

Abstract
The bacteriophage λ Red homologous recombination system has been studied over the past 50 years as a model system to define the mechanistic details of how organisms exchange DNA segments that share extended regions of homology. The λ Red system proved useful as a system to study because recombinants could be easily generated by co-infection of genetically marked phages. What emerged from these studies was the recognition that replication of phage DNA was required for substantial Red-promoted recombination in vivo, and the critical role that double-stranded DNA ends play in allowing the Red proteins access to the phage DNA chromosomes. In the past 16 years, however, the λ Red recombination system has gained a new notoriety. When expressed independently of other λ functions, the Red system is able to promote recombination of linear DNA containing limited regions of homology (∼50 bp) with the Escherichia coli chromosome, a process known as recombineering. This review explains how the Red system works during a phage infection, and how it is utilized to make chromosomal modifications of E. coli with such efficiency that it changed the nature and number of genetic manipulations possible, leading to advances in bacterial genomics, metabolic engineering, and eukaryotic genetics.
INTRODUCTION
Studies in the early 1950s showed that mutations in bacteriophage lambda could be generated and used in genetic crosses in Escherichia coli (1, 2). These pioneering studies eventually led to the establishment of a genetic linkage map of the linear λ chromosome. Just as importantly, however, these crosses demonstrated that λ chromosomes undergo genetic recombination. In time, λ phage biology would lend itself to the study of three different recombination systems in E. coli: the host RecABCD-dependent pathway of homologous recombination, the Int-Xis pathway of λ site-specific recombination, and λ’s own Red pathway of homologous recombination. It is the latter pathway that is the subject of this review.
Two important aspects of λ Red recombination will be emphasized in this review. The first is a description of the biochemical properties of the Red proteins and an overview of the proposed mechanisms of Red-promoted phage recombination. Two well-known models of Red recombination, the single-stranded (ss) DNA-annealing pathway and the RecA-assisted pathway, will be described, along with their shortcomings in reflecting the true nature of Red recombination during a phage infection. A recent model, the Replisome-Invasion/Template switch model of Red recombination, takes into account both the older and the more recent observations to propose a novel mechanism, whereby a λ Red-processed double-stranded (ds) DNA end invades a replication fork and captures one of the replisome polymerases (3). This model is particularly interesting because the λ Red proteins possess characteristics similar to recombinases from yeast and higher eukaryotes, making insights into its mechanism intriguingly relevant to other more complex systems.
The second aspect of the λ Red system reviewed here is the development over the past decade of a highly efficient method of chromosomal modification and gene replacement called “Recombineering” (recombinational engineering) that uses the λ Red proteins to manipulate bacterial chromosomes (and artificial chromosomes) with efficiencies that could not be achieved with restriction enzymes. Together with the use of site-specific recombination systems, such as Cre/loxP and Flp/FRT, counterselection cassettes, and the I-SceI meganuclease, any type of chromosomal manipulation can be easily achieved, including insertions, deletions, duplications, inversions, fusions, and single base-pair modifications. Previous discussions of Red recombineering protocols (4, 5, 6, 7) are included here for the sake of completeness. Proposed mechanisms for Red recombineering with oligonucleotides and short linear dsDNA substrates at the replication fork are also described. The phage lambda Red system ease for genetic manipulation of bacterial and phage chromosomes, bacterial artificial chromosomes (BACs), and plasmids has had profound effects in such diverse fields as bacterial genetics, metabolic engineering, and mammalian gene-targeting.
EARLY λ RED RECOMBINATION STUDIES
Discovery of λ red and gam Mutants
Following the identification of the E. coli recA gene (8), it was found that bacteriophage λ could recombine efficiently in recA-deficient hosts, revealing that λ encodes its own recombination system (9, 10, 11). This prompted a search for mutants of λ that were defective for homologous recombination. First evidence of such a mutant was found in a deletion mutant of a ɸ80-λ hybrid phage that was 100-fold defective for phage recombination in a recA host; the deletion was mapped to the central region of the hybrid (12). Two groups working independently (13, 14) isolated point mutants that were ∼100-fold down for recombination with a defective prophage in recA hosts. These mutations mapped to a central region of the linear λ chromosome near the cIII gene. This new recombination system was called red (for recombination defective) to distinguish these genes from the host rec recombination genes.
Shortly thereafter came the discovery of a gene important for the growth of λ red mutants in E. coli recA mutant hosts (15). This gene was known as the gamma gene (gam). The Gam protein was somehow linked to phage lambda replication, because mutants in λ gam failed to generate the “late mode” of DNA replication that leads to packageable concatemeric phage DNA (16). The growth defect and lack of concatemeric DNA of a λ gam mutant could be alleviated by a mutation in the recB gene of E. coli, which encodes the RecB subunit of the ATP-dependent DNA helicase/nuclease RecBCD (15, 16). It was surmised that the Gam protein was a function that inhibited the destructive capabilities of the RecBCD enzyme, which itself was an inhibitor of the late mode of λ DNA replication. This proposal was later verified by Karu et al. (17), who found that the purified Gam protein of λ inhibits the ATPase and exonuclease activities of RecBCD in vitro. These early studies generated the now well-accepted model that during a λ phage infection, multimers of λ DNA (the immediate precursor to packaging) are generated either by Red-promoted recombination of phage monomeric DNA species, or by the Gam-promoted (RecBCD-inhibited) rolling-circle mode of lambda DNA replication. In the absence of both red and gam genes, phage λ forms small plaques on a wild-type host and does not grow at all in a recA mutant host.
Comparisons of Red versus RecBCD Pathways of Recombination
Early studies comparing the E. coli host RecABCD versus λ Red systems showed that in wild-type host, λ red mutants were down ∼6- to 10-fold for growth and recombination (13), suggesting that the host RecABCD system was not as efficient for recombination with λ DNA as the Red system. The reason for this observation has to do with the RecBCD pathway signal sequence Chi (see below), which is absent in wild-type phage λ. Likewise, in tests where λ Red was asked to replace the E. coli recombination system, λ Red promoted low levels of conjugational and transductional recombination in recA hosts (18, 19). Kuzminov (20) suggested that this reflects the ability of λ Red to promote annealing of ssDNA intermediates generated in these pathways to the lagging strand of the replication fork, which is not efficient because of the long substrates (∼100 kb). (This was a keen insight at the time, given how the Red proteins are thought to act today—see below). When replication is completely blocked, Red cannot promote λ recombination in recA mutant hosts (21). When RecA is present, but RecBCD is absent, λ Red promotes host conjugational recombination at 10% the rate relative to the RecABCD system (22, 23). This pathway represented here is the RecA-assisted pathway of Red recombination, known to be active on nonreplicating phage substrates in E. coli (see below). The inability of the RecABCD and Red systems to fully complement each other reflects the differences in the substrates these recombination systems have evolved to work on (i.e., replicating phage genomes for λ Red versus dsDNA breaks following replication fork collapse and long linear pieces of the chromosome transferred during conjugation for RecABCD).
Historically, studies of λ phage red gam mutants recombining in wild-type hosts helped identify key steps in the mechanism of the E. coli RecBCD pathway (24, 25, 26). The main feature of the RecBCD pathway of recombination is the role of Chi sites (crossover hotspot instigator - short asymmetric sequence reading 5′ GCTGGTGG 3′) in modulating the dsDNA exonuclease activity of the enzyme (for review see references 27 and 28). RecBCD binds to dsDNA ends at the sites of DNA breaks and translocates along the DNA digesting both the 3′ and 5′ strands at the site of entry (29, 30, 31). After encountering a Chi sequence (from the right as shown above), RecBCD is modified so that the exonuclease activity on the 3′ strand is greatly suppressed, while digestion of the 5′ strand is slightly upregulated (29). Continued unwinding by Chi-modified RecBCD generates 3′-ssDNA that serves as a substrate for loading of the RecA protein (32, 33, 34). Since phage λ has no Chi sites in its chromosome, it cannot recombine efficiently via the RecBCD pathway. This lack of Chi results in a 10-fold decrease in λ red gam phage recombination (relative to wild-type λ) in E. coli. The residual recombination in this case is presumably the result of RecBCD acting on Chi-like sites. Single base-pair mutants of λ red gam phage that spontaneously generate a Chi site in their chromosomes gain the ability to grow and recombine efficiently in E. coli (25). This system was exploited by Stahl and colleagues to uncover the role of Chi acting as a hotspot in the RecBCD pathway of recombination (for review, see Stahl [35]).
In assays designed to detect the ability of recombination systems to promote gene replacement with linear DNA molecules, however, the λ Red system greatly outperformed the RecABCD system. When tested with small linear substrates between 2 and 3 kbp, λ Red promoted high rates of recombination whereas the host RecBCD system was inactive (23). This observation led to the development of the λ Red system as a tool for gene replacement (see below). Dabert and Smith (36) showed that appropriately positioned Chi sites allowed the host RecABCD system to promote gene replacement with linear fragments of 6.5 kb in length, although not with the frequency or the limited homology requirements exhibited by the λ Red system. Nonetheless, this RecABCD-based system might prove useful in bacteria other than E. coli, where λ Red may not work efficiently, but where the sequence of the cognate Chi site is known (see reference 37).
THE RED PROTEINS
λ Exonuclease
Prior to the discovery of the λ red mutants described above, λ Exo had already been identified as an exonuclease present in extracts of E. coli λ lysogens following UV induction (38, 39, 40). Purification and characterization of the exonuclease had identified an activity distinct from host exonucleases, one that digested the 5′-phosphorylated strand at dsDNA ends (41, 42), but bound weakly to nicked DNA (43). Its involvement in Red recombination came from the observation that many λ red recombination-deficient phage mutants did not produce this exonuclease (44). In addition, mutants in the red recombination genes were found to affect the structural properties of λ Exo, as measured by immunological assays (45, 46). Another correlation between the red genes and λ Exo was suggested by the fact that mutants that were thermosensitive for Red recombination also produced a thermosensitive exonuclease activity (46). These early studies, identifying λ Exo as a principal player in λ Red recombination, led to models suggesting that the role of the exonuclease was to generate ssDNA, which could then take part in DNA strand invasion or assimilation reactions. The actual role of the Beta protein took longer to propose, since its annealing activity was not observed until years later (as described below).
λ Exo (25.9 kDa, 226 amino acids) has a requirement for Mg2+, a pH optimum of 9.5, and shows a marked preference for substrates containing 5′-phosphates. Its rate of digestion on ssDNA is ∼1% or less than the rate on a blunt-ended dsDNA substrate. Predigested substrates containing 3′-ssDNA regions of 100 nucleotides or more are poor substrates for λ Exo (47). λ Exo is incapable of initiating digestion at nicked DNA, but has been shown to bind to such sites, leading to the assertion that the enzyme might have a role in ssDNA assimilation. Consistent with this idea, λ Exo has been shown to precisely trim the 5′-terminated single strand of a branched structure, suggesting a role in trimming the overlapping regions of recombination intermediates following synapsis (48), although this role has not been extensively characterized.
In a study by Mitsis and Kwagh (49), micrococcal nuclease (MN) protection experiments of λ Exo bound to a 30-bp dsDNA substrate with a 20-nucleotide 3′-ssDNA tail suggested that the enzyme is bound to 13 to 14 bp of the dsDNA region, with no protection of the ssDNA tail (the mock product of the enzymatic reaction). A slight enhancement of MN nicking was seen at the ssDNA-dsDNA junction. In studies at the single-molecule level (50), λ Exo was found to travel at a speed of 12 nucleotides (nt) per second, and was found to pause for variable times on the template during the digestion reaction. The biological significance for this pause, if any, is not known. Subramanian et al. (51) found the kcat for λ Exo to be 11.7 nt/s, in agreement with the turnover number determined by Perkings et al. (50). This study also found that WT λ Exo forms inert complexes with DNA molecules containing 5′-OH ends, consistent with the preference of λ Exo for substrates with 5′-phosphorylated ends (41). The authors characterized a λ Exo (R28A) mutant that was found to digest dsDNA poorly, irrespective of the phosphorylation of the 5′ end. The arginine-28 residue binds to phosphate in the crystal structure (see below), and is thought to play a role in positioning the enzyme bound to a dsDNA end. The effect of the mutation was to dramatically reduce the processivity of the enzyme with no change in the quaternary structure of the protein.
The crystal structure of λ Exo has been solved (52). The exonuclease exists as a trimer in solution and in the crystal structure. The trimer has a toroidal shape and a funnel-shaped central channel with openings of 30 Å on one side and 15 Å on the other. It is proposed that dsDNA enters the wider opening, where the 5′ and 3′ strands are separated. The 5′-ended strand is excised by one of the active sites within the trimer, while the 3′-ended ssDNA exits the narrower opening at the end of the central channel. The trimer is proposed to encircle the 3′-ending DNA strand in a “sliding clamp” configuration, thus explaining the highly processive nature of λ Exo (the enzyme degrades at least 3,000 nucleotides per binding event) (43).
The crystal structure of λ Exo complexed to DNA has also been determined (53) (see Fig. 1). The authors used a 12-bp blunt-ended DNA substrate complexed to λ Exo. The structure was crystallized with Ca2+ replacing Mg2+, to inhibit DNA degradation by the enzyme. However, within this complex, the scissile phosphate of the terminal nucleotide was distant from the active-site Ca2+ by 11 Å, suggesting the DNA was not fully inserted into the λ Exo trimer. Thus, they recrystallized the complex by using a 12-base DNA duplex containing a dinucleotide extension on the 5′ end, included a 5′-phosphate on the overhang, and used a nuclease-deficient K131A mutant that allowed them to use Mg2+ instead of Ca2+ as the bound cation. Key features of the λ Exo-DNA complex include the observation that Arg45 inserts into the minor groove of the DNA, perhaps acting as a rudder to keep the enzyme on track; a hydrophobic wedge (including Leu78) that unwinds DNA prior to cleavage, guiding the 2 nucleotides of the 5′-ended strand to one of the active sites in the trimer, while allowing the 3′-ended strand to pass through the central channel and out the back of the complex; and finally, a negatively charged pocket (including Arg-28) near the bottom of the central cavity that binds the 5′-phosphate group on the DNA, essentially “pulling” the DNA into position within the chamber. The authors suggest a ratchet mechanism for λ Exo, where following enzymatic cleavage and 5′ mononucleotide release, a new phosphate is exposed on the next nucleotide. The hydrophobic wedge, which is proposed to unwind 2 base pairs, is still intact following the 5′-mononucleotide release, but now with only 1 bp unwound. Binding of this new 5′-phosphate to the positively charged pocket at the back of the chamber moves the enzyme forward and helps unwind another base pair at the wedge, reinstating unwinding of 2 base pairs. This step also positions the next scissile bond in place at the active site. Thus, the components necessary for the processivity of λ Exo are seen at both the level of the quaternary structure (the trimer encircling the DNA) and within the monomer (in the role of the Arg-28 binding the 5′-phosphate). This model is consistent with the importance of Arg-28 in the processivity of λ Exo observed in vitro (51), as described above.
Figure 1.
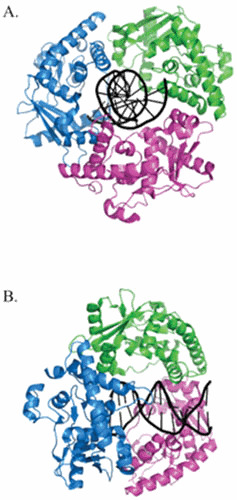
The trimeric structure of λ Exo. View of the λ Exonuclease trimer looking through the central channel (A) and the same view rotated 90° to the right (B). The three subunits are colored blue, green, and magenta. The dsDNA passes through the central channel of the trimer, is acted upon by one of three active sites, and exits out the back as ssDNA. The structures were generated by PyMol based on the coordinates described by Zhang et al. (53).
In comparisons of the structure of λ Exo with the RecE exonuclease from the Rac prophage (another 5′ → 3′-dsDNA exonuclease), Zhang and colleagues found that both enzymes share a toroidal structure with similarly sized central channels (54). Both enzymes also form oligomers (trimers for λ Exo and tetramers for RecE). Despite these similarities, λ Exo and RecE share no amino acid sequence homology. While they both belong to a superfamily of endonuclease-like enzymes (55), RecE sequence is more closely aligned to the RecB nuclease domain, especially near the active site. Surprisingly, the authors found that while λ Exo and RecE share a similar quaternary structure and positioning of their active sites, the subunits of each enzyme are packed into oligomers that are proposed to interact with a dsDNA end in opposite directions. In other words, if one aligns the central channels of each nuclease, the narrower side of the channel is formed by the N-terminal region of λ Exo and by the C-terminal region of RecE. This observation suggests that λ Exo and RecE evolved independently of one another to generate a common function for their respective phages.
λ Beta Protein
Early purifications of λ Exo had also revealed an associated protein called β protein (29.7 kDa, 261 amino acids), whose function was unknown at that time (56, 57). In addition, Radding and coworkers discovered that λ Beta and Exo form a 1:1 complex, although it has not yet been demonstrated whether the complex has any biological significance. Beta bound to Exo has no effect on the enzyme’s preference for dsDNA ends, pH optimum (9.5), KM (10 μM), Vmax, or dependency on Mg2+ for exonucleolytic activity, although Beta did have a 2-fold effect on the maximal binding of Exo to dsDNA ends (43). More recently, Tolun has reported that Beta decreased the extent of digestion of dsDNA by λ Exo, possibly by preventing λ Exo from rebinding to partially processed substrates (58). It has also been reported that, while Beta had no effect on the processivity of λ Exo, it did increase the pause time that the enzyme displays while digesting long dsDNA substrates (50). In investigations on whether λ Exo has a role in loading λ Beta onto ssDNA using gel mobility shift assays, more Beta was seen bound to ssDNA when present while λ Exo acted on a 2-kb linear dsDNA than when added after the λ Exo reaction was completed (K. Murphy, unpublished observations). A similar role for λ Exo in ssDNA loading of Beta has been reported by Tolun (58).
The first reported activity of the λ Beta protein was observed by Kmiec and Holloman (59), who found that Beta promoted renaturation of complementary single-stranded DNA. This function was later corroborated by Muniyappa and Radding (60), who, in addition, demonstrated a role for Beta in stimulating formation of joint molecules by RecA. Studies from Radding’s laboratory (61) have shown that Beta protein will bind to ssDNA faster if ssDNA ends are present. In addition, a Bet-oligo complex will bind tightly to a complementary oligo, but not to noncomplementary oligos. This complex is not formed by binding of Beta to annealed oligos, since, if the two oligos are annealed beforehand, Beta does not bind to the dsDNA product. The structure of this complex of Beta bound to two complementary DNA oligos is not known, but likely represents an important intermediate in models describing the mechanism of Beta-promoted annealing. Together with earlier studies from Radding’s laboratory on the properties of λ Exo (43, 48), the authors favored a model whereby Beta promotes renaturation by binding tighter to the nascent product of the renaturation reaction than it does to the single-stranded substrates, or to the dsDNA made from spontaneous annealing of the oligos. This model is based on a proposal by Hall and Kolodner based on similar properties exhibited by the RecT protein of Rac prophage (62).
λ Beta has been included in a group of proteins known as single-stranded DNA annealing proteins (SSAPs). SSAPs are present in both prokaryotes and eukaryotes and share similar structural and functional characteristics. These proteins promote recombination via RecA-dependent and independent pathways, form oligomeric rings and/or filaments in vitro, bind to ssDNA, and promote annealing of complementary ssDNA strands in an ATPase-independent fashion. (While some nonspecific single-stranded DNA binding proteins are able to promote annealing of two ssDNA molecules by melting out secondary structures in ssDNA (e.g., E. coli single-stranded DNA binding protein, SSB), the term SSAP is used here to describe those proteins that share the structural and functional characteristics described above). Using sensitive computational sequence analysis, the evolutionary history and classification of SSAPs by Iyer et al. (63) have shown that three distinct superfamilies of SSAPs exist. One superfamily is represented by λ Bet/RecT, another by the P22 Erf protein, and the third by the Rad52 family of proteins. Despite their similarities in biochemical functions and quaternary structure, there is no sequence similarity between the superfamilies, leading the authors to conclude that these proteins are evolutionarily distinct. That these proteins arose independently of each other, yet share the same functional characteristics and quaternary structures, reflects the high biological importance of these functions in recombinational and repair processes.
One of the extraordinary features of SSAPs, in general, is the highly suggestive ring-like structure of these proteins when observed under the electron microscope. These ring-like structures were first observed for the P22 Erf protein (essential recombination function) (64), the SSAP component of the P22 phage recombination system. Projections from the Erf rings were identified as the C-terminal domain of the protein (65). Analysis of Erf fragments generated by amber mutations revealed three different domains of the Erf protein: the N-terminal domain responsible for its ring-like quaternary structure, the C-terminal domain of unknown function, and the interdomain region responsible for stability of the ssDNA-binding activity. A similar configuration of these structural properties was reported for the λ Beta protein (66). In this study, the N-terminal fragment of Beta consisting of residues 1 to 130 was resistant to protease treatment in the absence of DNA, while residues 131 to 177 of Beta were more resistant to protease treatment in the presence of bound DNA. Using biotinylation of lysine residues and mass spectral analysis, the authors showed that the N-terminal 1 to 177 residues of Beta form a core DNA binding region. Consistent with this analysis, an N-terminal fragment of Beta (1–177) still bound to oligos as well as the full-length Beta. From the proteolysis experiments, the authors found that the N-terminal 30 amino acids of Beta protein become susceptible to protease treatment after binding DNA. An earlier study (67) has found that a 20-kDa N-terminal fragment of Beta (predicted to encode residues 1 to 184) was successfully cross-linked by photoactivation to a 36-base oligo, but not ones containing 27 or 17 bases, establishing a minimum size of ssDNA for the stable production of a Beta-ssDNA complex.
A study by Passy et al. (68) has shown that λ Beta exists in three distinct forms: small rings, large rings, and helical filaments. In the absence of DNA, Beta forms small rings (145 Å diameter) containing ∼12 subunits. Surrounding the central hole of ∼35 Å, there is a continuous ring of density that contains ∼12 projections extending out from it (presumably, as in the case for phage P22 Erf, these projections are formed by the C-terminal domain of the protein). In the presence of ssDNA, Beta forms larger rings of 15 to 16 subunits, structures not observed in the absence of ssDNA. Passy et al. (68) suggested that the small rings are converted directly or indirectly into large rings. The average large ring size was 185 Å in diameter, with a central hole of ∼75Å. When incubated with complementary ssDNA that could form dsDNA or dsDNA with ssDNA overhangs, Beta formed left-handed helical filaments with a variable pitch and a diameter similar to the large rings (∼200 Å). Blunt-ended linear dsDNA were poor substrates to make filaments; no filaments were found with Beta and circular dsDNA species. Rings were found early under annealing conditions, and on dsDNA substrates containing ssDNA overhangs, rings were often found associated with one end of a filament. On the contrary, fully annealed substrates were associated with filaments with no rings attached. These results suggest that the Beta rings bind ssDNA and initiate filament formation as part of the annealing process. A model was presented by Passy et al. (68) suggesting that large Beta rings are the structures that bind ssDNA to initiate annealing with a complementary strand. The ssDNA is thought to wrap around the ring as previously suggested for the P22 Erf recombinase (64). The annealing reaction then proceeds to generate a dsDNA that is supercoiled within the Beta helical filament. It may be that rings and filaments are two stable forms of the same Beta polymer, defined only by the type of DNA bound: single-stranded for rings and duplex DNA for filaments. Models of the ring and helical forms of Beta protein, complexed with ssDNA and dsDNA, respectively, are shown in Fig. 2.
Figure 2.
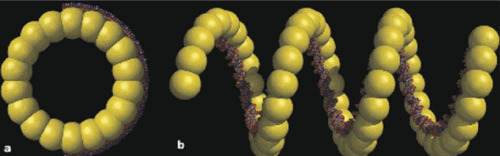
Models for λ Beta-DNA structures. (A) A large Beta ring (18 subunits) is shown with DNA wrapped around the outside of the ring, as previously suggested for P22 Erf (64). (B) After Beta-catalyzed annealing of complementary ssDNA strands, Beta-dsDNA filaments are formed. The authors estimate the Beta filament contains around 100 base pairs per supercoil turn of the DNA. Taken from Passy et al. (68), with permission.
A more recent study of Beta structure using atomic force microscopy showed, in the absence of DNA, that the rings were actually gapped ellipses with a right-handed helical structure (69). The authors measured 11 monomers of Beta per helical turn of the protein. The addition of ssDNA oligos disrupted these structures, but the addition of two complementary oligos promoted the formation of a stable complex, as seen before in Radding’s laboratory (61). Examination of this structure under the microscope revealed a left-handed structure with 14 Beta monomers per helical turn. Thus, DNA annealing changes both the handedness and curvature of the Beta helix. With 11 base pairs per Beta monomer, there are about 155 base pairs per helical turn of the filament. The authors describe an annealing model whereby the binding of ssDNA disrupts the right-handed Beta helix, promoting transient interactions between ssDNA molecules. Annealing between complementary ssDNA promotes binding of a second Beta monomer to form a stable complex that elongates to form a left-handed helix. The role of Beta is to facilitate both the initiation and propagation of the annealing event. It is noted that dsDNA is likely to be unwound to some degree within the complex, because dsDNA does not bind tightly to Beta in vitro.
As mentioned above, other SSAPs also form oligomeric rings. The Erf protein of phage P22, the Rac prophage RecT protein, and both yeast and human Rad52 proteins all form similar types of rings (64, 70, 71, 72). However, the binding modes can differ. RecT, for instance, forms large rings in the absence of ssDNA, and forms a filament on ssDNA (unlike Beta, which forms filaments on dsDNA) (73). Rad52 protein forms complexes with both ssDNA and dsDNA, although rings of Rad52 protein (like Beta rings) are only found on ssDNA (68).
One of the more interesting questions about the SSAP family of recombinases is what characteristics are shared between them and the RecA family of proteins that carry out strand invasion and strand exchange. In particular, how do λ Beta and RecT compare with the E. coli RecA protein? Hall and Kolodner have shown that the combination of RecT and RecE (AKA ExoVIII) can promote pairing and strand exchange between a linear dsDNA duplex and a ssDNA circle (62). Following the generation of ssDNA by RecE exonuclease acting on the linear DNA duplex, RecT started joint molecule formation by pairing the exposed linear ssDNA with the complementary circular ssDNA and then extending pairing beyond the ssDNA tail, generated by RecE, in effect catalyzing branch migration. This reaction was not simply the result of spontaneous branch migration, because a control reaction with histone H1 in place of RecT could promote pairing, but not strand exchange.
The λ Beta protein can also promote strand exchange. A study by Li et al. (74) showed that Beta was able to promote strand exchange between a 63-mer oligonucleotide and a 43-mer oligonucleotide annealed to M13 ssDNA. The 20 extra nucleotides of the “donor” 63-mer were complementary to the region within M13 DNA that was adjacent to the annealed 43-mer oligo. Beta was able to displace the bound shorter oligo and to drive branch migration, even when mismatches were present in the incoming longer oligo that would otherwise prevent spontaneous branch migration. In addition, there was a polarity that was absent from the spontaneous reaction, likely the result of greater binding of Beta to 3′ ends relative to 5′ ends. This polarity was not intrinsic to the reaction, because it disappeared in reactions with increasing concentrations of Beta protein. Thus, the annealing functions of these SSAPs have the ability to promote branch migration similar in nature, but not the extent, of the one promoted by RecA protein.
But what about strand invasion? The salient feature of recombination mediated by the RecA recombinase is the ability of the RecA-ssDNA nucleoprotein filament to search, find, and then invade a homologous duplex to promote strand exchange (75, 76). Studies have shown that both Beta and RecT have the ability to promote strand invasion, but limited in context and extent compared with RecA-promoted events. For instance, RecA can promote invasion and strand exchange between linear ssDNA and a linear homologous duplex; Beta and RecT cannot carry out this reaction. The strand invasion events reported for Beta and RecT occur between oligos and supercoiled dsDNA plasmids (62, 77). D-loop (displacement loops) structures are formed and are detected as species with altered mobilities using agarose gel electrophoresis. These reactions depend on the superhelicity of the target plasmid and low levels of GC base pairs (∼16% GC). D-loop formation with Beta is greatly inhibited by increasing the GC content to 37%, whereas for RecT, reactions did not occur when GC content exceeded 25% (62, 77). These results seem incompatible with Beta and RecT promoting strand invasion in vivo, given the 52% GC content exhibited by the E. coli chromosome (where these proteins have evolved), and the 50% GC content of phage λ.
There are aspects of these reactions, however, where a common theme between RecA-like proteins and SSAPs becomes apparent. For instance, strand invasion and recognition of homology by RecA and RecA-like eukaryotic homologs have been shown to principally involve the exchange of AT base pairs (78, 79, 80). Thus, perhaps most (if not all) DNA-pairing events in vitro are initiated at AT-rich sequences, and many of the proteins involved in various forms of DNA-pairing events share this common intrinsic property. The ability to further propagate the initial pairing event may involve additional features of a particular recombinase, like the ATPase activity associated with RecA and its homologs, and the partnership of SSAPs with their associated 5′ → 3′-dsDNA exonucleases. Other pairing activities promoted by RecT are reminiscent of RecA-pairing functions. For instance, RecT was found to promote unstacking of bases in ssDNA, unwinding of dsDNA, and aggregation between ssDNA and dsDNA substrates that was independent of homology, all trademarks of RecA-mediated pairing events in vitro (62).
Nonetheless, while they may share some intrinsic properties common among proteins evolved to promote pairing of DNA, both groups of proteins are biochemically distinct (60, 74, 81, 82). Given the lack of any sequence homology between RecA-like recombinases and SSAPs, it is likely that both groups of proteins evolved their functions independently, with any intrinsic similarities driven by the fact that they share common substrates (ssDNA and dsDNA). By whatever path λ Beta protein has evolved to promote recombination during a lambda infection, it and other phage-derived SSAPs share a common property allowing them to be exploited for use as a recombineering tool, a function clearly not shared by the RecA and RecA-like recombinases.
Anti-RecBCD Proteins
In the absence of Chi, the E. coli RecBCD enzyme remains a potent dsDNA exonuclease that has the ability to degrade incoming phage DNA. It is for this reason that lambda and other double-stranded phages encode anti-RecBCD functions like λ Gam, that prevent binding of RecBCD to dsDNA ends by interfering with the enzyme’s DNA-binding site (83, 84). A second method of protection from RecBCD is exemplified by functions like the T4 phage gene 2 protein, which caps dsDNA ends and directly interferes with RecBCD recognition of linear DNA (85). In still a third mechanism to deal with RecBCD, phage P22 of Salmonella enterica serovar Typhimurium produces a protein called Abc2 (anti-RecBCD) which hijacks RecBCD, modifying its activity such that it becomes part of the phage P22 recombination pathway (86, 87, 88). The common goal of all three mechanisms is to inactivate (or modify) the host system of recombination (RecBCD), allowing the phage recombination systems to act unimpeded during the phage infection. However, the lambda Gam protein, because of its direct effect on inhibiting the RecBCD enzyme, has been uniquely instrumental in allowing the λ Red and RecET recombination systems to promote recombineering events in vivo.
λ Gam (11.6 kDa, 98 amino acids) has been shown to inhibit all the known activities of the RecBCD enzyme (17, 89). It does this by binding to RecBCD and preventing the enzyme from binding to dsDNA ends (83, 84). In experiments where Gam complemented growth of a T4 2– phage, overexpression of the RecB subunit interfered with complementation, suggesting that Gam binds to the RecB subunit of the enzyme; overexpression of RecC and RecD had no effect in this test (90). In phage-plating assays, gam expression from a plasmid can mimic the effects of E. coli recBC mutant hosts (86, 89, 91). However, in other assays involving RecBCD-promoted conjugational recombination and restoration of UV resistance, plasmid-produced Gam only partially mimics the effects of a recBC mutation (89, 91). These observations are likely the result of the differential effects of residual levels of RecBCD in these assays. It has been proposed that there are low levels of RecBCD activity in cells expressing Gam from a plasmid, perhaps because of the inability of Gam to inhibit an ongoing RecBCD reaction, where the DNA-binding site for RecBCD is unavailable to Gam (17, 84), or because of insufficient Gam expression. The low levels of Gam may be required for cell viability, because Sergueev et al. (92) showed that Gam is lethal to E. coli when overexpressed from the PL operon via a defective prophage. This inviability is not due simply to total inactivation of RecBCD, because ΔrecBCD strains still retain 30% viability (93, 94). Interestingly, Gam expression can be beneficial for the cell: Gam-induced radioresistance occurs when Gam expression partially protects cells from the lethal effects of X-rays (95). It is believed that the resistance occurs because of the inhibition of DNA degradation following the production of multiple dsDNA breaks in vivo, allowing time and opportunity for the damaged DNA to be repaired (84, 95).
The crystallographic structure of Gam has been solved (83). It is an all-helical dimeric structure, with the two N-terminal H1 helices extending out from a dimerization domain (see Fig. 3). The authors favor a model of RecBCD inhibition whereby a conformational change in Gam takes place upon binding to RecBCD. The two long N-terminal helices of the dimer are highly hydrophobic and are proposed to be inserted into channels within the RecB and RecC subunits normally occupied by the 3′-ssDNA and 5′-ssDNA, respectively, of an unwound dsDNA substrate. Aromatic residues on the surface of the helices are proposed to interact with residues that normally interact with bases of the ssDNA. Gam is thus proposed to act as an ssDNA mimic, occupying the sites in RecBCD normally bound by the ssDNA strands of the unwound end. It is this inhibition of RecBCD that is required to see the high rates of gene replacement promoted by the Red and RecET phage recombination systems when using PCR products as substrates (see below).
Figure 3.
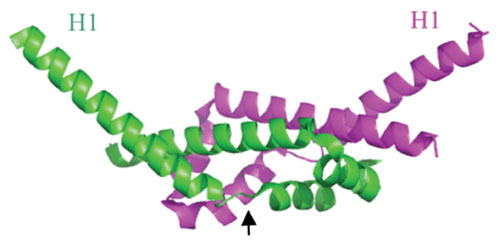
Ribbon diagram of the λ Gam protein dimer; chains A and B labeled green and magenta. It is an all α-helical protein with a dimerization domain (center region) and two protruding N-terminal helices (H1), sticking out at an angle of about 100° from each other. A proposed conformational change occurs upon binding of λ Gam to RecBCD, with the H1 helices rotating about 120° around the Gly-Ile-Pro hinge regions (denoted by arrow in the green subunit). The proposed conformation change places the H1 helices of each subunit into the ssDNA binding regions of RecB and RecCD, thus inhibiting binding of RecBCD to dsDNA ends. Structure generated by PyMol based on the coordinates described by Court et al. (83).
MECHANISMS OF λ RED RECOMBINATION
The Early Years
The pioneering work of Meselson and Weigle (96) and Kellenberger et al. (97) demonstrated that λ recombination may involve (at least some of the time) a “break-join” type mechanism, where, during an infection, DNA was proposed to physically break for unspecified reasons and then join with the similarly broken DNA of a coinfecting partner. An alternative proposal at the time was a “copy-choice” mechanism, where a recombinant was proposed to be fully synthesized using first one parental DNA as a template, then another. A third pathway proposed at the time was a hybrid of the first two called “break-copy,” where a broken chromosome would use a partner as a template to synthesize a full-length chromosome. (For a historical perspective of the beginnings of break-join and copy-choice mechanisms of recombination, see Tang [98]). In the Meselson and Weigle experiments, crosses were performed between unlabeled and C13- and N15-labeled phages, lysates were layered over cesium chloride gradients, and phage recombinants were separated from their parents by density-gradient centrifugation. The authors found recombinant phage in the unreplicated conserved (H/H) peak, indicating that portions of parental DNA could be found within recombinant phage particles, suggestive of a break-join mechanism. However, because the map positions of the markers were close to the right end of the chromosome, a break-copy mechanism could not be ruled out. In later experiments, using heavily labeled phage in both parents and markers associated with a wider central map interval, Meselson again found recombinants in the unreplicated (H/H) peak, supporting a break-join mechanism of λ recombination (99).
These experiments, however, were done before the three recombination systems acting in these crosses (the host rec system, and λ’s red and int pathways) had been identified. In fact, the markers that Meselson used in his later study occurred in a map interval where λ int is active. Thus, the break-join model for the mechanism of λ recombination described by Meselson (99) might not have been representative of the λ Red (or rec) system. In the years to follow, the genetic mutations defining the rec, red, and int recombination systems were discovered, and λ crosses were performed in hosts containing only one recombination system at a time (100). These experiments revealed that all three recombination systems could support break-join mechanism of λ recombination. About the same time, however, it was recognized that, in order to fully follow the path of parental DNA into recombinant progeny, it would be necessary to prevent any replication of phage DNA. Allowing the phage to replicate results in high titers of nonrecombinants (and descendants of recombinants), swamping out the small numbers of unreplicated recombinant phage (i.e., the ones that are best explained by a break-join mechanism). By preventing replication of the phage, one could then separate (conceivably) recombination events that were dependent on replication of the phage DNA from those that were not.
Stahl and colleagues designed many such experiments by performing λ crosses in hosts that were temperature sensitive for host and phage λ replication (101, 102). (These hosts were later identified as mutant in dnaB encoding the major replicative helicase.) Varying the temperatures at which phage crosses were carried out allowed limited (36°C) or more restricted (42°C) amounts of phage DNA replication. In later experiments, a double block to replication was used by employing both a dnaBts mutation in the host and a mutation in the λ replication function P (Pam) (103, 104). These crosses resulted in a severe (i.e., complete) block to phage DNA replication.
What became apparent from these and other studies was the following: (i) Curtailing replication severely inhibited the formation of recombinants generated by the λ Red system (102, 105). (ii) Recombination was necessary for the recovery of unreplicated phage (not including unabsorbed phage) (103, 104). (iii) In experiments where phage replication was modestly inhibited (e.g., by using a dnaBts strain at 36°C), slight changes in the density distributions of recombinant phage following sedimentation in cesium chloride gradients could be detected. These small density shifts were indicative of limited amounts of DNA synthesis (perhaps repair synthesis) in the recombinant phage DNA. The DNA synthesis occurred at different levels dependent upon what map interval in which the recombination took place (101, 102, 105). (iv) When one employed a more severe block to DNA replication (using a dnaBts host grown at 42°C and Pam phage), recombinants were limited to intervals in the right hand side of the lambda genetic map, in the region near the cohesive end site (cos) (106, 107, 108, 109). (v) The cos site, a phage sequence cut by the phage terminase enzyme to package phage genomes, could generate dsDNA ends to serve as an initiator of Red recombination (107). (vi) Double-stranded breaks provided by restriction enzymes also stimulated Red-promoted recombination among unreplicated phage (110, 111, 112). (vii) Removal of the host recombination system (recA) severely depleted recombination among unreplicated phage. Among the Red-promoted recombinants, crossovers were focused in the right end of the genetic map (106). (viii) A small amount of DNA synthesis was associated with unreplicated Red-promoted recombinant phage in the region of the crossover (109). In fact, this result is true for some of the recombinants even when full DNA replication is allowed, confirming the presence of a break-join type of recombination event (113).
