

Abstract
Background
Over their evolutionary history, corals have adapted to sea level rise and increasing ocean temperatures, however, it is unclear how quickly they may respond to rapid change. Genome structure and genetic diversity contained within may highlight their adaptive potential.
Results
We present chromosome-scale genome assemblies and linkage maps of the critically endangered Atlantic acroporids, Acropora palmata and A. cervicornis. Both assemblies and linkage maps were resolved into 14 chromosomes with their gene content and colinearity. Repeats and chromosome arrangements were largely preserved between the species. The family Acroporidae and the genus Acropora exhibited many phylogenetically significant gene family expansions. Macrosynteny decreased with phylogenetic distance. Nevertheless, scleractinians shared six of the 21 cnidarian ancestral linkage groups as well as numerous fission and fusion events compared to other distantly related cnidarians. Genetic linkage maps were constructed from one A. palmata family and 16 A. cervicornis families using a genotyping array. The consensus maps span 1,013.42 cM and 927.36 cM for A. palmata and A. cervicornis, respectively. Both species exhibited high genome-wide recombination rates (3.04 to 3.53 cM/Mb) and pronounced sex-based differences, known as heterochiasmy, with 2 to 2.5X higher recombination rates estimated in the female maps.
Conclusions
Together, the chromosome-scale assemblies and genetic maps we present here are the first detailed look at the genomic landscapes of the critically endangered Atlantic acroporids. These data sets revealed that adaptive capacity of Atlantic acroporids is not limited by their recombination rates. The sister species maintain macrosynteny with few genes with high sequence divergence that may act as reproductive barriers between them. In the Atlantic Acropora, hybridization between the two sister species yields an F1 hybrid with limited fertility despite the high levels of macrosynteny and gene colinearity of their genomes. Together, these resources now enable genome-wide association studies and discovery of quantitative trait loci, two tools that can aid in the conservation of these species.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-024-11025-3.
Keywords: Acropora, Coral, Genome, Chromosome, Ancestral linkage group, Linkage map, Recombination rate, Heterochiasmy, Hermaphrodite
Background
Corals are early branching metazoans with a long evolutionary history, first appearing in the fossil record 240 Mya, though phylogenomic analyses suggest the earliest scleractinians emerged around 425 Mya [1]. Several genome assemblies are now complete and reveal substantial similarities between early and late branching metazoans [2], with a particularly slow rate of genome rearrangement [3, 4] and mitochondrial mutation rate [5, 6] in the phylum Cnidaria (corals, hydrozoans, and jellyfish) when compared with other metazoans such as bilaterians. Over evolutionary time scales, corals have adapted to changing environments [7], but it is less clear how fast they may adapt to rapid changes. Aspects of adaptive capacity may include the structure of an organism’s genome, the genetic diversity contained within it, and the rate at which genetic diversity is recombined [8].
Corals have complex lifestyles: planktonic larvae settle and form sessile adult colonies via polyp budding and branch fragmentation [9–11]. During annual broadcast spawning events, adult colonies of the two Atlantic Acropora species, A. palmata and A. cervicornis, release egg/sperm bundles into the water column where they dissociate [12]. Self-fertilization is genet-specific and self-fertilizing genets occur at low frequency in the populations of both A. palmata and A. cervicornis [13–15]. Larvae develop for a few days in the water column before swimming towards the benthos where they settle and metamorphose [16]. Once a primary polyp has formed, symbiotic algae in the order Symbiodiniaceae colonize the coral tissue. Adult colonies of Atlantic acroporids most often harbor the species Symbiodinium ‘fitti’ [17]. Recruitment of sexually produced offspring into adult populations of these acroporids is now rare [18]. Populations of Atlantic acroporids have declined more than 80% in recent decades throughout the Atlantic and Caribbean due to anthropogenic impacts, infectious diseases, and temperature induced bleaching events [19, 20] leading to their current status as a federally listed threatened species under the US Endangered Species Act.
Genome assemblies are now available from all classes of cnidarians [21]. In Anthozoa, the Hexacorallia are represented by dozens of genomes from genera such as Acropora [22–24], Astrangia [25], Exaiptasia [26], Nematostella [3] and the Octocorallia by at least eight genomes from taxa such as Renilla [27], Dendronephthya [28], Xenia [29], and Heliopora [30]. Seven chromosome-resolved assemblies are published for scleractinian corals [22, 24, 31–33]. While most coral species are diploid, other ploidies exist (e.g. Pocillopora acuta [34],). The ancestral cnidarian chromosome number is seventeen [4], whereas coral genomes generally have fourteen chromosomes (2n = 28; [35]) and genome sizes are between 300 Mb – 1 Gb (eg., [22, 36–38]). The number of genes is typically 30,000–40,000 with some exceptions (e.g. Montipora capitata and Porites compressa in [34]).
Genetic diversity fuels adaptation by providing targets for selection (e.g. [39, 40]). Population genetic data indicate that corals are highly heterozygous and contain substantial genetic diversity over their large geographic ranges [41, 42], including the two Atlantic acroporids [43–49]. Hybridization and introgression among coral populations and species is facilitated by external fertilization of embryos and synchronized mass spawning events [7, 50, 51]. Indeed, the two Atlantic acroporids hybridize to form an F1 hybrid and backcrosses of the F1 hybrid into both parent species are observed at a low frequency [47].
Recombination allows for the separation of beneficial and detrimental alleles, such that selection may act upon them independently [52]. However, the role of recombination in adaptive evolution has been the subject of debate. While recombination has the capacity to create new, advantageous genetic combinations, it can also separate existing ones [53]. Recombination between adaptive loci may impede range expansions prompted by shifts in environmental conditions [54]. On the other hand, adaptive substitutions are correlated with higher recombination in several systems [8, 55, 56]. Further, recombination rate varies across individuals, across the genome, and across sexes [57, 58]. Global patterns of variation in recombination rates between males and females (heterochiasmy) across taxa suggest these differences may be adaptive [59]. Heterochiasmy in simultaneously hermaphroditic animals has been found in a limited number of studies published to date [60–62], and the recombination landscape of different sexes has only been studied in one other coral, Acropora millepora [60]. Here, we focus on the recombination landscape of two critically endangered sister species, Acropora palmata and A. cervicornis (Fig. 1). Both species are simultaneous hermaphrodites that reproduce sexually via gamete release and asexually via fragmentation [12]. Because these are endangered species, understanding their potential to adapt to changes is a pressing issue.
Fig. 1.

Sampling locations of Acropora palmata (A) and A. cervicornis (B). Both species are dominant reef-building corals of Caribbean and northwestern Atlantic reefs and are the only representative species of the genus Acropora in the region. Letter notation on the map indicates the geographic origin of A. palmata genome genet at Horseshoe Reef (HSR) and A. cervicornis genome genet near Grassy Key (GKR). Photos by IBB
One way to derive recombination rates is by building a genetic linkage map. Linkage maps can be generated from just one cross with many offspring or from few offspring across several families [63]. Because one biparental coral cross can generate hundreds of offspring, many recombination events can be cataloged among siblings from a few families, or even a single family, and used to order markers along a chromosome. Using a combination of long read, short read, Hi-C chromatin scaffolding, and linkage map anchoring of de novo assembled scaffolds, we report chromosome-level genome assemblies and genetic maps of the two Atlantic acroporid species, Acropora palmata (Lamarck, 1816) and A. cervicornis (Lamarck, 1816). With these assemblies and maps, we compare patterns of macrosynteny and gene colinearity at the whole genome level with Pacific acroporids and distant relatives and characterize the recombination landscapes in these sister species.
Methods
Sample collection and sequencing
Adult coral tissue was collected from the Acropora cervicornis genet M5 collected near Grassy Key (24.711783° N, 80.945966° W) and reared at the Coral Restoration Foundation Tavernier Nursery (CRF, 24.9822° N, 80.4363° W) and the A. palmata genet HS1 from Horseshoe Reef (25.1399° N, 80.2946° W) (Supplementary Table 1; [47]). High molecular weight genomic DNA (gDNA) was isolated from each coral tissue sample using the Qiagen DNeasy kit (Qiagen, Valencia, CA) with slight modifications described previously [64]. Paired-end 250 bp sequencing libraries (avg. insert size 550 nt) were constructed from 1.8–2 µg gDNA with the TruSeq DNA PCR-Free kit (Illumina, San Diego, CA) and sequenced on the Illumina HiSeq 2500 by the Genomics Core Facility at Pennsylvania State University. Additionally, coral tissue from A. palmata HS1 was collected by CRF in January of 2018, snap-frozen in liquid nitrogen and sent directly to Dovetail Genomics for DNA extraction followed by Chicago and Hi-C library preparation.
For the PacBio libraries, gamete bundles of A. cervicornis M5 (spawned 2015 and August 22, 2016 at the CRF nursery) and A. palmata HS1 (spawned August 20, 2016 at Horseshoe Reef) were collected during the annual coral spawn. Once the gamete bundles broke apart, sperm was separated from the eggs using a 100 μm filter and concentrated and washed with 0.2 μm filtered seawater through three rounds of centrifugation at 2,000 × g for 5 min at room temperature. The A. cervicornis sperm samples from 2015 were brought to a final concentration of 3 × 107 cells ml−1 after the addition of Cell Suspension Buffer and 2% agarose using the Bio-Rad CHEF Genomic DNA Plug Kits (Bio-Rad, Hercules, CA). Genomic DNA plugs were processed according to the manufacturer’s protocol and stored at 4 °C. The genomic DNA was extracted from the plugs in two ways, either using the QIAquick Gel Extraction kit (Qiagen) or by soaking the plugs overnight in 100 ul nuclease-free water at 4 °C followed by 1 h at -80 °C and recovered at 23,000 × g. Sperm samples of both species from 2016 were stored as 1 ml aliquots of concentrated sperm in 100% non-denatured ethanol at -20 °C until extraction. Genomic DNA was extracted using Nucleon Phytopure DNA extraction kit (Cytiva, Marlborough, MA) with the addition of RNase treatment and increased incubation time of 3 to 4 h at 65 °C during the cell lysis step. Genomic DNA elutions were combined and concentrated using the AMPure bead clean-up (final gDNA = 2 μg for A. cervicornis and 10 μg for A. palmata). Given the different final gDNA concentrations, PacBio libraries were prepared using a 20 kb size-selection protocol for A. palmata and a low input, no size selection protocol for A. cervicornis. Both libraries were sequenced on Sequel II by the Genomics Core Facility at Pennsylvania State University.
Because the initial A. cervicornis assembly exhibited low contiguity, an additional assembly was generated using Oxford Nanopore (ONT) long-read sequencing data. For the A. cervicornis ONT DNA library, coral tissue from the M5 genotype preserved in ethanol was provided by the Coral Restoration Foundation in 2021 and stored at -20 °C until extraction. Genomic DNA was extracted using the Qiagen MagAttract HMW DNA kit (MD, USA) following the manufacturer’s protocol. To further purify the gDNA, a salt-ethanol precipitation was performed. Briefly, 0.1 volumes of 3 M NaOAc (pH 5.2) were added to the DNA elution, followed by 3 volumes of 100% ethanol. The sample was centrifuged at approximately 20,000 × g for 1 h at 4 °C. The supernatant was then removed and the pellet was washed twice with cold 75% EtOH. The dried pellet was resuspended in Buffer AE (Qiagen, MD, USA) and long read libraries were generated using an Oxford Nanopore Ligation Sequencing Kit v10 (SQK-LSK110). Libraries were subsequently sequenced on an Oxford Nanopore PromethION flow cell (R9.4, FLO-PRO002) by the University of Wisconsin Biotechnology Center. Bases were called by the sequencing provider using Guppy v5.0.12 and delivered in fastq format.
K-mer genome size estimation
We removed low-quality bases (Phred score below 25) and adaptors from Illumina reads, discarding reads shorter than 50 bp, with cutadapt v1.6 [65]. Prior to genome assembly, 119-mer counting was performed on trimmed reads from each sample using jellyfish v2.2.10 [66] for the purpose of haploid genome size estimation. We utilized 119-mers because a k-mer length of 119 was identified as the optimal k-mer for de novo genome assembly from contamination filtered reads by KmerGenie v1.7048 [67] after testing a range of k-mers from 21 to 121. K-mer frequency histograms were analyzed using the GenomeScope2 web portal [68] and findGSE [69], which use a negative binomial and skew distribution model, respectively.
Contamination filtering of Illumina short read data
DNA extractions on the adult tissue used for Illumina sequencing were composed of the coral host and its associated microbial partners (algal symbionts and other microbes). To remove non-coral reads, we applied a modified series of filtering steps that compares sequence homology and GC content similar to process in BlobToolKit [70, 71] and described previously for A. cervicornis by Reich et al. [72]. Adaptor trimmed reads were initially assembled into contigs with SOAPdenovo2 v0.4 (parameters -K 95 –R) [73]. The contigs were compared to the genomes of the coral Acropora digitifera (NCBI: GCF_000222465.1; [74]), the symbiont Breviolum minutum (OIST: symbB.v1.0.genome.fa; [75]), and the NCBI nucleotide database (nt) using megablast (evalue 1e−5 threshold) [76]. Contigs with higher sequence similarity to non-cnidarians in the nt database were combined to make a local contamination database. Adaptor trimmed reads were then aligned with Bowtie2 v2.2.9 (parameters –q –fast; [77]) sequentially against the A. digitifera mitochondria (NBCI: KF448535.1), three concatenated Symbiodiniaceae genomes (Symbiodinium microadriaticum, Breviolum minutum, Fugacium kawagutii; [75, 78, 79], respectively) and the contamination database. Unaligned reads were extracted and used for short-read genome assembly described below.
Hybrid genome assembly of A. cervicornis and A. palmata
The trimmed and filtered short reads were assembled with SoapDeNovo-127mer v2.04 [73] using different k-mers for each species, A. palmata K = 99 and A. cervicornis K = 95. Contigs were filtered for additional symbiont contamination using megablast against the three Symbiodiniaceae genome assemblies described above. A surprising number of symbiont contigs, roughly 500,000 in each species assembly, were present despite our read contamination filtering [72]. The non-symbiont contigs were then assembled with PacBio long reads using the hybrid method DBG2OLC [80], k = 17 MinLen = 500 AdaptiveTh = 0.001 KmerCovTh = 2 MinOverlap = 20). PacBio reads were also assembled separately with Canu v1.5 [81], genomeSize = 400 m correctedErrorRate = 0.075 minReadLength = 500). The two assemblies (hybrid and PacBio only) were then combined using QuickMerge v0.2 [82], A. palmata = -hco 5.0 -c 1.5 -l 55000 -ml 1000; A. cervicornis = -hco 5.0 -c 1.5 -l 99500 -ml 1000) with the hybrid assembly as the reference and PacBio assembly as the query. Additional contig extension was performed with FinisherSC v2.1 [83]. Lastly, the assemblies were polished using Pilon v1.22 [84].
Hi-C scaffolding of hybrid Acropora palmata assembly
Our hybrid assembly of A. palmata was submitted to Dovetail Genomics for Hi-C analysis. They combined their proprietary HiRise scaffolding and Hi-C analysis (Supplementary Table 1), but the assembly was still far from chromosome-resolved (441 scaffolds, N50 = 6.8 Mb, and L50 = 16). In an effort to further improve the A. palmata genome assembly, we mapped the Hi-C paired-end reads separately back onto the Dovetail Genomics assembly with bwa-mem v 0.7.17 [85] with the mapping parameters -A1 -B4 -E50 -L0. We then followed the steps outlined by HiCExplorer v2.1.1 to create and correct a Hi-C contact matrix using default settings with a lower bin correction threshold of -1.5 [86]. This indicated there were more short range (< 20 kb) than long range (> 20 kb) contacts in the matrix. The corrected matrix was then used by HiCAssembler v1.1.1 [87] to further orient the scaffolds into pseudochromosomes with a minimum scaffold length set to 300,000 bp, a bin size of 15,000 and two iterations.
Nanopore assembly of Acropora cervicornis
PromethION data was trimmed and filtered with Porechop [88], resulting in a total of 94 Gb across 39.91 M reads of usable ONT data. With trimmed ONT data, metaFlye [89] was used to perform a long-read only metagenome assembly. Following the initial metaFlye assembly, which includes a long-read polishing step, the assembly was further polished in one round using hypo [90]. Illumina short read data from the M5 genet described above was trimmed using TrimGalore [91], and mapped to the preliminary assembly with bwa-mem [85] prior to use with hypo. ONT reads were then mapped to the assembly using minimap2 [92] and BAM files were sorted using samtools [93]. Using blastn [94], assemblies were searched against a custom database comprised of NCBI’s ref_euk_rep_genomes, ref_prok_rep_genomes, ref_viroids_rep_genomes, and ref_viruses_rep_genomes databases combined with dinoflagellate and Chlorella genomes [75, 95–98]. Using the mapping and blastn hits files, blobtools [99] was used to identify and isolate cnidarian contigs. Purge_dups [100] was utilized to identify and remove any remaining putative haplotigs in the respective assembly.
