

Abstract
Influenza poses a significant threat to the global economy and health. Inactivated virus vaccines were introduced in China for prevention in 2018. In this study, three pairs of hemagglutinin (HA) and neuraminidase (NA) gene sequences were obtained from three Swine influenza virus (IAV-S) inactivated vaccine strains that were marketed in China in 2018. Phylogenetic analysis was carried out with HA and NA gene sequences to investigate the relationship between vaccine use and virus genetic drift. The findings showed that the evolutionary rate of HA remained relatively stable from 2012 to 2017, with an average genetic distance of approximately 0.020731195. However, following the introduction of the swine influenza vaccine, there was a notable acceleration in the evolutionary rate of HA, accompanied by a significant increase in the genetic distance. In 2018, the value was 0.111750269, while in 2019 it was 0.176389393. In contrast, the evolution of NA was relatively smooth, with an average genetic distance of approximately 0.030386708. Finally, we demonstrated that commercial vaccines are weak neutralizers of wild strains through immunization experiments in animals. Thus, we have reason to believe that mutations in the virus favor virus evasion of vaccine immunity. Our findings suggest that vaccine use may significantly impact the evolution of the influenza virus by potentially stimulating mutations. The selection pressure of vaccine antibodies played a role in regulating the variation of IAV-S-H1N1.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-80457-4.
Keywords: Swine influenza virus, H1N1 subtype, Vaccine intervention, Genetic drift, Zoonosis
Subject terms: Vaccines, Virology
Introduction
In accordance with the World Health Organization (WHO), a zoonosis is defined as any disease or infection that can be transmitted naturally from vertebrates to humans or from humans to animals. These diseases have a serious impact on the lives and health of people. Examples include the 2009 “swine influenza” H1N1 outbreak, the 2013–2016 Ebola outbreak in West Africa and the 2019–2023 COVID-19 outbreak1–3. Swine influenza is an acute respiratory disease caused by the swine influenza virus (IAV-S). Pigs are widely considered " mixing vessels” for influenza viruses because of the presence of human and bird-like sialic acid receptors on pig cells that play an important role in the generating new viruses and facilitating their spread between different communities4,5. Multiple lineages of IAV-S, such as H1N1, H1N2, H3N2, are transmitted among pig populations6,7. Swine flu can affect pigs year-round, particularly in spring and autumn, and it can infect pigs of all ages.
The IAV-S was first isolated and identified by Shope in 1931, which was the classical H1N1 strain that caused the epidemic of swine flu in the United States in the early 20th century8. In 2009, a new pandemic strain, pdm09, which began in Mexico and was first reported by the United States, was a strain of H1N1 from pigs that spread rapidly through human-to-human contact to more than 30 countries9. IAV-S is endemic in many countries in North and South America, Europe, Asia and Africa. Classical swine influenza viruses caused high mortality in pigs in coastal cities of China. Two strains of H3N8 influenza viruses derived from European equine H3N8 influenza viruses of the early 1990s were isolated from pigs in central China in the period of 2005–2006. Infected pigs showed marked signs of respiratory disease, including depression and coughing. Pigs can be infected with different subtypes of swine influenza viruses, and new IAV-S may appear at any time through genetic recombination or mutation. People can be threatened with death from influenza viruses, and pigs can also be infected with avian and human influenza viruses10. The epidemic has had a profound impact on the global health. The IAV-S has a major impact on the pig industry and human public health.
IAV-S consists of eight genomic fragments of negative single-stranded RNA, each of which encodes at least one protein11. The polymerase complex, formed by two alkaline polymerases (PB1 and PB2) and one acidic polymerase (PA), surface glycoproteins (hemagglutinin (HA), neuraminidase (NA), and matrix (M)), nuclear proteins (NP), and nonstructural proteins (NS), are encoded by RNA fragments with the same name12. HA and NA are the two key surface glycoproteins of influenza. Previous studies have shown that multiple amino acid substitutions alter the stability of HA13, thus affecting the evolution of the virus. It is possible for influenza viruses to evade immunity through recombination-induced antigenic transfer, although this is a rare occurrence. When it does occur, it can result in devastating pandemic outbreaks14. The HA protein not only mediates virus-host membrane fusion, which is critical for virus entry, but he also acts as an antigenic glycoprotein on the surface of influenza viruses that can bind to antibodies15. The NA protein is a receptor-destroying enzyme that functions as a sialidase to promote the release of offspring viral particles by catalyzing the cleavage of terminal sialic acid residues in cells and viral proteins16. Previous studies have demonstrated that HA and NA are interacting functional antagonists that optimize the balance between HA binding affinity and NA enzyme activity to promote viral fitness, host specificity, transmissibility, infectivity, and virulence17,18. Therefore, according to the quasispecies theory, the evolution of IAV-S has a strong effect on adaptation, host range, virulence, and the appearance of new varieties, mainly due to point mutations and genomic remapping19. Although recombination is an important mechanism in the evolution of a virus, it is rare in IAV-S evasion of immunity20. We therefore focused on immune escape caused by amino acid mutations in the virus.
Vaccination with IAV-S is currently the main strategy for the prevention and control of this disease21,22. To mitigate the impact of viral infections on pig populations, China has approved the marketing of three commercial inactivated vaccines of the H1N1 subtype IAV-S since 2018. While vaccination can prevent epidemics and virus transmission, it may also influence the evolution of the virus. Previous studies have demonstrated that a variety of subtypes of IAV-S, including SIV H1N1, SIV H1N2, and EA SIV, are evolving in response to vaccine pressure, with the potential to evade the immune system23–26. The effect of swine influenza vaccines on virus evolution has not yet been studied in China. Therefore, it is of significance to systematically study the relationship between vaccine use and genomic diversity of the IAV-S.
