

Abstract
Depression is an episodic form of mental illness characterized by mood state transitions with poorly understood neurobiological mechanisms. Antidepressants reverse the effects of stress and depression on synapse function, enhancing neurotransmission, increasing plasticity, and generating new synapses in stress-sensitive brain regions. These properties are shared to varying degrees by all known antidepressants, suggesting that synaptic remodeling could play a key role in depression pathophysiology and antidepressant function. Still, it is unclear whether and precisely how synaptogenesis contributes to mood state transitions. Here, we review evidence supporting an emerging model in which depression is defined by a distinct brain state distributed across multiple stress-sensitive circuits, with neurons assuming altered functional properties, synapse configurations, and, importantly, a reduced capacity for plasticity and adaptation. Antidepressants act initially by facilitating plasticity and enabling a functional reconfiguration of this brain state. Subsequently, synaptogenesis plays a specific role in sustaining these changes over time.
Keywords: depression, stress, rapid-acting antidepressants, synaptic plasticity, dendritic spines, ketamine
1. INTRODUCTION
Depression is a chronic, recurrent psychiatric condition with an estimated lifetime prevalence of approximately 17% (Kessler et al. 2003). Importantly, depression is also a fundamentally episodic form of mental illness defined by discrete symptomatic periods, interposed between periods of wellness, and the temporal dynamics of mood state transitions vary across patients. In some individuals, depressive episodes can persist for months or years, while others cycle rapidly between depression and elevated mood (Post et al. 2003). Likewise, recovery duration is highly variable and difficult to predict. The neurobiological mechanisms driving the induction, remission, and recurrence of depressive episodes over time remain poorly understood, especially at the neural circuit level.
Converging evidence indicates that synaptic remodeling in stress-sensitive circuits plays an important role in the emergence of depressive episodes. In preclinical studies, chronic stress is associated with a reduction in the density of postsynaptic dendritic spines in prefrontal and hippocampal pyramidal neurons (McEwen 2007) (see the sidebar titled The Structure and Function of Dendritic Spines). These regressive synaptic effects are mediated in part by glucocorticoid stress hormones and related signaling molecules (Karst et al. 2005, Magariños et al. 1996). Glucocorticoids regulate synapse maturation in the developing brain (Liston & Gan 2011, Maras & Baram 2012) and facilitate learning-related plasticity across the life span, balancing synapse formation and pruning to enable learning, memory, and adaptation to changing environmental conditions (Joëls et al. 2006, Liston et al. 2013, McGaugh 2004). However, in chronic stress states, excessive glucocorticoid exposure disrupts this balance, leading to synapse loss and dendritic atrophy. Conversely, antidepressants increase synapse density, and some target molecular signaling pathways that promote synaptogenesis (Autry et al. 2011, Duman et al. 2016, Li et al. 2010). Still, it remains unclear whether and precisely how synaptogenesis contributes to the transition from depression to euthymic mood.
THE STRUCTURE AND FUNCTION OF DENDRITIC SPINES.
Dendritic spines are microscopic membrane protrusions in specific subtypes of neurons and usually (but not always) contain synapses. Early Golgi impregnation and more recent fixed-tissue and in vivo imaging studies have allowed investigators to quantify the effects of stress and antidepressants on spine density and to study spine dynamics across varying behavioral states (Kasai et al. 2003, Yuste & Bonhoeffer 2001). Spines are often classified by morphology as filopodia-like, thin, stubby, fenestrated, and mushroom (Kasai et al. 2003). The advent of transcranial two-photon imaging coupled with glutamate uncaging at single spines along dendritic segments has advanced our understanding of structure-function relationships. Glutamate sensitivity and AMPA receptor expression are highly correlated with large (mushroom) spine heads, while thin spines exhibit considerably lower sensitivity. Small or thin spines are easily generated and eliminated and may represent silent synapses, devoid of functional AMPA receptors (Matsuzaki et al. 2001). In breaking with the classification of major subtypes of spines, recent ultrastructural analysis suggests a continuum of spine morphologies based on specific parameters that are independently regulated (Ofer et al. 2021) and associated with distinct proteomic properties (Helm et al. 2021).
Here, we review evidence supporting an emerging model in which depression is defined by a distinct brain state distributed across multiple stress-sensitive circuits, with neurons assuming distinct functional properties, altered synapse configurations, and, importantly, a reduced capacity for plasticity and adaptation. Antidepressants act by facilitating plasticity, initiating a reconfiguration of this brain state. Subsequently, antidepressant-induced synapse formation—which follows and may be driven by changes in circuit function—plays a specific role in sustaining these changes over time. We review findings from studies of conventional monoamine-targeting antidepressants, ketamine, and emerging next-generation antidepressants. We build on a foundation elaborated in multiple reviews (Castrén & Hen 2013, Duman et al. 2016, Kavalali & Monteggia 2020) by focusing here on the relationship between chronic stress and antidepressant effects on circuit function and plasticity and in particular on emerging data implicating distinct mechanisms in initiating antidepressant effects and then sustaining them over time.
2. SYNAPSE DYSFUNCTION IN DEPRESSION PATHOPHYSIOLOGY
2.1. Synapse Dysfunction in Rodent Chronic Stress Models
Chronic stress is among the strongest and most extensively studied risk factors for depression, and psychosocial stressors may directly trigger depressive episodes in vulnerable individuals (Lupien et al. 2009). Much of what we know about the pathophysiology of depression comes from studies in rodents exposed to various forms of chronic stress (see Supplemental Discussion). These stress paradigms do not model depression per se, but they are useful for investigating how the brain responds to stress, understanding the neurobiological basis of individual differences in stress susceptibility, and identifying substrates of depression-related behaviors (Nestler & Hyman 2010).
While the adult brain was once thought to be essentially static, we now know that the brain responds adaptively to acute stressors and other threats, initiating a response (allostasis) aimed at mobilizing metabolic and cognitive resources and maximizing survival (McEwen 2007). With repeated stress exposure, these allostatic responses lead to longer-term changes in neuronal structure and function and synaptic remodeling (allostatic load), which are associated with the emergence of depression- and anxiety-related behaviors (McEwen 1998). Glucocorticoid stress hormones and related molecules play a key role in mediating stress effects on synaptic plasticity (Karst et al. 2005, Magariños et al. 1996). They are required for supporting synapse development early in life in both primary sensorimotor cortex and association cortex (Liston & Gan 2011), and circadian glucocorticoid oscillations support learning-related synaptic plasticity in adulthood (Liston et al. 2013). Indeed, psychosocial stress and glucocorticoids modulate synaptic plasticity in a variety of regions throughout the cortex (Liston & Gan 2011, Lu et al. 2021, McEwen 2007). However, as reviewed below, the effects of chronic stress vary by brain region and spine type (Figure 1; see the sidebar titled The Structure and Function of Dendritic Spines), with most showing net synapse loss but some showing increases in synapse density and potentiation—differences that may be related to regional variation in neuronal activity with implications for therapeutic interventions targeting synapse function. These changes provide a foundation for understanding the mechanisms by which antidepressants modulate mood state transitions, discussed in the following section.
Figure 1.
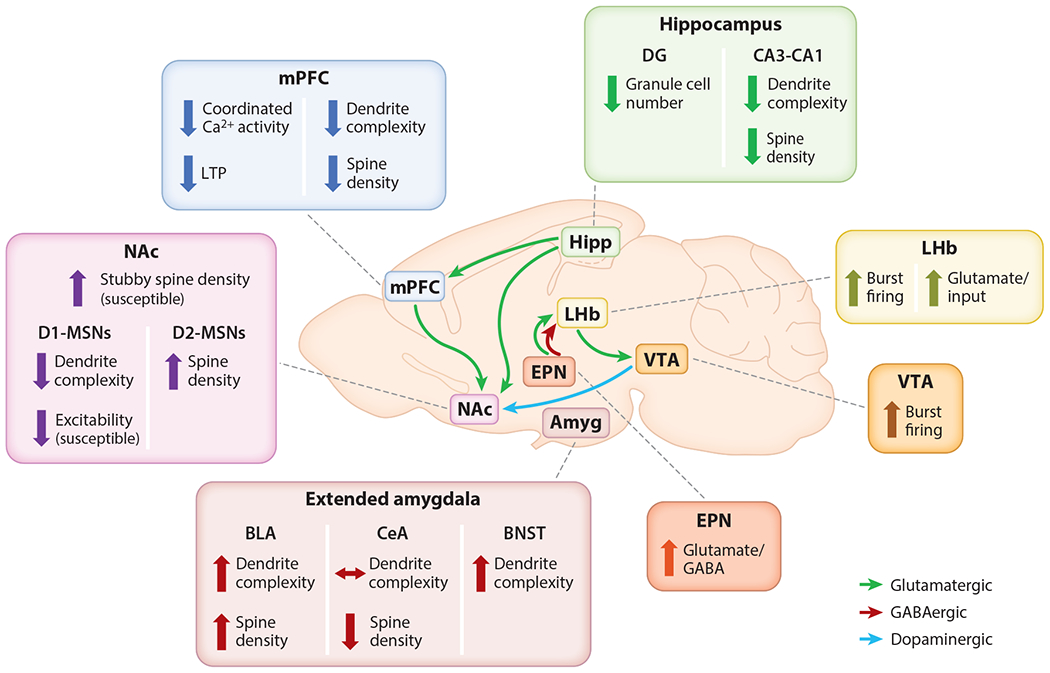
Regional synaptic effects of stress and depression models. Hipp and mPFC undergo dendritic atrophy and spine loss. NAc experiences an overall proliferation of stubby spines, but D1-MSNs may experience dendritic atrophy relative to D2-MSNs. Other regions, including amygdala and Amyg, EPN, VTA, and LHb, experience spine formation (spinogenesis) and increased excitability following stress. Abbreviations: Amyg, extended amygdala; BLA, basolateral amygdala; BNST, bed nucleus of the stria terminalis; Ca2+, calcium; CeA, central amygdala; D1-MSN, dopamine receptor 1 medium spiny neuron; D2-MSN, dopamine receptor 2 medium spiny neuron; DG, dentate gyrus; EPN, entopeduncular nucleus; GABA, γ-aminobutyric acid; Hipp, hippocampus; LHb, lateral habenula; LTP, long-term potentiation; mPFC, medial prefrontal cortex; NAc, nucleus accumbens; VTA, ventral tegmental area.