Linkage map construction
A full-sibling family was generated through a controlled cross between two Acropora palmata genets. Spawn was collected from two genets during the August 2018 spawning season in Curacao. Once egg-sperm bundles had broken apart, gametes were separated, and eggs were washed to remove any remaining self-sperm. The sperm from the genet designated as the sire was used to fertilize washed eggs from the genet designated as the dam. The resulting larvae were reared to 96 h post-fertilization in filtered seawater before preservation in individual 1.5 ml PCR tubes with 96% ethanol. A total of 105 full-sibling offspring were used in the construction of the genetic linkage map. Three to four polyps of each spawning parent were collected using coral cutters and preserved in 96% ethanol. For Acropora cervicornis, coral recruits from 16 families reared in a previous study until they first branched were used to construct a linkage map [15]. Samples of these recruits were preserved in 95% ethanol in 1.5 mL Eppendorf tubes and immediately placed into a -80° freezer until extraction.
For Acropora palmata larval offspring, high molecular weight DNA extractions followed the methods in Kitchen et al. [101]. Each larva was incubated in 12 μl of lysis solution (10.8 μl Buffer TL, 1 μl of Proteinase K, and 0.2 μl of 100 mg/ml RNAse A, all reagents from Omega BioTek) for 20 min at 55 °C. Next, 38 μl of Buffer TL and 50 μl of phenol/chloroform/isoamyl alcohol solution (25:24:1) was added to each sample and gently rocked for approximately 2 min. After centrifuging each sample for 10 min at 20,000 g, the top aqueous phase was removed and placed in a new tube. 50 μl of chloroform:isoamyl alcohol (24:1) was added to each sample and gently rocked for 2 min. Samples were centrifuged again at 10,000 rpm for 5 min and the top aqueous phase was again removed and placed into a new tube. The DNA was precipitated with 1.5 × volume of room-temperature isopropanol, 1/10 volume of 3 M sodium acetate (pH = 5.2) and 1 μl of glycogen (5 mg/ml) for 10 min at room temperature. Samples were then centrifuged at 20,000 g for 20 min and washed with 70% ice-cold ethanol. All supernatant was removed, and pellets were dried under a hood for approximately 30 min. Pellets were re-suspended in 30 μl of low TE buffer (10 mM Tris–HCl and 0.1 mM EDTA). Parental tissue was extracted using Qiagen DNeasy kit (Qiagen, Valencia, CA) following the modified protocol described in Kitchen et al. [101] and eluted in 100 μl of nuclease-free water.
Extracted samples were genotyped using the Applied Biosystems Axiom Coral Genotyping Array—550,962 (Thermo Fisher, Santa Clarita, CA, USA). The raw data were analyzed using the Axiom ‘Best Practices Workflow’ (BPW) with default settings (sample Dish QC ≥ 0.82, plate QC call rate ≥ 97; SNP call-rate cutoff ≥ 97; percentage of passing samples ≥ 95). The resulting genotyping files were converted to variant caller format (VCF) using the bcftools plugin affy2vcf [102] and filtered to represent only the recommended probeset identified by the Axiom BPW.
Acropora cervicornis recruits were sampled from the base of the colonies and DNA was extracted by Eurofins BioDiagnostics (WI, U.S.A) using LGC (Hoddesdon, UK) Sbeadex Animal DNA Purification Kits. Samples were run on two plates of the Applied Biosystems Axiom Coral Genotyping Array. Acropora cervicornis cross data was processed in the same manner as A. palmata, using the Axiom workflow and subsetting single nucleotide variants to only include recommended probes.
Acropora palmata and A. cervicornis linkage analysis was carried out using Lep-MAP3 [63] using the wrapper pipeline LepWrap [103]. Markers were first filtered for deviation from Mendelian inheritance and missing data via the Lep-MAP3 module ParentCall2. For A. cervicornis, the flag halfSibs = 1 was added to ParentCall2 to account for shared parentage among crosses. Recombination informative markers (here defined as those that were heterozygous in at least one parent) were next filtered using the Filtering2 module with a data tolerance of 0.0001. The remaining markers were assigned to 14 linkage groups (LGs) using an LG minimal size limit set to 5 markers using the module SeperateChromosomes2 and a logarithm of odds (LOD) score of 11 in A. palmata and 5 in A. cervicornis. For A. palmata, an informativeMask value of “123” was used and for A. cervicornis multi-family data, an informativeMask of “12” was used. Unassigned markers were iteratively added to existing LGs using a LOD limit of 2 and a LOD difference of 2. Markers were next ordered using the Kosambi mapping function as implemented in the module OrderMarkers2 with the identical limit set to 0.005, usePhysical = 1 0.1, 100 merge iterations, 3 phasing iterations, and the hyperPhaser parameter used to improve marker phasing. To remove markers at map edges that may erroneously inflate the map length, the last 10% of markers were trimmed if they fell more than 5% of the total centimorgan (cM) span away from the next nearest marker. After trimming, marker order was evaluated with a second round of OrderMarkers2 using the same parameters as previously described. Both paternal and maternal maps were generated and the option sexAverage = 1 was applied to include a sex-averaged consensus map. Average marker distance was calculated as the size of the linkage map in cM divided by the number of markers. As global orientation of a linkage group is arbitrary in Lep-MAP3, marker order was flipped for LGs in which the start of the genetic map (0 cM) corresponded to the end, rather than to the start of the physical map (the position 0 bp) of a given scaffold. To generate cleaned Marey maps, MareyMap Online [104] was used to remove aberrant markers and generate smoothed recombination maps using 2-degree polynomial LOESS estimation with a span of 0.25.
Linkage scaffolding of A. cervicornis Nanopore assembly
For A. cervicornis, no Hi-C data was available. As such, the A. cervicornis assembly was scaffolded using Lep-Anchor [105] with the linkage map generated by Lep-MAP3 [63]. To assist in orientation of contigs with markers, as well as placements of contigs without markers, minimap2 v2.24 [92] was used to generate a PAF file using the ONT data. Lep-Anchor was run via LepWrap and utilized default Lep-Anchor arguments, apart from setting the expected number of linkage groups to 14. Additionally, LepWrap implements the edge-trimming scripts for Lep-Anchor as was described above for Lep-MAP3.
Repeat identification, masking, and divergence analysis
For both assemblies, repetitive sequences were predicted with RepeatModeler v 1.0.11 [106], filtered for genuine genes based on blast similarity to the NCBI nr database or Acropora digitifera protein sequences (e-value ≤ 1e−5), combined with the Acropora TE consensus sequences in Repbase (n = 149), annotated separately against the invertebrate repeat database in CENSOR v4.2.29 [107] for “unknown” TEs, and soft masked using RepeatMasker v 4.0.7 [108]. We also ran the above series of steps on the genome assemblies of A. digitifera, A. tenuis and A. millepora to ensure comparable repeat estimates. The summary table for each species was generated using the buildSummary.pl utility script, and TE accumulation was calculated as the Kimura substitution level corrected for CpG content from the respective consensus sequence produced using the calcDivergence.pl and createRepeatLandscape.pl utility scripts in RepeatMasker. Kimura distance was converted to Jukes-Cantor distance using the formula JC = − 3/4*log(1 − 4*d/3), where d is the distance estimated by RepeatMasker. Assembly-free repeat identification, annotation and quantification was performed on 25% of the adapter-trimmed Illumina short-read data of each Atlantic species using dnaPipeTE v1.3.1 [109].
Gene prediction and annotation
For the A. palmata assembly, we used a combination of ab initio (GeneMark-ES v4.32; [110]) and reference-based tools (BRAKER v2.0; [111], PASA v2.1.0; [112], and exonerate v2.2.0; [113]) for gene prediction as previously described [114]. For BRAKER, RNAseq data produced on the Roche 454 GS FLX Titanium system was obtained from NCBI Bioproject PRJNA67695 [115] and mapped to the assembly using STARlong v2.5.3a [116] due to the average read lengths being greater than 300 bp. Gene models with read coverage greater than or equal to 90% were assigned as “BRAKER_HiQ'' predictions. The assembled A. palmata transcriptome from Polato et al. [115] was used as the input for PASA. Homology-based gene predictions were made with exonerate against all eukaryotic sequences in the UniProt database (n = 186,759), keeping predictions with at least 80% coverage. Gene predictions were combined with EVidenceModeler [112]. We also predicted tRNA sequences using tRNAscan_SE v1.3.1 [117]. The predicted genes were searched against the NCBI nr, UniProt Swiss-Prot and Trembl databases, and KEGG Automated Annotation Server. Blast-based searches were filtered by the top hit (e-value < 1e-5 threshold). GO annotations were extracted from UniProt of NCBI databases. Genes were also compared to OrthoDB v10.1 [118]. Gene annotation was assigned based on the e-value score < 1e-10 first to Swiss-Prot followed by Trembl and then NCBI. If no sequence homology was recovered, then the gene was annotated as a “hypothetical protein”. Gene predictions from the hybrid assembly were lifted over to the final Hi-C assembly using the UCSC liftOver process [119]. We also used homology-based prediction tool GeMoMA v1.6.1 [120] to map the A. palmata gene models to the Hi-C assembly. Liftover and GeMoMa predictions were combined with EVidenceModeler for the final gene set.
The original PacBio A. cervicornis assembly was annotated in a similar manner to A. palmata. However, the original assembly is superseded here by the ONT-based assembly. The ONT A. cervicornis LepWrap-scaffolded assembly was annotated using funannotate v1.8.13 [121] with RNAseq data obtained from four BioProjects available on NCBI SRA at the time of assembly (PRJNA222758, PRJNA423227, PRJNA529713, and PRJNA911752). All RNAseq data was adapter- and quality-trimmed using TrimGalore [91]. Briefly, funannotate train was run with a –max_intronlen of 100,000. Funannotate train is a wrapper that utilizes Trinity [122] and PASA [112] for transcript assembly. Upon completion of training, funannotate predict was run to generate initial gene predictions using the arguments –repeats2evm, –organism other, –max_intronlen 100,000, and –repeat_filter none. Additional transcript evidence from three sources (the initial A. cervicornis annotation described above, transcripts from Selwyn and Vollmer, [123], and the Osborne transcriptome, [124]) was provided to funannotate predict using the –transcript_evidence argument. Funannotate predict is a wrapper intended to separately run AUGUSTUS [125] and GeneMark [110] for gene prediction and EVidenceModeler [112] to combine gene models. Funannotate update was run to update annotations to be in compliance with NCBI formatting. For problematic gene models, funannotate fix was run to drop problematic IDs from the annotations. Finally, functional annotation was performed using funannotate annotate which annotates proteins using PFAM [126], InterPro [127], EggNog [128], UniProtKB [129], MEROPS [130], CAZyme [131], and GO [132].
Whole genome alignments and gene-level divergence
Genome assemblies of A. palmata, A. cervicornis M5 genet, and A. cervicornis K2 genet were aligned using minimap2 [92] with “asm5” setting for whole genome alignments, and the nucmer command within the mummer v4.0 package [133] with a minimum exact match length of 100 bp (-l 100), minimum cluster length of 500 (-c 500) and using all anchor positions (–maxmatch). To assess genome-scale synteny, the PAF alignments from minimap2 were plotted using both R package pafr v0.0.2 [134] and dotplotly [135]. The delta alignments from nucmer were visualized using the D-Genies web server [136]. Structural variants (insertions, deletions, tandem duplications and contractions, inversions and translocations) were identified from the whole genome alignments of A. palmata and A. cervicornis M5 genet using three tools: assemblytics [137], MUM&Co [138], and SVIM-asm [139]. Only MUM&Co and SVIM-asm were able to detect inversions and translocations.
To assess sequence divergence between the two species at the gene-level, rustybam [140] was used to split PAF alignments at gene coordinates and to calculate gene-level percent identity using matches and mismatches. Genes were considered outliers in sequence divergence if their percent identity was less the first quartile (Q1) minus three times the interquartile range (IQR). Enrichment analyses were performed using clusterProfiler v4.4.4 [141] with a custom database for A. palmata created with AnnotationForge v1.38.0 [142] to test for enrichment of gene ontology (GO) terms in the outlier gene set.
Orthologous gene identification and macrosynteny analysis
Genome completeness of each acroporid assembly was assessed using BUSCO v4.1.1 with the Metazoa odb10 orthologous gene set (n = 954 orthologues, [143]). To discover shared and unique gene families in A. cervicornis and A. palmata in relation to other species, OrthoFinder v2.5.2 [144] was run on the predicted proteins of each species listed in Supplementary Table 2 The species tree was constructed from a multiple sequence alignment with STAG and rooted by STRIDE in OrthoFinder v2.5.2 [144]. A presence/absence table of orthogroups, or sets of genes descended from a single gene in the last common ancestor of all the species being considered, was used to generate an UpSet intersecting set plot ( [145], as implemented in UpSetPlot [146]. The species tree from OrthoFinder was time-calibrated using r8s [147] with priors for Acropora (101 million years, [148]), Acroporidae (168 million years, [149]), Scleractinia (268 million years, [150]), and Anthozoa (541 million years, [151]) accessed via the Paleobiology Database [152]. CAFE 5 [153] was used to analyze time-calibrated phylogenies and the phylogenetic hierarchical orthogroups from OrthoFinder to identify gene families undergoing significant expansion or contraction in each node and tip. For nodes relevant to this study (Acroporidae, Acropora, Atlantic Acropora), we extracted significantly changing phylogenetic hierarchical orthogroups, as well as standard orthogroups unique to relevant nodes, and performed GO enrichment tests with clusterProfiler v4.4.4 [141] using a custom database for A. palmata created with AnnotationForge v1.38.0 [142].
Macrosyntenic patterns across the species with chromosome-resolved genome assemblies was assessed with Oxford Dot Plots (ODP, [154]), specifically mapping on the inferred ancestral linkage groups (ALGs) of sponge, cnidarian and bilaterians recently identified [2]. ODP runs an all-vs-all blast akin to OrthoFinder with diamond v2.0.15 [155] and identifies conserved syntenic gene arrangements between two genomes. Dot plots and ribbon diagrams were generated by ODP with default settings and restricting plotted scaffold length of 2 Mb to visualize conserved syntenic blocks across closely related or more distant taxa.
Results
Chromosome-scale genome assemblies of the Atlantic acroporids
To investigate the genomic conservation and divergence between the two Atlantic acroporids, we generated chromosome-scale genome assemblies for both species collected from the Florida Keys. For A. palmata (genet HS1, STAGdb ID HG0004), we used a hybrid assembly strategy that combined PacBio Sequel II long-reads with Illumina paired-end short reads to obtain an initial assembly with 2,043 scaffolds totaling to 304 Mb and an N50 of 282 kb (N50 is the minimum contig length to cover 50% of the genome). The assembly was further improved with Dovetail Chicago HiRise and Dovetail Hi-C data (all data used for genome assembly in both species described in Supplementary Table 1). After Hi-C scaffolding, the final 287 Mb haploid assembly was resolved into 14 pseudochromosomes (hereafter referred to as chromosomes, labeled Chr1—Chr14), a number consistent with the karyotype of A. palmata [156]. The A. palmata assembly has 406 scaffolds with an N50 of 18.66 Mb (Fig. 2A and Supplementary Table 3).
Fig. 2.

Atlantic acroporid genome assemblies. A Hi-C contact map of A. palmata genome resolved into 14 chromosomes using HiCAssembler [87]. B Dot plot visualization of colinear relationships of the 14 chromosomes/linkage groups between the sister species A. palmata (y-axis) and A. cervicornis (x-axis) using the D-genies web server [136]. The scale on each axis is in megabases (Mb). The points along the diagonal represent colinear genomic regions whereas those dots off the diagonal represent rearrangements (insertions, deletions, inversions and translocations). Yellow and purple boxes highlight two chromosomes, A. cervicornis LG6 and LG11, with complex rearrangements. (C, left) Comparison of A. cervicornis LG6 to A. palmata Chr4 reveals a 2.5 Mb inversion and 1.4 Mb translocation. (C, right) Complex rearrangements observed between A. cervicornis LG11 and A. palmata Chr 11, including a 0.765 Mb translocation. D Ribbon plot of syntenic orthologous genes conserved among scleractinians. The colored vertical links connect orthologous genes to the numbered chromosomes of the five species, represented by horizontal bars. Chromosome fusions or fissions are represented by crossing over of the colors that represent each ancestral linkage group. Chromosomal inversions were detected between Atlantic and Pacific acroporids (e.g. A. cervicornis L4, L6, and L12). Chromosomal changes were more numerous between Pacific than Atlantic acroporids. Comparing A. hyacinthus Chr 5, 10, 12 and 13 to all other acroporids indicates paracentric inversions of whole chromosome arms in this species
For A. cervicornis (genet M5, STAGdb ID HG0005), we initially used the same hybrid assembly strategy as for A. palmata relying on a combination of PacBio Sequel and Illumina short-read data (Supplementary Table 1). However, due to reduced high molecular weight genomic DNA available at the time, we were unable to size-select our PacBio library as we did for A. palmata, yielding shorter read lengths with an average read length and N50 of 3,238 bp and 4,394 bp, respectively, compared to 7,126 bp and 10,110 bp in A. palmata (Supplementary Table 1). Our first assembly was consequently less contiguous, with 4,382 scaffolds in 318 Mb and an N50 of 162 kb. We next turned to Oxford Nanopore PromethION (ONT) sequencing to generate additional long-read sequences but due to sample quality, the run produced an average read length of 2,366 bp, albeit with much higher overall data yield of 94.4 Gbp. Assembly of the high coverage ONT reads resulted in 6,381 contigs with an N50 of 711 Kb. To further resolve the A. cervicornis genome, we constructed a linkage map (described below) that was used to anchor and orient the ONT contigs into 14 linkage groups (LGs). These LGs correspond with high synteny to the Hi-C chromosomes assembled for A. palmata. Thus, the A. cervicornis LGs can be considered (pseudo)chromosomes. To better distinguish chromosomes for each species, we number the A. cervicornis chromosomes here as LG1—LG14. The final 305 Mb assembly was slightly more contiguous than A. palmata with a scaffold N50 of 20.05 Mb.