In this study, the sequences collected from three IAV-S vaccines were combined with the HA and NA sequences of wild-type IAV-S subtype H1N1 published from 1990 to 2020 in China. The impact of vaccine usage on virus evolution was analyzed by constructing an evolutionary tree and calculating the annual genetic distance of IAV-S gene evolution.
Results
The description analysis of HA genetic sequence of H1N1 subtype IAV-Ss in China During 1990–2020
According to the statistical data on the geographical distribution of the gene sequences we obtained from our database in the provinces of China show that the gene sequences mainly come from the east of China, and almost none in the west. (Fig. 1A) Among these, Guangdong contributed the highest number of gene sequences, followed by Hong Kong, Guangxi, Liaoning and Shandong. (Fig. 1B) Together, they constitute 69% of the total genetic sequences of IAV-S. Notably, these provinces are significant agricultural regions in China, and the number of IAV-S isolates is directly proportional to this. Hong Kong reported the first sequence of H1N1 influenza virus in China in 200127, and subsequent reports have increased. Therefore, it is crucial to pay close attention to the circulating IAV-S and timely prevention in these areas.
Fig. 1.

Geographic distribution of swine influenza virus (IAV-S) subtypes H1N1. (A) Geographic map showing the number of gene sequences we have collected for the strains isolated from H1N1 in each province of China. (B) The proportion of H1N1 subtypes IAV-S in different provinces of China. Clockwise according to the number of H1N1 subtype IAV-S. Provinces with less than 4% of flu viruses are not shown in the chart.
The phylogenetic analysis of HA and NA genetic sequences of H1N1 subtype IAV-S in China
We conducted phylogenetic analyses and constructed an evolutionary tree based on the collected HA and NA gene sequences of H1N1 IAV-S, along with isolated strains from 2012 onwards. The influenza viruses were classified according to the classical strains of the different branches: classical H1N1 (CS H1N1), Eurasian avian-like H1N1 (EA H1N1), triple reassortant H1N2 (TR H1N2), and 2009 pandemic H1N1 (2009/H1N1)28. In the phylogenetic tree, branch length represents the base substitution rate, which is used to measure the genomic distance between the ancestor and descendant viruses. According to the results (Fig. 2), following the release of the inactivated swine influenza H1N1 vaccine in 2018, the gene sequence of the HA protein of the strain isolated after 2018 diverged from that of the pre-2018 strain, forming a separate branch. This indicates that the HA gene of the post-2018 strain is significantly different from that of the pre-2018 strain. However, there were no significant differences in the evolutionary tree of NA. In conclusion, the high evolutionary rate of HA can be attributed to the intense evolutionary pressure it is subjected to.
Fig. 2.

Phylogenetic analyses of HA and NA genes of H1N1 swine influenza virus. (A) Phylogenetic tree of H1N1 HA gene. (B) Phylogenetic tree of the H1N1 NA gene. With reference to previous studies, the strains were classified into four main categories: TR H1N2 (purple), 2009 H1N1 (deep green), CS H1N1 (orange) and EA H1N1 (deep red). Based on the year of isolation of the strains, the strains were classified into three main categories,using the highlight marker: before the introduction of commercial vaccines (green for 2012-2014 and blue for 2015-2017) and after the introduction of commercial vaccines (yellow after 2018). Strains used in animal testing are shown in red and in an expanded graph.
Genetic distance of HA and NA around 2018
To assess the evolutionary rate of IAV-S in relation to vaccine use, we constructed an annual dataset based on the collection time and calculated the distance matrix between the series in the dataset. The data set comprises a series of points, each representing a genetic difference between the two strains in a given year. In 2018, the first batch of commercial inactivated vaccines against H1N1 subtype of IAV-S in China entered the market. We selected the HA and NA fragments of H1N1 subtype of IAV-S from 2009 to 2020 for analysis. The results demonstrated that the genetic distance of the HA gene was relatively substantial over the three-year period between 2009 and 2011 (with an average genetic distance observed in 2009–2010). The values were 0.111455838, 0.180529255, and 0.208795326, respectively (Supplementary Material 2), which was likely due to the influenza virus pandemic in 2009. The period from 2012 to 2017 was relatively smooth, and the evolutionary distance was relatively stable without large changes (Fig. 3A). After the swine influenza vaccine was launched in 2018, the evolutionary speed of IAV-S HA accelerated significantly, and the gene-protein genetic distance increased significantly (average genetic distance: 0.078178556). This increase was even more gradual from 2019 to 2020, reaching 0.111750269 and 0.176389393 (Supplementary Material 1). NA was relatively stable, and the annual genetic distance between gene sequences was basically 0 to 0.10000000(Fig. 3B), and there were also no notable changes in 2018. This result also confirmed that the vaccine pressure had certain influence on the evolution of the virus.
Fig. 3.
