

Abstract
Approximately 45% of the human genome is composed of transposable elements (TEs). Expression of these elements is tightly regulated during normal development. TEs may be expressed at high levels in embryonic stem cells but are epigenetically silenced in terminally differentiated cells. As part of the global “epigenetic dysregulation” that cells undergo during transformation from normal to cancer, TEs can lose epigenetic silencing and become transcribed, and, in some cases, active. Here, we summarize recent advances detailing the consequences of TE activation in cancer and describe how these understudied residents of our genome can both aid tumorigenesis and potentially be harnessed for anti-cancer therapies.
Keywords: Transposable elements, epigenetics, cancer, P53, LINEs, SINEs, LINE-1, ERVs, viral mimicry
Classes of transposable elements
Transposable elements (TEs), which compose approximately 45% of our genome, are globally dysregulated during cancer progression [1]. TEs are categorized into two general classes, the details of which have been extensively reviewed elsewhere [2–4]. Class I TEs, often referred to as retrotransposons, move throughout the genome via reverse-transcribed RNA intermediates while class II TEs, or DNA transposons, move autonomously throughout the genome as DNA segments (Figure 1a) [5]. Class I TEs include long-terminal repeat (LTR)-containing elements, such as endogenous retroviruses (ERVs). ERVs make up about 8% of the human genome and are the remnants of exogenous retroviruses that integrated into the germline millions of years ago. Like exogenous retroviruses, ERVs are composed of three genes, gag, pol, and env, flanked by LTRs [6]. The majority (90%) of these germline integrated ERV sequences have accumulated mutations over time or seen recombination between two LTRs, excising the provirus and leaving behind a solo LTR [6]. Class I TEs also include non-LTR transposons, such as long interspersed elements (LINEs) and short interspersed nuclear elements (SINEs, including Alu elements), which together make up about 27% of the human genome [7]. LINE-1 elements (L1) are the only known autonomously active retrotransposons in the human genome. SINEs are non-autonomous and require L1 proteins to facilitate their retrotransposition, as do another class of non-LTRs, SINE-VNTR-Alu elements (SVAs) (Figure 1a) [8–9].
Figure 1.
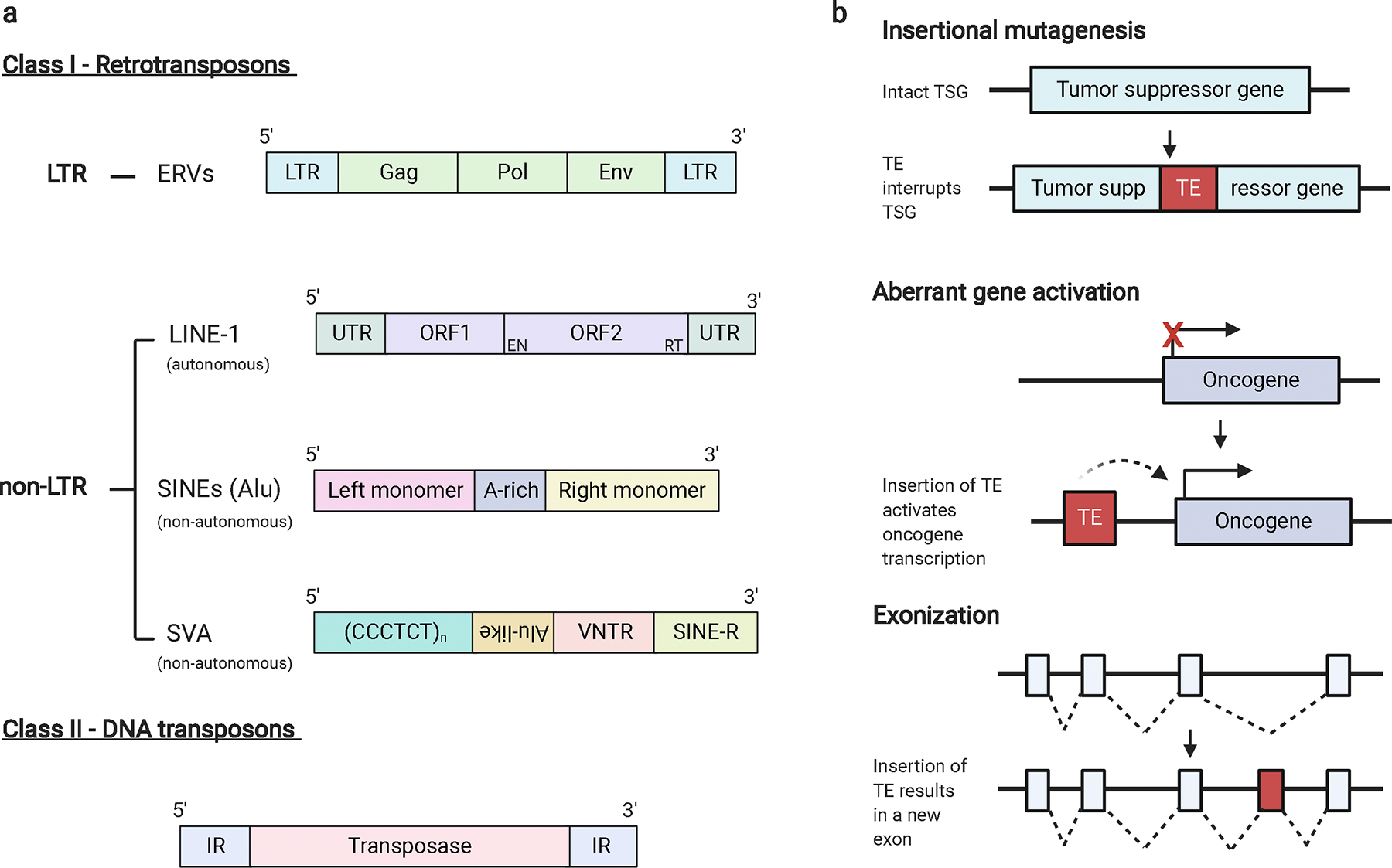
Mammalian TE classification and common mechanisms of mutagenesis.
a. TEs consist of 2 classes: class I (retrotransposons) and class II (DNA transposons). Class I TEs are further divided into those containing long-terminal repeats (LTRs) and those that do not (non-LTRs). ERVs are composed of gag, pol, and env genes flanked by LTRs. Non-LTRs consist of LINEs, SINEs, and SINE-VNTR-Alus (SVAs). LINE-1 elements consist of 2 open reading frames (ORFs) located between untranslated regions (UTRs). The second ORF encodes an endonuclease and reverse transcriptase. SINE elements, such as Alus, consist of a left-monomer and a right monomer separated by an A-rich region. SVAs contain a hexameric repeat of the sequence CCCTCT, an antisense Alu-like element, a variable number of tandem repeats (VNTR) region, and a SINE region (SINE-R). Class II transposons consist of a transposase flanked by inverted repeat (IR) regions. b. TE retrotransposition can promote oncogenesis via insertional mutagenesis (top), in which the insertion of a TE into a tumor suppressor gene (TSG) renders the gene inactive. Retrotransposed TEs can also act as promoters of oncogenes, rendering a normally silenced gene active (middle). TE mobilization can create new exons, a process called exonization, changing the resulting mRNA and protein composition (bottom).
During normal development, transcriptional regulation and expression of TEs is tightly controlled. ERVs may be expressed at high levels in embryonic stem cells (ESCs) but are silenced by DNA methylation and repressive histone modifications in terminally differentiated cells [10]. LINE and SINE elements are similarly silenced by DNA methylation, histone modifications, and RNA-mediated silencing [11]. As part of the global epigenetic dysregulation that cells undergo during transformation from normal to cancer, both LTR and non-LTR elements can lose epigenetic silencing and become transcribed, and, in some cases, active [12]. Despite considerable data showing dysregulation and increased expression of these elements, it is currently unclear whether they are benign “passengers” of tumorigenesis or whether they could play a role in tumor initiation and evolution. Epigenetic regulation of these elements is key to understanding their role in tumor initiation and progression.
Epigenetic regulation of TEs in healthy cells
If left unchecked, the constant mobilization of TEs throughout the genome would lead to rapid genetic instability. TE mobilization can lead to genetic disruption in a myriad of ways, including insertional mutagenesis, modification of regulatory sequences, inappropriate activation of gene transcription, and exonization, among others (Figure 1b) [5, 13–21]. To prevent this potentially mutagenic mobilization, most TEs are epigenetically silenced in somatic cells. Although evidence does not completely rule out the exaptation hypothesis, which postulates that a subset of TEs have been co-opted, or exapted, into regulatory or coding elements by the host organism to increase its fitness [19, 22–24]. However, the vast majority of TEs are epigenetically silenced via several mechanisms, including DNA methylation, histone modifications, and RNA-mediated silencing.
DNA methylation
DNA methylation, a process carried out by DNA methyltransferase (DNMT) enzymes, is an epigenetic silencing mark that predominantly involves the covalent addition of a methyl group to a cytosine base that precedes a guanine, termed a CpG dinucleotide [25]. Non-CpG methylation – methylation of cytosine bases followed by an adenine, thymine, or another cytosine base – has also been reported in the human genome, mostly in ESCs and brain tissue [26–29]. DNA methylation silences gene transcription by recruiting methyl-binding domain proteins to prevent the transcription machinery from accessing a particular stretch of DNA [30].
The majority of methylated cytosines in the genome are located in TEs, mediating their suppression [31]. All catalytic DNMT enzymes contribute to TE silencing. In mouse embryos deficient in the maintenance methyltransferase DNMT1, LTR transcript levels were elevated 50–100-fold [32]. Upon double knockout of the catalytic de novo DNMTs DNMT3A and DNMT3B in murine ESCs, methylation at cytosine residues preceding an adenosine (CpA sites) was entirely absent [33]. While DNA methylation of TEs is fairly stable in somatic cells, the epigenetic reprogramming that occurs in germ cells and pre-implantation embryos is associated with high levels of TE activity [31]. In a study of non-CpG methylation, Guo et al. reported evidence for strand-specific non-CpG methylation in LINEs and SINEs, but not LTRs [34].