2.1.1. Hippocampus.
The hippocampus was one of the first brain structures implicated as a target for regressive morphological changes in depression and following stress. Early reports found that glucocorticoid exposure induced cell death and atrophy (Mühlen & Ockenfels 1968). In multiple chronic stress models, hippocampal pyramidal cells exhibit apical dendritic atrophy, reductions in postsynaptic spine and AMPA receptor density, and deficits in excitatory neurotransmission and long-term potentiation (LTP) (Diamond & Rose 1994, Iñiguez et al. 2016, Magariños et al. 1996, Watanabe et al. 1992, Woolley et al. 1990). In the dentate gyrus, chronic stress interferes with neurogenesis (Gould et al. 1997, 1998; Snyder et al. 2011). At the molecular level, brain-derived neurotrophic factor (BDNF), a key regulator of neural growth and synapse formation, is implicated in the process of experience-dependent remodeling of synapses and dendrites (Poo 2001). BDNF levels are downregulated in the hippocampus following chronic stress (Shirayama et al. 2002), and deficiencies in activity-dependent BDNF signaling are sufficient to recapitulate the effects of stress on spine density, dendritic complexity, and memory (Chen et al. 2006, Soliman et al. 2010). Together, stress effects on synapse function and plasticity in the hippocampus may contribute to compromised fidelity of synaptic transmission at CA1 synapses and memory deficits (Conrad et al. 1996), as well as deficits in reward learning, social interaction, and stress resilience that are driven by hippocampal projections to the nucleus accumbens (NAc) and other areas (LeGates et al. 2018, Muir et al. 2020).
2.1.2. Medial prefrontal cortex.
As a mediator of cognitive control and regulator of the hypothalamic-pituitary-adrenal axis and stress response, the medial prefrontal cortex (mPFC) has been a focus of studies aimed at understanding structure-function relationships in depression and other stress-related psychiatric disorders. Stress impairs LTP and reduces excitability of mPFC projection neurons (Goldwater et al. 2009), and it is associated with synapse loss, reduced synaptic transmission, and reduced glutamate receptor expression (Li et al. 2010, Yuen et al. 2012). Structural changes are also apparent, including spine loss and dendritic retraction following chronic corticosterone (Radley et al. 2004, Wellman 2001), restraint (Liston et al. 2006), and variable stress (Radley et al. 2013). Spine loss is associated with impaired mPFC-dependent behaviors such as decision-making and attentional set shifting (Dias-Ferreira et al. 2009, Liston et al. 2006). The mPFC is also important for top-down control over structures that support aversive responding (e.g., paraventricular hypothalamus, amygdala). As such, regressive morphological changes in mPFC may be indicative of a withdrawal of that influence.
2.1.3. Amygdala.
The amygdalar complex regulates emotional and affective aspects of cognition and behavior and is implicated in depression and other stress-related disorders (Drevets et al. 1992, Sheline et al. 2001). In contrast to the hippocampus and prefrontal cortex, stress induces hyperexcitability and increases BDNF, spine formation, and dendritic arborization in the basolateral amygdala (BLA) (Rosenkranz et al. 2010; Vyas et al. 2002, 2003, 2004). This hypertrophy is not reversed by stress cessation, suggesting that enhanced synaptic connectivity of BLA principal neurons may support encoding of aversive stimuli and experiences (Vyas et al. 2004). Interestingly, elevated neurotrophic signaling persists in BLA even 21 days poststress (Lakshminarasimhan & Chattarji 2012). This could contribute to the chronic or recurring nature of disorders, including depression and posttraumatic stress disorder. Some neuroimaging studies also suggest amygdalar volume enlargements in depression (Drevets et al. 2008). Chronic isolation increases dendritic arborization in the bed nucleus of the stria terminalis (BNST) (Feldman et al. 1990), while central amygdala neurons appear to be resistant to stress-induced dendritic hypertrophy (Vyas et al. 2003). Importantly, distinct amygdala circuits are known to mediate varying aspects of behavior and may contribute to symptom heterogeneity in depression. While dendritic hypertrophy is common in dorsomedial prefrontal cortex–projecting and nonprojecting BLA neurons, increased spine size and density of mature mushroom spines (see the sidebar titled The Structure and Function of Dendritic Spines) is restricted to ventral hippocampus projectors (Zhang et al. 2019). It will be important to further parse stress effects on individual amygdala circuits.
2.1.4. Nucleus accumbens and ventral tegmental area.
The NAc and ventral tegmental area (VTA), which support reward-seeking behavior and salience processing, generally exhibit potentiated activity and spine formation (spinogenesis) following stress. In neuroimaging studies, depressed patients exhibit altered connectivity in related frontostriatal networks (Drysdale et al. 2017, Pizzagalli et al. 2009), and preclinical animal models have provided clues to the cell- and circuit-level mechanisms mediating these changes. Increased VTA excitability and BDNF signaling are key mediators of stress susceptibility. Both optogenetic inhibition and phasic (but not tonic) activation of VTA activity are sufficient to induce depression-related behavior (Chaudhury et al. 2013), suggesting a requirement for carefully tuned activity. Social defeat stress increases VTA activity and release of BDNF in NAc (Berton et al. 2006, Krishnan et al. 2007). Stress promotes spinogenesis broadly within the NAc of stress-susceptible mice (Christoffel et al. 2011), but cell type-specific effects may be more nuanced. Medium spiny neurons (MSNs), the main cell type within the NAc, can be subtyped by their expression of either dopamine receptor 1 or 2 (D1- and D2-MSNs, respectively). These populations support distinct behaviors (Hikida et al. 2010), raising the possibility of differential responses to stress. Indeed, stress is associated with reduced excitability and dendritic atrophy of D1- but not D2-MSNs (Fox et al. 2020, Francis et al. 2017, LeGates et al. 2018, Lim et al. 2012). Taken together, these studies show that chronic stress increases BDNF signaling and tonic excitability in VTA and promotes a net increase in immature stubby spine density across MSN types, while D1-MSNs may be prone to regressive structural changes.
2.1.5. Lateral habenula.
The lateral habenula (LHb) exhibits increased activity and potentiated presynaptic input following stress, suggesting that it may be a key driver of stress effects. The LHb processes negative valence information and reward prediction errors in humans (Salas et al. 2010) and rodents (Matsumoto & Hikosaka 2007). Deep brain stimulation of this structure reduces depression-like behaviors in rodents (Li et al. 2011) and depressed patients (Sartorius et al. 2010). Preclinical animal studies indicate that plasticity and activity within LHb or its inputs play a causal role in stress and depression models. Knockdown of plasticity-related genes or restoration of inhibitory signaling in LHb normalizes excitability and depression-related behavior (Lecca et al. 2016, Li et al. 2013) and is a process that also plays a critical role in mediating ketamine’s antidepressant effects (Yang et al. 2018). Aversive experiences enhance presynaptic excitatory input onto LHb (Li et al. 2011, Shabel et al. 2014), which may drive increased tonic and burst firing of VTA-projecting LHb neurons (Cerniauskas et al. 2019). Likewise, in zebrafish, repeated stress increases bursting activity in habenula neurons, which drives the emergence of depression-related behaviors through projections to the serotonergic raphe nucleus (Andalman et al. 2019). Together, these studies point to a potentiation of excitatory inputs to LHb as an important component of depression-related end points.