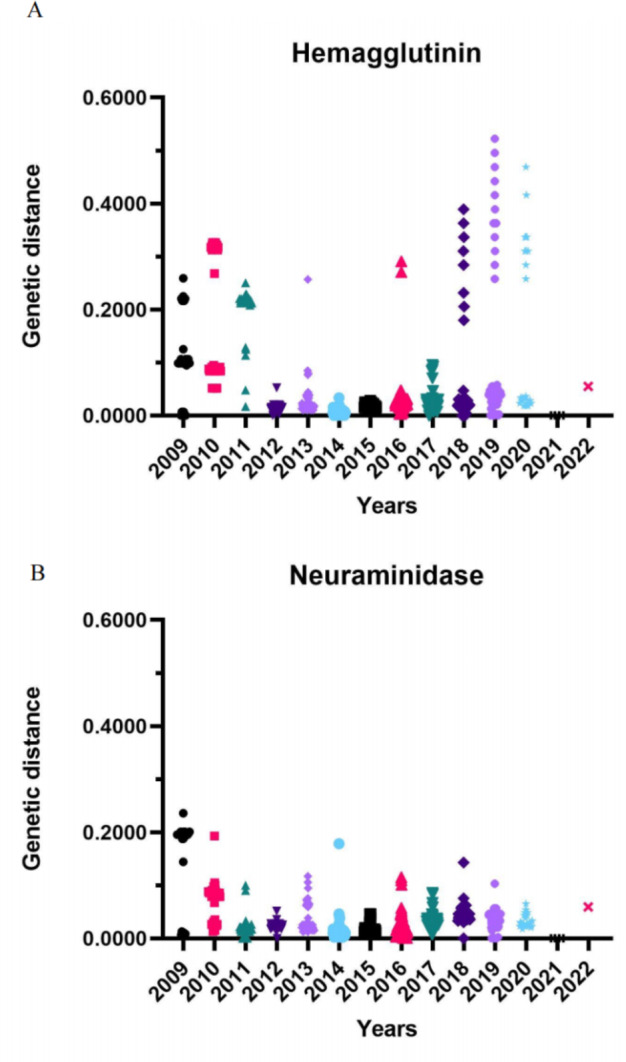
The genetic distance between HA sequence and NA sequence of H1N1 subtype swine influenza virus (IAV-S). Each representing a genetic difference between the two strains in a given year. (A) Distribution of genetic distance between HA sequences collected annually (excluding vaccine sequences). (B) Distribution of genetic distance (excluding vaccine sequences) between NA sequences collected annually.
HA gene mutations from 2010 to 2020
The nucleotide sequences were translated into the corresponding amino acid sequences and the collected post-2018 viral gene sequences were subjected to multiple sequence comparisons to go exploring the key sites of HA protein gene mutations. Our findings revealed that post-2018 influenza virus sequences exhibited a higher prevalence of amino acid mutations in the head region (aa63-aa268) of HA, including 64 K/V, 68 H/Q, 71 K/N, and 83 K/E, compared to the neck region, which demonstrated a relatively conservative pattern of mutation (Fig. 4A). By means of sequence comparison of the strains that emerged subsequent to 2018 with each of the three commerical vaccine strains (TJ strain, LN strain, and HLJ strain), it was possible to identify multiple sites exhibiting the same mutation profile. These included T65I, E83K, V137A, V158A, A214T, G239E, and M331L (Supplementary Material 3). The majority of these mutation sites are situated within the head region of the viral HA protein (Fig. 4B). The E83K mutation has been demonstrated to facilitate the evasion of vaccine-induced immunity, whereas the A214T mutation is situated within the antigen-binding region. The alteration of this site results in the modification of the virus’s original antigenic site29,30. Briefly, these continuous site mutations may lead to significant changes in viral proteins that favor viral escape vaccine immunity. Except for the superposition of point mutations, the main reason for the large genetic differences between sequences may be insertion mutations. These findings indicated that the use of inactivated vaccine affected the point mutation of HA sequence and thus the rate of virus evolution.
Fig. 4.

Amino acid mutation map of HA protein. (A)The amino acid sequence mutation labeling diagram of HA protein of H1N1 swine influenza virus. The size of the letter indicates the degree of conservation of the amino acid. A lar letter indicates that that amino acid at this site is more conservative. (B) 3D structural diagram of HA protein. Mutation sites are labelled using red.
Correlation analysis of HA and NA
In order to investigate the relationship between HA and NA, a scatter plot and a fitting curve were constructed, revealing a positive correlation between HA and NA. We performed a statistical analysis and found a significant correlation between the genetic distance evolution of HA and NA, with a p-value of less than 0.0001. But the correlation between the two was weak (R = 0.1807) (Fig. 5A). This result was also found by previous studies that genetic correlation between HA and NA was weak in IAV-S31.
Fig. 5.