What stands out as most pertinent from these early studies is the recognition of the role of replication in Red recombination. Although it was suggested in the early 1970s that λ Red recombination might occur best at replication forks or newly synthesized DNA (101, 114), the prevailing view was that the role of replication was to provide dsDNA ends to the λ Red functions, perhaps in the form of tips of rolling-circle replication intermediates, to act as initiators for a break-join type recombination reaction. From these and other studies emerged two classic pathways of bacteriophage λ Red recombination in phage biology: the RecA-dependent strand invasion pathway and the RecA-independent ssDNA annealing pathway (Fig. 4). The mechanistic details of these pathways have been supported by genetic studies using phage crosses in recA+ and recA− hosts as described above, and by biochemical studies on the structural and enzymatic properties of the λ Exo and Beta proteins. What follows is a description of these pathways, and a perspective on their roles during a normal lambda infection in wild-type cells.
Figure 4.
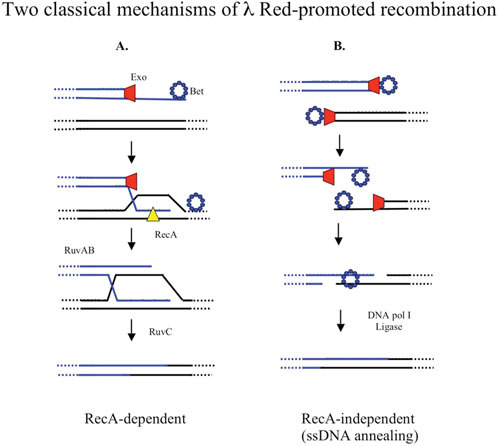
Two classic pathways of λ Red-promoted phage recombination. dsDNA ends of the phage chromosome are provided by the action of terminase. λ Exo (red trapezoid) binds to a dsDNA end and digests the 5′ strand, assisting Beta (blue ring made of small circles) to bind to the 3′-ssDNA tail. (A) The RecA-dependent pathway: In the absence of replication, Beta is replaced with RecA (yellow triangle) with the help of RecF pathway functions, which promotes invasion of the ssDNA into a homologous duplex. Recombination proceeds via branch migration, Holliday junction formation, and subsequent resolution of the intermediate by the host resolvasome, RuvABC. (B) The ssDNA annealing pathway: dsDNA ends are formed containing terminal redundancies, generated by the rolling-circle mode of replication and/or terminase cutting during the lytic infection. Exo and Beta process the ends as above. The Beta protein promotes annealing between the overlapping ssDNA ends, which are filled in by DNA polymerase I and ligated together to form a recombinant.
The RecA-Dependent Pathway of λ Red Recombination
In the RecA-dependent pathway of Red recombination, the role of the Exo and Beta proteins is to process a dsDNA end and to provide 3′-ssDNA for loading of RecA, with assistance by the host RecFOR functions. RecA promotes strand invasion of the ssDNA end into homologous duplex DNA (with no dsDNA ends in the recipient molecule having a role). From this point, the recombination steps follow the paradigm of the host-mediated recombination system of E. coli (shown in Fig. 4A), involving RuvABC-mediated branch migration and Holliday junction resolution (20, 115). The host RecBCD recombinase has no role in this pathway, because the anti-RecBCD λ Gam protein is provided along with the Red proteins.
Is this pathway biologically relevant during a wild-type phage infection in E. coli? λ phage grows well and recombines normally in a recA mutant. It was, in fact, the loss of growth and recombination that was used to identify Red as lambda’s recombination system (13, 14). As described above, the RecA dependency in this pathway is observable in replication-blocked crosses between two λ phages in a wild-type host. This dependency is relieved by replication of the phage. Thus, RecA is not required for recombination between replicating phage in a wild-type host. Its participation in a significant way in the Red-promoted recombination during a replicating lambda infection is possible, but unlikely. First, RecA normally needs to be loaded onto single-stranded DNA by the host RecBCD or RecFOR functions. Neither of these host functions is required for Red-promoted recombination when replication is allowed. (However, it cannot be ruled out that RecA is loaded on ssDNA by an unidentified process during a lambda infection, for example, in a process assisted by Beta or λ Orf protein; see below.) Furthermore, given the high affinity of Exo for DNA (a KM in the subnanomolar range) (49), its high expression levels as part of the PL operon, and the ability of λ Exo to load Beta onto the ssDNA it generates ([58]; Murphy, unpublished), a Red processed ssDNA end in vivo is most likely bound by the Beta protein, not RecA. Poteete has suggested that the RecA-dependent pathway is a “salvage pathway” (116) that occurs when a Red-processed Beta-bound ssDNA tail cannot find a homologous ssDNA partner (either present as a second Red-processed end, or within the context of a replication fork). In this case, the Beta protein has to be removed and replaced with RecA, with assistance from the RecFOR functions, to allow a strand-invasion type of recombination event.
If λ Exo provides a 3′-ssDNA end during a phage infection, why does the phage even need the Beta protein? As mentioned earlier, Red mutants are down 6- to 10-fold for growth and recombination, suggesting that the host RecA recombination system cannot fully substitute for the phage Red system. This result is due in part to the lack of Chi sites in λ DNA (see discussion in section “Comparisons of Red versus RecBCD Pathways of Recombination”). Also, Beta forms a complex with Exo in vitro, suggesting it might more efficiently utilize the 3′-ssDNA end generated by Exo, relative to RecA. Finally, the Red system may be mechanistically intertwined with phage DNA replication (in a way that the host recombination system is not) that presumably provides an advantage for the phage for growth and recombination following infection. What this advantage might be is unknown, but speculations on such mechanisms are discussed below (see discussion on replisome invasion models).
Red-Promoted ssDNA Annealing Pathway
The other classical pathway of Red recombination is the ssDNA annealing pathway of Red recombination (Fig. 4B). This pathway starts with two dsDNA breaks in different regions of the chromosome, one in each recombining parental phage (117). In this pathway of recombination, λ Red stitches together dsDNA ends using their overlapping sequences. The processive action of λ Exo degrades the 5′-ending strands at the dsDNA ends and loads λ Beta on the generated ssDNA ends. Beta then aligns and anneals the ssDNA substrates containing complementary overlaps. In this case, RecA is not required, because no strand invasion is necessary.
Might this pathway be active during a wild-type (replicating) λ infection? It has been proposed that lambda late replication generates dsDNA ends throughout the entire length of its chromosome via the rolling-circle mode of DNA replication (118). It was thus hypothesized that the variety of dsDNA ends generated during late infection represent the “non-allelic cut sites” that Red-promoted recombination splices into recombinants via the single-stranded DNA annealing pathway. The overlapping sequences required for ssDNA annealing might occur on the dsDNA ends of two different rolling-circle replication intermediates of opposite polarity (i.e., two circles replicating in opposite directions). This model of λ Red recombination is based largely on the in vitro properties of λ Exo and Beta proteins (41, 43, 48, 59, 60). In addition, key elements of this model were demonstrated by Red-dependent packaging of phage following transfection of recB spheroplasts with sheared (half-length) molecules of the λ chromosome (119). The authors used a recB host to prevent degradation of transfected DNA, and found that phage lacking λ Exo were down 400-fold (relative to wild type) for the formation of infectious centers. This generation of full-length λ chromosomes (from half-sized fragments) could best be explained by a Red-promoted ssDNA annealing reaction. Of note, however, was that Beta mutants were only down 10-fold in the same assay, an effect that may have resulted from the presence of RecA in these assays. The ssDNA annealing mechanism was also demonstrated in vivo in a study where a restriction enzyme cut was made in one of the two chromosomes during a replication-blocked λ cross in a recA host (117). The dsDNA end generated by the cut, along with a dsDNA end formed by cutting at the cos site (as part of the packaging mechanism), generated two chromosomes with overlapping DNA segments in vivo. Only when the cut with the restriction enzyme was made was significant Red-dependent recombination via ssDNA annealing observed.
However, experimental observations suggest that the ssDNA annealing pathway of Red recombination might be a minor pathway. First, in the Red-promoted ssDNA annealing reaction reported by Stahl et al. (117), only ∼ 0.1% of the incoming DNA was converted into the recombinant product in the recA host. As pointed out by Poteete (3), the low yield (and slow nature) of the ssDNA annealing reaction in vivo does not correspond well to the kinetics of a true λ phage infection and recombinant phage formation, suggesting that, while a pure single-stranded annealing reaction in the absence of replication can occur given proper substrates, it is not the major route of recombination supported by λ Red in vivo. Second, the ssDNA annealing reaction could not be reproduced in vitro using Red-containing extracts, purified λ Beta and Exo proteins, and linear DNA substrates, despite the processive action of λ Exo on these substrates, as monitored by gel mobility shift assays (A. R. Poteete and K. C. Murphy, unpublished observations). These observations contrast with the high rates of RecA-independent Red-dependent recombination observed in phage crosses when replication is allowed.
While the discussion above suggests that the ssDNA annealing pathway depicted in Fig. 4B may not be the optimal pathway for λ Red, this pathway may be more relevant to events mediated by the Rac prophage RecET recombination system. The Rac prophage recE gene encodes an 866-amino-acid protein, with the exonuclease function encoded by the last 260 residues. Most studies examining RecET recombination activity have only used this C-terminal 260-amino-acid domain (AKA, RecE-602). Fu et al. (120) have recently shown that the full-length RecE protein (along with RecT and λ Gam) can promote recombination between linear dsDNA species, via a proposed ssDNA annealing mechanism, at a much higher efficiency relative to either λ Red plus Gam, or RecE-602, RecT, and λ Gam. Thus, while the RecET system encoded by Rac prophage may have evolved to be more proficient than Red for a ssDNA annealing pathway of recombination, the λ Red system may be better designed for a pathway that involves invasions of existing replication forks (see below).
Replisome Invasion/Template Switch Model of λ Red Recombination
A new pathway of Red recombination, called the Replisome Invasion/Template Switch model (RITS) model, has been proposed (3). In this scheme, the replication fork plays a direct role in Red-mediated recombination as a target of an Exo-Beta-processed dsDNA end. The model was inspired by two observations. First, no significant Red-promoted recombination is observed in recA mutants in the absence of phage replication (101, 106, 112). Second, in ssDNA oligo-mediated recombineering, which requires λ Beta as the only phage-supplied function, there is a difference in the recombineering frequency when the oligo is targeted to the leading versus the lagging strand templates of a replication fork (the latter being favored). The difference is reported to be between 3- and 50-fold in E. coli, but can range much higher in other systems (103 to 104 fold in Mycobacterium smegmatis (121) and Pseudomonas aeruginosa (Murphy, unpublished). These differential rates of recombineering with oligos targeting the leading versus lagging strand templates suggest the replication fork as the target of a Beta protein-ssDNA complex.
In the study by Poteete (3), an in vivo physical assay employing a cross between a nonreplicating phage substrate and a replicating plasmid was used to test the RITS model; the substrates are depicted in Fig. 5. Recombination between the homologous regions of the phage and the plasmid (green box in Fig. 5) generates a crossover that places the two BamHI sites on the same DNA fragment. Following isolation of DNA from the cell and digestion with BamHI enzyme, the amount of the recombinant band is detected by Southern analysis (see legend to Fig. 5 for more details). The phage in this “physical” assay is used for delivery of the recombination substrate to the cells with high efficiency. Note that the recombining substrates are isolated directly from the cells and examined on polyacrylamide gels. Thus, the phage DNA is not packaged, and no viral progeny is produced. The study showed that the recombinant band: (i) is dependent on a dsDNA break and λ Red, but not RecA; (ii) is generated at a high frequency (30% of the incoming DNA); (iii) appears within 5 minutes following phage infection; and (iv) is inhibited by the presence of dideoxy nucleotides, revealing its replication dependency.
Figure 5.
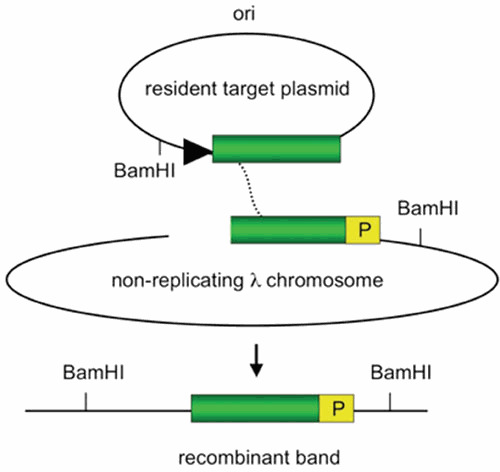
Substrates used to demonstrate Red-promoted replisome invasion. Recombination occurs between a replicating resident target plasmid (direction of replication shown by black triangle) and a nonreplicating λ chromosome. The homologous regions are denoted by the green box. The λ chromosome is delivered at high efficiency by infection, is inhibited from replicating by overexpression of the λ c1 repressor, and is cut in vivo by a chromosomally encoded PaeR7 restriction enzyme. DNA from the infected cells is isolated at different times after infection, cut with BamHI, and subjected to a Southern procedure. The amount of recombinant band (bottom) is detected by probing a Southern blot for sequences designated “P.” (Descriptions of substrates were derived from reference 3).
This study led to the development of the RITS model of λ Red phage recombination depicted in Fig. 6. In this model, λ Beta is loaded onto the ssDNA generated by λ Exo ([58]; Murphy, unpublished) and anneals the ssDNA to the lagging strand template of a replication fork. The model proposes that the incoming strand then serves as a new template for leading strand synthesis. The role of Beta in this model (so prominent in the ssDNA annealing pathway of Red recombination to anneal complimentary strands generated by λ Exo) is redefined here to anneal Exo-generated ssDNA to the lagging strand template of a replication fork and, in addition, to position the incoming strand to be captured by the leading strand DNA polymerase. In the RITS model, the template switch redirects the replication fork (like a train switching tracks) to the incoming substrate. How the replicative helicase is diverted on to the new substrate and how lagging strand synthesis on the new template is initiated (Fig. 6) were not specified in the initial description of the model (3).
Figure 6.
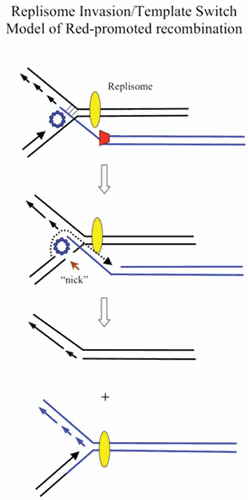
Replisome invasion/template switch model of λ recombination. The diagram depicts a recombination event between the tip of a rolling circle (circle not shown), and another replicon (either one of the replisomes of a theta-mode intermediate, or the replisome of a second rolling circle). (Top) A Red-processed dsDNA end (Beta bound to a ssDNA overhang generated by Exo) invades a replication fork and promotes annealing to the lagging strand template. (Middle) Beta captures the leading strand and promotes a template switch, such that the leading strand polymerase now uses the incoming strand as a template. (Bottom) Template switch (TS) model invokes a redirection of the replisome to the incoming strand. The template switch then connects one arm of the original replisome to the invading duplex (i.e., the recombination event). As before, red trapezoid, λ Exo; blue circles, λ Beta. Yellow oval represents the replisome.
New Replication Fork Model of λ Red Recombination
As an alternative to a redirected replisome, the Red-promoted recombination event could instead, like T4 phage-promoted recombination, initiate a new replication fork, which would travel in the opposite direction to the invaded fork shown in Fig. 7. In this scenario, the original fork is left unmodified. T4 (and yeast) can promote the establishment of new replication forks following recombinogenic 3′-strand invasion events, a process known as break-induced replication (BIR) or recombination-dependent DNA replication (RDR) (122, 123, 124, 125). (For an early suggestion that annealing of an enzyme-processed break might propagate a replication fork, see Mazin et al. [126].) The replication of the phage T4 linear chromosome is initiated from multiple origins and is independent of recombination. However, soon after infection, this origin-dependent replication mode terminates, and further replication of T4 DNA is promoted by recombination-dependent replication. The dsDNA ends of the linear chromosomes generated by origin-dependent replication become substrates for the UvsX recombinase, which promotes dsDNA end invasion into a second linear chromosome (or in a single infection, its terminally redundant end) to generate a D-loop. The D-loop is then a substrate for the assembly of a replisome. A large branched network of linear chromosomes is thus formed, followed by cutting off the branches by specialized endonucleases to form linear multimers, which are packaged into phage heads. The T4 chromosome carries its own set of replisome components to carry out this elaborate process; for reviews, see references 125 and 127.
Figure 7.
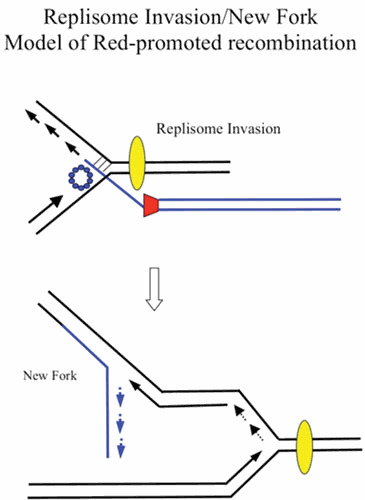
New replication fork model of λ recombination. As in Fig. 6, the annealing of the ssDNA generated by λ Exo anneals to an ssDNA region on the lagging strand template. In this model, however, the invaded replisome is not affected. Instead, the invasion of the incoming duplex initiates a new fork that travels in the opposite direction, with the annealed strand becoming the template for the new fork’s lagging strand. The incoming duplex is then connected to one arm of the fork (i.e., the recombination event). As before, red trapezoid, λ Exo; blue circles, λ Beta. Yellow oval represents the replisome.
However, if one imagines that λ Red promotes new replication forks by RDR, it likely does so by a different mechanism relative to that of T4 described above. One reason for this notion is that T4 promotes RDR by supplying its own replication system to initiate the process, whereas lambda relies on the host system for replicating its DNA. Whether the E. coli replication apparatus could be efficiently utilized to start new forks for replicating λ chromosomes, like the T4 system, is unknown. Second, in a T4 infection, annealing of the 3′ end is carried out by the UvsX protein, a recombinase capable of invading dsDNA substrates resulting in the formation of a D-loop. The displaced strand is critical for RDR, because it becomes the template for lagging strand synthesis of the new replisome. The invading 3′ strand then becomes the primer for the new leading strand (127). In the λ Red replisome invasion model, the 3′ end anneals to ssDNA on the lagging strand template. As such, no displaced strand is available for formation of a new lagging strand.
A model for how Red might promote formation of a new replication fork is presented in Fig. 7. The model suggests that, unlike T4, the strand with the annealing 3′ end (generated by Red on the end of an invading duplex) becomes the template strand for lagging strand synthesis in the new replication fork; the targeted fork continues unimpeded. This outcome is true whether the invading strand anneals to either the leading or lagging strand templates of the original fork, although the direction of the new replication fork would depend on which template strand is targeted for Beta-promoted annealing (see Fig. 8, courtesy of A. Kuzminov). Unlike studies with T4, however, details of this mechanism have not been rigorously tested in vivo or in vitro.
Figure 8.
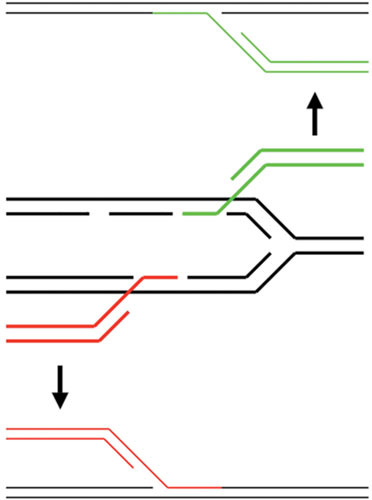
Diagram showing how the 3′-ssDNA tails of dsDNA substrates (generated by λ Exo acting on the dsDNA ends) could anneal to the ssDNA regions of a replication fork. The 3′-ssDNA tail on top (in green) anneals to an ssDNA region within the lagging strand template, while the 3′-ssDNA tail on bottom (in red) anneals to an ssDNA region within the leading strand template (a more infrequent event perhaps, due to lesser amounts of ssDNA expected on this template). In either case, the invading duplex becomes one prong of the new fork, with the annealed strand becoming the new lagging strand template.
Recombination-Dependent Replisome Disruption Model of λ Red Recombination
Finally, a third model for λ Red recombination involves a replisome invasion as described above for the RITS and Fork Initiation models, but with subsequent disruption of the replisome—the Recombination-dependent Replisome Disruption model (RRD). In this model, the act of invading the replisome and annealing the Red-generated 3′-ssDNA tail of the incoming dsDNA linear molecule triggers disassembly of the replisome, leaving behind a four-stranded structure. Such a structure could then be acted upon by a Holliday junction-type resolvase to generate an unreplicated chromosome, and a recombinant where the attacking duplex is now connected to the lagging strand arm of the replication fork. Details for how such an event may help the phage grow and recombine will be described elsewhere (unpublished data).
The Role of Red and Gam in Phage λ Replication
The role of the Red and Gam functions in λ DNA replication was examined directly by Enquist and Skalka (16). Using [H3] thymine incorporation as a measure of DNA synthesis, they found that Gam helps λ DNA to replicate by virtue of its inhibition of the host RecBCD enzyme, allowing phage DNA to replicate via the rolling-circle mode. As noted above, this process leads directly to multimeric forms of linear DNA (i.e., concatemers), which are the immediate precursors for DNA phage packaging into phage heads. As a result, the λ gam mutant accumulated only about half the amount of phage DNA as the wild type during a phage infection. This effect disappeared in a recB host, as expected given the role of Gam.
A little more puzzling, Enquist and Skalka (16) found that red mutants also showed about a 50% decrease in phage DNA equivalents relative to wild-type phage following infection (into either wild-type or recA hosts). They showed that rates of initial synthesis (primarily in the form of theta circles) were the same for wild type and the red mutant, suggesting that this decrease in phage replication was due to a decrease in the number (or size) of late-forming concatemeric DNA molecules. Nonetheless, the DNA generated by λ red mutants was not qualitatively modified relative to wild-type λ, because it was packaged into phage heads to generate infectious phage particles. The same could not be said of λ red gam mutants, which were able to replicate only via the theta mode of replication, generating a mixture of closed and nicked circular monomers. The explanation for this result is as follows: in the absence of Gam, rolling-circle intermediates are inhibited by RecBCD, and in the absence of Red, no circular dimers (or multimers) of λ DNA are formed. Since monomeric DNA is not a substrate for packaging, no viable phage progeny are generated.
Better and Freifelder (128) also studied the role of Red in λ phage DNA replication. Using an improved system that involved the use of a host containing an endA mutation to prevent nicking of circular monomers in their bacterial lysates, and by isolating the total pool of intracellular phage DNA molecules, they looked directly at DNA replication intermediates under the electron microscope. A key finding was that there was no difference between λ wild type and red mutants in the timing or numbers of rolling circles, suggesting that Red is not required for the generation of these types of replication intermediates. (In addition, unlike the findings of Enquist and Skalka [16], they did not find a reduction in DNA synthesized by the λ red mutant late in infection. The reason for this discrepancy is not known.) The authors also found that rolling-circle replication occurs throughout the λ life cycle, observable within 4 min after infection. As such, while these species predominate late in infection, they are not restricted from forming early. In agreement with the earlier studies, their analysis shows that the key role of Red for λ replication (which is most evident in the absence of Gam) is to promote circular multimers of λ DNA by recombination to generate a substrate suitable for packaging.
Other λ Functions That Influence the Mechanism of Red Recombination
The bet, exo, and gam functions discussed above are the only λ genes required for efficient Red-promoted recombination, because expression of these genes from plasmids in the absence of other λ functions provided wild-type levels of phage recombination and recombineering (23, 129). However, other λ functions can influence the outcome of recombination in the RecA-dependent Red-promoted pathway, as well as in host-promoted RecA-dependent events that are independent of the λ Red functions. Among these lambda phage accessory functions are the λ orf and rap genes (formerly ninB and ninG, respectively).
Sawitzke and Stahl (130) found that λ red int phage crosses recombining via the RecF pathway of recombination (the pathway present in recBCD sbcBC hosts) were not dependent on RecF. However, a dependency on RecF (and RecO and RecR) could be observed when the ninR region of phage lambda was deleted. They identified a small open reading frame responsible for this effect, and called it orf. The orf gene was cloned, producing a protein of 15 kDa that complemented the recombination defects of λ orf phage. The authors found that Orf expression from the plasmid does not suppress the conjugational deficiency of the recO mutant or the UV sensitivity of the recFOR mutant in E. coli (131). In a later study, these authors found that in the absence of replication, Orf is required for focusing recombination in the vicinity of the cut-site cos in both the RecF and λ Red pathways (132, 133). This focusing effect is not seen when λ recombines via the canonical RecF pathway (i.e., in the presence of RecF protein, the sbcB15 allele, and the absence of Orf). Since the role of RecFOR is thought to displace single-stranded DNA binding (SSB) protein and load RecA on to ssDNA regions of recombination intermediates (134, 135, 136, 137, 138, 139), Sawitzke and Stahl favor a model whereby Orf either displaces or competes with SSB, thus allowing RecA access to ssDNA during λ recombination. An additional role of Orf was proposed from these studies: to protect ssDNA from digestion by endogenous exonucleases.
In Red-mediated recombination of linear dsDNA substrates containing large regions of flanking homology into the E. coli chromosome, chromosomally encoded orf was found to compensate not only for mutations in recFOR, but in ruvAB and ruvC as well (116). Furthermore, orf expression complemented other host recFOR defects, including replication of a pSC101-derived plasmid and UV sensitivity. The complementation of UV sensitivity was not observed in a previous study (131), where the orf function was produced from a plasmid. In the study by Poteete (116), orf was expressed from the chromosome by using a controllable promoter at the galK locus. Thus, differences in the observed phenotypes in these studies could be due to the presence of a resident plasmid in the study by Sawitzke. In recBC sbcBC strains, plasmids exist as linear multimeric forms (140, 141), which might influence recombination/repair events involving dsDNA ends. Alternatively, the differences could be due to expression levels of Orf, as both studies found overexpression of orf is lethal to the cell.
If the normal role for RecFOR in λ orf crosses is to displace SSB and help load RecA onto ssDNA, and Orf can substitute for RecFOR in this capacity, then one might propose that the role of Orf is to bind SSB and help displace it from ssDNA during a RecA-dependent lambda recombination. A test of this notion would be that a mutation in recA that obviates the need of RecF in conjugational recombination (the srfA mutation, recA803 [134]), might also work to suppress the deficiencies caused by the absence of RecF and Orf in RecA-dependent Red-promoted recombination. This was found not to be the case, because the RecA803 strain (with no Orf) was unable to promote RecFOR-independent recombination via the Red pathway (116).
However, for RecA-dependent recombination via the Red pathway, it is Beta that must be removed from ssDNA for replacement by RecA, not SSB. An Orf-modified SSB may be a specialized modification that allows RecA to easily displace Beta without interference from SSB. In partial support of this proposal, Maxwell et al. (142) found that Orf binds to the E. coli SSB protein. The observation that λ Orf can suppress the recombination defects owing to mutations in ruvABC as well as recFOR is harder to explain, but may be a reflection of the pleiotropic roles SSB has in many replication, repair, and recombination processes (143). As suggested by Poteete (116), an Orf-modified SSB may inhibit a resolution event that does not lead to productive recombinants, or activate a RuvABC-independent pathway of resolution.
The study by Maxwell et al. (142) also determined the crystal structure of the Orf protein. The authors showed that Orf is an asymmetric dimer in the form of a ring with a funnel-like channel through its center. In addition, the study demonstrated that Orf binds to ssDNA. Using dsDNA substrates containing ssDNA gaps, the authors determined that ssDNA binds to a U-shaped cleft on the surface of the protein rather than going through the central channel (142). They suggested that a fluorescence quench observed when Orf binds ssDNA could indicate a conformational change that allows preferential binding of RecA or Beta. For RecA-dependent Red-promoted recombination, one would imagine that Orf’s role would be to bind ssDNA (perhaps guided there by an interaction with SSB) and to create a ssDNA conformation that favors binding of RecA over both Beta and SSB. In a recent study by Curtis et al. (144), deletion of the C-terminal 6 residues from Orf resulted in a mutant protein with reduced DNA binding affinity, an effect also seen with a W141F mutant. Truncation of the C-terminal 19 amino acids of Orf resulted in a protein that was unable to bind ssDNA, but left the protein in a largely unfolded state. The C terminus of the E. coli SSB protein is known to interact with numerous replication and recombination proteins (145). Interestingly, however, a 10-residue deletion of SSB’s C terminus did not disrupt the interaction between SSB and λ Orf, suggesting that some other region of SSB is responsible for binding to λ Orf.
In RecA-independent Red-promoted recombination (the ssDNA annealing pathway), the role of Orf is unclear; in fact, there may be no role at all, as orf is not required for Red-promoted recombination in freely replicating phage crosses (130, 146), or in Beta-promoted oligo-mediated recombineering events (see below). Overall, a complete description of how Orf mechanistically mediates Red recombination remains elusive.
Finally, the rap gene (formerly ninG) of phage λ was originally identified as a function that increased the level of RecABCD-promoted recombination between a phage and a plasmid (147) and, hence, received its name (recombination adept with plasmids). The rap gene is functionally analogous to the rusA gene of cryptic prophage in E. coli K-12 (148). It encodes a resolvase that can (along with recG) suppress the UV sensitivity of ruvA, ruvB, and ruvC mutants. The Rap resolvase has been shown to preferentially bind and nick artificial Holliday junctions, and other types of branched DNA structures, close to the position of strand crossovers. Duplex, partially duplex, and ssDNA substrates are not efficiently recognized by λ Rap (386). With branched substrates containing a dsDNA-ssDNA junction, Rap was found to nick in the duplex regions of the molecules. However, unlike resolvases such as RuvC from E. coli, phage T4 endonuclease VII and phage T7 endonuclease I, Rap did not promote symmetrical paired incisions in artificial Holliday junctions with fixed crossovers, or ones with limited homology (2 to 3 bp) at the core. It did, however, promote symmetrical cuts with structures containing larger regions of homology (11 to 12 bp) at the crossover point, with a preference for cutting between 5′G-C3′ dinucleotides (387). These cuts were nicks in duplex DNA, because they could be repaired by ligase. Nonetheless, Rap still produced cuts at other sites in these substrates, resulting in a moderate degree of asymmetry in its digestion pattern, often producing products with three arms (or even one arm), suggesting that some nicks were occurring opposite one another in one arm of these artificial Holliday junctions.
Like the orf gene described above, the rap function is not required for Red-promoted recombination under normal circumstances, but its effects can be observed with replication-blocked crosses. Under these conditions, the focusing of recombination near λ’s cos site (seen with wild-type λ) is not observed in the absence of orf or rap. It is believed that the λ Rap resolvase can focus recombination events in this region by resolving RecA-promoted strand invasion events before they have a chance to branch migrate toward other regions of the chromosome (133). The interdependence of Red recombination on Rap can be explained by the fact that the host RucC resolvase can participate in Red-promoted recombination. In assays measuring RecA-assisted Red-promoted recombination between a drug-resistance marker (drugR marker) flanked with ∼1.5-kb homologies to the host chromosome, recombination was down 35-fold in a recG ruvC mutant. This defect could be complemented by expression of the λ Rap protein (149).
GENE REPLACEMENT TECHNOLOGY
Nonreplicating Plasmids: Cointegrate Formation and Resolution
The ability to knockout a gene of interest is of central importance in the genetic analysis of bacteria, yeast, and higher eukaryotes. Historically, in bacteria, gene knockouts or replacements have involved the transformation with nonreplicative plasmids that carry the modified gene, a selectable marker, and often a counterselectable marker (e.g., sacB). Conditionally replicative plasmids include ColE1-derived plasmids that cannot replicate in polA strains (150, 151), a temperature-sensitive (repA mutant) pSC101 replicon (152), and phagemid-based vectors (153). Since the plasmid cannot replicate in the target host, a drug-resistant transformant is typically the result of cointegration of the plasmid into the recipient host chromosome, leaving both a wild-type and modified version of the gene of interest. Resolution of the cointegrant, found by screening for the loss of a counterselectable marker, results in either restoration of the wild-type gene or replacement of the gene with its modified version.
While the cointegrant scheme of gene replacement has been used successfully for many years in a variety of hosts, it is often a time-consuming endeavor. The homologous recombination step between the plasmid and the chromosome and the subsequent cointegrant resolution event do not occur at high frequencies. Many times the resolution event restores the wild-type allele at a high frequency, making the search for the rare gene replacement more difficult. Finally, cloning of the gene of interest and construction of the mutant allele in the nonreplicating vector is a prerequisite for gene replacement.
Recombination-Proficient Strains of E. coli
An alternative to the use of replication-deficient plasmids for gene replacement was the use of recombination-proficient E. coli strains that worked well for transformation with linear DNA (154, 155, 156, 157). The common genetic features of these strains included inactivation of the host RecBCD function, and upregulation of an alternative recombination pathway (either RecE or RecF pathways). These strains are genetically represented by ΔrecBCD sbcA (the RecE pathway) or ΔrecBCD sbcBC (the RecF pathway) (see references 20 and 158 for reviews). The sbc mutations are suppressors of recBC mutations that turn on alternate recombination pathways (159). The sbcA suppressor upregulates the recET genes of the Rac prophage, a defective lambdoid element found in strains of E. coli used in early studies of homologous recombination (160, 161). The recE and recT genes constitute the Rac prophage recombination system and are functionally equivalent to the λ Red functions Exo and Beta, respectively (162, 163). As discussed above, RecE is a 5′ → 3′-dsDNA exonuclease, while RecT is classified as an SSAP that forms rings, anneals complementary ssDNA, and promotes recombination. The use of the RecET genes for recombineering is discussed below.
Another suppressor of recBC deficiency is the sbcB15 mutation, a specific mutation in the xonA gene encoding Exonuclease I, a 3′-ssDNA exonuclease (164, 165) that inhibits DNA degradation but does not affect DNA binding of the enzyme (see below). Strains of this genetic background also spontaneously acquire null mutations in the sbcCD operon (166, 167), inactivating the ScbCD exonuclease (168), which results in healthier growth of the strains. The sbcB15 mutation activates the RecF pathway, which normally promotes recombination away from dsDNA ends, and allows it to catalyze recombination using dsDNA ends (169). In wild-type cells, the RecF pathway is known to be involved in the repair of blocked ssDNA gaps in replicated DNA (170) and in plasmid recombination (171). Components of this pathway (e.g., RecA, RecF, RecO, RecR) are also involved in the repair and reactivation of stalled replication forks (172, 173, 174). Interestingly, the sbcB15 mutation differs phenotypically from a xonA deletion (164). Zahradka et al. (175) found that the sbcB15 allele is a stronger suppressor of the recBC phenotype relative to ΔxonA, and is less affected by mutations in recF and recQ. These results suggest that the sbcB15 allele encodes an ExoI protein with an altered activity that promotes recombination. This supposition was confirmed by Thoms et al. (176), who showed that the sbcB15 allele encodes an ExoI A183V mutant protein, which retains only 1.6% of the activity of the wild-type protein, yet can still bind to 3′-ssDNA overhangs. The implication is that the SbcB15 protein (ExoI-A183V) binds to and protects 3′-ssDNA ends from degradation by other exonucleases, allowing the ends to survive and participate in RecFOR-mediated recombination events. The ability of the RecF pathway (in recBC sbcBC strains) to act upon dsDNA ends allowed this strain to be transformed with linear DNA for making chromosomal knockouts and mutations (154, 155, 157).
A third type of strain that has been used as a recipient for genetic modification by linear DNA carries the recD mutation (156, 177), which inactivates the exonuclease function of RecBCD, but leaves the RecBC recombinase intact (178). All three of these strains share the common features of inactivating the powerful RecBCD dsDNA exonuclease activity, while activating an endogenous recombination system.
The Onset of Red Recombineering
The use of recombination-proficient E. coli strains recBC sbcA, recBC sbcBC, and recD facilitated making mutants in this bacterium by a new approach, via transformation with a linear DNA species consisting of a drugR marker flanked by regions upstream and downstream of the targeted gene. Cointegrant formation and resolution events are not required to form a recombinant, but such reconstructions were limited to the specialized genetic backgrounds. Also, for investigators studying bacteria other than E. coli (including pathogenic species), the only pathway for targeted gene disruption was still the time-consuming use of nonreplicative plasmids (described above) carrying the truncated (or modified) gene of interest. It was recognized in 1998 (23) that E. coli cells expressing the λ Red system and the λ Gam protein should mimic the phenotypes of recombination-proficient recBC sbcA, recBC sbcBC, and recD mutants, in that the RecBCD enzyme was inhibited by λ Gam, and an alternative efficient recombination system (by λ Beta and Exo) was established. Since these functions had been supplied on plasmids and expressed in vivo (129), they could conceivably offer an easier method for gene replacement technology for bacterial species other than E. coli, including pathogenic species of bacteria. With that supposition, linear DNA fragments were constructed that contained drugR markers flanked by 1 kb of target DNA and electroporated into cells containing red and gam-expressing plasmids (23). The choice of 1-kb regions of homology reflected the belief at that time that dsDNA phage recombination systems work on DNA of such lengths (179, 180). In these experiments, expression of the Red system increased recombination yields 1 to 2 orders of magnitude relative to recBC sbcBC and recD hosts (23).
This study first demonstrated the use of PCR products as substrates for Red-promoted recombination. These substrates, however, contained 1 kb of flanking homology and the recombination observed was largely dependent on RecA. Thus, recombination with these substrates proceeded through the RecA-promoted pathway of λ Red recombination discussed above. In any case, λ Red + Gam expression not only provided a cellular environment for easy transformation of linear DNA substrates, but it also created a hyper-rec phenotype to a greater degree than the recombination-proficient strains it was trying to mimic. This result was unexpected.