DNA methylation levels of TEs are inversely correlated with the age of certain TEs. Ohtani et al. has shown that DNA methylation is typically found on younger TEs, specifically human ERVs, which are more CpG-rich than older ERVs, due to the relationship between CpG methylation and mutation rate [35–36]. A methylated cytosine (5-methylcytosine) can undergo spontaneous deamination to a thymine base, irreversibly changing the DNA sequence [37–38]. The longer an ERV element has been integrated into the genome, the more cytosine to thymine mutations it will have accumulated, thus decreasing the CpG content of that particular ERV. Interestingly, a recent study by Zhou et al. hypothesized an interplay between genome expansion, TE insertion, and the mutation of methylated CpG sites over time [39]. Upon observing a negative correlation between genome size and methylation of CpG sites within TEs, this whole-genome sequencing analysis of 53 organisms suggests that the spontaneous deamination of 5-methylcytosine bases to thymines may render a once epigenetically silenced TE active, ultimately leading to genomic expansion. Overall, DNA methylation plays an important role in silencing several classes of TEs in both developing and terminally differentiated mammalian cells.
Histone modifications
In addition to methylation of DNA, the tails of histones--the positively charged proteins around which DNA is wrapped to package our genome-- can be covalently modified to render a particular region of DNA more or less accessible to the cell’s transcription machinery. Unlike DNA methylation, histone methylation can be activating or suppressive depending on the location and degree to which a particular histone is methylated. Numerous other histone modifications exist, some of which include acetylation, phosphorylation, and ubiquitination [40]. Common repressive histone modifications, often associated with heterochromatin, are the trimethylation of lysines 9 and 27 on histone 3 (H3K9me3 and H3K27me3, respectively), which are frequently found on nucleosomes at TE loci [41–42]. Compared to younger, more recently acquired ERVs silenced by DNA methylation, the LTRs of some older ERVs are silenced by repressive histone modifications, including H3K9me2 and H3K9me3 [35]. In murine ESCs, LINE elements were enriched in dimethylation of arginine 3 on histone 4 (H4R3me2), marks associated with gene repression [43–44].
These silencing histone modifications are deposited by several groups of enzymes, often coming together to form multimeric complexes. Methylation of H3K9 is carried out by the H3-specific histone methyltransferase SETDB1, also known as the H3K9 methyltransferase ERG-associated protein with SET domain (ESET) and KMT1E [45–47]. In murine ESCs, SETDB1 is required for ERV silencing through interaction with Krüppel associated box (KRAB) associated protein 1 (KAP1, also called TRIM28) via the bridging KRAB zinc-finger protein 809 (ZFP809) [48–51]. This sequence-specific KRAB-ZFP/KAP1/SETDB1 axis mediates de novo histone methylation of ERVs in murine and human ESCs [52–53]. The number of KRAB-ZFPs is strongly correlated with the number of LTRs, suggesting KRAB-ZFPs and TEs may have co-evolved [54]. Indeed, the relationship between TEs and KRAB-ZFPs is often referred to as an evolutionary “arms race” [55]. KAP1 has also been implicated in silencing LINE-1 (L1) elements—the only functional, protein-coding retrotransposon in humans. L1 elements were initially shown to be silenced via DNA methylation in normal tissues as well as in human ESCs [56–57]. Recent work implicates the Human Silencing Hub (HUSH) complex with TRIM28/KAP1 in silencing of L1 elements [58].
Another group of protein complexes involved in histone methylation, specifically of H3K27, are the chromatin-modifier polycomb-repressive complexes 1 and 2 (PRC1 and PRC2) [41, 59–60]. In murine ESCs, Leeb and colleagues demonstrated that PRC1 and PRC2 act redundantly to stably suppress ERVs via the addition of H3K27me3 marks [61]. However, PRC2 alone likely plays a minor role in suppression of ERVs [35].
Other chromatin complexes have been implicated in mediating TE silencing. In murine ESCs, a chromatin-associated complex composed of death-domain associated protein (DAXX) and α-thalassemia/mental retardation X-linked (ATRX) has been shown to mediate TE repression by recruiting the H3K9 histone methyltransferase SUV39H [62]. Additionally, the chromatin remodeler SWI/SNF2-related, matrix associated, actin-dependent regulator of chromatin, subfamily A, member 6 (SMARCA6)—also known as lymphoid specific helicase (LSH)—contributes to L1 silencing in primary murine embryonic fibroblasts [63–64]. In murine ESCs, the SWI/SNF-like remodeler SMARCAD1 contributes to ERV silencing in an ATP-dependent manner [65–66]. Altogether, the silencing of TEs via a complex array of histone modifications deposited by several enzyme complexes illustrates the great lengths cells go to in order to silence these genomic elements.
RNA-mediated silencing
Although most TEs are epigenetically silenced by DNA methylation and histone modifications, RNA-mediating silencing of several TEs has been reported in mammals. Short (also referred to as small) interfering RNAs (siRNAs)—exogenously produced double stranded, non-coding RNAs complementary to a given mRNA sequence—can suppress L1 retrotransposition in human cell lines [67]. Depletion of DICER, a component of the RNA-induced silencing complex (RISC), results in a two-fold increase of L1 retrotransposition in these cell lines [67]. In murine ESCs harboring deletion of Dnmt1, TE repression was dependent upon DICER [68]. Micro-RNAs (miRNAs)– endogenously produced single-stranded non-coding RNAs that function similarly to siRNAs and are used to regulate normal cellular processes– have also been implicated in TE silencing. Hamdorf et al. identified miR-128 as a direct repressor of L1 retrotransposition in human cancer cells and induced pluripotent stem cells [69]. Aside from the RNA interference mediators siRNAs and miRNAs, PIWI-interacting RNAs (piRNA)—single-stranded RNAs that guide PIWI proteins to genomic sites to be silenced—have been implicated in L1 repression. piRNAs can restrict L1 retrotransposition in male mouse germ cells as well as in human induced pluripotent stem cells [70–75].
Silencing of TE transcription can also occur via Spen, the repressor protein of the long noncoding RNA Xist, which is involved in X chromosome inactivation [76]. In haploid mouse ESCs, Spen knockout resulted in a marked increase in ERV transcripts, especially from ERV-K. Loss of Spen was associated with a decrease in suppressive H3K9me3 marks along with a gain in activating H3K27ac and H3K4me3 marks. The RNA-binding domain of Spen binds to RNA transcripts derived from ERV-K in V6.5 murine ESCs.
Additional mechanisms of TE silencing have been identified, such as the cytoplasmic retroviral sensor TRIM5α, which restricts L1 retrotransposition via recognition of L1 ribonucleoprotein complexes [77]. L1 elements are also repressed by p53, a relationship discussed in detail below [78]. This extensive epigenetic silencing of TEs via DNA methylation, suppressive histone modifications, RNA-mediated silencing, and regulatory proteins prevents their mobilization, promoting the integrity and stability of the genome.
Epigenetic dysregulation of TEs in cancer
Due to the global epigenetic dysregulation that occurs during tumorigenesis, many TEs lose silencing and are transcribed in malignant cells. Cancer cells exhibit changes in the silencing DNA methylation mark, including global loss of methylation at regions that are silenced for genome stability, like TEs, and gain of methylation at the promoter regions of tumor suppressor genes [79]. A comparison of prostate cancer and normal cell lines found no significant differences in non-CpG methylation, suggesting that the hallmark methylation changes characteristic of cancer cells occur predominantly at CpG sites [80]. Histone modifications are likewise altered in cancer. Reduction of the repressive histone modification H4K20me3 is not only a hallmark of cancer but is also associated with hypomethylation of TEs [81].
One class of TEs that frequently escapes epigenetic silencing in cancer cells is ERVs. One potential explanation for ERV expression in malignancies was suggested in a recent RNA-seq analysis of 63 metastatic colorectal patients [82]. This study identified higher expression of the DNA demethylating enzymes TET2 and TET3 in tumors that had high levels of ERV expression, suggesting that demethylation may directly contribute to ERV transcription. These findings are supported by the numerous studies that have identified the expression of other human ERVs in cancers. ERV-K, a recently integrated ERV, is expressed in both hematologic and solid malignancies, including melanoma, breast cancer, ovarian cancer, pancreatic cancer, lymphoma, and prostate cancer [83–97]. In melanoma, expression of ERV-K may be due to increased transcriptional activity of the ERV-K promoter through demethylation of ERV-K LTRs [90]. ERV expression in ovarian cancer seems to be particularly common, as Wang-Johanning et al. have demonstrated the rare concurrent expression of multiple ERV surface envelope proteins--ERV-E, ERV-K, and ERV3 [87].
ERV-W is also frequently expressed in malignant cells. An ERV-targeted microarray study by Gimenez et al. revealed that the LTRs of six ERV-W loci upregulated in testicular cancer had methylation levels ranging from 0 – 30% in tumoral samples compared to 82 – 100% in DNA from adjacent healthy tissues [98]. The ERV-W env gene (syncytin-1), which facilitates trophoblast fusion and the formation of the placenta, plays a conflicting role in the pathogenesis and prognostic impact of several cancers [99–100]. Unlike many TEs that have been co-opted into regulatory elements in our genome, syncytin-1 is a unique case in which an ERV element has been adapted into a functional gene and is present only in eutherians (placental mammals) [99, 101]. Syncytin-1 expression has been detected in several leukemia and lymphoma cell lines as well as in patients with cutaneous T-cell lymphoma [102–103]. Syncytin-1 can promote invasion and metastasis via activation of endothelial-to-mesenchymal transition in endometrial carcinoma [104]. Although syncytin-1 promotes breast cancer-endothelial cell fusions, thereby promoting metastasis, it was also shown to be a positive prognostic indicator of recurrence-free survival in patients with breast cancer [105–106]. Two years later, the same group found that expression of syncytin-1 was correlated with a decrease in overall survival in rectal but not colon cancer patients, concluding that the prognostic impact of Syncytin-1 may vary according to the specific type of malignancy [107].
Lastly, several codogenic ERVs are expressed at varying levels in endometrial carcinoma and several of its prestages [108]. These numerous instances of ERV re-expression in various malignancies illustrate the consequences of the widespread epigenetic dysregulation of TEs in cancer.
The effect of TE expression on oncogenic pathways
The expression of some TEs in cancer due to global epigenetic dysregulation has oncogenic potential. TE transcription can drive expression of neighboring oncogenes in multiple cancer types [109]. A recent study demonstrated just how widespread the process of onco-exaptation—epigenetically reactivated TEs that function as cryptic promoters--is in cancer [109]. This study identified 129 onco-exaptation events involving 106 oncogenes across 3,864 tumors. At least one onco-exaptation event was identified in 49.7% of all the tumors with a prevalence of onco-exaptation between 10–80% across different cancer types. In a survey of Hodgkin’s Lymphoma cell lines, the expression of the transcription factor interferon regulatory factor 5 (IRF5) was driven by a normally dormant LTR located upstream of IRF5 [110]. Another Hodgkin’s Lymphoma study revealed that transcription of the proto-oncogene colony-stimulating factor 1 receptor (CSF1R) is initiated by an aberrantly activated LTR THE1B, a member of the MaLR family [111]. Lock et al. analyzed RNA-seq data from 101 diffuse large B-cell lymphoma patients and identified 98 TE-gene chimeric transcripts where TEs appeared to be functioning as the promoter [112]. A similar finding was detailed in a recent study by Jiang and Upton, in which TEs were found to serve as a frequent source of transcription factor binding sites and promoters in breast cancer [113].