Our assemblies of A. palmata (287.6 Mb) and A. cervicornis (305.4 Mb) were on the lower end of the predicted genome sizes from three different k-mer based tools that ranged from 290 to 354 Mb (Supplementary Table 4), and both assemblies are approximately 110 to 180 Mb smaller than genomes of other acroporids species assembled to date (Supplementary Table 3). When comparing estimates of genome completeness using BUSCO Metazoa v10 [143, 157], we identified 87.8% complete genes in A. palmata, compared with 93.1% in A. cervicornis (Supplementary Table 5). Both assemblies exhibit minimal remaining haplotig duplication, with 1.2% of BUSCO genes in A. palmata duplicated and 0.3% in A. cervicornis.
Recently, a genome assembly of another A. cervicornis genotype from the Florida Keys, genet K2 (STAGdb ID HG0582), was published [123]. Using minimap2 [92] whole genome alignments, we demonstrate that the two assemblies are mostly concordant (Supplementary Fig. 1). Both assemblies are similar in completeness according to BUSCO Metazoa v10 [143, 157] assessment with the M5 assembly (this study) showing 93.1% completeness and the K2 assembly showing 92.45% completeness, of which 0.30% and 0.42% are duplicated, respectively (Supplementary Table 5). The assemblies are similar in size, with the M5 assembly being 305 Mb in total length and the K2 assembly 307 Mb. The most notable difference is the gain in scaffold length, with a scaffold N50 of 20.051 Mb for the M5 assembly, compared with 2.8 Mb for the K2 assembly. The K2 assembly has more contiguous primary contigs, with a contig N50 of 2.711 Mb compared with 0.732 Mb in the M5 assembly. Some K2 contigs are split across multiple linkage groups in the M5 assembly (Supplementary Fig. 1). These regions may reflect novel structural variants between genets within the Florida population of Acropora cervicornis or may represent misassembly in either assembly. Hi-C scaffolding or additional ultra-long read sequencing should be performed to validate the structural variants between these A. cervicornis assemblies.
Genomic synteny is largely conserved in the sister species
Whole genome alignments of the two Atlantic acroporid genomes using minimap2 [92] and nucmer [133] revealed long stretches of colinear regions with interspersed rearrangements across the 14 chromosomes (Fig. 2B). As well as similarities, there were differences in physical lengths of chromosomes that resulted in different chromosome number/linkage group naming assignments for each species (see Supplementary Table 6). For example, the length of the corresponding syntenic chromosome pair of A. cervicornis LG2 was 4.87 Mb longer than A. palmata Chr3. Overall, we identified 10,532 structural variants (SV) totaling 33.02 Mb between the two assemblies using variant calling tools (Supplementary Table 7). An additional 1.4 Mb translocation was detected by whole genome alignment dot plots between A. cervicornis LG6 and A. palmata chromosome Chr4 (Fig. 2B and C). Dot plots also highlighted a large inversion of 2.5 Mb between the same syntenic chromosome pair (A. cervicornis LG6 and A. palmata Chr4) and numerous smaller SV types were identified near the middle of A. cervicornis LG11 and A. palmata Chr11 (Fig. 2C), a region that may correspond with the centromere.
At the gene level, percent identity between the two species is high, with orthologs maintaining a median nucleotide percent identity of 99.65%. A total of 536 genes exceeded the Q1-(3*IQR) threshold to be considered an outlier gene in terms of percent identity (Supplementary Table 8). These outlier genes exhibited no enrichment of molecular function or biological process GO terms but showed enrichment at two cellular process GO terms: spindle pole (GO:0000922, p.adjust = 0.03) and midbody (GO:0030406, p.adjust = 0.04). The one annotated gene with the lowest percent identity (83.83%) that is not associated with transposable elements is a homolog of the sulfatase-modifying factor 1 (SUMF1). Other notable outlier genes include genes associated with gamete compatibility and fertilization (SPAG1 and REJ).
Synteny and gene content in the genus Acropora
We then compared the genome architecture and gene content of the Caribbean acroporids to other acroporids. To predict gene models for each assembly, we used a combination of transcriptomic data and ab initio tools resulting in 31,827 and 34,013 genes in A. palmata and A. cervicornis, respectively (Supplementary Table 3). Combining our gene models with those of other acroporids with chromosome-resolved assemblies, we identified colinear (shared loci with the same arrangement on a given chromosome) and macrosyntenic (shared loci not necessarily in the same arrangement on a given chromosome) gene arrangements (Fig. 2D and Supplementary Fig. 2A). In accordance with the high degree of synteny at the whole genome level, 15,873 out of 17,243 one-to-one orthologs between A. palmata and A. cervicornis retained their colinearity (Supplementary Fig. 2). The number of orthologs that shared ordinal positions between A. cervicornis chromosomes and A. hyacinthus or A. millepora was 12,603 out of 13,000 and 12,075 out of 14,738, respectively. We found that the architecture of some chromosomes was largely unchanged at this scale of observation (e.g. A. cervicornis LG1 across acroporids, Fig. 2D). Nevertheless, several translocations and inversions were evident. Within the acroporids, interchromosomal translocations were observed in A. millepora with 85 genes of A. cervicornis LG8 located on Chr 14 of A. millepora and 132 genes of A. cervicornis LG5 located on A. millepora Chr 5 (Fig. 2D). Paracentric inversions of whole chromosome arms likely led to the A. hyacinthus Chrs 5, 10, 12 and 13 arrangements (Fig. 2D and Supplementary Fig. 2A).
Most scleractinians share six of 21 cnidarian ancestral linkage groups
Ancestral chromosomal fusions and rearrangements within the coral lineage were detected by mapping previously inferred ancestral linkage groups (ALGs) shared among sponges, cnidarians and bilaterians against our genomes [2]. We note changes in ancestral ALGs in the reporting of results below with fusions represented by the letter “x” (Supplementary Table 9). Of the 21 cnidarian- specific ALG arrangements, six (A1a, Ea, J1xQa, A1bxB3, NxA2, and B1xB2) were largely intact within the scleractinians (acroporids and Catalaphyllia), represented by LG7, LG13, LG12, LG11, LG14 and LG5 in A. cervicornis (Supplementary Fig. 2B and Supplementary Table 9). Interestingly, ALG Qb was lost from all cnidarian species surveyed here, with the exception of the jellyfish Cassiopea xamachana that largely retains the ancestral cnidarian ALG structure (Supplementary Table 9 and Supplementary Fig. 3). We identified seven cases of ALG fusions and one example of centric insertion within one of the acroporid chromosomes, represented by A. cervicornis LG10 (Supplementary Fig. 2B and Supplementary Table 9). A. millepora is the only acroporid species where a portion of ALG G fused with L.
Orthogroup analysis reveals gene family expansions in the acroporids
Expanding beyond the species with chromosome-resolved assemblies, we compared orthologous gene families, also known as orthogroups, shared among diverse cnidarian taxa, including representatives of the Hexacorallia and Octocorallia within Anthozoa and Hydrozoa and Scyphozoa within Medusozoa (Supplementary Table 2). We identified 2,403 conserved orthogroups among all cnidarians (Fig. 3). There are 165 unique orthogroups in Anthozoa enriched in the process angiogenesis (GO:0001525, p.adjust = 0.049) and 48 unique Scleractinia orthogroups enriched in growth factor binding (GO:0019838, p.adjust = 0.009), cell adhesion molecule binding (GO:0050839, p.adjust = 0.035) and D-inositol-3-phosphate glycosyltransferase activity (GO:0102710, p.adjust = 0.008). We further found 44 and 144 unique orthogroups in acroporids and Atlantic acroporids, respectively (Fig. 3). Only 39 of the 144 orthogroups shared between the Atlantic species were annotated, 12 of which were predicted as transposable elements.
Fig. 3.

Conservation of gene content among cnidarians. UpSet plot displaying the number of shared orthologous groups amongst selected taxonomic groups—Cnidaria (red), Anthozoa (blue), Hexacorallia (green), Scleractinia (teal), Acroporidae (green brown), Acropora (purple) and Atlantic (Caribbean) Acropora (yellow). The colored or black circles below the vertical bar chart indicate those species that belong to each intersection group. On the left, the bar chart represents the total number of orthologous groups identified in each taxon. Taxon labels in bold were assembled in this study. The species tree constructed from a multiple-sequence alignment of 1,011 single-copy orthogroups (348,712 amino acid positions) was inferred by STAG and rooted by STRIDE in OrthoFinder v2.5.2 [144]. The species tree was time-calibrated using r8s [147] with priors for Acropora (101 million years, [148]), Acroporidae (168 million years, [149]), Scleractinia (268 million years, [150]), and Anthozoa (541 million years, [151]) accessed via the Paleobiology Database [152]. Node values depict the number of significant (p < 0.05) gene family expansions ( +) and contractions (-) identified by CAFE 5 [153]. Node values are not depicted for nodes internal to Acropora
In addition to orthogroups unique to each clade, CAFE 5 [153] was used to identify shared phylogenetic hierarchical orthogroups undergoing significant expansion or contraction within each node and tip of the species tree. We found a total of 191 gene families undergoing a significant change at the node associated with the family Acroporidae (+ 185 expanding and -6 contracting), 139 in the genus Acropora (+ 119 and -20), and 434 in the Atlantic Acropora (+ 247 and -187) (Fig. 3). Within the significantly changing gene families of Acroporidae, there were predominantly expansions, with notable GO enrichment in transcription-related terms, such as the PRC1 and ASAP complexes (GO:0035102; p.adjust = 1.78e-18 and GO:0061574; p.adjust = 4.59e-08), transposition terms (GO:0032196; p.adjust = 6.82e-29, GO:0006313; p.adjust = 1.85e-19), and symbiont-containing vacuole membranes (GO:0020005; p.adjust = 1.53e-08, Supplementary Fig. 4).
Within the genus Acropora, gene families primarily expanded (Fig. 3), with GO enrichment highlighting terms associated with cell–cell adhesion (GO:0098609; p.adjust = 6.98e-05), cell recognition (GO:0008037; p.adjust = 5.92e-16), and nerve components (e.g. myelin sheath and potassium channel complex, GO:0043209; p.adjust = 2.45e-10 and GO:0034705; p.adjust = 2.81e-08, respectively) and processes (e.g. paranodal junction assembly, GO:0030913; p.adjust = 7.52e-21, Supplementary Fig. 5). Finally, Atlantic Acropora exhibit enrichment related to transcriptional complexes (PRC1 complex, GO:0035102; p.adjust = 2.11e-06), as well as terms related to transposition (GO:0032196; p.adjust = 1.00e-88 and GO:0006313; p.adjust = 7.51e-70), retrotransposition (GO:0044826; p.adjust = 1.18e-07), and viral response (e.g. viral capsid, GO:0019028; p.adjust = 9.27e-03, Supplementary Fig. 6).
Repetitive content is comparable among acroporids
Repetitive DNA plays a significant role in the size, organization and architecture of eukaryotic genomes [158]. To analyze transposable element (TE) content among the acroporid genome assemblies, we constructed species-specific repeat libraries for each assembly using a genome-guided approach with RepeatModeler [106]. To ensure that only bona fide repeats were included in our comparisons, we filtered out putative genes using a sequence similarity approach against the NCBI protein database or A. digitifera gene models. Using RepeatModeler and RepeatMasker, our analyses found a TE content of 16.69% in A. palmata and 18.91% in A. cervicornis (Supplementary Table 10). Using dnaPipeTE [109], an assembly-free method based on the Illumina short-reads, the total TE content was estimated to be 37.11% for A. palmata and 35.54% for A. cervicornis (Supplementary Fig. 7).
The dominant TEs were shared among the species we surveyed across methods. These TEs belong to DNA transposons superfamilies Tc/Mariner and hAT, long interspersed nuclear element (LINE) retrotransposon family Penelope and long terminal repeat (LTR) family Gypsy (Supplementary Table 10). The transposable activity of each repeat class was compared across species to determine if TE accumulation differed over their evolutionary past (Fig. 4B-F). Each species experienced a recent burst of DNA, LINE and LTR copies in their genomes, as evidenced by the increased genomic coverage of those classes with zero to very small genetic distances (Fig. 4B-F inset plots). Within the recent TE expansion, the Atlantic acroporids and A. millepora have a bimodal distribution of LTR transpositions, specifically those within the retrotransposon family Gypsy. Overall, however, few species-specific patterns emerged in the repeat landscapes of the acroporids.
Fig. 4.

Comparison of repetitive DNA among acroporid taxa. (A) Percentage of the genome attributed to the main transposable element classes [DNA transposons, long interspersed nuclear element (LINE), short interspersed nuclear element (SINE), long terminal repeat (LTR), rolling circle (RC) and other (satellites, simple repeats, and unclassified)] for each acroporid taxon. (B-F) Repeat landscapes of all transposable element classes except “other” for A. palmata (B), A. cervicornis (C), A. tenuis (D), A. digitifera (E) and A. millepora (F). The percentage of genome coverage (y-axis) of each repeat is shown relative to the Jukes-Cantor genetic distance observed between a given repetitive element and its respective consensus sequence. Individual repetitive elements were then summarized by their repeat class. The more recent repetitive element copies have lower Jukes-Cantor distance on the left side of the x-axis. The inset plot in each panel focuses on recent repeat insertions at a Jukes-Cantor distance below 0.05 (gray shaded region in full plot)
Genetic Maps
In A. palmata we assigned 2,114 informative markers to 14 linkage groups (LGs), representing the 14 chromosomes, with an average marker distance of 0.48 cM and a consensus, sex-average map length of 1,013.42 cM (Table 1). The gamete-specific maps exhibited heterochiasmy and varied in length, with a longer female map length (1,460.68 cM) than male map length (583.19 cM). At the chromosome-level, female map lengths were longer than male map lengths in all 14 chromosomes (Table 1). The genome-wide average recombination rate was higher in the female (5.49 cM/Mb) than in the male (2.19 cM/Mb) map (Table 1). The highest average recombination rate (7.00 cM/Mb) was in the female map associated with Chr11. The lowest average recombination rate (1.55 cM/Mb) was in the male map associated with Chr2.
Table 1.