2.2. Evidence for Synapse Dysfunction in Human Studies
Data from two sources provide evidence for depression-related synapse dysfunction and structural changes in humans. First, postmortem histological analyses show that pyramidal neurons are reduced in size (but not number) in the dorsolateral prefrontal and orbitofrontal cortex, with a corresponding reduction in cortical thickness (Rajkowska et al. 1999, 2007), while hippocampal granule cells are reduced in number, consistent with decreased neurogenesis (Boldrini et al. 2013). Subsequent electron microscopy, immunohistochemistry, and RNA sequencing studies confirmed a reduction in synapse number and related genes and proteins in prefrontal cortex, hippocampus, and NAc, among other regions (Duman & Aghajanian 2012, Kang et al. 2012, Labonté et al. 2017). Second, additional indirect evidence comes from clinical neuroimaging studies, which have identified numerous structural and functional alterations consistent with observations of synapse loss in preclinical and postmortem studies. These findings include hippocampal volume loss (Sheline et al. 1996); reduced prefrontal glutamate and γ-aminobutyric acid (GABA) levels as indexed by magnetic resonance spectroscopy (Hasler et al. 2007, Sanacora et al. 2004); reduced synapse density as indexed by positron emission tomography in PFC, anterior cingulate, and hippocampus (Holmes et al. 2019); and reduced functional connectivity in both chronic stress and depression in lateral prefrontal and other limbic networks (Liston et al. 2009, 2014; Lui et al. 2011). Of note, increased functional connectivity in the amygdala and other networks has also been repeatedly observed in depression (Drysdale et al. 2017, Hamilton & Gotlib 2008, Whitfield-Gabrieli & Ford 2012), consistent with preclinical studies showing that stress effects on synapse density and functional properties vary by region and may relate in part to differences in stress-induced activity states.
2.3. Summary
Chronic stress induces a distinct brain state characterized by widespread changes in the functional properties and synaptic connectivity of neurons in a distributed network of brain regions. Stress produces alterations in dendritic complexity, spine density, synapse number, and excitatory transmission. Although these changes encompass multiple brain regions, they are also region specific (Figure 1). Following stress, structures that mediate top-down control over depression-related behaviors and neuroendocrine systems such as the mPFC and hippocampus undergo dendritic retraction and spine loss and exhibit decreased activity in various contexts. Other areas that respond to aversive stimuli, including the amygdala, BNST, and LHb, show increased spine density and potentiated activity in chronic stress states. These contrasting effects suggest region-specific mechanisms and raise questions about how antidepressants might act across multiple structures to rescue or compensate for synaptic dysfunction. Importantly, chronic stress and excessive glucocorticoid exposure may contribute to the maintenance of a stress-induced brain state by disrupting synaptic remodeling dynamics and reducing the capacity for enduring plasticity.
3. SYNAPTIC PLASTICITY AS A THERAPEUTIC TARGET
Chronic stress induces a distinct brain state in which neurons assume distinct functional properties and alterations in synaptic connectivity. As we discuss below, antidepressants may act in part by reversing these effects on synapse function, initially facilitating a reconfiguration of brain state, then promoting synaptic changes that sustain the remitted state.
3.1. Monoaminergic Antidepressants
For decades, our understanding of antidepressant mechanisms was dominated by the early serendipitous discovery of the antidepressant properties of iproniazid, which were linked to its activity as a monoamine oxidase inhibitor (Pare & Sandler 1959). This discovery and the subsequent development of tricyclic and selective serotonin reuptake inhibitor (SSRI) antidepressants formed the basis of the monoamine hypothesis of depression, in which the key pathophysiological substrate was thought to be a deficiency of monoamine neuromodulation. However, this model could not explain why SSRIs and related antidepressants immediately potentiate monoamine availability, but the clinical response emerges over weeks or months (Hirschfeld 2000). Furthermore, stimulants and many other drugs that increase monoamine availability are not effective antidepressants.
Instead, conventional monoamine-targeting antidepressants are now understood to act at least in part by inducing synaptogenesis and enhancing synapse function [as well as neurogenesis and other forms of plasticity (for a review, see Castrén & Hen 2013)]. For example, chronic treatment with fluoxetine (an SSRI) enhances LTP and synaptic transmission (Bath et al. 2012, Wang et al. 2008), reverses stress effects on dendritic arborization and spine density (Bessa et al. 2009), and facilitates learning-related plasticity (Karpova et al. 2011). The long-term behavioral response to chronic fluoxetine requires BDNF (Chen et al. 2006), enhancing synaptic transmission in hippocampus (Bath et al. 2012) and reducing it in the NAc (Vialou et al. 2010), again underscoring the role of bidirectional effects on synapse function that vary by brain region. Importantly, however, unlike ketamine and other rapid-acting antidepressants reviewed below, conventional monoamine-targeting antidepressants generally modulate synaptic plasticity only after chronic treatment, and their effects on synaptogenesis are slow and relatively modest (Ampuero et al. 2010, Castrén & Hen 2013, Duman et al. 2016)—limitations that may explain the requirement for prolonged treatment, relatively high levels of treatment resistance, and the tendency for patients to relapse upon discontinuation.
3.2. Ketamine: Rapid Synaptogenesis in Depression-Related Circuits
Ketamine is an N-methyl-d-aspartate receptor (NMDAR) antagonist that was initially approved as an anesthetic agent and was clinically evaluated for antidepressant potential in the early 2000s (Berman et al. 2000). After positive results in multiple randomized controlled trials (Diazgranados et al. 2010, Murrough et al. 2013, Zarate et al. 2006), it is now used widely at subanesthetic doses as an antidepressant. These findings led to the 2019 US Food and Drug Administration approval of esketamine, an intranasal formulation of the (S) enantiomer of ketamine, marking a milestone in the translation of effective strategies for treatment-resistant depression. In addition to providing potentially life-saving treatment for severely depressed patients, the discovery of ketamine’s antidepressant properties opened new avenues for understanding depression pathophysiology for at least two reasons. First, ketamine acts much more rapidly than conventional monoamine-targeting antidepressants. In a recent meta-analysis, the average onset of symptom improvement was ~40 min postinfusion, peaking at ~24 h, and subsiding to placebo level after ~10–12 days (Kishimoto et al. 2016). Second, unlike essentially all commonly used antidepressant drugs before it, ketamine does not act by targeting monoaminergic neuromodulation and appears to engage multiple signaling mechanisms that are fundamentally different from conventional agents (see below). These unique properties have facilitated new experimental paradigms aimed at understanding the mechanisms that mediate mood transitions longitudinally and discovering new treatment targets.
Three key sources of evidence indicate that ketamine’s antidepressant effects involve synaptic plasticity. First, unlike conventional monoamine-targeting antidepressants, ketamine acts to modulate synapse function directly and rapidly—effects that are thought to explain its rapid onset. Its antidepressant effects have been linked to NMDAR antagonism in the hippocampus and prefrontal cortex, which activates the mammalian target of rapamycin (mTOR) pathway (Li et al. 2010). mTOR activation is required for subsequent increases in the expression of GluA1-containing α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR), postsynaptic density protein 95 (PSD95), and other synaptic proteins (Li et al. 2010). Interestingly, while infusing the selective mTOR inhibitor rapamycin directly into the prefrontal cortex blocked the antidepressant behavioral effects of ketamine in rats (Li et al. 2010), systemic pretreatment with rapamycin extended the duration of ketamine’s antidepressant effects in humans (Abdallah et al. 2020), which could be related to effects of systemic infusion on immune function and inflammation. These effects may also be mediated in part by hydroxynorketamine, a ketamine metabolite with antidepressant-like effects on behavior, AMPA signaling, and synapse function (Zanos et al. 2016), as well as NMDAR inhibition (Suzuki et al. 2017).
Second, ketamine’s antidepressant behavioral effects require BDNF and TrkB signaling (Figure 2), further reinforcing the hypothesis that synaptogenesis plays a key role. Ketamine enhances activity-dependent BDNF release by antagonizing NMDARs, deactivating eukaryotic elongation factor 2 (eEF2), and de-suppressing the translation of BDNF (Autry et al. 2011). Ketamine’s effects on both behavior and synapse function in turn require BDNF and TrkB signaling (Autry et al. 2011, Lin et al. 2021). Interestingly, ketamine, fluoxetine, and other antidepressant drugs—but not drugs lacking antidepressant properties—appear to bind directly to TrkB and promote the trafficking and clustering of TrkB on the cell surface, an effect that is potentiated by astrocyte-derived cholesterol (Casarotto et al. 2021). Antidepressants may accumulate at varying rates in the brain, differentially achieving concentrations capable of affecting TrkB signaling. This intriguing mechanism may be one way in which they induce their neuroplastic effects across timescales and brain regions.
Figure 2.