In addition to functioning as promoters, epigenetically reactivated TEs can act as enhancers of benign genes as well as oncogenes. In healthy cells, two phylogenetically conserved TEs in mammals are required for expression of the proopiomelanocortin (Pomc) gene in the hypothalamus of transgenic mice [114]. In cancer cells, TEs have been implicated in driving the expression of a novel isoform of IL-33 in colon cancer, an oncogenic truncated transcript of ERBB4 in anaplastic large-cell lymphoma, and an alternative transcript of the MET oncogene in bladder cancer [115–117]. Recently, TEs have been shown to act as enhancers in acute myeloid leukemia and chromic myeloid leukemia cell lines. Analysis of acute myeloid leukemia (AML) patients revealed TEs were more enriched for active transcription marks compared to normal tissues and a subset of these demonstrated enhancer activity [118]. Some ERVs contain STAT1 binding sites and act as interferon-inducible enhancers for a subset of interferon stimulated genes, important in promoting the inflammatory response, a process discussed in detail below [119–120].
At the protein level, TEs—particularly ERVs—have oncogenic potential. Inducible expression of ERV-K in healthy mice disrupts germ cell development, ultimately leading to carcinoma in situ, the precursor lesion of seminoma [121]. Other studies in several cancer types have demonstrated that knockdown of ERVs via short hairpin RNA (shRNA) or siRNA targeting reduces tumor growth in both cell lines and mouse xenograft models [88, 122–124]. In a syngeneic mouse model, renal cells expressing ERV-K env promoted the formation of pulmonary metastases [125]. Interestingly, ERV-K promotes intercellular fusion of melanoma cell lines, and syncytin (ERV-W env) is involved in the fusion of breast cancer cells to endothelial cells, allowing malignant cells to escape breast tissue and metastasize [126–128]. Similarly, syncytin-1 upregulation activates the epithelial-mesenchymal transition pathway in endometrial carcinoma patients [104]. Thus, TEs, specifically ERVs, can contribute to transformation and tumor growth by functioning as promoters, enhancers, and at the protein level [129].
Roles of LINE-1 (L1) in cancer
L1 retrotransposition in oncogenesis
As the only functional retrotransposon in humans, L1 plays a particularly important role in contributing to genomic instability in cancers and its expression is a hallmark of cancer [130]. L1 is transcribed as a bicistronic RNA that encodes an RNA binding protein, open reading frame 1 protein (ORF1p, also known as p40), and an endonuclease and reverse transcriptase (ORF2p) [5] (Figure 1a). L1 elements are usually epigenetically silenced in normal somatic tissues, but occasionally L1 promoters become active, resulting in L1 transcription and production of the ORF1p protein [5]. ORF1p is a nucleic acid-binding protein that is essential for retrotransposition of L1 elements in the genome. L1 elements can be amplified via the activity of ORF2p, which reverse transcribes L1 mRNA and inserts L1 DNA into a new position within the genome [131].
Retrotransposition of L1 elements has conflicting roles in oncogenesis. In some malignancies, L1 insertions are driver mutations, directly contributing to oncogenic transformation, while in others, they are benign passengers [132–133]. The first case in which a somatic L1 insertion was shown to be oncogenic was in a case of breast ductal adenocarcinoma, in which an L1 sequence was identified in an intron of the oncogene MYC [134]. The second case involved an L1 insertion that inactivated the tumor suppressor gene adenomatous polyposis coli (APC) in a colorectal cancer patient [12]. In addition to causing familial adenomatous polyposis (FAP), a heritable predisposition of developing colorectal cancer, driver mutations in APC occur in a majority of sporadic colon cancers [135]. Since then, oncogenic L1 insertions in hepatocellular carcinoma have been shown to activate the oncogenic β-catenin/Wnt signaling pathway as well as the poorly characterized transcription factor suppression of tumorigenicity 18 (ST18) [136]. L1 insertions have also been shown to initiate transcript variants in esophageal adenocarcinoma, bladder, and breast carcinomas [117, 137–138]. The most comprehensive study of L1 retrotransposition in cancers, which analyzed just under 3,000 genomes of cancers from 38 histological subtypes, identified several oncogenic roles of L1 retrotransposition, including chromosomal deletions encompassing tumor-suppressor genes, structural variations, and promotion of breakage-fusion cycles that trigger oncogene amplification [139].
There are also reports of LINE reactivation in premalignancies, suggesting that early retrotransposon activation may be a causal factor in some cancers [140]. For example, L1 insertions are present in all gastrointestinal cancers and their metastases as well as in the cancer precursor colonic adenomas [141]. However, while the previously mentioned studies demonstrate the oncogenic potential of L1 insertions, only approximately 1% of L1 insertions are thought to be tumor-initiating events [133]. Several passenger mutations involving L1 insertions have been identified in colorectal, breast, lung, prostate, and ovarian cancers [142–145]. Thus L1 retrotransposition is common across cancer subtypes, providing potential areas for new therapeutic development.
Regulation of L1 by P53
The tumor suppressor P53 is one of the key regulators preventing cancer and the TP53 gene that encodes this protein is the most commonly mutated gene in human cancers [146]. Around 90% of TP53 mutations in human cancers, called “hotspot” mutations, cluster in the DNA binding domain of the gene [147]. Mutated P53 may prevent binding to canonical targets while also promoting oncogenic transcription by binding to other loci with different transcriptional binding partners [146–147]. P53 transcriptionally represses TEs in model organisms, including fruit flies and zebrafish [148], and has conserved binding sites in ERV and L1 elements in humans [149]. In fact, 30% of the predicted binding sites for P53 are in ERVs. As P53 is mutated in more than 50% of cancers [147], P53-deficient malignant cells are predisposed to elevated expression of TEs, which can alter the stability and composition of the genome.
There is contradictory evidence regarding the relationship between P53 and L1. An agonistic relationship has been reported in a germ cell line, in which a P53 binding site in a recently acquired L1 element can increase transcription of that element [148]. Another study found that wild-type P53 increased the transcription of L1 elements compared to mutant P53 [149]. As P53 is active in somatic cells and strongly responsive to double-stranded breaks in chromatin, it was proposed that activated L1 transcription leads to the production of ORF2p, which induces double stranded breaks and would cause increased P53 activity [149]. This positive feedback loop created in response to DNA damage would ultimately cause the cell to undergo apoptosis (Figure 2a). In support of this proposed mechanism, Haoudi and colleagues showed that driving L1 expression induced apoptosis in the wild type P53 cell line G418R HCT-116, but not the mutant P53 G418R SW480 line, suggesting therapeutic potential for malignancies with wild-type P53 [150].
Figure 2.
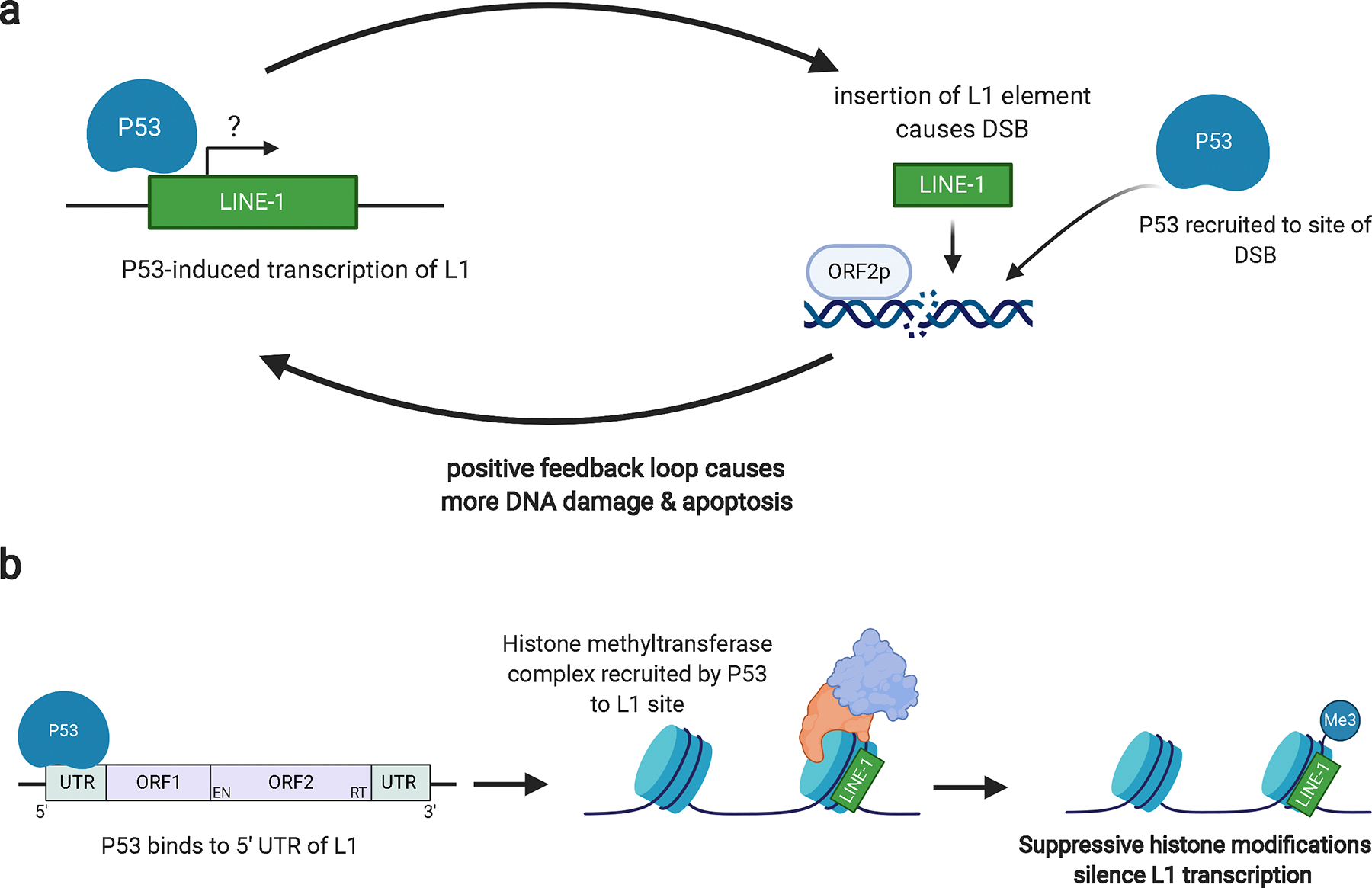
Models of the conflicting relationship between P53 and L1.
a. In an agonistic relationship, activation of P53 may induce L1 transcription, leading to the production of ORF1p and ORF2p proteins. ORF2p induces DSBs in order to promote L1 insertion. P53 is recruited to sites of DSBs and a positive feedback loop may be induced. The accumulation of severe DNA damage from several DSBs may cause the cell to undergo apoptosis. In cells with defective DNA replication and repair machinery, this damage will become irreparable, further driving the cell towards apoptosis. b. In an antagonistic relationship, P53 binding to the 5’ UTR of L1 elements recruits histone methyltransferase proteins. These epigenetic modifiers can then deposit suppressive histone modifications, silencing L1 transcription.