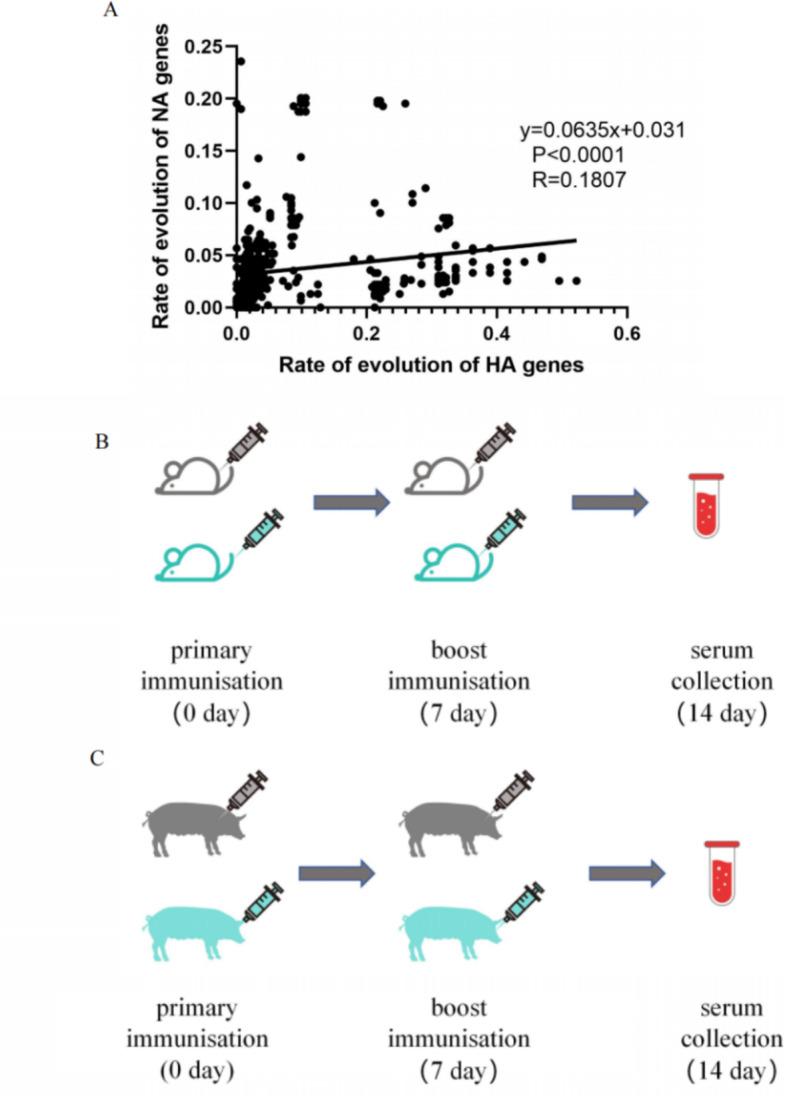
NA and HA amino acid correlation and animal experiment flowchart. (A)Amino acid Scatter diagram of correlation coefficient of rate of genetic evolution of H1N1 IAV-S HA and NA genes. Each point represents the rate of inheritance of a strain of virus. (B) Experimental protocols for mice experiments. (C) Experimental protocols for pig experiments.
6. Commercial vaccines have a weak ability to neutralise epidemic strains of swine influenza.
The effect of the vaccine on virus neutralization was determined by a Hemagglutinin inhibition (HI) assay and microneutralization assay using collected sera. HI experiments using sera from twice-immunized mice and pigs. The objective is to detect the ability of sera to neutralise IAV-S strains isolated in 2018 (A/swine/henan/SN13/2018) and 2023 (A/China/shandong/2023), respectively. It revealed that the commercial vaccine had a relatively weak ability to neutralize the H1N1 virus. The experimental procedure for animal immunisation is shown in Fig. 5B-C. The A/swine/Henan/SN13/2018 HI titer in the mice experiments were 13.3 (14 days) and the A/swine/China/Shandong/2023 HI results were 3.3 (14 days). (Fig. 6A) Similar results were found in the pig experiment. (Fig. 6B) With time migration, the vaccine-produced antibodies were less able to neutralise the IAV-S (A/swine/Henan/SN13/2018, A/swine/China/Shandong/2023) compared to the 2018 H1N1 strain. The results of the neutralization assay were similar to those of the HI assay. Sera immunized with commercial vaccines were protective against A/swine/Henan/SN13/2018 but less so against A/swine/China/Shandong/2023. (Fig. 6C) Based on these results, it can be surmised that under the pressure of the vaccine, the virus mutated to evade the vaccine’s action.
Fig. 6.
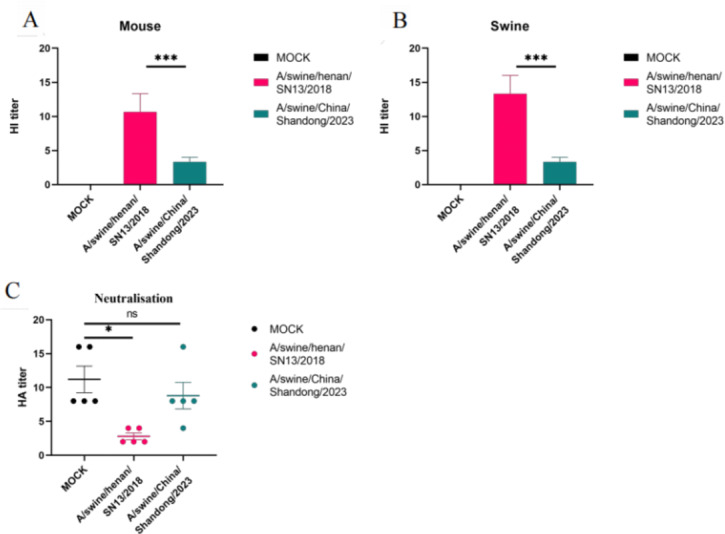
Histogram of hemagglutination inhibition assay and microneutralization assay. (A) Results of mice serum HI experiments; (B) Results of swine serum HI experiments. (C) Detection of neutralization effect of swine serum against A/swine/Henan/SN13/2018 and A/swine/China/Shandong/2023. Data in A-c are presented as mean values ± SEM. (***p < 0.001, **p < 0.01, *p < 0.05)
Discussion
In this study, we selected three vaccine strains that have been marketed in China since 2018. A comprehensive phylogenetic analysis was conducted on all published H1N1 subtype IAV-S HA, NA sequences and vaccine strain sequences from 1990 to 2020 to explore the relationship between vaccine use and viral genetic diversity. The influenza virus is composed of a single-stranded virus with eight anti-sense genomic RNA segments, and it will undergo genetic changes. This is why influenza viruses change or mutate over time32. Among them, HA is a major surface antigen of virus and also one of the main targets of host immune response. It plays a key role in the initial stages of infection: receptor binding and viral and cellular membrane fusion33,34. NA is another major surface glycoprotein and it serves as a key enzyme for influenza virus infection, replication, maturation, and delivery35,36. This also holds promise for more durable immunity induced by conventional vaccines because of the slower antigenic drift rate of NA37. The IAV gene encoding virus surface proteins HA and NA is the main target constituting neutralizing antibodies and is essential for virus evolution38. Influenza viruses can evolve through recombination and genetic mutation, which makes the study of the internal gene fragments extremely challenging39,40. The present study focuses on the HA and NA genes of H1N1 subtype IAV-S.
