 1999. Metabolic state of Zymomonas mobilis in glucose-, fructose-, and xylose-fed continuous cultures as analysed by 13C- and 31P-NMR spectroscopy. Arch Microbiol 171:371–385. [PubMed] 10.1007/s002030050724 [DOI] [PubMed] [Google Scholar]
- 240.Bhattacharya M, Fuhrman L, Ingram A, Nickerson KW, Conway T. 1995. Single-run separation and detection of multiple metabolic intermediates by anion-exchange high-performance liquid-chromatography and application to cell pool extracts prepared from Escherichia coli. Anal Biochem 232:98–106. [PubMed] 10.1006/abio.1995.9954 [DOI] [PubMed] [Google Scholar]
- 241.Castrillo JI, Hayes A, Mohammed S, Gaskell SJ, Oliver SG. 2003. An optimized protocol for metabolome analysis in yeast using direct infusion electrospray mass spectrometry. Phytochemistry 62:929–937. [PubMed] 10.1016/S0031-9422(02)00713-6 [DOI] [PubMed] [Google Scholar]
- 242.Mashego MR, Rumbold K, De Mey M, Vandamme E, Soetaert W, Heijnen JJ. 2007. Microbial metabolomics: past, present and future methodologies. Biotechnol Lett 29:1–16. [PubMed] 10.1007/s10529-006-9218-0 [DOI] [PubMed] [Google Scholar]
- 243.Kohlstedt M, Becker J, Wittmann C. 2010. Metabolic fluxes and beyond-systems biology understanding and engineering of microbial metabolism. Appl Microbiol Biotechnol 88:1065–1075. [PubMed] 10.1007/s00253-010-2854-2 [DOI] [PubMed] [Google Scholar]
- 244.Kim HU, Kim TY, Lee SY. 2008. Metabolic flux analysis and metabolic engineering of microorganisms. Mol Biosyst 4:113–120. [PubMed] 10.1039/B712395G [DOI] [PubMed] [Google Scholar]
- 245.Zamboni N, Fendt SM, Rühl M, Sauer U. 2009. (13)C-based metabolic flux analysis. Nat Protoc 4:878–892. [PubMed] 10.1038/nprot.2009.58 [DOI] [PubMed] [Google Scholar]
- 246.Price ND, Reed JL, Palsson BØ. 2004. Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat Rev Microbiol 2:886–897. [PubMed] 10.1038/nrmicro1023 [DOI] [PubMed] [Google Scholar]
- 247.Hong SH, Kim JS, Lee SY, In YH, Choi SS, Rih JK, Kim CH, Jeong H, Hur CG, Kim JJ. 2004. The genome sequence of the capnophilic rumen bacterium Mannheimia succiniciproducens. Nat Biotechnol 22:1275–1281. [PubMed] 10.1038/nbt1010 [DOI] [PubMed] [Google Scholar]
- 248.Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L. 2001. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292:929–934. [PubMed] 10.1126/science.292.5518.929 [DOI] [PubMed] [Google Scholar]
- 249.Yoon SH, Han MJ, Jeong H, Lee CH, Xia XX, Lee DH, Shim JH, Lee SY, Oh TK, Kim JF. 2012. Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol 13:R37. 10.1186/gb-2012-13-5-r37. [PubMed] 10.1186/gb-2012-13-5-r37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ishii N, Nakahigashi K, Baba T, Robert M, Soga T, Kanai A, Hirasawa T, Naba M, Hirai K, Hoque A, Ho PY, Kakazu Y, Sugawara K, Igarashi S, Harada S, Masuda T, Sugiyama N, Togashi T, Hasegawa M, Takai Y, Yugi K, Arakawa K, Iwata N, Toya Y, Nakayama Y, Nishioka T, Shimizu K, Mori H, Tomita M. 2007. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science 316:593–597. [PubMed] 10.1126/science.1132067 [DOI] [PubMed] [Google Scholar]
- 251.Keseler IM, Collado-Vides J, Gama-Castro S, Ingraham J, Paley S, Paulsen IT, Peralta-Gill M, Karp PD. 2005. EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res 33:D334–D337. [PubMed] 10.1093/nar/gki108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Keseler IM, Bonavides-Martinez C, Collado-Vides J, Gama-Castro S, Gunsalus RP, Johnson DA, Krummenacker M, Nolan LM, Paley S, Paulsen IT, Peralta-Gil M, Santos-Zavaleta A, Shearer AG, Karp PD. 2009. EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Res 37:D464–D470. [PubMed] 10.1093/nar/gkn751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Karp PD, Weaver D, Paley S, Fulcher C, Kubo A, Kothari A, Krummenacker M, Subhraveti P, Weerasinghe D, Gama-Castro S, Huerta AM, Muñiz-Rascado L, Bonavides-Martinez C, Weiss V, Peralta-Gil M, Santos-Zavaleta A, Schröder I, Mackie A, Gunsalus R, Collado-Vides J, Keseler IM, Paulsen I. 2014. The EcoCyc Database. In Kaper JB (ed), EcoSal Plus 2014. ASM Press, Washington, DC. 10.1128/ecosalplus.ESP-0009-2013. 10.1128/ecosalplus.ESP-0009-2013 [DOI] [Google Scholar]
- 254.Varma A, Palsson BO. 1993. Metabolic capabilities of Escherichia coli. II. Optimal growth patterns. J Theor Biol 165:503–522. 10.1006/jtbi.1993.1203 [DOI] [PubMed] [Google Scholar]
- 255.Varma A, Palsson BØ. 1994. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl Environ Microbiol 60:3724–3731. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Orth JD, Conrad TM, Na J, Lerman JA, Nam H, Feist AM, Palsson BØ. 2011. A comprehensive genome-scale reconstruction of Escherichia coli metabolism—2011. Mol Syst Biol 7:535. 10.1038/msb.2011.65. [PubMed] 10.1038/msb.2011.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Monk JM, Charusanti P, Aziz RK, Lerman JA, Premyodhin N, Orth JD, Feist AM, Palsson BØ. 2013. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc Natl Acad Sci USA 110:20338–20343. [PubMed] 10.1073/pnas.1307797110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Choi HS, Lee SY, Kim TY, Woo HM. 2010. In silico identification of gene amplification targets for improvement of lycopene production. Appl Environ Microbiol 76:3097–3105. [PubMed] 10.1128/AEM.00115-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Feist AM, Palsson BØ. 2008. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol 26:659–667. [PubMed] 10.1038/nbt1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Durot M, Bourguignon PY, Schachter V. 2009. Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol Rev 33:164–190. [PubMed] 10.1111/j.1574-6976.2008.00146.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.McCloskey D, Palsson BØ, Feist AM. 2013. Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol Syst Biol 9:661. 10.1038/msb.2013.18. [PubMed] 10.1038/msb.2013.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Garcia-Albornoz MA, Nielsen J. 2013. Application of genome-scale metabolic models in metabolic engineering. Ind Biotechnol 9:203–214. 10.1089/ind.2013.0011 [DOI] [Google Scholar]
- 263.Kim B, Kim WJ, Kim DI, Lee SY. 2015. Applications of genome-scale metabolic network model in metabolic engineering. J Ind Microbiol Biotechnol 42:339–348. [PubMed] 10.1007/s10295-014-1554-9 [DOI] [PubMed] [Google Scholar]
- 264.Orth J, Fleming R, Palsson B. 2010. Reconstruction and use of microbial metabolic networks: the core Escherichia coli metabolic model as an educational guide. In Karp PD (ed), EcoSal Plus 2010. ASM Press, Washington, DC. 10.1128/ecosalplus.10.2.1. 10.1128/ecosalplus.10.2.1 [DOI] [PubMed] [Google Scholar]
- 265.Thiele I, Palsson BØ. 2010. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc 5:93–121. [PubMed] 10.1038/nprot.2009.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Lee SY, Woo HM, Lee DY, Choi HS, Kim TY, Yun H. 2005. Systems-level analysis of genome-scale in silico metabolic models using MetaFluxNet. Biotechnol Bioprocess Eng 10:425–431. 10.1007/BF02989825 [DOI] [Google Scholar]
- 267.Weaver DS, Keseler IM, Mackie A, Paulsen IT, Karp PD. 2014. A genome-scale metabolic flux model of Escherichia coli K-12 derived from the EcoCyc database. BMC Syst Biol 8:79. 10.1186/1752-0509-8-79. [PubMed] 10.1186/1752-0509-8-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Edwards JS, Palsson BØ. 1998. How will bioinformatics influence metabolic engineering? Biotechnol Bioeng 58:162–169. [PubMed] 10.1002/(SICI)1097-0290(19980420)58:2/3<162::AID-BIT8>3.0.CO;2-J [DOI] [PubMed] [Google Scholar]