Later that year, Stewart and coworkers, using the E. coli Rac prophage RecET recombination system (181), recognized that PCR-generated fragments with as little as 42 bp of homology could also serve as efficient substrates for gene replacement in E. coli. Furthermore, the use of such small homologies revealed that this recombination was RecA-independent. The use of short homologies was a dramatic development, because flanking homologies could now be incorporated into PCR substrates by primer design, and thus the requirement for construction of plasmid-derived substrates containing long regions of homology was obviated. Zhang et al. (181) also showed that RecET could modify both plasmid and chromosomal DNA, and demonstrated the use of Flp and Cre with this new technology that could be used to make markerless gene deletions. The use of recET+ recombination also required inactivation of the RecBCD dsDNA exonuclease by coexpression of the λ gam gene.
The lambda Red system can also promote recombination with PCR-generated substrates containing short regions of homology. Datsenko and Wanner (182) showed that red and gam, under control of the PBAD promoter and expressed from a low copy number pSC101 replicon, promoted recombination with PCR substrates carrying 36-bp flanking homologies. They also described the use of FRT sites to generate markerless deletions. In the initial description of the Red system of gene replacement (23), the red genes were driven by the Plac promoter from the chromosome and induced with IPTG (isopropyl-β-d-thiogalactopyranoside), which promoted recombination with long homology substrates of 1 kb, but very poorly with PCR-generated substrates. However, when substituted with the stronger Ptac promoter, high levels of recombination were generated with PCR substrates as well, suggesting that high levels of the Red proteins are required for recombination with PCR substrates containing short homologies ([183]; unpublished observations). Don Court and his colleagues developed an expression system whereby the red and gam genes were produced from a defective prophage and driven by the strong PL promoter (184). The defective prophage had its lysis, replication, and structural genes removed and carried the temperature-sensitive repressor cI857. Thus, the red and gam genes are induced by incubation of cells at 42°C for 15 min. This system, with its high expression and tight control in the uninduced state, has the advantage to better control the levels of Red proteins within the “recombinogenic window,” thus preventing illegitimate events that might possibly occur in the uninduced state, especially when dealing with repetitive elements and large eukaryotic DNA cloned into BACs.
The high efficiency of Red and RecET-promoted recombination with such short regions of homology has allowed E. coli geneticists to perform PCR-mediated gene replacements that yeast geneticists have performed for years (185, 186, 187). This technology for engineering bacterial chromosomes using phage recombination systems has been termed recombineering (188, 189) and is now the common way to create gene knockouts in the E. coli chromosome. It allows investigators to alter single base pairs in their genes of interest, insert large regions of heterologous DNA, create small or large deletions, construct chromosomal transcriptional or translation fusions, and take control of gene expression by insertion of a controllable promoter into an endogenous operator. Together with site-specific systems of Cre and Flp, almost any imaginable reconstruction of the genome is possible, providing that the essential functions of E. coli are left intact.
RECOMBINEERING – METHODS AND MATERIALS
Sources of Red
To promote recombineering in E. coli, the λ red and gam functions can be expressed from a plasmid, a defective lambda prophage, or encoded within the bacterial chromosome. Recombineering protocols have been published previously (4, 5, 6, 7), and some of the details of these procedures differ in minor ways from the description presented below, in large part because of the source of the Red functions. Significant alterations between protocols are addressed here, although the reader should refer to these descriptions if using alternate sources of the Red functions.
The most popular (and practical) method is to express red and gam from a plasmid that can be later lost due to a heat-sensitive origin of replication. This setup allows one to cure the target cells of the Red/Gam-producing plasmid following gene replacement, by growth of the recombinant at 42°C (182, 183, 190). Alternatively, the plasmid can encode the counterselectable marker sacB to select for cells that lost the plasmid by outgrowth on sucrose-containing plates (191, 192). The expression of red should also be controllable, because constitutive expression of Red results in increased levels of spontaneous mutagenesis (183). Typically, controlled expression of red and gam is done by utilizing promoters Plac, PBAD, or the early PL promoter of phage λ. The repressors/activators associated with these promoters driving the red and gam genes (lacI, araC, and cI857 repressor, respectively) are also expressed from these plasmids, making them useful in a variety of E. coli and non-E. coli hosts. Table 1 lists details of some Red-producing plasmids, and sources for other vectors commonly used for Red recombineering containing other drugR markers.
Table 1.
Plasmids expressing λ red and gam
| Plasmid | Operon | Origin | DrugR marker | Reference | Source |
|---|---|---|---|---|---|
| pSIM5 | PL-gam-bet-exo | pSC101 | CamR | 190 | http://redrecombineering.ncifcrf.gov/strains--plasmids.html |
| pSIM9 | PL-gam-bet-exo | pRK2 | CamR | ||
| pKM208 | Ptac-gam-bet-exo | pSC101 | AmpR | 183 | www.addgene.org |
| pKD46 | PBAD-gam-bet-exo | pSC101 | AmpR | 182 | http://cgsc.biology.yale.edu/ |
| pRed/ET | PBAD-gam-bet-exo-recA | pSC101 | TetR | 388 | www.genebridges.com |
By and large, the choice of one or the other among these vectors (and their derivatives) depends on details of the strain under construction and investigator preference. The use of pSIM5 and its derivatives has been promoted because of its high expression and tight repression of the red and gam functions (190). In this study, side-by-side comparisons showed that the PBAD expression-vector pKD119 was down 10-fold relative to pSIM5 for recombineering of PCR-generated substrates. In similar comparisons, the Ptac-gam-red operon of pKM208 gave identical recombination frequencies relative to pSIM5 (unpublished observations). In addition, pKM208 has worked in pathogenic species of E. coli (e.g., EHEC) where pKD46 has failed to generate recombinants (unpublished observations), probably a result of the higher expression level of the red genes with Ptac relative to PBAD in this host. On the other hand, in E. coli K-12, PBAD-driven Red recombineering has been widely employed as a standard technique (193).
In some circumstances, however, it is beneficial to have the red and gam functions expressed from the chromosome. This is the case when the recombineering target is either a BAC or a resident plasmid. The Court laboratory has generated a number of strains where red and gam are expressed from their endogenous promoter (PL) within a defective prophage. The recombination functions are induced by heating the cells to 42°C for 15 min to inactivate the thermo-labile cI857 repressor. The heating step is limited to 15 min to prevent killing of the host by phage kil and gam functions. Once induced, the cells are highly recombinogenic, and can be used to modify BACs and plasmids with PCR substrates or single-stranded oligos (184, 188, 189). This setup can also be used to modify chromosomal genes. In this case, after the modification, the prophage can be removed by a subsequent recombination event, or the modified allele (if marked) can be transferred to a clean genetic background by P1 transduction.
Nonprophage chromosomally encoded red functions for recombineering have also been constructed. Strain KM22 contains a replacement of the recBCD genes of E. coli with a Plac-bet-exo operon (23). (The gam gene was not included since its target genes [recBCD] had been deleted.) A later strain (KM34) was constructed where the recBCD region was again deleted and replaced with the red function driven by the stronger Ptac promoter (unpublished observations). While both strains could perform gene replacement with substrates containing long regions of homology (∼1 kb), KM34 was ∼100-fold more proficient for recombineering with PCR substrates flanked by 40 bp of target homology (unpublished observations). This result revealed that higher levels of Red functions are necessary to promote efficient Red recombineering with short homologies (40 bp).
Recombineering Substrates
The real versatility of the λ Red recombineering technology is that PCR products can be used directly as recombination substrates in a one-step electroporation protocol. The basic steps of the procedure are listed in Fig. 9A. A drugR marker is used as the template for PCR, and primers are designed to generate a PCR product that contains the drugR marker (along with its regulatory region[s]) flanked by 40 to 50 bp of the target sequence. Typically, 20 bases on the 3′ end of the primers are complementary to the drug-resistance cassette, while 40 to 50 bases of the 5′ end of the primers contain target sequences (see Fig. 9B). One must take care not to use intact plasmids as a template for the PCR, because they will transform cells at high frequency and show up as false positives for gene replacement. To circumvent this problem, use gel-purified (ori–) plasmid fragments for templates or colony PCR where drugR markers are chromosomally located. Alternatively (or in addition), treat the PCR product with DpnI, which cuts only methylated DNA (i.e., the plasmid template) prior to electroporation. A drugR marker contained within a nonreplicating vector is also an option for a useful PCR template (182). A number of DNA templates to amplify drugR markers, along with suggested priming sequences, are listed in Table 2. Many of these genes are found within plasmids already present in most laboratories. The PCR substrates are then either ethanol precipitated or cleaned by column purification (e.g., Qiagen PCR purification kit), redissolved in 10 mM Tris-HCl, pH 8.0 (or water) and used directly in the electroporation protocol (see below).
Figure 9.
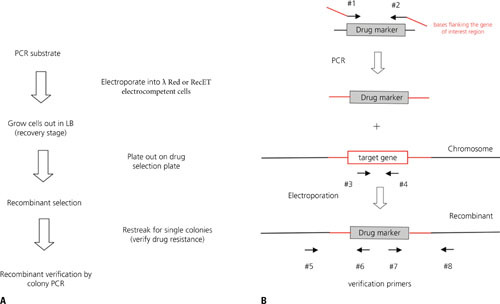
Gene replacement and verification of recombinants using recombineering. (A) Outline of the basic steps involved in recombineering. (B) Primer design for gene replacement and verification. The 3′ ends of primers 1 and 2 contain 20 bp for amplification of the drugR marker (including regulatory regions), while the 5′ ends of the primers contain 40 to 50 bp of sequence that flank the target gene (red lines). Primers 5 and 6 are used to verify the 5′ junction of the recombinant, and primers 7 and 8 are used to verify the 3′ junction of the recombinant. Primers 5 and 8 can be used to verify loss of wild-type sequence (either by agarose gel or restriction enzyme analysis). Alternatively, primers 3 and 4 are designed to generate an internal fragment of the target gene. This product should be absent in the recombinant.
Table 2.
Drug-resistance cassettes used for λ Red recombineering
| Antibiotic | Gene(s) | Primer pair | Drug conc. (μg/ml) | Cassette length (base pairs) |
|---|---|---|---|---|
| Ampicillin | Tn3 bla | 5′-CGCGGAACCCCTATTTGTTT-3′ 5′-GGTCTGACAGTTACCAATGC-3′ | 50 | 975 |
| Tetracycline | Tn10 tetRA | 5′-CTCGACATCTTGGTTACCGT-3′ 5′-CGCGGAATAACATCATTTGG-3′ | 7 | 1,996 |
| Kanamycin | Tn903 aph (type I) | 5′-CACGTTGTGTCTCAAAATCTC-3′ 5′-TACAACCAATTAACCAATTCTG-3′ | 20 | 944 |
| Kanamycin | Tn5 aph (type II) | 5′-TATGGACAGCAAGCGAACCG-3′ 5′-TCAGAAGAACTCGTCAAGAAG-3′ | 20 | 949 |
| Chloramphenicol | Tn9 cat | 5′-TGAGACGTTGATCGGCACGT-3′ 5′-ATTCAGGCGTAGCACCAGGC-3′ | 10 | 822 |
| Spectinomycin | Tn21 aadA | 5′-AAACGGATGAAGGCACGAA-3′ 5′-TTATTTGCCGACTACCTTGG-3′ | 20 | 1,080 |
| Gentamicin | Tn1696 aacC | 5′-CGAATCCATGTGGGAGTTTA-3′ 5′-TTAGGTGGCGGTACTTGGGT-3′ | 10 | 616 |
Preparation of Electrocompetent/Recombinogenic E. coli Cells
The basic steps for the preparation of recombinogenic/electrocompetent E. coli cells is that they are grown to mid-log phase in LB media, are induced for expression of the red and gam functions, and then washed extensively with ice-cold 10% glycerol (or water) to remove any trace of salt from the growth medium. The standard protocol in our laboratory uses the lacI-expressing pKM208 (AmpR) plasmid, which is a low copy number vector that contains a Ptac-gam-red operon (183). An overnight culture of cells containing pKM208 is grown at 30°C in LB containing 100 μg/ml ampicillin. A 100-μl aliquot of the overnight culture is inoculated into 20 ml of the same medium, and the culture is swirled at 30°C. (This volume is enough for 2 electroporations and can be scaled up as needed.) The 30°C temperature is important, because most of the Red-producing plasmids used for recombineering have temperature-sensitive origins of replication, allowing the cells to be cured of the plasmid following gene replacement by growth at 42°C. Also, cells containing the defective prophage system of Yu et al. (184) must be grown at 30°C to repress the otherwise lethal expression of the prophage functions. At a cell density of 107 cells/ml (A600 ∼ 0.05 to 0.1), IPTG is added to a final concentration of 1 mM. When the cell density reaches ∼108 cells/ml (A600 ∼ 0.5), the culture is moved to a 42°C water bath and swirled for an additional 15 minutes. This heat shock step is not absolutely necessary, but has been shown to increase recombination efficiency at some loci (unpublished observation); the mechanism of this effect is not understood. For the cells containing the defective prophage, this heating step is required, because it results in induction of the red and gam recombineering functions. The cells are then placed in an ice-water bath for 10 min, collected by centrifugation (6,000 rpm in SS34 Sorvall rotor), and resuspended in 2 ml of ice-cold 10% glycerol. This small volume allows for easy resuspension of the cells. The mixture is gently suspended with a 1-ml pipetman (no vortexing) and diluted with 18 ml of cold 10% glycerol. The cells are mixed by inverting the tube 3 to 4 times and collected by centrifugation as described before. The pellet, which should be handled with care so as not to loosen it, is collected immediately after centrifugation, resuspended in 1 ml of 10% glycerol and transferred to a sterile 1.5-ml microcentrifuge tube. The cells are spun at 10k for 1 min at 4°C, the supernatant carefully removed, and the pellet resuspended in 1 ml of 10% glycerol. The wash step is repeated once more. The cells are finally suspended in 90 μl of ice-cold 10% glycerol, placed on ice and used immediately. While cells can be frozen at this point for later use, the highest recombineering efficiencies come from the use of freshly prepared cells. In a similar protocol using a strain containing the defective prophage to produce Red and Gam functions, 3 ml of cells are grown in LB to 7 × 108, heat shocked at 42°C for 15 min, washed several times in cold water, and resuspended in 50 µl of water containing DNA substrates for electroporation (194, 195).
Electroporation and Plating
Electroporation cuvettes (0.1 cm) are cooled on ice at least 10 min prior to electroporation. Electrocompetent cells (50 μl) are mixed with 50 to 300 ng of DNA substrate in a sterile Eppendorf tube, transferred to the cooled cuvette, and kept on ice for an additional minute. In some instances, 1 to 20 ng of a circular plasmid control (containing a drugR marker distinct from the PCR substrate) is included in the mix as a means to measure electrocompetence. The cuvette is wiped dry and the cells are shocked at 1,800 V, 25 mF, and 200 ohms (parameters that are often included in a standard profile in electroporation controllers for E. coli cells using 0.1-cm cuvettes, e.g., BioRad Genepulser Xcell). The volume of DNA added to cells should be limited to reduce the salt content of the mixture. Typically, we use between 1 and 3 μl of DNA (in 10 mM Tris-HCl pH 8.0 or water) per 50 µl of cells. The time constant (τ) is the amount of time it takes for the initial voltage to drop to 37% of its initial value, and is an important parameter to monitor. It typically should be around 5 msec for E. coli cells using a 0.1-cm cuvette. If the sample is contaminated with salts, the time constant will typically fall, resulting in lower numbers of transformants. The volumes of cells used can be reduced to as much as 25 μl using a 0.1-cm cuvette. Alternatively, more cells (100 μl or more) can be used with wider-gap cuvettes (0.2 cm). If used, the 0.2-cm cuvettes require higher voltage for electroporation (see manufacturer’s suggestions).
Following the shock, the cells are collected with 0.5 ml osmotically neutral SOC, diluted into 2.5 ml SOB, and allowed to recover by rolling at 37°C for 1.5 to 2 h. We have also used LB (containing 5 mg/ml NaCl) for this purpose with little difference, although LB containing higher salt concentrations may be detrimental to survival of electroporated cells. Various aliquots of the culture (100 to 500 μl) are then plated on drug-selection plates at 37°C (or 30°C for the defective prophage system). Dilutions of the culture (100 μl of 10−4 and 10−5) are plated on LB plates to determine the total numbers of survivors. Recombineering frequencies are reported as recombinants per 108 survivors, or alternatively, as recombinants per competent cell. The competency of electroporation is determined as the frequency of recombinants per microgram of closed-circular plasmid DNA. The electrocompetence frequency of the cells should be on the order of 107 transformants per μg of supercoiled plasmid, or higher. Reported recombineering frequencies of 0.001 to 0.1% of electrocompetent cells should thus give rise to 10 to 1,000 recombinants per 100 ng of a dsDNA (PCR) substrate. The actual numbers of recombinants in any electroporation will vary depending on the fraction of total cells that are electrocompetent, the total numbers of cells that survive electroporation, and the amount of DNA that is electroporated. In addition, some loci in the chromosome are more recombinogenic than others, for reasons that are not completely understood (183).
Recombinant Selection and Verification
Following electroporation, the cells are grown out for a period of time to allow for in vivo expression of the drugR marker. This is typically 1 to 2 h, but overnight growth is sometimes necessary to achieve significant number of recombinants. One should use a drug concentration that is low enough for single-copy drug-resistant gene expression from the chromosome, but high enough to suppress background growth of nonrecombinants (see Table 2 for typical drug concentrations used in recombineering). For drug-resistant transformants, restreaking of the candidates on drug-containing plates is typically done to verify the resistant phenotype, because false positives are possible. In cases where one is selecting or screening for a recessive allele (e.g., sacB− or an auxotroph), extended outgrowth (∼4 to 5 h) is required to segregate the modified chromosome from wild-type chromosomes.
PCR is typically used to verify the chromosomal structure of the recombinant. Primers are used that target the drugR marker and a chromosomal region adjacent to the gene replacement, but not within the recombineering substrate (see Fig. 9B). Both the 3′ and 5′ junctions of the gene replacement should be examined. The primers should be selected to generate PCR products that are between 500 and 700 bp. These sizes are generally easy to generate by PCR and are readily distinguished from PCR artifacts (e.g., primer dimers). Alternatively (or in addition), one can use primers that flank the gene replacement. The PCR product of a true recombinant (compared with wild type) can be verified by a difference in gel mobility, altered restriction enzyme pattern, or sequencing. Finally, a set of primers amplifying an internal region of the target gene should be used to verify its absence in the gene knockout. This test is important because recombineering can sometimes occur in cells that have duplicated regions of their chromosomes, allowing both the target gene and a marked replacement to be present simultaneously. These duplicated regions can exist in a subpopulation of the cell culture, or arise as a consequence of an aberrant recombineering event. Such events occur rarely in E. coli, but quite often in M. smegmatis and Mycobacterium tuberculosis using the Che9c RecET system (unpublished observation).
RECOMBINEERING WITH SINGLE-STRANDED OLIGOS
In λ Red recombination, the role of λ Exo is thought to generate single-stranded DNA ends for the binding of the λ Beta protein. Ellis et al. (188) recognized that Beta alone might anneal a ssDNA substrate without the need for λ Exo activity. They tested this idea by electroporation of E. coli expressing only Beta and Gam with 70 nt single-stranded oligo (SSO) targeting an amber mutation within the galK gene. A frequency of 2 × 105 galK+ recombinants per 108 survivors of electroporation was found using 10 ng of a synthetic oligo. This rate is higher than that typically found with dsDNA PCR substrates. Amazingly, Ellis et al. (188) also found that a 70-nt oligo mediated deletion of a 3.3 kb region of chromosomal DNA. The deletion frequency occurred at the same frequency as changing a single base pair, a result leading to interesting speculations regarding the mechanism of Red recombineering (see “Mechanisms of Recombineering” below). Examining the homology length dependency, they found that when, instead of a 70-mer, a 40- to 60-nt oligo was used, the recombineering frequency dropped 5-fold, whereas, with a 30-nt oligo, the frequency dropped 44-fold. This same homology length dependency is also observed for recombineering with dsDNA substrates, and is consistent with the minimum length of ∼36 bases for efficient λ Beta binding to ssDNA (196).
Another key finding in the study by Ellis et al (188) was that oligos that targeted the lagging strand template generated recombinants at a frequency 2- to 50-fold higher relative to oligos that targeted the leading strand template. It was reasoned that the increased single-stranded nature of the discontinuous lagging strand template lends itself to more efficient annealing of ssDNA oligos by the λ Beta protein. The same finding had been seen earlier with transformation of ssDNA oligos into yeast cells (197, 198), with similar explanations. While there is evidence that transcription also may have role in the strand bias of gene targeting with oligos in yeast (199), replication is believed to be the predominant factor for this bias in a number of experimental systems examined (see reference 200 for review). This lagging strand bias is also observed in other bacteria, including M. tuberculosis and P. aeruginosa, when Beta-like functions from endogenous phages are used for SSO-mediated recombineering (201) (Murphy, unpublished). In these cases, the strand bias can be quite dramatic (103 to 104 fold) which may reflect a more restricted capability of targeting the leading strand template in these strains relative to E. coli. This strand bias of λ Beta-promoted oligo annealing led to the proposal that the λ Red proteins target the replication fork during oligo-mediated recombineering. Such mechanistic models are discussed in more detail below.
High rates of target gene alteration allow for screening of unselected mutations, where the mismatch amplification mutation assay (MAMA) PCR can be used to identify candidates that carry a 1-bp change (202, 203). In this scheme, one of the two primers in a PCR contains two 3′-terminal bases that do not pair with the wild-type target; no PCR product is generated. With a mutant target site, the 3′-terminal base anneals, but not the penultimate base, and a PCR product is produced, allowing one to identify the presence of the mutant allele in pools of candidates. When the PCR is performed with a common temperature for annealing and extension (60°C) and an extended number of cycles (40), MAMA-PCR was able to identify the desired change in the Brca2 gene contained on a BAC vector in 11 of 93 pools tested (203). Similarly, MAMA-PCR was also used to find single base pair changes in the M. smegmatis chromosome following phage Che9c RecET-promoted SSO-recombineering (121). In this case, positive clones were enriched by coelectroporation of the oligo with a circular HygR plasmid. The addition of hygromycin selects for cells that are electrocompetent within a population, thus enriching for cells that also take up the oligo. Selecting first for HygR, MAMA-PCR identified oligo-mediated changes in 3 to 5% of the M. smegmatis colonies.
When an oligo designed to create a base pair change anneals to its chromosomal target, it creates a mismatch, which is a substrate for the mismatch repair (MMR) system of E. coli (204, 205). Thus, MMR is predicted to be inhibitory to Red-promoted SSO-mediated recombineering. This prediction was fulfilled when Constantino and Court (206) showed that mutants of the E. coli MMR system increased SSO-mediated recombineering rates ∼100-fold, producing recombinants that approached 25% of the survivors of electroporation. The effect was dependent on the type of mismatch present. Oligos that created mismatches that are good substrates for MMR (G/G, T/C) showed 50- to 300-fold increases in recombineering rates in MMR-deficient hosts. Mismatches that are poorly repaired by the MMR system (C/C) gave high rates of Red-promoted SSO-mediated recombineering, whether MMR was defective or not. Thus, to avoid the use of MMR-deficient hosts, oligos that create a C/C mismatch can be used to obtain a very high frequency of oligo-mediated mutagenesis by Beta-promoted recombineering (206). For instance, an appropriately positioned tyrosine codon (TAC) or serine codon (TCA) could be easily converted to stop codons TAG and TGA, respectively. Alternatively, one can take advantage of the fact that multiple mismatches in a row (four to six) are not recognized by the MMR system (207). In a two-step method to generate a single base pair change using SSO-mediated recombineering, Yang and Sharon designed an oligo to make six consecutive changes in a small region of the target gene at high frequency (208). In the second recombineering step, all of them are changed back to the original sequence, with the exception of the desired base pair change. The changes are made in the wobble position of the coding regions of the target gene, and are thus silent mutations that do not alter the protein amino acid sequence.
Li et al. (209) examined the role of E. coli exonuclease functions in Red-promoted oligo-mediated recombineering in E. coli. They found that inactivation of the majority of exonucleases in the cell did not significantly alter the rate of oligo-mediated recombineering when saturating levels of the oligo were used (∼100 ng). However, a mutation that eliminated the 5′-3′ Exo activity of Pol I resulted in a 9-fold decrease in recombineering with a lagging strand oligo, and a 5-fold decrease with a leading strand oligo. By contrast, Poteete found no effect on Red-promoted oligo-mediated recombineering in strains containing either a knockout of the Pol I 5′-3′ Exo activity, or a strain containing an inactivation of the Klenow fragment (Pol I polymerase and its corresponding 3′-5′ Exo activity) (210). (However, the Pol I 5′-3′ Exo-deficient strain in this study did show a 6-fold reduction in recombineering with a dsDNA substrate.) The differences in the effects of Pol I 5′-3′ Exo- mutants in these studies is not known, but may have to do with variabilities of electroporation. The study by Li et al. reported recombinants as GalK+ transformants per 108 cell survivors, while Poteete reported strepR transformants per electroporated cell, using a coelectroporated plasmid to determine the transformation efficiency. Alternatively, the contrasting results with the Pol I mutants in these studies may have to do with differences in recombineering efficiencies between the two different targets sites (perhaps as a result of context effects).
The study by Li et al. (209) also examined the processing of oligos by cellular exonucleases. By using oligos that created mismatches in the wobble positions of the galK gene and employing a mismatch repair-deficient host, the authors were able to examine the loss of markers at different positions in the oligo owing to processing of the ends of the oligo by cellular exonucleases in vivo. (None of the mutations altered the amino acid residues of the GalK protein, except for the change that corrects the amber mutation.) In wild-type cells, markers were lost in the recombinants from both the 3′ and 5′ ends of the lagging strand (in 15% and 10% of the recombinants, respectively). The leading strand oligo experienced a greater loss of markers from the 3′ end (44% of the recombinants), while showing similar rates of loss from the 5′ end (13% of recombinants). With both oligos, a small percentage of recombinants showed loss of markers from both ends (2 to 7%). These results show that there is processing of the oligos in vivo, and, given the differences between the digestion patterns of lagging and leading strand oligos, exonucleolytic digestion is likely to occur while the oligo is annealed to ssDNA regions of the replication fork.
A host containing mutations of all four known ssDNA exonuclease functions (ExoI, RecJ, ExoIII, and ExoX) had only a minor effect on marker loss from the 3′ end of the leading strand oligo (when saturating levels of oligos were used); no major effects were observed with the lagging strand. However, recombinants generated in a strain mutant for the Pol I 5′-3′ Exo activity showed complete retention of markers from the 5′ end of the lagging strand oligo, with no effect on markers from the 3′ end; the rates of markers lost from the leading strand oligo were the same as wild-type cells. Clearly, among the exonucleases in vivo, the Pol I 5′-3′ Exo activity has the greatest effect on processing of oligos at the replication fork. This result was not unexpected given the role of Pol I at filling in the ssDNA gaps at the replication fork (211, 212, 213). The authors also examined mutations in the 3′-5′ proofreading exonuclease activity of dnaQ, which is part of the Pol III holoenzyme. Here, instead of retention of 3′ markers that might be expected, the authors found an increase in the loss of 5′ markers for both oligos (the effect was greater on the leading strand). The authors attributed this effect to the lower processivity of Pol III in the dnaQ mutant (214), which results in increased loading of Pol I (with its 5′-3′ Exo activity). Pol I would then promote 5′-marker loss as described for the lagging strand oligo.
Other notable points regarding Red-promoted oligo-mediated recombineering made by these investigators are the importance of homology at the ends of the oligos, especially the 5′ end. It is imagined that exonucleolytic processing of the 3′ end can be restored by the polymerase activity of Pol I. The same cannot be said of 5′ homology, which, when modified by bases to create mismatches following annealing, results in much greater loss of recombinant formation (about 100-fold) relative to the 3′ end. Finally, internal mismatches created by the oligo after annealing do not interfere with recombination levels as long as both 3′- and 5′-terminal homologies (∼20 bp) are present. These studies were all performed in a mismatch repair-deficient host.
In a study to optimize the conditions for Red-promoted oligo-mediated recombineering in E. coli, Sawitzke et al. (215) suggest the following rules: (i) use a 70-base oligo, (ii) target the lagging strand template, (iii) electroporate at least 100 ng of oligo (∼3,000 copies/bacterial cell), (iv) avoid mismatch repair correction, and (v) keep the altering base(s) in the center region of the oligo. The avoidance of MMR can be achieved by the inclusion of four to six mismatches at the wobble positions in the oligo surrounding the targeted base(s). These optimized conditions are good starting points for establishing oligo-mediated recombineering in other bacterial strains.
Recent results with Pseudomonas syringae and Legionella pneumophila have shown that oligo-mediated recombineering can occur in the absence of an exogenously expressed SSAP (216). The amount of oligo required is much higher (50-fold) relative to concentrations used for Beta-promoted oligo-mediated recombineering, and the frequencies are significantly lower (10- to 1,000-fold). An even lower, but measurable level of SSAP-independent oligo-mediated mutagenesis is evident with E. coli and Salmonella, demonstrating that oligo-mediated recombination can occur in the absence of any SSAP function in these bacteria. Interestingly, Dutra et al. (217) have found that E. coli missing three to four ssDNA exonucleases can easily be transformed with oligos in an SSAP-independent manner. This result suggests that one of the roles of the Beta protein in oligo-mediated recombineering (besides its annealing function) is to protect the oligo from cellular exonucleases. This idea is consistent with the high concentrations of oligos required for P. syringae oligo-mediated recombination, which presumably saturate the endogenous exonuclease functions in vivo, allowing SSAP-independent recombination to take place. In another study, the presence of carrier DNA (which contained no homology to the E. coli chromosome) resulted in higher rates of Beta-promoted recombineering when low concentrations of the targeting oligo were present (215), again suggesting that endogenous ssDNA exonucleases can interfere with oligo-mediated recombineering. Consistent with this interpretation, the carrier DNA effect was not seen in a strain missing four of its ssDNA exonucleases (RecJ, ExoI, ExoXII, and ExoX). The frequency of SSAP-independent oligo-mediated mutagenesis in L. pneumophila also showed an increase in a host deficient for endogenous nucleases RecJ and ExoVII (218). However, the lack of an effect on mutagenic frequencies with oligos that are blocked at their 3′ ends suggest a different mechanism than that observed with λ Beta-promoted recombineering (see “Mechanisms of Red Recombineering” below).
SITE-SPECIFIC RECOMBINATION SYSTEMS
The site-specific recombinases Flp and Cre have been used for the past 20 years to create insertions, deletions, inversions, and translocations in the chromosomes of bacteria, yeast, plants, Drosophila, and mammalian cells (219, 220, 221, 222, 223, 224, 225). These site-specific recombinases (SSR) act on pairs of their target sites. When on different DNAs, SSR systems promote insertion of one DNA molecule into the other. When target sites are on the same DNA, SSR systems can delete the intervening sequence (if the target sites are direct repeats) or invert the intervening sequence (if the target sites are in an inverted orientation). When combined with the efficiency of recombineering, the use of SSR target sites to manipulate the bacterial chromosome allows almost any genetic alteration to be made. In particular, the Cre-mediated deletion of drugR markers contained between two direct loxP sites (locus of crossover X in P1) has been most useful to date to create drugR marker-free gene deletions in bacterial chromosomes and episomes. The fact that Cre recombines with loxP at high efficiency, without the need of any host factors, allows this system to be used in virtually any cell. In fact, it’s been exploited most often in mammalian cell systems for the construction of conditional mouse mutations and knockouts (219, 223, 226). The uses of SSR systems in the generation of insertions and deletions into bacterial chromosomes are discussed below.
COUNTERSELECTION SCHEMES
Counterselection schemes (also known as negative selection) allow one to select for the absence of a gene, and replace it with virtually any possible modification desired (insertion, deletion, or substitution) without leaving behind any scar. One of the most popular and useful markers for this purpose is the sacB gene from Bacillus subtilis. The SacB protein is a levansucrase that transfers fructosyl residues from sucrose to various cellular constituents. Thus, the expression of sacB is lethal in E. coli (and many other bacteria) only in the presence of sucrose (191).
The use of sacB in such counterselection schemes has been used for many years, most often in nonreplicating plasmid-based classical gene replacement strategies to select for loss of a cointegrant. With recombineering technology, it has been used to make precise in-frame gene deletions in the chromosome, with no exogenous DNA left behind at the target site (183, 189). In practice, the sacB gene is typically linked to a drug selection marker, for example, the cat gene conferring resistance to chloramphenicol. The cat-sacB cassette is then used as a template in a PCR to generate a dsDNA recombineering substrate, where the cassette is flanked by 50 bp of target gene sequence (Fig. 10). The sequences can be selected to either delete the target gene, or to create a simple insertion. Once the recombinant is selected and verified (CamR and SucS), and demonstrated to still retain the Red-expressing plasmid (e.g., AmpR), a second recombineering event is performed to replace the cassette with a dsDNA fragment (or ∼70-mer oligo) that contains the desired mutation (red rectangle in Fig. 10). The drugR marker-free recombinant is selected on sucrose-containing plates (the counterselection step), verified by its sensitivity to chloramphenicol, and the presence of the modification is verified by sequencing.
Figure 10.
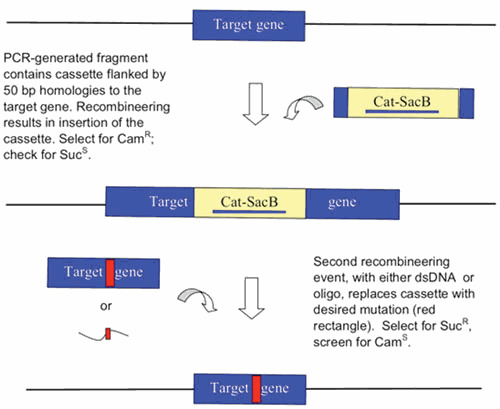
The use of sacB as a counterselection marker in recombineering. The cat-sacB cassette is used as a template for PCR to generate an amplicon that has the cassette flanked by 50 bp of target homology. The recombineering event can either insert the cassette into the target gene, or replace sequences within the target gene with the cassette. After selection for chloramphenicol resistance and verification of sucrose sensitivity, the modified strain is electroporated with either a dsDNA substrate or an oligo that contains the desired mutation (red rectangle). The modified strain is selected by resistance to sucrose and screened for sensitivity to chloramphenicol.
Recently, combining sacB into a cassette with tetA has increased the sensitivity of counterselection (227), eliminating the appearance of false positives to a large degree. Plating cells on plates containing both sucrose and fusaric acid increased the selectivity of counterselection by many orders of magnitude.
Other counterselection schemes have also been described. Wong et al. reported on a well-known scheme where the thyA gene is used as both a positive and a negative selection marker (228). The thyA gene encodes thymidylate synthase, an enzyme involved in the synthesis of dTTP from dUMP; it is essential for the synthesis of DNA. It requires tetrahydrofolate (THF) as a cofactor. THF is synthesized from dihydrofolate (DHF) by dihydrofolate reductase. Working in a thyA null strain, gene knockouts are generated by the replacement of a gene of interest with an exogenous thyA gene and selecting for the recombinant in growth media lacking thymine. The presence of a functional thyA exhausts the supply of THF (an essential cofactor), which cannot be replenished in the presence of trimethoprim, an inhibitor of dihydrofolate reductase. Thus, in a counterselection scheme, the loss of thyA is selected for by growth of the cells in the presence of thymine and trimethoprim. The downside of this counterselection scheme is the need to work in a thyA mutant genetic background.
Another useful counterselection scheme employs the galK gene, which encodes galactokinase, the enzyme catalyzing the first step in the utilization of galactose as a carbon source. Galactokinase converts galactose into galactose 1-phosphate. Warming et al. (229) took advantage of the fact that 2-deoxygalactose (DOC) is also a substrate for galactokinase, which when included in the media of a galK+ strain leads to the buildup of a toxic dead-end product, 2-deoxy-galactose-phosphate. Thus, in a ΔgalK genetic background, the galK gene can be used for both positive selection (growth in minimal media with galactose as the only carbon source) and counterselection (growth in the presence of DOC). This counterselection scheme has been used for recombineering of BACs in E. coli to construct deletions and point mutations (229, 230). The E. coli galK gene has recently been reported to work as a counterselection marker in mycobacteria (231). However, this galK scheme, like the use of thyA, is limited to cells containing a specific metabolic defect (in this case, a galK mutant). The ΔthyA or ΔgalK mutants are most useful as dedicated hosts for the manipulation of BACs by recombineering (228, 229, 230).
Another disadvantage of the thyA and galK counterselection schemes is that they require the use of minimal media in both the positive and negative selection steps. Thus, multiple washes of the cells are required before plating, and extended days of growth (2 to 3 days) are needed to obtain good-sized colonies. Other counterselection schemes have been developed that employ rich media, allowing for quicker selection of recombinants. The rpsL gene has been used as a counterselection marker for many years in classical gene-replacement schemes, and mostly used for recombineering of point mutations into BACs (232). This scheme takes advantage of a mutation in the endogenous rpsL locus that confers resistance to streptomycin. Overexpression of the wild-type rpsL gene from a gene replacement cassette suppresses streptomycin resistance, whereas loss of the cassette confers resistance to streptomycin. The rpsL gene is coexpressed with a drugR marker (typically, either kan or cat) to allow both positive and negative selection. Interestingly, a cassette containing rpsL and tetA (driven by the strong ompT promoter) has been reported to act synergistically for counterselection when residing on a BAC, exhibiting a negative selection rate on the order of 10−6 (∼20 times better than sacB) (233). This synergistic effect arises from the observation that overexpression of tetA alters E. coli membrane permeability, resulting in increased uptake of streptomycin.
A relatively new counterselection scheme combines the advantages of using one marker for both selection and counterselection, and does not require the use of minimal media. The E. coli tolC gene encodes an outer membrane pump for exporting toxic compounds from the cell. Outer surface loops of the TolC protein bind to and transport bacteriocins, including colicin E1, inside the cell (234). Mutants in tolC are tolerant to colicin E1. DeVito (235) demonstrated the use of tolC as both a selection marker (resistance to SDS) and a counterselection marker (resistance to colicin E1); the efficiency of counterselection with tolC ranged from 77 to 100%. Using recombineering with this scheme, DeVito sequentially and seamlessly deleted six of the seven 23S rRNA genes in E. coli. The only requirement for use of this system is that the host must be deleted of its endogenous tolC gene.