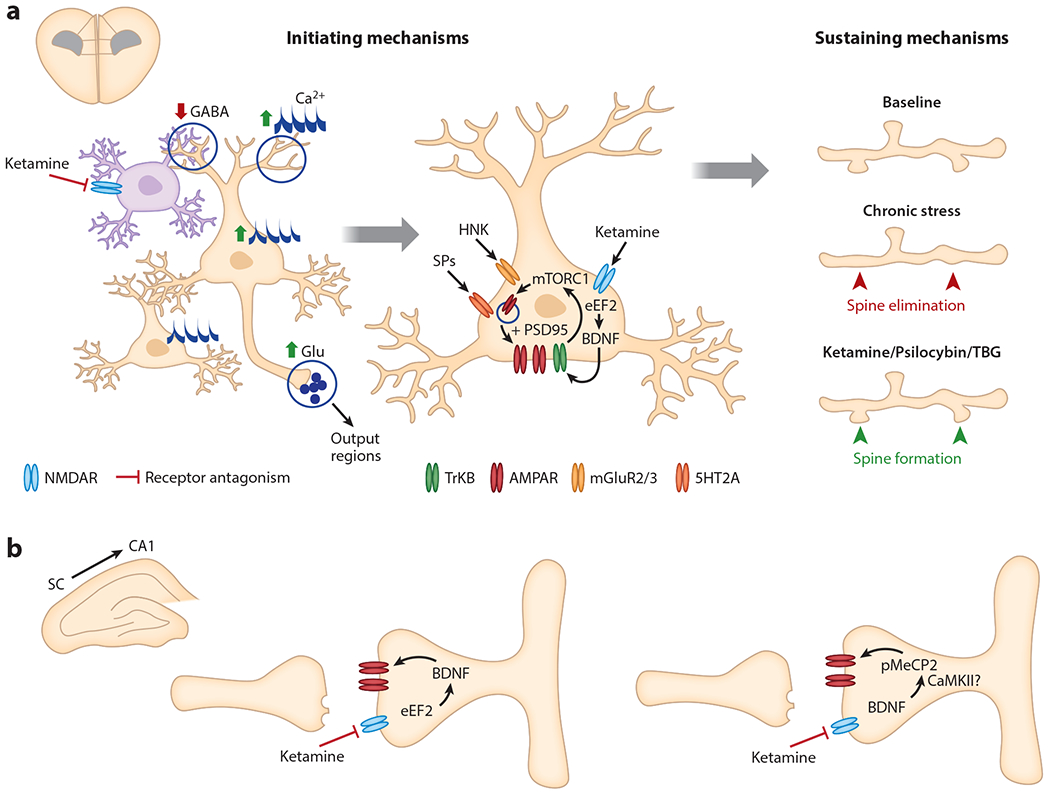
The initiating and sustaining mechanisms of rapid-acting antidepressants. (a) In the mPFC, ketamine acts to elevate glutamatergic tone of excitatory pyramidal neurons (tan), potentially through disinhibitory mechanisms involving tonically firing interneurons (purple). Ketamine leads to enhanced somatic and spine Ca2+ transients in layer 2/3 pyramidal cells within 1 h and increases multicellular ensemble activity within 3 h of administration (left). Ketamine, its metabolite HNK, and SPs each increase neurotrophic signaling and AMPAR insertion in mPFC excitatory neurons. The sustained antidepressant effects of ketamine and psychedelics involve targeted dendritic spinogenesis on excitatory projection neurons (right). Ketamine-induced spine formation is detected 12 h after administration and follows circuit reorganization. (b) In hippocampal SC to CA1 synapses, ketamine rapidly enhances BDNF release and increases postsynaptic glutamatergic transmission (left) while a sustained effect involves the delayed increase in pMeCP2 through a BDNF-dependent mechanism (right). Abbreviations: 5HT2A, 5-hydroxy-tryptamine receptor 2A; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor; BDNF, brain-derived neurotrophic factor; Ca2+, calcium; CaMKII, calcium-calmodulin kinase II; eEF2, eukaryotic elongation factor 2; GABA, γ-aminobutyric acid; Glu, glutamate; HNK, hydroxynorketamine; mGluR2/3, metabotropic glutamate receptor; mPFC, medial prefrontal cortex; mTORC1, mammalian target of rapamycin complex 1; NMDAR, N-methyl-d-aspartate receptor; pMeCP2, phosphorylated methyl-CpG-binding protein 2; PSD95, postsynaptic density protein 95; SC, Schaffer collateral; SP, serotonergic psychedelic; TBG, tabernanthalog; TrkB, tropomyosin receptor kinase B.
Third, these potent effects on activity-dependent BDNF and TrkB signaling are associated with direct and rapid effects on synapse formation. Ketamine increases the expression of synaptic proteins within 2 h after treatment and increases synapse density in prefrontal pyramidal neurons within 24 h, effects that require mTOR activation and BDNF (Lepack et al. 2014, Li et al. 2010). Two-photon imaging studies of spine remodeling in vivo indicate that these effects are driven by increased formation of new postsynaptic dendritic spines (with no effect on spine elimination), peaking 12–24 h after treatment, and ~20% of these new synapses persist for at least 2 weeks (Moda-Sava et al. 2019, Phoumthipphavong et al. 2016). Spectroscopy studies in humans have shown that ketamine rapidly increases prefrontal glutamate release (Abdallah et al. 2018; Milak et al. 2016, 2020) and modulates synaptic mGluR5 signaling (DeLorenzo et al. 2015, Esterlis et al. 2018). Structural MRI studies also provide indirect evidence that ketamine may reverse depression-related changes in neuronal structure (Abdallah et al. 2017).
3.3. Psychedelics and Psychedelic Analogs Target Synapse Dysfunction
A resurgence of interest in the therapeutic potential of psychedelic agents is heralding a second revolution in the development of novel antidepressants targeting synaptic plasticity. A recent randomized controlled trial showed that psilocybin—a mushroom-derived hallucinogen—had antidepressant benefits comparable to escitalopram (Carhart-Harris et al. 2021). These benefits were associated with enduring effects on amygdala function at least one month after treatment (Barrett et al. 2020). In two recent studies in mice (Hesselgrave et al. 2021, Shao et al. 2021), a single dose of psilocybin had antidepressant-like effects on learned helplessness and hedonic behavior (see Supplemental Discussion). These were associated with increased dendritic spine density in prefrontal layer 5 pyramidal neurons driven by increased spine formation rates and enhanced glutamatergic neurotransmission and persisted for at least 1 month after treatment.
Whether psilocybin-induced hallucinations (and ketamine-induced dissociative symptoms) are required for mediating effects on plasticity and behavior remains unclear and is a particularly pressing question for the field. In both studies (Hesselgrave et al. 2021, Shao et al. 2021), pretreatment with the 5-HT2AR antagonist ketanserin disrupted psilocybin’s effects on head twitching, a common behavioral screen for hallucinogenic activity in rodents, but not its effects on spine density or depression-related behaviors. As noted by the authors, this dose of ketanserin results in only partial blockade of 5-HT2AR and may be more rapidly cleared from the brain than psilocybin, so 5-HT2AR agonism may still be the mechanism by which psilocybin produces its antidepressant effects. Analogously, a newly described NMDAR positive allosteric modulator, GLYX-13 (Rapastinel), produced rapid and sustained antidepressant effects similar to ketamine in preclinical studies but without psychotomimetic side effects (Liu et al. 2017). Like ketamine, GLYX-13 rapidly activated the mTORC1 pathway and increased BDNF release and spinogenesis on mPFC layer 5 pyramidal neurons. Together, these convergent findings suggest that the antidepressant-like behavioral and plasticity effects of psilocybin and ketamine may be independent of psychotomimetic properties.
In addition to naturally occurring psychedelics, newly synthesized analogs of psychedelic compounds have shown therapeutic potential and again appear to act by promoting synaptogenesis. One such compound—termed tabernanthalog (TBG) and derived from ibogaine, a naturally-occurring psychedelic alkaloid—was designed and synthesized specifically to minimize hallucinogenic potential and other toxicities while preserving its 5-HT2AR agonism (Cameron et al. 2021). A single dose of TBG had antidepressant-like properties in the forced swim test and produced long-lasting therapeutic effects on cued reinstatement of conditioned heroin-seeking behavior. It also increased dendritic complexity in cultured cortical neurons and dendritic spine density in primary sensory cortex by promoting spine formation (Lu et al. 2021). Interestingly, the effects of TBG on both spine formation and forced swim test behavior 1 week after treatment were somewhat smaller than ketamine’s, consistent with a functional role for spine formation in mediating long-term antidepressant effects. Similarly promising results were obtained in a second recent report (Dong et al. 2021), which used a genetically encoded 5-HT sensor platform to identify a novel psychedelic analog (AAZ-A-154) with low hallucinogenic potential and potent antidepressant-like behavioral effects that were associated with dendritic outgrowth in cultured cells. In both cases, a critical next step will be to confirm that these drugs do not have hallucinogenic effects in humans and then test their therapeutic utility in carefully controlled, randomized trials.