The majority of evidence, however, suggests an antagonistic relationship between p53 and L1. Ardeljan et al. have shown that L1 retrotransposition is limited by the P53 DNA damage response and interferon signaling [12]. In the context of DNA damage, P53 represses the transcription of Alu elements [151]. The most recent study to investigate the relationship between P53 and L1 showed that P53 directly represses L1 in human cell lines via binding to the 5’ untranslated region (UTR) of L1 elements and promoting the addition of repressive histone modifications (Figure 2b) [152]. At the protein level, a comparison of ORF1p protein expression in wild-type and P53-mutant Wilms tumors revealed that L1 could be readily detected in P53 mutant tumors but not in P53 wild-type tumors [78]. Subsequent analysis of colon cancer samples in The Cancer Genome Atlas (TCGA) found that L1 transcripts were significantly elevated in tumors that harbored P53 loss [78]. A similar trend has been observed in TP53-deficient lung, pancreatic, and ovarian carcinoma patient samples [130].
Indirect evidence for the antagonistic relationship between P53 and L1 exists as well. In ovarian cancers, STIC lesions—the carcinoma in situ precursors to high grade serous ovarian cancer— are characterized by mutant TP53 along with demethylation and increased expression of L1 elements, which may drive genomic instability [153]. L1 ORF1p expression is also detectable in pancreatic intraepithelial neoplasias—the precursors of pancreatic ductal adenocarcinomas [130, 140]. Thus, the “guardian of the genome”, P53, plays a complicated role in the regulation of TEs, having been shown to act as an activator and a repressor of TEs. The mutation of TP53 in >50% of cancers may thus significantly affect TE transcription and retrotransposition, the effects of which are discussed below in the contexts of cell stress and tumorigenesis.
L1 retrotransposition induces cell stress via the DNA damage response
When L1 elements are active and able to retrotranspose, this can activate the DNA damage response and limit cell growth. Ardeljan et al. showed that overexpressing L1 in retinal pigment epithelium (RPE) cells (non-transformed) triggers a TP53-mediated G1 arrest and an interferon response [12]. L1 expression inhibited RPE clonogenic growth, which was rescued by knockdown of P53. However, when using a reporter assay to compare L1 insertion frequency in control and TP53-knockdown cells, no significant difference was found. Therefore, P53 restricts growth of these cells but not their retrotransposition potential. This study also demonstrated that L1 retrotransposition occurs in association with DNA replication; the DNA repair mechanisms that are coupled with replication reduce retrotransposon intermediates and it is the loss of these repair pathways that enhances L1 retrotransposition. This study utilized a knockout CRISPR screen in TP53-deficient cells and identified genes that influence the fitness of L1+ cells, including replication-coupled DNA repair pathways, replication stress signaling, and replication-fork restart factors. Due to the apoptotic response facilitated by the induction of L1 retrotransposition in P53-competent cells, activation of human L1 could be used as a therapeutic strategy with intact P53. By activating L1 retrotransposition, genomic instability is induced, leading to DNA damage and eventually apoptosis (Figure 2a).
In addition to this genomic instability, cancer cell damage may be further compounded through the combined used of antineoplastic compounds currently used in clinical trials, such as the DNA synthesis inhibitor mitomycin C [154] or the Ataxia Telangiectasia and Rad3 related (ATR) inhibitor ceralasertib, which inhibits DNA repair [155]. Findings from a companion study reported that LINE-1 retrotransposition is restricted by double-stranded break (DSB) repair and Fanconi Anemia factors that are active in S/G2 phase [156], suggesting a possible contribution of L1 retrotransposition to the pathogenesis of breast and ovarian cancers, a significant percentage of which are deficient in the DSB protein BRCA1.
L1 protein expression in human malignancies
Interestingly, pathologic analysis of L1 ORF1p expression in colon cancer points to a selective advantage for cells that lack L1 expression. In a heterogenous L1 expression in colon cancer, Ardeljan et al. found that both L1+ and L1− parts of the primary tumor both shared a BRAF (V600E) mutation, as well as numerous somatically acquired L1 insertions prior to the cessation of retrotransposition in the L1− component [12]. The L1− section, which derived from a L1+ lineage, and the loss of L1 expression were associated with an increased growth rate.
Pathologic analysis of L1 expression in gynecologic malignancies supports the hypothesis that L1 expression requires disabled P53 signaling. Ovarian serous carcinoma, a type of tumor that arises mainly from the epithelial layer of cells in the ovaries, can be high-grade serous carcinoma (HGSC) or low-grade serous carcinoma (LGSC). HGSC contributes to the majority of deaths as it is the most common subtype of ovarian cancer, contributing to around 70% of all ovarian cancers [157–158]. About 90% of HGSCs have mutations in the TP53 gene while displaying a much lower mutational load in other genes [159–161]. HGSCs originate from precursor lesions of the distal fallopian tubes that likely evolved in a stepwise manner from P53 signature lesions [162–163]. During carcinogenesis, L1 promoter regions often become hypomethylated leading to the expression of the L1 proteins. Accumulation of ORF1p along with the formation of the precursor STIC lesions is followed by progression to HGSC, in which L1 expression persists [153].
Two recent pathologic studies detailed early and frequent L1 protein expression in gynecologic malignancies. L1 ORF1p overexpression is likely established during the evolution of ovarian cancer lesions, which begin with the emergence of a mutant P53 signature and progress toward the precursor STIC stage. L1 overexpression increased as the lesions progressed toward malignancy and upon seeding to the ovary, the level of L1 overexpression in HGSC-concomitant STIC lesions neared that of HGSC [153]. Expression of L1 was found to be prevalent in all histological subtypes of ovarian cancer, especially in HGSCs, which showed the highest L1 expression compared to the other subtypes that were tested [164]. These differences reflect the fact that virtually all HGSCs harbor TP53 mutations and therefore exhibit a higher level of genomic rearrangement. L1 ORF1p expression was correlated with mutated P53 in HGSC and HGSCs had significantly higher expression of L1 compared to other ovarian cancer subtypes, which generally have wild type P53 [153, 164]. The overexpression of ORF1p in subtypes of ovarian carcinomas suggests the potential utility of ORF1p as biomarker for these malignancies [164].
Perturbation of TE epigenetic regulation for cancer therapy
The above examples serve as a direct illustration of the importance of epigenetic silencing of TEs in somatic cells, as their reactivation frequently leads to rapid genome instability via insertional mutagenesis, aberrant TE expression, and interruption of regulatory elements. However, recent work shows that therapeutic activation of TEs in cancers can actually initiate an anti-tumor immune response. Cancer cells resist the immune response in a process known as immune editing or immune evasion [165]. Initially, immune cells including natural killer cells and T-effector cells fight and kill cancer cells, but as cancers progress, malignant cells exhibit mechanisms of immune suppression. For example, tumors often express PD-L1, a ligand for the PD-1 protein on CD3 T-cells that inhibits killer T-cell action against tumor cells. T-regulatory cells that express CTLA-4 secrete cytokines to inhibit the action of T- and natural killer cells against tumors [165]. Recent advances utilizing therapies that reverse this evasion (specifically, inhibition of PD-1 and CTLA-4, termed “immune checkpoint blockade”) have shown durable responses for a proportion of patients with solid tumors [120]. Response often correlates with the amount of tumor infiltrating lymphocytes and the number of tumor neoantigens recognized by T-cells attacking the tumor [120].
Treating cancer cells with DNA methyltransferase inhibitors (DNMTi) “boosts” immune signaling from tumors through activation of type I and III interferon signaling induced by detection of double-stranded RNA (dsRNA) derived from TEs [167–169]. Low doses of the DNMTis Azacytidine (Aza) [170] and 5-aza-2’-deoxycytidine (Dac) [171] upregulate immune signaling, including the interferon response, cytokines, and antigen processing and presentation in breast, colon, lung, and ovarian cancer cell lines [167–169, 172]. DNMTis reduce global DNA methylation, which results in the upregulation of dsRNA, particularly dsRNA derived from inverted-repeat Alu elements [173]. The cytoplasmic dsRNA sensors TLR3 and MDA5 [168] detect these genetic elements and initiate a canonical interferon signaling pathway in a process termed “viral mimicry” (Figure 3). Studies have shown that the interferon response induced by DNMTi treatment was abrogated by inhibiting dsRNA sensors MDA5 and TLR3, proving that transcription of TE-derived dsRNA species caused the interferon response [168–169]. Subsequent work showed that the interferon response induced by DNMTi treatment can be increased by adding HDAC inhibitors (HDACi) [174], inhibitors of H3K9 methyltransferases [175], or Vitamin C, which is a cofactor for the TET DNA demethylases [176]. Treatment with DNMTis plus HDACis increased ERV expression in a mouse model of ovarian cancer, activating interferon signaling and recruiting CD8+ T cells to kill the tumors [177].
Figure 3.