In another scheme, mutant version of the pheS gene (A294S), which codes for the tRNA synthetase for phenylalanine, has also been used as a counterselectable marker (236, 237). The pheS (A294S) alpha subunit allows chlorophenylalanine to get incorporated into proteins, which is toxic for E. coli. Thus, the absence of pheS (A294G) gene can be selected for by the inclusion of chlorophenylalanine in the selection plates. This counterselection method was used by Li and Elledge (238) as a means to increase the efficiency of recombineering in the MAGIC method of in vivo cloning of plasmid constructs (see below).
Finally, a counterselection scheme based on the use of the E. coli toxin gene ccdB and its antidote gene ccdA has been used to make precise seamless deletions in E. coli (239). In this scheme, chromosomal insertions are made by the use of a ccdB-amp cassette (selecting for AmpR), while the ccdA antidote gene is supplied on the temperature-sensitive Red-producing plasmid (TetR). After the second recombineering event to replace the cassette is performed, the cells are plated at 42°C to select for the loss of the recombineering plasmid and the presence of the seamless chromosomal modification (i.e., cells that have not exchanged the cassette, and still have ccdB, will die because of the loss of ccdA).
There has been at least one report where the counterselection scheme using rpsL has not worked efficiently with recombineering. The problem may be associated with particular target sites, and not the counterselection scheme itself. Bird et al. (240) have reported that they attempted to modify BACs using an rpsL-neo cassette, and that a large portion of the counterselected strepR transformants (that should have contained a single base pair change) were instead intermolecular events between small repeat sequences that deleted the cassette from the BAC. They reasoned that the λ Red system (Beta + Exo) was promoting an intermolecular recombination event between these repeats, using the ssDNA annealing pathway pictured in Fig. 4B. They then tried using only the annealing function Beta to promote the second step (since oligo-mediated recombineering does not require the λ Exo) to prevent the Red system from promoting this undesired product. Consistent with their hypothesis, the intermolecular recombination events were suppressed when only Beta was used in the second step, and most of the recombinants were replacement of the cassette with oligo (i.e., the desired point mutation). Interestingly, the authors report using 40 pmol of an 100-mer oligo in their experiments, which is 8-fold higher than what is considered saturating (215). It may be that high concentrations of oligo promote replication fork disruption and subsequent dsDNA break formation, allowing Red to promote the intermolecular ssDNA annealing event. While there have been reports of oligos generating dsDNA breaks when they were transformed into eukaryotic cells (241), no reports of such breaks have been reported in E. coli.
All of these counterselection schemes can be used to make precise (seamless) deletions of the E. coli chromosome, BACs, or plasmids. Investigators who are targeting the chromosome and wish to maintain a wild-type (or pseudo-wild-type) background in their recombineering experiments should take advantage of sacB system, because this is a counterselection scheme that does not require the use of a particular genetic background. This may explain the wide popularity of sacB as a counterselection marker (i.e., the use of galK, thyA, rpsL, and tolC as counterselection markers requires the knockout or modification of these genes beforehand). However, for investigators who are targeting BACs or plasmids, these latter counterselection schemes may be more useful, given the high rate of spontaneous reversion of the sacB allele (∼10-4). In this case, strain backgrounds are used that optimize BAC or plasmid recombineering, such as DH10B derivatives (242). The genotypes for these strains include knockouts of recA (to prevent deleterious rearrangements), endA (for cleaner plasmid DNA minipreps), and mrr-hsdRMS-mcrABC (for decreased restriction activity leading to higher rates of DNA electroporation and transformation).
RECOMBINEERING PROTOCOLS
SSR-Mediated Markerless Gene Knockouts
The ease of use of recombineering makes it the obvious approach for the construction of gene (or operon) knockouts in bacterial chromosomes and BACs. Once primers arrive, the time required to construct a strain with a gene knockout can be as short as one day. The replacement of the target gene with the drugR marker allows easy selection of the gene replacement event. Subsequent removal of the drugR marker is often desired to target a second site in the chromosome with the same drugR marker, or in cases where the modified bacterium is to be used for animal studies. The most efficient way to remove a drugR marker is by flanking the resistance-encoding cassette with loxP or FRT sites, which allows one to precisely excise the resistance marker following gene replacement by expression of Cre or Flp recombinase, respectively.
The use of site-specific recombination (SSR) systems in recombineering is typified by the use of the Cre recombinase. One starts with a drugR marker that is flanked with loxP sites (the marker is said to be “floxed”); PCR templates have been constructed for these purposes (182). Alternatively, one could include the 34 bp loxP site in the 5′ end of the primer (in between the sequences at the 3′ ends that anneal to the drugR marker, and the 40 bp at the 5′ ends that contain the target site—see Fig. 11). After gene replacement, the Cre recombinase is supplied on either a plasmid with a temperature-sensitive origin of replication, or one that contains sacB for easy counterselection to remove the vector following gene eviction (see Table 3 for various Cre- and Flp-expressing plasmids). The expression of Cre recombinase irreversibly excises the drugR marker at high efficiency, generating a high percentage of cells in the population that have lost the drugR marker.
Figure 11.
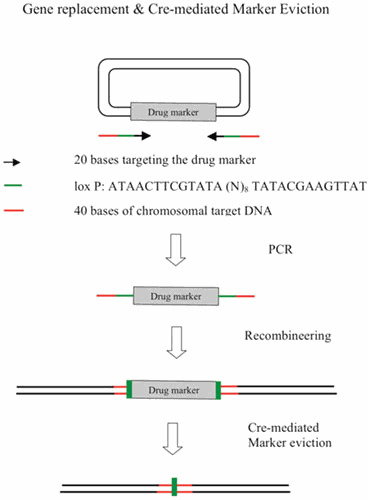
Gene replacement and Cre-mediated marker eviction (see text for details).
Table 3.
Cre- and Flp-expressing plasmids
| Plasmid | Features | Reference |
|---|---|---|
| pJW168 | PlacUV5-Cre, RepAts AmpR | 389 (Lucegen) |
| pBAD75Cre | PBAD-Cre, RepAts CamR | 390 |
| 1921-cICre | λ PR-Cre cI857 SpecR | 391 |
| pCreSacB | PgroEL-Cre, oriE, oriM, sacBR, KanR | Adrie J. C. Steyn (Univ. of Alabama) |
| pKM330 | PgroEL-Cre, oriE, oriM, sacBR, ZeoR | 392 (www.addgene.com) |
| pCP20 | λ PR-Flp, λ cI857, RepAts, AmpR, CamR | 393 (Coli Genetic Stock Center) |
| pCP20-Gm | λ PR-Flp, λ cI857, RepAts, GenR, CamR | 394 |
| pE-FLP | PE (P2 phage promoter)-Flp, RepAts, AmpR | 395 (www.addgene.com ) |
| loxP | ATAACTTCGTATA (N)8TATACGAAGTTAT | |
| FRT site | GAAGTTCCTATTC (N)8GTATAGGAACTTC |
After drugR marker eviction, there is a “scar” left over at the site of the excision event. The sequence will contain one loxP site, and flanking sequences from the template that were amplified by PCR. If marker eviction is reiterated many times in the same host, the scar becomes a repeat element that is distributed throughout the chromosome, which could potentially lead to undesired Cre-mediated chromosomal rearrangements. Thus, repetitive use of loxP sites is not recommended. Another concern is the polar effect the scar may have on downstream genes. It is often worthwhile to design the PCR primers for the original recombineering substrate so that, following excision, the scar creates an in-frame deletion devoid of any stop codons, to minimize effects on the expression of downstream genes. We have often designed our initial primers to generate a marked deletion that, following Cre-mediated marker eviction, contains the first and last 3 codons of the gene of interest interrupted by an in-frame scar. If one suspects a regulatory region imbedded in the 5′- and/or 3′-coding regions of the target gene, longer regions of the coding sequence may be left intact. In other cases, templates have been constructed that contain a promoter region within the scar, which allows for expression of downstream genes (182). However, if the target gene is within an operon, this scheme does not preserve the original strength and regulatory properties of the operon’s original promoter(s).
DrugR marker eviction is done by isolating the initial marked recombinant, transforming with a SSR-producing plasmid using a drugR marker different from that of the marked deletion and the Red-producing plasmid, allowing the cells to grow out and express the recombinase, then screening for cells that are sensitive to the drug. This event usually occurs at high frequency (because recombination between two direct repeats is irreversible). Marker-free recombinants can be found easily by plating an overnight culture for single colonies on drug-free media and stabbing the colonies on fresh plates (+/– antibiotic). Removal of the marker and curing of both the Red/ET- and SSR-producing plasmids can often be performed in one step. As an example, we have used the Cre-expressing plasmid (pKM330, a derivative of pCreSacB containing a zeocin-resistance marker) that also contains the sacB gene (see Table 3), to remove both the hygromycin marker in a targeted gene and the Che9 RecET-producing recombineering plasmid pJV53 (243) in M. smegmatis (K. C. Murphy, K. Papavinasasundaram, and C. M. Sassetti, unpublished data). The plasmid pKM330 contains the same origin of replication as the KanR RecET-producing plasmid pJV53, but contains a different drugR marker (ZeoR). Thus, by transformation with pKM330 and outgrowth in zeocin, pJV53 is lost via plasmid incompatibility, while the hygromycin-resistance marker is evicted following Cre-expression. By growing an overnight dilution of these cells nonselectively and plating on sucrose plates, cells that have lost the Cre-expressing plasmid (SucR), pJV53 (KanS), and the drugR marker (HygS) are easily found.
To create very large deletions in the E. coli chromosome one can place SSR sites at specific locations in the genome by recombineering, followed by transformation of a plasmid expressing the site-specific recombinase. In a variation of an earlier scheme where the Tn5 transposon delivered loxP sites randomly to various sites within the E. coli genome (244), Fukiya et al. (245) targeted loxP sites to specific regions of the chromosome using λ Red recombineering. Two different loxP sites (in direct orientation) are placed at genetic loci using two different drugR markers. The chromosomal positions of the loxP sites define the endpoints of the intended deletion (see Fig. 12). The PCR substrates are designed to place the drugR markers between the loxP sites within the chromosome, such that both markers are deleted following the site-specific recombination event (if so desired). Using this procedure, Fukiya et al. (245) deleted two regions of the E. coli chromosome greater that 100 kb in size with 100% efficiency. As described above for removal of a drugR marker, a loxP scar is left in place of the deleted sequence. Such a process allows one to define regions of chromosomes that contain nonessential functions.
Figure 12.
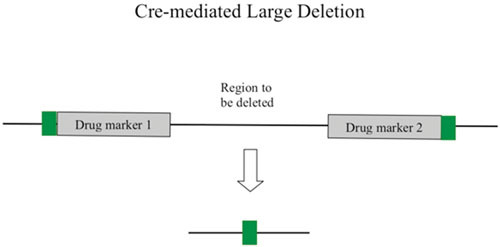
Cre-mediated large deletion (see text for details).
Precise Deletions
It is often desirable to leave no exogenous DNA at the site of a chromosomal deletion, as is the case when multiple deletions are to be made in the same strain, to minimize the effects on expression of downstream genes in an operon, or when the mutant strain is to be used for animal studies. The most common method is to use a counterselection cassette to mark the deletion (e.g., cat sacB), then perform a second replacement event with dsDNA containing the precise deletion (i.e., no exogenous DNA). If dsDNA substrates are used, the deletion can be designed ahead of time on a plasmid and liberated from the vector by restriction digestion (183). As an alternative, a 70-mer oligo can be used to replace the counterselection cassette, where 35 bases on each end of the oligo define the deletion endpoints. It was found that such oligos provide a high rate of Red-mediated deletion formation (188), which can be selected for by loss of the counterselection cassette.
Plasmids have been constructed that place sacB adjacent to a drugR marker (e.g., the cat-sacB cassette, where cat confers resistance to chloramphenicol) (246) or the npt-SacBR cassette (247) combining SacB expression with kanamycin resistance. Alternatively, the strain XTL298 contains the highly selective tetA-sacB counterselection cassette integrated into the chromosome at the araD locus (227). Plasmids (or strains) containing these cassettes can be used as templates for PCR to generate substrates for recombineering. The cat-sacB cassette is over 2.6 kb in size, so the PCR should be performed with cycling conditions that favor generation of large amplicons. This is typically done using mixtures of a high-fidelity polymerase and Taq polymerase (248, 249) and/or by adding 5 to 10 s to the extension time per cycle. The substrate is then electroporated into a λ Red-producing strain and transformants are selected for CamR. After isolation of the proper CamR SucS transformant, a second electroporation is performed with DNA containing the unmarked gene deletion sequence, which often consists of the first few codons of a gene fused to the last few codons of a gene. More of the coding sequence should be left intact if suspected regulatory regions for surrounding genes are imbedded within the coding regions of the target gene. Following electroporation, the cells are grown out for a longer period of time than usual (5 h or more, or overnight). In rich medium, E. coli contains multiple copies (4 to 8) of its chromosome, and only one of these is likely to be a substrate for recombineering. Thus, this long outgrowth step is required with sacB (and with most counterselection markers) to allow segregation of recombinant chromosomes from the nonrecombinant chromosomes (i.e., a sacB- chromosome would be lost on sucrose plates if it resides in the same cell with sacB+ chromosomes). Another consideration for this counterselection step is the spontaneous mutation rate of sacB+ to sacB- (SucS to SucR) which occurs at a frequency of ∼10-4. However, recombineering rates typically are equal to or exceed this reversion rate, and loss of the cat-sacB cassette can be easily detected by screening for the loss of the cat gene (CamS). Alternatively, the recently described tetA-sacB cassette has a spontaneous resistance rate on the order of ∼10−7 (227), making precise deletions using this cassette extremely easy to find.
Another way to create large unmarked deletions of the E. coli chromosome was first described by Posfai et al. (250) using suicide plasmids, and later modified by Kolisnychenko et al. (251) using recombineering. The key to this procedure is to flank the region to be deleted with repeat segments, and then induce a dsDNA break between these regions with the meganuclease I-SceI restriction enzyme. The I-SceI enzyme is a homing endonuclease from the mitochondria of Saccharomyces cerevisiae that recognizes an 18-bp recognition site, which does not exist in the E. coli genome sequence (252). Repair of the break by the host RecA-promoted recombination system (or λ Red) deletes the chromosome segment between the duplicated regions at high efficiency, because nonrepair is lethal to the cell. In a more efficient use of this scheme, recombineering is used to create the initial duplication as shown in Fig. 13. First, a PCR is performed with a template consisting of a drugR marker flanked by I-SceI sites. One of the primers (primer 1) contains a fusion of two 40-bp regions of the chromosome (regions X and Y in Fig. 13) and was generated by annealing two overlapping oligos followed by a DNA Pol I filling-in reaction. Once the drugR marker is crossed into the chromosome by recombineering, the cell is cured of the Red-producing plasmid and transformed with I-SceI-producing plasmid. (In later versions of this protocol, λ Red and I-SceI-expression were present on one plasmid under control of PBAD [253], or under control of different promoters [238, 254, 255]). Cutting of the I-SceI sites on the chromosome generates a dsDNA break, which is efficiently repaired by either the RecA homologous recombination pathway of E. coli or by λ Red. Recombination between the “Y” sequences shown in Fig. 13 generates a deletion of the chromosome, the endpoints of which are defined by the fusion sequence present in primer 1. It is this system that Kolisnychenko et al. (251) used to generate multiple deletions in the E. coli chromosome to produce a strain with a reduced genome (see below). This methodology of introducing a duplication and an I-SceI recognition site by a recombineering step, followed by an I-SceI-induced dsDNA break, was used for the construction of conditional lethal amber mutations, seamless deletions, point mutations, fusion tags, and insertion events in BACs with high efficiencies (253, 254, 256).
Figure 13.
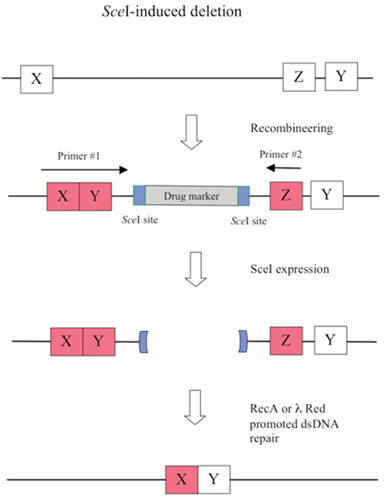
I-SceI-induced deletion (see text for details).
Inversions
Inversion of a sequence can be performed in a precise manner in two steps. In the first step, a counterselectable marker cassette (e.g., cat-sacB) is placed into the chromosome (selecting CamR) to replace the sequence to be inverted. Primers are then used that amplify the region to be inverted from wild-type DNA, with 50-bp flanks that place the desired sequence into the chromosome in an inverted orientation. Recombinants are selected by counterselection methods (e.g., sucrose-containing plates). In this scheme, an inversion of an E. coli chromosomal segment can be performed without the need to include exogenous DNA sequences (e.g., lox P sites).
Alternatively, inversions of DNA sequences within a bacterial chromosome can be done with the use of SSR sites as described above for deletions, with the exception that the loxP sites are placed in inverted orientation. In this case, the SSR event will invert the DNA sequence between the loxP sites. Since the recombinant still contains both loxP sites, the reaction is reversible. Thus, cells transformed with the Cre recombinase-producing plasmid will contain a 50% mixture of cells containing the parental and inverted orientation of the targeted sequence. Once the Cre-expressing plasmid is removed, PCR verification can be used to identify the desired orientation from isolated colonies.
Gap Repair
Schemes have been developed that take advantage of an increased rate of Red and RecET-promoted gap repair of transformed linear plasmids. In this repair event, recombination occurs between a linear plasmid and either another linear DNA species (via an annealing event) or between a linear plasmid and the chromosome (via a presumed replisome invasion event—see below). When occurring between two linear species, both the donor and the recipient possess terminal redundancies. This process was first described as an in vivo cloning method for PCR products (257). In this study, it was demonstrated that PCR products that shared terminal homologies to linear vectors could be cloned (without the need of restriction enzymes) by cotransformation of both species into cells expressing the RecET recombination system. Another study had previously suggested that DH5α hosts (without RecET) could also be used for this type of cloning (258), but only when chemically competent cells were employed. Furthermore, in direct comparisons, DH5α cells showed a 25-fold lower frequency of cloning relative to RecET-expressing hosts (257).
Using plasmids that overexpress the RecET and λ Red systems, Zhang et al. (259) demonstrated multiple examples of in vivo cloning. PCR primers were designed to amplify plasmid origins and selection markers, and contained 47 to 64 bases at the 5′ termini that were homologous to the intended target. The PCR products were electroporated into E. coli cells expressing RecET or λ Red, where the target sequences (ranging in size from 0.8 to 28 kb) resided either within the chromosome or on a BAC. By simply selecting for the drugR marker on the vector, between 30 and 100% of the vectors had picked up the insert via gap repair. Formation of nonrecombinant empty vectors is reduced by making sure there are no 5 nt or longer direct repeats in the terminal target homologies to promote their pairwise interaction. The authors further demonstrated the usefulness of this technology by coelectroporation of linear PCR-generated vectors and genomic DNA preparations from E. coli, yeast, and mammalian cells. While not quite as efficient with the more complex DNA samples, the authors still found in vivo cloning to work with 20 to 30% efficiency for the cloning of eukaryotic genes. The great advantage of this technique is that genes can be “levitated” out of complex mixtures without the need for a PCR. Instead, once recombined into the vector, the gene is amplified with high fidelity by the E. coli DNA replication system.
Red-promoted gap repair technology works well to exchange drugR markers and origins of replications between different plasmids. Datta et al. (190) have shown that different elements can be easily exchanged by electroporation of linearized plasmids into hosts containing the target sequences. For instance, a PCR is used to generate a linearized pBR322-based vector without its origin of replication, but with terminal sequences that will target a pSC101 ori. After recombineering, plasmid DNA is isolated and transformed into a polA strain of E. coli. This strain background does not support growth of pBR322-based origins (260), and thus colonies that arise will contain the new pSC101 ori. This manipulation allows a plasmid to be expressed at a different copy number relative to the original construct. DrugR markers can also be easily switched by a similar protocol of gap repair. Note, however, that the targets in these repair events are replicating, unlike ones described above for in vivo cloning, and likely involve a different mechanism of recombination.
Plasmid Recombineering
Alternatively, resident plasmids can be engineered by standard recombineering with PCR-based substrates as is done with the E. coli chromosome, although we have found, as others have reported (5, 261), that multimers of the plasmids are typically generated (although not always) in the recombineering process. It is noteworthy that these DNA forms are circular dimers, trimers, and tetramers containing mixed populations of parental and recombinant markers, and are not the same as the very large linear multimers observed with colE1 plasmids replicating in a host constitutive for Red, Gam, or Red + Gam expression (140, 262, 263). The plasmid multimers were not observed in cells induced for Red + Gam in the absence of added DNA. In other words, the multimers were dependent on a recombination event and not simply generated by the induction of the rolling-circle (sigma) mode of replication. The DNA substrate for recombinogenic multimeric plasmid formation could be either a dsDNA substrate or an oligo. It may be that, during outgrowth of the culture, dimers and higher multimers of the mutant plasmid (among an excess of unmodified parental plasmids) give the modified plasmid a competitive advantage, even when under selection. Such a phenomenon has been described in detail by Kuzminov (264). Alternatively, the multimerization may be a direct consequence of the recombineering event.
Digestion of these multimers with a single base pair cutter common to both plasmids, religation at low concentration, followed by transformation into recA hosts provides a way to obtain the monomeric recombinant plasmid with its new drugR marker. Finally, instead of using a drugR marker as an insert, one scheme to increase cloning efficiency is to provide the −35 region of a promoter on the ends of the recombineering fragment reading outward, which following recombineering, restores the expression of a promoterless selectable gene on the cloning vector, as reported by Yosef et al. (265).
Insertions
The insertion of large regions of foreign DNA into E. coli is one area of great interest, but until recently, has generally been performed by methods other than recombineering technology. These procedures take advantage of integrating large foreign DNA segments into chromosomal regions containing phage attachment sites (266), FRT sites (222), or transposon target sequences (267, 268, 269, 270). Alternatively, foreign DNA flanked by 5-kb regions of chromosomal DNA is inserted into the E. coli genome via an endogenous RecA-promoted pathway of recombination (271), although such a protocol requires construction of a plasmid containing the insertion flanked by chromosomal sequences beforehand.
Using recombineering, insertions of drugR markers of 1 to 2 kb in size have routinely been performed using λ Red and RecET. However, it has generally been observed (although often not reported) that larger sizes of DNA are less efficiently incorporated, if at all. This notion has been examined in more detail by Kuhlman and Cox (272), who measured recombineering efficiency as a function of insert size. The neo gene (conferring resistance to kanamycin) was embedded within various amounts of lacZ DNA into substrates containing 50 bp of flanking homology to a chromosomal target. Recombineering frequencies dropped dramatically when the insert size increased from 1 kb to 4.5 kb. The same effect was seen by Maresca et al. (273). Clearly, increased insert size does not favor efficient recombineering, which probably reflects on the mechanism of the process (discussed further below).
Large insertions using recombineering is most efficiently done by exploiting an in vivo cloning type of strategy discussed above, where both the insert and the target are linearized by digestion with the meganuclease I-SceI restriction enzyme. This procedure can be performed with targets such as plasmids, BACs, or the E. coli chromosome. For instance, the λ Red system was exploited by Li and Elledge (238) to develop an in vivo cloning method that facilitates the construction of plasmids for bacterial, yeast, and mammalian expression purposes. In this scheme, a pir-dependent donor plasmid (containing the insert) is transferred to a strain containing the recipient plasmid. The recipient host expresses both the I-SceI restriction enzyme and the λ Red recombination system. I-SceI cuts both the donor plasmid (liberating the insert) and the recipient plasmid (linearizing the plasmid at the target site), generating two linear species that share terminal homologies. The two linear fragments undergo Red-promoted in vivo recombination that combines the insert with the recipient plasmid, with the resulting loss of the I-SceI sites. To reduce the nonrecombinant background, the following genetic steps were taken: (i) the cells continue to express I-SceI (which selects against nonrecombinants), (ii) a pheS counterselection marker, which is excised following I-SceI-induced cleavage, is employed in the recipient plasmid, and (iii) a 20-bp lac operator sequence is added to the insert, which titrates out LacI repressor in the recipient, inducing a Plac-bla chromosomal operon to confer AmpR to a true recombinant. These genetic measures to ensure insertion efficiency resulted in ∼106-fold increase in the rate of accurate recombinational cloning, and represent a substantial advance in open reading frame expression technologies for high-throughput genomic and proteomic studies.
Another in vivo cloning method takes advantage of Red/RecET promoted repair of dsDNA breaks to integrate long DNA fragments (5 to 50 kb) into BACs. Rivero-Muller et al. (255) have described a recombineering protocol where large DNA fragments can be transferred between BACs. In the first step, an rpsL-kan counterselection module is integrated into a BAC of interest. Long (120 to 140 nt) primers are used to flank the rpsL-kan cassette with I-SceI sites, followed by 50 bp of sequence that are homologous to the endpoints of a desired insert sequence. Following this recombineering event, the BAC is purified and coelectroporated with a linear DNA containing the insert (a donor BAC that has been linearized by restriction digestion). The E. coli host contains a plasmid expressing both RecET and I-SceI, driven by PBAD and Ptet, respectively. The recipient BAC is linearized in vivo at the I-SceI site, while RecET promotes in vivo cloning by combining the two linear DNA species. A cloning efficiency of 69% is reported using this scheme. This protocol represented a large increase in cloning efficiency relative to a scheme where the recipient BAC was cut in vitro.
Finally, Kuhlman and Cox (272) described a method that allows site-specific integration of large synthetic constructs into the E. coli chromosome using the λ Red system (Fig. 14). An initial recombineering event inserts a drugR marker flanked by 25 bp of foreign DNA sequences within the E. coli chromosome. These 25 bp of foreign DNA define the endpoints of the insertion. Between the drugR marker and the endpoint sequences are I-SceI recognition sequences, positioned in such a way that expression of I-SceI endonuclease will release the drugR marker from the chromosome, leaving behind a dsDNA break containing foreign DNA at its ends. Following isolation of a clone containing the I-SceI-flanked drugR marker, a plasmid that contains the desired insertion sequence, also flanked by I-SceI sites, is electroporated into the host. In vivo expression of I-SceI and λ Red recombinase promotes recombination between the plasmid-liberated insert and the cut in the chromosome (Fig. 14). The half-site I-SceI sequences on the ends of the breaks are presumably trimmed in vivo, and do not interfere with the Red-promoted homologous recombination repair. This procedure allowed the authors to place 7-kb genetic constructs into six unique locations distributed symmetrically about the origin of replication.
Figure 14.
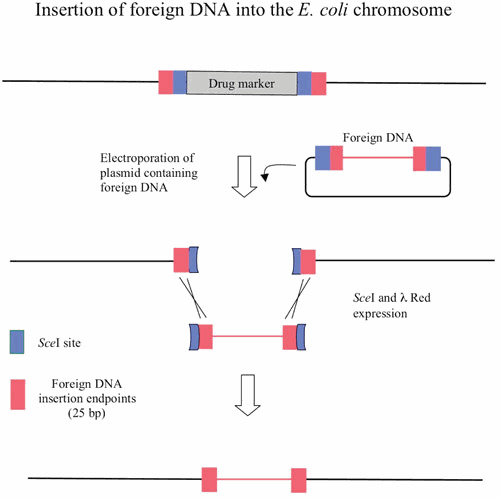
Insertion of foreign DNA into the E. coli chromosome (see text for details).
The authors claim that limited amount of homology for this event (25 bp) is used here to provide for efficient Red-promoted annealing event, without allowing for integration of the plasmid into I-SceI-uncut chromosomes. One imagines, however, that even at higher homologies, the repair of a broken chromosome by recombineering will proceed at a much higher rate than integration of the plasmid into the chromosome (an event not promoted by recombineering), because the cell will not survive unless the I-SceI-cut chromosome is repaired. In addition, continued expression of I-SceI would select against a simple integration event. Finally, note that for all these insertion events, the mechanism for the integration of these fragments likely does not require a replisome invasion, but merely annealing between the ends of the dsDNA breaks in vivo.
Duplications
A proposed strategy of generating a duplication in bacterial chromosomes using long homologies and phage transduction (274) should also be doable using shorter homologies and the λ Red recombination system. In such a scenario, a drugR marker is amplified by PCR with primers, where the location of the targeting sequences is reversed relative to those that would be expected to promote a simple insertion (Fig. 15). It is proposed that both ends of the recombineering fragment recombine independently with two different arms of a replication fork (an inherent requirement for annealing of Red-processed linear dsDNA ends to the template strands of the fork, given the antiparallel nature of dsDNA duplex). There are two models offered to explain duplication formation by λ Red. The first one involves the generation of new replication forks and is shown in Fig. 15. The first step is the annealing of a λ Exo-generated ssDNA tail of a linear dsDNA fragment to ssDNA regions of an invaded fork (also see Fig. 8). Red-promoted invasion of the sequences denoted by “A” in Fig. 15 generates the formation of a new replication fork (the green fork), which moves in the opposite direction relative to the original fork. In the same time frame, a second Red-promoted invasion event takes place between the other end of the recombineering fragment and the other arm of the original fork. A third replication fork is established (the red fork) now moving in the same direction as the original fork. These growing forks traveling in opposite directions would leave behind (in one of the chromosomes) the drugR marker (grey box) flanked by duplicated regions of the chromosome (blue boxes).
Figure 15.
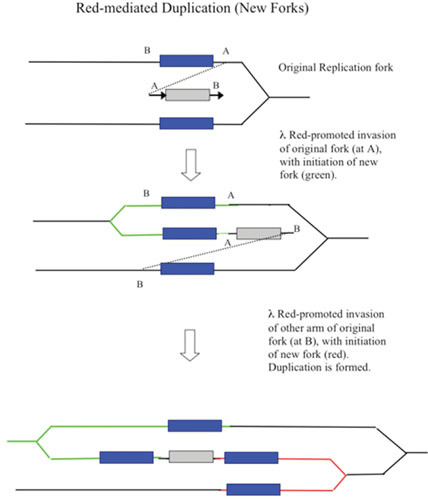
Red-mediated duplication with establishment of new forks (see text for details).
This mechanism involves both arms of the original fork and takes into account the idea that the replication fork is the target for Red recombineering. The first replisome invasion event at “A” in the mechanism depicted in Fig. 15 would likely occur by annealing of the Red-processed end (A) of the recombineering fragment to the lagging strand template, generating a fork that proceeds in the opposite direction of the original fork. The second event (by necessity) occurs by annealing to the leading strand template of the original fork. In this case, the annealing may require a ssDNA invasion step, because little ssDNA would be expected to be available for Beta-promoted annealing to the leading strand template. As such, this step may require the assistance of the host RecA protein. Supporting this assumption, Red-promoted duplications of E. coli chromosomal regions containing the lacZ gene were dependent on host recA function (275). The two events could be temporally reversed without affecting the final outcome. How these invasions may occur are shown more explicitly in Fig. 8, and thus are similar to the discussion above regarding how λ Red may promote the formation of new forks during phage infections in vivo.
An alternative to the formation of a new fork, a Red-promoted recombination event could lead to disruption of a fork, followed by a resolvase acting on the four-stranded junction. As seen in Fig. 16, the initial crossover occurs at position “A” and generates a fusion of the drugR marker (grey box) to the region to be duplicated (blue box), and a dsDNA break. A more detailed description of this proposed event is shown in Fig. 16 (top shaded panel). The ssDNA tail of the Red-processed end of the drugR marker (the “A” end) anneals to the lagging strand template of the replication fork. The four-stranded structure that forms, a gapped Holliday-type junction, is cut by a resolvase (e.g., RuvC – red star). Since gaps are present in the Holliday-type junction, only one cut is required to resolve this type of recombinational intermediate (see Fig. 16). The duplex invasion and subsequent cutting disrupts the replisome, fuses the drugR marker to the lagging strand (adjacent to the region to be duplicated), and releases a dsDNA break. The other product of resolvase action is repaired by PolI, which generates an intact unreplicating chromosome (not shown).
Figure 16.
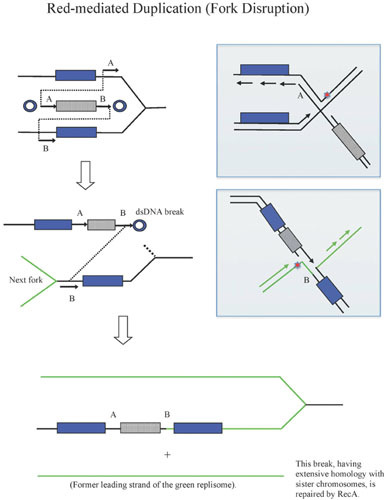
Red-mediated duplication with fork disruption (see text for details).
The dsDNA break formed in the first reaction is then repaired by a subsequent Red-promoted recombination event (at site B in Fig. 16), which occurs when another fork enters this region from the left (the green replisome). By necessity, the ssDNA tail of the Red-processed end of the break (the “B” end) would have to anneal to the leading strand template (see bottom shaded panel in Fig. 16). Again, the four-stranded structure formed could be resolved by a single cut (red star). Such an event would allow the incoming duplex to become the new leading strand of the green replisome. The cutting would also release the (former) green leading strand as a new dsDNA break. This break, however, carries extensive homology with sister chromosomes and could subsequently be repaired by the host RecABCD system, akin to what occurs following replication fork collapse, with subsequent restoration of the replication fork (276). In the recombinant, the drugR marker resides between the duplicated regions and serves as a selection marker for the presence of the duplication (i.e., a recombination event that resolves the duplication evicts the drugR marker). The duplication endpoints are defined by the sequences chosen for in “A” and “B” in the design of the primers. The first invasion event in this scheme could occur on either the leading or lagging strand templates, but by necessity, the second event must occur on the template opposite to the one used in the first event. In either case, the outcome is the same.
Thus, either the Red-promoted New Fork model or the Fork Disruption model could be used to explain duplication formation of the lac and dinB regions in an F’ factor described by Slechta et al. (277), the Red-mediated duplication of a region in lacIZY (278), as well as for the Red-promoted generation of blue colonies that contained a drugR marker in the lacZ gene in the study by Poteete (275). In the latter study, duplications were dependent on RecA, and curtailed 5- to 10-fold by mutations in RuvC, in support of the Fork Disruption model of Red-promoted duplications.
Reporter Fusions
The construction of transcriptional fusions of target genes to reporter functions is a critical step in the study of gene regulation in E. coli and other bacteria. Translational fusions (which create hybrid proteins) allow one to follow the fate of a particular protein with regard to its subcellular location, rate of degradation, and/or secretion. Classically, these fusions have been generated on plasmid constructs (or within plasmid-chromosome cointegrants) that place a reporter function (typically either β-galactosidase or luciferase) downstream of the ribosome binding site (RBS) for a transcriptional fusion, or within the reading frame of the target gene for a translational fusion. Classic methods used for the construction of reporter fusions typically involve the use of plasmid-chromosome integrants, phage attachment sites, and transposons (266, 279, 280, 281, 282, 283). Recombineering has made the construction of such chromosomal fusions a one-step protocol, and will replace the classical multistep protocols.
For transcriptional fusions, the basic protocol is to simply use recombineering to replace the target gene with a reporter function. For translational fusions, the coding region of the reporter function is fused to all or part of the coding region of the target gene. Using λ Red recombineering, Uzzau et al. (284) placed the FLAG tag sequences onto the C-terminal ends of a number of chromosomal genes in S. enterica serovar Typhimurium. This was done by incorporating the FLAG sequence into the primers that were used to amplify the kan or cam drugR markers by PCR. Ellermeier et al. (285) used recombineering to place FRT sites downstream of the promoters of a number of target genes. Transformation with a replication-deficient plasmid that contained a FRT site upstream of promoterless lacZY genes resulted in fusions of lacZ to the target promoter as a result of integration of the plasmid into the chromosome by SSR promoted by FLP, expressed from a helper plasmid with conditional replication origin. Translational fusions can also be generated in this way. However, this protocol requires both a recombineering step and a SSR event, and the use of a set of specialized nonreplicating plasmids.
The simplest method for the construction of transcriptional and translational fusions by recombineering is to use a PCR template where the reporter function lies adjacent to a drugR marker. Then, one only needs to perform a PCR and judiciously select the 50 bp at the 5′ ends of the primers to precisely place the reporter function behind the Shine-Dalgarno sequence (RBS) of the target gene (for transcriptional fusions), or fused to the coding sequence of the target gene. The drugR marker is placed downstream of the fusion construct and can be left intact or, if flanked by SSR sites, can be removed by an SSR-promoted eviction step. Gerlach et al. (286) have constructed a set of templates where the KanR-conferring aph gene is linked to a number of reporter functions including luciferase, lacZ, green fluorescent protein (GFP), DsRed, phoA, and HaloTag. The aph gene is flanked by FRT sites and can be excised with the Flp recombinase. The authors demonstrate the construction and analysis of transcriptional fusions of luciferase, β-galactosidase, and GFP to a number of genes in S. enterica serovar Typhimurium, as well as a translational fusion of luciferase to sseJ, a type III effector function. Transcriptional fusions using the luciferase construct were also demonstrated by recombineering in E. coli, S. enterica serovar Typhimurium, and Shigella flexneri. The same techniques can also be used with gene sequences cloned into BACs and cosmids to make transcriptional fusion that can be recombined back into the chromosomes of the eukaryotic organisms. Dolphin and Hope used recombineering to construct a marker-free gfp translational fusion in a Caenorhabditis elegans fosmid clone, which was subsequently used to generate transgenic worms for expression analysis (287). More recent descriptions of fosmid recombineering by this group describes the use of a strain that can induce higher copy numbers of recombineered BACs and fosmids following recombineering (288), and the use of complementary sets of constructs for the two-step generation of genetic fusions in C. elegans genomic clones (289).