3.4. Summary
Conventional monoamine-targeting antidepressants as well as ketamine, psychedelic compounds, and novel psychedelic analogs appear to act in part by enhancing synaptic transmission and promoting synaptogenesis in areas that lose synapses in chronic stress states. Although most studies have examined either chronic stress effects or antidepressant effects, a growing body of work (summarized in Supplemental Table 1) has examined both manipulations together and indicates that all three antidepressant classes have targeted rescue effects on the same functional and structural properties altered in chronic stress. Interestingly, although they have qualitatively similar effects on synapse function, those effects differ in degree and in the time required to achieve them: SSRIs have modest effects on synapse function and synaptogenesis that emerge slowly over weeks, while ketamine and psychedelics have rapid, potent effects after a single treatment, but they may not persist absent additional interventions. These differences may relate to their different therapeutic properties. Unbiased transcriptomic and proteomic screening studies are also identifying promising new therapeutic targets for promoting and sustaining synaptic remodeling (see the sidebar titled Discovering New Therapeutic Targets for Synapse Dysfunction). These findings raise several important questions: What is the relationship between stress and antidepressant effects? Is synaptogenesis required or merely correlated with antidepressant effects? Most importantly, if it is required, what purpose does it serve? Below, we review new data indicating that distinct mechanisms are responsible for initiating antidepressant effects versus sustaining them over time, that antidepressants act initially to facilitate a reconfiguration of the stress-induced brain state, and that antidepressant-induced synaptogenesis is critical specifically for sustaining that reconfiguration and maintaining remission.
DISCOVERING NEW THERAPEUTIC TARGETS FOR SYNAPSE DYSFUNCTION.
Several synaptic function–related genes are known to be downregulated in depression (Kang et al. 2007, 2012; Kim & Webster 2011, Labonté et al. 2017), including those associated with synaptic strength, vesicle transport, neurotransmission, spine growth, and axonal regeneration (Howard et al. 2019, Levey et al. 2021). Therefore, one path to new pharmacotherapies may lie in uncovering novel molecular mechanisms contributing to synaptic plasticity deficits. As a notable example, a transcriptional repressor, GATA1, which is upregulated in major depressive disorder, is sufficient to produce depressive-like behaviors, dendritic atrophy, and spine loss, suggesting a novel substrate for synaptic abnormalities in depression (Kang et al. 2012). The synaptogenic effects of antidepressants are likewise mediated by a host of molecular changes targeting protein synthesis, trafficking, scaffolding, and structural integrity of synapses. Some overlap has been reported in genetic variants associated with rapid antidepressant response (Guo et al. 2018) and psychedelic-induced changes in BDNF and synapse-related gene expression (Martin & Nichols 2018). Thus, large-scale unbiased genomic, transcriptomic, and proteomic studies have the potential to inform novel drug candidates with synaptic targets to sustain remission.
4. DEFINING MECHANISTIC ROLES FOR ANTIDEPRESSANT-INDUCED SYNAPSE FORMATION
Synapse loss and growth are closely associated, respectively, with the emergence of depression-related behaviors in chronic stress states and with the rescue of those behaviors after antidepressant treatment. Does synaptogenesis play a key causal role in mediating antidepressant effects? Emerging evidence from multiple sources indicates that distinct mechanisms are involved in initiating antidepressant effects, while others are involved in sustaining them over time.
We have shown that in multiple chronic stress models, the emergence of depression-related behavior is associated with spatially targeted, branch-specific elimination of postsynaptic dendritic spines on mPFC projection neurons and a reduction in coordinated, multicellular ensemble activity as measured by two-photon calcium imaging (Moda-Sava et al. 2019)—a conclusion supported by convergent findings in a recent study (Wilke et al. 2022). Ketamine rescued all three effects. One way of understanding causal relationships is to delineate the temporal sequence of these events. Unexpectedly, ketamine’s effects on behavior in the tail suspension test preceded those on spine formation, indicating that spine formation in PFC pyramidal neurons could not be required for initiating ketamine’s antidepressant effects. Interestingly, ketamine’s effects on ensemble activity in PFC microcircuits also preceded spine formation, suggesting that functional changes might be involved in initiating spine formation effects, while new synapses might be important for sustaining those effects over time. Consistent with this hypothesis, preservation of restored spines was strongly correlated with the long-term maintenance of ketamine’s effects on behavior in the tail suspension test (Moda-Sava et al. 2019).
To test this hypothesis directly, we used a recently developed optogenetic tool (Hayashi-Takagi et al. 2015)—activated synapse photoactivatable Rac1 (AS-PaRac1)—to selectively eliminate newly formed spines after ketamine treatment. This disrupted the effect of ketamine on synaptogenesis and interfered with the maintenance of ketamine’s effects on motivated escape behavior (Moda-Sava et al. 2019). Thus, synaptogenesis in prefrontal pyramidal neurons is not required for initiating ketamine’s antidepressant behavioral effects, but it is required for sustaining them over time.
If synaptogenesis is required only for sustaining ketamine’s antidepressant effects, how are they initiated? One potential mechanism involves rapid effects of ketamine on inhibitory interneurons to disinhibit hypoactive pyramidal cells (Li et al. 2010; Radley et al. 2006; Rajkowska et al. 1999, 2007; Yuen et al. 2012). Ketamine increases glutamatergic tone in the prefrontal cortex, likely through inhibition of tonically firing inhibitory interneurons (Homayoun & Moghaddam 2007, Moghaddam et al. 1997). These effects are mediated by GluN2B receptors on prefrontal somatostatin (SST) and parvalbumin (PV) interneurons, which are required for ketamine’s antidepressant behavioral effects (Gerhard et al. 2020). Ketamine directly inhibits the activity of prefrontal SST interneurons, leading to enhanced somatic and spine calcium transients in layer 2/3 pyramidal neurons within 1 h of administration (Ali et al. 2020) (Figure 2).
Dopamine signaling may also be involved in initiating ketamine’s behavioral effects. Chemogenetic activation of dopamine receptor 1 (Drd1)-expressing neurons mimicked the antidepressant effects of both systemic ketamine administration and local ketamine infusion into mPFC (Wu et al. 2021b). Drd1 blockade in mPFC prevented ketamine’s potentiation of glutamate-evoked spinogenesis ex vivo, while VTA inhibition also prevented ketamine-evoked spinogenesis 24 h after administration (Wu et al. 2021a). In another study, chronic stress disrupted correlated activity in PFC microcircuits, while ketamine had the opposite effect, through a mechanism that requires dopamine D2 receptors (Wilke et al. 2022). Together, these studies suggest that ketamine’s antidepressant behavioral effects may be initiated by rapid disinhibition of PFC projection neurons by SST and PV interneurons and by dopaminergic signaling, which is required for later effects on synaptogenesis. New synapses, in turn, are critical for sustaining these effects in the long term.
New evidence also points to distinct molecular mechanisms for initiating versus sustaining ketamine’s antidepressant effects (Figure 2). While mTOR, BDNF, and TrkB signaling are critical initiating processes (Autry et al. 2011, Li et al. 2010), ketamine’s sustained effects are transcription dependent and mediated by a delayed, BDNF-driven increase in hippocampal expression and phosphorylation of the transcriptional regulator MeCP2 (Kim et al. 2021). Phosphorylated methyl-CpG-binding protein 2 (pMeCP2), in turn, is required for the long-term maintenance of ketamine’s effects on synaptic strength and behavior (Kim et al. 2021).
5. CONCLUSIONS AND FUTURE DIRECTIONS
Available evidence supports an emerging model—consistent with other recent formulations (Castrén & Hen 2013, Duman & Aghajanian 2012, Kavalali & Monteggia 2020, Russo & Nestler 2013)—in which antidepressants act by enhancing plasticity in stress-sensitive circuits and promoting synaptogenesis. New synapses are hypothesized to play a specific role in sustaining antidepressant effects over time, prolonging remission, and preventing relapse. Chronic stress induces a shift in brain state in which neurons in distributed brain regions assume distinct functional properties that are associated with widespread alterations in synaptic connectivity. The most consistent findings indicate that chronic stress and depression are associated with neuronal hypoactivity, dendritic retraction, and synapse loss in the hippocampus and PFC, which may underlie deficits in memory, cognitive flexibility, attention, and cognitive control. Conversely, increased neural activity and spinogenesis have been described in the amygdala, dorsal striatum, VTA, and LHb, which may underlie alterations in anxiety, aversive learning, habit-driven behavior, and reward processing. In the NAc, another hub for reward processing, mixed effects of stress have been reported, associated in part with cell type. Importantly, with increased spine elimination, decreased spine formation, and decreased spine survival in chronic stress states, the capacity for enduring plasticity may be reduced.
Antidepressants enhance plasticity, which may initially facilitate a reconfiguration of brain state. Driven initially by enhanced BDNF, TrkB, and dopaminergic signaling and disinhibition of pyramidal cells by inhibitory interneurons, a variety of antidepressants (albeit on different time scales) reverse stress effects on neuronal activity in stress-sensitive circuits. Later, new synapses emerge, supported by pMeCP2 and D1R dopamine signaling, and synaptogenesis appears to be particularly important for sustaining remission.