- 269.Orth JD, Thiele I, Palsson BØ. 2010. What is flux balance analysis? Nat Biotechnol 28:245–248. [PubMed] 10.1038/nbt.1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Segre D, Vitkup D, Church GM. 2002. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci USA 99:15112–15117. [PubMed] 10.1073/pnas.232349399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Shlomi T, Berkman O, Ruppin E. 2005. Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc Natl Acad Sci USA 102:7695–7700. [PubMed] 10.1073/pnas.0406346102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ranganathan S, Suthers PF, Maranas CD. 2010. OptForce: an optimization procedure for identifying all genetic manipulations leading to targeted overproductions. PLoS Comput Biol 6:e1000744. 10.1371/journal.pcbi.1000744. 10.1371/journal.pcbi.1000744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Pharkya P, Burgard AP, Maranas CD. 2003. Exploring the overproduction of amino acids using the bilevel optimization framework OptKnock. Biotechnol Bioeng 84:887–899. [PubMed] 10.1002/bit.10857 [DOI] [PubMed] [Google Scholar]
- 274.Patil KR, Rocha I, Forster J, Nielsen J. 2005. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinformatics 6:308. 10.1186/1471-2105-6-308. [PubMed] 10.1186/1471-2105-6-308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.King ZA, Feist AM. 2013. Optimizing cofactor specificity of oxidoreductase enzymes for the generation of microbial production strains—OptSwap. Ind Biotechnol 9:236–246. 10.1089/ind.2013.0005 [DOI] [Google Scholar]
- 276.Park JM, Park HM, Kim WJ, Kim HU, Kim TY, Lee SY. 2012. Flux variability scanning based on enforced objective flux for identifying gene amplification targets. BMC Syst Biol 6:106. 10.1186/1752-0509-6-106. [PubMed] 10.1186/1752-0509-6-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Cotten C, Reed JL. 2013. Constraint-based strain design using continuous modifications (CosMos) of flux bounds finds new strategies for metabolic engineering. Biotechnol J 8:595–604. [PubMed] 10.1002/biot.201200316 [DOI] [PubMed] [Google Scholar]
- 278.Becker SA, Feist AM, Mo ML, Hannum G, Palsson BØ, Herrgard MJ. 2007. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc 2:727–738. [PubMed] 10.1038/nprot.2007.99 [DOI] [PubMed] [Google Scholar]
- 279.Schellenberger J, Que R, Fleming RMT, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J, Hyduke DR, Palsson BØ. 2011. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc 6:1290–1307. [PubMed] 10.1038/nprot.2011.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Lee DY, Yun H, Park S, Lee SY. 2003. MetaFluxNet: the management of metabolic reaction information and quantitative metabolic flux analysis. Bioinformatics 19:2144–2146. [PubMed] 10.1093/bioinformatics/btg271 [DOI] [PubMed] [Google Scholar]
- 281.Lee SY, Lee DY, Hong SH, Kim TY, Yun H, Oh YG, Park S. 2003. MetaFluxNet, a program package for metabolic pathway construction and analysis, and its use in large-scale metabolic flux analysis of Escherichia coli. Genome Inform 14:23–33. [PubMed] [PubMed] [Google Scholar]
- 282.Karp PD, Paley S, Romero P. 2002. The pathway tools software. Bioinformatics 18:S225–S232. [PubMed] 10.1093/bioinformatics/18.suppl_1.S225 [DOI] [PubMed] [Google Scholar]
- 283.McShan DC, Rao S, Shah I. 2003. PathMiner: predicting metabolic pathways by heuristic search. Bioinformatics 19:1692–1698. [PubMed] 10.1093/bioinformatics/btg217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Hatzimanikatis V, Li CH, Ionita JA, Henry CS, Jankowski MD, Broadbelt LJ. 2005. Exploring the diversity of complex metabolic networks. Bioinformatics 21:1603–1609. [PubMed] 10.1093/bioinformatics/bti213 [DOI] [PubMed] [Google Scholar]
- 285.Rahman SA, Advani P, Schunk R, Schrader R, Schomburg D. 2005. Metabolic pathway analysis web service (Pathway Hunter Tool at CUBIC). Bioinformatics 21:1189–1193. [PubMed] 10.1093/bioinformatics/bti116 [DOI] [PubMed] [Google Scholar]
- 286.Rodrigo G, Carrera J, Prather KJ, Jaramillo A. 2008. DESHARKY: automatic design of metabolic pathways for optimal cell growth. Bioinformatics 24:2554–2556. [PubMed] 10.1093/bioinformatics/btn471 [DOI] [PubMed] [Google Scholar]
- 287.Fenner K, Gao JF, Kramer S, Ellis L, Wackett L. 2008. Data-driven extraction of relative reasoning rules to limit combinatorial explosion in biodegradation pathway prediction. Bioinformatics 24:2079–2085. [PubMed] 10.1093/bioinformatics/btn378 [DOI] [PubMed] [Google Scholar]
- 288.Blum T, Kohlbacher O. 2008. MetaRoute: fast search for relevant metabolic routes for interactive network navigation and visualization. Bioinformatics 24:2108–2109. [PubMed] 10.1093/bioinformatics/btn360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Mithani A, Preston GM, Hein J. 2009. Rahnuma: hypergraph-based tool for metabolic pathway prediction and network comparison. Bioinformatics 25:1831–1832. [PubMed] 10.1093/bioinformatics/btp269 [DOI] [PubMed] [Google Scholar]
- 290.Pitkanen E, Jouhten P, Rousu J. 2009. Inferring branching pathways in genome-scale metabolic networks. BMC Syst Biol 3:103. 10.1186/1752-0509-3-103. [PubMed] 10.1186/1752-0509-3-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Heath AP, Bennett GN, Kavraki LE. 2010. Finding metabolic pathways using atom tracking. Bioinformatics 26:1548–1555. [PubMed] 10.1093/bioinformatics/btq223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Cho A, Yun H, Park JH, Lee SY, Park S. 2010. Prediction of novel synthetic pathways for the production of desired chemicals. BMC Syst Biol 4:35. 10.1186/1752-0509-4-35. [PubMed] 10.1186/1752-0509-4-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Moriya Y, Shigemizu D, Hattori M, Tokimatsu T, Kotera M, Goto S, Kanehisa M. 2010. PathPred: an enzyme-catalyzed metabolic pathway prediction server. Nucleic Acids Res 38:W138–W143. [PubMed] 10.1093/nar/gkq318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Carbonell P, Planson AG, Fichera D, Faulon JL. 2011. A retrosynthetic biology approach to metabolic pathway design for therapeutic production. BMC Syst Biol 5:122. 10.1186/1752-0509-5-122. [PubMed] 10.1186/1752-0509-5-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Carbonell P, Parutto P, Herisson J, Pandit SB, Faulon JL. 2014. XTMS: pathway design in an eXTended metabolic space. Nucleic Acids Res 42:W389–W394. [PubMed] 10.1093/nar/gku362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Campodonico MA, Andrews BA, Asenjo JA, Palsson BØ, Feist AM. 2014. Generation of an atlas for commodity chemical production in Escherichia coli and a novel pathway prediction algorithm, GEM-Path. Metab Eng 25:140–158. [PubMed] 10.1016/j.ymben.2014.07.009 [DOI] [PubMed] [Google Scholar]
- 297.Latendresse M, Krummenacker M, Karp PD. 2014. Optimal metabolic route search based on atom mappings. Bioinformatics 30:2043–2050. [PubMed] 10.1093/bioinformatics/btu150 [DOI] [PubMed] [Google Scholar]
- 298.Shin JH, Kim HU, Kim DI, Lee SY. 2013. Production of bulk chemicals via novel metabolic pathways in microorganisms. Biotechnol Adv 31:925–935. [PubMed] 10.1016/j.biotechadv.2012.12.008 [DOI] [PubMed] [Google Scholar]
- 299.Planson AG, Carbonell P, Grigoras I, Faulon JL. 2011. Engineering antibiotic production and overcoming bacterial resistance. Biotechnol J 6:812–825. [PubMed] 10.1002/biot.201100085 [DOI] [PubMed] [Google Scholar]
- 300.Medema MH, van Raaphorst R, Takano E, Breitling R. 2012. Computational tools for the synthetic design of biochemical pathways. Nat Rev Microbiol 10:191–202. [PubMed] 10.1038/nrmicro2717 [DOI] [PubMed] [Google Scholar]
- 301.Kroll J, Klinter S, Schneider C, Voss I, Steinbuchel A. 2010. Plasmid addiction systems: perspectives and applications in biotechnology. Microb Biotechnol 3:634–657. [PubMed] 10.1111/j.1751-7915.2010.00170.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Johnson IS. 1983. Human insulin from recombinant DNA technology. Science 219:632–637. [PubMed] 10.1126/science.6337396 [DOI] [PubMed] [Google Scholar]
- 303.Williams SG, Cranenburgh RM, Weiss AME, Wrighton CJ, Sherratt DJ, Hanak JAJ. 1998. Repressor titration: a novel system for selection and stable maintenance of recombinant plasmids. Nucleic Acids Res 26:2120–2124. [PubMed] 10.1093/nar/26.9.2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Lehnherr H, Maguin E, Jafri S, Yarmolinsky MB. 1993. Plasmid addiction genes of bacteriophage-P1 - doc, which causes cell-death on curing of prophage, and phd, which prevents host death when prophage is retained. J Mol Biol 233:414–428. [PubMed] 10.1006/jmbi.1993.1521 [DOI] [PubMed] [Google Scholar]