Genetic map summary statistics for Acropora palmata and Acropora cervicornis. Physical lengths, map length, and average recombination rates per chromosome for male, female, and sex-averaged maps of Acropora palmata and A. cervicornis. Mb = megabases, cM = centimorgan
| Chromosome | Length (Mb) | Number of Markers | Male Map Length (cM) | Female Map Length (cM) | Sex Averaged Map Length (cM) | Male Recombination Rate (cM/Mb) | Female Recombination Rate (cM/Mb) | Sex Averaged Recombination Rate (cM/Mb) | |
|---|---|---|---|---|---|---|---|---|---|
| Acropora palmata | Chr1 | 27.05 | 318 | 54.05 | 148.29 | 100.3 | 2 | 5.48 | 3.71 |
| Chr2 | 25.92 | 171 | 40.18 | 109.24 | 74.4 | 1.55 | 4.21 | 2.87 | |
| Chr3 | 21.9 | 155 | 42.18 | 116.88 | 79.18 | 1.93 | 5.34 | 3.62 | |
| Chr4 | 20.87 | 162 | 39.21 | 106.44 | 72.52 | 1.88 | 5.1 | 3.47 | |
| Chr5 | 20.54 | 162 | 36.65 | 95.01 | 65.39 | 1.78 | 4.63 | 3.18 | |
| Chr6 | 19.02 | 141 | 30.51 | 82.65 | 56.32 | 1.6 | 4.35 | 2.96 | |
| Chr7 | 18.66 | 143 | 30.61 | 102.66 | 66.01 | 1.64 | 5.5 | 3.54 | |
| Chr8 | 18.59 | 134 | 52.66 | 124.59 | 88.27 | 2.83 | 6.7 | 4.75 | |
| Chr9 | 17.67 | 129 | 47.71 | 96.2 | 71.22 | 2.7 | 5.44 | 4.03 | |
| Chr10 | 16.55 | 142 | 27.67 | 92.01 | 59.61 | 1.67 | 5.56 | 3.6 | |
| Chr11 | 16.42 | 113 | 51.94 | 115.02 | 81.74 | 3.16 | 7 | 4.98 | |
| Chr12 | 14.67 | 150 | 59.69 | 97.83 | 77.91 | 4.07 | 6.67 | 5.31 | |
| Chr13 | 14.61 | 112 | 28.92 | 79.95 | 53.63 | 1.98 | 5.47 | 3.67 | |
| Chr14 | 13.63 | 82 | 41.22 | 93.92 | 66.93 | 3.02 | 6.89 | 4.91 | |
| Acropora cervicornis | LG1 | 30.19 | 442 | 73.09 | 121.41 | 97.25 | 2.42 | 4.02 | 3.22 |
| LG2 | 26.77 | 495 | 50.62 | 96.61 | 73.61 | 1.89 | 3.61 | 2.75 | |
| LG3 | 25.26 | 356 | 46.01 | 90.6 | 68.3 | 1.82 | 3.59 | 2.7 | |
| LG4 | 20.97 | 433 | 54.48 | 89.59 | 72.03 | 2.6 | 4.27 | 3.44 | |
| LG5 | 20.93 | 562 | 53.8 | 108.28 | 81.04 | 2.57 | 5.17 | 3.87 | |
| LG6 | 20.56 | 340 | 27.42 | 76.07 | 51.75 | 1.33 | 3.7 | 2.52 | |
| LG7 | 20.05 | 305 | 33.67 | 87.65 | 60.66 | 1.68 | 4.37 | 3.03 | |
| LG8 | 18.96 | 310 | 44.65 | 94.95 | 69.8 | 2.36 | 5.01 | 3.68 | |
| LG9 | 18.53 | 340 | 48.51 | 78.94 | 63.73 | 2.62 | 4.26 | 3.44 | |
| LG10 | 18.31 | 256 | 39.53 | 88.94 | 64.24 | 2.16 | 4.86 | 3.51 | |
| LG11 | 17.26 | 170 | 19.07 | 61.6 | 40.34 | 1.1 | 3.57 | 2.34 | |
| LG12 | 15.82 | 231 | 43.29 | 81.06 | 62.17 | 2.74 | 5.12 | 3.93 | |
| LG13 | 15.29 | 277 | 20.43 | 71.73 | 46.08 | 1.34 | 4.69 | 3.01 | |
| LG14 | 14.96 | 342 | 47.38 | 105.34 | 76.36 | 3.17 | 7.04 | 5.1 |
The A. cervicornis linkage map was constructed with more offspring (154) from 16 families, and thus a greater number of informative markers were utilized in generating a consensus linkage map. In total, 4,859 markers were assigned to 14 linkage groups (LGs), with an average marker distance of 0.19 cM and a consensus map length of 927.36 cM (Table 1). Maps of A. cervicornis echoed the heterochiasmic maps of A. palmata, with a longer female map length (1,252.78 cM) than male map length (601.93 cM). As in A. palmata, for all 14 A. cervicornis LGs the female length was longer than the male length (Table 1). The genome-wide average recombination rate was higher in the female (4.41 cM/Mb) than in the male map (2.12 cM/Mb) (Table 1). The highest average recombination rate (7.04 cM/Mb) was in the female map associated with LG14. The lowest average recombination rate (1.10 cM/Mb) was in the male map associated with LG11.
Recombination landscapes were largely concordant between species, with similar recombination rates and centromere positions, as highlighted in Fig. 5 and Supplementary Fig. 8. However, one homologous chromosome pair (LG11/Chr11) exhibited large differences in map length in which the linkage map for A. palmata was almost twice as long as the map for A. cervicornis, despite similar physical size (115 cM vs. 61.6 cM in the female map). Female recombination rates were roughly two times as high as male rates in A. cervicornis and roughly 2.5 times as high in A. palmata.
Fig. 5.

Genetic maps for two homologous pairs of chromosomes of Acropora cervicornis and A. palmata. LG1 and Chr1 are homologous, as well as LG8 and Chr10. Percent SINE, LINE, LTR, and DNA repeats show putative centromere positions. Repeat content was calculated in 500 Kb sliding windows with 5 Kb steps. Note: A. palmata Chr1 and Chr10 x-axes indicating physical position are inverted due to the assembled sequence being reverse of the homologous chromosome in A. cervicornis
Discussion
Genome comparisons reveal chromosomal macrosynteny despite evolutionary divergence
Here, we present chromosome-scale genome assemblies of the Atlantic acroporids. Our assemblies are similar in quality, genome completeness and repetitive content to other published coral genomes but surpass many in genome contiguity. For example, the comparison of our A. cervicornis assembly with the recent genome assembly of another A. cervicornis genet from the Florida Keys [123] represents a sevenfold gain in contiguity (as measured by scaffold N50); otherwise the two genomes are remarkably similar in gene completeness and assembly size. Both species also possess the most common number of chromosomes amongst acroporids (2n = 28 in 72% of species surveyed; [35]), a value concordant with karyotyping in A. palmata [156]. The assembly of A. palmata is less complete than A. cervicornis, as assessed by BUSCO Metazoa v10 [143, 157]. The 87.8% completeness (Supplementary Table 5), compared with 93.1% in A. cervicornis, may be the result of small local mis-assemblies introduced during the Hi-C scaffolding process, incomplete polishing, or excessive purging of short, unscaffolded contigs. Nevertheless, the completeness of A. palmata is similar to other assemblies derived from similar data types and methods (i.e., short-read primary assembly with additional scaffolding [23, 36]).
Despite their smaller genome sizes, we found that the TE contents of A. palmata (16.69%) and A. cervicornis (18.01%, Supplementary Table 10), were similar to other acroporid species that had similar contig N50s and were analyzed using the same TE identification methods (namely RepeatModeler and RepeatMasker), ranging from 13.57% in A. digitifera to 19.62% in A. tenuis (Fig. 4A). These numbers are lower than previous estimates of 40% to 45% for acroporids using a different TE identification method on more fragmented assemblies [23]. Indeed, estimates from dnaPipeTE [109] (an assembly-free approach to TE content estimation) were comparatively much higher than those of RepeatModeler/RepeatMasker (Supplementary Fig. 7, Supplementary Table 10).
Between the Atlantic sister species, genomic synteny is largely conserved despite the relatively deep divergence time of approximately 5 million years [159, 160]. In a broader phylogenetic context, we found agreement with Ying et al. [161] and Shinzato et al. [23], wherein colinear relationships declined with phylogenetic distance within Acropora and in comparison with non-acroporids (Fig. 2D, Supplementary Fig. 2 and 3). For example, comparison of the acroporids, members of the complex clade of corals, with the coral Cataphyllia jardinei, which belongs to the robust coral clade, show macrosyntenic continuity within the 14 chromosomes (Supplementary Fig. 2A) but gene colinearity was mostly lost (Fig. 2D).
Structural variants were detected between A. palmata and A. cervicornis, but their presence should be independently confirmed due to the low marker density of the A. cervicornis linkage map. This linkage map was used for genome scaffolding and consisted of only 16 markers per Mb and contigs containing a single marker cannot be oriented correctly. Lep-Anchor [105] additionally utilizes long-read data to assist in contig orientation where linkage markers are sparse or absent, but in cases where long reads are too short to span repetitive regions, the correct orientation may still not be resolved. Long distance translocations and large-scale inversions may be more immune to these issues. Additionally, because of the presence of unbridged gaps from Hi-C and linkage scaffolding, break-ends may not be detected or supported by SV callers, despite being detected by alignment dot plots.
The two species discussed here naturally hybridize bidirectionally to form F1 hybrids [47], previously referred to as A. prolifera, and rare backcrosses of the F1 with both parent species have been documented. However, F2 generations have not been observed in genetic data from wild colonies [47, 50]. Given the paucity of later generation hybrids (backcrosses and F2s), the hybrids may undergo hybrid breakdown resulting in non-viable or less fit offspring. It is therefore assumed that some post-zygotic genetic mechanism, like differing genomic architectures or speciation genes, exists that represses reproduction between the parental species [50, 162]. For example, large structural variants can cause misalignment during F1 meiosis or death in F2 offspring due to the loss of gene copies required for survival [163]. Such structural variants (SVs) cause F2 sterility in interspecies hybrids of Drosophila [164], as well as F2 lethality in wild strains of Arabidopsis [165]. Although whole genome alignments between A. palmata and A. cervicornis demonstrate high levels of macrosynteny and conserved gene colinearity, some regions do exhibit large scale rearrangements (e.g., 2.5 Mb inversion on LG6/Chr4, Fig. 2B and C, Supplementary Table 7). SVs may be acting as a barrier to backcross and F2 offspring formation in the F1 hybrid adults and represent candidates for future studies of hybrid breakdown in this system. However, due to the linkage-based scaffolding used in the A. cervicornis assembly, such SVs should be validated using ultra long read or Hi-C sequencing as they may also represent assembly artifacts. Subsequently, population-level sequencing of A. palmata and A. cervicornis using long-read sequencing technologies could then be employed to assess if SVs represent fixed differences between species that could be linked to hybrid sterility.
In addition to putative SVs, speciation genes may also maintain the reproductive isolation between sister species [162]. Such genes may have a disproportionate effect in driving speciation and are represented by genes such as PRDM9 in mammals [166, 167] and NUP96 in Drosophila melanogaster and D. simulans [168]. Both PRDM9 and NUP96 cause sterility in F1 hybrids [166, 168]. In our analyses, we have identified genes with nucleotide divergence that significantly exceeds the genomic background (Supplementary Table 8). Such genes were enriched for the GO terms spindle pole (GO:0000922) and midbody (GO:0030406). The meiotic spindle is associated with meiotic drive leading to hybrid male sterility [169] and the formation and remnants of the meiotic midbody are important in developmental competency of mouse oocytes [170]. Such functions may, in part, drive the reproductive barriers isolating A. palmata and A. cervicornis.
Homologs of suREJ (sperm receptor for egg jelly) and SPAG1 (sperm-associated antigen 1) were also found amongst the most diverged genes between A. palmata and A. cervicornis (Supplementary Table 8). In sea urchins, suREJ is under positive selection and may be a mechanism to reduce gene flow between conspecifics [171]. Such a gene may play a role in reducing gene flow between A. palmata and A. cervicornis (as shown in choice/no choice hybridization crosses, [172]). However, suREJ has not been previously associated with hybrid breakdown or hybrid infertility in contrast to SPAG1 [173]. SPAG1 also supports the proper development of oocytes in mouse meiosis [174]. Given these functions in other organisms, SPAG1 may play a role in the bidirectional hybrid breakdown in the Atlantic Acropora species. Reciprocal crosses between A. palmata and A. cervicornis and F2 and backcrosses with the F1 hybrids may provide further insight into the role of SPAG1 in the reproductive isolation of the species.
Ancestral linkage groups and gene family expansions
Here, we found that over their 52—119 million years (Mya) of history [23], acroporids have retained conserved syntenic gene order to a high degree. While only a small sample size is available for comparison, the maintenance of chromosomal arrangements across deeply diverged coral lineages that split in the Devonian–Carboniferous, approximately 332–357 Mya [175], is surprising. Macrosyntenic patterns gradually degraded and chromosome numbers varied as we compared acroporids to more divergent species from Scleractinia, Actiniaria, Octocorallia, and Medusozoa (Supplementary Fig. 3).
Of the 21 cnidarian- specific ALG arrangements, six were largely intact within the scleractinians (acroporids and Catalaphyllia) and one ALG was lost from all but one cnidarian species surveyed. There were seven cases of ALG fusions and one example of centric insertion. A. millepora is the only acroporid species where a portion of ALG G fused with L. This fusion event in A. millepora presents an interesting target for further studies in light of the variable hybridization potential among Pacific species within the genus.
Comparison of gene families using presence/absence as well as phylogenetically informed expansions/contraction analyses of identified unique orthogroups at the family level and between the Pacific and Atlantic acroporids. Similar to a prior study [23], the acroporid-specific groups included overrepresentation of gene families involved in coral calcification (galaxin, matrix shell protein and skeletal organic matrix protein) and host-microbe interactions (prosaposin and toll-like receptor) relative to the other cnidarians. A minority of the orthogroups unique to the Atlantic species were annotated and those that were included transposable elements, suggesting numerous coding genes and/or repetitive element copies arose after gene flow stopped between the Atlantic and Pacific acroporids, approximately 2.8 Mya [176, 177]. Notable genes with lineage-specific duplications in the Atlantic acroporids include a gene involved with sperm function (OG0022455: cation channel sperm-associated protein 3), two genes involved in DNA replication (OG0022558: Serine/threonine-protein kinase Nek2 and OG0022391: replication protein A 70 kDa DNA-binding subunit C) and one gene in development (OG0022485: paired box protein). These duplicates should be assessed for functional differences in future studies exploring incompatibility between A. palmata and A. cervicornis.
In phylogenetically informed analyses, there was an enrichment of ontology terms relating to symbiont-containing vacuoles in the significantly expanding gene families in the Acroporidae. Symbiont-containing vacuole membranes are of crucial function to photosynthetic stony corals (e.g., [178]), of which the family Acroporidae is the most speciose and abundant [179]). It is possible that symbiont-associated gene families were crucial to the evolution and diversification of the family Acroporidae.
Within the genus Acropora, gene families that expanded were primarily related to cell–cell adhesion, cell recognition, and nerve components and processes. Acropora are unique in their highly complex and intricate morphologies and are the only coral genera to possess distinct axial and radial polyps [180–182]. The evolution of complex colony morphologies associated with the genus Acropora may have required neuron-related gene family expansions to help maintain cell-to-cell communication across increasingly complex skeleton morphologies. Retrotransposition terms were also found to be enriched in expanded gene families. Repeat elements represent a significant portion of Acropora genomes and these elements may still be dynamic in nature, despite not substantially altering genome size. In the absence of significant repeat expansions, further work is required to determine if retrotransposition drives copy number variants within populations of Acropora given the significant enrichment of terms associated with retrotransposition and viral response. Heat stress-dependent retrotransposition is prevalent in the coral symbiont Symbiodinium microadriaticum, and similar processes may be at play within the host species [183].
Heterochiasmy and recombination landscapes of the Caribbean acroporids
Heterochiasmy in A. palmata and A. cervicornis was among the most pronounced estimates observed in plants or animals [184]. Generally, recombination rates were higher in A. palmata, potentially due to differences in overall assembly length. The k-mer estimated genome sizes were similar (333 Mb in A. palmata and 331 Mb in A. cervicornis, Supplementary Table 4) but assembly sizes were more variable, with A. palmata being 287 Mb and A. cervicornis being 305 Mb. This would result in increased genome-wide A. palmata recombination rates simply due to assembly size. However, regardless of assembly sizes, genetic map lengths are greater in A. palmata (consensus map length 1013 cM) than in A. cervicornis (927 cM). Based on repeat density and local recombination rates (i.e. regions with elevated repeat content and suppressed recombination, as described in Hartley and O’Neill [185] and Schreiber et al. [186]), all chromosomes in both species appear to be metacentric or submetacentric (Fig. 5 and Supplementary Fig. 8), like in the Pacific acroporid, Acropora pruinosa [187]. Centromeric regions appear to be associated with long interspersed nuclear element (LINE) repeats, as shown by the prominent peaks in LINE content.
Within chromosomes, both species exhibit commonly observed local recombination landscapes (e.g., higher local recombination rates in females across whole chromosomes or higher recombination in males near telomeres; [58]). Twelve out of fourteen chromosomes exhibit recombination landscapes where local female rates are generally higher than male rates throughout the chromosome. Female maps exhibit marked declines in recombination around the presumed centromere while males show low, chromosome-wide recombination. However, in two cases, male local recombination rates are higher than female rates at one end of the chromosome, in telomeric regions (LG9/Chr9, LG8/Chr10, Fig. 5 and Supplementary Fig. 8).
Comparing recombination among the three acroporid species revealed numerous similarities among the Atlantic (A. palmata, A. cervicornis) and Pacific (A. millepora) corals. In all three acroporid linkage maps, the overall female map length was longer than the male length. However, the higher recombination in the female map in A. millepora was driven by only a subset of linkage groups [60]. In A. palmata and A. cervicornis, we find that the pattern is consistent across all chromosomes (Table 1, Fig. 5, and Supplementary Fig. 8).
The underlying causes of heterochiasmy are an active area of exploration in genetics [58, 188, 189]. Heterochiasmy was thought to be driven by the presence of sex chromosomes, but this is contradicted by similar patterns of heterochiasmy in simultaneous hermaphrodites that lack sex chromosomes (such as A. palmata and A. cervicornis), as well as species with environmental sex determination [57, 62, 190]. Other proposed explanations for heterochiasmy include genetic drift [191], haploid selection [188, 192], and female meiotic drive [184, 193]. Due to some consistency in patterns across divergent taxonomic groups, drift has been regarded as an unlikely explanation [58]. Wang et al. [60] concluded that haploid selection was the most likely culprit in the coral A. millepora at a time when theory for the role of meiotic drive had not yet been developed. There has been conflicting evidence for the role of haploid selection in animals, which have fewer expressed genes in sperm and eggs compared to plants [189, 194]. Higher recombination rates adjacent to centromeres in female maps suggest that meiotic drive may also be a potential mechanism for heterochiasmy in the Atlantic Acropora. The presence of drive may either serve to suppress or favor recombination in the female close to the centromeres depending on whether it occurs during Meiosis I or Meiosis II [184, 193]. During Meiosis I, selective pressure for increased recombination around the centromere is expected to reduce the spread of harmful drive alleles by decoupling them from the centromere [184].