This model raises several outstanding points. Perhaps most pressingly, our circuit-level understanding of the functional consequences of these changes and their relation to behavior is rudimentary. Chronic stress and antidepressants have consistent, opposing effects on synapse density, dendritic arborization, and neuronal activity, but it is unclear why the directionality of these effects varies by region. On a more fundamental level, we do not understand how these functional changes contribute to behavior. New approaches integrating single-cell and mesoscale calcium imaging, optogenetics, and multielectrode recordings with more interpretable behavioral paradigms probing decision-making, reward processing, motivation, cognitive flexibility, and reinforcement learning will be essential for linking these levels of analysis. Although they are outside the scope of this review, multiple recent studies are already advancing our understanding of the circuit- and network-level mechanisms mediating depression-related behavior and mood state transitions in both rodents (Chaudhury et al. 2013, Hultman et al. 2018, McGirr et al. 2017, Otis et al. 2017, Spellman et al. 2021, Tye et al. 2013, Warden et al. 2012) and human subjects (Kirkby et al. 2018, Scangos et al. 2021). Likewise, efforts to dissect the molecular, transcriptional, and epigenetic mechanisms underlying synapse loss in depression and activity-dependent synaptogenesis after antidepressant treatment will be critical for developing fundamentally new therapeutic approaches (Castrén & Hen 2013, Halldorsdottir & Binder 2017, Vialou et al. 2013). Finally, depression is a highly heterogeneous syndrome, not a unitary disease entity. Different pathophysiological mechanisms may be operational in subgroups of patients (Drysdale et al. 2017, Labonté et al. 2017, Williams 2016, Xia et al. 2018) and may contribute to individual differences in stress resilience and behavioral outcomes (Cerniauskas et al. 2019, Krishnan et al. 2007). This implies that the model we outlined here is oversimplified, and therapeutic interventions engaging different forms of plasticity in distinct brain regions may be required for achieving optimal outcomes across subtypes and individuals.
These questions notwithstanding, the model outlined above also has immediate implications for future studies. It suggests that interventions aimed at boosting plasticity, increasing synapse formation, and enhancing the survival of new synapses may be especially useful for sustaining remission and preventing relapse—a hypothesis that could be tested relatively easily in preclinical models. Such interventions could be especially important early during recovery when new synapses are more labile. This model also suggests strategies for tailoring existing interventions to achieve these goals—principles that are already informing the design of experimental neurostimulation therapies with notable success (Cole et al. 2020, Fox et al. 2012, Johansen-Berg et al. 2008, Mayberg et al. 2005).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funds to C.L. from the Rita Allen Foundation, the Hope for Depression Research Foundation, the Foundation for OCD Research, the National Institute of Mental Health, and the National Institute on Drug Abuse. P.K.P. was supported by a National Research Service Award F32 fellowship and a K99 Pathway to Independence Award from the National Institute of Mental Health.
DISCLOSURE STATEMENT
C.L. serves on the scientific advisory board of Delix Therapeutics and has formerly served as a consultant to Compass Group P.L.C. The other authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Abdallah CG, Averill LA, Gueorguieva R, Goktas S, Purohit P, et al. 2020. Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacology 45(6):990–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdallah CG, De Feyter HM, Averill LA, Jiang L, Averill CL, et al. 2018. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology 43(10):2154–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdallah CG, Jackowski A, Salas R, Gupta S, Sato JR, et al. 2017. The nucleus accumbens and ketamine treatment in major depressive disorder. Neuropsychopharmacology 42(8):1739–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali F, Gerhard DM, Sweasy K, Pothula S, Pittenger C, et al. 2020. Ketamine disinhibits dendrites and enhances calcium signals in prefrontal dendritic spines. Nat. Commun 11(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ampuero E, Rubio FJ, Falcon R, Sandoval M, Diaz-Veliz G, et al. 2010. Chronic fluoxetine treatment induces structural plasticity and selective changes in glutamate receptor subunits in the rat cerebral cortex. Neuroscience 169(1):98–108 [DOI] [PubMed] [Google Scholar]
- Andalman AS, Burns VM, Lovett-Barron M, Broxton M, Poole B, et al.2019. Neuronal dynamics regulating brain and behavioral state transitions. Cell 177(4):970–85.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, et al.2011. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475(7354):91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett FS, Doss MK, Sepeda ND, Pekar JJ, Griffiths RR. 2020. Emotions and brain function are altered up to one month after a single high dose of psilocybin. Sci. Rep 10(1):2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bath KG, Jing DQ, Dincheva I, Neeb CC, Pattwell SS, et al. 2012. BDNF Val66Met impairs fluoxetine-induced enhancement of adult hippocampus plasticity. Neuropsychopharmacology 37(5):1297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, et al. 2000. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47(4):351–54 [DOI] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, et al. 2006. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311(5762):864–68 [DOI] [PubMed] [Google Scholar]
- Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, et al. 2009. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol. Psychiatry 14(8):764–73 [DOI] [PubMed] [Google Scholar]
- Boldrini M, Santiago AN, Hen R, Dwork AJ, Rosoklija GB, et al. 2013. Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Neuropsychopharmacology 38(6):1068–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron LP, Tombari RJ, Lu J, Pell AJ, Hurley ZQ, et al. 2021.A non-hallucinogenic psychedelic analogue with therapeutic potential. Nature 589(7842):474–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carhart-Harris R, Giribaldi B, Watts R, Baker-Jones M, Murphy-Beiner A, et al. 2021. Trial of psilocybin versus escitalopram for depression. N. Engl. J. Med 384(15):1402–11 [DOI] [PubMed] [Google Scholar]
- Casarotto PC, Girych M, Fred SM, Kovaleva V, Moliner R, et al. 2021. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184(5):1299–313.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrén E, Hen R. 2013. Neuronal plasticity and antidepressant actions. Trends Neurosci. 36(5):259–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerniauskas I, Winterer J, de Jong JW, Lukacsovich D, Yang H, et al. 2019. Chronic stress induces activity, synaptic, and transcriptional remodeling of the lateral habenula associated with deficits in motivated behaviors. Neuron 104(5):899–915.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, et al. 2013. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 493(7433):532–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z-Y, Jing D, Bath KG, Ieraci A, Khan T, et al. 2006. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314(5796):140–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, et al. 2011. IκB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J. Neurosci 31(1):314–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole EJ, Stimpson KH, Bentzley BS, Gulser M, Cherian K, et al. 2020. Stanford accelerated intelligent neuromodulation therapy for treatment-resistant depression. Am. J. Psychiatry 177(8):716–26 [DOI] [PubMed] [Google Scholar]
- Conrad CD, Galea LAM, Kuroda Y, McEwen BS. 1996. Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine treatment. Behav. Neurosci 110(6):1321–34 [DOI] [PubMed] [Google Scholar]
- DeLorenzo C, DellaGioia N, Bloch M, Sanacora G, Nabulsi N,et al.2015.In vivo ketamine-induced changes in [11C]ABP688 binding to metabotropic glutamate receptor subtype 5. Biol. Psychiatry 77(3):266–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond DM, Rose GM. 1994. Stress impairs LTP and hippocampal-dependent memory. Ann. N. Y. Acad. Sci 746:411–14 [DOI] [PubMed] [Google Scholar]
- Dias-Ferreira E, Sousa JC, Melo I, Morgado P, Mesquita AR, et al. 2009. Chronic stress causes frontostriatal reorganization and affects decision-making. Science 325(5940):621–25 [DOI] [PubMed] [Google Scholar]
- Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, et al. 2010. A randomized add-on trial of an N-methyl-d-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry 67(8):793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Ly C, Dunlap LE, Vargas MV, Sun J, et al. 2021. Psychedelic-inspired drug discovery using an engineered biosensor. Cell 184(10):2779–92.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Furey ML. 2008. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct. Funct 213(1–2):93–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Videen TO, Price JL, Preskorn SH, Carmichael ST, Raichle ME. 1992. A functional anatomical study of unipolar depression. J. Neurosci 12(9):3628–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drysdale AT, Grosenick L, Downar J, Dunlop K, Mansouri F, et al. 2017. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat. Med 23(1):28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Aghajanian GK. 2012. Synaptic dysfunction in depression: potential therapeutic targets. Science 338(6103):68–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Aghajanian GK, Sanacora G, Krystal JH. 2016. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat. Med 22(3):238–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterlis I, DellaGioia N, Pietrzak RH, Matuskey D, Nabulsi N, et al. 2018. Ketamine-induced reduction in mGluR5 availability is associated with an antidepressant response: an [11C]ABP688 and PET imaging study in depression. Mol. Psychiatry 23(4):824–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman S, Conforti N, Saphier D. 1990. The preoptic area and bed nucleus of the stria terminalis are involved in the effects of the amygdala on adrenocortical secretion. Neuroscience 37(3):775–79 [DOI] [PubMed] [Google Scholar]