Bacteriophage Recombineering
It goes without saying that the type of construction for which the λ Red system should work best is the modification of the phage chromosomes. This is an important point because the great diversity and numbers of phages in the biosphere represent an untapped source of genetic material for studying viral biology, phage evolution, and methods to improve food processing (290, 291, 292). The methodology to modify phage DNA using λ Red was demonstrated by Oppenheim et al. (293), whereby a strain expressing the λ Red functions from a heat-inducible defective prophage was grown to mid-log phase, collected by centrifugation, and infected with the λ phage to be modified. Following a 15-min absorbance period, the cells were diluted back into culture and heat shocked at 42°C to induce the λ recombineering functions. After 15 min, the cells were collected by centrifugation and prepared for electrocompetence by washing in ice-cold water. The cells were electroporated with oligos (or PCR fragments) designed to generate point mutants, deletions, and gene replacements in the infecting phage chromosomes. Following lysis, genetic screens were used to identify the recombinant phage, which appear in the population at a frequency of approximately 2%. Oppenheim et al. (293) reported that mutations designed by the oligo (and genetically selected) were verified by sequencing, unaccompanied by other mutations. However, when examining unintended mutations accompanying the desired change in the cI repressor gene, deletions and base pair changes could be found at a rate 10 to 40 times higher than the rate expected for spontaneous mutations. The authors determined that it was not the recombineering process itself that generated these unintended mutants, but that they were due to errors in the synthesis of the oligos. These inadvertent mutations could be greatly alleviated by purifying the oligo by polyacrylamide gel electrophoresis.
Other phage systems have also been subject to modification by recombineering. The Che9c RecET recombineering system developed by van Kessel and Hatfull for mycobacteria (201) has been shown to promote genetic modification of phages from these hosts. In a technique called Bacterial Recombineering of Electroporated DNA (BRED) (294), phage DNA and recombineering substrates are coelectroporated into electrocompetent/recombinogenic M. smegmatis cells. The cells are allowed to grow for ∼2 h (before lysis) and then plated on a lawn of M. smegmatis to find infective centers. Plaques are collected and diagnostic PCR is used to identify those that contain mixtures of wild-type and recombinant phage. Mutant-containing plaques were found between 5 and 20% of the time, or higher when more sensitive PCR techniques were used. Positive signal plaques are replated to identify pure recombinant phage. Similar to the study by Oppenheim et al. with phage λ (293), multiple types of mutations can be generated in mycobacterial phages, including in-frame gene deletions, base pair changes, and addition of tags to phage open reading frames (for a recent review, see Marinelli et al. [294]).
Random Mutagenesis of Chromosomal Genes
Random mutagenesis of genes to find critical amino acid residues for protein activity or stability is often performed on plasmids. However, characterizations of the mutant protein produced from multicopy plasmids can often be misleading, because the copy number of the protein is usually higher relative to expression from a chromosomal (single-copy) locus. Mutant phenotypes may also be masked by overexpression of the mutant proteins when expressed in vivo. For this reason, it is best to characterize mutant phenotypes with genes expressed from their endogenous chromosomal loci. Mutation of plasmid-encoded genes, followed by transfer of interesting mutant alleles to the chromosome, can often be quite time consuming. In addition, the persistence of the mutant phenotype after transfer of the mutant gene to its chromosomal locus is not guaranteed.
Recombineering offers a way to generate random mutations of genes, and regions of interest, directly in the chromosome. This protocol has been described by De Lay and Cronan (295) in the isolation of three temperature-sensitive mutants of the acyl carrier protein (ACP) gene in E. coli. This protocol allowed the authors to isolate temperature-sensitive mutants of the gene for which previous attempts had failed. Mutagenic PCR of the target gene is carried out by standard protocols, as is a nonmutagenic PCR of a drugR marker (296) (see Fig. 17). Primers are selected for the PCRs that create a 20-bp overlap between the two PCR products, allowing one to perform overlap PCR with the two products (297). The overlap PCR product, which contains a mutant version of the target gene fused to the drugR marker, serves as a substrate for recombineering (see Fig. 17). Following transformation of the mutagenized target gene and outgrowth, cells are plated out on drug-supplemented plates to select for recombinants, and are then simultaneously (or subsequently) selected or screened for the desired mutant phenotype. It is best to test different sets of conditions for the PCR mutagenesis step beforehand, and employ the one that gives one mutation per targeted DNA region. This system is best suited for small genes, considering that the PCR products will typically be in the range of 2 kb for a 1-kb target gene. For longer genes, N-terminal or C-terminal regions of the gene could be targeted specifically, by placing the overlap sequences to the drugR marker at the 5′ or 3′ ends of the gene, respectively. Care should be taken in the ordering of error-free oligos, because the overlap sequence might contain elements of the promoter or termination signals of the gene of interest.
Figure 17.
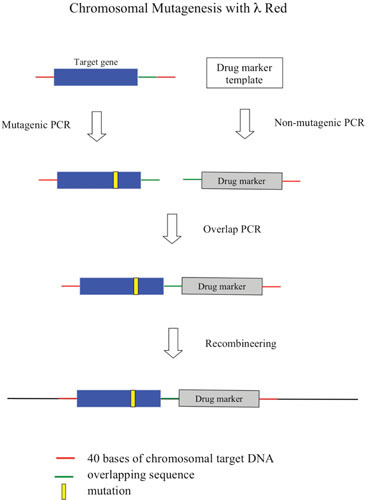
Chromosomal mutagenesis with λ Red (see text for details).
Genome Reduction
The ease of creating deletions of chromosomal DNA in bacteria by recombineering has prompted investigators to identify what may be considered a “core” bacterial genome, that is, a stably replicating genome that supports prolific growth of the bacteria under standard laboratory conditions. Most bacteria have in their genomes by-products of chromosomal evolution that include insertion sequence (IS) elements, transposons, cryptic prophages, no longer relevant functions, and inactivated genes. Some regions of the chromosome might be important for growth in specific environments, although they might be dispensable for growth in the laboratory. In addition, any region that encodes a redundant metabolic or regulatory function should be deletable without any adverse effects on growth or stability. The question arises: is it possible to get rid of bacterial junk and/or functionally redundant DNA without sacrificing both the growth and fitness of the organism? It has been proposed that bacteria with reduced genomes may be better suited as hosts for metabolic engineering studies, since unnecessary (wasteful) pathways could be avoided, leaving more energy and resources for the overproduction of a desired metabolite. Also, purification of a desired protein would be easier in a strain lacking unnecessary protein contaminants.
It was these considerations that motivated Blattner and Posfai to use the recombineering approach to generate a reduced genome of E. coli strain MG1655 (251, 298). They first identified genomic regions to delete by comparing the genomes of E. coli K-12 with five other E. coli species. Regions present in E. coli K-12 but absent from these other bacteria (termed strain-specific islands) were deleted by using a scheme outlined in Fig. 13. Using such a scheme, Posfai et al. (298) constructed a multiple deletion strain (MDS43) that contains a 15.3% reduction in genome content, representing a loss of 708,267 bp of DNA (743 genes). The similarly deleted MDS42 strain showed growth rates comparable to the parental strain MG1655, a loss of transposition events, increased electroporation efficiency, and greater stability of plasmids expressing proteins that would otherwise be unstable in MG1655. This last effect was due to the loss of IS-related insertion events in the target plasmid, allowing the MDS42 strain to express high levels of problematic proteins that could not be achieved in wild-type E. coli. In the most recent improvement in the endeavor, deletion of the three error-prone polymerases PolII, PolIV, and PolV from MDS42 resulted in a strain that was further reduced for spontaneous and induced mutagenesis, generating a high-fidelity strain for the mutation-free production of toxic proteins (299).
Mizoguchi et al. (300) generated a reduced genome strain that was deleted of 1 Mb of DNA (using a similar but different scheme to select regions for deletion). The reduced genome strain in this study (MGF-01) grew well in M9 minimal medium and reached cell densities 50% higher relative to the parent strain. MGF-01 and its wild-type counterpart were then engineered to overproduce threonine. The authors found that the MGF-01 strain was able to produce 2.4-fold higher levels of threonine relative to the parental strain, in large part due to its higher rate of glucose utilization and decreased production of acetate as a by-product.
The reduced genome strains MDS43 and MGF-01 were generated by recombineering techniques that allowed the investigators to precisely delete regions of the genome that had been previously selected, ones that would be reasonably assumed to not affect the growth rate or genomic stability of the bacterium. These efforts are in contrast to studies where reduced genomes are constructed by the manipulation of strains that have been previously targeted by Tn5 transposons carrying loxP sites. Once two transposons are residing in the same cell, the genetic region between them can be deleted by supplying Cre recombinase (245). However, in this procedure, the endpoints are generated randomly and loxP scars sites are left behind in the chromosome, two features not observed when genome reduction is performed with recombineering, as described above.
Promoter Engineering
Regulatable promoters are critical tools for the study of gene function. The ability to keep genes silent, yet subsequently turn them on by adding an inducer is critical for characterization of the role of specific genes in bacterial growth and pathogenesis. However, in fields such as metabolic engineering and synthetic biology, it is often desirable to have the expression of a given gene within an operon set at a particular level that optimizes the performance of a particular pathway or network. These fine-tuning adjustments can be done by screening a set of artificial promoters driving expression of a particular target gene (301). Sets of artificial promoters can be generated by mutagenic PCR. For instance, artificial promoters for Lactococcus lactis were generated by varying the spacer regions between the −10 and −35 regions of a L. lactis promoter consensus sequence; in addition, some consensus changes were generated as well (302). A total of 38 promoters were examined by measuring β-galactosidase expression in both L. lactis and E. coli, resulting in a library of gene expression variants covering three orders of magnitude in promoter output. Alper et al. (303) generated a set of 22 promoter mutants for E. coli, based on reproducible and homogeneous single-cell fluorescence distributions. They went on to characterize each of these promoters using three different criteria: GFP fluorescence per cell per hour, quantitative RT-PCR, and minimal inhibitory concentrations of chloramphenicol via cat gene expression. There was a good correlation between all three measurements of promoter variability, arguing for a valid library where the differences in gene expression are independent of the context in which they were measured. While initial characterization of these promoters was done on plasmids, having these promoters expressed from the chromosome is necessary to evaluate their effectiveness for use in strain engineering. However, transferring a library of promoters on plasmids to the chromosome by plasmid-integration techniques can be quite laborious (304).
Recombineering techniques have made feasible the generation and targeting of promoter libraries to particular genes on the chromosome. Alper et al. (303) transferred particular members of their promoter library to the E. coli chromosome by generating PCRs of both the neo gene (KanR) and particular promoter mutant constructs, and used overlap PCR to generate the recombineering cassette. The protocol is similar to the scheme shown in Fig. 17 (but without the use of mutagenic conditions in their initial PCR step). In some cases, they included the target gene as a third PCR to create a cassette with even longer homology. Meynial-Salles et al. (305) also employed the λ Red system to transfer a set of defined promoters to the E. coli chromosome driving expression of lacZ, but at the same time made further modifications to mRNA-stabilizing regions, the Shine-Dalgarno sequence (RBS), and the start codon. A diagram of their scheme is shown in Fig. 18. Using this technique, the authors found that five randomly selected colonies generated by recombineering varied in lacZ expression between 0.03 and 5.7 units/mg protein. These schemes to generate a defined promoter library within the chromosome at the target gene enable investigators to test a number of selected constructs, and then employ the optimized strain for production purposes without further manipulations.
Figure 18.
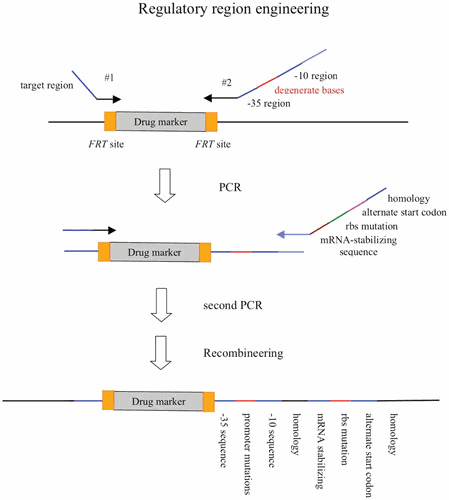
Regulatory region engineering (see text for details).
Increasing the Editing Efficiency of Recombineering with CRISPR-Cas9
In cases where recombineering is used to modify the chromosome without selection, the challenge has been to increase the frequency of the gene alteration to such a high level so that nonrecombinants (and/or escape mutants) represent a small percentage of the total number of colonies on the outgrowth plate. This has been achieved, for example, with oligo-mediated recombineering by the use of mismatch-repair-deficient hosts that increases the frequency of obtaining the directed mutation to virtually the theoretical limit (206) (as described above in “Recombineering with Single-Stranded Oligos”), albeit with the caveat that one is now working in a highly mutagenic background. A more recent solution to this problem has been the use of the CRISPR-Cas9 system, so widespread for the manipulation of mammalian chromosomes, which has been shown to increase the editing efficiency of recombineering by targeting nonrecombinants for destruction. While cutting mammalian chromosomes with RNA-guided Cas9 endonuclease allow the dsDNA break to be repaired by nonhomologous end joining (NHEJ), such repair mechanisms are not present (for the most part) in bacterial cells. Cutting all the chromosomes in E. coli, which leaves no uncut chromosome for recombinational repair, results in death of the cell. As such, CRISPR-Cas9 systems have been employed to increase the percentage of recombineering-promoted events by targeting the unmodified chromosomes. Jiang et al. (306) have shown that oligo-mediated recombineering events, targeting the rpsL gene in E. coli, resulted in 65% of the outgrowth colonies showing streptomycin resistance when the CRISPR-Cas9 system was employed to cut the unmodified rpsL gene. In addition, their results suggest that cutting at the rpsL locus resulted in a modest increase in the recombination rate (6.7%), suggesting that two mechanisms are in play to increase the editing efficiency: preventing outgrowth of nonrecombinants and dsDNA breaks stimulating Red recombination, which has been previously demonstrated (307). Other studies have shown that the combination of recombineering and CRISPR-Cas9 has allowed for high-frequency unmarked large deletions close to 20 kb and insertions up to 3 kb (308), as well as multigene editing without selection (309). These procedures, while requiring the generation and implementation of the CRISPR-CAS9 system, allow for markerless gene alterations without the use of antibiotic markers or counterselection schemes.
IMPACT OF RECOMBINEERING
E. coli Genomics
Not unexpectedly, one of the first genome-wide applications of recombineering has been construction of a library of gene knockouts of every nonessential function in E. coli (also known as the Keio collection) (193, 310). This collection followed the paradigm of the Saccharomyces Genome Deletion Project to create a single gene knockout collection for yeast (311, 312). This construction of the E. coli library was made feasible after the Red system was found to induce in E. coli a “yeast-like” proficiency of recombination with linear PCR products (23, 182, 184). In turn, the existence of this collection has enabled investigators to use genetics to begin to develop an interaction map of the E. coli proteome. By transferring members of a second library of CamR gene deletions into the KanR Keio collection by conjugational recombination, Typas et al. (313) screened for gene pairs that, when combined, generated a lethal phenotype, suggesting that the two genes are involved in redundant pathways, or that they represent avoidance-repair couples. Such a high-throughput genetic interaction assay had been previously performed with S. cerevisiae, but not for any prokaryotic cell.
Likewise, the use of recombineering technology allowed tandem affinity purification (TAP) or sequential protein affinity (SPA) tags to be fused to the C termini of 857 open reading frames in E. coli, enabling the Greenblatt and Emili laboratories to perform a large-scale analysis of protein interactions in vivo using affinity chromatography and mass spectrometry (314). The technology has been applied to the more than 1,000 orphaned (functionally unannotated) genes in E. coli to create a protein interaction network of nearly 6,000 physical interactions, with the putative assignment of many of these genes to functional classes (315). Chromosomal tagging of E. coli genes was also done by Watt et al. (316) who fused fluorescently labeled reporter functions to 23 genes using recombineering, then visually observed the cellular distribution of the corresponding proteins in live E. coli cells using fluorescence microscopy. Other examples of the application of recombineering technology to genomic analysis include the development of a systematic mutagenesis scheme for E. coli (317), a pipeline for the cloning and tagging of worm genes for genomic analysis of C. elegans (318), and a multiplex automated genome engineering technology for programming and accelerated evolution of E. coli (194). Clearly, besides the simplicity of creating single-gene knockouts or modifications, recombineering has enabled investigators to study E. coli on a genome-wide scale that was not previously possible.
Mouse Genetics
Over the past two decades, the use of the Cre-promoted site-specific recombination system of phage P1 has allowed investigators to manipulate mammalian genomes to insert, invert, or delete segments of chromosomal DNA with high specificity (219, 226, 319, 320, 321). This groundbreaking technology was followed by the addition of yet another innovative technology offered by bacteriophages for the manipulation of eukaryotic genomes—recombineering. The phage lambda Red and Rac prophage RecET systems have been employed to manipulate segments of mammalian genomes in BACs (189, 322, 323), allowing mouse gene replacement constructs to be generated in a shorter amount of time, with greater specificity, and without the need for restriction enzymes or ligation reactions. Restriction sites, promoters, terminators, loxP sites, or any other genetic element, incorporated into PCR products (either as part of a primer or amplified from the template), can be precisely recombined into specific regions of kilobase-sized mammalian DNA targets within BACs. The combination of both phage site-specific and homologous recombination systems has given the mouse geneticist the capability of assembling of virtually any type of construct desired. Most of the technical details described in this review for recombineering of BACs and chromosomes have been also applied in the modification of BACs used in the construction of eukaryotic targeting vectors. Recent technical reviews are currently available on the details of using recombineering for the construction of gene-targeting vectors (324), on recombineering-based procedures for the creation of Cre/loxP conditional knockouts (325), and on the construction of Cre-recombinase-expressing transgenic mice (326).
Microbial Pathogenesis
Moving the recombinogenic potential of the Red system to other bacterial species was one of the original goals in developing this system. Not surprisingly, recombineering with PCR-generated substrates with short flanking homologies has worked well in closely related species, like S. enterica serovar Typhimurium (327, 328, 329, 330, 331, 332, 333) and other Salmonella species (284, 334). Red recombineering with short homologies has also worked well in S. flexneri (335, 336) where its use for vaccine development has been instrumental (337, 338). Red-promoted gene replacement has also been reported for Klebsiella aerogenes (339). Pathogenic strains of E. coli, such as enterohemorrhagic E. coli (EHEC), enteropathogenic E. coli (EPEC), and uropathogenic E. coli (UPEC), are all amenable to Red-promoted PCR-mediated gene replacement (183, 340, 341, 342, 343). Further improvements for the engineering of deletions and fusion tags of pathogenic species of E. coli have recently been reported by Lee et al. (344). The use of I-SceI to liberate the recombineering fragment from a plasmid in vivo, together with the addition of a counterselectable marker (sacB) to that plasmid, promoted high efficiencies of Red recombineering for the modification of a number of pathogenic species of E. coli. The use of λ Red has also been reported for modification of chromosomal genes in P. aeruginosa (345), although a 3-step PCR to generate a recombineering substrate containing between 400 and 600 bp of homologous flanks is required. More recently, Liang and Liu have reported the use of the λ Red proteins in P. aeruginosa (PA) driven from a PBAD promoter in plasmid pKaraRed (346). They constructed seamless deletions by first replacing the chromosomal target with a sacB-bla cassette using small homology flanks (50 bp), followed by electroporation with PA chromosomal DNA that included a 1-kb homologous flank. The authors were able to construct seamless knockouts of numerous PA genes with efficiencies of approximately 90%.
The usefulness of the λ Red system for genetic engineering of the strains mentioned above relies on their relatedness to E. coli. In more distantly related bacteria, where the use of PCR products containing short flanking homologies is not very efficient, λ Red-driven recombineering of substrates containing longer flanking homologies (∼500 bp), generated by plasmid restriction digestion or overlap PCR, have successfully been used to construct gene replacements. Examples of organisms where such substrates have been used for gene targeting include Yersinia pestis (347, 348), Serratia marcescens (349), and Vibrio cholerae (350). It is possible that such recombination in these hosts is initiated by λ Red proteins at dsDNA ends, but carried to completion by the RecA-promoted host pathway.
Ultimately, to achieve high levels of PCR-mediated gene replacement in non-E. coli hosts, phage recombination systems from phage endogenous to those species will likely be required. This concept encouraged van Kessel and Hatfull (243) to examine the genomes of more than 3,000 mycobacterial phages for functions homologous to the λ Red and/or the Rac prophage recET functions. They found predicted recombination proteins to occur rarely in these genomes, but did find two phages (Che9c and Halo) to encode RecE-like homologs; Che9c also encoded a RecT homolog. The C terminus of the Che9c RecE homolog (gp-60) shares 28% identity with a RecB family nuclease domain, and the N-terminal two-thirds of Che9c RecT (gp-61) are 29% identical to RecT of Rac prophage. Che9c RecT also possesses a motif common to the RecT superfamily of ssDNA annealing proteins. Overproduction and characterization of the Che9c RecET proteins found RecE to contain a dsDNA exonuclease activity and RecT to bind preferentially to ssDNA, similar to λ Exo and Beta, respectively. Unlike λ, Che9c does not encode a Gam-like anti-RecBCD function. This may be important for development of high-frequency recombineering in mycobacteria, as M. smegmatis and M. tuberculosis encode two RecBCD-like functions (351). Knockout of one of them (MSMEG_1325, MSMEG_1327 and MSMEG_1328) in M. smegmatis resulted in a 10-fold increase in recombineering (Murphy, unpublished).
van Kessel and Hatfull (243) found that overexpression of the Che9c RecET proteins in M. smegmatis and M. tuberculosis (MTb) allowed the cells to be transformed with linear substrates containing drugR markers with ∼500 bases of flanking homology. PCR-generated substrates, containing a drugR marker flanked by 50 bp of target DNA, often give rise to drugR transformants of MTb, but not with an accuracy that makes it a reliable substrate for making gene replacements. We have found that the limited number of colonies obtained in MTb following transformation with small homology substrates is often the result of an illegitimate recombination event, yielding cells that contain the hygromycin resistance marker but still retain the wild-type locus of the target gene. Such illegitimate recombination events have been the hallmark of gene replacement schemes in MTb using suicide vectors as delivery vehicles. However, with the increased frequency of the Che9c RecET system acting on substrates containing 500-bp flanks, between 30 and 100% of the colonies are true gene replacements (Murphy et al., unpublished), verified by PCR analysis of the resulting DNA junctions between the drugR marker and the chromosome, and by the absence of the wild-type gene. The system has been used to place reporter tags on the C termini of chromosomal genes (352) and replace endogenous promoters with the controllable Ptet promoter (unpublished results). The Che9c RecT protein can also promote recombineering of M. smegmatis and M. tuberculosis with ssDNA oligos (121), making it a useful system for the verification of single-nucleotide polymorphisms (SNPs) that have been identified in drug-resistant MTb strains (353). One of the significant differences between Che9c RecT-promoted oligo-mediated recombineering and that promoted by λ Beta in E. coli is the very large difference in recombineering frequencies between oligos that target the leading versus the lagging strand template of the replication fork. In E. coli, this difference is typically between 3- and 50-fold, but it can reach 10,000-fold in M. smegmatis. The reasons for this large bias targeting one or the other strands of the replication fork in mycobacteria is not known, but may be a reflection of a mechanistic difference between the Beta and Che9c RecT functions.
In efforts to develop a recombineering system from Pseudomonas phages, Swingle et al. (354) have identified sequences in the chromosome of P. syringae B728a that are similar to the λ Red and RecET functions in E. coli. The authors found that the Pseudomonas RecT function promoted recombineering of an oligo targeting the rpsL allele at a frequency of 2.4 × 104 recombinants per 108 viable cells. They used 5 µg of the oligo, which contained three additional base pair changes adjacent to the one that conferred streptomycin resistance to avoid correction by the mismatch repair system. This rate is 25-fold higher that the RecT-independent rate, and 105-fold higher than the rate of spontaneous streptomycin resistance. The system can also promote recombineering of dsDNA substrates containing long homology flanks (∼500 bp), but not small (50 bp) flanks.
We have found similar results using the Orf-52 protein (a homolog of phage P22 Erf) from the D3 phage of P. aeruginosa (355). Targeting the rpsL allele with 100 ng of an oligo containing a 1-bp change, we obtained 103 streptomycin-resistant mutants per 108 viable cells (Murphy, unpublished). In the absence of Orf-52, no StrepR cells were found. Interestingly, similar to mycobacteria, the complementary oligo targeting the leading strand template gave no recombinants, indicating at least of 500-fold bias toward oligos that target the lagging strand template. We also found an accompanying exonuclease function, orf-51, which when expressed with orf-52, complemented the growth of λ gam red phage in recA host, indicating the presence of a bona fide recombination system. It failed, however, to promote recombineering of PCR substrates containing short (50 bp) regions of flanking homology. Thus, at this point, the Red functions expressed from pKaraRed seem to be best suited for recombineering in Pseudomonas species (346). Overall, however, as more phage genomes are sequenced and analyzed, phage SSAPs and exonucleases are likely to be identified to allow the manipulation of the genomes of pathogenic bacteria to create gene knockouts, identify critical virulence factors, and promote the development of vaccine strains.
Metabolic Engineering
Metabolic engineering is another discipline that recombineering technology has impacted in recent years. The metabolic engineer seeks to modify bacterial or yeast cells for the efficient overproduction of particular metabolites, to enhance the fitness of an organism, and/or to prevent the production of inhibitory side-products generated during the fermentation process. Genome reduction (discussed above) produced a strain that is highly transformable, genetically trimmed, and physically fit for metabolic engineering purposes. In other applications, recombineering has been used to knock out biosynthetic genes involved in the synthesis of natural products. These procedures often involve transfer of chromosomal DNA from species like Streptomyces or Mycobacteria into cosmids or BACs, followed by manipulation of DNA by recombineering (see Fig. 19 for the paradigm of recombineering in non-E. coli microbes). Modifications can include the addition of origins of transfer and phage attachment sites, allowing for the transfer and integration of the modified vector back into the original host (or a different species). For instance, a report by Gust et al. (356) describes recombineering of a Streptomyces coelicolor cosmid library in E. coli where the investigators identified a gene involved in geosmin biosynthesis. They replaced a segment of S. coelicolor DNA with an antibiotic resistance marker (flanked by FRT sites) and an oriTRK2 origin to allow transfer from E. coli back to S. coelicolor by conjugation. Once transferred, the cosmid clone, which cannot replicate in S. coelicolor, undergoes a double-crossover recombination event to generate a gene replacement in the S. coelicolor chromosome. The procedure has been used many times to make over 100 gene replacements in S. coelicolor.
Figure 19.
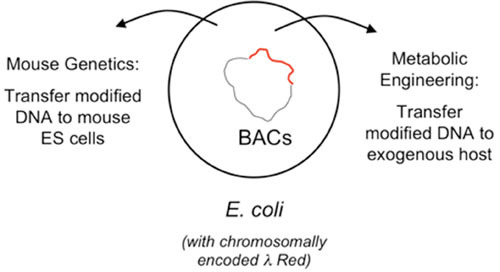
λ Red Recombineering of non-E. coli sequences. Exogenous DNAs (e.g., mycobacteria, mouse, or human DNA) are cloned into bacterial artificial chromosomes (BACs) for modifications, additions, or deletions. Schemes to recover the modified DNA for sending it back into the exogenous hosts are discussed in the text. ES cells, embryonic stem cells.
In another example of the use of λ Red in metabolic engineering, Wenzel et al. (357) rebuilt a biosynthetic gene cluster from myxobacterium Stigmatella aurantiaca using recombineering in E. coli. Myxobacteria are soil bacteria that are a rich source of natural products used for drug development. The modified myxobacterial gene cluster encodes a set of nonribosomal peptide synthetases (NPS) and a polyketide synthase (PKS) module that governs the synthesis of myxochromide S, a group of cyclic peptides with polyketide side chains. These investigators used recombineering to add a thioesterase domain to one NPS and inserted a toluic-acid-inducible promoter to drive expression of the gene cluster. To transfer the gene construct to Pseudomonas putida, they also incorporated an origin of transfer, a tetracycline resistance gene for selection purposes, and the trpE gene as a source of homology for integration into the P. putida chromosome. The engineered P. putida strain was found to produce a maximum yield of myxochromide S cyclic peptides that was five times greater than the original S. aurantiaca host from which the gene cluster was derived. More recently, Fu et al. (358) showed that the 30-kbp myxochromide S gene cluster, as well as the larger 58-kbp epothilone gene cluster from Sorangium cellulosum, could be modified for expression and transferred to heterologous hosts by transposition. By using recombineering to introduce components of the MycoMar transposase to the gene-cluster constructs, the authors report efficient transposition of both these large gene clusters to Myxococcus xanthus and P. putida.
It is expected that similar recombineering-based reconstructions like this one will become standard practice, where natural product genes and operons are combined into a single large DNA construct in E. coli, modified as needed, and combined with the elements to mobilize them to an alternative host by conjugation and transposition. In some cases, the λ Red functions may work directly in non-E. coli bacteria that may be useful for metabolic engineering, as was recently shown for the acid-tolerant Pantoea ananatis (359) and Agrobacterium tumefaciens (360). In other cases, RecT-like genes can be isolated from non-E. coli bacterial species, as was recently done for Lactobacillus reuteri and B. subtilis (361, 362). The L. reuteri RecT protein was used for development of an oligo-based recombineering protocol for lactic acid bacteria, which are commonly used for fermentation processes in the food and beverage industries. Likewise, Dong et al. (363) found a RecT-like function in Clostridium perfringens in an effort to develop a recombineering system for Clostridium acetobutylicum, a bacterium used for the production of organic solvents. An analysis of a number of different Beta- and RecT-like proteins from a variety of bacterial species shows they all can work in E. coli to promote oligo-mediated recombineering, albeit with varying degrees of success (364). Those phage annealases from bacteria more closely related to E. coli worked better than those from more divergent species, although there were exceptions, e.g., the EF2132* annealase from the Gram-positive Enterococcus faecalis worked as well as the λ Beta protein for oligo-mediated recombineering in E. coli. These data suggest that recombineering has the potential to be developed in many types of bacterial species. Consistent with this idea, recombineering systems for Photorhabdus luminescens and Xenorhabdus stockiae, lethal pathogens for insects, have recently been established by the utilization of phage genes within P. luminescens that are homologous to the λ red system (365).
Red recombineering has also been used to develop a laboratory-based methodology for accelerated evolution of E. coli (194). Multiplex Automated Genome Engineering (MAGE) is a process whereby cells expressing Red and Gam are electroporated with sets of degenerate oligos targeting multiple regions of the chromosome. As an example, Wang et al. (194) targeted the ribosome binding sites of 24 genes involved in the synthesis of lycopene. The oligos were designed to increase the translational efficiency of 20 genes previously implicated in lycopene synthesis, by modification of their ribosome binding sites. In addition, to increase flux through the targeted pathway, 4 genes in secondary pathways were targeted for inactivation by the insertion of a stop codon in their open reading frames. By repeated rounds of electroporation with the 24 oligos and subsequent outgrowth, MAGE was calculated to generate all the genetic variants predicted (∼15 billion). The recombineering with the oligos was performed in an E. coli mutS strain that expressed the λ Red and Gam functions from a prophage, and a plasmid (pAC-LYC) expressing a cyanobacterium lycopene cyclase (366), an enzyme required for the final steps of lycopene production. By screening colonies following 5 to 35 cycles of MAGE, the authors identified strains that contained higher concentrations of lycopene (as evidenced by the red color of the colonies); some of these strains showed a 5-fold increase in lycopene synthesis relative to the starting strain.
By coselecting for markers situated at different regions of the chromosome, Wang et al. (367) have shown that higher rates of oligo-mediated changes (in this case, the insertion of the 20 bp T7 promoter [5′-TAATACGACTCACTATAGGG-3′] at 12 different operons) could be obtained by coselection of a selectable marker (e.g., CamR) whose loci is near or within the chromosomal region of the targeted operon(s). With coselection, T7 promoter insertion events occurred within 25% of the cells after one round of MAGE, but only in 2 to 3% of the cells without coselection. The use of this methodology (called CoS-MAGE) resulted in the insertion of the T7 promoter in front of all 12 targeted operons, as well as 80 different strains containing different combinations of targeted operons. Some of these promoter insertions resulted in an increase in production of the desired metabolite (indole), while other combinations resulted in lower amounts relative to the starting strain. Promoter libraries such as these can be used to study epistatic interactions in gene networks. Overall, these recombineering-based approaches in metabolic engineering should continue to drive the synthesis of novel secondary metabolites, and the subsequent development of interesting new compounds and therapeutic agents. This coselection strategy has recently been applied to BAC recombineering, where the coelectroporation of a dsDNA substrate containing a selectable marker (KanR), and an unselectable oligo carrying a loxP site, was used to reduce the number of steps required to construct a conditional knockout gene-targeting vector (368).
MECHANISMS OF λ RED RECOMBINEERING
Recombineering with ssDNA Oligos
The difference in the frequencies of recombination when one uses an oligo that targets the lagging strand versus the leading strand of a replication fork indicates that the replication fork itself is the target of the Red-promoted oligo-mediated recombineering. This was noted by Ellis et al. (188), who proposed that the higher frequencies of recombineering with oligos containing the lagging strand sequence is a reflection of the higher amount of ssDNA target available on the lagging strand template. A model based on this observation has been described and is shown in Fig. 20. As shown in Fig. 20A to C, Beta is proposed to anneal the oligo to the lagging strand template between Okazaki fragments. Once annealed, the oligo is proposed to become incorporated into the lagging strand by the normal mechanisms that extend and ligate the Okazaki fragments via the actions of DNA polymerase I and ligase. The physical incorporation of the oligo into a target DNA was shown by Huen et al. (369) using a biotinylated oligo and isolating the recombinant plasmid with streptavidin-conjugated magnetic agarose beads. This assay had previously been employed by Radecke et al. (370) to demonstrate oligo uptake into plasmids during targeted gene alteration in eukaryotic cells.
Figure 20.
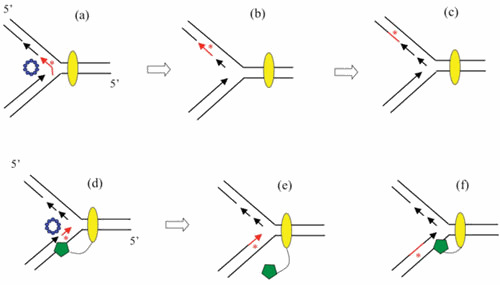
Beta-promoted Recombineering with ssDNA oligos. (A–C) Targeting of the lagging strand template. Beta promotes annealing of the oligo to a ssDNA region of the lagging strand template. Pol I and ligase promote filling in of the oligo and joining it with the surrounding Okazaki fragments to produce heteroduplex DNA (red asterisk). (D–F) Targeting of the leading strand template. Beta promotes annealing of the oligo to a ssDNA region of the leading strand template just ahead of the leading strand 3′ end. The leading strand polymerase (not shown) dissociates from its template, and because it is tethered to the clamp loader (green pentagon) of the replisome (denoted by yellow oval), it can reinitiate downstream at the 3′ end of the oligo. Annealing of the oligo creates either a mismatch, a small deletion, or small insertion (denoted by red asterisk), which must escape repair by the Mismatch Repair System of E. coli. The mutation is fixed via subsequent replication of the heteroduplex region.
While recombineering by oligos targeting the leading strand occurs at lower frequency, it does happen, and a model to address this eventuality is shown in Fig. 20D to F. In this scheme, Beta anneals the oligo to the leading strand template just ahead of the leading strand polymerase. Once Pol III reaches the 5′ end of the oligo, the polymerase is proposed to disengage from the template and to renew polymerization downstream by using the 3′ end of the oligo as the priming site. There are precedents for this model. Both leading and lagging strand polymerases are thought to be able to reinitiate polymerization downstream on their templates, since gaps can be found in both strands following encounters with lesions in their respective templates (reviewed by Wang [371]). Also, like the lagging strand polymerase, the leading strand polymerase (green pentagon in Fig. 20) is tethered to the clamp loader (372, 373), and, as a result, should have the capability to reengage its template following dissociation on encounter of the oligo. The annealing of the oligo to ssDNA regions of the replication fork is consistent with recent evidence (209) on the effects of Pol I 5′-3′ Exo- mutants on recombineering rates, and the differential processing of the 5′ ends of the oligos when annealed to either lagging or leading strand substrates in vivo (described above).
In an effort to increase the frequency of mutations introduced by oligos in the accelerated evolution process known as MAGE (see “Metabolic Engineering” section above), the Church laboratory has examined alterations in the protocol that increased the frequencies of oligo-mediated mutagenesis. They found that the use of an E. coli strain deleted of four ssDNA exonucleases (RecJ, ExoI, ExoVII, and ExoX) resulted in higher amounts of mean allele conversions using the Co-MAGE protocol (374). The implication is that more oligos were surviving the threat of degradation by these exonucleases prior to being annealed to the replication fork. While previous studies using a single oligo had shown that using large amounts of it precludes the need for this Exo-deficient strain (215), Mosberg et al. (374) believe that the benefit of having saturating levels of oligos is offset by the increased competition among many different oligos entering the cell during electroporation. Another improvement in the frequency of oligo-mediated recombineering was also seen by this group when they employed a dnaG mutant strain that possessed a decreased rate of priming (375), resulting in longer Okazaki fragments and increased accessibility of ssDNA on the lagging strand template. The combination of both the Exo-deficient strain and the DnaG mutant resulted in the highest levels of allelic conversion using the Co-MAGE technique.
Recombineering with dsDNA (PCR) Substrates
Little has shown that λ Exo acting on dsDNA digested the 5′ strand, leading to 3′-ssDNA overhangs that were proposed to be key intermediates in Red-promoted recombination (41). Given the role that dsDNA ends play in RecA-mediated dsDNA break repair events (20), one might imagine that λ Red might act on both ends of a linear dsDNA substrate to promote a double crossover event at its target site. A test of this idea came from a study designed to evaluate these 3′ overhangs in a Red-promoted dsDNA-mediated recombineering event. Yu et al. (376) generated dsDNA substrates (from overlapping oligos) that contained 3′-ssDNA overhangs. It was thought that a dsDNA substrate with 3′-ssDNA overhangs might mimic a λ Exo-generated intermediate (given the 5′-3′ exonuclease activity of the enzyme) and would thus require only the action of λ Beta to recombine. The assay was designed so that each oligo could not act alone to promote recombination, requiring the formation of dsDNA intermediate to promote recombination. As expected, this substrate did not require (and was not stimulated by) the action of λ Exo, and was active for recombineering. However, the rates of recombination were quite low (3 × 102 recombinants/108 survivors), suggesting this is not a major intermediate for Red-promoted recombineering of dsDNA substrates. As suggested by the authors, the low frequency of recombination with this substrate may be due to the absence of an Exo-promoted loading of Beta protein onto ssDNA. λ Exo may load Beta onto the ssDNA it generates in a quantitatively (or qualitatively) different way compared with Beta binding to a preformed substrate containing 3′-ssDNA overhangs. While this may not be a critical step for short sequences (ssDNA oligos), it may have a greater role for recombinogenic processing of dsDNA substrates. These experiments, however, could not address this question.