Phylogenetic analysis revealed a significant increase in the evolutionary rate of HA proteins from 2018 to 2020, with no major changes in NA evolution compared to pre-2018. Influenza virus evolutionary rate plummeted after 2021. This could be due to various factors, including the efficacy of the vaccine, changes in the environment, and the nature evolution of the virus. Based on the current research data, it has been demonstrated that the increasing number of approved inactivated vaccine strains has led to an increase in immune pressure on the influenza virus, which may increase the chances of strain mutation. And studies have also shown similar evolutionary changes in the HA gene after the Mexican H5N2 low-pathogenicity avian influenza vaccine41. Vaccine failures due to antigenic drift of field viruses have occurred in China, Indonesia, Egypt, and Vietnam, and previous studies have linked the H5N1 vaccine to resistant field strains in China, Egypt, Indonesia, Hong Kong and Vietnam42. The conclusions drawn in our study are consistent with these examples. It is therefore reasonable to assume that viruses are evolving under the pressure of vaccines and thus evading vaccine immunity.
The three vaccines in this study were the first three swine influenza H1N1 vaccines marketed in China, of which the TJ strain vaccine was widely used. In 2018, the swine influenza vaccine was first marketed in China, and the rate of evolution of the virus’ HA continued to accelerate after 2018 compared to before. However, after 2021, the evolutionary rate returned to the previous rate. This may be due to the fact that we collected fewer viral gene sequences in 2021. NA demonstrates a distinctive evolutionary pattern, showing no significant change in evolutionary distance following the introduction of vaccines. This suggests that the effect of the vaccine on the virus is more pronounced in the HA gene rather than NA gene. Therefore, we performed multiple sequence alignment to understand the mutation of HA under vaccine action.
By comparing collected wild-type virus genes with the TJ, LN, and HLJ vaccine virus genes, we found that there were multiple mutation sites of HA gene in TJ strain and HLJ strain, and the number of mutation site of HA1 is higher than that of HA2. This was also found by previous studies that the stem domain (HA2) evolved much more slowly than the head domain (HA1)43. Other studies have demonstrated that antigenic drift, resulting from a substantial number of site mutations, can occur. Furthermore, the occurrence of even a single amino acid substitution in the vicinity of the functional site of the HA head can give rise to a considerable degree of antigenic drift, without a corresponding significant alteration in the stem region44–46. This suggests that positive selection in the stem domain might not directly related to immune stress. Many mutation sites of HA1 such as S91R, D97N, N146D, S185T, E189G, S200P, S202I and A273T were detected47,48, while the mutation at HA2 was relatively rare.
However, compared with the LN strain, there were more unstable mutations, and more surprisingly, there were more mutation sites in HA2 than in HA1. We also found an increase in viral mutation sites after marketing of the vaccine, particularly concentrated in the receptor-binding region. There are also a large number of mutations at consecutive amino acid sites, potentially facilitating the virus in evading vaccine immunity. The results of animal immunization test and HI test show that the existing commercial vaccines exhibit a weak neutralizing ability against wild strains. It is reasonable to believe that mutations at these sites may beneficial for the virus to evade the immunity induced by vaccine. Furthermore, in addition to point mutations, insertion mutations have the potential to cause an increase in the genetic distance between viruses and promote viral evasion of immunity. Hence, there is a need to intensify the utilization of influenza virus vaccines and enhance monitoring of virus evolution. This approach will contribute to timelier and scientifically informed updates and usage of vaccine strains.
Previous studies have found that HA and NA have a co-evolutionary trend, that is, leading mutations occur in NA, followed by mutations in HA, and vice versa, and the mean time between successive mutations at these sites is also very similar42. However, the evolutionary correlation between HA and NA was found to be weak in our study. We considered the evolutionary correlation between HA and NA to be closely related to the host. This observation aligns with findings from previous studies31 that the co-evolution of HA and NA exhibits a strong correlation in humans but a weak correlation in avian and swine species.
Conclusions
Based on the results, it can be concluded that the use of IAV-S H1N1 vaccine may accelerate the evolutionary rate of the virus, leading to the emergence of strains capable of evading the immune response elicited by the vaccine. However, it is important to acknowledge that the short duration of vaccine marketing and the limited numbers of isolated strains make it challenging to determine whether there is a cumulative effect of the vaccine on virus evolution. Therefore, further studies will be conducted as more sequences of IAV-S H1N1 strains become available. Ongoing monitoring of vaccine use and evolution of the H1N1 subtype swine influenza virus will be crucial in predicting epidemic strains, updating and using vaccine strains in a more timely and effective manner. At the same time, the direction and focus of research on zoonotic diseases will be referenced.
Materials and methods
Sequence collection and alignment
The HA and NA genetic sequences of H1N1 subtype IAV-Ss isolated in China from 2009 to 2020 were searched from the Bacterial and Viral Bioinformatics Resource Center (https://www.bv-brc.org/) and Influenza Data database (GISAID; https://www.gisaid.org). A total of 567 HA and NA genetic sequences of H1N1 subtype IAV-Ss were downloaded. The nucleotide sequences were aligned using the MEGA software (version11.0.13) with muscle method. Then we compiled the geographic data (provinces of China) for all sequences. To minimize the impact of the 2009 influenza epidemic on the experimental results, gene sequences from 2012 onwards were selected as representatives for analysis. The study focused on three inactivated swine influenza vaccines available in 2018: A/swine/Tianjin/01/14 (TJ strain, EU004444), A/swine/Liaoning/32/2016(LN strain, HM754648) and A/swine/Heilongjiang/1130/2015(HLJ strain, KP404260).