- 305.Gazit E, Sauer RT. 1999. Stability and DNA binding of the Phd protein of the phage P1 plasmid addiction system. J Biol Chem 274:2652–2657. [PubMed] 10.1074/jbc.274.5.2652 [DOI] [PubMed] [Google Scholar]
- 306.Cranenburgh RM, Hanak JAJ, Williams SG, Sherratt DJ. 2001. Escherichia coli strains that allow antibiotic-free plasmid selection and maintenance by repressor titration. Nucleic Acids Res 29:e26. 10.1093/nar/29.5.e26. [PubMed] 10.1093/nar/29.5.e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Mccoy AJ, Adams NE, Hudson AO, Gilvarg C, Leustek T, Maurelli AT. 2006. L,L-Diaminopimelate aminotransferase, a trans-kingdom enzyme shared by Chlamydia and plants for synthesis of diaminopimelate/lysine. Proc Natl Acad Sci USA 103:17909–17914. [PubMed] 10.1073/pnas.0608643103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Voss I, Steinbuchel A. 2006. Application of a KDPG-aldolase gene-dependent addiction system for enhanced production of cyanophycin in Ralstonia eutropha strain H16. Metab Eng 8:66–78. [PubMed] 10.1016/j.ymben.2005.09.003 [DOI] [PubMed] [Google Scholar]
- 309.Song CW, Kim DI, Choi S, Jang JW, Lee SY. 2013. Metabolic engineering of Escherichia coli for the production of fumaric acid. Biotechnol Bioeng 110:2025–2034. [PubMed] 10.1002/bit.24868 [DOI] [PubMed] [Google Scholar]
- 310.Millard CS, Chao YP, Liao JC, Donnelly MI. 1996. Enhanced production of succinic acid by overexpression of phosphoenolpyruvate carboxylase in Escherichia coli. Appl Environ Microbiol 62:1808–1810. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Qian ZG, Xia XX, Lee SY. 2009. Metabolic engineering of Escherichia coli for the production of putrescine: a four carbon diamine. Biotechnol Bioeng 104:651–662. [PubMed] [DOI] [PubMed] [Google Scholar]
- 312.Qian ZG, Xia XX, Lee SY. 2011. Metabolic engineering of Escherichia coli for the production of cadaverine: a five carbon diamine. Biotechnol Bioeng 108:93–103. [PubMed] 10.1002/bit.22918 [DOI] [PubMed] [Google Scholar]
- 313.Koma D, Yamanaka H, Moriyoshi K, Ohmoto T, Sakai K. 2012. A convenient method for multiple insertions of desired genes into target loci on the Escherichia coli chromosome. Appl Microbiol Biotechnol 93:815–829. [PubMed] 10.1007/s00253-011-3735-z [DOI] [PubMed] [Google Scholar]
- 314.Choi YJ, Park JH, Kim TY, Lee SY. 2012. Metabolic engineering of Escherichia coli for the production of 1-propanol. Metab Eng 14:477–486. [PubMed] 10.1016/j.ymben.2012.07.006 [DOI] [PubMed] [Google Scholar]
- 315.Hanai T, Atsumi S, Liao JC. 2007. Engineered synthetic pathway for isopropanol production in Escherichia coli. Appl Environ Microbiol 73:7814–7818. [PubMed] 10.1128/AEM.01140-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Inokuma K, Liao JC, Okamoto M, Hanai T. 2010. Improvement of isopropanol production by metabolically engineered Escherichia coli using gas stripping. J Biosci Bioeng 110:696–701. [PubMed] 10.1016/j.jbiosc.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 317.Draths KM, Frost JW. 1995. Environmentally compatible synthesis of catechol from D-glucose. J Am Chem Soc 117:2395–2400. 10.1021/ja00114a003 [DOI] [Google Scholar]
- 318.Barker JL, Frost JW. 2001. Microbial synthesis of p-hydroxybenzoic acid from glucose. Biotechnol Bioeng 76:376–390. [PubMed] 10.1002/bit.10160 [DOI] [PubMed] [Google Scholar]
- 319.Kim B, Park H, Na D, Lee SY. 2014. Metabolic engineering of Escherichia coli for the production of phenol from glucose. Biotechnol J 9:621–629. [PubMed] 10.1002/biot.201300263 [DOI] [PubMed] [Google Scholar]
- 320.Zhang FZ, Carothers JM, Keasling JD. 2012. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat Biotechnol 30:354–359. [PubMed] 10.1038/nbt.2149 [DOI] [PubMed] [Google Scholar]
- 321.Schirmer A, Rude MA, Li XZ, Popova E, del Cardayre SB. 2010. Microbial biosynthesis of alkanes. Science 329:559–562. [PubMed] 10.1126/science.1187936 [DOI] [PubMed] [Google Scholar]
- 322.Kalscheuer R, Stolting T, Steinbuchel A. 2006. Microdiesel: Escherichia coli engineered for fuel production. Microbiology 152:2529–2536. [PubMed] 10.1099/mic.0.29028-0 [DOI] [PubMed] [Google Scholar]
- 323.Sun T, Miao LT, Li QY, Dai GP, Lu FP, Liu T, Zhang XL, Ma YH. 2014. Production of lycopene by metabolically-engineered Escherichia coli. Biotechnol Lett 36:1515–1522. [PubMed] 10.1007/s10529-014-1543-0 [DOI] [PubMed] [Google Scholar]
- 324.Yu JL, Xia XX, Zhong JJ, Qian ZG. 2014. Direct biosynthesis of adipic acid from a synthetic pathway in recombinant Escherichia coli. Biotechnol Bioeng 111:2580–2586. [PubMed] 10.1002/bit.25293 [DOI] [PubMed] [Google Scholar]
- 325.Rodrigues AL, Trachtmann N, Becker J, Lohanatha AF, Blotenberg J, Bolten CJ, Korneli C, Lima AOD, Porto LM, Sprenger GA, Wittmann C. 2013. Systems metabolic engineering of Escherichia coli for production of the antitumor drugs violacein and deoxyviolacein. Metabol Eng 20:29–41. [PubMed] 10.1016/j.ymben.2013.08.004 [DOI] [PubMed] [Google Scholar]
- 326.Balderas-Hernandez VE, Trevino-Quintanilla LG, Hernandez-Chavez G, Martinez A, Bolivar F, Gosset G. 2014. Catechol biosynthesis from glucose in Escherichia coli anthranilate-overproducer strains by heterologous expression of anthranilate 1,2-dioxygenase from Pseudomonas aeruginosa PAO1. Microb Cell Fact 13:136. 10.1186/s12934-014-0136-x. 10.1186/s12934-014-0136-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Lin Y, Shen X, Yuan Q, Yan Y. 2013. Microbial biosynthesis of the anticoagulant precursor 4-hydroxycoumarin. Nat Commun 4:2603. 10.1038/ncomms3603. [PubMed] 10.1038/ncomms3603 [DOI] [PubMed] [Google Scholar]
- 328.Koma D, Yamanaka H, Moriyoshi K, Sakai K, Masuda T, Sato Y, Toida K, Ohmoto T. 2014. Production of p-aminobenzoic acid by metabolically engineered Escherichia coli. Biosci Biotechnol Biochem 78:350–357. [PubMed] 10.1080/09168451.2014.878222 [DOI] [PubMed] [Google Scholar]
- 329.McKenna R, Nielsen DR. 2011. Styrene biosynthesis from glucose by engineered E. coli. Metab Eng 13:544–554. [PubMed] 10.1016/j.ymben.2011.06.005 [DOI] [PubMed] [Google Scholar]
- 330.McKenna R, Pugh S, Thompson B, Nielsen DR. 2013. Microbial production of the aromatic building-blocks (S)-styrene oxide and (R)-1,2-phenylethanediol from renewable resources. Biotechnol J 8:1465–1475. [PubMed] 10.1002/biot.201300035 [DOI] [PubMed] [Google Scholar]
- 331.Kang Z, Zhang CZ, Du GC, Chen J. 2014. Metabolic engineering of Escherichia coli for production of 2-phenylethanol from renewable glucose. Appl Biochem Biotechnol 172:2012–2021. [PubMed] 10.1007/s12010-013-0659-3 [DOI] [PubMed] [Google Scholar]
- 332.Rodriguez GM, Tashiro Y, Atsumi S. 2014. Expanding ester biosynthesis in Escherichia coli. Nat Chem Biol 10:259–265. [PubMed] 10.1038/nchembio.1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Lan EI, Dekishima Y, Chuang DS, Liao JC. 2013. Metabolic engineering of 2-pentanone synthesis in Escherichia coli. AIChE J 59:3167–3175. 10.1002/aic.14086 [DOI] [Google Scholar]
- 334.Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJY, Hanai T, Liao JC. 2008. Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng 10:305–311. [PubMed] 10.1016/j.ymben.2007.08.003 [DOI] [PubMed] [Google Scholar]
- 335.Baez A, Cho KM, Liao JC. 2011. High-flux isobutanol production using engineered Escherichia coli: a bioreactor study with in situ product removal. Appl Microbiol Biotechnol 90:1681–1690. [PubMed] 10.1007/s00253-011-3173-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Dekishima Y, Lan EI, Shen CR, Cho KM, Liao JC. 2011. Extending carbon chain length of 1-butanol pathway for 1-hexanol synthesis from glucose by engineered Escherichia coli. J Am Chem Soc 133:11399–11401. [PubMed] 10.1021/ja203814d [DOI] [PubMed] [Google Scholar]