- Fox MD, Buckner RL, White MP, Greicius MD, Pascual-Leone A. 2012. Efficacy of transcranial magnetic stimulation targets for depression is related to intrinsic functional connectivity with the subgenual cingulate. Biol. Psychiatry 72(7):595–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox ME, Chandra R, Menken MS, Larkin EJ, Nam H, et al. 2020. Dendritic remodeling of D1 neurons by RhoA/Rho-kinase mediates depression-like behavior. Mol. Psychiatry 25(5):1022–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis TC, Chandra R, Gaynor A, Konkalmatt P, Metzbower SR, et al. 2017. Molecular basis of dendritic atrophy and activity in stress susceptibility. Mol. Psychiatry 22(11):1512–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard DM, Pothula S, Liu R-J, Wu M, Li X-Y, et al. 2020. GABA interneurons are the cellular trigger for ketamine’s rapid antidepressant actions. J. Clin. Investig 130(3):1336–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, et al. 2009. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience 164(2):798–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E. 1997. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J. Neurosci 17(7):2492–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Tanapat P, McEwen BS, Flügge G, Fuchs E. 1998. Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. PNAS 95(6):3168–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Machado-Vieira R, Mathew S, Murrough JW, Charney DS, et al. 2018. Exploratory genome-wide association analysis of response to ketamine and a polygenic analysis of response to scopolamine in depression. Transl. Psychiatry 9(1):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldorsdottir T, Binder EB. 2017. Gene x environment interactions: from molecular mechanisms to behavior. Annu. Rev. Psychol 68:215–41 [DOI] [PubMed] [Google Scholar]
- Hamilton JP, Gotlib IH. 2008. Neural substrates of increased memory sensitivity for negative stimuli in major depression. Biol. Psychiatry 63(12):1155–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. 2007. Reduced prefrontal glutamate/glutamine and γ-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 64(2):193–200 [DOI] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, et al. 2015. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525(7569):333–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm MS, Dankovich TM, Mandad S, Rammner B, Jähne S, et al. 2021. A large-scale nanoscopy and biochemistry analysis of postsynaptic dendritic spines. Nat. Neurosci 24(8):1151–62 [DOI] [PubMed] [Google Scholar]
- Hesselgrave N, Troppoli TA, Wulff AB, Cole AB, Thompson SM. 2021. Harnessing psilocybin: antidepressant-like behavioral and synaptic actions of psilocybin are independent of 5-HT2R activation in mice. PNAS 118(17):e2022489118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikida T, Kimura K, Wada N, Funabiki K, Nakanishi S. 2010. Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron 66(6):896–907 [DOI] [PubMed] [Google Scholar]
- Hirschfeld RM. 2000. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 61(Suppl. 6):4–6 [PubMed] [Google Scholar]
- Holmes SE, Scheinost D, Finnema SJ, Naganawa M, Davis MT, et al. 2019. Lower synaptic density is associated with depression severity and network alterations. Nat. Commun 10(1):1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. 2007. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci 27(43):11496–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard DM, Adams MJ, Clarke T-K, Hafferty JD, Gibson J, et al. 2019. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci 22(3):343–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultman R, Ulrich K, Sachs BD, Blount C, Carlson DE, et al. 2018. Brain-wide electrical spatiotemporal dynamics encode depression vulnerability. Cell 173(1):166–80.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iñiguez SD, Aubry A, Riggs LM, Alipio JB, Zanca RM, et al. 2016. Social defeat stress induces depression-like behavior and alters spine morphology in the hippocampus of adolescent male C57BL/6 mice. Neurobiol. Stress 5:54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, Pu Z, Wiegert O, Oitzl MS, Krugers HJ. 2006. Learning under stress: How does it work? Trends Cogn. Sci 10(4):152–58 [DOI] [PubMed] [Google Scholar]
- Johansen-Berg H, Gutman DA, Behrens TEJ, Matthews PM, Rushworth MFS, et al. 2008. Anatomical connectivity of the subgenual cingulate region targeted with deep brain stimulation for treatment-resistant depression. Cereb. Cortex 18(6):1374–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Adams DH, Simen A, Simen BB, Rajkowska G, et al. 2007. Gene expression profiling in postmortem prefrontal cortex of major depressive disorder. J. Neurosci 27(48):13329–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, et al. 2012. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med 18(9):1413–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, et al. 2011. Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science 334(6063):1731–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst H, Berger S, Turiault M, Tronche F, Schütz G, Joëls M. 2005. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. PNAS 102(52):19204–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. 2003. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 26(7):360–68 [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Monteggia LM. 2020. Targeting homeostatic synaptic plasticity for treatment of mood disorders. Neuron 106(5):715–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Koretz D, et al. 2003. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 289(23):3095–105 [DOI] [PubMed] [Google Scholar]
- Kim J-W, Autry AE, Na ES, Adachi M, Björkholm C, et al. 2021. Sustained effects of rapidly acting antidepressants require BDNF-dependent MeCP2 phosphorylation. Nat. Neurosci 24(8):1100–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Webster MJ. 2011. Integrative genome-wide association analysis of cytoarchitectural abnormalities in the prefrontal cortex of psychiatric disorders. Mol. Psychiatry 16(4):452–61 [DOI] [PubMed] [Google Scholar]
- Kirkby LA, Luongo FJ, Lee MB, Nahum M, Van Vleet TM, et al. 2018. An amygdala-hippocampus subnetwork that encodes variation in human mood. Cell 175(6):1688–700.e14 [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Chawla JM, Hagi K, Zarate CA, Kane JM, et al. 2016. Single-dose infusion ketamine and non-ketamine N-methyl-d-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychol. Med 46(7):1459–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Han M-H, Graham DL, Berton O, Renthal W, et al. 2007. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131(2):391–404 [DOI] [PubMed] [Google Scholar]
- Labonté B, Engmann O, Purushothaman I, Menard C, Wang J, et al. 2017. Sex-specific transcriptional signatures in human depression. Nat. Med 23(9):1102–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshminarasimhan H, Chattarji S. 2012. Stress leads to contrasting effects on the levels of brain derived neurotrophic factor in the hippocampus and amygdala. PLOS ONE 7(1):e30481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecca S, Pelosi A, Tchenio A, Moutkine I, Lujan R, et al. 2016. Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression-like phenotypes in mice. Nat. Med 22:254–61 [DOI] [PubMed] [Google Scholar]
- LeGates TA, Kvarta MD, Tooley JR, Francis TC, Lobo MK, et al. 2018. Reward behaviour is regulated by the strength of hippocampus-nucleus accumbens synapses. Nature 564(7735):258–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepack AE, Fuchikami M, Dwyer JM, Banasr M, Duman RS. 2014. BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol 18(1):pyu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey DF, Stein MB, Wendt FR, Pathak GA, Zhou H, et al. 2021. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat. Neurosci 24:954–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, et al. 2011. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature 470(7335):535–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Zhou T, Liao L, Yang Z, Wong C, et al. 2013. βCaMKII in lateral habenula mediates core symptoms of depression. Science 341(6149):1016–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee B, Liu R-J, Banasr M, Dwyer JM, et al. 2010. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329(5994):959–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. 2012. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 487(7406):183–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P-Y, Ma ZZ, Mahgoub M, Kavalali ET, Monteggia LM. 2021.A synaptic locus for TrkB signaling underlying ketamine rapid antidepressant action. Cell Rep. 36(7):109513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston C, Chen AC, Zebley BD, Drysdale AT, Gordon R, et al. 2014. Default mode network mechanisms of transcranial magnetic stimulation in depression. Biol. Psychiatry 76:517–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston C, Cichon JM, Jeanneteau F, Jia Z, Chao MV, Gan W-B. 2013. Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat. Neurosci 16(6):698–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston C, Gan W-B. 2011. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. PNAS 108(38):16074–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston C, McEwen BS, Casey BJ. 2009. Psychosocial stress reversibly disrupts prefrontal processing and attentional control. PNAS 106(3):912–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, et al. 2006. Stress-induced alterations in pre-frontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci 26(30):7870–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R-J, Duman C, Kato T, Hare B, Lopresto D, et al. 2017. GLYX-13 produces rapid antidepressant responses with key synaptic and behavioral effects distinct from ketamine. Neuropsychopharmacology 42(6):1231–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Tjia M, Mullen B, Cao B, Lukasiewicz K, et al. 2021. An analog of psychedelics restores functional neural circuits disrupted by unpredictable stress. Mol. Psychiatry 10.1038/s41380-021-01159-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui S, Wu Q, Qiu L, Yang X, Kuang W, et al. 2011. Resting-state functional connectivity in treatment-resistant depression. Am. J. Psychiatry 168(6):642–48 [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. 2009. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci 10(6):434–45 [DOI] [PubMed] [Google Scholar]
- Magariños AM, McEwen BS, Flügge G, Fuchs E. 1996. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J. Neurosci 16(10):3534–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maras PM, Baram TZ. 2012. Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci. 35(5):315–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DA, Nichols CD. 2018. The effects of hallucinogens on gene expression. Curr. Top. Behav. Neurosci 36:137–58 [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O. 2007. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature 447(7148):1111–15 [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. 2001. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci 4(11):1086–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, et al. 2005. Deep brain stimulation for treatment-resistant depression. Neuron 45(5):651–60 [DOI] [PubMed] [Google Scholar]
- McEwen BS. 1998. Protective and damaging effects of stress mediators. N. Engl. J. Med 338(3):171–79 [DOI] [PubMed] [Google Scholar]
- McEwen BS. 2007. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol. Rev 87(3):873–904 [DOI] [PubMed] [Google Scholar]