Phylogenetic analysis
All available HA and NA genetic sequences of H1N1 subtype IAV-Ss, after being aligned, were analyzed using the Beast software (version 1.8.4) and were found the best model using jmodeltest 2.1.6 software. Phylogenetic trees were constructed using maximum likelihood (ML) method with GTR + I + G model. The bootstrap consensus tree, inferred from 1000 replicates, is taken to represent the evolutionary history of the taxa analyzed.
Calculation of genetic distance between sequences
The HA and NA genetic sequences were grouped into datasets based on collection year. The aligned genetic sequences calculated the genetic pairwise distance in each year by the MEGA software (version 11.0.13) using the Maximum Composite Likelihood model. The genetic data obtained were stored in matrix form. The results are presented in the form of a scatter plot, with the degree of difference in the genetic distance of each point expressed as a value representing the viral sequence.
4. Statistics of Amino Acid Types at Specific Sites.
The intercepted nucleotide sequence data sets of different years were converted into amino acid sequences using SnapGene software (version 6.0.2). We used the online website Weblogo (http://weblogo.berkeley.edu/) to draw the amino acid identification map. The mutated region of the viral amino acid is then analyzed to reflect the sequence preference at that location. And the 3D structure map of HA protein was constructed using SWISS-MODEL (https://swissmodel.expasy.org) to annotate the mutation sites.
Genetic correlation analysis between HA and NA genes
We conducted a correlation analysis of the annual genetic distances between the collected HA and NA genes of H1N1. At first, we calculated the annual genetic distance of the collected genes by using MEGA software (version 11.0.13). As the HA and NA gene sequences from the same year were extracted, pairwise scores under the same conditions could be obtained. Then we used SPSS software (version 18.0.0) to analyze the correlation between the genetic distances of HA and NA using Pearson correlation analysis. A p-value less than 0.05 was considered statistically significant.
Animal experiments
These experiments were conducted at Shandong First Medical University& Shandong Academy of Medical Sciences and approved by its ethics Committee. Two swine influenza viruses (TJ strain, A/swine/China/Shandong/2023(PP464144)), which are located in the same branch in the evolutionary tree as the strain of that year, were selected for animal immunization experiments to validate the impact of viral site mutations on the neutralization effect of commercial vaccines.
Before the experiments, we inoculated mice with β-propiolactone-inactivated wild swine influenza virus (H1N1) (A/swine/China/Shandong/2023 (PP464144)) to demonstrate that inactivated swine influenza viruses elicit immune responses in mice.The experiments used five 4-week-old female C57/BL6 mice. On day 0, intramuscular injection of β-propiolactone-inactivated wild swine influenza virus (H1N1) (A/swine/China/Shandong/2023 (PP464144)) administered, with mice receiving 200µL and pigs receiving 2 ml. On day 7, a second intramuscular injection of β-propiolactone-inactivated wild swine influenza virus (H1N1) (A/swine/China/Shandong/2023 (PP464144)) was administered. On days 14, blood was collected from mice and pigs, then centrifuged at 1000 rpm for 5 min to obtain serum. The results showed that the inactivated virus successfully induced immunity in mice (Supplementary Material 4). Mice and pigs will be injected with the commercial vaccine for subsequent experiments.
The animal experiments were conducted using 4-week-old female C57/BL6 mice and 4-weeks-old female pigs. The mice and pigs were divided into two groups of five animals each. (Fig. 5B-C) The first group received the commercial vaccine: on day 0, intramuscular injection of inactivated swine influenza H1N1 vaccine (TJ strain) (manufacturer: Wuhan Keqian Biology Co., Ltd) administered, with mice receiving 200µL and pigs receiving 2 ml. On day 7, another intramuscular injection of inactivated swine influenza H1N1 vaccine (TJ strain) was given again, and on days 14, blood was collected from mice and pigs, then centrifuged at 1000 rpm for 5 min to obtain serum. The second group, the blank control group, followed the same protocol as the commercial vaccine group but received a saline injection.
Hemagglutination inhibition assay
The hemagglutination inhibition (HI) assay was performed to evaluate HA functional antibodies capable of inhibiting erythrocyte agglutination. Sera were serially diluted 2-fold in V-bottom microtiter plates. An equal volume of IAV-S was added to each well and the erythrocytes were washed with PBS to prepare 0.5% chicken blood erythrocytes, which were stored at 4 °C and used within 24 h of preparation. Mix the plates with agitation and cover and allow the chicken blood erythrocytes to settle at room temperature for 20 min. The HI titer is determined from the inverse dilution of the last well of unagglutinated RBCs. Positive and negative serum controls are provided for each plate. Then, 50µL of 0.5% chicken blood erythrocytes were added and the mixture was incubated for 20 min at room temperature. The HI titter was calculated from the reciprocal of the highest dilution that completely inhibited erythrocyte hemagglutination.
Microneutralization assay
Cell-based microneutralization experiments were performed with reference to Tingting Li et al.49. Porcine serum from the experimental stage of the animal was diluted twofold with a virus mixture of 100 TCID50 and incubated for 1 h at room temperature. The serum-virus mixture was added to a 96-well plate of room MDCK cells. Adsorption was done for 1 h and then replaced in MEM medium containing trypsin. The HA assay of the supernatant was determined as evidence of neutralisation and the HA assay was performed according to previous experimental methods.