In the same study, when substrates containing 5′ overhangs were used, recombineering rates were elevated 500-fold, and the increase was dependent on the presence of both λ Exo and Beta. This result was unexpected given the 5′-3′ polarity of λ Exo digestion. The authors explained this observation by surmising that substrates with 5′ overhangs could be “filled-in” in vivo by DNA polymerase I. The blunt-ended dsDNA species would then be the preferred substrate for the action of λ Exo and Beta. Yu et al. (376) noted that one possibility for this observation is that the loading of λ Beta on to ssDNA generated by λ Exo would preclude any problems associated with ssDNA secondary structures that might interfere with efficient annealing of the ssDNA to target sites. Alternatively, the authors suggested that perhaps Exo acted on only one end of the dsDNA to generate full-length ssDNA, which would then anneal to ssDNA regions of the replication fork in the same manner as proposed earlier for oligos (188). Such a mechanism has recently been supported by other studies from the Church laboratory (see below). Overall, however, these studies did not lend much support to a model involving annealing of dsDNA substrates containing 3′ overhangs to their target molecules (i.e., replication forks) during recombineering events.
In a similar study, Muyrers et al. (377) generated dsDNA substrates containing 3′-ssDNA overhangs (the presumed intermediate) by presecting linear dsDNA substrates with T7 gene 6 exonuclease (a 5′-3′ exonuclease) or by treating substrates containing a triplet of phosphorothiolated nucleotides with RecE. The phosphorothiolated nucleotides were situated in such a way as to prevent extended digestion of the DNA by RecE beyond the region of homology, thus generating substrates with precise 3′-ssDNA overhangs. Neither of these substrates could be recombined in the presence of an annealing function (either RecT or λ Beta) in the absence of their cognate 5′-3′ exonucleases (RecE and λ Exo, respectively), suggesting that processing of both ends of a dsDNA substrate was not the major mechanism of Red or RecET recombineering. These studies also showed that the combined expression of individual components of λ Red or RecET recombineering systems (i.e., expression of RecE with λ Beta, or λ Exo with RecT) did not promote recombineering. Thus, both the λ Red and RecET recombination systems require interactions between their orthologous partners, again arguing for some critical interaction between the partners during recombination (e.g., loading of the SSAP on to ssDNA by the exonuclease function).
Red Recombineering via a ssDNA Intermediate
Recent work has given support to a model whereby λ Exo binds to only one end of the dsDNA substrate and processively degrades one of the ssDNAs of the duplex. Such an event would generate a ssDNA that could conceivably behave as a large oligo to anneal to ssDNA regions of the replication fork, as described above for short oligos. Mosberg et al. (378) generated a 1.2-kb lacZ::kan ssDNA substrate by using biotin capture and a DNA melting protocol, and demonstrated that this ssDNA species was capable of promoting recombineering at ∼15% of the rate of the corresponding dsDNA substrate. The authors speculated that the lower amounts of recombineering with ssDNA substrates of this size might be due to the presence of inhibitory secondary structure in the substrate, or the lack of Exo-assisted loading of Beta protein onto the ssDNA. They also performed ssDNA recombineering in a strain expressing only the λ Beta protein. While the rate with a dsDNA substrate fell 200-fold in this strain (relative to a Red-expressing strain), the ssDNA substrate remained highly recombinogenic, indicating that recombinants formed in this experiment with the purified ssDNA were not the result of contaminating dsDNA. In further support of the model, when these authors used a dsDNA substrate bearing mismatched bases in the flanking regions (where both bases differed from the wild-type sequence), 80% of the recombinants inherited mismatches from only one of the two strands of the DNA duplex. This result is best explained by a model encompassing a ssDNA intermediate as shown in Fig. 21.
Figure 21.
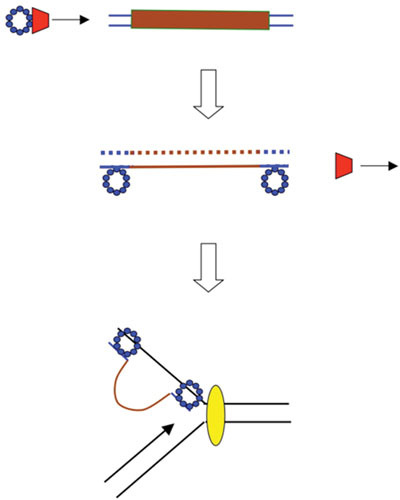
Red-promoted dsDNA recombineering via ssDNA intermediates. λ Exo binds to one end of a dsDNA substrate and degrades the 5′-ending strand. The long ssDNA generated by Exo is bound by Beta (exactly how and to what extent is not known). Beta then promotes annealing to ssDNA regions of the replication fork, much like the model in Fig. 20 for DNA oligos. The large nonhomologous ssDNA region encoding the drugR marker (brown line) is presumably stabilized by Beta bound to regions flanking the nonhomology. When the next replication fork passes through, the gene replacement is completed. This model was originally proposed by Yu et al. (376), and corroborated by the studies of Mosberg et al. (378).
The ssDNA intermediate model involves Exo-promoted digestion of only one of the two strands of a dsDNA substrate, the annealing of the ends of the ssDNA to the lagging strand template, and the presence of unpaired regions of ssDNA containing the drugR marker. A similar type of structure would be expected of an oligo designed to create a large deletion, with the exception that the large intervening unpaired region would be created in the template strand of a replication fork. Such structures with large unpaired regions are presumed to be stable long enough so that a subsequent replication fork would resolve the structure and form the recombinant via normal DNA synthesis.
This ssDNA intermediate model is also supported by observations of Maresca et al. (273), who took advantage of the fact that λ Exo requires a 5′-phosphate to efficiently degrade linear dsDNA. By generating PCR products that contain a 5′-phosphate on one end and a 5′-hydroxyl (or thiophosphate, which also makes ends resistant to λ Exo ) on the other, they ensured that λ Exo would efficiently degrade only one of the two strands of a dsDNA molecule (i.e., the one containing the 5′-phosphate). They show that, following electroporation into cells expressing λ Red and Gam functions, dsDNA molecules that generate a ssDNA species that can anneal to a lagging strand template are highly recombinogenic, whereas dsDNA substrates that generate ssDNA targeting the leading strand template are down 4- to 10-fold. These results, which are consistent with those of Mosberg et al. (378), suggest that λ Exo acts progressively on one of the two strands of a dsDNA substrate in vivo, generating a ssDNA intermediate that proceeds through a mechanism similar to that described for oligos. Maresca et al. (273) also showed that recombineering decreases dramatically when dsDNA insertions are increased from 1 to 4.5 kb, consistent with the limited processivity reported for λ Exo of ∼ 3 kb (43). The ssDNA intermediate model is also consistent with the experiments described by Lim et al. (379). In this study, substrates were constructed that contained flanking homology on either side of two drugR markers, and, in addition, between the two drugR markers. The authors show that recombinants most often retained only one of the two drugR markers, with only 10% of recombinants picking up both markers. Thus, homology internal to the dsDNA substrate was used more often than sequences at the ends of the DNA, consistent with a mechanism involving a ssDNA intermediate.
Finally, support for a ssDNA intermediate in Red-mediated recombineering of dsDNA comes from studies with phosphorothioate linkages incorporated into PCR substrates. Double-stranded DNA with phosphorothioate linkages are resistant to digestion by λ Exonuclease (380). Therefore, a PCR substrate containing phosphorothioates at the 5′ ends should be resistant to λ Exo, and be a poor substrate for recombineering. This is not the case. Mosberg et al. (374) have shown that the ssDNA exonuclease ExoVII can remove the terminal phosphorothioates, which then allows λ Exo to bind to the 5′-phosphate ends thus generated to initiate 5′-3′ degradation and promote recombineering. However, when the phosphorothioates were placed internally, in a region between the homologous ends and the drugR marker, neither wild-type E. coli or the xseA mutant (ExoVII deficient) could promote recombineering. Thus, while ExoVII can remove terminal phosphorothioates, it may not be able to gain access to λ Exo stalled at an internal phosphorothioate in vivo. In agreement with earlier results discussed above, internal phosphorothioates (in one strand only) were placed in such a way as to generate a full-length ssDNA intermediate (i.e., the other strand). This strand, which annealed to the lagging strand template of the replication fork, generated recombinants at a rate equivalent to unmodified substrates. On the other hand, internal phosphorothioates placed in such a way as to allow the survival of the strand that anneals to the leading strand template did not produce recombinants. These results are inconsistent with a model that degradation of both ends of linear dsDNA substrates is the principal mechanism of Red-promoted recombineering of PCR substrates. This conclusion applies to substrates with small regions of flanking homology (40 to 50 bp). Substrates containing longer regions of flanking homologies (e.g., 0.5 to 1 kb), which show a large RecA dependency, have not been tested using this type of methodology.
Red Recombineering with dsDNA with Single (or Limited) Base Pair Changes
The studies by Mosberg and Maresca (273, 378) show that recombineering principally follows through a ssDNA intermediate, although their data suggest at some low frequency, the mechanism may involve the presence of 5′-resected DNA at each end of a long dsDNA substrate. While these events may involve a combination of exonucleolytic processing of the ssDNA in vivo, and/or a primary recombination followed by a secondary event with the opposite strand in the same cell, other explanations are worth noting. One is offered by Court et al. (381), who provided a model of recombineering with dsDNA substrates that do not possess a large region of heterology relative to the target site (i.e., that do not include ∼1 kb drugR markers). The model is depicted in Fig. 22 and suggests an alternate (perhaps less favored) mechanism to that involving an ssDNA intermediate. In this scenario (Fig. 22A), the λ Exo-generated 3′-ssDNA region at one end of the DNA duplex is annealed by Beta to ssDNA regions of the lagging strand template of a replication fork. (This step was first suggested by Kuzminov, in his description of how the λ Red system could promote transduction in E. coli recA mutants [20]). This event causes the fork to stall, followed by fork regression that leads to the creation of the “chicken foot” structure (Fig. 22B). As the fork reverses, it is driven by a branch migration, which promotes annealing of the leading strand to the ssDNA of the recombineering substrate (that had previously been annealed to the lagging strand template). Extension of the leading strand by DNA polymerase I fills in the gap and ligase completes the formation of the chicken foot. Beta bound to the uninvolved end of the recombineering substrate protects this end from degradation by cellular nucleases (Fig. 22C). Reabsorption of the chicken foot reestablishes the replication fork structure, which should occur readily in the absence of any large heterology between the substrate and the chromosome, producing small mismatched regions in both arms of the fork (Fig. 22D). Upon subsequent replication of the chromosomal region, the changes encoded by the dsDNA substrate are stably incorporated into the chromosome, provided they are resistant to (or escape from) the mismatch repair system of E. coli. A region containing a long heterology (e.g., a drugR resistance marker) is not expected to branch migrate, and thus would not be able to proceed through the mechanism depicted in Fig. 22.
Figure 22.
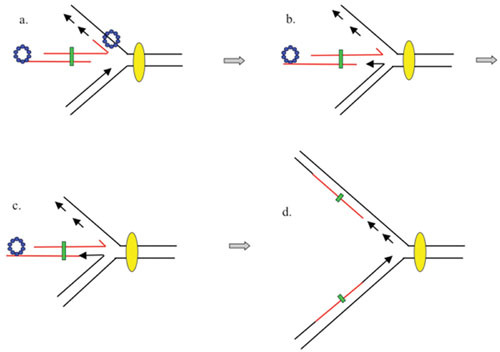
Proposed model for integration of dsDNA substrate with limited nonhomology into a replication fork. (A) Red-processed dsDNA end (Beta bound to a ssDNA overhang generated by Exo) invades a replication fork and promotes annealing to the lagging strand template. (B) The leading strand switches to the incoming duplex. Stalling and reversal of the fork generates a “chicken-foot” structure. (C) The leading strand polymerase fills in the gap previously generated by λ Exo, while DNA ligase connects the strands. (D) The Holliday junction branch migrates to the right and is reabsorbed, reestablishing a replication fork framework. The mutant base(s) form heteroduplexes (green boxes). Model taken from Court et al. (381).
Red Recombineering via the Template Switch Model
The template switch model of λ phage recombination presented by Poteete (depicted in Fig. 6) suggests that Beta anneals the incoming ssDNA tail of a Red-processed dsDNA end to the replication fork and, in addition, repositions the incoming ssDNA to be used as a template by a redirected leading strand DNA polymerase, as seen in Court’s model (see Fig. 22). In the template switch model, however, a ssDNA nick occurs in the leading strand template (by an unspecified endonuclease) leading to a splicing event between the incoming DNA and one prong of the replication fork; the fork is then redirected (Fig. 6). To apply this model to the recombineering of PCR substrates, the template switch model would require a second invasion event of the dsDNA end not involved in the primary replisome invasion, as suggested by Poteete (3). A recombinant in this case would inherit information from the 3′ overhangs generated by λ Exo at each end (that is, would acquire information from both strands) since the polymerase switch (at each end) copies the sequence from the 3′ overhangs in the process of generating the recombinant. This result, however, would be at odds with the data of Mosberg et al. (378), who found that the information from only one strand of a dsDNA substrate is found in 80% of their recombinants, consistent with a model involving a ssDNA intermediate.
However, the template switch model could be modified for recombineering of a PCR substrate as described in Fig. 23. In this scenario, the template switch and nick in the leading strand template take place, but the redirected polymerase promotes complete synthesis of the incoming ssDNA, regenerating a dsDNA end that can now be used in a second replisome invasion event. In effect, an ssDNA intermediate is involved, but it is incorporated differently than simply being annealed to the lagging strand template. λ Exo is expected to carry out the complete processing of a typical 1-kb PCR substrate in vivo given its processivity. Furthermore, this size DNA is similar to the amount of heteroduplex DNA that is generated by lambda recombination during a phage infection (180). In this scenario, since the incoming strand is copied, information from only the invading strand will be present in the recombinant, consistent with the data from Mosberg et al. (378).
Figure 23.
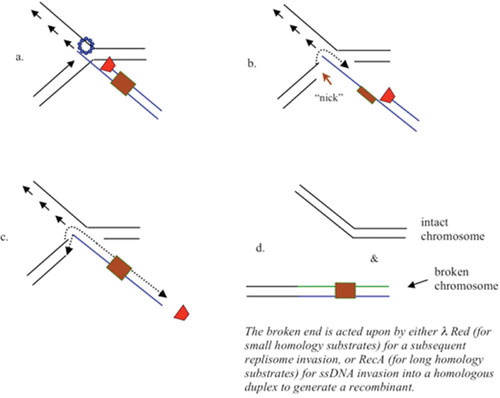
Template switch model of recombineering. (A) As before, a Red-processed dsDNA end (Beta bound to a ssDNA overhang generated by Exo) invades a replication fork and promotes annealing to the lagging strand template. (B) Beta captures the leading strand and promotes a template switch, such that the leading strand polymerase now uses the incoming strand as a template. A nick is introduced in the leading strand template by an unspecified nuclease. (C) The redirected polymerase completely resynthesizes the incoming strand, reestablishing a dsDNA end. The 3′ end of the invading strand is filled in and ligated to complete the recombination event. (D) The products of the reaction are an intact chromosome (after filling in and ligation) and the broken end containing the incoming substrate. This dsDNA end could be acted upon by the λ Red system and invade the leading strand template of another replisome (for small homology substrates), or by the RecA-dependent pathway for recombination (for long homology substrates) to complete the gene replacement.
The RecA Independence of Recombineering
The recombineering mechanisms described here, either with an oligo or a Red-processed dsDNA substrate, involve annealing of these substrates to ssDNA regions of a replication fork. As such, they do not require a RecA-mediated ssDNA invasion into duplex DNA chromosomal target sites. This is consistent with the RecA independence of recombineering reported for both oligos and PCR substrates with short regions of flanking homology (50 to 60 bp), as well as the RecA independence observed with lambda recombination in the presence of phage DNA replication. However, the original report on the use of λ Red for gene replacement, which used drugR markers containing ∼1 kb of flanking homology, showed a ∼50-fold dependence on RecA for efficient recombination (Murphy [23]), as have studies by Poteete et al. using similar sized substrates (149). The RecA dependence of recombineering in these studies may reflect both the larger size of the recombineering substrate and the higher level of homology with the target site. RecA-independent recombineering decreases with increasing size of the linear DNA (273), probably a reflection of the limited ability of λ Exo to make a complete ssDNA intermediate for dsDNA substrates 3 kb and greater in vivo. Perhaps the limited processing by λ Exo of substrates with 1-kb flanks, together with the greater amount of homology typical of RecA-mediated events, drive Red-promoted recombination of these substrates into the RecA-dependent pathway, as described in Fig. 4A.
As an alternative hypothesis, the longer recombineering substrates (∼3 kb) might involve a Red-promoted invasion of one end of the substrate, but perhaps a RecA-promoted event at the other end. In this scenario, diagrammed in Fig. 23, the first event involves a Red-promoted annealing of the λ Exo-processed end to the lagging strand template of a replication fork (a favored event as suggested by oligos that target the lagging strand template). This step is followed by a template switch to fill in the ssDNA gap created by λ. The other end of a long linear substrate (after processing by Red) would have to (by necessity) anneal to the leading strand template of a second replication fork (a disfavored event perhaps, due to the lack of ssDNA regions within this target, or the lack of nearby fork). Instead, because of the longer homologies available (e.g., 0.5 to 1 kb), this end may proceed via the RecA-promoted “salvage pathway” described by Poteete (116), which does not required a fork invasion event (Fig. 4A). In accordance with this model, the physical recombinant assay used to demonstrate the replisome invasion model (as seen in Fig. 6), where only one end is necessary to achieve a recombinant product, was totally RecA independent. On the other hand, as mentioned above, the Red-promoted duplication event of chromosomal regions containing the lacZ region, requiring both ends of a 3.5-kb linear DNA substrate, was largely RecA dependent (275).
SUMMARY
When Esther Lederberg discovered phage λ in 1950 (382), it was impossible for her to know how this phage would lend its recombination system to the manipulation of bacterial and mammalian DNA for so many purposes 60 years later. Recombineering technology has allowed investigators to make DNA constructs that otherwise would in many ways be impossible to piece together with restriction enzymes and ligases. Different schemes for generating altered chromosomes and BACs with λ Red and RecET, with the help of Cre/loxP, counterselection markers, and the I-SceI enzyme have been developed, with more advances likely forthcoming.
What is less understood is the mechanism of annealing that is so central to this process. While λ Exo’s ability to generate ssDNA in vivo seems an important first step in recombineering, it is still unclear how the SSAP binds ssDNA, how it finds the replication fork, and what the molecular details of the interactions are that position the processed ssDNA in the heart of a replication fork with such precision. Also, can the mechanism of recombineering help us decipher the mechanism of recombination that goes on during phage infection? These are questions to be addressed by further studies of a phage recombination system, still the best model for unraveling such molecular details. Besides the technology development purposes, the continued understanding of a system like λ Red is all the more relevant considering that its eukaryotic analog, the Rad52 protein, is an important component of most of the recombination pathways in yeast and human cells (71, 72, 383, 384, 385).
ACKNOWLEDGMENTS
I wish to thank Anthony Poteete, Martin Marinus, and Michael Volkert for helpful discussions. I especially thank Andrei Kuzminov for his critical reading and helpful suggestions on writing this review.
REFERENCES
- 1.Jacob F, Wollman E. 1954. Etude genetique d’un bacteriophage tempere d’Echerichia coli. I. Le systeme genetique du bacteriophage lambda. Ann Inst Pasteur 87:653–674. [PubMed] [Google Scholar]
- 2.Kaiser AD. 1955. A genetic study of the temperate coliphage. Virology 1:424–443. 10.1016/0042-6822(55)90036-2 [DOI] [PubMed] [Google Scholar]
- 3.Poteete AR. 2008. Involvement of DNA replication in phage lambda Red-mediated homologous recombination. Mol Microbiol 68:66–74. [PubMed] 10.1111/j.1365-2958.2008.06133.x [DOI] [PubMed] [Google Scholar]
- 4.Savage PJ, Leong JM, Murphy KC. 2006. Rapid allelic exchange in enterohemorrhagic Escherichia coli (EHEC) and other E. coli using lambda red recombination. Curr Protoc Microbiol Chapter 5:Unit5A.2. 10.1002/9780471729259.mc05a02s00. 10.1002/9780471729259.mc05a02s00 [DOI] [PubMed] [Google Scholar]
- 5.Sawitzke JA, Thomason LC, Costantino N, Bubunenko M, Datta S, Court DL. 2007. Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol 421:171–199. 10.1016/S0076-6879(06)21015-2 [DOI] [PubMed] [Google Scholar]
- 6.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. 2009. Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc 4:206–223. [PubMed] 10.1038/nprot.2008.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomason L, Court DL, Bubunenko M, Costantino N, Wilson H, Datta S, Oppenheim A. 2007. Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol Chapter 1:Unit 1.16. 10.1002/0471142727.mb0116s78. 10.1002/0471142727.mb0116s78 [DOI] [PubMed] [Google Scholar]
- 8.Clark AJ, Margulies AD. 1965. Isolation and characterization of recombination-deficient mutants of Escherichia coli K12. Proc Natl Acad Sci USA 53:451–459. [PubMed] 10.1073/pnas.53.2.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takano T. 1966. Behavoir of some episomal elements in a recombination-deficient mutant of Escherichia coli. Jpn J Microbiol 10:201–210. 10.1111/j.1348-0421.1966.tb00309.x [DOI] [Google Scholar]
- 10.van de Putte P, Zwenk H, Rorsch A. 1966. Properties of four mutants of Escherichia coli defective in genetic recombination. Mutat Res 3:381–392. [PubMed] 10.1016/0027-5107(66)90048-0 [DOI] [PubMed] [Google Scholar]
- 11.Brooks K, Clark AJ. 1967. Behavior of lambda bacteriophage in a recombination deficienct strain of Escherichia coli. J Virol 1:283–293. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franklin NC. 1967. Deletions and functions of the center of the f80-l phage genome. Evidence for a phage function promoting genetic recombination. Genetics 57:301–318. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Echols H, Gingery R. 1968. Mutants of bacteriophage λ defective for vegetative genetic recombination. J Mol Biol 34:239–249. [PubMed] 10.1016/0022-2836(68)90249-0 [DOI] [PubMed] [Google Scholar]
- 14.Signer ER, Weil J. 1968. Recombination in bacteriophage lambda. I. Mutants deficient in general recombination. J Mol Biol 34:261–271. [PubMed] 10.1016/0022-2836(68)90251-9 [DOI] [PubMed] [Google Scholar]
- 15.Zissler J, Singer E, Schaefer F. 1971. The role of recombination in the growth of bacteriophage λ. I. The Gamma gene, p 455–468. In Hershey AD (ed), The Bacteriophage Lambda. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 16.Enquist LW, Skalka A. 1973. Replication of bacteriophage λ DNA dependent on the function of host and viral genes. I. Interaction of red, gam and rec. J Mol Biol 75:185–212. [PubMed] 10.1016/0022-2836(73)90016-8 [DOI] [PubMed] [Google Scholar]
- 17.Karu AE, Sakaki Y, Echols H, Linn S. 1975. The gamma protein specified by bacteriophage gamma. Structure and inhibitory activity for the recBC enzyme of Escherichia coli. J Biol Chem 250:7377–7387. [PubMed] [PubMed] [Google Scholar]
- 18.Wackernagel W, Radding CM. 1974. Transformation and transduction of Escherichia coli: the nature of recombinants formed by Rec, RecF, and lambda Red, p 111–122. In Grell RF (ed), Mechanisms in Recombination. Plenum Press, New York, NY. 10.1007/978-1-4684-2133-0_11 [DOI] [Google Scholar]
- 19.Weisberg RA, Sternberg N. 1974. Transduction of recB2 host is promoted by lambda red+ function, p 107–109. In Grell RF (ed), Mechanisms in Recombination. Plenum Press, New York, NY. 10.1007/978-1-4684-2133-0_10 [DOI] [Google Scholar]
- 20.Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63:751–813. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stahl FW, Stahl MM, Malone RE. 1978. Red-mediated recombination of phage lambda in a recA –recB – host. Mol Gen Genet 159:207–211. 10.1007/BF00270895 [DOI] [Google Scholar]
- 22.Poteete AR, Volkert MR. 1988. Activation of recF-dependent recombination in Escherichia coli by bacteriophage lambda- and P22-encoded functions. J Bacteriol 170:4379–4381. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy KC. 1998. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol 180:2063–2071. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henderson D, Weil J. 1975. Recombination-deficient deletions in bacteriophage λ and their interactions with Chi mutations. Genetics 79:143–174. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lam ST, Stahl MM, McMilin KD, Stahl FW. 1974. Rec-mediated recombinational hot spot activity in bacteriophage lambda. II. A mutation which causes hot spot activity. Genetics 77:425–433. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stahl FW, Stahl MM. 1975. Rec-mediated recombinational hot spot activity in bacteriophage lambda. IV. Effect of heterology on Chi-stimulated crossing over. Mol Gen Genet 140:29–37. [PubMed] 10.1007/BF00268986 [DOI] [PubMed] [Google Scholar]
- 27.Dillingham MS, Kowalczykowski SC. 2008. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev 72:642–671. [PubMed] 10.1128/MMBR.00020-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers RS, Stahl FW. 1994. Chi and the RecBCD enzyme of Escherichia coli. Annu Rev Genet 28:49–70. [PubMed] 10.1146/annurev.ge.28.120194.000405 [DOI] [PubMed] [Google Scholar]
- 29.Dixon DA, Kowalczykowski SC. 1993. The recombination hotspot c is a regulatory sequence that acts by attenuating the nuclease activity of the E. coli RecBCD enzyme. Cell 73:87–96. 10.1016/0092-8674(93)90162-J [DOI] [PubMed] [Google Scholar]
- 30.Dixon DA, Kowalczykowski SC. 1995. Role of Escherichia coli recombination hotspot, c, in recABCD-dependent homologous pairing. J Biol Chem 270:16360–16370. [PubMed] 10.1074/jbc.270.27.16360 [DOI] [PubMed] [Google Scholar]
- 31.Taylor AF, Smith GR. 1980. Unwinding and rewinding of DNA by the RecBC enzyme. Cell 22:447–457. [PubMed] 10.1016/0092-8674(80)90355-4 [DOI] [PubMed] [Google Scholar]
- 32.Anderson DG, Churchill JJ, Kowalczykowski SC. 1999. A single mutation, RecB(D1080A,) eliminates RecA protein loading but not Chi recognition by RecBCD enzyme. J Biol Chem 274:27139–27144. [PubMed] 10.1074/jbc.274.38.27139 [DOI] [PubMed] [Google Scholar]
- 33.Anderson DG, Kowalczykowski SC. 1997. The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a chi-regulated manner. Cell 90:77–86. [PubMed] 10.1016/S0092-8674(00)80315-3 [DOI] [PubMed] [Google Scholar]
- 34.Arnold DA, Kowalczykowski SC. 2000. Facilitated loading of RecA protein is essential to recombination by RecBCD enzyme. J Biol Chem 275:12261–12265. [PubMed] 10.1074/jbc.275.16.12261 [DOI] [PubMed] [Google Scholar]
- 35.Stahl FW. 1998. Recombination in phage lambda: one geneticist’s historical perspective. Gene 223:95–102. [PubMed] 10.1016/S0378-1119(98)00246-7 [DOI] [PubMed] [Google Scholar]
- 36.Dabert P, Smith GR. 1997. Gene replacement with linear DNA fragments in wild type Escherichia coli: enhancement by chi sites. Genetics 145:877–889. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.el Karoui M, Ehrlich D, Gruss A. 1998. Identification of the lactococcal exonuclease/recombinase and its modulation by the putative Chi sequence. Proc Natl Acad Sci USA 95:626–631. [PubMed] 10.1073/pnas.95.2.626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korn D, Weissbach A. 1963. The effect of lysogenic induction on the deoxyribonucleases of Escherichia coli K12-lambda. I. Appearance of a new exonuclease activity. J Biol Chem 238:3390–3394. [PubMed] [PubMed] [Google Scholar]
- 39.Korn D, Weissbach A. 1964. Purification and properties of a deoxyribonucleic acid exonuclease associated with the formation of phage 434. J Biol Chem 239:3849–3857. [PubMed] [PubMed] [Google Scholar]
- 40.Radding CM. 1964. Nuclease activity in defective lysogens of phage lambda. Biochem Biophys Res Commun 15:8–12. [PubMed] 10.1016/0006-291X(64)90093-2 [DOI] [PubMed] [Google Scholar]
- 41.Little JW. 1967. An exonuclease induced by bacteriophage lambda. II. Nature of the enzymatic reaction. J Biol Chem 242:679–686. [PubMed] 10.1016/s0076-6879(67)12041-7 [DOI] [PubMed] [Google Scholar]
- 42.Little JW, Lehman IR, Kaiser AD. 1967. An exonuclease induced by bacteriophage lambda. I. Preparation of the crystalline enzyme. J Biol Chem 242:672–678. [PubMed] 10.1016/s0076-6879(67)12041-7 [DOI] [PubMed] [Google Scholar]
- 43.Carter DM, Radding CM. 1971. The role of exonuclease and beta protein of phage lambda in genetic recombination. II. Substrate specificity and the mode of action of lambda exonuclease. J Biol Chem 246:2502–2512. [PubMed] [PubMed] [Google Scholar]
- 44.Manly KF, Signer ER, Radding CM. 1969. Nonessential functions of bacteriophage lambda. Virology 37:177–188. [PubMed] 10.1016/0042-6822(69)90197-4 [DOI] [PubMed] [Google Scholar]
- 45.Radding CM. 1970. The role of exonuclease and beta protein of bacteriophage lambda in genetic recombination. I. Effects of red mutants on protein structure. J Mol Biol 52:491–499. [PubMed] 10.1016/0022-2836(70)90415-8 [DOI] [PubMed] [Google Scholar]
- 46.Shulman MJ, Hallick LM, Echols H, Signer ER. 1970. Properties of recombination-deficient mutants of bacteriophage lambda. J Mol Biol 52:501–520. [PubMed] 10.1016/0022-2836(70)90416-X [DOI] [PubMed] [Google Scholar]
- 47.Sriprakash KS, Lundh N, Huh M-O, Radding CM. 1975. The specificity of lambda exonuclease. Interactions with single-stranded DNA. J Biol Chem 250:5438–5445. [PubMed] [PubMed] [Google Scholar]
- 48.Cassuto E, Radding CM. 1971. Mechanism for the action of lambda exonuclease in genetic recombination. Nat New Biol 229:13–16. [PubMed] 10.1038/newbio229013a0 [DOI] [PubMed] [Google Scholar]
- 49.Mitsis PG, Kwagh JG. 1999. Characterization of the interaction of lambda exonuclease with the ends of DNA. Nucleic Acids Res 27:3057–3063. [PubMed] 10.1093/nar/27.15.3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perkins TT, Dalal RV, Mitsis PG, Block SM. 2003. Sequence-dependent pausing of single lambda exonuclease molecules. Science 301:1914–1918. [PubMed] 10.1126/science.1088047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subramanian K, Rutvisuttinunt W, Scott W, Myers RS. 2003. The enzymatic basis of processivity in lambda exonuclease. Nucleic Acids Res 31:1585–1596. [PubMed] 10.1093/nar/gkg266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kovall R, Matthews BW. 1997. Toroidal structure of lambda-exonuclease. Science 277:1824–1827. [PubMed] 10.1126/science.277.5333.1824 [DOI] [PubMed] [Google Scholar]
- 53.Zhang J, McCabe KA, Bell CE. 2011. Crystal structures of lambda exonuclease in complex with DNA suggest an electrostatic ratchet mechanism for processivity. Proc Natl Acad Sci USA 108:11872–11877. [PubMed] 10.1073/pnas.1103467108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, Xing X, Herr AB, Bell CE. 2009. Crystal structure of E. coli RecE protein reveals a toroidal tetramer for processing double-stranded DNA breaks. Structure 17:690–702. [PubMed] 10.1016/j.str.2009.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aravind L, Walker DR, Koonin EV. 1999. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res 27:1223–1242. [PubMed] 10.1093/nar/27.5.1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radding CM. 1966. Regulation of lambda exonuclease. I. Properties of lambda exonuclease purified from lysogens of lambda T11 and wild type. J Mol Biol 18:235–250. [PubMed] 10.1016/S0022-2836(66)80243-7 [DOI] [PubMed] [Google Scholar]
- 57.Radding CM, Shreffler DC. 1966. Regulation of lambda exonuclease. II. Joint regulation of exonuclease and a new lambda antigen. J Mol Biol 18:251–261. [PubMed] 10.1016/S0022-2836(66)80244-9 [DOI] [PubMed] [Google Scholar]
- 58.Tolun G. 2007. More than the sum of its parts: physical and mechanistic coupling in the phage lambda Red recombinase. PhD dissertation, University of Miami, Miami, FL. [Google Scholar]
- 59.Kmiec E, Holloman WK. 1981. Beta protein of bacteriophage lambda promotes renaturation of DNA. J Biol Chem 256:12636–12639. [PubMed] [PubMed] [Google Scholar]
- 60.Muniyappa K, Radding CM. 1986. The homologous recombination system of phage lambda. Pairing activities of beta protein. J Biol Chem 261:7472–7478. [PubMed] [PubMed] [Google Scholar]
- 61.Karakousis G, Ye N, Li Z, Chiu SK, Reddy G, Radding CM. 1998. The beta protein of phage lambda binds preferentially to an intermediate in DNA renaturation. J Mol Biol 276:721–731. [PubMed] 10.1006/jmbi.1997.1573 [DOI] [PubMed] [Google Scholar]
- 62.Hall SD, Kolodner RD. 1994. Homologous pairing and strand exchange promoted by the Escherichia coli RecT protein. Proc Natl Acad Sci USA 91:3205–3209. [PubMed] 10.1073/pnas.91.8.3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iyer LM, Koonin EV, Aravind L. 2002. Classification and evolutionary history of the single-strand annealing proteins, RecT, Redbeta, ERF and RAD52. BMC Genomics 3:8. 10.1186/1471-2164-3-8 [PubMed] 10.1186/1471-2164-3-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poteete AR, Sauer RT, Hendrix RW. 1983. Domain structure and quaternary organization of the bacteriophage P22 Erf protein. J Mol Biol 171:401–418. [PubMed] 10.1016/0022-2836(83)90037-2 [DOI] [PubMed] [Google Scholar]
- 65.Murphy KC, Casey L, Yannoutsos N, Poteete AR, Hendrix RW. 1987. Localization of a DNA-binding determinant in the bacteriophage P22 Erf protein. J Mol Biol 194:105–117. [PubMed] 10.1016/0022-2836(87)90719-4 [DOI] [PubMed] [Google Scholar]
- 66.Wu Z, Xing X, Bohl CE, Wisler JW, Dalton JT, Bell CE. 2006. Domain structure and DNA binding regions of beta protein from bacteriophage lambda. J Biol Chem 281:25205–25214. [PubMed] 10.1074/jbc.M512450200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mythili E, Muniyappa K. 1993. Formation of linear plasmid multimers promoted by the phage lambda Red-system in lon mutants of Escherichia coli. J Gen Microbiol 139:2387–2397. [PubMed] 10.1099/00221287-139-10-2387 [DOI] [PubMed] [Google Scholar]
- 68.Passy SI, Yu X, Li Z, Radding CM, Egelman EH. 1999. Rings and filaments of beta protein from bacteriophage lambda suggest a superfamily of recombination proteins. Proc Natl Acad Sci USA 96:4279–4284. [PubMed] 10.1073/pnas.96.8.4279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Erler A, Wegmann S, Elie-Caille C, Bradshaw CR, Maresca M, Seidel R, Habermann B, Muller DJ, Stewart AF. 2009. Conformational adaptability of Redbeta during DNA annealing and implications for its structural relationship with Rad52. J Mol Biol 391:586–598. [PubMed] 10.1016/j.jmb.2009.06.030 [DOI] [PubMed] [Google Scholar]
- 70.Shinohara A, Shinohara M, Ohta T, Matsuda S, Ogawa T. 1998. Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells 3:145–156. [PubMed] 10.1046/j.1365-2443.1998.00176.x [DOI] [PubMed] [Google Scholar]
- 71.Singleton MR, Wentzell LM, Liu Y, West SC, Wigley DB. 2002. Structure of the single-strand annealing domain of human RAD52 protein. Proc Natl Acad Sci USA 99:13492–13497. [PubMed] 10.1073/pnas.212449899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stasiak AZ, Larquet E, Stasiak A, Muller S, Engel A, Van Dyck E, West SC, Egelman EH. 2000. The human Rad52 protein exists as a heptameric ring. Curr Biol 10:337–340. [PubMed] 10.1016/S0960-9822(00)00385-7 [DOI] [PubMed] [Google Scholar]
- 73.Thresher RJ, Makhov AM, Hall SD, Kolodner R, Griffith JD. 1995. Electron microscopic visualization of RecT protein and its complexes with DNA. J Mol Biol 254:364–371. [PubMed] 10.1006/jmbi.1995.0623 [DOI] [PubMed] [Google Scholar]