- McGaugh JL. 2004. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu. Rev. Neurosci 27:1–28 [DOI] [PubMed] [Google Scholar]
- McGirr A, LeDue J, Chan AW, Xie Y, Murphy TH. 2017. Cortical functional hyperconnectivity in a mouse model of depression and selective network effects of ketamine. Brain 140(8):2210–25 [DOI] [PubMed] [Google Scholar]
- Milak MS, Proper CJ, Mulhem ST, Parter AL, Kegeles LS, et al. 2016. A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol. Psychiatry 21(3):320–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milak MS, Rashid R, Dong Z, Kegeles LS, Grunebaum MF, et al. 2020. Assessment of relationship of ketamine dose with magnetic resonance spectroscopy of Glx and GABA responses in adults with major depression: a randomized clinical trial. JAMA Netw. Open 3(8):e2013211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, et al. 2019. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364(6436):aat8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. 1997. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci 17(8):2921–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlen K, Ockenfels H. 1968. Morphologische Veränderungen im Diencephalon und Telencephalon nach Störungen des Regelkreises Adenohypophyse-Nebennierenrinde. Z. Zellforsch. Mikrosk. Anat 93:126–41 [PubMed] [Google Scholar]
- Muir J, Tse YC, Iyer ES, Biris J, Cvetkovska V, et al. 2020. Ventral hippocampal afferents to nucleus accumbens encode both latent vulnerability and stress-induced susceptibility. Biol. Psychiatry 88(11):843–54 [DOI] [PubMed] [Google Scholar]
- Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, et al. 2013. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am. J. Psychiatry 170(10):1134–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE. 2010. Animal models of neuropsychiatric disorders. Nat. Neurosci 13(10):1161–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofer N, Berger DR, Kasthuri N, Lichtman JW, Yuste R. 2021.Ultrastructural analysis of dendritic spine necks reveals a continuum of spine morphologies. Dev. Neurobiol 81(5):746–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Namboodiri VMK, Matan AM, Voets ES, Mohorn EP, et al. 2017. Prefrontal cortex output circuits guide reward seeking through divergent cue encoding. Nature 543(7643):103–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare CM, Sandler M. 1959. A clinical and biochemical study of a trial of iproniazid in the treatment of depression. J. Neurol. Neurosurg. Psychiatry 22:247–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoumthipphavong V, Barthas F, Hassett S, Kwan AC. 2016. Longitudinal effects of ketamine on dendritic architecture in vivo in the mouse medial frontal cortex. eNeuro 3(2):ENEURO.0133-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzagalli DA, Holmes AJ, Dillon DG, Goetz EL, Birk JL, et al. 2009. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am. J. Psychiatry 166(6):702–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo M-M. 2001. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci 2(1):24–32 [DOI] [PubMed] [Google Scholar]
- Post RM, Denicoff KD, Leverich GS, Altshuler LL, Frye MA, et al. 2003. Morbidity in 258 bipolar outpatients followed for 1 year with daily prospective ratings on the NIMH life chart method. J. Clin. Psychiatry 64(6):680–90 [DOI] [PubMed] [Google Scholar]
- Radley JJ, Anderson RM, Hamilton BA, Alcock JA, Romig-Martin SA. 2013. Chronic stress-induced alterations of dendritic spine subtypes predict functional decrements in an hypothalamo-pituitary-adrenal-inhibitory prefrontal circuit. J. Neurosci 33(36):14379–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, et al. 2006. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb. Cortex 16(3):313–20 [DOI] [PubMed] [Google Scholar]
- Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, et al. 2004. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience 125(1):1–6 [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, et al. 1999. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 45(9):1085–98 [DOI] [PubMed] [Google Scholar]
- Rajkowska G, O’Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. 2007. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology 32(2):471–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Venheim ER, Padival M. 2010. Chronic stress causes amygdala hyperexcitability in rodents. Biol. Psychiatry 67(12):1128–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Nestler EJ. 2013. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci 14(9):609–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Baldwin P, de Biasi M, Montague PR. 2010. BOLD responses to negative reward prediction errors in human habenula. Front. Hum. Neurosci 4:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, Wu Y-T, Appel M, et al. 2004. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch. Gen. Psychiatry 61(7):705–13 [DOI] [PubMed] [Google Scholar]
- Sartorius A, Kiening KL, Kirsch P, von Gall CC, Haberkorn U, et al. 2010. Remission of major depression under deep brain stimulation of the lateral habenula in a therapy-refractory patient. Biol. Psychiatry 67(2):e9–11 [DOI] [PubMed] [Google Scholar]
- Scangos KW, Makhoul GS, Sugrue LP, Chang EF, Krystal AD. 2021. State-dependent responses to intracranial brain stimulation in a patient with depression. Nat. Med 27(2):229–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabel SJ, Proulx CD, Piriz J, Malinow R. 2014. GABA/glutamate co-release controls habenula output and is modified by antidepressant treatment. Science 345(6203):1494–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L-X, Liao C, Gregg I, Davoudian PA, Savalia NK, et al. 2021. Psilocybin induces rapid and persistent growth of dendritic spines in frontal cortex in vivo. Neuron 109(16):2535–44.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA. 2001. Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol. Psychiatry 50(9):651–58 [DOI] [PubMed] [Google Scholar]
- Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. 1996. Hippocampal atrophy in recurrent major depression. PNAS 93(9):3908–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama Y, Chen AC-H, Nakagawa S, Russell DS, Duman RS. 2002. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci 22(8):3251–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. 2011. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 476(7361):458–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, et al. 2010. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science 327(5967):863–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman T, Svei M, Kaminsky J, Manzano-Nieves G, Liston C. 2021. Prefrontal deep projection neurons enable cognitive flexibility via persistent feedback monitoring. Cell 184(10):2750–66.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, Monteggia LM. 2017. Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546(7659):E1–3 [DOI] [PubMed] [Google Scholar]
- Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai H-C, et al. 2013. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 493(7433):537–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Feng J, Robison AJ, Nestler EJ. 2013. Epigenetic mechanisms of depression and antidepressant action. Annu. Rev. Pharmacol. Toxicol 53:59–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Robison AJ, Laplant QC, Covington HE 3rd, Dietz DM, et al. 2010. ΔFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat. Neurosci 13(6):745–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas A, Bernal S, Chattarji S. 2003. Effects of chronic stress on dendritic arborization in the central and extended amygdala. Brain Res. 965(1–2):290–94 [DOI] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Rao BSS, Chattarji S. 2002. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J. Neurosci 22(15):6810–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas A, Pillai AG, Chattarji S. 2004. Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience 128(4):667–73 [DOI] [PubMed] [Google Scholar]
- Wang J-W, David DJ, Monckton JE, Battaglia F, Hen R. 2008. Chronic fluoxetine stimulates maturation and synaptic plasticity of adult-born hippocampal granule cells. J. Neurosci 28(6):1374–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, et al. 2012. A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 492(7429):428–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Gould E, McEwen BS. 1992. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 588(2):341–45 [DOI] [PubMed] [Google Scholar]
- Wellman CL. 2001. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J. Neurobiol 49(3):245–53 [DOI] [PubMed] [Google Scholar]
- Whitfield-Gabrieli S, Ford JM. 2012. Default mode network activity and connectivity in psychopathology. Annu. Rev. Clin. Psychol 8:49–76 [DOI] [PubMed] [Google Scholar]
- Wilke SA, Lavi K, Byeon S, Donohue KC, Sohal VS. 2022. Convergence of clinically relevant manipulations on dopamine-regulated prefrontal activity underlying stress coping responses. Biol. Psychiatry 91(9):810–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LM. 2016. Precision psychiatry: a neural circuit taxonomy for depression and anxiety. Lancet Psychiatry 3(5):472–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, Gould E, McEwen BS. 1990. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 531(1–2):225–31 [DOI] [PubMed] [Google Scholar]
- Wu M, Minkowicz S, Dumrongprechachan V, Hamilton P, Kozorovitskiy Y. 2021a. Ketamine rapidly enhances glutamate-evoked dendritic spinogenesis in medial prefrontal cortex through dopaminergic mechanisms. Biol. Psychiatry 89(11):1096–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Minkowicz S, Dumrongprechachan V, Hamilton P, Xiao L, Kozorovitskiy Y. 2021b. Attenuated dopamine signaling after aversive learning is restored by ketamine to rescue escape actions. eLife 10:e64041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CH, Ma Z, Ciric R, Gu S, Betzel RF, et al. 2018. Linked dimensions of psychopathology and connectivity in functional brain networks. Nat. Commun 9(1):3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Cui Y, Sang K, Dong Y, Ni Z, et al. 2018. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 554(7692):317–22 [DOI] [PubMed] [Google Scholar]
- Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. 2012. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73(5):962–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. 2001. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu. Rev. Neurosci 24:1071–89 [DOI] [PubMed] [Google Scholar]
- Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, et al. 2016. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533(7604):481–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, et al. 2006. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63(8):856–64 [DOI] [PubMed] [Google Scholar]
- Zhang J-Y, Liu T-H, He Y, Pan H-Q, Zhang W-H, et al. 2019. Chronic stress remodels synapses in an amygdala circuit-specific manner. Biol. Psychiatry 85(3):189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.