- 337.Jantama K, Zhang X, Moore JC, Shanmugam KT, Svoronos SA, Ingram LO. 2008. Eliminating side products and increasing succinate yields in engineered strains of Escherichia coli C. Biotechnol Bioeng 101:881–893. [PubMed] 10.1002/bit.22005 [DOI] [PubMed] [Google Scholar]
- 338.Jung YK, Kim TY, Park SJ, Lee SY. 2010. Metabolic engineering of Escherichia coli for the production of polylactic acid and its copolymers. Biotechnol Bioeng 105:161–171. [PubMed] 10.1002/bit.22548 [DOI] [PubMed] [Google Scholar]
- 339.Harcum SW, Bentley WE. 1999. Heat-shock and stringent responses have overlapping protease activity in Escherichia coli. Implications for heterologous protein yield. Appl Biochem Biotechnol 80:23–37. [PubMed] 10.1385/ABAB:80:1:23 [DOI] [PubMed] [Google Scholar]
- 340.Corchero JL, Villaverde A. 1998. Plasmid maintenance in Escherichia coli recombinant cultures is dramatically, steadily, and specifically influenced by features of the encoded proteins. Biotechnol Bioeng 58:625–632. [PubMed] 10.1002/(SICI)1097-0290(19980620)58:6<625::AID-BIT8>3.0.CO;2-K [DOI] [PubMed] [Google Scholar]
- 341.Friehs K. 2004. Plasmid copy number and plasmid stability. Adv Biochem Eng Biotechnol 86:47–82. [PubMed] 10.1007/b12440 [DOI] [PubMed] [Google Scholar]
- 342.Hoch JA, Silhavy TJ. 1995. Two-Component Signal Transduction. ASM Press, Washington, DC. [Google Scholar]
- 343.Farmer WR, Liao JC. 2000. Improving lycopene production in Escherichia coli by engineering metabolic control. Nat Biotechnol 18:533–537. [PubMed] 10.1038/75398 [DOI] [PubMed] [Google Scholar]
- 344.Reitzer LJ, Magasanik B. 1985. Expression of glnA in Escherichia coli is regulated at tandem promoters. Proc Natl Acad Sci USA 82:1979–1983. 10.1073/pnas.82.7.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Cronan JE, Jr. 1997. In vivo evidence that acyl coenzyme A regulates DNA binding by the Escherichia coli FadR global transcription factor. J Bacteriol 179:1819–1823. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Ajikumar PK, Xiao WH, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G. 2010. Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science 330:70–74. [PubMed] 10.1126/science.1191652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Zhao J, Li Q, Sun T, Zhu X, Xu H, Tang J, Zhang X, Ma Y. 2013. Engineering central metabolic modules of Escherichia coli for improving beta-carotene production. Metab Eng 17:42–50. [PubMed] 10.1016/j.ymben.2013.02.002 [DOI] [PubMed] [Google Scholar]
- 348.Lv L, Ren YL, Chen JC, Wu Q, Chen GQ. 2015. Application of CRISPRi for prokaryotic metabolic engineering involving multiple genes, a case study: Controllable P(3HB-co-4HB) biosynthesis. Metab Eng 29:160–168. [PubMed] 10.1016/j.ymben.2015.03.013 [DOI] [PubMed] [Google Scholar]
- 349.Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. 2015. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol 81:2506–2514. [PubMed] 10.1128/AEM.04023-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Balderas-Hernandez VE, Sabido-Ramos A, Silva P, Cabrera-Valladares N, Hernandez-Chavez G, Baez-Viveros JL, Martinez A, Bolivar F, Gosset G. 2009. Metabolic engineering for improving anthranilate synthesis from glucose in Escherichia coli. Microb Cell Fact 8:19. 10.1186/1475-2859-8-19. [PubMed] 10.1186/1475-2859-8-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Connor MR, Cann AF, Liao JC. 2010. 3-Methyl-1-butanol production in Escherichia coli: random mutagenesis and two-phase fermentation. Appl Microbiol Biotechnol 86:1155–1164. [PubMed] 10.1007/s00253-009-2401-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Glebes TY, Sandoval NR, Gillis JH, Gill RT. 2015. Comparison of genome-wide selection strategies to identify furfural tolerance genes in Escherichia coli. Biotechnol Bioeng 112:129–140. [PubMed] 10.1002/bit.25325 [DOI] [PubMed] [Google Scholar]
- 353.Parekh S, Vinci VA, Strobel RJ. 2000. Improvement of microbial strains and fermentation processes. Appl Microbiol Biotechnol 54:287–301. [PubMed] 10.1007/s002530000403 [DOI] [PubMed] [Google Scholar]
- 354.Aldor IS, Krawitz DC, Forrest W, Chen C, Nishihara JC, Joly JC, Champion KM. 2005. Proteomic profiling of recombinant Escherichia coli in high-cell-density fermentations for improved production of an antibody fragment biopharmaceutical. Appl Environ Microbiol 71:1717–1728. [PubMed] 10.1128/AEM.71.4.1717-1728.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Qian ZG, Xia XX, Choi JH, Lee SY. 2008. Proteome-based identification of fusion partner for high-level extracellular production of recombinant proteins in Escherichia coli. Biotechnol Bioeng 101:587–601. [PubMed] 10.1002/bit.21898 [DOI] [PubMed] [Google Scholar]
- 356.He L, Xiao Y, Gebreselassie N, Zhang FZ, Antoniewicz MR, Tang YJJ, Peng LF. 2014. Central metabolic responses to the overproduction of fatty acids in Escherichia coli based on 13C-metabolic flux analysis. Biotechnol Bioeng 111:575–585. [PubMed] 10.1002/bit.25124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Wahl A, El Massaoudi M, Schipper D, Wiechert W, Takors R. 2004. Serial 13C-based flux analysis of an L-phenylalanine-producing E. coli strain using the sensor reactor. Biotechnol Prog 20:706–714. [PubMed] 10.1021/bp0342755 [DOI] [PubMed] [Google Scholar]
- 358.Hashiguchi K, Takesada H, Suzuki E, Matsui H. 1999. Construction of an L-isoleucine overproducing strain of Escherichia coli K-12. Biosci Biotechnol Biochem 63:672–679. [PubMed] 10.1271/bbb.63.672 [DOI] [PubMed] [Google Scholar]
- 359.Alper H, Jin YS, Moxley JF, Stephanopoulos G. 2005. Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab Eng 7:155–164. [PubMed] 10.1016/j.ymben.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 360.Fong SS, Burgard AP, Herring CD, Knight EM, Blattner FR, Maranas CD, Palsson BØ. 2005. In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol Bioeng 91:643–648. [PubMed] 10.1002/bit.20542 [DOI] [PubMed] [Google Scholar]
- 361.Pharkya P, Maranas CD. 2006. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab Eng 8:1–13. [PubMed] 10.1016/j.ymben.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 362.Chowdhury A, Zomorrodi AR, Maranas CD. 2014. k-OptForce: integrating kinetics with flux balance analysis for strain design. PLoS Comput Biol 10:e1003487. 10.1371/journal.pcbi.1003487. [PubMed] 10.1371/journal.pcbi.1003487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Ishihara K, Matsuoka M, Inoue Y, Tanase S, Ogawa T, Fukuda H. 1995. Overexpression and in vitro reconstitution of the ethylene-forming enzyme from Pseudomonas syringae. J Ferment Bioeng 79:205–211. 10.1016/0922-338X(95)90604-X [DOI] [Google Scholar]
- 364.Liu HW, Ramos KRM, Valdehuesa KNG, Nisola GM, Lee WK, Chung WJ. 2013. Biosynthesis of ethylene glycol in Escherichia coli. Appl Microbiol Biotechnol 97:3409–3417. [PubMed] 10.1007/s00253-012-4618-7 [DOI] [PubMed] [Google Scholar]
- 365.Niu W, Molefe MN, Frost JW. 2003. Microbial synthesis of the energetic material precursor 1,2,4-butanetriol. J Am Chem Soc 125:12998–12999. [PubMed] 10.1021/ja036391+ [DOI] [PubMed] [Google Scholar]
- 366.Li X, Cai Z, Li Y, Zhang Y. 2014. Design and construction of a non-natural malate to 1,2,4-butanetriol pathway creates possibility to produce 1,2,4-butanetriol from glucose. Sci Rep 4:5541. 10.1038/srep05541. [PubMed] 10.1038/srep05541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Alonso-Gutierrez J, Chan R, Batth TS, Adams PD, Keasling JD, Petzold CJ, Lee TS. 2013. Metabolic engineering of Escherichia coli for limonene and perillyl alcohol production. Metab Eng 19:33–41. [PubMed] 10.1016/j.ymben.2013.05.004 [DOI] [PubMed] [Google Scholar]
- 368.Yoneda H, Tantillo DJ, Atsumi S. 2014. Biological production of 2-butanone in Escherichia coli. ChemSusChem 7:92–95. [PubMed] 10.1002/cssc.201300853 [DOI] [PubMed] [Google Scholar]