- 74.Li Z, Karakousis G, Chiu SK, Reddy G, Radding CM. 1998. The beta protein of phage lambda promotes strand exchange. J Mol Biol 276:733–744. [PubMed] 10.1006/jmbi.1997.1572 [DOI] [PubMed] [Google Scholar]
- 75.Cox MM. 2007. Motoring along with the bacterial RecA protein. Nat Rev Mol Cell Biol 8:127–138. [PubMed] 10.1038/nrm2099 [DOI] [PubMed] [Google Scholar]
- 76.Roca AI, Cox MM. 1997. RecA protein: structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol 56:129–223. [PubMed] 10.1016/S0079-6603(08)61005-3 [DOI] [PubMed] [Google Scholar]
- 77.Rybalchenko N, Golub EI, Bi B, Radding CM. 2004. Strand invasion promoted by recombination protein beta of coliphage lambda. Proc Natl Acad Sci USA 101:17056–17060. [PubMed] 10.1073/pnas.0408046101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Folta-Stogniew E, O’Malley S, Gupta R, Anderson KS, Radding CM. 2004. Exchange of DNA base pairs that coincides with recognition of homology promoted by E. coli RecA protein. Mol Cell 15:965–975. [PubMed] 10.1016/j.molcel.2004.08.017 [DOI] [PubMed] [Google Scholar]
- 79.Gupta RC, Folta-Stogniew E, O’Malley S, Takahashi M, Radding CM. 1999. Rapid exchange of A:T base pairs is essential for recognition of DNA homology by human Rad51 recombination protein. Mol Cell 4:705–714. 10.1016/S1097-2765(00)80381-0 [DOI] [PubMed] [Google Scholar]
- 80.Gupta RC, Folta-Stogniew E, Radding CM. 1999. Human Rad51 protein can form homologous joints in the absence of net strand exchange. J Biol Chem 274:1248–1256. [PubMed] 10.1074/jbc.274.3.1248 [DOI] [PubMed] [Google Scholar]
- 81.Cox M, Lehman IR. 1987. Enzymes of general recombination. Annu Rev Biochem 56:229–262. [PubMed] 10.1146/annurev.bi.56.070187.001305 [DOI] [PubMed] [Google Scholar]
- 82.Muniyappa K, Shaner SL, Tang SS, Radding CM. 1984. Mechanism of the concerted action of RecA protein and helix destabilizing proteins in homologous recombination. Proc Natl Acad Sci USA 81:2757–2761. [PubMed] 10.1073/pnas.81.9.2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Court R, Cook N, Saikrishnan K, Wigley D. 2007. The crystal structure of lambda-Gam protein suggests a model for RecBCD inhibition. J Mol Biol 371:25–33. [PubMed] 10.1016/j.jmb.2007.05.037 [DOI] [PubMed] [Google Scholar]
- 84.Murphy KC. 2007. The lambda Gam protein inhibits RecBCD binding to dsDNA ends. J Mol Biol 371:19–24. [PubMed] 10.1016/j.jmb.2007.05.085 [DOI] [PubMed] [Google Scholar]
- 85.Oliver DB, Goldberg EB. 1977. Protection of parental T4 DNA from a restriction exonuclease by the product of gene 2. J Mol Biol 116:877–881. [PubMed] 10.1016/0022-2836(77)90276-5 [DOI] [PubMed] [Google Scholar]
- 86.Murphy KC. 1994. Biochemical characterization of P22 phage-modified Escherichia coli RecBCD enzyme. J Biol Chem 269:22507–22516. [PubMed] [PubMed] [Google Scholar]
- 87.Murphy KC. 2000. Bacteriophage P22 Abc2 protein binds to RecC increases the 5′ strand nicking activity of RecBCD and together with lambda bet, promotes Chi-independent recombination. J Mol Biol 296:385–401. [PubMed] 10.1006/jmbi.1999.3486 [DOI] [PubMed] [Google Scholar]
- 88.Murphy KC, Lewis LJ. 1993. Properties of Escherichia coli expressing bacteriophage P22 Abc (Anti-RecBCD) proteins, including inhibition of Chi activity. J Bacteriol 175:1756–1766. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murphy KC. 1991. Lambda Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme. J Bacteriol 173:5808–5821. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marsic N, Roje S, Stojiljkovic I, Salaj-Smic E, Trgovcevic Z. 1993. In vivo studies on the interaction of RecBCD enzyme and lambda Gam protein. J Bacteriol 175:4738–4743. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Friedman SA, Hays JB. 1986. Selective inhibition of Escherichia coli recBC activities by plasmid-encoded GamS function of phage lambda. Gene 43:255–263. [PubMed] 10.1016/0378-1119(86)90214-3 [DOI] [PubMed] [Google Scholar]
- 92.Sergueev K, Yu D, Austin S, Court D. 2001. Cell toxicity caused by products of the p(L) operon of bacteriophage lambda. Gene 272:227–235. [PubMed] 10.1016/S0378-1119(01)00535-2 [DOI] [PubMed] [Google Scholar]
- 93.Capaldo-Kimball F, Barbour SD. 1971. Involvement of recombination genes in growth and viability of Escherichia coli K-12. J Bacteriol 106:204–212. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miranda A, Kuzminov A. 2003. Chromosomal lesion suppression and removal in Escherichia coli via linear DNA degradation. Genetics 163:1255–1271. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trogovcevic Z, Rupp WD. 1975. Lambda bacteriophage gene produces and X-ray sensitivity of Escherichia coli: comparison of red-dependent and gam-dependent radioresistance. J Bacteriol 123:212–221. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meselson M, Weigle JJ. 1961. Chromosome brekage accompanying genetic recombination in bacteriophage. Proc Natl Acad Sci USA 47:857–868. [PubMed] 10.1073/pnas.47.6.857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kellenberger G, Zichichi ML, Weigle JJ. 1961. Exchange of DNA in the recombination of bacteriophage lambda. Proc Natl Acad Sci USA 47:869–878. [PubMed] 10.1073/pnas.47.6.869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tang RS. 1994. The return of copy-choice in DNA recombination. Bioessays 16:785–788. [PubMed] 10.1002/bies.950161102 [DOI] [PubMed] [Google Scholar]
- 99.Meselson M. 1964. On the mechanism of genetic recombination between DNA molecules. J Mol Biol 9:734–745. [PubMed] 10.1016/S0022-2836(64)80178-9 [DOI] [PubMed] [Google Scholar]
- 100.Kellenberger-Gujer G, Weisberg RA. 1971. Recombination in bacteriophage lambda I. Exchange of DNA promoted by phage and bacterial recombination mechanisms. In Hershey ADE (ed), The Bacteriophage Lambda. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 101.Stahl FW, McMilin KD, Stahl MM, Nozu Y. 1972. An enhancing role for DNA synthesis in formation of bacteriophage lambda recombinants. Proc Natl Acad Sci USA 69:3598–3601. [PubMed] 10.1073/pnas.69.12.3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stahl FW, Stahl MM. 1971. DNA synthesis associated with recombination, I. Recombination in a conditional DNA-negative host, p 431–442. In Hershey ADE (ed), The Bacteriophage Lambda. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 103.McMilin KD, Russo VE. 1972. Maturation and recombination of bacteriophage lambda DNA molecules in the absence of DNA duplication. J Mol Biol 68:49–55. [PubMed] 10.1016/0022-2836(72)90261-6 [DOI] [PubMed] [Google Scholar]
- 104.Stahl FW, McMilin KD, Stahl MM, Malone RE, Nozu Y, Russo VE. 1972. A role for recombination in the production of “free-loader” lambda bacteriophage particles. J Mol Biol 68:57–67. [PubMed] 10.1016/0022-2836(72)90262-8 [DOI] [PubMed] [Google Scholar]
- 105.Stahl FW, Stahl MM. 1971. DNA synthesis associated with recombination, II. Recombination between repressed chromosomes, p 443–453. In Hershey AD (ed), The Bacteriophage Lambda. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 106.Stahl FW, McMilin KD, Stahl MM, Crasemann JM, Lam S. 1974. The distribution of crossovers along unreplicated lambda bacteriophage chromosomes. Genetics 77:395–408. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stahl FW, Kobayashi I, Stahl MM. 1985. In phage λ, cos is a recombinator in the Red pathway. J Mol Biol 181:199–209. [PubMed] 10.1016/0022-2836(85)90085-3 [DOI] [PubMed] [Google Scholar]
- 108.Stahl FW, Kobayashi I, Stahl MM. 1982. Distance from cohesive end site cos determines the replication requirement for recombination in phage lambda. Proc Natl Acad Sci USA 79:6318–6321. [PubMed] 10.1073/pnas.79.20.6318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stahl FW, Stahl MM. 1986. DNA synthesis at the site of a Red-mediated exchange in phage lambda. Genetics 113:1–12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thaler DS, Stahl MM, Stahl FW. 1987. Evidence that the normal route of replication-allowed Red-mediated recombination involves double-chain ends. EMBO J 6:3171–3176. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thaler DS, Stahl MM, Stahl FW. 1987. Tests of the double-strand-break repair model for red-mediated recombination of phage lambda and plasmid lambda dv. Genetics 116:501–511. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Poteete AR, Fenton AC. 1993. Efficient double-strand break-stimulated recombination promoted by the general recombination systems of phages λ and P22. Genetics 134:1013–1021. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stahl FW, Fox MS, Faulds D, Stahl MM. 1990. Break-join recombination in phage lambda. Genetics 125:463–474. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Skalka A. 1974. A replicator’s view of recombination (and repair), p 421–432. In Grell RF (ed), Mechanisms in Genetic Recombination. Plenum Press, New York, NY. 10.1007/978-1-4684-2133-0_37 [DOI] [Google Scholar]
- 115.Kowalczykowski SC, Dixon DA, Eggleston AK, Lauer SD, Rehrauer WM. 1994. Biochemistry of homologous recombination in Escherichia coli. Mic Rev 58:401–465. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Poteete AR. 2004. Modulation of DNA repair and recombination by the bacteriophage lambda Orf function in Escherichia coli K-12. J Bacteriol 186:2699–2707. [PubMed] 10.1128/JB.186.9.2699-2707.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stahl MM, Thomason L, Poteete AR, Tarkowski T, Kuzminov A, Stahl FW. 1997. Annealing vs. invasion in phage lambda recombination. Genetics 147:961–977. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wilkins AS, Mistry J. 1974. Phage lambda’s generalized recombination system. Study of the intracellular DNA pool during lytic infection. Mol Gen Genet 129:275–293. [PubMed] 10.1007/BF00265693 [DOI] [PubMed] [Google Scholar]
- 119.Wackernagel W, Radding CM. 1973. Transfection by half-molecules and inverted molecules of λ DNA: requirement for exo and b-promoted recombination. Virology 52:425–432. [PubMed] 10.1016/0042-6822(73)90337-1 [DOI] [PubMed] [Google Scholar]
- 120.Fu J, Bian X, Hu S, Wang H, Huang F, Seibert PM, Plaza A, Xia L, Muller R, Stewart AF, Zhang Y. 2012. Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat Biotechnol 30:440–446. [PubMed] 10.1038/nbt.2183 [DOI] [PubMed] [Google Scholar]
- 121.van Kessel JC, Hatfull GF. 2008. Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: characterization of antimycobacterial drug targets. Mol Microbiol 67:1094–1107. [PubMed] 10.1111/j.1365-2958.2008.06109.x [DOI] [PubMed] [Google Scholar]
- 122.Kuzminov A. 2001. DNA replication meets genetic exchange: chromosomal damage and its repair by homologous recombination. Proc Natl Acad Sci USA 98:8461–8468. [PubMed] 10.1073/pnas.151260698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kraus E, Leung WY, Haber JE. 2001. Break-induced replication: a review and an example in budding yeast. Proc Natl Acad Sci USA 98:8255–8262. [PubMed] 10.1073/pnas.151008198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Llorente B, Smith CE, Symington LS. 2008. Break-induced replication: what is it and what is it for? Cell Cycle 7:859–864. [PubMed] 10.4161/cc.7.7.5613 [DOI] [PubMed] [Google Scholar]
- 125.Mosig G. 1998. Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu Rev Genet 32:379–413. [PubMed] 10.1146/annurev.genet.32.1.379 [DOI] [PubMed] [Google Scholar]
- 126.Mazin AV, Kuzminov AV, Dianov GL, Salganik RI. 1991. Mechanisms of deletion formation in Escherichia coli plasmids. II. Deletions mediated by short direct repeats. Mol Gen Genet 228:209–214. [PubMed] 10.1007/BF00282467 [DOI] [PubMed] [Google Scholar]
- 127.Kreuzer KN, Brister JR. 2010. Initiation of bacteriophage T4 DNA replication and replication fork dynamics: a review in the Virology Journal series on bacteriophage T4 and its relatives. Virol J 7:358. 10.1186/1743-422X-7-358. [PubMed] 10.1186/1743-422X-7-358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Better M, Freifelder D. 1983. Studies on the replication of Escherichia coli phage lambda DNA. I. The kinetics of DNA replication and requirements for the generation of rolling circles. Virology 126:168–182. [PubMed] 10.1016/0042-6822(83)90469-5 [DOI] [PubMed] [Google Scholar]
- 129.Poteete AR, Fenton AC. 1984. Lambda red-dependent growth and recombination of phage P22. Virology 134:161–167. [PubMed] 10.1016/0042-6822(84)90281-2 [DOI] [PubMed] [Google Scholar]
- 130.Sawitzke JA, Stahl FW. 1992. Phage lambda has an analog of Escherichia coli recO, recR and recF genes. Genetics 130:7–16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sawitzke JA, Stahl FW. 1994. The phage lambda orf gene encodes a trans-acting factor that suppresses Escherichia coli recO, recR, and recF mutations for recombination of lambda but not of E. coli. J Bacteriol 176:6730–6737. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sawitzke JA, Stahl FW. 1997. Roles for lambda Orf and Escherichia coli RecO, RecR and RecF in lambda recombination. Genetics 147:357–369. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tarkowski TA, Mooney D, Thomason LC, Stahl FW. 2002. Gene products encoded in the ninR region of phage lambda participate in Red-mediated recombination. Genes Cells 7:351–363. [PubMed] 10.1046/j.1365-2443.2002.00531.x [DOI] [PubMed] [Google Scholar]
- 134.Volkert MR, Hartke MA. 1984. Suppression of Escherichia coli recF mutations by recA-linked srfA mutations. J Bacteriol 157:498–506. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hegde SP, Qin MH, Li XH, Atkinson MA, Clark AJ, Rajagopalan M, Madiraju MV. 1996. Interactions of RecF protein with RecO, RecR, and single-stranded DNA binding proteins reveal roles for the RecF-RecO-RecR complex in DNA repair and recombination. Proc Natl Acad Sci USA 93:14468–14473. [PubMed] 10.1073/pnas.93.25.14468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Umezu K, Chi NW, Kolodner RD. 1993. Biochemical interaction of the Escherichia coli RecF, RecO, and RecR proteins with RecA protein and single-stranded DNA binding protein. Proc Natl Acad Sci USA 90:3875–3879. [PubMed] 10.1073/pnas.90.9.3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Umezu K, Kolodner RD. 1994. Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J Biol Chem 269:30005–30013. [PubMed] [PubMed] [Google Scholar]
- 138.Bork JM, Cox MM, Inman RB. 2001. The RecOR proteins modulate RecA protein function at 5′ ends of single-stranded DNA. EMBO J 20:7313–7322. [PubMed] 10.1093/emboj/20.24.7313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morimatsu K, Kowalczykowski SC. 2003. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol Cell 11:1337–1347. [PubMed] 10.1016/S1097-2765(03)00188-6 [DOI] [PubMed] [Google Scholar]
- 140.Cohen A, Clark AJ. 1986. Synthesis of linear plasmid multimers in Escherichia coli K-12. J Bacteriol 167:327–335. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kusano K, Nakayama K, Nakayama H. 1989. Plasmid-mediated lethality and plasmid multimer formation in an Escherichia coli recBC sbcBC mutant. Involvement of RecF recombination pathway genes. J Mol Biol 209:623–634. 10.1016/0022-2836(89)90000-4 [DOI] [PubMed] [Google Scholar]
- 142.Maxwell KL, Reed P, Zhang RG, Beasley S, Walmsley AR, Curtis FA, Joachimiak A, Edwards AM, Sharples GJ. 2005. Functional similarities between phage lambda Orf and Escherichia coli RecFOR in initiation of genetic exchange. Proc Natl Acad Sci USA 102:11260–11265. [PubMed] 10.1073/pnas.0503399102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Meyer RR, Laine PS. 1990. The single-stranded DNA-binding protein of Escherichia coli. Microbiol Rev 54:342–380. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Curtis FA, Reed P, Wilson LA, Bowers LY, Yeo RP, Sanderson JM, Walmsley AR, Sharples GJ. 2011. The C-terminus of the phage lambda Orf recombinase is involved in DNA binding. J Mol Recognit 24:333–340. [PubMed] 10.1002/jmr.1079 [DOI] [PubMed] [Google Scholar]
- 145.Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL. 2008. SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol 43:289–318. [PubMed] 10.1080/10409230802341296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Court D, Oppenheim AB. 1983. Phage lambda’s accessory genes, p 251–277. In Hendrix RW, Roberts JW, Stahl FW, Weisberg RA (ed), Lambda II. Cold Spring Harbor, Cold Spring Harbor, NY. [Google Scholar]
- 147.Hollifield WC, Kaplan EN, Huang HV. 1987. Efficient RecABC-dependent, homologous recombination between coliphage lambda and plasmids requires a phage ninR region gene. Mol Gen Genet 210:248–255. [PubMed] 10.1007/BF00325690 [DOI] [PubMed] [Google Scholar]
- 148.Mahdi AA, Sharples GJ, Mandal TN, Lloyd RG. 1996. Holliday junction resolvases encoded by homologous rusA genes in Escherichia coli K-12 and phage 82. J Mol Biol 257:561–573. [PubMed] 10.1006/jmbi.1996.0185 [DOI] [PubMed] [Google Scholar]
- 149.Poteete AR, Fenton AC, Wang HR. 2002. Recombination-promoting activity of the bacteriophage lambda Rap protein in Escherichia coli K-12. J Bacteriol 184:4626–4629. [PubMed] 10.1128/JB.184.16.4626-4629.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gutterson NI, Koshland DE, Jr. 1983. Replacement and amplification of bacterial genes with sequences altered in vitro. Proc Natl Acad Sci USA 80:4894–4898. [PubMed] 10.1073/pnas.80.16.4894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gay NJ. 1984. Construction and characterization of an Escherichia coli strain with a uncI mutation. J Bacteriol 158:820–825. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR. 1989. New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol 171:4617–4622. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Slater S, Maurer R. 1993. Simple phage-based system for generating allele replacements in Escherichia coli. J Bacteriol 175:4260–4262. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jasin M, Schimmel P. 1984. Deletion of an essential gene in Escherichia coli by site-specific recombination with linear DNA fragments. J Bacteriol 159:783–786. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Marinus MG, Carraway M, Frey AZ, Brown L, Arraj JA. 1983. Insertion mutations in the dam gene of Escherichia coli K-12. Molec Gen Genet 191:288–289. [PubMed] 10.1007/BF00327681 [DOI] [PubMed] [Google Scholar]
- 156.Russell CB, Thaler DS, Dahlquist FW. 1989. Chromosomal transformation of Escherichia coli recD strains with linearized plasmids. J Bacteriol 171:2609–2613. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Winans SC, Elledge SJ, Krueger JH, Walker GC. 1985. Site-directed insertion and deletion mutagenesis with cloned fragments in Escherichia coli. J Bacteriol 161:1219–1221. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Clark AJ, Sandler SJ. 1994. Homologous genetic recombination: the pieces begin to fall into place. Crit Rev Microbiol 20:125–142. [PubMed] 10.3109/10408419409113552 [DOI] [PubMed] [Google Scholar]
- 159.Barbour SD, Nagaishi H, Templin A, Clark AJ. 1970. Biochemical and genetic studies of recombination proficiency in Escherichia coli. II. Rec+ revertants caused by indirect suppression of rec- mutations. Proc Natl Acad Sci USA 67:128–135. [PubMed] 10.1073/pnas.67.1.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kaiser K, Murray NE. 1979. Physical characterisation of the “Rac prophage” in E. coli K12. Mol Gen Genet 175:159–174. [PubMed] 10.1007/BF00425532 [DOI] [PubMed] [Google Scholar]
- 161.Low B. 1973. Restoration by the rac locus of recombinant forming ability in recB - and recC - merozygotes of Escherichia coli K-12. Mol Gen Genet 122:119–130. [PubMed] 10.1007/BF00435185 [DOI] [PubMed] [Google Scholar]
- 162.Clark AJ, Sharma V, Brenowitz S, Chu CC, Sandler S, Satin L, Templin A, Berger I, Cohen A. 1993. Genetic and molecular analyses of the C-terminal region of the recE gene from the Rac prophage of Escherichia coli K-12 reveal the recT gene. J Bacteriol 175:7673–7682. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kushner SR, Nagaishi H, Clark AJ. 1974. Isolation of exonuclease VIII: the enzyme associated with sbcA indirect suppressor. Proc Natl Acad Sci USA 71:3593–3597. [PubMed] 10.1073/pnas.71.9.3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kushner SR, Nagaishi H, Clark AJ. 1972. Indirect suppression of recB and recC mutations by exonuclease I deficiency. Proc Natl Acad Sci USA 69:1366–1370. [PubMed] 10.1073/pnas.69.6.1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Templin A, Kushner SR, Clark AJ. 1972. Genetic analysis of mutations indirectly suppressing recB and recC mutations. Genetics 72:105–115. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 166.Lloyd RG, Buckman C. 1985. Identification and genetic analysis of sbcC mutations in commonly used recBC sbcB strains of Escherichia coli K-12. J Bacteriol 164:836–844. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Naom IS, Morton SJ, Leach DRF, Lloyd RG. 1989. Molecular organization of sbcC , a gene that affects genetic recombination and the viability of DNA palindromes in Escherichia coli K-12. Nucl Acids Res 17:8033–8045. [PubMed] 10.1093/nar/17.20.8033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Connelly JC, Leach DR. 1996. The sbcC and sbcD genes of Escherichia coli encode a nuclease involved in palindrome inviability and genetic recombination. Genes Cells 1:285–291. 10.1046/j.1365-2443.1996.23024.x [DOI] [PubMed] [Google Scholar]
- 169.Houri Z, Clark AJ. 1973. Genetic analysis of the recF pathway to genetic recombination in Escherichia coli K12: isolation and characterization of mutants. J Mol Biol 80:327–344. [PubMed] 10.1016/0022-2836(73)90176-9 [DOI] [PubMed] [Google Scholar]
- 170.Smith KC, Wang TV, Sharma RC. 1987. recA-dependent DNA repair in UV-irradiated Escherichia coli. J Photochem Photobiol B 1:1–11. [PubMed] 10.1016/1011-1344(87)80002-7 [DOI] [PubMed] [Google Scholar]
- 171.Kolodner R, Fishel RA, Howard M. 1985. Genetic recombination of bacterial plasmid DNA: effect of RecF pathway mutations on plasmid recombination in Escherichia coli. J Bacteriol 163:1060–1066. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Courcelle J, Hanawalt PC. 2001. Participation of recombination proteins in rescue of arrested replication forks in UV-irradiated Escherichia coli need not involve recombination. Proc Natl Acad Sci USA 98:8196–8202. [PubMed] 10.1073/pnas.121008898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cox MM. 2001. Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu Rev Genet 35:53–82. [PubMed] 10.1146/annurev.genet.35.102401.090016 [DOI] [PubMed] [Google Scholar]
- 174.Rangarajan S, Woodgate R, Goodman MF. 2002. Replication restart in UV-irradiated Escherichia coli involving pols II, III, V, PriA, RecA and RecFOR proteins. Mol Microbiol 43:617–628. [PubMed] 10.1046/j.1365-2958.2002.02747.x [DOI] [PubMed] [Google Scholar]
- 175.Zahradka K, Simic S, Buljubasic M, Petranovic M, Dermic D, Zahradka D. 2006. sbcB15 and DeltasbcB mutations activate two types of recf recombination pathways in Escherichia coli. J Bacteriol 188:7562–7571. [PubMed] 10.1128/JB.00613-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Thoms B, Borchers I, Wackernagel W. 2008. Effects of single-strand DNases ExoI, RecJ, ExoVII, and SbcCD on homologous recombination of recBCD+ strains of Escherichia coli and roles of SbcB15 and XonA2 ExoI mutant enzymes. J Bacteriol 190:179–192. [PubMed] 10.1128/JB.01052-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Shevell DE, Abou-Zamzam AM, Demple B, Walker GC. 1988. Construction of an Escherichia coli K-12 ada deletion by gene replacement in a recD strain reveals a second methyltransferase that repairs alkylated DNA. J Bacteriol 170:3294–3296. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Amundsen SK, Taylor AF, Chaudhury AM, Smith GR. 1986. recD: the gene for an essential third subunit of exonuclease V. Proc Natl Acad Sci USA 83:5558–5562. [PubMed] 10.1073/pnas.83.15.5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Botstein D, Matz MJ. 1970. A recombination function essential to the growth of bacteriophage P22. J Mol Biol 54:417–440. [PubMed] 10.1016/0022-2836(70)90119-1 [DOI] [PubMed] [Google Scholar]
- 180.Hill SA, Stahl MM, Stahl FW. 1997. Single-strand DNA intermediates in phage λ’s Red recombination pathway. Proc Natl Acad Sci USA 94:2951–2956. [PubMed] 10.1073/pnas.94.7.2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang Y, Buchholz F, Muyrers JP, Stewart AF. 1998. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet 20:123–128. [PubMed] 10.1038/2417 [DOI] [PubMed] [Google Scholar]
- 182.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645. [PubMed] 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Murphy KC, Campellone KG. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol 4:11. 10.1186/1471-2199-4-11 [PubMed] 10.1186/1471-2199-4-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA 97:5978–5983. [PubMed] 10.1073/pnas.100127597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullen C. 1993. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acid Res 21:3329–3330. [PubMed] 10.1093/nar/21.14.3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lorenz MC, Muir RS, Lim E, McElver J, Weber SC, Heitman J. 1995. Gene disruption with PCR products in Saccharomyces cerevisiae. Gene 158:113–117. [PubMed] 10.1016/0378-1119(95)00144-U [DOI] [PubMed] [Google Scholar]
- 187.Wach A, Brachat A, Pohlmann R, Philippsen P. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793–1808. [PubMed] 10.1002/yea.320101310 [DOI] [PubMed] [Google Scholar]
- 188.Ellis HM, Yu D, DiTizio T, Court DL. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci USA 98:6742–6746. [PubMed] 10.1073/pnas.121164898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Copeland NG, Jenkins NA, Court DL. 2001. Recombineering: a powerful new tool for mouse functional genomics. Nat Rev Genet 2:769–779. [PubMed] 10.1038/35093556 [DOI] [PubMed] [Google Scholar]
- 190.Datta S, Costantino N, Court DL. 2006. A set of recombineering plasmids for gram-negative bacteria. Gene 379:109–115. [PubMed] 10.1016/j.gene.2006.04.018 [DOI] [PubMed] [Google Scholar]
- 191.Gay P, LeCoq D, Steinmetz M, Ferrari E, Hoch J. 1983. Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. J Bacteriol 153:1424–1431. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chaveroche MK, Ghigo JM, d’Enfert C. 2000. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res 28:e97. 10.1093/nar/28.22.e97 [PubMed] 10.1093/nar/28.22.e97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. 10.1038/msb4100050 [PubMed] 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. 2009. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460:894–898. [PubMed] 10.1038/nature08187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wang HH, Xu G, Vonner AJ, Church G. 2011. Modified bases enable high-efficiency oligonucleotide-mediated allelic replacement via mismatch repair evasion. Nucleic Acids Res 39:7336–7347. [PubMed] 10.1093/nar/gkr183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mythili E, Kumar KA, Muniyappa K. 1996. Characterization of the DNA-binding domain of beta protein, a component of phage lambda red-pathway, by UV catalyzed cross-linking. Gene 182:81–87. [PubMed] 10.1016/S0378-1119(96)00518-5 [DOI] [PubMed] [Google Scholar]
- 197.Moerschell RP, Tsunasawa S, Sherman F. 1988. Transformation of yeast with synthetic oligonucleotides. Proc Natl Acad Sci USA 85:524–528. [PubMed] 10.1073/pnas.85.2.524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yamamoto T, Moerschell RP, Wakem LP, Komar-Panicucci S, Sherman F. 1992. Strand-specificity in the transformation of yeast with synthetic oligonucleotides. Genetics 131:811–819. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Liu L, Rice MC, Drury M, Cheng S, Gamper H, Kmiec EB. 2002. Strand bias in targeted gene repair is influenced by transcriptional activity. Mol Cell Biol 22:3852–3863. [PubMed] 10.1128/MCB.22.11.3852-3863.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Parekh-Olmedo H, Kmiec EB. 2007. Progress and prospects: targeted gene alteration (TGA). Gene Ther 14:1675–1680. [PubMed] 10.1038/sj.gt.3303053 [DOI] [PubMed] [Google Scholar]
- 201.van Kessel JC, Marinelli LJ, Hatfull GF. 2008. Recombineering mycobacteria and their phages. Nat Rev Microbiol 6:851–857. [PubMed] 10.1038/nrmicro2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cha RS, Zarbl H, Keohavong P, Thilly WG. 1992. Mismatch amplification mutation assay (MAMA): application to the c-H-ras gene. PCR Methods Appl 2:14–20. [PubMed] 10.1101/gr.2.1.14 [DOI] [PubMed] [Google Scholar]
- 203.Swaminathan S, Ellis HM, Waters LS, Yu D, Lee EC, Court DL, Sharan SK. 2001. Rapid engineering of bacterial artificial chromosomes using oligonucleotides. Genesis 29:14–21. [PubMed] 10.1002/1526-968X(200101)29:1<14::AID-GENE1001>3.0.CO;2-X [DOI] [PubMed] [Google Scholar]
- 204.Lobner-Olesen A, Skovgaard O, Marinus MG. 2005. Dam methylation: coordinating cellular processes. Curr Opin Microbiol 8:154–160. [PubMed] 10.1016/j.mib.2005.02.009 [DOI] [PubMed] [Google Scholar]
- 205.Modrich P. 1989. Methyl-directed DNA mismatch correction. J Biol Chem 264:6597–6600. [PubMed] [PubMed] [Google Scholar]
- 206.Costantino N, Court DL. 2003. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc Natl Acad Sci USA 100:15748–15753. [PubMed] 10.1073/pnas.2434959100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Parker BO, Marinus MG. 1992. Repair of DNA heteroduplexes containing small heterologous sequences in Escherichia coli. Proc Natl Acad Sci USA 89:1730–1734. [PubMed] 10.1073/pnas.89.5.1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yang Y, Sharan SK. 2003. A simple two-step, ‘hit and fix’ method to generate subtle mutations in BACs using short denatured PCR fragments. Nucleic Acids Res 31:e80. 10.1093/nar/gng080. [PubMed] 10.1093/nar/gng080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Li XT, Thomason LC, Sawitzke JA, Costantino N, Court DL. 2013. Bacterial DNA polymerases participate in oligonucleotide recombination. Mol Microbiol 88:906–920. [PubMed] 10.1111/mmi.12231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Poteete AR. 2013. Involvement of DNA replication proteins in phage lambda red-mediated homologous recombination. PLoS One 8:e67440. 10.1371/journal.pone.0067440. [PubMed] 10.1371/journal.pone.0067440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kuempel PL, Veomett GE. 1970. A possible function of DNA polymerase in chromosome replication. Biochem Biophys Res Commun 41:973–980. [PubMed] 10.1016/0006-291X(70)90180-4 [DOI] [PubMed] [Google Scholar]
- 212.Lehman IR, Uyemura DG. 1976. DNA polymerase I: essential replication enzyme. Science 193:963–969. [PubMed] 10.1126/science.781842 [DOI] [PubMed] [Google Scholar]
- 213.Okazaki R, Arisawa M, Sugino A. 1971. Slow joining of newly replicated DNA chains in DNA polymerase I-deficient Escherichia coli mutants. Proc Natl Acad Sci USA 68:2954–2957. [PubMed] 10.1073/pnas.68.12.2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Studwell PS, O’Donnell M. 1990. Processive replication is contingent on the exonuclease subunit of DNA polymerase III holoenzyme. J Biol Chem 265:1171–1178. [PubMed] [PubMed] [Google Scholar]
- 215.Sawitzke JA, Costantino N, Li XT, Thomason LC, Bubunenko M, Court C, Court DL. 2011. Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J Mol Biol 407:45–59. [PubMed] 10.1016/j.jmb.2011.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Swingle B, Markel E, Costantino N, Bubunenko MG, Cartinhour S, Court DL. 2009. Oligonucleotide recombination in Gram-negative bacteria. Mol Microbiol 75:138–148. [PubMed] 10.1111/j.1365-2958.2009.06976.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dutra BE, Sutera VA, Jr, Lovett ST. 2007. RecA-independent recombination is efficient but limited by exonucleases. Proc Natl Acad Sci USA 104:216–221. [PubMed] 10.1073/pnas.0608293104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Bryan A, Swanson MS. 2011. Oligonucleotides stimulate genomic alterations of Legionella pneumophila. Mol Microbiol 80:231–247. [PubMed] 10.1111/j.1365-2958.2011.07573.x [DOI] [PubMed] [Google Scholar]
- 219.Branda CS, Dymecki SM. 2004. Talking about a revolution: the impact of site-specific recombinases on genetic analyses in mice. Dev Cell 6:7–28. [PubMed] 10.1016/S1534-5807(03)00399-X [DOI] [PubMed] [Google Scholar]
- 220.Gilbertson L. 2003. Cre-lox recombination: Cre-ative tools for plant biotechnology. Trends Biotechnol 21:550–555. [PubMed] 10.1016/j.tibtech.2003.09.011 [DOI] [PubMed] [Google Scholar]
- 221.Huang LC, Wood EA, Cox MM. 1991. A bacterial model system for chromosomal targeting. Nucleic Acids Res 19:443–448. [PubMed] 10.1093/nar/19.3.443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Huang LC, Wood EA, Cox MM. 1997. Convenient and reversible site-specific targeting of exogenous DNA into a bacterial chromosome by use of the FLP recombinase: the FLIRT system. J Bacteriol 179:6076–6083. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Schnutgen F, Stewart AF, von Melchner H, Anastassiadis K. 2006. Engineering embryonic stem cells with recombinase systems. Methods Enzymol 420:100–136. [PubMed] 10.1016/S0076-6879(06)20007-7 [DOI] [PubMed] [Google Scholar]
- 224.Schweizer HP. 2003. Applications of the Saccharomyces cerevisiae Flp-FRT system in bacterial genetics. J Mol Microbiol Biotechnol 5:67–77. [PubMed] 10.1159/000069976 [DOI] [PubMed] [Google Scholar]
- 225.Siegal ML, Hartl DL. 2000. Application of Cre/loxP in Drosophila. Site-specific recombination and transgene coplacement. Methods Mol Biol 136:487–495. [PubMed] 10.1385/1-59259-065-9:487 [DOI] [PubMed] [Google Scholar]
- 226.Nagy A. 2000. Cre recombinase: the universal reagent for genome tailoring. Genesis 26:99–109. [PubMed] 10.1002/(SICI)1526-968X(200002)26:2<99::AID-GENE1>3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- 227.Li XT, Thomason LC, Sawitzke JA, Costantino N, Court DL. 2013. Positive and negative selection using the tetA-sacB cassette: recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res 41:e204. 10.1093/nar/gkt1075. [PubMed] 10.1093/nar/gkt1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wong QN, Ng VC, Lin MC, Kung HF, Chan D, Huang JD. 2005. Efficient and seamless DNA recombineering using a thymidylate synthase A selection system in Escherichia coli. Nucleic Acids Res 33:e59. 10.1093/nar/gni059. [PubMed] 10.1093/nar/gni059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. 10.1093/nar/gni035. [PubMed] 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hall RN, Meers J, Fowler E, Mahony T. 2012. Back to BAC: the use of infectious clone technologies for viral mutagenesis. Viruses 4:211–235. [PubMed] 10.3390/v4020211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Barkan D, Stallings CL, Glickman MS. 2011. An improved counterselectable marker system for mycobacterial recombination using galK and 2-deoxy-galactose. Gene 470:31–36. [PubMed] 10.1016/j.gene.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Heermann R, Zeppenfeld T, Jung K. 2008. Simple generation of site-directed point mutations in the Escherichia coli chromosome using Red(R)/ET(R) recombination. Microb Cell Fact 7:14. 10.1186/1475-2859-7-14. [PubMed] 10.1186/1475-2859-7-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Stavropoulos TA, Strathdee CA. 2001. Synergy between tetA and rpsL provides high-stringency positive and negative selection in bacterial artificial chromosome vectors. Genomics 72:99–104. [PubMed] 10.1006/geno.2000.6481 [DOI] [PubMed] [Google Scholar]
- 234.Nagel de Zwaig R, Luria SE. 1967. Genetics and physiology of colicin-tolerant mutants of Escherichia coli. J Bacteriol 94:1112–1123. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.DeVito JA. 2008. Recombineering with tolC as a selectable/counter-selectable marker: remodeling the rRNA operons of Escherichia coli. Nucleic Acids Res 36:e4. 10.1093/nar/gkm1084. [PubMed] 10.1093/nar/gkm1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kast P. 1994. pKSS--a second-generation general purpose cloning vector for efficient positive selection of recombinant clones. Gene 138:109–114. [PubMed] 10.1016/0378-1119(94)90790-0 [DOI] [PubMed] [Google Scholar]
- 237.Kast P, Hennecke H. 1991. Amino acid substrate specificity of Escherichia coli phenylalanyl-tRNA synthetase altered by distinct mutations. J Mol Biol 222:99–124. [PubMed] 10.1016/0022-2836(91)90740-W [DOI] [PubMed] [Google Scholar]
- 238.Li MZ, Elledge SJ. 2005. MAGIC, an in vivo genetic method for the rapid construction of recombinant DNA molecules. Nat Genet 37:311–319. [PubMed] 10.1038/ng1505 [DOI] [PubMed] [Google Scholar]
- 239.Wang H, Bian X, Xia L, Ding X, Muller R, Zhang Y, Fu J, Stewart AF. 2013. Improved seamless mutagenesis by recombineering using ccdB for counterselection. Nucleic Acids Res 42:e37. 10.1093/nar/gkt1339. [PubMed] 10.1093/nar/gkt1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bird AW, Erler A, Fu J, Heriche JK, Maresca M, Zhang Y, Hyman AA, Stewart AF. 2011. High-efficiency counterselection recombineering for site-directed mutagenesis in bacterial artificial chromosomes. Nat Methods 9:103–109. [PubMed] 10.1038/nmeth.1803 [DOI] [PubMed] [Google Scholar]