In bivalves, sessile organisms with similar reproductive strategies to corals, the global patterns of recombination are similar to the Atlantic Acropora with females exhibiting higher rates than those of males, with locally higher recombination rates at centromeres in females and locally higher recombination rates at telomeres in males [58]. We hypothesize that there may be bioenergetic constraints on the production of each gamete which limit the frequency of recombination in sperm, particularly in broadcast spawning marine organisms (such as corals and bivalves) that may produce billions of sperm but only few eggs in each reproductive event. In Acropora, a colony may produce thousands of gamete bundles containing eggs and sperm. Each bundle contains 4–6 eggs and ~ 100,000 to ~ 150,000 sperm cells [195]. This is in comparison with insects, which have higher male recombination rates when compared with females, and produce as few as 50 sperm cells per reproductive event and may produce fewer sperm in their entire lifetime than a single coral gamete bundle [196–199]. Additional hypotheses proposed by Sardell et al. also involve the differing life history traits (e.g., sex organ temperature or aging) of each sex. As the species discussed here are hermaphroditic, we do not think these hypotheses are likely to explain the heterochiasmy of the Atlantic acroporids.
The average genome-wide recombination rates for A. palmata and A. cervicornis (3.04 to 3.5 cM/Mb) are higher than the average recombination rates for animals (2.54 cM/Mb) [57]. Average recombination rates for the two species are similar to the rates for insects, crustaceans, and fish, but higher than the averages for groups such as birds, amphibians, reptiles, and mammals [57]. This may indicate a rapid response to selection in acroporids because the proportion of substitutions fixed by adaptive evolution is positively correlated with recombination rate [200]. Future work comparing recombination rates across coral populations and taxa would be valuable in clarifying the evolutionary consequences of these patterns.
The lifespan of corals may also play a role in their overall high recombination rate compared with other metazoans. Corals are amongst the longest living metazoans, with some species capable of living thousands of years (e.g., Acropora palmata [156]). Given that reef-building corals are sessile and can live for thousands of years but typically only reproduce once per year in mass spawning events (e.g., [201]), high recombination rates may maximize the frequency at which favorable gene combinations are produced [202, 203] to match changing environments.
The local and genome-wide recombination rates calculated from the genetic linkage maps for A. palmata and A. cervicornis provide novel insights into the recombination landscape of corals. The density of markers in this resource now opens the possibility for quantitative trait locus (QTL) analyses as well as more precise haplotype imputation and genetic association studies in these species (Fig. 5). QTL mapping allows for the identification of loci that have consistent, predictable effects on phenotype across individuals. In plants, this is frequently used to assist with breeding programs [204]. As populations of many corals have rapidly declined [205], such a tool could assist in the design of restoration approaches. Additionally, phasing and imputation software commonly used in genome-wide association studies (GWAS) such as BEAGLE [206], GLIMPSE2 [207], and SHAPEIT [208] take into account recombination rates across chromosomes to more accurately statistically phase and impute data. The generation of these assemblies and genetic maps now enables complex genetic association studies not previously possible in these threatened non-model organisms. With these data, we have also demonstrated the application of the Acropora SNP array [101] as a successful genotyping method for the generation of a genetic linkage map, which provides a cost-effective means for creating additional maps for the F1 hybrids of A. palmata and A. cervicornis.
Conclusions
The genomic resources presented here revealed that the adaptive capacity of endangered Atlantic Acropora corals is likely not hindered by their recombination rates, as both species exhibit high genome-wide recombination rates with prominent heterochiasmy between sexes in these simultaneous hermaphrodites. Moreover, the sister species exhibit remarkable levels of macrosynteny and gene colinearity with one another as well as with the Pacific species, especially considering the > 50 Mya history of the genus. Our assemblies suggest that, like many scleractinians, the Atlantic acroporid genomes consist of 14 chromosomes; a derived state compared to the last common ancestor of the Cnidaria which is proposed to have had 17 chromosomes [4]. The conserved number of haploid chromosomes among many, but not all, of the acroporids is 14 (2n = 28, [35]) and the high level of macrosynteny across the Acropora genus may enable these syngameons described above. In the Pacific, it has been suggested that hybridization acts as an evolutionary force driving speciation [209, 210]. However, in the Atlantic Acropora, hybridization between the two sister species yields an F1 hybrid with limited fertility [50] despite the high levels of macrosynteny and gene colinearity of their genomes. In this study, we highlight putative genes and gene families that may drive reproductive isolation of the two Atlantic sister species. Further experimental work is required to gather further support for these targets. Together, the chromosome-scale assemblies and genetic maps we present here are the first detailed look at the genomic landscapes of these critically endangered species. The availability of these genomic resources helps facilitate genome-wide association studies and discovery of quantitative trait loci which can aid in the conservation of endangered corals.
Supplementary Information
Additional file 1: Supplementary Table 1: Summary of sequencing libraries used for genome assembly of the Atlantic acroporids. All data and assemblies are available at NCBI BioProject PRJNA473816.
Additional file 2: Supplementary Table 2: Genomic resources used to compare gene content and syntenic arrangement. Genome assemblies and their predicted proteins were downloaded from sources noted below. Taxonomy is ranked as Subphylum, Class and Order.
Additional file 3: Supplementary Table 3: Assembly statistics for the genome assemblies of the acroporids compared in this study.
Additional file 4: Supplementary Table 4: K-mer genome size estimates. Three different k-mer based genome size prediction tools were used: GenomeScope2, findGSE and kmergenie. The first two tools used the k-mer distribution histogram generated by Jellyfish with a k-mer size of 119. bp = base pairs.
Additional file 5: Supplementary Table 5: Assembly statistics showing completeness and contiguity of Acropora cervicornis HG0582 [123] and HG0005 assemblies (this study). Mb = megabases. BUSCO = Benchmarking Universal Single-Copy Orthologs.
Additional file 6: Supplementary Table 6: Comparison of Acropora palmata Hi-C (pseudo) chromosomes that are homologous to A. cervicornis linkage groups. Mb = megabases.
Additional file 7: Supplementary Table 7: Number of structural variants identified between Acropora palmata and A. cervicornis genome assemblies. N/A = not applicable.
Additional file 8: Supplementary Table 8: Outlier genes between Acropora palmata and A. cervicornis in terms of sequence identity. Percent identity was calculated using rustybam [140] in windows corresponding with gene coordinates using whole genome PAF alignments derived from minimap2 [92]. Genes were considered outliers if their percent identity was below Q1 – (3*IQR).
Additional file 9: Supplementary Table 9: Tracing the 21 cnidarian ancestral linkage groups as defined by Simakov et al. 2022 in acroporid corals. x = chromosomal fusion. Acroporids share the same ALG architecture except for ALG L which is fused to G in A. millepora only. AHYA = Acropora hyacinthus , APAL = A. palmata , AMIL = A. millepora , ACER = A. cervicornis , CJAR = Catalaphyllia jardinei , NVEC = Nematostella vectensis , SSPP = Scolanthus sp. , XSPP = Xenia sp. , HVUL = Hydra vulgaris , HSYM = Hydractinia symbiolongicarpus , CXAM = Cassiopea xamachana.
Additional file 10: Supplementary Table 10: Summary of repeat content in Acropora palmata and A. cervicornis genome assemblies. Bolded transposable elements are abundant among all acroporids.
Additional file 11: Supplementary Fig. 1: Alignments of the two available Acropora cervicornis genome assemblies. Genet K2 assembly (STAGdb ID HG0582, Selwyn and Vollmer [123] is compared with the genet M5 assembly (STAGdb ID HG0005, this study). A) Assembly-assembly minimap2 alignments > 1Mbp and their chromosome locations, plotted with pafr. B) Dot plots comparing the HG0582 assembly with the 14 HG0005 chromosomes (plotted with dotplotly, [135]). Contigs are largely concordant with the exception of one large HG0582 contig that was split across HG0005 chromosomes.
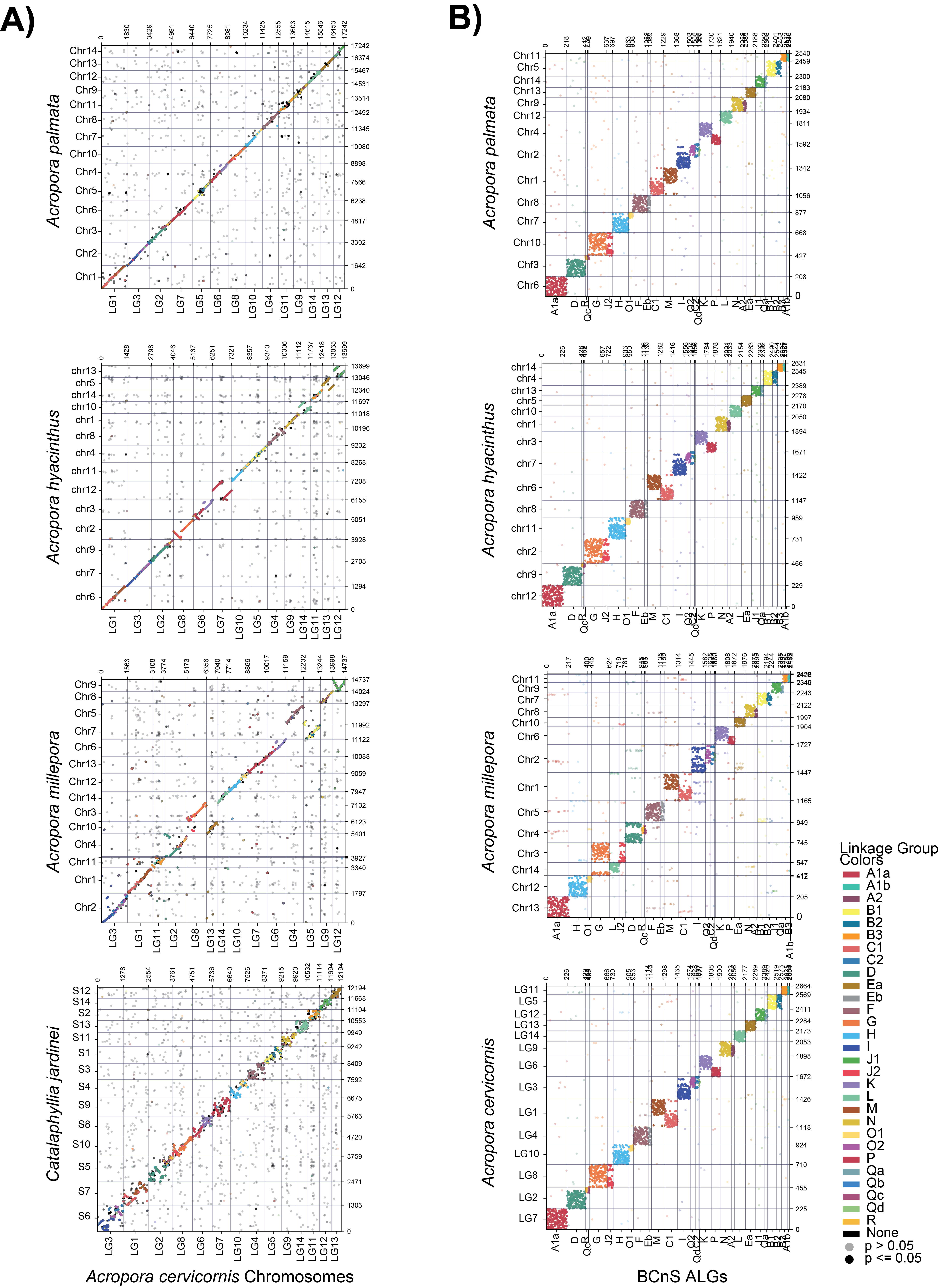{kind=link}
Additional file 12: Supplementary Fig. 2: Oxford dot plots of scleractinian chromosomes and 29 ancestral linkage groups. Scleractinian chromosomes are plotted against A. cervicornis chromosomes (A) and of acroporids chromosomes against 29 ancestral linkage groups (ALGs) of last common ancestor of sponges, cnidarians and bilaterians [2] (B).
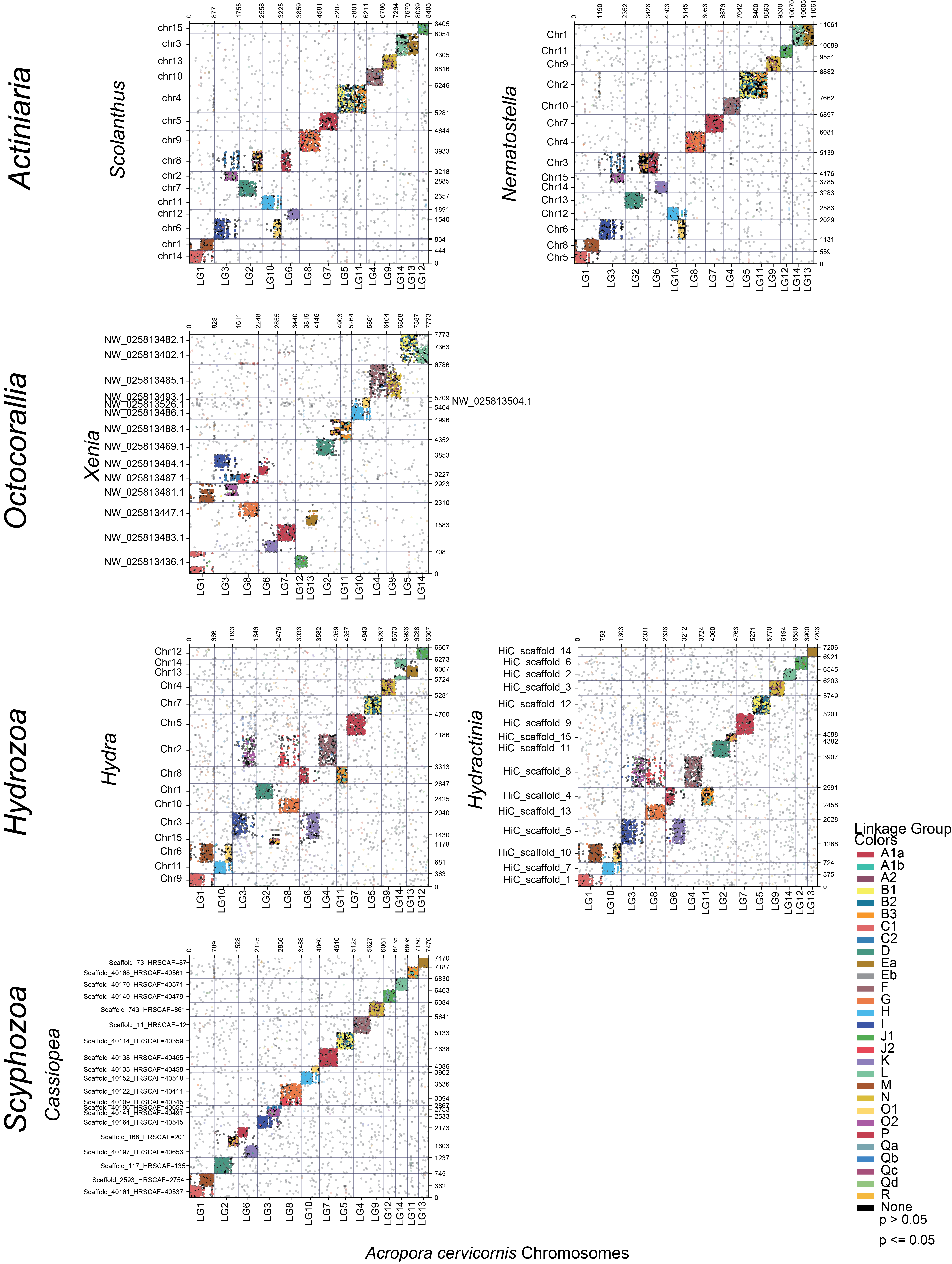{kind=link}
Additional file 13: Supplementary Fig. 3: Oxford dot plot of cnidarian assemblies against A. cervicornis chromosomes, arranged by taxonomic groups. Ancestral linkage groups proposed for the last common ancestor of Cnidaria, Bilateria and sponges [2] are indicated by color.
Additional file 14: Supplementary Fig. 4: Significantly enriched GO terms in expanding or contracting gene families of Acroporidae. Top 10 cell component, biological process, and molecular function GO terms associated with phylogenetically significant expand (orange) and contracting (blue) gene families for the family Acroporidae.
Additional file 15: Supplementary Fig. 5: Significantly enriched GO terms in expanding or contracting gene families of Acropora . Top 10 cell component, biological process, and molecular function GO terms associated with phylogenetically significant expand (orange) and contracting (blue) gene families for the genus Acropora.