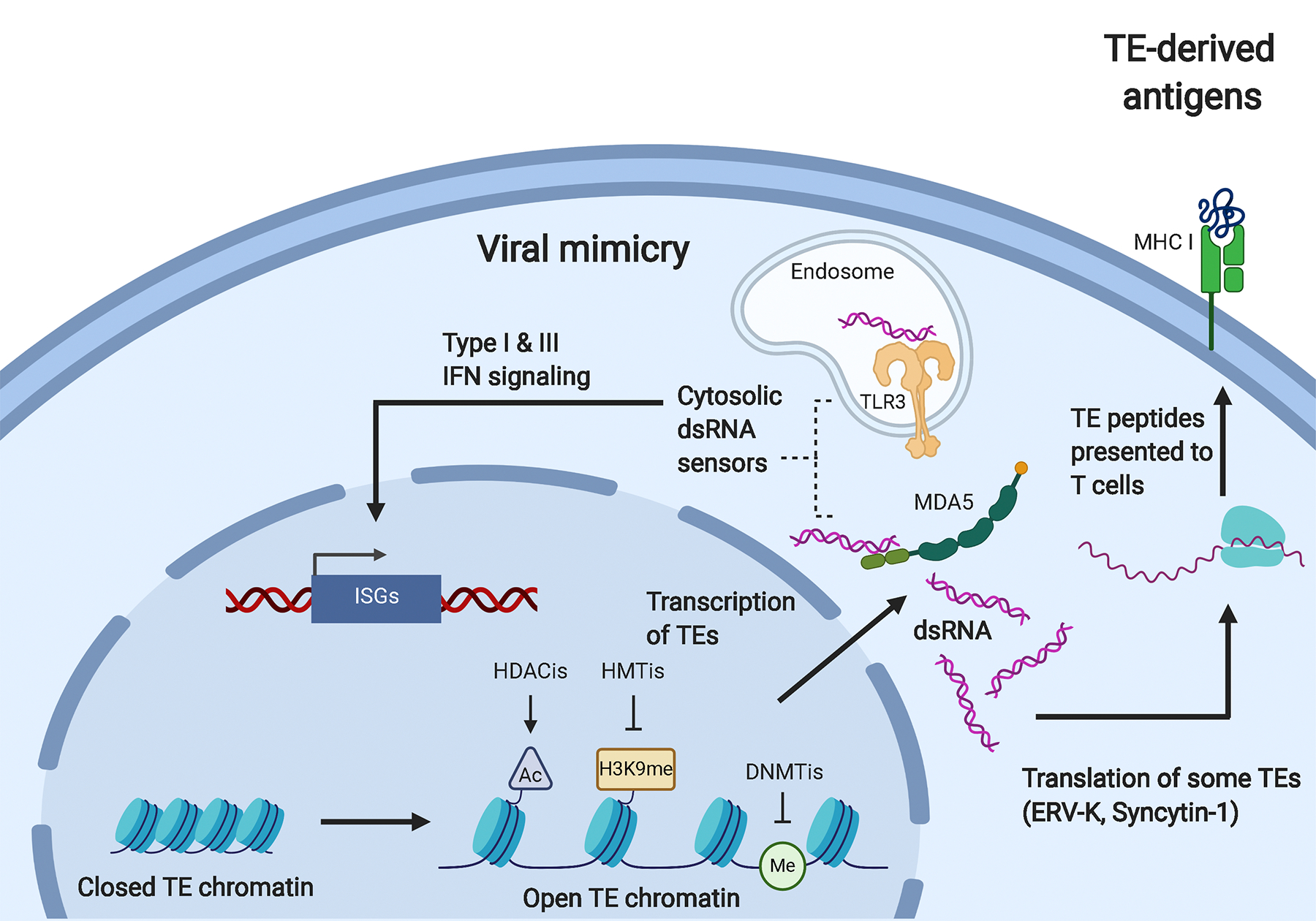
Anti-cancer therapeutic strategies using viral mimicry and TE-derived antigens.
Treatment of tumor cells with DNMTis, HDACis, and HMTis induces transcription of TEs. The dsRNA produced from TE transcription can be detected by the cytosolic dsRNA sensors MDA5 and TLR3. This detection induces type I and III interferon signaling, which results in the transcription of interferon-stimulated genes (ISGs) and induction of an inflammatory response, a process termed “viral mimicry.” The TE-derived dsRNA can also be translated into peptides, as is the case for ERV-K and Syncytin-1. These peptides can bind to MHC I molecules on the surface of the tumor where they can be recognized by T cells. Upon recognition of these tumor associated antigens, the tumor cells can be targeted and killed by immune cells.
In addition to induction of type I and III interferon signaling, viral mimicry has also been shown to occur through the loss of the H3K9 methyltransferase SETDB1 and the H3K4me1/2 demethylase LSD1 (also known as KDM1A). SETDB1 is known to be upregulated in several cancers and its loss in an AML cell line was shown to result in the transcription of dsRNA, promoting subsequent type I IFN signaling and apoptosis of AML cells [178]. Similarly, overexpression of LSD1 has been identified in multiple cancers [179–182]. Upon treatment of breast cancer cell lines with a catalytic inhibitor of LSD1, induction of viral mimicry was observed along with decreased expression of components of the dsRNA-degrading RISC complex [183–185]. This inhibition of LSD1 suppressed tumor growth and induced resistance to PD-1 blockade in vivo, further demonstrating the efficacy of using viral mimicry as a cancer therapy [184]. The HUSH complex has also been identified as a “gatekeeper” of type I IFN signaling via L1 repression [186]. Depletion of the HUSH complex in primary fibroblasts led to the induction of interferon stimulated genes (ISGs) via sensing of L1-derived dsRNA.
Pre-clinical and clinical studies testing epigenetic activation of TEs to induce immune signaling in cancer have shown promise. In mouse models, DNMTi treatment sensitized melanoma to subsequent anti-CTLA4 therapy [168, 187–188] and murine ovarian cancer to anti-PD-1 therapy [177]. The combination of Aza treatment with the depletion of the dsRNA-destabilizing enzyme ADAR1—which deaminates adenosine residues in RNA to inosines—has been shown to have synergistic anti-tumor effects in a xenograft mouse model of colorectal cancer [173]. A recent Phase Ib trial combining DNMTi treatment with anti-CTLA-4 in melanoma showed favorable results, including improved immune activation and anti-tumor activity [189]. This therapeutic exploitation of TEs to stimulate immune signaling is currently being tested in combination with checkpoint blockade therapy in clinical trials for melanoma, colorectal cancer, ovarian cancer, and kidney cancer, among others [189] (NCT01928576, NCT02961101, NCT03019003, NCT02811497, NCT02546986, NCT02397720, NCT02530463). Results from the METADUR trial-- a phase II trial that investigated the efficacy of CC-486, an oral form of Aza, with the T-cell survival promoter durvalumab (a monoclonal antibody against PD-1) in colorectal cancer, ovarian cancer, and breast cancer patients--were recently published [190]. It was found that viral mimicry was likely not induced as CC-486 did not penetrate the tumors, leading to no clinical responses from this trial. These data show the importance of developing epigenetic therapies with greater stability and improved pharmacokinetics to show efficacy in solid tumors.
Similarly, the cell cycle regulators cyclin-dependent kinases (CDKs) can also be therapeutically targeted to promote viral mimicry in cancer. As CDKs are integral for progression through the cell cycle, inhibitors of CDKs (CDKis) were initially developed to halt this process in rapidly dividing malignant cells [191–194]. Several studies have since demonstrated that inhibitors of CDK 4/6 also suppress the methyltransferase DNMT1 [195–197]. This inhibition of DNMT1 leads to hypomethylation of TEs, particularly ERVs, and can also be used to induce viral mimicry. While Cingöz and Goff have recently shown that CDKs are required for type I interferon production, the redundancy among CDK4/6 may be sufficient to overcome the post-transcriptional block of IFNβ induced by the CDK1/2/4 inhibitor R547 [198–199]. As inhibitors of CDK4/6 will selectively target rapidly proliferating cancer cells, this is another therapeutic strategy that can be used to boost immune signaling in tumor cells while sparing more quiescent healthy cells.
TEs as potential cancer antigens
In addition to the type I and III interferon response induced by dsRNA activation, a subset of TEs may be translated into proteins that can be presented as peptides and recognized by immune cells as tumor associated antigens (TAAs)—proteins expressed by tumor cells but are absent from normal cells [120, 200]. As discussed above, ERVs are frequently expressed in several cancer types. However, their foreign nature as a viral element presents an additional, and potentially exploitable, degree of antigenicity. The aberrant expression of ERVs in cancers has led several groups to pursue the development of ERV-targeted adaptive immune strategies, an approach made all the more promising by studies demonstrating the antigenicity of expressed ERVs in renal cell carcinoma [201–202]. In order for a T-cell to recognize an antigen on a target cell, the antigen must first be processed into a peptide fragment inside the target cell. This peptide fragment is then transported to the surface of the target cell, where it is presented to the immune system in the context of a major histocompatibility complex (MHC) molecule. The peptide-MHC complex is then recognized by a highly specific receptor on the surface of a T cell, which simultaneously binds both the antigen and the MHC molecule on the target cell [203].
Studies have not only identified ERV-specific T-cells in cancer patients, but have also shown that the corresponding TE-derived peptides are presented in the context of MHC class I molecules on tumors, confirming the potential of TEs as tumor-associated antigens (TAAs). A recent analysis of thousands of tumors from The Cancer Genome Atlas (TCGA) shows that TE expression in cancer is associated with immune infiltration and increased antigenicity [204]. The antigenic potential of these TAAs can be taken one step further by combining them with epigenetic therapeutics. A multi-omic analysis of glioblastoma cell lines treated with the DNMTi Dac identified 16 peptides from several TE subclasses that were expressed only in Dac-treated glioblastoma cells204. A study of myeloid malignancies identified greater numbers of ERV-specific T-cells in these patients compared to healthy donors205. These studies directly illustrate the potential use of TEs, specifically ERVs, as neoantigens induced via epigenetic manipulation.
Humoral immunity has also been studied, albeit less thoroughly than cell-mediated immunity, as antibodies against ERVs have been identified in several cancers. Antibodies against ERV-K have been detected in ovarian cancer and melanoma patients as well as in teratocarcinoma cell lines [87, 206–207]. Antibodies against ERV-E and ERV3 have also been identified in ovarian cancer patients [87]. These autologous humoral and T-cell responses to TEs that have been identified in cancer patients serve as validation for the development of adaptive immune strategies targeting TEs as novel cancer therapies.
Conclusion
As 45% of the human genome is composed of TEs, proper epigenetic silencing of these elements is integral to maintaining genome stability. In terminally differentiated cells, TE expression is inhibited via DNA methylation, suppressive histone modifications, and RNA-mediated mechanisms. In cancer cells, global epigenetic dysregulation of TEs leads to their expression, which can be oncogenic in some contexts. While the regulation of TEs is fairly well characterized, as is the expression of these elements in various malignancies, further exploration is needed in order to understand whether targeting TEs is a feasible therapeutic option. Recent work has shown that TE expression in cancer may be exploited via activation of the DNA damage response and innate immune signaling. Results from ongoing clinical trials combining epigenetic therapies that activate TEs with immune therapies will provide information on whether this combination can be effective in cancers. Further investigation is required in order to determine the feasibility of utilizing TEs as potential cancer antigens and/or biomarkers for specific cancers.