- 369.Niu W, Draths KM, Frost JW. 2002. Benzene-free synthesis of adipic acid. Biotechnol Prog 18:201–211. [PubMed] 10.1021/bp010179x [DOI] [PubMed] [Google Scholar]
- 370.Moon TS, Yoon SH, Lanza AM, Roy-Mayhew JD, Prather KLJ. 2009. Production of glucaric acid from a synthetic pathway in recombinant Escherichia coli. Appl Environ Microbiol 75:589–595. [PubMed] 10.1128/AEM.00973-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Moon TS, Dueber JE, Shiue E, Prather KLJ. 2010. Use of modular, synthetic scaffolds for improved production of glucaric acid in engineered E. coli. Metab Eng 12:298–305. [PubMed] 10.1016/j.ymben.2010.01.003 [DOI] [PubMed] [Google Scholar]
- 372.Martin VJJ, Pitera DJ, Withers ST, Newman JD, Keasling JD. 2003. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol 21:796–802. [PubMed] 10.1038/nbt833 [DOI] [PubMed] [Google Scholar]
- 373.Zurbriggen A, Kirst H, Melis A. 2012. Isoprene production via the mevalonic acid pathway in Escherichia coli (Bacteria). Bioenergy Res 5:814–828. 10.1007/s12155-012-9192-4 [DOI] [Google Scholar]
- 374.Kroll J, Klinter S, Steinbuchel A. 2011. A novel plasmid addiction system for large-scale production of cyanophycin in Escherichia coli using mineral salts medium. Appl Microbiol Biotechnol 89:593–604. [PubMed] 10.1007/s00253-010-2899-2 [DOI] [PubMed] [Google Scholar]
- 375.Frey KM, Oppermann-Sanio FB, Schmidt H, Steinbuchel A. 2002. Technical-scale production of cyanophycin with recombinant strains of Escherichia coli. Appl Environ Microbiol 68:3377–3384. [PubMed] 10.1128/AEM.68.7.3377-3384.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Hu XY, Shi QW, Yang T, Jackowski G. 1996. Specific replacement of consecutive AGG codons results in high-level expression of human cardiac troponin T in Escherichia coli. Protein Expr Purif 7:289–293. [PubMed] 10.1006/prep.1996.0041 [DOI] [PubMed] [Google Scholar]
- 377.Bentley WE, Mirjalili N, Andersen DC, Davis RH, Kompala DS. 1990. Plasmid-encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol Bioeng 35:668–681. [PubMed] 10.1002/bit.260350704 [DOI] [PubMed] [Google Scholar]
- 378.Chung H, Kim TY, Lee SY. 2012. Recent advances in production of recombinant spider silk proteins. Curr Opin Biotechnol 23:957–964. [PubMed] 10.1016/j.copbio.2012.03.013 [DOI] [PubMed] [Google Scholar]
- 379.Jiang M, Fang L, Pfeifer BA. 2013. Improved heterologous erythromycin A production through expression plasmid re-design. Biotechnol Prog 29:862–869. [PubMed] 10.1002/btpr.1759 [DOI] [PubMed] [Google Scholar]
- 380.Yomano LP, York SW, Ingram LO. 1998. Isolation and characterization of ethanol-tolerant mutants of Escherichia coli KO11 for fuel ethanol production. J Ind Microbiol Biotechnol 20:132–138. [PubMed] 10.1038/sj.jim.2900496 [DOI] [PubMed] [Google Scholar]
- 381.Lee JY, Yang KS, Jang SA, Sung BH, Kim SC. 2011. Engineering butanol-tolerance in Escherichia coli with artificial transcription factor libraries. Biotechnol Bioeng 108:742–749. [PubMed] 10.1002/bit.22989 [DOI] [PubMed] [Google Scholar]
- 382.Dunlop MJ, Dossani ZY, Szmidt HL, Chu HC, Lee TS, Keasling JD, Hadi MZ, Mukhopadhyay A. 2011. Engineering microbial biofuel tolerance and export using efflux pumps. Mol Syst Biol 7:487. 10.1038/msb.2011.21. [PubMed] 10.1038/msb.2011.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Shah AA, Wang C, Chung YR, Kim JY, Choi ES, Kim SW. 2013. Enhancement of geraniol resistance of Escherichia coli by MarA overexpression. J Biosci Bioeng 115:253–258. [PubMed] 10.1016/j.jbiosc.2012.10.009 [DOI] [PubMed] [Google Scholar]
- 384.Shah AA, Wang C, Yoon SH, Kim JY, Choi ES, Kim SW. 2013. RecA-mediated SOS response provides a geraniol tolerance in Escherichia coli. J Biotechnol 167:357–364. [PubMed] 10.1016/j.jbiotec.2013.07.023 [DOI] [PubMed] [Google Scholar]
- 385.Chubukov V, Mingardon F, Schackwitz W, Baidoo EE, Alonso-Gutierrez J, Hu Q, Lee TS, Keasling JD, Mukhopadhyay A. 2015. Acute limonene toxicity in Escherichia coli is caused by limonene-hydroperoxide and alleviated by a point mutation in alkyl hydroperoxidase (AhpC). Appl Environ Microbiol 81:4690–4696. [PubMed] 10.1128/AEM.01102-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Wörsdörfer B, Woycechowsky KJ, Hilvert D. 2011. Directed evolution of a protein container. Science 331:589–592. [PubMed] 10.1126/science.1199081 [DOI] [PubMed] [Google Scholar]
- 387.Khosla C, Keasling JD. 2003. Metabolic engineering for drug discovery and development. Nat Rev Drug Discov 2:1019–1025. [PubMed] 10.1038/nrd1256 [DOI] [PubMed] [Google Scholar]
- 388.Challis GL, Hopwood DA. 2003. Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci USA 100:14555–14561. [PubMed] 10.1073/pnas.1934677100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Staunton J, Weissman KJ. 2001. Polyketide biosynthesis: a millennium review. Nat Prod Rep 18:380–416. [PubMed] 10.1039/a909079g [DOI] [PubMed] [Google Scholar]
- 390.Watanabe K, Rude MA, Walsh CT, Khosla C. 2003. Engineered biosynthesis of an ansamycin polyketide precursor in Escherichia coli. Proc Natl Acad Sci USA 100:9774–9778. [PubMed] 10.1073/pnas.1632167100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Mutka SC, Carney JR, Liu YQ, Kennedy J. 2006. Heterologous production of epothilone C and D in Escherichia coli. Biochemistry 45:1321–1330. [PubMed] 10.1021/bi052075r [DOI] [PubMed] [Google Scholar]
- 392.Rawlings BJ. 2001. Type I polyketide biosynthesis in bacteria (part A—erythromycin biosynthesis). Nat Prod Rep 18:190–227. [PubMed] 10.1039/b009329g [DOI] [PubMed] [Google Scholar]
- 393.Yuzawa S, Kim W, Katz L, Keasling JD. 2012. Heterologous production of polyketides by modular type I polyketide synthases in Escherichia coli. Curr Opin Biotechnol 23:727–735. [PubMed] 10.1016/j.copbio.2011.12.029 [DOI] [PubMed] [Google Scholar]
- 394.Tsang AW, Horswill AR, Escalante-Semerena JC. 1998. Studies of regulation of expression of the propionate (prpBCDE) operon provide insights into how Salmonella typhimurium LT2 integrates its 1,2-propanediol and propionate catabolic pathways. J Bacteriol 180:6511–6518. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Haller T, Buckel T, Retey J, Gerlt JA. 2000. Discovering new enzymes and metabolic pathways: conversion of succinate to propionate by Escherichia coli. Biochemistry 39:4622–4629. [PubMed] 10.1021/bi992888d [DOI] [PubMed] [Google Scholar]
- 396.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. 2001. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 291:1790–1792. [PubMed] 10.1126/science.1058092 [DOI] [PubMed] [Google Scholar]
- 397.Dayem LC, Carney JR, Santi DV, Pfeifer BA, Khosla C, Kealey JT. 2002. Metabolic engineering of a methylmalonyl-CoA mutase-epimerase pathway for complex polyketide biosynthesis in Escherichia coli. Biochemistry 41:5193–5201. [PubMed] 10.1021/bi015593k [DOI] [PubMed] [Google Scholar]
- 398.Zhang HR, Boghigian BA, Pfeifer BA. 2010. Investigating the role of native propionyl-CoA and methylmalonyl-CoA metabolism on heterologous polyketide production in Escherichia coli. Biotechnol Bioeng 105:567–573. [PubMed] 10.1002/bit.22560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Pfeifer B, Hu ZH, Licari P, Khosla C. 2002. Process and metabolic strategies for improved production of Escherichia coli-derived 6-deoxyetythronolide B. Appl Environ Microbiol 68:3287–3292. [PubMed] 10.1128/AEM.68.7.3287-3292.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Lau J, Tran C, Licari P, Galazzo J. 2004. Development of a high cell-density fed-batch bioprocess for the heterologous production of 6-deoxyerythronolide B in Escherichia coli. J Biotechnol 110:95–103. [PubMed] 10.1016/j.jbiotec.2004.02.001 [DOI] [PubMed] [Google Scholar]
- 401.Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. 2007. Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat Prod Rep 24:162–190. [PubMed] 10.1039/B507395M [DOI] [PubMed] [Google Scholar]