- 241.Bonner M, Kmiec EB. 2009. DNA breakage associated with targeted gene alteration directed by DNA oligonucleotides. Mutat Res 669:85–94. [PubMed] 10.1016/j.mrfmmm.2009.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Liu P, Jenkins NA, Copeland NG. 2003. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13:476–484. [PubMed] 10.1101/gr.749203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.van Kessel JC, Hatfull GF. 2007. Recombineering in Mycobacterium tuberculosis. Nat Methods 4:147–152. [PubMed] 10.1038/nmeth996 [DOI] [PubMed] [Google Scholar]
- 244.Yu BJ, Sung BH, Koob MD, Lee CH, Lee JH, Lee WS, Kim MS, Kim SC. 2002. Minimization of the Escherichia coli genome using a Tn5-targeted Cre/loxP excision system. Nat Biotechnol 20:1018–1023. [PubMed] 10.1038/nbt740 [DOI] [PubMed] [Google Scholar]
- 245.Fukiya S, Mizoguchi H, Mori H. 2004. An improved method for deleting large regions of Escherichia coli K-12 chromosome using a combination of Cre/loxP and lambda Red. FEMS Microbiol Lett 234:325–331. [PubMed] [DOI] [PubMed] [Google Scholar]
- 246.Murphy KC, Campellone KG, Poteete AR. 2000. PCR-mediated gene replacement in Escherichia coli. Gene 246:321–330. [PubMed] 10.1016/S0378-1119(00)00071-8 [DOI] [PubMed] [Google Scholar]
- 247.Ried JL, Collmer A. 1987. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in Gram-negative bacteria by marker exchange-eviction mutagenesis. Gene 57:239–246. [PubMed] 10.1016/0378-1119(87)90127-2 [DOI] [PubMed] [Google Scholar]
- 248.Barnes WM. 1994. PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc Natl Acad Sci USA 91:2216–2220. [PubMed] 10.1073/pnas.91.6.2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Cheng S, Fockler C, Barnes WM, Higuchi R. 1994. Effective amplification of long targets from cloned inserts and human genomic DNA. Proc Natl Acad Sci USA 91:5695–5699. [PubMed] 10.1073/pnas.91.12.5695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Posfai G, Kolisnychenko V, Bereczki Z, Blattner FR. 1999. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res 27:4409–4415. [PubMed] 10.1093/nar/27.22.4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kolisnychenko V, Plunkett G, 3rd, Herring CD, Feher T, Posfai J, Blattner FR, Posfai G. 2002. Engineering a reduced Escherichia coli genome. Genome Res 12:640–647. [PubMed] 10.1101/gr.217202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Monteilhet C, Perrin A, Thierry A, Colleaux L, Dujon B. 1990. Purification and characterization of the in vitro activity of I-Sce I, a novel and highly specific endonuclease encoded by a group I intron. Nucleic Acids Res 18:1407–1413. [PubMed] 10.1093/nar/18.6.1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Herring CD, Glasner JD, Blattner FR. 2003. Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene 311:153–163. [PubMed] 10.1016/S0378-1119(03)00585-7 [DOI] [PubMed] [Google Scholar]
- 254.Herring CD, Blattner FR. 2004. Conditional lethal amber mutations in essential Escherichia coli genes. J Bacteriol 186:2673–2681. 10.1128/JB.186.9.2673-2681.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Rivero-Muller A, Lajic S, Huhtaniemi I. 2007. Assisted large fragment insertion by Red/ET-recombination (ALFIRE)--an alternative and enhanced method for large fragment recombineering. Nucleic Acids Res 35:e78. 10.1093/nar/gkm250. 10.1093/nar/gkm250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. [PubMed] 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 257.Oliner JD, Kinzler KW, Vogelstein B. 1993. In vivo cloning of PCR products in E. coli. Nucleic Acids Res 21:5192–5197. [PubMed] 10.1093/nar/21.22.5192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Bubeck P, Winkler M, Bautsch W. 1993. Rapid cloning by homologous recombination in vivo. Nucleic Acids Res 21:3601–3602. [PubMed] 10.1093/nar/21.15.3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zhang Y, Muyrers JP, Testa G, Stewart AF. 2000. DNA cloning by homologous recombination in Escherichia coli. Nat Biotechnol 18:1314–1317. [PubMed] 10.1038/82449 [DOI] [PubMed] [Google Scholar]
- 260.Kingsbury DT, Helinski DR. 1970. DNA polymerase as a requirement for the maintenance of the bacterial plasmid colicinogenic factor E1. Biochem Biophys Res Commun 41:1538–1544. [PubMed] 10.1016/0006-291X(70)90562-0 [DOI] [PubMed] [Google Scholar]
- 261.Thomason LC, Costantino N, Shaw DV, Court DL. 2007. Multicopy plasmid modification with phage lambda Red recombineering. Plasmid 58:148–158. [PubMed] 10.1016/j.plasmid.2007.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Poteete AR, Fenton AC, Murphy KC. 1988. Modulation of Escherichia coli RecBCD activity by the bacteriophage lambda Gam and P22 Abc functions. J Bacteriol 170:2012–2021. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Silberstein Z, Maor S, Berger I, Cohen A. 1990. Lambda Red-mediated synthesis of plasmid linear multimers in Escherichia coli K12. Mol Gen Genet 223:496–507. [PubMed] 10.1007/BF00264459 [DOI] [PubMed] [Google Scholar]
- 264.Kuzminov A. 1996. Mutant fixation via plasmid dimerization and its relation to human diseases. Trends Genet 12:246–249. [PubMed] 10.1016/0168-9525(96)30058-9 [DOI] [PubMed] [Google Scholar]
- 265.Yosef I, Bloushtain N, Shapira M, Qimron U. 2004. Restoration of gene function by homologous recombination: from PCR to gene expression in one step. Appl Environ Microbiol 70:7156–7160. [PubMed] 10.1128/AEM.70.12.7156-7160.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Haldimann A, Wanner BL. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393. [PubMed] 10.1128/JB.183.21.6384-6393.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Bao Y, Lies DP, Fu H, Roberts GP. 1991. An improved Tn7-based system for the single-copy insertion of cloned genes into chromosomes of gram-negative bacteria. Gene 109:167–168. [PubMed] 10.1016/0378-1119(91)90604-A [DOI] [PubMed] [Google Scholar]
- 268.Choi KH, Schweizer HP. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5:30. 10.1186/1471-2180-5-30 [PubMed] 10.1186/1471-2180-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Koch B, Jensen LE, Nybroe O. 2001. A panel of Tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into Gram-negative bacteria at a neutral chromosomal site. J Microbiol Methods 45:187–195. [PubMed] 10.1016/S0167-7012(01)00246-9 [DOI] [PubMed] [Google Scholar]
- 270.McKenzie GJ, Craig NL. 2006. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol 6:39. 10.1186/1471-2180-6-39. 10.1186/1471-2180-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Rong R, Slupska MM, Chiang JH, Miller JH. 2004. Engineering large fragment insertions into the chromosome of Escherichia coli. Gene 336:73–80. [PubMed] 10.1016/j.gene.2004.02.054 [DOI] [PubMed] [Google Scholar]
- 272.Kuhlman TE, Cox EC. 2009. Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res 38:e92. 10.1093/nar/gkp1193. [PubMed] 10.1093/nar/gkp1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Maresca M, Erler A, Fu J, Friedrich A, Zhang Y, Stewart AF. 2010. Single-stranded heteroduplex intermediates in lambda Red homologous recombination. BMC Mol Biol 11:54. 10.1186/1471-2199-11-54. [PubMed] 10.1186/1471-2199-11-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Galitski T, Roth JR. 1997. Pathways for homologous recombination between chromosomal direct repeats in Salmonella typhimurium. Genetics 146:751–767. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Poteete AR, Fenton AC, Nadkarni A. 2004. Chromosomal duplications and cointegrates generated by the bacteriophage lamdba Red system in Escherichia coli K-12. BMC Mol Biol 5:22. 10.1186/1471-2199-5-22. [PubMed] 10.1186/1471-2199-5-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kuzminov A. 1995. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol 16:373–384. [PubMed] 10.1111/j.1365-2958.1995.tb02403.x [DOI] [PubMed] [Google Scholar]
- 277.Slechta ES, Bunny KL, Kugelberg E, Kofoid E, Andersson DI, Roth JR. 2003. Adaptive mutation: general mutagenesis is not a programmed response to stress but results from rare coamplification of dinB with lac. Proc Natl Acad Sci USA 100:12847–12852. [PubMed] 10.1073/pnas.1735464100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Poteete AR. 2009. Expansion of a chromosomal repeat in Escherichia coli: roles of replication, repair, and recombination functions. BMC Mol Biol 10:14. 10.1186/1471-2199-10-14. [PubMed] 10.1186/1471-2199-10-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Hand NJ, Silhavy TJ. 2000. A practical guide to the construction and use of lac fusions in Escherichia coli. Methods Enzymol 326:11–35. [PubMed] 10.1016/S0076-6879(00)26044-8 [DOI] [PubMed] [Google Scholar]
- 280.Platt R, Drescher C, Park SK, Phillips GJ. 2000. Genetic system for reversible integration of DNA constructs and lacZ gene fusions into the Escherichia coli chromosome. Plasmid 43:12–23. [PubMed] 10.1006/plas.1999.1433 [DOI] [PubMed] [Google Scholar]
- 281.Silhavy TJ, Beckwith JR. 1985. Uses of lac fusions for the study of biological problems. Microbiol Rev 49:398–418. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Simons RW, Houman F, Kleckner N. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53:85–96. [PubMed] 10.1016/0378-1119(87)90095-3 [DOI] [PubMed] [Google Scholar]
- 283.Slauch JM, Silhavy TJ. 1991. Genetic fusions as experimental tools. Methods Enzymol 204:213–248. [PubMed] 10.1016/0076-6879(91)04011-C [DOI] [PubMed] [Google Scholar]
- 284.Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. 2001. Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci USA 98:15264–15269. [PubMed] 10.1073/pnas.261348198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Ellermeier CD, Janakiraman A, Slauch JM. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161. [PubMed] 10.1016/S0378-1119(02)00551-6 [DOI] [PubMed] [Google Scholar]
- 286.Gerlach RG, Holzer SU, Jackel D, Hensel M. 2007. Rapid engineering of bacterial reporter gene fusions by using Red recombination. Appl Environ Microbiol 73:4234–4242. [PubMed] 10.1128/AEM.00509-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Dolphin CT, Hope IA. 2006. Caenorhabditis elegans reporter fusion genes generated by seamless modification of large genomic DNA clones. Nucleic Acids Res 34:e72. 10.1093/nar/gkl352. [PubMed] 10.1093/nar/gkl352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Westenberg M, Bamps S, Soedling H, Hope IA, Dolphin CT. 2010. Escherichia coli MW005: lambda Red-mediated recombineering and copy-number induction of oriV-equipped constructs in a single host. BMC Biotechnol 10:27. 10.1186/1472-6750-10-27. [PubMed] 10.1186/1472-6750-10-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Hirani N, Westenberg M, Gami MS, Davis P, Hope IA, Dolphin CT. 2013. A simplified counter-selection recombineering protocol for creating fluorescent protein reporter constructs directly from C. elegans fosmid genomic clones. BMC Biotechnol 13:1. 10.1186/1472-6750-13-1. 10.1186/1472-6750-13-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Hatfull GF, Cresawn SG, Hendrix RW. 2008. Comparative genomics of the mycobacteriophages: insights into bacteriophage evolution. Res Microbiol 159:332–339. [PubMed] 10.1016/j.resmic.2008.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Norrby E. 2008. Nobel Prizes and the emerging virus concept. Arch Virol 153:1109–1123. [PubMed] 10.1007/s00705-008-0088-8 [DOI] [PubMed] [Google Scholar]
- 292.Rees CE, Dodd CE. 2006. Phage for rapid detection and control of bacterial pathogens in food. Adv Appl Microbiol 59:159–186. [PubMed] 10.1016/S0065-2164(06)59006-9 [DOI] [PubMed] [Google Scholar]
- 293.Oppenheim AB, Rattray AJ, Bubunenko M, Thomason LC, Court DL. 2004. In vivo recombineering of bacteriophage lambda by PCR fragments and single-strand oligonucleotides. Virology 319:185–189. [PubMed] 10.1016/j.virol.2003.11.007 [DOI] [PubMed] [Google Scholar]
- 294.Marinelli LJ, Piuri M, Swigonova Z, Balachandran A, Oldfield LM, van Kessel JC, Hatfull GF. 2008. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One 3:e3957. 10.1371/journal.pone.0003957. [PubMed] 10.1371/journal.pone.0003957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.De Lay NR, Cronan JE. 2006. Gene-specific random mutagenesis of Escherichia coli in vivo: isolation of temperature-sensitive mutations in the acyl carrier protein of fatty acid synthesis. J Bacteriol 188:287–296. [PubMed] 10.1128/JB.188.1.287-296.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Cadwell RC, Joyce GF. 1992. Randomization of genes by PCR mutagenesis. PCR Methods Appl 2:28–33. [PubMed] 10.1101/gr.2.1.28 [DOI] [PubMed] [Google Scholar]
- 297.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. [PubMed] 10.1016/0378-1119(89)90359-4 [DOI] [PubMed] [Google Scholar]
- 298.Posfai G, Plunkett G, 3rd, Feher T, Frisch D, Keil GM, Umenhoffer K, Kolisnychenko V, Stahl B, Sharma SS, de Arruda M, Burland V, Harcum SW, Blattner FR. 2006. Emergent properties of reduced-genome Escherichia coli. Science 312:1044–1046. [PubMed] 10.1126/science.1126439 [DOI] [PubMed] [Google Scholar]
- 299.Csorgo B, Feher T, Timar E, Blattner FR, Posfai G. 2012. Low-mutation-rate, reduced-genome Escherichia coli: an improved host for faithful maintenance of engineered genetic constructs. Microb Cell Fact 11:11. 10.1186/1475-2859-11-11. [PubMed] 10.1186/1475-2859-11-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Mizoguchi H, Sawano Y, Kato J, Mori H. 2008. Superpositioning of deletions promotes growth of Escherichia coli with a reduced genome. DNA Res 15:277–284. [PubMed] 10.1093/dnares/dsn019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Jensen PR, Hammer K. 1998. Artificial promoters for metabolic optimization. Biotechnol Bioeng 58:191–195. [PubMed] 10.1002/(SICI)1097-0290(19980420)58:2/3<191::AID-BIT11>3.0.CO;2-G [DOI] [PubMed] [Google Scholar]
- 302.Jensen PR, Hammer K. 1998. The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl Environ Microbiol 64:82–87. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. 2005. Tuning genetic control through promoter engineering. Proc Natl Acad Sci USA 102:12678–12683. [PubMed] 10.1073/pnas.0504604102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Solem C, Jensen PR. 2002. Modulation of gene expression made easy. Appl Environ Microbiol 68:2397–2403. [PubMed] 10.1128/AEM.68.5.2397-2403.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Meynial-Salles I, Cervin MA, Soucaille P. 2005. New tool for metabolic pathway engineering in Escherichia coli: one-step method to modulate expression of chromosomal genes. Appl Environ Microbiol 71:2140–2144. [PubMed] 10.1128/AEM.71.4.2140-2144.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. [PubMed] 10.1038/nbt.2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Thaler DS, Stahl MM, Stahl FW. 1987. Double-chain-cut sites are recombination hotspots in the Red pathway of phage lambda. J Mol Biol 195:75–87. [PubMed] 10.1016/0022-2836(87)90328-7 [DOI] [PubMed] [Google Scholar]
- 308.Pyne ME, Moo-Young M, Chung DA, Chou CP. 2015. Coupling the CRISPR/Cas9 system with lambda red recombineering enables simplified chromosomal gene replacement in Escherichia coli. Appl Environ Microbiol 81:5103–5114. [PubMed] 10.1128/AEM.01248-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. 2015. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol 81:2506–2514. [PubMed] 10.1128/AEM.04023-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Yamamoto N, Nakahigashi K, Nakamichi T, Yoshino M, Takai Y, Touda Y, Furubayashi A, Kinjyo S, Dose H, Hasegawa M, Datsenko KA, Nakayashiki T, Tomita M, Wanner BL, Mori H. 2009. Update on the Keio collection of Escherichia coli single-gene deletion mutants. Mol Syst Biol 5:335. 10.1038/msb.2009.92. [PubMed] 10.1038/msb.2009.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Shoemaker DD, Lashkari DA, Morris D, Mittmann M, Davis RW. 1996. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat Genet 14:450–456. [PubMed] 10.1038/ng1296-450 [DOI] [PubMed] [Google Scholar]
- 312.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906. [PubMed] 10.1126/science.285.5429.901 [DOI] [PubMed] [Google Scholar]
- 313.Typas A, Nichols RJ, Siegele DA, Shales M, Collins SR, Lim B, Braberg H, Yamamoto N, Takeuchi R, Wanner BL, Mori H, Weissman JS, Krogan NJ, Gross CA. 2008. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Methods 5:781–787. [PubMed] 10.1038/nmeth.1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, Davey M, Parkinson J, Greenblatt J, Emili A. 2005. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433:531–537. [PubMed] 10.1038/nature03239 [DOI] [PubMed] [Google Scholar]
- 315.Hu P, Janga SC, Babu M, Diaz-Mejia JJ, Butland G, Yang W, Pogoutse O, Guo X, Phanse S, Wong P, Chandran S, Christopoulos C, Nazarians-Armavil A, Nasseri NK, Musso G, Ali M, Nazemof N, Eroukova V, Golshani A, Paccanaro A, Greenblatt JF, Moreno-Hagelsieb G, Emili A. 2009. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol 7:e96. 10.1371/journal.pbio.1000096. 10.1371/journal.pbio.1000096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Watt RM, Wang J, Leong M, Kung HF, Cheah KS, Liu D, Danchin A, Huang JD. 2007. Visualizing the proteome of Escherichia coli: an efficient and versatile method for labeling chromosomal coding DNA sequences (CDSs) with fluorescent protein genes. Nucleic Acids Res 35:e37. 10.1093/nar/gkl1158. 10.1093/nar/gkl1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kang Y, Durfee T, Glasner JD, Qiu Y, Frisch D, Winterberg KM, Blattner FR. 2004. Systematic mutagenesis of the Escherichia coli genome. J Bacteriol 186:4921–4930. [PubMed] 10.1128/JB.186.15.4921-4930.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Sarov M, Schneider S, Pozniakovski A, Roguev A, Ernst S, Zhang Y, Hyman AA, Stewart AF. 2006. A recombineering pipeline for functional genomics applied to Caenorhabditis elegans. Nat Methods 3:839–844. [PubMed] 10.1038/nmeth933 [DOI] [PubMed] [Google Scholar]
- 319.Sauer B, Henderson N. 1988. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci USA 85:5166–5170. [PubMed] 10.1073/pnas.85.14.5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Sauer B, Henderson N. 1989. Cre-stimulated recombination at loxP-containing DNA sequences placed into the mammalian genome. Nucleic Acids Res 17:147–161. [PubMed] 10.1093/nar/17.1.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Metzger D, Chambon P. 2001. Site- and time-specific gene targeting in the mouse. Methods 24:71–80. [PubMed] 10.1006/meth.2001.1159 [DOI] [PubMed] [Google Scholar]
- 322.Muyrers JP, Zhang Y, Testa G, Stewart AF. 1999. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res 27:1555–1557. [PubMed] 10.1093/nar/27.6.1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56–65. [PubMed] 10.1006/geno.2000.6451 [DOI] [PubMed] [Google Scholar]
- 324.Lee SC, Wang W, Liu P. 2009. Construction of gene-targeting vectors by recombineering. Methods Mol Biol 530:15–27. [PubMed] 10.1007/978-1-59745-471-1_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Bouvier J, Cheng JG. 2009. Recombineering-based procedure for creating Cre/loxP conditional knockouts in the mouse. Curr Protoc Mol Biol Chapter 23:Unit 23.13. 10.1002/0471142727.mb2313s85. [PubMed] 10.1002/0471142727.mb2313s85 [DOI] [PubMed] [Google Scholar]
- 326.Parkitna JR, Engblom D, Schutz G. 2009. Generation of Cre recombinase-expressing transgenic mice using bacterial artificial chromosomes. Methods Mol Biol 530:325–342. [PubMed] 10.1007/978-1-59745-471-1_17 [DOI] [PubMed] [Google Scholar]
- 327.Chakravortty D, Hansen-Wester I, Hensel M. 2002. Salmonella pathogenicity island 2 mediates protection of intracellular Salmonella from reactive nitrogen intermediates. J Exp Med 195:1155–1166. [PubMed] 10.1084/jem.20011547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Clegg S, Hughes KT. 2002. FimZ Is a molecular link between sticking and swimming in Salmonella enterica serovar Typhimurium 1213. J Bacteriol 184:1209–1213. [PubMed] 10.1128/jb.184.4.1209-1213.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Freeman JA, Rappl CV, Kuhle V, Hensel M, Miller SI. 2002. SpiC Is required for translocation of Salmonella pathogenicity island 2 effectors and secretion of translocon proteins SseB and SseC. J Bacteriol 184:4971–4980. [PubMed] 10.1128/JB.184.18.4971-4980.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Havemann GD, Sampson EM, Bobik TA. 2002. PduA is a shell protein of polyhedral organelles involved in coenzyme B12-dependent degradation of 1,2-propanediol in Salmonella enterica serovar Typhimurium LT2. J Bacteriol 184:1253–1261. [PubMed] 10.1128/JB.184.5.1253-1261.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Price-Carter M, Tingey J, Bobik TA, Roth JR. 2001. The alternative electron acceptor tetrathionate supports B12-dependent anaerobic growth of Salmonella enterica serovar Typhimurium on ethanolamine or 1,2-propanediol. J Bacteriol 183:2463–2475. [PubMed] 10.1128/JB.183.8.2463-2475.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Stanley TL, Ellermeier CD, Slauch JM. 2000. Tissue-specific gene expression identifies a gene in the lysogenic phage Gifsy-1 that affects Salmonella enterica serovar Typhimurium survival in Peyer’s patches. J Bacteriol 186:4406–4413. 10.1128/JB.182.16.4406-4413.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Worlock AJ, Smith RL. 2002. ZntB is a novel Zn2+ transporter in Salmonella enterica serovar Typhimurium. J Bacteriol 184:4369–4373. 10.1128/JB.184.16.4369-4373.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Lu S, Killoran PB, Fang FC, Riley LW. 2002. The global regulator ArcA controls resistance to reactive nitrogen and oxygen intermediates in Salmonella enterica serovar Enteritidis. Infect Immun 70:451–461. [PubMed] 10.1128/IAI.70.2.451-461.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Zurawski DV, Mitsuhata C, Mumy KL, McCormick BA, Maurelli AT. 2006. OspF and OspC1 are Shigella flexneri type III secretion system effectors that are required for postinvasion aspects of virulence. Infect Immun 74:5964–5976. [PubMed] 10.1128/IAI.00594-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Runyen-Janecky L, Daugherty A, Lloyd B, Wellington C, Eskandarian H, Sagransky M. 2008. Role and regulation of iron-sulfur cluster biosynthesis genes in Shigella flexneri virulence. Infect Immun 76:1083–1092. [PubMed] 10.1128/IAI.01211-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Ranallo RT, Barnoy S, Thakkar S, Urick T, Venkatesan MM. 2006. Developing live Shigella vaccines using lambda Red recombineering. FEMS Immunol Med Microbiol 47:462–469. [PubMed] 10.1111/j.1574-695X.2006.00118.x [DOI] [PubMed] [Google Scholar]
- 338.Ranallo RT, Thakkar S, Chen Q, Venkatesan MM. 2007. Immunogenicity and characterization of WRSF2G11: a second generation live attenuated Shigella flexneri 2a vaccine strain. Vaccine 25:2269–2278. [PubMed] 10.1016/j.vaccine.2006.11.067 [DOI] [PubMed] [Google Scholar]
- 339.Janes BK, Pomposiello PJ, Perez-Matos A, Najarian DJ, Goss TJ, Bender RA. 2001. Growth inhibition caused by overexpression of the structural gene for glutamate dehydrogenase (gdhA) from Klebsiella aerogenes. J Bacteriol 183:2709–2714. [PubMed] 10.1128/JB.183.8.2709-2714.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Nevesinjac AZ, Raivio TL. 2005. The Cpx envelope stress response affects expression of the type IV bundle-forming pili of enteropathogenic Escherichia coli. J Bacteriol 187:672–686. [PubMed] 10.1128/JB.187.2.672-686.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Eto DS, Jones TA, Sundsbak JL, Mulvey MA. 2007. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PLoS Pathog 3:e100. 10.1371/journal.ppat.0030100. [PubMed] 10.1371/journal.ppat.0030100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Lindberg S, Xia Y, Sonden B, Goransson M, Hacker J, Uhlin BE. 2008. Regulatory Interactions among adhesin gene systems of uropathogenic Escherichia coli. Infect Immun 76:771–780. [PubMed] 10.1128/IAI.01010-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Wiles TJ, Dhakal BK, Eto DS, Mulvey MA. 2008. Inactivation of host Akt/protein kinase B signaling by bacterial pore-forming toxins. Mol Biol Cell 19:1427–1438. [PubMed] 10.1091/mbc.E07-07-0638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Lee DJ, Bingle LE, Heurlier K, Pallen MJ, Penn CW, Busby SJ, Hobman JL. 2009. Gene doctoring: a method for recombineering in laboratory and pathogenic Escherichia coli strains. BMC Microbiol 9:252. 10.1186/1471-2180-9-252. [PubMed] 10.1186/1471-2180-9-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Lesic B, Rahme LG. 2008. Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC Mol Biol 9:20. 10.1186/1471-2199-9-20. [PubMed] 10.1186/1471-2199-9-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Liang R, Liu J. 2010. Scarless and sequential gene modification in Pseudomonas using PCR product flanked by short homology regions. BMC Microbiol 10:209. 10.1186/1471-2180-10-209. [PubMed] 10.1186/1471-2180-10-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Derbise A, Lesic B, Dacheux D, Ghigo JM, Carniel E. 2003. A rapid and simple method for inactivating chromosomal genes in Yersinia. FEMS Immunol Med Microbiol 38:113–116. [PubMed] 10.1016/S0928-8244(03)00181-0 [DOI] [PubMed] [Google Scholar]
- 348.Sun W, Wang S, Curtiss R, 3rd. 2008. Highly efficient method for introducing successive multiple scarless gene deletions and markerless gene insertions into the Yersinia pestis chromosome. Appl Environ Microbiol 74:4241–4245. [PubMed] 10.1128/AEM.00940-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Rossi MS, Paquelin A, Ghigo JM, Wandersman C. 2003. Haemophore-mediated signal transduction across the bacterial cell envelope in Serratia marcescens: the inducer and the transported substrate are different molecules. Mol Microbiol 48:1467–1480. [PubMed] 10.1046/j.1365-2958.2003.03516.x [DOI] [PubMed] [Google Scholar]
- 350.Yamamoto S, Izumiya H, Morita M, Arakawa E, Watanabe H. 2009. Application of lambda Red recombination system to Vibrio cholerae genetics: simple methods for inactivation and modification of chromosomal genes. Gene 438:57–64. [PubMed] 10.1016/j.gene.2009.02.015 [DOI] [PubMed] [Google Scholar]
- 351.Sinha KM, Unciuleac MC, Glickman MS, Shuman S. 2009. AdnAB: a new DSB-resecting motor-nuclease from mycobacteria. Genes Dev 23:1423–1437. [PubMed] 10.1101/gad.1805709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Wei JR, Krishnamoorthy V, Murphy K, Kim JH, Schnappinger D, Alber T, Sassetti CM, Rhee KY, Rubin EJ. 2011. Depletion of antibiotic targets has widely varying effects on growth. Proc Natl Acad Sci USA 108:4176–4181. [PubMed] 10.1073/pnas.1018301108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Ioerger TR, O’Malley T, Liao R, Guinn KM, Hickey MJ, Mohaideen N, Murphy KC, Boshoff HI, Mizrahi V, Rubin EJ, Sassetti CM, Barry CE, 3rd, Sherman DR, Parish T, Sacchettini JC. 2013. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS One 8:e75245. 10.1371/journal.pone.0075245. 10.1371/journal.pone.0075245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Swingle B, Bao Z, Markel E, Chambers A, Cartinhour S. 2010. Recombineering using RecTE from Pseudomonas syringae. Appl Environ Microbiol 76:4960–4968. [PubMed] 10.1128/AEM.00911-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Kropinski AM. 2000. Sequence of the genome of the temperate, serotype-converting, Pseudomonas aeruginosa bacteriophage D3. J Bacteriol 182:6066–6074. [PubMed] 10.1128/JB.182.21.6066-6074.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci USA 100:1541–1546. [PubMed] 10.1073/pnas.0337542100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Wenzel SC, Gross F, Zhang Y, Fu J, Stewart AF, Muller R. 2005. Heterologous expression of a myxobacterial natural products assembly line in pseudomonads via red/ET recombineering. Chem Biol 12:349–356. [PubMed] 10.1016/j.chembiol.2004.12.012 [DOI] [PubMed] [Google Scholar]
- 358.Fu J, Wenzel SC, Perlova O, Wang J, Gross F, Tang Z, Yin Y, Stewart AF, Muller R, Zhang Y. 2008. Efficient transfer of two large secondary metabolite pathway gene clusters into heterologous hosts by transposition. Nucleic Acids Res 36:e113. 10.1093/nar/gkn499. 10.1093/nar/gkn499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Katashkina JI, Hara Y, Golubeva LI, Andreeva IG, Kuvaeva TM, Mashko SV. 2009. Use of the lambda Red-recombineering method for genetic engineering of Pantoea ananatis. BMC Mol Biol 10:34. 10.1186/1471-2199-10-34. [PubMed] 10.1186/1471-2199-10-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Hu S, Fu J, Huang F, Ding X, Stewart AF, Xia L, Zhang Y. 2014. Genome engineering of Agrobacterium tumefaciens using the lambda Red recombination system. Appl Microbiol Biotechnol 98:2165–2172. [PubMed] 10.1007/s00253-013-5412-x [DOI] [PubMed] [Google Scholar]
- 361.van Pijkeren JP, Britton RA. 2012. High efficiency recombineering in lactic acid bacteria. Nucleic Acids Res 40:e76. 10.1093/nar/gks147. [PubMed] 10.1093/nar/gks147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Sun Z, Deng A, Hu T, Wu J, Sun Q, Bai H, Zhang G, Wen T. 2015. A high-efficiency recombineering system with PCR-based ssDNA in Bacillus subtilis mediated by the native phage recombinase GP35. Appl Microbiol Biotechnol 99:5151–5162. [PubMed] 10.1007/s00253-015-6485-5 [DOI] [PubMed] [Google Scholar]
- 363.Dong H, Tao W, Gong F, Li Y, Zhang Y. 2014. A functional recT gene for recombineering of Clostridium. J Biotechnol 173:65–67. [PubMed] 10.1016/j.jbiotec.2013.12.011 [DOI] [PubMed] [Google Scholar]
- 364.Datta S, Costantino N, Zhou X, Court DL. 2008. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc Natl Acad Sci USA 105:1626–1631. [PubMed] 10.1073/pnas.0709089105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Yin J, Zhu H, Xia L, Ding X, Hoffmann T, Hoffmann M, Bian X, Muller R, Fu J, Stewart AF, Zhang Y. 2015. A new recombineering system for Photorhabdus and Xenorhabdus. Nucleic Acids Res 43:e36. 10.1093/nar/gku1336. [PubMed] 10.1093/nar/gku1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Cunningham FX, Jr, Sun Z, Chamovitz D, Hirschberg J, Gantt E. 1994. Molecular structure and enzymatic function of lycopene cyclase from the cyanobacterium Synechococcus sp strain PCC7942. Plant Cell 6:1107–1121. [PubMed] 10.1105/tpc.6.8.1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Wang HH, Kim H, Cong L, Jeong J, Bang D, Church GM. 2012. Genome-scale promoter engineering by coselection MAGE. Nat Methods 9:591–593. [PubMed] 10.1038/nmeth.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Newman RJ, Roose-Girma M, Warming S. 2015. Efficient conditional knockout targeting vector construction using co-selection BAC recombineering (CoSBR). Nucleic Acids Res 43:e124. 10.1093/nar/gkv600. 10.1093/nar/gkv600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Huen MS, Li XT, Lu LY, Watt RM, Liu DP, Huang JD. 2006. The involvement of replication in single stranded oligonucleotide-mediated gene repair. Nucleic Acids Res 34:6183–6194. [PubMed] 10.1093/nar/gkl852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Radecke S, Radecke F, Peter I, Schwarz K. 2006. Physical incorporation of a single-stranded oligodeoxynucleotide during targeted repair of a human chromosomal locus. J Gene Med 8:217–228. [PubMed] 10.1002/jgm.828 [DOI] [PubMed] [Google Scholar]
- 371.Wang TC. 2005. Discontinuous or semi-discontinuous DNA replication in Escherichia coli? Bioessays 27:633–636. [PubMed] 10.1002/bies.20233 [DOI] [PubMed] [Google Scholar]
- 372.Stukenberg PT, Turner J, O’Donnell M. 1994. An explanation for lagging strand replication: polymerase hopping among DNA sliding clamps. Cell 78:877–887. [PubMed] 10.1016/S0092-8674(94)90662-9 [DOI] [PubMed] [Google Scholar]
- 373.Onrust R, Finkelstein J, Turner J, Naktinis V, O’Donnell M. 1995. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. III. Interface between two polymerases and the clamp loader. J Biol Chem 270:13366–13377. 10.1074/jbc.270.22.13366 [DOI] [PubMed] [Google Scholar]
- 374.Mosberg JA, Gregg CJ, Lajoie MJ, Wang HH, Church GM. 2012. Improving lambda red genome engineering in Escherichia coli via rational removal of endogenous nucleases. PLoS One 7:e44638. 10.1371/journal.pone.0044638. [PubMed] 10.1371/journal.pone.0044638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Lajoie MJ, Gregg CJ, Mosberg JA, Washington GC, Church GM. 2012. Manipulating replisome dynamics to enhance lambda Red-mediated multiplex genome engineering. Nucleic Acids Res 40:e170. 10.1093/nar/gks751. [PubMed] 10.1093/nar/gks751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Yu D, Sawitzke JA, Ellis H, Court DL. 2003. Recombineering with overlapping single-stranded DNA oligonucleotides: testing a recombination intermediate. Proc Natl Acad Sci USA 100:7202–7212. [PubMed] 10.1073/pnas.1232375100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Muyrers JP, Zhang Y, Buchholz F, Stewart AF. 2000. RecE/RecT and Redalpha/Redbeta initiate double-stranded break repair by specifically interacting with their respective partners. Genes Dev 14:1971–1982. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 378.Mosberg JA, Lajoie MJ, Church GM. 2010. Lambda Red recombination in Escherichia coli occurs through a fully single-stranded intermediate. Genetics 186:791–799. [PubMed] 10.1534/genetics.110.120782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Lim SI, Min BE, Jung GY. 2008. Lagging strand-biased initiation of red recombination by linear double-stranded DNAs. J Mol Biol 384:1098–1105. [PubMed] 10.1016/j.jmb.2008.10.047 [DOI] [PubMed] [Google Scholar]
- 380.Liu XP, Liu JH. 2010. The terminal 5′ phosphate and proximate phosphorothioate promote ligation-independent cloning. Protein Sci 19:967–973. [PubMed] 10.1002/pro.374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Court DL, Sawitzke JA, Thomason LC. 2002. Genetic engineering using homologous recombination. Annu Rev Genet 36:361–388. [PubMed] 10.1146/annurev.genet.36.061102.093104 [DOI] [PubMed] [Google Scholar]
- 382.Lederberg EM. 1950. Lysogenicity in Escherichia coli K-12. Microb Genet Bull 1:5–9 (http://www.estherlederberg.com/LambdaP.html). [Google Scholar]
- 383.Bi B, Rybalchenko N, Golub EI, Radding CM. 2004. Human and yeast Rad52 proteins promote DNA strand exchange. Proc Natl Acad Sci USA 101:9568–9572. [PubMed] 10.1073/pnas.0403205101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Bassett CL, Kushner SR. 1984. Exonuclease I, III and V are required for stability of ColE1-related plasmids in Escherichia coli. J Bacteriol 157:661–664. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Mortensen UH, Lisby M, Rothstein R. 2009. Rad52. Curr Biol 19:R676–R677. [PubMed] 10.1016/j.cub.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 386.Sharples GJ, Corbett LM, McGlynn P. 1999. DNA structure specificity of Rap endonuclease. Nucleic Acids Res 27:4121–4127. [PubMed] 10.1093/nar/27.21.4121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Sharples GJ, Curtis FA, McGlynn P, Bolt EL. 2004. Holliday junction binding and resolution by the Rap structure-specific endonuclease of phage lambda. J Mol Biol 340:739–751. [PubMed] 10.1016/j.jmb.2004.05.030 [DOI] [PubMed] [Google Scholar]
- 388.Wang J, Sarov M, Rientjes J, Fu J, Hollak H, Kranz H, Xie W, Stewart AF, Zhang Y. 2006. An improved recombineering approach by adding RecA to lambda Red recombination. Mol Biotechnol 32:43–53. [PubMed] 10.1385/MB:32:1:043 [DOI] [PubMed] [Google Scholar]
- 389.Palmeros B, Wild J, Szybalski W, Le Borgne S, Hernandez-Chavez G, Gosset G, Valle F, Bolivar F. 2000. A family of removable cassettes designed to obtain antibiotic-resistance-free genomic modifications of Escherichia coli and other bacteria. Gene 247:255–264. [PubMed] 10.1016/S0378-1119(00)00075-5 [DOI] [PubMed] [Google Scholar]
- 390.Bigger BW, Tolmachov O, Collombet JM, Fragkos M, Palaszewski I, Coutelle C. 2001. An araC-controlled bacterial cre expression system to produce DNA minicircle vectors for nuclear and mitochondrial gene therapy. J Biol Chem 276:23018–23027. [PubMed] 10.1074/jbc.M010873200 [DOI] [PubMed] [Google Scholar]
- 391.Buchholz F, Ringrose L, Angrand PO, Rossi F, Stewart AF. 1996. Different thermostabilities of FLP and Cre recombinases: implications for applied site-specific recombination. Nucleic Acids Res 24:4256–4262. [PubMed] 10.1093/nar/24.21.4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Murphy KC, Papavinasasundaram K, Sassetti CM. 2015. Mycobacterial recombineering. Methods Mol Biol 1285:177–199. [PubMed] 10.1007/978-1-4939-2450-9_10 [DOI] [PubMed] [Google Scholar]
- 393.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. [PubMed] 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 394.Doublet B, Douard G, Targant H, Meunier D, Madec JY, Cloeckaert A. 2008. Antibiotic marker modifications of lambda Red and FLP helper plasmids, pKD46 and pCP20, for inactivation of chromosomal genes using PCR products in multidrug-resistant strains. J Microbiol Methods 75:359–361. [PubMed] 10.1016/j.mimet.2008.06.010 [DOI] [PubMed] [Google Scholar]
- 395.St-Pierre F, Cui L, Priest DG, Endy D, Dodd IB, Shearwin KE. 2013. One-step cloning and chromosomal integration of DNA. ACS Synth Biol 2:537–541. [PubMed] 10.1021/sb400021j [DOI] [PubMed] [Google Scholar]