Acknowledgements
This manuscript was supported by the National Cancer Institute under Award R21CA227259 (to KBC), an award from The Mathers Foundation (to KBC), and The Marlene and Michael Berman Endowed Fund for Ovarian Cancer Research (to KBC). The authors would like to acknowledge the Institute for Biomedical Sciences at the George Washington University for graduate student support and training. Figures were created with BioRender.com. The funding sources above had no involvement in or influence on the writing or preparation of this manuscript.
Abbreviations
- TE
transposable elements
- ERV
endogenous retrovirus
- LTR
long terminal repeat
- LINE
long interspersed element
- SINE
short interspersed nuclear element
- SVA
SINE-VNTR-Alu
- ESC
embryonic stem cell
- CpG
cytosine-phosphate-guanine
- DNMT
DNA methyltransferase
- H3K9me2/3
histone 3 lysine 9 di/trimethylation
- H3K27me3
histone 3 lysine 27 trimethylation
- LINE-1 (L1)
long interspersed (nuclear) element-1
- HUSH
human silencing hub
- PRC1/2
polycomb repressive complex 1/2
- siRNA
short/small interfering RNA
- RISC
RNA-induced silencing complex
- RISC
RNA-induced silencing complex
- miRNA
microRNA
- piRNA
PIWI-interacting RNA
- shRNA
short hairpin RNA
- AML
acute myeloid leukemia
- UTR
untranslated region
- TCGA
The Cancer Genome Atlas
- STIC
serous tubal intraepithelial carcinoma
- RPE
retinal pigment epithelium
- DSB
double-stranded break
- HGSC
high-grade serous carcinoma
- LGSC
low-grade serous carcinoma
- PD-1
programmed cell death protein-1
- CTLA-4
cytotoxic T-lymphocyte associated protein-4
- DNMTi
DNA methyltransferase inhibitor
- dsRNA
double-stranded RNA
- Aza
azacytidine
- Dac
5-aza-2’-deoxycytidine
- TLR3
toll-like receptor-3
- MDA5
melanoma differentiation-associated protein-5
- ISG
interferon-stimulated gene
- CDK
cyclin dependent kinase
- CDKi
cyclin dependent kinase inhibitor
- MHC
major histocompatibility complex
- TAA
tumor associated antigen
Footnotes
Conflicts of Interest
KBC is a consultant for ROME Therapeutics, Inc.
References
- 1.Bannert N & Kurth R Retroelements and the human genome: new perspectives on an old relation. Proc. Natl. Acad. Sci. U. S. A. 101 Suppl 2, 14572–14579 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Platt RN, Vandewege MW & Ray DA Mammalian transposable elements and their impacts on genome evolution. Chromosome Res. 26, 25–43 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levin HL & Moran JV Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 12, 615–627 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burns KH Our Conflict with Transposable Elements and Its Implications for Human Disease. Annu. Rev. Pathol. 15, 51–70 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Burns KH Transposable elements in cancer. Nat. Rev. Cancer 17, 415–424 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Blomberg J, Benachenhou F, Blikstad V, Sperber G & Mayer J Classification and nomenclature of endogenous retroviral sequences (ERVs): problems and recommendations. Gene 448, 115–123 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Kassiotis G Endogenous retroviruses and the development of cancer. J. Immunol. Baltim. Md 1950 192, 1343–1349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck CR, Garcia-Perez JL, Badge RM & Moran JV LINE-1 Elements in Structural Variation and Disease. Annu. Rev. Genomics Hum. Genet. 12, 187–215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ostertag EM, Goodier JL, Zhang Y & Kazazian HH Jr. SVA Elements Are Nonautonomous Retrotransposons that Cause Disease in Humans. Am. J. Hum. Genet. 73, 1444–1451 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grow EJ et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 522, 221–225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burns KH & Boeke JD Human transposon tectonics. Cell 149, 740–752 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ardeljan D et al. Cell fitness screens reveal a conflict between LINE-1 retrotransposition and DNA replication. Nat. Struct. Mol. Biol. 27, 168–178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazazian HH et al. Haemophilia A resulting from de novo insertion of L 1 sequences represents a novel mechanism for mutation in man. Nature 332, 164–166 (1988). [DOI] [PubMed] [Google Scholar]
- 14.Miki Y et al. Disruption of the APC Gene by a Retrotransposal Insertion of L1 Sequence in a Colon Cancer. Cancer Res. 52, 643–645 (1992). [PubMed] [Google Scholar]
- 15.Britten RJ Cases of ancient mobile element DNA insertions that now affect gene regulation. Mol. Phylogenet. Evol. 5, 13–17 (1996). [DOI] [PubMed] [Google Scholar]
- 16.Nekrutenko A & Li W-H Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 17, 619–621 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Brosius J The contribution of RNAs and retroposition to evolutionary novelties. Genetica 118, 99–116 (2003). [PubMed] [Google Scholar]
- 18.Piriyapongsa J, Polavarapu N, Borodovsky M & McDonald J Exonization of the LTR transposable elements in human genome. BMC Genomics 8, 291 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feschotte C The contribution of transposable elements to the evolution of regulatory networks. Nat. Rev. Genet. 9, 397–405 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conley AB, Piriyapongsa J & Jordan IK Retroviral promoters in the human genome. Bioinformatics 24, 1563–1567 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Chuong EB, Elde NC & Feschotte C Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 18, 71–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huda A, Mariño-Ramírez L & Jordan IK Epigenetic histone modifications of human transposable elements: genome defense versus exaptation. Mob. DNA 1, 2 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gould SJ & Vrba ES Exaptation—a Missing Term in the Science of Form. Paleobiology 8, 4–15 (1982). [Google Scholar]
- 24.Kidwell MG & Lisch DR Transposable elements and host genome evolution. Trends Ecol. Evol. 15, 95–99 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Jones PA Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Patil V, Ward RL & Hesson LB The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 9, 823–828 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He Y & Ecker JR Non-CG Methylation in the Human Genome. Annu. Rev. Genomics Hum. Genet. 16, 55–77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramsahoye BH et al. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. 97, 5237–5242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo JU et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 17, 215–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curradi M, Izzo A, Badaracco G & Landsberger N Molecular Mechanisms of Gene Silencing Mediated by DNA Methylation. Mol. Cell. Biol. 22, 3157–3173 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jansz N DNA methylation dynamics at transposable elements in mammals. Essays Biochem. 63, 677–689 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Walsh CP, Chaillet JR & Bestor TH Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20, 116–117 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Arand J et al. In Vivo Control of CpG and Non-CpG DNA Methylation by DNA Methyltransferases. PLOS Genet. 8, e1002750 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo W, Chung W-Y, Qian M, Pellegrini M & Zhang MQ Characterizing the strand-specific distribution of non-CpG methylation in human pluripotent cells. Nucleic Acids Res. 42, 3009–3016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohtani H, Liu M, Zhou W, Liang G & Jones PA Switching roles for DNA and histone methylation depend on evolutionary ages of human endogenous retroviruses. Genome Res. 28, 1147–1157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lavie L, Kitova M, Maldener E, Meese E & Mayer J CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2). J. Virol. 79, 876–883 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen JC, Rideout WM & Jones PA The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 22, 972–976 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoder JA, Walsh CP & Bestor TH Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13, 335–340 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Zhou W, Liang G, Molloy PL & Jones PA DNA methylation enables transposable element-driven genome expansion. Proc. Natl. Acad. Sci. 117, 19359–19366 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strahl BD & Allis CD The language of covalent histone modifications. Nature 403, 41–45 (2000). [DOI] [PubMed] [Google Scholar]
- 41.An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ernst J et al. Systematic analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He J et al. Transposable elements are regulated by context-specific patterns of chromatin marks in mouse embryonic stem cells. Nat. Commun. 10, 34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Girardot M et al. PRMT5-mediated histone H4 arginine-3 symmetrical dimethylation marks chromatin at G + C-rich regions of the mouse genome. Nucleic Acids Res. 42, 235–248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schultz DC, Ayyanathan K, Negorev D, Maul GG & Rauscher FJ SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16, 919–932 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang L et al. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene 21, 148–152 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Lohmann F et al. KMT1E mediated H3K9 methylation is required for the maintenance of embryonic stem cells by repressing trophectoderm differentiation. Stem Cells Dayt. Ohio 28, 201–212 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Matsui T et al. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464, 927–931 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Wolf D & Goff SP TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 131, 46–57 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Wolf D & Goff SP Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458, 1201–1204 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rowe HM et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Quenneville S et al. The KRAB-ZFP/KAP1 System Contributes to the Early Embryonic Establishment of Site-Specific DNA Methylation Patterns Maintained during Development. Cell Rep. 2, 766–773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rowe HM et al. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development 140, 519–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas JH & Schneider S Coevolution of retroelements and tandem zinc finger genes. Genome Res. 21, 1800–1812 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bruno M, Mahgoub M & Macfarlan TS The Arms Race Between KRAB-Zinc Finger Proteins and Endogenous Retroelements and Its Impact on Mammals. Annu. Rev. Genet. 53, 393–416 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Castro-Diaz N et al. Evolutionally dynamic L1 regulation in embryonic stem cells. Genes Dev. 28, 1397–1409 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Florl AR, Löwer R, Schmitz-Dräger BJ & Schulz WA DNA methylation and expression of LINE-1 and HERV-K provirus sequences in urothelial and renal cell carcinomas. Br. J. Cancer 80, 1312–1321 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robbez-Masson L et al. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. 28, 836–845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bracken AP, Dietrich N, Pasini D, Hansen KH & Helin K Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 20, 1123–1136 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferrari KJ et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 53, 49–62 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Leeb M et al. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 24, 265–276 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He Q et al. The DAXX/ATRX Complex Protects Tandem Repetitive Elements During DNA Hypomethylation by Promoting H3K9 Trimethylation. Cell Stem Cell 17, 273–286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang J et al. Lsh, an epigenetic guardian of repetitive elements. Nucleic Acids Res. 32, 5019–5028 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu W et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 24, 1613–1623 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sachs P et al. SMARCAD1 ATPase activity is required to silence endogenous retroviruses in embryonic stem cells. Nat. Commun. 