- 402.Gao X, Wang P, Tang Y. 2010. Engineered polyketide biosynthesis and biocatalysis in Escherichia coli. Appl Microbiol Biotechnol 88:1233–1242. [PubMed] 10.1007/s00253-010-2860-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Zhang WJ, Li YR, Tang Y. 2008. Engineered biosynthesis of bacterial aromatic polyketides in Escherichia coli. Proc Natl Acad Sci USA 105:20683–20688. [PubMed] 10.1073/pnas.0809084105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Stevens DC, Conway KR, Pearce N, Villegas-Penaranda LR, Garza AG, Boddy CN. 2013. Alternative sigma factor over-expression enables heterologous expression of a type II polyketide biosynthetic pathway in Escherichia coli. PLoS One 8:e64858. 10.1371/journal.pone.0064858. 10.1371/journal.pone.0064858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Yan YJ, Li Z, Koffas MAG. 2008. High-yield anthocyanin biosynthesis in engineered Escherichia coli. Biotechnol Bioeng 100:126–140. [PubMed] 10.1002/bit.21721 [DOI] [PubMed] [Google Scholar]
- 406.Watts KT, Lee PC, Schmidt-Dannert C. 2006. Biosynthesis of plant-specific stilbene polyketides in metabolically engineered Escherichia coli. BMC Biotechnol 6:22. 10.1186/1472-6750-6-22. [PubMed] 10.1186/1472-6750-6-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Lim CG, Fowler ZL, Hueller T, Schaffer S, Koffas MAG. 2011. High-yield resveratrol production in engineered Escherichia coli. Appl Environ Microbiol 77:3451–3460. [PubMed] 10.1128/AEM.02186-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Hwang EI, Kaneko M, Ohnishi Y, Horinouchi S. 2003. Production of plant-specific flavanones by Escherichia coli containing an artificial gene cluster. Appl Environ Microbiol 69:2699–2706. [PubMed] 10.1128/AEM.69.5.2699-2706.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Watts KT, Lee PC, Schmidt-Dannert C. 2004. Exploring recombinant flavonoid biosynthesis in metabolically engineered Escherichia coli. Chembiochem 5:500–507. [PubMed] 10.1002/cbic.200300783 [DOI] [PubMed] [Google Scholar]
- 410.Kaneko M, Hwang EI, Ohnishi Y, Horinouchi S. 2003. Heterologous production of flavanones in Escherichia coli: potential for combinatorial biosynthesis of flavonoids in bacteria. J Ind Microbiol Biotechnol 30:456–461. [PubMed] 10.1007/s10295-003-0061-1 [DOI] [PubMed] [Google Scholar]
- 411.Santos CNS, Koffas M, Stephanopoulos G. 2011. Optimization of a heterologous pathway for the production of flavonoids from glucose. Metab Eng 13:392–400. [PubMed] 10.1016/j.ymben.2011.02.002 [DOI] [PubMed] [Google Scholar]
- 412.Fowler ZL, Gikandi WW, Koffas MAG. 2009. Increased malonyl coenzyme A biosynthesis by tuning the Escherichia coli metabolic network and its application to flavanone production. Appl Environ Microbiol 75:5831–5839. [PubMed] 10.1128/AEM.00270-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Miyahisa I, Funa N, Ohnishi Y, Martens S, Moriguchi T, Horinouchi S. 2006. Combinatorial biosynthesis of flavones and flavonols in Escherichia coli. Appl Microbiol Biotechnol 71:53–58. [PubMed] 10.1007/s00253-005-0116-5 [DOI] [PubMed] [Google Scholar]
- 414.Leonard E, Yan YJ, Koffas MAG. 2006. Functional expression of a P450 flavonoid hydroxylase for the biosynthesis of plant-specific hydroxylated flavonols in Escherichia coli. Metab Eng 8:172–181. [PubMed] 10.1016/j.ymben.2005.11.001 [DOI] [PubMed] [Google Scholar]
- 415.Yan YJ, Chemler J, Huang LX, Martens S, Koffas MAG. 2005. Metabolic engineering of anthocyanin biosynthesis in Escherichia coli. Appl Environ Microbiol 71:3617–3623. [PubMed] 10.1128/AEM.71.7.3617-3623.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Katsuyama Y, Matsuzawa M, Funa N, Horinouch S. 2008. Production of curcuminoids by Escherichia coli carrying an artificial biosynthesis pathway. Microbiology 154:2620–2628. [PubMed] 10.1099/mic.0.2008/018721-0 [DOI] [PubMed] [Google Scholar]
- 417.Minami H, Kim JS, Ikezawa N, Takemura T, Katayama T, Kumagai H, Sato F. 2008. Microbial production of plant benzylisoquinoline alkaloids. Proc Natl Acad Sci USA 105:7393–7398. [PubMed] 10.1073/pnas.0802981105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Nakagawa A, Minami H, Kim JS, Koyanagi T, Katayama T, Sato F, Kumagai H. 2011. A bacterial platform for fermentative production of plant alkaloids. Nat Commun 2:326. 10.1038/ncomms1327. [PubMed] 10.1038/ncomms1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Chang MCY, Keasling JD. 2006. Production of isoprenoid pharmaceuticals by engineered microbes. Nat Chem Biol 2:674–681. [PubMed] 10.1038/nchembio836 [DOI] [PubMed] [Google Scholar]
- 420.Kim SW, Keasling JD. 2001. Metabolic engineering of the nonmevalonate isopentenyl diphosphate synthesis pathway in Escherichia coli enhances lycopene production. Biotechnol Bioeng 72:408–415. [PubMed] 10.1002/1097-0290(20000220)72:4<408::AID-BIT1003>3.0.CO;2-H [DOI] [PubMed] [Google Scholar]
- 421.Carter OA, Peters RJ, Croteau R. 2003. Monoterpene biosynthesis pathway construction in Escherichia coli. Phytochemistry 64:425–433. [PubMed] 10.1016/S0031-9422(03)00204-8 [DOI] [PubMed] [Google Scholar]
- 422.Reiling KK, Yoshikuni Y, Martin VJJ, Newman J, Bohlmann J, Keasling JD. 2004. Mono and diterpene production in Escherichia coli. Biotechnol Bioeng 87:200–212. [PubMed] 10.1002/bit.20128 [DOI] [PubMed] [Google Scholar]
- 423.Leonard E, Ajikumar PK, Thayer K, Xiao WH, Mo JD, Tidor B, Stephanopoulos G, Prather KLJ. 2010. Combining metabolic and protein engineering of a terpenoid biosynthetic pathway for overproduction and selectivity control. Proc Natl Acad Sci USA 107:13654–13659. [PubMed] 10.1073/pnas.1006138107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Zhou K, Qiao KJ, Edgar S, Stephanopoulos G. 2015. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat Biotechnol 33:377–383. [PubMed] 10.1038/nbt.3095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Ljungcrantz P, Carlsson H, Mansson MO, Buckel P, Mosbach K, Bulow L. 1989. Construction of an artificial bifunctional enzyme, beta-galactosidase galactose dehydrogenase, exhibiting efficient galactose channeling. Biochemistry 28:8786–8792. [PubMed] 10.1021/bi00448a016 [DOI] [PubMed] [Google Scholar]
- 426.Salles IM, Forchhammer N, Croux C, Girbal L, Soucaille P. 2007. Evolution of a Saccharomyces cerevisiae metabolic pathway in Escherichia coli. Metab Eng 9:152–159. [PubMed] 10.1016/j.ymben.2006.09.002 [DOI] [PubMed] [Google Scholar]
- 427.Liu Q, Wu KY, Cheng YB, Lu L, Xiao ET, Zhang YC, Deng ZX, Liu TG. 2015. Engineering an iterative polyketide pathway in Escherichia coli results in single-form alkene and alkane overproduction. Metab Eng 28:82–90. [PubMed] 10.1016/j.ymben.2014.12.004 [DOI] [PubMed] [Google Scholar]
- 428.Menendez-Bravo S, Comba S, Sabatini M, Arabolaza A, Gramajo H. 2014. Expanding the chemical diversity of natural esters by engineering a polyketide-derived pathway into Escherichia coli. Metab Eng 24:97–106. [PubMed] 10.1016/j.ymben.2014.05.002 [DOI] [PubMed] [Google Scholar]
- 429.Watanabe K, Wang CC, Boddy CN, Cane DE, Khosla C. 2003. Understanding substrate specificity of polyketide synthase modules by generating hybrid multimodular synthases. J Biol Chem 278:42020–42026. [PubMed] 10.1074/jbc.M305339200 [DOI] [PubMed] [Google Scholar]
- 430.Wilner OI, Weizmann Y, Gill R, Lioubashevski O, Freeman R, Willner I. 2009. Enzyme cascades activated on topologically programmed DNA scaffolds. Nat Nanotechnol 4:249–254. [PubMed] 10.1038/nnano.2009.50 [DOI] [PubMed] [Google Scholar]
- 431.Agapakis CM, Ducat DC, Boyle PM, Wintermute EH, Way JC, Silver PA. 2010. Insulation of a synthetic hydrogen metabolism circuit in bacteria. J Biol Eng 4:3. 10.1186/1754-1611-4-3. [PubMed] 10.1186/1754-1611-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Cai F, Sutter M, Bernstein SL, Kinney JN, Kerfeld CA. 2015. Engineering bacterial microcompartment shells: chimeric shell proteins and chimeric carboxysome shells. ACS Synth Biol 4:444–453. [PubMed] 10.1021/sb500226j [DOI] [PubMed] [Google Scholar]