Additional file 16: Supplementary Fig. 6: Significantly enriched GO terms in expanding or contracting gene families of the Atlantic Acropora . Top 10 cell component, biological process, and molecular function GO terms associated with phylogenetically significant expand (orange) and contracting (blue) gene families for the Atlantic Acropora species.
Additional file 17: Supplementary Fig. 7: Total repeat content estimates based on short-read sequence data of the Atlantic acroporids. Repeat classes are colored as red = DNA, green = LTR, yellow = LINE, blue = SINE, purple = Helitron, and gray = Other (i.e., satellites, simple repeats, unannotated TEs).
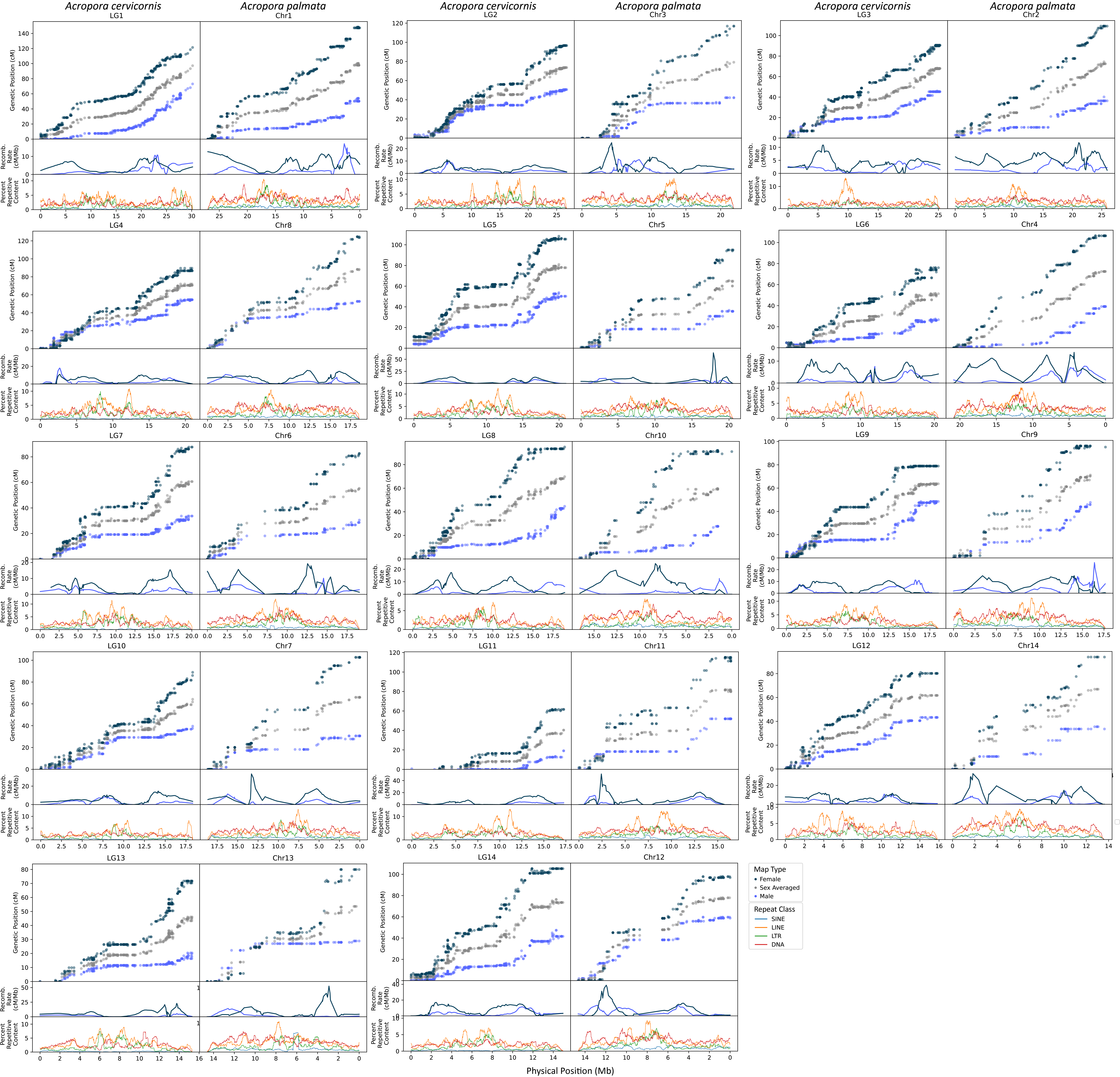{kind=link}
Additional file 18: Supplementary Fig. 8: Full linkage and recombination maps for all homologous chromosomes in the Atlantic acroporids. LINE content appears to peak in regions with low recombination rates, highlighting centromere positions.
Acknowledgements
The authors wish to thank the Caribbean Research and Management of Biodiversity (CARMABI) research station, the Coral Restoration Foundation (CRF), University of Wisconsin Biotechnology Center Next Generation Sequencing Core, Valerie Chamberland, Greg von Kustner, Meghann Devlin-Durante, Hannah G Reich, Kate L Vasquez-Kuntz, Cody Engelsma, Marina Villoch, Erich Bartels, Trinity L Conn, Erinn M Muller, Samuel Vohsen, and Abigail Clark for their invaluable contributions to the project. The co-authors would also like to acknowledge the Pennsylvania State University Huck Institutes’ Genomics Core Facility (RRID:SCR_023645) for use of Illumina HiSeq 2500 and Pacific BioSciences Sequel II sequencers.
Authors’ contributions
N.S.L. performed research, analyzed data, assembled A. cervicornis genome, constructed A. cervicornis linkage map, wrote the manuscript. S.A.K. performed research, analyzed data, assembled A. palmata genome, assembled first draft A. cervicornis genome, wrote the manuscript. K.H.S. performed research, analyzed data, generated A. palmata linkage map, wrote the manuscript. H.E., B.H.K, H.R.K analyzed data, edited manuscript. Z.D., C.C.O. performed research, edited manuscript. N.F. provided funding, edited manuscript. I.B.B. performed and supervised research, provided funding, wrote the paper, designed study, supervised the research. All authors reviewed the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. Research funded by the Revive and Restore Advanced Coral Toolkit Program funding to IBB, NOAA Restoration Center (#NA19NMF4630259) and Florida Fish & Wildlife Conservation Commission and Fish & Wildlife Research Institute (#21069) to Mote Marine Laboratory, and the National Science Foundation grant OCE-1537959 awarded to NDF and IBB. Dovetail Genomics partially funded the Hi-C assembly through an EOY Matching Funds Grant awarded to SAK and IBB. NSL and KHS were supported by the CBIOS (NIH T32 Kirschstein-NRSA: Computation, Bioinformatics, and Statistics) training program at The Pennsylvania State University (#T32GM102057). We acknowledge support by the Open Access publication fund of Alfred-Wegener-Institut Helmholtz-Zentrum für Polar- und Meeresforschung.
Data availability
Genome assemblies, gene predictions, functional annotations, and genetic maps are publicly available at https://zenodo.org/doi/10.5281/zenodo.12021086. NCBI genome accessions are GCA_025960835.2 for A. palmata, GCA_037043185.1 for A. cervicornis version 1, and GCA_041430625.1 for A. cervicornis version 2. All raw data and assemblies have been deposited in NCBI BioProject PRJNA473816 and all SRA accessions are provided in Supplementary Table 1.
Declarations
Ethics approval and consent to participate
Research was conducted under the following permits:
CRF-2016–020- 2016 spawning trip: M5 sperm collection only.
CRF-2017–009 and NOAA #FKNMS-2011–159-A4: 2017 spawning collection for the hybrid crosses.
CRF-2017–012 and NOAA #FKNMS-2011–159-A4: Horseshoe A. palmata fragment for the chromosome-level genome assembly at Dovetail.
NOAA #FKNMS-2019–012 A1, A2, A3, and A4, SAJ-2019–04431-(SP-GGM), PER00414: M5 ethanol-preserved tissue for Nanopore sequencing.
NOAA #FKNMS-2015–163 A2, A3: 2020 spawning of A. cervicornis and production of linkage map offspring.
CAR-2019–11, AN001_169, and AN001_172: spawning collections and export permits for A. palmata samples and production of linkage map.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’ s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Nicolas S Locatelli, Sheila A Kitchen and Kathryn H Stankiewicz contributed equally to this work.
References
- 1.Stolarski J, Kitahara MV, Miller DJ, Cairns SD, Mazur M, Meibom A. The ancient evolutionary origins of Scleractinia revealed by azooxanthellate corals. BMC Evol Biol. 2011;11:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simakov O, Bredeson J, Berkoff K, Marletaz F, Mitros T, Schultz DT, et al. Deeply conserved synteny and the evolution of metazoan chromosomes. Sci Adv. 2022;8:eabi5884. [DOI] [PMC free article] [PubMed]
- 3.Zimmermann B, Montenegro JD, Robb SMC, Fropf WJ, Weilguny L, He S, et al. Topological structures and syntenic conservation in sea anemone genomes. Nat Commun. 2023;14:8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimmermann B, Montenegro JD, Robb SMC, Fropf WJ, Weilguny L, He S, et al. Sea anemone genomes reveal ancestral metazoan chromosomal macrosynteny. 2023;:2020.10.30.359448.
- 5.Steele RE, David CN, Technau U. A genomic view of 500 million years of cnidarian evolution. Trends Genet. 2011;27:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shearer TL, Van Oppen MJH, Romano SL, Wörheide G. Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol Ecol. 2002;11:2475–87. [DOI] [PubMed] [Google Scholar]
- 7.Budd AF, Pandolfi JM. Evolutionary Novelty Is Concentrated at the Edge of Coral Species Distributions. Science. 2010;328:1558–61. [DOI] [PubMed] [Google Scholar]
- 8.Campos JL, Halligan DL, Haddrill PR, Charlesworth B. The Relation between Recombination Rate and Patterns of Molecular Evolution and Variation in Drosophila melanogaster. Mol Biol Evol. 2014;31:1010–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baird AH, Guest JR, Willis BL. Systematic and Biogeographical Patterns in the Reproductive Biology of Scleractinian Corals. Annu Rev Ecol Evol Syst. 2009;40:551–71. [Google Scholar]
- 10.Baums IB, Miller MW, Hellberg ME. Geographic Variation in Clonal Structure in a Reef-Building Caribbean Coral. Acropora palmata Ecological Monographs. 2006;76:503–19. [Google Scholar]
- 11.Harrison PL. Sexual reproduction of scleractinian corals. Coral reefs: an ecosystem in transition. 2011;59–85.
- 12.Szmant AM. Reproductive ecology of Caribbean reef corals. Coral Reefs. 1986;5:43–53. [Google Scholar]
- 13.Baums IB, Devlin-Durante MK, Polato NR, Xu D, Giri S, Altman NS, et al. Genotypic variation influences reproductive success and thermal stress tolerance in the reef building coral. Acropora palmata Coral Reefs. 2013;32:703–17. [Google Scholar]
- 14.Vasquez Kuntz KL, Kitchen SA, Conn TL, Vohsen SA, Chan AN, Vermeij MJA, et al. Inheritance of somatic mutations by animal offspring. Sci Adv. 2022;8:eabn0707. [DOI] [PMC free article] [PubMed]
- 15.Koch HR, Matthews B, Leto C, Engelsma C, Bartels E. Assisted sexual reproduction of Acropora cervicornis for active restoration on Florida’s Coral Reef. Front Mar Sci. 2022;9:959520.
- 16.Richmond R, Hunter C. Reproduction and recruitment of corals: comparisons among the Caribbean, the Tropical Pacific, and the Red Sea. Mar Ecol Prog Ser. 1990;60:185–203. [Google Scholar]
- 17.Baums IB, Devlin-Durante MK, LaJeunesse TC. New insights into the dynamics between reef corals and their associated dinoflagellate endosymbionts from population genetic studies. Mol Ecol. 2014;23:4203–15. [DOI] [PubMed] [Google Scholar]
- 18.Harper LM, Huebner LK, O’Cain ED, Ruzicka R, Gleason DF, Fogarty ND. Multi-year coral recruitment study across the Florida Reef Tract reveals boom-or-bust pattern among broadcast spawners and consistency among brooders. Mar Ecol Prog Ser. 2023;721:39–58. [Google Scholar]
- 19.Bruckner AW, Hill RL. Ten years of change to coral communities off Mona and Desecheo Islands, Puerto Rico, from disease and bleaching. Dis Aquat Org. 2009;87:19–31. [DOI] [PubMed] [Google Scholar]
- 20.Dudgeon SR, Aronson RB, Bruno JF, Precht WF. Phase shifts and stable states on coral reefs. Mar Ecol Prog Ser. 2010;413:201–16. [Google Scholar]
- 21.Holstein TW. The role of cnidarian developmental biology in unraveling axis formation and Wnt signaling. Dev Biol. 2022;487:74–98. [DOI] [PubMed] [Google Scholar]
- 22.Fuller ZL, Mocellin VJL, Morris LA, Cantin N, Shepherd J, Sarre L, et al. Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science. 2020;369:eaba4674. [DOI] [PubMed]
- 23.Shinzato C, Khalturin K, Inoue J, Zayasu Y, Kanda M, Kawamitsu M, et al. Eighteen Coral Genomes Reveal the Evolutionary Origin of Acropora Strategies to Accommodate Environmental Changes. Mol Biol Evol. 2021;38:16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.López-Nandam EH, Albright R, Hanson EA, Sheets EA, Palumbi SR. Mutations in coral soma and sperm imply lifelong stem cell renewal and cell lineage selection. Proc R Soc B: Biol Sci. 2023;290:20221766. [DOI] [PMC free article] [PubMed]
- 25.Stankiewicz KH. Coral biology and evolution through the lens of genomics and population genetics.
- 26.Baumgarten S, Simakov O, Esherick LY, Liew YJ, Lehnert EM, Michell CT, et al. The genome of Aiptasia, a sea anemone model for coral symbiosis. Proc Natl Acad Sci. 2015;112:11893–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang JB, Quattrini AM, Francis WR, Ryan JF, Rodríguez E, McFadden CS. A hybrid de novo assembly of the sea pansy (Renilla muelleri) genome. GigaScience. 2019;8:giz026. [DOI] [PMC free article] [PubMed]
- 28.Jeon Y, Park SG, Lee N, Weber JA, Kim H-S, Hwang S-J, et al. The Draft Genome of an Octocoral. Dendronephthya gigantea Genome Biol Evol. 2019;11:949–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu M, Zheng X, Fan CM, Zheng Y. Lineage dynamics of the endosymbiotic cell type in the soft coral Xenia. Nature. 2020;582:534–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ip JC-H, Ho M-H, Chan BKK, Qiu J-W. A draft genome assembly of reef-building octocoral Heliopora coerulea. Sci Data. 2023;10:381. [DOI] [PMC free article] [PubMed]
- 31.McKenna V, Archibald JM, Beinart R, Dawson MN, Hentschel U, Keeling PJ, et al. The Aquatic Symbiosis Genomics Project: probing the evolution of symbiosis across the tree of life. Wellcome Open Research. 2021;6:254. [Google Scholar]
- 32.Yu Y, Nong W, So WL, Xie Y, Yip HY, Haimovitz J, et al. Genome of elegance coral Catalaphyllia jardinei (Euphylliidae). Front Mar Sci. 2022;9:991391.
- 33.Thomas L, Underwood JN, Rose NH, Fuller ZL, Richards ZT, Dugal L, et al. Spatially varying selection between habitats drives physiological shifts and local adaptation in a broadcast spawning coral on a remote atoll in Western Australia. Sci Adv. 2022;8:eabl9185. [DOI] [PMC free article] [PubMed]
- 34.Stephens TG, Lee J, Jeong Y, Yoon HS, Putnam HM, Majerová E, et al. High-quality genome assemblies from key Hawaiian coral species. GigaScience. 2022;11:giac098. [DOI] [PMC free article] [PubMed]
- 35.Kenyon JC. Models of reticulate evolution in the coral genus Acropora based on chromosome numbers: parallels with plants. Evolution. 1997;51:756–67. [DOI] [PubMed] [Google Scholar]
- 36.Prada C, Hanna B, Budd AF, Woodley CM, Schmutz J, Grimwood J, et al. Empty Niches after Extinctions Increase Population Sizes of Modern Corals. Curr Biol. 2016;26:3190–4. [DOI] [PubMed] [Google Scholar]
- 37.Pootakham W, Sonthirod C, Naktang C, Kongjandtre N, Putchim L, Sangsrakru D, et al. De novo Assembly of the Brain Coral Platygyra sinensis Genome. Front Mar Sci. 2021;8:732650.
- 38.Bongaerts P, Cooke IR, Ying H, Wels D, den Haan S, Hernandez-Agreda A, et al. Morphological stasis masks ecologically divergent coral species on tropical reefs. Curr Biol. 2021;31:2286–98. [DOI] [PubMed] [Google Scholar]
- 39.Torda G, Quigley KM. Drivers of adaptive capacity in wild populations: Implications for genetic interventions. Front Mar Sci. 2022;9:947989.