Ethics declaration
All experiments were conducted in compliance with national and local regulations. All animal experiments were conducted at Shandong First Medical University& Shandong Academy of Medical Sciences and approved by its ethics Committee (project identifier: 2023097). Mice experiments were conducted in our approved SPF animal facility at Shandong First Medical University& Shandong Academy of Medical Sciences. Pig experiments were conducted in a normal environment. All animal experiments comply with China’s Regulations on the Administration of Laboratory Animals and ARRIVE guidelines.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Bin Fu and Cuilian Yu from Shandong First Medical University (Jinan, China) for their kind help in the data analysis of experimental samples.
Author contributions
Zhao Wang: experimental idea, experimental design, review and revision of the article. Xinkun Zhao: article writing, experimental design, experimental implementation, data analysis and processing. Mingshuai Shen: article writing, experimental design. Kezhou Wang: experimental idea, experimental design. Li Cui: experimental implementation, data analysis. Cun Liu: experimental design. Jieshi Yu: article Revision and experimental design. Guisheng Wang: data analysis. Mihajlo Erdeljan: article Revision. Shumin Chen:Evolutionary tree mapping and guidance on experimental protocols.
Funding
This work was supported by the National Natural Science Funds (32102750), Shandong Province Pig Industry Technology System (SDAIT-08-17), The Major Scientific and Technological Innovation Project(2023CXGC010705), Innovation Project of Shandong Academy of Medical Sciences and Technology Bureau “20 Colleges and Universities” (2021GXRC011).
Data availability
The data that support the study results are available upon reasonable request. Corresponding authors should be contacted if data from this study are required.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Xinkun Zhao and Mingshuai Shen
References
- 1.Dawood, F. S. et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect. Dis.12 (9), 687–695 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Gebreyes, W. A. et al. The global one health paradigm: challenges and opportunities for tackling infectious diseases at the human, animal, and environment interface in low-resource settings. PLoS Negl. Trop. Dis.8 (11), e3257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Estimating excess mortality. Due to the COVID-19 pandemic: a systematic analysis of COVID-19-related mortality, 2020-21. Lancet399 (10334), 1513–1536 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito, T. et al. Molecular basis for the generation in pigs of influenza a viruses with pandemic potential. J. Virol.72 (9), 7367–7373 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma, W., Kahn, R. E. & Richt, J. A. The pig as a mixing vessel for influenza viruses: human and veterinary implications. J. Mol. Genet. Med.3 (1), 158–166 (2008). [PMC free article] [PubMed] [Google Scholar]
- 6.Qiao, C. et al. Novel triple reassortant H1N2 influenza viruses bearing six internal genes of the pandemic 2009/H1N1 influenza virus were detected in pigs in China. J. Clin. Virol.61 (4), 529–534 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Anderson, T. K. et al. Swine Influenza A viruses and the tangled relationship with humans. Cold Spring Harb Perspect. Med.11(3). (2021). [DOI] [PMC free article] [PubMed]
- 8.Schultz-Cherry, S., Olsen, C. W. & Easterday, B. C. History of Swine influenza. Curr. Top. Microbiol. Immunol.370, 21–28 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Smith, G. J. et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza a epidemic. Nature459 (7250), 1122–1125 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Kong, W., Ye, J., Guan, S., Liu, J. & Pu, J. Epidemic status of swine influenza virus in China. Indian J. Microbiol.54 (1), 3–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das, K., Aramini, J. M., Ma, L. C., Krug, R. M. & Arnold, E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol.17 (5), 530–538 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouvier, N. M. & Palese, P. The biology of influenza viruses. Vaccine26 (Suppl 4), D49–53 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russell, C. J., Hu, M. & Okda, F. A. Influenza Hemagglutinin Protein Stability, activation, and pandemic risk. Trends Microbiol.26 (10), 841–853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu, N. C. & Wilson, I. A. Influenza hemagglutinin structures and antibody recognition. Cold Spring Harb Perspect. Med.10 (8), a038778 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varghese, J. N., Laver, W. G. & Colman, P. M. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 a resolution. Nature303 (5912), 35–40 (1983). [DOI] [PubMed] [Google Scholar]
- 16.Colman, P. M. & Ward, C. W. Structure and diversity of influenza virus neuraminidase. Curr. Top. Microbiol. Immunol.114, 177–255 (1985). [DOI] [PubMed] [Google Scholar]
- 17.Kosik, I. & Yewdell, J. W. Influenza Hemagglutinin and neuraminidase: Yin⁻Yang proteins Coevolving to thwart immunity. Viruses11(4). (2019). [DOI] [PMC free article] [PubMed]
- 18.Gao, R. et al. New insights into the neuraminidase-mediated hemagglutination activity of influenza A(H3N2) viruses. Antiviral Res.218, 105719 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domingo, E., de Ávila, A. I., Gallego, I., Sheldon, J. & Perales, C. Viral fitness: history and relevance for viral pathogenesis and antiviral interventions. Pathog Dis.77(2). (2019). [DOI] [PubMed]
- 20.Bergmann, M., García-Sastre, A. & Palese, P. Transfection-mediated recombination of influenza a virus. J. Virol.66 (12), 7576–7580 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nichol, K. L. & Treanor, J. J. Vaccines for seasonal and pandemic influenza. J. Infect. Dis.194 (Suppl 2), S111–118 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Yu, J. et al. A recombinant chimeric influenza virus vaccine expressing the consensus H3 hemagglutinin elicits broad hemagglutination inhibition antibodies against divergent swine H3N2 influenza viruses. Vaccine41 (42), 6318–6326 (2023). [DOI] [PubMed] [Google Scholar]