10, 1335 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fukuda K & Shinkai Y SETDB1-Mediated Silencing of Retroelements. Viruses 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang N & Kazazian HH L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat. Struct. Mol. Biol. 13, 763–771 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Berrens RV et al. An endosiRNA-Based Repression Mechanism Counteracts Transposon Activation during Global DNA Demethylation in Embryonic Stem Cells. Cell Stem Cell 21, 694–703.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamdorf M et al. miR-128 represses L1 retrotransposition by binding directly to L1 RNA. Nat. Struct. Mol. Biol. 22, 824–831 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Newkirk SJ et al. Intact piRNA pathway prevents L1 mobilization in male meiosis. Proc. Natl. Acad. Sci. U. S. A. 114, E5635–E5644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goodier JL & Kazazian HH Retrotransposons Revisited: The Restraint and Rehabilitation of Parasites. Cell 135, 23–35 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Saleh A, Macia A & Muotri AR Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol. 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tóth KF, Pezic D, Stuwe E & Webster A The piRNA Pathway Guards the Germline Genome Against Transposable Elements. Adv. Exp. Med. Biol. 886, 51–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Slotkin RK & Martienssen R Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Marchetto MCN et al. Differential L1 regulation in pluripotent stem cells of humans and apes. Nature 503, 525–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carter AC et al. Spen links RNA-mediated endogenous retrovirus silencing and X chromosome inactivation. eLife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Volkmann B et al. Human TRIM5α senses and restricts LINE-1 elements. Proc. Natl. Acad. Sci. 117, 17965–17976 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wylie A et al. p53 genes function to restrain mobile elements. Genes Dev. 30, 64–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sharma S, Kelly TK & Jones PA Epigenetics in cancer. Carcinogenesis 31, 27–36 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Truong M, Yang B, Wagner J, Desotelle J & Jarrard DF Analysis of Promoter Non-CG Methylation in Prostate Cancer. Epigenomics 5, 65–71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fraga MF et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37, 391–400 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Mendis SR et al. Comprehensive transcriptome analysis reveals link between epigenetic dysregulation, endogenous retrovirus expression and immunogenicity in metastatic colorectal carcinoma. J. Clin. Oncol. 37, 3535–3535 (2019). [Google Scholar]
- 83.Löwer R, Löwer J & Kurth R The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc. Natl. Acad. Sci. U. S. A. 93, 5177–5184 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang-Johanning F et al. Expression of human endogenous retrovirus k envelope transcripts in human breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 7, 1553–1560 (2001). [PubMed] [Google Scholar]
- 85.Wang-Johanning F et al. Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene 22, 1528–1535 (2003). [DOI] [PubMed] [Google Scholar]
- 86.Büscher K et al. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 65, 4172–4180 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Wang-Johanning F et al. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int. J. Cancer 120, 81–90 (2007). [DOI] [PubMed] [Google Scholar]
- 88.Serafino A et al. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp. Cell Res. 315, 849–862 (2009). [DOI] [PubMed] [Google Scholar]
- 89.Reiche J, Pauli G & Ellerbrok H Differential expression of human endogenous retrovirus K transcripts in primary human melanocytes and melanoma cell lines after UV irradiation. Melanoma Res. 20, 435–440 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Stengel S, Fiebig U, Kurth R & Denner J Regulation of human endogenous retrovirus-K expression in melanomas by CpG methylation. Genes. Chromosomes Cancer 49, 401–411 (2010). [DOI] [PubMed] [Google Scholar]
- 91.Katoh I et al. Activation of the Long Terminal Repeat of Human Endogenous Retrovirus K by Melanoma-Specific Transcription Factor MITF-M. Neoplasia N. Y. N 13, 1081–1092 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schanab O et al. Expression of human endogenous retrovirus K is stimulated by ultraviolet radiation in melanoma. Pigment Cell Melanoma Res. 24, 656–665 (2011). [DOI] [PubMed] [Google Scholar]
- 93.Schmitt K, Reichrath J, Roesch A, Meese E & Mayer J Transcriptional Profiling of Human Endogenous Retrovirus Group HERV-K(HML-2) Loci in Melanoma. Genome Biol. Evol. 5, 307–328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Singh S et al. Human endogenous retrovirus K (HERV-K) rec mRNA is expressed in primary melanoma but not in benign naevi or normal skin. Pigment Cell Melanoma Res. 26, 426–428 (2013). [DOI] [PubMed] [Google Scholar]
- 95.Reis BS et al. Prostate cancer progression correlates with increased humoral immune response to a human endogenous retrovirus GAG protein. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 19, 6112–6125 (2013). [DOI] [PubMed] [Google Scholar]
- 96.Jäger M et al. Alternate-locus aware variant calling in whole genome sequencing. Genome Med. 8, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li M et al. Down-regulation of human endogenous retrovirus type K (HERV-K) viral env RNA in pancreatic cancer cells decreases cell proliferation and tumor growth. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 23, 5892–5911 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gimenez J et al. Custom human endogenous retroviruses dedicated microarray identifies self-induced HERV-W family elements reactivated in testicular cancer upon methylation control. Nucleic Acids Res. 38, 2229–2246 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mi S et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785–789 (2000). [DOI] [PubMed] [Google Scholar]
- 100.Blond JL et al. An envelope glycoprotein of the human endogenous retrovirus HERV-W is expressed in the human placenta and fuses cells expressing the type D mammalian retrovirus receptor. J. Virol. 74, 3321–3329 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dupressoir A, Lavialle C & Heidmann T From ancestral infectious retroviruses to bona fide cellular genes: role of the captured syncytins in placentation. Placenta 33, 663–671 (2012). [DOI] [PubMed] [Google Scholar]
- 102.Sun Y et al. Expression of syncytin in leukemia and lymphoma cells. Leuk. Res. 34, 1195–1202 (2010). [DOI] [PubMed] [Google Scholar]
- 103.Maliniemi P et al. Expression of Human Endogenous Retrovirus-W Including Syncytin-1 in Cutaneous T-Cell Lymphoma. PLoS ONE 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu C et al. Upregulation of syncytin-1 promotes invasion and metastasis by activating epithelial-mesenchymal transition-related pathway in endometrial carcinoma. OncoTargets Ther. 12, 31–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bjerregaard B, Holck S, Christensen IJ & Larsson L-I Syncytin is involved in breast cancer-endothelial cell fusions. Cell. Mol. Life Sci. 63, 1906–1911 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Larsson L-I, Holck S & Christensen IJ Prognostic role of syncytin expression in breast cancer. Hum. Pathol. 38, 726–731 (2007). [DOI] [PubMed] [Google Scholar]
- 107.Larsen JM et al. Syncytin immunoreactivity in colorectal cancer: Potential prognostic impact. Cancer Lett. 280, 44–49 (2009). [DOI] [PubMed] [Google Scholar]
- 108.Agafonov DE, Huang Y, Grote M & Sprinzl M Efficient suppression of the amber codon in E. coli in vitro translation system. FEBS Lett. 579, 2156–2160 (2005). [DOI] [PubMed] [Google Scholar]
- 109.Jang HS et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet. 51, 611–617 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Babaian A et al. Onco-exaptation of an endogenous retroviral LTR drives IRF5 expression in Hodgkin lymphoma. Oncogene 35, 2542–2546 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Lamprecht B et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat. Med. 16, 571–579 (2010). [DOI] [PubMed] [Google Scholar]
- 112.Lock FE et al. Distinct isoform of FABP7 revealed by screening for retroelement-activated genes in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U. S. A. 111, E3534–E3543 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiang J-C & Upton KR Human transposons are an abundant supply of transcription factor binding sites and promoter activities in breast cancer cell lines. Mob. DNA 10, 16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lam DD et al. Partially Redundant Enhancers Cooperatively Maintain Mammalian Pomc Expression Above a Critical Functional Threshold. PLoS Genet. 11, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lock FE et al. A novel isoform of IL-33 revealed by screening for transposable element promoted genes in human colorectal cancer. PloS One 12, e0180659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Scarfò I et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood 127, 221–232 (2016). [DOI] [PubMed] [Google Scholar]
- 117.Wolff EM et al. Hypomethylation of a LINE-1 Promoter Activates an Alternate Transcript of the MET Oncogene in Bladders with Cancer. PLOS Genet. 6, e1000917 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zeng Y et al. Characterization of functional transposable element enhancers in acute myeloid leukemia. Sci. China Life Sci. 63, 675–687 (2020). [DOI] [PubMed] [Google Scholar]
- 119.Chuong EB, Elde NC & Feschotte C Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 351, 1083–1087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Topalian SL, Drake CG & Pardoll DM Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Galli UM et al. Human endogenous retrovirus rec interferes with germ cell development in mice and may cause carcinoma in situ, the predecessor lesion of germ cell tumors. Oncogene 24, 3223–3228 (2005). [DOI] [PubMed] [Google Scholar]
- 122.Li M et al. Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 23, 5892–5911 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mangeney M, Pothlichet J, Renard M, Ducos B & Heidmann T Endogenous retrovirus expression is required for murine melanoma tumor growth in vivo. Cancer Res. 65, 2588–2591 (2005). [DOI] [PubMed] [Google Scholar]
- 124.Zhou F et al. Activation of HERV-K Env protein is essential for tumorigenesis and metastasis of breast cancer cells. Oncotarget 7, 84093–84117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kraus B et al. Vaccination directed against the human endogenous retrovirus-K envelope protein inhibits tumor growth in a murine model system. PloS One 8, e72756 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huang G, Li Z, Wan X, Wang Y & Dong J Human endogenous retroviral K element encodes fusogenic activity in melanoma cells. J. Carcinog. 12, 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bjerregaard B, Holck S, Christensen IJ & Larsson L-I Syncytin is involved in breast cancer-endothelial cell fusions. Cell. Mol. Life Sci. CMLS 63, 1906–1911 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mortensen K, Lichtenberg J, Thomsen PD & Larsson L-I Spontaneous fusion between cancer cells and endothelial cells. Cell. Mol. Life Sci. CMLS 61, 2125–2131 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bannert N, Hofmann H, Block A & Hohn O HERVs New Role in Cancer: From Accused Perpetrators to Cheerful Protectors. Front. Microbiol. 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rodić N et al. Long Interspersed Element-1 Protein Expression Is a Hallmark of Many Human Cancers. Am. J. Pathol. 184, 1280–1286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Babushok DV & Kazazian HH Progress in understanding the biology of the human mutagen LINE-1. Hum. Mutat. 28, 527–539 (2007). [DOI] [PubMed] [Google Scholar]
- 132.Rodić N & Burns KH Long Interspersed Element–1 (LINE-1): Passenger or Driver in Human Neoplasms? PLOS Genet. 9, e1003402 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cajuso T et al. Retrotransposon insertions can initiate colorectal cancer and are associated with poor survival. Nat. Commun. 10, 4022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Morse B, Rotherg PG, South VJ, Spandorfer JM & Astrin SM Insertional mutagenesis of the myc locus by a LINE-1 sequence in a human breast carcinoma. Nature 333, 87–90 (1988). [DOI] [PubMed] [Google Scholar]