- 433.Parsons JB, Frank S, Bhella D, Liang MZ, Prentice MB, Mulvihill DP, Warren MJ. 2010. Synthesis of empty bacterial microcompartments, directed organelle protein incorporation, and evidence of filament-associated organelle movement. Mol Cell 38:305–315. [PubMed] 10.1016/j.molcel.2010.04.008 [DOI] [PubMed] [Google Scholar]
- 434.Choudhary S, Quin MB, Sanders MA, Johnson ET, Schmidt-Dannert C. 2012. Engineered protein nano-compartments for targeted enzyme localization. PLoS One 7:e33342. 10.1371/journal.pone.0033342. [PubMed] 10.1371/journal.pone.0033342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Lawrence AD, Frank S, Newnham S, Lee MJ, Brown IR, Xue WF, Rowe ML, Mulvihill DP, Prentice MB, Howard MJ, Warren MJ. 2014. Solution structure of a bacterial microcompartment targeting peptide and its application in the construction of an ethanol bioreactor. ACS Synth Biol 3:454–465. [PubMed] 10.1021/sb4001118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Fehér T, Planson AG, Carbonell P, Fernández-Castané A, Grigoras I, Dariy E, Perret A, Faulon JL. 2014. Validation of RetroPath, a computer-aided design tool for metabolic pathway engineering. Biotechnol J 9:1446–1457. [PubMed] 10.1002/biot.201400055 [DOI] [PubMed] [Google Scholar]
- 437.Bauer S, Shiloach J. 1974. Maximal exponential-growth rate and yield of Escherichia coli obtainable in a bench-scale fermentor. Biotechnol Bioeng 16:933–941. [PubMed] 10.1002/bit.260160707 [DOI] [PubMed] [Google Scholar]
- 438.Lee SY. 1996. High cell-density culture of Escherichia coli. Trends Biotechnol 14:98–105. [PubMed] 10.1016/0167-7799(96)80930-9 [DOI] [PubMed] [Google Scholar]
- 439.Riesenberg D, Guthke R. 1999. High-cell-density cultivation of microorganisms. Appl Microbiol Biotechnol 51:422–430. [PubMed] 10.1007/s002530051412 [DOI] [PubMed] [Google Scholar]
- 440.Shiloach J, Fass R. 2005. Growing E. coli to high cell density. A historical perspective on method development. Biotechnol Adv 23:345–357. [PubMed] 10.1016/j.biotechadv.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 441.Jensen EB, Carlsen S. 1990. Production of recombinant human growth-hormone in Escherichia coli - expression of different precursors and physiological-effects of glucose, acetate, and salts. Biotechnol Bioeng 36:1–11. [PubMed] 10.1002/bit.260360102 [DOI] [PubMed] [Google Scholar]
- 442.Rezaei M, Zarkesh-Esfahani SH. 2012. Optimization of production of recombinant human growth hormone in Escherichia coli. J Res Med Sci 17:681–685. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 443.Jung G, Denefle P, Becquart J, Mayaux JF. 1988. High-cell density fermentation studies of recombinant Escherichia coli strains expressing human interleukin-1-beta. Ann Inst Pasteur Microbiol 139:129–146. [PubMed] 10.1016/0769-2609(88)90100-7 [DOI] [PubMed] [Google Scholar]
- 444.Nausch H, Huckauf J, Koslowski R, Meyer U, Broer I, Mikschofsky H. 2013. Recombinant production of human interleukin 6 in Escherichia coli. PLoS One 8:e54933. 10.1371/journal.pone.0054933. [PubMed] 10.1371/journal.pone.0054933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Lee SY, Chang HN. 1995. Production of poly(3-hydroxybutyric acid) by recombinant Escherichia coli strains - genetic and fermentation studies. Can J Microbiol 41:207–215. [PubMed] 10.1139/m95-189 [DOI] [PubMed] [Google Scholar]
- 446.Lee SY, Yim KS, Chang HN, Chang YK. 1994. Construction of plasmids, estimation of plasmid stability, and use of stable plasmids for the production of poly(3-hydroxybutyric acid) by recombinant Escherichia coli. J Biotechnol 32:203–211. [PubMed] 10.1016/0168-1656(94)90183-X [DOI] [PubMed] [Google Scholar]
- 447.Korz DJ, Rinas U, Hellmuth K, Sanders EA, Deckwer WD. 1995. Simple fed-batch technique for high cell-density cultivation of Escherichia coli. J Biotechnol 39:59–65. 10.1016/0168-1656(94)00143-Z [DOI] [PubMed] [Google Scholar]
- 448.van Haveren J, Scott EL, Sanders J. 2008. Bulk chemicals from biomass. Biofuels Bioprod Biorefining-Biofpr 2:41–57. 10.1002/bbb.43 [DOI] [Google Scholar]
- 449.Yang FX, Hanna MA, Sun RC. 2012. Value-added uses for crude glycerol-a byproduct of biodiesel production. Biotechnol Biofuels 5:1–10. [PubMed] 10.1186/1754-6834-5-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Kim BS, Lee SC, Lee SY, Chang YK, Chang HN. 2004. High cell density fed-batch cultivation of Escherichia coli using exponential feeding combined with pH-stat. Bioprocess Biosyst Eng 26:147–150. [PubMed] 10.1007/s00449-003-0347-8 [DOI] [PubMed] [Google Scholar]
- 451.Martinez-Gomez K, Flores N, Castaneda HM, Martinez-Batallar G, Hernandez-Chavez G, Ramirez OT, Gosset G, Encarnacion S, Bolivar F. 2012. New insights into Escherichia coli metabolism: carbon scavenging, acetate metabolism and carbon recycling responses during growth on glycerol. Microb Cell Fact 11:46. 10.1186/1475-2859-11-46. 10.1186/1475-2859-11-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Lee SY. 1998. Poly(3-hydroxybutyrate) production from xylose by recombinant Escherichia coli. Bioprocess Eng 18:397–399. 10.1007/s004490050462 [DOI] [Google Scholar]
- 453.Vinuselvi P, Lee SK. 2012. Engineered Escherichia coli capable of co-utilization of cellobiose and xylose. Enzyme Microb Technol 50:1–4. [PubMed] 10.1016/j.enzmictec.2011.10.001 [DOI] [PubMed] [Google Scholar]
- 454.Lee J, Lee SY, Park S. 1997. Fed-batch culture of Escherichia coli W by exponential feeding of sucrose as a carbon source. Biotechnol Tech 11:59–62. 10.1007/BF02764454 [DOI] [Google Scholar]
- 455.Lee JW, Choi S, Park JH, Vickers CE, Nielsen LK, Lee SY. 2010. Development of sucrose-utilizing Escherichia coli K-12 strain by cloning beta-fructofuranosidases and its application for L-threonine production. Appl Microbiol Biotechnol 88:905–913. [PubMed] 10.1007/s00253-010-2825-7 [DOI] [PubMed] [Google Scholar]
- 456.Kim JR, Kim SH, Lee SY, Lee PC. 2013. Construction of homologous and heterologous synthetic sucrose utilizing modules and their application for carotenoid production in recombinant Escherichia coli. Bioresour Technol 130:288–295. [PubMed] 10.1016/j.biortech.2012.11.148 [DOI] [PubMed] [Google Scholar]
- 457.Bokinsky G, Peralta-Yahya PP, George A, Holmes BM, Steen EJ, Dietrich J, Lee TS, Tullman-Ercek D, Voigt CA, Simmons BA, Keasling JD. 2011. Synthesis of three advanced biofuels from ionic liquid-pretreated switchgrass using engineered Escherichia coli. Proc Natl Acad Sci USA 108:19949–19954. [PubMed] 10.1073/pnas.1106958108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Lee SY, Chang HN. 1993. High cell-density cultivation of Escherichia coli W using sucrose as a carbon source. Biotechnol Lett 15:971–974. 10.1007/BF00131766 [DOI] [Google Scholar]
- 459.Jahreis K, Bentler L, Bockmann J, Hans S, Meyer A, Siepelmeyer J, Lengeler JW. 2002. Adaptation of sucrose metabolism in the Escherichia coli wild-type strain EC3132. J Bacteriol 184:5307–5316. [PubMed] 10.1128/JB.184.19.5307-5316.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Challener C. 2014. Genomatica BDO process recognised. Chem Ind 78:6-6. [Google Scholar]
- 461.Barton NR, Burgard AP, Burk MJ, Crater JS, Osterhout RE, Pharkya P, Steer BA, Sun J, Trawick JD, Van Dien SJ, Yang TH, Yim H. 2015. An integrated biotechnology platform for developing sustainable chemical processes. J Ind Microbiol Biotechnol 42:349–360. [PubMed] 10.1007/s10295-014-1541-1 [DOI] [PubMed] [Google Scholar]
- 462.Burk MJ. 2010. Sustainable production of industrial chemicals from sugars. Int Sugar J 112:30–35. [Google Scholar]
- 463.Burgard AP, Pharkya P, Maranas CD. 2003. OptKnock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng 84:647–657. [PubMed] 10.1002/bit.10803 [DOI] [PubMed] [Google Scholar]
- 464.Danneil A, Flickinger E, Graefje H, Plass R, Schnur R. 1975. Production of very pure butanediol. US patent 3,891,511.