- 23.López-Valiñas, Á. et al. Identification and characterization of Swine Influenza Virus H1N1 variants generated in Vaccinated and Nonvaccinated, Challenged pigs. Viruses13 (10), 2087 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.López-Valiñas, Á. et al. Genetic diversification patterns in swine influenza a virus (H1N2) in vaccinated and nonvaccinated animals. Front. Cell. Infect. Microbiol.13, 1258321 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.López-Valiñas, Á. et al. Vaccination against swine influenza in pigs causes different drift evolutionary patterns upon swine influenza virus experimental infection and reduces the likelihood of genomic reassortments. Front. Cell. Infect. Microbiol.13, 1111143 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murcia, P. R. et al. Evolution of an eurasian avian-like influenza virus in naïve and vaccinated pigs. PLoS Pathog. 8 (5), e1002730 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong, J. Y. et al. Analysis of potential changes in seriousness of influenza A and B viruses in Hong Kong from 2001 to 2011. Epidemiol. Infect.143 (4), 766–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, H. et al. Prevalence, Genetics and Evolutionary properties of eurasian avian-like H1N1 swine influenza viruses in Liaoning. Viruses14 (3), 643 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Badar, N. et al. Evolutionary analysis of seasonal influenza a viruses in Pakistan 2020–2023. Influenza Other Respir Viruses. 18 (2), e13262 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang, Q. et al. Molecular epidemiology and evolution of A(H1N1)pdm09 and H3N2 virus during winter 2012–2013 in Beijing, China. Infect. Genet. Evol.26, 228–240 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Jang, J. & Bae, S. E. Comparative Co-evolution Analysis between the HA and NA genes of Influenza A Virus. Virol. (Auckl). 9, 1178122x18788328 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labella, A. M. & Merel, S. E. Influenza. Med. Clin. North. Am.97 (4), 621–645 (2013). x. [DOI] [PubMed] [Google Scholar]
- 33.Russell, R. J. et al. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. U S A. 105 (46), 17736–17741 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao, R. et al. Human Monoclonal Antibody Derived from Transchromosomic Cattle Neutralizes Multiple H1 Clades of Influenza A Virus by Recognizing a Novel Conformational Epitope in the Hemagglutinin Head Domain. J Virol 94(22). (2020). [DOI] [PMC free article] [PubMed]
- 35.McAuley, J. L., Gilbertson, B. P., Trifkovic, S., Brown, L. E. & McKimm-Breschkin, J. L. Influenza Virus Neuraminidase structure and functions. Front. Microbiol.10, 39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao, R., Sheng, Z., Sreenivasan, C. C., Wang, D. & Li, F. Influenza A virus antibodies with antibody-dependent Cellular cytotoxicity function. Viruses12(3). (2020). [DOI] [PMC free article] [PubMed]
- 37.Johansson, B. E., Bucher, D. J. & Kilbourne, E. D. Purified influenza virus hemagglutinin and neuraminidase are equivalent in stimulation of antibody response but induce contrasting types of immunity to infection. J. Virol.63 (3), 1239–1246 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shao, W., Li, X., Goraya, M. U., Wang, S. & Chen, J. L. Evolution of Influenza A Virus by Mutation and Re-assortment. Int. J. Mol. Sci.18(8). (2017). [DOI] [PMC free article] [PubMed]
- 39.Reid, A. H. & Taubenberger, J. K. The origin of the 1918 pandemic influenza virus: a continuing enigma. J. Gen. Virol.84 (Pt 9), 2285–2292 (2003). [DOI] [PubMed] [Google Scholar]
- 40.He, C. Q. et al. Homologous recombination evidence in human and swine influenza a viruses. Virology380 (1), 12–20 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Lee, C. W., Senne, D. A. & Suarez, D. L. Effect of vaccine use in the evolution of Mexican lineage H5N2 avian influenza virus. J. Virol.78 (15), 8372–8381 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swayne, D. E. Impact of vaccines and vaccination on global control of avian influenza. Avian Dis.56 (4 Suppl), 818–828 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Kudo, T., Ohkoshi, A. & Horikoshi, K. Molecular cloning and expression of a xylanase gene of alkalophilic Aeromonas sp. 212 in Escherichia coli. J. Gen. Microbiol.131 (10), 2825–2830 (1985). [DOI] [PubMed] [Google Scholar]
- 44.Lewis, N. S. et al. Antigenic and genetic evolution of equine influenza A (H3N8) virus from 1968 to 2007. J. Virol.85 (23), 12742–12749 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koel, B. F. et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science342 (6161), 976–979 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Koel, B. F. et al. Antigenic variation of clade 2.1 H5N1 virus is determined by a few amino acid substitutions immediately adjacent to the receptor binding site. mBio5 (3), e01070–e01014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu, B. et al. Molecular evolution and characterization of hemagglutinin and neuraminidase of influenza A(H1N1)pdm09 viruses isolated in Beijing, China, during the 2017–2018 and 2018–2019 influenza seasons. Arch. Virol.166 (1), 179–189 (2021). [DOI] [PubMed] [Google Scholar]
- 48.El Moussi, A. et al. Genetic diversity of HA1 domain of heammaglutinin gene of influenza A(H1N1)pdm09 in Tunisia. Virol. J.10, 150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li, T. et al. Identification of a cross-neutralizing antibody that targets the receptor binding site of H1N1 and H5N1 influenza viruses. Nat. Commun.13 (1), 5182 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the study results are available upon reasonable request. Corresponding authors should be contacted if data from this study are required.