- 135.Fodde R The APC gene in colorectal cancer. Eur. J. Cancer Oxf. Engl. 1990 38, 867–871 (2002). [DOI] [PubMed] [Google Scholar]
- 136.Shukla R et al. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 153, 101–111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lin L et al. Multiple forms of genetic instability within a 2-Mb chromosomal segment of 3q26.3–q27 are associated with development of esophageal adenocarcinoma. Genes. Chromosomes Cancer 45, 319–331 (2006). [DOI] [PubMed] [Google Scholar]
- 138.Cruickshanks HA & Tufarelli C Isolation of cancer-specific chimeric transcripts induced by hypomethylation of the LINE-1 antisense promoter. Genomics 94, 397–406 (2009). [DOI] [PubMed] [Google Scholar]
- 139.Rodriguez-Martin B et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet. 52, 306–319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ishak CA & De Carvalho DD Reactivation of Endogenous Retroelements in Cancer Development and Therapy. Annu. Rev. Cancer Biol. 4, 159–176 (2020). [Google Scholar]
- 141.Ewing AD et al. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Res. 25, 1536–1545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Solyom S et al. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 22, 2328–2338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lee E et al. Landscape of Somatic Retrotransposition in Human Cancers. Science 337, 967–971 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Asch HL et al. Comparative expression of the LINE-1 p40 protein in human breast carcinomas and normal breast tissues. Oncol. Res. 8, 239–247 (1996). [PubMed] [Google Scholar]
- 145.Iskow RC et al. Natural Mutagenesis of Human Genomes by Endogenous Retrotransposons. Cell 141, 1253–1261 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hussain SP & Harris CC Molecular epidemiology of human cancer: contribution of mutation spectra studies of tumor suppressor genes. Cancer Res. 58, 4023–4037 (1998). [PubMed] [Google Scholar]
- 147.Baugh EH, Ke H, Levine AJ, Bonneau RA & Chan CS Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 25, 154–160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Levine AJ, Ting DT & Greenbaum BD P53 and the defenses against genome instability caused by transposons and repetitive elements. BioEssays 38, 508–513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Harris CR et al. p53 responsive elements in human retrotransposons. Oncogene 28, 3857–3865 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Haoudi A, Semmes OJ, Mason JM & Cannon RE Retrotransposition-Competent Human LINE-1 Induces Apoptosis in Cancer Cells With Intact p53. J. Biomed. Biotechnol. 2004, 185–194 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Leonova KI et al. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc. Natl. Acad. Sci. 110, E89–E98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tiwari B et al. p53 directly represses human LINE1 transposons. Genes Dev. 34, 1439–1451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pisanic TR et al. Long Interspersed Nuclear Element 1 Retrotransposons Become Deregulated during the Development of Ovarian Cancer Precursor Lesions. Am. J. Pathol. 189, 513–520 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Al Bayyat G, Arreaza-Kaufman D, Venkateswaran N, Galor A & Karp CL Update on pharmacotherapy for ocular surface squamous neoplasia. Eye Vis. 6, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee J et al. Results from a phase I, open-label study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer (NCT02630199). J. Clin. Oncol. 38, 3503–3503 (2020). [Google Scholar]
- 156.Mita P et al. BRCA1 and S Phase DNA Repair Pathways Restrict LINE-1 Retrotransposition in Human Cells. Nat. Struct. Mol. Biol. 27, 179–191 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vang R, Shih I-M & Kurman RJ OVARIAN LOW-GRADE AND HIGH-GRADE SEROUS CARCINOMA: Pathogenesis, Clinicopathologic and Molecular Biologic Features, and Diagnostic Problems. Adv. Anat. Pathol. 16, 267–282 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kurman RJ Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann. Oncol. 24, x16–x21 (2013). [DOI] [PubMed] [Google Scholar]
- 159.Kindelberger DW et al. Intraepithelial Carcinoma of the Fimbria and Pelvic Serous Carcinoma: Evidence for a Causal Relationship. Am. J. Surg. Pathol. 31, 161–169 (2007). [DOI] [PubMed] [Google Scholar]
- 160.Kuhn E et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—evidence supporting the clonal relationship of the two lesions. J. Pathol. 226, 421–426 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bell D et al. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee Y et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 211, 26–35 (2007). [DOI] [PubMed] [Google Scholar]
- 163.Bowtell DD et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 15, 668–679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xia Z et al. Expression of L1 retrotransposon open reading frame protein 1 in gynecologic cancers. Hum. Pathol. 92, 39–47 (2019). [DOI] [PubMed] [Google Scholar]
- 165.Schreiber RD, Old LJ & Smyth MJ Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 331, 1565–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 166.Topalian SL, Drake CG & Pardoll DM Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Li H et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 5, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chiappinelli KB et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Roulois D et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 162, 961–973 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tsai H-C et al. Transient Low Doses of DNA Demethylating Agents Exert Durable Anti-tumor Effects on Hematological and Epithelial Tumor Cells. Cancer Cell 21, 430–446 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kaminskas E et al. Approval Summary: Azacitidine for Treatment of Myelodysplastic Syndrome Subtypes. Clin. Cancer Res. 11, 3604–3608 (2005). [DOI] [PubMed] [Google Scholar]
- 172.Karpf AR et al. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc. Natl. Acad. Sci. 96, 14007–14012 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mehdipour P et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature (2020) doi: 10.1038/s41586-020-2844-1. [DOI] [PubMed] [Google Scholar]
- 174.Brocks D et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 49, 1052–1060 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu M et al. Dual inhibition of DNA and histone methyltransferases increases viral mimicry in ovarian cancer cells. Cancer Res. 78, 5754–5766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liu M et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc. Natl. Acad. Sci. U. S. A. 113, 10238–10244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Stone ML et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl. Acad. Sci. U. S. A. 114, E10981–E10990 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cuellar TL et al. ---Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia--. J. Cell Biol. 216, 3535–3549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lv T et al. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PloS One 7, e35065 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhao Z-K et al. Overexpression of lysine specific demethylase 1 predicts worse prognosis in primary hepatocellular carcinoma patients. World J. Gastroenterol. 18, 6651–6656 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jie D et al. Positive expression of LSD1 and negative expression of E-cadherin correlate with metastasis and poor prognosis of colon cancer. Dig. Dis. Sci. 58, 1581–1589 (2013). [DOI] [PubMed] [Google Scholar]
- 182.Yu Y et al. High expression of lysine-specific demethylase 1 correlates with poor prognosis of patients with esophageal squamous cell carcinoma. Biochem. Biophys. Res. Commun. 437, 192–198 (2013). [DOI] [PubMed] [Google Scholar]
- 183.Karakaidos P, Verigos J & Magklara A LSD1/KDM1A, a Gate-Keeper of Cancer Stemness and a Promising Therapeutic Target. Cancers 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sheng W et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–563.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pratt AJ & MacRae IJ The RNA-induced Silencing Complex: A Versatile Gene-silencing Machine. J. Biol. Chem. 284, 17897–17901 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tunbak H et al. The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nat. Commun. 11, 5387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chiappinelli KB, Zahnow CA, Ahuja N & Baylin SB Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 76, 1683–1689 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Strick R, Strissel PL, Baylin SB & Chiappinelli KB Unraveling the molecular pathways of DNA-methylation inhibitors: human endogenous retroviruses induce the innate immune response in tumors. OncoImmunology 5, e1122160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Giacomo AMD et al. Guadecitabine plus ipilimumab in unresectable melanoma: the NIBIT-M4 clinical trial. Clin. Cancer Res. (2019) doi: 10.1158/1078-0432.CCR-19-1335. [DOI] [PubMed] [Google Scholar]
- 190.Taylor K et al. An open-label, phase II multicohort study of an oral hypomethylating agent CC-486 and durvalumab in advanced solid tumors. J. Immunother. Cancer 8, e000883 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sherr CJ & Roberts JM CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13, 1501–1512 (1999). [DOI] [PubMed] [Google Scholar]
- 192.Carnero A Targeting the cell cycle for cancer therapy. Br. J. Cancer 87, 129–133 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dickson MA & Schwartz GK Development of cell-cycle inhibitors for cancer therapy. Curr. Oncol. 16, 36–43 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Xu H et al. Recent advances of highly selective CDK4/6 inhibitors in breast cancer. J. Hematol. Oncol.J Hematol Oncol 10, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bourdeau V & Ferbeyre G CDK4-CDK6 inhibitors induce autophagy-mediated degradation of DNMT1 and facilitate the senescence antitumor response. Autophagy 12, 1965–1966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Acevedo M et al. A CDK4/6-Dependent Epigenetic Mechanism Protects Cancer Cells from PML-induced Senescence. Cancer Res. 76, 3252–3264 (2016). [DOI] [PubMed] [Google Scholar]
- 197.Goel S et al. CDK4/6 inhibition triggers anti-tumor immunity. Nature 548, 471–475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Cingöz O & Goff SP Cyclin-dependent kinase activity is required for type I interferon production. Proc. Natl. Acad. Sci. 115, E2950–E2959 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.DePinto W et al. In vitro and in vivo activity of R547: a potent and selective cyclin-dependent kinase inhibitor currently in phase I clinical trials. Mol. Cancer Ther. 5, 2644–2658 (2006). [DOI] [PubMed] [Google Scholar]
- 200.Krishnamurthy J et al. Genetic Engineering of T Cells to Target HERV-K, an Ancient Retrovirus on Melanoma. Clin. Cancer Res. 21, 3241–3251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Takahashi Y et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J. Clin. Invest. 118, 1099–1109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cherkasova E et al. Detection of an Immunogenic HERV-E Envelope with Selective Expression in Clear Cell Kidney Cancer. Cancer Res. 76, 2177–2185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Germain RN MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 76, 287–299 (1994). [DOI] [PubMed] [Google Scholar]
- 204.Kong Y et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 10, 5228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Saini SK et al. Human endogenous retroviruses form a reservoir of T cell targets in hematological cancers. Nat. Commun. 11, 5660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Boller K, Janssen O, Schuldes H, Tönjes RR & Kurth R Characterization of the antibody response specific for the human endogenous retrovirus HTDV/HERV-K. J. Virol. 71, 4581–4588 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hahn S et al. Serological response to human endogenous retrovirus K in melanoma patients correlates with survival probability. AIDS Res. Hum. Retroviruses 24, 717–723 (2008). [DOI] [PubMed] [Google Scholar]