- 40.Mathur S, Mason AJ, Bradburd GS, Gibbs HL. Functional genomic diversity is correlated with neutral genomic diversity in populations of an endangered rattlesnake. Proc Natl Acad Sci. 2023;120:e2303043120. [DOI] [PMC free article] [PubMed]
- 41.Baums IB, Boulay JN, Polato NR, Hellberg ME. No gene flow across the Eastern Pacific Barrier in the reef-building coral Porites lobata. Mol Ecol. 2012;21:5418–33. [DOI] [PubMed] [Google Scholar]
- 42.Voolstra CR, Hume BCC, Armstrong EJ, Mitushasi G, Porro B, Oury N, et al. Disparate genetic divergence patterns in three corals across a pan-Pacific environmental gradient highlight species-specific adaptation. NPJ Biodivers. 2023;2:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baums IB, Miller MW, Hellberg ME. Regionally isolated populations of an imperiled Caribbean coral. Acropora palmata Mol Ecol. 2005;14:1377–90. [DOI] [PubMed] [Google Scholar]
- 44.Drury C, Dale KE, Panlilio JM, Miller SV, Lirman D, Larson EA, et al. Genomic variation among populations of threatened coral: Acropora cervicornis. BMC Genomics. 2016;17:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drury C, Schopmeyer S, Goergen E, Bartels E, Nedimyer K, Johnson M, et al. Genomic patterns in Acropora cervicornis show extensive population structure and variable genetic diversity. Ecol Evol. 2017;7:6188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Devlin-Durante MK, Baums IB. Genome-wide survey of single-nucleotide polymorphisms reveals fine-scale population structure and signs of selection in the threatened Caribbean elkhorn coral, Acropora palmata. PeerJ. 2017;5:e4077. [DOI] [PMC free article] [PubMed]
- 47.Kitchen SA, Ratan A, Bedoya-Reina OC, Burhans R, Fogarty ND, Miller W, et al. Genomic variants among threatened Acropora corals. G3 Genes, Genomes, Genetics. 2019;9:1633–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Canty SWJ, Fox G, Rowntree JK, Preziosi RF. Genetic structure of a remnant Acropora cervicornis population. Sci Rep. 2021;11:3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.García-Urueña R, Kitchen SA, Schizas NV. Fine scale population structure of Acropora palmata and Acropora cervicornis in the Colombian Caribbean. PeerJ. 2022;10:e13854. [DOI] [PMC free article] [PubMed]
- 50.Vollmer SV, Palumbi SR. Hybridization and the evolution of reef coral diversity. Science. 2002;296:2023–5. [DOI] [PubMed] [Google Scholar]
- 51.Fogarty ND, Lowenberg M, Ojima MN, Knowlton N, Levitan DR. Asymmetric conspecific sperm precedence in relation to spawning times in the Montastraea annularis species complex (Cnidaria: Scleractinia). J Evol Biol. 2012;25:2481–8. [DOI] [PubMed] [Google Scholar]
- 52.Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78:737–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otto SP, Lenormand T. Resolving the paradox of sex and recombination. Nat Rev Genet. 2002;3:252–61. [DOI] [PubMed] [Google Scholar]
- 54.Eriksson M, Rafajlović M. The Effect of the Recombination Rate between Adaptive Loci on the Capacity of a Population to Expand Its Range. Am Nat. 2021;197:526–42. [DOI] [PubMed] [Google Scholar]
- 55.Castellano D, Coronado-Zamora M, Campos JL, Barbadilla A, Eyre-Walker A. Adaptive Evolution Is Substantially Impeded by Hill-Robertson Interference in Drosophila. Mol Biol Evol. 2016;33:442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grandaubert J, Dutheil JY, Stukenbrock EH. The genomic determinants of adaptive evolution in a fungal pathogen. Evolution Letters. 2019;3:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stapley J, Feulner PGD, Johnston SE, Santure AW, Smadja CM. Variation in recombination frequency and distribution across eukaryotes: patterns and processes. Biological Sciences: Philosophical Transactions of the Royal Society B; 2017. p. 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sardell JM, Kirkpatrick M. Sex Differences in the Recombination Landscape. Am Nat. 2020;195:361–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cooney CR, Mank JE, Wright AE. Constraint and divergence in the evolution of male and female recombination rates in fishes. Evolution. 2021;75:2857–66. [DOI] [PubMed] [Google Scholar]
- 60.Wang S, Zhang L, Meyer E, Matz MV. Construction of a high-resolution genetic linkage map and comparative genome analysis for the reef-building coral Acropora millepora. Genome Biol. 2009;10:1–17. [DOI] [PMC free article] [PubMed]
- 61.Li H, Liu X, Zhang G. A Consensus Microsatellite-Based Linkage Map for the Hermaphroditic Bay Scallop (Argopecten irradians) and Its Application in Size-Related QTL Analysis. PLoS One. 2012;7:e46926. [DOI] [PMC free article] [PubMed]
- 62.Theodosiou L, McMillan WO, Puebla O. Recombination in the eggs and sperm in a simultaneously hermaphroditic vertebrate. Proc Natl Acad Sci. 2016;283:20161821. [DOI] [PMC free article] [PubMed]
- 63.Rastas P. Lep-MAP3: robust linkage mapping even for low-coverage whole genome sequencing data. Bioinformatics. 2017;33:3726–32. [DOI] [PubMed] [Google Scholar]
- 64.Baums I, Kitchen S. Acropora DNA extraction with Qiagen DNeasy tissue kit. 2020.
- 65.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetJournal. 2011;17:10–2. [Google Scholar]
- 66.Marcais G, Kingsford C. Jellyfish: A fast k-mer counter. Tutorialis e Manuais. 2012;1:1–8. [Google Scholar]
- 67.Chikhi R, Medvedev P. Informed and automated k-mer size selection for genome assembly. Bioinformatics. 2014;30:31–7. [DOI] [PubMed] [Google Scholar]
- 68.Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11:1432. [DOI] [PMC free article] [PubMed]
- 69.Sun H, Ding J, Piednoël M, Schneeberger K. findGSE: estimating genome size variation within human and Arabidopsis using k-mer frequencies. Bioinformatics. 2018;34:550–7. [DOI] [PubMed] [Google Scholar]
- 70.Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front Genet. 2013;4:237. [DOI] [PMC free article] [PubMed]
- 71.Challis R, Richards E, Rajan J, Cochrane G, Blaxter M. BlobToolKit – Interactive Quality Assessment of Genome Assemblies. G3 Genes|Genomes|Genetics. 2020;10:1361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reich H, Kitchen SA, Fogarty ND, Baums IB. Genomic variation of an endosymbiotic dinoflagellate (Symbiodinium ’fitti’) among closely related coral hosts. Mol Ecol. 2021;00:1–15. [DOI] [PubMed] [Google Scholar]
- 73.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:2047–217X. [DOI] [PMC free article] [PubMed]
- 74.Shinzato C, Shoguchi E, Kawashima T, Hamada M, Hisata K, Tanaka M, et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature. 2011;476:320–3. [DOI] [PubMed] [Google Scholar]
- 75.Shoguchi E, Shinzato C, Kawashima T, Gyoja F, Mungpakdee S, Koyanagi R, et al. Draft assembly of the symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr Biol. 2013;23:1399–408. [DOI] [PubMed] [Google Scholar]
- 76.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin S, Cheng S, Song B, Zhong X, Lin X, Li W, et al. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science. 2015;350:691–4. [DOI] [PubMed] [Google Scholar]
- 79.Aranda M, Li Y, Liew YJ, Baumgarten S, Simakov O, Wilson MC, et al. Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci Rep. 2016;6:39734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ye C, Hill CM, Wu S, Ruan J, Ma Z. DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci Rep. 2016;6:31900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chakraborty M, Baldwin-Brown JG, Long AD, Emerson JJ. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016;44:e147. [DOI] [PMC free article] [PubMed]
- 83.Lam K-K, LaButti K, Khalak A, Tse D. FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics. 2015;31:3207–9. [DOI] [PubMed] [Google Scholar]
- 84.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLOS ONE. 2014;9:e112963. [DOI] [PMC free article] [PubMed]
- 85.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013.
- 86.Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun. 2018;9:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Renschler G, Richard G, Valsecchi CIK, Toscano S, Arrigoni L, Ramírez F, et al. Hi-C guided assemblies reveal conserved regulatory topologies on X and autosomes despite extensive genome shuffling. Genes Dev. 2019;33:1591–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wick RR, Judd LM, Gorrie CL, Holt KE. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb Genom. 2017;3:e000132. [DOI] [PMC free article] [PubMed]
- 89.Kolmogorov M, Bickhart DM, Behsaz B, Gurevich A, Rayko M, Shin SB, et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods. 2020;17:1103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kundu R, Casey J, Sung W-K. HyPo: Super Fast & Accurate Polisher for Long Read Genome Assemblies. bioRxiv. 2019.
- 91.Krueger F, James F, Ewels P, Afyounian E, Schuster-Boeckler B. FelixKrueger/TrimGalore: v0.6.7. 2021. 10.5281/zenodo.5127899.
- 92.Li H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: Architecture and applications. BMC Bioinformatics. 2009;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shoguchi E, Beedessee G, Tada I, Hisata K, Kawashima T, Takeuchi T, et al. Two divergent Symbiodinium genomes reveal conservation of a gene cluster for sunscreen biosynthesis and recently lost genes. BMC Genomics. 2018;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shoguchi E, Beedessee G, Hisata K, Tada I, Narisoko H, Satoh N, et al. A New Dinoflagellate Genome Illuminates a Conserved Gene Cluster Involved in Sunscreen Biosynthesis. Genome Biol Evol. 2021;13:evaa235. [DOI] [PMC free article] [PubMed]
- 97.Hamada M, Schröder K, Bathia J, Kürn U, Fraune S, Khalturina M, et al. Metabolic co-dependence drives the evolutionarily ancient Hydra–Chlorella symbiosis. eLife. 2018;7:e35122. [DOI] [PMC free article] [PubMed]
- 98.Beedessee G, Kubota T, Arimoto A, Nishitsuji K, Waller RF, Hisata K, et al. Integrated omics unveil the secondary metabolic landscape of a basal dinoflagellate. BMC Biol. 2020;18:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Laetsch DR, Blaxter ML. BlobTools: Interrogation of genome assemblies. F1000Res. 2017;6:1287. [Google Scholar]
- 100.Guan D, Guan D, McCarthy SA, Wood J, Howe K, Wang Y, et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020;36:2896–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kitchen SA, Von Kuster G, Kuntz KLV, Reich HG, Miller W, Griffin S, et al. STAGdb: a 30K SNP genotyping array and Science Gateway for Acropora corals and their dinoflagellate symbionts. Sci Rep. 2020;10:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Genovese G. gtc2vcf. GitHub. https://github.com/freeseek/gtc2vcf. Accessed 31 May 2024.
- 103.Dimens PV. pdimens/LepWrap: 4.0.1. 2022.
- 104.Siberchicot A, Bessy A, Guéguen L, Marais GA. MareyMap Online: A User-Friendly Web Application and Database Service for Estimating Recombination Rates Using Physical and Genetic Maps. Genome Biol Evol. 2017;9:2506–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rastas P. Lep-Anchor: automated construction of linkage map anchored haploid genomes. Bioinformatics. 2020;36:2359–64. [DOI] [PubMed] [Google Scholar]
- 106.Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA. 2020;117:9451–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jurka J, Klonowski P, Dagman V, Pelton P. CENSOR–a program for identification and elimination of repetitive elements from DNA sequences. Comput Chem. 1996;20:119–21. [DOI] [PubMed] [Google Scholar]
- 108.Smit A, Hubley R, Green P. RepeatMasker Open-4.0.
- 109.Goubert C. Assembly-Free Detection and Quantification of Transposable Elements with dnaPipeTE. Methods Mol Biol. 2023;2607:25–43. [DOI] [PubMed] [Google Scholar]
- 110.Brůna T, Lomsadze A, Borodovsky M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom Bioinform. 2020;2:lqaa026. [DOI] [PMC free article] [PubMed]
- 111.Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform. 2021;3:lqaa108. [DOI] [PMC free article] [PubMed]
- 112.Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Slater GSC, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brückner A, Barnett AA, Bhat P, Antoshechkin IA, Kitchen SA. Molecular evolutionary trends and biosynthesis pathways in the Oribatida revealed by the genome of Archegozetes longisetosus. Acarologia. 2022;62:532–73. [Google Scholar]
- 115.Polato NR, Vera JC, Baums IB. Gene Discovery in the Threatened Elkhorn Coral: 454 Sequencing of the Acropora palmata Transcriptome. PLoS One. 2011;6:e28634. [DOI] [PMC free article] [PubMed]
- 116.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kriventseva EV, Kuznetsov D, Tegenfeldt F, Manni M, Dias R, Simão FA, et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019;47:D807–D811. [DOI] [PMC free article] [PubMed]
- 119.Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Brief Bioinform. 2013;14:144–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Keilwagen J, Hartung F, Grau J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. In: Kollmar M, editor. Gene Prediction: Methods and Protocols. New York, NY: Springer; 2019. p. 161–77. [DOI] [PubMed] [Google Scholar]
- 121.Palmer JM, Stajich J. nextgenusfs/funannotate: funannotate v1.8.13 (Version 1.8.13). Zenodo. 2020. 10.5281/zenodo.1134477.
- 122.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Selwyn JD, Vollmer SV. Whole genome assembly and annotation of the endangered Caribbean coral Acropora cervicornis. G3 Genes|Genomes|Genetics. 2023. 10.1093/g3journal/jkad232. [DOI] [PMC free article] [PubMed]
- 124.Osborne CC. Velveteenie/Caribbean_Acropora_Transcriptomes: v1_10.02.2023. 2023.
- 125.Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–W439. [DOI] [PMC free article] [PubMed]
- 126.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009;37:D211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, et al. EggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Boutet E, Lieberherr D, Tognolli M, Schneider M, Bansal P, Bridge AJ, et al. Uniprotkb/swiss-prot, the manually annotated section of the uniprot knowledgebase. In: Methods in Molecular Biology. 2016. p. 23–54. [DOI] [PubMed]
- 130.Rawlings ND, Barrett AJ, Bateman A. MEROPS: The peptidase database. Nucleic Acids Res. 2009;38:D227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huang L, Zhang H, Wu P, Entwistle S, Li X, Yohe T, et al. DbCAN-seq: A database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 2018;46:D516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput Biol. 2018;14:e1005944. [DOI] [PMC free article] [PubMed]
- 134.David Winter MC Kate Lee. pafr: Read, Manipulate and Visualize “Pairwise mApping Format” Data. 2020.
- 135.Poorten T. dotPlotly. GitHub. https://github.com/tpoorten/dotPlotly. Accessed 31 May 2024.
- 136.Cabanettes F, Klopp C. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ. 2018;6:e4958. [DOI] [PMC free article] [PubMed]
- 137.Nattestad M, Schatz MC. Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics. 2016;32:3021–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.O’Donnell S, Fischer G. MUM&Co: accurate detection of all SV types through whole-genome alignment. Bioinformatics. 2020;36:3242–3. [DOI] [PubMed] [Google Scholar]
- 139.Heller D, Vingron M. SVIM-asm: structural variant detection from haploid and diploid genome assemblies. Bioinformatics. 2021;36:5519–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vollger MR, Hulselmans G. mrvollger/rustybam: v0.1.33. 2023.
- 141.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). 2021;2:100141. [DOI] [PMC free article] [PubMed]
- 142.Marc Carlson HP. AnnotationForge. 2017.
- 143.Manni M, Berkeley MR, Seppey M, Zdobnov EM. BUSCO: Assessing Genomic Data Quality and Beyond. Curr Protoc. 2021;1:e323. [DOI] [PubMed]
- 144.Emms DM, Kelly S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lex A, Gehlenborg N, Strobelt H, Vuillemot R, Pfister H. UpSet: Visualization of Intersecting Sets. IEEE Trans Visual Comput Graphics. 2014;20:1983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nothman J. UpSetPlot. GitHub. https://github.com/jnothman/UpSetPlot. Accessed 31 May 2024.
- 147.Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19:301–2. [DOI] [PubMed] [Google Scholar]
- 148.Baron-Szabo RC, Schlagintweit F, Rashidi K. Coral fauna across the Cretaceous-Paleogene boundary at Zagros and Sistan Suture zones and Yazd Block of Iran. Swiss Journal of Palaeontology. 2023;142:7. [Google Scholar]
- 149.Alloiteau J. Monographie des madreporaires fossiles de Madagascar. Annales Geologiques de Madagascar. 1958;25:1–218. [Google Scholar]
- 150.Rigby JK, Jiasong F, Wei Z. Sphinctozoan Sponges from the Permian Reefs of South China. J Paleontol. 1989;63:404–39. [Google Scholar]
- 151. Berg HC, Cruz EL. Map and table describing fossil collections and related samples in the Ketchikan and Prince Rupert quadrangles, southeastern Alaska. U.S. Geological Survey; 1982.