- 465.Dhingra V, Rao KV, Narasu ML. 2000. Current status of artemisinin and its derivatives as antimalarial drugs. Life Sci 66:279–300. 10.1016/S0024-3205(99)00356-2 [DOI] [PubMed] [Google Scholar]
- 466.Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MCY, Withers ST, Shiba Y, Sarpong R, Keasling JD. 2006. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440:940–943. [PubMed] 10.1038/nature04640 [DOI] [PubMed] [Google Scholar]
- 467.Westfall PJ, Pitera DJ, Lenihan JR, Eng D, Woolard FX, Regentin R, Horning T, Tsuruta H, Melis DJ, Owens A, Fickes S, Diola D, Benjamin KR, Keasling JD, Leavell MD, McPhee DJ, Renninger NS, Newman JD, Paddon CJ. 2012. Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc Natl Acad Sci USA 109:111–118. [PubMed] 10.1073/pnas.1110740109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D, Leavell MD, Tai A, Main A, Eng D, Polichuk DR, Teoh KH, Reed DW, Treynor T, Lenihan J, Fleck M, Bajad S, Dang G, Dengrove D, Diola D, Dorin G, Ellens KW, Fickes S, Galazzo J, Gaucher SP, Geistlinger T, Henry R, Hepp M, Horning T, Iqbal T, Jiang H, Kizer L, Lieu B, Melis D, Moss N, Regentin R, Secrest S, Tsuruta H, Vazquez R, Westblade LF, Xu L, Yu M, Zhang Y, Zhao L, Lievense J, Covello PS, Keasling JD, Reiling KK, Renninger NS, Newman JD. 2013. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496:528–532. [PubMed] 10.1038/nature12051 [DOI] [PubMed] [Google Scholar]
- 469.Pitera DJ, Paddon CJ, Newman JD, Keasling JD. 2007. Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab Eng 9:193–207. [PubMed] 10.1016/j.ymben.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 470.Ma SM, Garcia DE, Redding-Johanson AM, Friedland GD, Chan R, Batth TS, Haliburton JR, Chivian D, Keasling JD, Petzold CJ, Lee TS, Chhabra SR. 2011. Optimization of a heterologous mevalonate pathway through the use of variant HMG-CoA reductases. Metab Eng 13:588–597. [PubMed] 10.1016/j.ymben.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 471.Anthony JR, Anthony LC, Nowroozi F, Kwon G, Newman JD, Keasling JD. 2009. Optimization of the mevalonate-based isoprenoid biosynthetic pathway in Escherichia coli for production of the anti-malarial drug precursor amorpha-4,11-diene. Metab Eng 11:13–19. [PubMed] 10.1016/j.ymben.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 472.Newman JD, Marshall J, Chang M, Nowroozi F, Paradise E, Pitera D, Newman KL, Keasling JD. 2006. High-level production of amorpha-4,11-diene in a two-phase partitioning bioreactor of metabolically engineered Escherichia coli. Biotechnol Bioeng 95:684–691. [PubMed] 10.1002/bit.21017 [DOI] [PubMed] [Google Scholar]
- 473.Tsuruta H, Paddon CJ, Eng D, Lenihan JR, Horning T, Anthony LC, Regentin R, Keasling JD, Renninger NS, Newman JD. 2009. High-level production of amorpha-4,11-diene, a precursor of the antimalarial agent artemisinin, in Escherichia coli. PloS One 4:e4489. 10.1371/journal.pone.0004489. [PubMed] 10.1371/journal.pone.0004489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Chang MCY, Eachus RA, Trieu W, Ro DK, Keasling JD. 2007. Engineering Escherichia coli for production of functionalized terpenoids using plant P450s. Nat Chem Biol 3:274–277. [PubMed] 10.1038/nchembio875 [DOI] [PubMed] [Google Scholar]
- 475.Paddon CJ, Keasling JD. 2014. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat Rev Microbiol 12:355–367. [PubMed] 10.1038/nrmicro3240 [DOI] [PubMed] [Google Scholar]
- 476.Zhang X, Wang X, Shanmugam KT, Ingram LO. 2011. L-Malate production by metabolically engineered Escherichia coli. Appl Environ Microbiol 77:427–434. [PubMed] 10.1128/AEM.01971-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Vemuri GN, Eiteman MA, Altman E. 2002. Succinate production in dual-phase Escherichia coli fermentations depends on the time of transition from aerobic to anaerobic conditions. J Ind Microbiol Biotechnol 28:325–332. [PubMed] 10.1038/sj.jim.7000250 [DOI] [PubMed] [Google Scholar]
- 478.Zhu Y, Eiteman MA, DeWitt K, Altman E. 2007. Homolactate fermentation by metabolically engineered Escherichia coli strains. Appl Environ Microbiol 73:456–464. [PubMed] 10.1128/AEM.02022-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Mazumdar S, Blankschien MD, Clomburg JM, Gonzalez R. 2013. Efficient synthesis of L-lactic acid from glycerol by metabolically engineered Escherichia coli. Microb Cell Fact 12:7. 10.1186/1475-2859-12-7. 10.1186/1475-2859-12-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Saini M, Wang ZW, Chiang CJ, Chao YP. 2014. Metabolic engineering of Escherichia coli for production of butyric acid. J Agric Food Chem 62:4342–4348. [PubMed] 10.1021/jf500355p [DOI] [PubMed] [Google Scholar]
- 481.Vuoristo KS, Mars AE, Sangra JV, Springer J, Eggink G, Sanders JP, Weusthuis RA. 2015. Metabolic engineering of itaconate production in Escherichia coli. Appl Microbiol Biotechnol 99:221–228. [PubMed] 10.1007/s00253-014-6092-x [DOI] [PubMed] [Google Scholar]
- 482.Okamoto S, Chin T, Hiratsuka K, Aso Y, Tanaka Y, Takahashi T, Ohara H. 2014. Production of itaconic acid using metabolically engineered Escherichia coli. J Gen Appl Microbiol 60:191–197. [PubMed] 10.2323/jgam.60.191 [DOI] [PubMed] [Google Scholar]
- 483.Ohta K, Beall DS, Mejia JP, Shanmugam KT, Ingram LO. 1991. Genetic improvement of Escherichia coli for ethanol production: chromosomal integration of Zymomonas mobilis genes encoding pyruvate decarboxylase and alcohol dehydrogenase II. Appl Environ Microbiol 57:893–900. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Jarboe LR, Grabar TB, Yomano LP, Shanmugan KT, Ingram LO. 2007. Development of ethanologenic bacteria, p 237–261. In Olsson L (ed), Biofuels, vol 108. Springer, Berlin. [PubMed] 10.1007/10_2007_068 [DOI] [PubMed] [Google Scholar]
- 485.Altaras NE, Cameron DC. 1999. Metabolic engineering of a 1,2-propanediol pathway in Escherichia coli. Appl Environ Microbiol 65:1180–1185. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Soucaille P, Voelker F, Figge R. 2012. Micro-organisms for the production of 1,2-propanediol obtained by a combination of evolution and rational design. US patent 8,298,807.
- 487.Cervin MA, Soucaille P, Valle F. 2010. Process for the biological production of 1,3-propanediol with high yield. US patent 7,745,184.
- 488.Shen CR, Lan EI, Dekishima Y, Baez A, Cho KM, Liao JC. 2011. Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl Environ Microbiol 77:2905–2915. [PubMed] 10.1128/AEM.03034-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Bestvater T, Louis P, Galinski EA. 2008. Heterologous ectoine production in Escherichia coli: by-passing the metabolic bottle-neck. Saline Systems 4:12. 10.1186/1746-1448-4-12. 10.1186/1746-1448-4-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Gu P, Yang F, Su T, Li F, Li Y, Qi Q. 2014. Construction of an L-serine producing Escherichia coli via metabolic engineering. J Ind Microbiol Biotechnol 41:1443–1450. [PubMed] 10.1007/s10295-014-1476-6 [DOI] [PubMed] [Google Scholar]
- 491.Lee M, Smith GM, Eiteman MA, Altman E. 2004. Aerobic production of alanine by Escherichia coli aceF ldhA mutants expressing the Bacillus sphaericus alaD gene. Appl Microbiol Biotechnol 65:56–60. [PubMed] 10.1007/s00253-004-1560-3 [DOI] [PubMed] [Google Scholar]
- 492.Ying HX, He X, Li Y, Chen KQ, Ouyang PK. 2014. Optimization of culture conditions for enhanced lysine production using engineered Escherichia coli. Appl Biochem Biotechnol 172:3835–3843. [PubMed] 10.1007/s12010-014-0820-7 [DOI] [PubMed] [Google Scholar]
- 493.Yuan P, Cao W, Wang Z, Chen K, Li Y, Ouyang P. 2015. Enhancement of L-phenylalanine production by engineered Escherichia coli using phased exponential L-tyrosine feeding combined with nitrogen source optimization. J Biosci Bioeng 120:36–40. [PubMed] 10.1016/j.jbiosc.2014.12.002 [DOI] [PubMed] [Google Scholar]
- 494.Tribe DE, Pittard J. 1979. Hyperproduction of tryptophan by Escherichia coli - genetic manipulation of the pathways leading to tryptophan formation. Appl Environ Microbiol 38:181–190. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Song CW, Lee J, Ko YS, Lee SY. 2015. Metabolic engineering of Escherichia coli for the production of 3-aminopropionic acid. Metab Eng 30:121–129. [PubMed] 10.1016/j.ymben.2015.05.005 [DOI] [PubMed] [Google Scholar]
- 496.Shen CR, Liao JC. 2008. Metabolic engineering of Escherichia coli for 1-butanol and 1-propanol production via the keto-acid pathways. Metab Eng 10:312–320. [PubMed] 10.1016/j.ymben.2008.08.001 [DOI] [PubMed] [Google Scholar]
- 497.Oliver JW, Machado IM, Yoneda H, Atsumi S. 2013. Cyanobacterial conversion of carbon dioxide to 2,3-butanediol. Proc Natl Acad Sci USA 110:1249–1254. [PubMed] 10.1073/pnas.1213024110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Park SY, Kim B, Lee S, Oh M, Won JI, Lee J. 2014. Increased 2,3-butanediol production by changing codon usages in Escherichia coli. Biotechnol Appl Biochem 61:535–540. [PubMed] 10.1002/bab.1216 [DOI] [PubMed] [Google Scholar]
- 499.Zhou J, Wang C, Yang L, Choi ES, Kim SW. 2015. Geranyl diphosphate synthase: an important regulation point in balancing a recombinant monoterpene pathway in Escherichia coli. Enzyme Microb Technol 68:50–55. [PubMed] 10.1016/j.enzmictec.2014.10.005 [DOI] [PubMed] [Google Scholar]
- 500.Wang C, Yoon SH, Jang HJ, Chung YR, Kim JY, Choi ES, Kim SW. 2011. Metabolic engineering of Escherichia coli for alpha-farnesene production. Metab Eng 13:648–655. [PubMed] 10.1016/j.ymben.2011.08.001 [DOI] [PubMed] [Google Scholar]
- 501.Kajiwara S, Fraser PD, Kondo K, Misawa N. 1997. Expression of an exogenous isopentenyl diphosphate isomerase gene enhances isoprenoid biosynthesis in Escherichia coli. Biochem J 324:421–426. [PubMed] 10.1042/bj3240421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Rodrigues AL, Becker J, de Souza Lima AO, Porto LM, Wittmann C. 2014. Systems metabolic engineering of Escherichia coli for gram scale production of the antitumor drug deoxyviolacein from glycerol. Biotechnol Bioeng 111:2280–2289. [PubMed] 10.1002/bit.25297 [DOI] [PubMed] [Google Scholar]
- 503.Leonard E, Yan Y, Fowler ZL, Li Z, Lim CG, Lim KH, Koffas MA. 2008. Strain improvement of recombinant Escherichia coli for efficient production of plant flavonoids. Mol Pharm 5:257–265. [PubMed] 10.1021/mp7001472 [DOI] [PubMed] [Google Scholar]
- 504.Lin Z, Xu Z, Li Y, Wang Z, Chen T, Zhao X. 2014. Metabolic engineering of Escherichia coli for the production of riboflavin. Microb Cell Fact 13:104. 10.1186/s12934-014-0104-5. 10.1186/s12934-014-0104-5 [DOI] [PMC free article] [PubMed] [Google Scholar]