- 152.Peters SE, McClennen M. The Paleobiology Database application programming interface. Paleobiology. 2016;42:1–7. [Google Scholar]
- 153.Mendes FK, Vanderpool D, Fulton B, Hahn MW. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics. 2021;36:5516–8. [DOI] [PubMed] [Google Scholar]
- 154.Schultz DT, Haddock SHD, Bredeson JV, Green RE, Simakov O, Rokhsar DS. Ancient gene linkages support ctenophores as sister to other animals. Nature. 2023;618:110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60. [DOI] [PubMed] [Google Scholar]
- 156.Devlin-Durante MK, Miller MW, Group CAR, Precht WF, Baums IB. How old are you? Genet age estimates in a clonal animal. Mol Ecol. 2016;25:5628–46. [DOI] [PubMed] [Google Scholar]
- 157.Manni M, Berkeley MR, Seppey M, Sim~ Ao FA, Zdobnov EM. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol. 2021;38:4647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Klaus JS, McNeill DF, Budd AF, Coates AG. Neogene reef coral assemblages of the Bocas del Toro region, Panama: the rise of Acropora palmata. Coral Reefs. 2012;31:191–203. [Google Scholar]
- 160.Wallace CC. Acroporidae of the Caribbean. Geologica Belgica. 2012.
- 161.Ying H, Cooke I, Sprungala S, Wang W, Hayward DC, Tang Y, et al. Comparative genomics reveals the distinct evolutionary trajectories of the robust and complex coral lineages. Genome Biol. 2018;19:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wu C-I, Ting C-T. Genes and speciation. Nat Rev Genet. 2004;5:114–22. [DOI] [PubMed] [Google Scholar]
- 163.Zhang L, Reifová R, Halenková Z, Gompert Z. How Important Are Structural Variants for Speciation? Genes (Basel). 2021;12:1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Masly JP, Jones CD, Noor MAF, Locke J, Orr HA. Gene Transposition as a Cause of Hybrid Sterility in Drosophila. Science. 2006;313:1448–50. [DOI] [PubMed] [Google Scholar]
- 165.Bikard D, Patel D, Le Metté C, Giorgi V, Camilleri C, Bennett MJ, et al. Divergent Evolution of Duplicate Genes Leads to Genetic Incompatibilities Within A. thaliana. Science. 2009;323:623–6. [DOI] [PubMed] [Google Scholar]
- 166.Mukaj A, Piálek J, Fotopulosova V, Morgan AP, Odenthal-Hesse L, Parvanov ED, et al. Prdm9 Intersubspecific Interactions in Hybrid Male Sterility of House Mouse. Mol Biol Evol. 2020;37:3423–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ponting CP. What are the genomic drivers of the rapid evolution of PRDM9? Trends Genet. 2011;27:165–71. [DOI] [PubMed] [Google Scholar]
- 168.Presgraves DC, Balagopalan L, Abmayr SM, Orr HA. Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature. 2003;423:715–9. [DOI] [PubMed] [Google Scholar]
- 169.McDermott SR, Noor MAF. The role of meiotic drive in hybrid male sterility. Philos Trans R Soc Lond B Biol Sci. 2010;365:1265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jung GI, Londoño-Vásquez D, Park S, Skop AR, Balboula AZ, Schindler K. An oocyte meiotic midbody cap is required for developmental competence in mice. Nat Commun. 2023;14:7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pujolar JM, Pogson GH. Positive Darwinian selection in gamete recognition proteins of Strongylocentrotus sea urchins. Mol Ecol. 2011;20:4968–82. [DOI] [PubMed] [Google Scholar]
- 172.Fogarty ND, Vollmer SV, Levitan DR. Weak Prezygotic Isolating Mechanisms in Threatened Caribbean Acropora Corals. PLoS One. 2012;7:e30486. [DOI] [PMC free article] [PubMed]
- 173.Hayashida K, Kohno S. Hybrid male sterility is caused by mitochondrial DNA deletion. Mol Biol Rep. 2009;36:1365–9. [DOI] [PubMed] [Google Scholar]
- 174.Huang C, Wu D, Khan FA, Jiao X, Guan K, Huo L. The GTPase SPAG-1 orchestrates meiotic program by dictating meiotic resumption and cytoskeleton architecture in mouse oocytes. MBoC. 2016;27:1776–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Quattrini AM, Rodríguez E, Faircloth BC, Cowman PF, Brugler MR, Farfan GA, et al. Palaeoclimate ocean conditions shaped the evolution of corals and their skeletons through deep time. Nat Ecol Evol. 2020;4:1531–8. [DOI] [PubMed] [Google Scholar]
- 176.van Oppen MJH, McDonald BJ, Willis B, Miller DJ. The Evolutionary History of the Coral Genus Acropora (Scleractinia, Cnidaria) Based on a Mitochondrial and a Nuclear Marker: Reticulation, Incomplete Lineage Sorting, or Morphological Convergence? Mol Biol Evol. 2001;18:1315–29. [DOI] [PubMed] [Google Scholar]
- 177.O’Dea A, Lessios HA, Coates AG, Eytan RI, Restrepo-Moreno SA, Cione AL, et al. Formation of the Isthmus of Panama. Sci Adv. 2016;2:e1600883. [DOI] [PMC free article] [PubMed]
- 178.Barott KL, Venn AA, Perez SO, Tambutté S, Tresguerres M. Coral host cells acidify symbiotic algal microenvironment to promote photosynthesis. Proc Natl Acad Sci. 2015;112:607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wallace CC, Willis BL. Systematics of the Coral Genus Acropora: Implications of New Biological Findings for Species Concepts. Annu Rev Ecol Syst. 1994;25:237–62. [Google Scholar]
- 180.Randall RH. Morphologic diversity in the scleractinian genus Acropora. In: Proc 4th Inter Coral Reef Symp. 1981.
- 181.Veron J, Wallace CC. Scleractinia of Eastern Australia, Part 5: Acroporidae. Aust Inst Mar Sci Monogr Ser. 1984;6:485. [Google Scholar]
- 182.Wallace CC. Staghorn corals of the world: a revision of the coral genus Acropora (Scleractinia; Astrocoeniina; Acroporidae) worldwide, with emphasis on morphology, phylogeny and biogeography. CSIRO publishing; 1999.
- 183.Chen JE, Cui G, Wang X, Liew YJ, Aranda M. Recent expansion of heat-activated retrotransposons in the coral symbiont Symbiodinium microadriaticum. ISME J. 2018;12:639–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Brandvain Y, Coop G. Scrambling Eggs: Meiotic Drive and the Evolution of Female Recombination Rates. Genetics. 2012;190:709–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hartley G, O’Neill RJ. Centromere Repeats: Hidden Gems of the Genome. Genes (Basel). 2019;10:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Schreiber M, Gao Y, Koch N, Fuchs J, Heckmann S, Himmelbach A, et al. Recombination Landscape Divergence Between Populations is Marked by Larger Low-Recombining Regions in Domesticated Rye. Mol Biol Evol. 2022;39:msac131. [DOI] [PMC free article] [PubMed]
- 187.Taguchi T, Tagami E, Mezaki T, Vacarizas JM, Canon KL, Avila TN, et al. Karyotypic mosaicism and molecular cytogenetic markers in the scleractinian coral Acropora pruinosa Brook, 1982 (Hexacorallia, Anthozoa, Cnidaria). Coral Reefs. 2020;39:1415–25. [Google Scholar]
- 188.Lenormand T. The Evolution of Sex Dimorphism in Recombination. Genetics. 2003;163:811–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mank JE. The evolution of heterochiasmy: the role of sexual selection and sperm competition in determining sex-specific recombination rates in eutherian mammals. Genetics Research. 2009;91:355–63. [DOI] [PubMed] [Google Scholar]
- 190.Miles LG, Isberg SR, Glenn TC, Lance SL, Dalzell P, Thomson PC, et al. A genetic linkage map for the saltwater crocodile (Crocodylus porosus). BMC Genomics. 2009;10:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nei M. Linkage modifications and sex difference in recombination. Genetics. 1969;63:681–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lenormand T, Dutheil J. Recombination Difference between Sexes: A Role for Haploid Selection. PLoS Biol. 2005;3:e63. [DOI] [PMC free article] [PubMed]
- 193.Haig D. Games in tetrads: segregation, recombination, and meiotic drive. Am Nat. 2010;176:404–13. [DOI] [PubMed] [Google Scholar]
- 194.Joseph SB, Kirkpatrick M. Haploid selection in animals. Trends Ecol Evol. 2004;19:592–7. [Google Scholar]
- 195.Paxton CW, Baria MVB, Weis VM, Harii S. Effect of elevated temperature on fecundity and reproductive timing in the coral Acropora digitifera. Zygote. 2016;24:511–6. [DOI] [PubMed] [Google Scholar]
- 196.Pearcy M, Delescaille N, Lybaert P, Aron S. Team swimming in ant spermatozoa. Biol Lett. 2014;10:20140308. [DOI] [PMC free article] [PubMed]
- 197.Esfandi K, He XZ, Wang Q. Sperm allocation strategies in a sperm heteromorphic insect. Current Zoology. 2020;66:285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wedell N, Cook PA. Butterflies tailor their ejaculate in response to sperm competition risk and intensity. Proc Biol Sci. 1999;266:1033. [Google Scholar]
- 199.Metzler S, Schrempf A, Heinze J. Individual- and ejaculate-specific sperm traits in ant males. J Insect Physiol. 2018;107:284–90. [DOI] [PubMed] [Google Scholar]
- 200.Cavassim MIA, Andersen SU, Bataillon T, Schierup MH. Recombination Facilitates Adaptive Evolution in Rhizobial Soil Bacteria. Mol Biol Evol. 2021;38:5480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Babcock RC, Bull GD, Harrison PL, Heyward AJ, Oliver JK, Wallace CC, et al. Synchronous spawnings of 105 scleractinian coral species on the Great Barrier Reef. Mar Biol. 1986;90:379–94. [Google Scholar]
- 202.Stapley J, Feulner PGD, Johnston SE, Santure AW, Smadja CM. Recombination: the good, the bad and the variable. Philos Trans R Soc Lond B Biol Sci. 2017;372:20170279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Uecker H, Hermisson J. The Role of Recombination in Evolutionary Rescue. Genetics. 2016;202:721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kulwal PL. Trait Mapping Approaches Through Linkage Mapping in Plants. Adv Biochem Eng Biotechnol. 2018;164:53–82. [DOI] [PubMed] [Google Scholar]
- 205.Hughes TP, Kerry JT, Baird AH, Connolly SR, Dietzel A, Eakin CM, et al. Global warming transforms coral reef assemblages. Nature. 2018;556:492–6. [DOI] [PubMed] [Google Scholar]
- 206.Browning BL, Zhou Y, Browning SR. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am J Hum Genet. 2018;103:338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rubinacci S, Hofmeister RJ, Sousa da Mota B, Delaneau O. Imputation of low-coverage sequencing data from 150,119 UK Biobank genomes. Nat Genet. 2023;55:1088–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Delaneau O, Zagury J-F, Robinson MR, Marchini JL, Dermitzakis ET. Accurate, scalable and integrative haplotype estimation. Nat Commun. 2019;10:5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Willis BL, van Oppen MJH, Miller DJ, Vollmer SV, Ayre DJ. The Role of Hybridization in the Evolution of Reef Corals. Annu Rev Ecol Evol Syst. 2006;37:489–517. [Google Scholar]
- 210.Richards ZT, van Oppen MJH, Wallace CC, Willis BL, Miller DJ. Some Rare Indo-Pacific Coral Species Are Probable Hybrids. PLoS One. 2008;3:e3240. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Table 1: Summary of sequencing libraries used for genome assembly of the Atlantic acroporids. All data and assemblies are available at NCBI BioProject PRJNA473816.
Additional file 2: Supplementary Table 2: Genomic resources used to compare gene content and syntenic arrangement. Genome assemblies and their predicted proteins were downloaded from sources noted below. Taxonomy is ranked as Subphylum, Class and Order.
Additional file 3: Supplementary Table 3: Assembly statistics for the genome assemblies of the acroporids compared in this study.
Additional file 4: Supplementary Table 4: K-mer genome size estimates. Three different k-mer based genome size prediction tools were used: GenomeScope2, findGSE and kmergenie. The first two tools used the k-mer distribution histogram generated by Jellyfish with a k-mer size of 119. bp = base pairs.
Additional file 5: Supplementary Table 5: Assembly statistics showing completeness and contiguity of Acropora cervicornis HG0582 [123] and HG0005 assemblies (this study). Mb = megabases. BUSCO = Benchmarking Universal Single-Copy Orthologs.
Additional file 6: Supplementary Table 6: Comparison of Acropora palmata Hi-C (pseudo) chromosomes that are homologous to A. cervicornis linkage groups. Mb = megabases.
Additional file 7: Supplementary Table 7: Number of structural variants identified between Acropora palmata and A. cervicornis genome assemblies. N/A = not applicable.
Additional file 8: Supplementary Table 8: Outlier genes between Acropora palmata and A. cervicornis in terms of sequence identity. Percent identity was calculated using rustybam [140] in windows corresponding with gene coordinates using whole genome PAF alignments derived from minimap2 [92]. Genes were considered outliers if their percent identity was below Q1 – (3*IQR).
Additional file 9: Supplementary Table 9: Tracing the 21 cnidarian ancestral linkage groups as defined by Simakov et al. 2022 in acroporid corals. x = chromosomal fusion. Acroporids share the same ALG architecture except for ALG L which is fused to G in A. millepora only. AHYA = Acropora hyacinthus , APAL = A. palmata , AMIL = A. millepora , ACER = A. cervicornis , CJAR = Catalaphyllia jardinei , NVEC = Nematostella vectensis , SSPP = Scolanthus sp. , XSPP = Xenia sp. , HVUL = Hydra vulgaris , HSYM = Hydractinia symbiolongicarpus , CXAM = Cassiopea xamachana.
Additional file 10: Supplementary Table 10: Summary of repeat content in Acropora palmata and A. cervicornis genome assemblies. Bolded transposable elements are abundant among all acroporids.
Additional file 11: Supplementary Fig. 1: Alignments of the two available Acropora cervicornis genome assemblies. Genet K2 assembly (STAGdb ID HG0582, Selwyn and Vollmer [123] is compared with the genet M5 assembly (STAGdb ID HG0005, this study). A) Assembly-assembly minimap2 alignments > 1Mbp and their chromosome locations, plotted with pafr. B) Dot plots comparing the HG0582 assembly with the 14 HG0005 chromosomes (plotted with dotplotly, [135]). Contigs are largely concordant with the exception of one large HG0582 contig that was split across HG0005 chromosomes.
Additional file 12: Supplementary Fig. 2: Oxford dot plots of scleractinian chromosomes and 29 ancestral linkage groups. Scleractinian chromosomes are plotted against A. cervicornis chromosomes (A) and of acroporids chromosomes against 29 ancestral linkage groups (ALGs) of last common ancestor of sponges, cnidarians and bilaterians [2] (B).
Additional file 13: Supplementary Fig. 3: Oxford dot plot of cnidarian assemblies against A. cervicornis chromosomes, arranged by taxonomic groups. Ancestral linkage groups proposed for the last common ancestor of Cnidaria, Bilateria and sponges [2] are indicated by color.
Additional file 14: Supplementary Fig. 4: Significantly enriched GO terms in expanding or contracting gene families of Acroporidae. Top 10 cell component, biological process, and molecular function GO terms associated with phylogenetically significant expand (orange) and contracting (blue) gene families for the family Acroporidae.
Additional file 15: Supplementary Fig. 5: Significantly enriched GO terms in expanding or contracting gene families of Acropora . Top 10 cell component, biological process, and molecular function GO terms associated with phylogenetically significant expand (orange) and contracting (blue) gene families for the genus Acropora.
Additional file 16: Supplementary Fig. 6: Significantly enriched GO terms in expanding or contracting gene families of the Atlantic Acropora . Top 10 cell component, biological process, and molecular function GO terms associated with phylogenetically significant expand (orange) and contracting (blue) gene families for the Atlantic Acropora species.
Additional file 17: Supplementary Fig. 7: Total repeat content estimates based on short-read sequence data of the Atlantic acroporids. Repeat classes are colored as red = DNA, green = LTR, yellow = LINE, blue = SINE, purple = Helitron, and gray = Other (i.e., satellites, simple repeats, unannotated TEs).
Additional file 18: Supplementary Fig. 8: Full linkage and recombination maps for all homologous chromosomes in the Atlantic acroporids. LINE content appears to peak in regions with low recombination rates, highlighting centromere positions.
Data Availability Statement
Genome assemblies, gene predictions, functional annotations, and genetic maps are publicly available at https://zenodo.org/doi/10.5281/zenodo.12021086. NCBI genome accessions are GCA_025960835.2 for A. palmata, GCA_037043185.1 for A. cervicornis version 1, and GCA_041430625.1 for A. cervicornis version 2. All raw data and assemblies have been deposited in NCBI BioProject PRJNA473816 and all SRA accessions are provided in Supplementary Table 1.